 Grace M. Bidgood
Grace M. Bidgood Narelle Keating1,2
Narelle Keating1,2 Karen Doggett
Karen Doggett Sandra E. Nicholson
Sandra E. Nicholson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 June 2024
Sec. Cytokines and Soluble Mediators in Immunity
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1419951
This article is part of the Research TopicSOCS: Regulation of the Immune System at a Whole New LevelView all 10 articles
The Suppressor of Cytokine Signaling (SOCS) family proteins are important negative regulators of cytokine signaling. SOCS1 is the prototypical member of the SOCS family and functions in a classic negative-feedback loop to inhibit signaling in response to interferon, interleukin-12 and interleukin-2 family cytokines. These cytokines have a critical role in orchestrating our immune defence against viral pathogens and cancer. The ability of SOCS1 to limit cytokine signaling positions it as an important immune checkpoint, as evidenced by the detection of detrimental SOCS1 variants in patients with cytokine-driven inflammatory and autoimmune disease. SOCS1 has also emerged as a key checkpoint that restricts anti-tumor immunity, playing both a tumor intrinsic role and impacting the ability of various immune cells to mount an effective anti-tumor response. In this review, we describe the mechanism of SOCS1 action, focusing on the role of SOCS1 in autoimmunity and cancer, and discuss the potential for new SOCS1-directed cancer therapies that could be used to enhance adoptive immunotherapy and immune checkpoint blockade.
Cytokines are critical regulators of numerous cellular functions, including cell survival, proliferation, differentiation and chemotaxis, and are essential for growth, haematopoiesis, and innate and adaptive immunity. Cytokines direct cellular responses by binding to membrane-bound receptor complexes and activating the intracellular JAK-STAT (JAnus Kinase-Signal Transducers and Activators of Transcription) signaling pathway. The four mammalian JAK tyrosine kinases (JAK1, JAK2, JAK3 and TYK2) are constitutively associated with various receptor intracellular domains and become activated via trans or autophosphorylation of the JAK activation loop following cytokine engagement and receptor oligomerization. Activated JAKs then phosphorylate tyrosine residues within the receptor cytoplasmic domains, providing a docking site for Src homology 2 (SH2) domain-containing proteins, including the STATs (1). There are seven mammalian STAT proteins, STAT1–4, STAT5A, STAT5B, and STAT6 that dock to the receptors via their SH2 domains. The receptor associated JAKs then phosphorylate tyrosine residues in the STAT proteins, inducing a conformational change in pre-existing STAT dimers, and enabling nuclear translocation and a transcriptional response (2–4) (Figure 1A). In addition to the STATs, cytokines activate various other pathways, notably the PI3K/AKT (6) and Ras/MAPK (7) signaling cascades. Many cytokines are pleiotropic and redundant in nature, however, by activating various receptor-JAK-STAT combinations, individual cytokines can direct discrete cellular responses.
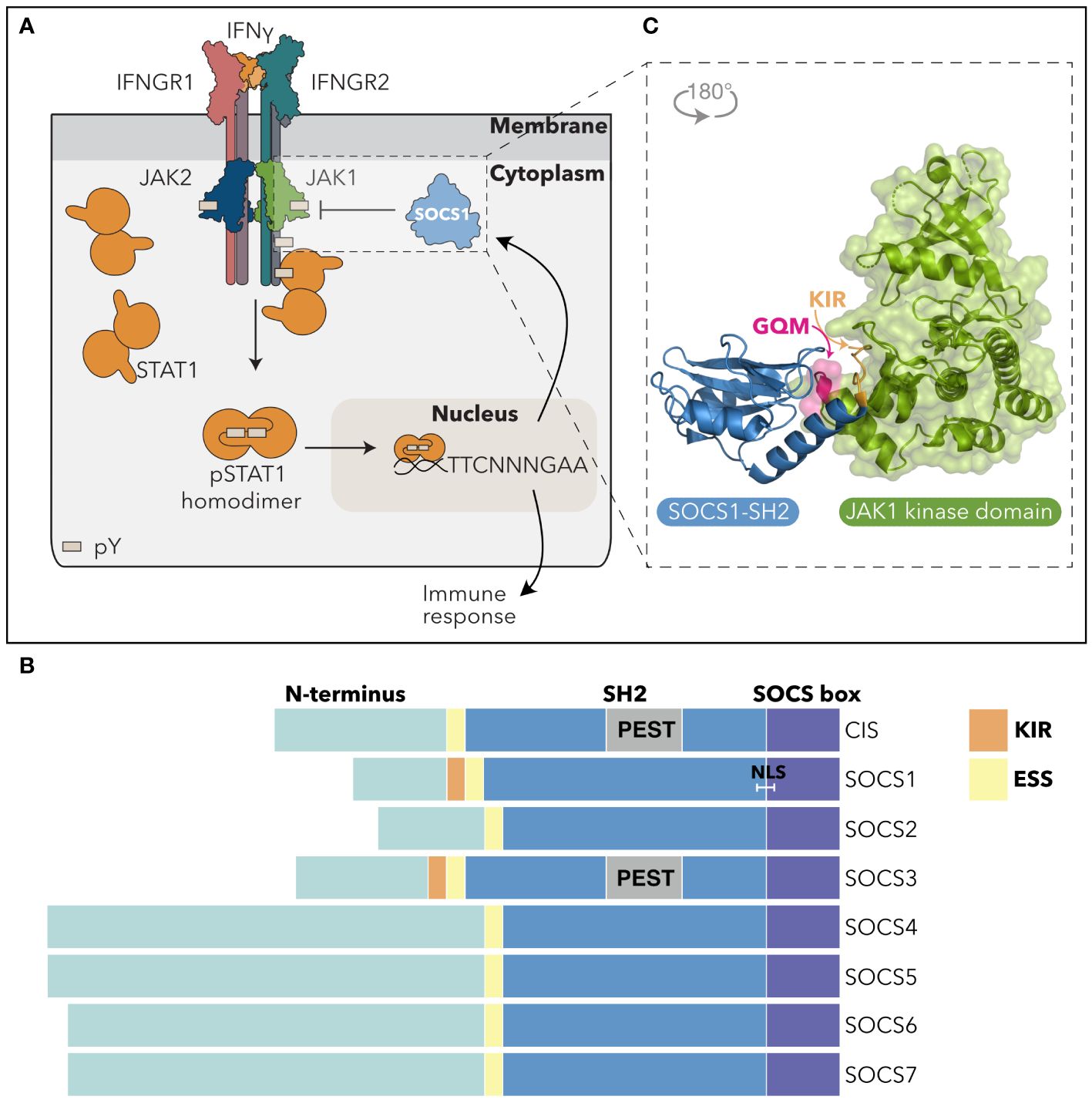
Figure 1 SOCS1 inhibits the IFNγ-driven JAK-STAT pathway. (A) SOCS1 negatively regulates IFNγ signaling. Upon engagement with the IFNGR complex, IFNγ induces tyrosine-phosphorylation and activation of JAK1 and JAK2. Activated JAK1/2 phosphorylate intracellular Tyr motifs in the IFNGR1, leading to STAT1 recruitment via the STAT1-SH2 domain and its subsequent phosphorylation by JAK. pSTAT1 dimers undergo a conformation change and translocate to the nucleus, binding to gamma interferon activation sites (GAS) to regulate transcription and drive a cellular response. SOCS1 is an IFNγ-response gene and is induced to inhibit IFNγ signaling in a classic negative feedback loop. pY = phosphotyrosine; TTCNNNGAA = IFNγ-activated promoter sequences. (B) SOCS domain architecture. The SOCS domain architecture consists of an unstructured N-terminal region of variable length (teal), a central SH2 domain (blue) and a C-terminal SOCS box motif (purple). SOCS1 and SOCS3 are distinguished by a KIR that precedes the ESS and SH2 domain region. The CIS and SOCS3 SH2 domains contain a PEST insertion (grey), while SOCS1 contains a putative NLS. (C) SOCS1 inhibition of JAK. Non-canonical binding of the SOCS1-SH2 domain (blue) to the JAK-GQM motif (pink), enables blocking of JAK-enzymatic activity via the SOCS1-KIR (orange). PDB: 6C7Y (5). SH2, Src-Homology 2 domain; ESS, Extended SH2 Sequence; KIR, Kinase Inhibitory Region; PEST, sequence rich in proline (P), glutamic acid (E), serine (S), and threonine (T); NLS*, nuclear localisation signal.
Excessive cytokine signaling can lead to inflammation and myeloproliferative disease, often with disastrous consequences, and signaling is therefore tightly regulated to maintain an appropriate cellular and systemic response. The JAK-STAT pathway is regulated at multiple levels, including by receptor trafficking, phosphatases, Protein-inhibitors of Activated STAT (PIAS) proteins, and the Suppressor Of Cytokine Signaling (SOCS) proteins (8–11).
The SOCS protein family consists of eight proteins, CIS (Cytokine-Inducible SH2-containing protein) and SOCS1–7, that are characterized by an N-terminal region of varying length, a central Src homology 2 (SH2) domain and a C-terminal SOCS box (Figure 1B). CIS was discovered in 1995 by Yoshimura and colleagues as a regulator of interleukin (IL)-3 and erythropoietin (EPO) signaling (12), while SOCS1 was independently discovered in 1997 by three different groups through its capacity to suppress IL-6 signaling [SOCS1 (13)], recognition by an anti-STAT3 antibody [SSI-1 (14)], and binding to the JAK2 kinase domain [JAB (15)]. The homology between CIS and SOCS1 led to the discovery of the SOCS and greater SOCS box family via a conserved C-terminal motif referred to as the “SOCS box” (9).
The SOCS N-terminal regions are predicted to be unstructured (16) and vary in length and sequence, effectively dividing the SOCS proteins into those with a short (CIS, SOCS1–3) or long (SOCS4–7) N-terminal region (Figure 1B). To date, there are no clear functional roles for the N-terminal regions, as at least in structure-function studies, deletion of the N-terminus has no impact on SOCS function (17–19).
As in other SH2-containing proteins, the SOCS-SH2 domains recognise linear phosphotyrosine (pTyr) motifs within their target proteins, with binding selectivity dictated by residues flanking the pTyr and with a preference for a hydrophobic residue in the +3 position relative to the Tyr (5, 20–23). Structurally, the SOCS-SH2 domain displays the canonical SH2-fold of three central β-sheets flanked by two α-helices (22, 24). However, there are a few distinguishing features, most notably an additional α-helix termed the extended SH2 subdomain (ESS) located N-terminal to the conserved SH2 sequence. The ESS interacts with residues either side of the SOCS-pTyr-binding (BC) loop, stabilizing the interaction with pTyr (20, 25). In addition, the CIS and SOCS3-SH2 domains contain an unstructured PEST motif insertion (rich in proline, glutamic acid, serine and threonine) that at least in SOCS3, appears to regulate protein stability (20).
SOCS1 and SOCS3 have a unique ability to directly inhibit JAK enzymatic activity. This was first demonstrated by Endo and colleagues for SOCS1 (15), with the activity subsequently shown to be mediated by a short “kinase inhibitory region” or “KIR” that preceded the ESS and SH2 domain (19, 25). Structural and biophysical characterization of the SOCS: JAK complexes revealed a direct interaction between the SOCS1/3-ESS and BC loop and a GQM motif present in JAK1, JAK2 and TYK2 (but not JAK3) that enabled the SOCS-KIR to act as a pseudosubstrate, partially blocking the substrate binding groove on JAK and subsequent JAK enzymatic activity (5, 26) (Figure 1C). The SOCS1-SH2 domain is thought to interact with the phosphorylated JAK activation loop (25), and at least under some conditions, SH2 interaction with phosphorylated JAK is required for inhibition of kinase activity (27). However, given the structural constraints it seems unlikely that SOCS1 can simultaneously interact with a single JAK molecule via both the substrate binding groove and the activation loop (5), thus raising some interesting questions about the stoichiometry of the SOCS1:JAK complexes that are yet to be resolved.
The SOCS box contains two motifs, the BC box and Cul5 box that interact respectively, with the adaptor proteins Elongin B and C, and the E3 ubiquitin ligase scaffold protein Cullin-5. Together with Ring Box 2 (RBX2), this forms an E3 ligase complex that mediates the ubiquitination and proteasomal degradation of SH2-bound targets (28–30). The SOCS proteins therefore function as substrate receptors for a Cullin-5 RING ligase (CRL5) complex. However, Cullin-5 interaction with the SOCS1 and SOCS3-SOCS boxes is relatively weak compared to other SOCS family members (100 and 10-fold lower affinity, respectively) (31), suggesting that SOCS1 and SOCS3 predominantly act as negative regulators through their ability to directly inhibit JAK catalytic activity (5).
Finally, SOCS1 has been reported to have a nuclear localization signal (NLS) sequence that bridges the SH2 domain and SOCS box, consistent with various reports that place SOCS1 in the nucleus (32–34). However, how nuclear localization impacts SOCS1 regulation of cytokine signaling remains unclear.
SOCS1 is a critical negative regulator of signaling in response to type I, II and III IFN and IL-2, IL-4, IL-7, IL-12, IL-13, IL-15, and IL-21 (35–43). SOCS1 is constitutively expressed in the thymus where it plays a vital role in T cell development and homeostasis (44–46). However, under infectious challenge or during inflammatory disease, SOCS1 expression is induced in response to various cytokines, including those known to be regulated by SOCS1, enabling it to act in a classic negative feedback loop (Table 1).
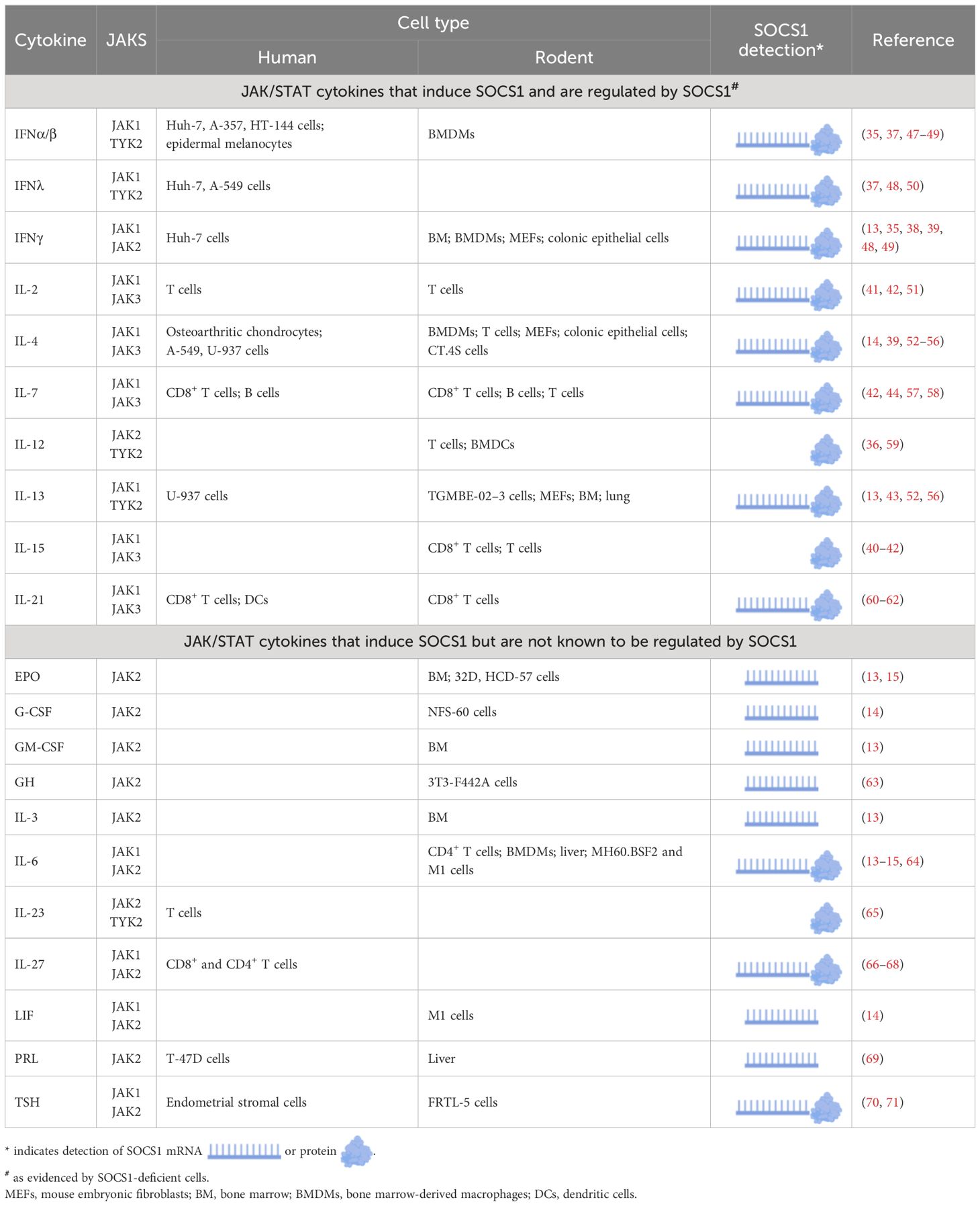
Table 1 SOCS1 induction in response to various cytokines.
The importance of SOCS1 in limiting spontaneous inflammation is evident from mouse models, where homozygous deletion of the Socs1 gene (Socs1-/-) results in perinatal lethality around 3-weeks of age, due to fatty degeneration and necrosis of the liver, and immune infiltration of multiple organs (46, 72). The multi-organ inflammation and lethality results from hyperresponsiveness to inflammatory cytokines and can be rescued by compound deletion of Ifng or treatment with neutralising anti-IFNγ antibodies (38, 45, 73). Mice lacking both Socs1 and Ifng eventually develop a fatal inflammatory disease at around 6-months of age, indicating the involvement of other cytokine pathways (74). Socs1-/- lethality is similarly rescued by deletion of Rag2 (no T, B or NK cells), Stat6 (IL-4) or components of type I IFN signaling, IFNAR1 and TYK2 (35, 45, 47, 75). Lethality is also partially rescued by deletion of Stat4 (IL-12), with mice succumbing to inflammatory disease at 1–2 months of age (36).
In addition to an enhanced response to IFNγ, the pathology in Socs1-/- neonates is associated with elevated serum levels of IFNγ, most likely due to excessive production by T cells in response to IL-1, IL-2 and IL-12 (36, 45, 75, 76). The inflammatory disease is no doubt compounded by a deficiency in peripheral Foxp3+ regulatory and γδ T cells in the Socs1-/- mice (77, 78). Additionally, SOCS1 has been shown to limit dendritic cell (DC) maturation and antigen-presentation through inhibition of IFNγ and IL-4 (79, 80).
The KIR and SH2 binding to pTyr are both required for SOCS1 to inhibit signaling; mice bearing single point mutations in either domain (KIR:F59A; SH2:R105A) phenocopy full Socs1 deletion, dying shortly after birth (27). In contrast, mice lacking the SOCS1-SOCS box survive, although they develop a multi-organ inflammatory disease with age (81). This is consistent with weak Cullin-5 interaction with the SOCS1-SOCS box (31), and further evidence that SOCS1 does not rely on its E3 ubiquitin ligase activity to inhibit cytokine signaling.
The apparent selective regulation of IFN, IL-12 and IL-2 family signaling by SOCS1 is intriguing. SOCS1 directly inhibits JAK1, JAK2 and TYK2 activity through the KIR, and the SOCS1-SH2 domain has been proposed to interact with the phosphorylated activation loops of all four JAK catalytic domains (5, 25). Given that various combinations of JAK1, JAK2 and TYK2 are associated with most cytokine receptors (either singly or in combination), it is unclear how SOCS1 selectively regulates IFN, IL-12 and IL-2 family signaling, although this may simply reflect greater induction of SOCS1 protein by these cytokines (Table 1). To date, no receptor pTyr residues have been identified as high affinity SOCS1-SH2 binders, despite pTyr interaction being required for SOCS1 inhibition of signaling (5, 27). In contrast, SOCS3 binds with high affinity to phosphorylated tyrosines within the gp130, IL-12, leptin and G-CSF receptors (82–87), providing a clear mechanism underlying the selective SOCS3 regulation of the corresponding cytokine pathways.
In addition to being induced by JAK/STAT signaling, SOCS1 expression is down-regulated by a well-characterised microRNA, miR-155. MiR-155 is derived from a non-coding transcript referred to as the B cell integration cluster and is upregulated in various immune cells in response to inflammatory stimuli and infection, with multiple targets in addition to SOCS1 [reviewed in (88, 89)]. An elegant study by Lu and colleagues (90) mutated the miR-155 binding site in the Socs1 mRNA 3’UTR, partially recapitulating the effects of miR-155 deletion to reduce disease severity in the autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis, and limit the expansion of NK cells in response to murine cytomegalovirus (MCMV) infection (90).
Cytokines and inflammatory mediators are strongly implicated in the pathogenesis of autoimmune diseases. For example, type I and type II IFN (91, 92), and IL-12 (93) are associated with systemic lupus erythematosus (SLE), and the IL-12-driven transcription factor STAT4 is a dominant genetic risk allele (93, 94). Exacerbated IFNγ and IL-12 responses are also known to drive chronic joint inflammation in rheumatoid arthritis (95–97). Importantly, SOCS1 inhibits the cellular response to these key inflammatory mediators of SLE and RA.
Inborn errors of immunity arising in SOCS1 have been reported in patients presenting with a range of clinical phenotypes, including immune dysregulation with multi-system autoimmunity, autoimmune diseases such as SLE and chronic autoimmune cytopenia, and malignancy (98–104). The majority of reported inborn errors of immunity in SOCS1 are private (not reported in the general population), heterozygous, autosomal dominant and loss-of-function variants (98–100, 102, 103) (Figure 2). The lack of homozygosity implies the SOCS1 gene is indispensable, consistent with the lethal phenotype of Socs1-/- mice (46, 72). Given the majority of variants result in loss of protein, it implies the dose of SOCS1 is important, accounting for the autosomal dominance and consistent with the inflammatory disease in aging Socs1+/- mice (105). Two rare SOCS1 missense variants have also been reported in cis (P50L, A76G), however, they appeared to have little impact on SOCS1 function (104).
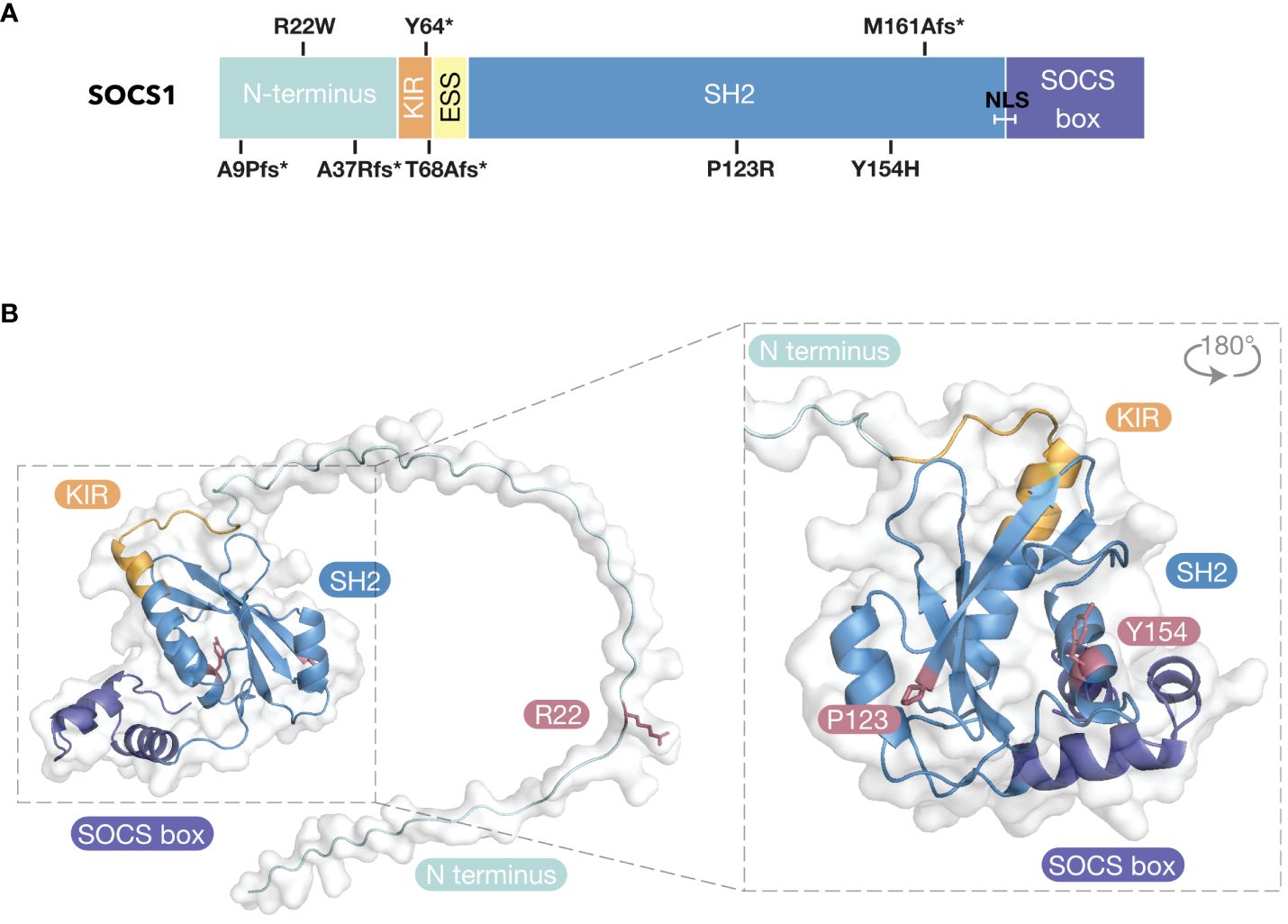
Figure 2 Inborn Errors of Immunity of SOCS1. (A) SOCS1 variants mapped to domain architecture. * = stop codon; fs, frameshift; SH2, Src-Homology 2; KIR, Kinase Inhibitory Region; ESS, Extended SH2 Sequence; NLS, Nuclear Localization Sequence. (B) SOCS1 missense variants mapped to predicted structure of SOCS1 (AlphaFold 2.0 database).
Patient mutations within the SOCS1-KIR (Y64>stop, T68A with a frameshift resulting in a stop codon: fs>stop) and the SH2 domain (M161Afs>stop, P123R, Y154H) result in loss of SOCS1 expression. The two unrelated patients that harbored the Y64>stop variant presented with Hyper IgE-like syndrome with eczema (P1) and eosinophilic allergic alveolitis (P2) (98). The patient carrying the T68>fs variant presented with severe dermatitis, recurrent skin infections and psoriatic arthritis (P3) (99). P4 and P5 carried the M161A frameshift variant (M161Afs>stop) and were from unrelated families. P4 presented with common variable immunodeficiency (CVID)-like phenotype (98) and P5 presented with Evans syndrome (100). Four patients within the same family were heterozygous for Y154H and presented with a range of autoimmune diseases including SLE (P6), immune thrombocytopenia (ITP; P7), psoriasis (P8 and P9) and spondyloarthritis, autoimmune hepatitis and pancreatitis (P9) (100). Two patients within the same family were heterozygous for P123R and presented with severe ITP (P10) and ITP thyroiditis polyarthritis (P11) (100). The remaining five patients (P12–17) carried SOCS1 variants that clustered within the N-terminal domain (A9Pfs>stop, R22W, A37Rfs>stop) (100, 102). These patients suffered from various autoimmune diseases, including Evans syndrome, coeliac disease, psoriasis, SLE, and multisystem inflammatory syndrome in children (MIS-C). While the KIR and SH2 missense variants likely disrupted the SOCS1 structure (thus resulting in loss of SOCS1 expression), the R22W variant may be the first evidence that the N-terminal region of SOCS1 is required for SOCS1 inhibitory activity (Figure 2).
For diseases such as SLE and eosinophilia, where the key cytokines driving pathology are known [type I IFN (106); IL-2 family cytokines (107), respectively], it is clear how loss of SOCS1 results in pathogenesis. Similarly, the dermatitis seen in P3 only responded to treatment with dual IL4Rα and IL17A blockade (99), making an obvious link to SOCS1 regulation of the IL-4 pathway. However, diseases such as CVID and Evans syndrome have unknown causes and the patient’s pathology arises from dysregulation of multiple immune pathways, as do other inborn errors of immunity, such as STAT1 gain-of-function variants. In addition, some family members harboring SOCS1 variants were asymptomatic, indicating SOCS1 haploinsufficiency does not have complete clinical penetrance. Despite this, SOCS1 haploinsufficiency has complete cellular penetrance. Cells derived from all individuals with loss-of-function SOCS1 variants consistently had augmented IFNγ, IL-2 and IL-4 signaling responses, with increased STAT1, STAT5 and STAT6 phosphorylation, respectively (98–103). Consequently, the etiology of SOCS1 haploinsufficiency still remains unclear (100). Current treatment of SOCS1 haploinsufficient patients largely relies on immunosuppressive drugs such as corticosteroids or JAK-inhibitors to lower systemic inflammation (98–103), however, given the potential side-effects associated with long-term treatment, more targeted approaches are required.
It is evident that SOCS1 inhibition of cytokine signaling is required to maintain immune homeostasis and resolve inflammatory signaling. Given the importance of inflammation and the immune response in cancer initiation and development (108), and the contribution of hyperactive JAKs to myeloproliferative disorders and lymphoid cancers (109), it is not surprising that a growing body of work has highlighted a role for SOCS1 in cancer.
In many cases, silencing of the SOCS1 gene and/or reduced mRNA or protein levels, is associated with cancer versus normal tissue (110–112). Hypermethylation of the SOCS1 gene has been reported in multiple cancers and has been linked to cell growth in hepatocellular, pancreatic, oesophageal and gastric cancer (113–116). For instance, in hepatocellular carcinoma, hypermethylation of the SOCS1 promoter region was observed in 65% of patient samples, with restoration of SOCS1 levels inhibiting cell growth in vitro (113, 117). In addition to gene silencing, high levels of microRNAs such as miR-19a/b or miR-155 correlated with low SOCS1 expression in cancer cell lines and primary tumors (118–122). Although miRNAs have many targets, Jiang et al. (120), demonstrated that SOCS1 siRNA recapitulated the oncogenic properties of miR-155 in breast cancer, while restoration of SOCS1 attenuated the impact of miR-155.
Several somatic SOCS1 mutations have been associated with B cell lymphomas, such as Diffuse large B cell lymphoma (DBCL) and Hodgkin lymphoma (123–125). Detrimental mutations were associated with the germinal centre B (GCB) cell-like subtype of DBCL (123), consistent with IL-4-JAK-STAT6 signaling in this subtype (126) being regulated by SOCS1. In addition, a patient with an inborn error of immunity in SOCS1 (A9P>fs) also presented with Hodgkin lymphoma (as well as psoriasis and Coeliac disease) (100).
A polymorphism in the SOCS1 promoter region (-1478 CA>del) has been reported to increase SOCS1 expression in atopic asthma (127), and has been associated with breast and gastric cancer, as well as with a poorer outcome in colon cancer (128–131). The effect of this polymorphism remains unclear, although it does not appear to be linked to tumor suppression.
Although some of the growth inhibitory effects of SOCS1 in cancer can be attributed to regulation of JAK/STAT signaling, SOCS1 has also been reported to target other pathways, including via MET receptor tyrosine kinase, p21, p53 and NF-kB (132–139). Consistent with its nuclear localization, SOCS1 interaction with the p53 tumor suppressor has been proposed to promote p53 activation and cellular senescence (136, 137). In addition, SOCS1-mediated ubiquitination and degradation of the NF-kB p65 subunit limits NF-kB-driven transcription (138, 139). Hence reduced SOCS1 levels would potentially limit p53 activity and potentiate NF-kB-driven cell growth.
The concept of SOCS1 as a tumor suppressor is reviewed in more detail by Ilangumaran and colleagues (this research topic) (140). The remainder of this review will focus on SOCS1 regulation of anti-tumor immunity.
Type II IFN (IFNγ) and to a lesser extent the type I IFNs, drive a transcriptional program in cancer cells that results in apoptotic cell death, in addition to influencing anti-tumor immunity. This includes secretion of chemoattractants, activation of cytotoxic T cells and enhancement of antigen presentation (141–148). However, IFNγ also drives expression of molecules on cancer cells such as programmed death-ligand (PD-L)1 and PD-L2 that inhibit T cell function, contributing to immune evasion (149). The impact of these opposing effects is likely to depend on the strength and persistence of signaling, as well as the cancer context and tumor microenvironment (TME).
Antibody therapies that target the immune checkpoints PD-L1/PD1 and CTLA4 have revolutionized cancer treatment (immune checkpoint blockade; ICB) (150). However, many patients remain refractory to treatment or develop resistance (151). The importance of the IFNγ pathway (and its regulation by SOCS1) was established by a series of seminal studies that used genetic screens and analysis of patient samples to show that defects in IFNγ pathway components (Ifngr1, Ifngr2, Jak1, Jak2, Stat1, Irf1) or amplification of the negative regulators Socs1 and PIAS4, conferred resistance to ICB (152–157).
Consistent with this, depletion of Socs1 in a lung cancer cell line that was resistant to ICB, restored the IFNγ response and sensitized tumours to anti-PD1 therapy (158). In another study, Dhainaut and colleagues (159) used spatial transcriptomics coupled with functional genomics to understand the impact of individual genes within tumor lesions. Although loss of Socs1 increased CD4+ and CD8+ T cell infiltration into the tumor lesions, it also promoted tumor growth, likely via IFNγ-driven expression of PD-L1 inhibiting T cell function. Critically, anti-PD-L1 blockade preferentially depleted Socs1 deficient lesions (159).
A study by Song et al. (160), linked cancer-specific defects in microRNA processing to reduced miR-155 levels, with a corresponding increase in SOCS1 that suppressed IFNγ responses and increased resistance to T cell killing. This was associated with decreased PD-L1, antigen presentation and secretion of the T cell-attractant chemokines CXCL9 and CXCL10 (160). In a second study, high miR-155 expression in tumors correlated with improved anti-tumor immune profiles and outcomes in breast cancer patients. This was connected to miR-155 suppression of SOCS1 enhancing IFN-driven CXCL9, CXCL10 and CXCL11 production and immune infiltration, sensitizing to ICB (161). More recently, House and colleagues (162) identified IRF-I as a key negative regulator of CXCL9 production in cancer and myeloid cells through induction of SOCS1 and subsequent inhibition of IFNγ signaling.
These studies highlighted a link between reduced SOCS1 and (i) increased expression of immune checkpoints, thus sensitizing tumors to ICB, and (ii) IFN-driven chemokine production that increased immune cell infiltration into the TME. Furthermore, they suggest that upregulation of PD-L1 in response to reduced SOCS1 levels could potentially improve patient responses to ICB.
While global genetic deletion revealed the critical role of SOCS1 in immune cell development (42, 44, 45, 163), the gross defects in neonatal mice confounded the study of mature immune cell function, particularly in the context of tumor immunity. Subsequent studies have revealed a key inhibitory role for SOCS1 in multiple immune subsets that coordinate to mount an effective anti-tumor immune response.
A genome wide CRISPR screen identified SOCS1 as a negative regulator of proliferation and cytotoxicity in primary human T cells (164). Socs1 was later identified as a non-redundant inhibitor of antigen-experienced CD4+ T cell proliferation and effector function in mice (165). Inactivation of Socs1 in CD4+ T cells maintained proliferation, while inactivation in CD8+ T cells enhanced survival and effector function; adoptive transfer of Socs1-deficient CD4+ and CD8+ cells together giving greater therapeutic benefit. Similar results were obtained in human CD19-chimeric antigen receptor (CAR)-T cells, highlighting the unique roles of SOCS1 in CD4+ vs CD8+ T cells, and the synergistic potential of targeting SOCS1 in adoptive CAR-T cell immunotherapy (165). A second genetic screen, this time in CD8+ cells, identified Socs1 as a key checkpoint that not only restricted CD8+ T cell expansion, but also infiltration of CD8+ cells into the TME (166). The authors went on to evaluate SOCS1 deletion in primary human T cells and tumor infiltrating lymphocytes, demonstrating enhanced IL-2-driven STAT5 and IL-12-STAT4 activation, associated with increased IFNγ production and reduced tumor growth in an immunodeficient mouse model (166).
SOCS1 expression in DCs restricted antigen presentation and the magnitude of the resulting adaptive immune response, with reduction of SOCS1 in DCs promoting T cell-mediated anti-tumor immunity (167–170). Socs1-/- DCs induced a stronger T helper (TH)1 response, associated with increased DC production of IFNγ (168). Conditional deletion of Socs1 in myeloid cells resulted in a significantly reduced B16F10 tumor burden and suppressed DSS/DHM-induced colon cancer growth. In co-cultures, DCs derived from these mice induced higher IFNγ production from CD4+ T cells and enhanced CD8+ T cell cytotoxic activity (171). These studies highlight a cell-intrinsic role for SOCS1 in macrophages and DCs that restricts myeloid potentiation of the anti-tumor T cell response.
SOCS1 clearly has specific roles in CD4+ and CD8+ T cells, DCs and macrophages that limit anti-tumor immunity, in addition to a tumor intrinsic role that also impacts immune cell recognition and killing. Taken together, this positions SOCS1 as a potentially powerful negative regulator of the anti-tumor immune response (Figure 3).
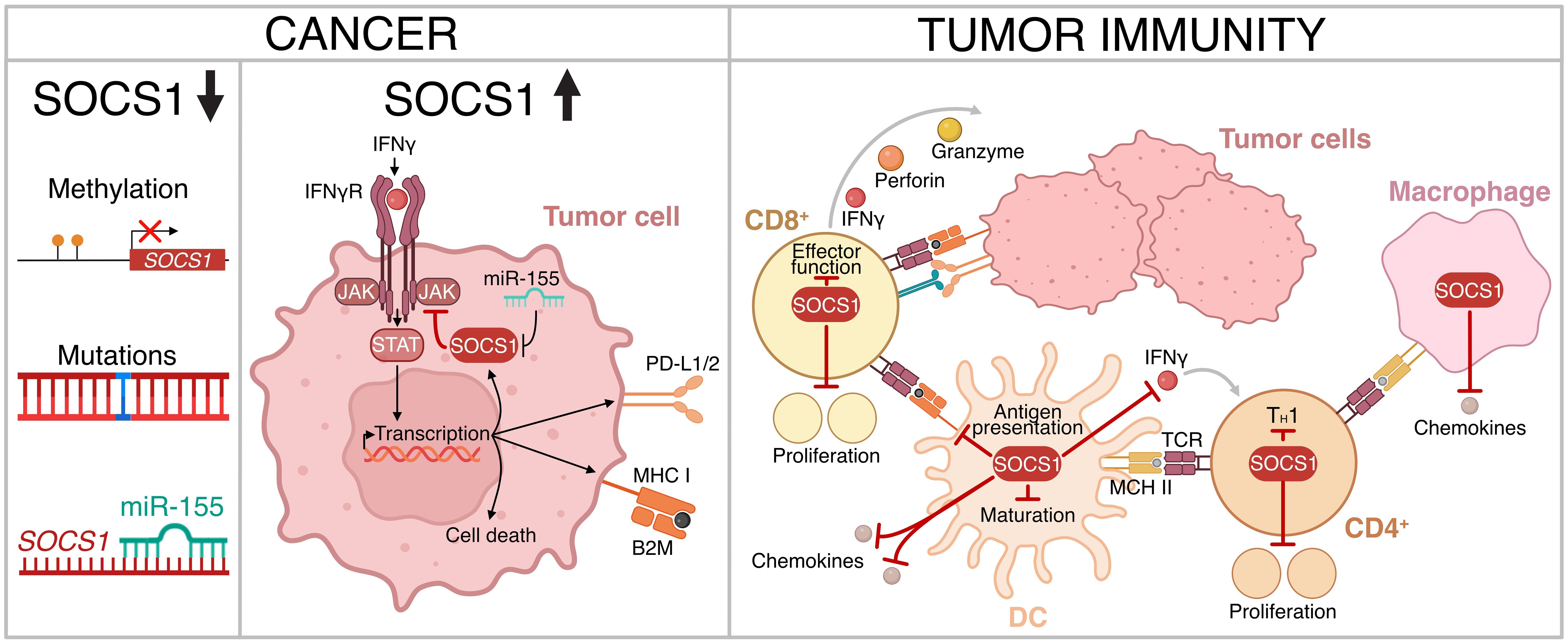
Figure 3 The role of SOCS1 in regulating tumor immunity. Downregulation of expression or loss of SOCS1 function has been observed in certain cancer subtypes (left panel). SOCS1 can confer resistance to immune checkpoint blockade via inhibition of IFNγ signaling and down-regulation of immune checkpoints PD-L1/2 (center panel). In addition, SOCS1 limits anti-tumor immunity through various immune cell types (right panel). Figure created with BioRender.com.
Cytokines such as the IFNs are an important aspect of our immune defence and are critical for the elimination of infectious pathogens and transformed or cancerous cells. As the key negative regulator of many of these pathways, SOCS1 is a bona-fide intracellular immune checkpoint, with loss of SOCS1 resulting in unrestrained cytokine responses associated with autoimmunity and other inflammatory diseases. Correspondingly, elevation of SOCS1 levels in both cancer and immune cells, limits the effectiveness of cytokines that drive anti-tumor immunity. Both scenarios present attractive opportunities for therapeutic intervention.
SOCS1 deficiency has been implicated in the pathogenesis of lupus, uveitis, and asthma, highlighting the role of SOCS1 in regulating immune homeostasis (52, 77, 172–176). Identifying individuals with inflammatory disease associated with genetic inactivation of SOCS1 may help stratify patients for treatment with JAK inhibitors. SOCS1 has the potential to dampen signaling from all IFNs, in addition to IL-2 family cytokines and IL-12. This represents an overlapping spectrum of activities compared to the current JAK inhibitors which include JAK-specific and pan-inhibitors (177). Strategies that increase SOCS1 levels or mimic its activity to reduce cytokine signaling may have utility as an alternative to or in combination with JAK inhibitors and would have a broader spectrum of activity than biologics targeting single receptor chains or cytokines.
Cell penetrating-SOCS1 peptidomimetics that mimic the activity of the SOCS1 kinase inhibitory region (KIR) have successfully been used to treat lupus-like disease, uveitis, and experimental autoimmune encephalitis (model of multiple sclerosis) in rodent models (178, 179). Additionally, cell penetrating forms of recombinant SOCS1 protein have been shown to suppress cytokine signaling (180). These studies highlight the potential of enhancing SOCS1 levels and/or developing drugs that mimic SOCS1 activity in the treatment of autoimmune disease. Alternative approaches to increase SOCS1 levels, such as antagomirs that target miR-155, may also have some utility given the recent advances in RNA delivery systems (181).
SOCS1 has been reported to have both positive and negative effects in the context of cancer control. However, although reduced SOCS1 expression correlated with poor prognosis in a variety of solid cancers, in many instances the relationship between SOCS1 levels and cancer etiology remains unclear. In addition, a complex mix of driver and co-operating mutations together with the localised inflammatory milieux may result in varying SOCS1 levels. This is often further complicated by an inability to distinguish between intrinsic tumor expression of SOCS1 and expression in the surrounding cellular environment. The growing application of single cell sequencing and spatial omics should advance our understanding of how SOCS1 expression impacts disease progression in specific cancer and cellular contexts.
CRISPR-Cas9 gene editing in whole genome screening has been a powerful discovery-tool in many fields, as illustrated by the studies discussed in this review. One of the striking findings to emerge, in addition to the importance of IFNγ signaling in maintaining the response to ICB, is the potential synergy to be gained by reducing SOCS1 in tumors in combination with ICB. SOCS1 is also a critical checkpoint in immune cells, limiting aspects of immune cell development as well as effector cell function. Interestingly, tumor-specific, and immune-cell reduction of SOCS1 both lead to increased chemokine secretion and lymphocyte recruitment and infiltration into the TME. Reducing SOCS1 levels to sustain IFNγ signaling may therefore be a useful therapeutic approach to reinvigorate lymphocyte function in an immunosuppressive TME.
However, it is challenging to target an intracellular protein, particularly one that is part of a closely related family and relies on a common modality of binding to pTyr for its mechanism of action. One interesting example of modulating SOCS1 levels, is the lipid nanoparticle (LNP) delivery of SOCS1-targeting small interfering RNA (siRNA) to bone marrow-derived DCs. Vaccination with OVA-specific DCs containing Socs1 siRNA prior to challenge with OVA-bearing B16F10 melanoma cells, successfully suppressed tumor growth through increased cytokine secretion and antigen presentation (182).
The most obvious and perhaps feasible application for reduced SOCS1 levels is the field of adoptive T cell therapy. Adoptive T cell therapies genetically modify a patient’s T cells to express receptors for tumor antigens (CAR-T cells), prior to expansion and infusion back into the patient (183). CAR-T cells have shown impressive clinical efficacy in haematological malignancies, with six FDA-approved CARs now in the clinic. However, CAR-T cells are much less effective in solid tumors, most likely due to an immunosuppressive TME (184–189). An alternative approach involves expansion of a patient’s TILs prior to adoptive transfer, with the first TIL-therapy recently approved by the FDA for melanoma (lifileucel; Amtagvi, Iovance Biotherapeutics) (190). The studies discussed here highlight the potential for inactivation of SOCS1 in CD8+ and/or CD4+ CAR-T cells or TILs, to overcome T cell exhaustion and improve proliferation, survival, and effector function in adoptive T cell immunotherapy. Going forward it will be critical to evaluate potential exacerbation of adverse effects, such as cytokine release syndrome, as a result of SOCS1 deletion (191).
In summary, SOCS1 is an important immune checkpoint and an attractive target in both autoimmunity and cancer. Its intrinsic roles in both tumor and immune cells make it a central player that restricts IFNγ-driven killing and facilitates escape from PD1/CTLA4 blockade. The challenge will be to find innovative ways to target SOCS1 in the different disease contexts. We look forward to watching the next chapter unfold with interest.
GMB: Data curation, Writing – original draft, Writing – review & editing. NK: Writing – original draft, Writing – review & editing. KD: Supervision, Writing – review & editing. SEN: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. GMB was supported by an Australian government Research Training Program Scholarship. NK was supported in part by an Australian government Research Training Program Scholarship and a Fulbright Scholarship. This review was written with support in part from NHMRC Ideas Grant 2011761, Victorian State Government Operational Infrastructure Support and the Australian Government NHMRC Independent Research Institutes Infrastructure Support Scheme (IRIISS).
We acknowledge the Wurundjeri people of the Kulin nation as the traditional owners and guardians of the land on which the work was performed.
SEN and KD receive research funding from a pharmaceutical partner to target SOCS proteins in disease. SEN has been awarded research funding from WEHI Ventures to target SOCS proteins in disease. The funders had no role in the content or writing of the manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity. (2012) 36:503–14. doi: 10.1016/j.immuni.2012.03.013
2. Shuai K, Schindler C, Prezioso VR, Darnell JE Jr. Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science. (1992) 258:1808–12. doi: 10.1126/science.1281555
3. Braunstein J, Brutsaert S, Olson R, Schindler C. STATs dimerize in the absence of phosphorylation. J Biol Chem. (2003) 278:34133–40. doi: 10.1074/jbc.M304531200
4. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. (2002) 3:651–62. doi: 10.1038/nrm909
5. Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun. (2018) 9:1558. doi: 10.1038/s41467-018-04013-1
6. Hjortsberg L, Lindvall C, Corcoran M, Arulampalam V, Chan D, Thyrell L, et al. Phosphoinositide 3-kinase regulates a subset of interferon-alpha-stimulated genes. Exp Cell Res. (2007) 313:404–14. doi: 10.1016/j.yexcr.2006.10.022
7. Katsoulidis E, Li Y, Yoon P, Sassano A, Altman J, Kannan-Thulasiraman P, et al. Role of the p38 mitogen-activated protein kinase pathway in cytokine-mediated hematopoietic suppression in myelodysplastic syndromes. Cancer Res. (2005) 65:9029–37. doi: 10.1158/0008-5472.CAN-04-4555
8. Liu B, Mink S, Wong KA, Stein N, Getman C, Dempsey PW, et al. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat Immunol. (2004) 5:891–8. doi: 10.1038/ni1104
9. Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, et al. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A. (1998) 95:114–9. doi: 10.1073/pnas.95.1.114
10. Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci. (2008) 13:4925–32. doi: 10.2741/3051
11. Cendrowski J, Mamińska A, Miaczynska M. Endocytic regulation of cytokine receptor signaling. Cytokine Growth Factor Rev. (2016) 32:63–73. doi: 10.1016/j.cytogfr.2016.07.002
12. Yoshimura A, Ohkubo T, Kiguchi T, Jenkins NA, Gilbert DJ, Copeland NG, et al. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. (1995) 14:2816–26. doi: 10.1002/j.1460-2075.1995.tb07281.x
13. Starr R, Willson TA, Viney EM, Murray LJL, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature. (1997) 387:917–21. doi: 10.1038/43206
14. Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature. (1997) 387:924–9. doi: 10.1038/43219
15. Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. (1997) 387:921–4. doi: 10.1038/43213
16. Feng ZP, Chandrashekaran IR, Low A, Speed TP, Nicholson SE, Norton RS. The N-terminal domains of SOCS proteins: a conserved region in the disordered N-termini of SOCS4 and 5. Proteins. (2012) 80:946–57. doi: 10.1002/prot.23252
17. Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M, et al. Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J. (1999) 18:375–85. doi: 10.1093/emboj/18.2.375
18. Narazaki M, Fujimoto M, Matsumoto T, Morita Y, Saito H, Kajita T, et al. Three distinct domains of SSI-1/SOCS-1/JAB protein are required for its suppression of interleukin 6 signaling. Proc Natl Acad Sci U S A. (1998) 95:13130–4. doi: 10.1073/pnas.95.22.13130
19. Sasaki A, Yasukawa H, Suzuki A, Kamizono S, Syoda T, Kinjyo I, et al. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells. (1999) 4:339–51. doi: 10.1046/j.1365-2443.1999.00263.x
20. Babon JJ, McManus EJ, Yao S, DeSouza DP, Mielke LA, Sprigg NS, et al. The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol Cell. (2006) 22:205–16. doi: 10.1016/j.molcel.2006.03.024
21. Zadjali F, Pike AC, Vesterlund M, Sun J, Wu C, Li SS, et al. Structural basis for c-KIT inhibition by the suppressor of cytokine signaling 6 (SOCS6) ubiquitin ligase. J Biol Chem. (2011) 286:480–90. doi: 10.1074/jbc.M110.173526
22. Bullock AN, Debreczeni JÉ, Edwards AM, Sundström M, Knapp S. Crystal structure of the SOCS2–elongin C–elongin B complex defines a prototypical SOCS box ubiquitin ligase. Proc Natl Acad Sci U S A. (2006) 103:7637–42. doi: 10.1073/pnas.0601638103
23. Krebs DL, Uren RT, Metcalf D, Rakar S, Zhang JG, Starr R, et al. SOCS-6 binds to insulin receptor substrate 4, and mice lacking the SOCS-6 gene exhibit mild growth retardation. Mol Cell Biol. (2002) 22:4567–78. doi: 10.1128/MCB.22.13.4567-4578.2002
24. Waksman G, Kominos D, Robertson SC, Pant N, Baltimore D, Birge RB, et al. Crystal structure of the phosphotyrosine recognition domain SH2 of v-src complexed with tyrosine-phosphorylated peptides. Nature. (1992) 358:646–53. doi: 10.1038/358646a0
25. Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. (1999) 18:1309–20. doi: 10.1093/emboj/18.5.1309
26. Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol. (2013) 20:469–76. doi: 10.1038/nsmb.2519
27. Doggett K, Keating N, Dehkhoda F, Bidgood GM, Meza Guzman LG, Leong E, et al. The SOCS1 KIR and SH2 domain are both required for suppression of cytokine signaling in vivo. Cytokine. (2023) 165:156167. doi: 10.1016/j.cyto.2023.156167
28. Kamura T, Sato S, Haque D, Liu L, Kaelin WG Jr., Conaway RC, et al. The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. (1998) 12:3872–81. doi: 10.1101/gad.12.24.3872
29. Zhang J-G, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci U S A. (1999) 96:2071–6. doi: 10.1073/pnas.96.5.2071
30. Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. (2004) 18:3055–65. doi: 10.1101/gad.1252404
31. Babon JJ, Sabo JK, Zhang J-G, Nicola NA, Norton RS. The SOCS box encodes a hierarchy of affinities for Cullin5: implications for ubiquitin ligase formation and cytokine signalling suppression. J Mol Biol. (2009) 387:162–74. doi: 10.1016/j.jmb.2009.01.024
32. Koelsche C, Strebovsky J, Baetz A, Dalpke AH. Structural and functional analysis of a nuclear localization signal in SOCS1. Mol Immunol. (2009) 46:2474–80. doi: 10.1016/j.molimm.2009.05.020
33. Baetz A, Koelsche C, Strebovsky J, Heeg K, Dalpke AH. Identification of a nuclear localization signal in suppressor of cytokine signaling 1. FASEB J. (2008) 22:4296–305. doi: 10.1096/fj.08-116079
34. Gielen V, Sykes A, Zhu J, Chan B, Macintyre J, Regamey N, et al. Increased nuclear suppressor of cytokine signaling 1 in asthmatic bronchial epithelium suppresses rhinovirus induction of innate interferons. J Allergy Clin Immunol. (2015) 136:177–88.e11. doi: 10.1016/j.jaci.2014.11.039
35. Gingras S, Parganas E, de Pauw A, Ihle JN, Murray PJ. Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. J Biol Chem. (2004) 279:54702–7. doi: 10.1074/jbc.M411043200
36. Eyles JL, Metcalf D, Grusby MJ, Hilton DJ, Starr R. Negative regulation of interleukin-12 signaling by suppressor of cytokine signaling-1. J Biol Chem. (2002) 277:43735–40. doi: 10.1074/jbc.M208586200
37. Blumer T, Coto-Llerena M, Duong FHT, Heim MH. SOCS1 is an inducible negative regulator of interferon λ (IFN-λ)-induced gene expression in vivo. J Biol Chem. (2017) 292:17928–38. doi: 10.1074/jbc.M117.788877
38. Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. (1999) 98:597–608. doi: 10.1016/S0092-8674(00)80047-1
39. Chinen T, Kobayashi T, Ogata H, Takaesu G, Takaki H, Hashimoto M, et al. Suppressor of cytokine signaling-1 regulates inflammatory bowel disease in which both IFNgamma and IL-4 are involved. Gastroenterology. (2006) 130:373–88. doi: 10.1053/j.gastro.2005.10.051
40. Ilangumaran S, Ramanathan S, Ning T, La Rose J, Reinhart B, Poussier P, et al. Suppressor of cytokine signaling 1 attenuates IL-15 receptor signaling in CD8+ thymocytes. Blood. (2003) 102:4115–22. doi: 10.1182/blood-2003-01-0175
41. Ilangumaran S, Ramanathan S, La Rose J, Poussier P, Rottapel R. Suppressor of cytokine signaling 1 regulates IL-15 receptor signaling in CD8+CD44high memory T lymphocytes 1. J Immunol. (2003) 171:2435–45. doi: 10.4049/jimmunol.171.5.2435
42. Cornish AL, Chong MM, Davey GM, Darwiche R, Nicola NA, Hilton DJ, et al. Suppressor of cytokine signaling-1 regulates signaling in response to interleukin-2 and other gamma c-dependent cytokines in peripheral T cells. J Biol Chem. (2003) 278:22755–61. doi: 10.1074/jbc.M303021200
43. Fukuyama S, Nakano T, Matsumoto T, Oliver BG, Burgess JK, Moriwaki A, et al. Pulmonary suppressor of cytokine signaling-1 induced by IL-13 regulates allergic asthma phenotype. Am J Respir Crit Care Med. (2009) 179:992–8. doi: 10.1164/rccm.200806-992OC
44. Chong MM, Cornish AL, Darwiche R, Stanley EG, Purton JF, Godfrey DI, et al. Suppressor of cytokine signaling-1 is a critical regulator of interleukin-7-dependent CD8+ T cell differentiation. Immunity. (2003) 18:475–87. doi: 10.1016/S1074-7613(03)00078-5
45. Marine J-C, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, et al. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. (1999) 98:609–16. doi: 10.1016/S0092-8674(00)80048-3
46. Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, et al. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci U S A. (1998) 95:14395–9. doi: 10.1073/pnas.95.24.14395
47. Fenner JE, Starr R, Cornish AL, Zhang J-G, Metcalf D, Schreiber RD, et al. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat Immunol. (2006) 7:33–9. doi: 10.1038/ni1287
48. Liu B, Chen S, Guan Y, Chen L. Type III interferon induces distinct SOCS1 expression pattern that contributes to delayed but prolonged activation of jak/STAT signaling pathway: implications for treatment non-response in HCV patients. PloS One. (2015) 10:e0133800. doi: 10.1371/journal.pone.0133800
49. Lesinski GB, Zimmerer JM, Kreiner M, Trefry J, Bill MA, Young GS, et al. Modulation of SOCS protein expression influences the interferon responsiveness of human melanoma cells. BMC Cancer. (2010) 10:142. doi: 10.1186/1471-2407-10-142
50. Wei H, Wang S, Chen Q, Chen Y, Chi X, Zhang L, et al. Suppression of interferon lambda signaling by SOCS-1 results in their excessive production during influenza virus infection. PloS Pathog. (2014) 10:e1003845. doi: 10.1371/journal.ppat.1003845
51. Sporri B, Kovanen PE, Sasaki A, Yoshimura A, Leonard WJ. JAB/SOCS1/SSI-1 is an interleukin-2-induced inhibitor of IL-2 signaling. Blood. (2001) 97:221–6. doi: 10.1182/blood.V97.1.221
52. McCormick SM, Gowda N, Fang JX, Heller NM. Suppressor of cytokine signaling (SOCS)1 regulates interleukin-4 (IL-4)-activated insulin receptor substrate (IRS)-2 tyrosine phosphorylation in monocytes and macrophages via the proteasome. J Biol Chem. (2016) 291:20574–87. doi: 10.1074/jbc.M116.746164
53. He Q, Sun C, Lei W, Ma J. SOCS1 regulates apoptosis and inflammation by inhibiting IL-4 signaling in IL-1β-stimulated human osteoarthritic chondrocytes. BioMed Res Int. (2017) 2017:4601959. doi: 10.1155/2017/4601959
54. Whyte CS, Bishop ET, Rückerl D, Gaspar-Pereira S, Barker RN, Allen JE, et al. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J Leukoc Biol. (2011) 90:845–54. doi: 10.1189/jlb.1110644
55. Dickensheets H, Vazquez N, Sheikh F, Gingras S, Murray PJ, Ryan JJ, et al. Suppressor of cytokine signaling-1 is an IL-4-inducible gene in macrophages and feedback inhibits IL-4 signaling. Genes Immun. (2007) 8:21–7. doi: 10.1038/sj.gene.6364352
56. Hebenstreit D, Luft P, Schmiedlechner A, Regl G, Frischauf AM, Aberger F, et al. IL-4 and IL-13 induce SOCS-1 gene expression in A549 cells by three functional STAT6-binding motifs located upstream of the transcription initiation site. J Immunol. (2003) 171:5901–7. doi: 10.4049/jimmunol.171.11.5901
57. Corfe SA, Rottapel R, Paige CJ. Modulation of IL-7 thresholds by SOCS proteins in developing B lineage cells. J Immunol. (2011) 187:3499–510. doi: 10.4049/jimmunol.1100424
58. Ghazawi FM, Faller EM, Parmar P, El-Salfiti A, MacPherson PA. Suppressor of cytokine signaling (SOCS) proteins are induced by IL-7 and target surface CD127 protein for degradation in human CD8 T cells. Cell Immunol. (2016) 306–307:41–52. doi: 10.1016/j.cellimm.2016.07.002
59. Evel-Kabler K, Song XT, Aldrich M, Huang XF, Chen SY. SOCS1 restricts dendritic cells' ability to break self tolerance and induce antitumor immunity by regulating IL-12 production and signaling. J Clin Invest. (2006) 116:90–100. doi: 10.1172/JCI26169
60. Gagnon J, Ramanathan S, Leblanc C, Ilangumaran S. Regulation of IL-21 signaling by suppressor of cytokine signaling-1 (SOCS1) in CD8(+) T lymphocytes. Cell Signal. (2007) 19:806–16. doi: 10.1016/j.cellsig.2006.10.003
61. Strengell M, Lehtonen A, Matikainen S, Julkunen I. IL-21 enhances SOCS gene expression and inhibits LPS-induced cytokine production in human monocyte-derived dendritic cells. J Leukoc Biol. (2006) 79:1279–85. doi: 10.1189/jlb.0905503
62. Chen Y, Yu F, Jiang Y, Chen J, Wu K, Chen X, et al. Adoptive transfer of interleukin-21-stimulated human CD8+ T memory stem cells efficiently inhibits tumor growth. J Immunother. (2018) 41:274–83. doi: 10.1097/CJI.0000000000000229
63. Adams TE, Hansen JA, Starr R, Nicola NA, Hilton DJ, Billestrup N. Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J Biol Chem. (1998) 273:1285–7. doi: 10.1074/jbc.273.3.1285
64. Diehl S, Anguita J, Hoffmeyer A, Zapton T, Ihle JN, Fikrig E, et al. Inhibition of th1 differentiation by IL-6 is mediated by SOCS1. Immunity. (2000) 13:805–15. doi: 10.1016/S1074-7613(00)00078-9
65. Garg A, Rawat P, Spector SA. Interleukin 23 produced by myeloid dendritic cells contributes to T-cell dysfunction in HIV type 1 infection by inducing SOCS1 expression. J Infect Dis. (2014) 211:755–68. doi: 10.1093/infdis/jiu523
66. Schneider R, Yaneva T, Beauseigle D, El-Khoury L, Arbour N. IL-27 increases the proliferation and effector functions of human naïve CD8+ T lymphocytes and promotes their development into Tc1 cells. Eur J Immunol. (2011) 41:47–59. doi: 10.1002/eji.201040804
67. Wang H, Li Z, Yang B, Yu S, Wu C. IL-27 suppresses the production of IL-22 in human CD4+ T cells by inducing the expression of SOCS1. Immunol Letters. (2013) 152:96–103. doi: 10.1016/j.imlet.2013.05.001
68. Liu H, Rohowsky-Kochan C. Interleukin-27-mediated suppression of human Th17 cells is associated with activation of STAT1 and suppressor of cytokine signaling protein 1. J Interferon Cytokine Res. (2011) 31:459–69. doi: 10.1089/jir.2010.0115
69. Pezet A, Favre H, Kelly PA, Edery M. Inhibition and restoration of prolactin signal transduction by suppressors of cytokine signaling. J Biol Chem. (1999) 274:24497–502. doi: 10.1074/jbc.274.35.24497
70. Park ES, Kim H, Suh JM, Park SJ, Kwon O-Y, Kim YK, et al. Thyrotropin induces SOCS-1 (Suppressor of cytokine signaling-1) and SOCS-3 in FRTL-5 thyroid cells. Mol Endocrinol. (2000) 14:440–8. doi: 10.1210/mend.14.3.0433
71. Aghajanova L, Stavreus-Evers A, Lindeberg M, Landgren BM, Sparre LS, Hovatta O. Thyroid-stimulating hormone receptor and thyroid hormone receptors are involved in human endometrial physiology. Fertil Steril. (2011) 95:230–7, 7.e1–2. doi: 10.1016/j.fertnstert.2010.06.079
72. Naka T, Matsumoto T, Narazaki M, Fujimoto M, Morita Y, Ohsawa Y, et al. Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proc Natl Acad Sci U S A. (1998) 95:15577–82. doi: 10.1073/pnas.95.26.15577
73. Bullen DV, Darwiche R, Metcalf D, Handman E, Alexander WS. Neutralization of interferon-gamma in neonatal SOCS1-/- mice prevents fatty degeneration of the liver but not subsequent fatal inflammatory disease. Immunology. (2001) 104:92–8. doi: 10.1046/j.1365-2567.2001.01294.x
74. Metcalf D, Mifsud S, Di Rago L, Nicola NA, Hilton DJ, Alexander WS. Polycystic kidneys and chronic inflammatory lesions are the delayed consequences of loss of the suppressor of cytokine signaling-1 (SOCS-1). Proc Natl Acad Sci U S A. (2002) 99:943–8. doi: 10.1073/pnas.022628499
75. Naka T, Tsutsui H, Fujimoto M, Kawazoe Y, Kohzaki H, Morita Y, et al. SOCS-1/SSI-1-deficient NKT cells participate in severe hepatitis through dysregulated cross-talk inhibition of IFN-gamma and IL-4 signaling in vivo. Immunity. (2001) 14:535–45. doi: 10.1016/S1074-7613(01)00132-7
76. Chong MMW, Metcalf D, Jamieson E, Alexander WS, Kay TWH. Suppressor of cytokine signaling-1 in T cells and macrophages is critical for preventing lethal inflammation. Blood. (2005) 106:1668–75. doi: 10.1182/blood-2004-08-3049
77. Collins EL, Jager LD, Dabelic R, Benitez P, Holdstein K, Lau K, et al. Inhibition of SOCS1–/– lethal autoinflammatory disease correlated to enhanced peripheral foxp3+ Regulatory T cell homeostasis. J Immunol. (2011) 187:2666–76. doi: 10.4049/jimmunol.1003819
78. Fujimoto M, Naka T, Nakagawa R, Kawazoe Y, Morita Y, Tateishi A, et al. Defective thymocyte development and perturbed homeostasis of T cells in STAT-induced STAT inhibitor-1/suppressors of cytokine signaling-1 transgenic mice. J Immunol. (2000) 165:1799–806. doi: 10.4049/jimmunol.165.4.1799
79. Hanada T, Yoshida H, Kato S, Tanaka K, Masutani K, Tsukada J, et al. Suppressor of cytokine signaling-1 is essential for suppressing dendritic cell activation and systemic autoimmunity. Immunity. (2003) 19:437–50. doi: 10.1016/S1074-7613(03)00240-1
80. Jackson SH, Yu CR, Mahdi RM, Ebong S, Egwuagu CE. Dendritic cell maturation requires STAT1 and is under feedback regulation by suppressors of cytokine signaling. J Immunol. (2004) 172:2307–15. doi: 10.4049/jimmunol.172.4.2307
81. Zhang JG, Metcalf D, Rakar S, Asimakis M, Greenhalgh CJ, Willson TA, et al. The SOCS box of suppressor of cytokine signaling-1 is important for inhibition of cytokine action in vivo. Proc Natl Acad Sci U.S.A. (2001) 98:13261–5. doi: 10.1073/pnas.231486498
82. Babon JJ, Varghese LN, Nicola NA. Inhibition of IL-6 family cytokines by SOCS3. Semin Immunol. (2014) 26:13–9. doi: 10.1016/j.smim.2013.12.004
83. Hörtner M, Nielsch U, Mayr LM, Heinrich PC, Haan S. A new high affinity binding site for suppressor of cytokine signaling-3 on the erythropoietin receptor. Eur J Biochem. (2002) 269:2516–26. doi: 10.1046/j.1432-1033.2002.02916.x
84. Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, et al. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem. (2000) 275:40649–57. doi: 10.1074/jbc.M007577200
85. Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I, Yoshimura A. CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J Biol Chem. (2000) 275:29338–47. doi: 10.1074/jbc.M003456200
86. Schmitz J, Weissenbach M, Haan S, Heinrich PC, Schaper F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J Biol Chem. (2000) 275:12848–56. doi: 10.1074/jbc.275.17.12848
87. Lehmann U, Schmitz J, Weissenbach M, Sobota RM, Hortner M, Friederichs K, et al. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J Biol Chem. (2003) 278:661–71. doi: 10.1074/jbc.M210552200
88. Hu J, Huang S, Liu X, Zhang Y, Wei S, Hu X. miR-155: an important role in inflammation response. J Immunol Res. (2022) 2022:7437281. doi: 10.1155/2022/7437281
89. Mahesh G, Biswas R. MicroRNA-155: A master regulator of inflammation. J Interferon Cytokine Res. (2019) 39:321–30. doi: 10.1089/jir.2018.0155
90. Lu LF, Gasteiger G, Yu IS, Chaudhry A, Hsin JP, Lu Y, et al. A single miRNA-mRNA interaction affects the immune response in a context- and cell-type-specific manner. Immunity. (2015) 43:52–64. doi: 10.1016/j.immuni.2015.04.022
91. Oke V, Gunnarsson I, Dorschner J, Eketjäll S, Zickert A, Niewold TB, et al. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Ther. (2019) 21:107. doi: 10.1186/s13075-019-1878-y
92. Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol. (2014) 192:5459–68. doi: 10.4049/jimmunol.1002795
93. Ueno H. The IL-12-STAT4 axis in the pathogenesis of human systemic lupus erythematosus. Eur J Immunol. (2020) 50:10–6. doi: 10.1002/eji.201948134
94. Hagberg N, Joelsson M, Leonard D, Reid S, Eloranta ML, Mo J, et al. The STAT4 SLE risk allele rs7574865[T] is associated with increased IL-12-induced IFN-γ production in T cells from patients with SLE. Ann Rheum Dis. (2018) 77:1070–7. doi: 10.1136/annrheumdis-2017-212794
95. Manoury-Schwartz B, Chiocchia G, Bessis N, Abehsira-Amar O, Batteux F, Muller S, et al. High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J Immunol. (1997) 158:5501–6. doi: 10.4049/jimmunol.158.11.5501
96. Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. (2003) 198:1951–7. doi: 10.1084/jem.20030896
97. Schulze-Koops H, Kalden JR. The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best Pract Res Clin Rheumatol. (2001) 15:677–91. doi: 10.1053/berh.2001.0187
98. Körholz J, Gabrielyan A, Sowerby JM, Boschann F, Chen LS, Paul D, et al. One gene, many facets: multiple immune pathway dysregulation in SOCS1 haploinsufficiency. Front Immunol. (2021) 12:680334. doi: 10.3389/fimmu.2021.680334
99. Gruber C, Lee A, Buta S, Khattri S, Gottlieb AB, Frost JM, et al. IL4Rα and IL17A blockade rescue autoinflammation in SOCS1 haploinsufficiency. J Clin Immunol. (2023) 44:36. doi: 10.1007/s10875-023-01635-z
100. Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun. (2020) 11:5341. doi: 10.1038/s41467-020-18925-4
101. Michniacki TF, Walkovich K, DeMeyer L, Saad N, Hannibal M, Basiaga ML, et al. SOCS1 haploinsufficiency presenting as severe enthesitis, bone marrow hypocellularity, and refractory thrombocytopenia in a pediatric patient with subsequent response to JAK inhibition. J Clin Immunol. (2022) 42:1766–77. doi: 10.1007/s10875-022-01346-x
102. Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol. (2020) 146:1194–200.e1. doi: 10.1016/j.jaci.2020.07.033
103. Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. (2020) 583:90–5. doi: 10.1038/s41586-020-2265-1
104. Du Y, Brodeur KE, Hsu E, Chen L, Chen Q, Liu M, et al. In cis "benign" SOCS1 variants linked to enhanced interferon signaling and autoimmunity. J Autoimmun. (2023) 140:103119. doi: 10.1016/j.jaut.2023.103119
105. Fujimoto M, Tsutsui H, Xinshou O, Tokumoto M, Watanabe D, Shima Y, et al. Inadequate induction of suppressor of cytokine signaling-1 causes systemic autoimmune diseases. Int Immunol. (2004) 16:303–14. doi: 10.1093/intimm/dxh030
106. Ohl K, Tenbrock K. Inflammatory cytokines in systemic lupus erythematosus. J BioMed Biotechnol. (2011) 2011:432595. doi: 10.1155/2011/432595
107. Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. (2013) 13:9–22. doi: 10.1038/nri3341
108. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
109. Chen E, Staudt LM, Green AR. Janus kinase deregulation in leukemia and lymphoma. Immunity. (2012) 36:529–41. doi: 10.1016/j.immuni.2012.03.017
110. Beaurivage C, Champagne A, Tobelaim WS, Pomerleau V, Menendez A, Saucier C. SOCS1 in cancer: An oncogene and a tumor suppressor. Cytokine. (2016) 82:87–94. doi: 10.1016/j.cyto.2016.01.005
111. Sobah ML, Liongue C, Ward AC. SOCS proteins in immunity, inflammatory diseases, and immune-related cancer. Front Med (Lausanne). (2021) 8:727987. doi: 10.3389/fmed.2021.727987
112. Sharma J, Larkin J 3rd. Therapeutic implication of SOCS1 modulation in the treatment of autoimmunity and cancer. Front Pharmacol. (2019) 10:324. doi: 10.3389/fphar.2019.00324
113. Yoshikawa H, Matsubara K, Qian G-S, Jackson P, Groopman JD, Manning JE, et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet. (2001) 28:29–35. doi: 10.1038/ng0501-29
114. Fukushima N, Sato N, Sahin F, Su GH, Hruban RH, Goggins M. Aberrant methylation of suppressor of cytokine signalling-1 (SOCS-1) gene in pancreatic ductal neoplasms. Br J Cancer. (2003) 89:338–43. doi: 10.1038/sj.bjc.6601039
115. Sugase T, Takahashi T, Serada S, Nakatsuka R, Fujimoto M, Ohkawara T, et al. Suppressor of cytokine signaling-1 gene therapy induces potent antitumor effect in patient-derived esophageal squamous cell carcinoma xenograft mice. Int J Cancer. (2017) 140:2608–21. doi: 10.1002/ijc.30666
116. Souma Y, Nishida T, Serada S, Iwahori K, Takahashi T, Fujimoto M, et al. Antiproliferative effect of SOCS-1 through the suppression of STAT3 and p38 MAPK activation in gastric cancer cells. Int J Cancer. (2012) 131:1287–96. doi: 10.1002/ijc.27350
117. Nagai H, Kim YS, Konishi N, Baba M, Kubota T, Yoshimura A, et al. Combined hypermethylation and chromosome loss associated with inactivation of SSI-1/SOCS-1/JAB gene in human hepatocellular carcinomas. Cancer Lett. (2002) 186:59–65. doi: 10.1016/S0304-3835(02)00244-6
118. Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci U S A. (2008) 105:12885–90. doi: 10.1073/pnas.0806202105
119. Mignacca L, Saint-Germain E, Benoit A, Bourdeau V, Moro A, Ferbeyre G. Sponges against miR-19 and miR-155 reactivate the p53-Socs1 axis in hematopoietic cancers. Cytokine. (2016) 82:80–6. doi: 10.1016/j.cyto.2016.01.015
120. Jiang S, Zhang H-W, Lu M-H, He X-H, Li Y, Gu H, et al. MicroRNA-155 functions as an oncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. (2010) 70:3119–27. doi: 10.1158/0008-5472.CAN-09-4250
121. Zhang W, Ji W, Zhao X. MiR-155 promotes anaplastic thyroid cancer progression by directly targeting SOCS1. BMC Cancer. (2019) 19:1093. doi: 10.1186/s12885-019-6319-4
122. Huang C, Li H, Wu W, Jiang T, Qiu Z. Regulation of miR-155 affects pancreatic cancer cell invasiveness and migration by modulating the STAT3 signaling pathway through SOCS1. Oncol Rep. (2013) 30:1223–30. doi: 10.3892/or.2013.2576
123. Schif B, Lennerz JK, Kohler CW, Kreuz M, Bentink S, Melzner I, et al. SOCS1 mutation subtypes predict divergent outcomes in diffuse large B-cell lymphoma (DLBCL) patients. Oncotarget. (2012) 4:35–47. doi: 10.18632/oncotarget.774
124. Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. (2012) 109:3879–84. doi: 10.1073/pnas.1121343109
125. Mottok A, Renné C, Willenbrock K, Hansmann M-L, Bräuninger A. Somatic hypermutation of SOCS1 in lymphocyte-predominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood. (2007) 110:3387–90. doi: 10.112/blood-2007-03-082511
126. Lu X, Nechushtan H, Ding F, Rosado MF, Singal R, Alizadeh AA, et al. Distinct IL-4-induced gene expression, proliferation, and intracellular signaling in germinal center B-cell-like and activated B-cell-like diffuse large-cell lymphomas. Blood. (2005) 105:2924–32. doi: 10.1182/blood-2004-10-3820
127. Harada M, Nakashima K, Hirota T, Shimizu M, Doi S, Fujita K, et al. Functional polymorphism in the suppressor of cytokine signaling 1 gene associated with adult asthma. Am J Respir Cell Mol Biol. (2007) 36:491–6. doi: 10.1165/rcmb.2006-0090OC
128. Ayyildiz T, Dolar E, Oral B, Erturk B, Haktanir AE, Adim SB, et al. SOCS-1 1478 CA/del gene polymorphism affects survival in colorectal carcinoma. Niger J Clin Pract. (2022) 25:239–47. doi: 10.4103/njcp.njcp_1309_21
129. Hartavi M, Olmez OF, Oral B, Cubukcu E, Nak SG. The SOCS-1 -1478CA/del functional polymorphism (rs33989964) is associated with gastric cancer but is unrelated to overall survival. Mol Biol Rep. (2023) 50:3489–92. doi: 10.1007/s11033-023-08296-6
130. Hartavi M, Kurt E, Oral B, Olmez OF, Cubukcu E, Deligonul A, et al. The SOCS-1 -1478CA/del polymorphism is not associated with colorectal cancer or age at onset in Turkish subjects. Asian Pac J Cancer Prev. (2013) 14:7583–6. doi: 10.7314/APJCP.2013.14.12.7583
131. Paeiz H, Salehi Z, Mashayekhi F, Saeidi Saedi H, Mirzanejad L. The importance of SOCS1 - 1478 CA/del polymorphism and expression in breast cancer: a case-control study in the north of Iran. Breast Cancer Res Treat. (2023) 202:389–95. doi: 10.1007/s10549-023-07070-3
132. Gui Y, Yeganeh M, Donates YC, Tobelaim WS, Chababi W, Mayhue M, et al. Regulation of MET receptor tyrosine kinase signaling by suppressor of cytokine signaling 1 in hepatocellular carcinoma. Oncogene. (2015) 34:5718–28. doi: 10.1038/onc.2015.20
133. Gui Y, Khan MGM, Bobbala D, Dubois C, Ramanathan S, Saucier C, et al. Attenuation of MET-mediated migration and invasion in hepatocellular carcinoma cells by SOCS1. World J Gastroenterol. (2017) 23:6639–49. doi: 10.3748/wjg.v23.i36.6639
134. Yeganeh M, Gui Y, Kandhi R, Bobbala D, Tobelaim WS, Saucier C, et al. Suppressor of cytokine signaling 1-dependent regulation of the expression and oncogenic functions of p21CIP1/WAF1 in the liver. Oncogene. (2016) 35:4200–11. doi: 10.1038/onc.2015.485
135. Khan MGM, Boufaied N, Yeganeh M, Kandhi R, Petkiewicz S, Sharma A, et al. SOCS1 deficiency promotes hepatocellular carcinoma via SOCS3-dependent CDKN1A induction and NRF2 activation. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15030905
136. Calabrese V, Mallette FA, Deschênes-Simard X, Ramanathan S, Gagnon J, Moores A, et al. SOCS1 links cytokine signaling to p53 and senescence. Mol Cell. (2009) 36:754–67. doi: 10.1016/j.molcel.2009.09.044
137. Saint-Germain E, Mignacca L, Huot G, Acevedo M, Moineau-Vallée K, Calabrese V, et al. Phosphorylation of SOCS1 inhibits the SOCS1-p53 tumor suppressor axis. Cancer Res. (2019) 79:3306–19. doi: 10.1158/0008-5472.CAN-18-1503
138. Strebovsky J, Walker P, Lang R, Dalpke AH. Suppressor of cytokine signaling 1 (SOCS1) limits NFkappaB signaling by decreasing p65 stability within the cell nucleus. FASEB J. (2011) 25:863–74. doi: 10.1096/fj.10-170597
139. Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. (2003) 12:1413–26. doi: 10.1016/S1097-2765(03)00490-8
140. Ilangumaran S, Bobbala D, Ramanathan S. SOCS1: regulator of T cells in autoimmunity and cancer. Curr Top Microbiol Immunol. (2017) 410:159–89. doi: 10.1007/82_2017_63
141. Lu C, Klement JD, Ibrahim ML, Xiao W, Redd PS, Nayak-Kapoor A, et al. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J Immunother Cancer. (2019) 7:157. doi: 10.1186/s40425-019-0635-8
142. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. (2014) 20:1301–9. doi: 10.1038/nm.3708
143. Yu R, Zhu B, Chen D. Type I interferon-mediated tumor immunity and its role in immunotherapy. Cell Mol Life Sci. (2022) 79:191. doi: 10.1007/s00018-022-04219-z
144. Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev. (2018) 63:40–7. doi: 10.1016/j.ctrv.2017.11.007
145. Bhat P, Leggatt G, Waterhouse N, Frazer IH. Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. (2017) 8:e2836–e. doi: 10.1038/cddis.2017.67
146. Maraskovsky E, Chen WF, Shortman K. IL-2 and IFN-gamma are two necessary lymphokines in the development of cytolytic T cells. J Immunol. (1989) 143:1210–4. doi: 10.4049/jimmunol.143.4.1210
147. Curtsinger JM, Agarwal P, Lins DC, Mescher MF. Autocrine IFN-γ promotes naive CD8 T cell differentiation and synergizes with IFN-α to stimulate strong function. J Immunol. (2012) 189:659–68. doi: 10.4049/jimmunol.1102727
148. Xu X, Fu XY, Plate J, Chong AS. IFN-gamma induces cell growth inhibition by Fas-mediated apoptosis: requirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. (1998) 58:2832–7.
149. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
150. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369
151. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. (2016) 315:1600–9. doi: 10.1001/jama.2016.4059
152. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. (2017) 547:413–8. doi: 10.1038/nature23270
153. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
154. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discovery. (2017) 7:188–201. doi: 10.1158/2159-8290.CD-16-1223
155. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. (2016) 167:397–404.e9. doi: 10.1016/j.cell.2016.08.069
156. Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired IFNγ resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun. (2017) 8:15440. doi: 10.1038/ncomms15440
157. Lawson KA, Sousa CM, Zhang X, Kim E, Akthar R, Caumanns JJ, et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature. (2020) 586:120–6. doi: 10.1038/s41586-020-2746-2
158. Bullock BL, Kimball AK, Poczobutt JM, Neuwelt AJ, Li HY, Johnson AM, et al. Tumor-intrinsic response to IFNγ shapes the tumor microenvironment and anti-PD-1 response in NSCLC. Life Sci Alliance. (2019) 2. doi: 10.1101/531236
159. Dhainaut M, Rose SA, Akturk G, Wroblewska A, Nielsen SR, Park ES, et al. Spatial CRISPR genomics identifies regulators of the tumor microenvironment. Cell. (2022) 185:1223–39.e20. doi: 10.1016/j.cell.2022.02.015
160. Song T-Y, Long M, Zhao H-X, Zou M-W, Fan H-J, Liu Y, et al. Tumor evolution selectively inactivates the core microRNA machinery for immune evasion. Nat Commun. (2021) 12:7003. doi: 10.1038/s41467-021-27331-3
161. Wang J, Wang Q, Guan Y, Sun Y, Wang X, Lively K, et al. Breast cancer cell-derived microRNA-155 suppresses tumor progression via enhancing immune cell recruitment and antitumor function. J Clin Invest. (2022) 132. doi: 10.1172/JCI157248
162. House IG, Derrick EB, Sek K, Chen AXY, Li J, Lai J, et al. CRISPR-Cas9 screening identifies an IRF1-SOCS1-mediated negative feedback loop that limits CXCL9 expression and antitumor immunity. Cell Rep. (2023) 42:113014. doi: 10.1016/j.celrep.2023.113014
163. Ramanathan S, Gagnon J, Leblanc C, Rottapel R, Ilangumaran S. Suppressor of cytokine signaling 1 stringently regulates distinct functions of IL-7 and IL-15 in vivo during T lymphocyte development and homeostasis. J Immunol. (2006) 176:4029–41. doi: 10.4049/jimmunol.176.7.4029
164. Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell. (2018) 175:1958–71.e15. doi: 10.1016/j.cell.2018.10.024
165. Sutra Del Galy A, Menegatti S, Fuentealba J, Lucibello F, Perrin L, Helft J, et al. In vivo genome-wide CRISPR screens identify SOCS1 as intrinsic checkpoint of CD4(+) T(H)1 cell response. Sci Immunol. (2021) 6:eabe8219. doi: 10.1101/2021.04.12.439455
166. Schlabach MR, Lin S, Collester ZR, Wrocklage C, Shenker S, Calnan C, et al. Rational design of a SOCS1-edited tumor-infiltrating lymphocyte therapy using CRISPR/Cas9 screens. J Clin Invest. (2023) 133. doi: 10.1101/2023.09.05.555798
167. Shen L, Evel-Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. (2004) 22:1546–53. doi: 10.1038/nbt1035
168. Hanada T, Tanaka K, Matsumura Y, Yamauchi M, Nishinakamura H, Aburatani H, et al. Induction of hyper th1 cell-type immune responses by dendritic cells lacking the suppressor of cytokine signaling-1 gene1. J Immunol. (2005) 174:4325–32. doi: 10.4049/jimmunol.174.7.4325
169. He M, Chen X, Luo M, Ouyang L, Xie L, Huang Z, et al. Suppressor of cytokine signaling 1 inhibits the maturation of dendritic cells involving the nuclear factor kappa B signaling pathway in the glioma microenvironment. Clin Exp Immunol. (2020) 202:47–59. doi: 10.1111/cei.13476
170. Zheng Y, Hu B, Xie S, Chen X, Hu Y, Chen W, et al. Dendritic cells infected by Ad-sh-SOCS1 enhance cytokine-induced killer (CIK) cell immunotherapeutic efficacy in cervical cancer models. Cytotherapy. (2017) 19:617–28. doi: 10.1016/j.jcyt.2017.01.008
171. Hashimoto M, Ayada T, Kinjyo I, Hiwatashi K, Yoshida H, Okada Y, et al. Silencing of SOCS1 in macrophages suppresses tumor development by enhancing antitumor inflammation. Cancer Sci. (2009) 100:730–6. doi: 10.1111/j.1349-7006.2009.01098.x
172. Sukka-Ganesh B, Larkin 3J. Therapeutic potential for targeting the suppressor of cytokine signalling-1 pathway for the treatment of SLE. Scand J Immunol. (2016) 84:299–309. doi: 10.1111/sji.12475
173. He C, Yu CR, Sun L, Mahdi RM, Larkin J 3rd, Egwuagu CE. Topical administration of a suppressor of cytokine signaling-1 (SOCS1) mimetic peptide inhibits ocular inflammation and mitigates ocular pathology during mouse uveitis. J Autoimmun. (2015) 62:31–8. doi: 10.1016/j.jaut.2015.05.011
174. Qiu LJ, Xu K, Liang Y, Cen H, Zhang M, Wen PF, et al. Decreased SOCS1 mRNA expression levels in peripheral blood mononuclear cells from patients with systemic lupus erythematosus in a Chinese population. Clin Exp Med. (2015) 15:261–7. doi: 10.1007/s10238-014-0309-2
175. Li J, Zhao S, Yi M, Hu X, Li J, Xie H, et al. Activation of JAK-STAT1 signal transduction pathway in lesional skin and monocytes from patients with systemic lupus erythematosus. Zhong Nan Da Xue Xue Bao Yi Xue Ban. (2011) 36:109–15. doi: 10.3969/j.issn.1672-7347.2011.02.003
176. Ramírez-Vélez G, Medina F, Ramírez-Montaño L, Zarazúa-Lozada A, Hernández R, Llorente L, et al. Constitutive phosphorylation of interferon receptor A-associated signaling proteins in systemic lupus erythematosus. PloS One. (2012) 7:e41414. doi: 10.1371/journal.pone.0041414
177. Shawky AM, Almalki FA, Abdalla AN, Abdelazeem AH, Gouda AM. A comprehensive overview of globally approved JAK inhibitors. Pharmaceutics. (2022) 14. doi: 10.3390/pharmaceutics14051001
178. Mujtaba MG, Flowers LO, Patel CB, Patel RA, Haider MI, Johnson HM. Treatment of mice with the suppressor of cytokine signaling-1 mimetic peptide, tyrosine kinase inhibitor peptide, prevents development of the acute form of experimental allergic encephalomyelitis and induces stable remission in the chronic relapsing/remitting form. J Immunol. (2005) 175:5077–86. doi: 10.4049/jimmunol.175.8.5077
179. Sharma J, Collins TD, Roach T, Mishra S, Lam BK, Mohamed ZS, et al. Suppressor of cytokine signaling-1 mimetic peptides attenuate lymphocyte activation in the MRL/lpr mouse autoimmune model. Sci Rep. (2021) 11:6354. doi: 10.1038/s41598-021-86017-4
180. DiGiandomenico A, Wylezinski LS, Hawiger J. Intracellular delivery of a cell-penetrating SOCS1 that targets IFN-gamma signaling. Sci Signal. (2009) 2:ra37. doi: 10.1126/scisignal.1162191
181. Paoletti A, Ly B, Cailleau C, Gao F, de Ponfilly-Sotier MP, Pascaud J, et al. Liposomal antagomiR-155–5p restores anti-inflammatory macrophages and improves arthritis in preclinical models of rheumatoid arthritis. Arthritis Rheumatol. (2024) 76:18–31. doi: 10.1002/art.42665
182. Yu Z, Wu M, Huang Y, Wang Y, Chen Y, Long Q, et al. Single-component lipid nanoparticles for engineering SOCS1 gene-silenced dendritic cells to boost tumor immunotherapy. Biomater Sci. (2022) 11:263–77. doi: 10.1039/D2BM01549H
183. Met Ö, Jensen KM, Chamberlain CA, Donia M, Svane IM. Principles of adoptive T cell therapy in cancer. Semin Immunopathol. (2019) 41:49–58. doi: 10.1007/s00281-018-0703-z
184. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
185. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
186. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
187. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
188. Mardiana S, Solomon BJ, Darcy PK, Beavis PA. Supercharging adoptive T cell therapy to overcome solid tumor-induced immunosuppression. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aaw2293
189. Yong CSM, Dardalhon V, Devaud C, Taylor N, Darcy PK, Kershaw MH. CAR T-cell therapy of solid tumors. Immunol Cell Biol. (2017) 95:356–63. doi: 10.1038/icb.2016.128
190. Sarnaik AA, Hamid O, Khushalani NI, Lewis KD, Medina T, Kluger HM, et al. Lifileucel, a tumor-infiltrating lymphocyte therapy, in metastatic melanoma. J Clin Oncol. (2021) 39:2656–66. doi: 10.1200/JCO.21.00612
Keywords: SOCS1, cytokine, JAK-STAT, immunotherapy, autoimmunity, cancer
Citation: Bidgood GM, Keating N, Doggett K and Nicholson SE (2024) SOCS1 is a critical checkpoint in immune homeostasis, inflammation and tumor immunity. Front. Immunol. 15:1419951. doi: 10.3389/fimmu.2024.1419951
Received: 19 April 2024; Accepted: 28 May 2024;
Published: 14 June 2024.
Edited by:
Raymond P. Donnelly, United States Food and Drug Administration, United StatesReviewed by:
Akihiko Yoshimura, Keio University, JapanCopyright © 2024 Bidgood, Keating, Doggett and Nicholson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra E. Nicholson, c25pY2hvbHNvbkB3ZWhpLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.