 Shruthi Kannan
Shruthi Kannan Rolando A. Vedia
Rolando A. Vedia Jeffrey J. Molldrem
Jeffrey J. Molldrem- 1Department of Hematopoietic Biology and Malignancy, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Evolution of Cancer, Leukemia, and Immunity Post Stem cEll transplant (ECLIPSE), Therapeutics Discovery Division, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 3Department of Stem Cell Transplantation and Cellular Therapy, Division of Cancer Medicine, UT MD Anderson Cancer Center, Houston, TX, United States
This mini review summarizes the immunobiology of myelodysplastic syndromes, specifically focusing on the interactions between immune cells, cytokines, and dysplastic cells within the tumor microenvironment in the bone marrow. We elucidate in detail how immune dysregulation and evasion influence the initiation and progression of myelodysplastic syndromes, as well as resistance to therapy and progression to AML. In addition, we highlight a range of therapeutic strategies, including the most recent breakthroughs and experimental therapies for treating MDS. Finally, we address the existing knowledge gaps in the understanding of the immunobiology of MDS and propose future research directions, promising advancements toward enhancing clinical outcomes and survival for patients with MDS.
Introduction
Myelodysplastic syndromes (MDS) are a complex and heterogeneous group of hematologic malignancies, characterized by dysfunctional hematopoiesis leading to cytopenias, reduced survival, and an increased risk of transformation to acute myeloid leukemias (AML) (1, 2). The annual age-adjusted incidence for MDS is 4 per 100,000 Americans, and increases substantially with increasing age, with median onset of 70 years (3). Additional risk factors for MDS include previous exposure to environmental toxins and disorders like autoimmune disorders or congenital predisposition syndromes like Fanconi anemia or germline variants in hematopoietic stem cells (HSCs) (1, 2). Depending on disease severity, MDS patients can be grouped into lower-risk and higher-risk groups. MDS patients can be categorized into lower-risk and higher-risk groups, depending on disease severity (1). Lower-risk MDS patients are much less likely to develop AML and have fewer mutations, lower myeloblast counts, with mutations in splicing factor genes such as SF3B1, that tends to improve survival. In contrast, higher-risk MDS patients carry more mutations, including TP53 mutations which are associated with worse outcomes, more myeloblasts, and a higher risk of progression to AML (1, 2). The International Prognostic Scoring System (IPSS) and IPSS-R (Revised) categorize patients based on their risk level, percentage of myeloblasts, genetic abnormalities, and cytopenias (1). The Molecular International Prognostic Scoring System for Myelodysplastic Neoplasms (IPSS-M) was recently created to evaluate the prognostic and survival impacts of somatic mutations associated with development of MDS (4).
Despite three approved therapies and allogeneic stem cell transplantation (alloSCT), currently the only potentially curative strategy, the overall 5-year survival rate for MDS patients remains low at 31%. Moreover, not all patients are eligible for alloSCT, which is associated with increased morbidity and graft versus host disease (3). Hypomethylating agents (HMAs), particularly 5-azacytidine and 5-aza- 2’-deoxycytidine (decitabine), have been the standard of care for MDS patients, since their approval in 2004 and 2006, respectively. While some patients with MDS have shown clinical improvement with these agents, many do not respond positively to HMAs or experience a relapse after an initial positive response (5).
MDS can be caused by multiple molecular as well as non-molecular pathophysiologies. Mutations tend to accumulate in the HSCs of the bone marrow, which lead to uncontrolled proliferation, evasion of cellular apoptosis and thereby result in dysfunctional hematopoiesis and MDS (6). Patients usually present with a median of three somatic mutations, with 90% of patients exhibiting at least one. While several of these are driver mutations that determine prognosis and treatment choices, others have neutral or unknown prognoses (7). Interestingly, mutations in genes such as SF3B1 are associated with better prognosis, while others like TP53, DNMT3A and ASXL1 are associated with worse outcomes.
TP53 mutations are of particular concern as they represent one of the highest-risk subsets of MDS patients with significantly reduced survival, complex karyotypes, heterogeneity in allelic states, drug resistance, and clinical outcomes (8, 9). Clonal dynamic modeling has shown that the persistence of these TP53-aberrant clones is the primary driver of malignant hematopoiesis and constitutes measurable residual disease (MRD), leading to relapse and treatment resistance (9).
An overview of the most mutated genes in MDS, their function, and immune connection is described in Table 1.
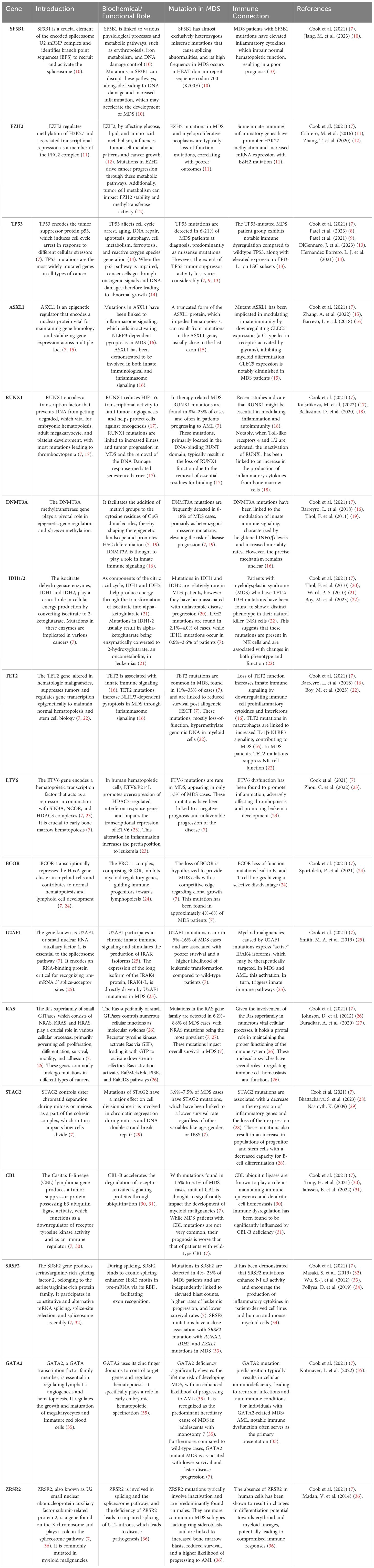
Table 1. presents a compilation of frequently mutated genes in MDS, along with their functions, the prevalence of mutations in MDS, their connection to the immune system, and corresponding references.
High-resolution genome scanning and high-throughput sequencing have led to discovery of mutations in genes involved in methylation, transcription, cell signaling, histone modification, RNA splicing, and other pathways that may contribute to MDS progression, demonstrating that a better understanding of MDS genetics can target abnormalities and improve patient outcomes (37). Some of the non-molecular mechanisms which are implicated in the progression of MDS include the regulation of the immune system, the diversity of T-cells and their receptors, factors in the bone marrow microenvironment, abnormalities in methylation and other epigenetic markers, cell signaling molecules, and the length of telomeres (37).
Understanding the immune system’s key role in the pathogenesis of MDS is crucial, as it has a pivotal role in the development progression of the disease, and the risk of transformation to AML. The dysregulation and evasion of the immune system directly leads to impaired hematopoiesis and bone marrow failure, which is characteristic of MDS, shaping the microenvironment of the disease and influencing clinical responses such as resistance to HMAs. Studying the complex interaction of immune cells, cytokines and MDS stem cells, alongside elucidating disease mechanisms, also offers novel therapeutic targets for improving patient responses. MDS prevalence is expected to keep rising due to an aging population and the increasing number of people surviving chemotherapy or radiation treatment for cancer (4). Considering recent genomic, immunologic, and sequencing advances and the rising number of MDS cases, a comprehensive mechanistic approach is needed to understand MDS immune dysfunction. This mini review paper examines and condenses the immune components of MDS, encompassing pathophysiological mechanisms, treatment approaches, recent breakthroughs, and future research avenues, aiming to enhance understanding and guide future research towards improving patient outcomes and refining therapeutic strategies.
Evidence for immune dysfunction in MDS
In recent years, increasing evidence has underscored the intricate interplay between immunological dysfunction and the pathogenesis of MDS, revealing insights into its multifaceted etiology and identifying potential targets for therapies.
It is becoming increasingly observed that immune-related occurrences are closely associated with MDS patients, such as cytopenias and autoimmune disorders that emerge before, during, or after MDS and bone marrow failure diagnosis (38). DNA-damaging treatments for autoimmune disorders, notably systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), have shown to increase the risk of MDS and other myeloid malignancies. Analysis of the molecular genetics of these MDS patients revealed that mutations in genes like RUNX1, WT1, BCOR, and TP53 were associated with typical autoinflammatory symptoms, a higher likelihood of developing AML, lower survival rates, and a worse prognosis (39). Co-occurring mutations in TET2, IDH1, IDH2 and SRSF2 were linked to the development of systemic inflammatory and autoimmune diseases, some of which are also present in MDS patients (39).
Other rheumatologic and autoimmune disorders closely linked to MDS are VEXAS syndrome, leukocytoclastic vasculitis, pan vasculitis, autoimmune hemolytic anemia, and immune thrombocytopenia (38, 40). Patients with VEXAS syndrome, a monogenic disease caused by UBA1 gene mutation, present with systemic autoinflammatory disease associated with MDS (41). Most vasculitis cases linked to MDS are idiopathic, and several cases have autoantibodies, which are frequently observed in low-risk MDS patients without excessive blasts (42).
The precise relationship between MDS and autoimmune disorders presents a significant and unresolved conundrum, raising questions about whether they co- occur, influence each other’s pathophysiology, or arise because of treatment for one another (38). Previous studies have focused on the prevalence of rheumatologic symptoms in MDS patients rather than on the role of autoinflammation in the development of MDS in patients with rheumatologic diseases. The factors that cause MDS’s autoimmune and rheumatologic symptoms warrant further studies to better understand its immune component.A less common form of MDS, hypocellular MDS, affects 10-15% of MDS patients and is characterized by a significant reduction in bone marrow cellularity (30%) (43). Compared to normal/hypercellular MDS, these patients are generally younger, exhibit more severe cytopenias, with higher transfusion dependence, and display lower bone marrow blast percentages (44). These patients also share cytogenetic abnormalities that suggest a common underlying pathogenesis, which might differ from the more cellular forms of MDS (45). Research indicates that using immunosuppressive therapies, particularly those utilizing anti-thymocyte globulin (ATG) to eliminate T cells, can be an effective method for treating this particular patient population (46). This indicates that the immunological characteristics of hypocellular MDS may significantly influence the mechanism and duration of treatment responses, similar to autoimmune conditions (47).
Hypocellular myelodysplastic syndrome (MDS) shares pathophysiological traits with nonmalignant bone marrow failure conditions like acquired aplastic anemia (AA) and paroxysmal nocturnal hemoglobinuria (PNH), which are linked to T cell-mediated autoimmunity and are associated with an increased risk of MDS and progressing to AML (48, 49). The transition from primary AA to secondary MDS can be indicated by the presence of PNH clones in hypocellular MDS (48). It is therefore very crucial to consider various immunological characteristics and features of hypocellular MDS, as they are likely to be pivotal in shaping disease progression.
Large granular lymphocytic leukemia (LGL) is a proliferation of cytotoxic T lymphocytes (CTL) that has been shown to co-exist alongside MDS clones. Likely due to a shared role of aging in these disorders, there seems to be a unique connection between MDS and LGL (50). Patients with both MDS/LGL show common MDS characteristics like abnormal cytogenetics, dysplasia of myeloid cells in the bone marrow, ringed sideroblasts and the existence of highly clonal somatic myeloid mutations (51). The evolution of MDS may be caused by repeated DNA damage due to excessive proliferation of large granular lymphocytes, immune suppression of myelopoiesis, and a myeloid escape clone (52).
Felty’s syndrome, characterized by a triad of RA, splenomegaly and granulocytopenia symptoms, has also been shown to co-occur with MDS and has been linked to increased incidence of MDS (53). The fact that alloSCT remains the only potentially curative regimen for MDS patients, suggests that one of the major components driving responses in MDS patients is the immune component.
While immune evasion mechanisms in MDS are not fully understood, the condition is associated with widespread immune cell dysfunction, as well as abnormal expression of checkpoints, cytokines, and inflammatory signaling pathways (54). Immune evasion in MDS is complex, varied, and dynamic, as evidenced by the coexistence and reciprocal transition of immune hyperfunction and immune repression (54, 55). The upregulation of immune checkpoints (e.g., PD-1/PD-L1, CTLA-4, TIM-3, TIGIT, CD47) on T cells in MDS creates an immune-evasive tumor microenvironment, leading to reduced cytotoxicity, impaired NK cell function, and T-cell exhaustion (54, 55). Abnormal levels of growth factors, chemokines, and cytokines, such as GM-CSF, TNF-α, TGF-β, IL-6, IL-8, IL-32, and IFN-γ, have been associated with immune evasion and worse outcomes in MDS (56, 57). Myeloid-derived suppressor cells (MDSCs) help tumor cells avoid immune detection by secreting immunosuppressive cytokines such as IL-10, IL-1β, and TGF-β, especially in high-risk MDS (58).
Several immune cells such as MDSCs, Tregs, NK cells, macrophages, monocytes and neutrophils also show abnormal regulation in MDS patients, including altered numbers, impaired function and abnormal cytokine secretion (55). The impaired ability of dendritic cells to stimulate T cells also contributes to the evasion of immune surveillance and progression to AML (55). The pathophysiology of MDS also involves persistent innate immunity and inflammatory signaling pathways, such as TLR signaling (55).
Overall, the increasing body of evidence about immunological dysfunction in MDS enriches our understanding of its causes and suggests the transformative capacity of immunomodulatory therapies in revolutionizing treatment for this complex disease.
Treatments with immunomodulatory effects used to treat MDS
With mounting evidence that the immune system plays a major role in MDS, ongoing research is prioritizing immune manipulation to improve treatment outcomes. Treatments for MDS that have immunomodulatory effects can be categorized into two groups: those that specifically target T lymphocytes and other cellular components of the immune system and those that modulate cytokines and their pathways to stimulate an immune response against malignant clones and myeloblasts. Figure 1 describes an overview of treatments that have been commonly used to treat myelodysplastic syndromes that have been shown to have immunomodulatory effects.
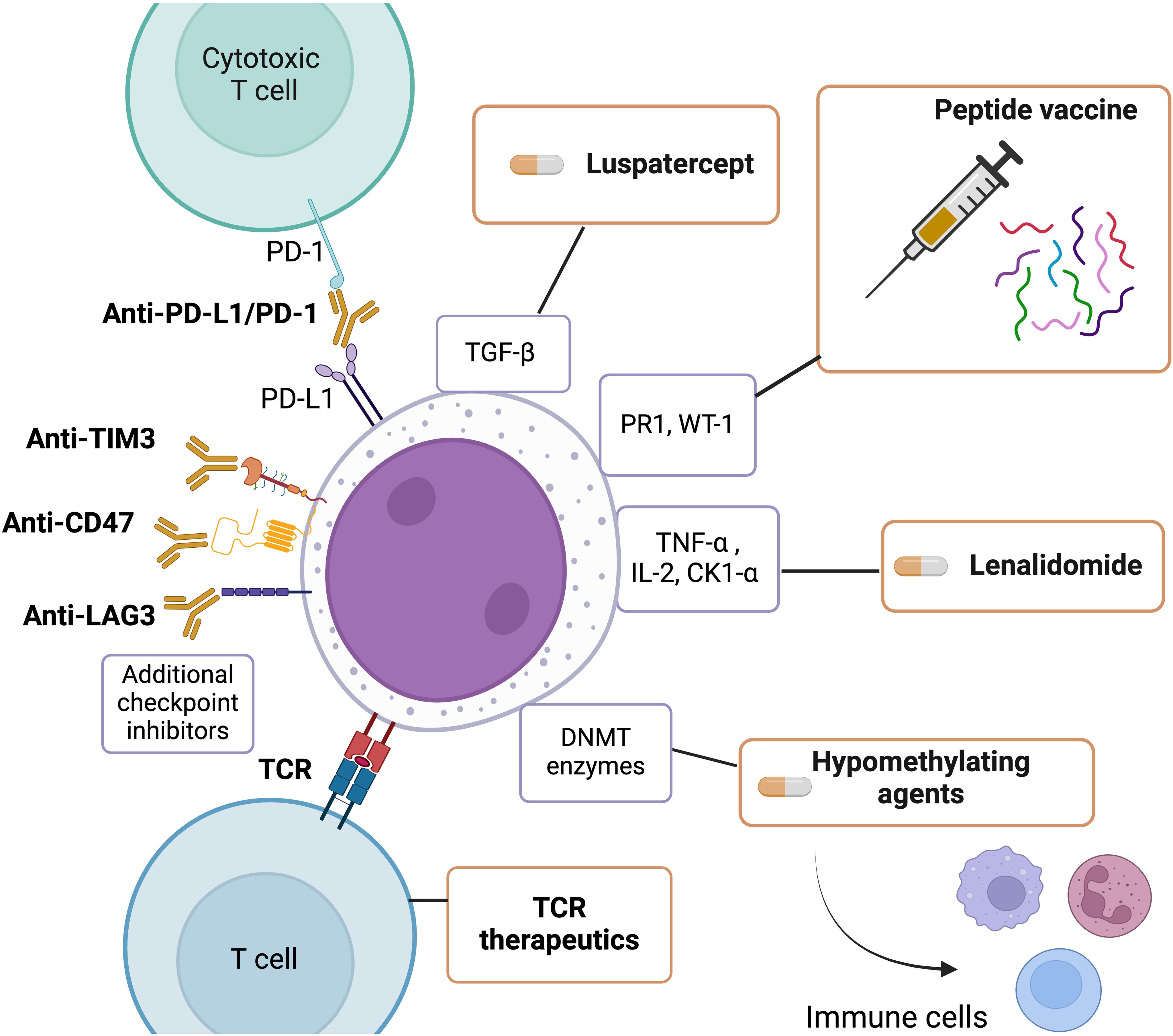
Figure 1. An overview of treatments commonly used to treat myelodysplastic syndrome with immunomodulatory effects. This Figure was created using BioRender.com.
Targeting the cellular component of the immune system
Hypomethylating agents (HMAs) have been the first line of treatment for MDS for years as they reverse integrate into nucleic acids and inhibit DNMT enzymes, ultimately leading to DNA demethylation and reprogramming of tumor cells (59). HMAs increase non-self-recognition and cytotoxic T cell activity against cancerous cells (59). Some patients may initially respond to HMAs but can quickly acquire resistance. Patients with primary resistance to HMAs do not improve after 4-6 treatment cycles, while secondary resistance patients respond but eventually relapse. HMA resistance mechanisms are being studied but much still needs to be elucidated. However, it is evident that HMAs affect the immune system cells and the bone marrow niche (59). Previous research has shown that decitabine treatment increases CD38 expression on CD8+ and CD4+ T cells and is negatively correlated with reduced IFNγ production and T cell function (60). HMAs have also been shown to induce regulatory T cells (61) and have an activating effect on NK cells (62). Furthermore, studies have shown that decitabine can stimulate dendritic cells (63), and treatment with HMA reduces the presence of myeloid-derived suppressor cells (MDSCs) (64). HMAs have also been shown to boost anti-tumor immune responses by upregulating various genes associated with immunomodulatory pathways (65). Understanding how hypomethylating agents upregulate these genes can help identify MDS-expressed self-antigens in order to develop effective immune based treatments.
Likely due to high disease burden, low mutation burden, immunosuppressive tumor microenvironment, and moderate overexpression of immune checkpoint co-stimulatory receptors on T cells and ligands on leukemic cells, immune checkpoint therapies alone are ineffective in hematologic malignancies (65). Combination approaches of immune checkpoint inhibitors (anti-PD1/PD-L1 or anti-CTLA4 antibodies) in conjunction with HMAs are being explored in MDS (66–68). HMAs increase inhibitory checkpoint molecule expression, suppressing the immune response leading to T cell exhaustion, which can be reversed by PD-1/PD-L1 blockade (65). These combination therapies may also benefit from HMAs’ ability to overcome checkpoint inhibitor therapy’s immune evasion mechanisms, such as MHC downregulation, tumor antigen expression, and co-stimulatory ligand expression (65).
Several MDS clinical trials are testing additional immune checkpoint inhibitors like anti-TIM3, anti-CD47, and anti-LAG3 (69). Anatolia, an anti-TIM3 antibody, demonstrated clinical efficacy in Phase 1B trials involving higher-risk MDS patients when combined with HMA therapies (70). Magrolimab, an anti-CD47 antibody, has also shown synergistic effects when used in combination with azacytidine and other hypomethylating agents in these patients (71). PD-1H, a protein expressed by hematopoietic cells that acts as a homolog of PD-1 or PD-L1, and IRAK4, which is elevated in MDS patients with splicing factor mutations, are also potential immunological targets (69). In addition to immune checkpoint inhibition, adoptive cellular therapies such as bi-specific T-cell engager molecules (BiTEs), CAR-T, and CAR-NK cell therapies have shown minimal safety and efficacy in MDS patients (69). A deep comprehension of the interplay between patient immune response and MDS clones is needed to understand the lack of efficacy of these agents in MDS.
Peptide vaccines, such as the PR1 and WT1 antigen-specific combination, have been studied in a clinical trial for AML/MDS (72–74). Most patients exhibited a CD8+ T-cell response specific to PR1 or WT1, however, these vaccines have shown limited or transient efficacy. Advanced MDS stages are likely to be less conducive to eliciting an immune response through peptide vaccines. Optimal conditions may involve reduced tumor load after cytoreduction when the disease is in remission or post alloSCT (72).
In its early stages, T cell receptor (TCR) therapeutics are a developing focus in MDS research, requiring further study to enhance their effectiveness and safety in clinical settings. Initial research indicates that personalized adoptive cell therapy can select, immunize, and expand T cells outside the body against MDS patient-specific tumor cell neoantigens and neopeptides for therapeutic purposes.
It has been demonstrated that a subset of MDS patients (especially those that present with autoimmunity) recover from pancytopenia after receiving immunosuppressive therapies like ATG and cyclosporine (75). These interventions are especially successful in the early stages of MDS and can restore hematopoiesis and reduce transfusion dependence (75). Studies have shown that these cytopenias are caused by T lymphocytes suppressing hematopoietic progenitors, driven by predominant clonal T cells (75, 76). Effective hematopoiesis is recovered in patients who respond well to such immunosuppressive therapies, as evidenced by the loss of these clonally dominant T lymphocytes and the re-emergence of polyclonal T cells, which normalize the T-cell repertoire (76).
Several Toll-like receptor (TLR) signaling pathway components are elevated in MDS patients, and aberrant TLR signaling has been connected to ineffective hematopoiesis, a disease hallmark, and depletion of bone marrow CD34+ cells. As a result, drugs that block increased TLR signaling have been evaluated for possible therapeutic uses (77, 78). In leukemias, TLR agonists are also being studied for their possible anti-tumor effects, which include inducing cell death and differentiation by strengthening the immune system’s defense against leukemia cells (78).
Targeting cytokines
MDS patients with chromosomal abnormality deletion 5q are treated with lenalidomide, an immunomodulator that impairs ribosomal proteins to induce cell apoptosis (79). In several clinical trials, MDS patients receiving lenalidomide achieved transfusion independence for at least 26 weeks, compared to placebo treated patients (80). Lenalidomide has been demonstrated to suppress the synthesis of pro-inflammatory cytokines, particularly TNFα, which is known to suppress hematopoiesis and act as an immune regulator (81). In this manner, lenalidomide acts as an immune regulator for hematopoiesis in MDS, alongside having direct effects on myeloid cells and lymphocytes by increasing IL2 (81). Lenalidomide has also been found to induce the ubiquitination of CK1α by the E3 ubiquitin ligase CUL4–RBX1–DDB1–CRBN (known as CRL4CRBN), which results in CK1α degradation. Since CK1α targets TP53 for degradation, lenalidomide can induce TP53 activation and growth inhibition (79). MDS del(5q) patients who develop resistance to lenalidomide tend to acquire TP53 mutations. CK1α is also a negative regulator of Wnt signaling, and lenalidomide treatment also results in increased levels of β- catenin (79). Both these mechanisms lead to further disease progression.
Abnormal TGFβ signaling in MDS, characterized by reduced (inhibitory) I-SMADs and elevated TGFβ ligands like activin and GDF11, results in the activation of the TGFβ pathway, which impairs late-stage erythropoiesis consistent with morphologic erythroid dysplasia found in most MDS cases (82). Luspatercept, which decreases SMAD2 and SMAD3 signaling during erythropoiesis by binding to the TGFβ superfamily ligands, is a recently approved drug for MDS patients. Luspatercept is often given to MDS patients with SF3B1 mutations or those with ring sideroblasts (83). A randomized, double-blind clinical trial found that luspatercept improved transfusion independence for at least 8 weeks compared to placebo treatment (84).
Other MDS therapies that have been investigated include the use of venetoclax, which targets the Bcl2 anti-apoptotic protein, in combination with 5-aza for relapsed/refractory MDS (85). Venetoclax caused a long-lasting response in the “CMP” pattern MDS, a HSC architecture consistent during HMA therapy and resistance. These studies suggest that molecular profiling may help treat MDS drug resistance (86). Due to elevated NEDD8 and NAE protein levels, in MDS patient samples, researchers have investigated using pevonedistat, a NEDDylation pathway drug, combined with 5-aza as a treatment option. This combination was well tolerated in elderly AML patients who were unfit for high-dose induction therapy, and it is now being targeted forHMA-resistant MDS patients (87–89). Several current clinical trials are investigating the effectiveness of signal transduction inhibitors like KRAS inhibitors and FLT3 inhibitors, pathways that are frequently altered in MDS (5). Several MDS patients have IDH1/2 mutations, so isocitrate dehydrogenase inhibitors are being studied alone and with HMAs (5). These drugs cannot cure MDS because IDH1/2 mutations tend to be subclonal but combining them with agents that target these mutations’ collaborative pathways may be beneficial (90). The effects on the immune component with these therapies needs to be further studied and characterized.
Future directions
Various combinations of established and innovative targeted drugs, alone or with HMAs, have shown efficacy in randomized trials for MDS patients, with a significant associated immune component. However, the research for new and effective treatments for MDS has been hampered by various obstacles (4).
One major challenge in studying the immunobiology of MDS is the lack of robust mouse models and PDX models that accurately recapitulate disease features because primary human MDS cells do not grow well in xenograft mice (4). The recent creation of mouse models like MISTRG, allows better engraftment of patient derived MDS stem and progenitor cells (69). MDS also represents a spectrum of diseases, from low risk to high-risk features, and a single GEMM model is often insufficient in capturing all the features of this complex disease. Additional better validated mouse models will be needed to provide more reliable pre-clinical data for understanding the disease and developing innovative treatments (4).
Artificial intelligence and machine learning have facilitated the analysis of molecular abnormalities, pathological features, and clinical variables in biological sciences alongside advanced technologies such as long-read sequencing, immunopeptidomics, single-cell sequencing advancements, and differential expression analysis. These advancements can provide more accurate estimations of a patient’s survival and prognosis at any stage of their disease progression, incorporating a greater level of precision medicine into MDS alongside the integration of MDS assays (1, 2).
With these developments, MDS research will certainly benefit from an understanding of the pathophysiology and mechanisms of early-stage/lower-risk patients with few symptoms and a low mutational burden. Lower-risk MDS patients usually receive treatment with an emphasis on managing their symptoms, including anemia, neutropenia, thrombocytopenia, and other related conditions. Sequencing has shown that aberrant MDS cells in the immunophenotypically-defined HSC compartment can drive secondary AML progression. Furthermore, it has been observed that most HSCs are already mutated at the onset of MDS (5). Directing therapeutic interventions towards these specific cells may offer the optimal approach to enhance the prognosis of patients with MDS.
Similarly, future research should focus on hypocellular MDS patients and those with an autoimmune response. The immune microenvironment in MDS and the presence of antigen- specific T cell clones in MDS, particularly in immunoreactive cases, will also be better understood with the aid of advanced technologies. We can comprehend the antigens that T-cells in the MDS microenvironment encounter and react to when presented on HLA molecules, potentially leading to the discovery of novel neoantigens exclusive to malignant MDS cells. T cells may also have autoimmunity to myeloid precursors and contribute to cytopenia’s, which needs to be further studied.
The bone marrow microenvironment’s MDSC population should also be studied alongside T cells. MDSCs regulate most cancers’ immune systems, so it is important to study their role in the bone marrow microenvironment and distinguish them from other myeloid elements to understand their unique role in MDS (91).
Intensive focus on MDS neoantigen research in high-risk or advanced-stage MDS patients could be particularly advantageous, as tumors with higher mutational burden are typically more responsive to T cells (92). Regarding this, a deeper comprehension of the dysregulation of antigen presentation in MDS may help us figure out how to enhance the accuracy of target and therapeutic antigen presentation.
In conclusion, the intricate and multifaceted nature of MDS demands a multipronged investigative approach that delves deeper into the underlying mechanisms driving these disorders. Mechanism-based studies in clinical trials that characterize immune effects on myelodysplastic clones or precursors need to be carried out, as mounting evidence suggests that immune dysregulation plays a pivotal role in driving pathogenesis and progression of MDS. Collaborative efforts to target the immune component hold promise for novel therapeutic strategies aimed at altering the natural history of MDS, offering renewed hope for improved outcomes and quality of life of affected individuals.
Author contributions
SK: Writing – review & editing, Writing – original draft. RV: Writing – review & editing. JM: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sekeres MA, Taylor J. Diagnosis and treatment of myelodysplastic syndromes. JAMA. (2022) 328:872. doi: 10.1001/jama.2022.14578
2. Garcia-Manero G. Myelodysplastic syndromes: 2023 update on diagnosis, risk- stratification, and Management. Am J Hematol. (2023) 98:1307–25. doi: 10.1002/ajh.26984
3. Zeidan AM, Shallis RM, Wang R, Davidoff A, Ma X. Epidemiology of myelodysplastic syndromes: Why characterizing the beast is a prerequisite to taming it. Blood Rev. (2019) 34:1–15. doi: 10.1016/j.blre.2018.09.001
4. Merz AM, Sébert M, Sonntag J, Kubasch AS, Platzbecker U, Adès L. Phase to phase: Navigating drug combinations with hypomethylating agents in higher-risk MDS trials for optimal outcomes. Cancer Treat Rev. (2024) 123:102673. doi: 10.1016/j.ctrv.2023.102673
5. Rodriguez-Sevilla JJ, Adema V, Garcia-Manero G, Colla S. Emerging treatments for myelodysplastic syndromes: Biological Rationales and Clinical Translation. Cell Rep Med. (2023) 4:100940. doi: 10.1016/j.xcrm.2023.100940
6. Woll P-S, Kjällquist U, Chowdhury O, Doolittle H, Wedge DC, Thongjuea S, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell. (2014) 25(6):794–808. doi: 10.1016/j.ccr.2014.03.036
7. Cook MR, Karp JE, Lai C. The spectrum of genetic mutations in myelodysplastic syndrome: Should we update prognostication? eJHaem. (2021) 3:301–13. doi: 10.1002/jha2.317
8. Patel SA, Cerny J, Gerber WK, Ramanathan M, Ediriwickrema A, Tanenbaum B, et al. Prognostic heterogeneity and Clonal Dynamics within distinct subgroups of myelodysplastic syndrome and acute myeloid leukemia with tp53 disruptions. eJHaem. (2023) 4:1059–70. doi: 10.1002/jha2.791
9. Patel SA, Lloyd MR, Cerny J, Shi Q, Simin K, Ediriwickrema A, et al. Clinico-genomic profiling and clonal dynamic modeling of tp53-aberrant myelodysplastic syndrome and acute myeloid leukemia. Leukemia Lymphoma. (2021) 62:3348–60. doi: 10.1080/10428194.2021.1957869
10. Jiang M, Chen M, Liu Q, Jin Z, Yang X, Zhang W. SF3B1 mutations in myelodysplastic syndromes: A potential therapeutic target for modulating the entire disease process. Front Oncol. (2023) 13:1116438. doi: 10.3389/fonc.2023.1116438
11. Cabrero M, Wei Y, Yang H, Ganan-Gomez I, Bohannan Z, Colla S, et al. Down-regulation of EZH2 expression in myelodysplastic syndromes. Leukemia Res. (2016) 44:1–7. doi: 10.1016/j.leukres.2016.02.009
12. Zhang T, Gong Y, Meng H, Li C, Xue L. Symphony of epigenetic and metabolic regulation—interaction between the histone methyltransferase EZH2 and metabolism of tumor. Clin Epigenet. (2020) 12:72. doi: 10.1186/s13148-020-00862-0
13. DiGennaro J, Sallman DA. TP53-mutated myelodysplastic syndrome and acute myeloid leukemia: Current guidelines, therapies, and future considerations. Acta Haematologica. (2023) 147:175–85. doi: 10.1159/000535628
14. Hernández Borrero LJ, El-Deiry WS. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim Biophys Acta (BBA) - Rev Cancer. (2021) 1876:188556. doi: 10.1016/j.bbcan.2021.188556
15. Zhang A, Wang S, Ren Q, Wang Y, Jiang Z. Prognostic value of ASXL1 mutations in patients with myelodysplastic syndromes and Acute myeloid leukemia: A meta-analysis. Asia-Pacific J Clin Oncol. (2022) 19:e183-e194. doi: 10.1111/ajco.13897
16. Barreyro L, Chlon TM, Starczynowski DT. Chronic immune response dysregulation in MDS pathogenesis. Blood. (2018) 132:1553–60. doi: 10.1182/blood-2018-03-784116
17. Kaisrlikova M, Vesela J, Kundrat D, Votavova H, Dostalova Merkerova M, Krejcik Z, et al. RUNX1 mutations contribute to the progression of MDS due to disruption of antitumor cellular defense: A study on patients with lower-risk MDS. Leukemia. (2022) 36:1898–906. doi: 10.1038/s41375-022-01584-3
18. Bellissimo DC, Chen C, Zhu Q, Bagga S, Lee C-T, He B, et al. Runx1 negatively regulates inflammatory cytokine production by neutrophils in response to toll-like receptor signaling. Blood Adv. (2020) 4:1145–58. doi: 10.1182/bloodadvances.2019000785
19. Thol F, Winschel C, Ludeking A, Yun H, Friesen I, Damm F, et al. Rare occurrence of DNMT3A mutations in myelodysplastic syndromes. Haematologica. (2011) 96:1870–3. doi: 10.3324/haematol.2011.045559
20. Thol F, Weissinger EM, Krauter J, Wagner K, Damm F, Wichmann M, et al. IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica. (2010) 95:1668–74. doi: 10.3324/haematol.2010.025494
21. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. (2010) 17:225–34. doi: 10.1016/j.ccr.2010.01.020
22. Boy M, Bisio V, Zhao L-P, Guidez F, Schell B, Lereclus E, et al. Myelodysplastic syndrome associated TET2 mutations affect NK cell function and genome methylation. Nat Commun. (2023) 14:588. doi: 10.1038/s41467-023-36193-w
23. Zhou C, Uluisik R, Rowley JW, David C, Jones CL, Scharer CD, et al. Germline ETV6 mutation promotes inflammation and disrupts lymphoid development of early hematopoietic progenitors. Exp Hematol. (2022) 112–113:24–34. doi: 10.1016/j.exphem.2022.06.002
24. Sportoletti P, Sorcini D, Falini B. BCOR gene alterations in hematologic diseases. Blood. (2021) 138:2455–68. doi: 10.1182/blood.2021010958
25. Smith MA, Choudhary GS, Pellagatti A, Choi K, Bolanos LC, Bhagat TD, et al. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid Malignancies. Nat Cell Biol. (2019) 21:640–50. doi: 10.1038/s41556-019-0314-5
26. Johnson DS, Chen YH. Ras family of small GTPases in immunity and inflammation. Curr Opin Pharmacol. (2012) 12:458–63. doi: 10.1016/j.coph.2012.02.003
27. Buradkar A, Bezerra E, Coltro G, Lasho TL, Finke CM, Gangat N, et al. Landscape of RAS pathway mutations in patients with myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes: A study of 461 molecularly annotated patients. Leukemia. (2020) 35:644–9. doi: 10.1038/s41375-020-0889-7
28. Ashish Bhattacharya S, Sakuma M, Walter W, Haferlach T, Malinverni R, Buschbeck M. P707: Mutations in the cohesin gene STAG2 are associated with reduced inflammatory gene expression and an increase in hematopoietic stem cell populations in myelodysplastic syndrome patients. HemaSphere. (2023) 7(Suppl):e67131fc. doi: 10.1097/01.hs9.0000969732.67131.fc
29. Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet. (2009) 43:525–58. doi: 10.1146/annurev-genet-102108-134233
30. Tong H, Li X, Zhang J, Gong L, Sun W, Calderon V, et al. Ubiquitin ligases CBL and CBL-B maintain the homeostasis and immune quiescence of dendritic cells. Front Immunol. (2021) 12:757231. doi: 10.3389/fimmu.2021.757231
31. Janssen E, Peters Z, Alosaimi MF, Smith E, Milin E, Stafstrom K, et al. Immune dysregulation caused by homozygous mutations in CBLB. J Clin Invest. (2022) 132(20):e154487. doi: 10.1172/jci154487
32. Masaki S, Ikeda S, Hata A, Shiozawa Y, Kon A, Ogawa S, et al. Myelodysplastic syndrome-associated SRSF2 mutations cause splicing changes by altering binding motif sequences. Front Genet. (2019) 10:338. doi: 10.3389/fgene.2019.00338
33. Wu S-J, Kuo Y-Y, Hou H-A, Li L-Y, Tseng M-H, Huang C-F, et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood. (2012) 120:3106–11. doi: 10.1182/blood-2012-02-412296
34. Pollyea DA, Harris C, Rabe JL, Hedin BR, De Arras L, Katz S, et al. Myelodysplastic syndrome-associated spliceosome gene mutations enhance innate immune signaling. Haematologica. (2019) 104(9):e388-e392. doi: 10.3324/haematol.2018.214155
35. Kotmayer L, Romero-Moya D, Marin-Bejar O, Kozyra E, Català A, Bigas A, et al. GATA2 deficiency and MDS/AML: Experimental strategies for disease modelling and future therapeutic prospects. Br J Haematology. (2022) 199:482–95. doi: 10.1111/bjh.18330
36. Madan V, Kanojia D, Jia L, Okamoto R, Sato-Otsubo A, Kohlmann A, et al. ZRSR2 mutations cause dysregulated RNA splicing in MDS. Blood. (2014) 124:4609–9. doi: 10.1182/blood.v124.21.4609.4609
37. Visconte V, Tiu RV, Rogers HJ. Pathogenesis of myelodysplastic syndromes: An overview of molecular and non-molecular aspects of the disease. Blood Res. (2014) 49:216. doi: 10.5045/br.2014.49.4.216
38. Barcellini W, Fattizzo B. Immune phenomena in myeloid neoplasms: an “Egg or chicken“ Question. Front Immunol. (2021) 12:751630. doi: 10.3389/fimmu.2021.751630
39. Watad A, Kacar M, Bragazzi NL, Zhou Q, Jassam M, Taylor J, et al. Somatic mutations and the risk of undifferentiated autoinflammatory disease in MDS: An under-recognized but prognostically important complication. Front Immunol. (2021) 12:610019. doi: 10.3389/fimmu.2021.610019
40. Kim Y-E, Ahn SM, Oh JS, Hong S, Lee C-K, Yoo B, et al. Incidence of and risk factors for myelodysplastic syndrome in patients with rheumatologic diseases. Rheumatology. (2023) 63(5):1305–12. doi: 10.1093/rheumatology/kead374
41. Templé M, Kosmider O. VEXAS syndrome: A novelty in MDS Landscape. Diagnostics. (2022) 12:1590. doi: 10.3390/diagnostics12071590
42. Braun T, Fenaux P. Myelodysplastic syndromes (MDS) and autoimmune disorders (AD): Cause or consequence? Best Pract Res Clin Haematology. (2013) 26:327–36. doi: 10.1016/j.beha.2013.09.003
43. Hasserjian RP, Orazi A, Brunning R, Germing U, Le Beau MM, Porwit A, et al. Myelodysplastic syndromes: overview. In: Swerdlow SHCE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, et al, editors. WHO classification of tumors of haematopoietic and lymphoid tissues. IARC, Lyon (France (2017). p. 98–106.
44. Huang T-C, Ko B-S, Tang J-L, Hsu C, Chen C-Y, Tsay W, et al. Comparison of hypoplastic myelodysplastic syndrome (MDS) with normo-/hypercellular MDS by international prognostic scoring system, Cytogenetic and genetic studies. Leukemia. (2007) 22:544–50. doi: 10.1038/sj.leu.2405076
45. Bono E, McLornan D, Travaglino E, Gandhi S, Gallì A, Khan AA, et al. Clinical, histopathological and molecular characterization of hypoplastic myelodysplastic syndrome. Leukemia. (2019) 33:2495–505. doi: 10.1038/s41375-019-0457-1
46. Komrokji RS, Mailloux AW, Chen D-T, Sekeres MA, Paquette R, Fulp WJ, et al. A phase II multicenter rabbit anti-thymocyte globulin trial in patients with myelodysplastic syndromes identifying a novel model for response prediction. Haematologica. (2014) 99:1176–83. doi: 10.3324/haematol.2012.083345
47. Calabretto G, Attardi E, Teramo A, Trimarco V, Carraro S, Mossuto S, et al. Hypocellular myelodysplastic syndromes (H-MDS): From clinical description to immunological characterization in the Italian multi-center experience. Leukemia. (2022) 36:1947–1950. doi: 10.1038/s41375-022-01592-3
48. Durrani J, Maciejewski JP. Idiopathic aplastic anemia vs hypocellular myelodysplastic syndrome. Hematology. (2019) 2019:97–104. doi: 10.1182/hematology.2019000019
49. Sun L, Babushok DV. Secondary myelodysplastic syndrome and leukemia in acquired aplastic anemia and paroxysmal nocturnal hemoglobinuria. Blood. (2020) 136:36–49. doi: 10.1182/blood.2019000940
50. Durrani J, Awada H, Kishtagari A, Visconte V, Kerr C, Adema V, et al. Large granular lymphocytic leukemia coexists with myeloid clones and myelodysplastic syndrome. Leukemia. (2019) 34:957–62. doi: 10.1038/s41375-019-0601-y
51. Malcovati L, Cazzola M. The shadowlands of MDS: Idiopathic cytopenias of undetermined significance (ICUS) and clonal hematopoiesis of indeterminate potential (chip). Hematology. (2015) 2015:299–307. doi: 10.1182/asheducation-2015.1.299
52. Sloand EM, Melenhorst JJ, Tucker ZC, Pfannes L, Brenchley JM, Yong A, et al. T-cell immune responses to wilms tumor 1 protein in myelodysplasia responsive to immunosuppressive therapy. Blood. (2011) 117:2691–9. doi: 10.1182/blood-2010-04-277921
53. Lopez MA. Myelodysplastic syndromes differential diagnosis. Rare Disease Advisor (2023). Available at: https://www.rarediseaseadvisor.com/disease-info-pages/myelodysplastic-syndromes-differential-diagnosis/.
54. Rodriguez-Sevilla JJ, Colla S. T-cell dysfunctions in myelodysplastic syndromes. Blood. (2024) 143:1329–43. doi: 10.1182/blood.2023023166
55. Peng X, Zhu X, Di T, Tang F, Guo X, Liu Y, et al. The Yin-yang of immunity: Immune dysregulation in myelodysplastic syndrome with different risk stratification. Front Immunol. (2022) 13:994053. doi: 10.3389/fimmu.2022.994053
56. Banerjee T, Calvi LM, Becker MW, Liesveld JL. Flaming and fanning: The spectrum of inflammatory influences in myelodysplastic syndromes. Blood Rev. (2019) 36:57–69. doi: 10.1016/j.blre.2019.04.004
57. Pardanani A, Finke C, Lasho TL, Al-Kali A, Begna KH, Hanson CA, et al. IPSS-independent prognostic value of plasma CXCL10, IL-7 and IL-6 levels in myelodysplastic syndromes. Leukemia. (2011) 26:693–9. doi: 10.1038/leu.2011.251
58. Teodorescu P, Pasca S, Dima D, Tomuleasa C, Ghiaur G. Targeting the Microenvironment in MDS: The final frontier. Front Pharmacol. (2020) 11:1044. doi: 10.3389/fphar.2020.01044
59. Stomper J, Rotondo JC, Greve G, Lübbert M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia. (2021) 35:1873–89. doi: 10.1038/s41375-021-01218-0
60. Zhao C, Jia B, Wang M, Schell TD, Claxton DF, Ehmann WC, et al. Multi-dimensional analysis identifies an immune signature predicting response to decitabine treatment in elderly patients with AML. Br J Haematology. (2019) 188:674–84. doi: 10.1111/bjh.16228
61. Goodyear OC, Dennis M, Jilani NY, Loke J, Siddique S, Ryan G, et al. Azacitidine augments expansion of regulatory T cells after allogeneic stem cell transplantation in patients with acute myeloid leukemia (AML). Blood. (2012) 119:3361–9. doi: 10.1182/blood-2011-09-377044
62. Sohlberg E, Pfefferle A, Andersson S, Baumann BC, Hellström-Lindberg E, Malmberg K-J. Imprint of 5-azacytidine on the natural killer cell repertoire during systemic treatment for high-risk myelodysplastic syndrome. Oncotarget. (2015) 6:34178–90. doi: 10.18632/oncotarget.6213
63. Kwon Y-R, Kim HJ, Sohn M-J, Lim J-Y, Park K-S, Lee S, et al. Effects of decitabine on allogeneic immune reactions of donor lymphocyte infusion via activation of dendritic cells. Exp Hematol Oncol. (2020) 9:22. doi: 10.1186/s40164-020-00178-y
64. Triozzi PL, Aldrich W, Achberger S, Ponnazhagan S, Alcazar O, Saunthararajah Y. Differential effects of low-dose decitabine on immune effector and suppressor responses in melanoma-bearing mice. Cancer Immunology Immunotherapy. (2012) 61:1441–1450. doi: 10.1007/s00262-012-1204-x
65. Daver N, Boddu P, Garcia-Manero G, Yadav SS, Sharma P, Allison J, et al. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia. (2018) 32:1094–105. doi: 10.1038/s41375-018-0070-8
66. Zeidan AM, Knaus HA, Robinson TM, Towlerton AMH, Warren EH, Zeidner JF, et al. A Multi-center phase I trial of Ipilimumab in patients with myelodysplastic syndromes following hypomethylating agent failure. Clin Cancer Res. (2018) 24:3519–27. doi: 10.1158/1078-0432.ccr-17-3763
67. Zeidan AM, Boss I, Beach CL, Copeland WB, Thompson E, Fox BA, et al. A randomized phase 2 trial of azacitidine with or without durvalumab as first-line therapy for higher-risk myelodysplastic syndromes. Blood Adv. (2022) 6:2207–18. doi: 10.1182/bloodadvances.2021005487
68. Chien KS, Kim K, Nogueras-Gonzalez GM, Borthakur G, Naqvi K, Daver NG, et al. Phase II study of azacitidine with pembrolizumab in patients with intermediate-1 or higher-risk myelodysplastic syndrome. Br J Haematology. (2021) 195:378–87. doi: 10.1111/bjh.17689
69. Bewersdorf JP, Xie Z, Bejar R, Borate U, Boultwood J, Brunner AM, et al. Current landscape of translational and clinical research in myelodysplastic syndromes/neoplasms (MDS): Proceedings from the 1st International Workshop on MDS (iwmds) of the International Consortium for MDS (icMDS). Blood Rev. (2023) 60:101072. doi: 10.1016/j.blre.2023.101072
70. Brunner AM, Esteve J, Porkka K, Knapper S, Traer E, Scholl S, et al. Efficacy and safety of Sabatolimab (MBG453) in combination with hypomethylating agents (HMAS) in patients (PTS) with very high/high-risk myelodysplastic syndrome (VHR/hr-MDS) and Acute myeloid leukemia (AML): Final analysis from A phase IB study. Blood 138(Supplement. (2021) 1):244–4. doi: 10.1182/blood-2021-146039
71. Chao MP, Takimoto CH, Feng DD, McKenna K, Gip P, Liu J, et al. Therapeutic targeting of the macrophage immune checkpoint CD47 in myeloid Malignancies. Front Oncol. (2020) 9:1380. doi: 10.3389/fonc.2019.01380
72. Gera K, Chauhan A, Castillo P, Rahman M, Mathavan A, Mathavan A, et al. Vaccines: A promising therapy for myelodysplastic syndrome. J Hematol Oncol. (2024) 17:4. doi: 10.1186/s13045-023-01523-4
73. Rezvani K, Yong ASM, Mielke S, Savani BN, Musse L, Superata J, et al. Leukemia-associated antigen specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid Malignancies. Blood. (2007) 110:287–7. doi: 10.1182/blood.v110.11.287.287
74. Qazilbash MH, Wieder E, Thall PF, Wang X, Rios R, Lu S, et al. PR1 peptide vaccine induces specific immunity with clinical responses in myeloid Malignancies. Leukemia. (2016) 31:697–704. doi: 10.1038/leu.2016.254
75. Parikh AR, Olnes MJ, Barrett AJ. Immunomodulatory treatment of myelodysplastic syndromes: Antithymocyte globulin, cyclosporine, and alemtuzumab. Semin Hematol. (2012) 49:304–11. doi: 10.1053/j.seminhematol.2012.07.004
76. Kochenderfer JN, Kobayashi S, Wieder ED, Su C, Molldrem JJ. Loss of T- lymphocyte clonal dominance in patients with myelodysplastic syndrome responsive to immunosuppression. Blood. (2002) 100:3639–45. doi: 10.1182/blood-2002-01-0155
77. Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol. (2011) 186:5367–75. doi: 10.4049/jimmunol.1003438
78. Paracatu LC, Schuettpelz LG. Contribution of aberrant toll like receptor signaling to the pathogenesis of myelodysplastic syndromes. Front Immunol. (2020) 11:1236. doi: 10.3389/fimmu.2020.01236
79. Krönke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature. (2015) 523(7559):183–8. doi: 10.1038/nature14610
80. Sekeres MA, Maciejewski JP, Giagounidis AA, Wride K, Knight R, Raza A, et al. Relationship of treatment-related cytopenias and response to lenalidomide in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. (2008) 26:5943–9. doi: 10.1200/JCO.2007.15.5770
81. Kotla V, Goel S, Nischal S, Heuck C, Vivek K, Das B, et al. Mechanism of action of lenalidomide in hematological Malignancies. J Hematol Oncol. (2009) 2:36. doi: 10.1186/1756-8722-2-36
82. Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al. Transforming growth factor- β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. (2014) 20:408–14. doi: 10.1038/nm.3512
83. Chan O, Komrokji RS. Luspatercept in the treatment of lower-risk myelodysplastic syndromes. Future Oncol. (2021) 17:1473–81. doi: 10.2217/fon-2020-1093
84. Fenaux P, Platzbecker U, Mufti GJ, Garcia-Manero G, Buckstein R, Santini V, et al. [amp]]hellip; List, A Luspatercept in patients with lower-risk myelodysplastic syndromes. F. New Engl J Med. (2020) 382(2):140–51. doi: 10.1056/nejmoa1908892
85. Bazinet A, Darbaniyan F, Jabbour E, Montalban-Bravo G, Ohanian M, Chien K, et al. Azacitidine plus venetoclax in patients with high- risk myelodysplastic syndromes or chronic myelomonocytic leukaemia: phase 1 results of a single-centre, dose-escalation, dose-expansion, phase 1-2 study. Lancet Haematol. (2022) 9:e756–65. doi: 10.1016/S2352-3026(22)00216-2
86. Ganan-Gomez I, Yang H, Ma F, Montalban-Bravo G, Thongon N, Marchica V, et al. Stem cell architecture drives myelodysplastic syndrome progression and predicts response to venetoclax-based therapy. Nat Med. (2022) 28:557–67. doi: 10.1038/s41591-022-01696-4
87. Hua S, Feng T, Yin L, Wang Q, Shao X. NEDD9 overexpression: prognostic and guidance value in acute myeloid leukaemia. J Cell Mol Med. (2021) 25:9331–9. doi: 10.1111/jcmm.16870
88. Swords RT, Coutre S, Maris MB, Zeidner JF, Foran JM, Cruz J, et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood. (2018) 131:1415–24. doi: 10.1182/blood-2017-09-805895
89. Moyo TK, Watts JM, Skikne BS, Mendler JH, Klimek VM, Chen S-C, et al. Preliminary results from a phase II study of the combination of pevonedistat and azacitidine in the treatment of MDS and MDS/MPN after failure of DNA methyltransferase inhibition. Blood. (2019) 134:4236–6. doi: 10.1182/blood-2019-130003
90. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Campbell P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. J. Blood. (2013) 122:3616–27. doi: 10.1182/blood-2013-08-518886
91. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
Keywords: myelodysplastic neoplasms, myelodysplastic syndromes, immune dysfunction, immunomodulatory therapies, immunobiology
Citation: Kannan S, Vedia RA and Molldrem JJ (2024) The immunobiology of myelodysplastic neoplasms: a mini-review. Front. Immunol. 15:1419807. doi: 10.3389/fimmu.2024.1419807
Received: 18 April 2024; Accepted: 27 August 2024;
Published: 16 September 2024.
Edited by:
Shyam Patel, University of Massachusetts Medical School, United StatesReviewed by:
Vadim V. Sumbayev, University of Kent, United KingdomCopyright © 2024 Kannan, Vedia and Molldrem. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey J. Molldrem, am1vbGxkcmVAbWRhbmRlcnNvbi5vcmc=