 Noah Watson
Noah Watson Sivaraman Kuppuswamy
Sivaraman Kuppuswamy William Luke Ledford
William Luke Ledford Sangeetha Sukumari-Ramesh
Sangeetha Sukumari-Ramesh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 10 July 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1419685
Histone deacetylases (HDACs) are critical regulators of inflammatory gene expression, and the efficacy of pan-HDAC inhibitors has been implicated in various disease conditions. However, it remains largely unclear how HDACs precisely regulate inflammation. To this end, evaluating the isoform-specific function of HDACs is critical, and the isoform-specific targeting could also circumvent the off-target effects of pan-HDAC inhibitors. This review provides an overview of the roles of HDAC3, a class I HDAC isoform, in modulating inflammatory responses and discusses the molecular mechanisms by which HDAC3 regulates inflammation associated with brain pathology, arthritis, cardiovascular diseases, lung pathology, allergic conditions, and kidney disorders. The articles also identify knowledge gaps in the field for future studies. Despite some conflicting reports, the selective inhibition of HDAC3 has been demonstrated to play a beneficial role in various inflammatory pathologies. Exploring the potential of HDAC3 inhibition to improve disease prognosis is a promising avenue requiring further investigation.
Histone acetylation is a post-translational histone modification that regulates diverse functions, such as protein-protein interactions, DNA recognition, protein stability, and gene expression (1, 2). The interplay between histone acetyltransferases (HATs) and histone deacetylases (HDACs) dynamically modulates the acetylation status of histones, resulting in structural changes to chromatin and thus transcriptional regulation (1, 2). Enhanced HAT activity facilitates chromatin relaxation and gene transcription (3–5), whereas HDACs remove acetyl groups from histones, causing chromatin condensation and gene repression (6). In line with this, the inhibition of HDAC activity causes histone hyperacetylation and transcriptional activation of genes. DNA expression microarrays indicate that the effect of HDAC inhibitors [HDACi (s)] on gene expression is not global but rather limited to a subset of genes (1–7%) (7–11).
HDACs are a family of proteins, with 18 HDAC isoforms currently identified in humans (12). Based on the sequence homology, HDACs are classified into class I (HDAC1, 2, 3 and 8), class IIa (HDAC4, 5, 7 and 9), class IIb (HDAC6 and 10), class III (SIRT1–7) and class IV (HDAC11). The enzymatic activity of class I, II, and IV HDACs requires zinc metal, whereas sirtuins or class III require nicotine adenine dinucleotide as a co-factor (13). Class I HDACs are primarily nuclear and play an important role in cell survival, apoptosis, proliferation, and differentiation (14, 15). Class II HDACs are found in the nucleus and cytoplasm (16, 17). Besides deacetylating histones, HDACs can modulate the acetylation status of nuclear, cytosolic, and mitochondrial non-histone proteins (18), including transcription factors, affecting their structure, stability, interactions, function, and signaling. Hence, HDACs can regulate a wide range of cellular processes.
HDACs have earned much attention in the field of immunology, as they are implicated in various innate and adaptive immune responses, including the synthesis and release of cytokines (19, 20). To this end, the acetylation status of histones and non-histone proteins can affect the expression of inflammatory genes. Also, broad-spectrum HDAC inhibitors exert anti-inflammatory effects and reduce inflammation and disease severity in a wide range of conditions (21). Though HDACs are attractive targets due to the existing clinical applicability of HDAC inhibitors in various disorders (22–24), the use of pan-HDAC inhibitors in clinical trials is associated with several adverse effects, such as fatigue, nausea/vomiting, and diarrhea (25). To alleviate the unwanted side effects of pan-HDAC inhibition, emerging research focuses on targeting individual isoforms of HDACs. The selective targeting of isoforms will also help elucidate the precise mechanism by which HDACs regulate diverse disease processes, which remains largely unclear and controversial.
Histone deacetylase 3 (HDAC3), a class I HDAC isoform, is unique among class I HDACs as it carries nuclear export and localization signals and can shuttle between the nucleus and cytoplasm (26). Owing partly to its non-nuclear localization, HDAC3 acts beyond as a co-repressor (27). Also, HDAC3 exerts enzymatic and non-enzymatic functions. The enzymatic activity of HDAC3 is an important mechanism regulating gene transcription. HDAC3 has often been purified as part of a complex that contains a co-repressor, NCoR1 (nuclear receptor co-repressor), or its homolog NCoR2 (SMRT; silencing mediator of retinoic and thyroid receptors) (28–30). HDAC3 is the prominent HDAC associated with NCoR1 and SMRT (31, 32), which regulates transcriptional repression. Hence, HDAC3 could have distinct functions compared to other class I HDACs. HDAC3 requires interaction with the deacetylase activating domain (DAD) within SMRT or NCoR1 for its enzymatic activity (33). It has been documented that inositol tetraphosphate facilitates the interaction between HDAC3 and DAD (34). Binding to inositol tetraphosphate and DAD triggers a conformational change in HDAC3, allowing substrates to access the catalytic site (34, 35). Global deletion of HDAC3 is embryonically lethal, but mice with mutations in the DAD of both NCoR1 and SMRT live to adulthood despite undetectable deacetylase activity in the embryo (36, 37) suggesting that non-enzymatic activity of HDAC3 drives the growth or survival of embryos.
There is a strong body of research connecting HDAC3 to the inflammatory response. Therefore, evaluating the therapeutic efficacy of HDAC3 inhibition in various inflammatory disease contexts is an ongoing and emerging area of research interest. Functionally, HDAC3 is crucial for the induction of pro-inflammatory gene expression in macrophages in response to inflammatory stimulus, lipopolysaccharide (38). The anti-inflammatory cytokine-mediated stimulation of macrophages into alternate activation involves epigenetic mechanisms (39, 40), and macrophages lacking HDAC3 are phenotypically similar to IL-4-induced alternatively activated macrophages (39). Also, HDAC3 is demonstrated to mediate mitochondrial adaptations to drive IL-1β dependent inflammation in macrophages through non-histone deacetylation (41). Besides, HDAC3 expression was found to be upregulated during various inflammatory settings (38, 42, 43), and selective HDAC3 inhibition modulates inflammation in multiple pathologies. Herein, we provide an overview of the functional roles of HDAC3 regarding inflammation associated with various disease conditions.
Neuroinflammation, the inflammatory response in the CNS, is characterized by glial activation, and upregulation, and secretion of inflammatory mediators such as cytokines, chemokines, and reactive oxygen species. The degree of neuroinflammation depends on the type, duration, and severity of insult or injury. In the uninjured brain, microglia, the inflammatory cells of the CNS, actively survey the brain microenvironment for non-functional neurons and serve as the sentinels of infection. Upon a brain insult or injury, microglia undergo activation, resulting in transcriptional and phenotypical changes with the release of various cytokines, chemokines, and reactive oxygen species. Activated microglia/macrophages polarize to a pro-inflammatory M1 phenotype or an anti-inflammatory M2 phenotype (44) and exhibit migratory and phagocytic potential, contributing to disease progression or repair. Microglia respond to both systemic and brain pathologies. Neuroinflammation also often results in the recruitment of peripheral cells to the brain, further aggravating or alleviating the neuroinflammatory cascade depending on the stage or type of neuropathological condition. In general, the microglia-mediated immune response or the transient activation of microglia is regarded as an intrinsic mechanism to protect or repair the brain. However, neuropathological conditions often result in chronic activation of microglia, culminating in neuronal death, neurodegeneration, and neurological and cognitive decline.
Growing evidence suggests that HDAC3 could be a potential target to modulate neuroinflammation. For instance, the use of a broad-spectrum HDAC inhibitor, valproic acid, modulated microglial polarization towards M2 phenotype and improved outcomes post-traumatic spinal cord injury (45). Valproic acid-mediated neuroprotection was associated with the inhibition of HDAC3 expression and activity in the lesioned spinal cord as well as upregulation of STAT1 - NF-κB P65 interaction, thereby attenuating NF-κB P65 DNA binding (45). As per the study, HDAC3 could serve as a critical modulator of STAT1 - NF-κB P65 interaction and neuroinflammation by regulating the acetylation status of both STAT1 and NF-κB P65 (45). Also, in ischemia/reperfusion-induced brain injury, HDAC3-mediated regulation of NF-κB p65 acetylation in microglia has been demonstrated to play a role in neuroinflammation (46). Mechanistically, it was demonstrated that HDAC3-mediated cGAS transcription and neuroinflammation in ischemia/reperfusion-induced brain injury were associated with NF-κB p65 deacetylation (46). Furthermore, a pan-HDAC inhibitor, Belinostat, attenuated neuroinflammation in an experimental model of autoimmune encephalomyelitis by increasing the acetylation status of NF-κB P65 with a reduction in the expression of HDAC3 (47). In line with this observation, a selective inhibitor of HDAC3, RGFP966, reduced demyelination in a cuprizone-induced demyelination model and improved neurological behavior (48). The study also showed that RGFP966 significantly reduced M1-like microglia/macrophage activation and the levels of proinflammatory cytokines, such as TNF-α, IL-1β, and iNOS. The neuroprotection conferred by RFGP966 in the mouse model of cuprizone was attributed to modulating P2X7R/STAT3/NF-κB p65/NLRP3 signaling pathways (48). Another study documented an increased HDAC3 expression in microglia/macrophages in a mouse model of ischemic stroke and RGFP966-mediated reduction in brain damage by attenuating AIM2 expression, possibly via modulating STAT1 acetylation (49). Though RGFP966 is being widely used for selectively targeting HDAC3, a study used BRD3308 to selectively inhibit HDAC3 in a mouse model after intraventricular hemorrhage. BRD3308 reduced neuroinflammation and microglial pyroptosis, with the modulation of the PPARγ/NLRP3/GSDMD pathway after intraventricular hemorrhage (50).
Consistent with the role of HDAC3 in neuroinflammatory responses, HDAC3 inhibition attenuated the expression of proinflammatory cytokines in repeatedly LPS-challenged human monocytes and M1 macrophages (51). Also, HDAC3 inhibition using RGFP966 reduced LPS-induced primary microglial activation (52). In the presence of LPS, RGFP966 modulated the expression of proteins involved in the TLR pathway and the phosphorylation of STAT3 and STAT5 in primary microglia (52). Taken together, the data indicate a crucial role for HDAC3 in regulating the acetylation status of transcription factors such as NF-κB p65, STAT1, and STAT3 and, hence, neuroinflammation.
Rheumatoid arthritis (RA) is an autoimmune disease characterized by a chronic state of unknown etiology and progressive damage to the cartilage that can lead to lifelong disability (53). Dysregulated immune function is a contributing factor in RA pathogenesis and disease progression. Both local and systemic immune abnormalities occur in association with RA (54). During the course of the disease, fibroblast-like synoviocytes (FLS) exhibit abnormal activation, which is associated with altered expression of major histocompatibility complex (MHC)-II, pro-inflammatory cytokines, adhesion molecules, proangiogenic factors, and matrix-degrading enzymes (55–57), contributing to synovial inflammation and joint damage (58). Furthermore, the pathogenesis of RA is closely related to the abnormal activation of FLS (59).
Histone deacetylases play roles in RA progression, and HDAC inhibitors exhibited therapeutic efficacy and anti-inflammatory effects in animal models of RA (60–65). A study showed that the pan-HDAC inhibitors, Trichostatin A and ITF2357, suppressed IL-6 production induced by IL-1β, TNF-α, and TLR ligands in RA-FLS (66). Moreover, a class I HDAC inhibitor, MS-275, and a pan HDAC inhibitor, Suberoylanilide Hydroxamic Acid, attenuated the inflammatory response in LPS-induced human RA synovial fibroblastic E11 cells (67). Notably, the effects of pan-HDAC inhibitors in reducing inflammatory gene expression in RA-FLS were recapitulated by HDAC3 inhibition (68). It has been demonstrated that STAT1 and phosphorylated STAT1 levels are elevated in RA-FLS, contributing to inflammation (69, 70). Also, STAT1 hyperacetylation is a prerequisite for STAT1 dephosphorylation and inactivation (71). Notably, the genetic inhibition of HDAC3 attenuated IL-1β-induced STAT1 phosphorylation in RA-FLS, implicating a critical role of HDAC3 in RA-FLS activation and associated inflammation (68). However, the inhibition of HDAC3/6 did not affect the acetylation status of STAT1 in the presence of IL-1β in RA-FLS (68), suggesting a novel mechanism by which HDAC3 regulates STAT1 phosphorylation, warranting investigation.
Extracellular cold-inducible RNA-binding protein (CIRP) is a novel pro-inflammatory molecule involved in various inflammatory diseases. In patients with RA, increased CIRP levels are found in the serum and synovial fluid, and elevated CIRP levels in the synovial fluid correlate with disease activity (72). A recent study has reported that human CIRP induced the proliferation, migration, and invasion of RA-FLS and released IL-1β and IL-33 from RA-FLS (73). Moreover, the inhibition of CIRP significantly reduced the abnormal activation of RA-FLS and arthritis severity in adjuvant arthritis in rats (73). Per the same study, the knockdown of TLR4 inhibited extracellular CIRP-induced RA-FLS activation and HDAC3 expression in RA-FLS, suggesting a role of CIRP-TLR4-HDAC3 signaling in RA-associated synovial inflammation. Also, both genetic and pharmacological inhibition of HDAC3 suppressed extracellular CIRP-induced abnormal activation of RA-FLS in vitro and RGFP966 treatment attenuated arthritis severity of adjuvant arthritis in rats (73), implicating a crucial role of HDAC3 in RA-associated synovial inflammation. Therefore, further studies are highly warranted to establish the precise role of HDAC3 in RA pathology and the mechanism by which HDAC3 regulates synovial inflammation in RA.
Apart from RA-FLS, the other cell type that plays a crucial role in the RA pathophysiology is peripheral blood mononuclear cells (PBMC), which could release abnormal levels of inflammatory cytokines (74). A recent study postulated that before the development of synovitis in RA patients, systemic autoimmunity is initiated, resulting in cells such as monocytes from the peripheral blood infiltrating into the synovial tissue or joint fluid, causing inflammation (75). Also, the cytokines released from PBMCs can induce the differentiation of helper T cells towards Th1, Th2, Th17, and Treg cells, thereby modulating inflammation in RA (76). Consistent with the emerging role of epigenetic mechanisms in regulating cytokine release in RA (77), changes in HDAC activity in PBMCs from RA have been reported. However, there are inconsistencies between studies. For instance, a global increase in HDAC activity in PBMCs in RA and the efficacy of selective inhibition of HDAC3 in attenuating IL-6 release from RA PBMCs have been reported (78). On the contrary, another study reported a reduction in total HDAC activity and HDAC3 activity with an increase in total histone H3 acetylation in PBMCs from RA patients compared to healthy subjects (79). Despite these conflicting observations, the balance between HDAC and HAT activity was significantly altered in RA PBMCs, implicating further a potential role of histone acetylation in the pathophysiology of RA (79). Therefore, additional investigation is highly required to establish the precise functional role of HDAC3 and epigenetic mechanisms in PBMC-associated pathology in RA.
Osteoarthritis (OA) is another common form of arthritis in which HDAC3 plays a role. The expression of HDAC3 was shown to be higher in degraded cartilage compared to non-degraded cartilage. Furthermore, HDAC3 expression increased when primary human chondrocytes (PHCs) were stimulated with IL-1β (80), and the genetic inhibition of HDAC3 in PHCs augmented cartilage-specific genes and reduced the expression of a hypertrophy-related gene (80).
Overall, HDAC3 could be an efficient target to improve outcomes after RA and OA. However, further studies are highly warranted to elucidate the efficacy of selective HDAC3 inhibition and the mechanism by which HDAC3 regulates the development and progression of RA and OA.
Atherosclerosis is the major underlying pathology of cardiovascular diseases (CVD), the leading cause of morbidity and mortality globally (81). Atherosclerosis is a chronic inflammatory disease arising from an imbalance in lipid metabolism and a maladaptive immune response. Macrophages play a key role in the progression and regression of atherosclerotic cardiovascular disease (82). Increased expression of pro-inflammatory cytokines, such as IL-1β, could regulate the expression of cholesterol efflux protein ABCA1 in macrophages, thereby promoting foam cell formation and development of atherosclerosis (83). Several studies have explored the therapeutic potential of HDAC inhibitors in CVD. Interestingly, myeloid-specific conditional deletion of HDAC3 shifted macrophages to an anti-inflammatory phenotype with improved lipid accumulation and plaque stability in a mouse model of atherosclerosis (84). Apart from regulating macrophage phenotype, HDAC3 plays an important role in regulating the adhesion of monocytes to the sites of inflammation (85). To this end, knockdown to HDAC3 attenuated TNF-α–mediated VCAM-1 expression in human primary endothelial cells (Human Umbilical Vein Endothelial Cells; HUVECs) and monocyte adhesion to the activated HUVECs. Also, in humans, HDAC3 was the sole HDAC upregulated in ruptured lesions and its expression inversely correlated with plaque-stabilizing TGF-β (84).
HDAC3 regulates endothelial function in normal physiology and pathology. Lentiviral-mediated knockdown of HDAC3 in endothelial cells reduced cell survival, suggesting that HDAC3 plays a critical role in endothelial cell survival in vitro (86). In a mouse model of type 2 diabetes mellitus (T2DM), HDAC3 activity, but not protein expression, was found to be increased in endothelial cells (87). Moreover, treatment with RGFP966 alleviated T2DM-associated endothelial dysfunction and the knockdown of Nrf2 abolished HDAC3 inhibition-induced endothelial protection in T2DM both in vitro and in vivo (87). Also, HDAC3 has been shown to regulate the expression of immune modulator galectin-9 in HUVECs (88). Notably, HDAC3 knockdown in endothelial cells reduced IFN-γ-induced expression of galectin-9, whereas overexpression of HDAC3 induced the interaction between IFN response factor 3 (IRF3) and phosphoinositol 3-kinase (PI3K) leading to IRF3 phosphorylation and galectin-9 expression (88). Evidence suggested that HDAC3 could serve as a scaffold protein facilitating PI3K/IRF3 interaction and regulating galectin-9 expression in endothelial cells (88).
Endothelial to mesenchymal transition (EndMT) contributes to multiple vasculopathies, including atherosclerosis, and facilitates the transition from vascular inflammation to plaque formation (89, 90). Of note, HDAC3 expression was upregulated in atherosclerotic plaque in a mouse model of atherosclerosis and regulated the induction of EndMT (91). Functionally, the pharmacological inhibition of HDAC3 in a mouse model of atherosclerosis reduced atherosclerotic lesions and inhibited EndMT, whereas the genetic overexpression of HDAC3 induced EndMT in HUVECs (91). Also, HDAC3 modulated the gene expression of IL-6, ICAM-1, and MCP-1 in HUVECs and the number of monocytes attached to HUVECs in the presence of inflammatory stimuli (91). These findings suggest a critical role of HDAC3 in vascular inflammation and the induction of EndMT.
Myocardial infarction (MI) is a common and life-threatening condition in which a blockage in the coronary artery leads to oxygen deprivation, injury, and cell death. The cell death can cause a high degree of inflammation as macrophages are recruited to the injury. In a rat model of ischemia-reperfusion injury, treatment with RGFP966 alleviated inflammatory response, oxidative stress, and injury in myocardial tissue, possibly by reducing the levels of cyclin-dependent kinase-2 (92), further implicating the efficacy of HDAC3 inhibition in reducing inflammation.
Though the aforementioned studies point out the potential of HDAC3 inhibition in alleviating cardiovascular inflammation, a contradictory finding documents an inverse association between the expression of HDAC3 and NF-κB/p65 in ox-LDL-induced HUVECs (93). As per the study, the overexpression of HDAC3 attenuated the levels of TNF-α and IL-1β in the arterial tissue in a mouse model of atherosclerosis (93). Due to this discrepancy between observations, additional investigation is necessary to validate the efficacy of HDAC3 inhibition for cardiovascular diseases.
While many studies reported a reduction in the proinflammatory response when HDAC3 was inhibited, some studies have indicated the inhibition of HDAC3 can augment inflammation. For instance, in chronic obstructive pulmonary disease (COPD), a pathological condition resulting from inhalation of air pollutants and cigarette smoke, pulmonary macrophages secrete a large and varied number of inflammatory factors. Using a model of human alveolar macrophages, it has been shown that acute cigarette smoke exposure is associated with reduced total nuclear HDAC activity and nuclear HDAC3 protein expression (94). Also, siRNA-mediated knockdown of HDAC3 in the in vitro model of alveolar macrophages augmented LPS-induced release of IL-1β and IL-8, possibly implicating a negative regulatory role of HDAC3 in inflammation, but mechanistic studies are yet to be conducted. Given the role of HDAC3 in modulating the acetylation status and the nuclear export of NF-κB, the study postulated that NF-κB signaling could be a possible mechanism by which HDAC3 regulates pulmonary inflammation (94). Also, Ergosterol treatment switched macrophage polarization to M2 phenotype with an increase in HDAC3 expression and a reduction in acetyl NF-κB/p65 in COPD models (95), suggesting that ergosterol-mediated protection of COPD is associated with HDAC3-mediated deacetylation.
Acute lung injury is characterized by damage to alveolar epithelial cells and capillary endothelial cells, causing refractory hypoxemia and acute respiratory distress syndrome. Nimbolide, a chemical constituent of Azadirachta indica, improved endotoxin-induced acute respiratory distress syndrome by inhibiting TNF-α mediated nuclear translocation of both NF-κB and HDAC3 (96). Furthermore, RGFP966 reduced the levels of proinflammatory cytokines in a model of inflammatory lung disease. Using an in vitro approach, the study also reported that the anti-inflammatory effects of RGFP966 are attributed to the modulation of NF-κB transcriptional activity but not NF-κB p65 acetylation or localization (97). Furthermore, Th2 cytokine-driven pulmonary inflammation was limited in mice lacking HDAC3 in macrophages (39), which implies that inhibition of HDAC3 can be targeted to attenuate lung inflammation.
Neutrophilic airway inflammation is associated with reduced total HDAC activity in blood monocytes (98), implicating a possible role of epigenetic mechanisms in the disease pathology. However, there was no change in HDAC3 gene expression levels.
Taken together, the current data implicate opposing roles of HDAC3 in pulmonary inflammation, with inhibition being either deleterious or beneficial. Further research using alternate approaches is necessary to validate the findings and elucidate the mechanism by which HDAC3 regulates pulmonary pathologies.
HDAC3 has been implicated in playing a role in diabetic-related inflammation, which can interfere with insulin signaling and glucose homeostasis. In-vitro studies have shown that genetic knock-down of HDAC3 restored glucose-stimulated insulin secretion and reduced caspase-3 activity in beta cells in the presence of cytokines (99). Also, HDAC3 inhibition improved pancreatic β cell function and plasma glucose levels in a rat model of type 2 diabetes (100). Interestingly, increased HDAC3 activity and mRNA expression were observed in the PBMCs of type 2 diabetic patients in comparison with control subjects and HDAC3 activity positively correlated with proinflammatory markers, fasting plasma glucose, and insulin resistance (101). These findings suggest a critical role of HDAC3 in inflammation and other complications associated with diabetes.
Inflammation and apoptosis are the key mechanisms responsible for diabetic osteoporosis (102). In a streptozotocin (STZ) model of diabetes, HDAC 1 and 3 expression in femoral heads was found to be upregulated (103). Puerarin (PU), an isoflavone, improved STZ-induced blood glucose levels and osteoporosis with a reduction in inflammation and apoptosis in rats (103). Also, PU reduced STZ-mediated upregulation of HDAC1 and 3 expressions in femoral heads, suggesting a possible mechanism by which Puerarin (PU) improved diabetes-related complications. In line with this finding, inhibition of HDAC1/3 attenuated inflammation and cell death in fructose-treated cells (103). However, further studies need to be conducted to find which isoform of HDAC, among HDAC1 and 3, is responsible for PU-mediated effects.
RGFP966 has been shown to have beneficial effects in diabetic cardiomyopathy (DCM) in mice, causing a reduction in diabetes-induced cardiac oxidative stress, inflammation, fibrosis, hypertrophy, and insulin resistance (104). Notably, HDAC3 activity and phosphorylated extracellular regulated kinases 1 and 2 (ERK1/2), an indicator of cardiac hypertrophy, were upregulated in diabetic hearts (104). Additionally, the level of a nuclear ERK1/2 phosphatase, DUSP5 (dual specificity phosphatase 5), was decreased in diabetic hearts (104). Mechanistically, RGFP966 treatment augmented DUSP5 expression, modulated ERK1/2 signaling, and prevented DCM in mice (104).
Diabetes enhances the risk of stroke and its recurrence. RGFP966 treatment conferred protection against cerebral ischemia/reperfusion injury in diabetic mice by modulating brain oxidative stress, apoptosis, and autophagy (105). Also, RGFP966 treatment has been shown to have beneficial effects in some liver pathologies, particularly diabetes-induced liver damage. In diabetic mice, RGFP966 treatment reduced hepatic inflammation, fibrosis, and oxidative stress (106). These protective effects were associated with enhanced signaling of Nrf2 (106), an antioxidant and anti-inflammatory transcription factor. However, further studies are warranted to ensure the safe use of HDAC3 inhibitors for hepatic pathology, as liver-specific deletion of HDAC3 resulted in fatty liver in mice (107). Also, HDAC3 genetic deletion rescued palmitate-induced reduction in the expression of genes related to fatty acid oxidation in C2C12 myotubes, further implicating the role of HDAC3 in fatty acid metabolism (108).
Histone acetylation and deacetylation play roles in allergic inflammation (109). Of note, HDAC3 expression was upregulated in a mouse model of triphasic cutaneous anaphylaxis (triphasic cutaneous reaction; TpCR) (110). Moreover, HDAC3 regulated the expression of monocyte chemoattractant protein 1 (MCP1; a mediator of monocyte recruitment) and allergic skin inflammation in vivo (110). Besides, the suppressor of cytokine signaling 1 (SOCS1), a protein with contradictory roles in inflammation, regulated the expression of HDAC3 and allergic inflammation (111). Notably, antigen stimulation enhanced the expression of SOCS1, HDAC3, and HDAC6, in RBL2H3 basophilic leukemia cells (111). Furthermore, SOCS1 increased the interaction between high-affinity IgE receptor (FcϵRIβ) and HDAC3 in an antigen-independent manner, implicating a critical role of SOCS1/HDAC3 signaling in allergic inflammation (111).
The levels of hyaluronic acid (HA), a major component of the extracellular matrix, are elevated in allergic reactions in vivo and the increase in HA correlates with the influx of inflammatory cells (112). Despite the altered levels of HDAC3 in allergic response, it was postulated that HDAC3 may regulate allergic inflammation by modulating the production of low or high-molecular-weight HA (43). Furthermore, a study examining the role of HDAC3 in allergic rhinitis (AR) demonstrated decreased levels of multiple pro-inflammatory cytokines, and reduced allergic responses in mice upon RGFP966 treatment (113).
Altogether, various studies reveal a crucial role of HDAC3 in allergic pathologies. However, since the differences in the functional roles of HA of varying sizes are controversial, further studies are highly required to elucidate the pro or anti-allergic effects of high and low-molecular-weight HA and the mechanism by which HDAC3 regulates HA production and allergic inflammation.
Inflammation plays a major role in chronic kidney disease (CKD), as it can lead to fibrosis and renal damage. It has been demonstrated that in a mouse model of kidney fibrosis, HDAC3 protein expression in the kidney was elevated and the deletion of HDAC3 via a genetic approach (CAG-Cre+) reduced the renal expression of TNF-α and fibrosis in a mouse model of kidney fibrosis (114). Also, in a rat model of hyperuricemia-induced fibrosis, the depletion of HDAC3 via a genetic approach blunted renal fibrosis (115). Altogether, the data implicate a key role of HDAC3 in renal fibrosis and associated inflammation. In line with this, treatment with RGFP966 attenuated kidney fibrosis in mice (114). Though the mechanism by which HDAC3 regulates fibrosis is yet to be defined, based on in vitro studies, it was postulated that hyperacetylation at Lys122 could reduce the transcription activity of NF-κB upon HDAC3 deletion (114), which further implicates a critical role of NF-κB acetylation in inflammatory disease conditions.
Acute kidney injury (AKI) and CKD are two distinct pathologies, but AKI can progress to CKD, characterized by various pathological events, including inflammation (116). However, the precise mechanism by which AKI progresses to CKD is largely unknown. In a mouse model of AKI-CKD, HDAC3 was found to be elevated in the kidney, and HDAC3 conditional deletion attenuated renal ferroptosis, and fibrosis (117). Consistent with this finding, RGFP966 treatment reduced renal ferroptosis and fibrosis in AKI-CKD mice, with a modulation in the expression of GPX4, a master regulator of ferroptosis, implicating a key role of HDAC3 in AKI-CKD transition (117).
HDAC3 has emerged as a pivotal regulator of a wide range of inflammatory conditions and has been demonstrated to play a role in immune cell differentiation and inflammatory gene expression. However, despite the emerging interest in targeting HDAC3 to modulate inflammation and disease pathologies, the precise mechanism by which HDAC3 regulates inflammatory gene expression profiles remains enigmatic. Mechanistic studies mostly employed in vitro approaches and focused on, and implied, to a large extent, the role of transcription factors such as NF-kB and STAT1 and their post-translational modification by HDAC3 in modulating inflammation (Figure 1). To gain further mechanistic insights, cell-specific functional studies using transgenic or conditional knock-out animals and in vivo studies employing unbiased proteomic and transcriptomic approaches are critical and required. Analyzing the enzymatic and nonenzymatic functions of HDAC3 and developing additional selective inhibitors or activators of HDAC3 could also be helpful in further defining its role in various pathological conditions. Despite some conflicting reports, the selective inhibition of HDAC3 has been demonstrated to play a beneficial role in various inflammatory pathologies. The selective inhibition of HDACs could also circumvent the off-target effects of pan-HDAC inhibitors. Notably, RGFP966 (10 mg/kg) selectively inhibited HDAC3 over other HDACs in mice (118), and its systemic administration (10 mg/kg) daily for 14 days did not induce significant toxic effects on the mouse brain and major organs (119), implicating its suitability for therapeutic purposes. However, clinical studies have yet to be conducted evaluating its safe use and efficacy in humans. Besides its therapeutic potential, HDAC3 activity or expression is altered in several pathological conditions, implicating its potential to serve as a diagnostic and prognostic marker of inflammation, warranting investigation. Overall, continued research into the interplay between HDAC3 and inflammation holds promise for advancing our understanding of inflammatory diseases and developing more effective treatment strategies.
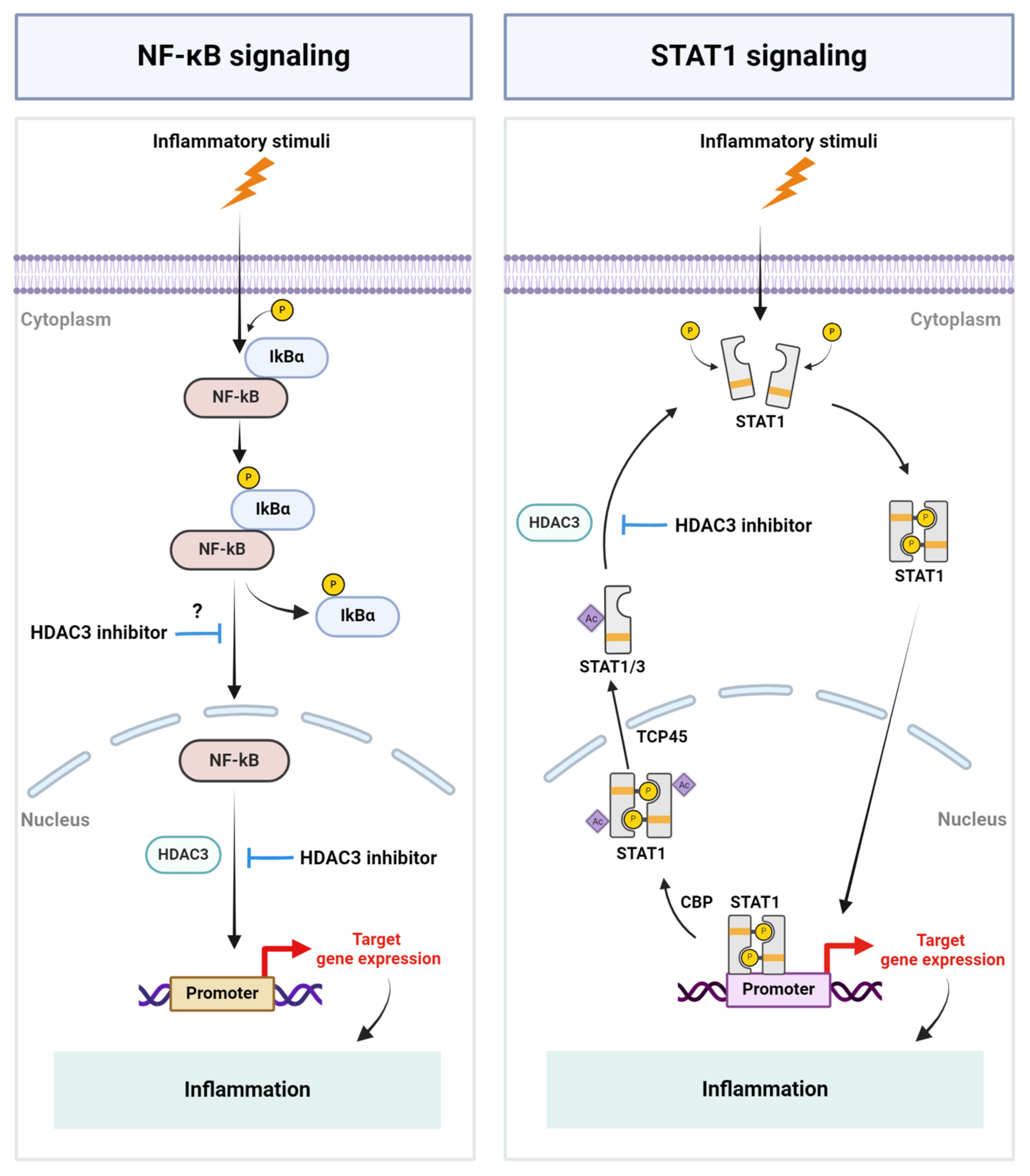
Figure 1 Schematic representation of key mechanisms by which HDAC3 regulates inflammation. Inflammatory stimuli cause the activation of the NF-kB signaling pathway and HDAC3-mediated NF-kB DNA-binding, resulting in the expression of NF-kB-regulated genes, including inflammatory genes. HDAC3 regulates STAT signaling by modulating the acetylation status of STAT1 and STAT3. P, Phosphate group; IkBα, I kappa B alpha; Ac, Acetyl group; CBP, CREB binding protein; TCP45, T cell protein tyrosine phosphatase 45. Created with BioRender.com.
NW: Writing – original draft, Writing – review & editing. SK: Writing – original draft, Writing – review & editing. WL: Writing – original draft, Writing – review & editing. SS-R: Conceptualization, Funding acquisition, Project administration, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Institutes of Health, R01NS107853, R03AG077460 and R01NS132794 to SS-R.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. (1999) 98:285–94. doi: 10.1016/S0092-8674(00)81958-3
2. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. (2000) 403:41–5. doi: 10.1038/47412
3. Norton VG, Marvin KW, Yau P, Bradbury EM. Nucleosome linking number change controlled by acetylation of histones H3 and H4. J Biol Chem. (1990) 265:19848–52. doi: 10.1016/S0021-9258(17)45450-0
4. Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. (1993) 72:73–84. doi: 10.1016/0092-8674(93)90051-Q
5. Hebbes TR, Thorne AW, Crane-Robinson C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. (1988) 7:1395–402. doi: 10.1002/embj.1988.7.issue-5
6. Rundlett SE, Carmen AA, Kobayashi R, Bavykin S, Turner BM, Grunstein M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci USA. (1996) 93:14503–8. doi: 10.1073/pnas.93.25.14503
7. Zhu WG, Lakshmanan RR, Beal MD, Otterson GA. DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Res. (2001) 61:1327–33.
8. Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. (2000) 92:1210–6. doi: 10.1093/jnci/92.15.1210
9. Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther. (2003) 2:151–63.
10. Mariadason JM, Corner GA, Augenlicht LH. Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Res. (2000) 60:4561–72.
11. Chambers AE, Banerjee S, Chaplin T, Dunne J, Debernardi S, Joel SP, et al. Histone acetylation-mediated regulation of genes in leukaemic cells. Eur J Cancer. (2003) 39:1165–75. doi: 10.1016/S0959-8049(03)00072-8
12. Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. (2007) 26:5541–52. doi: 10.1038/sj.onc.1210620
13. Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. (2006) 75:435–65. doi: 10.1146/annurev.biochem.74.082803.133500
14. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 128:693–705. doi: 10.1016/j.cell.2007.02.005
15. Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci USA. (2007) 104:17335–40. doi: 10.1073/pnas.0706487104
16. Grozinger CM, Schreiber SL. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci USA. (2000) 97:7835–40. doi: 10.1073/pnas.140199597
17. McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. (2000) 408:106–11. doi: 10.1038/35040593
18. Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. (2005) 363:15–23. doi: 10.1016/j.gene.2005.09.010
19. Carta S, Tassi S, Semino C, Fossati G, Mascagni P, Dinarello CA, et al. Histone deacetylase inhibitors prevent exocytosis of interleukin-1beta-containing secretory lysosomes: role of microtubules. Blood. (2006) 108:1618–26. doi: 10.1182/blood-2006-03-014126
20. Licciardi PV, Karagiannis TC. Regulation of immune responses by histone deacetylase inhibitors. ISRN Hematol. (2012) 2012:690901. doi: 10.5402/2012/690901
21. Adcock IM. HDAC inhibitors as anti-inflammatory agents. Br J Pharmacol. (2007) 150:829–31. doi: 10.1038/sj.bjp.0707166
22. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. (2009) 10:32–42. doi: 10.1038/nrg2485
23. Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. (2011) 32:335–43. doi: 10.1016/j.it.2011.04.001
24. Guo FQ, Li XJ, Chen LY, Yang H, Dai HY, Wei YS, et al. Study of relationship between inflammatory response and apoptosis in perihematoma region in patients with intracerebral hemorrhage. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. (2006) 18:290–3.
25. Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical toxicities of histone deacetylase inhibitors. Pharm (Basel). (2010) 3:2751–67. doi: 10.3390/ph3092751
26. Yang WM, Tsai SC, Wen YD, Fejer G, Seto E. Functional domains of histone deacetylase-3. J Biol Chem. (2002) 277:9447–54. doi: 10.1074/jbc.M105993200
27. Karagianni P, Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene. (2007) 26:5439–49. doi: 10.1038/sj.onc.1210612
28. Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, et al. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. (2003) 22:1336–46. doi: 10.1093/emboj/cdg120
29. Goodson M, Jonas BA, Privalsky MA. Corepressors: custom tailoring and alterations while you wait. Nucl Recept Signal. (2005) 3:e003. doi: 10.1621/nrs.03003
30. Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet. (2010) 11:109–23. doi: 10.1038/nrg2736
31. Li J, Wang J, Wang J, Nawaz Z, Liu JM, Qin J, et al. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. (2000) 19:4342–50. doi: 10.1093/emboj/19.16.4342
32. Wen YD, Perissi V, Staszewski LM, Yang WM, Krones A, Glass CK, et al. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc Natl Acad Sci USA. (2000) 97:7202–7. doi: 10.1073/pnas.97.13.7202
33. Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. (2001) 21:6091–101. doi: 10.1128/MCB.21.18.6091-6101.2001
34. Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. (2012) 481:335–40. doi: 10.1038/nature10728
35. Arrar M, Turnham R, Pierce L, de Oliveira CA, McCammon JA. Structural insight into the separate roles of inositol tetraphosphate and deacetylase-activating domain in activation of histone deacetylase 3. Protein Sci. (2013) 22:83–92. doi: 10.1002/pro.2190
36. Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. (2008) 30:61–72. doi: 10.1016/j.molcel.2008.02.030
37. You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. (2013) 20:182–7. doi: 10.1038/nsmb.2476
38. Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci USA. (2012) 109:E2865–74. doi: 10.1073/pnas.1121131109
39. Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. (2011) 25:2480–8. doi: 10.1101/gad.175950.111
40. Chen S, Yang J, Wei Y, Wei X. Epigenetic regulation of macrophages: from homeostasis maintenance to host defense. Cell Mol Immunol. (2020) 17:36–49. doi: 10.1038/s41423-019-0315-0
41. Chi Z, Chen S, Xu T, Zhen W, Yu W, Jiang D, et al. Histone deacetylase 3 couples mitochondria to drive IL-1beta-dependent inflammation by configuring fatty acid oxidation. Mol Cell. (2020) 80:43–58.e7. doi: 10.1016/j.molcel.2020.08.015
42. Ziesche E, Kettner-Buhrow D, Weber A, Wittwer T, Jurida L, Soelch J. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-kappaB. Nucleic Acids Res. (2013) 41:90–109. doi: 10.1093/nar/gks916
43. Kim Y, Eom S, Park D, Kim H, Jeoung D. The hyaluronic acid-HDAC3-miRNA network in allergic inflammation. Front Immunol. (2015) 6:210. doi: 10.3389/fimmu.2015.00210
44. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
45. Chen S, Ye J, Chen X, Shi J, Wu W, Lin W, et al. Valproic acid attenuates traumatic spinal cord injury-induced inflammation via STAT1 and NF-kappaB pathway dependent of HDAC3. J Neuroinflamm. (2018) 15:150. doi: 10.1186/s12974-018-1193-6
46. Liao Y, Cheng J, Kong X, Li S, Li X, Zhang M, et al. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics. (2020) 10:9644–62. doi: 10.7150/thno.47651
47. Shen Y, Yang R, Zhao J, Chen M, Chen S, Ji B, et al. The histone deacetylase inhibitor belinostat ameliorates experimental autoimmune encephalomyelitis in mice by inhibiting TLR2/MyD88 and HDAC3/NF-kappaB p65-mediated neuroinflammation. Pharmacol Res. (2022) 176:105969. doi: 10.1016/j.phrs.2021.105969
48. Sun W, Zhang N, Liu B, Yang J, Loers G, Siebert HC, et al. HDAC3 inhibitor RGFP966 ameliorated neuroinflammation in the cuprizone-induced demyelinating mouse model and LPS-stimulated BV2 cells by downregulating the P2X7R/STAT3/NF-kappaB65/NLRP3 activation. ACS Chem Neurosci. (2022) 13:2579–98. doi: 10.1021/acschemneuro.1c00826
49. Zhang MJ, Zhao QC, Xia MX, Chen J, Chen YT, Cao X, et al. The HDAC3 inhibitor RGFP966 ameliorated ischemic brain damage by downregulating the AIM2 inflammasome. FASEB J. (2020) 34:648–62. doi: 10.1096/fj.201900394RRR
50. Li Y, Liu C, Wang G, Wang H, Liu X, Huang C, et al. HDAC3 inhibitor (BRD3308) modulates microglial pyroptosis and neuroinflammation through PPARgamma/NLRP3/GSDMD to improve neurological function after intraventricular hemorrhage in mice. Neuropharmacology. (2023) 237:109633. doi: 10.1016/j.neuropharm.2023.109633
51. Ghiboub M, Zhao J, Li Yim AYF, Schilderink R, Verseijden C, van Hamersveld PHP. HDAC3 mediates the inflammatory response and LPS tolerance in human monocytes and macrophages. Front Immunol. (2020) 11:550769. doi: 10.3389/fimmu.2020.550769
52. Xia M, Zhao Q, Zhang H, Chen Y, Yuan Z, Xu Y, et al. Proteomic analysis of HDAC3 selective inhibitor in the regulation of inflammatory response of primary microglia. Neural Plast. (2017) 2017:6237351. doi: 10.1155/2017/6237351
53. Lin YJ, Anzaghe M, Schulke S. Update on the pathomechanism, diagnosis, and treatment options for rheumatoid arthritis. Cells. (2020) 9(4). doi: 10.3390/cells9040880
54. Kim JW, Suh CH. Systemic manifestations and complications in patients with rheumatoid arthritis. J Clin Med. (2020) 9(6). doi: 10.3390/jcm9062008
55. Bucala R, Ritchlin C, Winchester R, Cerami A. Constitutive production of inflammatory and mitogenic cytokines by rheumatoid synovial fibroblasts. J Exp Med. (1991) 173:569–74. doi: 10.1084/jem.173.3.569
56. Lafyatis R, Remmers EF, Roberts AB, Yocum DE, Sporn MB, Wilder RL. Anchorage-independent growth of synoviocytes from arthritic and normal joints. Stimulation by exogenous platelet-derived growth factor and inhibition by transforming growth factor-beta and retinoids. J Clin Invest. (1989) 83:1267–76. doi: 10.1172/JCI114011
57. Ritchlin C, Dwyer E, Bucala R, Winchester R. Sustained and distinctive patterns of gene activation in synovial fibroblasts and whole synovial tissue obtained from inflammatory synovitis. Scand J Immunol. (1994) 40:292–8. doi: 10.1111/j.1365-3083.1994.tb03465.x
58. Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. (2020) 16:316–33. doi: 10.1038/s41584-020-0413-5
59. Kondo N, Kuroda T, Kobayashi D. Cytokine networks in the pathogenesis of rheumatoid arthritis. Int J Mol Sci. (2021) 22:10922. doi: 10.3390/ijms222010922
60. Chung YL, Lee MY, Wang AJ, Yao LF. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol Ther. (2003) 8:707–17. doi: 10.1016/S1525-0016(03)00235-1
61. Joosten LA, Leoni F, Meghji S, Mascagni P. Inhibition of HDAC activity by ITF2357 ameliorates joint inflammation and prevents cartilage and bone destruction in experimental arthritis. Mol Med. (2011) 17:391–6. doi: 10.2119/molmed.2011.00058
62. Lin HS, Hu CY, Chan HY, Liew YY, Huang HP, Lepescheux L, et al. Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br J Pharmacol. (2007) 150:862–72. doi: 10.1038/sj.bjp.0707165
63. Nasu Y, Nishida K, Miyazawa S, Komiyama T, Kadota Y, Abe N, et al. Trichostatin A, a histone deacetylase inhibitor, suppresses synovial inflammation and subsequent cartilage destruction in a collagen antibody-induced arthritis mouse model. Osteoarthritis Cartilage. (2008) 16:723–32. doi: 10.1016/j.joca.2007.10.014
64. Nishida K, Komiyama T, Miyazawa S, Shen ZN, Furumatsu T, Doi H, et al. Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAF1/Cip1) expression. Arthritis Rheum. (2004) 50:3365–76. doi: 10.1002/art.20709
65. Saouaf SJ, Li B, Zhang G, Shen Y, Furuuchi N, Hancock WW, et al. Deacetylase inhibition increases regulatory T cell function and decreases incidence and severity of collagen-induced arthritis. Exp Mol Pathol. (2009) 87:99–104. doi: 10.1016/j.yexmp.2009.06.003
66. Grabiec AM, Korchynskyi O, Tak PP, Reedquist KA. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann Rheum Dis. (2012) 71:424–31. doi: 10.1136/ard.2011.154211
67. Choo QY, Ho PC, Tanaka Y, Lin HS. Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatol (Oxford). (2010) 49:1447–60. doi: 10.1093/rheumatology/keq108
68. Angiolilli C, Kabala PA, Grabiec AM, Van Baarsen IM, Ferguson BS, Garcia S, et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann Rheum Dis. (2017) 76:277–85. doi: 10.1136/annrheumdis-2015-209064
69. Yoshida S, Arakawa F, Higuchi F, Ishibashi Y, Goto M, Sugita Y, et al. Gene expression analysis of rheumatoid arthritis synovial lining regions by cDNA microarray combined with laser microdissection: up-regulation of inflammation-associated STAT1, IRF1, CXCL9, CXCL10, and CCL5. Scand J Rheumatol. (2012) 41:170–9. doi: 10.3109/03009742.2011.623137
70. Kasperkovitz P, Verbeet N, Smeets T, Van Rietschoten J, Kraan M, Van Der Pouw Kraan T, et al. Activation of the STAT1 pathway in rheumatoid arthritis. Annals of the rheumatic diseases. (2004) 63(3):233–239. doi: 10.1136%2Fard.2003.013276
71. Kramer OH, Knauer SK, Greiner G, Jandt E, Reichardt S, Guhrs KH, et al. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. (2009) 23:223–35. doi: 10.1101/gad.479209
72. Yoo IS, Lee SY, Park CK, Lee JC, Kim Y, Yoo SJ, et al. Serum and synovial fluid concentrations of cold-inducible RNA-binding protein in patients with rheumatoid arthritis. Int J Rheum Dis. (2018) 21:148–54. doi: 10.1111/1756-185X.12892
73. Yao F, Zhao Y, Yu Q, Hu W, Lin Y, Chen Y, et al. Extracellular CIRP induces abnormal activation of fibroblast-like synoviocytes from patients with RA via the TLR4-mediated HDAC3 pathways. Int Immunopharmacol. (2024) 128:111525. doi: 10.1016/j.intimp.2024.111525
74. Wang Y, Xie X, Zhang C, Su M, Gao S, Wang J, et al. Rheumatoid arthritis, systemic lupus erythematosus and primary Sjogren’s syndrome shared megakaryocyte expansion in peripheral blood. Ann Rheum Dis. (2022) 81:379–85. doi: 10.1136/annrheumdis-2021-220066
75. Song X, Zhang Y, Zhao L, Fan J, Peng T, Ma Y, et al. Analyzation of the peripheral blood mononuclear cells atlas and cell communication of rheumatoid arthritis patients based on single-cell RNA-seq. J Immunol Res. (2023) 2023:6300633. doi: 10.1155/2023/6300633
76. Kosmaczewska A, Swierkot J, Ciszak L, Wiland P. The role of Th1, Th17, and Treg cells in the pathogenesis of rheumatoid arthritis including anti-inflammatory action of Th1 cytokines. Postepy Hig Med Dosw (Online). (2011) 65:397–403. doi: 10.5604/17322693.948971
77. Ferraccioli G, Carbonella A, Gremese E, Alivernini S. Rheumatoid arthritis and Alzheimer’s disease: genetic and epigenetic links in inflammatory regulation. Discovery Med. (2012) 14:379–88.
78. Gillespie J, Savic S, Wong C, Hempshall A, Inman M, Emery P, et al. Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum. (2012) 64:418–22. doi: 10.1002/art.33382
79. Li Y, Zhou M, Lv X, Song L, Zhang D, He Y, et al. Reduced activity of HDAC3 and increased acetylation of histones H3 in peripheral blood mononuclear cells of patients with rheumatoid arthritis. J Immunol Res. (2018) 2018:7313515. doi: 10.1155/2018/7313515
80. Meng F, Li Z, Zhang Z, Yang Z, Kang Y, Zhao X, et al. MicroRNA-193b-3p regulates chondrogenesis and chondrocyte metabolism by targeting HDAC3. Theranostics. (2018) 8:2862–83. doi: 10.7150/thno.23547
81. Roth GA, Huffman MD, Moran AE, Feigin V, Mensah GA, Naghavi M, et al. Global and regional patterns in cardiovascular mortality from 1990 to 2013. Circulation. (2015) 132:1667–78. doi: 10.1161/CIRCULATIONAHA.114.008720
82. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. (2013) 13:709–21. doi: 10.1038/nri3520
83. Xu F, Shen L, Chen H, Wang R, Zang T, Qian J, et al. circDENND1B Participates in the Antiatherosclerotic Effect of IL-1beta Monoclonal Antibody in Mouse by Promoting Cholesterol Efflux via miR-17-5p/Abca1 Axis. Front Cell Dev Biol. (2021) 9:652032. doi: 10.3389/fcell.2021.652032
84. Hoeksema MA, Gijbels MJ, Van den Bossche J, van der Velden S, Sijm A, Neele AE, et al. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. (2014) 6:1124–32. doi: 10.15252/emmm.201404170
85. Inoue K, Kobayashi M, Yano K, Miura M, Izumi A, Mataki C, et al. Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule-1 expression. Arterioscler Thromb Vasc Biol. (2006) 26:2652–9. doi: 10.1161/01.ATV.0000247247.89787.e7
86. Zampetaki A, Zeng L, Margariti A, Xiao Q, Li H, Zhang Z, et al. Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation. (2010) 121:132–42. doi: 10.1161/CIRCULATIONAHA.109.890491
87. Huang S, Chen G, Sun J, Chen Y, Wang N, Dong Y, et al. Histone deacetylase 3 inhibition alleviates type 2 diabetes mellitus-induced endothelial dysfunction via Nrf2. Cell Commun Signal. (2021) 19:35. doi: 10.1186/s12964-020-00681-z
88. Alam S, Li H, Margariti A, Martin D, Zampetaki A, Habi O, et al. Galectin-9 protein expression in endothelial cells is positively regulated by histone deacetylase 3. J Biol Chem. (2011) 286:44211–7. doi: 10.1074/jbc.M111.242289
89. Li Y, Lui KO, Zhou B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol. (2018) 15:445–56. doi: 10.1038/s41569-018-0023-y
90. Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. (2015) 125:4514–28. doi: 10.1172/JCI82719
91. Chen L, Shang C, Wang B, Wang G, Jin Z, Yao F, et al. HDAC3 inhibitor suppresses endothelial-to-mesenchymal transition via modulating inflammatory response in atherosclerosis. Biochem Pharmacol. (2021) 192:114716. doi: 10.1016/j.bcp.2021.114716
92. Song K, Li L, Quan Q, Wei Y, Hu S. Inhibited histone deacetylase 3 ameliorates myocardial ischemia-reperfusion injury in a rat model by elevating microRNA-19a-3p and reducing cyclin-dependent kinase 2. IUBMB Life. (2020) 72:2696–709. doi: 10.1002/iub.2402
93. Wang J, Xu X, Li P, Zhang B, Zhang J. HDAC3 protects against atherosclerosis through inhibition of inflammation via the microRNA-19b/PPARgamma/NF-kappaB axis. Atherosclerosis. (2021) 323:1–12. doi: 10.1016/j.atherosclerosis.2021.02.013
94. Winkler AR, Nocka KN, Williams CM. Smoke exposure of human macrophages reduces HDAC3 activity, resulting in enhanced inflammatory cytokine production. Pulm Pharmacol Ther. (2012) 25:286–92. doi: 10.1016/j.pupt.2012.05.003
95. Sun X, Liu Y, Feng X, Li C, Li S, Zhao Z, et al. The key role of macrophage depolarization in the treatment of COPD with ergosterol both in vitro and in vivo. Int Immunopharmacol. (2020) 79:106086. doi: 10.1016/j.intimp.2019.106086
96. Pooladanda V, Thatikonda S, Bale S, Pattnaik B, Sigalapalli DK, Bathini NB, et al. Nimbolide protects against endotoxin-induced acute respiratory distress syndrome by inhibiting TNF-alpha mediated NF-kappaB and HDAC-3 nuclear translocation. Cell Death Dis. (2019) 10:81. doi: 10.1038/s41419-018-1247-9
97. Leus NG, van der Wouden PE, van den Bosch T, Hooghiemstra WTR, Ourailidou ME, Kistemaker LE, et al. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-kappaB p65 transcriptional activity. Biochem Pharmacol. (2016) 108:58–74. doi: 10.1016/j.bcp.2016.03.010
98. Gunawardhana LP, Gibson PG, Simpson JL, Powell H, Baines KJ. Activity and expression of histone acetylases and deacetylases in inflammatory phenotypes of asthma. Clin Exp Allergy. (2014) 44:47–57. doi: 10.1111/cea.12168
99. Chou DH, Holson EB, Wagner FF, Tang AJ, Maglathlin RL, Lewis TA, et al. Inhibition of histone deacetylase 3 protects beta cells from cytokine-induced apoptosis. Chem Biol. (2012) 19:669–73. doi: 10.1016/j.chembiol.2012.05.010
100. Lundh M, Galbo T, Poulsen SS, Mandrup-Poulsen T. Histone deacetylase 3 inhibition improves glycaemia and insulin secretion in obese diabetic rats. Diabetes Obes Metab. (2015) 17:703–7. doi: 10.1111/dom.12470
101. Sathishkumar C, Prabu P, Balakumar M, Lenin R, Prabhu D, Anjana RM, et al. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin Epigenet. (2016) 8:125. doi: 10.1186/s13148-016-0293-3
102. Yaturu S, Humphrey S, Landry C, Jain SK. Decreased bone mineral density in men with metabolic syndrome alone and with type 2 diabetes. Med Sci Monit. (2009) 15:CR5–9.
103. Guo CJ, Xie JJ, Hong RH, Pan HS, Zhang FG, Liang YM. Puerarin alleviates streptozotocin (STZ)-induced osteoporosis in rats through suppressing inflammation and apoptosis via HDAC1/HDAC3 signaling. BioMed Pharmacother. (2019) 115:108570. doi: 10.1016/j.biopha.2019.01.031
104. Xu Z, Tong Q, Zhang Z, Wang S, Zheng Y, Liu Q, et al. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin Sci (Lond). (2017) 131:1841–57. doi: 10.1042/CS20170064
105. Zhao B, Yuan Q, Hou JB, Xia ZY, Zhan LY, Li M, et al. Inhibition of HDAC3 ameliorates cerebral ischemia reperfusion injury in diabetic mice in vivo and in vitro. J Diabetes Res 2019. (2019) 2019:8520856. doi: 10.1155/2019/8520856
106. Zhang J, Xu Z, Gu J, Jiang S, Liu Q, Zheng Y, et al. HDAC3 inhibition in diabetic mice may activate Nrf2 preventing diabetes-induced liver damage and FGF21 synthesis and secretion leading to aortic protection. Am J Physiol Endocrinol Metab. (2018) 315:E150–62. doi: 10.1152/ajpendo.00465.2017
107. Sun Z, Miller RA, Patel RT, Chen J, Dhir R, Wang H, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med. (2012) 18:934–42. doi: 10.1038/nm.2744
108. Lee SJ, Choi SE, Lee HB, Song MW, Kim YH, Jeong JY, et al. A class I histone deacetylase inhibitor attenuates insulin resistance and inflammation in palmitate-treated C2C12 myotubes and muscle of HF/HFr diet mice. Front Pharmacol. (2020) 11:601448. doi: 10.3389/fphar.2020.601448
109. Bhavsar P, Ahmad T, Adcock IM. The role of histone deacetylases in asthma and allergic diseases. J Allergy Clin Immunol. (2008) 121:580–4. doi: 10.1016/j.jaci.2007.12.1156
110. Kim Y, Kim K, Park D, Lee E, Lee H, Lee YS, et al. Histone deacetylase 3 mediates allergic skin inflammation by regulating expression of MCP1 protein. J Biol Chem. (2012) 287:25844–59. doi: 10.1074/jbc.M112.348284
111. Noh K, Kim M, Kim Y, Kim H, Kim H, Byun J, et al. miR-122-SOCS1-JAK2 axis regulates allergic inflammation and allergic inflammation-promoted cellular interactions. Oncotarget. (2017) 8:63155–76. doi: 10.18632/oncotarget.v8i38
112. Ghosh S, Samarasinghe AE, Hoselton SA, Dorsam GP, Schuh JM. Hyaluronan deposition and co-localization with inflammatory cells and collagen in a murine model of fungal allergic asthma. Inflammation Res. (2014) 63:475–84. doi: 10.1007/s00011-014-0719-3
113. Zhang W, Sun X, Ba G, Tang R, Lin H. RGFP966, a selective HDAC3 inhibitor, ameliorates allergic and inflammatory responses in an OVA-induced allergic rhinitis mouse model. Int Immunopharmacol. (2021) 93:107400. doi: 10.1016/j.intimp.2021.107400
114. Wang Y, Jiao B, Hu Z, Wang Y. Critical Role of histone deacetylase 3 in the regulation of kidney inflammation and fibrosis. Kidney Int. (2024) 105:775–90. doi: 10.1016/j.kint.2024.01.010
115. Hu L, Yang K, Mai X, Wei J, Ma C. Depleted HDAC3 attenuates hyperuricemia-induced renal interstitial fibrosis via miR-19b-3p/SF3B3 axis. Cell Cycle. (2022) 21:450–61. doi: 10.1080/15384101.2021.1989899
116. Tanemoto F, Mimura I. Therapies targeting epigenetic alterations in acute kidney injury-to-chronic kidney disease transition. Pharm (Basel). (2022) 15(2). doi: 10.3390/ph15020123
117. Zhang L, Chen F, Dong J, Wang R, Bi G, Xu D, et al. HDAC3 aberration-incurred GPX4 suppression drives renal ferroptosis and AKI-CKD progression. Redox Biol. (2023) 68:102939. doi: 10.1016/j.redox.2023.102939
118. Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, et al. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci USA. (2013) 110:2647–52. doi: 10.1073/pnas.1213364110
Keywords: HDAC3, inflammation, histone deacetylases, epigenetic mechanisms, HDAC
Citation: Watson N, Kuppuswamy S, Ledford WL and Sukumari-Ramesh S (2024) The role of HDAC3 in inflammation: mechanisms and therapeutic implications. Front. Immunol. 15:1419685. doi: 10.3389/fimmu.2024.1419685
Received: 18 April 2024; Accepted: 10 June 2024;
Published: 10 July 2024.
Edited by:
Jixin Zhong, Huazhong University of Science and Technology, ChinaReviewed by:
Aaron Winkler, Pfizer, United StatesCopyright © 2024 Watson, Kuppuswamy, Ledford and Sukumari-Ramesh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sangeetha Sukumari-Ramesh, c3JhbWVzaEBhdWd1c3RhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.