 Jun Wang
Jun Wang Hongzhou Lu
Hongzhou Lu Qian Li
Qian Li- 1The Third People’s Hospital of Shenzhen (National Clinical Research Center for Infectious Diseases) and The Second Affiliated Hospital of Southern University of Science and Technology, Shenzhen, Guangdong, China
- 2Clinical Research Center, The Fifth People’s Hospital of Wuxi, Jiangnan University, Wuxi, Jiangsu, China
Hepatitis B Virus (HBV) is a stealthy and insidious pathogen capable of inducing chronic necro-inflammatory liver disease and hepatocellular carcinoma (HCC), resulting in over one million deaths worldwide per year. The traditional understanding of Chronic Hepatitis B (CHB) progression has focused on the complex interplay among ongoing virus replication, aberrant immune responses, and liver pathogenesis. However, the dynamic progression and crucial factors involved in the transition from HBV infection to immune activation and intrahepatic inflammation remain elusive. Recent insights have illuminated HBV’s exploitation of the sodium taurocholate co-transporting polypeptide (NTCP) and manipulation of the cholesterol transport system shared between macrophages and hepatocytes for viral entry. These discoveries deepen our understanding of HBV as a virus that hijacks hepatocyte metabolism. Moreover, hepatic niche macrophages exhibit significant phenotypic and functional diversity, zonal characteristics, and play essential roles, either in maintaining liver homeostasis or contributing to the pathogenesis of chronic liver diseases. Therefore, we underscore recent revelations concerning the importance of hepatic niche macrophages in the context of viral hepatitis. This review particularly emphasizes the significant role of HBV-induced metabolic changes in hepatic macrophages as a key factor in the transition from viral infection to immune activation, ultimately culminating in liver inflammation. These metabolic alterations in hepatic macrophages offer promising targets for therapeutic interventions and serve as valuable early warning indicators, shedding light on the disease progression.
1 Introduction
Chronic Hepatitis B (CHB) affects metabolic processes, potentially contributing to the development of Metabolic Dysfunction-Associated Fatty Liver Disease (MASLD). This dual pathology intensifies further liver injury, increasing the risks of cirrhosis and liver cancer, and imposing a significant disease burden (1–4). Although CHB and MASLD share common risk factors for liver fibrosis and cirrhosis, particularly host metabolic factors, the causal link between hepatitis B virus (HBV) infection and MASLD remains elusive (1, 2). This is largely due to an incomplete understanding of how HBV impacts host metabolic processes and subsequently influences immunopathogenesis.
The inflammatory responses induced by the HBV are capable to exacerbate hepatic steatosis through various mechanisms. HBV DNA transcription can lead to metabolic reprogramming in the liver, accelerating processes such as hepatic regeneration, inflammation, and fibrosis (5). The HBV X (HBx) protein activates signaling pathways such as Phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) and Toll-like receptors (TLRs), which influence hepatic lipogenesis, the conversion of cholesterol to bile acids (BAs), and hepatic lipid homeostasis, thereby contributing to hepatic steatosis (6–9). Additionally, HBV pre-S1 binding to NTCP) may alter hepatic cholesterol metabolism, leading to hepatic steatosis (10). Despite these findings collectively support the notion that metabolism plays a significant role in liver inflammation in CHB, the specific details of the intermediate mechanisms connecting intrahepatic metabolic change with immune infiltration during CHB remain unclear.
Growing evidence has reinforced the notion that HBV can manipulate metabolic processes by cross-talking with macrophages. Basically, HBV was known as a stealthy virus which can hijack the host’s BAs metabolic pathway. This can occur either by directly sequestering them through binding to the NTCP receptor or by being taken up by a subtype of macrophages via the cholesterol transport system to hepatocytes for viral entry (10). Consequently, the overloaded lipid transport system leads to compensatory up-regulation of BAs and dysfunctional cholesterol metabolism, resulting in the accumulation of lipid metabolites. This accumulation gradually triggers an intensified inflammatory response within hepatic macrophages (11, 12). With consistent exposure to lipid metabolites, hepatic macrophages are likely to undergo significant phenotypic and functional changes, primarily within the vicinity of the liver portal area, bile ducts, or lipid-accumulating regions (13, 14). For instance, a specific subset of liver macrophages, Trem2+ macrophages, has been found to shift toward a pro-inflammatory state in CHB-ACLF (14). Additionally, hepatic macrophages can release chemokines such as C-X-C motif chemokine ligand 9 and 10 (CXCL9 and CXCL10), which attract lymphocytes and monocytes/macrophages, thereby further contributing to liver inflammation (15). Hence, liver macrophages act as a conduit and potentially connect HBV-induced metabolic shifts with immune infiltrates exhibiting zonation characteristics.
The current consensus recognizes that hepatic niche macrophages, comprising resident macrophages, Kupffer cells (KCs), and monocyte-derived macrophages (MDMs) and their various subsets, play a crucial role in maintaining hepatic homeostasis by participating in various processes, including metabolic regulation, modulation of inflammation, promotion of immune tolerance, support for immunologic cell death, and facilitation of tissue repair in the physiological state (16–18). Despite researchers having a profound understanding of these issues, discussions regarding the involvement of hepatic macrophages in CHB progression have been still prominent, particularly in relation to three questions: 1) whether they promote immune activation or immune tolerance during CHB, 2) how they perceive hepatocyte stress or directly detect pathogens, and 3) what are their intrahepatic roles in HBV infection, metabolic hijacking and immune inflammation?
This review provides a contemporary insight into the knowledge accumulated in recent years. We emphasize the novel perspectives for intrahepatic metabolic alterations during HBV entry and replication, and shedding light on the multifaceted functions of hepatic macrophages during CHB processes, including bridge between HBV-Mediated Metabolic Change with Intrahepatic Inflammation.
2 Metabolic changes and characteristics during HBV entry and intrahepatic replication
2.1 Novel route for HBV entry: macrophage-mediated reverse cholesterol transport enhances HBV targeting in hepatocytes
Although HBV is known to enter hepatocytes through NTCP, the mechanism by which a single HBV particle can establish chronic liver infection after intravenous injection, evading scavenger cells in the circulation or spleen, has remained a puzzle (10, 19). Surprisingly, Esser K. et al., utilizing an ex vivo liver perfusion model, discovered that HBV, in association with lipoproteins, exhibited a tendency to be taken up by liver macrophages, leading to its accumulation in recycling endosomes before being re-secreted by macrophages, this time in association with free cholesterol obtained from the endocytosed lipoproteins. Subsequently, in the same ex vivo model, HBV was taken up by hepatocytes along with macrophage-derived neutral lipids (20). These findings support the conclusion that HBV efficiently targets hepatocytes not solely through a routine viral-specific host cell approach but also by exploiting neighboring cells in the liver. This research provides compelling evidence that HBV associates with lipoproteins, using the cholesterol transport pathway through macrophages and their cargo delivery system to reach hepatocytes (Figure 1).
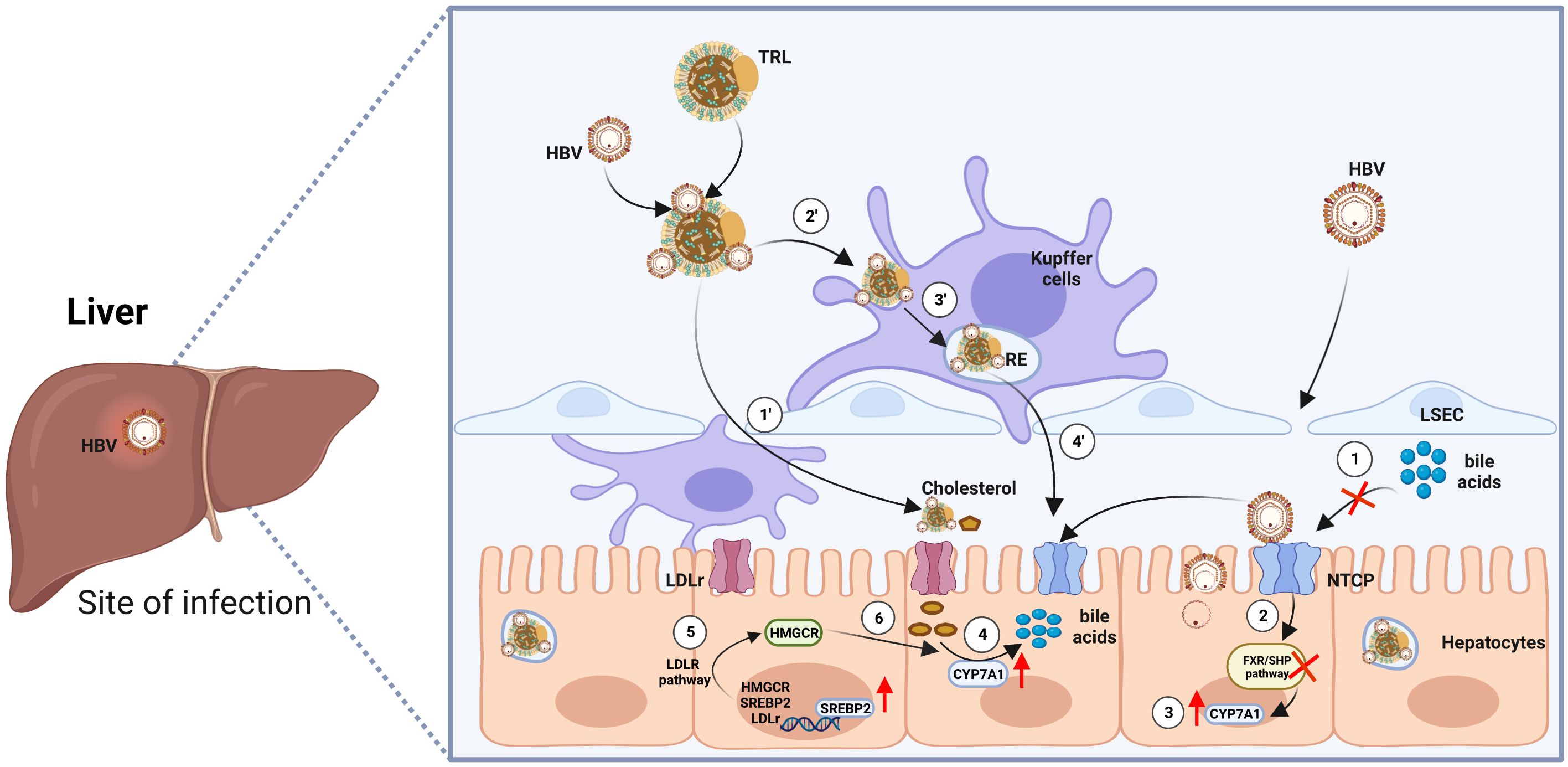
Figure 1 Model of HBV entry into hepatocytes via two routes. First Route: ① Enveloped hepatitis B virus (HBV) particles circulate within the sinusoidal blood, traversing the fenestrae of sinusoidal endothelial cells to access the space of Disse. Eventually, they reach the integral membrane protein receptor NTCP, which concurrently impedes the transport of bile acids. ② Repressed bile acid transport inhibits the farnesoid X receptor/small heterodimer partner (FXR/SHP) signaling pathway, leading to the compulsory upregulation of the transcription factor Cytochrome P450 Family 7 Subfamily A Member 1 (CYP7A1) ③. ④ Increased CYP7A1 accelerates the reversed transport of cholesterol and promotes the synthesis of bile acids. ⑤ The low density lipoprotein receptor (LDLR) pathway is activated, further promoting the process of bile acid synthesis. Second Route: ① HBV became entrapped within serum triglyceride-rich lipoproteins (TRL), traverse the endothelial cell layer to access the space of Disse. Once there, they are ensnared through electrostatic interactions with HSPG (heparan sulfate proteoglycan) before ultimately binding with NTCP (sodium taurocholate co-transporting polypeptide). ② ③ When associated with lipoproteins, HBV is preferentially internalized by liver macrophages into recycling endosomes. ④ Subsequently, HBV particles enter hepatocytes. Cholesterol esters of endocytosed lipoproteins undergo hydrolysis by the endosomal acid lipase, liberating free cholesterol. This free cholesterol is then conveyed to the cell membrane, released from the macrophage, and can bind to extracellular lipid receptors, facilitating additional uptake into hepatocytes. The figure was created by BioRender (BioRender.com).
However, echoing Cheng Y’s remarks, a fundamental question remains unresolved: How does HBV interact with Apolipoprotein E (ApoE)-rich lipoproteins (21)? Given the high efficacy of HBV in exploiting macrophages for trans-infection of hepatocytes, we posit that HBV may engage in a molecular interaction with ApoE, which could potentially involve their protein structures. To address this question, we performed docking simulations involving Hepatitis B core antigen (HBcAg) (Figure 2A), Hepatitis B surface antigen (HBsAg) (Figure 2B), Hepatitis B e antigen (HBeAg) (Figure 2C) proteins, and ApoE molecules. The top 15 weighted scores of the “balance” calculation, calculated from the interface root mean square deviation (IRMSD) method by the Cluspro2 online analysis platform, are normalized and presented as a heatmap (Figure 2D). In contrast to HBsAg, HBcAg, and HBeAg have a more robust binding with ApoE. This kind of conjunction could happen in HBV-uptaking macrophages.
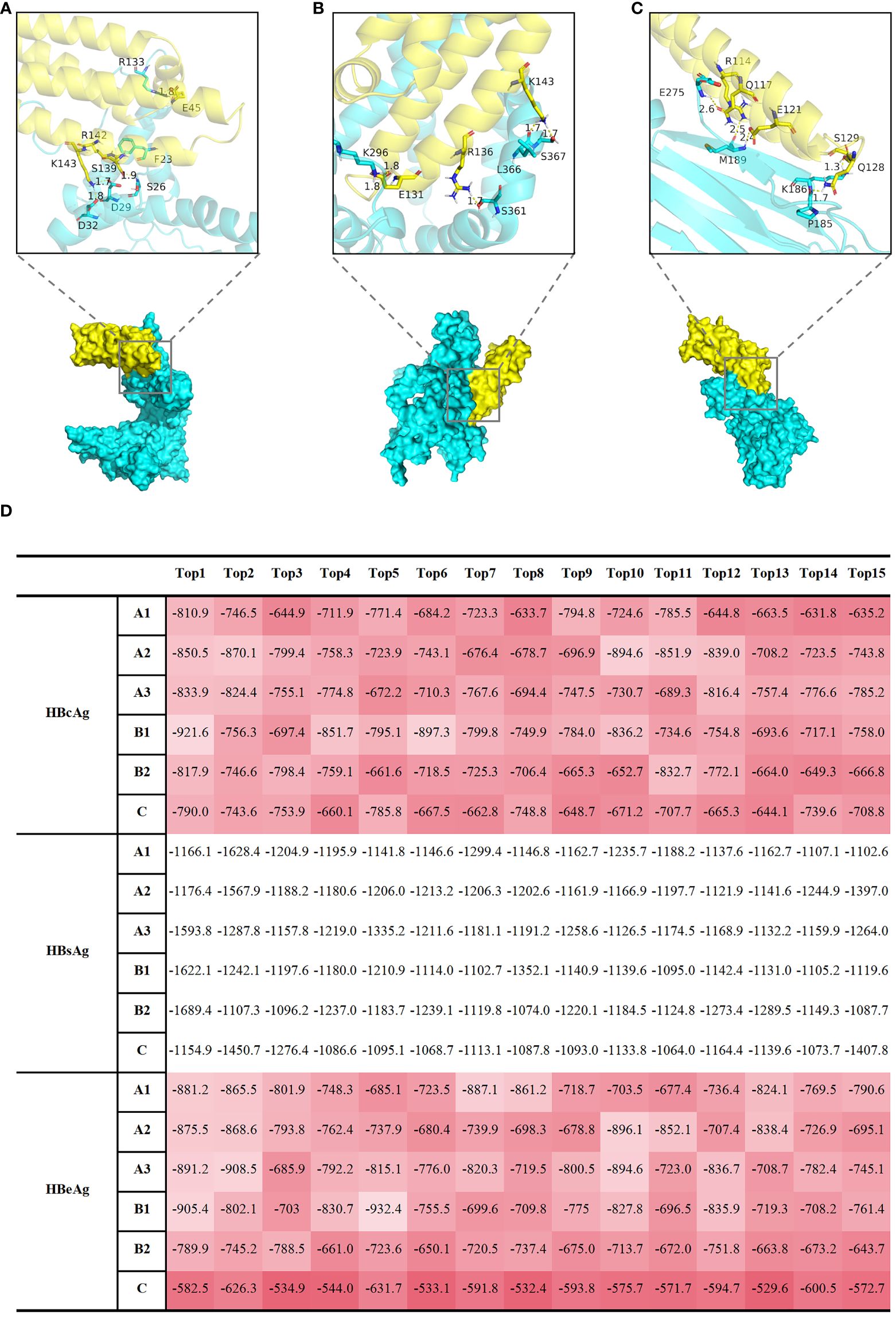
Figure 2 Computational docking analyses were performed to examine the interactions between HBcAg, HBsAg, and HBeAg proteins with APOE molecules, shedding light on potential molecular associations. Display of docking results between representative HBcAg (protein ID, Q9E6S6) (A), HBsAg (protein ID, Q9E6S4) (B), HBeAg (protein ID, P0C6H2) (C) proteins, and APOE (protein ID, P02649) molecules. The protein IDs were obtained from UniProt (https://www.uniprot.org/). The yellow protein represents APOE, and the Cyan protein represents HBV. Local graphs depict the Top 5 polar bonds with the closest distances in the docking results. Red denotes oxygen (O) atoms, and blue denotes nitrogen (N) atoms. Numbers indicate the distances between atoms forming polar bonds (unit: angstroms, Å). (D) The table displays the top 15 results of the 'balance' calculation weight scores, obtained through the Cluspro2 online analysis platform using the interface root mean square deviation (IRMSD) method. A1, A2, A3, B1, B2, C represent different HBV subtypes, each uniquely identified by specific protein IDs obtained from UniProt (https://www.uniprot.org/). The protein IDs for HBeAg are as follows: Q91C37 (A1 subgenotype), P0C692 (A2 subgenotype), Q4R1S0 (A3 subgenotype), P0C699 (B1 subgenotype), P0C6G7 (B2 subgenotype), P0C6H2 (C subgenotype); The protein IDs for HBsAg are as follows: P31873 (A1 subgenotype), O91534 (A2 subgenotype), Q4R1S6 (A3 subgenotype), Q9QBF0 (B1 subgenotype), Q9QAB7 (B2 subgenotype), Q9E6S4 (C subgenotype); The protein IDs for HBcAg are as follows: I7JHV6 (A1 subgenotype), P0C696 (A2 subgenotype), P0C697 (A3 subgenotype), P0C677 (B1 subgenotype), Q9QAB9 (B2 subgenotype), Q9E6S6 (C subgenotype); The protein IDs for AOPE is P02649. A higher value, represented by a darker red shade in (D), corresponds to a stronger polar bond energy, suggesting a more favorable and robust binding between the two proteins.
2.2 Altered lipid metabolism in CHB livers induces hepatocyte damage by activating DAMPs and PAMPs
As depicted in Figure 1, lipid metabolism is altered from the beginning of HBV entry. However, hepatocytes damage with the abnormal elevation of alanine transaminase (ALT) only happened during hepatitis phase of CHB infection. As shown in the study by Allen et al., treatment with BAs did not lead to increased caspase 3 activity in mouse hepatocytes or the release of ALT into the culture medium. This compelling observation strongly supports the notion that BAs do not directly induce apoptosis or necrosis in hepatocytes (22, 23). Furthermore, early growth response protein 1 (Egr1), a critical regulator of various genes, particularly those involved in pro-inflammatory cytokine production and pathways associated with pathogen-associated molecular patterns (PAMPs), was found to be upregulated (22). Additional evidence indicates that BAs need to enter and accumulate within hepatocytes to stimulate cytokines expression, and this effect is not mediated by cytokines membrane receptors (24, 25). Taken together, these findings, observed in both humans and mice, support the hypothesis that when BAs accumulate within hepatocytes, they initiate liver injury by triggering an inflammatory response, leading to the production of pathophysiologically relevant concentrations of chemokines and adhesion molecules, including Monocyte Chemoattractant Protein-1 (MCP-1/Ccl2), Macrophage inflammatory protein-2 (MIP-2/Cxcl2), and Intercellular cell adhesion molecule-1 (ICAM-1) (24) of which lay the foundation for the infiltration of monocytes/macrophages.
Moreover, once these BAs accumulate within the cell, they trigger endoplasmic reticulum (ER) stress and induce mitochondrial damage (25–27). The detection of mitochondrial damage in hepatocytes treated with BAs implies that these damaged mitochondria might release damage-associated molecular patterns (DAMPs), subsequently activating PAMPs, such as TLRs. Notably, TLR9 is a receptor residing within the ER and endosomes, serving as an intracellular DNA sensor. Previous research has shown that mitochondrial DNA can activate TLR9 and stimulate the expression of inflammatory cytokines (27). The involvement of TLR9 is further substantiated by observations that bile acid-induced cytokines responses are diminished in mouse hepatocytes when mitochondria are protected by agents such as cyclosporine A (27). Based on this evidence, we endorse the idea that progressive lipid metabolism alteration induced by HBV infection initiates PAMPs and DAMPs, thereby shaping a pro-inflammatory intrahepatic micro-environment, without direct damage to hepatocytes (Figure 3).
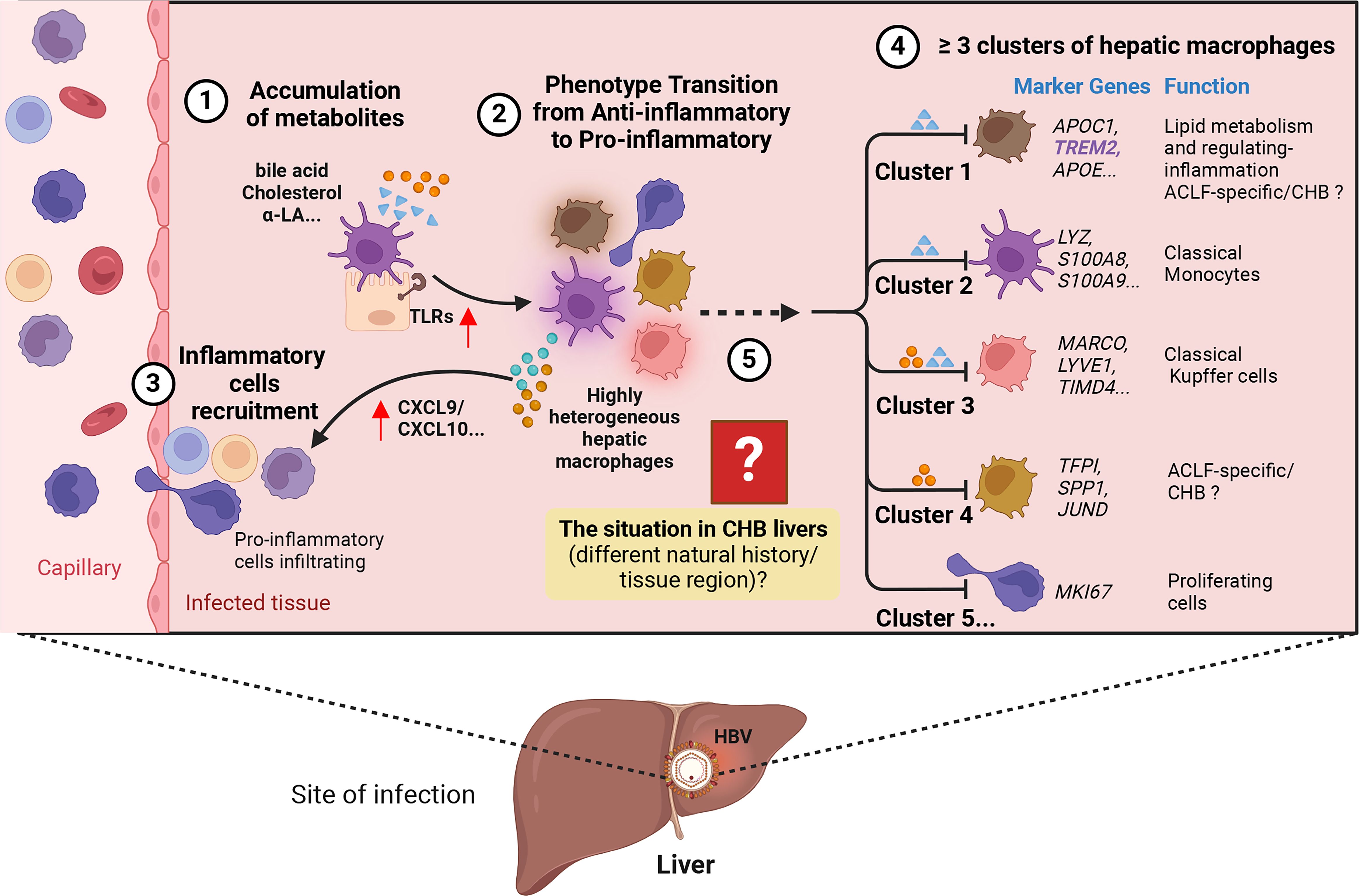
Figure 3 Hypothesis on the process of how HBV induces liver injury. ① Accumulation of metabolites, including bile acids and cholesterol, in the liver. ② Metabolism alteration promotes the expression of pro-inflammatory signaling like Toll-like receptors (TLRs) in hepatocytes, leading to the transition of highly heterogeneous hepatic macrophages into a pro-inflammatory phenotype characterized by increased expression of pro-inflammatory cytokines and activation of specific signaling pathways. ③ More inflammatory cells are recruited into the liver through the release of chemokines like CXCL9/10. ④ Within the highly heterogeneous hepatic macrophages, the presence of at least five potential clusters can be identified in both physiological and pathological states of the liver (e.g., Acute-on-Chronic Liver Failure, ACLF; Chronic hepatitis B, CHB…). Each cluster is associated with specific marker genes and potential functions. ⑤ The phenotypic and functional transitions may exhibit variation across different stages of CHB and diverse liver regions. The figure was created by BioRender (BioRender.com).
2.3 Characteristics of intrahepatic metabolic changes during CHB: spatial and temporal specificity
With our advancing understanding of liver anatomy and the liver micro-environment, Guilliams M. et al., emphasized that the liver has a dynamic complexity that has not yet been adequately described (28). We now recognize the liver as a complex organ characterized by a large number of microscopic functional units, including hepatic (classic) lobules, portal lobules, and liver acini. These acini can be divided into three zones based on their proximity to portal canals and adjacent central veins, contributing to uneven blood flow in these three zones. Questions about the relationship between liver blood flow and hepatic metabolism were raised by Macdonald AC as early as 1979 (29). In line with earlier perspectives, Esser K, et al., also explored the deposition of HBV in the perisinusoidal space of Disse, where HBV can bind to its receptor on hepatocytes or even be directly transported to hepatocytes by disrupting the reverse cholesterol transport (20). The precise metabolic alterations induced by HBV in different regions of the liver, various cell types, and at different disease stages remain largely unknown.
Most recently, Li J. et al., conducted an analysis of PBMC transcriptomics from patients in five distinct disease stages, including acute-on-chronic liver failure (ACLF), acute-on-chronic hepatic dysfunction (ACHD), liver cirrhosis (LC), CHB, and normal controls (NC). Their study aimed to demonstrate the presence of immune-metabolism disorders during the development of HBV-ACLF. Notably, they reported increased expression of metabolic genes related to lipid metabolism, fatty acid metabolism, oxygen homeostasis, and autophagy in the PBMC transcriptomics of CHB patients compared to healthy controls (HC) (13). Subsequently, Peng B. et al., characterized the immune micro-environment in the livers of patients with ACLF and also investigated the role of lipid metabolism. They employed single-cell RNA-sequencing (scRNA-seq) to examine liver non-parenchymal cells (NPCs) and PBMCs from healthy controls, cirrhosis patients, and ACLF patients. It’s important to note that well-defined natural history disease stages of CHB patients were not included for further comparison (14).
In 2021, Sun Z. et al., conducted a study comparing serum BA profiles among different groups: CHB patients with normal ALT (CHB-NALT), CHB patients with abnormal ALT (CHB-AALT), and healthy controls (HCs). They employed Ultra-high-performance liquid-chromatography and analyzed transcriptomic data of hepatic gene expression. The findings revealed a significantly higher percentage of conjugated BAs and primary BAs in CHB patients, even in the absence of apparent liver injury. Moreover, CHB-AALT group exhibited higher levels of serum BA species, including glycolithocholic acid (GLCA), taurochenodeoxycholic acid (TDCA), taurolithocholic acid (TLCA), and taurochenodeoxycholic acid (TUCDA) compared to CHB-NALT (30). Clearly, there is a distinct metabolic fingerprint between the inflammatory (hepatitis) and non-inflammatory (non-hepatitis) phases. However, the specific metabolic changes that occur at each of the four natural infection history stages, and the factors that determine the transition between these stages, remain largely unknown.
Despite the rapid advancements in omics-sequencing methods, including spatial transcriptomics and spatial proteomics, there is still a lack of evidence regarding the spatial (tissue/cell-specific) and temporal (different disease stages) metabolic changes within the liver tissues in CHB.
3 Limitations of current studies on the mechanisms of HBV-mediated metabolic changes: are hepatocytes the sole targets?
Current studies suggest that hepatocytes are the primary targets of HBV-induced intrahepatic metabolic changes. Are hepatocytes the only targets of HBV-induced intrahepatic metabolic changes? Shin HJ. et al., demonstrated that the HBx promotes gluconeogenesis through the nitric oxide (NO)/c-Jun N-terminal kinases (JNK) pathway (31). Li H. et al., verified that HBV upregulates glycolysis (32). In addition, Liu B. et al., found that HBx-mediated NF-E2-related factor 2 (Nrf2) activation promotes the pentose phosphate pathway (PPP) by stimulating glucose-6-phosphate dehydrogenase (G6PD) expression (33). Furthermore, accumulating evidence from studies using HBV-replicating cell lines and mouse models have shown that HBV can promote fatty acid synthesis through various mechanisms, and is a potential trigger of liver steatosis (6–8, 24, 33–36). Additionally, HBV infection also impacts other hepatic metabolic signaling pathways, such as nucleic acid metabolism and vitamin metabolism (37, 38).
Notably, the identification of the bona fide receptor for HBV, confirmed through stable HBV infection models and human hepatocytes, has revealed that the viral entry mechanisms for HBV (19). However, it remains unclear whether and what extent HBV can influence cellular metabolism via NTCP. Oehler N. et al., subsequently demonstrate that the binding of HBV to NTCP limits its function, subsequently promoting compensatory BA synthesis and cholesterol provision (39). Patman G. et al., further support that HBV infection alters bile acid metabolism (40).
In other words, traditional studies on how HBV infection affects metabolic changes has predominantly relied on hepatoma cell lines, HBV transgenic mice, or peripheral non-tumor tissues after surgical resection. It’s worth noting that hepatocyte-specific transporters are known to be altered or lost in cell lines, mice models, and primary human hepatocyte cultures (Table 1). The narrow tissue and host tropism of HBV has constrained studies on interactions established by HBV in human hepatic niche (41, 42).
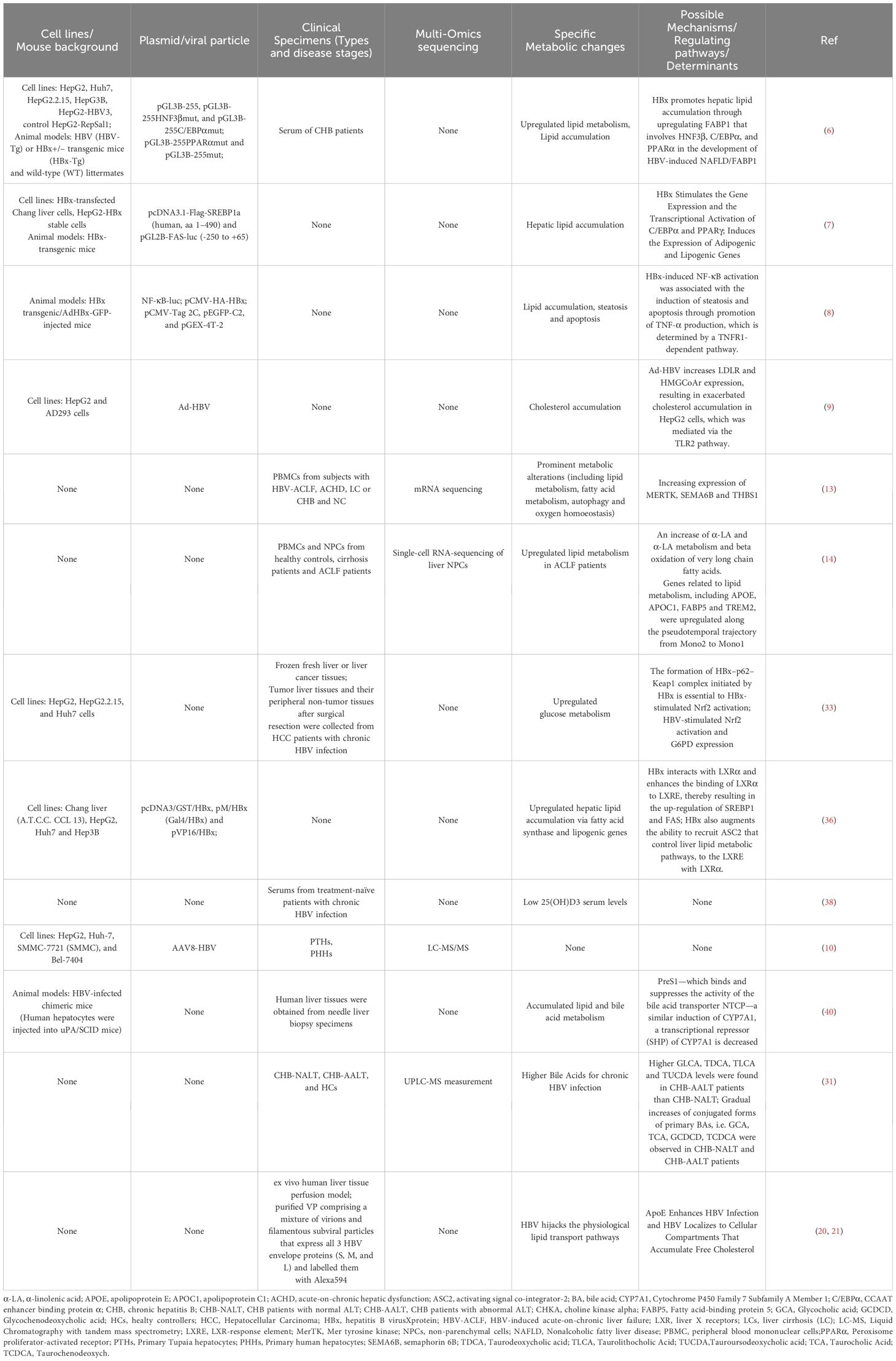
Table 1 Summary of HBV induced metabolic alterations via cells lines, animal models, and clinical specimens.
4 Function of hepatic macrophages niche: shaping intrahepatic micro-environment for immune tolerance or infiltration
4.1 Viral replication magnitude: a contradictory determinant of immune tolerance and immune activation
It is known that KCs prefer to reside in the liver, and they are enriched with an array of pattern recognition receptors (PRRs) on cellular surface, including TLRs and nucleotide-binding oligomerization domain-like receptors (NLRs). This localization and receptor expression make them first-line responders to pathogens such as HBV. Liver macrophages are also capable of directly recognizing HBV (43, 44). However, the long-standing debate as to whether HBV induces immune activation or promotes liver tolerance remains unresolved.
Liu J. et al., demonstrated that HBV stimuli induce KC-mediated T cell tolerance through the secretion of IL-10 in a TLR2-dependent manner (45). Li’s research further supported the concept that KCs Support HBV-Mediated CD8+ T cell exhaustion via the TLR2 signaling pathway (46). Suzanne Faure-Dupuy et al., reported that exposure to HBV or HBV-producing cells during differentiation and activation led to distinct responses. Pro-inflammatory macrophages (namely M1-MDMs) secreted lower levels of IL-6 and IL-1β, while anti-inflammatory macrophages (namely M2-MDMs)secreted higher levels of IL-10 when exposed to HBV during activation (47). In conclusion, their findings suggest that KCs play a role in promoting the secretion of anti-inflammatory cytokines such as IL-10 and TGF-β, contributing to transient activation by secreting IL-6, and potentially increasing the expression of inhibitory checkpoint factors (e.g., PD-1/PD-L1) (48). These results raise the possibility that HBV-driven limited activation of hepatic macrophages could be a contributing factor to the establishment and persistence of infections in vivo.
However, Cheng X. et al., provided evidence that HBV has evolved to evade the innate immunity of hepatocytes but activates macrophages during infection (49). Cheng’s research underscored that macrophage could detect HBV, but this typically requires exposure to high HBV titers. This observation may help explain the prolonged “window period” observed during acute infection and why HBV tends to establish chronic infections. In the most recent study, it was found that the magnitude of viral replication and the specific anti-viral immune responses should ideally dictate the extent of inflammation. However, it’s important to note that a direct correlation between these factors is not consistently observed in patients with chronic viral hepatitis (50).
In summary, HBV-mediated hepatic macrophages have the potential to induce either an anti-inflammatory or pro-tolerogenic differentiation. This phenotypic/functional switch may either support or inhibit further liver pathogenesis in CHB. Nevertheless, the precise mechanisms responsible for chronic liver inflammation and the key factors influencing the various stages of the disease’s natural history are yet to be fully elucidated (Figure 3).
4.2 Hepatic macrophage niche are attracted to sites where lipid metabolites are exposed for clearance
Single-cell and spatial transcriptomic technologies have unveiled an underappreciated heterogeneity among liver macrophages, prompting us to reconsider the role of macrophages in liver homeostasis and disease (17, 18). Previous research has categorized five hepatic macrophage clusters with distinct transcriptomes, including the MARCOhi cluster, Trem2hi cluster, S100A8hi cluster, MMP19hi cluster, and MKi67hi cluster in ACLF livers (13). Guilliams M, et al., further provided a more comprehensive depiction of the human liver atlas and identified two subsets of the Trem2hi cluster (mature and immature), which they later named Lipid-associated macrophages (LAMs) (18). Importantly, a notion supported by the induction of LAMs marker genes through the treatment of murine bone marrow macrophages with lipids in vitro. Besides, Ramachandran P. et al., demonstrated location of LAMs around the bile ducts in the healthy mouse, human, and macaque liver. However, in the presence of steatosis, LAMs are preferentially recruited to the steatotic regions of the liver (51). Guilliams M’s research suggests that LAMs can be induced by local lipid exposure (18).
Trem2 signaling has been recognized as a crucial sensor of metabolic pathology, responding to disruptions in tissue-level lipid homeostasis and giving rise to a novel and conserved Trem2+LAM subset (52–54). In accordance with these studies, Trem2+macrophages were found to express lipid metabolism genes marked by APOC1, APOE, GPNMB, and SPP1, as well as pro-inflammatory genes including IL18, CCL5, CCL18, among others (55). Additionally, the first identified scar-associated macrophages in liver cirrhosis, known as Trem2+CD9+macrophages, were found to originate from circulating monocytes and expand in the context of liver fibrosis. These Trem2+CD9+macrophages displayed pro-fibrogenic effects, contributing to tissue repair following liver injury (55). As metabolic alterations play a pivotal role in reshaping the hepatic micro-environment niche, including the recruitment of Trem2+monocytes/macrophages and the activation of Trem2+monocytes/macrophages toward a pro-inflammatory state, the precise phenotype and functional changes in the micro-environment of CHB livers, as well as variations across disease stages, remain unclear.
In a recent study, Dib L. et al., demonstrated that LAMs undergo a transition to an inflammatory state in human atherosclerosis. This transition revealed the presence of PLIN2hi/TREM1hi macrophages, which are associated with TLR-dependent inflammation and linked to cerebrovascular events (53). Peng B. et al., shed light on the immune-suppressive role of Trem2+monocytes/macrophages in the context of late-stage ACLF livers (14). Notably, their study provided further evidence that the accumulation of α-linolenic acid (α-LA) could enhance the expression of Trem2 on monocytes/macrophages in an inflammatory environment. This phenomenon was demonstrated through in vitro experiments involving LPS and α-LA stimulation on CD14+cells, suggesting that increased levels of unsaturated free fatty acids (FFAs) and disturbances in α-LA metabolism may serve as a bridge facilitating the differentiation and activation of Trem2+monocytes/macrophages in ACLF livers (14).
In the context of CHB, the binding of HBV pre-S1 to NTCP hinders bile acid uptake and compensatory upregulates the expression of cholesterol synthesis genes, including 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase and the LDL receptor. Consequently, this process is associated with the promotion of TLR signaling and inflammasome activation in hepatic macrophages (12, 13), suggesting it’s bridge role between enhancing lipid metabolites and pro-inflammation. However, HBV exploits NTCP in hepatocytes, disrupting the cholesterol transport pathway between macrophages and their target hepatocytes (10). Whether the specific type of hepatic macrophages involved, such as whether it is LAMs, known to be activated by local lipid exposure, and their role in eliminating excess metabolites and metabolic intermediates, triggering phenotype and functional reprogramming of the hepatic infiltrating microenvironment, remains unknown and is worthy of attention (Figure 4).
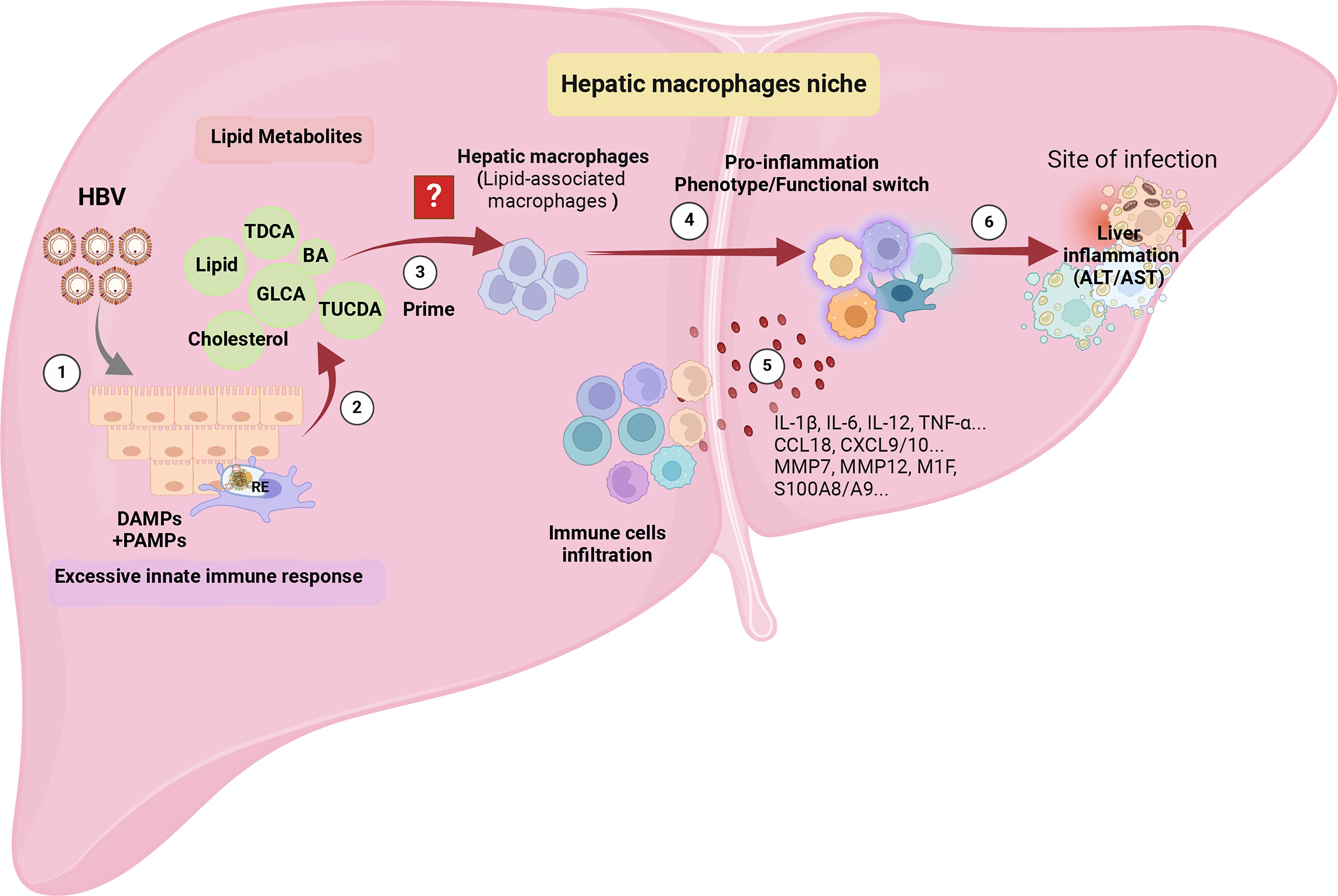
Figure 4 The potential immunopathology of how HBV shapes hepatic macrophage niches, ultimately leading to liver inflammation. ① Persistent HBV stimulation induces abnormal lipid metabolism, characterized by the accumulation of metabolites such as bile acids and cholesterol in the liver microenvironment. ② The aberrant lipid metabolism triggers an innate immune response by releasing PAMPs, such as the up-regulation of TLRs’ expression in hepatocytes, and DAMPs released by damaged hepatocyte mitochondria. ③ The accumulation of lipids recruits a specific cluster of macrophages, possibly Lipid-Associated Macrophages (LAMs), identified by the expression of Trem2, triggering a transition into a pro-inflammatory phenotype. ④ The activated LAMs exhibit a distinctive transcriptomic profile with elevated expression of IL-1β, IL-6, IL-12, TNF-α, CCL18, CXCL9/10, MMP7, MMP12, M1F, S100A8/A9, leading to the recruitment of more inflammatory cells. ⑤ Deepening liver inflammation occurs as a result of increased infiltration of inflammatory cells into the liver. The figure was created by BioRender (BioRender.com).
4.3 Pro-inflammatory hepatic macrophages, orchestrating increased liver infiltration and exacerbating liver inflammation
Traditional research categorized liver macrophages into “M1” or “M2” subsets, with a focus on their phenotype shifts and functional changes. M1 macrophages are associated with a pro-inflammatory effect, producing cytokines such as tumor necrosis factor (TNF)-α, IL-1β, and reactive oxygen species (ROS), which contribute to liver inflammation and injury as the disease progresses. In contrast, M2 macrophages exhibit an anti-inflammatory phenotype. It’s important to note that liver macrophages niche is highly plastic and can rapidly transition from a homeostatic state to a pro-inflammatory state in response to viral infection (56, 57).
Accordingly, we hypotheses that, the prolonged disruption in lipid and cholesterol efflux between hepatic macrophages and hepatocytes during the immune tolerance stage of CHB patients leads to the accumulation of excessive cholesterol and lipid metabolites. This, in turn, exacerbates pro-inflammatory signaling through both PAMPs and DAMPs within host cells. Consequently, this process reshapes the macrophage niche, inducing a transition in the immune micro-environment from macrophage-mediated tolerance to immune activation (58).
In Do TH’s research, Trem2+macrophages display heightened lipid metabolism profiles alongside pro-inflammatory profiles, resulting in the up regulation of pro-inflammatory chemokines, cytokines, MMPs, and S100 proteins to further recruit and activate immune cells (55). In Dib L’s research, once the macrophages shift into a pro-inflammatory phenotype (PLIN2hi/Trem1hi), they exhibit a unique chemokine signature with transcripts for pro-atherogenic CCR2 ligands (CCL2 and CCL7), which play a non-redundant role in atherogenesis and monocyte recruitment (51). Aberrant immune metabolism in the liver of CHB leads to pro-inflammatory macrophage activation. This, in turn, sustains the production of pro-inflammatory cytokines, including Chemokine (C-X-C motif) ligand 9/10 (CXCL-9/CXCL-10), and others, which are essential for recruiting lymphocytes and monocytes/macrophages to the liver tissue (15, 58). These studies provide further support for the concept that pro-inflammatory hepatic macrophages play a central role in enhancing liver infiltration and exacerbating liver inflammation (Figure 4) (19).
5 Future perspectives
CHB is characterized by distinct and dynamic disease progression stages, yet the mechanism governing host immune responses, particularly within the liver, and the development of liver pathology throughout these stages remain elusive. In this context, we highlight the pivotal role of hepatic macrophages in both recognizing HBV through innate immunity and evading HBV immune surveillance through various mechanisms. However, the specific trigger responsible for shifting the immune tolerance state of the CHB-infected liver towards an immune-activated state, ultimately resulting in liver inflammation and liver injury, remains unidentified.
As the body of evidence regarding the involvement of HBV in metabolic processes grows, it becomes increasingly clear that HBV employs a dual strategy for entering target cells (10, 13, 21). Besides, our review initially emphasizes the significant role of HBV-induced metabolic changes in hepatic macrophages as a key factor in the transition from viral infection to immune activation, ultimately leading to liver inflammation. We propose that metabolic alterations in specific hepatic macrophages subsets are crucial for CHB liver inflammation and disease progression. Our concept presents new potential intervention strategies for achieving a functional cure in CHB: early blockade of HBV-induced metabolic changes and inflammation; modulation of the pro-inflammatory phenotype of liver-resident macrophages by targeting specific markers or genes; and targeted metabolic therapy with antiviral and anti-inflammatory effects.
However, current studies on the mechanisms of HBV-mediated metabolic changes and intrahepatic inflammation have been conducted either in hepatoma cell lines like HepG2 and Huh7, HBV transgenic mice, or peripheral non-tumor tissues after surgical resection. These in vitro experiments are insufficient to accurately reflect the levels of HBV-mediated metabolic and inflammatory changes (8, 24, 31–33, 36–38). Although the rapid advancement of omics technologies, multi-omics studies on clinical liver biopsy samples have compensated for the lack of observations on real clinical samples, existing research still lacks monitoring of samples from different stages of CHB’s natural history, as well as pathological and omics-level monitoring of different spatial or anatomical regions (13, 14, 17, 18, 51–55).
Our latest insights underscore that HBV induces significant alterations in metabolic pathways, particularly within the interplay between hepatocytes and the hepatic macrophage efflux system during HBV infection (13, 21). This dynamic interaction serves as a critical link connecting HBV infection and pro-inflammatory activation. While this hypothesis is promising, it necessitates further experimental validation. Several unresolved issues warrant additional emphasis: 1) The specific metabolic profiles for distinct cell clusters, particularly hepatocytes and macrophages at different stages of CHB, remain un-characterized. 2) The degree of liver injury and the extent of changes in key cellular metabolic factors have not yet be established. 3) In-depth understanding of spatial zonation into CHB liver by multi-omics and subsequent validations are required.
Therefore, future studies should prioritize diverse cohorts representing different stages of CHB natural history. Combining multi-omics sequencing technologies to analyze these abundant clinical samples is essential for a comprehensive understanding and control of changes at the genomic, proteomic, metabolomic, and other levels. Bioinformatic analyses should focus on characterizing cell cluster-specific metabolic profiles within the liver and examining correlations between metabolic changes and key biomarkers associated with immune activation or liver injury. Comprehensive omics-sequence analyses and subsequent research are crucial to substantiate these findings, aiming to elucidate the spatial and temporal dynamics of intrahepatic metabolites and immune-pathological profiles. Additionally, effective animal models of inflammation progression and in vitro experiments are needed to complement omics findings and confirm our conclusion: the potential role of hepatic macrophages in activating metabolic changes and inflammatory responses that ultimately result in liver injury.
Author contributions
JW: Conceptualization, Writing – original draft, Writing – review & editing. HL: Writing – review & editing. QL: Conceptualization, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the China’s National Key R&D Programs (No. 2023YFC2306700), National Science Foundation of China (No. 82301973); the Chinesisch-Deutsches Mobilitats programm (No. M-0569); Basic and Applied Basic Research Fund of Guangdong Province: Regional Joint Fund-Youth Fund Project (No. 2021A1515110831; No. 2022A1515111163); the China Post-Doctoral Science Foundation (No. 2022M721473), the Shenzhen Third People’s Hospital project (No. 21250G1001; No. 22240G1005), Shenzhen Science and Technology Innovations Committee (No. JSGGZD20220822095200001, No. JCYJ20220530163406014) and the Top Talent Support Program for young and middle-aged people of Wuxi Health Committee (No.BJ2023093). Research Projects of Wuxi Health Committee (No. M202203) and the Wuxi Science and Technology Fund Project (No. Y20222008).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Huang SC, Liu CJ. Chronic hepatitis B with concurrent metabolic dysfunction-associated fatty liver disease: Challenges and perspectives. Clin Mol Hepatol. (2023) 29:320–31. doi: 10.3350/cmh.2022.0422
2. Charatcharoenwitthaya P, Pongpaibul A, Kaosombatwattana U, Bhanthumkomol P, Bandidniyamanon W, Pausawasdi N, et al. The prevalence of steatohepatitis in chronic hepatitis B patients and its impact on disease severity and treatment response. Liver Int. (2017) 37:542–51. doi: 10.1111/liv.13271
3. Choi HSJ, Brouwer WP, Zanjir WMR, de Man RA, Feld JJ, Hansen BE, et al. Nonalcoholic steatohepatitis is associated with liver-related outcomes and all-cause mortality in chronic hepatitis B. Hepatology. (2020) 71:539–48. doi: 10.1002/hep.30857
4. Lee YB, Ha Y, Chon YE, Kim MN, Lee JH, Park H, et al. Association between hepatic steatosis and the development of hepatocellular carcinoma in patients with chronic hepatitis B. Clin Mol Hepatol. (2019) 25:52–64. doi: 10.3350/cmh.2018.0040
5. Tourkochristou E, Assimakopoulos SF, Thomopoulos K, Marangos M, Triantos C. NAFLD and HBV interplay - related mechanisms underlying liver disease progression. Front Immunol. (2022) 13:965548. doi: 10.3389/fimmu.2022.965548
6. Wu YL, Peng XE, Zhu YB, Yan XL, Chen WN, Lin X. Hepatitis B virus X protein induces hepatic steatosis by enhancing the expression of liver fatty acid binding protein. J Virol. (2015) 90:1729–40. doi: 10.1128/JVI.02604-15
7. Kim KH, Shin HJ, Kim K, Choi HM, Rhee SH, Moon HB, et al. Hepatitis B virus X protein induces hepatic steatosis via transcriptional activation of SREBP1 and PPARgamma. Gastroenterology. (2007) 132:1955–67. doi: 10.1053/j.gastro.2007.03.039
8. Kim JY, Song EH, Lee HJ, Oh YK, Choi KH, Yu DY, et al. HBx-induced hepatic steatosis and apoptosis are regulated by TNFR1- and NF-kappaB-dependent pathways. J Mol Biol. (2010) 397:917–31. doi: 10.1016/j.jmb.2010.02.016
9. Li YJ, Zhu P, Liang Y, Yin WG, Xiao JH. Hepatitis B virus induces expression of cholesterol metabolism-related genes via TLR2 in HepG2 cells. World J Gastroenterol. (2013) 19:2262–9. doi: 10.3748/wjg.v19.i14.2262
10. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. (2012) 13:1. doi: 10.7554/eLife.00049
11. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. (2021) 18:45–56. doi: 10.1038/s41423-020-00558-8
12. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. (2015) 15:104–16. doi: 10.1038/nri3793
13. Li J, Liang X, Jiang J, Yang L, Xin J, Shi D, et al. Chinese Group on the Study of Severe Hepatitis B (COSSH). PBMC transcriptomics identifies immune-metabolism disorder during the development of HBV-ACLF. Gut. (2022) 71:163–75. doi: 10.1136/gutjnl-2020-323395
14. Peng B, Li H, Liu K, Zhang P, Zhuang Q, Li J, et al. Intrahepatic macrophage reprogramming associated with lipid metabolism in hepatitis B virus-related acute-on-chronic liver failure. J Transl Med. (2023) 21:419. doi: 10.1186/s12967-023-04294-1
15. Cao S, Liu M, Sehrawat TS, Shah VH. Regulation and functional roles of chemokines in liver diseases. Nat Rev Gastroenterol Hepatol. (2021) 18:630–47. doi: 10.1038/s41575-021-00444-2
16. Barreby E, Aouadi M. To be or not to be a hepatic niche macrophage. Immunity. (2022) 55:198–200. doi: 10.1016/j.immuni.2022.01.008
17. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. (2018) 9:4383. doi: 10.1038/s41467-018-06318-7
18. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. (2022) 185:379–396.e38. doi: 10.1016/j.cell.2021.12.018
19. Nkongolo S, Ni Y, Lempp FA, Kaufman C, Lindner T, Esser-Nobis K, et al. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J Hepatol. (2014) 60:723–31. doi: 10.1016/j.jhep.2013.11.022
20. Esser K, Cheng X, Wettengel JM, Lucifora J, Hansen-Palmus L, Austen K, et al. Hepatitis B virus targets lipid transport pathways to infect hepatocytes. Cell Mol Gastroenterol Hepatol. (2023) 16:201–21. doi: 10.1016/j.jcmgh.2023.03.011
21. Cheng Y, Cheng T, Jin DY. Reverse cholesterol transport of macrophages repurposed for hepatitis B virus entry. Cell Mol Gastroenterol Hepatol. (2023) 16:317–8. doi: 10.1016/j.jcmgh.2023.05.004
22. Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. (2011) 178:175–86. doi: 10.1016/j.ajpath.2010.11.026
23. Zhang Y, Hong JY, Rockwell CE, Copple BL, Jaeschke H, Klaassen CD. Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver Int. (2012) 32:58–69. doi: 10.1111/j.1478-3231.2011.02662.x
24. Cai SY, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight. (2017) 2:e90780. doi: 10.1172/jci.insight.90780
25. Yerushalmi B, Dahl R, Devereaux MW, Gumpricht E, Sokol RJ. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology. (2001) 33:616–26. doi: 10.1053/jhep.2001.22702
26. Bochkis IM, Rubins NE, White P, Furth EE, Friedman JR, Kaestner KH. Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat Med. (2008) 14:828–36. doi: 10.1038/nm.1853
27. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
28. Guilliams M, Scott CL. Liver macrophages in health and disease. Immunity. (2022) 55:1515–29. doi: 10.1016/j.immuni.2022.08.002
29. Macdonald AC, Marble AE, Perkins JG. Hepatic blood flow and metabolism. Arch Surg. (1979) 114:616–22. doi: 10.1001/archsurg.1979.01370290066014
30. Sun Z, Huang C, Shi Y, Wang R, Fan J, Yu Y, et al. Distinct bile acid profiles in patients with chronic hepatitis B virus infection reveal metabolic interplay between host, virus and gut microbiome. Front Med (Lausanne). (2021) 4:708495. doi: 10.3389/fmed.2021.708495
31. Shin HJ, Park YH, Kim SU, Moon HB, Park DS, Han YH, et al. Hepatitis B virus X protein regulates hepatic glucose homeostasis via activation of inducible nitric oxide synthase. J Biol Chem. (2011) 286:29872–81. doi: 10.1074/jbc.M111.259978
32. Li H, Zhu W, Zhang L, Lei H, Wu X, Guo L, et al. The metabolic responses to hepatitis B virus infection shed new light on pathogenesis and targets for treatment. Sci Rep. (2015) 5:8421. doi: 10.1038/srep08421
33. Liu B, Fang M, He Z, Cui D, Jia S, Lin X, et al. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. (2015) 6:e1980. doi: 10.1038/cddis.2015.322
34. Chen G, Liang G, Ou J, Goldstein JL, Brown MS. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc Natl Acad Sci U S A. (2004) 101:11245–50. doi: 10.1073/pnas.0404297101
35. Seo JB, Moon HM, Kim WS, Lee YS, Jeong HW, Yoo EJ, et al. Activated liver X receptors stimulate adipocyte differentiation through induction of peroxisome proliferator-activated receptor gamma expression. Mol Cell Biol. (2004) 24:3430–44. doi: 10.1128/MCB.24.8.3430-3444.2004
36. Kim K, Kim KH, Kim HH, Cheong J. Hepatitis B Virus X protein induces lipogenic transcription factor SREBP1 and fatty acid synthase through the activation of nuclear receptor LXRalpha. Biochem J. (2008) 416:219–30. doi: 10.1042/BJ20081336
37. Zheng G, Fu Y, He C. Nucleic acid oxidation in DNA damage repair and epigenetics. Chem Rev. (2014) 114:4602–20. doi: 10.1021/cr400432d
38. Farnik H, Bojunga J, Berger A, Allwinn R, Waidmann O, Kronenberger B, et al. Low vitamin D serum concentration is associated with high levels of hepatitis B virus replication in chronically infected patients. Hepatology. (2013) 58:1270–6. doi: 10.1002/hep.v58.4
39. Oehler N, Volz T, Bhadra OD, Kah J, Allweiss L, Giersch K, et al. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology. (2014) 60:1483–93. doi: 10.1002/hep.v60.5
40. Patman G. Hepatitis: HBV infection alters bile acid metabolism gene profile. Nat Rev Gastroenterol Hepatol. (2014) 11:332. doi: 10.1038/nrgastro.2014.62
41. Du Y, Broering R, Li X, Zhang X, Liu J, Yang D, et al. In vivo mouse models for hepatitis B virus infection and their application. Front Immunol. (2021) 12:766534. doi: 10.3389/fimmu.2021.766534
42. Allweiss L, Dandri M. Experimental in vitro and in vivo models for the study of human hepatitis B virus infection. J Hepatol. (2016) 64:S17–31. doi: 10.1016/j.jhep.2016.02.012
43. Hösel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M, et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology. (2009) 50:1773–82. doi: 10.1002/hep.23226
44. Boltjes A, van Montfoort N, Biesta PJ, Op den Brouw ML, Kwekkeboom J, van der Laan LJ, et al. Kupffer cells interact with hepatitis B surface antigen in vivo and in vitro, leading to proinflammatory cytokine production and natural killer cell function. J Infect Dis. (2015) 211:1268–78. doi: 10.1093/infdis/jiu599
45. Liu J, Yu Q, Wu W, Huang X, Broering R, Werner M, et al. TLR2 stimulation strengthens intrahepatic myeloid-derived cell-mediated T cell tolerance through inducing kupffer cell expansion and IL-10 production. J Immunol. (2018) 200:2341–51. doi: 10.4049/jimmunol.1700540
46. Li M, Sun R, Xu L, Yin W, Chen Y, Zheng X, et al. Kupffer cells support hepatitis B virus-mediated CD8+ T cell exhaustion via hepatitis B core antigen-TLR2 interactions in mice. J Immunol. (2015) 195:3100–9. doi: 10.4049/jimmunol.1500839
47. Faure-Dupuy S, Delphin M, Aillot L, Dimier L, Lebossé F, Fresquet J, et al. Hepatitis B virus-induced modulation of liver macrophage function promotes hepatocyte infection. J Hepatol. (2019) 71:1086–98. doi: 10.1016/j.jhep.2019.06.032
48. You Q, Cheng L, Kedl RM, Ju C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology. (2008) 48:978–90. doi: 10.1002/hep.v48:3
49. Cheng X, Xia Y, Serti E, Block PD, Chung M, Chayama K, et al. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology. (2017) 66:1779–93. doi: 10.1002/hep.29348
50. Tan-Garcia A, Wai LE, Zheng D, Ceccarello E, Jo J, Banu N, et al. Intrahepatic CD206+ macrophages contribute to inflammation in advanced viral-related liver disease. J Hepatol. (2017) 67:490–500. doi: 10.1016/j.jhep.2017.04.023
51. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
52. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell. (2019) 178:686–698.e14. doi: 10.1016/j.cell.2019.05.054
53. Dib L, Koneva LA, Edsfeldt A, Zurke YX, Sun J, Nitulescu M, et al. Lipid-associated macrophages transition to an inflammatory state in human atherosclerosis, increasing the risk of cerebrovascular complications. Nat Cardiovasc Res. (2023) 2:656–72. doi: 10.1038/s44161-023-00295-x
54. Calmus Y, Poupon R. Shaping macrophage’s function and innate immunity by bile acids: mechanisms and implication in cholestatic liver diseases. Clin Res Hepatol Gastroenterol. (2014) 38:550–6. doi: 10.1016/j.clinre.2014.07.007
55. Do TH, Ma F, Andrade PR, Teles R, de Andrade Silva BJ, Hu C, et al. TREM2 macrophages induced by human lipids drive inflammation in acne lesions. Sci Immunol. (2022) 7:eabo2787. doi: 10.1126/sciimmunol.abo2787
56. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
57. Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol. (2016) 13:316–27. doi: 10.1038/cmi.2015.104
Keywords: HBV, hepatic macrophage niches, liver inflammation, metabolism, lipid metabolism (fatty acids)
Citation: Wang J, Lu H and Li Q (2024) Hepatic macrophage niche: a bridge between HBV-mediated metabolic changes with intrahepatic inflammation. Front. Immunol. 15:1414594. doi: 10.3389/fimmu.2024.1414594
Received: 09 April 2024; Accepted: 02 July 2024;
Published: 18 July 2024.
Edited by:
Mohamed Mahdi, University of Debrecen, HungaryReviewed by:
Feng Liu, Peking University People’s Hospital, ChinaTuo Shao, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2024 Wang, Lu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qian Li, bGlxaWFuMTk5MDA4MDFAaG90bWFpbC5jb20=; Hongzhou Lu, bHVob25nemhvdUBzenN5LnN1c3RlY2guZWR1LmNu