 Biswanath Majumder
Biswanath Majumder Nishanth Belugali Nataraj
Nishanth Belugali Nataraj Leela Maitreyi†‡
Leela Maitreyi†‡
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 19 July 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1414376
Mismatch repair proficient (MMRp) tumors of colorectal origin are one of the prevalent yet unpredictable clinical challenges. Despite earnest efforts, optimal treatment modalities have yet to emerge for this class. The poor prognosis and limited actionability of MMRp are ascribed to a low neoantigen burden and a desert-like microenvironment. This review focuses on the critical roadblocks orchestrated by an immune evasive mechanistic milieu in the context of MMRp. The low density of effector immune cells, their weak spatiotemporal underpinnings, and the high-handedness of the IL-17-TGF-β signaling are intertwined and present formidable challenges for the existing therapies. Microbiome niche decorated by Fusobacterium nucleatum alters the metabolic program to maintain an immunosuppressive state. We also highlight the evolving strategies to repolarize and reinvigorate this microenvironment. Reconstruction of anti-tumor chemokine signaling, rational drug combinations eliciting T cell activation, and reprograming the maladapted microbiome are exciting developments in this direction. Alternative vulnerability of other DNA damage repair pathways is gaining momentum. Integration of liquid biopsy and ex vivo functional platforms provide precision oncology insights. We illustrated the perspectives and changing landscape of MMRp-CRC. The emerging opportunities discussed in this review can turn the tide in favor of fighting the treatment dilemma for this elusive cancer.
One and half decades after the ‘immuno-oncology tsunami’ that hit the clinical development landscape and shifted the momentum of treatment modalities, multiple new immunotherapy agents are now on the horizon. This progress raised optimism for many late-stage cancers for which treatment options are heavily exhausted. However, a second breakthrough remains elusive. The overall response rate (ORR) also remains static (20–40%) for all cancers. A rapid surge of therapeutic targets for developing new checkpoint blockades and other immune-agonist classes is on the ground (1, 2). There is also a new wave of developing RNA-based cancer vaccines to provide deep, durable memory responses (3). One vaccine candidate in recent time showed encouraging outcomes in lethal pancreatic adenocarcinoma (4). Aligned with this momentum, we witness an explosion of combination trials that aim to enable better survival outcomes (5, 6).
Colorectal cancer (CRC) is considered a global epidemic; over the decades, it has emerged as a pivotal cancer type affecting the younger population and is associated with late-stage detection and poor overall survival (OS) (7). CRC individuals with DNA mismatch repair deficiency (MMRd) positively respond to immune checkpoint blockade (ICB). A striking response failure in mismatch repair-proficient (MMRp) patients shows the diminishing return of the same therapy. In CRC, most of the success stories for ICB, thus far, have been limited only to tumors that manifest MMRd. However, this subtype is represented by only 15% of all CRCs and is far behind endometrial cancers (30%) and gastric cancers (20%) (8). For MMRp, ORR is not very different (10–15%) between TMB high and low CRC (9, 10). This realization prompted looking for effective treatment options for the MMRp subtype. The trend implies the necessity of addressing unmet needs both at personalized and population levels. At the heart of this challenge is the underlying complexity of the tumor microenvironment and its unpredictable dynamic immune milieu that form a barrier to effective therapy (11).
In this review, we discuss the changing clinical landscape of MMRp-dependent cancer indications (mainly CRC) and their uniquely hostile tumor microenvironment that hinders the success of current immune-based interventions. Both conceptual progress and clinical translation are illustrated in the light of rapidly evolving spatial biology contexts like tertiary lymphoid structures and gut microbiomes. We discuss the perspectives and challenges of biomarker-guided treatment selections for MMRp agonist cancers. We also highlight the alternatively actionable DNA repair pathways as emerging vulnerabilities to combat the treatment dilemma. Finally, we presented significant progress on the horizon of patient-derived functional ex vivo platforms that raised the hope of bridging the critical mechanistic gaps between drug pipelines and informed clinical decisions.
A pan-genomic analysis from the 100000 Genome Cancer Program integrating genomic and clinical data revealed the highest enrichment of specific DNA. It deciphered MMR signatures in MSI-high (i.e. MMRd) colon adenocarcinoma and uterine corpus endometrial carcinoma. MMRp in this spectrum showed a negative association with survival compared to MMRd patients. Germline variants of MMR in this study found their link with the onset of colon adenocarcinoma at an early age (12).
The intertwining intratumor heterogeneity (ITH) with TMB and TILs contextually represents a complex biology. ITH, either primary, adaptive or acquired during treatments, is considered a spatiotemporal bottleneck for a high response rate and duration of response. ITH encompasses genetic, phenotypic and dynamic tumor microenvironmental milieu and orchestrates therapy resistance. It also leads to the evolution of new resistant clones or the expansion of drug-tolerant persisters (13). Deciphering this ITH through the lens of MMRp and developing strategies to combat ITH mutations in tolerant cells are vital for adopting rational intervention. The study in the autochthonous mice model of lung and colon cancers highlighted that high TMB and MMRd do not guarantee immunogenic tumor infiltrating lymphocytes (TILs) and a positive response to checkpoint blockade. The subclonal escape of T cell response in these tumors was orchestrated by an immune-mediated increase in clonal diversity (14).
Instead of relying explicitly on genomics, the gradient of TME modulators like chemokines, neoangiogenesis, blood vessels, nutrients, and oxygen, along with ECM stiffness in time and space, play pivotal roles (15–17). Echoing this realization, the immune milieu is thought to collectively provide an actionable dynamic niche that interacts with the drugs and make the tumors reactive to ICB (18, 19). CRC show properties of reversible (mutation-independent) drug tolerance where recurrence is imminent after tumor cells are relieved from therapy pressure. Interestingly, barcoding and mathematical modeling suggested that equipotent clonal complexity is maintained for all cells throughout this process without any temporospatial loss. Under such conditions, tumors mimic a developmentally programmed diapause state at transcriptomic and signaling levels to overcome environmental turbulence (20). Indeed, drug-tolerant and disseminating tumor cells are, in general, notorious immune evaders that take advantage of being unnoticed by the immune radar to escape the primary sites and survive as silent perpetrators (21, 22).
Although MMRp and MMRd are binary molecular classes, recent profiling identified an intermediate category, i.e. heterogenous MMR or MMRh. The clonal overlap of MMRp and MMRd distinguishes it from the two classical subtypes. Gene expression analysis of CRC identified 14.5% of MMRd and 4.5% of MMRp cases as shared with this MMRh. The MMRh subclass allegedly evolves from double MMR gene loss. It is mechanistically linked to high TMB, TILs, and CD8 exhaustion phenotypes. High TMB (70 mut/mb) is attributable to higher subclonal variants. Genes associated with the MAPK pathway, antigen presentation and IFN-γ signaling pathway were significantly upregulated in MMRh class compared to MMRp (23). Moreover, 6-thioguanine and TMZ-induced enrichment of MMRd clones in MMRp tumors yielded encouraging outcomes. In two isogenic mice CT26 cell lines of MMRp (Mlh1+/+) and MMRd (Mlh1-/-) backgrounds, cross-complementing MMRp tumors selectively with MMRd clones rescued the immune surveillance program. MMRp clones, challenged with at least 50% MMRd cells, elicited tumor rejection. Both chemical induction and clonal competition strategies were able to underpin a heterogeneous MMR context of improved anti-tumor immune reactivity (24). This study in mice cell lines of CT-26 with MMRp backbone affirmed that reconstitution of MMRp clones with MMRd powered them to eliminate the MMRp fraction. The clonal and sub-clonal contexts of these two studies highlighted the differences in experimental approaches and interpretations of results. Specific TMB/neoantigens low subclones of mice tumors can evade an immune attack due to defects in cross-priming or active interference by dysfunctional T cells or immune ignorance. Moreover, these tumors are thought to acquire immunogenicity during in vivo repropagation at the clonal level but not at the sub-clonal level. As a result, the rapid contraction of MMRp clones was attainable (25, 26). This divergent clonal journey revealed the dynamicity of the ecological and evolutionary landscape of MMRp (27).
Further dissection of MMR status at the molecular level sheds light on the key regulatory elements driving transcriptomic machinery. Mutations (indels) in diverse CRC samples revealed that MSI-high CRC largely harbor gained enhancers that selectively offer the privilege of recurrent growth of these tumors through increased affinity for putative transcription factor, e.g. Forkhead Box D4 (FOXD4) and target gene overexpression that is regulated by these enhancers (28). Some of these genes have been implicated in chemoresistance, unrestricted oncogenic EGFR signaling, regulation of proliferation and apoptosis in primary CRC tumors and in established MSI cell lines. In the MSS cohort, the occurrence of enhancer indels was found to be at a much lower rate. Compared to MSS, MSI-high CRC has shown 50% more gained enhancers at TGTTT(Tn). It was linked to H3K27ac enrichment. A panel of 10 different FOX- transcription factors (FOX-TFs), encompassing FOXP2, FOXC1, FOXD3, FOXM1, FOXJ3, FOXA1, FOXO1, FOXO3, FOXG1 and FOXA2, presented the consensus sequence, a signature motif at indel alleles, and confirmed the binding affinity of FOXs. However, due to the degenerative nature of this consensus motif, findings did not specify the dominance of any single FOX member from the family in the enhancer activation. Instead, it proposed additional studies to fill this gap, elucidate cooperative interaction with other factors and inter-tumor heterogeneity, if any (29). This also implies the need for further delineation of other enhancers and super-enhancers to gauge their differential impact on the oncogenic driver alterations, making MMRd and MMRp tumors more vulnerable to therapies (30).
About 30% CRC are hereditary and germline predisposition affects CRC susceptibility. About 5%–7% of CRC cases are caused by germline mutations. Classic hereditary CRC syndromes are mainly due to germline mutations in APC, MUTYH, and mutations in genes encoding four mismatch repair enzymes, namely MSH2, MSH6, PMS2 and MLH1 (31, 32). Pathogenic somatic mutations (predominantly biallelic) in coding regions of one of these four mismatch repair enzymes lead to the development of MMR deficiency of CRC and eventually give rise to an MSI phenotype. Interestingly, Lynch syndrome (LS) or hereditary nonpolyposis, CRC, represents one-third of these MMRd, is an early onset CRC, therefore suggesting precedence of germline mutations (33). Similarly, tracking the germline defects in the MMRp genes provided significant screening opportunities for CRC of hereditary background. DNA mismatch repair protein O6-methylguanine DNA methyltransferase (MGMT) is frequently detected in CRC. Its epigenetic inactivation in somatic clusters is prevalent. However, elucidating whether the same defects are perpetuated in the germline background, particularly in MMRp, showed non-confirmatory results. No promoter methylation linked to constitutive MGMT inactivation was confirmed. Indeed, two rare heterozygous germline variants were detected in 4 families. Further segregation of these variants in neoplastic lesions in the affected family suggested that more data are needed to establish their link to MMRp in familial CRC (34). Although most CRCs by default are MSS, and MMRp represents low TMB, the study showed that 7.5% of colon and 9.5% of rectal cancers of this background also had high TMB (defined by >10 mutations per Mb). Interestingly, KRAS mutations and gene mutations involved in DNA damage repair (DDR) machinery and epigenetic modifiers were high in TMB-high MSS tumors. These findings suggest that molecular alterations are potential triggers of TMB in CRC (35). Like MMRh, these subsets open new opportunities for target dependency and vulnerabilities to gauge their promise in differentiated intervention.
A hostile MMRp context of tumor-immune microenvironments (TiME) is a formidable therapeutic challenge in achieving clinical success. Low TMB and immunological ignorance are two critical hallmarks of the MMRp tumor ecosystem. Further complexity of a tangled network emanating from dysbiosis of microbiome and problematic metabolomics has the propensity to escalate the suppressiveness of the microenvironment to diverse therapy regimens. Understanding the dynamicity and dimensionality of this misdirected microenvironment and its immune restrictive milieu is pivotal to developing novel rationale interventions that can mount an all-out attack on these tumors (Figure 1).
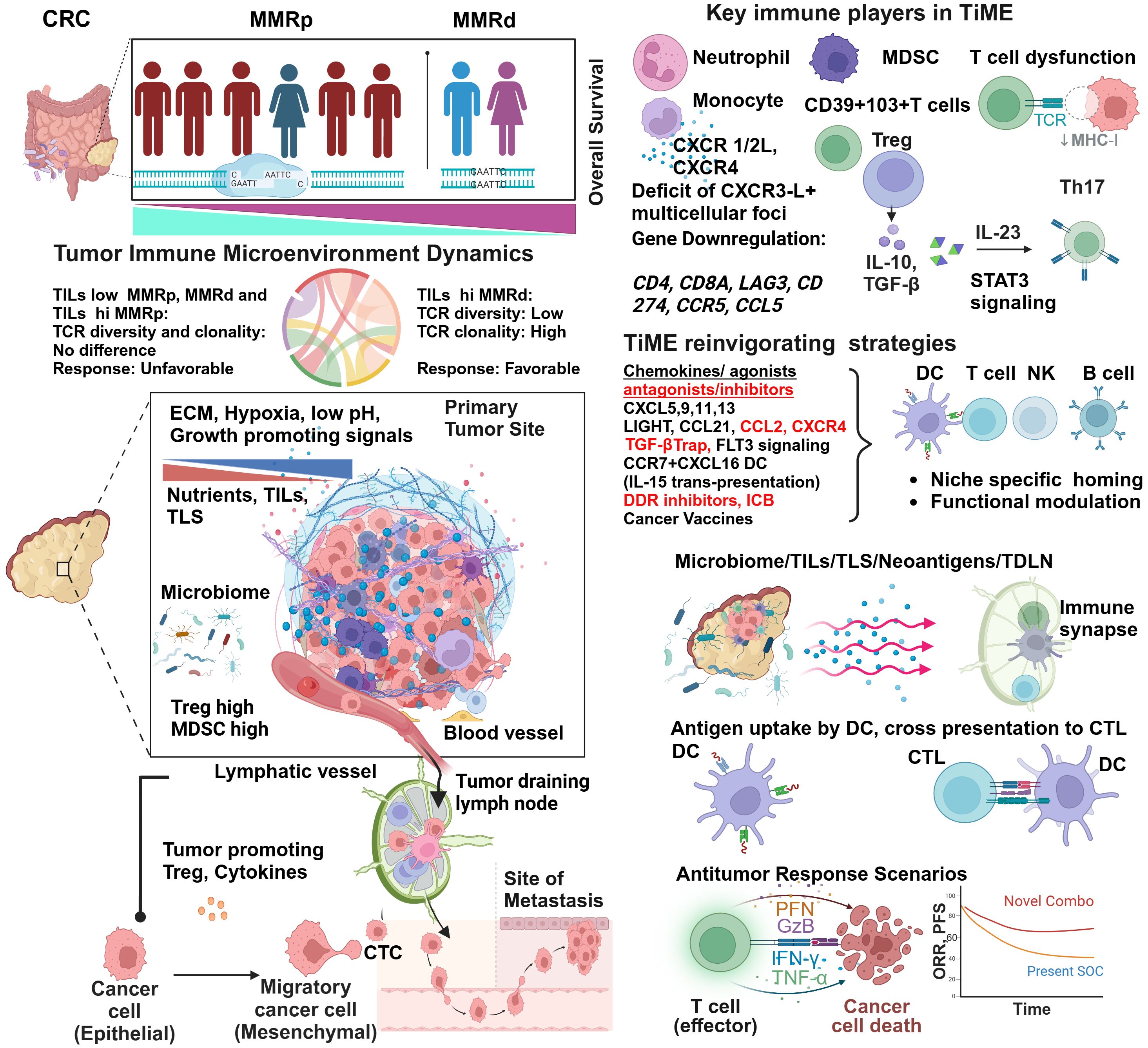
Figure 1 Tumor immune microenvironment of MMRp displays mechanistic constrains: potential strategies of reinvigorating. Despite representing more than three-fourths of the entire CRC population, the majority of MMRp CRC belongs to high-risk and poor prognoses. The TiME of CRC has multiple mechanistic barriers that hamper the therapy success. High TCR diversity and low TILs density in the margin and core at primary CRC sites are associated with desmoplastic stroma, growth-promoting oncogenic signaling, poor blood vessel density and patterns leading to oxygen and nutrients deprivation. Poor microbiome context and active involvement of Treg and MDSC in a traditionally low TMB milieu critically orchestrate immune evasion of disseminating tumor cells to distant sites through unguarded blood vessels and immunologically skewed TDLN’s surveillance. The phenotypic analysis of TILs confirms the presence of immune cell types of suppressive functions and corresponding cytokines and chemokine networks that protect the tumor from immune attacks. Several agonists and antagonists of chemokines, TGF-β-targeted therapy, and vaccines can act in concert with other strategies to reinvigorate and stabilize TILs and TLS via niche-specific recruitments of the anti-tumor immune army. Finally, augmenting neoantigen load and DC functionalities cross prime CD8+T cells. In totality, other immune and non-immune targets present in the TiME provide an opportunity to rationally target this challenging microenvironment in clinical settings and improve the response.
One critical differentiator in response to ICB between MMRd and MMRp is TILs density and its proximity (core and invasive margin) to target tumors. While a higher number of MMRp CRC patients are ascribed to be TILs deprived, i.e. they have lower TILs density, inter-tumor TILs heterogeneity (30–90%) in MMRd is not uncommon (Figure 1). This contexture includes a spatial heterogeneity of CD3+ and CD8+ TILs in the invasive margin and tumor core. Interestingly, a diminished TILs footprint in MMRd acts to underpin MMRp+ CRC response to ICB, a feature paralleling their functional TiME orientation (36).
It is intriguing to note that not only the TILs density and distribution but TCR repertoire and clonal landscape in MMRp CRC are also different from its MMRd counterpart. While T cell clonality and the richness of TCR repertoire have similar imprints in MMRp with TILs low and TILs high context, a sharp contrast contradicts this signature in MMRd tumors. In MMRd, higher T cell clonality was observed to be matched with lower TCR richness in TILs high tumors while comparing them with TILs low tumors within the same MMR class (Figure 1). It is imperative to note that under high TMB, T cells are clonally expanded in MMRd with high TILs. The constraints of low TMB in MMRp made both TILs low and TILs high tumor uniform in their clonal expansion program, and they maintained similar TCR diversity (37). These findings show that T cell clonal dynamics in TMB with a low and high background may reciprocally impact immunosurveillance. Studies in other cancers claimed that global TMB alone is not a perfect proxy for the foreignness of antigens. An evolutionarily persistent TMB, due to single or multiple copy regions per cell, provides a better response opportunity by creating a bottleneck for tumors that is difficult for them to overcome (38).
The organ specific immune atlas is emerging as an important platform to capture deeper phenotypic perspectives and insights. Single-cell RNA sequencing revealed the existence of a distinct immune hub in a spatially defined CRC cancer-immune network. A shared myeloid-rich inflammatory immune hub in tumors below the colonic lumen and CXCR3-ligand positive anti-tumor multicellular foci accompanied by activated T cells in MMRd tumors contextually distinguish them from MMRp (39). Like spatial TILs, quantitative analysis of global TILs in MMRp is a logical step in defining their prognostic impact. In high power field (HPF) quantification of TILs, based on a threshold set as >3 (high) vs <3 (low), five years of recurrence-free survival was observed to be higher in MMRp CRC with high TILs (94.6%) compared to their low TILs counterpart (77.9%). More importantly, in multivariate analysis using stages and TILs as key discriminators along with MMRp status, the higher stage with high TILs resulted in a similar relapse-free survival (RFS) to that of the lower stage alone without impacting OS (40).
Reactive TILs are not the only immune subset that encounters tumors. Multiple evasion points that perturb the TiME are contextually intertwined.
In the case of conventional antigen-presenting cells (APC), gene defects mainly govern β2 microglobulins (B2M) inactivation and HLA class 1 dysregulation in CRC. ICB under these circumstances increases γδT cell subsets with PD1, killer-cell immunoglobulin-like receptors (KIR) and cytotoxic activation markers in TME. These players, in concerts, determine the preferential retention of ICB responsiveness in cell lines of MMRd and MMRp backgrounds as well as patient-derived organoids that are defective of B2M gene and show concomitant loss of HLA class1 presentation machinery. HT-29 CRC cell lines of MMRp lineage retain B2M function compared to MMRd lines (HCT-15 and Lovo), where B2M gene defects (HLA -1 antigen presentation loss) in HT-15 cell lines instigate the most profound ICB response orchestrated mainly by PD1+ γδT cells in a coculture based drug reactivity assay (measuring Caspase3/7). The reintroduction of B2M genes in MMRd cell lines resulted in a loss of tumor killing by γδT cells in response to ICB under similar conditions. Further delineation of MMRd clinical CRC samples by multiplex spatial immune profiling suggested a remarkable increase in γδT cells in B2M defective cases. MMRd Patient-derived organoids with B2M loss elicited a better response by PD1+ restricted γδT cells (41).
Preclinical models of MMRp CRC (accounting for 95% of all mCRC) revealed that these tumors preferentially spread to the liver following orthotopic implantation but are restricted when heterotopically implanted in a subcutaneous site known for its context deficit and poor vascularization. This complementary liver metastasis model importantly recapitulated the paucity of CD8 and DC, consequently maintaining the non-responsiveness to immune checkpoint blockade (ICB). Combined treatment of Feline McDonough sarcoma (FMS)-like tyrosine kinase 3 ligand (Flt3L) plus ICB therapy improved survival by enhancing dendritic cell infiltration (42) (Figure 1). Indeed, whole-genome analyses of metastatic colorectal cancers from a pan-cancer Hartwig database of 2256 MMRp samples confirmed that only 1.6% of these samples clonally showed B2M and concurrent loss of heterogenicity (LOH), limiting their prognostication impact (43). In another study, pexidartinib, a CSF-1R–directed tyrosine kinase inhibitor (TKI), in combination with durvalumab (anti–PDL-1) in CRC and other cancers, resulted in limited efficacy. Pexidartinib impaired the development and functionalities of DCs due to the inhibition of FLT3 signaling ex vivo and in vivo (44). These findings illustrate the importance of maintaining active FLT3 signaling to achieve reasonable response (Figure 1).
Interestingly, in MMRd CRC, B2M mutations that canonically disrupt antigen presentation machinery showed paradoxical outcomes. It prevented disease recurrence, metastasis and helped manage prolonged survival. In this case, NK cell mediated inhibitory effects in the absence of HLA 1 defect prevented metastatic spread (45, 46). Unlike CRC, in endometrial cancer, defects in T cell activation signaling due to JAK1 mutation turn the tumors immune inert (47). Phase I/II multicenter study of autologous DC with Avelumab in mCRC for pharmacodynamics (pD), safety and efficacy showed well-tolerated outcomes but a modest 6-month PFS (only for 11% of patients). Interestingly, the rewiring of lipid metabolism against glutamine and glucose utilization and the generating reactive oxygen species (ROS) in response to this combination contributed to longitudinal progression. There is an urgent call for tailoring novel therapies to target this dependency as a vulnerable checkpoint (48).
In recent years, conceptual progress and clinical promises of tertiary lymphoid structures (TLS), a specialized tumor-immune microenvironmental niche, have attracted attention (49). They have a concerted influence on priming/amplification//licensing itineraries in TiME. The TLS army involves diverse lineage-specific subsets like plasma cells/B cells, different DCs like conventional DC (cDC), follicular DC (fDC) and other myeloid and lymphoid-derived cell types. In CRC, TIL, TLS and their abundance are mainly elucidated in MMRd (50). For MMRp, the ongoing efforts dissect niche-dependent immune evasion and design rationale therapies that can enrich the TLS footprint and redefine ICB response. In parallel, microbiome-immune crosstalk in eliciting anti-tumor response is appreciated in CRC and implicated the roles of TLS (51). From the qualitative and quantitative perspectives, the size, composition and spatiotemporal dynamics of TLS and non-TLS immune hubs like TILs and lymphonets promise new therapeutic modalities (52). A 56-marker multiplex IHC-driven cellular classifier (CODEX) at the invasive front identified CD4+PDL-1-positive cells in the granulocyte neighborhood as the only positive prognostic marker in high-risk advanced-stage CRC. In contrast, the lack of inter-compartment connectivity in TiME contributes to unfavorable outcomes (53). At preclinical levels, however, there are limitations to the potential human translation of TLS. One reason is that besides wide gaps in TME, mice tumors exhibit rapid and aggressive growth. This property inherently restricts the scope of mature TLS formation within a defined temporal neogenesis window. The cells that populate an immature or suppressive TLS, e.g., Breg, Treg and MDSC, can also perpetuate in the MMRp (54–56).
Chemokines, released by tumor cells and other cell types like stromal fibroblasts and endothelial cells, act as chemo-attractants. Through engaging cognate receptors, they recruit immune cells that are anti-tumor or immune suppressive (pro-tumor) in functions. Multiple interactive chemokine axes also influence therapy outcomes. These depend on the types of chemokine ligands, cognate receptors on the target cells and specific TME contexts (57, 58).
Mechanistically, the reconstituted chemokines milieu could be a critical orchestrator of reinvigorating the depressed TLS and TiME. Spatially delineated biomarkers or ‘biopatterns’ shed light on the niche-specific recruitment and interactions of immune cells in TME. A comprehensive knowledge of their contexts is important for microenvironment-guided therapy selection (58, 59). The MMRp tumor-immune niche is the home of several suppressive immune subsets. Monocytes, neutrophils, MDSC, Tregs and Th17 cells are lead players in this domain and are responsible for maintaining a tumor-friendly suppressive network (Figure 1 and Table 1).
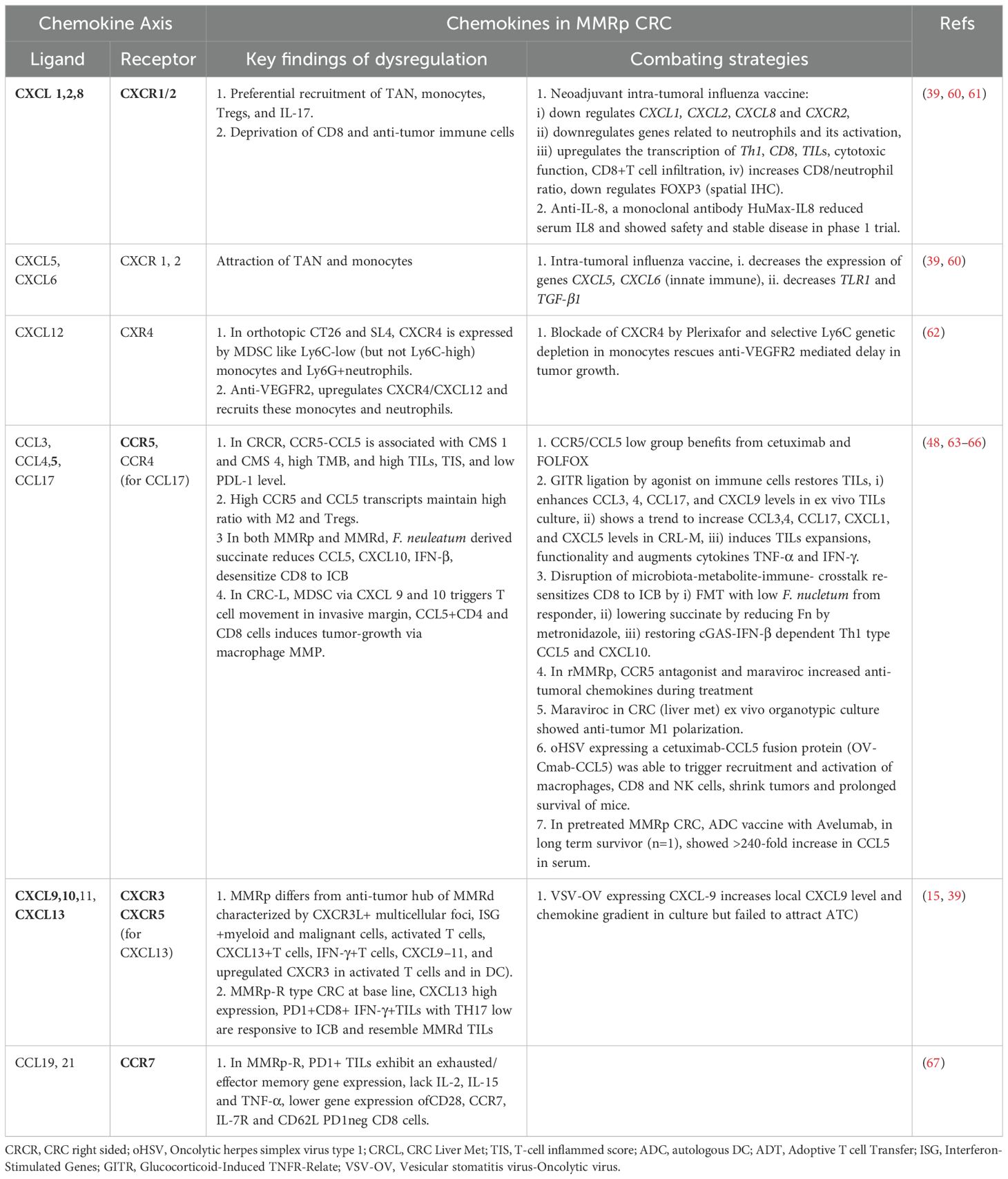
Table 1 Key Chemokines, their receptors, functions and combating strategies in MMRp CRC.
Preferential recruitments of tumor-associated neutrophils (TAN), monocytes, other myeloid- derived suppressor cells (MDSCs), Tregs, and Th17 functionally cooperate through chemokines like CXCR1/2 and CXCR4-CXCL12. In this network, IL-10, TGF-β, IL-10, IL-23, and STAT3 signaling worsen the prognosis (58, 67–70). Their interplay hampers the prospects of therapies. For example, anti-VEGFR treatment in MMRp mice orthotopic model triggers a positive feedback loop that further upregulates CXCR4-CCL12 and recruit monocytes and neutrophils in the TME (62).
In a hyperpolarized TME, CCR5 and CCL5 in MMRp are tightly associated with the right-side colon, poor prognosis-related consensus molecular subtypes 1 and 4 (CMS 1 and CMS 4), high TMB, and high TILs. Different myeloid and lymphoid subsets like M1, M2 macrophages, B cells, CD4, CD8, T regs and NK cells, in coordination with PDL-1, CTLA-4 and PARP, orchestrate a depressed immune network. It is evident that CCR5/CCL5 low group benefits from targeted therapy of cetuximab and FOLFOX (71). Paradoxically, past research showed a poor prognostic link to CCR5. The study showed a marginal improvement in specific combinations. However, due to the unavailability of data from retrospective analysis, confounding effects cannot be ruled out (71). Collectively, these modalities can guide the homing of immune cells in MMRp tumors known for traditionally lacking reactive TLS footprints. As illustrated in Figure 1 and Table 1, TiME of MMRp selective tumors has deficits in TILs and TLS. It shows a distinct bias for Treg and MDSC (mainly monocytes and TAN). Moreover, the immunosuppressive cytokines that MDSCs augment, facilitate epithelial-mesenchymal transition (EMT), increase the propensity for distant metastasis of disseminated tumor cells and confer failure to therapies (67, 72, 73). In the invasive margin of CRC liver metastasis (CRC-L), CCL5+CD4 and CD8 cells, recruited by myeloid-derived CXCL9 and CXCL10, attract macrophages promoting invasion and tumor growth by MMP (65). However, the same CCL5 in T cells is impaired under succinate high microenvironment created by F. nucleatum and mediate ICB nonresponse (66). These findings suggest the contrasting roles of CCL5-CCR5 axis in different TME contexts.
A number of therapeutic options are emerging on this horizon to fill the vacuum. The key modulators are chemokine agonists and antagonists that reciprocally orchestrate niche-specific recruitment and modulation dynamics. Functionally active chemokine axes, their dysregulation in MMRp and mechanistic interventions are illustrated in Table 1. A rational approach in MMRp can unlock the potential of reinvigorating its TiME. Retaining or reconstructing niche specific chemokine networks led by A) CXCR3 and its ligands CXCL9, 10, 11, CXCR5 and its ligand CXCL13, B) CXCR2 and its ligand CXCL5,6, C) CCR4 and its ligand CCL17, and D: CCR5 and its ligands CCL5, and additionally CCR7 hold promise in this space (reviewed in 57,58). Chemokines like CCL1, CCL2, CCL8, CCL12, and their receptors (e.g. CXCR1/2, CXCR4) reciprocally facilitate the homing of myeloid suppressors (monocytes, TAN, MDSC) and Treg. The drugs or antagonists targeting their actions can rejuvenate the immune reactive interface (58, 65, 73).
Neoadjuvant intra-tumoral influenza vaccine in MMRp CRC showed downregulation of pro-tumor chemokine genes, TGF-β genes. It concomitantly upregulated genes involved in Th1, CD8, increased TILs and cytotoxic function. The same vaccine decreased the Treg transcription factor FOXP3 at the protein level (60). Autologous dendritic cell (ADC) vaccine with Avelumab showed a decline in serum CCL2 level in pretreated MMRp CRC and a 240-fold increase of serum CCL5 in a long-term survivor (48). Inhibition of CXCR4 by Plerixafor or selective Ly6C targeted genetic ablation in monocytes rescues mice from anti-VEGFR2 induced tumor progression (62). CCR5 antagonist maraviroc has been tested in preclinical ex vivo tumor culture. In an independent clinical study, it demonstrated anti-tumor macrophage repolarization and anti-tumor chemokine augmentation, respectively (65, 74). CXCR3/5 ligands selectively recruit cells like DC, CD8 and T helper 1 as part of a niche-specific homing program (39, 58, 75). Therapies rely on agonists that drive the targeted enrichment of chemokines like CXCL9, 10, 11 and 13 and facilitate CD8 recruitment. For example, Glucocorticoid-Induced TNFR-Related (GITR) ligation by its agonist enhanced CCL3, CCL4, CCL17, and CXCL9 levels in CRC derived TILs in ex vivo culture. It induced TILs expansions, functionality and augmented proinflammatory cytokine (e.g. TNF-α, IFN-γ) (64). Gut microbiota adds a layer of criticality to this interface. Disruption of microbiota-metabolite-immune- crosstalk with low F. nucleatum (Fn) from responder or reducing F. nucleatum by metronidazole diminished the local succinic acid in TME and re-sensitized CD8 to ICB. This intervention also restored cGAS-IFN-β dependent CCL5 and CXCL10 following their decrease by high succinate (66). Anti-IL-8 antibody reduced serum IL-8 in phase 1 trial (61). Oncolytic virus expressing CXCL-9 restored local chemokine gradient but failed to recruit adoptive T cells (ATC) in culture (15).
Chemokines function as important modulators of TLS in both MMRd, MMRp scenarios (76, 77). Chemokines like LIGHT, LTa, CCL21, and APC activating agonists for TLR4 and CD40 are critical druggable targets to boost TLS (78). CCR7+ CXCL16+DC mediated trans-presentation of IL-15 to effector-like CTLs in perivascular niches orchestrate their survival and expansion. This survival and proliferation signaling loop averts an irreversible terminal differentiation of CTLs into the hypofunctional or tolerant state and maximizes the quality of response (79), (Figure 1 and Table 1).
IL-15 trans-presentation, TGF-β-Trap with anti-EGFR, DDR inhibitors and cancer vaccines are also under active development to overcome the outstanding challenges (48, 60, 79–81). While mechanistically compatible TILs in such scenarios may provide a milieu for immune-based interventions, other tumor intrinsic evasion strategies can still be a barrier that avert T cell-mediated attack of tumors (22). For example, perforins and granzymes are two critical polarized cytotoxic effector molecules released from activated NK and T cells. Perforins act as a port of entry for granzymes. However, tumor cells manipulate their inherent ESCRT-mediated membrane trafficking strategy to repair these pores and, therefore, block the entry of granzyme (82).
At the systemic level, a compromised immune activation network signals a prospective disease that is often advanced beyond primary sites. A recent study also proposed that preserving the tumor-draining lymph nodes (TDLN) may benefit anti-tumor immune reactions. Based on CRC data, dissection of immune phenotypic profiling showed differentiated TILs and TCR repertoire dynamics in lymph nodes (LN). This profiling separated MMRd from MMRp. In general, lymph node lymphocytes (LNL) show an intermediate functional state when compared with peripheral blood (lowest) and intratumor TILs (richest in tumor-reactive TILs). Stage-dependent TIL analysis also showed higher TILs in early-stage MMRd compared to matched early-stage or late-stage MMRp. Cytotoxicity-related genes also maintain similar enrichment patterns in MSI-H/MMRd cohorts. In this continuum, shared TCRs analysis of TILs showed their lower percentage in the proximal LN (pLN) of MMRp compared to MMRd. These data show the potential benefit of avoiding excessive non-metastasis LN dissection in MMRd (83, 84). CRC from MMRp origin contains neoantigen reactive autologous TILs co-expressing CD39 + 103+ T cell subsets in the CMS4 (less immunogenic) context. These T cells are known for promoting a paracrine TGF-β signaling loop and have the worst prognosis. Further delineation of checkpoint status targeting TGF-β and its trap with PDL-1, in this context, expected to reinvigorate TIL effector functions (69, 85, 86). However, an anti-PD-L1:TGF-β trap fusion protein directly targeting MSS-positive metastatic CRC failed to control the recurrence of ctDNA and, instead, elevated the level of ctDNA (86). Other strategies of dual targeting TGF-β with EGFR (e.g. BCA 101) are under development. Its combination with ICB in preclinical in vitro coculture assay using PBMC in EGFR-insensitive human colon cancer cell line HCT-116 (MSI hi) showed synergy with a high TGF-β footprint. Immune-reconstituted human colon cancer HT-29 (MSS) in mice xenograft model mechanistically elicited a potent immune-mediated anti-tumor response upon BCA 101 exposure (80) and Figure 1. More studies intersecting the TGF-β crosstalk in suppressive MMRp are needed to boost the quality of responses. KRAS mutant CRC and similar cancers were portrayed as undruggable until recently. A lymph node-targeted KRAS mutant peptide vaccine with a CpG oligo adjuvant (Amph-CpG-7909) in the AMPIFY-201 trial tested this therapy on 5 CRC patients, all from MMRp background. In 84% of cases, it showed T cell response ex vivo. Of this response, 54% involved both CD4 and CD8-specific T cells. In 84% of cases, there was a decline in biomarker (ct-DNA level) from the baseline, and in 24% (3 Ca-Pancreas and 3 CRC) cases, total clearance of biomarkers was achieved (87) and Figure 1. One outstanding question is what should be an ideal therapy plan for CRC patients with preexisting autoimmune conditions. IL-17-IL-23 axis is a clinical target in multiple autoimmune disorders (88). Therefore, a rational combination of these agents in MMRp patients with existing and new I-O and non-IO agents deserves evaluation through proper trials.
Among different theories, a complex interplay of intrinsic and extrinsic factors and cellular plasticity governs the initiation, maintenance and progression of cancers (89). The cancer risk mapping in higher mammals identified new attributes independent of body size and age that were thought to accommodate more cancer-causing mutations or “bad luck” mutations, initially coined by Tomasetti and Vogelstein (90). These risks include diets and loss of the gut microbiome homeostasis (91). New insights highlighted the clouds of complex systemic landscape in the frontiers of cancer hallmark. Besides metabolic alteration, ageing and obesity, tissue macro and microenvironment, myeloid dysfunction and other physiological dysregulation, genetic and environmental factors, this conceptual progress reiterates microbiome as a lead dimension (92).
In recent years, the role of the microbiome in redefining novel immune therapy has fascinated clinicians and researchers alike. Physiological decoding of the microbiome and its metabolite derivatives (e.g., amino acids and short-chain fatty acids) in MMRd and MMRp identified functionally distinct footprints. For example, enrichment of Bacteroides fragilis and sulfidogenic Fusobacterium nucleatum (Fn) were profound in MMRd. In contrast, Bacteroides fragilis was deprived in the MMRp tumor-microbiome interface. Both these species are linked to a maladapted metabolic landscape in the gut niche (93, 94). To further narrow down at the level of clades, a recent study identified that clade 2 of the Fn subspecies animalis (Fna) strain is predominant behind intra-tumoral loads following heavy colonization in the CRC niche (95). Fusobacterium nucleatum gets the upper hand in an MMRp ecosystem. It confers resistance to chemotherapy, promotes Wnt signaling, binds to TIGIT through its Fap2 component, and activates inhibitory cytokine-producing Treg and M2 macrophages. TLR4-NFkB signaling under such conditions is impaired in chemo-resistant CRC due to the upregulation of autophagy and anti-apoptotic signals (96, 97). Among different metabolites, bacteria-derived inosine acts via A2A adenosine receptor (A2AR) in Th1 cells, facilitates T cell and DC crosstalk and increases the metabolic fitness of CD8+T cells to trigger tumor killing (98). It serves as an alternative carbon source where there is a restriction of glucose availability to CD8 (99). The chronic inflammation due to a low-fibre diet and altered bacterial interaction with archaea insults the gut ecosystem, shifting a balance to dysbiosis (100, 101). This property also orchestrated a metabolomics bias where MMRd tumor had more association with host protective amino acid biosynthesis (102).
Loss of resistance to the colonization of harmful invading bacteria, a new hub created by them in the vicinity of the tumor and inside tumor core including immune cells challenge the host protective anti-tumor immune function. Th17 cells help maintain homeostasis (eubiosis) in a normal gut. However, damage caused by bacterial invasion on gut epithelia triggers the loss of IL-17RA. The systemic spread of Th17 cells and B cells to distal organs facilitates tumor promotion via Dual oxidase 2 (DUOX2). This study showed compartmentalized and context-dependent roles of IL-17 signaling (103). Specific cellular contexts of the IL-1 receptor (IL-1R) also determine the impact of microbial induced IL1 signaling on CRC pathogenesis. While IL-1R deletion in epithelial cells blocks CRC progression independent of inflammation, the same defects in the T cells and the myeloid cells (mainly neutrophil) restrict and exacerbate tumor growth and progression, respectively, following the microbial invasion in tumors (104). The targeted ablation of source bacteria can block the compensatory loop. Similarly, in the mice inflammation model, CD4-driven IL-10 production through macrophages augments IL-17 production (105). Fn is a dominant player in this paradigm. It drives a shift that augments formate production. Aryl hydrocarbon receptor (AhR) signaling promotes invasion and cancer stem cell properties in in vitro co-culture of Fn with CRC cells under this condition. In mice, Fn injection increases the Th17 cell expansion and tumor growth (106). Since IL-17 low MMRp tumors favor ICB outcome, similar cross-talk, and context are expected to persist in their suppressive TME. The γδT-17 cells represent another subset that drives an MDSC bias in CRC (67, 107, Figure 2).
Systems-level diversity of the gut microbiome describes their influences in shaping CRC tumor niche. The 16S rRNA gene sequencing of dMMR (n=29) and pMMR(n=201) in tumors (T) and matched adjacent normal (N) tissues deciphered critical differences in their diversity both at alpha and beta levels. Overall, species diversity of gut microbiome (alpha diversity) was higher in the MMRd-T niche than in MMRp-T and MMRd-N. This comparative profiling showed significant differences (beta diversity) between MMRd and MMRp (Figure 2). Secondary Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis confirmed microbiota-related glycan metabolism, vitamins and nucleotide biosynthesis, active cell death, and defects in DNA repair machinery in MMRd tumors. Indeed, these properties favored PFS and OS upon immunomodulator exposure in MMRd and outperformed MMRp, where microbiota relies predominantly on lipid metabolism (108).
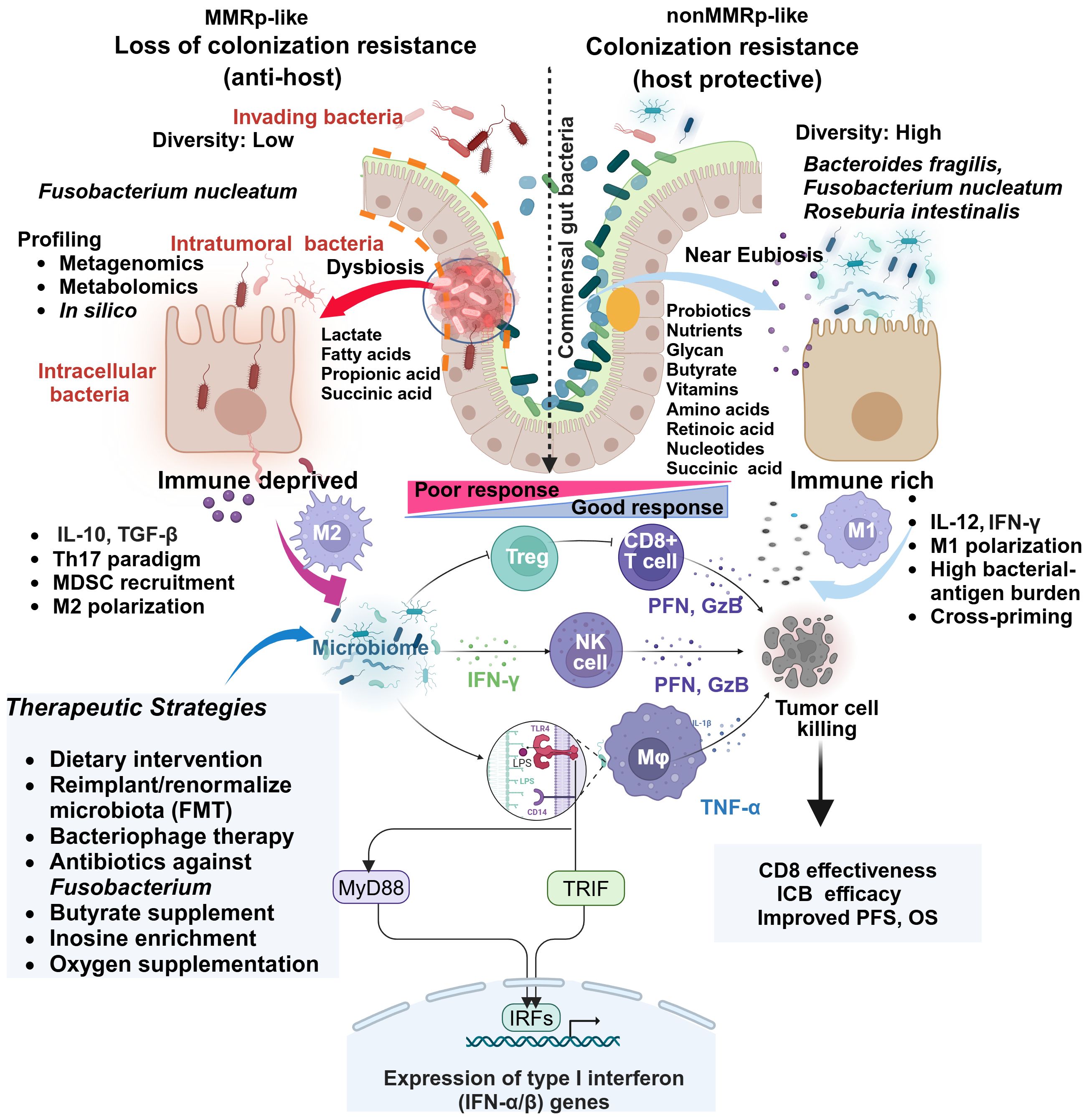
Figure 2 Comparative gut microbiome profiles in MMRp and non MMRp tumor hosts underline multiple contextual constraints and explain barriers to therapy success. The global loss of protective gut resident commensal bacteria makes the border porous for invading bacteria and supports their colonization. Different next-generation sequencing platforms and in silico analysis enable determining the high load of such bacteria and dissecting the loss of diversity in MMRp interface. This maladaptation promotes the metabolic bias in the microenvironment characterized by the overproduction of lactate, propionate, long-chain fatty acids and concomitant loss of glycans, short-chain fatty acids like butyrates, vitamins, amino acids, retinoic acids, and nucleic acids. In such conditions, intra-tumoral and intracellular bacteria facilitate the polarization of immune cells like MDSC, Treg and M2 macrophages, creating a suppressive paracrine cytokine loop. This polarization indicates a sharp contrast with MMRd, where a permissive metabolic footprint favors the preservation of cytokines like IL-12 and IFN-γ, bacterial antigen presentation by M1 macrophages. Under this condition, the interaction of bacterial LPS with TLR in macrophages triggers a signaling pathway via canonical myeloid differentiation primary response 88 (MyD88) and TIR domain-containing adaptor inducing interferon-β (TRIF) that engages IRFs and produces type 1 IFN. Fn-mediated altered cytokines and other anti-apoptotic mechanisms confer resistance by orchestrating an M1 toM2 paradigm shift. Macrophages (M1), CD8, and NK-mediated production of anti-tumor cytotoxic effectors like perforin (PFN) and granzyme-B (GzB) elicit tumor-killing effects. TGF-β, IL-10 and IL-17 impair immune-effector function in MMRp. Multiple strategies focusing on improving the hostile tumor-microbiome interface in MMRp can reverse the suppressive state. Adapted from “Keystone Gut Microbiota Species Provide Colonization Resistance to Invading Bacteria” by BioRender.com (2021). Retrieved from, https://app.biorender.com/biorender-templates.
In a permissive MMRd ecosystem, microbiota supports vitamin A metabolite and retinoic acid accumulation. It galvanizes nucleic acid and protein breakdown machinery in a heterogenous gut immune-interaction network. The accumulation of lactate and other short-chain organic acids like propanoic acid, owing to microbiota depletion in MMRp, makes the tumors immunosuppressive (109) (Figure 2). Lactic acid-producing bacteria Lactobacillus iners rewire host tumor metabolic pathway in cervical cancer and confer chemo- and radiotherapy resistance. A similar L-Lactate producing bacterial population reduces recurrence-free survival (RFS) in colorectal adenocarcinoma (110). Targeting the metabolic hardwire that regulates local oxygen levels or reduces hypoxia will provide insights into their therapeutic prospects (111, 112). For example, antagonists targeting immune-specific CD73 or genetic deletion of A2AR, reversing lactate and hypoxia-induced immune suppression are in clinical development (113, 114). Therefore, combining such agents with approved immune or non-immune therapies may boost the anti-tumor response in MMRp. A spatial metabolomics landscape of the tumors also adds an interactive milieu in this context (115). Extending its crosstalk with the microbial metabolomic network will further define the metabolic vulnerabilities in MMRp and other similar tumors.
While the proximal and distal gut microbiota define the fate of tumors, intra-tumoral bacteria in such scenarios pose a serious health challenge. Integrated metagenomics and metabolomic profiling further expand the scope of ecosystem-level deep mining and shed light on undetected metabolites (116, 117). Pan cancer profiling of intra-tumoral bacterial hubs helped elucidate their distinct indication-specific composition. It also confirmed their intracellular presence, which covers tumor and immune cells. For example, Firmicutes and Bacteroidetes phyla were the two most abundant species in a cohort of 22 CRC samples (118). From these perspectives, the distinct orientation of dysbiosis and its polymorphic microbiome spectrum are suspected to drive toxin-induced mutagenesis in the gut ecosystem (119). CRC patient-derived fecal gavage has been associated with inducing GI tract carcinogenesis in germ-free mice (120). Contrary to this, CRC with MMRp persuasively displayed a hostile metabolic microenvironment that facilitates disease progression and therapy resistance. The conceptual progress in tumor-microbiome interactions also sheds new light on the presence of intertumoral bacteria and their geospatial micro-niche in the tumor ecosystem. New single-cell RNA sequencing technology has mitigated the low biomass challenge and improved the robustness of capturing the tumor microbiome diversity. The preferentially high bacterial population density in vasculature deprived (i.e. CD34 negative) and Ki67 negative pockets with suppressive immune contexture in CRC forms the basis for the non-random heterogeneity of microbiota (121). This profiling added valuable knowledge about resources in such tumor ecosystems and can gauge potential benefits from complementary therapy.
Intra-tumor microbiome maps of CRC (and GI) have been developed in a pan-cancer study. It revealed an MSI-MSS distinction of their communities. It also showed poor survival after ICB in the F. nucleatum high group in the case of NSCLC (109, 122). However, there is a paucity of knowledge on the premises of MMRp (102, 118). As a dysregulated immune-microbiome interplay in MMRp CRC corrupts TME, reinstating a patient-friendly microbiota context and relevant therapeutic strategies potentiate a better therapy response in MMRp (109, 122, summarized in Figure 2 and Table 2). Further studies on the intracellular bacterial population in MMRp+ CRC and its spatial distribution in an immune-excluded context coupled with metabolic programming will offer valuable insights and perspectives. The comprehensive landscape of tumor-associated bacteria highlights the scope of fine-tuning therapeutic intervention at the tumor-microbiome interface.
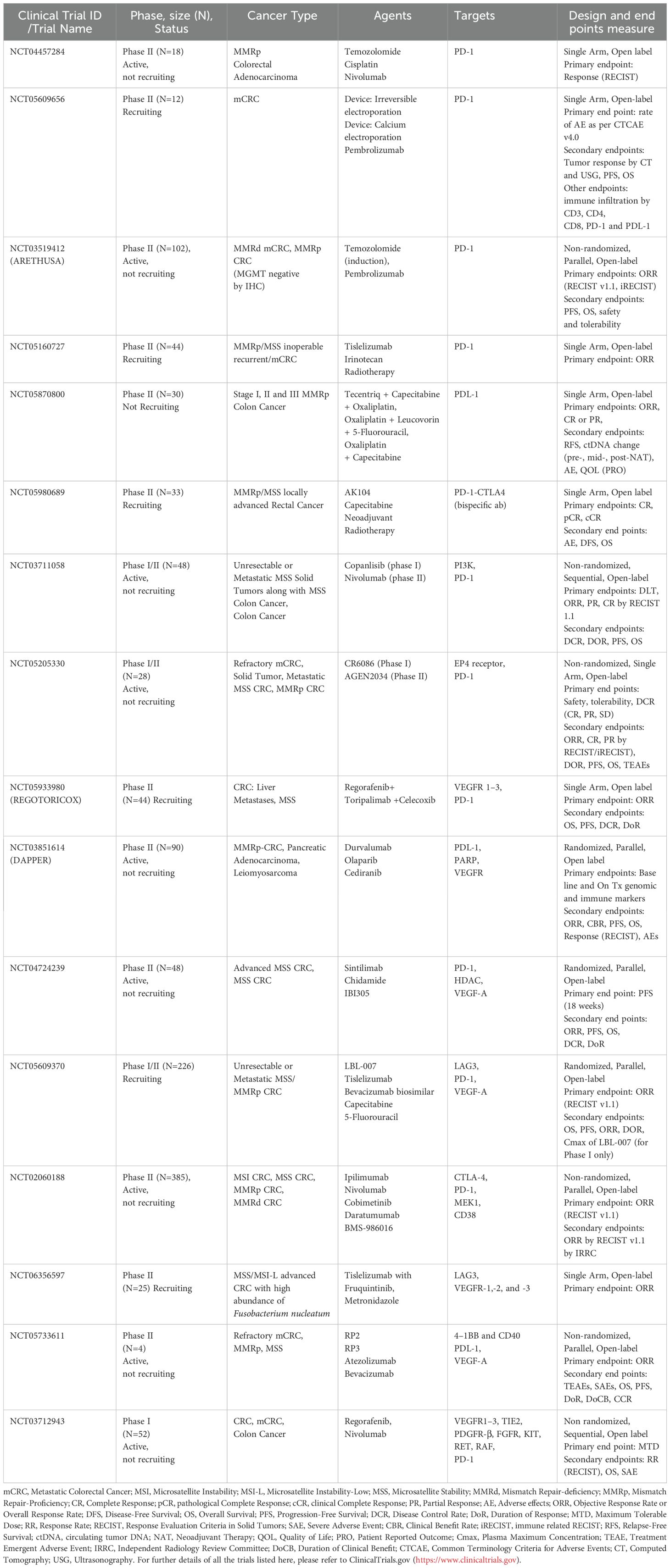
Table 2 Ongoing clinical trials that include MMRp positive CRC patients.
Several strategies leveraged diverse aspects of microbiomes and their perturbation in preclinical and clinical settings and identified their potentials and limitations.
The probiotic bacterium Clostridium butyricum inhibits Wnt signaling, reduces the risk of colon cancer development and boosts anti-microbial macrophage function while sparing inflammation-induced tissue damage (123, 124). Faecal microbiota analysis of CRC patients revealed that Roseburia intestinalis, a probiotic species, and the metabolite (butyrate) generated by it, protect mice (CT-26 and MC 38) and human hosts from gut inflammation and damage. They also unleash CD8-induced anti-PD-1 efficacy in MSI-low/MMRp (CT-26) orthotopic mice. The substantial depletion of this species was observed in patient-derived stools compared to healthy individuals. Its transfer to mice from healthy humans inhibited tumor growth (125). A conversation between Group 3 innate lymphoid cells (ILC3s) and T cells through the engagement of MHCII complex following supplementation of microbiota orchestrate type 1 innate immune response and responsiveness to PD1 inhibition in mice. Lack of MHC-II in ILC3s or microbiota harvested from ILC3s dysregulated subjects failed to elicit immune therapy response following their transfer to mice (126).
Similar systemic clearance was reported when Fn was targeted by silver nanoparticle-bound M13 phage (Ag-M13). Reduction of Fn-induced MDSC and reinvigoration of APC functions were reclaimed under this therapy condition in mice models. Ag-M13 acted in synergy with ICB or chemo-agents (127). In general, antibiotics also have precedence in interfering with ICB (128). The dose, sequence, spectrum, limited and emergency-only use of antibiotics can overcome this challenge. Target-specific antibiotics and bacteriophages make their way to better deal with this situation (66, 129). Through modulating specific chemokine production, gut microbiota helps infiltrate the anti-tumor T cells to the tumor sites and improves the survival opportunities (130). Bifidogenic live bacterial products complementing the microbiome, tested initially in renal cell carcinoma (RCC), would add an interesting vertical in this direction (131). More importantly, several vaccines targeting Fn and Bf in colon cancer are under preclinical development. Depending on the risk association, these vaccine candidates will be used for therapeutic or prophylactic purposes (132).
Analysis of drug-metabolome association in allogeneic hematopoietic cell transplant (alloHCT) recipients among cancer patients may benefit these populations. Longitudinally tracked fecal microbial species revealed a substantial loss of alpha diversity or dysbiosis. It also gathered information regarding a reversal under different medications. An in silico computational prediction model mirrors the in vitro measurement of antibacterial activity and patient clinical outcomes (133).
Antibiotics can reduce bacterial loads that pose a threat to prognosis and response to therapy. For example, F. nucleatum in CRC accumulates succinic acids. This high succinic acid in tumor hinders response to PD1 inhibition by obstructing CD8 cells. Both FMT from the responder and antibiotic metronidazole overcome this restrain (66). Likewise, by enhancing the safe and effective local delivery, liposomal antibiotic administration in mice targeting F. nucleatum elicited cytotoxic T cell response through increasing the immunogenic neoantigen burden of bacterial origin. This modulation further helps in T cell priming and recognition of antigen-naive and reactive tumors (134) (Figure 2). In clinical CRC, before surgical resection, eliminating anaerobic bacteria load upon antibiotics treatment improved disease-free survival (DFS) by 25.5%.
Antibiotics can lead to the vertical loss of healthy intestinal flora, but probiotic and fecal microbial transplantation (FMT) can compensate for the antibiotic-induced loss of gut microbiota. Current limitations within this realm involve compatibility, stability, unknown composition, kinetics, and dynamics, which could be ethical concerns. For an amenable resolution of these concerns, instead of adopting a blanket use, individualized assessment of gut health and other supporting methods like physical activities are important factors that can improve the outcomes (135–137). Reimposing anti-dysbiotic barriers requires coordinated approaches. Supplementing the gut ecosystem with niche-modifying commensal species prevents colonization by invaders. It prevents the accumulation of metabolically challenging pathogenic microbes and releases bacterial antigens to boost the pro-immunogenic immune network (137) and Figure 2.
Both immune and targeted therapy rely on the individualized selection of patients to maximize benefit from a given treatment modality. Integrating biomarkers that predict response is of pivotal importance in this context. This also provide information pertaining to resistance and help designing rational therapeutics.
Collectively, 85% of CRCs are of MMRp type (138). Overall, 10% CRC and 5% metastatic CRC of MMRp status show a response to ICB (33). Combining anti-PD1 with novel anti-CTLA4 in heavily pretreated (median prior line: 4) CRC with MSS status showed promising safety and efficacy (ORR 24%). In case of no history of liver metastases/ablation of liver metastases without recurrence, a better outcome was achieved (n=24, ORR 42%, and DCR 96%). This response included a patient with SD (RECIST 1.1) who showed ongoing metabolic complete response (mCR) by PET after CEA normalization. For all responder cases, metastatic sites spanned soft tissue, peritoneum, retroperitoneum, pleural effusions, bone, lungs, and lymph nodes. Responder mutation profile confirmed RAS mutations (4 KRAS, 1 NRAS), no BRAF mutations, a high TMB (TMB=10) in one case, one case of CPS >50%, and no single POLE mutations cases (139). Both MSS CRC with and without liver metastasis showed benefits from ICB, where liver metastasis conferred more frequent resistance (140). This implies the urgent requirement to improve the overall response to this therapy in MMRp tumors, reducing its gap with MMRd. Indeed, challenging the current response rate for all modalities with new and more effective treatment regimens is a continuous process and needs innovative, rational approaches integrated with multimodal diagnostics and predictive tools.
From oncoimmune perspectives, however, response predictive gene signatures revealed that a preexisting immunoreactive profile does not explicitly depend on suppressive tumor immune microenvironment represented by spatial CD8 and IFN-γ and colocalized PDL-1/IDO1 checkpoint genes. Irrespective of IL-17 low or high niche states, the IL-17 low MMRp landscape mimicked a primary CRC responsive to ICB. In the same study, a panel of immunomodulatory genes (precisely, LAG-3, CD8A, CD4, CD274) showed similar expression patterns between MMRp and MMRd responder cohorts. However, it indicated reciprocal downregulation in the MMRp non-responder cohort (67). Rationally targeting the IL-23/Stat3/IL-17 signaling axis in IL-17 high MRRp+ CRC may offer a mechanistic basis for overcoming adaptive resistance to ICB. Analysis of TCGA data and cell line profiling of MMRp from CRC revealed that high expression of immunoglobulin superfamily 6 (IGSF6) is correlative with infiltration of CD4+ T cells, CD8+ T cells, CD68+ macrophages and conferred sensitivity to immunotherapy and chemotherapy (141). Lineage tracking elucidated an interesting new role of CD4 cells in both providing help to cytotoxic CD8 cells and directly acting as cytotoxic killer cells. This observation unlocks a new gate for understanding its implication of targeting coinhibitory receptor LAG-3 that mechanistically crosstalk with MHC class II (142–145). Interestingly, a first-in-human multicohort safety and efficacy study of anti-LAG-3 antibody MK4280 (favezelimab) with pembrolizumab in CRC that progressed on two prior lines following combination (2C+5), ORR was 6.3% (4PR, 1CR by RECIST). In contrast, the median duration of response (DOR) and OS were 10.6 months and 8.3 months, respectively. Both these endpoints were better compared to monotherapy. In particular, patients with PDL-1 status >1 combined positive score (CPS) showed a prominent response (146).
Beating the current ORR across therapeutic modalities in MMRp is a formidable challenge. It warrants smart and novel vulnerability mapping strategies. There is an increasing interest in understanding ostensibly dysfunctional immune contexture. Fine-tuning the T cell pre-exhaustion dynamics is critical for preventing their final differentiation into terminally exhausted T (TET) cells (147, 148). TET cells present an irreversible phenotype and frustrate immune intervention strategies like PDL-1 blockade. For poorly immunogenic MMRp tumors, targeting other potentially actionable MMRp and non-MMRp vulnerabilities would exert similar mileage. New therapeutic developments leveraging new immunomodulators are in the preclinical pipelines to potentiate this paradigm shift (Table 1).
DNA damage response (DDR) as an overtly orchestrated system has multiple actively operating networks like class 1 defects in double-strand break (DSB) and replication repair (BRCA 1 and BRCA 2 mutations), class 2 defects in signaling (ATM, ATR, CHK1, CHK2) and class 3 defects (MMR) leading to high TMB (81). Understanding the therapeutic opportunities of targeting each of these defects in colorectal cancers has gained momentum in recent years. Several modalities targeting them are under active development (149). Dissecting the diverse facets of counter-regulation and coregulation of their interactive molecular circuits in governing the protection of the tumor cells against cytotoxic insults offers novel opportunities for turning the MMRp tumors vulnerable to emerging therapies (Figure 3).
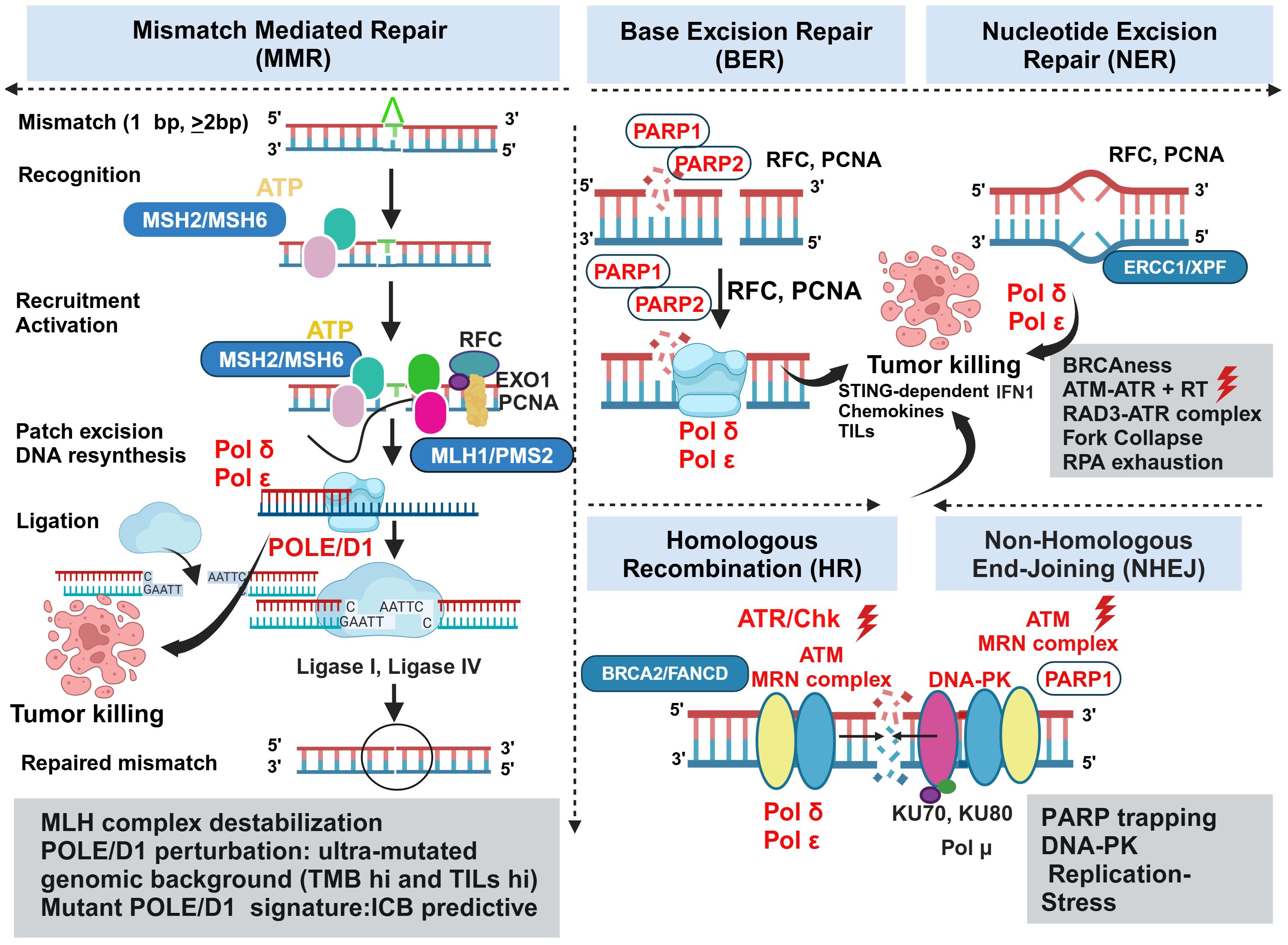
Figure 3 Vulnerabilities and alternative actionabilities in MMRp tumors decipher the key biomarkers and molecular targets in DDR machinery. MMRp tumors efficiently bypass key base pair mismatches using a repair mechanism that recruits repair proteins in the recognition-activation-resynthesis-ligation cascade. Although MLH complex destabilization and PLOEed perturbation are key actionable areas, the limited options in this class of MMR system highlight the need to search for parallel alternative targets involved in base excision repair (BER), nucleotide excision repair (NER), homologous recombination (HR) and nonhomologous end joining (NHEJ). Biomarkers for each repair category are presented in boxes. Therapeutic targets and their perturbations are indicated in red. POLE/D1, PARP, ATM, ATR, and Chk are key targets for which drugs are either under clinical development or approved for one or more indications. ATR and concurrent radiation can act in synergy to induce STING-dependent IFN1 production, and deliver conditional lethal hits leading to the killing of the tumors. Key steps of molecular mechanisms are depicted in the figure and mentioned in corresponding boxes. MSH, MutS homologs; MLH, MutL protein homolog; PMS2, Postmeiotic segregation Increased 2; EXO1, exonuclease 1; RFC, Replication factor C; PCNA, proliferating cell nuclear antigen; POLε, DNA polymerase epsilon POLδ, polymerase delta; XP-F, xeroderma pigmentosum; ERCC1, excision repair cross-complementation group1; FMCD2, Fanconi anemia group D2; MRN, Mre11-Rad50-Nbs1.
Pathogenic missense mutations in proofreading enzyme polymerase epsilon (POLE) - at DNA binding and catalytic sites, largely operating in gastric cancer and CRC, have been implicated in perturbing MMR efficiency and generated ultra-mutated genomic landscape illuminated with both high TMB and TILs predictive for ICB response. From these perspectives, POLE/D1-like molecules provide a unique example of alternative and complementary target biology and raise the hope of benefiting MMRp patients to ICB (150). Mice CT26 tumors harboring Pole P286R mutant clones showed better response to ICB mono and combination therapies by a 3-fold increase in CD3 infiltrations. However, they extended modest survival in the patient tumor-derived xenograft (PDX) model. These may be due to the absence of human-specific stable immune contexture in PDX. A mutant POLE/POLED1 signature outperforms traditional approaches in stratifying patients likely to benefit from ICB. These data further suggest that a pathogenic mutation affecting the fidelity of DNA repair enzyme can boost anti-tumor immunity of ICB (151).
Nonredundant and alternative DNA repair pathways spatially and temporally converse and converge to avoid human replication protein A (RPA) exhaustion followed by “replication catastrophe” and cell death. Ataxia telangiectasia and Rad3 related (ATR) protects the cells from this vulnerability (152, 153). In contrast, their defects lead to synthetic lethality. BRCA 1 and BRCA 2 are two targets for which there is interest in developing biomarker-guided DNA repair agents. These agents perturb classical non-homologous end joining (NHEJ) and other complementary repair systems like homologous recombination (HR) and alternate end joining (alt EJ). Molecules that leverage the target biology of PARP, Ataxia-telangiectasia mutated (ATM), ATR, CHK1 and WEE 1 are either in trials or under development (154) and Figure 3. Study showed that concurrent radiation and inhibitor of ATM, a DNA damage repair protein, elicits tumor growth inhibition mainly by augmenting STING-dependent IFN1 production and chemokines critical for immune infiltration. In CT 26 and MC38 mice models, ceralasertib, a potent ATR inhibitor, showed no direct effects on tumor killing, which is typical for this class of agents. Instead, it induces immunomodulating effects on proliferating CD8+T cells when intermittent dosing was applied in contrast to continuous dosing. It changes monocyte-MDSC (M-MDSC) and TAM dynamics and increases DC in mice TME. Type 1 IFN (IFN1) is augmented in cancer patients upon ceralasertib therapy (155). Therapy-induced upregulation of PDL-1 and MHC1 on the tumor surface further offers a temporal window of sequential PDL-1 inhibition in combination with anti-ATM agent (156). Although this study was done in the mice model of HNSCC, it perceivably reciprocates the same mechanism of action in the MMRp-like context where poor immunogenicity is a confounding factor. Preclinical studies using multiple in vivo mice models deciphered the involvement of ATR-mediated DNA repairing machinery in radiation-resistant CRC. This defect impaired DC-mediated tumor antigen cross-presentation via upregulation of CD47 (‘eat me not signal’) and PDL-1. It drives further crosstalk through the cognate engagement of PD-1 and SIRPα signaling cascade. A rationale combination of RT with anti-SIRPα and anti-PDL-1 targeting this axis resulted in a complete response in primary and abscopal tumors in a STING-dependent manner. These data imply the mechanistic link between ATR inhibition in inducing anti-tumor response when the DNA repair pathway confers RT resistance (157). ATR- ATR-Checkpoint Kinase 1 (Chk) also surged as a viable target aiming to overturn MMRp-driven therapy constraints. DNA alkylating agent MNNG induced MeG/T mismatch lesion by inhibiting Chk1 signaling. N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) orchestrated the fork collapse and DSB in embryonic stem cells in the absence of ATR-Chk1 activation. It also perturbed their ability to handle replication stress and led to rapid induction of apoptosis. However, a transient S phase checkpoint in Hela cells under MNNG pressure and the active state of ATR-Chk1 induced G2 arrest (158).
Another critical barrier that obstructs successful therapy outcomes is linked to chemotherapy (CT) and RT-induced DNA damage. Subsequent evasion of this response by a compensatory repair mechanism is mediated by the DNA-dependent protein kinase (DNA-PK). Further dissection of this network revealed that both NHEJ and HR could happen sequentially. In that case, DNA-PK and MRN/CtlP coordinate in this event (159). The open-label, phase I trial of peposertib (formerly M3814), an inhibitor of DNA-PK, showed tolerance in a cohort of 31 solid tumor patients. However, only modest outcomes (stable diseases) were observed in 12 patients for >12 weeks (160). Nevertheless, the targetability of DNA-PK has been established in multiple in vitro preclinical studies, including studies that demonstrated the druggability of its catalytic domain subunits using small molecule inhibitors (161). Learning from molecular biology harnessed DNA-PK mediated excessive end resection to the non-propagating quiescent G0 phase. However, it was not evident in the G1 or G2 phase of the cell cycle owing to the detachment of FBXL12, a ubiquitylation-promoting factor that targets KU70/KU80 subunits of DNA-PK only in G0 (162) and Figure 3.
In addition to the germline and somatic coding mutations in key MMR enzymes, transcriptional silencing of MLH1 through promoter hypermethylation (MLH1 methylation) was observed in 10–20% of all CRC cases. This MLH1 methylation is one of the main causes of sporadic CRC (162). Promoter hypermethylation in hMLH1 gene is associated with microsatellite instability and BRAF mutations, accompanied in some cases by somatic loss of the wild-type allele (163, 164). However, the status and impact of MLH1 methylation are less explored in MMRp from a translational perspective (27). Epigenetic readers, writers and erasers/degraders represent an active cluster for therapeutic development. Their roles in MMRp tumors of diverse indications still need to be fully elucidated. N6-methyladenosine (m6A) METTL3/METTL14, the writer constituents of methyltransferase complex (MTC), impeded TILs recruitment in MMRp CRC. Targeted silencing of this axis augmented STAT1-mediated IFN-γ production and elicited anti-tumor effects (165). Analysis of TCGA data, tissue microarray, RNA-Seq and preclinical mice experiments using MC38 (MDSC rich), CT26 (immune inflamed, MMRp), and CD34/immune reconstituted humanized immune CRC xenograft mice models deciphered that m6A reader YTHDF1 had an inverse correlation with IFN-γ gene signature. Indeed, perturbation of YTHDF1 by gene silencing averted resistance to anti-PD1 therapy by inhibiting MDSC infiltration and boosting cytotoxic CD8 functions in MMRp-positive CRC (166).
As DDR based therapeutics are gaining rapid momentum in the targeted oncology arena, there are outstanding challenges related to their target biology validation, structure-based drug design and selectivity. Multiple DDR targets have high sequence homology. For instance, DNA-PK, ATM and ATR share similar sequences; therefore, there is more likelihood of off-target effects. Cryo-electron microscopy (Cryo-EM) enables structural resolutions of ATM and ATR. However, it needs other coactivating proteins like RPA for ATR and MRN for ATM. This knowledge gap currently hinders structure-based DDR drug design (167). Mechanistically, when more than one repair modalities operate, they tend to diminish the efficacy of the selected agent. Besides redundancy and limited biomarkers for target specificity selection, toxicity, target loss and target resistance are leading drivers of efficacy loss. ATM and ATR axis can be used as salvage therapy in PARP inhibitor refractory tumors or expanded for HRP tumors (168). Uncertain actionability with limited knowledge of the microenvironment context also poses a challenge. Some targets, like BRCA mutations, have a low prevalence (5%) in CRC, mostly confined to MSI-H. Even in BRCA mutant cancers, the tumors can escape inflammation-driven immune attacks using lesser-known mechanisms that are both tumor-intrinsic and tumor-immune microenvironment-regulated in nature (169). Although PARP inhibitors are at the forefront of DDR driven therapies, PARP-trapping by proximity ligation assay in BRCA1 mutant breast cancer (CaBr) showed both efficacy and off-target bone marrow toxicity following PARP inhibitor monotherapy and poses a challenge for combination (170). The recent withdrawal of late-line PARP inhibitors for multiple indications warrants further learning of the root causes (171). For all these targets, pharmacological dosing and additional mechanisms of actions involving immune-mediated and direct killing are not elaborated in most of the investigations. Similarly, there is scope to gain more insights into whether ATR at continuous dose is inferior to intermittent (holiday) doses.
The clinical CRC world is equipped with robust selection biomarkers like KRAS mutations and MSI/MMR. Current clinical guidelines recommend testing the MMR status of all CRC samples irrespective of the clinical stages of the disease. PDL-1 genetic variation (del, polysomy, amplification) is more frequent in MMRd compared to MMRp in new CRC at the time of diagnosis and is linked to poor prognosis (172). High congruence (99%) of MMRp proteins was observed between IHC and MSI molecular testing based on a large sample size of >3K and inter-site cross-validation (173). Discordance was observed when samples were collected using different methods; sample volumes varied, and different training methods were used to handle the tissues (174. Pancreatic and endometrial cancers with Lynch Syndrome (LS) were the two areas of high discordance between MSI and MSH (175). An AI-guided classifier achieved a performance score that appreciated the clinical benchmark (95% sensitivity for MMRp/MSS and MMRd) without taking help from any manual annotation steps (176). MMRp and MMRd binary paradigm has been shifted in recent years (27). For an ongoing process like MMR, a robust, specific and sensitive assay incorporating multiple inputs and its validation will reduce the false detection of MMRd due to the unrelated presence of high TMB.
Liquid biopsy (ctDNA) has emerged as a valuable tool to longitudinally monitor treatment outcomes or tumor progression non-invasively. In patients with advanced GI cancers, ctDNA accelerated enrolment (doubled compared to regular biopsy) by shortening screening duration three times without any negative impact on treatment outcomes (177, 178). Moreover, clonal landscaping from 2000 patient-derived liquid biopsy samples identified several actionable driver alterations (177). Further, a study of 445 CRC patients with stage 2 disease (2:1 randomization) evaluated liquid biopsy-guided management. A ctDNA-positive result at 4 and 7-weeks post-surgery prompted a chemotherapy decision. In contrast, patients who were ctDNA-negative were spared from CT. This study elucidated that ctDNA-guided 2 years of recurrence-free survival was non-inferior to non-ctDNA-guided standard clinicopathological criteria (93.5% and 92.4%, respectively; 95% CI, -4.1 to 6.2 (noninferiority margin, -8.5 percentage points). These data indicate the promise of this approach in managing adjuvant chemo treatment without enhancing the risk of recurrence-free survival (179).
As illustrated in Figure 4, liquid biopsy can be used as an alternative or complementary tool along with other multi-omics platforms like functional genomics, epigenomics and spatial biology insights in a systems biology context to a) predict response and a potential relapse in the clonally biased immune evaded TME, b) expand the strategic window to combat clinical recurrence and, c) provide a synthetic lethality screen to underpin clinically actionable drug targets for these advanced, primary treatment failure conditions (181–184).
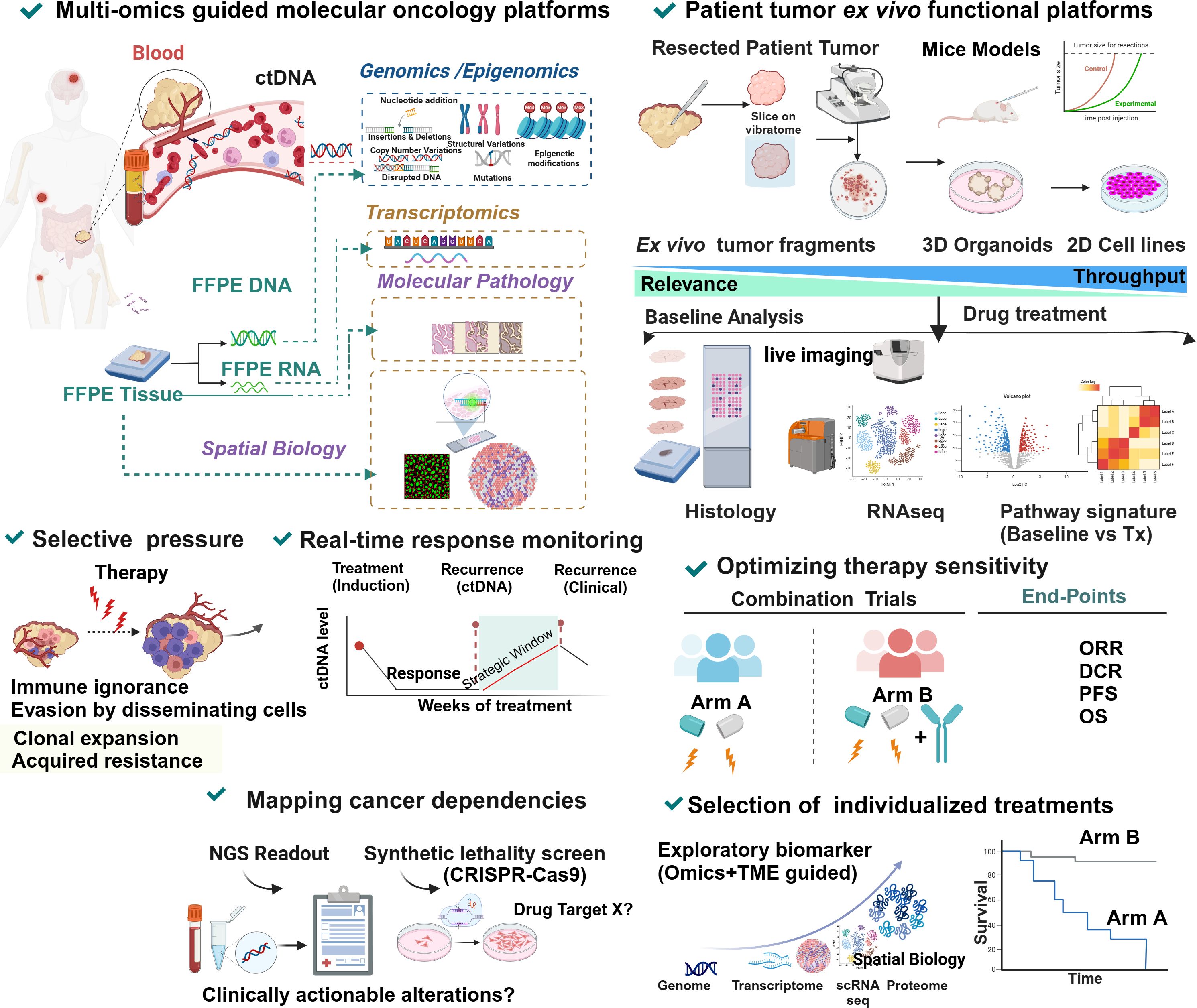
Figure 4 Precision oncology molecular multi-omics and functional platforms in predicting recurrence, response and guiding rational combinations in MMRp. Information obtained from systematic and multilayered molecular profiling of patient tumors converging genomics, epigenomics and proteomics from a longitudinal analysis of liquid biopsy and clinical biopsied samples (fresh unfixed or fixed tissues) provide critical spatiotemporal dynamic contexts of biomarkers, signatures and tumor-immune interface. Finally, it helps predict the recurrence risk, including therapy-driven or therapy-independent recurrence and clonal expansion. Change in ctDNA levels in serum is a reliable predictor that informs about a prospective clinical recurrence and, therefore, opens a strategic window in guiding the treatment plan ahead of recurrence. Multiple synthetic lethality screens like CRISPER knockout and conditional lethality decipher the pathway dependency and oncogenic addictions for delineating the druggable targets (180). Functional prediction platforms led by microenvironment-guided drug sensitivity screens actively leverage information from contextually relevant phenotypic readouts in a mechanistic setting. This clinical avatar works in coordination with molecular oncology modalities where clinically meaningful evidence of actionability is available and can provide an alternative solution when such biomarker information is absent or not translatable. An integrative cross-functional approach uses multiple live systems covering 2D cell lines, 3D organoids and non-dissociated tumor slices depending on the requirements and availability. Mice models can still evaluate the systems-level modulation of drugs and their synergy. These models show the advantage of obtaining data from real-world diverse assays using live cultures focusing on drug reactivity and functional modulation patterns in time and space. The provision is there to integrate the outputs into a predictive score. The clinical relevance, speed and scalability are not uniform across the platforms. Microenvironment-guided selection of optimal therapy combination in trials led by such assay outputs takes informed decisions by integrating multi-omics and spatial biology context at single-cell levels. The platform-guided selection has the power to improve response rate and differentiate superior combinations and synergy.
These approaches harmonize in guiding critical treatment decisions for naïve, relapsed or refractory conditions and therefore open new trial/therapy opportunities where regular tissue biopsy is not feasible or there is an urgent need for molecular guided (e.g. in CRC, KRAS/NRAS/BRAFV600E/MSI) treatment decisions (177, 185). An earlier systematic review in 2018 highlighted a relatively low clinical actionability that has remained a bottleneck for decades in selecting NGS-guided rationale therapies. It benefits only a minor (10%) percentage of patients representing indications like NSCLC, melanoma and RCC from the frontline ICB (186). However, recent findings from the MSK-IMPACT assay that used OncoKB 2017 and 2022 versions showed overall evidence-driven improvement of clinical actionability for existing SOCs and IO agents. The platforms increased the enrolment in clinical trials for new agents. Likewise, the actionability scaled from 8.9% to 31.6% (187). This study also exposed the gaps where non-actionable alterations (i.e., cases where no response predictive or treatment selection biomarkers are available) are confined mainly to TP53 (43%), KRAS (19%) and CDKN2A (12.2%). More importantly, only MSI-H and TMB high showed a rise in the actionability trajectory.
For non-actionable, more precisely, cases where no drug-matching biomarkers are available, liquid biopsy-based ex vivo functional filters can directly inform drug sensitivity (188). Indeed, ctDNA-based approaches do not readily provide critical spatial biology insights during diagnosis and treatments and are not ready to replace conventional biopsy.
Further validation of liquid biopsy through clinical trials may save time and resources in late-phase development by its informed integration for DDR ATM, BRCA 1,2 related mutation profiling (189). CTCs outbound for a clustered migration have a much higher potential to metastasize than solitary CTCs. Intercellular compartments (nanolumenal) concentrating growth-promoting ligands facilitate high metastatic potential (190). Phenotypes or molecular signatures that differentiate the CTC-derived oligoclonal precursors open new avenues to tailor next-generation precision medicine solutions in relevant landscapes.
Success and failure of oncology and immunooncology drug trials hinge on three main verticals- a: profound insights into target biology and anti-target ‘avoidome’, b: preclinical platforms, and c: time of the decision to go for the trials (191). The upfront limitation of preclinical platforms is that the models that filter a drug for late-stage nominations are highly porous due to a lack of critical contexts mechanistically mirroring a patient’s tumor immune microenvironment. The traditional in vitro and in vivo mice tumor models remain the backbone of discovery research for decades. Recent in vitro cell line panels helped identify potential combinations based on targeted drug screens for CRC and other indications (192). Similarly, mice data demonstrated the feasibility of parallel response modeling of multiple agents in tandem to accelerate this screening phase (193). However, in I-O, their independent contributions are not consistent. This constraint also limits the biomarker-guided patient selections. These perspectives prompted the development and validation of other alternative platforms that can reduce dependency on conventional systems if not completely replaced. Integrating molecular signature and spatial TiME contexts adds powerful predictive insights (194) and Figure 4. They also minimize the trial failure rates. PDX, syngeneic mice and their ex vivo 3D culture counterparts, when integrated and complemented with multiplex readouts, could help advance our understanding of metastatic diseases and bridge the critical knowledge gaps (195). Reverse translation of mice data and forward translation of the short-term ex vivo data synergistically may augment their predictive power of immune checkpoint response (196).
In recent years, patient-derived 3D organoids, organ-on-chips, and non-dissociated tumor fragments have emerged as functionally relevant platforms to screen drugs in TME settings that are close to the real world (Figure 4). They guide prioritizing novel combinations based on specific vulnerabilities (197). Studies showed that these models can be adopted rapidly. They can faithfully predict ICB drug reactivity and clinical outcomes based on parameters and scores generated using the assays and their discriminatory contributions (198, 199) and Figure 4. Two independent utility studies highlighted that ex vivo response prediction can signal a positive correlation in the case of liquid cancers. Kornauth et al. reported an image-based single-cell functional precision medicine (scFPM) n-of-one approach. In 54% of cases it showed >1.3-fold enhanced PFS after median follow-up for 23.9 months. Exceptionally, 40% of the responders showed a three-times longer duration of response than expected. A second study using a multi-omics ex vivo platform in a functional precision medicine tumor board (FPMTB) reported 97% actionability. This study also reported 59% ORR and linkage of IL-15 overexpression with resistance to FLT3 inhibitor and instructed to alloHSCT for 5 patients (200, 201). A recent ex vivo study with 101 bone marrow samples from 79 eligible patients further informed on the variability of the drug sensitivity. It defined patient stratification based on actionable multiplex pathology inputs and image-based deep learning (202). CRC-driven models are well represented in this new era of ex vivo technology (65, 155, 203–206) and Figure 4.
In CRC, peritoneal metastasis is associated with the lowest survival rate. Therapies that can improve the OS are limited for this condition. Narasimhan et al. described a medium-throughput ex vivo ‘peritoniods’ model for addressing this challenge (207). The model showed a 68% (19/28) success rate of stability. Most patients whose ascites were used were prediagnosed with the worst prognosis CMS 4/MSS. Drug sensitivity testing using this model led to a decision impact on two patients. One of the patients who had multiple rounds of treatment failure showed partial response (PR) to the gemcitabine–capecitabine combination arm three months post-therapy. Notably, regorafenib failed to show sensitivity against any sample (207). The observed lack of response to regorafenib may be due to compromised angiogenic and stromal contexts in ex vivo ascites (207–209). EGFR inhibitor osimertinib and HDAC inhibitor vorinostat also displayed higher sensitivity. However, regulatory barriers to off-label therapy prohibited testing them on the patients. Another patient received ‘off-label’ Vandetanib on compassionate grounds, but it was too late to attain the expected benefit. These findings demonstrated the value of ascites-based ex vivo organoids in informed treatment selection in clinically challenging scenarios.
Although not all ex vivo platforms are high throughput, they provide suitable substrates to test multiple drug arms in parallel. In evaluating five drug combinations in parallel, an ex vivo organotypic slice culture identified potential benefits only from the combination of Src inhibitor and MEK inhibitor. In 29 surgically resected samples from MMRp CRC in this study, the baseline level of phosphorylated Src was used as a biomarker coupled with wild-type KRAS G12 (210). Mechanistically, an independent study delineated the consequence of inhibiting the RAF-MEK-ERK axis. The death of KRAS mutant CRC organoids was observed upon exposure to low doses of RAF and ERK inhibitors in combination (211). A similar screen may help prioritize rational synergy in MMRp CRC. The microorganospheres (MOS) derived from CRC patient biopsy enabled rapid testing with a turnaround time of 8–14 days. Moreover, testing the immunotherapy agent in this MOS predicted sensitivity with 75% accuracy and resistance (NPV) with 75% accuracy (212). Encouraging results were obtained from the co-culture of tumor-immune organoids of CRC from MMRd and MMRp backgrounds. This study differentiated the CD8-driven killing of CRC organoids upon ICB. Further elucidation of this response revealed the selective engagement of MHC-I and cytotoxic effector molecules (203). Indeed, this study independently predicted better efficacy of ICB in MMRd associated CRC and lent credence to this platform for systems-level testing of similar IO modalities and preserving MoA (Figure 4). The realization of ex vivo response modeling is gaining momentum, particularly after the inspiring outcomes from dostarlimab trial in CRC from MMRd class (213). It galvanizes the efforts of looking for similar benefits in other cancers.
Unlike cell lines of primary origins and, to some extent, patient-derived organoids and PDX, short-term explant slices largely retain genomic fidelity without introducing any new driver or pathogenic variants (214–218). In this context, RNAseq data demonstrated intra and inter-tumor variability in the retention of clonal and immune landscapes in GBM. Despite underlying heterogeneity, patient baseline and corresponding non-dissociated tumor explant fragments matched better than their primary cell line counterparts (219). Metabolically, In vivo and PDEx models demonstrated the preferential utilization of glucose over L-glutamine. This background gives these models an edge over cell line-based in vitro culture (220). Indeed, metabolic heterogeneity in cancer is a dynamic paradigm and depends on factors that regulate local oxygen and nutrient gradients, tumor cell density, stromal composition, exosome dynamics, and ECM stiffness. In temporal settings, therapy-induced changes and metastasis also adapt altered metabolic programs (17, 19).
PDEx maintains angiogenesis gene signature and blood vessel phenotypes in ex vivo culture (205, 221). However, their functional impact still needs to be elucidated. Hasselluhn et al. demonstrated the active maintenance of critical cell types such as tumor epithelial, fibroblast, myeloid, T cells and blood vessel density in short-term ex vivo culture. In this system, the paracrine cascade of hedge hogde (HH) activation promoted angiosuppression by inhibiting WNT and VEGFR2 signaling. Inhibition of Smoothened (SMO) in this background induced angiogenic switching. Subsequently, the explants regained vascular level hypersprouting following inhibition of the HH pathway target (222). Experimental evidence also supports the ability of the organoids and tumor fragments to gauge radiosensitivity. Organotypic brain slice culture (OBSC) using a multiparametric algorithm led to developing a sensitivity score that normalizes off-target effects (223). Besides retaining the tumor, immune and genomic fidelity, efforts to recreate or retain microbial interface in ex vivo culture have also recently gained momentum (224).
The deliverables of ex vivo response prediction rely on a battery of complementary, multimodal and cross-functional assays and integrated scores (Figure 4). Real-time monitoring of drug reactivity in PDEx system needs quantitative evaluation using baseline and proper vehicle control to capture optimal assay peak and corresponding delta. An integrative model of preferably label-free spatial live imaging, in contrast to terminal snapshots, complementary cross-functional bioassays, multiplex spatial omics at baseline and on treatments, adds more edges to the robustness and multimodality perspectives in the current systems (225–227). As tumor agonistic approaches, these functional tools rapidly gain momentum in late oncology drug developments and leverage forward and reverse translation for rationale combinations. In previous studies, the number of samples with matched clinical outcomes was either indirect or limited. Further trials aim to understand the clinical utility of these functional screening platforms (228). The decision to co-culturing tumor with immune compartments like PBMC and TILs must be carefully informed based on the target indications and candidate drugs. For greater acceptability, current limitations of ex vivo models, like variability, must be overcome (229, 230) and Table 3. Best practices for tissue procurements for IO trials are evolving. Similar guidance can be implemented to improve ex vivo prediction (231). New regulatory guidelines have classified the ex vivo systems as laboratory-developed tests (LTD) and medical devices (MD). Therefore, these tests are under the high-risk category and subject to more stringent validation and quality control requirements (228, 232).
CRC is a challenging late-stage neoplasm for which there is a pressing need to develop more effective therapies. This review delves into MMRp pathophysiology, current progress in its complex molecular landscape, immune contexts, metabolic state and biomarkers. These are the major pain points for cracking the therapy barriers and beating the overall low response rate of the existing drugs. Transformative approaches are vital to overturn the global mortality rates tied to MMRp. About 1–10% of MMRp is MSI-H. They maintain high TILs compared to the MMRp-MSS group and resemble MMRd (27). Cross-learning from MMRd enriches our knowledge. Enhancing the scope of adaptive therapy for MMRp tumors appreciates strategies undertaken for MMRd cancers (233). The MSI-MMRp subgroup with high TILs is uniquely positioned in this spectrum.
Deciphering the unique TiME in MMRp tumors and addressing its low TMB is crucial for rational renormalization. However, the key obstacles include a restrained anti-tumor immune paradigm, a deficient or depressed TILs-TLS state, and an overwhelming TGF-β/Th17 signaling bias that explicitly fosters immune suppression (234). MMRp TiME is highly decorated with tumor-promoting chemokines and cytokines networks presented by CXCL 1–8, CXCR4, IL-10 and their divergent downstream cascades. They choreograph the tumor-homing and retention of the immunosuppressors like monocytes, TAN, TAM and Treg cells (62) (Figure 1 and Table 1). Capturing a comprehensive chemokine footprint can help rejuvenate immune modulation and suppress tumor-promoting functions. Multiple strategies are emerging to reconstruct the chemokine network. Small molecules and antibodies to target pro-tumor ligands, oncolytic virus-expressing chemokine ligands and neoadjuvant vaccines are few that have raised optimism (59, 60), (Figures 1, 2; Table 1).
We discussed the impact of a skewed gut microbiome on treatment resistance and immune evasion. Changes in microbiome diversity leads to an environment high in lactate and other tumor-protecting metabolites (66, 109, 110). This microbiome-immune cross-talk disrupts immune cell function through TGF-β and Th17 signaling, and hinders key immune activation cascades. We illustrated the strategies to combat this hostile TiME (66) (Figures 1, 2; Tables 1, 2). The critical insights gained from these approaches will shape future research and clinical practices.
Indeed, the gut microbiome poses promises and, at the same time, personalization challenges in treating CRC. The abundance of F. nucleatum in CRC impairs ICB effects on CD8 through local succinate build-up (66). Eliminating tumor-invading F. nucleatum and other toxicogenic species and restoring healthy microbiota showed early promise across MMR classes (66, 68, 127, 129, 132, 134). However, safety and efficacy data from more extensive trials is needed before routinely adopting this modalities. Several studies demonstrated the untapped potential of molecularly alternative vulnerabilities. Targeting specific enzymes like POLE/D1 and other complementary MMR-independent repair mechanisms like ATM/ATR/DNA-PK and PARP may offer new treatment avenues in this direction (Figure 3).
Resources are emerging to prioritize molecular oncology precision through leveraging omics and multi-omics platforms, including liquid biopsy for response monitoring in advanced CRC (189). This can enhance actionable omics-guided predictive biomarkers. In heterogeneous and dynamic settings like CRC, genomics data alone may not be enough to determine therapy efficacy in the MMRp context. Complementary approaches like liquid biopsy and TME-guided functional platforms can be stand-alone options and conform to positive changes in trials and management. Ex vivo tumor models like 3D organoids, slices, and ascites showed the potential to understand therapy resistance and optimal treatment decisions for CRC (197, 198, 203, 235). Insights from spatial biology integrated with AI/ML open a new frontier in personalized combination selection (Figure 4; Table 3). Contrasting responses to ICB in MMRp and MMRd have been demonstrated using CRC-derived organoids (203). Studies are investigating the predictive strength of ex vivo models (65, 228, 236). However, the current status indicates that further clinical validation and refinement are essential before the widespread adoption of these functional screens. Space limit prevents detailed discussion of all the subtopics here.
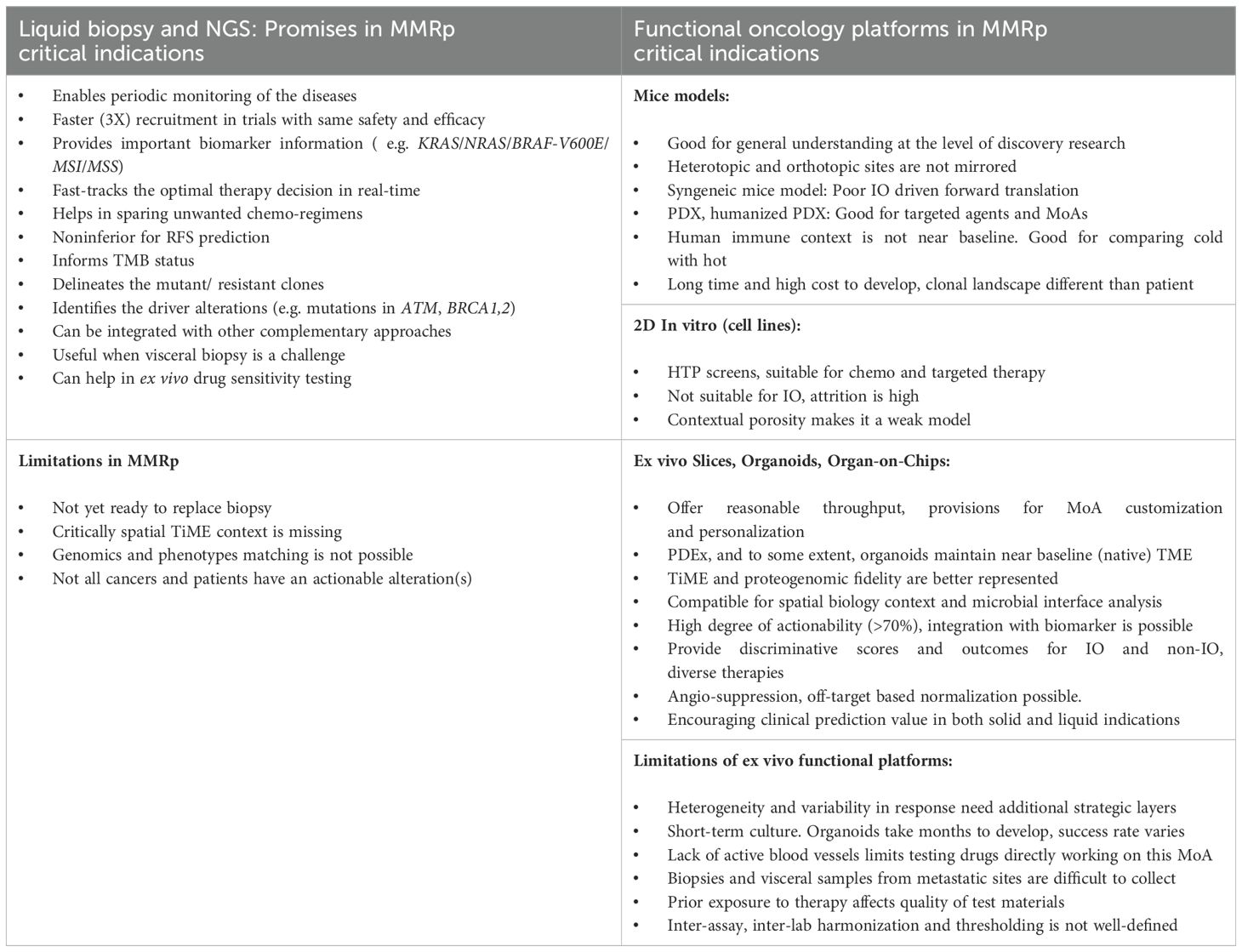
Table 3 Key strengths and limitations of precision oncology molecular and functional platforms.
Ongoing trials pave the way for exploring rational combinations in MMRp, often with promising results. For example, a triple combination of PD1 inhibitor, HDAC inhibitor, and anti-VEGF agent significantly improved PFS and ORR. RNA sequencing confirmed high CD8 infiltration in the triplet arm cohort (237). Neoadjuvant DC vaccine boosted the immune profile (60). Other ongoing trials that leverage combinatorial opportunities in MMRp-positive CRC are summarized in Table 2. A collaborative framework that continues to engage clinical experts and translational scientists on this premise will reduce the time required to develop new therapeutic modalities. Finally, the outlook evolving from ongoing MMRp research sets to transform the journey toward precision oncology.
BM: Conceptualization, Writing – original draft, Writing – review & editing, Visualization. LM: Writing – review & editing. NN: Writing – review & editing. SD: Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We acknowledge the immense support provided by Dr. Balasubramanian V during preparation and revision of this article. Authors express their gratitude to the editor and reviewers for their critical insights and constructive suggestions to improve the manuscript. All the graphical illustrations were created with BioRender.com.
BM, LM and NN are employees of Bugworks Research Inc. and hold shares of the company. SD is co-founder of Bugworks Research Inc and holds equities. BM serves as an honorary scientific advisor of Praesidia Biotherapeutics, Bangalore, India and a consultant of Aryastha Life Sciences, Hyderabad, India.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Roy D, Roy D, Gilmour C, Patnaik S, Wang LL. Combinatorial blockade for cancer immunotherapy: targeting emerging immune checkpoint receptors. Front Immunol. (2023) 14:1264327. doi: 10.3389/fimmu.2023.1264327
2. Saez–Ibanez AR, Upadhaya S, Campbell J. Immuno–oncology clinical trials take a turn beyond PD1/PDL1 inhibitors. Nat Rev Drug Discovery. (2023) 22:442–3. doi: 10.1038/d41573–023–00066–0
3. Chehelgerdi M, Chehelgerdi M. The use of RNA–based treatments in the field of cancer immunotherapy. Mol Cancer. (2023) 22:106. doi: 10.1186/s12943-023-01807-w
4. Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. (2023) 618:144–50. doi: 10.1038/s41586-023-06063-y
5. Ledford H. Cocktails for cancer with a measure of immunotherapy. Nature. (2016) 532:162–4. doi: 10.1038/532162a
6. Upadhaya S, Neftelinov ST, Hodge J, Campbell J. Challenges and opportunities in the PD1/PDL1 inhibitor clinical trial landscape. Nat Rev Drug Discovery. (2022) 21:482–3. doi: 10.1038/d41573-022-00030-4
7. Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin. (2023) 73:233–54. doi: 10.3322/caac.21772
8. Cortes–Ciriano I, Lee S, Park WY, Kim TM, Park PJ. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun. (2017) 8:15180. doi: 10.1038/ncomms15180
9. Sharma P, Hu–Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
10. Valero C, Lee M, Hoen D, Zehir A, Berger MF, Seshan VE, et al. Response rates to anti–PD–1 immunotherapy in microsatellite–stable solid tumors with 10 or more mutations per megabase. JAMA Oncol. (2021) 7:739–43. doi: 10.1001/jamaoncol.2020.7684
11. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. (2020) 52:17–35. doi: 10.1016/j.immuni.2019.12.011
12. Sosinsky A, Ambrose J, Cross W, Turnbull C, Henderson S, Jones L, et al. Insights for precision oncology from the integration of genomic and clinical data of 13,880 tumors from the 100,000 Genomes Cancer Programme. Nat Med. (2024) 30:279–89. doi: 10.1038/s41591-023-02682-0
13. Marusyk A, Janiszewska M, Polyak K. Intratumor heterogeneity: the rosetta stone of therapy resistance. Cancer Cell. (2020) 37:471–84. doi: 10.1016/j.ccell.2020.03.007
14. Westcott PMK, Muyas F, Hauck H, Smith OC, Sacks NJ, Ely ZA, et al. Mismatch repair deficiency is not sufficient to elicit tumor immunogenicity. Nat Genet. (2023) 55:1686–95. doi: 10.1038/s41588-023-01499-4
15. Eckert EC, Nace RA, Tonne JM, Evgin L, Vile RG, Russell SJ. Generation of a tumor–specific chemokine gradient using oncolytic vesicular stomatitis virus encoding CXCL9. Mol Ther Oncolytics. (2019) 16:63–74. doi: 10.1016/j.omto.2019.12.003
16. Kalli M, Poskus MD, Stylianopoulos T, Zervantonakis IK. Beyond matrix stiffness: targeting force–induced cancer drug resistance. Trends Cancer. (2023) 9:937–54. doi: 10.1016/j.trecan.2023.07.006
17. Demicco M, Liu XZ, Leithner K, Fendt SM. Metabolic heterogeneity in cancer. Nat Metab. (2024) 6:18–38. doi: 10.1038/s42255-023-00963-z
18. Boedtkjer E, Pedersen SF. The acidic tumor microenvironment as a driver of cancer. Annu Rev Physiol. (2020) 82:103–26. doi: 10.1146/annurev-physiol-021119-034627
19. Jin MZ, Jin WL. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. (2020) 5:166. doi: 10.1038/s41392-020-00280-x
20. Rehman SK, Haynes J, Collignon E, Brown KR, Wang Y, Nixon AML, et al. Colorectal cancer cells enter a diapause–like DTP state to survive chemotherapy. Cell. (2021) 184:226–242.e21. doi: 10.1016/j.cell.2020.11.018
21. Adam–Artigues A, Valencia Salazar LE, Aguirre–Ghiso JA. Immune evasion by dormant disseminated cancer cells: A Fermi paradox? Cancer Cell. (2024) 42:13–5. doi: 10.1016/j.ccell.2023.12.017
22. Goddard ET, Linde MH, Srivastava S, Klug G, Shabaneh TB, Iannone S, et al. Immune evasion of dormant disseminated tumor cells is due to their scarcity and can be overcome by T cell immunotherapies. Cancer Cell. (2024) 42:119–134.e12. doi: 10.1016/j.ccell.2023.12.011
23. Berrino E, Aquilano MC, Valtorta E, Amodio V, Germano G, Gusmini M, et al. Unique patterns of heterogeneous mismatch repair protein expression in colorectal cancer unveil different degrees of tumor mutational burden and distinct tumor microenvironment features. Mod Pathol. (2023) 36:100012. doi: 10.1016/j.modpat.2022.100012
24. Amodio V, Lamba S, Chilà R, Cattaneo CM, Mussolin B, Corti G, et al. Genetic and pharmacological modulation of DNA mismatch repair heterogeneous tumors promotes immune surveillance. Cancer Cell. (2023) 41:196–209.e5. doi: 10.1016/j.ccell.2022.12.003
25. Westcott PMK, Sacks NJ, Schenkel JM, Ely ZA, Smith O, Hauck H, et al. Low neoantigen expression and poor T–cell priming underlie early immune escape in colorectal cancer. Nat Cancer. (2021) 2:1071–85. doi: 10.1038/s43018-021-00247-z
26. Gejman RS, Chang AY, Jones HF, DiKun K, Hakimi AA, Schietinger A, et al. Rejection of immunogenic tumor clones is limited by clonal fraction. Elife. (2018) 7:e41090. doi: 10.7554/eLife.41090
27. Xu Y, Liu K, Li C, Li M, Zhou X, Sun M, et al. Microsatellite instability in mismatch repair proficient colorectal cancer: clinical features and underlying molecular mechanisms. EBioMedicine. (2024) 103:105142. doi: 10.1016/j.ebiom.2024.105142
28. Shimada K, Bachman JA, Muhlich JL, Mitchison TJ. shinyDepMap, a tool to identify targetable cancer genes and their functional connections from Cancer Dependency Map data. Elife. (2021) 10:e57116. doi: 10.7554/eLife.57116
29. Hung S, Saiakhova A, Faber ZJ, Bartels CF, Neu D, Bayles I, et al. Mismatch repair–signature mutations activate gene enhancers across human colorectal cancer epigenomes. Elife. (2019) 8:e40760. doi: 10.7554/eLife.40760
30. Zhou RW, Xu J, Martin TC, Zachem AL, He J, Ozturk S, et al. A local tumor microenvironment acquired super–enhancer induces an oncogenic driver in colorectal carcinoma. Nat Commun. (2022) 13:6041. doi: 10.1038/s41467-022-33377-8
31. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. (2000) 343:78–85. doi: 10.1056/NEJM200007133430201
32. Yurgelun MB, Kulke MH, Fuchs CS, Allen BA, Uno H, Hornick JL, et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J Clin Oncol. (2017) 35:1086–95. doi: 10.1200/JCO.2016.71.0012
33. Overman MJ, Ernstoff MS, Morse MA. Where we stand with immunotherapy in colorectal cancer: toxicity management. Asco Educ Book. (2019) 38:239–47. doi: 10.1200/EDBK_200821
34. Belhadj S, Moutinho C, Mur P, Setien F, Llinàs–Arias P, Pérez–Salvia M, et al. Germline variation in O6–methylguanine–DNA methyltransferase (MGMT) as cause of hereditary colorectal cancer. Cancer Lett. (2019) 447:86–92. doi: 10.1016/j.canlet.2019.01.019
35. Voutsadakis IA. High tumor mutation burden (TMB) in microsatellite stable (MSS) colorectal cancers: Diverse molecular associations point to variable pathophysiology. Cancer Treat Res Commun. (2023) 36:100746. doi: 10.1016/j.ctarc.2023.100746
36. Yoon HH, Shi Q, Heying EN, Muranyi A, Bredno J, Ough F, et al. Intertumoral heterogeneity of CD3+ and CD8+ T–cell densities in the microenvironment of DNA mismatch–repair–deficient colon cancers: implications for prognosis. Clin Cancer Res. (2019) 25:125–33. doi: 10.1158/1078-0432.CCR-18-1984
37. Kim JK, Chen CT, Keshinro A, Khan A, Firat C, Vanderbilt C, et al. Intratumoral T–cell repertoires in DNA mismatch repair–proficient and –deficient colon tumors containing high or low numbers of tumor–infiltrating lymphocytes. Oncoimmunology. (2022) 11:2054757. doi: 10.1080/2162402X.2022.2054757
38. Niknafs N, Balan A, Cherry C, Hummelink K, Monkhorst K, Shao XM, et al. Persistent mutation burden drives sustained anti–tumor immune responses. Nat Med. (2023) 29:440–9. doi: 10.1038/s41591-022-02163-w
39. Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell. (2021) 184:4734–4752.e20. doi: 10.1016/j.cell.2021.08.003
40. Jimenez–Rodriguez RM, Patil S, Keshinro A, Shia J, Vakiani E, Stadler Z, et al. Quantitative assessment of tumor–infiltrating lymphocytes in mismatch repair proficient colon cancer. Oncoimmunology. (2020) 9:1841948. doi: 10.1080/2
41. de Vries NL, van de Haar J, Veninga V, Chalabi M, Ijsselsteijn ME, van der Ploeg M, et al. γδ T cells are effectors of immunotherapy in cancers with HLA class I defects. Nature. (2023) 613:743–50. doi: 10.1038/s41586-022-05593-1
42. Ho WW, Gomes–Santos IL, Aoki S, Datta M, Kawaguchi K, Talele NP, et al. Dendritic cell paucity in mismatch repair–proficient colorectal cancer liver metastases limits immune checkpoint blockade efficacy. Proc Natl Acad Sci U S A. (2021) 118:e2105323118. doi: 10.1073/pnas.2105323118
43. Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan–cancer whole–genome analyses of metastatic solid tumours. Nature. (2019) 575:210–6. doi: 10.1038/s41586-019-1689-y
44. Voissière A, Gomez–Roca C, Chabaud S, Rodriguez C, Nkodia A, Berthet J, et al. The CSF–1R inhibitor pexidartinib affects FLT3–dependent DC differentiation and may antagonize durvalumab effect in patients with advanced cancers. Sci Transl Med. (2024) 16:eadd1834. doi: 10.1126/scitranslmed.add1834
45. Tikidzhieva A, Benner A, Michel S, Formentini A, Link KH, Dippold W, et al. Microsatellite instability and Beta2–Microglobulin mutations as prognostic markers in colon cancer: results of the FOGT–4 trial. Br J Cancer. (2012) 106:1239–45. doi: 10.1038/bjc.2012.53
46. Barrow P, Richman SD, Wallace AJ, Handley K, Hutchins GGA, Kerr D, et al. Confirmation that somatic mutations of beta–2 microglobulin correlate with a lack of recurrence in a subset of stage II mismatch repair deficient colorectal cancers from the QUASAR trial. Histopathology. (2019) 75:236–46. doi: 10.1111/his.13895
47. Albacker LA, Wu J, Smith P, Warmuth M, Stephens PJ, Zhu P, et al. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PloS One. (2017) 12:e0176181. doi: 10.1371/journal.pone.0176181
48. Español–Rego M, Fernández–Martos C, Elez E, Foguet C, Pedrosa L, Rodríguez N, et al. A Phase I–II multicenter trial with Avelumab plus autologous dendritic cell vaccine in pre–treated mismatch repair–proficient (MSS) metastatic colorectal cancer patients, GEMCAD 1602 study. Cancer Immunol Immunother. (2023) 72:827–40. doi: 10.1007/s00262–022–03283–5
49. Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. (2006) 7:344–53. doi: 10.1038/ni1330
50. Fridman WH, Meylan M, Petitprez F, Sun CM, Italiano A, Sautès–Fridman C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol. (2022) 19:441–57. doi: 10.1038/s41571-022-00619-z
51. Overacre–Delgoffe AE, Bumgarner HJ, Cillo AR, Burr AHP, Tometich JT, Bhattacharjee A, et al. Microbiota–specific T follicular helper cells drive tertiary lymphoid structures and anti–tumor immunity against colorectal cancer. Immunity. (2021) 54:2812–2824.e4. doi: 10.1016/j.immuni.2021.11.003
52. Gaglia G, Burger ML, Ritch CC, Rammos D, Dai Y, Crossland GE, et al. Lymphocyte networks are dynamic cellular communities in the immunoregulatory landscape of lung adenocarcinoma. Cancer Cell. (2023) 41:871–886.e10. doi: 10.1016/j.ccell.2023.03.015
53. Schürch CM, Bhate SS, Barlow GL, Phillips DJ, Noti L, Zlobec I, et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell. (2020) 182:1341–1359.e19. doi: 10.1016/j.cell.2020.07.005
54. Fridman WH, Meylan M, Pupier G, Calvez A, Hernandez I, Sautès–Fridman C. Tertiary lymphoid structures and B cells: An intratumoral immunity cycle. Immunity. (2023) 56:2254–69. doi: 10.1016/j.immuni.2023.08.009
55. Li S, Mirlekar B, Johnson BM, Brickey WJ, Wrobel JA, Yang N, et al. STING–induced regulatory B cells compromise NK function in cancer immunity. Nature. (2022) 610:373–80. doi: 10.1038/s41586-022-05254-3
56. Zhang H, AbdulJabbar K, Moore DA, Akarca A, Enfield KSS, Jamal–Hanjani M, et al. Spatial positioning of immune hotspots reflects the interplay between B and T cells in lung squamous cell carcinoma. Cancer Res. (2023) 83:1410–25. doi: 10.1158/0008-5472.CAN-22-2589
57. Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity. (2021) 54:859–74. doi: 10.1016/j.immuni.2021.01.012
58. Mempel TR, Lill JK, Altenburger LM. How chemokines organize the tumour microenvironment. Nat Rev Cancer. (2024) 24:28–50. doi: 10.1038/s41568-023-00635-w
59. Shiao SL, Gouin KH 3rd, Ing N, Ho A, Basho R, Shah A, et al. Single–cell and spatial profiling identify three response trajectories to pembrolizumab and radiation therapy in triple negative breast cancer. Cancer Cell. (2024) 42:70–84.e8. doi: 10.1016/j.ccell.2023.12.012
60. Gögenur M, Balsevicius L, Bulut M, Colak N, Justesen TF, Fiehn AK, et al. Neoadjuvant intratumoral influenza vaccine treatment in patients with proficient mismatch repair colorectal cancer leads to increased tumor infiltration of CD8+ T cells and upregulation of PD–L1: a phase 1/2 clinical trial. J Immunother Cancer. (2023) 11:e006774. doi: 10.1136/jitc–2023–006774
61. Bilusic M, Heery CR, Collins JM, Donahue RN, Palena C, Madan RA, et al. Phase I trial of HuMax–IL8 (BMS–986253), an anti–IL–8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J Immunother Cancer. (2019) 7:240. doi: 10.1186/s40425-019-0706-x
62. Jung K, Heishi T, Incio J, Huang Y, Beech EY, Pinter M, et al. Targeting CXCR4–dependent immunosuppressive Ly6Clow monocytes improves antiangiogenic therapy in colorectal cancer. Proc Natl Acad Sci U S A. (2017) 114:10455–60. doi: 10.1073/pnas.1710754114
63. Tian L, Xu B, Chen Y, Li Z, Wang J, Zhang J, et al. Specific targeting of glioblastoma with an oncolytic virus expressing a cetuximab-CCL5 fusion protein via innate and adaptive immunity. Nat Cancer. (2022) 3:1318–35. doi: 10.1038/s43018-022-00448-0
64. Rakké YS, Campos Carrascosa L, van Beek AA, de Ruiter V, van Gemerden RS, Doukas M, et al. GITR ligation improves anti–PD1–mediated restoration of human MMR–proficient colorectal carcinoma tumor–derived T cells. Cell Mol Gastroenterol Hepatol. (2023) 15:77–97. doi: 10.1016/j.jcmgh.2022.09.007
65. Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez–Carmona M, et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti–CCR5 therapy in cancer patients. Cancer Cell. (2016) 29:587–601. doi: 10.1016/j.ccell.2016.03.005
66. Jiang SS, Xie YL, Xiao XY, Kang ZR, Lin XL, Zhang L, et al. Fusobacterium nucleatum–derived succinic acid induces tumor resistance to immunotherapy in colorectal cancer. Cell Host Microbe. (2023) 31:781–797.e9. doi: 10.1016/j.chom.2023.04.010
67. Llosa NJ, Luber B, Tam AJ, Smith KN, Siegel N, Awan AH, et al. Intratumoral adaptive immunosuppression and type 17 immunity in mismatch repair proficient colorectal tumors. Clin Cancer Res. (2019) 25:5250–9. doi: 10.1158/1078-0432.CCR-19-0114
68. Perez LG, Kempski J, McGee HM, Pelzcar P, Agalioti T, Giannou A, et al. TGF–β signaling in Th17 cells promotes IL–22 production and colitis–associated colon cancer. Nat Commun. (2020) 11:2608. doi: 10.1038/s41467-020-16363-w
69. Massagué J, Sheppard D. TGF–β signaling in health and disease. Cell. (2023) 186:4007–37. doi: 10.1016/j.cell.2023.07.036
70. McFarlane AJ, Fercoq F, Coffelt SB, Carlin LM. Neutrophil dynamics in the tumor microenvironment. J Clin Invest. (2021) 131:e143759. doi: 10.1172/JCI143759
71. Battaglin F, Baca Y, Millstein J, Yang Y, Xiu J, Arai H, et al. CCR5 and CCL5 gene expression in colorectal cancer: comprehensive profiling and clinical value. J Immunother Cancer. (2024) 12:e007939. doi: 10.1136/jitc-2023-007939
72. Wang Y, Ding Y, Deng Y, Zheng Y, Wang S. Role of myeloid–derived suppressor cells in the promotion and immunotherapy of colitis–associated cancer. J Immunother Cancer. (2020) 8:e000609. doi: 10.1136/jitc-2020-000609
73. Günther K, Leier J, Henning G, Dimmler A, Weissbach R, Hohenberger W, et al. Prediction of lymph node metastasis in colorectal carcinoma by expression of chemokine receptor CCR7. Int J Cancer. (2005) 116:726–33. doi: 10.1002/ijc.21123
74. Haag GM, Springfeld C, Grün B, Apostolidis L, Zschäbitz S, Dietrich M, et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite–stable metastatic colorectal cancer – The PICCASSO phase I trial. Eur J Cancer. (2022) 167:112–22. doi: 10.1016/j.ejca.2022.03.017
75. Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, et al. Biomolecular network reconstruction identifies T–cell homing factors associated with survival in colorectal cancer. Gastroenterology. (2010) 138:1429–40. doi: 10.1053/j.gastro.2009.10.057
76. Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, et al. Unique ectopic lymph node–like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am J Pathol. (2011) 179:37–45. doi: 10.1016/j.ajpath.2011.03.007
77. Meylan M, Petitprez F, Becht E, Bougoüin A, Pupier G, Calvez A, et al. Tertiary lymphoid structures generate and propagate anti–tumor antibody–producing plasma cells in renal cell cancer. Immunity. (2022) 55:527–541.e5. doi: 10.1016/j.immuni.2022.02.001
78. Fridman WH, Zitvogel L, Sautès–Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
79. Di Pilato M, Kfuri–Rubens R, Pruessmann JN, Ozga AJ, Messemaker M, Cadilha BL, et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell. (2021) 184:4512–4530.e22. doi: 10.1016/j.cell.2021.07.015
80. Boreddy SR, Nair R, Pandey PK, Kuriakose A, Marigowda SB, Dey C, et al. BCA101 is a tumor–targeted bifunctional fusion antibody that simultaneously inhibits EGFR and TGFβ Signaling to durably suppress tumor growth. Cancer Res. (2023) 83:1883–904. doi: 10.1158/0008-5472.CAN-21-4425
81. Hopkins JL, Lan L, Zou L. DNA repair defects in cancer and therapeutic opportunities. Genes Dev. (2022) 36:278–93. doi: 10.1101/gad.349431.122
82. Ritter AT, Shtengel G, Xu CS, Weigel A, Hoffman DP, Freeman M, et al. ESCRT–mediated membrane repair protects tumor–derived cells against T cell attack. Science. (2022) 376:377–82. doi: 10.1126/science.abl3855
83. Delclaux I, Ventre KS, Jones D, Lund AW. The tumor–draining lymph node as a reservoir for systemic immune surveillance. Trends Cancer. (2024) 10:28–37. doi: 10.1016/j.trecan.2023.09.006
84. Inamori K, Togashi Y, Fukuoka S, Akagi K, Ogasawara K, Irie T, et al. Importance of lymph node immune responses in MSI-H/dMMR colorectal cancer. JCI Insight. (2021) 6:e137365. doi: 10.1172/jci.insight.137365
85. van den Bulk J, Verdegaal EME, Ruano D, Ijsselsteijn ME, Visser M, van der Breggen R, et al. Neoantigen–specific immunity in low mutation burden colorectal cancers of the consensus molecular subtype 4. Genome Med. (2019) 11:87. doi: 10.1186/s13073-019-0697-8
86. Morris VK, Overman MJ, Lam M, Parseghian CM, Johnson B, Dasari A, et al. Bintrafusp alfa, an anti–PD–L1:TGF–β trap fusion protein, in patients with ctDNA–positive, liver–limited metastatic colorectal cancer. Cancer Res Commun. (2022) 2:979–86. doi: 10.1158/2767-9764.CRC-22-0194
87. Pant S, Wainberg ZA, Weekes CD, Furqan M, Kasi PM, Devoe CE, et al. Lymph–node–targeted, mKRAS–specific amphiphile vaccine in pancreatic and colorectal cancer: the phase 1 AMPLIFY–201 trial. Nat Med. (2024) 30:531–42. doi: 10.1038/s41591-023-02760-3
88. Frieder J, Kivelevitch D, Menter A. Secukinumab: a review of the anti–IL–17A biologic for the treatment of psoriasis. Ther Adv Chronic Dis. (2018) 9:5–21. doi: 10.1177/2040622317738910
89. Jassim A, Rahrmann EP, Simons BD, Gilbertson RJ. Cancers make their own luck: theories of cancer origins. Nat Rev Cancer. (2023) 23:710–24. doi: 10.1038/s41568-023-00602-5
90. Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. (2015) 347:78–81. doi: 10.1126/science.1260825
91. Vincze O, Colchero F, Lemaître JF, Conde DA, Pavard S, Bieuville M, et al. Cancer risk across mammals. Nature. (2022) 601:263–7. doi: 10.1038/s41586-021-04224-5
92. Swanton C, Bernard E, Abbosh C, André F, Auwerx J, Balmain A, et al. Embracing cancer complexity: Hallmarks of systemic disease. Cell. (2024) 187:1589–616. doi: 10.1016/j.cell.2024.02.009
93. Kim JM, Oh YK, Kim YJ, Oh HB, Cho YJ. Polarized secretion of CXC chemokines by human intestinal epithelial cells in response to Bacteroides fragilis enterotoxin: NF–kappa B plays a major role in the regulation of IL–8 expression. Clin Exp Immunol. (2001) 123:421–7. doi: 10.1046/j.1365-2249.2001.01462.x
94. Chung L, Orberg ET, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis Toxin Coordinates a Pro–carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe. (2018) 23:421. doi: 10.1016/j.chom.2018.02.004
95. Zepeda–Rivera M, Minot SS, Bouzek H, Wu H, Blanco–Míguez A, Manghi P, et al. A distinct Fusobacterium nucleatum clade dominates the colorectal cancer niche. Nature. (2024) 628:424–32. doi: 10.1038/s41586-024-07182-w
96. Yu TC, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. (2017) 170:548–563.e16. doi: 10.1016/j.cell.2017.07.008
97. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. (2015) 42:344–55. doi: 10.1016/j.immuni.2015.01.010
98. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome–derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. (2020) 369:1481–9. doi: 10.1126/science.abc3421
99. Wang T, Gnanaprakasam JNR, Chen X, Kang S, Xu X, Sun H, et al. Inosine is an alternative carbon source for CD8+–T–cell function under glucose restriction. Nat Metab. (2020) 2:635–47. doi: 10.1038/s42255-020-0219-4
100. Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, et al. A dietary fiber–deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell. (2016) 167:1339–1353.e21. doi: 10.1016/j.cell.2016.10.043
101. Coker OO, Wu WKK, Wong SH, Sung JJY, Yu J. Altered gut archaea composition and interaction with bacteria are associated with colorectal cancer. Gastroenterology. (2020) 159:1459–1470.e5. doi: 10.1053/j.gastro.2020.06.042
102. Hale VL, Jeraldo P, Chen J, Mundy M, Yao J, Priya S, et al. Distinct microbes, metabolites, and ecologies define the microbiome in deficient and proficient mismatch repair colorectal cancers. Genome Med. (2018) 10:78. doi: 10.1186/s13073-018-0586-6
103. Chandra V, Li L, Le Roux O, Zhang Y, Howell RM, Rupani DN, et al. Gut epithelial Interleukin–17 receptor A signaling can modulate distant tumors growth through microbial regulation. Cancer Cell. (2024) 42:85–100.e6. doi: 10.1016/j.ccell.2023.12.006
104. Dmitrieva–Posocco O, Dzutsev A, Posocco DF, Hou V, Yuan W, Thovarai V, et al. Cell–type–specific responses to interleukin–1 control microbial invasion and tumor–elicited inflammation in colorectal cancer. Immunity. (2019) 50:166–180.e7. doi: 10.1016/j.immuni.2018.11.015
105. Brockmann L, Tran A, Huang Y, Edwards M, Ronda C, Wang HH, et al. Intestinal microbiota–specific Th17 cells possess regulatory properties and suppress effector T cells via c–MAF and IL–10. Immunity. (2023) 56:2719–2735.e7. doi: 10.1016/j.immuni.2023.11.003
106. Ternes D, Tsenkova M, Pozdeev VI, Meyers M, Koncina E, Atatri S, et al. The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat Metab. (2022) 4:458–75. doi: 10.1038/s42255-022-00558-0
107. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. γδT17 cells promote the accumulation and expansion of myeloid–derived suppressor cells in human colorectal cancer. Immunity. (2014) 40:785–800. doi: 10.1016/j.immuni.2014.03.013
108. Jin M, Wu J, Shi L, Zhou B, Shang F, Chang X, et al. Gut microbiota distinct between colorectal cancers with deficient and proficient mismatch repair: A study of 230 CRC patients. Front Microbiol. (2022) 13:993285. doi: 10.3389/fmicb.2022.993285
109. Li J, Guo Y, Liu J, Guo F, Du L, Yang Y, et al. Depicting the landscape of gut microbial–metabolic interaction and microbial–host immune heterogeneity in deficient and proficient DNA mismatch repair colorectal cancers. J Immunother Cancer. (2023) 11:e007420. doi: 10.1136/jitc-2023-007420
110. Colbert LE, El Alam MB, Wang R, Karpinets T, Lo D, Lynn EJ, et al. Tumor–resident Lactobacillus iners confer chemoradiation resistance through lactate–induced metabolic rewiring. Cancer Cell. (2023) 41:1945–1962.e11. doi: 10.1016/j.ccell.2023.09.012
111. Colombani T, Eggermont LJ, Hatfield SM, Rogers ZJ, Rezaeeyazdi M, Memic A, et al. Oxygen–generating cryogels restore T cell mediated cytotoxicity in hypoxic tumors. Adv Funct Mater. (2021) 31:2102234. doi: 10.1002/adfm.202102234
112. Hatfield SM, Sitkovsky MV. Antihypoxic oxygenation agents with respiratory hyperoxia to improve cancer immunotherapy. J Clin Invest. (2020) 130:5629–37. doi: 10.1172/JCI137554
113. Augustin RC, Leone RD, Naing A, Fong L, Bao R, Luke JJ. Next steps for clinical translation of adenosine pathway inhibition in cancer immunotherapy. J Immunother Cancer. (2022) 10:e004089. doi: 10.1136/jitc-2021-004089
114. Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat Commun. (2021) 12:3236. doi: 10.1038/s41467-021-23331-5
115. Coy S, Wang S, Stopka SA, Lin JR, Yapp C, Ritch CC, et al. Single cell spatial analysis reveals the topology of immunomodulatory purinergic signaling in glioblastoma. Nat Commun. (2022) 13:4814. doi: 10.1038/s41467-022-32430-w
116. Ternes D, Karta J, Tsenkova M, Wilmes P, Haan S, Letellier E. Microbiome in colorectal cancer: how to get from meta–omics to mechanism? Trends Microbiol. (2020) 28:401–23. doi: 10.1016/j.tim.2020.01.001
117. Puig–Castellví F, Pacheco–Tapia R, Deslande M, Jia M, Andrikopoulos P, Chechi K, et al. Advances in the integration of metabolomics and metagenomics for human gut microbiome and their clinical applications. TrAC – Trends Analytical Chem. (2023) 167. doi: 10.1016/j.trac.2023.117248
118. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science. (2020) 368:973–80. doi: 10.1126/science.aay9189
119. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
120. Wong SH, Zhao L, Zhang X, Nakatsu G, Han J, Xu W, et al. Gavage of fecal samples from patients with colorectal cancer promotes intestinal carcinogenesis in germ–free and conventional mice. Gastroenterology. (2017) 153:1621–1633.e6. doi: 10.1053/j.gastro.2017.08.022
121. Galeano Niño JL, Wu H, LaCourse KD, Kempchinsky AG, Baryiames A, Barber B, et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature. (2022) 611:810–7. doi: 10.1038/s41586-022-05435-0
122. Battaglia TW, Mimpen IL, Traets JJH, van Hoeck A, Zeverijn LJ, Geurts BS, et al. A pan–cancer analysis of the microbiome in metastatic cancer. Cell. (2024) 187:2324–2335.e19. doi: 10.1016/j.cell.2024.03.021
123. Chen D, Jin D, Huang S, Wu J, Xu M, Liu T, et al. Clostridium butyricum, a butyrate–producing probiotic, inhibits intestinal tumor development through modulating Wnt signaling and gut microbiota. Cancer Lett. (2020) 469:456–67. doi: 10.1016/j.canlet.2019.11.019
124. Schulthess J, Pandey S, Capitani M, Rue–Albrecht KC, Arnold I, Franchini F, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity. (2019) 50:432–445.e7. doi: 10.1016/j.immuni.2018.12.018
125. Kang X, Liu C, Ding Y, Ni Y, Ji F, Lau HCH, et al. Roseburia intestinalis generated butyrate boosts anti–PD–1 efficacy in colorectal cancer by activating cytotoxic CD8+ T cells. Gut. (2023) 72:2112–22. doi: 10.1136/gutjnl-2023-330291
126. Goc J, Lv M, Bessman NJ, Flamar AL, Sahota S, Suzuki H, et al. Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell. (2021) 184:5015–5030.e16. doi: 10.1016/j.cell.2021.07.029
127. Dong X, Pan P, Zheng DW, Bao P, Zeng X, Zhang XZ. Bioinorganic hybrid bacteriophage for modulation of intestinal microbiota to remodel tumor–immune microenvironment against colorectal cancer. Sci Adv. (2020) 6:eaba1590. doi: 10.1126/sciadv.aba1590
128. Wilson BE, Routy B, Nagrial A, Chin VT. The effect of antibiotics on clinical outcomes in immune–checkpoint blockade: a systematic review and meta–analysis of observational studies. Cancer Immunol Immunother. (2020) 69:343–54. doi: 10.1007/s00262-019-02453-2
129. Zheng DW, Dong X, Pan P, Chen KW, Fan JX, Cheng SX, et al. Phage–guided modulation of the gut microbiota of mouse models of colorectal cancer augments their responses to chemotherapy. Nat BioMed Eng. (2019) 3:717–28. doi: 10.1038/s41551-019-0423-2
130. Cremonesi E, Governa V, Garzon JFG, Mele V, Amicarella F, Muraro MG, et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut. (2018) 67:1984–94. doi: 10.1136/gutjnl-2016-313498
131. Dizman N, Meza L, Bergerot P, Alcantara M, Dorff T, Lyou Y, et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: a randomized phase 1 trial. Nat Med. (2022) 28:704–12. doi: 10.1038/s41591-022-01694-6
132. Holt RA. Oncomicrobial vaccines: The potential for a Fusobacterium nucleatum vaccine to improve colorectal cancer outcomes. Cell Host Microbe. (2023) 31:141–5. doi: 10.1016/j.chom.2022.11.014
133. Nguyen CL, Markey KA, Miltiadous O, Dai A, Waters N, Sadeghi K, et al. High–resolution analyses of associations between medications, microbiome, and mortality in cancer patients. Cell. (2023) 186:2705–2718.e17. doi: 10.1016/j.cell.2023.05.007
134. Wang M, Rousseau B, Qiu K, Huang G, Zhang Y, Su H, et al. Killing tumor–associated bacteria with a liposomal antibiotic generates neoantigens that induce anti–tumor immune responses. Nat Biotechnol. (2023). doi: 10.1038/s41587-023-01957-8
135. Subbiah V, Pant S. Translating immuno–onco–microbiome–based therapeutics: precision bugs for immune checkpoint drugs! Ann Oncol. (2023) 34:503–6. doi: 10.1016/j.annonc.2023.03.002
136. Ting NL, Lau HC, Yu J. Cancer pharmacomicrobiomics: targeting microbiota to optimise cancer therapy outcomes. Gut. (2022) 71:1412–25. doi: 10.1136/gutjnl-2021-326264
137. Ispoglou T, McCullough D, Windle A, Nair S, Cox N, White H, et al. Addressing cancer anorexia–cachexia in older patients: Potential therapeutic strategies and molecular pathways. Clin Nutr. (2024) 43:552–66. doi: 10.1016/j.clnu.2024.01.009
138. Alese OB, Wu C, Chapin WJ, Ulanja MB, Zheng–Lin B, Amankwah M, et al. Update on emerging therapies for advanced colorectal cancer. Am Soc Clin Oncol Educ Book. (2023) 43:e389574. doi: 10.1200/EDBK_389574
139. Bullock A, Grossman J, Fakih M, Lenz H, Gordon M, Margolin K, et al. LBA O–9 Botensilimab, a novel innate/adaptive immune activator, plus balstilimab (anti–PD–1) for metastatic heavily pretreated microsatellite stable colorectal cancer. Ann Oncol. (2022) n3. S4. doi: 10.1016/j.annonc.2022.04.453
140. Wang C, Sandhu J, Ouyang C, Ye J, Lee PP, Fakih M. Clinical response to immunotherapy targeting programmed cell death receptor 1/programmed cell death ligand 1 in patients with treatment–resistant microsatellite stable colorectal cancer with and without liver metastases. JAMA Netw Open. (2021) 4:e2118416. doi: 10.1001/jamanetworkopen.2021.18416
141. Rong YM, Xu YC, Chen XC, Zhong ME, Tan YX, Liang YF, et al. IGSF6 is a novel biomarker to evaluate immune infiltration in mismatch repair–proficient colorectal cancer. Sci Rep. (2023) 13:20368. doi: 10.1038/s41598-023-47739-9
142. Oh DY, Fong L. Cytotoxic CD4+ T cells in cancer: Expanding the immune effector toolbox. Immunity. (2021) 54:2701–11. doi: 10.1016/j.immuni.2021.11.015
143. Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. (2018) 564:268–72. doi: 10.1038/s41586-018-0694-x
144. Aggarwal V, Workman CJ, Vignali DAA. LAG–3 as the third checkpoint inhibitor. Nat Immunol. (2023) 24:1415–22. doi: 10.1038/s41590-023-01569-z
145. Mellman I, Chen DS, Powles T, Turley SJ. The cancer–immunity cycle: Indication, genotype, and immunotype. Immunity. (2023) 56:2188–205. doi: 10.1016/j.immuni.2023.09.011
146. Garralda E, Sukari A, Lakhani NJ, Patnaik A, Lou Y, Im SA, et al. A first–in–human study of the anti–LAG–3 antibody favezelimab plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. ESMO Open. (2022) 7:100639. doi: 10.1016/j.esmoop.2022.100639
147. Shakiba M, Zumbo P, Espinosa–Carrasco G, Menocal L, Dündar F, Carson SE, et al. TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion. J Exp Med. (2022) 219:e20201966. doi: 10.1084/jem.20201966
148. Dada H, Dustin ML. Goldilocks and the three TILs. J Exp Med. (2022) 219:e20212269. doi: 10.1084/jem.20212269
149. Mauri G, Arena S, Siena S, Bardelli A, Sartore–Bianchi A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann Oncol. (2020) 31:1135–47. doi: 10.1016/j.annonc.2020.05.027
150. Rousseau B, Bieche I, Pasmant E, Hamzaoui N, Leulliot N, Michon L, et al. PD–1 blockade in solid tumors with defects in polymerase epsilon. Cancer Discovery. (2022) 12:1435–48. doi: 10.1158/2159-8290.CD-21-0521
151. Ma X, Riaz N, Samstein RM, Lee M, Makarov V, Valero C, et al. Functional landscapes of POLE and POLD1 mutations in checkpoint blockade–dependent antitumor immunity. Nat Genet. (2022) 54:996–1012. doi: 10.1038/s41588-022-01108-w
152. Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. (2013) 155:1088–103. doi: 10.1016/j.cell.2013.10.043
153. Toledo L, Neelsen KJ, Lukas J. Replication catastrophe: when a checkpoint fails because of exhaustion. Mol Cell. (2017) 66:735–49. doi: 10.1016/j.molcel.2017.05.001
154. Cleary JM, Aguirre AJ, Shapiro GI, D'Andrea AD. Biomarker–guided development of DNA repair inhibitors. Mol Cell. (2020) 78:1070–85. doi: 10.1016/j.molcel.2020.04.035
155. Hardaker EL, Sanseviero E, Karmokar A, Taylor D, Milo M, Michaloglou C, et al. The ATR inhibitor ceralasertib potentiates cancer checkpoint immunotherapy by regulating the tumor microenvironment. Nat Commun. (2024) 15:1700. doi: 10.1038/s41467-024-45996-4
156. Jin WJ, Zangl LM, Hyun M, Massoud E, Schroeder K, Alexandridis RA, et al. ATM inhibition augments type I interferon response and antitumor T–cell immunity when combined with radiation therapy in murine tumor models. J Immunother Cancer. (2023) 11:e007474. doi: 10.1136/jitc-2023-007474
157. Hsieh RC, Krishnan S, Wu RC, Boda AR, Liu A, Winkler M, et al. ATR–mediated CD47 and PD–L1 up–regulation restricts radiotherapy–induced immune priming and abscopal responses in colorectal cancer. Sci Immunol. (2022) 7:eabl9330. doi: 10.1126/sciimmunol.abl9330
158. Gupta D, Lin B, Cowan A, Heinen CD. ATR–Chk1 activation mitigates replication stress caused by mismatch repair–dependent processing of DNA damage. Proc Natl Acad Sci U S A. (2018) 115:1523–8. doi: 10.1073/pnas.1720355115
159. Deshpande RA, Myler LR, Soniat MM, Makharashvili N, Lee L, Lees–Miller SP, et al. DNA–dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci Adv. (2020) 6:eaay0922. doi: 10.1126/sciadv.aay0922
160. van Bussel MTJ, Awada A, de Jonge MJA, Mau–Sørensen M, Nielsen D, Schöffski P, et al. A first–in–man phase 1 study of the DNA–dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br J Cancer. (2021) 124:728–35. doi: 10.1038/s41416-020-01151-6
161. Fok JHL, Ramos–Montoya A, Vazquez–Chantada M, Wijnhoven PWG, Follia V, James N, et al. AZD7648 is a potent and selective DNA–PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat Commun. (2019) 10:5065. doi: 10.1038/s41467-019-12836-9
162. Fowler FC, Chen BR, Zolnerowich N, Wu W, Pavani R, Paiano J, et al. DNA–PK promotes DNA end resection at DNA double strand breaks in G0 cells. Elife. (2022) 11:e74700. doi: 10.7554/eLife.74700
163. Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. (2006) 38:787–93. doi: 10.1038/ng1834
164. Suter CM, Martin DI, Ward RL. Germline epimutation of MLH1 in individuals with multiple cancers. Nat Genet. (2004) 36:497–501. doi: 10.1038/ng1342
165. Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, et al. m6 A RNA methyltransferases METTL3/14 regulate immune responses to anti–PD–1 therapy. EMBO J. (2020) 39:e104514. doi: 10.15252/embj.2020104514
166. Bao Y, Zhai J, Chen H, Wong CC, Liang C, Ding Y, et al. Targeting m6A reader YTHDF1 augments antitumour immunity and boosts anti–PD–1 efficacy in colorectal cancer. Gut. (2023) 72:1497–509. doi: 10.1136/gutjnl-2022-328845
167. Rao Q, Liu M, Tian Y, Wu Z, Hao Y, Song L, et al. Cryo–EM structure of human ATR–ATRIP complex. Cell Res. (2018) 28:143–56. doi: 10.1038/cr.2017.158
168. Priya B, Ravi S, Kirubakaran S. Targeting ATM and ATR for cancer therapeutics: Inhibitors in clinic. Drug Discovery Today. (2023) 28:103662. doi: 10.1016/j.drudis.2023.103662
169. van Vugt MATM, Parkes EE. When breaks get hot: inflammatory signaling in BRCA1/2–mutant cancers. Trends Cancer. (2022) 8:174–89. doi: 10.1016/j.trecan.2021.12.003
170. Hopkins TA, Ainsworth WB, Ellis PA, Donawho CK, DiGiammarino EL, Panchal SC, et al. PARP1 trapping by PARP inhibitors drives cytotoxicity in both cancer cells and healthy bone marrow. Mol Cancer Res. (2019) 17:409–19. doi: 10.1158/1541-7786.MCR-18-0138
171. Lee JY, Lee YY, Park JY, Shim SH, Kim SI, Kong TW, et al. Major clinical research advances in gynecologic cancer in 2022: highlight on late–line PARP inhibitor withdrawal in ovarian cancer, the impact of ARIEL–4, and SOLO–3. J Gynecol Oncol. (2023) 34:e51. doi: 10.3802/jgo.2023.34.e51
172. Yang L, Liu S, He W, Xiong Z, Xia L. Characterisation of tumor microenvironment and prevalence of CD274/PD–L1 genetic alterations difference in colorectal Cancer. BMC Cancer. (2023) 23:221. doi: 10.1186/s12885-023-10610-1
173. Guyot D'Asnières De Salins A, Tachon G, Cohen R, Karayan–Tapon L, Junca A, Frouin E, et al. Discordance between immunochemistry of mismatch repair proteins and molecular testing of microsatellite instability in colorectal cancer. ESMO Open. (2021) 6:100120. doi: 10.1016/j.esmoop.2021.100120
174. Fornaro L, Lonardi S, Catanese S, Nappo F, Pietrantonio F, Pellino A, et al. Concordance of microsatellite instability and mismatch repair status in paired biopsies and surgical specimens of resectable gastroesophageal adenocarcinoma: time for a call to action. Gastric Cancer. (2023) 26:958–68. doi: 10.1007/s10120-023-01411-3
175. Matsubayashi H, Oishi T, Sasaki K, Abe M, Kiyozumi Y, Higashigawa S, et al. Discordance of microsatellite instability and mismatch repair immunochemistry occurs depending on the cancer type. Hum Pathol. (2023) 135:54–64. doi: 10.1016/j.humpath.2022.12.016
176. Echle A, Ghaffari Laleh N, Quirke P, Grabsch HI, Muti HS, Saldanha OL, et al. Artificial intelligence for detection of microsatellite instability in colorectal cancer–a multicentric analysis of a pre–screening tool for clinical application. ESMO Open. (2022) 7:100400. doi: 10.1016/j.esmoop.2022.100400
177. Nakamura Y, Taniguchi H, Ikeda M, Bando H, Kato K, Morizane C, et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat Med. (2020) 26:1859–64. doi: 10.1038/s41591-020-1063-5
178. Nakamura Y, Yoshino T. Clinical utility of analyzing circulating tumor DNA in patients with metastatic colorectal cancer. Oncologist. (2018) 23:1310–8. doi: 10.1634/theoncologist.2017-0621
179. Tie J, Cohen JD, Lahouel K, Lo SN, Wang Y, Kosmider S, et al. Circulating tumor DNA analysis guiding adjuvant therapy in stage II colon cancer. N Engl J Med. (2022) 386:2261–72. doi: 10.1056/NEJMoa2200075
180. Pacini C, Duncan E, Gonçalves E, Gilbert J, Bhosle S, Horswell S, et al. A comprehensive clinically informed map of dependencies in cancer cells and framework for target prioritization. Cancer Cell. (2024) 42:301–16.e9. doi: 10.1016/j.ccell.2023.12.016
181. Jin H, Wang L, Bernards R. Rational combinations of targeted cancer therapies: background, advances and challenges. Nat Rev Drug Discovery. (2023) 22:213–34. doi: 10.1038/s41573-022-00615-z
182. Wang JM, Hong R, Demicco EG, Tan J, Lazcano R, Moreira AL, et al. Deep learning integrates histopathology and proteogenomics at a pan–cancer level. Cell Rep Med. (2023) 4:101173. doi: 10.1016/j.xcrm.2023.101173
183. Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. (2020) 20:662–80. doi: 10.1038/s41568-020-0285-7
184. Galon J, Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity. (2020) 52:55–81. doi: 10.1016/j.immuni.2019.12.018
185. Pascual J, Attard G, Bidard FC, Curigliano G, De Mattos–Arruda L, Diehn M, et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: a report from the ESMO Precision Medicine Working Group. Ann Oncol. (2022) 33:750–68. doi: 10.1016/j.annonc.2022.05.520
186. Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open. (2019) 2:e192535. doi: 10.1001/jamanetworkopen.2019.2535
187. Suehnholz SP, Nissan MH, Zhang H, Kundra R, Nandakumar S, Lu C, et al. Quantifying the expanding landscape of clinical actionability for patients with cancer. Cancer Discovery. (2024) 14:49–65. doi: 10.1158/2159-8290.CD-23-0467
188. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, et al. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. (2014) 345:216–20. doi: 10.1126/science.1253533
189. Pal A, Shinde R, Miralles MS, Workman P, de Bono J. Applications of liquid biopsy in the Pharmacological Audit Trail for anticancer drug development. Nat Rev Clin Oncol. (2021) 18:454–67. doi: 10.1038/s41571-021-00489-x
190. Wrenn ED, Yamamoto A, Moore BM, Huang Y, McBirney M, Thomas AJ, et al. Regulation of collective metastasis by nanolumenal signaling. Cell. (2020) 183:395–410.e19. doi: 10.1016/j.cell.2020.08.045
191. Fraser JS, Murcko MA. Structure is beauty, but not always truth. Cell. (2024) 187:517–20. doi: 10.1016/j.cell.2024.01.003
192. Jaaks P, Coker EA, Vis DJ, Edwards O, Carpenter EF, Leto SM, et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature. (2022) 603:166–73. doi: 10.1038/s41586-022-04437-2
193. Tatarova Z, Blumberg DC, Korkola JE, Heiser LM, Muschler JL, Schedin PJ, et al. A multiplex implantable microdevice assay identifies synergistic combinations of cancer immunotherapies and conventional drugs. Nat Biotechnol. (2022) 40:1823–33. doi: 10.1038/s41587-022-01379-y
194. van Renterghem AWJ, van de Haar J, Voest EE. Functional precision oncology using patient–derived assays: bridging genotype and phenotype. Nat Rev Clin Oncol. (2023) 20:305–17. doi: 10.1038/s41571-023-00745-2
195. Wakefield L, Agarwal S, Tanner K. Preclinical models for drug discovery for metastatic disease. Cell. (2023) 186:1792–813. doi: 10.1016/j.cell.2023.02.026
196. Sharma P, Goswami S, Raychaudhuri D, Siddiqui BA, Singh P, Nagarajan A, et al. Immune checkpoint therapy–current perspectives and future directions. Cell. (2023) 186:1652–69. doi: 10.1016/j.cell.2023.03.006
197. Letai A, Bhola P, Welm AL. Functional precision oncology: Testing tumors with drugs to identify vulnerabilities and novel combinations. Cancer Cell. (2022) 40:26–35. doi: 10.1016/j.ccell.2021.12.004
198. Voabil P, de Bruijn M, Roelofsen LM, Hendriks SH, Brokamp S, van den Braber M, et al. An ex vivo tumor fragment platform to dissect response to PD–1 blockade in cancer. Nat Med. (2021) 27:1250–61. doi: 10.1038/s41591-021-01398-3
199. Basak NP, Jaganathan K, Das B, Muthusamy O, M R, Malhotra R, et al. Tumor histoculture captures the dynamic interactions between tumor and immune components in response to anti–PD1 in head and neck cancer. Nat Commun. (2024) 15:1585. doi: 10.1038/s41467-024-45723-z
200. Kornauth C, Pemovska T, Vladimer GI, Bayer G, Bergmann M, Eder S, et al. Functional precision medicine provides clinical benefit in advanced aggressive hematologic cancers and identifies exceptional responders. Cancer Discovery. (2022) 12:372–87. doi: 10.1158/2159-8290.CD-21-0538
201. Malani D, Kumar A, Brück O, Kontro M, Yadav B, Hellesøy M, et al. Implementing a functional precision medicine tumor board for acute myeloid leukemia. Cancer Discovery. (2022) 12:388–401. doi: 10.1158/2159-8290.CD-21-0410
202. Kropivsek K, Kachel P, Goetze S, Wegmann R, Festl Y, Severin Y, et al. Ex vivo drug response heterogeneity reveals personalized therapeutic strategies for patients with multiple myeloma. Nat Cancer. (2023) 4:734–53. doi: 10.1038/s43018-023-00544-9
203. Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, et al. Generation of tumor–reactive T cells by co–culture of peripheral blood lymphocytes and tumor organoids. Cell. (2018) 174:1586–1598.e12. doi: 10.1016/j.cell.2018.07.009
204. Kasper SH, Morell–Perez C, Wyche TP, Sana TR, Lieberman LA, Hett EC. Colorectal cancer–associated anaerobic bacteria proliferate in tumor spheroids and alter the microenvironment. Sci Rep. (2020) 10:5321. doi: 10.1038/s41598-020-62139-z
205. Majumder B, Baraneedharan U, Thiyagarajan S, Radhakrishnan P, Narasimhan H, Dhandapani M, et al. Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity. Nat Commun. (2015) 6:6169. doi: 10.1038/ncomms7169
206. Brijwani N, Jain M, Dhandapani M, Zahed F, Mukhopadhyay P, Biswas M, et al. Rationally co–targeting divergent pathways in KRAS wild–type colorectal cancers by CANscript technology reveals tumor dependence on Notch and Erbb2. Sci Rep. (2017) 7:1502. doi: 10.1038/s41598-017-01566-x
207. Narasimhan V, Wright JA, Churchill M, Wang T, Rosati R, Lannagan TRM, et al. Medium–throughput drug screening of patient–derived organoids from colorectal peritoneal metastases to direct personalized therapy. Clin Cancer Res. (2020) 26:3662–70. doi: 10.1158/1078-0432.CCR-20-0073
208. Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández–Mateos J, Khan K, et al. Patient–derived organoids model treatment response of metastatic gastrointestinal cancers. Science. (2018) 359:920–6. doi: 10.1126/science.aao2774
209. Avolio M, Trusolino L. Rational treatment of metastatic colorectal cancer: A reverse tale of men, mice, and culture dishes. Cancer Discovery. (2021) 11:1644–60. doi: 10.1158/2159-8290.CD-20-1531
210. Gavert N, Zwang Y, Weiser R, Greenberg O, Halperin S, Jacobi O, et al. Ex vivo organotypic cultures for synergistic therapy prioritization identify patient–specific responses to combined MEK and Src inhibition in colorectal cancer. Nat Cancer. (2022) 3:219–31. doi: 10.1038/s43018-021-00325-2
211. Ozkan–Dagliyan I, Diehl JN, George SD, Schaefer A, Papke B, Klotz–Noack K, et al. Low–dose vertical inhibition of the RAF–MEK–ERK cascade causes apoptotic death of KRAS mutant cancers. Cell Rep. (2020) 31:107764. doi: 10.1016/j.celrep.2020.107764
212. Ding S, Hsu C, Wang Z, Natesh NR, Millen R, Negrete M, et al. Patient–derived micro–organospheres enable clinical precision oncology. Cell Stem Cell. (2022) 29:905–917.e6. doi: 10.1016/j.stem.2022.04.006
213. Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola–Essel M, et al. PD–1 blockade in mismatch repair–deficient, locally advanced rectal cancer. N Engl J Med. (2022) 386:2363–76. doi: 10.1056/NEJMoa2201445
214. Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. (2015) 521:43–7. doi: 10.1038/nature14415
215. Ben–David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, et al. Patient–derived xenografts undergo mouse–specific tumor evolution. Nat Genet. (2017) 49:1567–75. doi: 10.1038/ng.3967
216. Muraro MG, Muenst S, Mele V, Quagliata L, Iezzi G, Tzankov A, et al. Ex–vivo assessment of drug response on breast cancer primary tissue with preserved microenvironments. Oncoimmunology. (2017) 6:e1331798. doi: 10.1080/2162402X.2017.1331798
217. Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M, et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature. (2017) 545:187–92. doi: 10.1038/nature22081
218. Chen X, Sifakis EG, Robertson S, Neo SY, Jun SH, Tong L, et al. Breast cancer patient–derived whole–tumor cell culture model for efficient drug profiling and treatment response prediction. Proc Natl Acad Sci U S A. (2023) 120:e2209856120. doi: 10.1073/pnas.2209856120
219. LeBlanc VG, Trinh DL, Aslanpour S, Hughes M, Livingstone D, Jin D, et al. Single–cell landscapes of primary glioblastomas and matched explants and cell lines show variable retention of inter– and intratumor heterogeneity. Cancer Cell. (2022) 40:379–392.e9. doi: 10.1016/j.ccell.2022.02.016
220. Muir A, Danai LV, Vander Heiden MG. Microenvironmental regulation of cancer cell metabolism: implications for experimental design and translational studies. Dis Model Mech. (2018) 11:dmm035758. doi: 10.1242/dmm.035758
221. Kumar N, Prasad P, Jash E, Jayasundar S, Singh I, Alam N, et al. cAMP regulated EPAC1 supports microvascular density, angiogenic and metastatic properties in a model of triple negative breast cancer. Carcinogenesis. (2018) 39:1245–53. doi: 10.1093/carcin/bgy090
222. Hasselluhn MC, Decker–Farrell AR, Vlahos L, Thomas DH, Curiel–Garcia A, Maurer HC, et al. Tumor explants elucidate a cascade of paracrine SHH, WNT, and VEGF signals driving pancreatic cancer angiosuppression. Cancer Discovery. (2024) 14:348–61. doi: 10.1158/2159-8290.CD-23-0240
223. Mann B, Zhang X, Bell N, Adefolaju A, Thang M, Dasari R, et al. A living ex vivo platform for functional, personalized brain cancer diagnosis. Cell Rep Med. (2023) 4:101042. doi: 10.1016/j.xcrm.2023.101042
224. Shah P, Fritz JV, Glaab E, Desai MS, Greenhalgh K, Frachet A, et al. A microfluidics–based in vitro model of the gastrointestinal human–microbe interface. Nat Commun. (2016) 7:11535. doi: 10.1038/ncomms11535
225. Heninger E, Kosoff D, Rodems TS, Sethakorn N, Singh A, Gungurthi H, et al. Live cell molecular analysis of primary prostate cancer organoids identifies persistent androgen receptor signaling. Med Oncol. (2021) 38:135. doi: 10.1007/s12032-021-01582-y
226. Liu R, Rizzo S, Whipple S, Pal N, Pineda AL, Lu M, et al. Evaluating eligibility criteria of oncology trials using real–world data and AI. Nature. (2021) 592:629–33. doi: 10.1038/s41586-021-03430-5
227. Liu CJ, Smith JT, Wang Y, Ouellette JN, Rogers JD, Oliner JD, et al. Assessing cell viability with dynamic optical coherence microscopy. BioMed Opt Express. (2024) 15:1408–17. doi: 10.1364/BOE.509835
228. Morand du Puch CB, Vanderstraete M, Giraud S, Lautrette C, Christou N, Mathonnet M. Benefits of functional assays in personalized cancer medicine: more than just a proof–of–concept. Theranostics. (2021) 11:9538–56. doi: 10.7150/thno.55954
229. LeSavage BL, Suhar RA, Broguiere N, Lutolf MP, Heilshorn SC. Next–generation cancer organoids. Nat Mater. (2022) 21:143–59. doi: 10.1038/s41563-021-01057-5
230. Klein SG, Alsolami SM, Steckbauer A, Arossa S, Parry AJ, Ramos Mandujano G, et al. A prevalent neglect of environmental control in mammalian cell culture calls for best practices. Nat BioMed Eng. (2021) 5:787–92. doi: 10.1038/s41551-021-00775-0
231. Gastman B, Agarwal PK, Berger A, Boland G, Broderick S, Butterfield LH, et al. Defining best practices for tissue procurement in immuno–oncology clinical trials: consensus statement from the Society for Immunotherapy of Cancer Surgery Committee. J Immunother Cancer. (2020) 8:e001583. doi: 10.1136/jitc-2020-001583
232. Singhal U, Horrow C, Kesselheim AS, Morgan TM. Modernizing federal oversight of laboratory–developed tests – toward safety, validity, and utility. N Engl J Med. (2023) 389:1735–7. doi: 10.1056/NEJMp2307585
233. Le DT, Hubbard–Lucey VM, Morse MA, Heery CR, Dwyer A, Marsilje TH, et al. A blueprint to advance colorectal cancer immunotherapies. Cancer Immunol Res. (2017) 5:942–9. doi: 10.1158/2326-6066.CIR-17-0375
234. Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. (2011) 71:1263–71. doi: 10.1158/0008-5472.CAN-10-2907
235. Roelofsen LM, Voabil P, de Bruijn M, Herzig P, Zippelius A, Schumacher TN, et al. Protocol for ex vivo culture of patient–derived tumor fragments. STAR Protoc. (2023) 4:102282. doi: 10.1016/j.xpro.2023.102282
236. Williams ST, Wells G, Conroy S, Gagg H, Allen R, Rominiyi O, et al. Precision oncology using ex vivo technology: a step towards individualised cancer care? Expert Rev Mol Med. (2022) 24:e39. doi: 10.1017/erm.2022.32
Keywords: colorectal cancer, MMRp, tumor immune microenvironment, gut microbiota, therapeutic vulnerability, predictive biomarkers, precision medicine, functional platforms
Citation: Majumder B, Nataraj NB, Maitreyi L and Datta S (2024) Mismatch repair-proficient tumor footprints in the sands of immune desert: mechanistic constraints and precision platforms. Front. Immunol. 15:1414376. doi: 10.3389/fimmu.2024.1414376
Received: 08 April 2024; Accepted: 17 June 2024;
Published: 19 July 2024.
Edited by:
William Vermi, ASST Spedali Civili di Brescia, ItalyReviewed by:
Valentina Angerilli, University of Padua, ItalyCopyright © 2024 Majumder, Nataraj, Maitreyi and Datta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Biswanath Majumder, Ymlzd2FuYXRoQGJ1Z3dvcmtzcmVzZWFyY2guY29t
†These authors have contributed equally to this work
‡ORCID: Biswanath Majumder, orcid.org/0009-0002-7030-7414
Nishanth Belugali Nataraj, orcid.org/0000-0003-2970-6739
Leela Maitreyi, orcid.org/0009-0004-2260-5059
Santanu Datta, orcid.org/0000-0001-5335-5276
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.