 Pegah Abdollahi
Pegah Abdollahi Hanne Marie Norseth
Hanne Marie Norseth Fredrik Schjesvold
Fredrik Schjesvold- Oslo Myeloma Center, Department of Hematology, Oslo University Hospital, Oslo, Norway
Multiple myeloma (MM) is a hematological cancer marked by plasma cell accumulation in the bone marrow. Despite treatment advancements, MM remains incurable in most patients. MM-associated immune dysregulation fosters disease progression, prompting research into immunotherapy to combat the disease. An area of immunotherapy investigation is the design of myeloma vaccine therapy to reverse tumor-associated immune suppression and elicit tumor-specific immune responses to effectively target MM cells. This article reviews vaccine immunotherapy for MM, categorizing findings by antigen type and delivery method. Antigens include idiotype (Id), tumor-associated (TAA), tumor-specific (TSA), and whole tumor lysate. Myeloma vaccination has so far shown limited clinical efficacy. However, further studies are essential to optimize various aspects, including antigen and patient selection, vaccine timing and sequencing, and rational combinations with emerging MM treatments.
1 Introduction
Multiple myeloma (MM) is a hematological malignancy characterized by the accumulation of clonal plasma cells in the bone marrow (BM) and secretion of monoclonal immunoglobulins, leading to the hallmark symptoms known as CRAB (hypercalcemia, renal insufficiency, anemia, and bone lesions). Despite the advent of numerous new treatments that have significantly improved outcomes, MM remains largely incurable, and the disease continues to have a fatal outcome for most patients in advanced stages (1). Increasing research attention have been placed on immunotherapy that targets MM cells utilizing the host immune system to eliminate the malignant cells in the BM (2). Myeloma is associated with immune dysregulation due to ineffective antigen presentation and effector cell dysfunction creating an immunosuppressive milieu that fosters disease progression. An area of investigation is the design of myeloma vaccine therapy (2). Cancer vaccination is a therapeutic approach designed to activate the immune system to recognize and combat cancer cells. These vaccines can serve as preventive measures, known as prophylactic vaccines, or as treatment options for individuals already diagnosed with cancer, referred to as therapeutic vaccines (3). The choice of antigen targeted by any cancer vaccine is critical to its clinical efficacy. Tumor antigens are classified into two broad categories: tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs) (4). TAAs are self-antigens that are either preferentially or abnormally expressed in tumor cells but may also be present at some level in normal cells. TSAs, also known as neoantigens, include antigens that are encoded solely by cancer cells and are tumor-specific, eliciting high-affinity T cell response (5). Vaccines are categorized into shared and personalized vaccines. Shared antigens are public antigens and can be presented by a relatively common human leukocyte antigen (HLA) allele in patients. Such vaccines are promising candidates for off-the-shelf immunotherapy. On the other hand, personalized cancer vaccines have recently gained more attention due to advancements in high-throughput gene sequencing, mass spectrometry (MS) and bioinformatics, which enable the identification of HLA-bound peptides and the prediction of unique personalized neoantigens (6). In addition, cancer vaccines can be delivered through various platforms such as peptide vaccines, RNA vaccines, DNA vaccines, viral vaccines, and antigen-presenting cell (APC)/dendritic cell (DC) vaccines (7). Today, most vaccines under clinical investigation involve the delivery of tumor antigens in combination with an adjuvant or other costimulatory factors. Adjuvants are essential components of cancer vaccines as they enhance immune responses by activating innate immune pathways. Various adjuvants, such as Toll-like receptor (TLR) agonists and cytokines are used to improve the efficacy of cancer vaccines (3). For more detailed information on cancer vaccines, please refer to references 3-7.
This article aims to review major findings in vaccine immunotherapy against MM, categorized based on the type of antigens and means of their delivery either as peptide-, protein-, DNA- or DC-based vaccines. The discussed tumor antigens encompass idiotype (Id) antigens, TAA, TSA, and whole tumor lysate. In MM, malignant plasma cells secrete a monoclonal immunoglobulin (paraprotein) containing tumor-specific antigenic determinants known as Id. Ids are formed through gene rearrangement during B-cell maturation and somatic hypermutation (8). Although Id antigens are a subset of TSA, a dedicated section to these antigens is included in this review due to the historical significance of investigating vaccines against myeloma Id antigens.
2 Idiotype vaccine
2.1 Id protein/peptide- or DNA-based vaccine
One of the initial investigations of Id-vaccines goes back to a study in 1995 by Kwak et al., where they immunized a healthy sibling BM donor with myeloma immunoglobulin from the plasma of the recipient. Detection of a lymphoproliferative response, with recovery of a recipient CD4+ T-cell line with unique specificity for myeloma Id, was a proof of concept that immunization with Id may represent a new strategy for enhancing the specific antitumor effect (9). Later, more studies investigated the potential of Id vaccination as a treatment strategy (10, 11). A cohort of five patients with stage I-III MM underwent repeated immunization with autologous serum M-component, administered in alum as an adjuvant. While successful in three patients, the elicited immune response was modest in magnitude and short-lived (10). Subsequent studies introduced immunogenic carrier proteins such as keyhole limpet hemocyanin (KLH) (12) or filamentous phage (13) with or without adjuvants such as granulocyte-macrophage colony-stimulating factor (GM-CSF) (12, 14, 15), interleukin (IL)-2 (12), IL-12 (14) to enhance anti-tumor immunity of Id proteins. Even though these strategies were capable of evoking tumor-specific immune responses, the clinical response was modest (11, 15). For instance, in the study by Osterborg et al., Id vaccine was used together with GM-CSF in five stage II MM patients. All patients developed an Id-specific T-cell immunity, but a significant reduction in M-component concentration was noted in only one individual (15). Massaia et al., vaccinated patients with minimal residual disease (MRD) following high-dose chemotherapy (HDT) using the Id protein with KLH and a low dose of the adjuvant cytokines GM-CSF or IL-2. An idiotype-specific T-cell response was documented in 75% of patients with no decrease in tumor burden (12). Another study explored the utilization of Id-specific protein conjugated to KLH with low doses of GM-CSF as maintenance therapy in the first remission post-HDT and peripheral blood progenitor cell infusion. A retrospective case-matched analysis revealed similar survival and progression free survival (PFS) durations between the vaccine and control groups who were treated with interferon (IFN)-α and/or dexamethasone as maintenance therapy (16).
More efforts were employed where Id DNA-based vaccine encoding patient-specific single chain variable fragment, or Id linked to fragment C (FrC) of tetanus toxin was investigated in a non-randomized phase I clinical trial. Fourteen patients received vaccine initiated at least 6 months post-autologous stem cell transplant (ASCT)/HDT. Over the 52-week study period, serum paraprotein was undetectable, decreased or remained stable for ten patients (71%), whilst ongoing complete response (CR)/partial response (PR) was maintained for 11 patients (79%). Moreover, median time to progression (TTP) was 38.0 months for 13 patients. However, due to prior ASCT/HDT, distinguishing vaccine effects from delayed treatment response was challenging in this study because CR, PR and stable disease (SD) were already achieved in seven, six and one patients at onset of vaccination, post-ASCT/HDT (8). In one of the most recent studies published by Qazilbash et al., another strategy was applied in Id vaccination. In this randomized phase 2 trial, a prime-and-boost strategy was employed by collecting T lymphocytes from patients that had been vaccine primed in vivo with patient-specific Id-KLH, followed by ex vivo activation of the T cells with CD3/CD28 magnetic beads and reinfusing these cells after HDT and autologous hematopoietic stem cell transplantation (AHCT) with subsequent booster doses of the assigned vaccine. Even though the vaccine led to robust immune response which persisted up to +180 days post–AHCT, there was no clinical benefit in the group that was vaccinated with Id-KLH compared to the control group with only KLH (17). Table 1 shows the summary of trials using Id protein/peptide- or DNA-based Id vaccine.
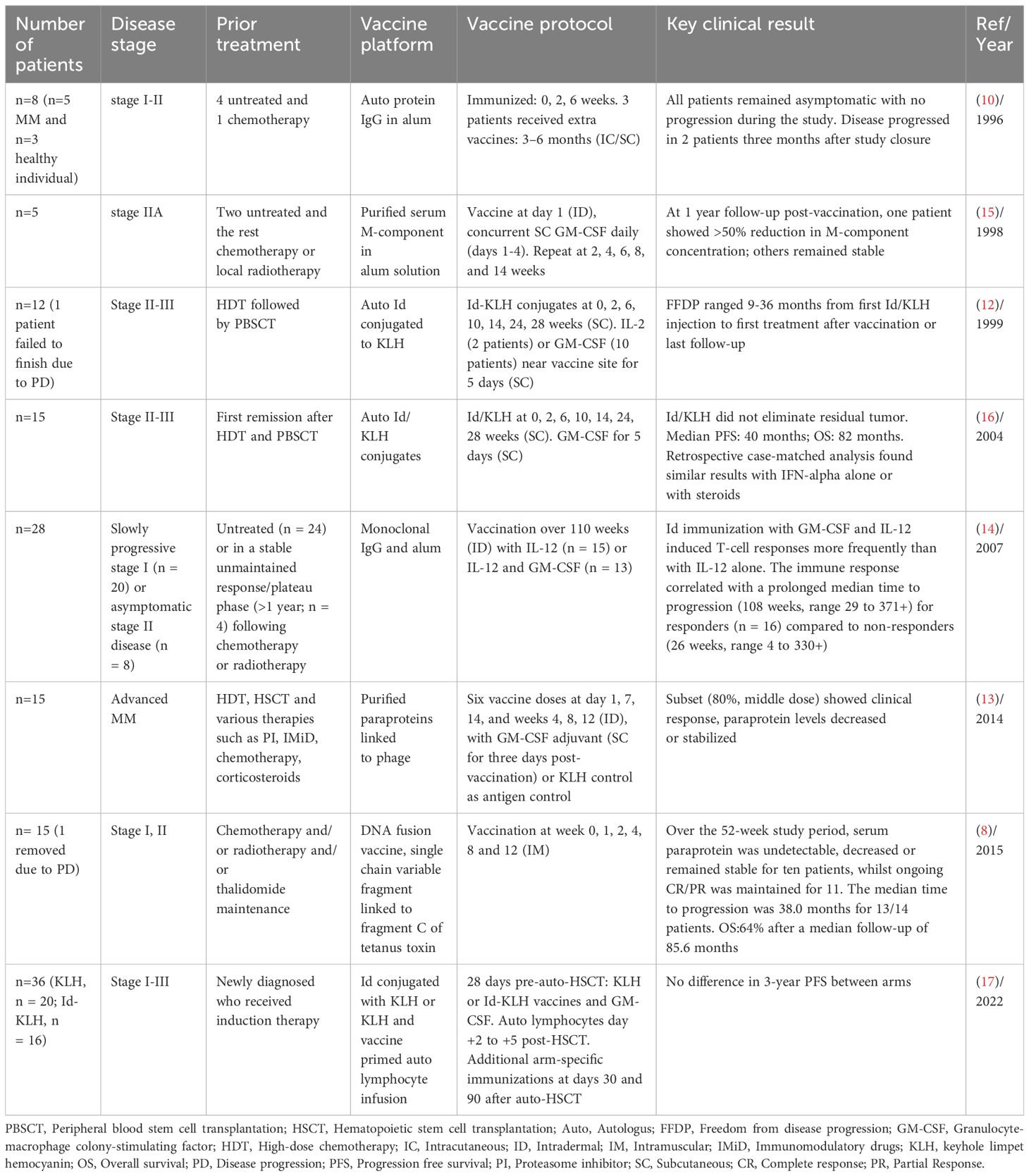
Table 1 Summary of trials using non-DC-based Id vaccines in MM.
2.2 DC-based Id vaccines
The professional APCs, DCs, play a critical role in the initiation and regulation of innate and adaptive immune responses. By exploiting their ability to potentiate host effector and memory CD8 T cell responses critical for anti-tumor immunity, DC vaccines have emerged as one of the leading strategies for cancer immunotherapy (18). DC-based vaccines were first tested in context of Id antigen in MM. Historically, Id DC-based vaccines have been tested in over a decade in MM trials with no clear clinical benefit. Several optimization methods have been proposed for the generation, route of administration, and timing of vaccination. The original methodology involved the isolation of DCs from peripheral blood harvested by leukapheresis and subsequent density gradient centrifugations (19, 20). Subsequently, the addition of cytokines, namely GM-CSF and IL-4, became standard practice for culturing either adherent mononuclear peripheral blood cells (21, 22), CD34+ (23) or CD14+ cells (24, 25). Various maturation protocols were suggested following Id-pulsing to enhance effectiveness, including exposure to tumor-necrosis factor alpha (TNF-α) alone (26, 27), TNF-α and IL1-B (28), CD40 ligand (29) or a combination of TNF-α, IL-6, IL-1, and prostaglandin E2 (30).
Different vaccination routes, including subcutaneous (23, 28), intradermal (24, 26), intravenous (20, 22, 27), and intranodal (29) have been investigated. In a phase I/II trial, Curti et al. compared Id-pulsed DCs through sequential subcutaneous and intravenous routes. Each patient served as their own control, undergoing sequential subcutaneous and intravenous administrations. Results showed that subcutaneous administration induced a more robust T-cell response, suggesting its effectiveness in eliciting immunological responses compared to intravenous infusion (25). Additionally, the vaccines were tested in diverse patient groups, encompassing those with advanced myeloma (21, 23, 31), following chemotherapy (23, 31), individuals with no pre-treatment (30), those with lower tumor burdens following ASCT (19, 27) and smoldering MM (SMM) (29). As mentioned, despite extensive efforts, the Id-DC based vaccines did not yield satisfactory clinical outcomes.
A study in 1998 by Wen et al. is one of the first trials of a DC-based Id vaccine against MM, and involved a 43-year-old patient with advanced-stage refractory myeloma. DCs were isolated from the mononuclear cells by a series of density gradient centrifugations. Although the clinical response was limited, this study confirmed the functional abilities of ex-vivo-generated myeloma DCs by producing Id-specific immune responses (31). Later some optimization was applied to the DC generation by culturing the adherent mononuclear cell with cytokines such as GM-CSF and IL-4. In 1999, two patients with advanced refractory MM received an autologous DC vaccine loaded with Id antigen and KLH, accompanied by GM-CSF as an adjuvant. The results showed that both patients developed an Id-specific T-cell proliferative response, characterized by the production of interferon gamma (IFN-γ). However, there was no clinical benefit in these two cases (21). Nevertheless, considering that these patients had advanced disease, it was anticipated that the vaccination might not exert an optimal effect on this particular group. Therefore, additional trials have been conducted on cohorts of patients with less compromised immune systems or lower tumor burdens, with the anticipation that this might yield enhanced efficacy of the vaccine. Six myeloma patients with early-stage/early-relapse were vaccinated with autologous DCs (intravenously) prepared as in the previous study and loaded with Id antigens and KLH. However, only a period of stable Id serum levels was observed for 3 patients and 2 patients experienced disease progression post-vaccination (22). In another study, DC-based Id vaccination was tested following HDT and peripheral blood progenitor cell transplantation (PBPCT) in 26 patients where lower tumor burden was expected. Notably, only four patients developed an Id-specific proliferative T cell response. Three of these immune responders were in CR at the time of vaccination. A total of 17 patients were alive at a median follow-up of 30 months after transplantation. These findings suggested that a lower tumor burden might lead to better immune responses. However, attributing these outcomes solely to the vaccine was challenging due to the absence of a control arm for comparison (20). Investigation of DC-based Id vaccination was also performed in untreated stage-I patients. In this trial, nine patients received autologous monocyte-derived DCs pulsed with Id and KLH. The vaccine led to T-cell proliferation in 56% of patients. As for other studies, the clinical response was modest, with slight reductions in M protein observed in three patients (30).
Efforts to enhance efficacy of DC-based Id vaccination have led to optimizations in DC generation. In early studies with DC-based vaccination, immature DCs were pulsed with the antigen. However, immature DCs have suboptimal ability to activate T cells. Therefore, DCs exposed to antigens in the presence of maturation-inducing cytokines was introduced. This was first tested in five patients in stable partial remission after HDT. Furthermore, it was proposed to switch the administration route from intravenous to subcutaneous to overcome potential limitations linked to intravenous vaccination such as the potential accumulation of DCs in organs like the lung, liver, and spleen. Even with modifications to DC generation and a shift in the administration route, the clinical benefits were not remarkable. Moreover, attributing the clinical response solely to DC vaccination remained challenging, especially given the timing initiated four months after chemotherapy/auto-HCT (28).
Despite exploring optimizations in DC generation, administration routes, vaccination timing, and addition of immunogenic carrier protein/adjuvants, no remarkable results were observed. Bendandi et al. hypothesized that the lack of clinical significance could be explained by a deficiency in both quantity and quality of DC obtained from MM patients. Therefore, they performed a pilot study in which 4 MM patients received allogeneic dendritic cells (alloDC) followed by Id-KLH. The vaccine only induced detectable anti-KLH effector T- and B-cell responses without inducing T-cell proliferation against tumor-specific Id. From a clinical standpoint the results were not impressive with one patient with SD after stopping vaccination, while 3 of them progressed within study follow-up (24). Table 2 shows the summary of trials using DC-based Id vaccine in MM.
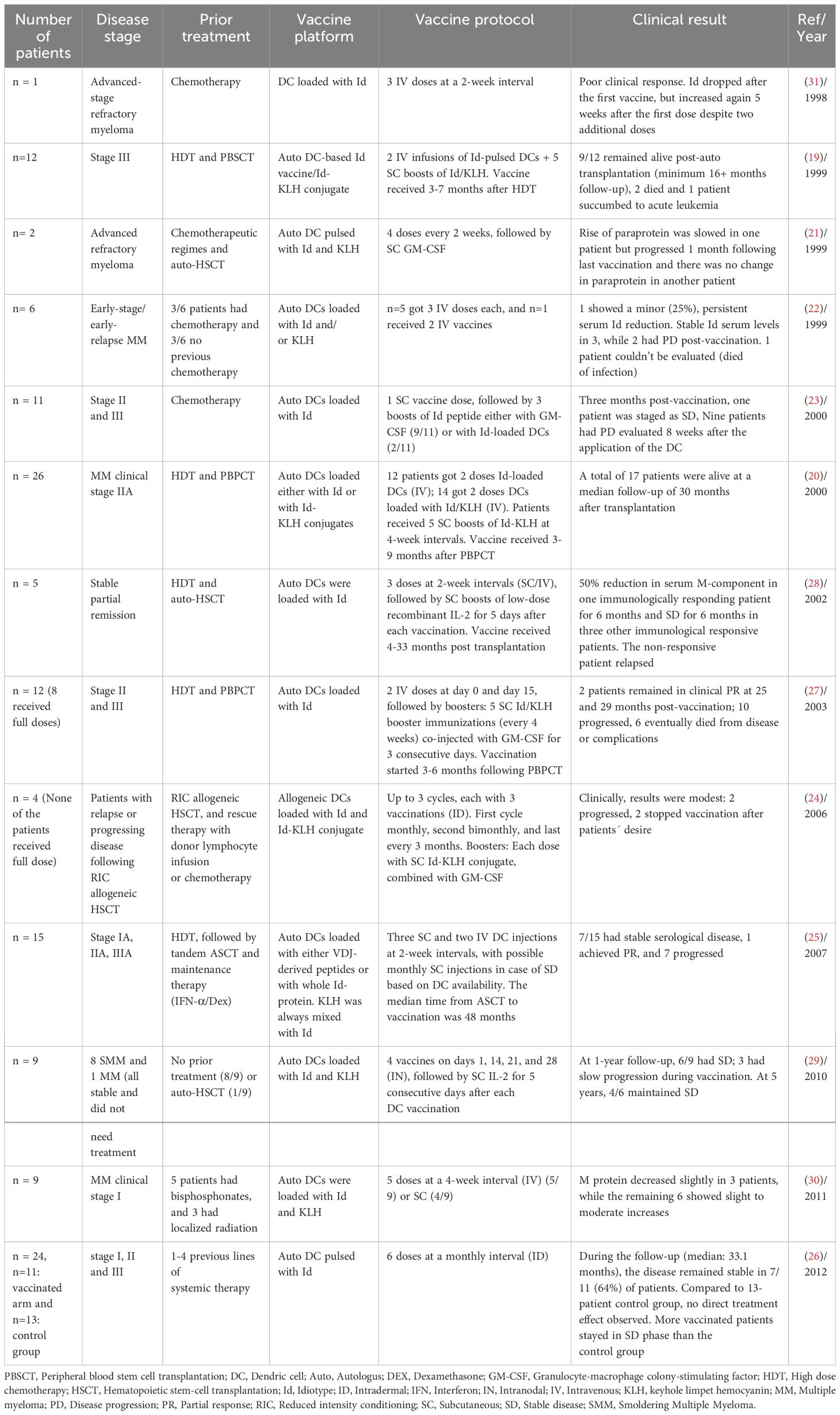
Table 2 Summary of trials using DC-based Id vaccine in MM.
3 Tumor-associated antigen
3.1 Peptide-based TAA vaccine
Although Id vaccination induces detectable immune responses, it fails to show clinical efficacy. This can possibly be attributed to the inhibitory effect of high levels of circulating Id proteins on T cells which leads to tolerance, and the fact that Id protein is a weak antigen (8, 29). Vaccines with TAA have been developed as an alternative to Id vaccines. Various TAAs are studied in MM both in clinical, pre-clinical and in vitro setting, such as Mucin-1 (MUC1), Receptor for hyaluronan-mediated motility (RHAMM), B-cell lymphoma 2 (BCL-2) family, Wilms tumor 1 (WT1), X-box binding protein 1 (XBP1), syndecan-1 (CD138), CS1 (SLAM7), cancer testis antigens (CTA), Dickkopf-1 (DKK1), methylmalonate-semialdehyde dehydrogenase (MMSA-1), heat shock protein (HSP), telomerase reverse transcriptase (hTERT) and survivin. Table 3 shows the summary of trials using TAA as an antigen in MM.
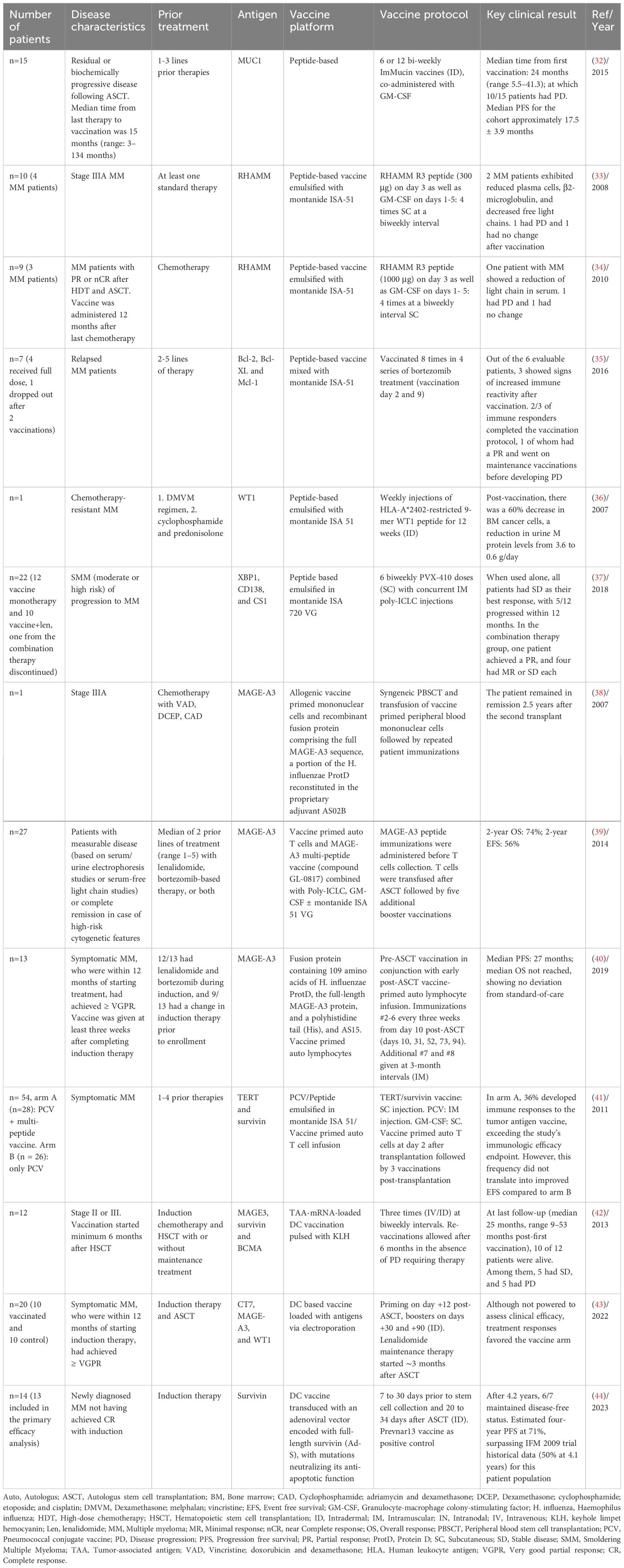
Table 3 Summary of trials using tumor-associated antigen (TAA) as an antigen in MM.
The glycoprotein MUC1 is found in various cancers, including MM. ImMucin, a 21-mer synthetic long-peptide vaccine was tested in a phase I/II study in 15 myeloma patients. ImMucin induced significant T-cell responses and increased antibody titers against MUC1. However, clinical efficacy was suboptimal with median PFS of 17.5 ± 3.9 months (32).
RHAMM-derived peptide R3 was investigated in a phase I/II study as another potential immunogenic antigen for hematologic malignancies, including MM. The initial study with a 300 μg vaccine in 10 patients with positive HLA-A2, including 4 with MM, showed positive clinical and immunological responses. Among MM patients receiving the 300 μg vaccine, 3 out of 4 showed increased specific CD8+ T cells and 2/4 showed reductions in clonal markers. However, the limited sample size warrants caution in drawing definitive conclusions (33, 34).
BCL-2 family protein was investigated in a phase 1 trial as another target antigen in MM. Patients with relapsed MM received vaccinations with peptides from Bcl-2, Bcl-XL, and Mcl-1 mixed with montanide ISA-51 as an adjuvant together with bortezomib. Due to peptide HLA-restriction, patients received peptides according to their HLA-A-positivity, i.e., HLA-A1, HLA-A2 and/or HLA-A3. Among 7 patients, 3 demonstrated vaccination-induced peptide antigen-specific T-cell responses compared to the baseline (before vaccination). However, small sample size hindered a conclusive assessment of clinical efficacy (35). The WT1 gene, linked to childhood renal tumor Wilms tumor, plays dual roles as an oncogene and as a key component of certain normal cellular processes. It is highly expressed in hematopoietic malignancies and solid cancers, including myeloma, where its expression increases with disease progression (45, 46). Studies have identified various epitopes in WT1 that can elicit WT1-specific cytotoxic T lymphocyte (CTL) responses in a human HLA-restricted manner. Therefore, WT1 peptide-based immunotherapy could be an option for patients with malignant diseases [30, 33]. In a case study by Tsuboi et al., a 57-year-old chemotherapy-resistant MM patient received weekly intradermal vaccinations with HLA-A*2402-restricted 9-mer WT1 peptide and montanide ISA51 as an adjuvant. Post-vaccination, there was a 60% decrease in BM cancer cells, a reduction in urine M protein levels from 3.6 to 0.6 g/day. Additionally, there was a notable shift in C-X-C chemokine receptor type (CXCR)4 positive cells, suggesting effective migration of WT1-specific CTLs to the tumor site (36).
Multi-peptide vaccines have also been investigated to combat limitations of single antigen peptide-based vaccination, including potential resistance by antigenic escape and downregulation of target antigens. PVX-410 is a peptide-based vaccine, which consists of 4, 9-mer peptides from three different antigens including XBP1, CD138, and CS1. In one study, 22 moderate to high-risk SMM patients positive for HLA-A2 were administered with PVX-410 subcutaneously and an adjuvant, poly-ICLC, with or without lenalidomide. PVX-410 consistently generated specific, durable immune responses, particularly enhanced with lenalidomide. However, overall clinical responses were still modest. When used alone, all patients had SD as their best response, with 5/12 progressing within 12 months. In the combination therapy group, one patient achieved a PR, and four had minimal response or SD (37).
DKK1, MMSA-1, and HSP have been investigated as TAAs with potential for vaccine therapy against MM. DKK1, a Wingless-related integration site (Wnt)/β-catenin signaling inhibitor, induces peptide-specific CTLs that recognize and lyse myeloma cells with positive HLA-A*0201 (47). MMSA-1 is a membrane protein specifically expressed in MM cells. When combined with DKK1 in a vaccine, it enhanced CTL responses in HLA-A*0201-restricted manner, improved survival, and alleviated bone destruction in pre-clinical model (1). Tumor cell-derived HSPs, like gp96, have been explored as TAAs in myeloma in mice. Pooled HSPs, including gp96 from established murine myeloma cell lines, showed promise as an off-the-shelf vaccine effectively treating mice with large myeloma tumor burdens with HSP combined with anti-B7H1 or anti–IL-10 monoclonal antibodies (48). However, further studies are needed to confirm their efficacy in clinical setting.
Moreover, CTA antigens, initially discovered in melanoma patients are targets for immunotherapy due to their limited expression on normal tissues. A study by Andrade et al., indicated that the expression of more than six CTA antigens on myeloma cells holds prognostic value, being associated with shorter overall survival (OS) in MM patients (49, 50). Prominent CTA antigens like NY-ESO-1, LAGE-1, MAGE-A1, MAGE-A2, MAGE-A3, and MAGE-C1 (CT-7) have demonstrated the potential to stimulate T-cell responses, making them candidates for cancer immunotherapy (38). Among the CTA antigens, MAGE-A3 have been tested in a clinical setting. In a case study a healthy donor, who was the patient’s identical twin, was immunized with MAGE-A3 protein along with AS02B as an adjuvant. Following a syngeneic peripheral blood stem cell transplant, primed donor cells were transferred to the patient. Subsequently, the patient received additional immunizations with MAGE-A3, along with a second transfusion of peripheral bone marrow mononuclear cells. The MAGE-A3 immunizations were well-tolerated and resulted in the development of robust MAGE-A3-specific antibodies, CTLs, and T-helper responses in both twins. Interestingly, the CTL response targeted a previously unknown HLA-A*6801 binding MAGE-A3 peptide, which remained detectable in the patient more than a year after the last immunization. Multiple T-helper cellular responses were detected with the dominant response to an HLA-DR11 restricted MAGE-A3 epitope. Encouragingly, the patient remained in remission 2.5 years after the second transplant. This case study suggests the potential efficacy of this personalized immunotherapeutic approach for treating MAGE-A3-positive MM patients (38). In another phase II study, the safety and efficacy of ex vivo expanded autologous T cells primed in vivo using a MAGE-A3 multi-peptide vaccine combined with Poly-ICLC and GM-CSF were evaluated in twenty-seven myeloma patients. The vaccine includes two HLA-A2–restricted class I epitopes and a promiscuous class II epitope. Results showed that MAGE-A3–specific CD8 T cells were detected in 7 out of 8 assessable patients with HLA-A2+. Furthermore, vaccine-specific T cells capable of producing cytokines were generated in 19 out of 25 patients. The 2-year OS was 74% and the 2-year event-free survival (EFS) was 56% (39). A similar study was performed by Cohen et al. where recombinant MAGE-A3 with AS15 immunostimulant were administered to 13 MM subjects pre- and post-ASCT in conjunction with early post-SCT vaccine-primed autologous lymphocyte infusion. The combination immunotherapy resulted in high-titer humoral immunity and durable and robust, antigen-specific CD4+ T-cell responses in all subjects. However, median PFS was 27 months, and median OS was not reached, suggesting no differences from standard-of-care (40).
Other CTAs tested in pre-clinical settings have been suggested as possible immunotherapy targets in MM. Spontaneous NY-ESO-1 antibodies and specific CD8+ T cells have been detected in vivo in NY-ESO-1 positive MM cases (51, 52). In a pre-clinical model, a complex vaccine named NACH, based on NY-ESO-1-alum-CpG ODN-HH2 was investigated. The Alum-CpG ODN-HH2 combinational adjuvant used in the NACH vaccine is a novel immune adjuvant. The vaccine demonstrated promising anti-tumor effects and immunogenicity in prophylactic and therapeutic models of murine MM. The NACH vaccine inhibited tumor growth, leading to a significant extension of survival time in mice. In the therapeutic model, seven out of ten tumor-bearing mice in the NACH vaccine group survived up to day 90, while all mice in the control group died within 40 days (53). LAGE-1a, another CTA antigen, frequently expressed in MM patients, shares high mRNA sequence similarity with NY-ESO-1. In silico analysis identified seven peptides present in both LAGE-1a and NY-ESO-1, recognized by T lymphocytes in different tumors. Therefore, it was hypothesized that an anti-NY-ESO-1 vaccine could potentially benefit MM patients with tumors that express LAGE-1a but not NY-ESO-1 (54). MAGE-C1 (55, 56), SPAN-Xb (57, 58), and SP-17 (49, 59) are other CTA antigens capable of eliciting CTL responses, rendering them targets for vaccine-based immunotherapy MM.
Similar to Id vaccines, the practical incorporation of peptide-based tumor-TAA vaccines into clinical practice have not occurred. A study by Rapoport A.P et al. compared immunogenicity of the rate-limiting catalytic subunit of the telomerase complex called hTERT (60) and anti-apoptotic protein survivin as a target antigen vaccine to pneumococcal conjugate vaccine in 54 MM patients. Patients positive for HLA-A2 (including any A2 allele) were assigned to arm A where they received hTERT/survivin vaccine. The study suggested that although this multi-peptide tumor antigen vaccine had a higher immune response frequency than reported for idiotype vaccines, TAA still falls short of that induced by microbial vaccines. This suggests that achieving a more substantial immune response to cancer vaccines may be necessary for significant long-term clinical benefits (41).
3.2 DC-based TAA
DC-based vaccines using TAA associated antigens have also been assessed as alternatives to Id antigens. Twelve stage II or III myeloma patients with minimum PR after induction chemotherapy and HCT/ASCT were vaccinated. DCs were generated from adherent CD14+ cells loaded with MAGE3, survivin and BCMA via electroporation and were pulsed with or without KLH. Notably, all patients developed strong anti-KLH T-cell responses, which indicates immune recovery after high-dose melphalan, and in two patients, vaccine-specific T cells were detected in delayed-type hypersensitivity biopsies. At last follow-up, 10 of the 12 patients were alive at a median follow-up of 25 months (range 9–53 months) after the first vaccination. Of these patients, 5 had SD and 5 had progressive disease (42). In another study, mRNA electroporation of DCs was conducted, utilizing Langerhans DCs loaded with CT7, MAGE-A3, and WT1. These Langerhans-type DC, derived from CD34+ hematopoietic progenitor cells, were considered potentially more potent stimulators of CTLs against tumor antigens in comparison to monocyte-derived DCs in vitro. The patients were randomized to receive either the vaccine within 100 days after ASCT or were placed in the control group without the vaccine. While the study was not powered to assess clinical efficacy, treatment responses favored the arm receiving the vaccine (43).
In one recently published study, Locke and colleagues designed a DC-based vaccine targeting anti-apoptotic protein survivin in a phase 1 clinical trial. DCs were engineered via adenoviral vector to express a version full-length survivin (Ad-S) containing two mutations that neutralize the anti-apoptotic function of wild-type survivin. Thirteen newly diagnosed MM patients who did not achieve CR with induction therapy were vaccinated 7 to 30 days prior to stem cell collection and 20 to 34 days after ASCT. The vaccine in combination with ASCT was well tolerated, with only minor adverse effects noted. Notably, a remarkable 85% of patients exhibited either a T-cell response or an antibody response against survivin. Seven patients exhibited enhanced clinical responses at day +90, all linked to survivin-specific immune responses. Impressively, after a median follow-up of 4.2 years, six out of these seven patients maintained a disease-free status. The estimated four-year PFS was 71%, surpassing historical data from the IFM 2009 trial with approximately PFS of 50% at 4.1 years for this patient population (44). Survivin has emerged as a notable TAA in MM. Among the 8 ongoing trials in MM, two are focused on developing vaccines targeting survivin. One such vaccine is named TXSVN, utilizing a weakened form of a live Salmonella bacterial strain genetically modified to produce survivin. The second vaccine is a peptide-based formulation known as SVN53-67/M57-KLH, derived from the survivin protein (Table 4).
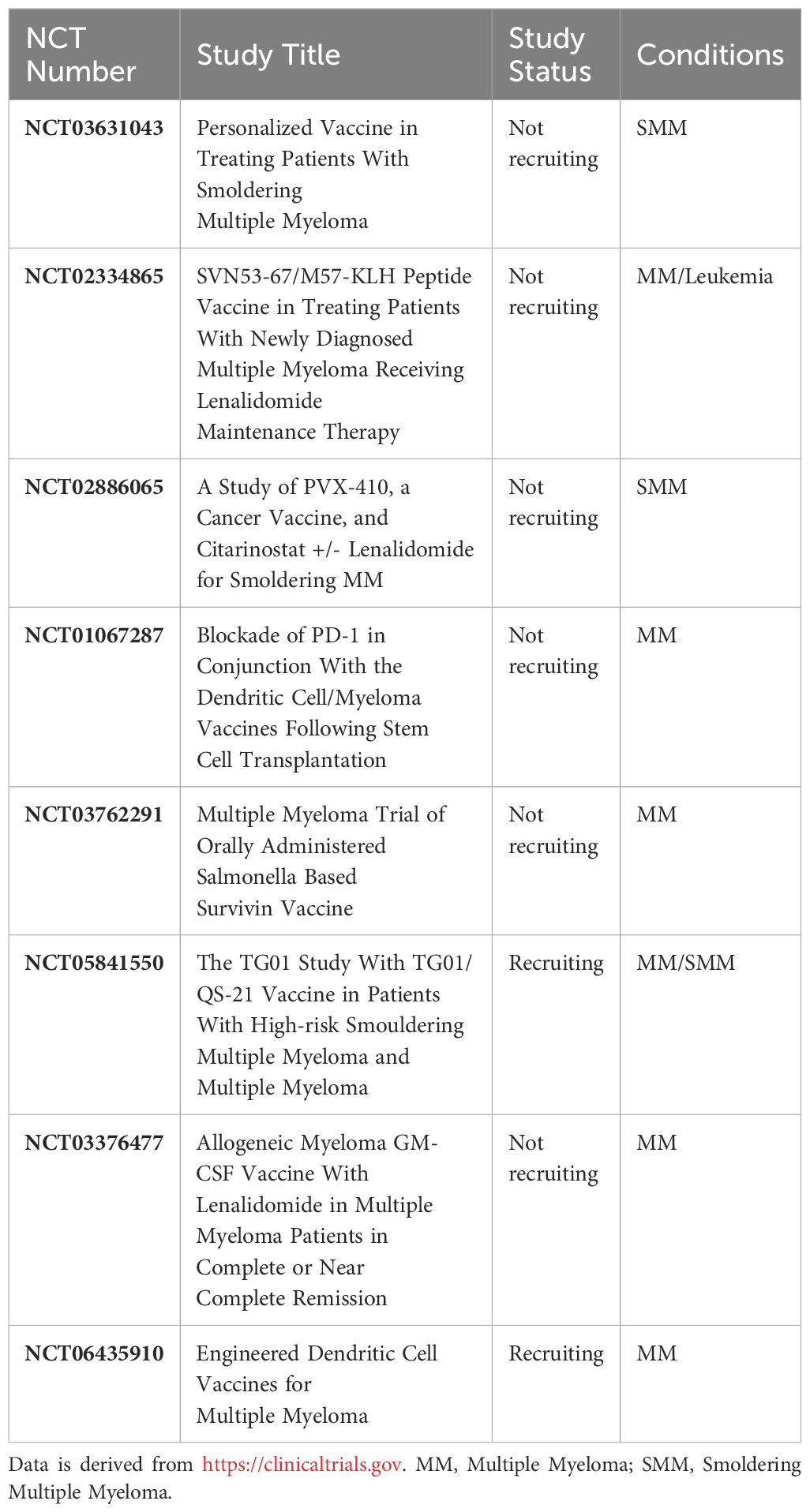
Table 4 List of ongoing vaccine trials against MM.
4 Tumor-specific antigen
4.1 Peptide-based TSA vaccine
While certain studies on TAAs show promise, the overall assessment is challenging due to the limited patient numbers in each clinical trial. Further research with larger cohorts is necessary for conclusive findings on the efficacy of TAA-based immunotherapies. Moreover, due to their status as non-mutated self-antigens, in general TAAs may exhibit low immunogenicity attributed to the influence of central T cell tolerance. Therefore, TSAs, also called neoantigens, appear as another type of target antigens for vaccine immunotherapy (61, 62).
Somatic mutations in cancer cells can give rise to novel protein sequences that can be presented by APCs as neoantigens to the host immune system. Tumor neoantigens represent excellent targets for immunotherapy, due to their specific expression in cancer tissue (63). The landscape of neoantigens in 184 MM patients, as identified through next-generation sequencing, revealed shared neoantigens in NRAS, KRAS, and Interferon regulatory factor 4 (IRF4) genes in relapsed patients and in KRAS in newly diagnosed patients supporting the possibility of neoantigen-based vaccines in MM patients with such mutations (63). Perturbations in the MAPK pathway, particularly mutations in KRAS, NRAS, and BRAF, are observed in a significant percentage of MM cases, with RAS mutations detected in up to 70% of relapsed/refractory cases (64). These mutations, associated with MAPK activation, may impact prognosis, contributing to transitions from precursor conditions to myeloma and from intramedullary to extramedullary disease with increasing prevalence as the disease progresses (64). Targeting shared neoantigens, such as those associated with RAS mutations, is being explored in clinical trials, exemplified by the ongoing study by our group using the TG01 cancer vaccine in high-risk SMM or MM patients (Table 4) with measurable disease after ≥1 line of treatment. TG01, composed of 7 synthetic peptides mimicking mutated forms of the RAS protein, has demonstrated promising results in patients with pancreatic cancer. The primary endpoint of the TG01-study is safety, with key secondary endpoints including immunological response to the vaccine, overall response rate, OS and PFS (65).
Another neoantigen-based vaccine is PGV-001. PGV-001 is a personalized genomic vaccine which targets up to 10 predicted personal tumor neoantigens based on patient’s HLA profile. PGV-001 was tested in 13 patients with multiple cancer types in the adjuvant setting including three patients with MM who had undergone ASCT. Vaccine peptides were administered over the course of 27 weeks with poly-ICLC and a tetanus helper peptide. One patient was lost to follow-up, but among the remaining 12 patients with a mean follow-up of 925 days, four showed no evidence of disease, 4 received subsequent lines of therapy, and 4 have passed away. Notably, only two of the deceased patients had documented recurrence of their malignancy. The details of the patients and which of them were MM is not described in this abstract. Immune monitoring of immunogenicity is ongoing. However, initial analysis demonstrated induction of neoantigen-specific CD4 and CD8 T cell expansion (66).
As noted earlier, TSA prompt the activation of high-avidity T cells, given their lack of thymic selection and central tolerance. While there has been significant focus on Id, showing limited clinical efficacy in myeloma due to its abundance, research on other neoantigens than Ids in myeloma remains limited. Further studies are essential to evaluate their potential as target antigens in myeloma vaccine therapy (61).
5 Whole tumor antigen
5.1 DC-based vaccine
Single antigen-specific vaccines are vulnerable to immune evasion due to downregulation of antigen expression. Alternatively, whole cell targets have also been explored as a strategy, which seek to establish a polyclonal immunologic response. Such vaccines could potentially include a broad array of antigens, including neoantigens (2). This is particularly relevant in myeloma, where a significant tumor mutation burden exists. A study on 664 newly diagnosed myeloma patients found mean somatic and missense mutation loads of 405.84 and 63.90 mutations per patient, respectively. There was a positive relationship between mutation and neoantigen burdens (67). Additionally, high-dose melphalan therapy, a standard treatment before ASCT, is associated with a high mutational burden and possibly more neoantigens at relapse in myeloma patients (68).
Rosenblatt et al. developed a tumor vaccine by chemically fusing patient-derived myeloma cells with autologous DCs. This vaccine offered the advantage of potentially presenting a diverse range of myeloma-associated antigens within the context of DC-mediated co-stimulation. Fusion cells were created by co-culturing DCs and myeloma cells with polyethylene glycol. The DC fusion vaccine was tested in 17 patients, the majority of whom had advanced disease. Vaccination was well-tolerated and resulted in the expansion of lymphocytes that reacted with the patient’s own myeloma cells in most evaluated patients, as well as documented humoral responses. Patients with advanced disease experienced disease stabilization, with three patients showing ongoing SD at 12, 25, and 41 months, respectively. In the current study, regulatory T cell levels remained stable throughout the vaccination period (69). The same vaccine was further tested in patients following ASCT with the hypothesis that autologous transplantation provides an optimal setting for a fusion vaccine due to the enhanced immunologic environment resulting from tumor cytoreduction and regulatory T-cell depletion. The study achieved promising outcomes with 47% of patients achieving a CR/near CR (nCR) as best response and 78% of patients achieving at least a VGPR. Notably, nearly 35% of CRs occurred greater than 100 days post-transplant, after undergoing vaccination. Although delayed effects of chemotherapy may be observed, the significant number of late responses in the absence of maintenance therapy might be suggestive of a vaccine-mediated effect (70). Based on these results the group was encouraged to examine the efficacy of the fusion vaccine in conjunction with lenalidomide as maintenance therapy after auto-HCT. However, DC/MM fusion vaccination with lenalidomide did not result in a statistically significant increase in CR rates at 1-year post-transplant but was associated with a significant increase in circulating MM–reactive lymphocytes indicative of tumor-specific immunity (71).
The same group conducted another clinical trial involving MM patients who received an anti-programmed cell death protein 1 (PD-1) antibody (Pidilizumab) in combination with a DC/myeloma fusion cell vaccine following autologous transplantation with the aim to overcome the immunosuppressive milieu by which tumor cells evade host immunity. This trial involved 22 patients and showed that this combination could induce anti-tumor immune responses, and, in a subgroup of patients, led to the complete eradication of measurable disease after transplant. After the transplantation, regulatory T cell levels decreased significantly and remained low throughout the immunotherapy period. Six patients achieved a best response of VGPR, and six patients reached nCR/CR. The median PFS from the transplant was 19 months, with ongoing follow-up (Table 4) (72). Other whole cell lysates such as GVAX and DCOne vaccine has been tested as off-the shelf vaccines with a diverse antigen repertoire. MM-GVAX is a vaccine, in which two established heterogeneous myeloma cell lines, H929 and U266, are administered with the K562 cell line (GVAX®) engineered to overexpress GM-CSF. H929 exhibited t(4;14), and a mutated NRAS, and U266 has several mutations involving the BRAF and TP53 pathways. Patients enrolled were serum/urine immunofixation positive and maintained nCR for at least 4 months. In the publication from 2021, of 15 patients, 8 (53.3%) had deepened treatment response and achieved true CR. The median OS was 7.8 years. MM-GVAX triggered clonal T-cell expansion and cytokine responses that have remained durable up to 7 years in all patients. This trial is ongoing (Table 4) (73, 74). DCOne is another off-the shelf tumor vaccine, which is derived from a human acute myeloid leukemia (AML) cell line that can be differentiated to the phenotype of fully functional DCs, and endogenously expresses TAA. These antigens are presented alongside HLA molecules and various co-stimulatory molecules critical for T-cell activation. Although originally engineered as a cancer vaccine in AML, DCOne expresses tumor antigens found in a variety of hematologic malignancies, including MM. MM with the DCOne vaccine resulted in the expansion of activated CD8+ T cells expressing interferon-γ and perforin. Further, co-culture of patient’s tumor cells with peripheral blood mononuclear cells and DCOne induced cytotoxic T-lymphocyte-mediated killing of autologous MM cells, highlighting the therapeutic potential of DCOne in myeloma (2). Table 5 shows the summary of trials using whole tumor antigen in MM.
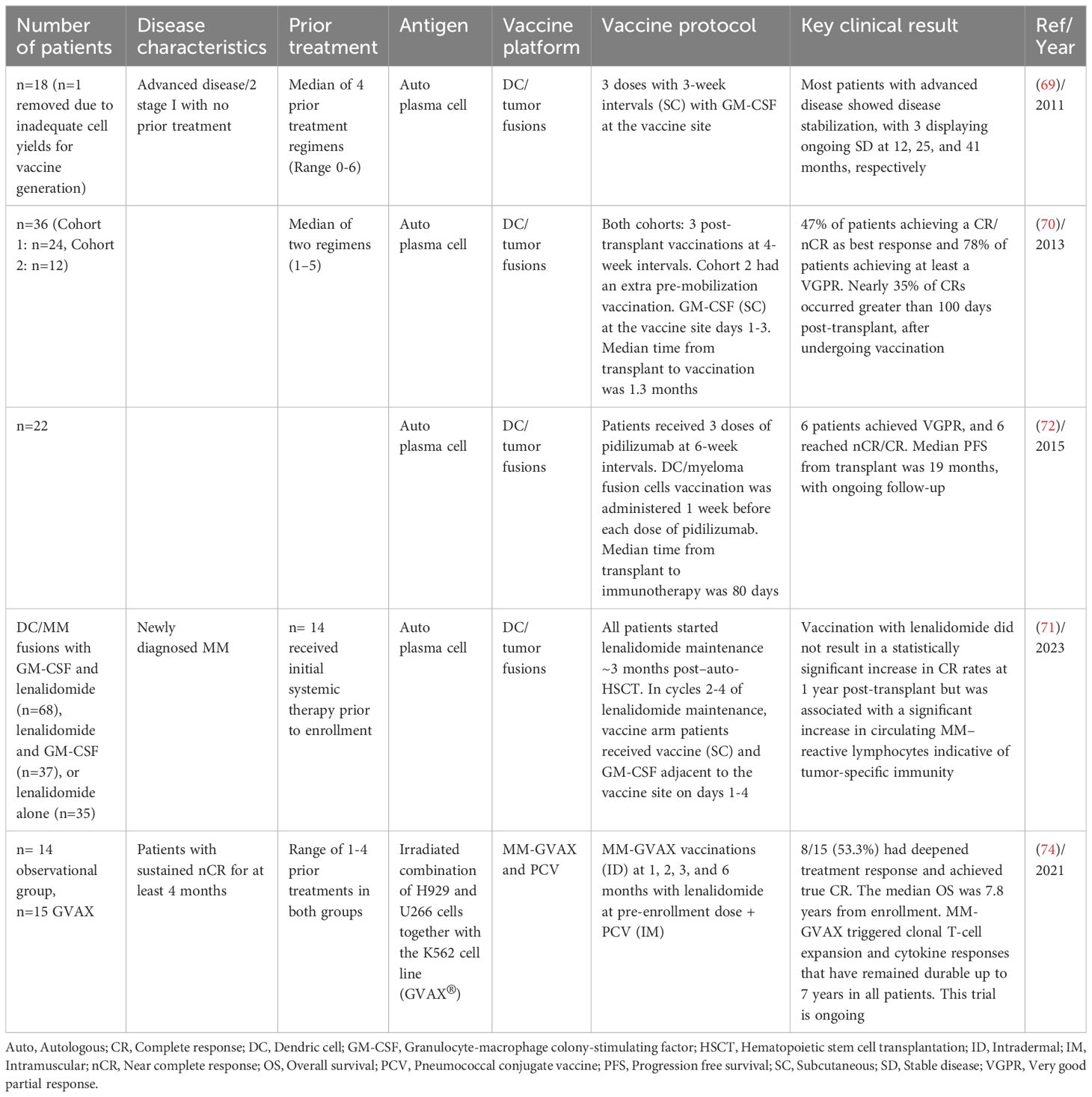
Table 5 Summary of trials using whole tumor antigen in MM.
6 Conclusion and future perspective
Despite extensive research, tumor vaccine has not yet been incorporated into myeloma therapy. The predominant emphasis has been on Id vaccines for many years which has yielded occasional immunological responses but with limited clinical efficacy. Subsequent investigations have explored diverse antigenic targets in clinical trials. Nevertheless, the widespread testing of various antigens in early-phase trials with restricted patient cohorts poses challenges in establishing definitive conclusions. Moreover, direct comparisons between studies even with the same antigen type are hindered by variations in patient characteristics such as age, cytogenetics/risk factors, and the disease stage at which vaccination occurred. Furthermore, a significant number of vaccine studies have been conducted during periods when chemotherapy constituted the standard of care and there was more focus on using vaccine alone to eradicate tumor. However, the combination of vaccination with newly established treatments like IMiDs, proteasome inhibitors, and new generation immunotherapies such as chimeric antigen receptor T (CAR-T) cells and bispecific antibodies has not yet been addressed.
Generally, the intricate nature of vaccines coupled with the complexity of myeloma as a disease, presents challenges for integrating vaccines into myeloma treatment strategies. Vaccine therapy is complex and requires thorough experimentation. This includes selecting the tumor antigen, formulating the vaccine, determining the delivery vehicle (62), deciding on the route and frequency of administration, establishing the timing of vaccination in addition to accounting for the complexities of the tumor microenvironment in MM (Figure 1). Optimizing all these factors collectively may be necessary to improve clinical efficacy.
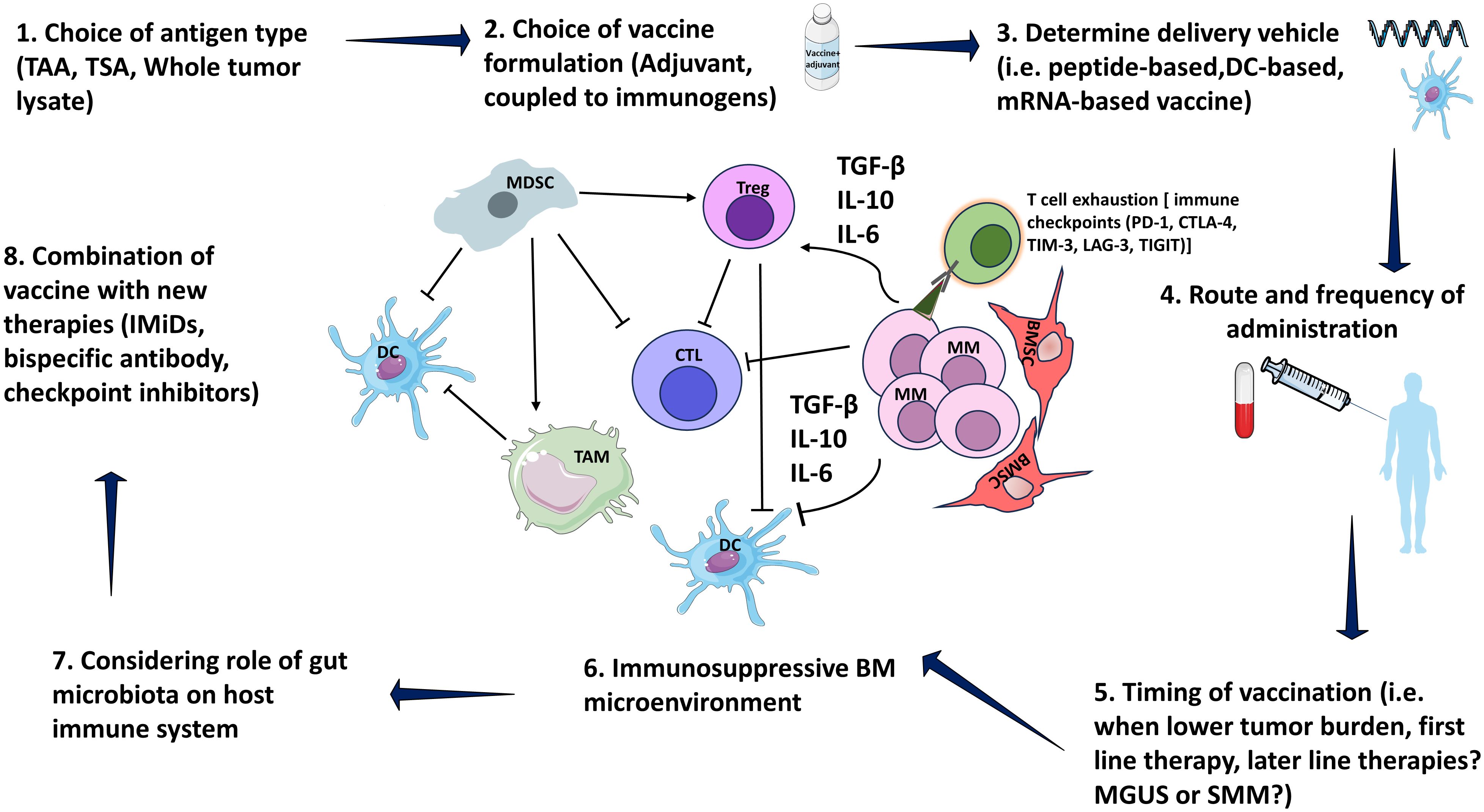
Figure 1 Consideration in designing vaccine. MM bone marrow microenvironment is consisted of immunosuppressive elements including MDSCs, TAM and Tregs. MDSCs stimulate TAM as well as Tregs via IL-10. MDSCs also inhibit CTLs via IL-10. Tregs inhibit CTL, and DC function by direct cellular interactions and via secretion of suppressive cytokines, such as TGF-β and IL-10. Moreover, MM cells secrete several cytokines including IL-6, TGF-β, and IL-10 that inhibit DCs, CTLs, and stimulate Tregs. Myeloma patients express multiple immune checkpoint receptors, including PD-1, CTLA-4, TIM-3, LAG-3, TIGIT. Terminal T cell exhaustion is associated with the loss of cytotoxicity by CD4+ and CD8+ T cells subsets that produce IFN-γ, a critical cytokine for tumor immunity. MM, Multiple myeloma; MGUS, Monoclonal gammopathy of undetermined significance; SMM, Smoldering MM; BM, Bone marrow; DC, Dendritic cells; BMSC, Bone marrow stromal cells; CTL, Cytotoxic T lymphocyte; MDSCs, myeloid-derived suppressor cells; TAM, Tumor-associated macrophages; Treg, Regulatory T-cells; PD-1, programmed cell death domain protein 1; CTLA-4, Cytotoxic T-lymphocyte-associated protein 4; TIM-3, T cell immunoglobulin mucin-3; LAG-3, lymphocyte activation gene 3; TIGIT, T cell immunoglobulin and ITIM domain; Interleukin (IL)-10; TGF-β1, transforming growth factor-β1; TAA, tumor-associated antigen; TSA, tumor-specific antigen. Parts of figures were used/adapted from pictures provided by Servier Medical Art (Servier; https://smart.servier.com/), licensed under a Creative Commons Attribution 4.0 Unported License (https://creativecommons.org/licenses/by/4.0/).
6.1 Antigen selection
Ideally, the target antigen must be immunogenic, highly expressed in tumor, low or absent in normal tissues and tumor-specific to avoid off-target effects (2). Several antigens, including Ids, TAAs, and TSAs, have been explored in myeloma research. Despite efforts to enhance their immunogenicity, especially for Ids by coupling vaccinations with immunogens and adjuvants, there has been no remarkable clinical efficacy (51). The study on TSAs other than Ids is still limited. Predicting TSA as targets is both time-consuming and costly, requiring advanced methods to predict neoantigens capable of eliciting neoantigen-specific T cell responses. Moreover, accurate HLA typing is crucial for reliably predicting immunogenic antigens and improving therapeutic efficacy, adding another layer of complexity to vaccine development. This is due to the polymorphism of human HLA alleles, which encompass over 24,000 unique gene complexes (6). Although there is strong rationale for the use of patient-specific vaccines that express multiple antigens such as whole tumor lysate, there is potential concern for the reestablishment of tumor tolerance over time facilitating disease progression (70). This could partly explain the delayed adoption of whole tumor lysate vaccine in myeloma clinical settings. Gut microbiota antigens possess inherent strong immunogenic properties, but remain an unexplored area in myeloma research. The presence of highly similar antigenic epitopes between TAAs and microbial antigens suggests the potential for a robust cross-reacting CD8+ T cell response. Specifically, T cell memory induced by specific microbial antigens may translate into anti-cancer T cell memory, exerting long-term control over cancer growth. Examples include molecular mimicry observed between MAGE-A10 and cytomegalovirus. This revelation opens avenues for exploring the cross-reactivity between myeloma-associated TAAs and microbial antigens, offering new insights for enhancing the efficacy of myeloma vaccine therapy (62).
6.2 Platform for vaccine delivery
Peptide-based vaccination has emerged as a promising method for eliciting antitumor T-cell responses. However, its effectiveness remains limited, even when the peptide is co-delivered with a potent adjuvant. This may be attributed to cancer-induced dysfunction in DC, which play an important role in influencing the quantity and quality of antitumor immunity. Consequently, the delivery of tumor antigens through DC-based peptide approaches is gaining recognition as a potential strategy in cancer immunotherapy (75). Since the inception of DC use in myeloma therapy, numerous studies have focused on optimizing DC generation, particularly utilizing idiotype as a source of antigens. Some studies have demonstrated the heightened potency of CD34+ hematopoietic progenitor cell (HPC)-derived Langerhans-type DCs to monocyte derived DCs in stimulating CTL (43). However, their clinical application in myeloma studies remains limited. In addition, there has been huge focus on ex-vivo generation of DCs derived from myeloma patients. Shinde et al. generated DCs from MM and healthy donor samples. While both MM-DCs and healthy donor-derived DCs showed mature phenotypes, MM-DCs exhibited lower migratory capacity and cytokine secretion, making them less effective in inducing an anti-MM response (76). Furthermore, efforts to enhance the delivery of antigens to DCs are being explored through various methods. These include pulsing DCs with antigen peptide pools and employing gene editing techniques such as electroporation (42), lipid nanoparticles (77), or viral vectors to improve antigen presentation (78). The COVID-19 pandemic led to notable advancements in mRNA vaccines, presenting an opportunity for their application in antitumor therapy. These vaccines offer robust cellular and humoral immunity, surpassing conventional pathogen or protein-based vaccines. Furthermore, mRNA vaccines offer advantages such as rapid development, safety, fewer side effects, and flexibility, thereby facilitating the implementation of personalized vaccine strategies, particularly in the context of TSAs. The utilization of mRNA vaccines for antigen delivery represents a novel approach warranting investigation for its potential in myeloma therapy (79, 80).
6.3 Route and timing of administration
The route of administration and the optimal duration of vaccination remain topics of ongoing debate, with insufficient comparisons in clinical trials. Another factor that has been overlooked in myeloma is the timing of tumor vaccine administration. In a large number of published clinical studies, vaccines are administered shortly after systemic anticancer treatment. This timing poses a challenge as natural immunity may be suppressed by chemotherapy at the time of vaccination (26). One suggested approach that has been explored in numerous trials involves the potential benefits of vaccination after ASCT (25, 43, 70–72), particularly when a state of deep response has been achieved. The engraftment of cellular immune compartments at this stage provides an opportunity to shape antitumor immunity. However, challenges to this approach include delayed immune reconstitution, with T cell numbers taking at least 3 months to return to normal, leading to suboptimal responses to vaccine therapy (40, 43). To address this issue, some studies have investigated pre-ASCT vaccination, followed by the early post-ASCT transfer of vaccine-primed T cells in a lymphodepleted environment. Post-transplant booster vaccinations were then administered to expand vaccine-specific T cells. Despite these efforts, this strategy did not yield favorable clinical outcomes (39–41). Nevertheless, these studies are limited, and more investigation is needed before drawing any conclusive results. Another potential approach is to target patients at an early stage in their disease course, particularly those with monoclonal gammopathy of undetermined significance (MGUS) or SMM. However, implementing this strategy would require treating a substantial number of patients and conducting long-term follow-up to establish clinical benefits. As of now, conclusive studies supporting this hypothesis are lacking (81). Three ongoing trials are currently investigating vaccines in patients with SMM, including our study called TG-01 targeting RAS mutation, PVX-410 (a multi-peptide cancer vaccine from XBP1, CD138, and CS1), and a personalized cancer vaccine made from an individual’s blood and bone marrow (Table 4).
6.4 Vaccine-extrinsic factors
Vaccine-extrinsic factors, including the tumor microenvironment and dysfunctional host immune responses in MM underscores the complexities of eliciting effective immune responses (43, 82). The MM tumor microenvironment is populated by immune-suppressive cells, including myeloid-derived suppressor cells, regulatory T cells, and tumor-associated macrophages. Additionally, inadequate antigen presentation, resistance to natural killer (NK) cell lysis, T-cell exhaustion and/or senescence, is associated with poorer outcomes after ASCT and characterizes multiple relapsed disease (43, 82). Myeloma-derived cytokines, like transforming growth factor (TGF)-β, IL-6 and IL-10, contribute to immune dysfunction (83, 84). In addition, vaccination effectiveness decreases with age due to T cell unresponsiveness resulting from age-related immune system changes (85). This is particularly significant for MM, with the average age at diagnosis being 70. Another extrinsic factor, which has been scarcely studied in myeloma patients, is the potential effect of gut microbiota on the efficacy of tumor vaccines. Radojević et al. reported a correlation between the composition of the gut microbiota and the immunogenicity of DCs by analyzing the fecal microbiota composition of 14 healthy donors, along with the phenotype and cytokines produced by monocyte-derived DCs [61]. Another study by Calcinotto et al. indicated that Prevotella heparinolytica promotes the differentiation of Th17 cells colonizing the gut and migrating to the BM of transgenic Vk*MYC mice, where they favor progression of MM (86). This subject is complex, as many factors can potentially affect gut microbiota including treatments like antibiotics which is commonly administered to myeloma patients.
6.5 Combination of vaccine with new generation therapies
It should be noted that most tumor vaccines alone do not directly eliminate tumor lesions, but tumor vaccines are potentially promising in eliminating MRD (62). Therefore, further trials are needed to define the role of vaccination in the era of new pharmacologic therapies such as immune checkpoint inhibitors (ICI), antibody-drug conjugates, bispecific antibodies, and CAR-T cells. Durable responses to these treatments are infrequent, but combination strategies with vaccines to prime anti-MM immunity offer an approach to boost responses or eliminate the MRD. For example, combination of vaccine with ICI seems reasonable approach but the safety concerns in the absence of a clear signal of improved efficacy have become a major obstacle to the clinical application of ICI in MM (43, 87). Vaccination prior to bispecific antibody constructs may augment therapeutic potency by creating pools of antigen-experienced T cells. Vaccines can also promote the expansion and efficacy of chimeric antigen receptor T cells. In addition, it should be noted that although bispecific antibodies and CAR-T currently dominate the MM immunotherapy landscape, cancer vaccines offer distinct advantages. Unlike CAR-T cells or bispecific antibodies, which primarily target cell surface tumor-specific antigens, cancer vaccines have the potential to target intracellular antigens. This capability presents an opportunity to address a broader range of tumor antigens (88).
6.6 Conclusion
In conclusion, even though vaccination against MM has so far not yielded very impressive clinical results, further studies focusing on selection of the appropriate antigen, patient population, optimization of timing and sequencing of vaccine, and identification of rational combinations are warranted. These elements, in combination with other immune-directed interventions to overcome the immunosuppressive activity of the tumor microenvironment may exert a greater benefit with improved and durable clinical responses.
Author contributions
PA: Conceptualization, Writing – original draft, Writing – review & editing. HN: Conceptualization, Writing – original draft, Writing – review & editing. FS: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We would like to acknowledge Ranveig Braathen (Group leader at Oslo University Hospital) and Anne Marit Sponaas (Researcher at Norwegian University of Science and Technology) for their invaluable contributions and insightful comments on the manuscript. We acknowledge the use of ChatGPT version 3.5, which was employed at times to refine the language.
Conflict of interest
HN: Honoraria: Janssen, BMS and Sanofi. FS: Honoraria: Amgen, BMS, Takeda, Abbvie, Janssen, Novartis, SkyliteDX, Oncopeptides, Sanofi, Schain, Pfizer, Daiki-Sankyo; Consultancy: GSK, Celgene, Takeda, Janssen, Oncopeptides, Sanofi.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lu C, Meng S, Jin Y, Zhang W, Li Z, Wang F, et al. A novel multi-epitope vaccine from MMSA-1 and DKK1 for multiple myeloma immunotherapy. Br J Haematol. (2017) 178:413–26. doi: 10.1111/bjh.14686
2. Leaf RK, Stroopinsky D, Pyzer AR, Kruisbeek AM, van Wetering S, Washington A, et al. DCOne as an allogeneic cell-based vaccine for multiple myeloma. J Immunother. (2017) 40:315–22. doi: 10.1097/CJI.0000000000000185
3. Kaczmarek M, Poznanska J, Fechner F, Michalska N, Paszkowska S, Napierala A, et al. Cancer vaccine therapeutics: limitations and effectiveness-A literature review. Cells. (2023) 12:2159. doi: 10.3390/cells12172159
4. Paston SJ, Brentville VA, Symonds P, Durrant LG. Cancer vaccines, adjuvants, and delivery systems. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.627932
5. Buonaguro L, Tagliamonte M. Selecting target antigens for cancer vaccine development. Vaccines-Basel. (2020) 8:615. doi: 10.3390/vaccines8040615
6. Fan T, Zhang MN, Yang JX, Zhu ZA, Cao WL, Dong CY. Therapeutic cancer vaccines: advancements, challenges, and prospects. Signal Transduct Target Ther. (2023) 8:450. doi: 10.1038/s41392-023-01674-3
7. Janes ME, Gottlieb AP, Park KS, Zhao ZM, Mitragotri S. Cancer vaccines in the clinic. Bioeng Transl Med. (2024) 9:e10588. doi: 10.1002/btm2.10588
8. McCann KJ, Godeseth R, Chudley L, Mander A, Di Genova G, Lloyd-Evans P, et al. Idiotypic DNA vaccination for the treatment of multiple myeloma: safety and immunogenicity in a phase I clinical study. Cancer Immunol Immunother. (2015) 64:1021–32. doi: 10.1007/s00262-015-1703-7
9. Kwak LW, Taub DD, Duffey PL, Bensinger WI, Bryant EM, Reynolds CW, et al. Transfer of myeloma idiotype-specific immunity from an actively immunised marrow donor. Lancet. (1995) 345:1016–20. doi: 10.1016/S0140-6736(95)90757-2
10. Bergenbrant S, Yi Q, Osterborg A, Bjorkholm M, Osby E, Mellstedt H, et al. Modulation of anti-idiotypic immune response by immunization with the autologous M-component protein in multiple myeloma patients. Br J Haematol. (1996) 92:840–6. doi: 10.1046/j.1365-2141.1996.419959.x
11. Osterborg A, Henriksson L, Mellstedt H. Idiotype immunity (natural and vaccine-induced) in early stage multiple myeloma. Acta Oncol. (2000) 39:797–800. doi: 10.1080/028418600750063523
12. Massaia M, Borrione P, Battaglio S, Mariani S, Beggiato E, Napoli P, et al. Idiotype vaccination in human myeloma: generation of tumor-specific immune responses after high-dose chemotherapy. Blood. (1999) 94:673–83. doi: 10.1182/blood.V94.2.673
13. Roehnisch T, Then C, Nagel W, Blumenthal C, Braciak T, Donzeau M, et al. Phage idiotype vaccination: first phase I/II clinical trial in patients with multiple myeloma. J Transl Med. (2014) 12:119. doi: 10.1186/1479-5876-12-119
14. Hansson L, Abdalla AO, Moshfegh A, Choudhury A, Rabbani H, Nilsson B, et al. Long-term idiotype vaccination combined with interleukin-12 (IL-12), or IL-12 and granulocyte macrophage colony-stimulating factor, in early-stage multiple myeloma patients. Clin Cancer Res. (2007) 13:1503–10. doi: 10.1158/1078-0432.CCR-06-1603
15. Osterborg A, Yi Q, Henriksson L, Fagerberg J, Bergenbrant S, Jeddi-Tehrani M, et al. Idiotype immunization combined with granulocyte-macrophage colony-stimulating factor in myeloma patients induced type I, major histocompatibility complex-restricted, CD8- and CD4-specific T-cell responses. Blood. (1998) 91:2459–66. doi: 10.1182/blood.V91.7.2459
16. Coscia M, Mariani S, Battaglio S, Di Bello C, Fiore F, Foglietta M, et al. Long-term follow-up of idiotype vaccination in human myeloma as a maintenance therapy after high-dose chemotherapy. Leukemia. (2004) 18:139–45. doi: 10.1038/sj.leu.2403181
17. Qazilbash MH, Saini NY, Cha SC, Wang Z, Stadtmauer EA, Baladandayuthapani V, et al. A randomized phase 2 trial of idiotype vaccination and adoptive autologous T-cell transfer in patients with multiple myeloma. Blood. (2022) 139:1289–301. doi: 10.1182/blood.2020008493
18. Fu C, Zhou L, Mi QS, Jiang A. DC-based vaccines for cancer immunotherapy. Vaccines (Basel). (2020) 8:706. doi: 10.3390/vaccines8040706
19. Reichardt VL, Okada CY, Liso A, Benike CJ, Stockerl-Goldstein KE, Engleman EG, et al. Idiotype vaccination using dendritic cells after autologous peripheral blood stem cell transplantation for multiple myeloma–a feasibility study. Blood. (1999) 93:2411–9. doi: 10.1182/blood.V93.7.2411
20. Liso A, Stockerl-Goldstein KE, Auffermann-Gretzinger S, Benike CJ, Reichardt V, van Beckhoven A, et al. Idiotype vaccination using dendritic cells after autologous peripheral blood progenitor cell transplantation for multiple myeloma. Biol Blood Marrow Transplant. (2000) 6:621–7. doi: 10.1016/S1083-8791(00)70027-9
21. Cull G, Durrant L, Stainer C, Haynes A, Russell N. Generation of anti-idiotype immune responses following vaccination with idiotype-protein pulsed dendritic cells in myeloma. Br J Haematol. (1999) 107:648–55. doi: 10.1046/j.1365-2141.1999.01735.x
22. Lim SH, Bailey-Wood R. Idiotypic protein-pulsed dendritic cell vaccination in multiple myeloma. Int J Cancer. (1999) 83:215–22. doi: 10.1002/(ISSN)1097-0215
23. Titzer S, Christensen O, Manzke O, Tesch H, Wolf J, Emmerich B, et al. Vaccination of multiple myeloma patients with idiotype-pulsed dendritic cells: immunological and clinical aspects. Br J Haematol. (2000) 108:805–16. doi: 10.1046/j.1365-2141.2000.01958.x
24. Bendandi M, Rodriguez-Calvillo M, Inoges S, Lopez-Diaz de Cerio A, Perez-Simon JA, Rodriguez-Caballero A, et al. Combined vaccination with idiotype-pulsed allogeneic dendritic cells and soluble protein idiotype for multiple myeloma patients relapsing after reduced-intensity conditioning allogeneic stem cell transplantation. Leuk Lymphoma. (2006) 47:29–37. doi: 10.1080/10428190500272473
25. Curti A, Tosi P, Comoli P, Terragna C, Ferri E, Cellini C, et al. Phase I/II clinical trial of sequential subcutaneous and intravenous delivery of dendritic cell vaccination for refractory multiple myeloma using patient-specific tumour idiotype protein or idiotype (VDJ)-derived class I-restricted peptides. Br J Haematol. (2007) 139:415–24. doi: 10.1111/j.1365-2141.2007.06832.x
26. Zahradova L, Mollova K, Ocadlikova D, Kovarova L, Adam Z, Krejci M, et al. Efficacy and safety of Id-protein-loaded dendritic cell vaccine in patients with multiple myeloma–phase II study results. Neoplasma. (2012) 59:440–9. doi: 10.4149/neo_2012_057
27. Reichardt VL, Milazzo C, Brugger W, Einsele H, Kanz L, Brossart P. Idiotype vaccination of multiple myeloma patients using monocyte-derived dendritic cells. Haematologica. (2003) 88:1139–49. doi: 10.3324/%x
28. Yi Q, Desikan R, Barlogie B, Munshi N. Optimizing dendritic cell-based immunotherapy in multiple myeloma. Br J Haematol. (2002) 117:297–305. doi: 10.1046/j.1365-2141.2002.03411.x
29. Yi Q, Szmania S, Freeman J, Qian J, Rosen NA, Viswamitra S, et al. Optimizing dendritic cell-based immunotherapy in multiple myeloma: intranodal injections of idiotype-pulsed CD40 ligand-matured vaccines led to induction of type-1 and cytotoxic T-cell immune responses in patients. Br J Haematol. (2010) 150:554–64. doi: 10.1111/j.1365-2141.2010.08286.x
30. Rollig C, Schmidt C, Bornhauser M, Ehninger G, Schmitz M, Auffermann-Gretzinger S. Induction of cellular immune responses in patients with stage-I multiple myeloma after vaccination with autologous idiotype-pulsed dendritic cells. J Immunother. (2011) 34:100–6. doi: 10.1097/CJI.0b013e3181facf48
31. Wen YJ, Ling M, Bailey-Wood R, Lim SH. Idiotypic protein-pulsed adherent peripheral blood mononuclear cell-derived dendritic cells prime immune system in multiple myeloma. Clin Cancer Res. (1998) 4:957–62.
32. Carmon L, Avivi I, Kovjazin R, Zuckerman T, Dray L, Gatt ME, et al. Phase I/II study exploring ImMucin, a pan-major histocompatibility complex, anti-MUC1 signal peptide vaccine, in multiple myeloma patients. Br J Haematol. (2015) 169:44–56. doi: 10.1111/bjh.13245
33. Schmitt M, Schmitt A, Rojewski MT, Chen J, Giannopoulos K, Fei F, et al. RHAMM-R3 peptide vaccination in patients with acute myeloid leukemia, myelodysplastic syndrome, and multiple myeloma elicits immunologic and clinical responses. Blood. (2008) 111:1357–65. doi: 10.1182/blood-2007-07-099366
34. Greiner J, Schmitt A, Giannopoulos K, Rojewski MT, Gotz M, Funk I, et al. High-dose RHAMM-R3 peptide vaccination for patients with acute myeloid leukemia, myelodysplastic syndrome and multiple myeloma. Haematologica. (2010) 95:1191–7. doi: 10.3324/haematol.2009.014704
35. Jorgensen NG, Ahmad SM, Abildgaard N, Straten PT, Svane IM, Andersen MH, et al. Peptide vaccination against multiple myeloma using peptides derived from anti-apoptotic proteins: a phase I trial. Stem Cell Investig. (2016) 3:95. doi: 10.21037/sci
36. Tsuboi A, Oka Y, Nakajima H, Fukuda Y, Elisseeva OA, Yoshihara S, et al. Wilms tumor gene WT1 peptide-based immunotherapy induced a minimal response in a patient with advanced therapy-resistant multiple myeloma. Int J Hematol. (2007) 86:414–7. doi: 10.1007/BF02983998
37. Nooka AK, Wang ML, Yee AJ, Kaufman JL, Bae J, Peterkin D, et al. Assessment of safety and immunogenicity of PVX-410 vaccine with or without lenalidomide in patients with smoldering multiple myeloma: A nonrandomized clinical trial. JAMA Oncol. (2018) 4:e183267. doi: 10.1001/jamaoncol.2018.3267
38. Szmania S, Gnjatic S, Tricot G, Stone K, Zhan F, Moreno A, et al. Immunization with a recombinant MAGE-A3 protein after high-dose therapy for myeloma. J Immunother. (2007) 30:847–54. doi: 10.1097/CJI.0b013e318158fcff
39. Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Xu YY, Kalos M, et al. Combination immunotherapy after ASCT for multiple myeloma using MAGE-A3/Poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin Cancer Res. (2014) 20:1355–65. doi: 10.1158/1078-0432.CCR-13-2817
40. Cohen AD, Lendvai N, Nataraj S, Imai N, Jungbluth AA, Tsakos I, et al. Autologous lymphocyte infusion supports tumor antigen vaccine–induced immunity in autologous stem cell transplant for multiple myeloma. Cancer Immunol Res. (2019) 7:658–69. doi: 10.1158/2326-6066.CIR-18-0198
41. Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Fang HB, Cai L, et al. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood. (2011) 117:788–97. doi: 10.1182/blood-2010-08-299396
42. Hobo W, Strobbe L, Maas F, Fredrix H, Greupink-Draaisma A, Esendam B, et al. Immunogenicity of dendritic cells pulsed with MAGE3, Survivin and B-cell maturation antigen mRNA for vaccination of multiple myeloma patients. Cancer Immunol Immunother. (2013) 62:1381–92. doi: 10.1007/s00262-013-1438-2
43. Chung DJ, Sharma S, Rangesa M, DeWolf S, Elhanati Y, Perica K, et al. Langerhans dendritic cell vaccine bearing mRNA-encoded tumor antigens induces antimyeloma immunity after autotransplant. Blood Adv. (2022) 6:1547–58. doi: 10.1182/bloodadvances.2021005941
44. Freeman CL, Atkins R, Varadarajan I, Menges M, Edelman J, Baz R, et al. Survivin dendritic cell vaccine safely induces immune responses and is associated with durable disease control after autologous transplant in patients with myeloma. Clin Cancer Res. (2023) 29:4575–85. doi: 10.1158/1078-0432.CCR-22-3987
45. Li Z, Oka Y, Tsuboi A, Fujiki F, Harada Y, Nakajima H, et al. Identification of a WT1 protein-derived peptide, WT1, as a HLA-A 0206-restricted, WT1-specific CTL epitope. Microbiol Immunol. (2008) 52:551–8. doi: 10.1111/j.1348-0421.2008.00069.x
46. Hatta Y, Takeuchi J, Saitoh T, Itoh T, Ishizuka H, Iriyama N, et al. WT1 expression level and clinical factors in multiple myeloma. J Exp Clin Cancer Res. (2005) 24:595–9.
47. Qian J, Xie J, Hong S, Yang J, Zhang L, Han X, et al. Dickkopf-1 (DKK1) is a widely expressed and potent tumor-associated antigen in multiple myeloma. Blood. (2007) 110:1587–94. doi: 10.1182/blood-2007-03-082529
48. Qian J, Hong S, Wang S, Zhang L, Sun L, Wang M, et al. Myeloma cell line–derived, pooled heat shock proteins as a universal vaccine for immunotherapy of multiple myeloma. Blood. (2009) 114:3880–9. doi: 10.1182/blood-2009-06-227355
49. Zaleska J, Giannopoulos K. ImImmunotherapeutical approaches for multiple myeloma. Acta Haematol Polonica. (2012) 43:68–74. doi: 10.1016/S0001-5814(12)31007-4
50. Andrade VC, Vettore AL, Felix RS, Almeida MS, Carvalho F, Oliveira JS, et al. Prognostic impact of cancer/testis antigen expression in advanced stage multiple myeloma patients. Cancer Immun. (2008) 8:2.
51. Szmania S, Tricot G, van Rhee F. NY-ESO-1 immunotherapy for multiple myeloma. Leuk Lymphoma. (2006) 47:2037–48. doi: 10.1080/10428190600742292
52. van Rhee F, Szmania SM, Zhan F, Gupta SK, Pomtree M, Lin P, et al. NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood. (2005) 105:3939–44. doi: 10.1182/blood-2004-09-3707
53. Wang H, Huang W, Gao H, Liu TT. NY-ESO-1 protein vaccine combining alum, cpG ODN, and HH2 complex adjuvant induces protective and therapeutic anti-tumor responses in murine multiple myeloma. Onco Targets Ther. (2020) 13:8069–77. doi: 10.2147/OTT.S255713
54. de Carvalho F, Vettore AL, Inaoka RJ, Karia B, Andrade VC, Gnjatic S, et al. Evaluation of LAGE-1 and NY-ESO-1 expression in multiple myeloma patients to explore possible benefits of their homology for immunotherapy. Cancer Immun. (2011) 11:1. doi: 10.1158/1424-9634.DCL-1.11.1
55. Jungbluth AA, Ely S, DiLiberto M, Niesvizky R, Williamson B, Frosina D, et al. The cancer-testis antigens CT7 (MAGE-C1) and MAGE-A3/6 are commonly expressed in multiple myeloma and correlate with plasma-cell proliferation. Blood. (2005) 106:167–74. doi: 10.1182/blood-2004-12-4931
56. Anderson LD Jr., Cook DR, Yamamoto TN, Berger C, Maloney DG, Riddell SR. Identification of MAGE-C1 (CT-7) epitopes for T-cell therapy of multiple myeloma. Cancer Immunol Immunother. (2011) 60:985–97. doi: 10.1007/s00262-011-1009-3
57. Wang Z, Zhang Y, Liu H, Salati E, Chiriva-Internati M, Lim SH. Gene expression and immunologic consequence of SPAN-Xb in myeloma and other hematologic Malignancies. Blood. (2003) 101:955–60. doi: 10.1182/blood-2002-06-1930
58. Frank C, Hundemer M, Ho AD, Goldschmidt H, Witzens-Harig M. Cellular immune responses against the cancer-testis antigen SPAN-XB in healthy donors and patients with multiple myeloma. Leuk Lymphoma. (2008) 49:779–85. doi: 10.1080/10428190801911688
59. Chiriva-Internati M, Wang Z, Salati E, Bumm K, Barlogie B, Lim SH. Sperm protein 17 (Sp17) is a suitable target for immunotherapy of multiple myeloma. Blood. (2002) 100:961–5. doi: 10.1182/blood-2002-02-0408
60. Thalmensi J, Pliquet E, Liard C, Escande M, Bestetti T, Julithe M, et al. Anticancer DNA vaccine based on human telomerase reverse transcriptase generates a strong and specific T cell immune response. Oncoimmunology. (2016) 5:e1083670. doi: 10.1080/2162402X.2015.1083670
61. Xie N, Shen G, Gao W, Huang Z, Huang C, Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduct Target Ther. (2023) 8:9. doi: 10.1038/s41392-022-01270-x
62. Zhang L, Xiang Y, Li Y, Zhang J. Gut microbiome in multiple myeloma: Mechanisms of progression and clinical applications. Front Immunol. (2022) 13:1058272. doi: 10.3389/fimmu.2022.1058272
63. Perumal D, Imai N, Lagana A, Finnigan J, Melnekoff D, Leshchenko VV, et al. Mutation-derived neoantigen-specific T-cell responses in multiple myeloma. Clin Cancer Res. (2020) 26:450–64. doi: 10.1158/1078-0432.CCR-19-2309
64. Shirazi F, Jones RJ, Singh RK, Zou J, Kuiatse I, Berkova Z, et al. Activating KRAS, NRAS, and BRAF mutants enhance proteasome capacity and reduce endoplasmic reticulum stress in multiple myeloma. Proc Natl Acad Sci USA. (2020) 117:20004–14. doi: 10.1073/pnas.2005052117
65. Monsen H, Norseth HM, Remen N, Abrahamsen I, Lysén A, Schjesvold F. Pb2118: tg01-study: A phase 1/2 study with tg01 immunotherapy vaccination. Hemasphere. (2023) 7:e51467aa. doi: 10.1097/01.HS9.0000975244.51467.aa
66. Marron TU, Saxena M, Bhardwaj N, Meseck M, Rubinsteyn A, Finnigan J, et al. Abstract LB048: An adjuvant personalized neoantigen peptide vaccine for the treatment of Malignancies (PGV-001). Cancer Res. (2021) 81:LB048–LB. doi: 10.1158/1538-7445.AM2021-LB048
67. Miller A, Asmann Y, Cattaneo L, Braggio E, Keats J, Auclair D, et al. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. (2017) 7:e612. doi: 10.1038/bcj.2017.94
68. Samur MK, Roncador M, Aktas Samur A, Fulciniti M, Bazarbachi AH, Szalat R, et al. High-dose melphalan treatment significantly increases mutational burden at relapse in multiple myeloma. Blood. (2023) 141:1724–36. doi: 10.1182/blood.2022017094
69. Rosenblatt J, Vasir B, Uhl L, Blotta S, Macnamara C, Somaiya P, et al. Vaccination with dendritic cell/tumor fusion cells results in cellular and humoral antitumor immune responses in patients with multiple myeloma. Blood. (2011) 117:393–402. doi: 10.1182/blood-2010-04-277137
70. Rosenblatt J, Avivi I, Vasir B, Uhl L, Munshi NC, Katz T, et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin Cancer Res. (2013) 19:3640–8. doi: 10.1158/1078-0432.CCR-13-0282
71. Chung DJ, Shah N, Wu J, Logan B, Bisharat L, Callander N, et al. Randomized phase II trial of dendritic cell/myeloma fusion vaccine with lenalidomide maintenance after upfront autologous hematopoietic cell transplantation for multiple myeloma: BMT CTN 1401. Clin Cancer Res. (2023) 29:4784–96. doi: 10.1158/1078-0432.CCR-23-0235
72. Rosenblatt J, Avivi I, Binyamini N, Uhl L, Somaiya P, Stroopinsky D, et al. Blockade of PD-1 in combination with dendritic cell/myeloma fusion cell vaccination following autologous stem cell transplantation is well tolerated, induces anti-tumor immunity and may lead to eradication of measureable disease. Blood. (2015) 126:4218–. doi: 10.1182/blood.V126.23.4218.4218
73. Ohmine K, Uchibori R. Novel immunotherapies in multiple myeloma. Int J Hematol. (2022) 115:799–810. doi: 10.1007/s12185-022-03365-1
74. Biavati L, Huff CA, Ferguson A, Sidorski A, Stevens MA, Rudraraju L, et al. An allogeneic multiple myeloma GM-CSF-secreting vaccine with lenalidomide induces long-term immunity and durable clinical responses in patients in near complete remission. Clin Cancer Res. (2021) 27:6696–708. doi: 10.1158/1078-0432.CCR-21-1916
75. Salem ML. The use of dendritic cells for peptide-based vaccination in cancer immunotherapy. Methods Mol Biol. (2014) 1139:479–503. doi: 10.1007/978-1-4939-0345-0_37
76. Shinde P, Fernandes S, Melinkeri S, Kale V, Limaye L. Compromised functionality of monocyte-derived dendritic cells in multiple myeloma patients may limit their use in cancer immunotherapy. Sci Rep. (2018) 8:5705. doi: 10.1038/s41598-018-23943-w
77. Vavassori V, Ferrari S, Beretta S, Asperti C, Albano L, Annoni A, et al. Lipid nanoparticles allow efficient and harmless ex vivo gene editing of human hematopoietic cells. Blood. (2023) 142:812–26. doi: 10.1182/blood.2022019333
78. Cornel AM, van Til NP, Boelens JJ, Nierkens S. Strategies to genetically modulate dendritic cells to potentiate anti-tumor responses in hematologic Malignancies. Front Immunol. (2018) 9:982. doi: 10.3389/fimmu.2018.00982
79. Wang BL, Pei JL, Xu SN, Liu J, Yu JM. Recent advances in mRNA cancer vaccines: meeting challenges and embracing opportunities. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1246682
80. Yao RH, Xie CY, Xia XJ. Recent progress in mRNA cancer vaccines. Hum Vacc Immunother. (2024) 20:2307187. doi: 10.1080/21645515.2024.2307187
81. Rhee F. Idiotype vaccination strategies in myeloma: how to overcome a dysfunctional immune system. Clin Cancer Res. (2007) 13:1353–5. doi: 10.1158/1078-0432.CCR-06-2650
82. Soekojo CY, Ooi M, de Mel S, Chng WJ. Immunotherapy in multiple myeloma. Cells. (2020) 9:601. doi: 10.3390/cells9030601
83. Uckun FM. Overcoming the immunosuppressive tumor microenvironment in multiple myeloma. Cancers (Basel). (2021) 13:2018. doi: 10.3390/cancers13092018
84. Neumeister P, Schulz E, Pansy K, Szmyra M, Deutsch AJ. Targeting the microenvironment for treating multiple myeloma. Int J Mol Sci. (2022) 23:7627. doi: 10.3390/ijms23147627
85. Gravekamp C, Chandra D. Aging and cancer vaccines. Crit Rev Oncog. (2013) 18:585–95. doi: 10.1615/CritRevOncog.v18.i6
86. Calcinotto A, Brevi A, Chesi M, Ferrarese R, Garcia Perez L, Grioni M, et al. Microbiota-driven interleukin-17-producing cells and eosinophils synergize to accelerate multiple myeloma progression. Nat Commun. (2018) 9:4832. doi: 10.1038/s41467-018-07305-8
87. Boussi LS, Avigan ZM, Rosenblatt J. Immunotherapy for the treatment of multiple myeloma. Front Immunol. (2022) 13:1027385. doi: 10.3389/fimmu.2022.1027385
Keywords: multiple myeloma (MM), immunotherapy, vaccine, tumor-associated antigen (TAA), tumor-specific antigen (TSA), idiotype (Id)
Citation: Abdollahi P, Norseth HM and Schjesvold F (2024) Advances and challenges in anti-cancer vaccines for multiple myeloma. Front. Immunol. 15:1411352. doi: 10.3389/fimmu.2024.1411352
Received: 02 April 2024; Accepted: 17 July 2024;
Published: 01 August 2024.
Edited by:
Rena Feinman, Hackensack Meridian Health, United StatesReviewed by:
Rafael Gongora, University of Salamanca, SpainCansu Cimen Bozkus, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2024 Abdollahi, Norseth and Schjesvold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pegah Abdollahi, cGVnYWJkQG91cy1oZi5ubw==