 Yumeng Wang
Yumeng Wang Xintian Shou2†*
Xintian Shou2†*- 1Department of Traditional Chinese Medicine, Beijing Tsinghua Changgung Hospital, School of Clinical Medicine, Tsinghua University, Beijing, China
- 2Cardiovascular Diseases Center, Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 3Department of Cardiovascular, Dongfang Hospital, Beijing University of Chinese Medicine, Beijing, China
Ischemic heart disease (IHD) can trigger responses from the innate immune system, provoke aseptic inflammatory processes, and result in the recruitment and accumulation of neutrophils. Excessive recruitment of neutrophils is a potential driver of persistent cardiac inflammation. Once recruited, neutrophils are capable of secreting a plethora of inflammatory and chemotactic agents that intensify the inflammatory cascade. Additionally, neutrophils may obstruct microvasculature within the inflamed region, further augmenting myocardial injury in the context of IHD. Immune-related molecules mediate the recruitment process of neutrophils, such as immune receptors and ligands, immune active molecules, and immunocytes. Non-immune-related molecular pathways represented by pro-resolving lipid mediators are also involved in the regulation of NR. Finally, we discuss novel regulating strategies, including targeted intervention, agents, and phytochemical strategies. This review describes in as much detail as possible the upstream molecular mechanism and external intervention strategies for regulating NR, which represents a promising therapeutic avenue for IHD.
1 Introduction
Neutrophils, representing the predominant fraction of granulocytes in the majority of mammalian species, serve an integral function within the innate immune defense mechanism. They are short-lived, nonspecific, and highly mobile, enabling entry into the tissue where other cells/molecules cannot invade. Aseptic inflammation is the main response of innate immunity and the necessary condition to trigger and drive vascular diseases. After the onset of aseptic inflammation, neutrophils are the first leukocytes to be recruited to the site of inflammation and subsequently promote the immune response (1). A moderate immune response coordinates a beneficial dynamic homeostatic response and contributes to tissue repair, while an exaggerated response can cause additional damage. Ischemic heart diseases (IHD), such as myocardial infarction (MI), myocardial ischemia-reperfusion(I/R) injury, and heart failure (HF), can initiate innate immune responses, induce aseptic inflammation (2), and culminate in the mobilization and proliferation of neutrophils (3).
Neutrophil recruitment (NR) is pivotal in mediating the aseptic inflammatory response associated with IHD (3). Pathological recruitment of neutrophils is triggered by changes in the surface of endothelial cells caused by inflammatory mediator (including histamine, cysteinyl leukotrienes, and cytokines) stimulation (4–7). Once recruited, neutrophils can release various inflammatory and chemotactic mediators that cause cell damage and in turn further facilitate the recruitment process; it can also block small blood vessels at the site of inflammation, thereby impeding blood flow (8, 9). Animal studies and clinical studies have demonstrated that increased neutrophil counts correlate with the severity of coronary artery damage in patients with coronary artery disease (CAD) (10, 11). Neutrophil-to-leukocyte ratio (NLR) has been shown to be an independent predictor of outcome in patients with stable CAD, as well as a predictor of short- and long-term mortality in patients with acute coronary syndrome (ACS), ST-segment elevation myocardial infarction (STEMI), and heart transplantation (12–14). Consequently, the recruitment and subsequent accumulation of neutrophils may serve as key determinants of sustained cardiac inflammation in the context of IHD. Therapeutic modulation and inhibition of these processes may prove beneficial in attenuating myocardial inflammation and impeding the advancement of IHD pathology.
Recent studies have predominantly directed attention toward elucidating the global contribution of neutrophils to ischemic heart disease pathophysiology. In contrast, the mechanisms governing neutrophil recruitment have been comparatively underexplored, largely attributable to the pronounced heterogeneity in initiating signals for neutrophil recruitment across diverse tissue (15, 16). In recent years, the upstream molecular mechanism and external intervention strategies for regulating NR have attracted the attention of many scholars, and many satisfactory results have been obtained. However, the findings have not been systematically summarized. This paper intends to summarize the specific process and influence of NR in IHD, as well as the molecular mechanism of regulating NR and effective intervention measures, in order to provide ideas for the study of ischemic heart disease from this perspective.
2 Cascade reactions of NR in IHD
NR is considered to be a series of cascade reactions: tethering, rolling, adhesion, crawling, and transmigration. When cardiac ischemia occurs, aseptic inflammation induces inflammatory mediators to stimulate endothelial cells. P-selectin and E-selectin bind to their glycosylated ligands, allowing neutrophils to be tethered and trapped in fast-flowing blood and subsequently roll with the flow along the vascular endothelium. Chemokines activate neutrophils by binding to their receptors. Then, neutrophils express integrins (LFA1 and MAC1) binding to endothelial surface adhesion molecule (ICAM-1/ICAM-2), promoting stable adhesion of neutrophils to vascular endothelial cells. Finally, neutrophils crawl at the endothelial cell-cell junction, release protease to digest the endothelial basement membrane (EBM), and complete vertical transmigration through paracellular (between endothelial cells) and transcellular (through endothelial cells). The process of transmigration also requires the involvement of integrins, CAM, and multiple connexins.
3 Role of NR in IHD
3.1 Atherosclerosis
Neutrophils are the main cell group interacting with atherosclerotic endothelium, as well as the main cell component of atherosclerotic lesions. Clinical and experimental investigations have consistently demonstrated a marked presence of neutrophils within aortic plaques, particularly concentrated in the shoulder regions where inflammatory activity is notably heightened (17, 18). NR plays an important role in atherosclerosis and plaque rupture. First, neutrophils recruited in the plaque will release granular protein, which will be deposited in endothelial cells and vascular walls to adhere to monocytes, activate macrophages, release proinflammatory factors, and thus aggravate the inflammatory response. Secondly, the proteolytic enzyme released by granular protein can weaken the fibrous cap and promote plaque rupture. Finally, myeloperase (MPO) released by granular protein induces vascular endothelial cell apoptosis by reducing the utilization of NO, leading to plaque erosion (19). In addition, cholesterol crystals in plaques also trigger neutrophils to release neutrophil extracellular traps (NETs), which indirectly promote NR in atherosclerotic plaques by promoting the release of macrophages and activating T-helper 17 (TH17) cells (Figure 1).
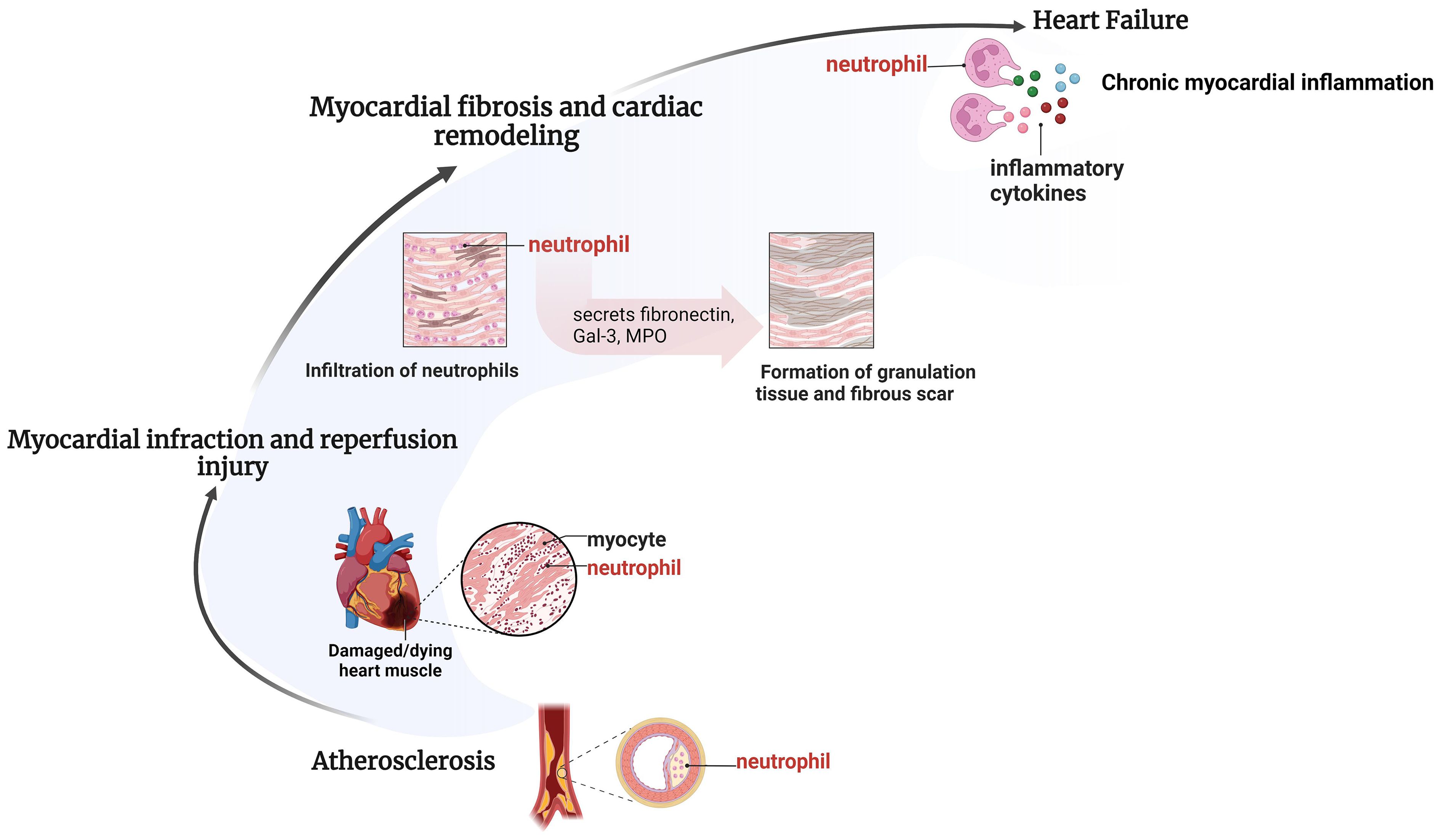
Figure 1 Pathological processes of different IHD involving neutrophils. Neutrophil recruitment takes place in various pathological processes of different IHD. Neutrophil recruitment enhances atherosclerotic plaque formation, exacerbates inflammatory responses and myocardial injury during myocardial infarction and reperfusion injury, speeds up myocardial fibrosis and ventricular remodeling, resulting in persistent chronic inflammation and facilitating the onset of late-stage heart failure. Created by BioRender.
3.2 Myocardial infarction
Myocardial infarction can quickly activate the innate immune pathway, and trigger a strong inflammatory response in the early stage. The necrosis of myocardial cells and stroma will promote the occurrence of damage related molecule pattern (DAMP) (20), which can be recognized by toll-like receptor (TLR), and promote the activation of innate immune cells and inflammation (21). The inflammatory response caused a marked upregulation of chemokines in the infarcted myocardium, which facilitated the onset of the NR. Meanwhile, ROS mediated by myocardial ischemia can also promote NR. The process of neutrophil recruitment changed with time after myocardial infarction. Histological examination showed that neutrophils appeared 12–24 hours after the ischemic attack and reached the peak on the third day, which suggested that neutrophils were recruited most in the early stage (1–3 days) after myocardial infarction and were mainly N1 polarized proinflammatory neutrophils. From the 5th to the 7th day, the proportion of N2 polarized anti-inflammatory neutrophils increased (22, 23). After 7 days, neutrophils began to disappear from the infarcted area, and lymphocytes and monocytes gradually increased (24). It should be noted that the transmigration of neutrophils through the endothelium, the core step of NR, occurs in both ischemic and border regions.
3.3 Myocardial ischemia-reperfusion injury
Neutrophils are the main type of inflammatory cells in myocardial I/R injury. The pathological mechanism of NR in myocardial I/R injury is consistent with that of myocardial infarction, as both involve aseptic inflammatory reactions triggered by the activation of innate immune pathways. Myocardial ischemia mainly affects the myocardial tissue in the central necrotic area of the occluded coronary artery perfusion area. The process of reperfusion limits the infarct size of the central necrotic area but starts molecular events that lead to lethal reperfusion injury in the border area (25). After permanent coronary artery occlusion, neutrophil infiltration is mainly limited to the edge of the infarcted area. The process of reperfusion changes the distribution of neutrophils, making them recruit in the infarcted area of the myocardium (24). Neutrophils infiltrate into the infarcted area, producing a large number of reactive oxygen species (ROS) and proteolytic enzymes (26), aggravating the damage of myocardial tissue, which in turn maximally recruited and activated more neutrophils, forming a vicious circle.
3.4 Heart failure
Acute inflammation is an essential mechanism for heart repair and defense after injury. Systemic inflammation has been recognized as a pathobiological feature of acute and chronic heart failure, associated with the development, progression, and complications of HF, and predicted adverse outcomes independently of other clinical parameters such as left ventricular ejection fraction (27). The timely clearance of neutrophils is the first critical step in the regression of inflammation during the healing process after MI-induced myocardial injury. The production of locally endogenous peptides/bioactive lipids by neutrophil terminates NR, which is a marker of inflammation resolution. However, if neutrophils are not cleared on time and remain in a state of continuous recruitment, chronic inflammation and advanced heart failure will occur (28). The innate immune system is active in patients with chronic heart failure, accompanied by a persistent chronic inflammatory response. Neutrophils recruited into the myocardium release more pro-inflammatory factors than anti-inflammatory factors, which can further exacerbate myocardial damage and cardiac decline. It has been shown that higher neutrophil levels in the first 12 hours after acute myocardial infarction predict the development of chronic HF (29).
3.5 Myocardial fibrosis and cardiac remodeling
Inflammation plays a key role in poor ventricular remodeling and decreased cardiac function after injury (30). Overactive inflammatory signaling can aggravate myocardial damage and lead to poor cardiac remodeling. The N2-type neutrophils express anti-inflammatory molecules in the late stage of cardiac remodeling, and their proportion increases continuously after myocardial infarction, reaching nearly 20% on the 7th day (23). On day 3 of myocardial infarction, neutrophils initiate apoptosis, reduce inflammatory signaling, and begin to help ECM reorganize. Neutrophils recruited to the infarct area remove dead cells and matrix debris by phagocytosis in preparation for scarring in the area. On days 5 to 7 of myocardial infarction, neutrophils produce ECM proteins needed for scarring, including fibronectin, Gal-3, and fibrinogen (31). In addition, the heme MPO released by the primary granules of neutrophils can promote the deposition of myocardial collagen in the atrium and ventricle, which is the key medium for myocardial remodeling. Recruitment of neutrophils leads to increased concentrations of MPO, and the oxidant produced by MPO can lead to impaired myocardial function and poor ventricular remodeling after myocardial infarction (32, 33). Therefore, over recruitment of neutrophils leads to excessive myocardial collagen deposition and ECM remodeling, leading to the formation of mature scars, promoting myocardial fibrosis, and compromising organ compliance (Figure 1).
4 Molecular mechanisms regulating NR in IHD
4.1 Immune-related molecular mechanisms
4.1.1 Immune receptors and ligands
Immune receptors and their corresponding ligands are instrumental in mediating the homing of neutrophils to ischemic myocardial tissue. For example, transmembrane protein receptor dectin-1 can promote the expression of chemokine CXCL1 and granulocyte colony-stimulating factor (G-CSF) in macrophages through the spleen tyrosine kinase (Syk)/nuclear factor kappa-B(NF-κB) signaling pathway, and can also regulate the production of interleukin (IL) 23 and IL-1β, affecting neutrophil recruitment and myocardial I/R injury (34). The upregulation of NF-κB p65 expression in ECs increases the expression of adhesion molecules, leading to robust infiltration of neutrophils and disorganization/degradation of ECM in the myocardial ischemic border zone (35). TLR can affect signal transduction of its downstream protein myeloid differentiation primary response gene 88(MyD88) or TIR domain-containing adaptor inducing interferon (TRIF) -β, initiating neutrophil recruitment in the I/R myocardium (36). In addition, in animal models of heart failure, fibrinogen (FN) can act as a ligand for TLR2 and TLR4, promote the process of neutrophil adhesion by regulating TLR and cardiac endothelial cell adhesion molecules, and participate in neutrophil recruitment (30) (Table 1; Figure 2).
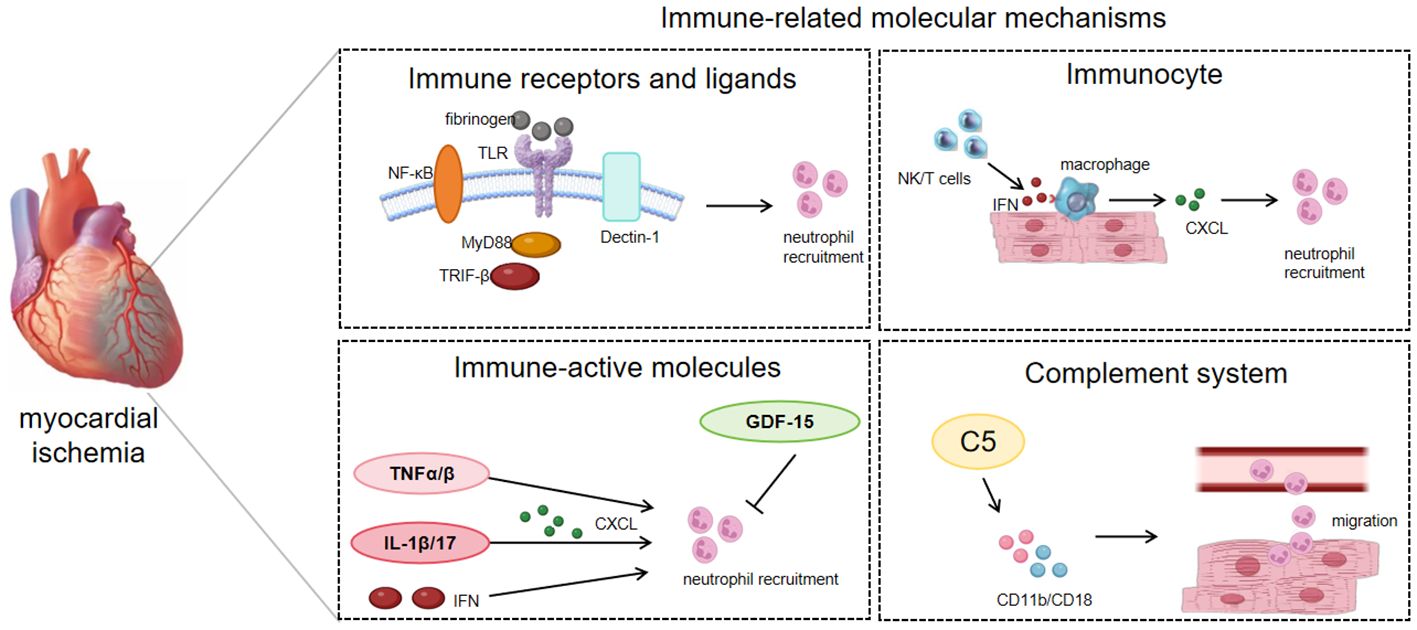
Figure 2 Immune-related regulatory mediators of NR in IHD pathophysiology.
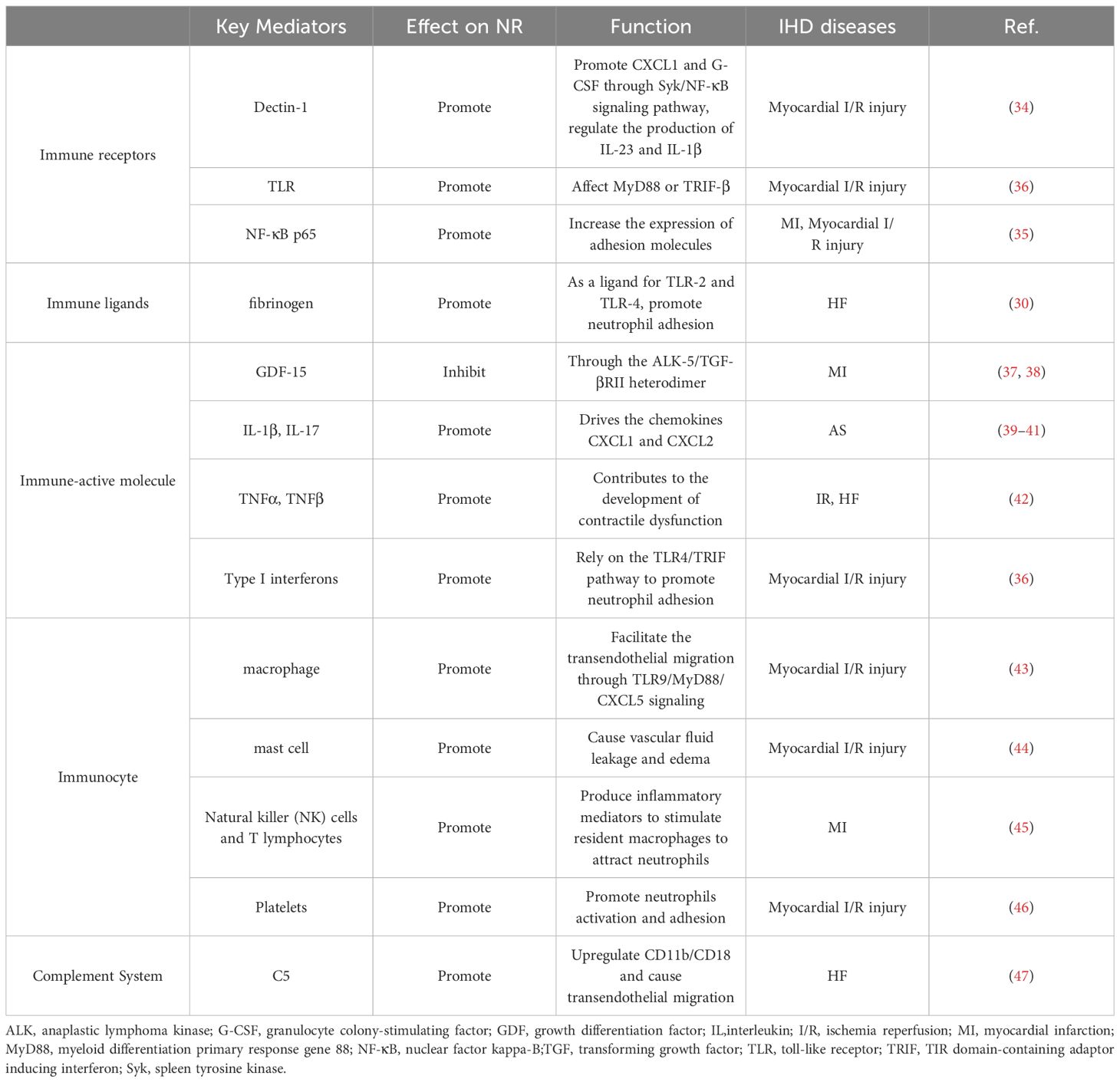
Table 1 Immune-related regulatory mediators of NR in IHD pathophysiology.
4.1.2 Immune-active molecule
Many immune-active molecules, encompassing a diverse array of cytokines, exert influence over the process of neutrophil recruitment in the context of IHD. Growth differentiation factor (GDF)-15, a transforming growth factor beta (TGF-β)-related cytokine, is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice and inhibits NR process in mice through anaplastic lymphoma kinase (ALK)-5/TGF-βRII heterodimer (37, 38). IL-1β, secreted by macrophages, upregulates the T-cell-derived cytokine IL-17, which drives the chemokines CXCL1 and CXCL2 to promote NR and amplify the inflammatory response (39). Tumor necrosis factor (TNF) α and TNFβ also enhance the activation and recruitment of neutrophils in the myocardium, thereby further damaging the myocardium (42). Type I interferons can rely on the TLR4/TRIF pathway to promote neutrophil adhesion to coronary endothelial cells, thus coordinating neutrophil recruitment to injured myocytes (36).
4.1.3 Immunocyte
Various subtypes of macrophages are primarily responsible for encouraging the recruitment of neutrophils in IHD. The cytokine interleukin-1β (IL-1β) secreted by macrophages can up-regulate the T-cell-derived cytokine IL-17, which drives the chemokines CXCL1 and CXCL2 to promote neutrophil recruitment and amplify inflammation (43). Heart-resident CCR2+ macrophages facilitate the transendothelial migration of neutrophils into injured hearts through TLR9/MyD88/CXCL5 signaling (43). In addition, the process of degranulation and mediator release of mast cells contributes to the inflammatory response, causing vascular fluid leakage and edema, and promoting NR (44). Natural killer (NK) cells and T lymphocytes produce inflammatory mediators such as IFN-γ, which stimulate resident macrophages to attract neutrophils to cardiac tissue (45). Platelets can also aggravate myocardial I/R damage by promoting neutrophil activation and adhesion (46).
4.1.4 Complement system
When myocardial ischemia occurs, inflammation stimulates complement activation and accelerates neutrophil adherence. The strong neutrophil attraction of C5a causes neutrophils to adhere firmly to endothelial cells by upregulating CD11b/CD18 (Mac-1) and causing transendothelial migration (48). In addition, C5a also stimulates neutrophils to produce superoxide, which aggravates the occurrence of oxidative stress (49). Therefore, inhibition of C5 may reduce neutrophil infiltration during myocardial I/R injury in a manner that provides cardiac protection (47) (Table 1; Figure 2).
4.2 Non-immune-related molecular mechanisms
Numerous pro-resolving lipid mediators that are produced by neutrophils in turn affect their own recruitment. For example, lipid toxin A4, a dual anti-inflammatory and pro-resolution lipid mediator, induces changes in the phosphorylation of cytoskeletal proteins, leading to inhibition of neutrophil migration (50, 51). The protectins can also play a similar role in inhibiting neutrophil infiltration (52). Annexins are a widely distributed class of calcium-dependent phospholipid binding proteins. Annexin 1 inhibits neutrophil adhesion and migration in a mitogen-activated protein kinase (MAPK)-dependent manner via formyl peptide receptor (FPR) signaling (53–55), and its mimics exhibit the same cardioprotective effect through modulation of immune cell activation in myocardial infarction (56).
Various proteins released in physiological or pathological states affect the recruitment process of neutrophils and become biomarkers of inflammatory response in IHD. Angiopoietin-2 (Angpt2), recognized as a biomarker of poor outcome in IHD, plays a critical role in MI. In the acute phase of MI, endothelial-derived Angpt2 antagonized Angpt1/Tie2 signaling, which was greatly involved in increased adhesion molecular expression, enhanced neutrophil infiltration, and intensified inflammatory response (35). S100A8∕A9, a kind of Ca2+ binding protein, has become a valuable biomarker and treatment target to detect and modulate neutrophil involvement in myocardial infarction. Studies have shown that its blockers can significantly reduce neutrophil infiltration in infarcted hearts (57). Developmental endothelial site-1 (DEL-1) is an anti-inflammatory glycoprotein whose deficiency exacerbates pressure overload-induced heart failure by promoting neutrophil infiltration and the formation of neutrophil extracellular traps (58). transcriptional co-activator yes-associated protein (YAP) reduced cardiomyocyte (CM) necrosis and neutrophil infiltration after I/R stress (59).
Some biological processes can also affect NR processes in IHD. Adipose tissue lipolysis and cardiomyocyte lipid accumulation augmented cardiac inflammation. Inhibition of adipose tissue lipolysis reduces lipid droplet accumulation, attenuates neutrophil infiltration, and improves cardiac function (60). Exocytosis is also a factor affecting NR. Studies have shown that neutrophil exocytosis inhibitors (Nexinhib20) not only help reduce the exocytosis process, but also inhibit neutrophil adhesion and activation of β2 integrin, thereby reducing neutrophil recruitment (61). By enhancing M2 macrophage-induced efferocytosis of apoptotic neutrophils, mesenchymal stem cells markedly decrease neutrophil number and improve cardiac repair after myocardial I/R in rats (62). In addition, enhancing vascular integrity and inhibiting the high permeability of cardiac microvascular endothelium (CME) contributed to reducing the expression of vascular cell adhesion molecule 1 (VCAM-1) on CME cells, leading to reduced neutrophil infiltration (63).
5 Interventions to regulate NR
5.1 Targeted intervention
Targeted inhibition of key mediators within the cascade reactions weakens NR. Both MPO-knockout models (64) and MPO inhibitors (65) lead to reduced neutrophils recruitment, and improved ventricular function and remodeling in myocardial infarction. The absence of NR adhesion molecules also inhibited NR (66). Receptor antagonists of cytokines and chemokines, such as IL-1 receptor antagonist (67) and NLRP3-inflammasome inhibitor (68), reduce the extent of neutrophil infiltration and myocardial infarction size. Targeted regulation of related proteins or genes in the molecular mechanism of NR directly inhibits the NR process. Studies have shown that ferrostatin-1 (Fer-1), as a specific inhibitor of ferroptosis, can reduce the level of pro-ferroptotic hydroperoxy-arachidonoyl-phosphatidylethanolamine, reduce cardiomyocyte apoptosis and block neutrophil recruitment (36).
Targeting adenosine helps to regulate the NR process. Adenosine A2b receptor (Adora2b), as a G-protein-coupled receptor, its activation inhibits neutrophils from releasing TNF-α, which in turn reduces NR and reduces I/R damage (42, 69, 70). The adenosine analogue AMP579 alleviates heart damage by inhibiting neutrophil activation and excessive vascular adhesion (71, 72). Targeted inhibitors of adenosine hydrolase help to inhibit NR. Targeting PDE4B (the hydrolase of cyclic adenosine monophosphate) inhibits neutrophil-endothelial cell interaction and expression of cell adhesion molecules, neutrophil cardiac infiltration, and release of proinflammatory cytokines, exerting cardioprotection in acute myocardial infarction (73).
In ischemic heart disease animal models, targeted depletion of complement significantly leads to a considerable reduction in neutrophil numbers (74). The C3 inhibitor, sCR1, reduces the infarct size and minimizes the accumulation of neutrophils in the infarct area, possibly by reducing C5a production, promoting adhesion receptor expression, and promoting chemotactic processes (75, 76). Anti-C5 therapy in the setting of MI/R significantly inhibits cell apoptosis, necrosis, and neutrophil infiltration (77). The recombinant human C5a receptor antagonist CGS 32359 inhibits surgical I/R injury after coronary occlusion and reduces the extent of high-risk myocardial infarction associated with reduced neutrophil aggregation (78).
5.2 Agents intervention
Several clinically used drugs have been found to either directly or indirectly regulate NR (Table 2). Metoprolol has been shown to inhibit neutrophil migration in an Adrenergic receptor (ADR) β1-dependent manner (79). During the early stages of neutrophil recruitment, it inhibits the structural and functional changes necessary for the effective engagement of circulating platelets, resulting in erratic intravascular dynamics, weakened neutrophil-platelet interactions, and blunted inflammation. Trimetazidine has been shown to inhibit neutrophil accumulation after myocardial ischemia and reperfusion (80). Empagliflozin prevented the permeation of neutrophils into the myocardium and therefore suppressed the transcription of these inflammatory cytokines (81). Metformin, as a first-line drug for hypoglycemia, can also inhibit NR in IHD. It has been shown to reduce myocardial neutrophil activity and cardiac remodeling after myocardial infarction through the AMPK pathway, thereby alleviating myocardial injury (82). Colchicine has been shown to inhibit neutrophil proliferation through inflammatory signaling pathways and reduce microvascular obstruction after myocardial I/R injury (83).
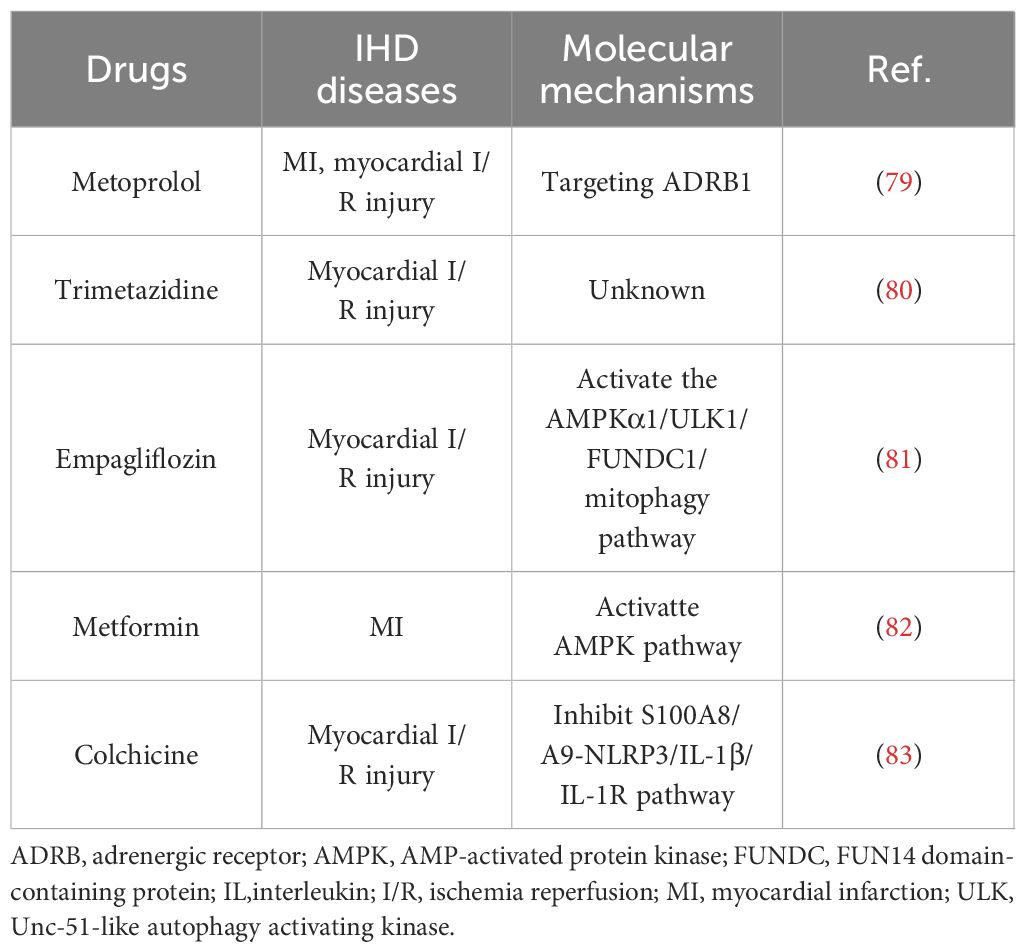
Table 2 Agents interventions to regulate NR.
5.3 Phytochemical strategies
Herbal medicines have been widely utilized alone or as a supplement and alternative therapy to treat various disorders in East Asia because of their reduced toxicity, fewer side effects, and cheaper cost (84). Many herbal ingredients may act synergistically to protect against myocardial injury in IHD through suppressing NR, such as salvianolic acid b, stigmasterol, resveratrol (85), tetrandrine (86), baicalin (87), silibinin (88), 2-methoxycinnamaldehyde (89), celastrol (90), et al. (Table 3). These phytochemicals, derived from different herbs, alleviated the inflammatory response of IHD to varying degrees by inhibiting NR through different molecular pathways.
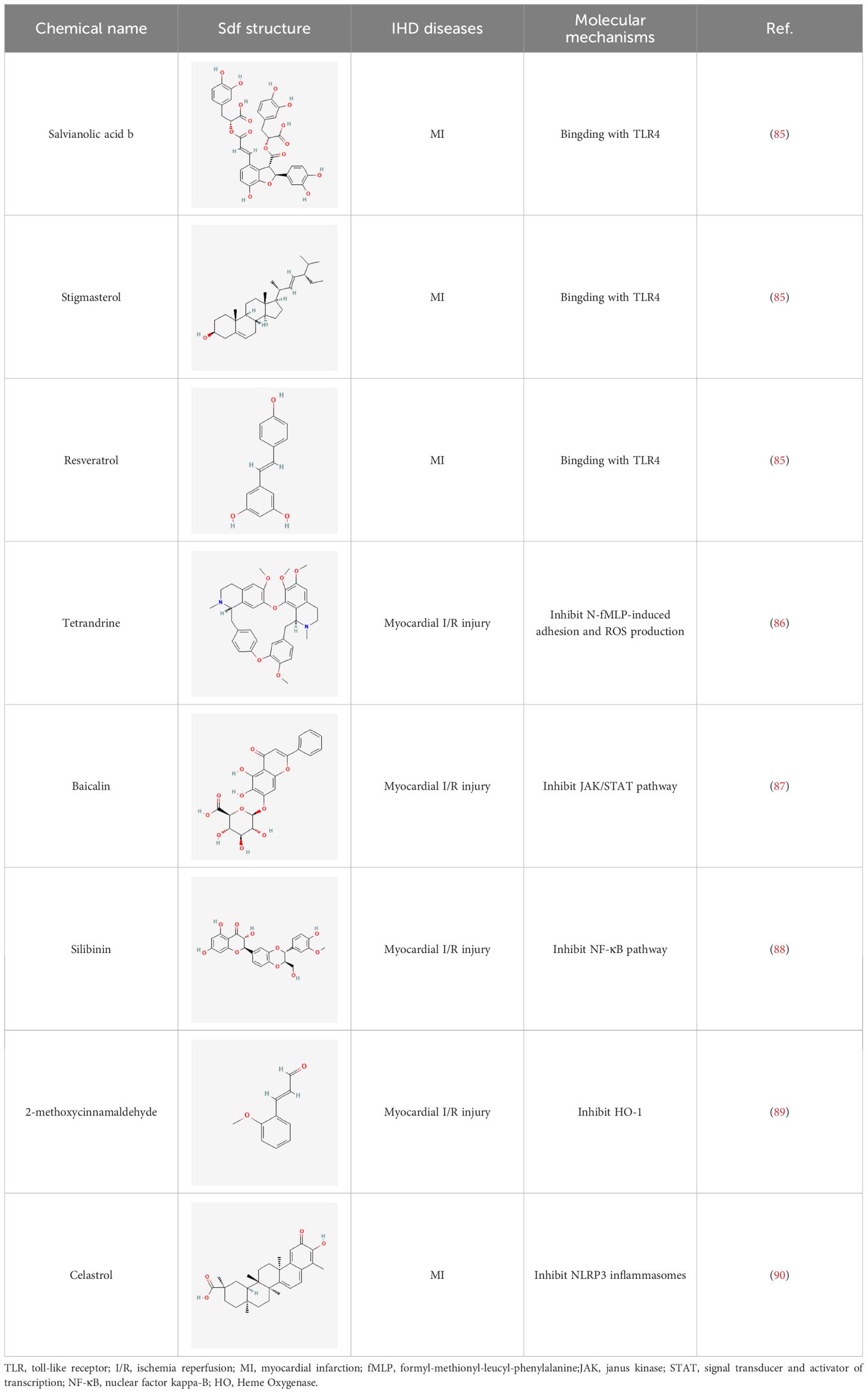
Table 3 Phytochemical strategies to regulate NR.
6 Concluding remarks and Perspectives
Vast efforts have been continuously engaged over the past decades in the search for cardioprotective pharmacological agents in preventing immunoinflammatory response. Neutrophil recruitment is an important part of the immunoinflammatory response, which acts as a double-edged sword in the pathogenesis, progression, and healing of IHD. In the early stages of cardiac ischemia, the moderate inflammatory response caused by neutrophil recruitment lays the foundation for subsequent healing and repair by clearing dead myocardium and stromal debris. However, the occurrence of NR can promote the release of various inflammatory and chemotactic mediators, leading to cell damage and aggravating inflammatory response to myocardial injury. Therefore, how to inhibit the specific parts of the innate immune system that cause damage without affecting the healing process of ischemic myocardium and infarcted myocardium has become the research frontier of intervention in ischemic heart disease from the perspective of neutrophil recruitment. This review mainly discusses and summarizes the biological information of NR in the context of myocardial ischemia, which will contribute to a better understanding of their effects in the pathophysiology of IHD, and provide new avenues and methods for the prevention and treatment of IHD.
Author contributions
YW: Writing – original draft. XS: Funding acquisition, Methodology, Writing – original draft. YWu: Methodology, Writing – review & editing. DL: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Beijing Natural Science Foundation [7222284 and 7244504].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
2. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. (2011) 17:1391–401. doi: 10.1038/nm.2507
3. Zheng Y, Gardner SE, Clarke MCH. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler Thromb Vasc Biol. (2011) 31:2781–6. doi: 10.1161/ATVBAHA.111.224907
4. Mócsai A, Walzog B, Lowell CA. Intracellular signalling during neutrophil recruitment. Cardiovasc Res. (2015) 107:373–85. doi: 10.1093/cvr/cvv159
5. Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. (2011) 17:1381–90. doi: 10.1038/nm.2514
6. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
7. Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol. (2011) 32:452–60. doi: 10.1016/j.it.2011.06.008
8. Chatelain P, Latour JG, Tran D, de Lorgeril M, Dupras G, Bourassa M. Neutrophil accumulation in experimental myocardial infarcts: relation with extent of injury and effect of reperfusion. Circulation. (1987) 75:1083–90. doi: 10.1161/01.CIR.75.5.1083
9. Hausenloy DJ, Chilian W, Crea F, Davidson SM, Ferdinandy P, Garcia-Dorado D, et al. The coronary circulation in acute myocardial ischaemia/reperfusion injury: a target for cardioprotection. Cardiovasc Res. (2019) 115:1143–55. doi: 10.1093/cvr/cvy286
10. Strassheim D, Dempsey EC, Gerasimovskaya E, Stenmark K, Karoor V. Role of inflammatory cell subtypes in heart failure. J Immunol Res. (2019) 2019:2164017. doi: 10.1155/2019/2164017
11. Sharma K, Patel AK, Shah KH, Konat A. Is neutrophil-to-lymphocyte ratio a predictor of coronary artery disease in western Indians? Int J Inflam. (2017) 2017:4136126. doi: 10.1155/2017/4136126
12. Meeuwsen JAL, Wesseling M, Hoefer IE, de Jager SCA. Prognostic value of circulating inflammatory cells in patients with stable and acute coronary artery disease. Front Cardiovasc Med. (2017) 4:44. doi: 10.3389/fcvm.2017.00044
13. Dogan I, Karaman K, Sonmez B, Celik S, Turker O. Relationship between serum neutrophil count and infarct size in patients with acute myocardial infarction. Nucl Med Commun. (2009) 30:797–801. doi: 10.1097/MNM.0b013e32832e3a16
14. Budzianowski J, Pieszko K, Burchardt P, Rzeźniczak J, Hiczkiewicz J. The role of hematological indices in patients with acute coronary syndrome. Dis Markers. (2017) 2017:3041565. doi: 10.1155/2017/3041565
15. Petri B, Phillipson M, Kubes P. The physiology of leukocyte recruitment: an in vivo perspective. J Immunol. (2008) 180:6439–46. doi: 10.4049/jimmunol.180.10.6439
16. Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. (2010) 10:427–39. doi: 10.1038/nri2779
17. Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, Itoh A, et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. (2002) 106:2894–900. doi: 10.1161/01.cir.0000042674.89762.20
18. Rotzius P, Thams S, Soehnlein O, Kenne E, Tseng C-N, Björkström NK, et al. Distinct infiltration of neutrophils in lesion shoulders in ApoE-/- mice. Am J Pathol. (2010) 177:493–500. doi: 10.2353/ajpath.2010.090480
19. Soehnlein O, Weber C. Myeloid cells in atherosclerosis: initiators and decision shapers. Semin Immunopathol. (2009) 31:35–47. doi: 10.1007/s00281–009-0141-z
20. Arslan F, Smeets MB, O’Neill LAJ, Keogh B, McGuirk P, Timmers L, et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation. (2010) 121:80–90. doi: 10.1161/CIRCULATIONAHA.109.880187
21. Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M, et al. Alarmins: awaiting a clinical response. J Clin Invest. (2012) 122:2711–9. doi: 10.1172/JCI62423
22. Vafadarnejad E, Rizzo G, Krampert L, Arampatzi P, Arias-Loza A-P, Nazzal Y, et al. Dynamics of cardiac neutrophil diversity in murine myocardial infarction. Circ Res. (2020) 127:e232–49. doi: 10.1161/CIRCRESAHA.120.317200
23. Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, et al. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. (2016) 110:51–61. doi: 10.1093/cvr/cvw024
24. Williams FM. Neutrophils and myocardial reperfusion injury. Pharmacol Ther. (1996) 72:1–12. doi: 10.1016/s0163–7258(96)00090–3
25. Timmers L, Pasterkamp G, de Hoog VC, Arslan F, Appelman Y, de Kleijn DPV. The innate immune response in reperfused myocardium. Cardiovasc Res. (2012) 94:276–83. doi: 10.1093/cvr/cvs018
26. Jaeschke H, Smith CW. Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol. (1997) 61:647–53. doi: 10.1002/jlb.61.6.647
27. Murphy SP, Kakkar R, McCarthy CP, Januzzi JL. Inflammation in heart Failure: JACC state-of-the-art review. J Am Coll Cardiol. (2020) 75:1324–40. doi: 10.1016/j.jacc.2020.01.014
28. Kain V, Halade GV. Role of neutrophils in ischemic heart failure. Pharmacol Ther. (2020) 205:107424. doi: 10.1016/j.pharmthera.2019.107424
29. Rashidi F, Rashidi A, Golmohamadi A, Hoseinzadeh E, Mohammadi B, Mirzajani H, et al. Does absolute neutrophilia predict early congestive heart failure after acute myocardial infarction? A cross-sectional study. South Med J. (2008) 101:19–23. doi: 10.1097/SMJ.0b013e31815d3e11
30. Valiente-Alandi I, Potter SJ, Salvador AM, Schafer AE, Schips T, Carrillo-Salinas F, et al. Inhibiting fibronectin attenuates fibrosis and improves cardiac function in a model of heart failure. Circulation. (2018) 138:1236–52. doi: 10.1161/CIRCULATIONAHA.118.034609
31. Daseke MJ, Valerio FM, Kalusche WJ, Ma Y, DeLeon-Pennell KY, Lindsey ML. Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Res Cardiol. (2019) 114:37. doi: 10.1007/s00395–019-0746-x
32. Vasilyev N, Williams T, Brennan M-L, Unzek S, Zhou X, Heinecke JW, et al. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation. (2005) 112:2812–20. doi: 10.1161/CIRCULATIONAHA.105.542340
33. Mollenhauer M, Friedrichs K, Lange M, Gesenberg J, Remane L, Kerkenpaß C, et al. Myeloperoxidase mediates postischemic arrhythmogenic ventricular remodeling. Circ Res. (2017) 121:56–70. doi: 10.1161/CIRCRESAHA.117.310870
34. Fan Q, Tao R, Zhang H, Xie H, Lu L, Wang T, et al. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation. (2019) 139:663–78. doi: 10.1161/CIRCULATIONAHA.118.036044
35. Lee S-J, Lee C-K, Kang S, Park I, Kim YH, Kim SK, et al. Angiopoietin-2 exacerbates cardiac hypoxia and inflammation after myocardial infarction. J Clin Invest. (2018) 128:5018–33. doi: 10.1172/JCI99659
36. Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. (2019) 129:2293–304. doi: 10.1172/JCI126428
37. Artz A, Butz S, Vestweber D. GDF-15 inhibits integrin activation and mouse neutrophil recruitment through the ALK-5/TGF-βRII heterodimer. Blood. (2016) 128:529–41. doi: 10.1182/blood-2016–01-696617
38. Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med. (2011) 17:581–8. doi: 10.1038/nm.2354
39. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
40. Griffin GK, Newton G, Tarrio ML, Bu D, Maganto-Garcia E, Azcutia V, et al. IL-17 and TNF-α sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol. (2012) 188:6287–99. doi: 10.4049/jimmunol.1200385
41. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. (2015) 349:316–20. doi: 10.1126/science.aaa8064
42. Schulz R, Aker S, Belosjorow S, Heusch G. TNFalpha in ischemia/reperfusion injury and heart failure. Basic Res Cardiol. (2004) 99:8–11. doi: 10.1007/s00395–003-0431-x
43. Li W, Hsiao H-M, Higashikubo R, Saunders BT, Bharat A, Goldstein DR, et al. Heart-resident CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight. (2016) 1:87315. doi: 10.1172/jci.insight.87315
44. Bhattacharya K, Farwell K, Huang M, Kempuraj D, Donelan J, Papaliodis D, et al. Mast cell deficient W/Wv mice have lower serum IL-6 and less cardiac tissue necrosis than their normal littermates following myocardial ischemia-reperfusion. Int J Immunopathol Pharmacol. (2007) 20:69–74. doi: 10.1177/039463200702000108
45. Hoyer FF, Nahrendorf M. Interferon-γ regulates cardiac myeloid cells in myocardial infarction. Cardiovasc Res. (2019) 115:1815–6. doi: 10.1093/cvr/cvz143
46. Khandoga A, Biberthaler P, Enders G, Axmann S, Hutter J, Messmer K, et al. Platelet adhesion mediated by fibrinogen-intercelllular adhesion molecule-1 binding induces tissue injury in the postischemic liver. vivo Transplant. (2002) 74:681–8. doi: 10.1097/00007890–200209150–00016
47. Tourki B, Halade G. Leukocyte diversity in resolving and nonresolving mechanisms of cardiac remodeling. FASEB J. (2017) 31:4226–39. doi: 10.1096/fj.201700109R
48. Foreman KE, Glovsky MM, Warner RL, Horvath SJ, Ward PA. Comparative effect of C3a and C5a on adhesion molecule expression on neutrophils and endothelial cells. Inflammation. (1996) 20:1–9. doi: 10.1007/BF01487740
49. Tyagi S, Klickstein LB, Nicholson-Weller A. C5a-stimulated human neutrophils use a subset of beta2 integrins to support the adhesion-dependent phase of superoxide production. J Leukoc Biol. (2000) 68:679–86. doi: 10.1046/j.1365-2516.2000.00429.x
50. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. (2008) 8:349–61. doi: 10.1038/nri2294
51. Fu T, Mohan M, Brennan EP, Woodman OL, Godson C, Kantharidis P, et al. Therapeutic potential of lipoxin A4 in chronic inflammation: focus on cardiometabolic disease. ACS Pharmacol Transl Sci. (2020) 3:43–55. doi: 10.1021/acsptsci.9b00097
52. Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol. (2010) 177:1576–91. doi: 10.2353/ajpath.2010.100322
53. Perucci LO, Sugimoto MA, Gomes KB, Dusse LM, Teixeira MM, Sousa LP. Annexin A1 and specialized proresolving lipid mediators: promoting resolution as a therapeutic strategy in human inflammatory diseases. Expert Opin Ther Targets. (2017) 21:879–96. doi: 10.1080/14728222.2017.1364363
54. D’Amico M, Di Filippo C, La M, Solito E, McLean PG, Flower RJ, et al. Lipocortin 1 reduces myocardial ischemia-reperfusion injury by affecting local leukocyte recruitment. FASEB J. (2000) 14:1867–9. doi: 10.1096/fj.99–0602fje
55. La M, D’Amico M, Bandiera S, Di Filippo C, Oliani SM, Gavins FN, et al. Annexin 1 peptides protect against experimental myocardial ischemia-reperfusion: analysis of their mechanism of action. FASEB J. (2001) 15:2247–56. doi: 10.1096/fj.01–0196com
56. Chen J, Oggero S, Cecconello C, Dalli J, Hayat H, Hjiej Andaloussi A, et al. The Annexin-A1 mimetic RTP-026 promotes acute cardioprotection through modulation of immune cell activation. Pharmacol Res. (2023) 198:107005. doi: 10.1016/j.phrs.2023.107005
57. Mareş RG, Sabău AH, Cocuz IG, Tomuţ ME, Szabo IA, Szőke AR, et al. S100A8∕A9 is a valuable biomarker and treatment target to detect and modulate neutrophil involvement in myocardial infarction. Rom J Morphol Embryol. (2023) 64:151–8. doi: 10.47162/RJME.64.2.04
58. Zhao M, Zheng Z, Yin Z, Zhang J, Peng S, Liu J, et al. DEL-1 deficiency aggravates pressure overload-induced heart failure by promoting neutrophil infiltration and neutrophil extracellular traps formation. Biochem Pharmacol. (2023) 218:115912. doi: 10.1016/j.bcp.2023.115912
59. Chen J, Ma Q, King JS, Sun Y, Xu B, Zhang X, et al. aYAP modRNA reduces cardiac inflammation and hypertrophy in a murine ischemia-reperfusion model. Life Sci Alliance. (2020) 3:e201900424. doi: 10.26508/lsa.201900424
60. Bottermann K, Kalfhues L, Nederlof R, Hemmers A, Leitner LM, Oenarto V, et al. Cardiomyocyte p38 MAPKα suppresses a heart-adipose tissue-neutrophil crosstalk in heart failure development. Basic Res Cardiol. (2022) 117:48. doi: 10.1007/s00395–022-00955–2
61. Liu W, Cronin CG, Cao Z, Wang C, Ruan J, Pulikkot S, et al. Nexinhib20 inhibits neutrophil adhesion and β2 integrin activation by antagonizing rac-1-guanosine 5’-triphosphate interaction. J Immunol. (2022) 209:1574–85. doi: 10.4049/jimmunol.2101112
62. Zhang Z, Tian H, Yang C, Liu J, Zhang H, Wang J, et al. Mesenchymal stem cells promote the resolution of cardiac inflammation after ischemia reperfusion via enhancing efferocytosis of neutrophils. J Am Heart Assoc. (2020) 9:e014397. doi: 10.1161/JAHA.119.014397
63. Zhang H, Kim H, Park BW, Noh M, Kim Y, Park J, et al. CU06–1004 enhances vascular integrity and improves cardiac remodeling by suppressing edema and inflammation in myocardial ischemia-reperfusion injury. Exp Mol Med. (2022) 54:23–34. doi: 10.1038/s12276–021-00720-w
64. Askari AT, Brennan M-L, Zhou X, Drinko J, Morehead A, Thomas JD, et al. Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction. J Exp Med. (2003) 197:615–24. doi: 10.1084/jem.20021426
65. Ali M, Pulli B, Courties G, Tricot B, Sebas M, Iwamoto Y, et al. Myeloperoxidase inhibition improves ventricular function and remodeling after Experimental myocardial infarction. JACC Basic Transl Sci. (2016) 1:633–43. doi: 10.1016/j.jacbts.2016.09.004
66. Granger DN, Schmid-Schönbein GW. Physiology and pathophysiology of leukocyte adhesion. USA: Oxford University Press (1995).
67. Suzuki K, Murtuza B, Smolenski RT, Sammut IA, Suzuki N, Kaneda Y, et al. Overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia-reperfusion injury associated with reduction in apoptosis. Circulation. (2001) 104:I308–I3. doi: 10.1161/hc37t1.094871
68. Gao R-F, Li X, Xiang H-Y, Yang H, Lv C-Y, Sun X-L, et al. The covalent NLRP3-inflammasome inhibitor Oridonin relieves myocardial infarction induced myocardial fibrosis and cardiac remodeling in mice. Int Immunopharmacol. (2021) 90:107133. doi: 10.1016/j.intimp.2020.107133
69. Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. (1996) 98:2854–65. doi: 10.1172/JCI119114
70. Koeppen M, Harter PN, Bonney S, Bonney M, Reithel S, Zachskorn C, et al. Adora2b signaling on bone marrow derived cells dampens myocardial ischemia-reperfusion injury. Anesthesiology. (2012) 116:1245–57. doi: 10.1097/ALN.0b013e318255793c
71. Budde JM, Velez DA, Zhao Z, Clark KL, Morris CD, Muraki S, et al. Comparative study of AMP579 and adenosine in inhibition of neutrophil-mediated vascular and myocardial injury during 24 h of reperfusion. Cardiovasc Res. (2000) 47:294–305. doi: 10.1016/s0008–6363(00)00115–2
72. Nakamura M, Zhao ZQ, Clark KL, Velez DV, Guyton RA, Vinten-Johansen J. A novel adenosine analog, AMP579, inhibits neutrophil activation, adherence and neutrophil-mediated injury to coronary vascular endothelium. Eur J Pharmacol. (2000) 397:197–205. doi: 10.1016/s0014–2999(00)00234-x
73. Wan Q, Xu C, Zhu L, Zhang Y, Peng Z, Chen H, et al. Targeting PDE4B (Phosphodiesterase-4 subtype B) for cardioprotection in acute myocardial infarction via neutrophils and microcirculation. Circ Res. (2022) 131:442–55. doi: 10.1161/CIRCRESAHA.122.321365
74. Arumugam TV, Magnus T, Woodruff TM, Proctor LM, Shiels IA, Taylor SM. Complement mediators in ischemia-reperfusion injury. Clin Chim Acta. (2006) 374:33–45. doi: 10.1016/j.cca.2006.06.010
75. Zacharowski K, Otto M, Hafner G, Marsh HC, Thiemermann C. Reduction of myocardial infarct size with sCR1sLe(x), an alternatively glycosylated form of human soluble complement receptor type 1 (sCR1), possessing sialyl Lewis x. Br J Pharmacol. (1999) 128:945–52. doi: 10.1038/sj.bjp.0702889
76. Shandelya SM, Kuppusamy P, Herskowitz A, Weisfeldt ML, Zweier JL. Soluble complement receptor type 1 inhibits the complement pathway and prevents contractile failure in the postischemic heart. Evidence that complement activation is required for neutrophil-mediated reperfusion injury. Circulation. (1993) 88:2812–26. doi: 10.1161/01.cir.88.6.2812
77. Vakeva AP, Agah A, Rollins SA, Matis LA, Li L, Stahl GL. Myocardial infarction and apoptosis after myocardial ischemia and reperfusion: role of the terminal complement components and inhibition by anti-C5 therapy. Circulation. (1998) 97:2259–67. doi: 10.1161/01.cir.97.22.2259
78. Riley RD, Sato H, Zhao ZQ, Thourani VH, Jordan JE, Fernandez AX, et al. Recombinant human complement C5a receptor antagonist reduces infarct size after surgical revascularization. J Thorac Cardiovasc Surg. (2000) 120:350–8. doi: 10.1067/mtc.2000.107281
79. García-Prieto J, Villena-Gutiérrez R, Gómez M, Bernardo E, Pun-García A, García-Lunar I, et al. Neutrophil stunning by metoprolol reduces infarct size. Nat Commun. (2017) 8:14780. doi: 10.1038/ncomms14780
80. Williams FM, Tanda K, Kus M, Williams TJ. Trimetazidine inhibits neutrophil accumulation after myocardial ischaemia and reperfusion in rabbits. J Cardiovasc Pharmacol. (1993) 22:828–33. doi: 10.1097/00005344–199312000–00008
81. Cai C, Guo Z, Chang X, Li Z, Wu F, He J, et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion through activating the AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. (2022) 52:102288. doi: 10.1016/j.redox.2022.102288
82. Soraya H, Rameshrad M, Mokarizadeh A, Garjani A. Metformin attenuates myocardial remodeling and neutrophil recruitment after myocardial infarction in rat. Bioimpacts. (2015) 5:3–8. doi: 10.15171/bi.2015.02
83. Tan Y, Bao X, Li Y, Song G, Lu H, Sun X, et al. Colchicine attenuates microvascular obstruction after myocardial ischemia-reperfusion injury by inhibiting the proliferation of neutrophil in bone marrow. Cardiovasc Drugs Ther. (2023). doi: 10.1007/s10557–023-07528-y
84. Wang Z, Qi F, Cui Y, Zhao L, Sun X, Tang W, et al. An update on Chinese herbal medicines as adjuvant treatment of anticancer therapeutics. Biosci Trends. (2018) 12:220–39. doi: 10.5582/bst.2018.01144
85. Jiang F, Zeng Z, Zhou X, Tan M, Zhang W, Li M, et al. Transcriptomic analysis uncovers immunogenic characteristics of ferroptosis for myocardial infarction and potential therapeutic prediction of chinese herbs. Evid Based Complement Alternat Med. (2022) 2022:4918343. doi: 10.1155/2022/4918343
86. Shen YC, Chen CF, Sung YJ. Tetrandrine ameliorates ischaemia-reperfusion injury of rat myocardium through inhibition of neutrophil priming and activation. Br J Pharmacol. (1999) 128:1593–601. doi: 10.1038/sj.bjp.0702958
87. Xu M, Li X, Song L. Baicalin regulates macrophages polarization and alleviates myocardial ischaemia/reperfusion injury via inhibiting JAK/STAT pathway. Pharm Biol. (2020) 58:655–63. doi: 10.1080/13880209.2020.1779318
88. Chen Y-H, Lin H, Wang Q, Hou J-W, Mao Z-J, Li Y-G. Protective role of silibinin against myocardial ischemia/reperfusion injury-induced cardiac dysfunction. Int J Biol Sci. (2020) 16:1972–88. doi: 10.7150/ijbs.39259
89. Hwa JS, Jin YC, Lee YS, Ko YS, Kim YM, Shi LY, et al. 2-methoxycinnamaldehyde from Cinnamomum cassia reduces rat myocardial ischemia and reperfusion injury in vivo due to HO-1 induction. J Ethnopharmacol. (2012) 139:605–15. doi: 10.1016/j.jep.2011.12.001
Keywords: neutrophil recruitment, immunoinflammation, ischemic heart disease, aseptic inflammation, innate immunity
Citation: Wang Y, Shou X, Wu Y and Li D (2024) Immuno-inflammatory pathogenesis in ischemic heart disease: perception and knowledge for neutrophil recruitment. Front. Immunol. 15:1411301. doi: 10.3389/fimmu.2024.1411301
Received: 02 April 2024; Accepted: 14 June 2024;
Published: 10 July 2024.
Edited by:
Vinay Kumar, The Pennsylvania State University, United StatesReviewed by:
Hina Sultana, University of North Carolina System, United StatesSuman Asalla, The Ohio State University, United States
Copyright © 2024 Wang, Shou, Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Li, ZG9uZ2Rvbmc4NzExMDNAMTYzLmNvbQ==; Xintian Shou, c2hvdXhpbnRpYW5AMTYzLmNvbQ==
†These authors have contributed equally to this work