 Qianhui Zhang1†
Qianhui Zhang1† Miao Huang1,2†Elizabeth Rosalind Thomas3Lu Wang1
Miao Huang1,2†Elizabeth Rosalind Thomas3Lu Wang1 Jia Liu1Xiang Li2
Jia Liu1Xiang Li2 Jiesi Luo2
Jiesi Luo2 Wenjun Zou1*
Wenjun Zou1* Jianming Wu2,4*
Jianming Wu2,4*- 1State Key Laboratory of Southwestern Chinese Medicine Resources, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 2School of Basic Medical Sciences, Southwest Medical University, Luzhou, China
- 3Department of Microbiology, North Eastern Indira Gandhi Regional Institute of Health and Medical Sciences, Shillong, India
- 4Education Ministry Key Laboratory of Medical Electrophysiology, Medical Key Laboratory for Drug Discovery and Druggability Evaluation of Sichuan Province, Luzhou Key Laboratory of Activity Screening and Druggability Evaluation for Chinese Materia Medica, Luzhou, China
Primary immune thrombocytopenia (ITP) is an acquired autoimmune disorder characterized by the destruction of platelets. Although it was long believed that the critical role of autoantibodies in platelet destruction, primarily through the Fc-dependent platelet clearance pathway, recent findings indicate that the significance of the Fc-independent platelet clearance pathway mediated by hepatocytes, thus shedding light on a previously obscure aspect of ITP pathogenesis. Within this context, the desialylation of platelets has emerged as a pivotal biochemical marker. Consequently, targeting platelet desialylation emerges as a novel therapeutic strategy in the pathogenesis of ITP. Notably, prevailing research has largely focused on antiplatelet antibodies and the glycosylation-associated mechanisms of platelet clearance, while comprehensive analysis of platelet desialylation remains scant. In response, we retrospectively discuss the historical progression, inducing factors, generation process, and molecular regulatory mechanisms underlying platelet desialylation in ITP pathogenesis. By systematically evaluating the most recent research findings, we contribute to a comprehensive understanding of the intricate processes involved. Moreover, our manuscript delves into the potential application of desialylation regulatory strategies in ITP therapy, heralding novel therapeutic avenues. In conclusion, this manuscript not only fills a critical void in existing literature but also paves the way for future research by establishing a systematic theoretical framework. By inspiring new research ideas and offering insights into the development of new therapeutic strategies and targeted drugs, our study is poised to significantly advance the clinical management of ITP.
Graphical Abstract The role of platelet desialylation in primary immune thrombocytopenia.
1 Introduction
Primary immune thrombocytopenia (ITP), an acquired autoimmune bleeding disorder, displays diverse characteristics of heterogeneity (1), chronicity (2), asymptomatic nature (3), and severity (4). As the global population ages, there is a discernible increase in the quantity of ITP cases. The reported incidence of primary ITP ranges from 1.9 to 6.4 per 100,000 per year in children (5) and 2 to 10 per 100,000 per year in adults (6). Notably, there is a higher prevalence among individuals over 60 years of age, contributing to the growing health concern associated with this condition. Additionally, women of childbearing age exhibit a slightly elevated prevalence compared to men in the same age group, with rates ranging approximately from 9.5 to 23.6 per 100,000 per year (7). Beyond the physical manifestation, patients grappling with ITP face not only the imminent risk of severe or fatal bleeding events but also content with a myriad of challenges, including a high disease burden and unfavorable prognosis. These factors contribute significantly to mental stress and psychological burden. A retrospective analysis highlights the economic impact, revealing that drug therapy for ITP incurs costs in hundreds of millions of dollars annually, translating to an average yearly expense of approximately $28,000 per ITP patient in the United States (8). Despite notable advancements in ITP treatment, the disease’s incidences remain elevated, and there exists a lack of standardization in both diagnosis and treatment protocols. Currently, there are no standardized guidelines for the diagnosis and treatment of ITP, which is a diagnosis of exclusion. This means that the disease is identified by the absence of other causes or conditions that could explain the reduction in platelet count. In addition to reduced platelets, the rest of the blood cell counts are normal. The diagnosis of ITP can only be confirmed by other secondary factors of thrombocytopenia except medical history, physical examination, blood count, and peripheral blood smear microscopy (9–11). Nevertheless, this limited approach introduces a risk of misdiagnosis, underscoring the critical need for precise clinical diagnosis and individualized treatment strategies. Addressing these challenges is imperative for enhancing patient outcomes and refining the overall management of ITP.
However, a lack of a clear understanding of the pathogenesis of ITP directly hinders the attainment of precise diagnosis and personalized treatment strategies. Current research suggests that the pathogenesis may be linked to immune system dysregulation and the generation of autoantibodies targeting platelets. In other words, the body’s immune system erroneously identifies its platelets as foreign substances, leading to the production of antibodies that attack these platelets. This process results in excessive platelet destruction and clearance (12). Prior investigations have proposed that platelet destruction caused by platelet autoantibodies primarily occurs through the Fc-dependent platelet clearance pathway (FcγR). In this pathway, the Fc segment of autoantibodies in ITP patients binds to the Fc receptor on the surfaces of macrophages or hepatic macrophages (Kupffer cells) located in the hepatosplenic reticuloendothelial system. This forms an antibody-receptor complex triggering platelet destruction and clearance by immune cells. Simultaneously, it inhibits platelet aggregation and adhesion, ultimately leading to thrombocytopenia (13, 14). Apart from the platelet destruction mediated by FcγR, recent research has shown a growing interest in hepatocyte-mediated Fc-independent platelet clearance pathway. In this process, platelet autoantibodies induce the desialylation of sialic acid from the platelet surface. Desialylated platelets are then recognized and cleared by the Ashwell-Morell receptor (AMR) present in hepatocytes (15, 16). Platelet desialylation emerges as a critical biochemical marker influencing both platelet count and functionality. Mounting evidence indicates that platelet desialylation plays a pivotal role in ITP pathogenesis, with the process of platelet destruction likely correlated with the production of platelet desialylation and its molecular regulatory mechanisms (17–19). Subsequent observations revealed a gradual recovery in peripheral platelet counts in mice as sialidase levels increased (16). Clinical studies demonstrated that patients with infection-induced thrombocytopenia exhibit significantly higher levels of platelet desialylation (16, 20). Additionally, the use of the sialidase inhibitor Oseltamivir to inhibit platelet desialylation could significantly alleviate the phenomenon of low platelet counts (21, 22), thus suggesting a broader mechanism of platelet desialylation. Altered desialylation in patients with ITP not only impairs platelet count and functionality but also induces increased apoptosis, as evidenced by heightened caspase-9 activation, particle formation, and a reduction in platelet lifespan. These factors collectively contribute to a decrease in platelet count and compromised function (23–26). The aforementioned discoveries suggest that the liver’s phagocytosis of desialylated platelets signifies a major pathway for platelet breakdown, significantly contributing to the development of ITP.
Therefore, in this manuscript, we synthesized the latest domestic and international research findings to delineate the historical progression of platelet desialylation and its triggering factors. We subsequently conducted an in-depth analysis of the generation process of platelet desialylation and its molecular regulatory mechanisms. Finally, we explore the potential applications of sialylation regulation strategies in the treatment of ITP. Furthermore, we propose some prospective views and strategic recommendations that can inform the clinical treatment of ITP by elucidating the role of platelet desialylation in the pathogenesis of the condition. These recommendations include methods to reduce viral and bacterial infections, along with their associated antigenic stimulation of the immune system. We also explore approaches to modulate the expression of anti-platelet antibodies and the formation process of sialic acid-glycoprotein complexes. Additionally, we delve into the investigation of specific targets and biomarkers associated with platelet desialylation to mitigate its impact on platelet function. The overarching goal of these efforts is to offer more beneficial targets and strategies for clinical management and drug development of ITP, to avoid unnecessary or ineffective treatments. By facilitating a precise diagnosis and individualized treatment for ITP patients, we aspire to enhance treatment outcomes and improve the quality of life for those affected by ITP.
2 Historical progression of platelet desialylation
Over the past few decades, significant breakthroughs have been made in understanding the process of platelet desialylation and its impact on the development and progression of hematological diseases (Figure 1). Research from as early as the 1960s first noted that platelets in ITP patients had reduced sialic acid content, leading to their excessive destruction and clearance (17). This observation sparked in-depth studies into the mechanisms of platelet desialylation, particularly the Fc-independent pathways of platelet clearance. In 2003, Hoffmeister et al. made a groundbreaking discovery, revealing that platelets stored in vitro and then transfused into humans were quickly cleared by the liver. Further studies revealed that platelets stored for less than 2 hours lost β-galactose (β-Gal) on the membrane glycoprotein GPIbα, thereby exposing N-acetyl glucosamine (N-GlcNAc) recognition sites. This led to the rapid recognition and clearance of these platelets by αMβ2 macrophages in the liver (27, 28). This finding answered long-standing questions about why stored platelets were rapidly cleared after transfusion. However, subsequent studies showed that blocking the N-GlcNAc/αMβ2 pathway did not resolve the issue of liver clearance of stored platelets. A breakthrough came in 2008 when Grewal found that pneumococcal-induced sepsis led to the release of neuraminidase (NEUs) by the bacteria, which caused platelet desialylation and exposed β-Gal, allowing the platelets to be recognized and cleared by the AMR (29). This discovery not only shed light on a novel function of the AMR but also offered a fresh perspective on the platelet clearance pathway’s mechanism. Studies also indicated that long-term storage (over 48 hours) of platelets led to the recognition of β-Gal on GPIbα by the AMR, potentially leading to platelet desialylation (18, 30, 31). Additionally, desialylation of the von Willebrand factor (vWF) receptor on platelets accelerated their clearance (32). In 2015, Li et al. found that anti-GPIbα antibodies in ITP patients trigger the surface translocation of NEU1, leading to platelet desialylation and subsequent recognition and clearance by the AMR. This process could be blocked by sialidase inhibitors (15). Further research discovered that the binding of desialylated platelets to the AMR was discovered to activate the JAK2/STAT3 signaling pathway, promoting the production of thrombopoietin (TPO) production and thereby regulating thrombopoiesis (33). With an improved understanding of platelet desialylation mechanisms, researchers have developed more accurate diagnostic methods and therapeutic approaches, including sialic acid level testing (34), immunomodulators (35), immunoglobulin therapies (36), and a few novel medications (37). These advancements aim to alleviate symptoms and enhance the quality of life for patients with ITP.
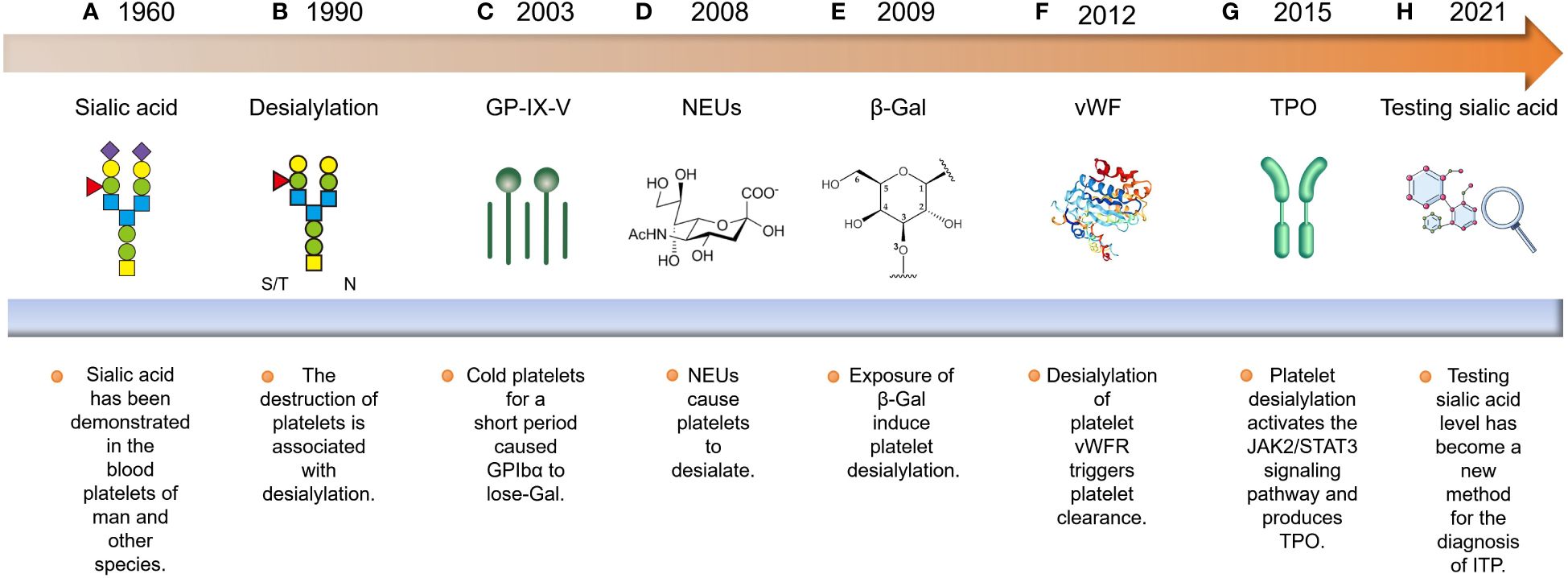
Figure 1 Historical progression of platelet desialylation: (A, B) The discovery of platelet desialylation can be traced back to the 1960s. (C) Subsequently, research has elucidated its significant impact on the pathogenesis of ITP, particularly in relation to the exposure of GPIbα to N-GlcNAC recognition sites, which are cleared by αMβ2 recognition. (D, E) Furthermore, researchers found that neuraminidases released by Streptococcus pneumoniae caused platelet desialylation, exposing β-Gal. This exposed residue is recognized and cleared by the AMR. (F) Additionally, desialylation of the vWFR on platelets accelerated their clearance. (G, H) With an in-depth understanding of the mechanisms governing platelet desialylation, relevant diagnostic and therapeutic methods have emerged. These include sialic acid level detection, immunosuppressant administration, and immunoglobulin therapy.
Hence, reviewing the research on platelet desialylation not only provides references for the treatment of ITP but also offers hope for enhancing the quality of life for these patients.
3 Inducing factors for platelet desialylation
The factors contributing to platelet desialylation primarily encompass aging under normal physiological conditions, autoimmune system disorders in pathological circumstances, exposure to foreign antigens and autoantigens, and assaults by pathogen-derived NEUs. These elements collectively play a significant role in the generation of platelet desialylation (Figure 2).
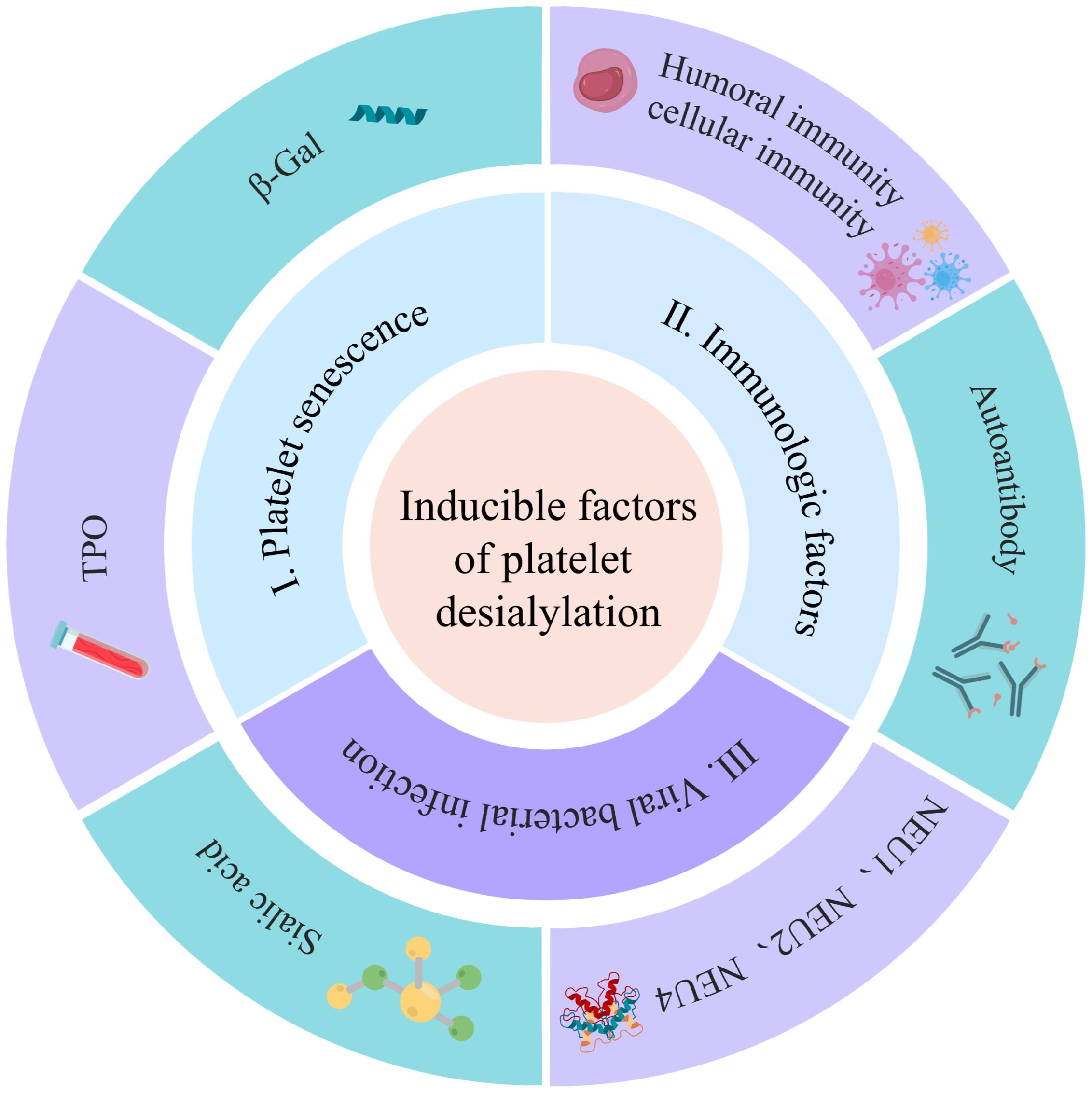
Figure 2 Inducing factors for platelet desialylation: The factors contributing to platelet desialylation include immune factors, viral and bacterial infection, and platelet senescence. The immune factors predominantly encompass platelet autoantibodies, humoral immunity, and cellular immunity. Additionally, foreign antigens and self-platelet antigens carried by the viruses can trigger cross-activation of B cells and T cells, leading to epitope spreading and subsequent platelet desialylation. Simultaneously, pathogen-derived NEUs serve as pivotal regulators, contributing significantly to the induction of platelet desialylation.
The factors contributing to platelet desialylation are diverse and intricate among which are (i) Aging platelets: Under normal physiological conditions, aging platelets shed sialic acid residue from the surface glycoproteins during circulation. This exposes the subterminal β-Gal, rendering targets for recognition, and clearance by the AMR. This process also induces an up-regulation of TPO, a significant factor in platelet desialylation (33). The influence on platelet desialylation is noteworthy; (ii) Abnormal disorders of the autoimmune system: In pathological states, autoimmune system disorders in ITP patients result in the generation of corresponding antibodies such as anti-GPIIb/IIIa, anti-GPIbα, anti-GPIb/IX and anti-GPIa/IIa antibody. These antibodies may induce platelet desialylation, subsequently cleared by excessive phagocytosis by the monocyte-macrophage system (38–40). The impact of abnormal autoimmune system disorders on platelet desialylation is substantial; (iii) Viral and bacterial infections: Plays a crucial role in platelet desialylation. Antigens carried by viruses, combined with native platelet antigens, lead to cross-activation of T and B cells, epitope spreading, and eventually platelet desialylation (40). Concurrently, NEUs originating from pathogens modify the sialic acids on platelet surfaces, triggering platelet desialylation (37). NEUs, characterized by a mushroom-like tetrameric glycoprotein structure, are prevalent in viruses, bacteria, and mammalian cells (41). NEUs are categorized into four types ranging from NEU1 to NEU4 (Table 1). Among them, NEU1, NEU2, and NEU4 are associated with platelet desialylation and are predominantly found on resting platelets. Specifically aggregated by GPIbα activated by vWF, these enzymes catalyze the sialic acid moiety, cleaving the sialic acid residues at the end of the sialic acid glycoprotein complex. This process induces platelet desialylation, creating a positive feedback loop of “platelet activation-desialylation”, ultimately resulting in platelet clearance in the liver (52). Hence, the inhibition of NEUs in the pathogenesis of ITP may contribute to thrombocytopenia. Reducing viral and bacterial infections, along with inhibiting NEUs during the pathogenesis of ITP, holds the potential for ameliorating thrombocytopenia.
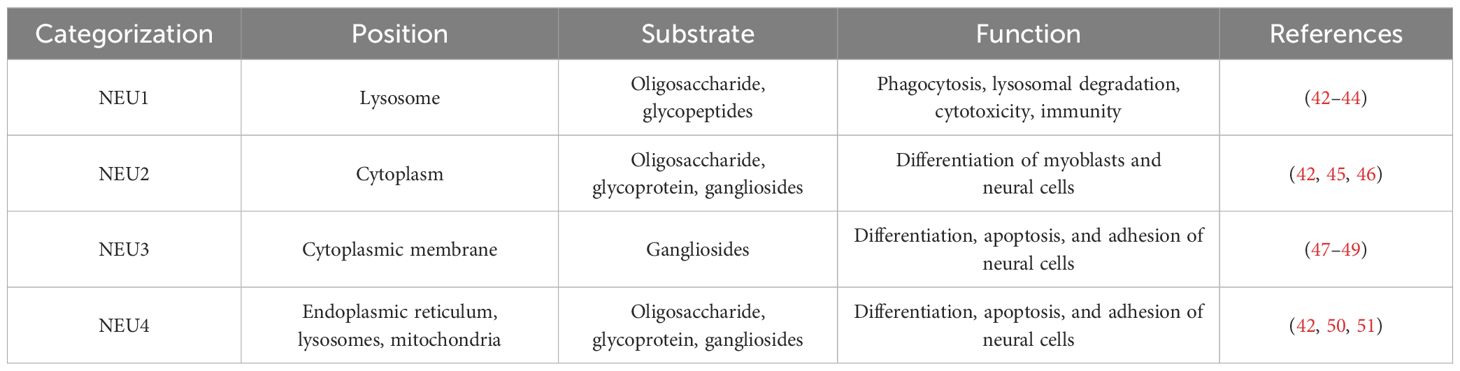
Table 1 Types of Sialidases.
The aforementioned studies suggest a crucial role for immune factors in platelet desialylation, with abnormalities in the autoimmune system identified as a primary predisposing factor for this phenomenon. Furthermore, specific viral and bacterial infections, along with the aging platelets, have been linked to platelet desialylation. In conclusion, a thorough exploration of these predisposing factors is essential for shedding light on the role of platelet desialylation in the pathogenesis of ITP.
4 The process of generation of platelet desialylation
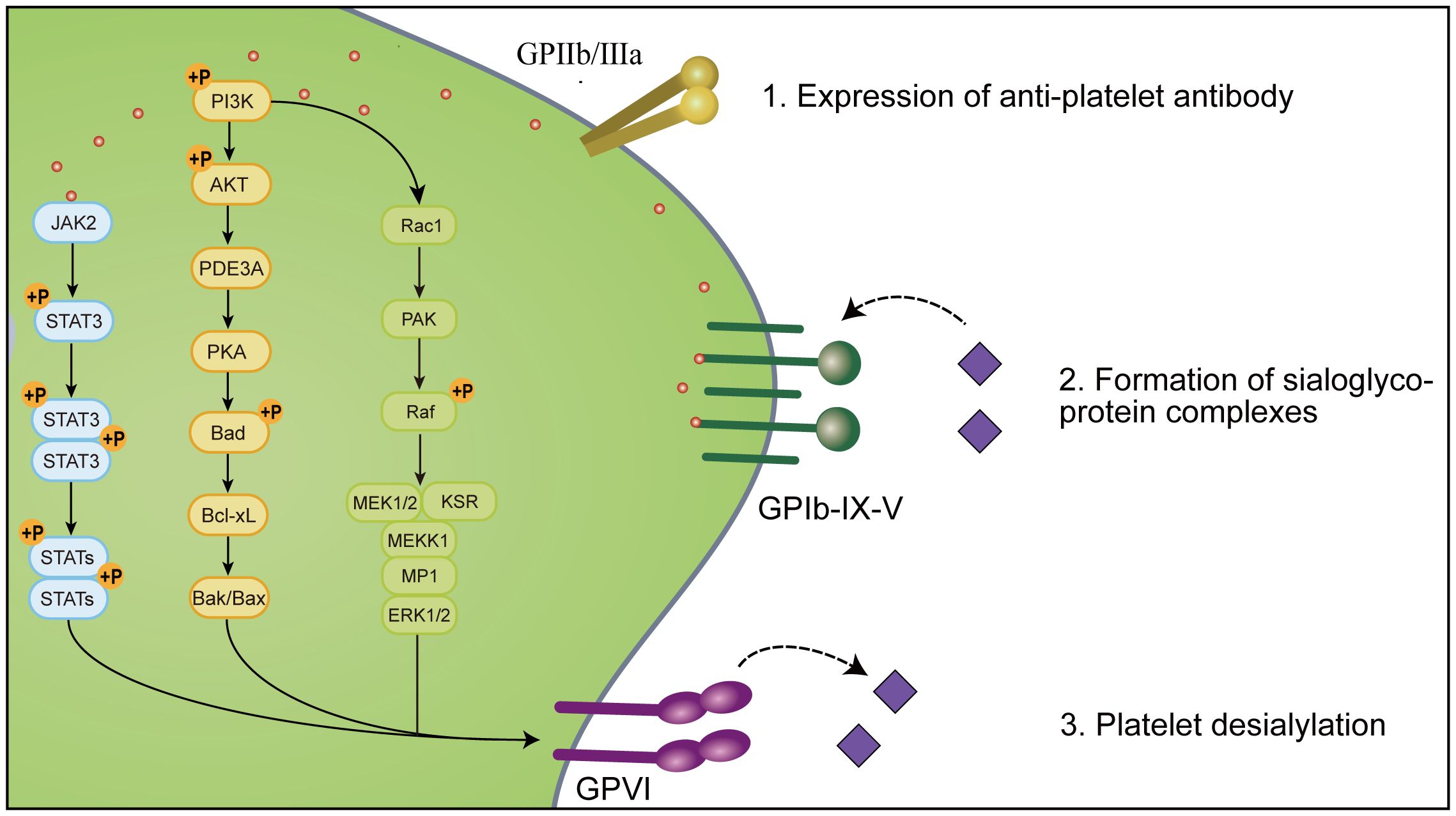
Platelet desialylation is a sophisticated biochemical process comprised of three pivotal stages (Figure 3). Initially, there is an expression of relevant antiplatelet antibodies like GPIa/IIa, GPIbα, and GPIb/IX. Subsequently, specific antibodies interact with sialic acid molecules via their specific domains, forming sialic acid-glycoprotein complexes that facilitate mutual recognition and adhesion of platelets to other cells or molecules. Ultimately, platelet desialylation is triggered by NEUs, which removes the terminal sialic acid residues from the sialic acid-glycoprotein complexes. This process leads to the identification of desialylated platelets, subsequently recognized and cleared by the AMR through an Fc-independent platelet clearance pathway.
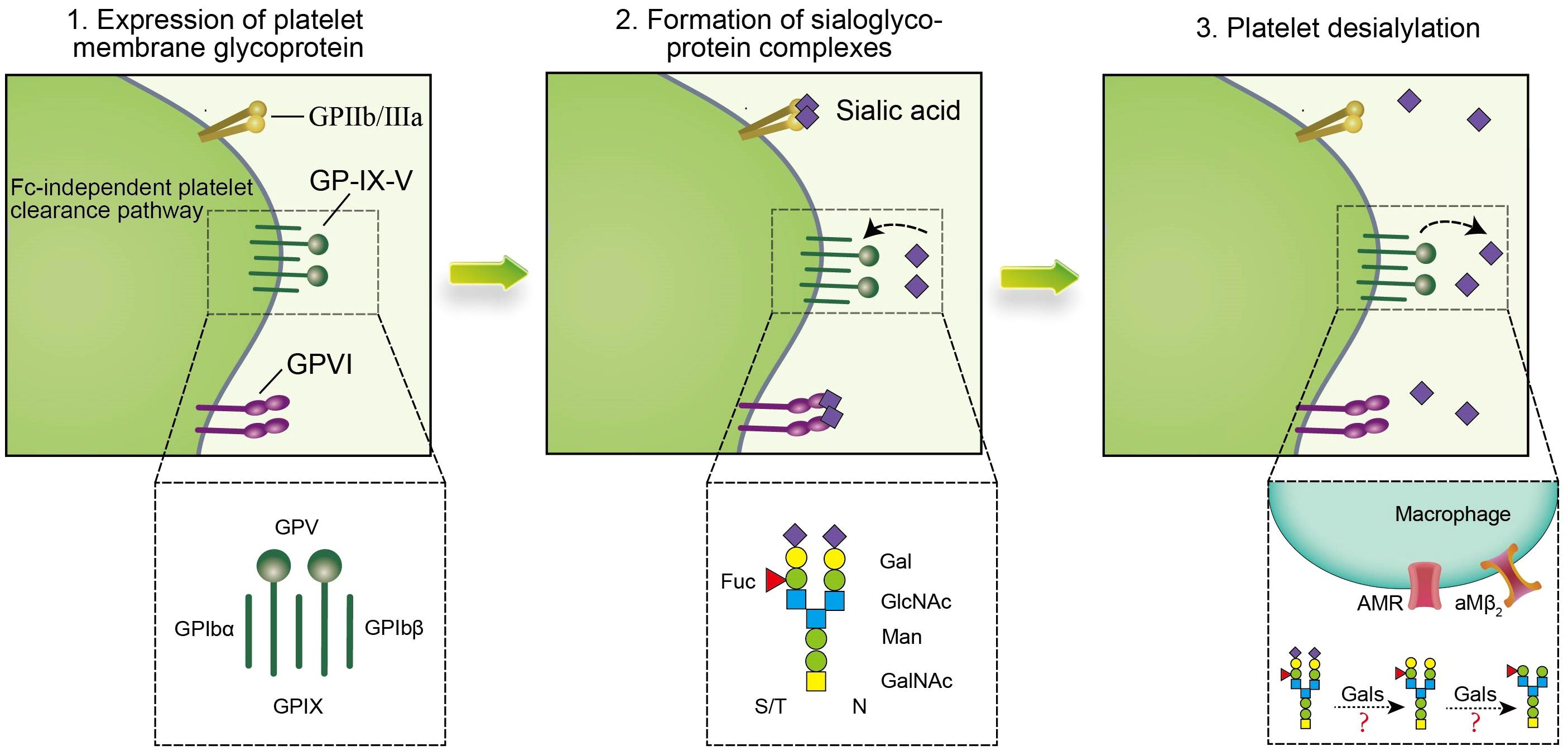
Figure 3 Platelet desialylation involves three key steps: 1. Expression of anti-platelet antibodies, such as anti-GPIIb/IIIa antibodies, anti-GP-IX-V antibodies and anti-GPVI antibodies, in which anti-GPIbα antibodies clear platelets through the Fc-independent platelet clearance pathway, thereby activating immune cells and affecting platelet function and survival. In addition, the abnormal expression or mutation of these anti-platelet antibodies may lead to thrombocytopenia and impaired function, and promote the excessive clearance of platelets by the immune system, thus exacerbating the disease of ITP; 2. Specific binding sites (such as β-Gal and N-GlcNAc) on the terminal sugar chains of platelet surface glycoproteins form sialoglycoprotein complexes with sialic acid molecules to change the structure and function of platelet glycoproteins, thereby affecting platelet biological activity and function; 3. Gals may mediate platelet desialylation, resulting in removal of terminal sialic acid, thereby exposing β-Gal and N-GlcNAc on the surface of platelets, allowing them to be recognized and cleared by specific receptors of the immune system such as AMR and αMβ2. Together, these steps constitute the complex mechanism of platelet desialylation in ITP, and its abnormality may lead to impaired and reduced platelet function, which in turn affects the blood coagulation process.
4.1 Expression of antiplatelet antibodies
The expression of antiplatelet antibodies represents an initial step in the process of platelet desialylation, and their levels significantly correlate with the pathogenesis of ITP. These antibodies encompass class I antibodies targeting human leukocyte antigens and those directed at glycoproteins. Notably, glycoproteins targeting antibodies are predominantly localized within the megakaryocyte system, exhibiting specificity as autoantibodies.
Categorized based on the antigenic target site, these antibodies include various types such as anti-GPIa/IIa, anti-GPIbα, anti-GPIb/IX, and anti-GPIIb/IIIa antibodies (53, 54). In the majority of ITP patients, anti-GPIIb/IIIa antibodies are present in 70% to 80% of cases, while anti-GPIbα antibodies are found in 20% to 40% of cases. A minority of ITP patients may have both or exhibit antibodies against other glycoproteins (38). Among these, the anti-GPIIb/IIIa antibody primarily mediates platelet clearance through the FcγR pathway. The binding of GPIIb/IIIa to the Fc segment of its specific antibody activates the macrophage’s tyrosine kinase, binding to the low-affinity FcγRIIA or FcγRIIIA on the macrophage’s surface. This results in platelet removal through both direct phagocytosis and complement-mediated phagocytosis in the spleen (7, 55).
In contrast, most studies reported that anti-GPIbα antibodies predominantly utilize an Fc-independent pathway for platelet clearance. When GPIbα binds to the F(ab)2 segment of its specific antibody, it further stimulates platelet activation through the GPIbα-mediated signaling pathway. This induction leads to the removal of sialic acid residues from the platelet surface, exposing subterminal β-Gal, which is recognized and cleared by AMR (24, 56, 57). Overall, anti-platelet antibodies such as GPIbα play a critical role in platelet desialylation. Abnormal expression or mutation of these antibodies can result in thrombocytopenia and impaired function, contributing to the immune system’s excessive clearance of platelets and exacerbating the condition of ITP.
4.2 Formation of sialic acid-glycoprotein complexes
The formation of sialic acid-glycoprotein complexes refers to the binding of specific glycoproteins on the surface of platelets to sialic acid molecules. This process significantly influences platelet function and biological activity, and it is a key mechanism in the desialylation of platelets in ITP. During the pathogenesis of ITP, specific binding sites on the terminal glycan chains of platelet glycoproteins, such as β-Gal and N-GlcNAc, can bind to sialic acid molecules, forming “sialic acid-glycoprotein complexes”.
This complex formation alters the structure and function of the platelet glycoproteins, thereby impacting the biological activity and lifespan of platelets within the body. Taking GPIIb/IIIa (αIIbβ3) and GPIb-IX-V as examples, these glycoproteins play central roles in both physiological and pathological processes of platelets. GPIIb/IIIa, a key factor in platelet adhesion and aggregation (58, 59), and GPIb-IX-V, which serves as a receptor for vWF and is involved in platelet adhesion and activation following vascular injury, are frequently targeted (60, 61). After binding with sialic acid, these glycoproteins may influence the recognition and clearance mechanisms of platelets, contributing to the pathogenesis of ITP. It has been discovered that GPIIb/IIIa promotes NEU1 surface translocation and platelet desialylation in GPIIb/IIIa-containing ITP patients relies on FcγRIIA signaling on the surface of splenic macrophages rather than the platelet activation pathway (7).
Thus, understanding the binding mechanism between platelet surface glycoproteins and sialic acid, namely, the formation of sialic acid-glycoprotein complexes, is crucial for exploring the role of platelet desialylation in the pathogenesis of ITP.
4.3 Platelet desialylation
Initially, galactosidases (Gals) may catalyze the removal of sialic acid residues from the terminal end of the sialic acid-glycoprotein complex, initiating the process of platelet desialylation. Subsequently, desialylation leads to the sequential exposure of β-Gal and N-GlcNAC on the sialic acid-glycoprotein complex, which is then recognized and cleared by the AMR or αMβ2 in the liver. Ultimately, deglycosylation results in the exposure of N-GlcNAC, which may be recognized and cleared through carbohydrate receptors, further mediating their uptake by macrophages.
Notably, the AMR, the principal lectin expressed on hepatocytes, is a highly conserved transmembrane hetero-oligomeric glycoprotein complex. The AMR recognizes, and scavenges circulating desialylated platelet glycoproteins (62). It has been established that the AMR facilitates platelet clearance by binding β-Gal, triggering the production of hepatic TPO via the JAK2-STAT3 pathway (33). While the AMR does regulate platelet clearance to a certain degree, the mechanisms by which it participates in the clearance of desialylated platelets have not been fully elucidated. Further research is needed to explore these mechanisms in greater detail.
Additionally, the integrin αMβ2 (also known as CD11b/CD18, CR3, or MAC-1), situated on the surface of Kupffer cells, is another receptor that recognizes and binds desialylated platelets as large clusters of non-covalently bound heterodimeric transmembrane glycoproteins. It has been discovered that Kupffer cells can rightfully identify and engage in the phagocytosis of desialylated platelets through αMβ2 (63). Furthermore, cold-degalactosylated platelets are cleared by the MAC-1. Recently, it was revealed that macrophage galactose lectin (MGL) from Kupffer cells aids in the clearance of desialylated platelets through collaboration with the AMR (62). These findings imply that platelet desialylation is a crucial element of the “glycan-agglutinin” mechanism, referring to platelet clearance mediated by glycan-agglutinin binding (64, 65).
In summary, the desialylation process of platelets encompasses several crucial stages, among which anti-GPIIb/IIIa antibodies and anti-GPIbα antibodies may be expressed, which play a significant role in the activation and aggregation of platelets. Furthermore, the formation of the sialic acid-glycoprotein complex and platelet desialylation are indispensable in the pathogenesis of ITP. Aberrations in these processes have the potential to lead to desialylation and structural disarray of platelets, thereby impacting the coagulation cascade.
5 Molecular regulatory mechanisms of platelet desialylation in ITP
The molecular regulatory process of platelet desialylation involves the coordinated regulation of several key signaling pathways, including PI3K/AKT, JAK/STAT, and MAPK signaling pathways (Figure 4). A comprehensive understanding of these signaling pathways and their interconnections is crucial to grasp the role of platelet desialylation in the pathogenesis of ITP.
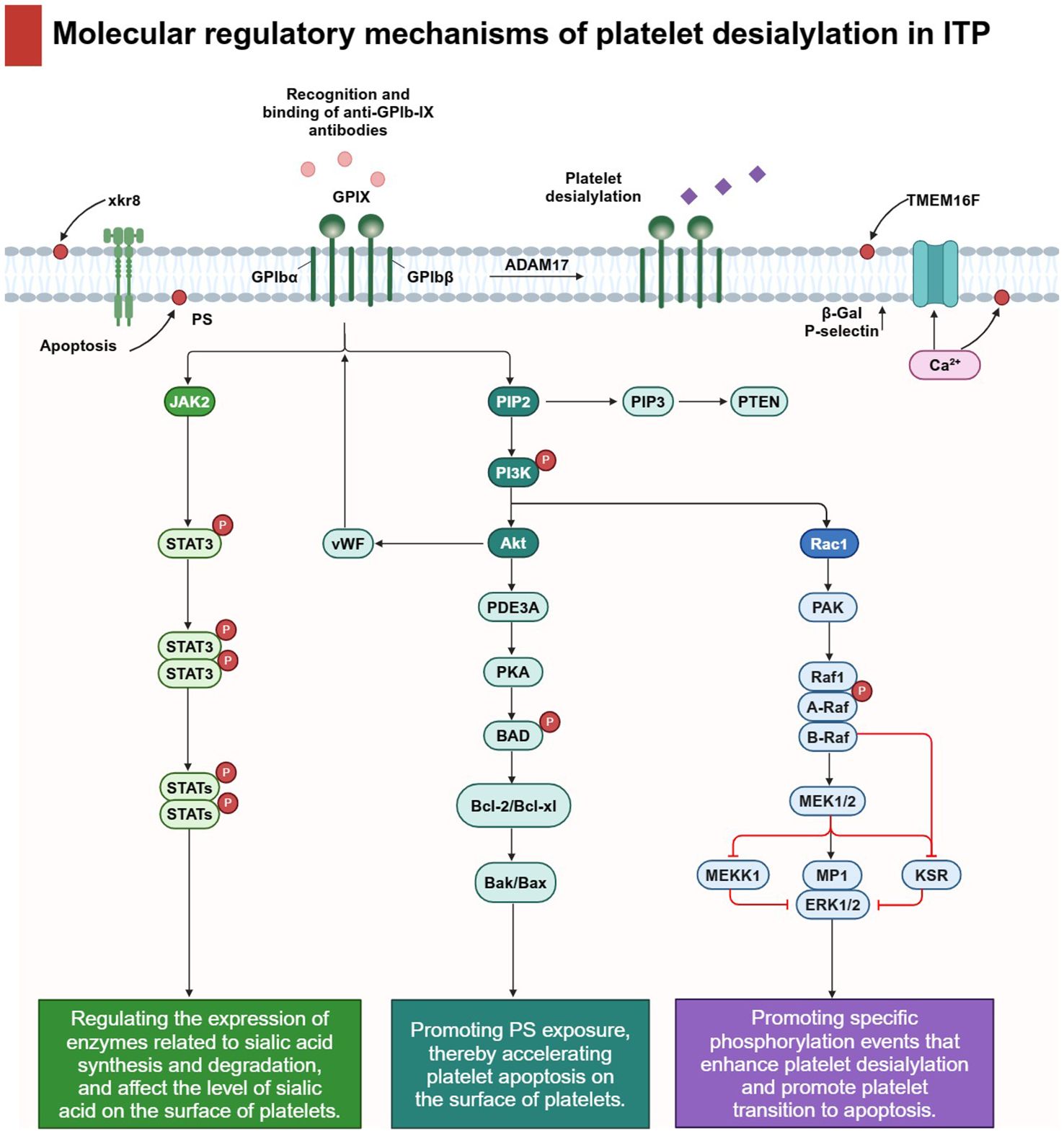
Figure 4 Molecular regulatory mechanisms of platelet desialylation in ITP: The molecular regulatory mechanism of platelet desialylation involves the regulation of several key signaling pathways. In particular, the recognition and binding of anti-GPIb-IX antibodies triggers cascade activation of downstream signals, thereby regulating multiple signaling pathways closely related to platelet function. For example, the PI3K/AKT signaling pathway plays a central role in platelet desialylation, accelerating platelet apoptosis by inhibiting PKA activity and promoting PS exposure. Secondly, JAK/STAT signaling pathway directly affects the level of sialic acid on the surface of platelets and accelerates the degradation process of sialic acid by regulating the expression of enzymes related to sialic acid synthesis and degradation. Finally, activated MAPK signaling pathways enhance platelet desialylation and promote platelet transition to apoptosis by promoting specific phosphorylation events. Activation of these signaling pathways not only promotes biochemical changes in desialylation, but also leads to significant changes in platelet function. Under the action of metalloproteinase ADAM17, the sialic acid on the cell surface is clipped and activates the anti-apoptotic signaling pathway within the cell by interacting with receptors on the cell surface. In addition, through the synergic action of calcium-activated TMEM16F and xkr8 molecules, PS is redistributed from the inner lobules of the plasma membrane to the outer lobules, and this change further leads to increased expression of β-Gal and P-selectin, which intensifies platelet activation and clearance.
5.1 PI3K/AKT signaling pathway
The PI3K/AKT signaling pathway is a critical cellular pathway with significant implications for the pathogenesis of ITP, playing a pivotal role in the generation of platelet desialylation.
In the context of platelet desialylation, the PI3K/AKT signaling pathway contributes to maintaining platelet stability, inhibiting apoptosis, and promoting cell survival. Activation of PI3K kinase leads to the generation of the intracellular secondary signaling molecule PIP3, subsequently activating the AKT kinase. Activated AKT then mediates platelet desialylation through various pathways. Research has suggested that the decreased platelet counts observed in ITP patients with anti-GPIbα antibodies may be attributed to the activation of the PI3K pathway induced by the anti-GPIbα antibody, leading to AKT activation, a downstream effector of PI3K (66). Subsequently, AKT induces platelet apoptosis by diminishing the activity of protein kinase A (PKA), regulated by phosphodiesterase (PDE3A), and simultaneously instigates platelet activation through the AKT pathway. The exposure of apoptotic and activated platelets to phosphatidylserine (PS) on the membrane surface allows recognition, phagocytosis, and subsequent elimination of platelets by Kupffer cells, resulting in decreased platelet count. Suppression or genetic ablation of AKT or AKT-regulated apoptotic signaling, or the obstruction of PS exposure, can protect platelets from clearance. Inhibiting the biological activity of PDE3A, PKA, PS, etc., or conducting gene knockdown of relevant proteins could potentially mitigate antibody-induced platelet removal and augment the platelet count. Another investigation revealed that Akt not only interacts with the cytoplasmic structural domain of GPIbα but also transmits vWF-GPIbα interaction signaling, culminating in platelet activation (67, 68). Furthermore, the PI3K/AKT signaling pathway may potentially interact with other crucial signaling pathways to accelerate platelet desialylation. For instance, the interaction of AKT with other proteins in the PI3K signaling pathway could trigger the release of calcium ions from platelets and activate the Transmembrane Protein 16F (TMEM16F), thereby influencing the aggregation and adhesion properties of platelets (66, 69). Consequently, the suppression of AKT-mediated apoptosis mitigates platelet clearance in vivo, presenting a potential novel therapeutic approach for treating ITP.
These findings underscore the pivotal regulatory function of the PI3K/AKT signaling pathway in platelet desialylation, thereby impacting the pathogenesis of ITP through its influence on platelet activation and apoptosis.
5.2 JAK/STAT signaling pathway
In the pathogenesis of ITP, the JAK/STAT signaling pathway may regulate platelet desialylation through diverse mechanisms. Initially, its activation may potentially influence the expression of key enzymes involved in sialic acid synthesis, consequently leading to reduced sialic acid production. Subsequently, aberrant JAK/STAT signaling may enhance sialidase activity, accelerating sialic acid degradation and resulting in reduced sialic acid on the platelet surface. Ultimately, this pathway may also govern the expression of genes associated with the biosynthesis and degradation of sialic acid, thereby adjusting the concentration of sialic acid on the platelet surface at the transcriptional level, consequently influencing the process of platelet desialylation. Observations indicate that the AMR interacts with desialylated platelets, modulating the production of TPO via the JAK2 signaling pathway and STAT3 activation, both in vitro and in vivo (33, 70). This discovery offers new perspectives and a deeper understanding of the prevalent adverse clinical effects associated with JAK1/2 inhibition in ITP. Another study found that IL-1β could induce an autocrine loop of IFN-β in hematopoietic cells, activating the JAK/STAT signaling pathway and further mediating the surface translocation of NEU1 on MKs in patients with ITP, leading to platelet desialylation and decreased platelet production. The JAK1/2 inhibitor Baricitinib has demonstrated its ability to reverse platelet desialylation and dysfunction both in vitro and in vivo (71).
The above-mentioned results highlight the significant function of the JAK/STAT signaling pathway in platelet desialylation. It not only regulates the signaling aspects of platelet desialylation but also influences the sialic acid content on the platelet surface by modulating NEU expression.
5.3 MAPK signaling pathway
In platelets, the MAPK signaling pathway serves as a regulator for various cellular processes, including platelet differentiation and apoptosis. Consequently, this pathway might also be associated with platelet sialic acid metabolism and desialylation. Chen et al. observed significantly elevated expression levels of MAPK and mTOR, along with their phosphorylated proteins, in the platelets of ITP patients compared to healthy individuals. This suggests that the activation of the MAPK signaling pathway in the platelets of ITP patients might be associated with platelet desialylation and apoptosis (72). Furthermore, the activation of cytoplasmic phospholipase A2, mediated by p38 MAPK, resulted in the release of arachidonic acid from membrane phospholipids, causing the shedding of 14–3-3ζ from Bad. Subsequently, 14–3-3ζ, bound to the cytoplasmic region of GPIbα, initiates platelet desialylation and apoptosis (73, 74). The aforementioned findings indicate that the MAPK signaling pathway could play a pivotal role in the process of platelet desialylation, providing valuable insights into the modulating of this process and enhancing our understanding of the pathogenesis of ITP. However, the precise influence of this signaling pathway on the process of platelet desialylation remains to be clarified, necessitating further research in the future.
In conclusion, the molecular regulatory mechanisms of platelet desialylation involve the modulation of multiple signaling pathways. These regulatory mechanisms are crucial for maintaining platelet stability, functionality, and lifespan. Aberrant activation or impairment of these signaling pathways could lead to a decrease in platelet count and abnormal function. Hence, a comprehensive exploration of the molecular regulatory mechanisms of platelet desialylation is anticipated to provide new pathways and strategies for the accurate diagnosis and personalized therapy of ITP.
6 The potential application of sialylation modulation strategies in the treatment of ITP
The treatment strategy for ITP should adhere to the principles of stratification and personalization to ensure sustained platelet counts at safe levels and reduce incidents of bleeding while minimizing adverse effects. Glucocorticoids remain the primary therapeutic agent for ITP. However, a staggering 98% of patients undergoing prolonged hormonal therapy experience side effects, leading to 38% discontinuing or reducing their medication due to intolerance. Additionally, some patients exhibit no response to glucocorticoids or experience a disease relapse upon cessation of hormonal treatment (75, 76). Furthermore, certain strategies such as the administration of rituximab (77, 78) and splenectomy (79, 80) aim to manage ITP by reducing platelet destruction. However, there remains a subset of patients for whom these treatments are ineffective or who encounter postoperative recurrence issues (81). In recent years, the emergence of sialidase inhibitors has opened up novel therapeutic avenues for ITP patients, showing enhanced efficacy in clinical management (Table 2). Currently, primary sialidase inhibitors include Oseltamivir and 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA), which may enhance platelet production by inhibiting platelet desialylation, consequently augmenting platelet counts in ITP patients.
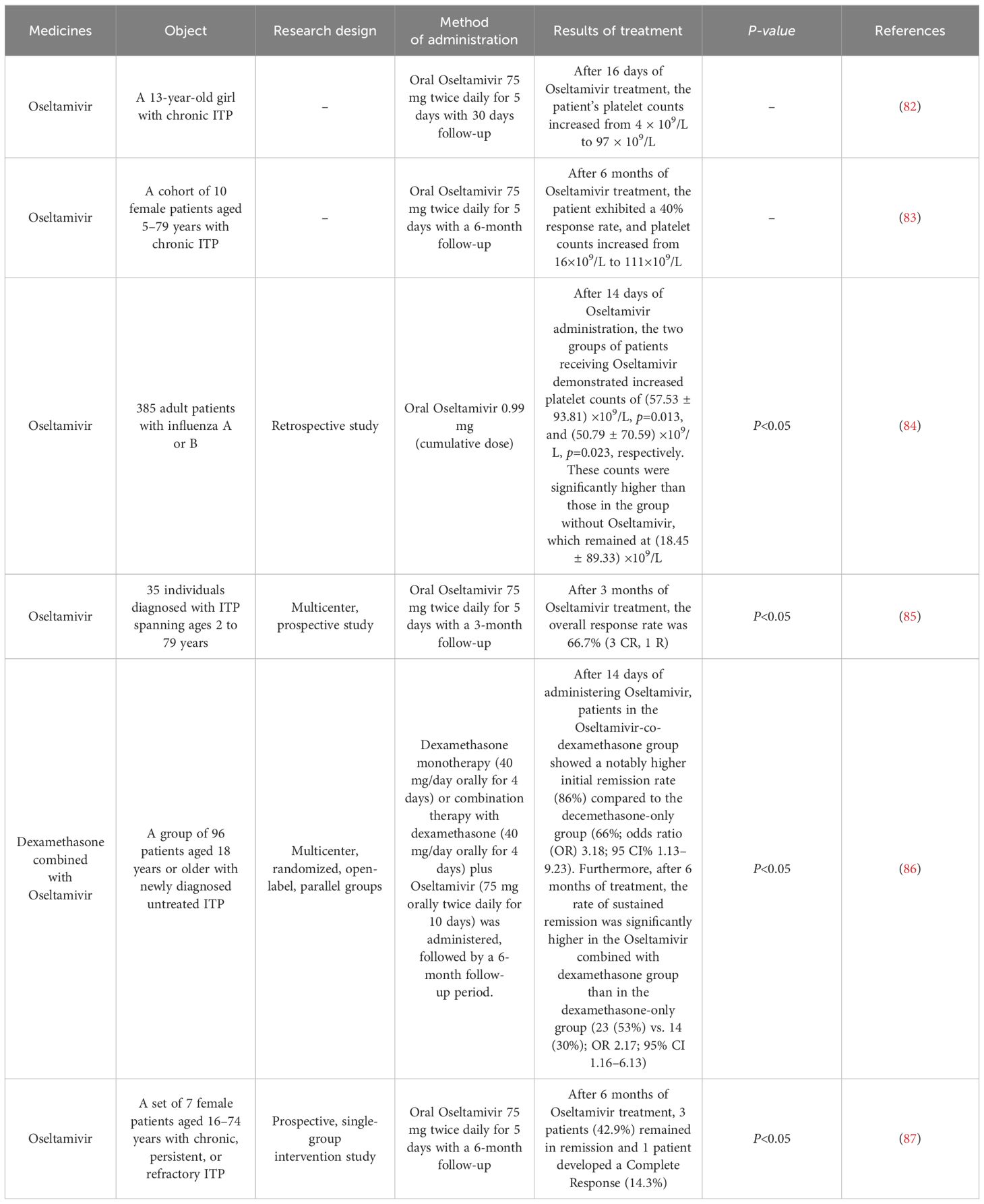
Table 2 Therapeutic Effect of Sialidase Inhibitors.
Oseltamivir is a specific sialidase inhibitor widely utilized in clinical settings for the prophylaxis and therapy of Influenza A or B virus infection, stands out among these inhibitors (88). Compared to existing ITP treatment drugs, Oseltamivir mitigates the immune cells’ onslaught on platelets by interfering with the structure of sialic acids on their surface. Meanwhile, this oral therapy is clinically readily available and more cost-effective compared to intravenous immunoglobulin therapy and most second-line therapies. Furthermore, re-administration of Oseltamivir during relapse resulted in higher remission rates. Multiple independent retrospective studies indicate that Oseltamivir may mitigate the destruction of peripheral platelets by inhibiting platelet desialylation and restoring platelet counts in ITP patients (82–84, 89, 90). Combining Oseltamivir with other therapeutic approaches has the potential to optimize current first-line therapy, potentially enhancing platelet responses to improve the long-term prognosis of ITP patients. In a clinical study, four out of six patients with refractory ITP who were administered Oseltamivir in conjunction with other ITP medications achieved remission and exhibited higher initial response rates (85). A recently published Phase II trial investigating the efficacy of Oseltamivir coupled with dexamethasone for treating ITP in Lancet Hematology indicated that the group treated with the Oseltamivir-dexamethasone combination demonstrated significantly higher effectiveness in treating ITP compared to the group treated with dexamethasone alone (86). Although additional studies have explored the potential of Oseltamivir as a novel target therapy in ITP, more comprehensive large-scale clinical trials are required to systematically assess its long-term effectiveness and side effects (such as nausea, vomiting, headache, muscle pain and drowsiness). Substantial evidence-based medicine is needed to validate the safety and effectiveness of Oseltamivir as a first-line therapeutic choice for ITP.
DANA, a synthetic inhibitor of mammalian, bacterial, and viral sialidases, has been investigated as a potential therapeutic agent for ITP. Several in vivo experiments utilizing a mouse model of ITP showed that DANA effectively decreased platelet breakdown by inhibiting platelet desialylation (91, 92). These promising results suggest that DANA holds potential as a viable therapeutic strategy for ITP, providing valuable insights for future clinical investigations and the advancement of related pharmaceuticals.
The aforementioned findings propose that aberrant desialylation alterations in ITP could present novel therapeutic targets. Consequently, inhibiting desialylation using sialidase inhibitors is expected to emerge as a fresh therapeutic approach for treating patients diagnosed with ITP. However, further clinical investigations are necessary to confirm the safety and long-term efficacy of these medications, as well as to determine the optimal methods for their utilization.
7 Conclusions and perspective
The classical mechanisms of ITP mainly involve autoantibodies and the destruction of platelets mediated by the immune system (93, 94). However, recent research indicates that the Fc-independent pathway of platelet clearance also plays a part in the pathogenesis of ITP, with desialylation recognized as a new mechanism for Fc-independent platelet clearance. NEUs facilitate platelet desialylation, leading to Fc-independent platelet clearance in patients with ITP via the AMR.
Given the complexity of the biological processes driving the pathogenesis of ITP, potential directions for future research towards mitigating platelet desialylation could include the following four areas: (1) reducing the aging of circulating platelets and the stimulation of the immune system due to viral and bacterial infections and their associated antigens; (2) regulating the expression of antiplatelet antibodies and the formation of the sialic acid-glycoprotein complex to prevent platelet reduction and dysfunction caused by their abnormal expression or mutation; (3) through comprehensive research of platelet desialylation, exploring specific targets and conducting subsequent experiments to regulate the degree of platelet desialylation using AMR inhibitors and gene knockout techniques, to reduce the production of antiplatelet antibodies; (4) identifying specific biomarkers associated with platelet desialylation, such as those that could modulate NEU activity based on variations in platelet counts.
In conclusion, the process of platelet desialylation significantly impacts the pathogenesis of ITP. Therefore, a comprehensive exploration of the factors causing platelet desialylation, the underlying biochemistry and molecular regulation of this process, and the application of desialylation regulation strategies for the treatment of ITP may help develop new therapeutic approaches and techniques for this disease. It will also facilitate the accurate diagnosis and individualized treatment of ITP patients to enhance treatment effectiveness and improve their overall well-being while reducing the burden of immune-related platelet disorders on the healthcare system.
Author contributions
QZ: Conceptualization, Investigation, Methodology, Writing – original draft. MH: Conceptualization, Investigation, Validation, Writing – original draft. ET: Validation, Writing – review & editing. LW: Investigation, Validation, Writing – review & editing. JAL: Investigation, Validation, Writing – review & editing. XL: Methodology, Software, Writing – review & editing. JSL: Software, Supervision, Writing – review & editing. WZ: Conceptualization, Supervision, Writing – review & editing. JW: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the State Key Laboratory of Southwestern Chinese Medicine Resources [Grant number: SKLTCM202313 and SKLTCM2022028], National Natural Science Foundation of China [82074129, 82204666 and 82374073], Sichuan Science and Technology Program [2023NSFSC0691, 2023NSFSC2155, 2022YFS0635, 2022YFS0635-B1 and 2022JDJQ0061], Natural Science Foundation of Sichuan Province (No. 2024NSFSC0610).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AMR, Ashwell-Morell receptor; DANA, 2-deoxy-2,3-didehydro-N-acetylneuraminic acid; ITP, Primary immune thrombocytopenia; MKs, Megakaryocytes; NEUs, Neuraminidases; PDE3A, Phosphodiesterase; PKA, Protein kinase A; PS, Phosphatidylserine; TMEM16F, Transmembrane protein 16F; TPO, Thrombopoietin; vWF, von Willebrand factor; β-Gal, β-galactose; N-GlcNAC, N-acetyl glucosamine.
References
1. Cao J, Ji L, Zhan Y, Shao X, Xu P, Wu B, et al. MST4 kinase regulates immune thrombocytopenia by phosphorylating STAT1-mediated M1 polarization of macrophages. Cell Mol Immunol. (2023) 20:1413–27. doi: 10.1038/s41423–023-01089–8
2. Malik A, Sayed A, Han P, Tan M, Watt E, Constantinescu-Bercu A, et al. The role of CD8+ T-cell clones in immune thrombocytopenia. BLOOD. (2023) 141:2417–29. doi: 10.1182/blood.2022018380
3. Mahajna H, Verstockt B, Bergemalm D, Castiglione F, Rodriguez-Moranta F, Savarino E, et al. Idiopathic Thrombocytopaenic Purpura associated with Inflammatory Bowel Disease: a multicentre ECCO CONFER case series. J Crohns Colitis. (2023) 17:722–7. doi: 10.1093/ecco-jcc/jjac179
4. Jiang Y, Cheng Y, Ma S, Li T, Chen Z, Zuo X, et al. Systemic lupus erythematosus-complicating immune thrombocytopenia: From pathogenesis to treatment. J Autoimmun. (2022) 132:102887. doi: 10.1016/j.jaut.2022.102887
5. Terrell DR, Beebe LA, Vesely SK, Neas BR, Segal JB, George JN. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol. (2010) 85:174–80. doi: 10.1002/ajh.21616
6. Moulis G, Palmaro A, Montastruc J-L, Godeau B, Lapeyre-Mestre M, Sailler L. Epidemiology of incident immune thrombocytopenia: a nationwide population-based study in France. Blood. (2014) 124:3308–15. doi: 10.1182/blood-2014–05-578336
7. Zheng S, Ahmadi Z, Leung H, Wong R, Yan F, Perdomo J, et al. Antiplatelet antibody predicts platelet desialylation and apoptosis in immune thrombocytopenia. Haematologica. (2022) 107:2195–205. doi: 10.3324/haematol.2021.279751
8. Deuson R, Danese M, Mathias SD, Schoonen M, Fryzek J. The burden of immune thrombocytopenia in adults: evaluation of the thrombopoietin receptor agonist romiplostim. J Med Eco. (2012) 15:956–76. doi: 10.3111/13696998.2012.688902
9. Miltiadous O, Hou M, Bussel JB. Identifying and treating refractory ITP: difficulty in diagnosis and role of combination treatment. Blood. (2020) 135:472–90. doi: 10.1182/blood.2019003599
10. Visweshwar N, Ayala I, Jaglal M, Killeen R, Sokol L, Laber DA, et al. Primary immune thrombocytopenia: a “diagnosis of exclusion”? Blood Coagul Fibrinol. (2022) 33:289–94. doi: 10.1097/MBC.0000000000001144
11. Mingot-Castellano ME, Bastida JM, Caballero-Navarro G, Entrena Ureña L, González-López TJ, González-Porras JR, et al. Novel therapies to address unmet needs in ITP. Pharm (Basel). (2022) 15:779. doi: 10.3390/ph15070779
12. Weitz I, Liebman H. Complement in immune thrombocytopenia (ITP): The role of complement in refractory ITP. Br J Haematol. (2023) 203:96–100. doi: 10.1111/bjh.19070
13. Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood. (2009) 113:6511–21. doi: 10.1182/blood-2009–01-129155
14. Chen Y, Hu J, Chen Y. Platelet desialylation and TFH cells-the novel pathway of immune thrombocytopenia. Exp Hematol Oncol. (2021) 10:21. doi: 10.1186/s40164–021-00214–5
15. Li J, van der Wal DE, Zhu G, Xu M, Yougbare I, Ma L, et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun. (2015) 6:7737. doi: 10.1038/ncomms8737
16. Tao L, Zeng Q, Li J, Xu M, Wang J, Pan Y, et al. Platelet desialylation correlates with efficacy of first-line therapies for immune thrombocytopenia. J Hematol Oncol. (2017) 10:46. doi: 10.1186/s13045–017-0413–3
17. Aakhus AM, Stavem P, Hovig T, Pedersen TM, Solum NO. Studies on a patient with thrombocytopenia, giant platelets and a platelet membrane glycoprotein Ib with reduced amount of sialic acid. Br J Haematol. (1990) 74:320–9. doi: 10.1111/j.1365-2141.1990.tb02590.x
18. Sørensen AL, Rumjantseva V, Nayeb-Hashemi S, Clausen H, Hartwig JH, Wandall HH, et al. Role of sialic acid for platelet life span: exposure of beta-galactose results in the rapid clearance of platelets from the circulation by asialoglycoprotein receptor-expressing liver macrophages and hepatocytes. Blood. (2009) 114:1645–54. doi: 10.1182/blood-2009–01-199414
19. Roeser A, Lazarus A, Mahévas M. B cells and antibodies in refractory immune thrombocytopenia. Br J Haematol. (2023) 203:43–53. doi: 10.1111/bjh.18773
20. Li M, Li X, Fan K, Yu Y, Gong J, Geng S, et al. Platelet desialylation is a novel mechanism and a therapeutic target in thrombocytopenia during sepsis: an open-label, multicenter, randomized controlled trial. J Hematol Oncol. (2017) 10:104. doi: 10.1186/s13045–017-0476–1
21. Josefsson EC, Gebhard HH, Stossel TP, Hartwig JH, Hoffmeister KM. The macrophage alphaMbeta2 integrin alphaM lectin domain mediates the phagocytosis of chilled platelets. J Biol Chem. (2005) 280:18025–32. doi: 10.1074/jbc.M501178200
22. Muthiah C, Lian Q, Benz S, Szabo A, Hoffmeister K, Botero JP, et al. An extensive database analysis demonstrates significant increase in platelet quantity in unselected hospitalized patients following treatment with oseltamivir. Haematologica. (2024) 10:283731. doi: 10.3324/haematol.2023.283731
23. Bussel JB, Soff G, Balduzzi A, Cooper N, Lawrence T, Semple JW. A review of romiplostim mechanism of action and clinical applicability. Drug Des Devel Ther. (2021) 15:2243–68. doi: 10.2147/DDDT.S299591
24. Quach ME, Dragovich MA, Chen W, Syed AK, Cao W, Liang X, et al. Fc-independent immune thrombocytopenia via mechanomolecular signaling in platelets. Blood. (2018) 131:787–96. doi: 10.1182/blood-2017–05-784975
25. Zeng DF, Chen F, Wang S, Chen SL, Xu Y, Shen MQ, et al. Autoantibody against integrin α(v) β(3) contributes to thrombocytopenia by blocking the migration and adhesion of megakaryocytes. J Thromb Haemost. (2018) 16:1843–56. doi: 10.1111/jth.14214
26. Qi J, You T, Pan T, Wang Q, Zhu L, Han Y. Downregulation of hypoxia-inducible factor-1α contributes to impaired megakaryopoiesis in immune thrombocytopenia. Thromb Haemost. (2017) 117:1875–86. doi: 10.1160/TH17–03-0155
27. Hoffmeister KM, Felbinger TW, Falet H, Denis CV, Bergmeier W, Mayadas TN, et al. The clearance mechanism of chilled blood platelets. Cell. (2003) 112:87–97. doi: 10.1016/s0092–8674(02)01253–9
28. Hoffmeister KM, Josefsson EC, Isaac NA, Clausen H, Hartwig JH, Stossel TP. Glycosylation restores survival of chilled blood platelets. Science. (2003) 301:1531–4. doi: 10.1126/science.1085322
29. Grewal PK, Uchiyama S, Ditto D, Varki N, Le DT, Nizet V, et al. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat Med. (2008) 14:648–55. doi: 10.1038/nm1760
30. Rumjantseva V, Grewal PK, Wandall HH, Josefsson EC, Sørensen AL, Larson G, et al. Dual roles for hepatic lectin receptors in the clearance of chilled platelets. Nat Med. (2009) 15:1273–80. doi: 10.1038/nm.2030
31. Rumjantseva V, Hoffmeister KM. Novel and unexpected clearance mechanisms for cold platelets. Transfus Apher Sci. (2010) 42:63–70. doi: 10.1016/j.transci.2009.10.008
32. Jansen AJG, Josefsson EC, Rumjantseva V, Liu QP, Falet H, Bergmeier W, et al. Desialylation accelerates platelet clearance after refrigeration and initiates GPIbα metalloproteinase-mediated cleavage in mice. Blood. (2012) 119:1263–73. doi: 10.1182/blood-2011–05-355628
33. Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falet H, et al. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med. (2015) 21:47–54. doi: 10.1038/nm.3770
34. Ramírez-López A, Román M, Manzano E, Acuña P, Arias-Salgado E, Salces M, et al. The importance of platelet glycoside residues in the haemostasis of patients with immune thrombocytopaenia. J Clin Med. (2021) 10:1661. doi: 10.3390/jcm10081661
35. Lv Y, Shi H, Liu H, Zhou L. Current therapeutic strategies and perspectives in refractory ITP: What have we learned recently? Front Immunol. (2022) 13:953716. doi: 10.3389/fimmu.2022.953716
36. Park J, Choi H, Jung K, Lee K, Shim J, Park K, et al. Production of recombinant human IgG1 Fc with beneficial N-glycosylation pattern for anti-inflammatory activity using genome-edited chickens. Commun Biol. (2023) 6:589. doi: 10.1038/s42003–023-04937–5
37. Provan D, Newland A. Investigational drugs for immune thrombocytopenia. Expert Opin Investig Drugs. (2022) 31:715–27. doi: 10.1080/13543784.2022.2075340
38. Li J, Callum JL, Lin Y, Zhou Y, Zhu G, Ni H. Severe platelet desialylation in a patient with glycoprotein Ib/IX antibody-mediated immune thrombocytopenia and fatal pulmonary hemorrhage. Haematologica. (2014) 99:e61–63. doi: 10.3324/haematol.2013.102897
39. Zhang L, Zhang M, Du X, Cheng Y, Cheng G. Safety and efficacy of eltrombopag plus pulsed dexamethasone as first-line therapy for immune thrombocytopenia. Br J Haematol. (2020) 189:369–78. doi: 10.1111/bjh.16327
40. Audia S, Mahévas M, Nivet M, Ouandji S, Ciudad M, Bonnotte B. Immune thrombocytopenia: recent advances in pathogenesis and treatments. Hemasphere. (2021) 5:e574. doi: 10.1097/HS9.0000000000000574
41. Qiu J, Liu X, Li X, Zhang X, Han P, Zhou H, et al. CD8(+) T cells induce platelet clearance in the liver via platelet desialylation in immune thrombocytopenia. Sci Rep. (2016) 6:27445. doi: 10.1038/srep27445
42. van der Wal DE, Davis AM, Mach M, Marks DC. The role of neuraminidase 1 and 2 in glycoprotein Ibα-mediated integrin αIIbβ3 activation. Haematologica. (2020) 105:1081–94. doi: 10.3324/haematol.2019.215830
43. Gorelik A, Illes K, Mazhab-Jafari MT, Nagar B. Structure of the immunoregulatory sialidase NEU1. Sci Adv. (2023) 9:eadf8169. doi: 10.1126/sciadv.adf8169
44. Chen QQ, Liu K, Shi N, Ma G, Wang P, Xie HM, et al. Neuraminidase 1 promotes renal fibrosis development in male mice. Nat Commun. (2023) 14:1713. doi: 10.1038/s41467–023-37450–8
45. Mozzi A, Mazzacuva P, Zampella G, Forcella ME, Fusi PA, Monti E. Molecular insight into substrate recognition by human cytosolic sialidase NEU2. Proteins. (2012) 80:1123–32. doi: 10.1002/prot.24013
46. Dai W, Wu G, Liu K, Chen Q, Tao J, Liu H, et al. Lactate promotes myogenesis via activating H3K9 lactylation-dependent up-regulation of Neu2 expression. J Cachexia Sarcopenia Muscle. (2023) 14:2851–65. doi: 10.1002/jcsm.13363
47. Howlader MA, Li C, Zou C, Chakraberty R, Ebesoh N, Cairo CW. Neuraminidase-3 is a negative regulator of LFA-1 adhesion. Front Chem. (2019) 7:791. doi: 10.3389/fchem.2019.00791
48. Karhadkar TR, Chen W, Pilling D, Gomer RH. Inhibitors of the sialidase NEU3 as potential therapeutics for fibrosis. Int J Mol Sci. (2022) 24:239. doi: 10.3390/ijms24010239
49. Yang WH, Westman JS, Heithoff DM, Sperandio M, Cho JW, Mahan MJ, et al. Neu3 neuraminidase induction triggers intestinal inflammation and colitis in a model of recurrent human food-poisoning. Proc Natl Acad Sci U.S.A. (2021) 118:e2100937118. doi: 10.1073/pnas.2100937118
50. Zhang X, Dou P, Akhtar ML, Liu F, Hu X, Yang L, et al. NEU4 inhibits motility of HCC cells by cleaving sialic acids on CD44. Oncogene. (2021) 40:5427–40. doi: 10.1038/s41388–021-01955–7
51. Bosmann HB, Myers MW, Dehond D, Ball R, Case KR. Mitochondrial autonomy. Sialic acid residues on the surface of isolated rat cerebral cortex and liver mitochondria. J Cell Biol. (1972) 55:147–60. doi: 10.1083/jcb.55.1.147
52. Ma JY, Chen ZP, Gu H, Fu LL, Wu RH. Research progress of desialylation in immune thrombocytopenia. Chin J Exp Hematol. (2018) 26:928–32. doi: CNKI:SUN:XYSY.0.2018-03-053
53. Kim D. Recent advances in treatments of adult immune thrombocytopenia. Blood Res. (2022) 57:112–9. doi: 10.5045/br.2022.2022038
54. Marín-Quílez A, Díaz-Ajenjo L, Di Buduo C, Zamora-Cánovas A, Lozano M, Benito R, et al. Inherited thrombocytopenia caused by variants in crucial genes for glycosylation. Int J Mol Sci. (2023) 24:5109. doi: 10.3390/ijms24065109
55. Xie XJ, Chen ZP. Research progress on the pathogenesis of pediatric immune thrombocytopenia. Chin J Pract Pediatr. (2021) 36:90–4. doi: 10.19538/j.ek2021020603
56. Li J, Karakas D, Xue F, Chen Y, Zhu G, Yucel YH, et al. Desialylated platelet clearance in the liver is a novel mechanism of systemic immunosuppression. Res (Wash D C). (2023) 6:236. doi: 10.34133/research.0236
57. Amini S, Porcelijn L, Sobels A, Kartachova M, de Haas M, Zwaginga J, et al. Anti-glycoprotein antibodies and sequestration pattern of indium-labeled platelets in immune thrombocytopenia. Blood Adv. (2022) 6:1797–803. doi: 10.1182/bloodadvances.2021004822
58. Pillois X, Peters P, Segers K, Nurden AT. In silico analysis of structural modifications in and around the integrin αIIb genu caused by ITGA2B variants in human platelets with emphasis on Glanzmann thrombasthenia. Mol Genet Genomic Med. (2018) 6:249–60. doi: 10.1002/mgg3.365
59. Botero JP, Lee K, Branchford BR, Bray PF, Freson K, Lambert MP, et al. Glanzmann thrombasthenia: genetic basis and clinical correlates. Haematologica. (2020) 105:888–94. doi: 10.3324/haematol.2018.214239
60. Urbanus RT, van der Wal DE, Koekman CA, Huisman A, van den Heuvel DJ, Gerritsen HC, et al. Patient autoantibodies induce platelet destruction signals via raft-associated glycoprotein Ibα and Fc RIIa in immune thrombocytopenia. Haematologica. (2013) 98:e70–2. doi: 10.3324/haematol.2013.087874
61. Li C, Piran S, Chen P, Lang S, Zarpellon A, Jin JW, et al. The maternal immune response to fetal platelet GPIbα causes frequent miscarriage in mice that can be prevented by intravenous IgG and anti-FcRn therapies. J Clin Invest. (2011) 121:4537–47. doi: 10.1172/JCI57850
62. Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Castanheira FVES, et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J Exp Med. (2020) 217:e20190723. doi: 10.1084/jem.20190723
63. Zhang X-H, Wang Q-M, Zhang J-M, Feng F-E, Wang F-R, Chen H, et al. Desialylation is associated with apoptosis and phagocytosis of platelets in patients with prolonged isolated thrombocytopenia after allo-HSCT. J Hematol Oncol. (2015) 8:116. doi: 10.1186/s13045–015-0216–3
64. Hoffmeister KM. The role of lectins and glycans in platelet clearance. J Thromb Haemost. (2011) 9 Suppl 1:35–43. doi: 10.1111/j.1538-7836.2011.04276.x
65. Wang Y, Chen W, Zhang W, Lee-Sundlov MM, Casari C, Berndt MC, et al. ODesialylation of -glycans on glycoprotein Ibα drives receptor signaling and platelet clearance. Haematologica. (2021) 106:220–9. doi: 10.3324/haematol.2019.240440
66. Chen M, Yan R, Zhou K, Li X, Zhang Y, Liu C, et al. Akt-mediated platelet apoptosis and its therapeutic implications in immune thrombocytopenia. Proc Natl Acad Sci U.S.A. (2018) 115:E10682–91. doi: 10.1073/pnas.1808217115
67. Ruan J, Sun R, Wang J, Sui X, Qu H, Yuan D, et al. Gene mutations in the PI3K/Akt signaling pathway were related to immune thrombocytopenia pathogenesis. MEDICINE. (2023) 102:e32947. doi: 10.1097/MD.0000000000032947
68. Yin H, Stojanovic A, Hay N, Du X. The role of Akt in the signaling pathway of the glycoprotein Ib-IX induced platelet activation. Blood. (2008) 111:658–65. doi: 10.1182/blood-2007–04-085514
69. Quach ME, Chen W, Li R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood. (2018) 131:1512–21. doi: 10.1182/blood-2017–08-743229
70. Reusswig F, Fazel Modares N, Brechtenkamp M, Wienands L, Krüger I, Behnke K, et al. Efficiently restored thrombopoietin production by Ashwell-Morell receptor and IL-6R induced Janus kinase 2/signal transducer and activator of transcription signaling early after partial hepatectomy. Hepatology. (2021) 74:411–27. doi: 10.1002/hep.31698
71. Zhao P, An Z, Fu HX, Liu HX, Yang L, Qu QY, et al. S296: IL-1B signalling induces, and baricitinib alleviates NEU1-mediated megakaryocyte desialylation in immune thrombocytopenia. HemaSphere. (2023) 7:e73161e9. doi: 10.1097/01.HS9.0000968096.73161.e9
72. Chen Y, Luo L, Zheng Y, Zheng Q, Zhang N, Gan D, et al. Association of platelet desialylation and circulating follicular helper T cells in patients with thrombocytopenia. Front Immunol. (2022) 13:810620. doi: 10.3389/fimmu.2022.810620
73. Gitz E, Koekman CA, van den Heuvel DJ, Deckmyn H, Akkerman JW, Gerritsen HC, et al. Improved platelet survival after cold storage by prevention of glycoprotein Ibα clustering in lipid rafts. Haematologica. (2012) 97:1873–81. doi: 10.3324/haematol.2012.066290
74. van der Wal D, Gitz E, Du V, Lo K, Koekman C, Versteeg S, et al. Arachidonic acid depletion extends survival of cold-stored platelets by interfering with the [glycoprotein Ibα-14–3-3ζ] association. Haematologica. (2012) 97:1514–22. doi: 10.3324/haematol.2011.059956
75. Hu XY, Zhao T, Zhang SY, Le LX, Song PF, Li J. Drug combination treatment for primary immune thrombocytopenia in adults. Chin Med Pharm. (2023) 13:45–48 + 85. doi: 10.20116/j.issn2095–0616.2023.14.10
76. Hamed E, Ibrahim A, Meabed M, Khalaf A, El Demerdash D, Elgendy M, et al. Therapeutic outcomes of high dose-dexamethasone versus prednisolone. PHARMACEUTICALS. (2023) 16:1215. doi: 10.3390/ph16091215
77. Mageau A, Bonnotte B, Ebbo M, Dossier A, Galicier L, Souchaud-Debouverie O, et al. Efficacy and safety of two rituximab biosimilars for treating immune thrombocytopenia: a reference-product matched study. PLATELETS. (2023) 34:2200848. doi: 10.1080/09537104.2023.2200848
78. Sanfilippo K, Cuker A. TPO-RAs and ITP remission: cause or coincidence? BLOOD. (2023) 141:2790–1. doi: 10.1182/blood.2023020243
79. Canales-Herrerias P, Crickx E, Broketa M, Sokal A, Chenon G, Azzaoui I, et al. High-affinity autoreactive plasma cells disseminate through multiple organs in patients with immune thrombocytopenic purpura. J Clin Invest. (2022) 132:e153580. doi: 10.1172/JCI153580
80. Mendoza A, Alvarez-Román M, Monzón-Manzano E, Acuña P, Arias-Salgado E, Rivas-Pollmar I, et al. Study of platelet kinetics in immune thrombocytopenia to predict splenectomy response. Br J Haematol. (2023) 204:315–23. doi: 10.1111/bjh.19145
81. Cantoni S, Carpenedo M, Nichelatti M, Sica L, Rossini S, Milella M, et al. Clinical relevance of antiplatelet antibodies and the hepatic clearance of platelets in patients with immune thrombocytopenia. Blood. (2016) 128:2183–5. doi: 10.1182/blood-2016–03-708388
82. Bigot P, Auffret M, Gautier S, Weinborn M, Ettahar N-K, Coupé P. Unexpected platelets elevation in a patient with idiopathic thrombocytopenia treated with oseltamivir for influenza infection. Fundam Clin Pharmacol. (2016) 30:483–5. doi: 10.1111/fcp.12213
83. Shao L, Wu Y, Zhou H, Qin P, Ni H, Peng J, et al. Successful treatment with oseltamivir phosphate in a patient with chronic immune thrombocytopenia positive for anti-GPIb/IX autoantibody. Platelets. (2015) 26:495–7. doi: 10.3109/09537104.2014.948838
84. Shaim H, McCaffrey P, Trieu JA, DeAnda A, Yates SG. Evaluating the effects of oseltamivir phosphate on platelet counts: a retrospective review. Platelets. (2020) 31:1080–4. doi: 10.1080/09537104.2020.1714576
85. Revilla N, Corral J, Miñano A, Mingot-Castellano ME, Campos RM, Velasco F, et al. Multirefractory primary immune thrombocytopenia; targeting the decreased sialic acid content. Platelets. (2019) 30:743–51. doi: 10.1080/09537104.2018.1513476
86. Sun L, Wang J, Shao L, Yuan C, Zhao H, Li D, et al. Dexamethasone plus oseltamivir versus dexamethasone in treatment-naive primary immune thrombocytopenia: a multicentre, randomised, open-label, phase 2 trial. Lancet Haematol. (2021) 8:e289–98. doi: 10.1016/S2352–3026(21)00030–2
87. Colunga-Pedraza P, Pena-Lozano S, Sanchez-Rendon E, de la Garza-salazar F, Colunga-Pedraza J, Gomez-De Leon A, et al. Oseltamivir as rescue therapy for persistent, chronic, or refractory immune thrombocytopenia: a case series and review of the literature. J Thromb Thromb. (2022) 54:360–6. doi: 10.1007/s11239–022-02651–3
88. Hanula R, Bortolussi-Courval É, Mendel A, Ward BJ, Lee TC, McDonald EG. Evaluation of oseltamivir used to prevent hospitalization in outpatients with influenza: A systematic review and meta-analysis. JAMA Intern Med. (2023) 184:18–27. doi: 10.1001/jamainternmed.2023.0699
89. Álvarez-Román MT, Rivas Pollmar MI, Bernardino JI, Lozano ML, Martín-Salces M, Fernández-Bello I, et al. Thrombopoietin receptor agonists in conjunction with oseltamivir for immune thrombocytopenia. AIDS. (2016) 30:1141–2. doi: 10.1097/QAD.0000000000001036
90. Jansen AJG, Peng J, Zhao H-G, Hou M, Ni H. Sialidase inhibition to increase platelet counts: A new treatment option for thrombocytopenia. Am J Hematol. (2015) 90:E94–95. doi: 10.1002/ajh.23953
91. van Der Wal DE, Zhu G, Li J, Vadasz B, Issaka Y, Lang S, et al. Desialylation: A novel platelet clearance mechanism and a potential new therapeutic target in anti-GPIb antibody mediated thrombocytopenia. Blood. (2012) 120:265–5. doi: 10.1182/blood.V120.21.265.265
92. Zhang Y, Albohy A, Zou Y, Smutova V, Pshezhetsky AV, Cairo CW. Identification of selective inhibitors for human neuraminidase isoenzymes using C4,C7-modified 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA) analogues. J Med Chem. (2013) 56:2948–58. doi: 10.1021/jm301892f
93. Broome CM, Röth A, Kuter DJ, Scully M, Smith R, Wang J, et al. Safety and efficacy of classical complement pathway inhibition with sutimlimab in chronic immune thrombocytopenia. Blood Adv. (2023) 7:987–96. doi: 10.1182/bloodadvances.2021006864
Keywords: platelets, sialic acid, primary immune thrombocytopenia, pathogenesis, apoptosis
Citation: Zhang Q, Huang M, Thomas ER, Wang L, Liu J, Li X, Luo J, Zou W and Wu J (2024) The role of platelet desialylation as a biomarker in primary immune thrombocytopenia: mechanisms and therapeutic perspectives. Front. Immunol. 15:1409461. doi: 10.3389/fimmu.2024.1409461
Received: 30 March 2024; Accepted: 03 June 2024;
Published: 24 June 2024.
Edited by:
Joseph T.Y. Lau, University at Buffalo, United StatesReviewed by:
Karin Hoffmeister, Versiti Blood Research Institute, United StatesGuoyun Chen, University of Tennessee Health Science Center (UTHSC), United States
Copyright © 2024 Zhang, Huang, Thomas, Wang, Liu, Li, Luo, Zou and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianming Wu, amlhbm1pbmd3dUBzd211LmVkdS5jbg==; Wenjun Zou, em91d2VuanVuQGNkdXRjbS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship