 Rhea Nickerson
Rhea Nickerson Christina S. Thornton
Christina S. Thornton Brent Johnston
Brent Johnston Amy H. Y. Lee
Amy H. Y. Lee Zhenyu Cheng
Zhenyu Cheng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 28 June 2024
Sec. Microbial Immunology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1405376
Pseudomonas aeruginosa is a highly adaptable opportunistic pathogen capable of exploiting barriers and immune defects to cause chronic lung infections in conditions such as cystic fibrosis. In these contexts, host immune responses are ineffective at clearing persistent bacterial infection, instead driving a cycle of inflammatory lung damage. This review outlines key components of the host immune response to chronic P. aeruginosa infection within the lung, beginning with initial pathogen recognition, followed by a robust yet maladaptive innate immune response, and an ineffective adaptive immune response that propagates lung damage while permitting bacterial persistence. Untangling the interplay between host immunity and chronic P. aeruginosa infection will allow for the development and refinement of strategies to modulate immune-associated lung damage and potentiate the immune system to combat chronic infection more effectively.
Pseudomonas aeruginosa is a highly adaptable Gram-negative bacterium which is prevalent in the environment and causes a wide range of opportunistic infections in humans. P. aeruginosa can infect diverse tissues, including the lung, where it can cause acute pneumonia or chronic infections that exacerbate the pathology of conditions including cystic fibrosis (CF), non-CF bronchiectasis (NCFB) and chronic obstructive pulmonary disease (COPD) (1–4). P. aeruginosa is one of the most common and fatal multi-drug resistant bacteria mediating acute pneumonia in hospital settings (5–7). In the context of chronic infections, P. aeruginosa is the most prevalent pathogen in adult persons with CF (pwCF), with recent registry data in the USA, UK, and Canada reporting up to 40-50% are chronically colonized, despite significant therapeutic advances over the last decade (8–11). Chronic P. aeruginosa infection is associated with adverse outcomes and greater mortality in pwCF. P. aeruginosa’s role in other chronic lung conditions remains understudied, though reports are beginning to highlight its importance in the pathology and outcomes of these diseases as well. P. aeruginosa is able to chronically colonize the lungs of COPD patients, in a similar manner to pwCF, where it has been associated with increased respiratory symptoms and inflammatory responses in the airway (2, 4). Similar findings have been reported for NCFB, where P. aeruginosa colonization has been recorded in up to 50% of adults (4, 12). In these cohorts, P. aeruginosa was more likely to cause persistent infections than other common lung pathogens, and was also associated with a greater rate of hospitalizations (12). Taken together, much of the pathogenicity of chronic P. aeruginosa infection stems from maladaptive hyper-inflammatory host immune responses, which fail to clear infection and contribute to self-perpetuating lung damage. However, therapeutic interventions targeting chronic P. aeruginosa are still broadly limited to antibiotic-mediated eradication efforts, which frequently fail in chronic settings due to bacterial adaptations such as biofilm formation and increasing prevalence of multi-drug resistance (13). Better understanding the dysregulation of host immunity in chronic P. aeruginosa infection will lead to the development of novel therapies to modulate immune-associated lung damage and potentiate the immune system to combat chronic infection more effectively.
This review aims to outline key contexts for chronic P. aeruginosa infection, including host factors that influence its transition to chronicity; elements of the immune response which are either protective or pathological; and strategies to modulate chronic P. aeruginosa infection, including antibiotics, vaccines, immunomodulatory therapies, and cystic fibrosis transmembrane conductance regulator (CFTR) modulators.
As an opportunistic pathogen, P. aeruginosa rarely causes infection in healthy, immunocompetent individuals, but frequently capitalizes on immune dysfunction or mechanical disruption for colonization. Reflecting this, P. aeruginosa is not a common cause of community-acquired pneumonia but is among the leading causes of hospital-acquired pneumonia, where it frequently causes more severe illness than other bacterial pathogens due to its array of virulence factors (5, 14–16). P. aeruginosa can exploit patients who are immunocompromised due to other health concerns, such as those undergoing immunotherapy, chemotherapy, or receiving mechanical ventilation (14). The most common outcomes of acute respiratory infection with P. aeruginosa are either bacterial clearance, by the host immune response or antibiotic treatment –or, in the event of a failure to control infection, dissemination of bacteria resulting in sepsis, or respiratory failure as an outcome of uncontrolled infection or excessive immune-mediated damage secondary to infection (16). However, in these settings, acute infections with P. aeruginosa rarely transition to a state of chronicity.
Chronic infections with P. aeruginosa occur in select circumstances, most often in patients with structural chronic lung conditions, such as CF, NCFB, and COPD. These conditions are united by defects in lung innate immunity, such as impairment of the mucociliary elevator, mucus buildup, damage to the lung epithelial barrier, and/or baseline inflammation, which render the lungs more susceptible to infection (Figure 1).
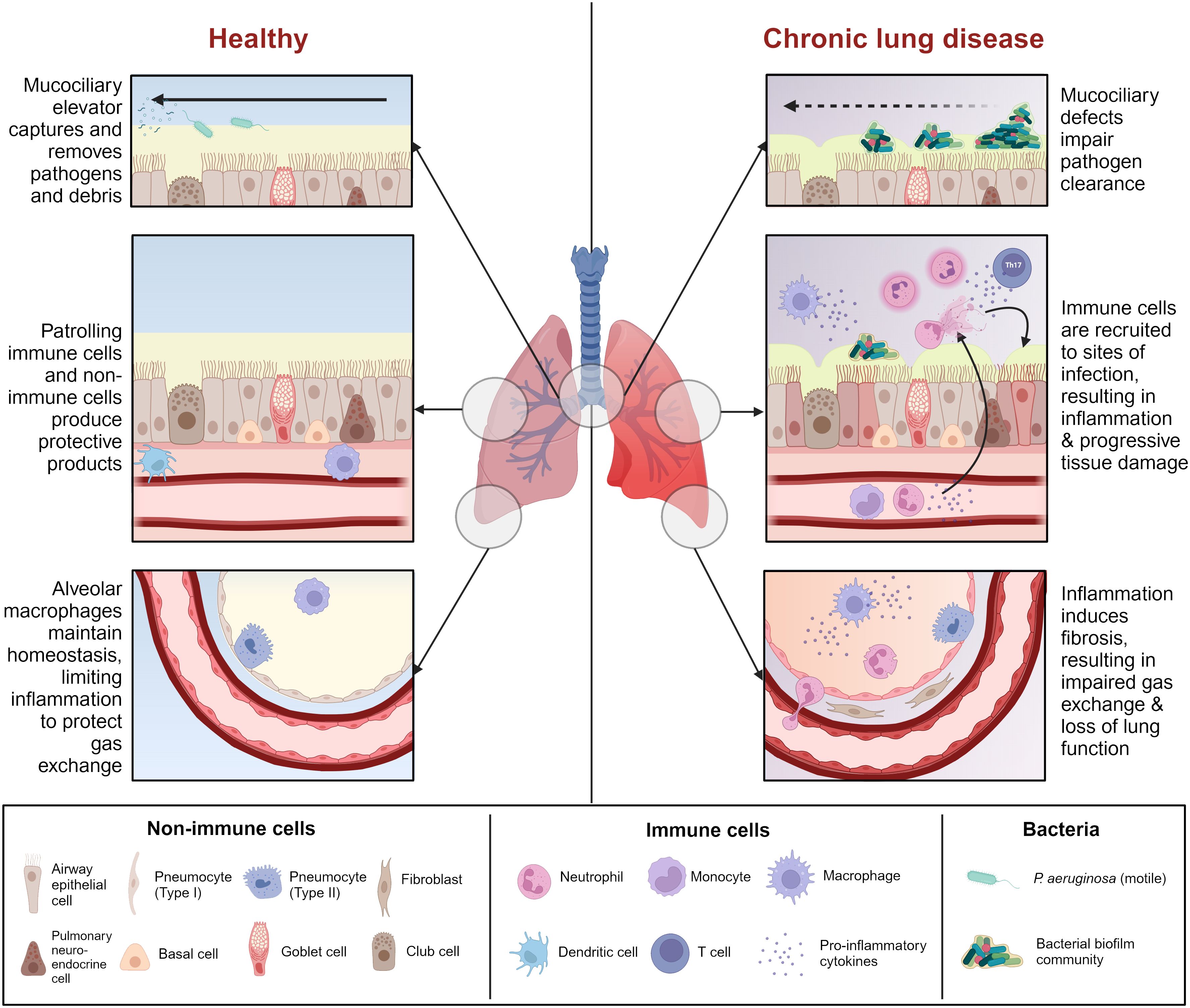
Figure 1 Comparison between healthy airway and chronic lung disease. Chronic lung disease is characterized by mucociliary defects (i.e., mucus dehydration, ciliary dysfunction) which impair initial clearance of pathogens from the airway. Reduced pathogen clearance promotes immune cell recruitment and activation, resulting in a cascade of inflammation and tissue damage which renders the airway more susceptible to infection. Some chronic lung diseases are characterized by pre-existing inflammation which heightens this effect. Prolonged unresolving inflammation leads to tissue remodeling and fibrosis, which affects gas exchange in the alveoli, ultimately leading to lung failure. Figure created with BioRender.com.
CF is the most common autosomal recessive genetic disease in the Caucasian population, affecting approximately 70,000 individuals worldwide (17). CF results from mutations in the cystic fibrosis transmembrane conductance receptor (CFTR), a chloride channel that coordinates ion and fluid transport across the epithelium. CFTR is found throughout the body, and although CF affects numerous organs including the pancreas, intestine, liver, reproductive tract, and sweat glands, the primary cause of morbidity and mortality in CF is respiratory failure (18). In the lungs, CFTR regulates both chloride ion absorption and secretion in distinct cell types to fine-tune the volume and hydration of airway surface liquid (ASL) (19). Mutations in CFTR result in reduced ASL volume and mucus dehydration in the CF airway, leading to mucus plugging and attenuated mucociliary clearance –preventing the clearance of pathogens from the lungs (20, 21). Mucus dehydration and accumulation have been shown to provide a unique niche that promotes biofilm formation by pathogens such as P. aeruginosa, allowing the transition towards a chronic phenotype (22, 23). CFTR mutations may also predispose pwCF to sterile inflammation and impair innate immune cell function, rendering them even more susceptible to infection (24–26). Establishment of chronic bacterial infection results in a self-perpetuating inflammatory response characterized by intense neutrophil recruitment and production of inflammatory mediators, which leads to irreversible airway damage and fibrosis (27). PwCF are classically colonized by specific pathogens, with Staphylococcus aureus predominating in children, but transitioning to P. aeruginosa as the dominant colonizing pathogen once individuals reach adolescence. Several adaptive features, including P. aeruginosa’s ability to better adjust to the progressively diseased lung environment and evade host immunity and/or directly target S. aureus to outcompete it, contribute to dominance of P. aeruginosa in the adult CF lung (28). Other classical CF pathogens include Achromobacter xylosoxidans, Burkholderia cepacia complex, Haemophilus influenzae, and Stenotrophomonas maltophilia (29, 30). However, it is becoming increasingly recognized that chronic respiratory infections in pwCF are polymicrobial, and that CF disease intrinsically perturbs the airway microbiome (31, 32). Recent metagenomic work has demonstrated that the airway microbial communities of pwCF are distinct from healthy individuals, beginning as early as 4 years old, and even in the absence of classical CF pathogens (32). Airway microbial load and composition are unique to each individual with CF, although patterns emerge based on the presence of classical CF pathogens, and the stage of lung disease (32–36). Airway commensal communities have been observed to fragment over time, correlating with the emergence and takeover of CF pathogens such as P. aeruginosa, S. aureus, and S. maltophilia. Similar trends in disruption of airway commensal diversity by pathogens like P. aeruginosa are beginning to be observed for other diseases such as NCFB. Only in end-stage lung disease do bacterial monocultures of dominant CF pathogens like P. aeruginosa take over in the lungs (32). However, it is the sheer prevalence of P. aeruginosa in the CF population, as well as its strong association with worsened outcomes, that has prompted significant research into chronic P. aeruginosa infection.
Other conditions that involve enhanced susceptibility to chronic P. aeruginosa infection also involve defects in the lung mucus layer, mucociliary elevator defects, and inflammatory lung damage. NCFB refers to a wide range of disease etiologies (more than 50 currently recognized) which result in inflammation, dilation, and irreversible thickening of the bronchial walls (37). These airway architectural changes are often accompanied by mucus accumulation, chronic infection of the airways by pathogens like P. aeruginosa, and, similar to CF, persistent neutrophilic inflammation which perpetuates a vicious cycle of airway damage (38). Only 38% of bronchiectasis cases have identifiable causes, which can include congenital causes, such as primary ciliary dyskinesis; genetic causes like alpha-1 antitrypsin deficiency; autoimmune conditions including systemic lupus erythematosus and rheumatoid arthritis; environmental or lifestyle-related causes, including long-term smoking or exposure to pollutants; or post-infectious causes, such as prior infection with various respiratory pathogens, including COVID-19 (38–40). Several idiopathic conditions such as diffuse panbronchiolitis also proceed to bronchiectasis in late-stage disease (41). Following disease onset, NCFB patients are more susceptible to respiratory infections with several key pathogens, of which P. aeruginosa and H. influenzae are the most prevalent (39). A recent study utilizing the European EMBARC registry found that in most southern European countries, over 50% of adults with NCFB were colonized with P. aeruginosa within a one-year period, making it the most common pathogen in this population (39). Recent culture-independent work has shown that a distinct cluster of NCFB patients are characterized by a Pseudomonas-dominant microbiome, characterized by lower alpha diversity and positive correlation with clinical features such as exacerbations (42, 43). As in CF, P. aeruginosa infection is more common in adults than children (44, 45). P. aeruginosa infection is considered a major cause of morbidity in all bronchiectasis patients, defined by increased exacerbations in respiratory pathology, reduced lung function, and increased mortality, particularly when infection becomes chronic (12, 46).
COPD is characterized by progressive airflow limitation, airway inflammation, emphysema, and chronic mucus production resulting from airway narrowing (47). COPD is caused by prolonged exposure to external toxic irritants, such as cigarette smoke and air pollutants, with a variety of genetic and environmental risk factors (47). There are multiple subtypes of COPD that interestingly correlate with different airway pathogen prevalences (48). P. aeruginosa is less common in COPD than in CF or NCFB, with an overall prevalence estimated to be approximately 4%, increasing to 13% in patients with advanced airway obstruction (2, 49, 50). However, COPD affects approximately 100 times as many individuals as CF, and is increasing in incidence, underscoring that P. aeruginosa infections in COPD are still a significant concern (51). P. aeruginosa has been shown to cause both acute infections leading to disease exacerbations, and persistent long-term infections in a subset of patients (52). COPD exacerbations caused by P. aeruginosa are generally more severe, and associated with higher levels of hospitalization and mortality than exacerbations caused by other identifiable pathogens (51). Generally, the COPD patients who fail to clear P. aeruginosa and retain long-term infections are more severely-ill than the patients with short-term infections (53). COPD patients with persistent P. aeruginosa infection followed over years exhibited clonal diversification of a stable infection that evolved toward characteristics such as hypermutability, increased antibiotic resistance, reduced protease production, loss of motility, and biofilm production, all signs of bacterial evolution toward chronicity (2, 54).
The question of differentiating acute from chronic P. aeruginosa infection is one that remains challenging and sometimes leaves past experimental work in the context of “chronic” infection difficult to interpret. Chronic infection has historically been defined in clinical settings by repeated detection of P. aeruginosa culture-positive samples over various timeframes, often during routine testing, although there is no set standard (55). The most commonly-used guidelines are the Leeds criteria, which define chronic infection as greater than 50% P. aeruginosa-positive monthly cough swabs/sputum cultures within a 12-month period (56). While practical for clinical use, this definition does not actually distinguish persistent P. aeruginosa infection from recurrent acute infections in susceptible individuals. Given the association between chronic infection and bacterial adaptations such as antibiotic resistance, biofilm formation, and immune evasion, it is important to understand how chronic P. aeruginosa infections develop, and to be able to identify when infections transition into chronicity.
The development of chronic P. aeruginosa infection often follows a similar trajectory and is associated with various adaptations to the lung environment. Most of the work charting the evolution of chronic P. aeruginosa over time has been done in the CF context; however, recent findings in NCFB and COPD patients are generally converging with this work, despite a few important differences. Initial infection with P. aeruginosa commonly begins in childhood for pwCF (57). The disparate etiologies of NCFB mean that it can begin in childhood or adulthood, with current findings indicating that P. aeruginosa infections are more common in adults with NCFB. COPD affects primarily adults, and so it can be assumed that P. aeruginosa infections are also acquired in adulthood for COPD. P. aeruginosa can be acquired from environmental sources, as it is a ubiquitous environmental pathogen found in soil and water. However, P. aeruginosa can also be acquired from patient-to-patient transmission, which occurs more commonly in CF than NCFB or COPD (58). The P. aeruginosa strains able to cause patient-to-patient transmission are called clonal or epidemic strains, with the Liverpool epidemic strain (LES) being the most notable example (58). Most of these clonal strains have similar adaptations including enhanced biofilm formation and high levels of antibiotic resistance which give them colonization advantages.
Because of the strong association between P. aeruginosa infection and worsened lung function decline, initial P. aeruginosa infections are heavily treated with antibiotics and often successfully cleared. This widespread eradication therapy has led to significant decreases in P. aeruginosa infection rates in children and adolescents with CF, shifting the median age of chronic P. aeruginosa acquisition in this group (8, 57). However, these eradication efforts often eventually fail (59) as P. aeruginosa adapts within the lung to evade antibiotic clearance (60). Related to this, concerns are increasing regarding the rising prevalence of MDR P. aeruginosa strains, including clonal strains which more readily establish chronic infections due to their multi-drug resistance (61). When eradication efforts fail, this often begins an “intermittent” phase of infection, characterized by cycles of colonization, treatment, and recolonization, during which time microbiological culture samples may vary. Eventually, intermittently-colonizing P. aeruginosa acquires mutations and undergoes lifestyle changes which protect the bacteria from both host and antibiotic clearance, resulting in chronic infection (62).
Chronically-infected individuals are generally infected with a single strain of P. aeruginosa which persists and genetically diversifies over a prolonged period of time, leading to significant spatiotemporal diversity within different regions of the lung (58, 60, 63). Occasionally, individuals can be superinfected with new P. aeruginosa strains which usually outcompete or are outcompeted by the resident population (58). Longitudinal tracking of P. aeruginosa populations over a ten-year period has shown that during the early stages of chronic infection, the P. aeruginosa population rapidly diversifies and gains a wide range of mutations, but over time, these genetically diverse lineages converge upon specific chronicity-associated phenotypes which allow the bacteria to resist antibiotics, evade immune clearance, and survive in the lung environment (63). Chronicity-associated phenotypes include mucoidy resulting from overproduction of exopolysaccharides such as alginate or Psl/Pel, which favor biofilm formation, and related loss of flagellum and pili resulting in loss of motility and transition to a sessile lifestyle (55, 62, 64). Chronicity is also associated with reduced production of exoproducts such as proteases and some key virulence factors such as the type III secretion system (T3SS), but overexpression of other virulence factors such as Cif which dysregulates endocytic recycling of CFTR, reducing chloride secretion across the epithelium (65, 66). Other chronicity-related phenotypes include increased efflux pump expression; changes in lipopolysaccharide (LPS) structure which reduce antigenicity; metabolic changes allowing for survival in hypoxic mucus microenvironments and use of different nutrients available in the lung; mutation of quorum sensing regulators such as LasR; and emergence of hypermutators and persisters (Figure 2) (63, 67–69). However, it is important to note that although these chronicity-associated phenotypes become enriched on a population level over time within the lung, this population is composed of spatially and temporally heterogenous subpopulations which are phenotypically diverse according to the needs of their specific lung niche. Given this heterogeneity, most of these chronicity-associated phenotypes, such as mucoidy, reduced motility, altered metabolism, and reduced virulence factor expression, are reversible, allowing P. aeruginosa to adapt effectively to changing selective pressures within the lung.
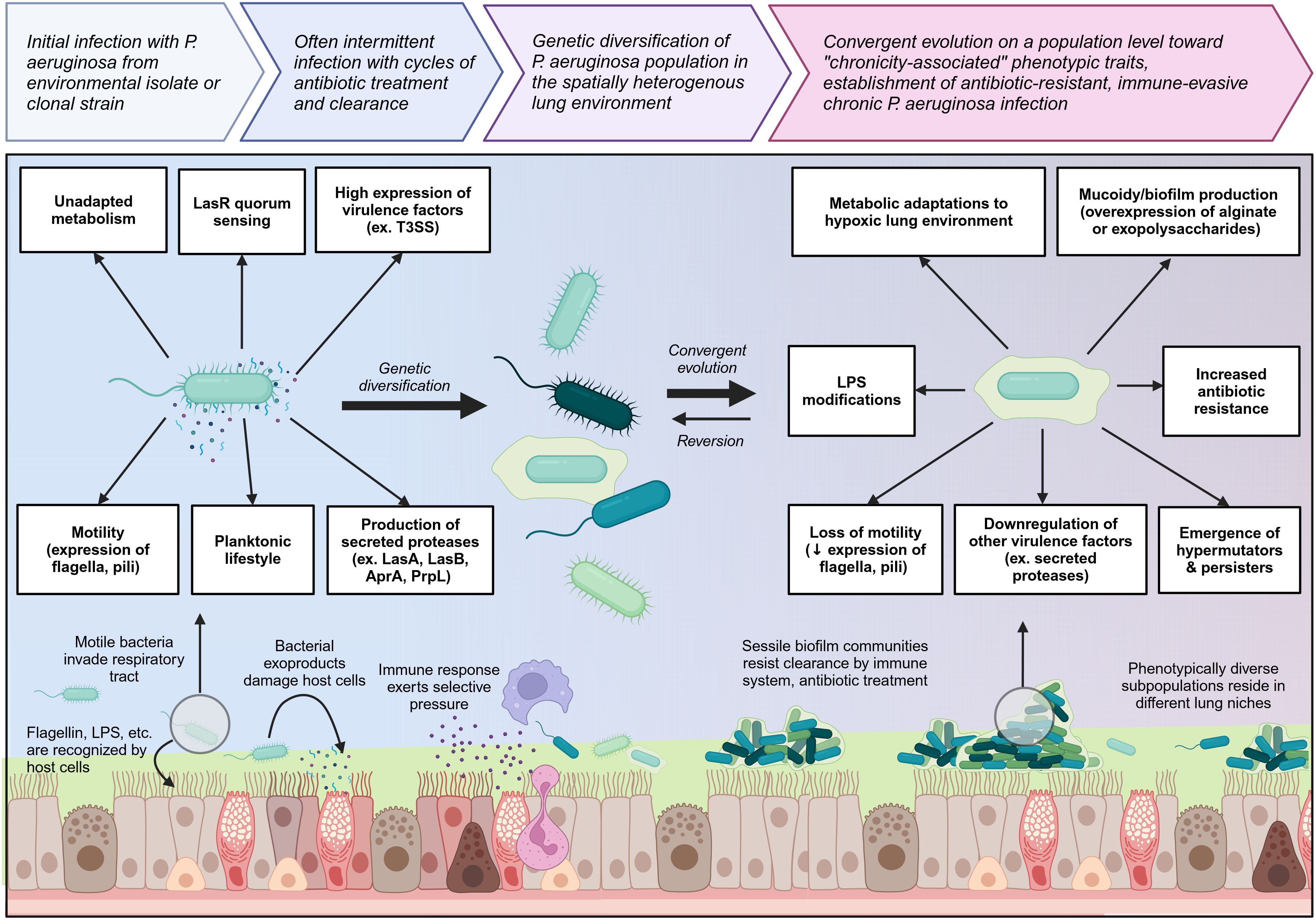
Figure 2 Overview of the transition from initial colonizing P. aeruginosa to chronic lung-adapted P. aeruginosa. P. aeruginosa generally enters the lung as a motile planktonic bacterium expressing flagella and pili and producing high levels of virulence factors and exoproducts such as secreted proteases which are immunogenic and damage the lung tissue. This prototypical acute phenotype is outlined on the left. Initial infection can occur with environmental isolates or clonal strains which may already have some degree of lung adaptation. As P. aeruginosa adapts to the lung environment in chronic lung diseases such as cystic fibrosis, it genetically diversifies and then converges on a population level toward a set of chronicity-associated phenotypes highlighted on the right. It loses key virulence factors and becomes less immunogenic. Transition to a sessile biofilm lifestyle as well as mucoidy increase resistance to both immune and antibiotic clearance. Biofilm communities undergo clonal diversification to give rise to hypermutators and persisters which further increase P. aeruginosa resilience within the lung niche. Created with BioRender.com.
Recent advances in sequencing technologies, bioinformatics methods, and even sample collection strategies have revealed new complexities to the ways P. aeruginosa evolves and diversifies during chronic infections. All of these complex factors shape the interplay between P. aeruginosa and host immunity in important ways, but the scope of these topics exceeds the focus of this review. Many of the topics mentioned briefly above, as well as many more we were unable to cover, have recently been thoroughly reviewed elsewhere (57, 58, 60, 69–71).
Chronic infection with P. aeruginosa is associated with significant lung damage and worsened outcomes, which is greatly compounded by a sustained and dysregulated immune response to infection, in addition to damage mediated directly by bacterial infection itself. The immune response to chronic P. aeruginosa is predominantly characterized by sustained neutrophilic infiltration resulting in lung damage caused by release of neutrophil effector molecules, but also involves a wide range of other cellular players such as macrophages and specific T helper cell subsets, which play various roles including contributing to this vicious cycle of neutrophilic inflammation, producing damaging mediators of their own, or preventing the formation of an effectively-balanced immune response (72). A better understanding of this dysregulated inflammatory response to infection will aid in the further development of targeted immunomodulatory therapies. Notably, it is important to recognize that much of the work investigating these dysregulated responses to chronic P. aeruginosa infection has been done in the context of CF, where the CFTR defect may also directly impact the functions of some immune cells, though this remains controversial (26). More work needs to be done investigating chronic P. aeruginosa infection in other contexts such as COPD and NCFB. Challenges in effectively modelling chronic P. aeruginosa infection using animal models, both within and outside the context of CF, have also added layers of complexity to unraveling context- and model-specific responses to infection (73). This section aims to introduce key pathways important for sensing and responding to P. aeruginosa infection, followed by the roles of innate and adaptive immune cells in mediating bacterial clearance, host damage, and regulating the immune response (Figure 3).
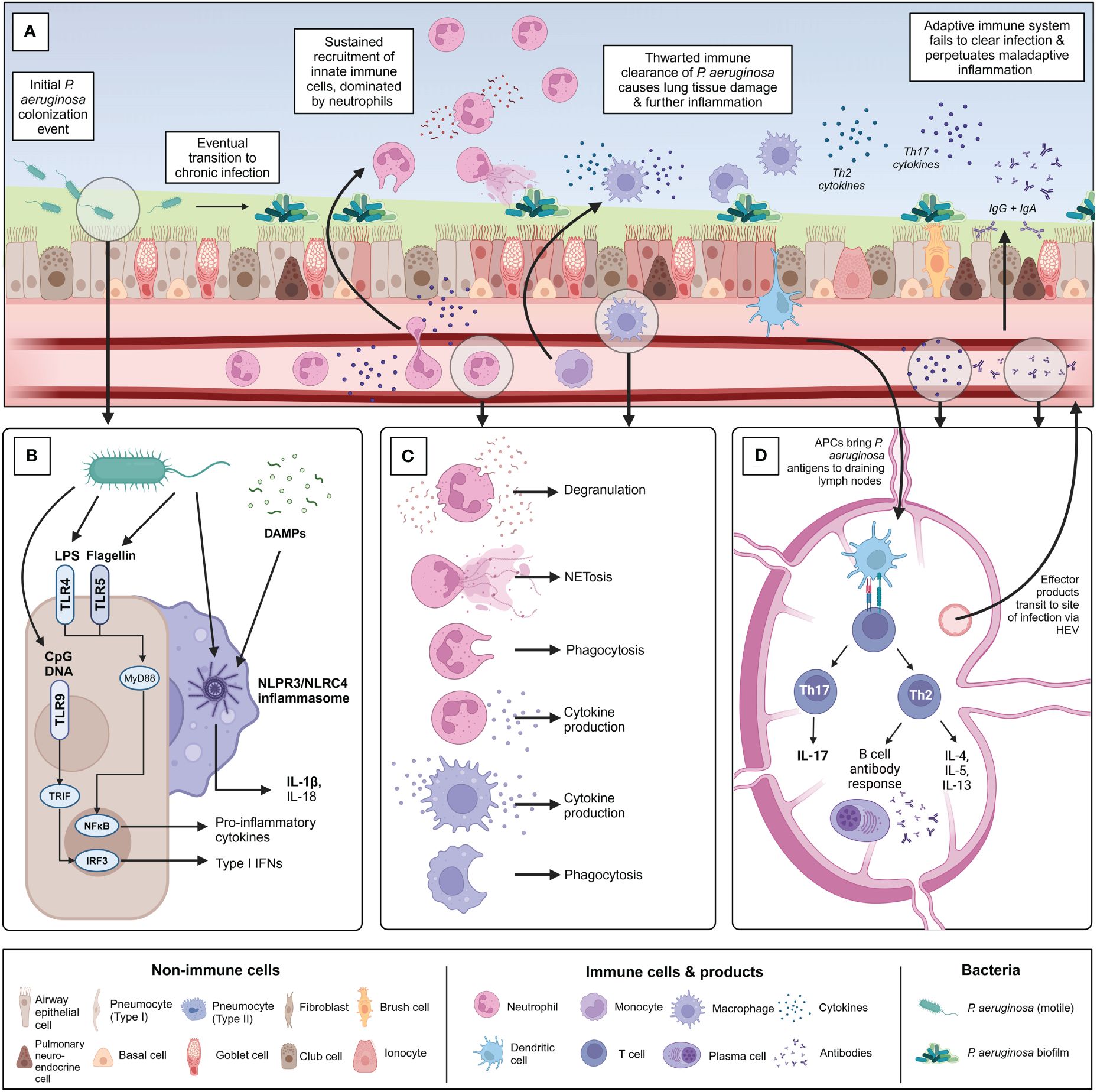
Figure 3 Overview of the immune response to chronic P. aeruginosa infection within the lung. (A) Summary of the timeline of chronic P. aeruginosa infection establishment and key immune players involved. Following initial potentially intermittent colonization, P. aeruginosa adapts to the diseased lung environment and establishes a biofilm community. (B) Overview of the primary pattern recognition receptors and pathogen-associated molecular patterns associated with initial recognition of P. aeruginosa by sentinel airway epithelial cells and alveolar macrophages. (C) Following pathogen recognition, innate immune cells are recruited to the site of infection. Chronic P. aeruginosa infection is characterized by neutrophilia and extensive neutrophil-mediated lung damage because of degranulation, NETosis, and cytokine production. Monocyte-derived macrophages are recruited from circulation and polarized into inflammatory “M1” macrophages at the site of infection. Interstitial macrophages and alveolar macrophages also contribute to the macrophage involvement in infection. Macrophages also contribute to production of pro-inflammatory cytokines. Phagocytosis by both macrophages and neutrophils is thwarted by chronic bacterial adaptations. (D) Antigen-presenting cells such as dendritic cells are activated within the lung and travel to local draining lymph nodes to activate adaptive T and B-cell mediated immunity. Naïve CD4+ T cells are predominantly polarized into Th2 and Th17 populations in chronic P. aeruginosa infection. Th2 cells produce characteristic cytokines IL-4, IL-5, and IL-13 and support the formation of the B cell antibody response. Robust IgG and IgA antibody production occurs in response to chronic P. aeruginosa infection but is largely ineffectual at clearing infection or protecting against re-infection and may contribute to further inflammation by immune complex formation. Th17 cells produce Il-17 family cytokines which indirectly support neutrophil maturation and recruitment, further contributing to detrimental neutrophil-mediated lung damage. Figure created using BioRender.com.
Various germline-encoded pathways are utilized by non-immune cells such as airway epithelial cells, and innate immune cells such as macrophages and neutrophils, in the early sensing of P. aeruginosa as it enters the respiratory tract. The pathways governing initial recognition of P. aeruginosa are vital for directing the subsequent inflammatory response in both acute and chronic infection, and there are several ways these pathways become dysregulated when infection becomes chronic.
When P. aeruginosa enters the lung, the first cells it encounters will likely be airway epithelial cells, as well as various sentinel immune cells, from dendritic cells (DCs) and mast cells in the upper airways, to alveolar macrophages in the alveoli. These various cell types play critical roles in the initial recognition and shaping of the inflammatory response to pathogens, which occurs in the first minutes, hours, and days post-infection. Sustained pathogen recognition signaling in chronic infection can help skew and sustain dysregulated host inflammatory responses over a much longer time frame. Our host cells possess a range of pattern recognition receptors (PRRs), which equip them to recognize and respond to conserved microbial pathogen-associated molecular patterns (PAMPs) as well as host cell-derived damage-associated molecular patterns (DAMPs). Patterns of PRR expression vary between cell types. Airway epithelial cells, for example, express several toll-like receptors (TLRs), including cell-surface TLR2, TLR4, and TLR5, which can recognize P. aeruginosa lipoproteins, LPS, and flagellin, respectively, as well as endosomal TLR9, which can recognize P. aeruginosa unmethylated CpG DNA (74–76). It is important to note that no single TLR deficiency significantly compromises the immune response to P. aeruginosa, although mice deficient in multiple TLRs do experience enhanced susceptibility to infection (77, 78). TLR activation results in signal transduction through MyD88 and TRIF. MyD88 signaling leads to liberation of NFĸB, as well as assembly of AP-1 downstream of the mitogen-activated protein (MAP) kinase pathways, ultimately resulting in transcription of key cytokines and chemokines which coordinate the host inflammatory response (76). Mice selectively deficient for MyD88 in lung epithelial cells were more susceptible to acute P. aeruginosa infection, with reduced recruitment of neutrophils and impaired early bacterial control post-infection (79). MyD88 was also required to control chronic P. aeruginosa infection in a murine model, where complete deletion of MyD88 resulted in severe and lethal lung pathology following infection (80, 81). TLR signal transduction can also occur independent of the MyD88 pathway via TRIF-dependent mechanisms which have also been shown to be important for host defense against P. aeruginosa infection (82, 83). TRIF activation of IRF3/7 leading to IFN-β production is commonly implicated in antiviral protection, but IRF3/7 have also shown importance in protection against acute lung P. aeruginosa infection in mice, where IRF3-deficient mice had reduced lung bacterial clearance, lower neutrophil recruitment, and altered cytokine profiles (84).
TLR activation in both immune and non-immune cells shapes the immune response to P. aeruginosa in various ways. TLR activation induces airway epithelial cells to secrete crucial antimicrobial molecules into the mucus layer, including β-defensins, lysozyme, lactoferrin, cathelidicin LL-37, and surfactant proteins A and D (85). TLR signaling can also directly potentiate innate immune cell functions such as phagocytosis and degranulation. Perhaps most importantly, TLR activation results in the production of key pro-inflammatory cytokines such as TNF, IL-6, pro-IL-1β, pro-IL-18, IL-12, and IL-23; as well as chemokines such as CXCL8, CXCL1, CXCL2, CXCL10, CCL2, and CCL5 (86). These molecules coordinate the recruitment and activation of key immune cell populations. In chronic infection, sustained elevated levels of these cytokines are associated with worsened lung function, demonstrating that a robust unresolving inflammatory response to P. aeruginosa is both unable to clear infection and drives lung damage, as will be discussed further below.
In addition to TLR signaling, inflammasome assembly downstream of nucleotide oligomerization domain (NOD)-like receptors (NLRs) also plays an important role in the response to P. aeruginosa infection, particularly in chronic lung inflammatory conditions like CF. There are various inflammasomes, but the two most well-characterized in P. aeruginosa infection are the NLRP3 inflammasome, which assembles in response to oxidative stress-induced PAMPs and DAMPs, and the NLRC4 inflammasome, which assembles in response to bacterial PAMPs such as the T3SS and flagellin (87–89).
Inflammasome assembly in the cytoplasm leads to the activation of caspase-1 which cleaves pro-IL-1β and pro-IL-18 produced downstream of NFĸB to generate mature IL-1β and IL-18 (90). Inflammasome assembly can also lead to a form of inflammatory cell death termed pyroptosis, which contributes to P. aeruginosa killing by macrophages and neutrophils, but can also cause pathological inflammation if overactivated (91, 92). Innate immune cells such as alveolar macrophages and neutrophils are important producers of IL-1β via inflammasome assembly in response to P. aeruginosa infection, with airway epithelial cells implicated as playing a lesser role (93). IL-1β is an important cytokine in both acute and chronic P. aeruginosa infection, where it has been associated with successful control of infection in acute settings, and deleterious pathology in chronic settings (88, 94–96). High IL-1β levels in sputum produced by pwCF have been associated with P. aeruginosa colonization and worsened lung function (97, 98). In the early stages of infection, IL-1β produced by macrophages binds IL-1R on airway epithelial cells and stimulates them to produce neutrophil-recruiting chemokines such as CXCL8, a cascade which can be amplified with chronic infection. Later in infection, IL-1β signaling also triggers the activation of IL-17A secreting T helper 17 (Th17) cells, modulating the Th17/T regulatory cell (Treg) balance, which will be discussed in greater detail below (99). Some studies have reported that the NLRP3 inflammasome is the primary contributor to IL-1β and IL-18-dependent inflammation in P. aeruginosa infection and CF, with the NLRC4 inflammasome playing a lesser or even counter-regulatory role (95, 100). Other reports have instead suggested that chronic P. aeruginosa isolates from CF are less able to activate inflammasome signaling and that this may be a mechanism of immune evasion that promotes bacterial persistence (101, 102). This stands in contrast to studies which have reported benefits of blocking inflammasome activation in both acute and chronic P. aeruginosa and CF (92, 95, 96), indicating that the timing and relative contribution of inflammasome signaling to chronic P. aeruginosa infection within and outside the context of CF remains to be fully understood.
As described above, PRR signaling pathways drive production of pro-inflammatory cytokines and other antimicrobial mediators, as well as recruitment of immune cells to infection sites. The activity of these pathways typically promotes effective clearance of P. aeruginosa in acute infection –but what about chronic infection?
As briefly noted above, some studies have reported PRR signaling defects in contexts such as CF which may make it easier for P. aeruginosa to gain a foothold in the lungs of these patients, such as defects in the ability of CF airway epithelial cells to respond to pathogens due to reduced cell surface expression of TLR4 (103, 104). Whether these defects are an intrinsic consequence of CFTR mutation, or an adaptive or tolerogenic response to chronic airway inflammation in pwCF, is a topic that remains controversial. Similar findings have been reported for other chronic lung conditions like COPD, where airway epithelial cell TLR4 expression has been shown to be downregulated by cigarette smoke exposure (105).
P. aeruginosa may also have mechanisms to exploit PRR signaling pathways to improve bacterial persistence. For example, TLR5-mediated recognition of flagellin has been reported in pwCF to mediate the formation of granulocytic myeloid-derived suppressor cells (MDSCs). These MDSCs are capable of suppressing Th17 responses, discussed in more detail below, which may indirectly limit neutrophil activation and recruitment. This suggests that flagellated P. aeruginosa may be able to exploit TLR5 signaling to suppress antibacterial immune responses which may be relevant for the establishment of chronic infection (106). Interestingly, in the patients profiled in this study, the presence of MDSCs actually correlated with improved lung function, which suggests some benefit of these anti-inflammatory MDSCs in controlling chronic inflammation. However, these patients primarily possessed flagellated nonmucoid P. aeruginosa isolates, in contrast to the mucoid non-flagellated isolates more common in adult pwCF with established disease. This suggests that targeting TLR5 signaling may be useful for P. aeruginosa in initially establishing chronic infection, but no longer beneficial once the bacteria have established chronicity and are able to transition to a nonmotile, biofilm-living phenotype.
Once P. aeruginosa establishes chronic infection, it undergoes adaptations which render it less susceptible to recognition by PRRs, leading to immune evasion which favors persistence. Chronic P. aeruginosa isolates often undergo LPS modifications such as loss of O-antigen and lipid A modifications, and downregulate expression of flagella, pili, and T3SS which prevent TLR5 and NLRC4 inflammasome-mediated recognition (107). Encasement in a biofilm matrix may also hide PAMPs from PRRs, and bacteria in lung biofilm communities have been visualized to sit on top of airway epithelial cells rather than in contact with them like planktonic bacteria, further reducing contacts and chances for recognition (22, 108). However, despite these immune evasive adaptations, the inflammatory response to chronic P. aeruginosa infection is robust and characterized by extensive release of DAMPs in addition to PAMPs which drive vicious cycles of inflammatory mediator production and immune cell recruitment, which will be detailed below.
The epithelial lining of the lung is comprised of diverse cell types whose composition varies between the upper and lower respiratory tract. The trachea, bronchi, and bronchioles are lined by a pseudostratified epithelium dominated by ciliated cells, with occasional secretory cells including club, neuroendocrine, and goblet cells, as well as basal progenitor cells located below the epithelium (85). The tight junctions between airway epithelial cells provide a critical mechanical barrier which protects the lung parenchyma from the contents of the airway lumen (85). As previously mentioned, the luminal surface of the airway epithelium is coated in a thin layer of pericellular fluid which allows for effective ciliary beating, with a layer of protective mucus on top that entraps particles and pathogens to be removed by this mucociliary elevator (85). This mucus barrier is maintained by fluids, mucins, and host defense proteins produced by the epithelial and secretory cells of the airway epithelium. Disruptions to normal mucociliary clearance, as described above for conditions like CF, NCFB, and COPD render the lung susceptible to colonization by opportunistic pathogens like P. aeruginosa.
Mucociliary clearance is perhaps underrecognized as one of the principal structural immune defense mechanisms in the lung. Modelling mucociliary clearance is still a difficult task, though numerous strategies have been developed (109–111). However, it appears that there are still important gaps in effectively modelling mucociliary clearance in disease contexts such as CF, COPD, and NCFB, as well as modelling the mechanistic effects of infection with pathogens such as P. aeruginosa and biofilm formation on already-impaired mucociliary clearance in these conditions (109). Work has been done to demonstrate that the composition and concentration of airway mucus in CF influences the biophysical properties of P. aeruginosa biofilms, making them more robust and able to withstand physical and chemical challenges (112). From a therapeutic point of view, impaired mucociliary clearance and mucus accumulation in diseases such as CF limit the penetration and functionality of anti-P. aeruginosa antibiotics (113, 114), and targeting mucus specifically with mucolytic therapies has been shown to both improve the function of antibiotics like tobramycin and improve mucociliary transport (115). It has long been recognized that mucociliary clearance is an important readout for the efficacy of CF therapies (116), as we are now seeing with new highly effective CFTR modulator therapies (HEMT). Even partially restoring mucociliary clearance with HEMT is significantly impacting the landscape of lung infections in pwCF (117), as will be discussed further in section 4.4. Overall, the relationship between infection and mucociliary clearance warrants further exploration from a mechanistic point of view.
Moving from structural barriers to specific cell types, airway epithelial cells themselves play crucial roles in initiating and propagating the immune response to P. aeruginosa infection. As described above, PRR-mediated recognition of P. aeruginosa stimulates airway epithelial cells to produce cytokines and chemokines such as TNF, IL-6, IL-1β, CXCL8, CCL2, CCL5, and CXCL1 which lead to significant recruitment and activation of neutrophils, monocytes, and other immune cell populations. Airway epithelial cells also respond to cytokine production by other cell types, such as IL-1β production by alveolar macrophages, furthering cycles of inflammatory cytokine production which, in acute infection, resolve with pathogen clearance and the ensuing clean-up, but in chronic infection, perpetuate as a consequence of unresolved pathogen and damage-associated signals.
Airway epithelial cells also undergo remodeling in response to chronic infection as they are battered by excessive inflammation. They undergo significant bystander damage from neutrophil products such as reactive oxygen species (ROS) and neutrophil elastase (NE), which will be detailed below. Destruction of lung epithelium results in release of danger signals and leakage of profibrotic mediators such as growth factors and coagulation components which activate underlying fibroblasts to drive lung fibrosis (118–120). In these ways, continual lung injury over time results in disrupted homeostatic responses and tissue repair mechanisms, leading to irreversible lung damage and functional decline.
Macrophages are the primary innate immune population resident within the lung. The lung is populated by two main macrophage populations at steady-state: alveolar macrophages located in the lumen of the airways, and interstitial macrophages found in the lung parenchyma. During infection, monocytes are recruited from the circulation in large numbers and polarized by the lung inflammatory milieu to differentiate into different macrophage subsets, which exist on a continuum from the prototypical inflammatory “M1” macrophages to anti-inflammatory/tissue-repair/regulatory “M2” macrophages (121). These macrophage populations play numerous key roles in infection, including phagocytosis of pathogens and debris; production of cytokines to shape the immune milieu; and tissue remodeling and repair.
Amongst the first innate immune cells to respond to P. aeruginosa infection in the lung are tissue-resident alveolar macrophages, which can begin to mount anti-pathogen responses in the first few hours following infection. Alveolar macrophages are a unique population of macrophages that have been described as the “sentinels” of the lung. Under healthy conditions, they are responsible for the maintenance of homeostasis through phagocytosis of dead cells and debris to help clear inflammatory stimuli from the lung, protecting the thin alveolar barrier where gas exchange takes place (122, 123). Alveolar macrophages also play an important role in surfactant homeostasis (121). Dysregulated surfactant homeostasis contributes to various chronic lung diseases, including CF, where it is associated with increased susceptibility to infection (124, 125). In infection, once alveolar macrophages are activated by pathogens and cytokine signals from nearby epithelial cells, they release a network of cytokines and chemokines that contribute to the recruitment and activation of neutrophils, monocytes, dendritic cells, natural killer (NK) cells, and later T cells within sites of infection (126, 127). Alveolar macrophages can also directly phagocytose and kill P. aeruginosa, which will be described in greater detail below. While neutrophils are the major cell type responsible for driving pathology in P. aeruginosa infection, as will be detailed below, alveolar macrophages play crucial roles in regulating neutrophil lifespan, and phagocytosing dying neutrophils to limit neutrophil-induced tissue damage (128). Dysregulations in the ability of alveolar macrophages to regulate neutrophil homeostasis during chronic infection can drive further pathology.
The specific role of interstitial macrophages in chronic P. aeruginosa is less well-characterized, as they are a highly heterogenous population originating from various sources (121). Interstitial macrophages have only recently been discovered to participate in lung fibrosis (129), which may be important at late stages of chronic infection and chronic lung disease.
During chronic infection, monocyte-derived macrophages are continually recruited from the circulation in a CCR2-dependent manner, as tissue-resident alveolar macrophages are diminished, gradually replacing both alveolar and interstitial macrophages over time (130). This population has been shown to contribute to excessive neutrophil recruitment through CXCL2 production and drive detrimental tissue remodeling and fibrosis through production of high levels of TGF-β (130). Use of CCR2 inhibitors to block recruitment of monocyte-derived macrophages in a chronically inflamed CF model reduced both neutrophilic infiltration and TGF-β levels (130). This emphasizes that better understanding the contribution of specific macrophage subsets to chronic inflammation will allow for more specific targeting of detrimental populations.
The various macrophage populations described above recognize and kill P. aeruginosa via several pathways. As mentioned, macrophages express numerous PRRs, including TLRs and NLRs, which allow them to sense and respond to pathogens like P. aeruginosa (74). Via these pathways macrophages produce a wide range of cytokines that shape the response to infection, allowing for the recruitment of neutrophils and later other immune cells that are instrumental to bacterial control (126, 131). Airway macrophages are an important source of numerous pro-inflammatory cytokines and chemokines including IL-1α, IL-1β, IL-6, TNF-α, IFN-γ, IL-18, IL-4, IL-23, CXCL8, CCL2, CCL3, CCL4, and CCL20, amongst others (132–134). Not all macrophages produce the same cytokines in the same frequencies. Macrophages are polarized by cytokine signals received from airway epithelial cells to amplify specific cytokine responses (122, 135). The cytokine-producing arm of macrophages has been shown to be essential for protection in various murine P. aeruginosa infection models. Macrophage cytokine production has been correlated with effective neutrophil recruitment in acute infection (134, 136). Case studies in pwCF and murine models of chronic P. aeruginosa infection have shown that this same cytokine production and ability to recruit other immune cells can be detrimental, showcasing that responses that are protective early in infection can become pathogenic later (121, 130, 137). In acute infection, this recruitment of immune cells leads to effective bacterial clearance and infection resolution, whereas in chronic infection, recruited immune cells are unable to clear infection and instead perpetuate inflammatory signaling and damage within the lung.
One of the most important functions of macrophages is phagocytosis. Phagocytic killing is crucial for the control of P. aeruginosa. Individuals with genetic deficiencies in phagocyte responses are predisposed to P. aeruginosa infection (138), and depletion of phagocytes in animal models increases susceptibility to and lethality of infection (139). Once bacteria such as P. aeruginosa are phagocytosed, they are killed intracellularly within phagolysosomes by NADPH oxidase-dependent ROS production (140). Like many Gram-negative pathogens, P. aeruginosa is susceptible to opsonic phagocytosis. Briefly, phagocytes such as macrophages express antibody-binding Fc receptors and complement binding receptors which allow them to recognize opsonized extracellular P. aeruginosa (141). Other proteins such as surfactant A and C are reported to bind P. aeruginosa and assist in this process (142, 143). However, alveolar macrophages tend to express relatively low levels of opsonic receptors, suggesting that this may not be their primary pathway of P. aeruginosa clearance in lung infections (144). Macrophages are also capable of killing P. aeruginosa by non-opsonic phagocytosis, which may play an important role in controlling bacteria at early stages of colonization (145). Non-opsonic phagocytosis can be mediated by a variety of different receptors (144, 146). One key element that macrophages and other phagocytes use to recognize P. aeruginosa is flagella, both via TLR5-mediated recognition of flagellin as well as recognition of specifically swimming motility itself (145, 147, 148). While flagella expression is important for initial invasion and colonization of the lung, likely via binding to host cell Muc1 mucin in the upper airway (149, 150), loss of flagella is a common occurrence in chronic P. aeruginosa infection as bacteria transition to a nonmotile biofilm lifestyle. Loss of flagella in chronic P. aeruginosa isolates has been shown to increase bacterial resistance to phagocytosis by over a hundred-fold (145, 151). In addition, shifting of P. aeruginosa to the mucoid phenotype, a common adaptation in chronic infections, also prevents phagocytosis (152–155). Alginate production by mucoid P. aeruginosa has been shown to inhibit receptor-ligand interactions important for phagocytosis in human and murine macrophages (156). P. aeruginosa strains overexpressing the exopolysaccharide Psl limit complement deposition by preventing access to the bacterial outer membrane (155). Persister cells, a metabolically dormant subpopulation of antibiotic-tolerant bacteria prevalent in chronic infection, also resist C5b complement deposition, are engulfed at a ten-to-one-hundred-fold lower rate following opsonization, and even resist killing once engulfed (157). These various adaptations of chronic P. aeruginosa to avoid and modulate macrophage phagocytosis indicate that this may represent an initially effective mechanism for early control of infection, suggesting that improving the ability of macrophages to phagocytose and clear P. aeruginosa in chronic infections could be a therapeutic avenue.
One additional confounder when considering the role of macrophages in chronic P. aeruginosa infection within the specific context of CF, is the controversy over whether CFTR defects result in intrinsic macrophage defects that predispose these patients to infection. Early studies reported that CF macrophages had phagocytic defects, and once CFTR expression was identified in macrophages, studies suggested that loss of CFTR resulted in defects in their phagolysosome acidification (158–161). However, these findings have been contradicted by recent studies demonstrating that various populations of CF macrophages do not have defects in phagocytosis or phagolysosomal acidification, as well as results suggesting that macrophage defects in CF are instead a consequence of chronic inflammation (162, 163). Even more recent work in pwCF receiving HEMT have reported that CFTR modulators are able to potentiate CF macrophage function, restoring their ability to phagocytose and effectively kill P. aeruginosa as well as altering their inflammatory profiles (164–166). Whether this indicates that CFTR modulators are acting directly on intrinsic CFTR-mediated defects in macrophages or are mediating improved macrophage function by reducing chronic lung inflammation is still unclear. A recent study demonstrated that CFTR modulators were in fact able to enhance phagocytosis in both CF and non-CF macrophages, suggesting that the previously reported ability of CFTR modulators to improve macrophage function may not be CF-specific (167). Better understanding how macrophage function is modulated in the context of chronic P. aeruginosa-infection-associated inflammation will be important for the development and rational use of novel therapies.
Neutrophils are the earliest circulating cells to respond upon P. aeruginosa infection in the airway, able to arrive at sites of infection within minutes to hours, and they play an essential role in controlling acute infection (168). Neutropenic individuals are far more susceptible to P. aeruginosa infection, and neutrophil depletion in mice renders them lethally susceptible to extremely low inocula of P. aeruginosa (138, 168, 169), underscoring their importance. Neutrophils have also been highlighted as central players in the pathology of chronic lung diseases including CF (170) and COPD (171, 172). The interplay between neutrophil dysregulation in these diseases and P. aeruginosa susceptibility is, as will be discussed below, complex and likely bidirectional, with disease-specific neutrophil impairment leading to enhanced P. aeruginosa susceptibility, and P. aeruginosa infection enhancing neutrophil-mediated lung damage and disease progression.
Neutrophils are recruited to sites of infection by inflammatory cytokines, particularly the chemokine CXCL8 [murine homologues are keratinocyte chemoattractant (KC) and macrophage-inflammatory protein 2 (MIP-2)], which binds CXCR1 and CXCR2 receptors on neutrophils (173–175). Neutrophil transmigration from circulation into the lung also requires the CD18 integrins, loss of which increases susceptibility to acute P. aeruginosa pneumonia (176). Other soluble mediators important for the migration and activation of neutrophils include cytokines TNF and IL-1β, complement anaphylatoxin C5a, and lipid mediators like leukotriene B4 (LTB4) (170). IL-17 cytokines, namely IL-17A, also play an important indirect role in neutrophil accumulation and activation by inducing local airway epithelial cells to produce more neutrophil mobilizing factors (177, 178).
At sites of infection, neutrophils are activated by a variety of signals, including recognition of bacterial PAMPs by PRRs, as previously mentioned; cytokines and chemokines, such as CXCL8, which in addition to mediating neutrophil migration, also activates neutrophil effector functions such as degranulation and respiratory burst; and opsonins such as complement proteins and antibodies, which tag bacteria for phagocytosis (179, 180).
Neutrophils can kill P. aeruginosa through a variety of mechanisms, including phagocytosis, degranulation, and the production of neutrophil extracellular traps (NETs). Neutrophil phagocytosis occurs via mechanisms like those described above for macrophages. Neutrophils engulf bacteria into phagosomes, which fuse with lysosomes to form phagolysosomes, where NADPH oxidase-generated ROS and antimicrobial granule contents come together to kill internalized bacteria (181). In addition to phagocytic killing, neutrophils release a variety of antimicrobials and ROS into the extracellular environment through the process of degranulation. Neutrophil granule contents which are capable of inhibiting P. aeruginosa include antimicrobial peptides, such as α-defensins and cathelicidin LL-37; proteases such as neutrophil elastase (NE) and matrix metalloprotease-9 (MMP-9); other enzymes like myeloperoxidase (MPO) and lysozyme; and other soluble antimicrobials like lactoferrin (182, 183). These products, particularly NE and ROS, have been extensively associated with tissue damage and lung pathology in chronic P. aeruginosa infections in CF, COPD, and NCFB (170). Neutrophils can also undergo a unique form of inflammatory cell death termed NETosis where, in response to pathogens such as P. aeruginosa, neutrophils condense and extrude their cellular DNA coated in granule-derived antimicrobial proteins, such as NE, to ‘trap’ extracellular bacteria (184, 185). These NETs also play crucial roles in neutrophil-mediated lung pathology in chronic infection, which will be discussed in detail below. In addition to these direct killing mechanisms, neutrophils can also indirectly shape the immune milieu. Though not historically considered major cytokine producers, neutrophils do also produce a range of cytokines and chemokines, such as TNF, IL-1β, IFNγ, IL-4, IL-10, IL-13, IL-17, CXCL8, and CXCL9/10, which contribute to shaping the inflammatory response to infection (178, 186–189). Neutrophils, primarily by virtue of their large numbers, have been identified as major producers of IL-1β in the lung milieu of pwCF, downstream of NLRP3 inflammasome activation (96). Targeting neutrophil-mediated cytokine production in neutrophilic diseases may be another method to offset maladaptive inflammation (96).
Despite the importance of neutrophil-mediated killing mechanisms for clearance of P. aeruginosa in acute infection, many of these products have non-specific ‘off-target’ effects on host tissues which, in conditions like chronic infection, prevent effective immune clearance, and ultimately even heighten lung damage and contribute significantly to lung pathology. The neutrophil product NE has been shown to cleave the complement receptor CR1 expressed on neutrophils (190), as well as the opsonin iC3b bound to the surface of P. aeruginosa (191). This results in an opsonin-receptor mismatch which impairs the complement-mediated phagocytosis of opsonized P. aeruginosa, the stimulation of ROS production, and ultimately the ability of neutrophils to effectively kill opsonized P. aeruginosa (191). Potentially as a consequence of this thwarted phagocytosis, and other mechanisms, neutrophilia is a common hallmark of chronic P. aeruginosa infection, with neutrophils commonly observed surrounding biofilms in high numbers (170, 192, 193). Biofilm formation is one of several bacterial strategies which allows P. aeruginosa to resist many neutrophil killing mechanisms (152, 155). Unfortunately, neutrophils have also been shown to unwittingly promote P. aeruginosa biofilm formation. P. aeruginosa has been reported to co-opt extracellular DNA produced by NETosis for its biofilm extracellular polymeric substance (EPS) (194). Furthermore, excessive NETosis has been associated with increased mucus viscosity, further hampering mucociliary clearance of bacteria, and ultimately exacerbating lung injury in CF as well as NCFB (185, 195–197). Several reviews provide greater detail on the detrimental roles of NETosis in CF and chronic P. aeruginosa infection (170, 198–201). Neutrophils can also become dysregulated by chronic inflammatory contexts. For example, neutrophils isolated from pwCF chronically infected with P. aeruginosa exhibit prolonged survival (202–204). Neutrophil apoptosis is a tightly regulated process which limits the release of neutrophil contents to prevent damage to the surrounding tissue. Apoptosis is coordinated with non-inflammatory clearance (efferocytosis) by phagocytes to successfully resolve inflammatory responses (205). In COPD, cigarette smoke has been shown to impar the ability of alveolar macrophages to effectively phagocytose apoptotic neutrophils (206). In conditions of chronic infection and continuous inflammatory signaling, it is likely that the dysregulation of neutrophil activation and survival leads to a viciously intensifying inflammatory cycle that is unable to resolve. This central role of neutrophils in coordinating the maladaptive response to pathogens such as P. aeruginosa in chronic lung disease has led to significant efforts to therapeutically modulate neutrophil activity and products, which will be discussed in a later section.
Neutrophils and macrophages represent the main innate immune populations involved in the response to P. aeruginosa in the lung. However, there are other immune cells which play supporting roles, or whose roles in P. aeruginosa infection, particularly chronic infection, are still being characterized.
Dendritic cells (DCs) are the professional antigen-presenting cells of the immune system, functioning to bridge the innate and adaptive responses by activating and differentiating naïve T cells into effector subsets. This has relevance for chronic P. aeruginosa infection as T cell polarization toward Th2 and Th17 immunity has been associated with increased pathology and worsened outcomes, for distinct reasons which will be discussed further below (207–209). Limited work has investigated the roles of DCs in chronic P. aeruginosa infection, but they are known to be activated and express co-stimulatory molecules CD80 and CD86 in the lung and regional lymph nodes in response to P. aeruginosa (210). Furthermore, P. aeruginosa appears able to modulate DC subpopulations, resulting in a skewing of the resting conventional DC (cDC) population toward cDC2s that support a Th2 response rather than cDC1s that promote Th1 and bacterial clearance (211). This fits into the existing paradigm of Th2-skewed immunity in chronic P. aeruginosa and suggests that DCs may be contributing to this later T cell imbalance. P. aeruginosa quorum sensing molecules have also been shown to impair the ability of DCs to induce T cell proliferation, which could also impair the generation of an effective adaptive response (212).
Other granulocytic cell populations, namely mast cells and eosinophils, play important roles in some chronic airway conditions, such as asthma and COPD, and as such have also been investigated for potential roles in CF and chronic lung inflammation associated with P. aeruginosa (213, 214). Eosinophils were found to be activated and contribute to lung destruction in CF, though they play a far more minor role than neutrophils owing at least in part to their much smaller population (215). In bronchiectasis, eosinophilic inflammation was associated with P. aeruginosa infection (216). Eosinophils have been recognized to play an important role in a subtype of COPD (217, 218), although eosinophilic COPD appears less-associated with P. aeruginosa infection than neutrophilic COPD (48). Overall, eosinophils appear to play only a minor role in chronic P. aeruginosa infection in the context of CF or other diseases. Mast cells also respond to P. aeruginosa, and may contribute to the inflammatory cascade by producing cytokines and chemokines such as CXCL8 and CCL20 to further recruit neutrophils and monocyte-derived macrophages to the respiratory tract (219–221). However, the involvement of these cells is also understudied.
Outside the myeloid compartment, various innate lymphoid populations have also been implicated in the response to P. aeruginosa lung infection. Natural killer (NK) cells are most commonly associated with protection against viral infection and cancer but have also been reported to kill some bacteria (222). NK cells can be recovered from bronchoalveolar lavage fluid (BALF) following P. aeruginosa lung infection, suggesting recruitment in response to infection, and NK cell depletion has resulted in increased mortality and P. aeruginosa lung bacterial burden in acute murine models (222, 223). NK cells have recently been demonstrated to directly kill extracellular P. aeruginosa through release of granzymes B and H which damage the bacterial membrane and induce ROS production resulting in bacterial death (224). Work remains to be done to assess whether NK cells play a meaningful role in chronic P. aeruginosa infection and whether they could be therapeutically harnessed.
Innate lymphoid cells (ILCs) are a plastic group of innate lymphocytes that reside in tissues, particularly at mucosal surfaces, where they function primarily as cytokine secretors (225). ILCs are divided into subsets that mirror conventional T helper cell subsets, with ILC1s producing Th1 cytokines, ILC2s mirroring Th2s, and ILC3s generating Th17 cytokines (225). ILCs play a broad spectrum of roles, from protecting against pathogenic threats including viruses, bacteria, and helminths; regulating microbiota crosstalk; promoting allergic and autoinflammatory reactions; and mediating tissue maintenance and repair (226). ILC2s are the predominant ILC subtype found in the lung; however, it is ILC3s, with their ability to produce IL-17 and IL-22, which have been implicated in the response to lung P. aeruginosa infection (227). One study found that ILC3s were responsible for the majority of innate immune cell production of IL-17 in the lungs in response to P. aeruginosa infection, which in this model was identified as critical in preventing the transition to established chronic infection (228). Another study utilizing Rag/γ-chain-/- mice deficient in T cells, NK cells, and ILCs had an increased P. aeruginosa load compared to classical Rag-/- knockout mice (lacking only T and B cells), suggesting a potential contribution of innate lymphocytes to infection control (229). However, the contributions of ILCs to P. aeruginosa infection and chronic lung disease remain understudied.
Unconventional immune cell populations, such as γδ T cells and natural killer T (NKT) cells, have also been associated with P. aeruginosa infection. γδ T cells are a rare population of innate-like T cells which are resident in tissues and mucosal sites, including the lung (230). γδ T cells have been implicated in the response to acute P. aeruginosa infection, as γδ T cell-deficient mice exhibited enhanced susceptibility to intranasal P. aeruginosa challenge characterized by increased bacterial load, reduced survival, delayed neutrophil recruitment, and enhanced production of specific pro-inflammatory cytokines IL-1β, IL-6, and TNF (231). This effect did not appear to be mediated primarily by IL-17, as γδ T cells were not major IL-17 producers in this model (231). This stands in contrast to other reports which found that IL-17 production by γδ T cells was important for neutrophil chemotaxis and elimination of P. aeruginosa during acute murine lung infection, as well as for B cell stimulation resulting in antibody production (232, 233). Extrapolating these results to chronic infection, γδ T cells may play a role in modulating the cytokine milieu of the inflamed airway, potentially to detrimental effect. Another unconventional T cell subset which straddles the border of innate and adaptive immunity is the NKT cell. These cells recognize lipid and glycolipid antigens rather than peptides. Reports on the role of NKT cells in P. aeruginosa infection are conflicting and appear to differ based on experimental conditions including P. aeruginosa strain and mouse background. Jα18-/- mice deficient in NKT cells were reported to have impaired clearance of P. aeruginosa strain D4, but not PAO1 or PAK (231, 234, 235). These conflicting results may depend, in part, on host genetic background, and differing levels of NKT cells. Indeed, one study found that NKT cells played an important role in P. aeruginosa clearance from the lungs of BALB/c mice, which have greater NKT cell frequencies, but had no significant role in C57BL/6 mice (236). This strain-dependent difference was also associated with different NKT cell cytokine profiles as well as differential impacts on neutrophil recruitment (236), again highlighting the importance of specific cytokine production and modulation of neutrophils in shaping immune responses to P. aeruginosa. The roles of γδ T cells and NKT cells in human P. aeruginosa infection and CF are not known. However, NKT cell activation does not appear to be impaired by CFTR dysfunction in pwCF (237).
The adaptive immune system also plays crucial roles in P. aeruginosa infection, particularly in chronic infection. Early work established the relevance of adaptive immunity to P. aeruginosa infection by demonstrating that Rag2-/- mice, which are devoid of classical T and B cells, had an impaired ability to clear P. aeruginosa from the lungs, resulting in decreased survival (235). The ability of B cells to respond with robust antibody production in chronic P. aeruginosa infection has been long-established; however, the reasons behind the limited protective ability of this response remain to be fully elucidated. T cell responses, particularly CD4+ T cell responses, have also been shown to play critical roles in shaping the cytokine milieu in chronic P. aeruginosa infection and chronic lung disease more broadly. Untangling the relationship between adaptive immune responses and chronic P. aeruginosa infection has proven somewhat challenging, as experimental models of chronic infection have various limitations. It remains difficult to establish consistent and long-lasting chronic infections in murine models (238, 239), which makes it difficult to study adaptive populations that require at least a week to mount a primary response.
Various T cell subsets, specifically CD4+ T helper (Th) subsets, have been implicated in the pathology of chronic P. aeruginosa infection, particularly in the context of CF. Several early studies implicated Th2 skewing as a major component of the maladaptive inflammatory response and susceptibility to pathogens such as P. aeruginosa which occur in pwCF (207, 240). Later, with the discovery of Th17 cells, this paradigm was broadened to implicate both populations in shaping inappropriate responses to P. aeruginosa infection in chronic lung disease (209).
Th2 cells are polarized in the presence of IL-4 and upregulate the transcription factor GATA-3. Th2 cells produce the cytokines IL-4, IL-5, and IL-13 (241). Th2 immunity is associated with combatting extracellular pathogens, particularly parasites, but also contributes to allergic airway diseases such as asthma (242). Early studies demonstrated that a Th2-dominated immune response was associated with chronic P. aeruginosa infection in pwCF compared to uncolonized pwCF (207, 240). Even outside the specific context of CF, chronic P. aeruginosa infection was more severe in Th2-skewed BALB/c mice than Th1-skewed C3H/HeN mice (243). High production of prototypical Th1 cytokines, namely IFNγ, correlated with improved lung function in pwCF, and resistance to P. aeruginosa re-infection in murine models is associated with a shift toward a Th1-dominant response (208). Given these observations, the question becomes: what factors are influencing this skewing of T helper cell subsets? Some work has implicated bacterial products as playing a direct role in skewing T cell immunity. For example, N-acyl homoserine lactone quorum sensing (QS) molecules produced by P. aeruginosa have been shown to suppress Th1 cytokines and proliferation, skewing the balance toward Th2 cytokines and Th2 immunity (212, 244). Recently, the P. aeruginosa protease elastase B (LasB) was reported to induce Th2-associated genes through activation of epithelial growth factor family members (245). But this representation of a Th2 vs. Th1 skewing is an oversimplification which fails to account for the heterogeneity of cytokine responses and other immune polarizations, particularly the contribution of Th17 immunity, which has more recently been described to also play an important role in chronic P. aeruginosa lung infection.
Th17 cells are polarized in response to TGF-β, IL-6, IL-21, and IL-23, and defined by the transcription factor RORγt (246). Th17 cells produce IL-17 family cytokines, primarily IL-17A, which is a proinflammatory cytokine involved in granulopoesis, as well as recruitment and activation of neutrophils. IL-17A stimulates the production of several other proinflammatory cytokines and chemokines, namely TNF, IL-6, GM-CSF, and CXCL8 (247, 248). Th17 responses are associated with protection from extracellular pathogens such as bacteria; however, they can also lead to inflammation and autoimmunity. Studies have reported that Th17-induced cytokines are heightened in sputum of P. aeruginosa-colonized pwCF, and that levels of IL-17A are also increased in pwCF undergoing exacerbations (209, 249, 250). Heightened levels of Th2 and Th17 cytokines also correlated with subsequent P. aeruginosa infection in pwCF, suggesting that Th2-Th17 immunity may be a risk factor for (re-) infection as well as a potential marker of ongoing infection (249). However, IL-17 may not play a solely detrimental role. In a murine model of chronic P. aeruginosa lung infection, IL-17 was reported to be required for infection control (228). In this model, loss of IL-17RA increased 2-week infection rates from 25% in wild-type (WT) animals to 100% in IL-17RA-/- mice and increased overall bacterial loads. However, this model surprisingly noted no significant differences in neutrophilic infiltration with loss of IL-17RA. By contrast, in a mouse model of COPD and P. aeruginosa infection, treatment with neutralizing antibodies targeting IL-17A significantly reduced P. aeruginosa bacterial burden, induced significantly less neutrophil infiltration, and improved lung function (251). Administration of exogenous IL-17A instead exacerbated P. aeruginosa-mediated inflammation and lung dysfunction. The differences between these two models may stem from differences in basic infection context: healthy mice as compared to mice with existing chronic inflammatory lung disease. In the latter case, IL-17 may mediate pathologic responses by amplifying a harmful inflammatory cascade, rather than initiating a protective response in the short-term leading to infection resolution, as has been suggested by others (228). Taken together, this work suggests that IL-17 may play a protective role in preventing the transition from acute or intermittent infection to chronic infection by mediating a protective neutrophilic response; however, once chronic infection has been established, robust production of IL-17, primarily mediated by Th17 cells, plays a detrimental role in driving lung pathology and a non-resolving immune response.
Other considerations implicated in maladaptive T cell immunity to P. aeruginosa include the reported impairment of regulatory T cell (Treg) function in chronic P. aeruginosa infection. Some studies show this occurs independently of CF (252). Cross-regulation between Th17 and Treg lineage commitment does imply that a Th17-skewed immune response would negatively regulate inducible Tregs, leading to a lessened Treg response (253). Other reports suggest that Treg dysfunction is a feature of CF disease which may be affected by CFTR mutations, as CFTR potentiator/corrector therapies appear to affect Treg function (254, 255). Most likely, the combination of both factors leads to greater impacts on Tregs, as reported by one study which showed that Tregs from pwCF or Cftr-/- mice had an impaired ability to suppress conventional T cells, and that this effect was enhanced by P. aeruginosa infection. This study also observed that chronic P. aeruginosa decreased overall Treg levels in pwCF (256). Loss of Tregs appeared to primarily impact the memory compartment and may prevent rebalancing of the immune response in chronic infection (256).
Several outstanding questions remain regarding the roles of T cell immunity in chronic P. aeruginosa infection. Are chronic lung diseases, such as CF, skewed toward Th2/Th17 immunity, and poor Treg responsiveness, prior to and independently of bacterial infection, or does infection with pathogens such as P. aeruginosa drive this response? Is a Th17 response protective in acute infection and maladaptive in chronic infection, and if so, how and when does this shift occur? Would improving Treg function allow for better control of dysregulated inflammatory responses to chronic P. aeruginosa? These questions, and others, highlight that the T cell response to chronic P. aeruginosa infection is complex and still in the process of being untangled.
B cells respond robustly to chronic P. aeruginosa infection through production of high levels of P. aeruginosa-specific IgG and IgA antibodies (Abs) and immune complex formation. However, this robust humoral response is not associated with bacterial clearance or clinical improvement and has even in some cases been suggested to be detrimental (257, 258). Though B cell responses were some of the earliest characterized responses to chronic lung P. aeruginosa infection (259, 260), they remain poorly understood to this day (257).
Early studies reported P. aeruginosa-specific Abs were increased in the presence of the mucoid phenotype and associated with a poor prognosis in CF (261). High serum anti-P. aeruginosa Ab levels are associated with persistent infection, to the degree that they have been suggested as a diagnostic tool, indicating that a robust Ab response is not protective or able to clear infection (262, 263). PwCF appear to generate high levels of circulating IgG as well as high levels of primarily IgA serotypes in the ASL lining the lung (257). The formation of immune complexes (ICs) in both sputum and serum has long been reported during chronic P. aeruginosa lung infection, primarily in pwCF (259, 264). However, studies have not identified definitively whether IC formation is detrimental. Early work reported that ICs stimulated greater inflammation via complement activation, as well as worse clinical status in patients; however, other longitudinal studies failed to find correlations (265–268). Others have suggested the efficacy of the Ab response to P. aeruginosa may be impaired by various factors, including the inflammatory CF lung environment, chronic exposure to P. aeruginosa antigens, and phenotypic changes in P. aeruginosa including LPS modifications, loss of flagella, overproduction of alginate, and biofilm lifestyle (257). Other factors may include impaired Ab transport in dehydrated ASL and entrapment in thickened biofilms and mucus, as well as increased degradation by host or bacterial proteases which are plentiful in chronically infected lungs (269–271). Several groups have shown anti-P. aeruginosa Abs developed in the chronic inflammatory context of CF may have lower avidity or affinity, which negatively impacts their efficacy as opsonins to stimulate phagocytosis, thus preventing bacterial clearance (272–275). A reduced ability of phagocytes to kill opsonized P. aeruginosa, both from loss of phagocytic capacity due to chronic inflammatory stimuli or intrinsic defects, as well as evasion methods of P. aeruginosa, also limits the functionality of Ab responses. Finally, while Abs may not be able to clear chronic P. aeruginosa, they still play an important role by protecting against dissemination from local sites to systemic infection. Overall, B cell responses to P. aeruginosa are robust but fail to clear chronic infection, which has implications for development of Ab-based therapies as well as vaccines, discussed in the following section.
Historically, treatment of chronic P. aeruginosa has relied on antibiotics prescribed upon initial detection of infection (i.e., for eradication) or pulmonary exacerbation in conditions such as CF, COPD, and NCFB. Given the known association between P. aeruginosa infection in pwCF and poor outcomes, current clinical guidelines recommend chronic suppressive nebulized antibiotics to control P. aeruginosa (276). However, antibiotic use is becoming fraught with the global rise of antimicrobial resistance, which is increasingly being found in P. aeruginosa isolates from chronic lung infections (277). Additionally, antibiotics are frequently unsuccessful in clearing P. aeruginosa infection in chronic lung diseases, particularly in adults. Once infections transition to a chronic state, they become increasingly resistant to antibiotics through adaptations such as biofilm formation, hypermutability, and the formation of persister subpopulations, as discussed previously. The limited efficacy of both systemic and nebulized antimicrobials indicates a need for novel approaches to both prevent and clear chronic P. aeruginosa infections in individuals with chronic lung disease.
However, clearance of infection is not the sole metric by which to reduce pathology and improve outcomes in these patients. As described above, the immune system contributes substantially to lung damage and morbidity associated with chronic P. aeruginosa infection, and so represents an additional target to reduce the disease burden of chronic infections in these patients. This section will discuss several key approaches to modulate infection, including methods designed to treat or prevent infection itself by targeting P. aeruginosa, immunomodulatory therapies designed to reduce the pathology of infection, and finally therapies designed to correct the innate defects, specifically in CF, which predispose individuals to chronic P. aeruginosa infection. Insights from CFTR modulators in terms of changes to immune function and bacterial colonization are beginning to shed new light on the relationship between inflammation and chronic P. aeruginosa infection which may have broader applicability in non-CF contexts.
Two of the earliest antibiotics proven effective for pwCF were inhaled tobramycin and oral azithromycin, which are used in approximately two-thirds of pwCF with chronic P. aeruginosa infection, most often in combination (278). These antibiotics are also frequently used to treat P. aeruginosa in COPD and NCFB. Tobramycin is an aminoglycoside antibiotic which binds the 30s subunit of the bacterial ribosome to inhibit translation initiation (279). Inhaled long-term use of tobramycin appears to improve lung function and reduce exacerbation rates in patients, likely by helping to keep P. aeruginosa under check (280). Azithromycin belongs to the macrolide class of antibiotics and operates by binding to the 50s subunit of the bacterial ribosome and inhibiting translation, although it is largely prescribed in pwCF at non-bactericidal doses for its immunomodulatory function and other mechanisms of action, which will be discussed in section 4.3, rather than anti-P. aeruginosa activity (281). Another common antibiotic used in combination with tobramycin is inhaled aztreonam, a beta-lactam antibiotic which inhibits bacterial cell wall synthesis (282, 283). Novel antibiotics as well as combination therapies are still being robustly investigated in CF and other chronic P. aeruginosa infection contexts, which have recently been summarized by others (284). Clinical decisions regarding antibiotic treatment in pwCF are beginning to shift in the era of CFTR modulators to account for improvements in lung function, reduced ability to acquire sputum samples, and the changing landscape of bacterial infections (285).
Development of a vaccine to protect against P. aeruginosa would represent a significant advance for pwCF and other chronic lung conditions. Vaccine development efforts for P. aeruginosa have been ongoing for over half a century but have been hindered by a multiple factors, including the plasticity of P. aeruginosa owing in part to its large genome, its diversity of virulence factors and serotypes, the complexity of its pathogenesis, and its interactions with the lung environment and immune system (128). Hundreds of vaccines have been developed pre-clinically, but comparatively few have reached clinical trials (128, 284).
A variety of potential antigenic targets have been investigated using numerous vaccine strategies. LPS has been one of the most well-investigated targets due to its immunogenicity and surface accessibility; however, early vaccines ran into challenges with toxicity, and later vaccines have faced difficulties in ensuring protection against different, mutable P. aeruginosa serotypes (286, 287). Flagellar antigens, as well as pili, have also been vaccine targets, given their importance for P. aeruginosa virulence and initial colonization. Vaccines solely targeting flagellar antigens elicited some protective antibodies in early-stage clinical trials, but not in sufficient proportions of participants (288). Various outer membrane porins, particularly OprF, have also been investigated as antigenic targets (289). Alginate, an exopolysaccharide which is often overproduced in chronic infections, has also been targeted, with early vaccines inducing the development of opsonophagocytic Abs against alginate which could theoretically potentiate phagocytic responses (290). However, low immunogenicity has led to efforts to enhance the immunogenic properties of alginate, such as conjugation with other antigens to transform it into a T-cell dependent antigen (128). Targeting the T3SS of P. aeruginosa, particularly its key component PcrV, has shown some efficacy in reducing airway inflammation and damage in pwCF in a stage II clinical trial (291, 292). Overall, these more recent approaches have transitioned to the use of multiple antigenic targets in combination with various adjuvants to maximize the breadth and types of protection (293–295). Whole-cell killed and live-attenuated bacterial vaccine approaches have been less studied, but several are still under development. Mucosal administration of paraformaldehyde-killed P. aeruginosa induced protective immune responses in rat lungs (296). Alternative methods of P. aeruginosa inactivation have been recently used, including H2O2 which was less toxic and resulted in better epitope retention than paraformaldehyde, leading to more effective IgG protection (297). Live-attenuated vaccine strategies have also been effective at inducing multifactorial humoral and cellular immune protection, but some concerns remain about residual virulence (298, 299). Novel vaccine delivery strategies, such as DNA and RNA vaccines, are also being considered against P. aeruginosa (289, 300). Various mRNA vaccine candidates are showing promising results, including PcrV and OprF-I vaccines which were recently shown to be able to induce Th1-skewed immunity, confer broad protection, and reduce bacterial load in murine P. aeruginosa infection models (289).
Future directions for vaccine development will require a better understanding of what the most effective correlates for immune protection against P. aeruginosa are. Humoral immune responses are well-induced by many of these vaccine strategies, but patients with chronic P. aeruginosa infection also have robust Ab responses that fail to provide significant protection. Our understanding of T-cell mediated immunity to P. aeruginosa is also incomplete: Th2 immunity appears maladaptive in chronic infection, with Th1 responses perhaps being a better target. Th17 immunity may be protective in acute infection but pathogenic in chronic infection, leaving the question of whether it would be an effective correlate of vaccine protection up in the air. Overall, acquiring a greater understanding of immunity to P. aeruginosa infection, particularly adaptive immunity, in the context of chronic lung conditions like CF where patients may have inherently dysregulated immune responses, will be critical to developing effective vaccines. Recognizing the variability of protein antigens, as well as their loss during chronic infection –for example, loss of flagella, pili, T3SS—and shielding within biofilms should also be considered when thinking about developing vaccines for populations at risk of chronic P. aeruginosa infection. Different routes of administration, such intranasal or inhaled vaccination, may be necessary to induce protection at mucosal sites, which will be important particularly for P. aeruginosa lung infections (298, 299). Several reviews have recently been written which go into greater detail on the topic of vaccine development against P. aeruginosa (128, 284, 301).
Immunomodulatory therapies are intended to target the pathology associated with chronic P. aeruginosa infection, rather than treat the infection itself (Figure 4). Their importance is highlighted by the significant work reviewed above, which indicates that the immune response to chronic P. aeruginosa infection often causes more lung damage and pathology than the infection itself. Neutrophil granule products, reactive oxygen species, and other immune products cause significant direct damage to airway epithelial cells, and inflammatory cytokine production drives further inflammatory processes forward –all of these represent potential targets for immunomodulatory therapies to limit this vicious cycle of inflammation.
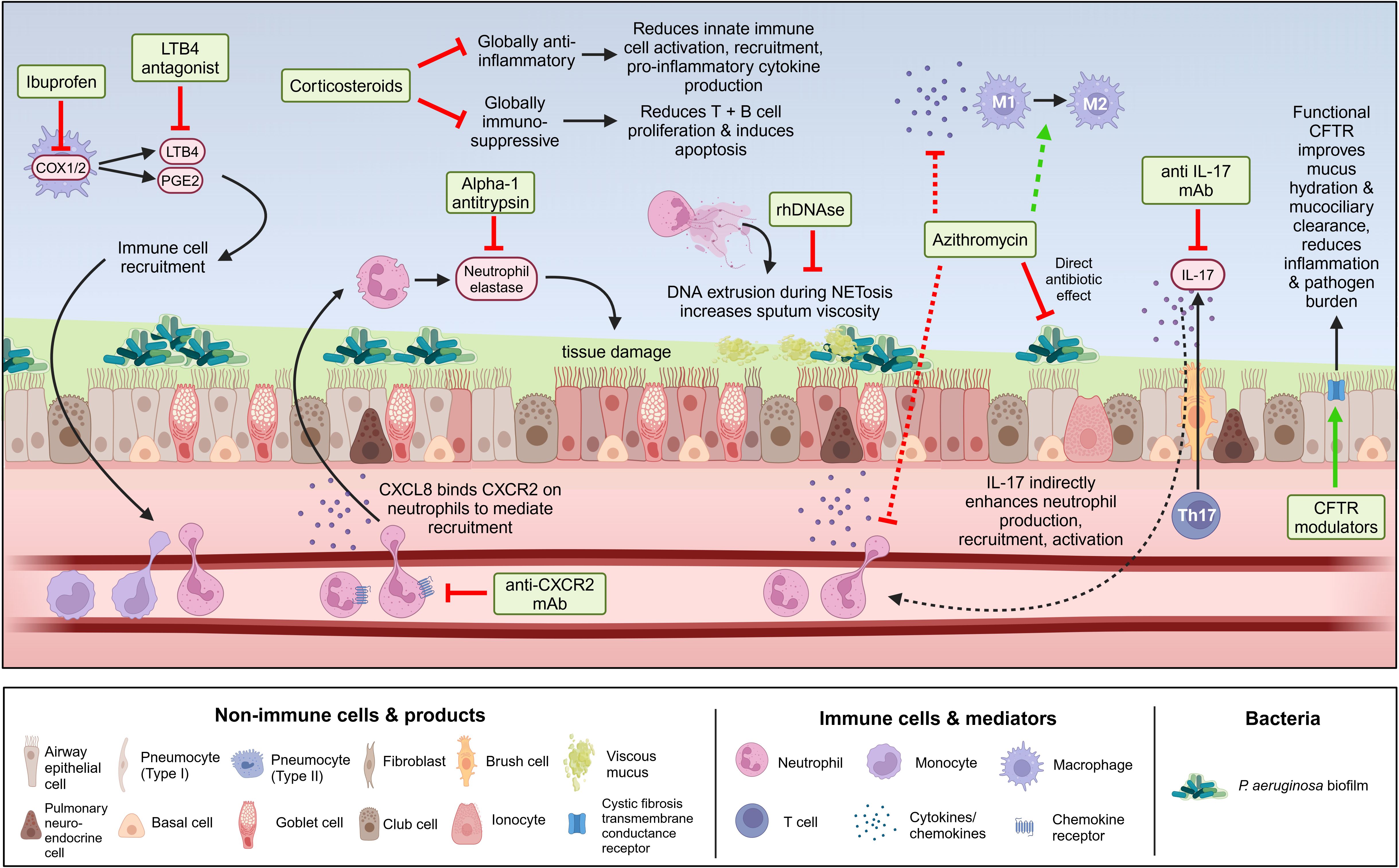
Figure 4 Immunomodulatory therapies available and under development for chronic P. aeruginosa infection in the context of chronic lung diseases, particularly CF. Beginning with non-specific therapies, corticosteroids have broad anti-inflammatory, immunosuppressive, anti-proliferative, and vasoconstrictive effects that can reduce the progression of inflammation-driven lung damage in chronic lung disease, but also increase the risk of infections such as P. aeruginosa. High-dose ibuprofen acts on the COX-1 and COX-2 receptors to inhibit prostaglandin (ex. PGE2) and leukotriene (ex. LTB4) synthesis, reducing immune cell migration into the airways. Azithromycin is a macrolide antibiotic which, in addition to directly inhibiting bacterial growth, has been reported to have various immunomodulatory functions on innate immune cells like macrophages and neutrophils. Moving into more targeted therapies, alpha-1-antitrypsin is an antiprotease naturally produced in the human lung which restricts neutrophil elastase activity and can be given supplementarily to reduce lung damage. Recombinant human DNAse (rhDNAse) targets enhanced DNA concentrations in sputum because of NET release by neutrophils to reduce sputum viscosity and improve pulmonary function. LTB4 antagonists block the neutrophil chemoattractant and activator LTB4 but caused pulmonary exacerbations in clinical trials. Anti-CXCR2 monoclonal antibodies (mAbs) block the CXCR2 receptor on neutrophils preventing binding of the neutrophil chemoattractant and activator CXCL8. Anti-IL-17 mAbs block IL-17 function to limit neutrophil proliferation and activation. Finally, CFTR modulators, by correcting CFTR dysfunction indirectly modulate the immune system by improving overall lung function, reducing pathogen burden, and reducing inflammation. Figure created in BioRender.com.
The first immunomodulatory therapies used in CF and other chronic lung conditions were broad spectrum anti-inflammatories, such as corticosteroids. Corticosteroid use is generally limited to inhaled and short-term, given the risk of adverse side effects (302), and recent studies have shown they have limited benefits in CF (303). Inhaled corticosteroids have been associated with increased risk of P. aeruginosa infection in other chronic lung conditions, such as COPD, due to their broadly immunosuppressive effects (304, 305). Despite this, corticosteroids are still broadly used in COPD (51, 171). By contrast, ibuprofen is a non-steroidal anti-inflammatory drug which has been shown to slow the progression of lung damage in pwCF, associated with its ability to reduce neutrophil migration into the airways by inhibiting leukotriene and prostaglandin production (302). However, uptake in ibuprofen use has been variable in adult CF clinics, potentially limited by concerns regarding side effects (i.e., nephrotoxicity) and the subsequent clinical monitoring associated with long-term high-dose use (302). High-dose ibuprofen is utilized more in pediatric CF patients (306), as they generally have fewer pre-existing comorbidities and additional medications. Azithromycin, described above in the antibiotic section, is notable as it has several postulated mechanisms of action outside of its antibiotic function. The first includes anti-virulence effects, such as suppression of quorum sensing, protein translation, and secretion of virulence effectors (307). Azithromycin has also been reported to have broad immunomodulatory properties. Azithromycin has been shown to reduce neutrophil levels and inflammatory markers such as myeloperoxidase in the serum of pwCF uninfected with P. aeruginosa (308) as well as patients with bronchiolitis obliterans syndrome (309), indicating that it may have beneficial immunomodulatory effects even in the absence of chronic P. aeruginosa infection. Azithromycin is also used for its immunomodulatory effects in COPD and NCFB (51, 310). Preclinical studies have indicated that azithromycin may inhibit various pro-inflammatory signaling pathways, including NFκB, STAT1, and inflammasome activation, resulting in reduced production of pro-inflammatory cytokines such as TNF and IL-1β and shifted macrophage polarization (311–313). However, other studies have countered the clinical relevance of these findings, reporting that long-term azithromycin use is still primarily effective in pwCF infected with P. aeruginosa, suggesting that its immunomodulatory benefits may come second to benefits derived from direct interference with bacterial growth (314–316). Another clinical trial reported that, in addition to azithromycin’s benefits being restricted to the short-term, long-term usage resulted in increased prevalence of macrolide resistance in other organisms (such as S. aureus) (317), which raises concerns about its long-term use for non-antibiotic purposes.
Immunomodulatory therapies targeting specific problematic aspects of the inflammatory response to chronic infection are more appealing. Because much chronic P. aeruginosa pathology is driven by neutrophils, several strategies have been developed to either target soluble effectors involved in neutrophil recruitment, or neutrophil granule products themselves. Starting with the top of the cascade, antagonists have been developed to block CXCR2, the receptor expressed on neutrophils which binds CXCL8 (amongst other chemokines) to mediate neutrophil migration. Clinical trial results indicated that while use of a CXCR2 antagonist lowered neutrophil numbers in the lungs of pwCF, it had no effect on sputum neutrophil levels, indicating that effects may be localized or that CXCR1 (present in humans but not mice) may compensate (318). Use of a similar CXCR2 antagonist was effective in improving lung function in COPD, indicating that it may be a disease-agnostic treatment (319). Another approach involves neutralizing Abs targeting IL-17, which significantly reduced neutrophil infiltration, lowered proinflammatory cytokine production, and reduced tissue damage in murine lungs during chronic P. aeruginosa infection (320, 321). The success of anti-IL-17 Abs like secukinumab in autoimmune diseases have set a precedent for potential translatability (322). Other approaches have been developed to target neutrophil products like NE, which damages the lung epithelium. Alpha 1-antitrypsin (AAT) is an antiprotease naturally produced in the healthy lung to restrain protease activity. In CF, AAT levels are dysregulated, allowing for excess protease activity (323, 324). Hereditary AAT deficiency is also a rare causative agent of COPD (325). Inhaled AAT was found to decrease neutrophilic inflammation and directly impair NE function in P. aeruginosa infection without compromising the host’s ability to clear bacteria (326, 327). PwCF treated with inhaled AAT had decreased airway inflammation, measured by parameters including neutrophil infiltration and pro-inflammatory cytokine production (328). Completed trials have not reported increases in lung function, but trials were generally short-term (i.e. up to one month). Studies conducted over longer timeframes may demonstrate the ability of AAT to limit long-term lung function decline. More recently, AAT was demonstrated to directly bind NE embedded in human NETs to limit NET-mediated damage to the epithelial barrier (329). An example of a targeted therapy that has recently entered successful clinical use is recombinant human DNAse (rhDNAse). rhDNAse is currently used to treat pwCF by targeting enhanced DNA concentrations in CF sputum due to NET release by neutrophils. Studies have shown that rhDNAse reduced sputum viscosity, improved lung function, and lowered pulmonary exacerbations; however, clinical trials showed only mild decreases in factors such as NE and CXCL8, with no changes in neutrophils or other inflammatory cytokines in the short term. Long-term treatment over 3 years did prevent increases in several inflammatory mediators, resulting in guideline based recommendations (330–333).
There is a crucial balance to be struck in the use of immunomodulatory therapies in chronic P. aeruginosa infection. Successful immunomodulatory therapies must be able to limit inflammation without blunting host immune responses to the degree that the host is vulnerable to new infections or loses the ability to control P. aeruginosa infection on the local level, resulting in infectious exacerbation and/or systemic spread. One example of this concern was demonstrated in a phase II clinical trial of a receptor antagonist for LTB4, a potent neutrophil chemoattractant and activator. This trial demonstrated a statistically significant increase in pulmonary exacerbations in patients receiving the LTB4 receptor antagonist amelubant (52-55% in the treatment group versus 33-43% in the placebo), resulting in premature termination of the trial (334). A better understanding of the complex immune processes involved in susceptibility to P. aeruginosa infection as well as those involved in lung damage and pathology, is necessary for the careful and rational design of novel immunomodulatory therapies.
The successful introduction of HEMT to a large proportion of the CF population, estimated to be more than 90% in some countries (335), has dramatically improved both pulmonary and extra-pulmonary health. However, structural lung damage with infections persists.
CFTR modulators fall into 2 functional categories: potentiators, which enhance the channel opening probability of CFTR, and correctors, which improve CFTR folding and trafficking (3, 336). Amongst CFTR modulators, two are considered highly effective. Ivacaftor, targeting the Gly551Asp gating mutation to increase channel time, was the first available HEMT released in 2012 but available only to ~5% of pwCF. Approved in 2019, Elexacaftor-Tezacaftor-Ivacaftor (ETI) is an HEMT relevant to many pwCF given its efficacy for at least one copy of the F508del CFTR mutation (and approved by the FDA for other specific rare mutations), accounting for ≥90% of individuals (337). Recent data from a compassionate access program has also demonstrated real-world efficacy across non-F508del mutations.
CFTR modulators have been reported to affect lung inflammation and immune parameters in various ways. Some in vitro studies have suggested that CFTR modulators can directly modulate inflammatory responses in CFTR-deficient cells. For example, CFTRF508del epithelial cells treated with lumacaftor/ivacaftor exhibited less exuberant inflammatory responses, decreased production of CXCL8, and enhanced tissue repair following P. aeruginosa exposure (338, 339). Animal models have allowed for tracking of the timing of therapy initiation, demonstrating that administration of ivacaftor to CFTRG551D ferrets in utero partially protected animals from multiorgan pathologies caused by CF, and led to reduced mucus accumulation and lung bacterial infections. Withdrawal of therapy at any age led to the rapid onset of pathology including airway inflammation (340). More recently, studies have begun to be published investigating the effects of ETI therapy, though much of the significant work in this area is yet to come. Some studies have reported positive immunomodulatory effects, including significant decreases in blood levels of inflammatory cytokines CXCL8, IL-6, and IL-17A, as well as normalization of peripheral blood immune cell composition (341), even in pwCF with advanced disease (342). However, another study reported no significant decrease in blood levels of CXCL8 and IL-6 following 6 months of ETI treatment, as well as no overall changes in the functionality of blood neutrophils and monocytes collected from these patients (343).
Numerous studies have also evaluated the effects of CFTR modulator therapy on bacterial colonization in pwCF, particularly with P. aeruginosa. Results from the GOAL study indicated that 1 year of ivacaftor treatment significantly reduced the odds of P. aeruginosa culture positivity (by 35%), as well as the odds of P. aeruginosa being mucoid (by 23%), indicating a reduction in chronic infection burden (344). These changes may correspond with improved mucociliary clearance also observed in this study population, indicating some repair of this essential lung defense mechanism (117). However, another study by Harris et al. looking at subpopulations within the GOAL study, contrastingly observed minimal changes in P. aeruginosa colonization or bacterial load 6 months following ivacaftor therapy (345). They also reported overall minimal shifts in microbial communities, although, importantly, younger patients and patients without P. aeruginosa infection experienced greater shifts, suggesting that ivacaftor-induced changes in lung function may have greater ramifications for microbial colonization in patients with less-severe lung disease (345). A study in a separate cohort by Hisert et al. reported ivacaftor significantly reduced sputum concentrations of P. aeruginosa during the first year of therapy; however, patients still failed to eradicate their chronic P. aeruginosa infections and bacterial density rebounded after the first year, suggesting that ivacaftor therapy may not be sufficient to clear chronic P. aeruginosa, especially in patients with more severe lung disease (346). Recent studies investigating ETI therapy have shown more promising results. A study investigating 124 subjects receiving ETI therapy for at least 1 year reported that P. aeruginosa colonization decreased from approximately 54% of patients to 30% of patients (347), which has been corroborated by other studies (341). In a German CF cohort, it was recently shown that 21 months of ETI therapy overall decreased P. aeruginosa detection in throat swabs and sputum from approximately 40% to 23%, although it was less effective in older patients and those with lower baseline lung function (348), as previously reported for ivacaftor. However, the PROMISE study by Nichols et al. reported that while ETI therapy significantly decreased the sputum bacterial density of pathogens such as P. aeruginosa, S. aureus, and other species, those pathogens were still detectable by PCR despite sputum becoming culture-negative, indicating that infections still persisted at lower levels (349). This suggests that culture-based methods may not be sensitive enough to detect persisting pathogens. However, ETI treatment did increase overall bacterial diversity in sputum, which has also been reported by others (349, 350). ETI therapy has been shown to restructure the airway microbiome and metabolome, changing the metabolic niche in airway mucus to disfavor CF pathogens and decreasing the ratio of classical CF pathogens to anaerobes (350). Initiation of ETI therapy has also been shown to lead to a conversion in CF airway microbiomes from highly divergent and individualized before therapy toward a more similar and even community composition one year following therapy, suggesting a shift toward a healthier airway microbiome composition (351). Overall, these results suggest that HEMTs, particularly ETI therapy, have the potential to alter chronic P. aeruginosa infection in pwCF, decreasing but not entirely clearing P. aeruginosa from the lungs because of changes to the airway niche via improvements in CFTR function.
Overall, HEMTs, particularly with the approval of ETI therapy, are changing the landscape of CF disease and chronic P. aeruginosa infection and may potentially have broader impacts in non-CF chronic lung disease in future. Currently, trials are ongoing to confirm the safety of modulators in the infant, and even prenatal, populations of pwCF –those most likely to receive the greatest benefit from therapy in the form of attenuated development of structural lung disease and subsequent infections. However, there still exists a significant population of adult pwCF with established disease, including chronic inflammation and chronic infection, as well as a subset of patients (around 10%) who are ineligible for existing CFTR modulators. CFTR modulators have had variable success, thus far, at eradicating or lessening the burden of chronic P. aeruginosa infection in pwCF, with less success in those with more established lung disease (which often occurs because of P. aeruginosa infection itself). These populations would still benefit from the development of novel immunomodulatory therapies, and a deeper understanding of the host immune mechanisms rendering them susceptible to chronic infection with opportunistic pathogens like P. aeruginosa.
The opportunistic pathogen P. aeruginosa has demonstrated a remarkable ability to colonize the lungs of individuals with chronic lung disease, particularly CF, as well as COPD and NCFB. Chronic infection leads to a robust yet ineffective inflammatory response that causes extensive and often irreversible lung damage leading to eventual lung failure. Chronic P. aeruginosa infection, for this reason, is strongly associated with worsened outcomes in individuals with chronic lung diseases. The inefficacy of the immune response to chronic P. aeruginosa is still not fully understood, although several key players have been identified. Neutrophils are recruited and sustained in high numbers during chronic infection via cytokine signaling which is skewed toward a particular proinflammatory milieu indicative of Th2 and Th17 immunity. Phagocytic and cytotoxic killing of P. aeruginosa is thwarted by various bacterial adaptations such as biofilm formation, LPS antigen modifications, loss of virulence factors such as flagella, and formation of persister cells, but also by impaired immune function in cells such as macrophages and neutrophils which may be secondary to chronic inflammatory stimuli and, in the case of CF, CFTR-intrinsic defects. B cells produce high levels of anti-P. aeruginosa IgG and IgA that also fail to provide appropriate protection. However, many questions remain. What are the roles of numerous other cell types in chronic P. aeruginosa infection? What key differences between acute and chronic infection render a response protective in one context but pathological in the other? Will differences between chronic P. aeruginosa infection in different chronic lung diseases require different treatment approaches?
With greater understanding of how the immune response to chronic P. aeruginosa is dysregulated, and how dysregulated host immunity may contribute to susceptibility to chronic P. aeruginosa infection, comes a greater ability to develop therapies able to modulate immunity against P. aeruginosa –either by dampening harmful inflammatory mediators such as neutrophil effectors, or by finding ways to boost immunity against P. aeruginosa, for example, through vaccine development. Many novel approaches to treat chronic P. aeruginosa and its associated inflammation have been developed, but comparatively few have found success, which may be a consequence of our still-incomplete knowledge. Even in the era of CFTR modulators, which can partially correct CFTR dysfunction and restore crucial aspects of lung function in pwCF, P. aeruginosa remains a threat. Current CFTR modulators appear unable to fully clear P. aeruginosa from patients already colonized, though they may reduce bacterial burden by improving mucociliary clearance and altering the lung microenvironment. However, CFTR modulator therapies are also generating new insights which will continue to advance our understanding of chronic P. aeruginosa respiratory infections, both in CF and other chronic lung diseases. As the landscape of chronic P. aeruginosa infection changes, with pwCF living longer lives and the prevalence of other chronic lung disease such as COPD and NCFB increasing, it is becoming both more conceivable and more pivotal to untangle the complex roles of host immunity in chronic P. aeruginosa infection.
RN: Conceptualization, Writing – original draft, Writing – review & editing. CT: Writing – review & editing. BJ: Writing – review & editing. AL: Writing – review & editing. ZC: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The host-Pseudomonas aeruginosa interaction projects in the Cheng lab are supported by the Natural Science and Engineering Research Council (NSERC) of Canada Discovery Grants (grant number: RGPIN/04912-2016) and Canadian Institutes of Health Research Project Grant (grant number: PJT165970).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Desai H, Eschberger K, Wrona C, Grove L, Agrawal A, Grant B, et al. Bacterial colonization increases daily symptoms in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. (2014) 11:303–9. doi: 10.1513/AnnalsATS.201310-350OC
2. Martínez-Solano L, Macia MD, Fajardo A, Oliver A, Martinez JL. Chronic Pseudomonas aeruginosa Infection in chronic obstructive pulmonary disease. Clin Infect Dis. (2008) 47:1526–33. doi: 10.1086/593186
3. Ribeiro CMP, Higgs MG, Muhlebach MS, Wolfgang MC, Borgatti M, Lampronti I, et al. Revisiting host-pathogen interactions in cystic fibrosis lungs in the era of CFTR modulators. Int J Mol Sci. (2023) 24:5010. doi: 10.3390/ijms24055010
4. Wilson R, Aksamit T, Aliberti S, De Soyza A, Elborn JS, Goeminne P, et al. Challenges in managing Pseudomonas aeruginosa in non-cystic fibrosis bronchiectasis. Respir Med. (2016) 117:179–89. doi: 10.1016/j.rmed.2016.06.007
5. Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol. (2013) 34:1–14. doi: 10.1086/668770
6. Fujitani S, Sun H-Y, Yu VL, Weingarten JA. Pneumonia due to Pseudomonas aeruginosa. Chest. (2011) 139:909–19. doi: 10.1378/chest.10-0166
7. Reyes J, Komarow L, Chen L, Ge L, Hanson BM, Cober E, et al. Global epidemiology and clinical outcomes of carbapenem-resistant Pseudomonas aeruginosa and associated carbapenemases (POP): a prospective cohort study. Lancet Microbe. (2023) 4:e159–70. doi: 10.1016/S2666-5247(22)00329-9
8. Cystic Fibrosis Foundation. 2021 Patient registry Annual Data Report. Bethesda, Maryland: Cystic Fibrosis Foundation (2022).
9. Nährlich L, Burkhart M, Wosniok J. German Cystic Fibrosis Registry Annual Report 2021. Bonn, Germany: Mukoviszidose e.V. & Mukoviszidose Institut gGmbH (2022).
10. Cystic Fibrosis Canada. The Canadian Cystic Fibrosis Registry 2021 Annual Data Report. Toronto, Canada: Cystic Fibrosis Canada (2023).
11. Cystic Fibrosis Trust. UK Cystic Fibrosis Registry 2021 Annual Data Report. UK: Cystic Fibrosis Trust (2022).
12. McDonnell MJ, Jary HR, Perry A, MacFarlane JG, Hester KLM, Small T, et al. Non cystic fibrosis bronchiectasis: A longitudinal retrospective observational cohort study of Pseudomonas persistence and resistance. Respir Med. (2015) 109:716–26. doi: 10.1016/j.rmed.2014.07.021
13. King P, Citron DM, Griffith DC, Lomovskaya O, Dudley MN. Effect of oxygen limitation on the in vitro activity of levofloxacin and other antibiotics administered by the aerosol route against Pseudomonas aeruginosa from cystic fibrosis patients. Diagn Microbiol Infect Dis. (2010) 66:181–6. doi: 10.1016/j.diagmicrobio.2009.09.009
14. Reynolds D, Kollef M. The epidemiology and pathogenesis and treatment of pseudomonas aeruginosa infections: An update. Drugs. (2021) 81:2117–31. doi: 10.1007/s40265-021-01635-6
15. Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014. Infect Control Hosp Epidemiol. (2016) 37:1288–301. doi: 10.1017/ice.2016.174
16. Vincent J-L, Sakr Y, Singer M, Martin-Loeches I, MaChado FR, Marshall JC, et al. Prevalence and outcomes of infection among patients in Intensive Care Units in 2017. JAMA. (2020) 323:1478. doi: 10.1001/jama.2020.2717
17. Ciofu O, Tolker-Nielsen T, Jensen PØ, Wang H, Høiby N. Antimicrobial resistance, respiratory tract infections and role of biofilms in lung infections in cystic fibrosis patients. Adv Drug Delivery Rev. (2015) 85:7–23. doi: 10.1016/j.addr.2014.11.017
18. Campodónico VL, Gadjeva M, Paradis-Bleau C, Uluer A, Pier GB. Airway epithelial control of Pseudomonas aeruginosa infection in cystic fibrosis. Trends Mol Med. (2008) 14:120–33. doi: 10.1016/j.molmed.2008.01.002
19. Lei L, Traore S, Romano Ibarra GS, Karp PH, Rehman T, Meyerholz DK, et al. CFTR-rich ionocytes mediate chloride absorption across airway epithelia. J Clin Invest. (2023) 133:e171268. doi: 10.1172/JCI171268
20. Rubin BK. Mucus structure and properties in cystic fibrosis. Paediatr Respir Rev. (2007) 8:4–7. doi: 10.1016/j.prrv.2007.02.004
21. Mall MA. Role of cilia, mucus, and airway surface liquid in mucociliary dysfunction: lessons from mouse models. J Aerosol Med Pulm Drug Delivery. (2008) 21:13–24. doi: 10.1089/jamp.2007.0659
22. Su S, Hassett DJ. Anaerobic Pseudomonas aeruginosa and other obligately anaerobic bacterial biofilms growing in the thick airway mucus of chronically infected cystic fibrosis patients: an emerging paradigm or “Old Hat”? Expert Opin Ther Targets. (2012) 16:859–73. doi: 10.1517/14728222.2012.708025
23. Matsui H, Wagner VE, Hill DB, Schwab UE, Rogers TD, Button B, et al. A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci. (2006) 103:18131–6. doi: 10.1073/pnas.0606428103
24. Alexis NE, Muhlebach MS, Peden DB, Noah TL. Attenuation of host defense function of lung phagocytes in young cystic fibrosis patients. J Cyst Fibros. (2006) 5:17–25. doi: 10.1016/j.jcf.2005.11.001
25. Bonfield T, Chmiel JF. Impaired innate immune cells in cystic fibrosis: Is it really a surprise? J Cyst Fibros. (2017) 16:433–5. doi: 10.1016/j.jcf.2017.06.001
26. Ratner D, Mueller C. Immune responses in cystic fibrosis: Are they intrinsically defective? Am J Respir Cell Mol Biol. (2012) 46:715–22. doi: 10.1165/rcmb.2011-0399RT
27. Cohen-Cymberknoh M, Kerem E, Ferkol T, Elizur A. Airway inflammation in cystic fibrosis: molecular mechanisms and clinical implications. Thorax. (2013) 68:1157–62. doi: 10.1136/thoraxjnl-2013-203204
28. Pernet E, Guillemot L, Burgel P-R, Martin C, Lambeau G, Sermet-Gaudelus I, et al. Pseudomonas aeruginosa eradicates Staphylococcus aureus by manipulating the host immunity. Nat Commun. (2014) 5:5105. doi: 10.1038/ncomms6105
29. Salsgiver EL, Fink AK, Knapp EA, LiPuma JJ, Olivier KN, Marshall BC, et al. Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis. Chest. (2016) 149:390–400. doi: 10.1378/chest.15-0676
30. Gannon AD, Darch SE. Same game, different players: emerging pathogens of the CF lung. mBio. (2021) 12:e01217–20. doi: 10.1128/mBio.01217-20
31. LiPuma JJ. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev. (2010) 23:299–323. doi: 10.1128/CMR.00068-09
32. Pienkowska K, Pust M-M, Gessner M, Gaedcke S, Thavarasa A, Rosenboom I, et al. The cystic fibrosis upper and lower airway metagenome. Microbiol Spectr. (2023) 11:e03633–22. doi: 10.1128/spectrum.03633-22
33. Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci. (2012) 109:5809–14. doi: 10.1073/pnas.1120577109
34. Coburn B, Wang PW, Diaz Caballero J, Clark ST, Brahma V, Donaldson S, et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci Rep. (2015) 5:10241. doi: 10.1038/srep10241
35. Cuthbertson L, Walker AW, Oliver AE, Rogers GB, Rivett DW, Hampton TH, et al. Lung function and microbiota diversity in cystic fibrosis. Microbiome. (2020) 8:45. doi: 10.1186/s40168-020-00810-3
36. Carmody LA, Caverly LJ, Foster BK, Rogers MAM, Kalikin LM, Simon RH, et al. Fluctuations in airway bacterial communities associated with clinical states and disease stages in cystic fibrosis. PloS One. (2018) 13:e0194060. doi: 10.1371/journal.pone.0194060
37. Whitters D, Stockley R. Immunity and bacterial colonisation in bronchiectasis. Thorax. (2012) 67:1006–13. doi: 10.1136/thoraxjnl-2011-200206
38. Macfarlane L, Kumar K, Scoones T, Jones A, Loebinger MR, Lord R. Diagnosis and management of non-cystic fibrosis bronchiectasis. Clin Med. (2021) 21:e571–7. doi: 10.7861/clinmed.2021-0651
39. Chalmers JD, Polverino E, Crichton ML, Ringshausen FC, De Soyza A, Vendrell M, et al. Bronchiectasis in Europe: data on disease characteristics from the European Bronchiectasis registry (EMBARC). Lancet Respir Med. (2023) 11:637–49. doi: 10.1016/S2213-2600(23)00093-0
40. José RJ, Manuel A, Gibson-Bailey K, Lee L. Post COVID-19 bronchiectasis: a potential epidemic within a pandemic. Expert Rev Respir Med. (2020) 14:1183–4. doi: 10.1080/17476348.2020.1804366
41. Poletti V. Diffuse panbronchiolitis. Eur Respir J. (2006) 28:862–71. doi: 10.1183/09031936.06.00131805
42. Mac Aogáin M, Narayana JK, Tiew PY, Ali NABM, Yong VFL, Jaggi TK, et al. Integrative microbiomics in bronchiectasis exacerbations. Nat Med. (2021) 27:688–99. doi: 10.1038/s41591-021-01289-7
43. Rosenboom I, Thavarasa A, Richardson H, Long MB, Wiehlmann L, Davenport CF, et al. Sputum metagenomics of people with bronchiectasis. ERJ Open Res. (2024) 10:01008–2023. doi: 10.1183/23120541.01008-2023
44. King P. The pathophysiology of bronchiectasis. Int J Chron Obstruct Pulmon Dis. (2009) 4: 411–9. doi: 10.2147/COPD.S6133
45. Miao X-Y, Ji X-B, Lu H-W, Yang J-W, Xu J-F. Distribution of major pathogens from sputum and bronchoalveolar lavage fluid in patients with noncystic fibrosis bronchiectasis: A systematic review. Chin Med J (Engl). (2015) 128:2792–7. doi: 10.4103/0366-6999.167360
46. Choi H, Chalmers JD. Bronchiectasis exacerbation: a narrative review of causes, risk factors, management and prevention. Ann Transl Med. (2023) 11:25–5. doi: 10.21037/atm-22-3437
47. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. (2022) 399:2227–42. doi: 10.1016/S0140-6736(22)00470-6
48. Wang Z, Liu H, Wang F, Yang Y, Wang X, Chen B, et al. A refined view of airway microbiome in chronic obstructive pulmonary disease at species and strain-levels. Front Microbiol. (2020) 11:1758. doi: 10.3389/fmicb.2020.01758
49. Lieberman D, Lieberman D. Pseudomonal infections in patients with COPD: epidemiology and management. Am J Respir Med. (2003) 2:459–68. doi: 10.1007/BF03256673
50. Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PloS One. (2011) 6:e16384. doi: 10.1371/journal.pone.0016384
51. Hassett DJ, Borchers MT, Panos RJ. Chronic obstructive pulmonary disease (COPD): Evaluation from clinical, immunological and bacterial pathogenesis perspectives. J Microbiol. (2014) 52:211–26. doi: 10.1007/s12275-014-4068-2
52. Murphy TF. Pseudomonas aeruginosa in adults with chronic obstructive pulmonary disease. Curr Opin Pulm Med. (2009) 15:138–42. doi: 10.1097/MCP.0b013e328321861a
53. Döring G, Parameswaran IG, Murphy TF. Differential adaptation of microbial pathogens to airways of patients with cystic fibrosis and chronic obstructive pulmonary disease. FEMS Microbiol Rev. (2011) 35:124–46. doi: 10.1111/j.1574-6976.2010.00237.x
54. Maciá MD, Blanquer D, Togores B, Sauleda J, Pérez JL, Oliver A. Hypermutation is a key factor in development of multiple-antimicrobial resistance in Pseudomonas aeruginosa strains causing chronic lung infections. Antimicrob Agents Chemother. (2005) 49:3382–6. doi: 10.1128/AAC.49.8.3382-3386.2005
55. Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Høiby N, et al. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol. (2012) 10:841–51. doi: 10.1038/nrmicro2907
56. Lee TWR, Brownlee KG, Conway SP, Denton M, Littlewood JM. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J Cyst Fibros. (2003) 2:29–34. doi: 10.1016/S1569-1993(02)00141-8
57. Blanchard AC, Waters VJ. Opportunistic pathogens in cystic fibrosis: epidemiology and pathogenesis of lung infection. J Pediatr Infect Dis Soc. (2022) 11:S3–S12. doi: 10.1093/jpids/piac052
58. Parkins MD, Somayaji R, Waters VJ. Epidemiology, biology, and impact of clonal Pseudomonas aeruginosa infections in cystic fibrosis. Clin Microbiol Rev. (2018) 31:e00019–18. doi: 10.1128/CMR.00019-18
59. Blanchard AC, Horton E, Stanojevic S, Taylor L, Waters V, Ratjen F. Effectiveness of a stepwise Pseudomonas aeruginosa eradication protocol in children with cystic fibrosis. J Cyst Fibros. (2017) 16:395–400. doi: 10.1016/j.jcf.2017.01.007
60. Rossi E, La Rosa R, Bartell JA, Marvig RL, Haagensen JAJ, Sommer LM, et al. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat Rev Microbiol. (2021) 19:331–42. doi: 10.1038/s41579-020-00477-5
61. Oliver A, Mulet X, López-Causapé C, Juan C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist Update. (2015) 21–22:41–59. doi: 10.1016/j.drup.2015.08.002
62. Moradali MF, Ghods S, Rehm BHA. Pseudomonas aeruginosa lifestyle: a paradigm for adaptation, survival, and persistence. Front Cell Infect Microbiol. (2017) 7:39. doi: 10.3389/fcimb.2017.00039
63. Bartell JA, Sommer LM, Haagensen JAJ, Loch A, Espinosa R, Molin S, et al. Evolutionary highways to persistent bacterial infection. Nat Commun. (2019) 10:629. doi: 10.1038/s41467-019-08504-7
64. Bianconi I, Jeukens J, Freschi L, Alcalá-Franco B, Facchini M, Boyle B, et al. Comparative genomics and biological characterization of sequential Pseudomonas aeruginosa isolates from persistent airways infection. BMC Genomics. (2015) 16:1105. doi: 10.1186/s12864-015-2276-8
65. Cullen L, Weiser R, Olszak T, Maldonado RF, Moreira AS, Slachmuylders L, et al. Phenotypic characterization of an international Pseudomonas aeruginosa reference panel: strains of cystic fibrosis (CF) origin show less in vivo virulence than non-CF strains. Microbiology. (2015) 161:1961–77. doi: 10.1099/mic.0.000155
66. Stanton BA. Effects of Pseudomonas aeruginosa on CFTR chloride secretion and the host immune response. Am J Physiol-Cell Physiol. (2017) 312:C357–66. doi: 10.1152/ajpcell.00373.2016
67. Mulcahy LR, Burns JL, Lory S, Lewis K. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J Bacteriol. (2010) 192:6191–9. doi: 10.1128/JB.01651-09
68. Bianconi I, D’Arcangelo S, Esposito A, Benedet M, Piffer E, Dinnella G, et al. Persistence and microevolution of Pseudomonas aeruginosa in the cystic fibrosis lung: a single-patient longitudinal genomic study. Front Microbiol. (2019) 9:3242. doi: 10.3389/fmicb.2018.03242
69. Camus L, Vandenesch F, Moreau K. From genotype to phenotype: adaptations of Pseudomonas aeruginosa to the cystic fibrosis environment. Microb Genomics. (2021) 7:1–19. doi: 10.1099/mgen.0.000513
70. Thornton CS, Acosta N, Surette MG, Parkins MD. Exploring the cystic fibrosis lung microbiome: making the most of a sticky situation. J Pediatr Infect Dis Soc. (2022) 11:S13–22. doi: 10.1093/jpids/piac036
71. Cramer N, Klockgether J, Tümmler B. Microevolution of Pseudomonas aeruginosa in the airways of people with cystic fibrosis. Curr Opin Immunol. (2023) 83:102328. doi: 10.1016/j.coi.2023.102328
72. Williams BJ, Dehnbostel J, Blackwell TS. Pseudomonas aeruginosa : Host defence in lung diseases. Respirology. (2010) 15:1037–56. doi: 10.1111/j.1440-1843.2010.01819.x
73. Semaniakou A, Croll RP, Chappe V. Animal models in the pathophysiology of cystic fibrosis. Front Pharmacol. (2019) 9:1475. doi: 10.3389/fphar.2018.01475
74. Raoust E, Balloy V, Garcia-Verdugo I, Touqui L, Ramphal R, Chignard M. Pseudomonas aeruginosa LPS or flagellin are sufficient to activate TLR-dependent signaling in murine alveolar macrophages and airway epithelial cells. PloS One. (2009) 4:e7259. doi: 10.1371/journal.pone.0007259
75. Feuillet V, Medjane S, Mondor I, Demaria O, Pagni PP, Galán JE, et al. Involvement of Toll-like receptor 5 in the recognition of flagellated bacteria. Proc Natl Acad Sci U.S.A. (2006) 103:12487–92. doi: 10.1073/pnas.0605200103
76. McIsaac SM, Stadnyk AW, Lin T-J. Toll-like receptors in the host defense against Pseudomonas aeruginosa respiratory infection and cystic fibrosis. J Leukoc Biol. (2012) 92:977–85. doi: 10.1189/jlb.0811410
77. Ramphal R, Balloy V, Jyot J, Verma A, Si-Tahar M, Chignard M. Control of Pseudomonas aeruginosa in the lung requires the recognition of either lipopolysaccharide or flagellin. J Immunol Baltim Md 1950. (2008) 181:586–92. doi: 10.4049/jimmunol.181.1.586
78. Ramphal R, Balloy V, Huerre M, Si-Tahar M, Chignard M. TLRs 2 and 4 are not involved in hypersusceptibility to acute Pseudomonas aeruginosa lung infections. J Immunol Baltim Md 1950. (2005) 175:3927–34. doi: 10.4049/jimmunol.175.6.3927
79. Anas AA, Van Lieshout MHP, Claushuis TAM, De Vos AF, Florquin S, De Boer OJ, et al. Lung epithelial MyD88 drives early pulmonary clearance of Pseudomonas aeruginosa by a flagellin dependent mechanism. Am J Physiol-Lung Cell Mol Physiol. (2016) 311:L219–28. doi: 10.1152/ajplung.00078.2016
80. Mackowiak C, Marchiol T, Paljetak HC, Fauconnier L, Palomo J, Secher T, et al. Chronic Pseudomonas aeruginosa lung infection is IL-1R independent, but relies on MyD88 Signaling. ImmunoHorizons. (2021) 5:273–83. doi: 10.4049/immunohorizons.2000095
81. Power MR, Peng Y, Maydanski E, Marshall JS, Lin T-J. The development of early host response to Pseudomonas aeruginosa lung infection is critically dependent on myeloid differentiation factor 88 in mice. J Biol Chem. (2004) 279:49315–22. doi: 10.1074/jbc.M402111200
82. Björkbacka H, Fitzgerald KA, Huet F, Li X, Gregory JA, Lee MA, et al. The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiol Genomics. (2004) 19:319–30. doi: 10.1152/physiolgenomics.00128.2004
83. Power MR, Li B, Yamamoto M, Akira S, Lin T-J. A Role of Toll-IL-1 receptor domain-containing adaptor-inducing IFN-β in the host response to Pseudomonas aeruginosa lung infection in mice. J Immunol. (2007) 178:3170–6. doi: 10.4049/jimmunol.178.5.3170
84. Carrigan SO, Junkins R, Yang YJ, MacNeil A, Richardson C, Johnston B, et al. IFN regulatory factor 3 contributes to the host response during Pseudomonas aeruginosa lung infection in mice. J Immunol. (2010) 185:3602–9. doi: 10.4049/jimmunol.0903429
85. Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. (2015) 16:27–35. doi: 10.1038/ni.3045
86. Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
87. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. (2007) 204:3235–45. doi: 10.1084/jem.20071239
88. Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat Commun. (2015) 6:6201. doi: 10.1038/ncomms7201
89. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci. (2010) 107:3076–80. doi: 10.1073/pnas.0913087107
90. De Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol. (2014) 6:a016287. doi: 10.1101/cshperspect.a016287
91. Lu F, Lan Z, Xin Z, He C, Guo Z, Xia X, et al. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J Cell Physiol. (2020) 235:3207–21. doi: 10.1002/jcp.29268
92. Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest. (2013) 123:1630–7. doi: 10.1172/JCI66142
93. Tang A, Sharma A, Jen R, Hirschfeld AF, Chilvers MA, Lavoie PM, et al. Inflammasome-mediated IL-1β production in humans with cystic fibrosis. PloS One. (2012) 7:e37689. doi: 10.1371/journal.pone.0037689
94. Mijares LA, Wangdi T, Sokol C, Homer R, Medzhitov R, Kazmierczak BI. Airway epithelial MyD88 restores control of Pseudomonas aeruginosa murine infection via an IL-1–dependent pathway. J Immunol. (2011) 186:7080–8. doi: 10.4049/jimmunol.1003687
95. Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat Commun. (2016) 7:10791. doi: 10.1038/ncomms10791
96. McElvaney OJ, Zaslona Z, Becker-Flegler K, Palsson-McDermott EM, Boland F, Gunaratnam C, et al. Specific inhibition of the NLRP3 inflammasome as an antiinflammatory strategy in cystic fibrosis. Am J Respir Crit Care Med. (2019) 200:1381–91. doi: 10.1164/rccm.201905-1013OC
97. McElvaney OJ, Gunaratnam C, Reeves EP, McElvaney NG. A specialized method of sputum collection and processing for therapeutic interventions in cystic fibrosis. J Cyst Fibros. (2019) 18:203–11. doi: 10.1016/j.jcf.2018.06.001
98. Petrocheilou A, Moudaki A, Kaditis AG. Inflammation and infection in cystic fibrosis: update for the clinician. Children. (2022) 9:1898. doi: 10.3390/children9121898
99. Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, et al. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell–iTreg cell balance. Nat Immunol. (2015) 16:286–95. doi: 10.1038/ni.3099
100. Tan Q, Ai Q, He Y, Li F, Yu J. P. aeruginosa biofilm activates the NLRP3 inflammasomes in vitro. Microb Pathog. (2022) 164:105379. doi: 10.1016/j.micpath.2021.105379
101. Huus KE, Joseph J, Zhang L, Wong A, Aaron SD, Mah T-F, et al. Clinical isolates of Pseudomonas aeruginosa from chronically infected cystic fibrosis patients fail to activate the inflammasome during both stable infection and pulmonary exacerbation. J Immunol. (2016) 196:3097–108. doi: 10.4049/jimmunol.1501642
102. Phuong MS, Hernandez RE, Wolter DJ, Hoffman LR, Sad S. Impairment in inflammasome signaling by the chronic Pseudomonas aeruginosa isolates from cystic fibrosis patients results in an increase in inflammatory response. Cell Death Dis. (2021) 12:241. doi: 10.1038/s41419-021-03526-w
103. John G, Yildirim AÖ, Rubin BK, Gruenert DC, Henke MO. TLR-4–mediated innate immunity is reduced in cystic fibrosis airway cells. Am J Respir Cell Mol Biol. (2010) 42:424–31. doi: 10.1165/rcmb.2008-0408OC
104. John G, Chillappagari S, Rubin BK, Gruenert DC, Henke MO. Reduced surface toll-like receptor-4 expression and absent interferon-γ–inducible protein-10 induction in cystic fibrosis airway cells. Exp Lung Res. (2011) 37:319–26. doi: 10.3109/01902148.2011.569968
105. MacRedmond RE, Greene CM, Dorscheid DR, McElvaney NG, O’Neill SJ. Epithelial expression of TLR4 is modulated in COPD and by steroids, salmeterol and cigarette smoke. Respir Res. (2007) 8:84. doi: 10.1186/1465-9921-8-84
106. Rieber N, Brand A, Hector A, Graepler-Mainka U, Ost M, Schäfer I, et al. Flagellin induces myeloid-derived suppressor cells: implications for Pseudomonas aeruginosa infection in cystic fibrosis lung disease. J Immunol. (2013) 190:1276–84. doi: 10.4049/jimmunol.1202144
107. Cigana C, Curcurù L, Leone MR, Ieranò T, Lorè NI, Bianconi I, et al. Pseudomonas aeruginosa exploits Lipid A and muropeptides modification as a strategy to lower innate immunity during cystic fibrosis lung infection. PloS One. (2009) 4:e8439. doi: 10.1371/journal.pone.0008439
108. Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest. (2002) 109:317–25. doi: 10.1172/JCI13870
109. Vanaki S M, Holmes D, Saha SC, Chen J, Brown RJ, Jayathilake PG. Muco-ciliary clearance: A review of modelling techniques. J Biomech. (2020) 99:109578. doi: 10.1016/j.jbiomech.2019.109578
110. Benam KH, Vladar EK, Janssen WJ, Evans CM. Mucociliary defense: emerging cellular, molecular, and animal models. Ann Am Thorac Soc. (2018) 15:S210–5. doi: 10.1513/AnnalsATS.201806-439AW
111. Antunes MB, Cohen NA. Mucociliary clearance – a critical upper airway host defense mechanism and methods of assessment. Curr Opin Allergy Clin Immunol. (2007) 7:5–10. doi: 10.1097/ACI.0b013e3280114eef
112. Rouillard KR, Kissner WJ, Markovetz MR, Hill DB. Effects of mucin and DNA concentrations in airway mucus on Pseudomonas aeruginosa biofilm recalcitrance. mSphere. (2022) 7:e00291–22. doi: 10.1128/msphere.00291-22
113. Müller L, Murgia X, Siebenbürger L, Börger C, Schwarzkopf K, Sewald K, et al. Human airway mucus alters susceptibility of Pseudomonas aeruginosa biofilms to tobramycin, but not colistin. J Antimicrob Chemother. (2018) 73:2762–9. doi: 10.1093/jac/dky241
114. Rouillard KR, Markovetz MR, Kissner WJ, Boone WL, Plott LM, Hill DB. Altering the viscoelastic properties of mucus-grown Pseudomonas aeruginosa biofilms affects antibiotic susceptibility. Biofilm. (2023) 5:100104. doi: 10.1016/j.bioflm.2023.100104
115. Rouillard KR, Esther CP, Kissner WJ, Plott LM, Bowman DW, Markovetz MR, et al. Combination treatment to improve mucociliary transport of Pseudomonas aeruginosa biofilms. PloS One. (2024) 19:e0294120. doi: 10.1371/journal.pone.0294120
116. Donaldson SH, Corcoran TE, Laube BL, Bennett WD. Mucociliary clearance as an outcome measure for cystic fibrosis clinical research. Proc Am Thorac Soc. (2007) 4:399–405. doi: 10.1513/pats.200703-042BR
117. Donaldson SH, Laube BL, Corcoran TE, Bhambhvani P, Zeman K, Ceppe A, et al. Effect of ivacaftor on mucociliary clearance and clinical outcomes in cystic fibrosis patients with G551D-CFTR. JCI Insight. (2018) 3:e122695. doi: 10.1172/jci.insight.122695
118. Warheit-Niemi HI, Hult EM, Moore BB. A pathologic two-way street: how innate immunity impacts lung fibrosis and fibrosis impacts lung immunity. Clin Transl Immunol. (2019) 8:e1065. doi: 10.1002/cti2.1065
119. Smith RS, Kelly R, Iglewski BH, Phipps RP. The Pseudomonas autoinducer N -(3-oxododecanoyl) homoserine lactone induces cyclooxygenase-2 and prostaglandin E2 production in human lung fibroblasts: implications for inflammation. J Immunol. (2002) 169:2636–42. doi: 10.4049/jimmunol.169.5.2636
120. Perfetto B, Donnarumma G, Criscuolo D, Paoletti I, Grimaldi E, Tufano MA, et al. Bacterial components induce cytokine and intercellular adhesion molecules-1 and activate transcription factors in dermal fibroblasts. Res Microbiol. (2003) 154:337–44. doi: 10.1016/S0923-2508(03)00084-6
121. Bruscia EM, Bonfield TL. Cystic fibrosis lung immunity: the role of the macrophage. J Innate Immun. (2016) 8:550–63. doi: 10.1159/000446825
122. Bissonnette EY, Lauzon-Joset J-F, Debley JS, Ziegler SF. Cross-talk between alveolar macrophages and lung epithelial cells is essential to maintain lung homeostasis. Front Immunol. (2020) 11:583042. doi: 10.3389/fimmu.2020.583042
123. Neupane AS, Willson M, Chojnacki AK, Castanheira FVES, Morehouse C, Carestia A, et al. Patrolling alveolar macrophages conceal bacteria from the immune system to maintain homeostasis. Cell. (2020) 183:110–125.e11. doi: 10.1016/j.cell.2020.08.020
124. Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med. (2010) 61:105–19. doi: 10.1146/annurev.med.60.041807.123500
125. Green FHY, Mustafa Al-Saiedy M, Gunasekara L, Bjornson C, Yang A, Chiu A, et al. Surfactant dysfunction in cystic fibrosis: Mechanisms and reversal with a cyclodextrin drug. 7.3 Cystic Fibrosis. Eur Respir Soc. (2016) 48:OA1494. doi: 10.1183/13993003.congress-2016.OA1494
126. Hashimoto S, Pittet JF, Hong K, Folkesson H, Bagby G, Kobzik L, et al. Depletion of alveolar macrophages decreases neutrophil chemotaxis to Pseudomonas airspace infections. Am J Physiol. (1996) 270:L819–828. doi: 10.1152/ajplung.1996.270.5.L819
127. Cheung DO, Halsey K, Speert DP. Role of pulmonary alveolar macrophages in defense of the lung against Pseudomonas aeruginosa. Infect Immun. (2000) 68:4585–92. doi: 10.1128/IAI.68.8.4585-4592.2000
128. Sainz-Mejías M, Jurado-Martín I, McClean S. Understanding Pseudomonas aeruginosa–host interactions: the ongoing quest for an efficacious vaccine. Cells. (2020) 9:2617. doi: 10.3390/cells9122617
129. Gu Y, Lawrence T, Mohamed R, Liang Y, Yahaya BH. The emerging roles of interstitial macrophages in pulmonary fibrosis: A perspective from scRNA-seq analyses. Front Immunol. (2022) 13:923235. doi: 10.3389/fimmu.2022.923235
130. Öz HH, Cheng E-C, Di Pietro C, Tebaldi T, Biancon G, Zeiss C, et al. Recruited monocytes/macrophages drive pulmonary neutrophilic inflammation and irreversible lung tissue remodeling in cystic fibrosis. Cell Rep. (2022) 41:111797. doi: 10.1016/j.celrep.2022.111797
131. Kooguchi K, Hashimoto S, Kobayashi A, Kitamura Y, Kudoh I, Wiener-Kronish J, et al. Role of alveolar macrophages in initiation and regulation of inflammation in Pseudomonas aeruginosa pneumonia. Infect Immun. (1998) 66:3164–9. doi: 10.1128/IAI.66.7.3164-3169.1998
132. Brennan S, Sly PD, Gangell CL, Sturges N, Winfield K, Wikstrom M, et al. Alveolar macrophages and CC chemokines are increased in children with cystic fibrosis. Eur Respir J. (2009) 34:655–61. doi: 10.1183/09031936.00178508
133. Sagel SD, Chmiel JF, Konstan MW. Sputum biomarkers of inflammation in cystic fibrosis lung disease. Proc Am Thorac Soc. (2007) 4:406–17. doi: 10.1513/pats.200703-044BR
134. McClellan SA, Huang X, Barrett RP, Van Rooijen N, Hazlett LD. Macrophages restrict Pseudomonas aeruginosa growth, regulate polymorphonuclear neutrophil influx, and balance pro- and anti-inflammatory cytokines in BALB/c mice. J Immunol. (2003) 170:5219–27. doi: 10.4049/jimmunol.170.10.5219
135. Kannan S, Huang H, Seeger D, Audet A, Chen Y, Huang C, et al. Alveolar epithelial type II cells activate alveolar macrophages and mitigate P. aeruginosa infection. PloS One. (2009) 4:e4891. doi: 10.1371/journal.pone.0004891
136. Singh S, Barr H, Liu Y-C, Robins A, Heeb S, Williams P, et al. Granulocyte-macrophage colony stimulatory factor enhances the pro-inflammatory response of interferon-γ-treated macrophages to Pseudomonas aeruginosa infection. PloS One. (2015) 10:e0117447. doi: 10.1371/journal.pone.0117447
137. Meyer M, Huaux F, Gavilanes X, Van Den Brûle S, Lebecque P, Lo Re S, et al. Azithromycin reduces exaggerated cytokine production by M1 alveolar macrophages in cystic fibrosis. Am J Respir Cell Mol Biol. (2009) 41:590–602. doi: 10.1165/rcmb.2008-0155OC
138. Andrews T, Sullivan KE. Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev. (2003) 16:597–621. doi: 10.1128/CMR.16.4.597-621.2003
139. Kurahashi K, Sawa T, Ota M, Kajikawa O, Hong K, Martin TR, et al. Depletion of phagocytes in the reticuloendothelial system causes increased inflammation and mortality in rabbits with Pseudomonas aeruginosa pneumonia. Am J Physiol-Lung Cell Mol Physiol. (2009) 296:L198–209. doi: 10.1152/ajplung.90472.2008
140. Cifani N, Pompili B, Anile M, Patella M, Diso D, Venuta F, et al. Reactive-oxygen-species-mediated P. aeruginosa killing is functional in human cystic fibrosis macrophages. PloS One. (2013) 8:e71717. doi: 10.1371/journal.pone.0071717
141. Flannagan RS, Jaumouillé V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol Mech Dis. (2012) 7:61–98. doi: 10.1146/annurev-pathol-011811-132445
142. Glasser SW, Senft AP, Whitsett JA, Maxfield MD, Ross GF, Richardson TR, et al. Macrophage dysfunction and susceptibility to pulmonary Pseudomonas aeruginosa infection in surfactant protein C-deficient mice. J Immunol. (2008) 181:621–8. doi: 10.4049/jimmunol.181.1.621
143. Mariencheck WI, Savov J, Dong Q, Tino MJ, Wright JR. Surfactant protein A enhances alveolar macrophage phagocytosis of a live, mucoid strain of P. aeruginosa. Am J Physiol-Lung Cell Mol Physiol. (1999) 277:L777–86. doi: 10.1152/ajplung.1999.277.4.L777
144. Heale J, Pollard AJ, Stokes RW, Simpson D, Tsang A, Massing B, et al. Two distinct receptors mediate nonopsonic phagocytosis of different strains of Pseudomonas aeruginosa. J Infect Dis. (2001) 183:1214–20. doi: 10.1086/319685
145. Amiel E, Lovewell RR, O’Toole GA, Hogan DA, Berwin B. Pseudomonas aeruginosa evasion of phagocytosis is mediated by loss of swimming motility and is independent of flagellum expression. Infect Immun. (2010) 78:2937–45. doi: 10.1128/IAI.00144-10
146. Pollard AJ, Currie A, Rosenberger CM, Heale J-P, Finlay BB, Speert DP. Differential post-transcriptional activation of human phagocytes by different Pseudomonas aeruginosa isolates: Phagocytosis of Pseudomonas by human macrophages. Cell Microbiol. (2004) 6:639–50. doi: 10.1111/j.1462-5822.2004.00388.x
147. Lovewell RR, Collins RM, Acker JL, O’Toole GA, Wargo MJ, Berwin B. Step-wise loss of bacterial flagellar torsion confers progressive phagocytic evasion. PloS Pathog. (2011) 7:e1002253. doi: 10.1371/journal.ppat.1002253
148. Mahenthiralingam E, Speert DP. Nonopsonic phagocytosis of Pseudomonas aeruginosa by macrophages and polymorphonuclear leukocytes requires the presence of the bacterial flagellum. Infect Immun. (1995) 63:4519–23. doi: 10.1128/iai.63.11.4519-4523.1995
149. Lillehoj EP, Hyun SW, Kim BT, Zhang XG, Lee DI, Rowland S, et al. Muc1 mucins on the cell surface are adhesion sites for Pseudomonas aeruginosa. Am J Physiol-Lung Cell Mol Physiol. (2001) 280:L181–7. doi: 10.1152/ajplung.2001.280.1.L181
150. Lillehoj EP, Kim BT, Kim KC. Identification of Pseudomonas aeruginosa flagellin as an adhesin for Muc1 mucin. Am J Physiol-Lung Cell Mol Physiol. (2002) 282:L751–6. doi: 10.1152/ajplung.00383.2001
151. Luzar MA, Thomassen MJ, Montie TC. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: relationship to patient clinical condition. Infect Immun. (1985) 50:577–82. doi: 10.1128/iai.50.2.577-582.1985
152. Cabral DA, Loh BA, Speert DP. Mucoid Pseudomonas aeruginosa resists nonopsonic phagocytosis by human neutrophils and macrophages. Pediatr Res. (1987) 22:429–31. doi: 10.1203/00006450-198710000-00013
153. Krieg DP, Helmke RJ, German VF, Mangos JA. Resistance of mucoid Pseudomonas aeruginosa to nonopsonic phagocytosis by alveolar macrophages in vitro. Infect Immun. (1988) 56:3173–9. doi: 10.1128/iai.56.12.3173-3179.1988
154. Leid JG, Willson CJ, Shirtliff ME, Hassett DJ, Parsek MR, Jeffers AK. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-γ-mediated macrophage killing. J Immunol. (2005) 175:7512–8. doi: 10.4049/jimmunol.175.11.7512
155. Mishra M, Byrd MS, Sergeant S, Azad AK, Parsek MR, McPhail L, et al. Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization: Psl and the innate immune response towards P. aeruginosa Cell Microbiol. (2012) 14:95–106. doi: 10.1111/j.1462-5822.2011.01704.x
156. Rowe WJ, Lebman DA, Ohman DE. Mechanism of resistance to phagocytosis and pulmonary persistence in mucoid Pseudomonas aeruginosa. Front Cell Infect Microbiol. (2023) 13:1125901. doi: 10.3389/fcimb.2023.1125901
157. Hastings CJ, Himmler GE, Patel A, Marques CNH. Immune response modulation by Pseudomonas aeruginosa persister cells. mBio. (2023) 14:e00056–23. doi: 10.1128/mbio.00056-23
158. Painter RG, Valentine VG, Lanson NA, Leidal K, Zhang Q, Lombard G, et al. CFTR expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry. (2006) 45:10260–9. doi: 10.1021/bi060490t
159. Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol. (2006) 8:933–44. doi: 10.1038/ncb1456
160. Xu Y, Krause A, Hamai H, Harvey B-G, Worgall TS, Worgall S. Proinflammatory phenotype and increased caveolin-1 in alveolar macrophages with silenced CFTR mRNA. PloS One. (2010) 5:e11004. doi: 10.1371/journal.pone.0011004
161. Tarique AA, Sly PD, Holt PG, Bosco A, Ware RS, Logan J, et al. CFTR-dependent defect in alternatively-activated macrophages in cystic fibrosis. J Cyst Fibros. (2017) 16:475–82. doi: 10.1016/j.jcf.2017.03.011
162. Law SM, Stanfield SJ, Hardisty GR, Dransfield I, Campbell CJ, Gray RD. Human cystic fibrosis monocyte derived macrophages display no defect in acidification of phagolysosomes when measured by optical nanosensors. J Cyst Fibros. (2020) 19:203–10. doi: 10.1016/j.jcf.2019.09.003
163. Leuer L, Krill A, Wilkens H, Wagenpfeil G, Bischoff M, Meier C, et al. The phagocytosis of blood leukocytes from cystic fibrosis patients is not impaired in general. Lung. (2020) 198:235–9. doi: 10.1007/s00408-019-00290-9
164. Barnaby R, Koeppen K, Nymon A, Hampton TH, Berwin B, Ashare A, et al. Lumacaftor (VX-809) restores the ability of CF macrophages to phagocytose and kill Pseudomonas aeruginosa. Am J Physiol-Lung Cell Mol Physiol. (2018) 314:L432–8. doi: 10.1152/ajplung.00461.2017
165. Zhang S, Shrestha CL, Kopp BT. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci Rep. (2018) 8:17066. doi: 10.1038/s41598-018-35151-7
166. Zhang S, Shrestha CL, Robledo-Avila F, Jaganathan D, Wisniewski BL, Brown N, et al. Cystic fibrosis macrophage function and clinical outcomes after elexacaftor/tezacaftor/ivacaftor. Eur Respir J. (2023) 61:2102861. doi: 10.1183/13993003.02861-2021
167. Aridgides DS, Mellinger DL, Gwilt LL, Hampton TH, Mould DL, Hogan DA, et al. Comparative effects of CFTR modulators on phagocytic, metabolic and inflammatory profiles of CF and nonCF macrophages. Sci Rep. (2023) 13:11995. doi: 10.1038/s41598-023-38300-9
168. Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect Immun. (2009) 77:5300–10. doi: 10.1128/IAI.00501-09
169. Hirche TO, Benabid R, Deslee G, Gangloff S, AChilefu S, Guenounou M, et al. Neutrophil elastase mediates innate host protection against. Pseudomonas aeruginosa J Immunol. (2008) 181:4945–54. doi: 10.4049/jimmunol.181.7.4945
170. Khan MA, Ali ZS, Sweezey N, Grasemann H, Palaniyar N. Progression of cystic fibrosis lung disease from childhood to adulthood: neutrophils, neutrophil extracellular trap (NET) formation, and NET degradation. Genes. (2019) 10:183. doi: 10.3390/genes10030183
171. Meijer M, Rijkers GT, Van Overveld FJ. Neutrophils and emerging targets for treatment in chronic obstructive pulmonary disease. Expert Rev Clin Immunol. (2013) 9:1055–68. doi: 10.1586/1744666X.2013.851347
172. Ponce-Gallegos M, Ramírez-Venegas A, Falfán-Valencia R. Th17 profile in COPD exacerbations. Int J Chron Obstruct Pulmon Dis. (2017) 12:1857–65. doi: 10.2147/COPD.S136592
173. Morohashi H, Miyawaki T, Nomura H, Kuno K, Murakami S, Matsushima K, et al. Expression of both types of human interleukin-8 receptors on mature neutrophils, monocytes, and natural killer cells. J Leukoc Biol. (1995) 57:180–7. doi: 10.1002/jlb.57.1.180
174. Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ. CXC Chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun. (2000) 68:4289–96. doi: 10.1128/IAI.68.7.4289-4296.2000
175. Zarbock A, Deem TL, Burcin TL, Ley K. Gαi2 is required for chemokine-induced neutrophil arrest. Blood. (2007) 110:3773–9. doi: 10.1182/blood-2007-06-094565
176. Mizgerd JP, Horwitz BH, Quillen HC, Scott ML, Doerschuk CM. Effects of CD18 deficiency on the emigration of murine neutrophils during pneumonia. J Immunol Baltim Md 1950. (1999) 163:995–9.
177. Mizunoe S, Shuto T, Suzuki S, Matsumoto C, Watanabe K, Ueno-Shuto K, et al. Synergism between interleukin (IL)-17 and toll-like receptor 2 and 4 signals to induce IL-8 expression in cystic fibrosis airway epithelial cells. J Pharmacol Sci. (2012) 118:512–20. doi: 10.1254/jphs.11240FP
178. Brodlie M, McKean MC, Johnson GE, Anderson AE, Hilkens CMU, Fisher AJ, et al. Raised interleukin-17 is immunolocalised to neutrophils in cystic fibrosis lung disease. Eur Respir J. (2011) 37:1378–85. doi: 10.1183/09031936.00067110
179. Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The balancing act of neutrophils. Cell Host Microbe. (2014) 15:526–36. doi: 10.1016/j.chom.2014.04.011
180. Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol. (1994) 56:559–64. doi: 10.1002/jlb.56.5.559
181. Gierlikowska B, Stachura A, Gierlikowski W, Demkow U. Phagocytosis, degranulation and extracellular traps release by neutrophils—the current knowledge, pharmacological modulation and future prospects. Front Pharmacol. (2021) 12:666732. doi: 10.3389/fphar.2021.666732
182. Leid JG, Kerr M, Selgado C, Johnson C, Moreno G, Smith A, et al. Flagellum-mediated biofilm defense mechanisms of Pseudomonas aeruginosa against host-derived lactoferrin. Infect Immun. (2009) 77:4559–66. doi: 10.1128/IAI.00075-09
183. Cole AM, Thapa DR, Gabayan V, Liao H-I, Liu L, Ganz T. Decreased clearance of Pseudomonas aeruginosa from airways of mice deficient in lysozyme M. J Leukoc Biol. (2005) 78:1081–5. doi: 10.1189/jlb.0205073
184. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
185. Yoo D, Floyd M, Winn M, Moskowitz SM, Rada B. NET formation induced by Pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase–DNA and neutrophil elastase–DNA complexes. Immunol Lett. (2014) 160:186–94. doi: 10.1016/j.imlet.2014.03.003
186. Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol. (2014) 5:508. doi: 10.3389/fimmu.2014.00508
187. Taylor PR, Bonfield TL, Chmiel JF, Pearlman E. Neutrophils from F508del cystic fibrosis patients produce IL-17A and express IL-23 - dependent IL-17RC. Clin Immunol. (2016) 170:53–60. doi: 10.1016/j.clim.2016.03.016
188. Kasama T, Miwa Y, Isozaki T, Odai T, Adachi M, Kunkel S. Neutrophil-derived cytokines: potential therapeutic targets in inflammation. Curr Drug Target -Inflamm Allergy. (2005) 4:273–9. doi: 10.2174/1568010054022114
189. Yang F, Feng C, Zhang X, Lu J, Zhao Y. The diverse biological functions of neutrophils, beyond the defense against infections. Inflammation. (2017) 40:311–23. doi: 10.1007/s10753-016-0458-4
190. Berger M, Sorensen RU, Tosi MF, Dearborn DG, Döring G. Complement receptor expression on neutrophils at an inflammatory site, the Pseudomonas-infected lung in cystic fibrosis. J Clin Invest. (1989) 84:1302–13. doi: 10.1172/JCI114298
191. Tosi MF, Zakem H, Berger M. Neutrophil elastase cleaves C3bi on opsonized pseudomonas as well as CR1 on neutrophils to create a functionally important opsonin receptor mismatch. J Clin Invest. (1990) 86:300–8. doi: 10.1172/JCI114699
192. Marteyn BS, Burgel P-R, Meijer L, Witko-Sarsat V. Harnessing neutrophil survival mechanisms during chronic infection by Pseudomonas aeruginosa: novel therapeutic targets to dampen inflammation in cystic fibrosis. Front Cell Infect Microbiol. (2017) 7:243. doi: 10.3389/fcimb.2017.00243
193. Bjarnsholt T, Jensen PØ, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, et al. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr Pulmonol. (2009) 44:547–58. doi: 10.1002/ppul.21011
194. Walker TS, Tomlin KL, Worthen GS, Poch KR, Lieber JG, Saavedra MT, et al. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect Immun. (2005) 73:3693–701. doi: 10.1128/IAI.73.6.3693-3701.2005
195. Marcos V, Zhou-Suckow Z, Önder Yildirim A, Bohla A, Hector A, Vitkov L, et al. Free DNA in cystic fibrosis airway fluids correlates with airflow obstruction. Mediators Inflammation. (2015) 2015:1–11. doi: 10.1155/2015/408935
196. Rada B. Interactions between neutrophils and Pseudomonas aeruginosa in cystic fibrosis. Pathogens. (2017) 6:10. doi: 10.3390/pathogens6010010
197. Keir HR, Shoemark A, Dicker AJ, Perea L, Pollock J, Giam YH, et al. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: an international, observational, multicohort study. Lancet Respir Med. (2021) 9:873–84. doi: 10.1016/S2213-2600(20)30504-X
198. Jo A, Kim DW. Neutrophil extracellular traps in airway diseases: pathological roles and therapeutic implications. Int J Mol Sci. (2023) 24:5034. doi: 10.3390/ijms24055034
199. Skopelja-Gardner S, Theprungsirikul J, Lewis KA, Hammond JH, Carlson KM, Hazlett HF, et al. Regulation of Pseudomonas aeruginosa-mediated neutrophil extracellular traps. Front Immunol. (2019) 10:1670. doi: 10.3389/fimmu.2019.01670
200. Cheng OZ, Palaniyar N. NET balancing: a problem in inflammatory lung diseases. Front Immunol. (2013) 4:1. doi: 10.3389/fimmu.2013.00001
201. Martínez-Alemán SR, Campos-García L, Palma-Nicolas JP, Hernández-Bello R, González GM, Sánchez-González A. Understanding the Entanglement: neutrophil extracellular traps (NETs) in cystic fibrosis. Front Cell Infect Microbiol. (2017) 7:104. doi: 10.3389/fcimb.2017.00104
202. Dibbert B, Weber M, Nikolaizik WH, Vogt P, Schöni MH, Blaser K, et al. Cytokine-mediated Bax deficiency and consequent delayed neutrophil apoptosis: A general mechanism to accumulate effector cells in inflammation. Proc Natl Acad Sci. (1999) 96:13330–5. doi: 10.1073/pnas.96.23.13330
203. McKeon DJ, Condliffe AM, Cowburn AS, Cadwallader KC, Farahi N, Bilton D, et al. Prolonged survival of neutrophils from patients with F508 CFTR mutations. Thorax. (2008) 63:660–1. doi: 10.1136/thx.2008.096834
204. Moriceau S, Lenoir G, Witko-Sarsat V. In cystic fibrosis homozygotes and heterozygotes, neutrophil apoptosis is delayed and modulated by diamide or roscovitine: evidence for an innate neutrophil disturbance. J Innate Immun. (2010) 2:260–6. doi: 10.1159/000295791
205. Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res. (2009) 43:25–61. doi: 10.1007/s12026-008-8049-6
206. Noda N, Matsumoto K, Fukuyama S, Asai Y, Kitajima H, Seki N, et al. Cigarette smoke impairs phagocytosis of apoptotic neutrophils by alveolar macrophages via inhibition of the histone deacetylase/Rac/CD9 pathways. Int Immunol. (2013) 25:643–50. doi: 10.1093/intimm/dxt033
207. Moser C, Kjaergaard S, Pressler T, Kharazmi A, Koch C, Hoiby N. The immune response to chronic Pseudomonas aeruginosa lung infection in cystic fibrosis patients is predominantly of the Th2 typeNote. APMIS. (2000) 108:329–35. doi: 10.1034/j.1600-0463.2000.d01-64.x
208. Moser C, Jensen PO, Kobayashi O, Hougen HP, Song Z, Rygaard J, et al. Improved outcome of chronic Pseudomonas aeruginosa lung infection is associated with induction of a Th1-dominated cytokine response. Clin Exp Immunol. (2002) 127:206–13. doi: 10.1046/j.1365-2249.2002.01731.x
209. Tiringer K, Treis A, Fucik P, Gona M, Gruber S, Renner S, et al. A Th17- and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. (2013) 187:621–9. doi: 10.1164/rccm.201206-1150OC
210. Damlund DSM, Christophersen L, Jensen PØ, Alhede M, Høiby N, Moser C. Activation of pulmonary and lymph node dendritic cells during chronic Pseudomonas aeruginosa lung infection in mice. APMIS. (2016) 124:500–7. doi: 10.1111/apm.12530
211. Brassard J, Roy J, Lemay A-M, Beaulieu M-J, Bernatchez E, Veillette M, et al. Exposure to the gram-negative bacteria Pseudomonas aeruginosa influences the lung dendritic cell population signature by interfering with CD103 expression. Front Cell Infect Microbiol. (2021) 11:617481. doi: 10.3389/fcimb.2021.617481
212. Skindersoe ME, Zeuthen LH, Brix S, Fink LN, Lazenby J, Whittall C, et al. Pseudomonas aeruginosa quorum-sensing signal molecules interfere with dendritic cell-induced T-cell proliferation. FEMS Immunol Med Microbiol. (2009) 55:335–45. doi: 10.1111/j.1574-695X.2008.00533.x
213. Levine H, Cohen-Cymberknoh M, Klein N, Hoshen M, Mussaffi H, Stafler P, et al. Reversible airway obstruction in cystic fibrosis: Common, but not associated with characteristics of asthma. J Cyst Fibros. (2016) 15:652–9. doi: 10.1016/j.jcf.2016.01.003
214. Ye SC, Desai S, Karlsen E, Kwong E, Wilcox PG, Quon BS. Association between elevated peripheral blood eosinophil count and respiratory outcomes in adults with cystic fibrosis. J Cyst Fibros. (2022) 21:1048–52. doi: 10.1016/j.jcf.2022.03.009
215. Koller DY, Gotz M, Eichler I, Urbanek R. Eosinophilic activation in cystic fibrosis. Thorax. (1994) 49:496–9. doi: 10.1136/thx.49.5.496
216. Shoemark A, Shteinberg M, De Soyza A, Haworth CS, Richardson H, Gao Y, et al. Characterization of eosinophilic bronchiectasis: a European multicohort study. Am J Respir Crit Care Med. (2022) 205:894–902. doi: 10.1164/rccm.202108-1889OC
218. David B, Bafadhel M, Koenderman L, De Soyza A. Eosinophilic inflammation in COPD: from an inflammatory marker to a treatable trait. Thorax. (2021) 76:188–95. doi: 10.1136/thoraxjnl-2020-215167
219. Lin T-J, Garduno R, Boudreau RTM, Issekutz AC. Pseudomonas aeruginosa activates human mast cells to induce neutrophil transendothelial migration via mast cell-derived IL-1α and β. J Immunol. (2002) 169:4522–30. doi: 10.4049/jimmunol.169.8.4522
220. Lin T-J, Maher LH, Gomi K, McCurdy JD, Garduno R, Marshall JS. Selective Early production of CCL20, or macrophage inflammatory protein 3α, by human mast cells in response to. Pseudomonas aeruginosa Infect Immun. (2003) 71:365–73. doi: 10.1128/IAI.71.1.365-373.2003
221. Sun G, Liu F, Lin T-J. Identification of Pseudomonas aeruginosa -induced genes in human mast cells using suppression subtractive hybridization: up-regulation of IL-8 and CCL4 production. Clin Exp Immunol. (2005) 142:199–205. doi: 10.1111/j.1365-2249.2005.02909.x
222. Broquet A, Roquilly A, Jacqueline C, Potel G, Caillon J, Asehnoune K. Depletion of natural killer cells increases mice susceptibility in a Pseudomonas aeruginosa pneumonia model. Crit Care Med. (2014) 42:e441–50. doi: 10.1097/CCM.0000000000000311
223. Wesselkamper SC, Eppert BL, Motz GT, Lau GW, Hassett DJ, Borchers MT. NKG2D Is critical for NK cell activation in host defense against Pseudomonas aeruginosa respiratory infection. J Immunol. (2008) 181:5481–9. doi: 10.4049/jimmunol.181.8.5481
224. Feehan DD, Jamil K, Polyak MJ, Ogbomo H, Hasell M, Li SS, et al. Natural killer cells kill extracellular Pseudomonas aeruginosa using contact-dependent release of granzymes B and H. PloS Pathog. (2022) 18:e1010325. doi: 10.1371/journal.ppat.1010325
225. Artis D, Spits H. The biology of innate lymphoid cells. Nature. (2015) 517:293–301. doi: 10.1038/nature14189
226. Eberl G, Colonna M, Di Santo JP, McKenzie ANJ. Innate lymphoid cells: A new paradigm in immunology. Science. (2015) 348:aaa6566. doi: 10.1126/science.aaa6566
227. Hoffmann JP, Kolls JK, McCombs JE. Regulation and function of ILC3s in pulmonary infections. Front Immunol. (2021) 12:672523. doi: 10.3389/fimmu.2021.672523
228. Bayes HK, Ritchie ND, Evans TJ. Interleukin-17 is required for control of chronic lung infection caused by Pseudomonas aeruginosa. Infect Immun. (2016) 84:3507–16. doi: 10.1128/IAI.00717-16
229. Villeret B, Ghinnagow R, Kheir S, Born-Bony M, Kolls JK, Garcia-Verdugo I, et al. Pseudomonas aeruginosa lung infection subverts lymphocytic responses through IL-23 and IL-22 post-transcriptional regulation. Int J Mol Sci. (2022) 23:8427. doi: 10.3390/ijms23158427
230. Ribot JC, Lopes N, Silva-Santos B. γδ T cells in tissue physiology and surveillance. Nat Rev Immunol. (2021) 21:221–32. doi: 10.1038/s41577-020-00452-4
231. Omar T, Ziltener P, Chamberlain E, Cheng Z, Johnston B. Mice lacking γδ T cells exhibit impaired clearance of Pseudomonas aeruginosa lung infection and excessive production of inflammatory cytokines. Infect Immun. (2020) 88:e00171–20. doi: 10.1128/IAI.00171-20
232. Pan T, Tan R, Li M, Liu Z, Wang X, Tian L, et al. IL17-producing γδ T cells may enhance humoral immunity during pulmonary Pseudomonas aeruginosa infection in mice. Front Cell Infect Microbiol. (2016) 6:170. doi: 10.3389/fcimb.2016.00170
233. Liu J, Qu H, Li Q, Ye L, Ma G, Wan H. The responses of γδ T-cells against acute Pseudomonas aeruginosa pulmonary infection in mice via interleukin-17. Pathog Dis. (2013) 68:44–51. doi: 10.1111/2049-632X.12043
234. Kinjo T, Nakamatsu M, Nakasone C, Yamamoto N, Kinjo Y, Miyagi K, et al. NKT cells play a limited role in the neutrophilic inflammatory responses and host defense to pulmonary infection with Pseudomonas aeruginosa. Microbes Infect. (2006) 8:2679–85. doi: 10.1016/j.micinf.2006.07.016
235. Nieuwenhuis EES, Matsumoto T, Exley M, Schleipman RA, Glickman J, Bailey DT, et al. CD1d-dependent macrophage-mediated clearance of Pseudomonas aeruginosa from lung. Nat Med. (2002) 8:588–93. doi: 10.1038/nm0602-588
236. Benoit P, Sigounas VY, Thompson JL, Van Rooijen N, Poynter ME, Wargo MJ, et al. The role of CD1d-restricted NKT cells in the clearance of Pseudomonas aeruginosa from the lung is dependent on the host genetic background. Infect Immun. (2015) 83:2557–65. doi: 10.1128/IAI.00015-15
237. Rzemieniak SE, Hirschfeld AF, Victor RE, Chilvers MA, Zheng D, Van Den Elzen P, et al. Acidification-dependent activation of CD1d-restricted natural killer T cells is intact in cystic fibrosis. Immunology. (2010) 130:288–95. doi: 10.1111/j.1365-2567.2009.03234.x
238. Facchini M, De Fino I, Riva C, Bragonzi A. Long term chronic Pseudomonas aeruginosa airway infection in mice. J Vis Exp. (2014) 85:51019. doi: 10.3791/51019
239. Reyne N, McCarron A, Cmielewski P, Parsons D, Donnelley M. To bead or not to bead: A review of Pseudomonas aeruginosa lung infection models for cystic fibrosis. Front Physiol. (2023) 14:1104856. doi: 10.3389/fphys.2023.1104856
240. Hartl D, Griese M, Kappler M, Zissel G, Reinhardt D, Rebhan C, et al. Pulmonary TH2 response in Pseudomonas aeruginosa–infected patients with cystic fibrosis. J Allergy Clin Immunol. (2006) 117:204–11. doi: 10.1016/j.jaci.2005.09.023
241. Lloyd CM, Snelgrove RJ. Type 2 immunity: Expanding our view. Sci Immunol. (2018) 3:eaat1604. doi: 10.1126/sciimmunol.aat1604
242. Akdis CA, Arkwright PD, Brüggen M-C, Busse W, Gadina M, Guttman-Yassky E, et al. Type 2 immunity in the skin and lungs. Allergy. (2020) 75:1582–605. doi: 10.1111/all.14318
243. Moser C, Johansen HK, Song Z, Hougen HP, Rygaard J, Høiby N. Chronic Pseudomonas aeruginosa lung infection is more severe in Th2 responding BALB/c mice compared to Th1 responding C3H/HeN mice. APMIS Acta Pathol Microbiol Immunol Scand. (1997) 105:838–42.
244. Telford G, Wheeler D, Williams P, Tomkins PT, Appleby P, Sewell H, et al. The Pseudomonas aeruginosa quorum-sensing signal molecule N -(3-oxododecanoyl)- l -homoserine lactone has immunomodulatory activity. Infect Immun. (1998) 66:36–42. doi: 10.1128/IAI.66.1.36-42.1998
245. Agaronyan K, Sharma L, Vaidyanathan B, Glenn K, Yu S, Annicelli C, et al. Tissue remodeling by an opportunistic pathogen triggers allergic inflammation. Immunity. (2022) 55:895–911.e10. doi: 10.1016/j.immuni.2022.04.001
246. Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol. (2007) 19:652–7. doi: 10.1016/j.coi.2007.07.020
247. Jones CE, Chan K. Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene- α, and granulocyte–colony-stimulating factor by human airway epithelial cells. Am J Respir Cell Mol Biol. (2002) 26:748–53. doi: 10.1165/ajrcmb.26.6.4757
248. Laan M, Cui ZH, Hoshino H, Lötvall J, Sjöstrand M, Gruenert DC, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol Baltim Md 1950. (1999) 162:2347–52.
249. Decraene A, Willems-Widyastuti A, Kasran A, De Boeck K, Bullens DM, Dupont LJ. Elevated expression of both mRNA and protein levels of IL-17A in sputum of stable cystic fibrosis patients. Respir Res. (2010) 11:177. doi: 10.1186/1465-9921-11-177
250. Cockx M, Gouwy M, Van Damme J, Struyf S. Chemoattractants and cytokines in primary ciliary dyskinesia and cystic fibrosis: key players in chronic respiratory diseases. Cell Mol Immunol. (2018) 15:312–23. doi: 10.1038/cmi.2017.118
251. Ding F, Han L, Fu Q, Fan X, Tang R, Lv C, et al. IL-17 Aggravates Pseudomonas aeruginosa airway infection in acute exacerbations of chronic obstructive pulmonary disease. Front Immunol. (2022) 12:811803. doi: 10.3389/fimmu.2021.811803
252. Ding F-M, Zhu S-L, Shen C, Ji X-L, Zhou X. Regulatory T cell activity is partly inhibited in a mouse model of chronic Pseudomonas aeruginosa lung infection. Exp Lung Res. (2015) 41:44–55. doi: 10.3109/01902148.2014.964351
253. Gao W, Thompson L, Zhou Q, Putheti P, Fahmy TM, Strom TB, et al. Treg versus Th17 lymphocyte lineages are cross-regulated by LIF versus IL-6. Cell Cycle. (2009) 8:1444–50. doi: 10.4161/cc.8.9.8348
254. Westhölter D, Beckert H, Straßburg S, Welsner M, Sutharsan S, Taube C, et al. Pseudomonas aeruginosa infection, but not mono or dual-combination CFTR modulator therapy affects circulating regulatory T cells in an adult population with cystic fibrosis. J Cyst Fibros. (2021) 20:1072–9. doi: 10.1016/j.jcf.2021.05.001
255. Westhölter D, Raspe J, Uebner H, Pipping J, Schmitz M, Straßburg S, et al. Regulatory T cell enhancement in adults with cystic fibrosis receiving Elexacaftor/Tezacaftor/Ivacaftor therapy. Front Immunol. (2023) 14:1107437. doi: 10.3389/fimmu.2023.1107437
256. Hector A, Schäfer H, Pöschel S, Fischer A, Fritzsching B, Ralhan A, et al. Regulatory T-cell impairment in cystic fibrosis patients with chronic Pseudomonas infection. Am J Respir Crit Care Med. (2015) 191:914–23. doi: 10.1164/rccm.201407-1381OC
257. Mauch RM, Jensen PØ, Moser C, Levy CE, Høiby N. Mechanisms of humoral immune response against Pseudomonas aeruginosa biofilm infection in cystic fibrosis. J Cyst Fibros. (2018) 17:143–52. doi: 10.1016/j.jcf.2017.08.012
258. Mauch RM, Nørregaard LL, Ciofu O, Levy CE, Høiby N. IgG avidity to Pseudomonas aeruginosa over the course of chronic lung biofilm infection in cystic fibrosis. J Cyst Fibros. (2018) 17:356–9. doi: 10.1016/j.jcf.2017.09.004
259. Schiøtz PO, Nietsen H, Høiby N, Glikmann G, Svehag S -E. Immune complexes in the sputum of patients with cystic fibrosis suffering from chronic Pseudomonas aeruginosa lung infection. Acta Pathol Microbiol Scand [C]. (1978) 86C:37–40. doi: 10.1111/j.1699-0463.1978.tb02555.x
260. Kronborg G, Fomsgaard A, Galanos C, Freudenberg MA, Høiby N. Antibody responses tolipid Acore, and O sugars of the Pseudomonas aeruginosa lipopolysaccharide in chronically infected cystic fibrosis patients. J Clin Microbiol. (1992) 30:1848–55. doi: 10.1128/jcm.30.7.1848-1855.1992
261. Hoiby N, Flensborg EW, Beck B, Friis B, Jacobsen SV, Jacobsen L. Pseudomonas aeruginosa infection in cystic fibrosis. Diagnostic and prognostic significance of Pseudomonas aeruginosa precipitins determined by means of crossed immunoelectrophoresis. Scand J Respir Dis. (1977) 58:65–79.
262. Silva Filho LVRFD, Ferreira FDA, Reis FJC, Britto MCAD, Levy CE, Clark O, et al. Pseudomonas aeruginosa infection in patients with cystic fibrosis: scientific evidence regarding clinical impact, diagnosis, and treatment. J Bras Pneumol. (2013) 39:495–512. doi: 10.1590/S1806-37132013000400015
263. Mauch RM, Levy CE. Serum antibodies to Pseudomonas aeruginosa in cystic fibrosis as a diagnostic tool: A systematic review. J Cyst Fibros. (2014) 13:499–507. doi: 10.1016/j.jcf.2014.01.005
264. McFarlane H, Holzel A, Brenchley P, Allan JD, Wallwork JC, Singer BE, et al. Immune complexes in cystic fibrosis. BMJ. (1975) 1:423–8. doi: 10.1136/bmj.1.5955.423
265. Moss RB, Hsu YP, Lewiston NJ, Curd JG, Milgrom H, Hart S, et al. Association of systemic immune complexes, complement activation, and antibodies to Pseudomonas aeruginosa lipopolysaccharide and exotoxin A with mortality in cystic fibrosis. Am Rev Respir Dis. (1986) 133:648–52. doi: 10.1164/arrd.1986.133.4.648
266. Lagacé J, Mercier J, Fréchette M, Fournier D, Dubreuil M, Lamarre A, et al. Circulating immune complexes, antibodies to Pseudomonas aeruginosa, and pulmonary status in cystic fibrosis. J Clin Lab Immunol. (1989) 30:7–11.
267. Disis ML, McDonald TL, Colombo JL, Kobayashi RH, Angle CR, Murray S. Circulating immune complexes in cystic fibrosis and their correlation to clinical parameters. Pediatr Res. (1986) 20:385–90. doi: 10.1203/00006450-198605000-00002
268. Wisnieski JJ, Todd EW, Fuller RK, Jones PK, Dearborn DG, Boat TF, et al. Immune complexes and complement abnormalities in patients with cystic fibrosis. Increased mortality associated with circulating immune complexes and decreased function of the alternative complement pathway. Am Rev Respir Dis. (1985) 132:770–6. doi: 10.1164/arrd.1985.132.4.770
269. Bojanowski CM, Lu S, Kolls JK. Mucosal immunity in cystic fibrosis. J Immunol. (2021) 207:2901–12. doi: 10.4049/jimmunol.2100424
270. Sloane AJ, Lindner RA, Prasad SS, Sebastian LT, Pedersen SK, Robinson M, et al. Proteomic analysis of sputum from adults and children with cystic fibrosis and from control subjects. Am J Respir Crit Care Med. (2005) 172:1416–26. doi: 10.1164/rccm.200409-1215OC
271. Fick RB, Baltimore RS, Squier SU, Reynolds HY. IgG proteolytic activity of Pseudomonas aeruginosa in cystic fibrosis. J Infect Dis. (1985) 151:589–98. doi: 10.1093/infdis/151.4.589
272. Eichler I, Joris L, Hsu YP, Van Wye J, Bram R, Moss R. Nonopsonic antibodies in cystic fibrosis. Pseudomonas aeruginosa lipopolysaccharide-specific immunoglobulin G antibodies from infected patient sera inhibit neutrophil oxidative responses. J Clin Invest. (1989) 84:1794–804. doi: 10.1172/JCI114364
273. Polanec J, Patzer J, Grzybowski J, Strukelj M, P Pavelic Z. Amount and avidity of IgG antibodies to Pseudomonas aeruginosa exotoxin A antigen in cystic fibrosis patients. Pathol Oncol Res. (1997) 3:26–9. doi: 10.1007/BF02893348
274. Ciofu O, Petersen TD, Jensen P, Hoiby N. Avidity of anti-P aeruginosa antibodies during chronic infection in patients with cystic fibrosis. Thorax. (1999) 54:141–4. doi: 10.1136/thx.54.2.141
275. McQuillan K, Gargoum F, Murphy MP, McElvaney OJ, McElvaney NG, Reeves EP. Targeting IgG autoantibodies for improved cytotoxicity of bactericidal permeability increasing protein in cystic fibrosis. Front Pharmacol. (2020) 11:1098. doi: 10.3389/fphar.2020.01098
276. Mogayzel PJ, Naureckas ET, Robinson KA, Brady C, Guill M, Lahiri T, et al. Cystic fibrosis foundation pulmonary guideline. Pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann Am Thorac Soc. (2014) 11:1640–50. doi: 10.1513/AnnalsATS.201404-166OC
277. Cogen JD, Nichols DP, Goss CH, Somayaji R. Drugs, drugs, drugs: current treatment paradigms in cystic fibrosis airway infections. J Pediatr Infect Dis Soc. (2022) 11:S32–9. doi: 10.1093/jpids/piac061
278. Nichols DP, Singh PK, Baines A, Caverly LJ, Chmiel JF, Gibson RL, et al. Testing the effects of combining azithromycin with inhaled tobramycin for P. aeruginosa in cystic fibrosis: a randomised, controlled clinical trial. Thorax. (2022) 77:581–8. doi: 10.1136/thoraxjnl-2021-217782
279. Reyhanoglu G, Reddivari AKR. Tobramycin (2024). Treasure Island (FL: StatPearls Publishing. Available at: http://www.ncbi.nlm.nih.gov/books/NBK551695/ (Accessed February 8, 2024).
280. Smith S, Rowbotham NJ. Inhaled anti-pseudomonal antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst Rev. (2022) 2022:166. doi: 10.1002/14651858.CD001021.pub4
281. Sandman Z, Iqbal OA. Azithromycin (2024). Treasure Island (FL: StatPearls Publishing. Available at: http://www.ncbi.nlm.nih.gov/books/NBK557766/ (Accessed February 8, 2024).
282. Flume PA, Clancy JP, Retsch-Bogart GZ, Tullis DE, Bresnik M, Derchak PA, et al. Continuous alternating inhaled antibiotics for chronic pseudomonal infection in cystic fibrosis. J Cyst Fibros. (2016) 15:809–15. doi: 10.1016/j.jcf.2016.05.001
283. DrugBank. Aztreonam. Edmonton, Alberta, Canada: DrugBank (2024). Available at: https://go.drugbank.com/drugs/DB00355.
284. Elmassry MM, Colmer-Hamood JA, Kopel J, San Francisco MJ, Hamood AN. Anti-Pseudomonas aeruginosa vaccines and therapies: an assessment of clinical trials. Microorganisms. (2023) 11:916. doi: 10.3390/microorganisms11040916
285. Elborn JS, Blasi F, Burgel P-R, Peckham D. Role of inhaled antibiotics in the era of highly effective CFTR modulators. Eur Respir Rev. (2023) 32:220154. doi: 10.1183/16000617.0154-2022
286. Pennington JE, Reynolds HY, Wood RE, Robinson RA, Levine AS. Use of a Pseudomonas aeruginosa vaccine in patients with acute leukemia and cystic fibrosis. Am J Med. (1975) 58:629–36. doi: 10.1016/0002-9343(75)90498-2
287. Cryz SJ, Fürer E, Cross AS, Wegmann A, Germanier R, Sadoff JC. Safety and immunogenicity of a Pseudomonas aeruginosa O-polysaccharide toxin A conjugate vaccine in humans. J Clin Invest. (1987) 80:51–6. doi: 10.1172/JCI113062
288. Döring G, Meisner C, Stern M, for the Flagella Vaccine Trial Study Group. A double-blind randomized placebo-controlled phase III study of a Pseudomonas aeruginosa flagella vaccine in cystic fibrosis patients. Proc Natl Acad Sci. (2007) 104:11020–5. doi: 10.1073/pnas.0702403104
289. Wang X, Liu C, Rcheulishvili N, Papukashvili D, Xie F, Zhao J, et al. Strong immune responses and protection of PcrV and OprF-I mRNA vaccine candidates against Pseudomonas aeruginosa. NPJ Vaccines. (2023) 8:76. doi: 10.1038/s41541-023-00672-4
290. Pier GB, Saunders JM, Ames P, Edwards MS, Auerbach H, Goldfarb J, et al. Opsonophagocytic killing antibody to Pseudomonas aeruginosa mucoid exopolysaccharide in older noncolonized patients with cystic fibrosis. N Engl J Med. (1987) 317:793–8. doi: 10.1056/NEJM198709243171303
291. Yang F, Gu J, Yang L, Gao C, Jing H, Wang Y, et al. Protective efficacy of the trivalent Pseudomonas aeruginosa vaccine candidate PcrV-OprI-Hcp1 in murine pneumonia and burn models. Sci Rep. (2017) 7:3957. doi: 10.1038/s41598-017-04029-5
292. Milla CE, Chmiel JF, Accurso FJ, VanDevanter DR, Konstan MW, Yarranton G, et al. Anti-PcrV antibody in cystic fibrosis: A novel approach targeting Pseudomonas aeruginosa airway infection. Pediatr Pulmonol. (2014) 49:650–8. doi: 10.1002/ppul.22890
293. Weimer ET, Ervin SE, Wozniak DJ, Mizel SB. Immunization of young African green monkeys with OprF epitope 8–OprI–type A- and B-flagellin fusion proteins promotes the production of protective antibodies against nonmucoid Pseudomonasaeruginosa. Vaccine. (2009) 27:6762–9. doi: 10.1016/j.vaccine.2009.08.080
294. Hassan R, El-Naggar W, Abd El-Aziz AM, Shaaban M, Kenawy HI, Ali YM. Immunization with outer membrane proteins (OprF and OprI) and flagellin B protects mice from pulmonary infection with mucoid and nonmucoid Pseudomonas aeruginosa. J Microbiol Immunol Infect. (2018) 51:312–20. doi: 10.1016/j.jmii.2016.08.014
295. Hashemi FB, Behrouz B, Irajian G, Laghaei P, Korpi F, Fatemi MJ. A trivalent vaccine consisting of “flagellin A+B and pilin” protects against Pseudomonas aeruginosa infection in a murine burn model. Microb Pathog. (2020) 138:103697. doi: 10.1016/j.micpath.2019.103697
296. Cripps AW, Dunkley ML, Clancy RL. Mucosal and systemic immunizations with killed Pseudomonas aeruginosa protect against acute respiratory infection in rats. Infect Immun. (1994) 62:1427–36. doi: 10.1128/iai.62.4.1427-1436.1994
297. Fan Y, Mu Y, Lu L, Tian Y, Yuan F, Zhou B, et al. Hydrogen peroxide-inactivated bacteria induces potent humoral and cellular immune responses and releases nucleic acids. Int Immunopharmacol. (2019) 69:389–97. doi: 10.1016/j.intimp.2019.01.055
298. Cabral MP, Correia A, Vilanova M, Gärtner F, Moscoso M, García P, et al. A live auxotrophic vaccine confers mucosal immunity and protection against lethal pneumonia caused by Pseudomonas aeruginosa. PloS Pathog. (2020) 16:e1008311. doi: 10.1371/journal.ppat.1008311
299. Kamei A, Coutinho-Sledge YS, Goldberg JB, Priebe GP, Pier GB. Mucosal vaccination with a multivalent, live-attenuated vaccine induces multifactorial immunity against Pseudomonas aeruginosa Acute Lung Infection. Infect Immun. (2011) 79:1289–99. doi: 10.1128/IAI.01139-10
300. Gong Q, Ruan MD, Niu MF, Qin CL, Hou Y, Guo JZ. Immune efficacy of DNA vaccines based on oprL and oprF genes of Pseudomonas aeruginosa in chickens. Poult Sci. (2018) 97:4219–27. doi: 10.3382/ps/pey307
301. Killough M, Rodgers A, Ingram R. Pseudomonas aeruginosa: recent advances in vaccine development. Vaccines. (2022) 10:1100. doi: 10.3390/vaccines10071100
302. Lands LC, Stanojevic S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst Rev. (2016). doi: 10.1002/14651858.CD001505.pub4
303. McElvaney OJ, Heltshe SL, Odem-Davis K, West NE, Sanders DB, Fogarty B, et al. Adjunctive systemic corticosteroids for pulmonary exacerbations of cystic fibrosis. Ann Am Thorac Soc. (2023). doi: 10.1513/AnnalsATS.202308-673OC
304. Shafiek H, Verdú J, López-Causapé C, Iglesias A, Ramon-Clar L, Juan C, et al. Inhaled corticosteroid use and its association with Pseudomonas aeruginosa infection in COPD. Eur Respir J. (2021) 58:PA798. doi: 10.1183/13993003.congress-2021.PA798
305. Cerón-Pisa N, Shafiek H, Martín-Medina A, Verdú J, Jordana-Lluch E, Escobar-Salom M, et al. Effects of inhaled corticosteroids on the innate immunological response to Pseudomonas aeruginosa infection in patients with COPD. Int J Mol Sci. (2022) 23:8127. doi: 10.3390/ijms23158127
306. Konstan MW, VanDevanter DR, Sawicki GS, Pasta DJ, Foreman AJ, Neiman EA, et al. Association of high-dose ibuprofen use, lung function decline, and long-term survival in children with cystic fibrosis. Ann Am Thorac Soc. (2018) 15:485–93. doi: 10.1513/AnnalsATS.201706-486OC
307. Tateda K, Comte R, Pechere J-C, Köhler T, Yamaguchi K, Van Delden C. Azithromycin inhibits quorum sensing in Pseudomonas aeruginosa. Antimicrob Agents Chemother. (2001) 45:1930–3. doi: 10.1128/AAC.45.6.1930-1933.2001
308. Ratjen F, Saiman L, Mayer-Hamblett N, Lands LC, Kloster M, Thompson V, et al. Effect of azithromycin on systemic markers of inflammation in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa. Chest. (2012) 142:1259–66. doi: 10.1378/chest.12-0628
309. Verleden GM, Vanaudenaerde BM, Dupont LJ, Van Raemdonck DE. Azithromycin reduces airway neutrophilia and interleukin-8 in patients with bronchiolitis obliterans syndrome. Am J Respir Crit Care Med. (2006) 174:566–70. doi: 10.1164/rccm.200601-071OC
310. Chalmers JD, Boersma W, Lonergan M, Jayaram L, Crichton ML, Karalus N, et al. Long-term macrolide antibiotics for the treatment of bronchiectasis in adults: an individual participant data meta-analysis. Lancet Respir Med. (2019) 7:845–54. doi: 10.1016/S2213-2600(19)30191-2
311. Cigana C, Assael BM, Melotti P. Azithromycin selectively reduces tumor necrosis factor alpha levels in cystic fibrosis airway epithelial cells. Antimicrob Agents Chemother. (2007) 51:975–81. doi: 10.1128/AAC.01142-06
312. Haydar D, Cory TJ, Birket SE, Murphy BS, Pennypacker KR, Sinai AP, et al. Azithromycin polarizes macrophages to an M2 phenotype via inhibition of the STAT1 and NF-κB signaling pathways. J Immunol. (2019) 203:1021–30. doi: 10.4049/jimmunol.1801228
313. Gualdoni GA, Lingscheid T, Schmetterer KG, Hennig A, Steinberger P, Zlabinger GJ. Azithromycin inhibits IL-1 secretion and non-canonical inflammasome activation. Sci Rep. (2015) 5:12016. doi: 10.1038/srep12016
314. Nichols DP, Odem-Davis K, Cogen JD, Goss CH, Ren CL, Skalland M, et al. Pulmonary outcomes associated with long-term azithromycin therapy in cystic fibrosis. Am J Respir Crit Care Med. (2020) 201:430–7. doi: 10.1164/rccm.201906-1206OC
315. Doull I. Cystic fibrosis 2019: Year in review. Paediatr Respir Rev. (2020) 35:95–8. doi: 10.1016/j.prrv.2020.04.001
316. Saiman L. Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. (2010) 303:1707. doi: 10.1001/jama.2010.563
317. Samson C, Tamalet A, Thien HV, Taytard J, Perisson C, Nathan N, et al. Long-term effects of azithromycin in patients with cystic fibrosis. Respir Med. (2016) 117:1–6. doi: 10.1016/j.rmed.2016.05.025
318. Moss RB, Mistry SJ, Konstan MW, Pilewski JM, Kerem E, Tal-Singer R, et al. Safety and early treatment effects of the CXCR2 antagonist SB-656933 in patients with cystic fibrosis. J Cyst Fibros. (2013) 12:241–8. doi: 10.1016/j.jcf.2012.08.016
319. Kirsten AM, Förster K, Radeczky E, Linnhoff A, Balint B, Watz H, et al. The safety and tolerability of oral AZD5069, a selective CXCR2 antagonist, in patients with moderate-to-severe COPD. Pulm Pharmacol Ther. (2015) 31:36–41. doi: 10.1016/j.pupt.2015.02.001
320. Lorè NI, Cigana C, Riva C, De Fino I, Nonis A, Spagnuolo L, et al. IL-17A impairs host tolerance during airway chronic infection by Pseudomonas aeruginosa. Sci Rep. (2016) 6:25937. doi: 10.1038/srep25937
321. Hsu D, Taylor P, Fletcher D, Van Heeckeren R, Eastman J, Van Heeckeren A, et al. Interleukin-17 pathophysiology and therapeutic intervention in cystic fibrosis lung infection and inflammation. Infect Immun. (2016) 84:2410–21. doi: 10.1128/IAI.00284-16
322. Patel DD, Lee DM, Kolbinger F, Antoni C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann Rheum Dis. (2013) 72:iii116–23. doi: 10.1136/annrheumdis-2012-202371
323. Birrer P. Proteases and antiproteases in cystic fibrosis: pathogenetic considerations and therapeutic strategies. Respiration. (1995) 62:25–8. doi: 10.1159/000196490
324. Suter S, Chevallier I. Proteolytic inactivation of alpha 1-proteinase inhibitor in infected bronchial secretions from patients with cystic fibrosis. Eur Respir J. (1991) 4:40–9.
325. Rahaghi FF, Miravitlles M. Long-term clinical outcomes following treatment with alpha 1-proteinase inhibitor for COPD associated with alpha-1 antitrypsin deficiency: a look at the evidence. Respir Res. (2017) 18:105. doi: 10.1186/s12931-017-0574-1
326. Cantin AM, Woods DE. Aerosolized prolastin suppresses bacterial proliferation in a model of chronic Pseudomonas aeruginosa lung infection. Am J Respir Crit Care Med. (1999) 160:1130–5. doi: 10.1164/ajrccm.160.4.9807166
327. McElvaney NG, Hubbard RC, Birrer P, Crystal RG, Chernick MS, Frank MM, et al. Aerosol α1 -antitrypsin treatment for cystic fibrosis. Lancet. (1991) 337:392–4. doi: 10.1016/0140-6736(91)91167-S
328. Griese M, Latzin P, Kappler M, Weckerle K, Heinzlmaier T, Bernhardt T, et al. 1-Antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur Respir J. (2006) 29:240–50. doi: 10.1183/09031936.00047306
329. Hudock KM, Collins MS, Imbrogno MA, Kramer EL, Brewington JJ, Ziady A, et al. Alpha-1 antitrypsin limits neutrophil extracellular trap disruption of airway epithelial barrier function. Front Immunol. (2023) 13:1023553. doi: 10.3389/fimmu.2022.1023553
330. Jones AP, Wallis C, Kearney CE, The Cochrane Collaboration. Dornase alfa for cystic fibrosis. In: Cochrane Database of Systematic Reviews. John Wiley & Sons, Ltd, Chichester, UK (2003). p. CD001127. doi: 10.1002/14651858.CD001127
331. Suri R, Marshall LJ, Wallis C, Metcalfe C, Bush A, Shute JK. Effects of recombinant human DNAse and hypertonic saline on airway inflammation in children with cystic fibrosis. Am J Respir Crit Care Med. (2002) 166:352–5. doi: 10.1164/rccm.2110015
332. Konstan MW, Ratjen F. Effect of dornase alfa on inflammation and lung function: Potential role in the early treatment of cystic fibrosis. J Cyst Fibros. (2012) 11:78–83. doi: 10.1016/j.jcf.2011.10.003
333. Paul K, Rietschel E, Ballmann M, Griese M, Worlitzsch D, Shute J, et al. Effect of Treatment with dornase alpha on airway inflammation in patients with cystic fibrosis. Am J Respir Crit Care Med. (2004) 169:719–25. doi: 10.1164/rccm.200307-959OC
334. Schmitt-Grohé S, Zielen S. Leukotriene receptor antagonists in children with cystic fibrosis lung disease: anti-inflammatory and clinical effects. Pediatr Drugs. (2005) 7:353–63. doi: 10.2165/00148581-200507060-00004
335. Lopes-Pacheco M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol. (2020) 10:1662. doi: 10.3389/fphar.2019.01662
336. Taylor-Cousar JL, Robinson PD, Shteinberg M, Downey DG. CFTR modulator therapy: transforming the landscape of clinical care in cystic fibrosis. Lancet. (2023) 402:1171–84. doi: 10.1016/S0140-6736(23)01609-4
337. Cuevas-Ocaña S, Laselva O, Avolio J, Nenna R. The era of CFTR modulators: improvements made and remaining challenges. Breathe. (2020) 16:200016. doi: 10.1183/20734735.0016-2020
338. Ruffin M, Roussel L, Maillé É, Rousseau S, Brochiero E. Vx-809/Vx-770 treatment reduces inflammatory response to Pseudomonas aeruginosa in primary differentiated cystic fibrosis bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L635–41. doi: 10.1152/ajplung.00198.2017
339. Adam D, Bilodeau C, Sognigbé L, Maillé É, Ruffin M, Brochiero E. CFTR rescue with VX-809 and VX-770 favors the repair of primary airway epithelial cell cultures from patients with class II mutations in the presence of Pseudomonas aeruginosa exoproducts. J Cyst Fibros Off J Eur Cyst Fibros Soc. (2018) 17:705–14. doi: 10.1016/j.jcf.2018.03.010
340. Sun X, Yi Y, Yan Z, Rosen BH, Liang B, Winter MC, et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci Transl Med. (2019) 11:eaau7531. doi: 10.1126/scitranslmed.aau7531
341. Sheikh S, Britt RD, Ryan-Wenger NA, Khan AQ, Lewis BW, Gushue C, et al. Impact of elexacaftor–tezacaftor–ivacaftor on bacterial colonization and inflammatory responses in cystic fibrosis. Pediatr Pulmonol. (2023) 58:825–33. doi: 10.1002/ppul.26261
342. Dhote T, Martin C, Regard L, Pesenti L, Kanaan R, Carlier N, et al. Normalisation of circulating neutrophil counts after 12 months of elexacaftor-tezacaftor-ivacaftor in patients with advanced cystic fibrosis. Eur Respir J. (2023) 61:2202096. doi: 10.1183/13993003.02096-2022
343. Schmidt H, Höpfer LM, Wohlgemuth L, Knapp CL, Mohamed AOK, Stukan L, et al. Multimodal analysis of granulocytes, monocytes, and platelets in patients with cystic fibrosis before and after Elexacaftor–Tezacaftor–Ivacaftor treatment. Front Immunol. (2023) 14:1180282. doi: 10.3389/fimmu.2023.1180282
344. Heltshe SL, Mayer-Hamblett N, Burns JL, Khan U, Baines A, Ramsey BW, et al. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis. (2015) 60:703–12. doi: 10.1093/cid/ciu944
345. Harris JK, Wagner BD, Zemanick ET, Robertson CE, Stevens MJ, Heltshe SL, et al. Changes in airway microbiome and inflammation with ivacaftor treatment in patients with cystic fibrosis and the G551D mutation. Ann Am Thorac Soc. (2020) 17:212–20. doi: 10.1513/AnnalsATS.201907-493OC
346. Hisert KB, Heltshe SL, Pope C, Jorth P, Wu X, Edwards RM, et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am J Respir Crit Care Med. (2017) 195:1617–28. doi: 10.1164/rccm.201609-1954OC
347. Beck MR, Hornick DB, Pena TA, Singh SB, Wright BA. Impact of elexacaftor/tezacaftor/ivacaftor on bacterial cultures from people with cystic fibrosis. Pediatr Pulmonol. (2023) 58:1569–73. doi: 10.1002/ppul.26362
348. Dittrich A-M, Sieber S, Naehrlich L, Burkhart M, Hafkemeyer S, Tümmler B. Use of elexacaftor/tezacaftor/ivacaftor leads to changes in detection frequencies of Staphylococcus aureus and Pseudomonas aeruginosa dependent on age and lung function in people with cystic fibrosis. Int J Infect Dis. (2024) 139:124–31. doi: 10.1016/j.ijid.2023.11.013
349. Nichols DP, Morgan SJ, Skalland M, Vo AT, Van Dalfsen JM, Singh SBP, et al. Pharmacologic improvement of CFTR function rapidly decreases sputum pathogen density, but lung infections generally persist. J Clin Invest. (2023) 133:e167957. doi: 10.1172/JCI167957
350. Sosinski LM, CM H, Neugebauer KA, Ghuneim L-AJ, Guzior DV, Castillo-Bahena A, et al. A restructuring of microbiome niche space is associated with Elexacaftor-Tezacaftor-Ivacaftor therapy in the cystic fibrosis lung. J Cyst Fibros. (2022) 21:996–1005. doi: 10.1016/j.jcf.2021.11.003
Keywords: Pseudomonas aeruginosa, chronic lung disease, inflammation, neutrophils, innate immune response, adaptive immune response
Citation: Nickerson R, Thornton CS, Johnston B, Lee AHY and Cheng Z (2024) Pseudomonas aeruginosa in chronic lung disease: untangling the dysregulated host immune response. Front. Immunol. 15:1405376. doi: 10.3389/fimmu.2024.1405376
Received: 22 March 2024; Accepted: 14 June 2024;
Published: 28 June 2024.
Edited by:
Jean-Michel Sallenave, INSERM U1152 Physiopathologie et Epidémiologie des Maladies Respiratoires, FranceReviewed by:
Simon Rousseau, McGill University, CanadaCopyright © 2024 Nickerson, Thornton, Johnston, Lee and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenyu Cheng, emhlbnl1LmNoZW5nQGRhbC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.