 Laure Migayron
Laure Migayron Sylvie Bordes2
Sylvie Bordes2 Julien Seneschal
Julien Seneschal Katia Boniface
Katia Boniface- 1Univ. Bordeaux, CNRS, Immuno ConcEpT, UMR 5164, Bordeaux, France
- 2R&D Department, SILAB, Brive-la-Gaillarde, France
- 3CHU de Bordeaux, Dermatology and Pediatric Dermatology, National Reference Center for Rare Skin Disorders, Hôpital Saint-André, UMR 5164, Bordeaux, France
Chronic inflammatory skin diseases are multifactorial diseases that combine genetic predisposition, environmental triggers, and metabolic disturbances associated with abnormal immune responses. From an immunological perspective, the better understanding of their physiopathology has demonstrated a large complex network of immune cell subsets and related cytokines that interact with both epidermal and dermal cells. For example, in type-1-associated diseases such as alopecia areata, vitiligo, and localized scleroderma, recent evidence suggests the presence of a type-2 inflammation that is well known in atopic dermatitis. Whether this type-2 immune response has a protective or detrimental impact on the development and chronicity of these diseases remains to be fully elucidated, highlighting the need to better understand its involvement for the management of patients. This mini-review explores recent insights regarding the potential role of type-2-related immunity in alopecia areata, vitiligo, and localized scleroderma.
Introduction
The characterization of the diversity of immune cell subsets has extended our understanding of the complexity of the mechanisms driving the development and recurrence of chronic inflammatory disorders and hastened the subsequent use of targeted therapies. Three major types of innate and adaptive cell-mediated effector immunity have been identified (1, 2). While these immune responses are primarily involved in protection against pathogens, their aberrant activation can also be harmful and lead to the development of autoimmunity or to inflammatory or allergic diseases (1, 3).
Type-1 immunity mainly involves innate lymphoid type-1 cells (ILC1), natural killer (NK) cells, CD4 Th1 and cytotoxic CD8 Tc1 cells, mainly inducing interferon (IFN)-γ and tumor necrosis factor (TNF)-α (1). Besides its protective role against intracellular pathogens such as viruses, it is also implicated in inflammatory diseases such as alopecia areata (AA), localized scleroderma (LS), and vitiligo.
Type-2-associated immune cells include ILC2, Th2 cells, eosinophils, mast cells, basophils, and alternatively activated macrophages, which are known to release cytokines like interleukin (IL)-4, IL-5, IL-13, or IL-31 (3–6). These cells are critical for the defense of the organism against extracellular pathogens (i.e. helminth parasites) and for the maintenance of tissue homeostasis (tissue regeneration and wound repair) (7). However, besides its protective role, pathogenic activation of type-2 immune response contributes to the development of allergic and inflammatory diseases such as asthma, allergic rhinitis, and atopic dermatitis (AD) (5, 8, 9). AD is characterized by skin barrier dysfunction contributing to an aberrant sensitization to environmental allergens (10). In AD, type-2 cytokines like IL-4 and IL-13 are directly implicated in the impairment of epidermal barrier integrity observed in AD lesions by inhibiting the synthesis of key structural proteins such as filaggrin, loricrin, honerin, and involucrin (11–14). The role of type-2 inflammation in AD, and especially that of IL-4/IL-13, is exemplified by the efficacy of therapies targeting these cytokines, e.g. the anti-IL-4Rα antibody dupilumab and the anti-IL-13 antibodies tralokinumab and lebrikizumab for moderate to severe AD (15–30). However, the pathophysiology of AD is more complex with heterogenous phenotypes underlying different endotypes, with the involvement of type-1 (e.g. Th1 cells) and/or type-3 (Th17 and Th22 cells) immune cell subsets (31–35). Likewise, an increasing body of evidence has shown that type-2-associated immune response may also play a role in the development of type1 or type-3-related skin diseases, hence increasing the complexity of disease pathogenesis and patient stratification. This mini-review examines recent insights into the role of type-2 inflammation in type-1-associated skin inflammatory diseases with a focus on LS, AA, and vitiligo (Figure 1).
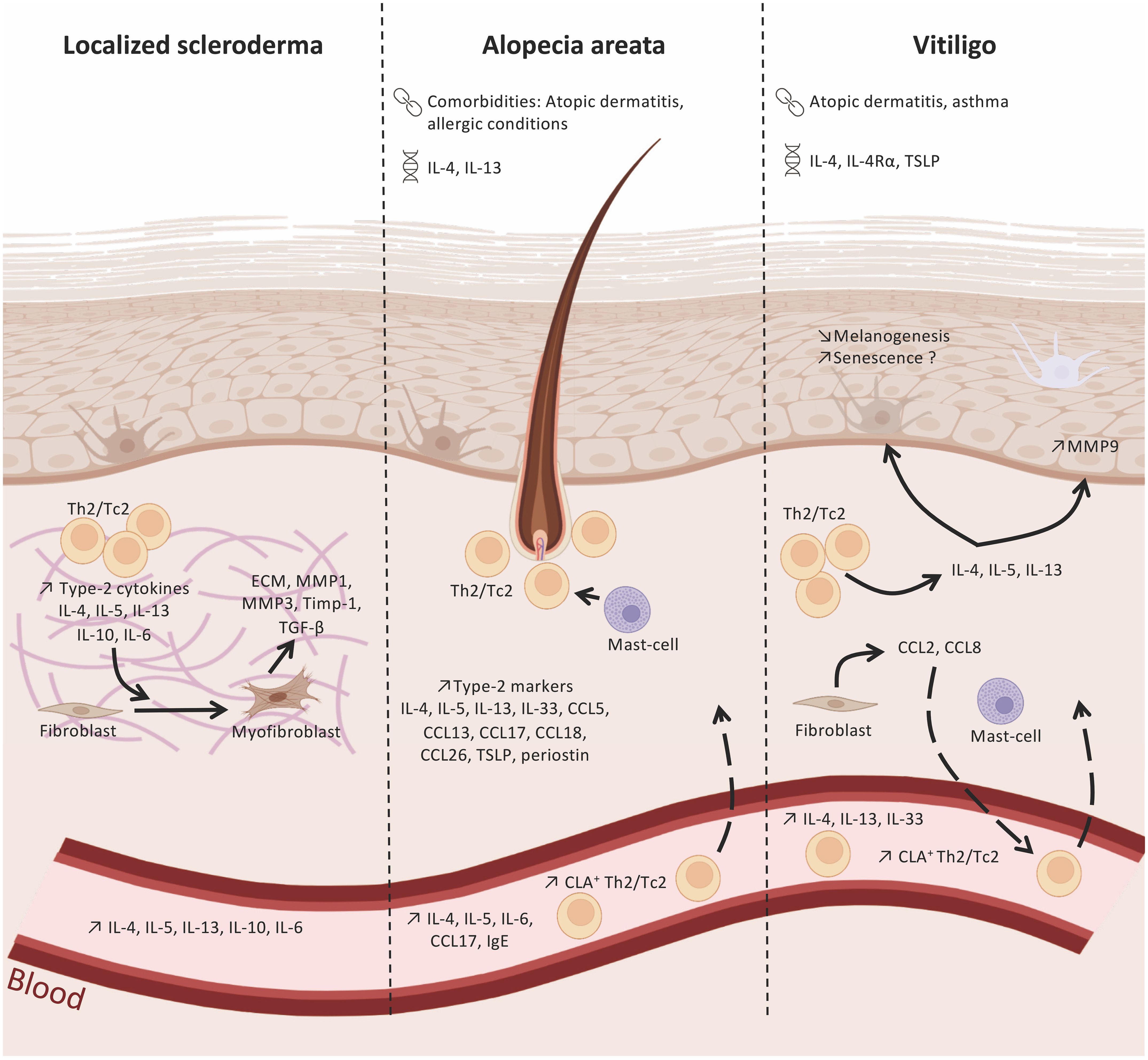
Figure 1 Type-2 immunity in localized scleroderma, alopecia areata and vitiligo. Polymorphims with IL-4/IL-13 genes have been identified in alopecia areata (AA) and vitiligo. In addition, these two diseases are associated with atopic dermatitis or allergic conditions. Type-2 immunity cells and markers found in the skin and blood of patients withlocalized scleroderma (LS), AA, and vitiligo, suggesting their role in the immune network of these pathologies. In LS, type-2 cytokines released by Th2 and Tc2 cell subsets (e.g. IL-4, IL-5, IL-13) infiltrating LS lesions promote the differentiation of fibroblasts into myofibroblasts and the production of pro-fibrotic factors, like TGF-β. AA skin lesions display elevated levels of type-2 cytokines and chemokines released by epidermal, dermal, and immune cells that will contribute to the recruitment of Th2/Tc2 and my influence hair loss. The type-2 environment in vitiligo skin may regulate melanogenesis and the loss melanocytes. : comorbidities;
: comorbidities;  polymorphisms. Created with BioRender.com.
polymorphisms. Created with BioRender.com.
Localized scleroderma
LS is a rare autoimmune skin disorder characterized by inflammation and fibrosis of the skin, with dense collagen deposition in the dermis and underlying connective tissues (36). Inflammatory patches and/or bands of thickened skin develop on the head and neck region, trunk and extremities. Morphea is the most frequent subtype of LS with onset between 40 and 50 years of age (37). LS is classified into five main types according to the extent and depth of fibrosis: limited, generalized, linear, deep and mixed (37, 38). Its pathogenesis is based on genetic predisposition combined with external triggers such as trauma, repeated friction, and surgery, that induce aberrant inflammatory and profibrotic responses, fibroblasts being a critical factor during the development of the disease (39–41). During the early inflammatory stage of LS, CD4+ T cells, macrophages, and eosinophils infiltrate the skin and adjacent blood vessels (36, 42, 43). Both Th1 and Th17 responses seem implicated in this primary stage, with an increased release of chemokine (C-X-C motif) ligand (CXCL)9/10, IFN-γ, TNF-α, IL-23, IL-17 and transforming growth factor (TGF)-β (36, 44). CXCL9 and CXCL10 serum levels correlate with the disease activity (45, 46). Interestingly, Werner et al. recently identified clusters of inflammatory fibroblasts prone to release CXCL12 or CXCL9/10 in LS lesions. The same study also demonstrated the crosstalk between fibroblasts and infiltrated immune cells (e.g. macrophages and T cells) to perpetuate inflammatory signals in lesions (47). Indeed, inflammatory fibrosis was shown to be dependent on CXCL9 and its receptor CXCR3 in a mouse model of skin fibrosis, thereby confirming the involvement of a type-1 immune response in the early phase of skin fibrosis (48). In addition, the increased expression of several adhesion molecules by endothelial cells, such as vascular cell adhesion molecule 1 (VCAM1), Intercellular Adhesion Molecule1 (ICAM1) and E-selectin, contributes to the recruitment of immune cells in the lesional areas (49).
Fibrosis is a key mechanism defining LS lesions and is characterized by excessive deposition of extracellular matrix (ECM) components such as collagen in the tissue. TGF-β is considered as a major profibrotic factor owing to its effects on fibroblast proliferation, differentiation, migration, and the production of extracellular cellular matrix components (50, 51). However, clinical trials blocking TGF-β produced conflicting results (52, 53).
It has been postulated that as the disease progresses, a shift occurs to a type-2 immune response that is associated with the development of skin fibrosis. Type-2-related cytokines (IL-4, IL-5, IL-6 and IL-13) are increased in the serum and skin of LS patients, and IL-13 serum levels correlate with the number of lesions in LS (54–56). Such type-2 immunity appears to be associated with the fibrotic/sclerotic stage of the disease (36). In vitro studies showed that IL-4 and IL-13 induce an excessive production of ECM components such as collagen, periostin, proteoglycan synthesis, and fibronectin by scleroderma and/or normal fibroblasts (57–61). These cytokines also stimulate the production of TGF-β and the synthesis of matrix metalloproteinase (MMP)1, MMP3 and TIMP-1 (a tissue inhibitor of MMP), as well as the proliferation of fibroblasts and their differentiation in myofibroblasts (62–64). Interestingly, the inhibition of type-2 signaling prevents the development of cutaneous fibrosis in vivo (65–67). A phase II clinical trial is ongoing to test the efficacy of dupilumab in localized scleroderma patients (NCT04200755).
Alopecia areata
AA is a chronic non-scarring hair loss condition affecting 0.5–2% of the population and resulting from an autoimmune response targeting the hair follicle (68). AA is predominantly driven by a type-1 inflammatory response associated with the production of IFNγ by antigen-specific CD8+ NKG2D+ Tc1 and CD4+ Th1 cells in response to an environmental trigger, such as stress, viral infection, or trauma. This induces the collapse of the immune privilege of the hair follicle leading to its growth arrest (69). IFNγ also contributes to the increased inflammation through the induction of CXCL9/10 by the hair follicle epithelium, leading to the recruitment of CXCR3+ T cells to the bulb (70).
Recent data also suggest the contribution of the type-2 immune response in AA pathogenesis. From a clinical perspective, AA is associated with atopic dermatitis and allergic conditions, and an atopic background increases the risk of developing it (71–75). Loss-of-function mutations in the gene encoding filaggrin are associated with the severity of AA in patients with a history of AD, and genetic studies identified the association of AA with polymorphisms for the genes encoding IL-4 and IL-13 (76–78). An increase in mast cells with a pro-inflammatory phenotype in the perifollicular area of AA patients was reported. These mast cells display an increased degranulation activity and could interact with CD8+ T cells to provide co-stimulatory signals (via 4–1BBL, OX40L, ICAM1) and possibly to present neo-autoantigens (79). In addition, AA skin lesions display an increase in type-2-related cytokines and chemokines, including IL-4, IL-5, IL-13, IL-33, chemokine (C-C motif) ligand (CCL)-5, CCL13, CCL17, CCL18, CCL26, TSLP and periostin (80–83). Interestingly, after intralesional corticosteroid injection, a downregulation of CCL18 was associated with a clinical improvement (82). Levels of IL-4, IL-5, IL-6, IL-13, CCL13, CCL17, CCL22, CCL26, and IgE are also increased in AA patients’ sera (83–88). Czarnowicki et al. observed an increase in circulating skin-homing cutaneous lymphocyte-associated antigen (CLA)+ Th2 and CLA+ Tc2 cell subsets in AA patients compared to healthy controls that correlated with disease activity. In contrast, IFNγ was associated with the chronicity of the disease (89). All these data highlight the putative role of Th2 cells in disease pathogenesis.
However, the use of dupilumab in AA led to conflicting results, some investigations showing a significant improvement while others reporting exacerbation or new onset of the disease (90–100). Patients with an atopic background and high IgE levels exhibited a better response to dupilumab (101). Recent data suggest that non-atopic AA patients display an increase in circulating Tc1 cells while AA patients with concomitant AD show a skewed Th2 profile (102). In addition, the infiltration of CCR4+ Th2 cells around the hair bulb in skin lesions is more extensive in AA patients with AD (102). Altogether, these data suggest that as in AD, different clinical phenotypes and related endotypes likely define AA patients.
Vitiligo
Vitiligo, the most common depigmenting skin disease, is defined by a type-1 skewed immune bias, with the involvement of IFNα, IFNγ, TNFα, CXCL9, CXCL10, and CXCL16 (103–105). Melanocyte loss results from the cytotoxic activity of CD8 T cells and detachment of melanocytes from the basal layer of the epidermis in response to the cytokine microenvironment (106). The perilesional skin of vitiligo patients is characterized by the infiltration of CXCR3+ NKG2D+ melanocyte-specific resident memory CD8 T cells and recirculating memory T cells producing IFNγ and TNFα (107–112). These type-1 cytokines impair the expression of genes involved in melanocyte adhesion, function, and melanogenesis (113, 114). IFNγ and TNFα also induce melanocyte detachment through the production of MMP9 by keratinocytes, which cleaves E-cadherin, a transmembrane glycoprotein important for melanocyte adhesion (115). In addition, these cytokines amplify the local inflammation through the release of CXCL9/10 by epidermal cells (105, 107). The type-1 inflammation is not restricted to the perilesional skin but concerns also the nonlesional skin of vitiligo patients (116).
Recent data suggest the involvement of a more complex cytokine network in disease pathogenesis with the involvement of type-2 cytokines. Epidemiological studies demonstrated the association of vitiligo with atopic diseases driven by a type-2 immune response, like AD or asthma (117–120). Genome wide association studies (GWAS) identified TSLP gene polymorphism in patients with vitiligo (121). Despite the absence of evidence from GWAS, smaller genetic studies identified polymorphisms of the gene coding for IL-4 as a risk factor for developing vitiligo (122, 123). These polymorphisms correlate with an increase in IL-4 and IgE levels in the serum of vitiligo patients. IL-4 receptor (IL-4R)-α and TSLP gene polymorphisms are associated with an increased susceptibility to vitiligo, reinforcing the putative role of type-2 cytokines in vitiligo (121, 124, 125). In addition, IL-4, IL-13, and IL-33 levels are increased in the serum of vitiligo patients (126–129). An increase in mast cells in vitiligo lesions was reported (130, 131). Czarnowocki et al. reported an increase in both circulating skin-homing CLA+ T cells producing IFNγ or IL-13 in patients. IL-13 levels decreased with vitiligo duration, suggesting its potential role in the early stages of the disease (132). We recently showed that vitiligo skin T cells produce both type-1 and type-2 cytokines, and in particular IL-13 (105). In addition, levels of chemokines that can be associated with a type-2 immune response, such as CCL5, CCL18, CXCL12, or CXCL16, are increased in vitiligo perilesional skin (104, 105, 133). A recent study in a mouse vitiligo model induced by the inoculation of melanoma cells, depletion of regulatory T cells, and excision of the tumor showed that IFNγ induces the secretion of CCL2 and CCL8 by dermal fibroblasts through JAK2/STAT1 signaling, resulting in type-2 cell attraction (134). Indeed, CCL2 is implicated in Th2 polarization and CCL8 in the recruitment of Th2 cells (135, 136). These data suggest the interconnected role of type-1 and type-2 immune responses in the inflammatory environment observed in vitiligo.
So far, the potential impact of type-2-related cytokines on melanocytes has received little attention. IL-4 and IL-13 were reported to inhibit melanogenesis (137, 138). Moreover, IL-13 induces the production of matrix metalloproteinase (MMP)-9 by keratinocytes (115, 139) and may therefore contribute to melanocyte loss in vitiligo together with IFNγ and TNFα. In addition, melanocytes and fibroblasts present a senescence pattern in vitiligo skin (140–142). IFNγ and TNF-α were shown to induce senescence in melanocytes (143, 144), and it would be interesting to evaluate the impact of type-2 cytokines in senescence in vitiligo given that IL-13 can promote senescence in submandibular glands (145). Nonetheless, type-2-related cytokines may also be protective in some subclinical subsets of vitiligo, since dupilumab induced or worsened vitiligo in AD patients (100, 146–148).
Conclusion
Accumulating evidence is underlining the complexity of the cellular and cytokine network involved in the pathogenesis and flares of chronic autoimmune and inflammatory skin diseases. This diversity is likely linked to subclinical phenotypes and associated endotypes, as shown in AD. The role of the type-2 immune response is well characterized in atopic dermatitis and other type-2-related skin diseases. Recent data emphasize its role in other inflammatory skin disorders like vitiligo, AA and LS, which may explain the efficacy of small molecules like JAK inhibitors that target multiple cytokine pathways. In addition, the efficacy of emerging treatments targeting the type-2 response is being investigated, especially the IL-4/IL-13 axis in scleroderma and more recently in AA. The findings may be promising in a clinical subset of patients. Future studies will undoubtedly further decipher the role of the type-2 immune response in these diseases and provide insights into how they are involved in their pathogenesis and how to stratify patients. This may provide much needed guidance on choosing the most appropriate targeted therapy for patients.
Author contributions
LM: Writing – original draft, Writing – review & editing. KB: Writing – original draft, Writing – review & editing. SB: Writing – review & editing. BC: Writing – review & editing. JS: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank R. Cooke for copyediting.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. (2015) 135:626–35. doi: 10.1016/j.jaci.2014.11.001
2. Tuzlak S, Dejean AS, Iannacone M, Quintana FJ, Waisman A, Ginhoux F, et al. Repositioning TH cell polarization from single cytokines to complex help. Nat Immunol. (2021) 22:1210–7. doi: 10.1038/s41590-021-01009-w
3. Stott B, Lavender P, Lehmann S, Pennino D, Durham S, Schmidt-Weber CB. Human IL-31 is induced by IL-4 and promotes TH2-driven inflammation. J Allergy Clin Immunol. (2013) 132:446–454.e5. doi: 10.1016/j.jaci.2013.03.050
4. Zhu J. T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine. (2015) 75:14–24. doi: 10.1016/j.cyto.2015.05.010
5. Beck LA, Cork MJ, Amagai M, De Benedetto A, Kabashima K, Hamilton JD, et al. Type 2 inflammation contributes to skin barrier dysfunction in atopic dermatitis. JID Innov. (2022) 2:100131. doi: 10.1016/j.xjidi.2022.100131
6. Kato A. Group 2 innate lymphoid cells in airway diseases. Chest. (2019) 156:141–9. doi: 10.1016/j.chest.2019.04.101
7. Anthony RM, Rutitzky LI, Urban JF, Stadecker MJ, Gause WC. Protective immune mechanisms in helminth infection. Nat Rev Immunol. (2007) 7:975–87. doi: 10.1038/nri2199
8. Maspero J, Adir Y, Al-Ahmad M, Celis-Preciado CA, Colodenco FD, Giavina-Bianchi P, et al. Type 2 inflammation in asthma and other airway diseases. ERJ Open Res. (2022) 8:00576–2021. doi: 10.1183/23120541.00576-2021
9. Gandhi NA, Bennett BL, Graham NMH, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discovery. (2016) 15:35–50. doi: 10.1038/nrd4624
10. Bieber T. Interleukin-13: Targeting an underestimated cytokine in atopic dermatitis. Allergy. (2020) 75:54–62. doi: 10.1111/all.13954
11. Furue M. Regulation of filaggrin, loricrin, and involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: pathogenic implications in atopic dermatitis. Int J Mol Sci. (2020) 21:5382. doi: 10.3390/ijms21155382
12. Howell MD, Fairchild HR, Kim BE, Bin L, Boguniewicz M, Redzic JS, et al. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J Invest Dermatol. (2008) 128:2248–58. doi: 10.1038/jid.2008.74
13. Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, DeBenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. (2009) 124:R7–12. doi: 10.1016/j.jaci.2009.07.012
14. Kim K, Kim H, Sung GY. An interleukin-4 and interleukin-13 induced atopic dermatitis human skin equivalent model by a skin-on-A-chip. Int J Mol Sci. (2022) 23:2116. doi: 10.3390/ijms23042116
15. Silverberg JI, Guttman-Yassky E, Thaçi D, Irvine AD, Stein Gold L, Blauvelt A, et al. Two phase 3 trials of lebrikizumab for moderate-to-severe atopic dermatitis. N Engl J Med. (2023) 388:1080–91. doi: 10.1056/NEJMoa2206714
16. Koskeridis F, Evangelou E, Ntzani EE, Kostikas K, Tsabouri S. Treatment with dupilumab in patients with atopic dermatitis: systematic review and meta-analysis. J Cutan Med Surg. (2022) 26:613–21. doi: 10.1177/12034754221130969
17. Wollenberg A, Blauvelt A, Guttman-Yassky E, Worm M, Lynde C, Lacour JP, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol. (2021) 184:437–49. doi: 10.1111/bjd.19574
18. Duggan S. Tralokinumab: first approval. Drugs. (2021) 81:1657–63. doi: 10.1007/s40265-021-01583-1
19. Paller AS, Flohr C, Cork M, Bewley A, Blauvelt A, Hong HCH, et al. Efficacy and safety of tralokinumab in adolescents with moderate to severe atopic dermatitis: the phase 3 ECZTRA 6 randomized clinical trial. JAMA Dermatol. (2023) 159:596–605. doi: 10.1001/jamadermatol.2023.0627
20. Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. (2016) 375:2335–48. doi: 10.1056/NEJMoa1610020
21. Simpson EL, Paller AS, Siegfried EC, Boguniewicz M, Sher L, Gooderham MJ, et al. Efficacy and safety of dupilumab in adolescents with uncontrolled moderate to severe atopic dermatitis: A phase 3 randomized clinical trial. JAMA Dermatol. (2020) 156:44–56. doi: 10.1001/jamadermatol.2019.3336
22. Hosseini-Ashrafi M, Clayton TH, Herring M, Herety N, Arkwright PD. Real world outcomes of children treated with dupilumab for moderate-to severe atopic dermatitis: A single centre retrospective observational UK study. Clin Exp Dermatol. (2024) 8:llae013. doi: 10.1093/ced/llae013
23. Müller S, Maintz L, Bieber T. Treatment of atopic dermatitis: Recently approved drugs and advanced clinical development programs. Allergy. (2024) 8. doi: 10.1111/all.16009
24. Simpson EL, Lockshin B, Lee LW, Chen Z, Daoud M, Korotzer A. Real-world effectiveness of dupilumab in adult and adolescent patients with atopic dermatitis: 2-year interim data from the PROSE registry. Dermatol Ther (Heidelb). (2024) 14:261–70. doi: 10.1007/s13555-023-01061-4
25. Pezzolo E, Schena D, Gambardella A, Rossi M, Barei F, Calzavara Pinton P, et al. Survival, efficacy and safety of tralokinumab after 32 and 52 weeks of treatment for moderate-to-severe atopic dermatitis in adults: A multicentre real-world study. J Eur Acad Dermatol Venereol. (2024) 38:e11–3. doi: 10.1111/jdv.19382
26. Ferrucci S, Barei F, Tavecchio S, Marzano AV, Zussino M, Naldi L, et al. Assessment of patient-reported outcomes at 24 weeks of treatment with tralokinumab for atopic dermatitis: a multicentric real-life experience. J Dermatolog Treat. (2023) 34:2285243. doi: 10.1080/09546634.2023.2285243
27. Yosipovitch G, Lio PA, Rosmarin D, Serra-Baldrich E, Legat FJ, Casillas M, et al. Lebrikizumab improved itch and reduced the extent of itch interference on sleep in patients with moderate-to-severe atopic dermatitis: two randomized, placebo-controlled, phase III trials. Br J Dermatol. (2024) 190:289–91. doi: 10.1093/bjd/ljad435
28. Kimura N, Ponda P. Lebrikizumab monotherapy for the treatment of moderate to severe atopic dermatitis. J Allergy Clin Immunol Pract. (2023) 11:2957–60. doi: 10.1016/j.jaip.2023.06.042
29. Bernardo D, Bieber T, Torres T. Lebrikizumab for the treatment of moderate-to-severe atopic dermatitis. Am J Clin Dermatol. (2023) 24:753–64. doi: 10.1007/s40257-023-00793-5
30. Paller AS, Flohr C, Eichenfield LF, Irvine AD, Weisman J, Soung J, et al. Safety and efficacy of lebrikizumab in adolescent patients with moderate-to-severe atopic dermatitis: A 52-week, open-label, phase 3 study. Dermatol Ther (Heidelb). (2023) 13:1517–34. doi: 10.1007/s13555-023-00942-y
31. Czarnowicki T, He H, Canter T, Han J, Lefferdink R, Erickson T, et al. Evolution of pathologic T-cell subsets in patients with atopic dermatitis from infancy to adulthood. J Allergy Clin Immunol. (2020) 145:215–28. doi: 10.1016/j.jaci.2019.09.031
32. Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. (2019) 143:1–11. doi: 10.1016/j.jaci.2018.10.032
33. Bakker DS, Nierkens S, Knol EF, Giovannone B, Delemarre EM, van der Schaft J, et al. Confirmation of multiple endotypes in atopic dermatitis based on serum biomarkers. J Allergy Clin Immunol. (2021) 147:189–98. doi: 10.1016/j.jaci.2020.04.062
34. Wu Y, Gu C, Wang S, Yin H, Qiu Z, Luo Y, et al. Serum biomarker-based endotypes of atopic dermatitis in China and prediction for efficacy of dupilumab. Br J Dermatol. (2023) 188:649–60. doi: 10.1093/bjd/ljad032
35. Suzuki T, Kondo S, Ogura Y, Otsuka M, Tokura Y. How do classical subtypes correspond to endotypes in atopic dermatitis? Int J Mol Sci. (2023) 25:265. doi: 10.3390/ijms25010265
36. Papara C, De Luca DA, Bieber K, Vorobyev A, Ludwig RJ. Morphea: the 2023 update. Front Med (Lausanne). (2023) 10:1108623. doi: 10.3389/fmed.2023.1108623
37. Knobler R, Geroldinger-Simić M, Kreuter A, Hunzelmann N, Moinzadeh P, Rongioletti F, et al. Consensus statement on the diagnosis and treatment of sclerosing diseases of the skin, Part 1: Localized scleroderma, systemic sclerosis and overlap syndromes. J Eur Acad Dermatol Venereol. (2024). doi: 10.1111/jdv.19912
38. Knobler R, Moinzadeh P, Hunzelmann N, Kreuter A, Cozzio A, Mouthon L, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, Part 1: localized scleroderma, systemic sclerosis and overlap syndromes. J Eur Acad Dermatol Venereol. (2017) 31:1401–24. doi: 10.1111/jdv.14458
39. Snarskaya ES, Vasileva KD. Localized scleroderma: actual insights and new biomarkers. Int J Dermatol. (2022) 61:667–74. doi: 10.1111/ijd.15811
40. Grabell D, Hsieh C, Andrew R, Martires K, Kim A, Vasquez R, et al. The role of skin trauma in the distribution of morphea lesions: a cross-sectional survey of the Morphea in Adults and Children cohort IV. J Am Acad Dermatol. (2014) 71:493–8. doi: 10.1016/j.jaad.2014.04.009
41. Jacobe H, Ahn C, Arnett FC, Reveille JD. Major histocompatibility complex class I and class II alleles may confer susceptibility to or protection against morphea: findings from the Morphea in Adults and Children cohort. Arthritis Rheumatol. (2014) 66:3170–7. doi: 10.1002/art.38814
42. Fleischmajer R, Perlish JS, Reeves JR. Cellular infiltrates in scleroderma skin. Arthritis Rheumatol. (1977) 20:975–84. doi: 10.1002/art.1780200410
43. Walker D, Susa JS, Currimbhoy S, Jacobe H. Histopathological changes in morphea and their clinical correlates: Results from the Morphea in Adults and Children Cohort V. J Am Acad Dermatol. (2017) 76:1124–30. doi: 10.1016/j.jaad.2016.12.020
44. Mirizio E, Liu C, Yan Q, Waltermire J, Mandel R, Schollaert KL, et al. Genetic signatures from RNA sequencing of pediatric localized scleroderma skin. Front Pediatr. (2021) 9:669116. doi: 10.3389/fped.2021.669116
45. O’Brien JC, Rainwater YB, Malviya N, Cyrus N, Auer-Hackenberg L, Hynan LS, et al. Transcriptional and cytokine profiles identify CXCL9 as a biomarker of disease activity in morphea. J Invest Dermatol. (2017) 137:1663–70. doi: 10.1016/j.jid.2017.04.008
46. Magee KE, Kelsey CE, Kurzinski KL, Ho J, Mlakar LR, Feghali-Bostwick CA, et al. Interferon-gamma inducible protein-10 as a potential biomarker in localized scleroderma. Arthritis Res Ther. (2013) 15:R188. doi: 10.1186/ar4378
47. Werner G, Sanyal A, Mirizio E, Hutchins T, Tabib T, Lafyatis R, et al. Single-cell transcriptome analysis identifies subclusters with inflammatory fibroblast responses in localized scleroderma. Int J Mol Sci. (2023) 24:9796. doi: 10.3390/ijms24129796
48. Richmond JM, Patel D, Watanabe T, Chen HW, Martyanov V, Werner G, et al. CXCL9 links skin inflammation and fibrosis through CXCR3-dependent upregulation of col1a1 in fibroblasts. J Invest Dermatol. (2023) 143:1138–1146.e12. doi: 10.1016/j.jid.2022.11.025
49. Sartori-Valinotti JC, Tollefson MM, Reed AM. Updates on morphea: role of vascular injury and advances in treatment. Autoimmune Dis. (2013) 2013:467808. doi: 10.1155/2013/467808
50. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. (2016) 12:325–38. doi: 10.1038/nrneph.2016.48
51. Ong CH, Tham CL, Harith HH, Firdaus N, Israf DA. TGF-β-induced fibrosis: A review on the underlying mechanism and potential therapeutic strategies. Eur J Pharmacol. (2021) 911:174510. doi: 10.1016/j.ejphar.2021.174510
52. Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheumatol. (2007) 56:323–33. doi: 10.1002/art.22289
53. Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. (2015) 125:2795–807. doi: 10.1172/JCI77958
54. Ihn H, Sato S, Fujimoto M, Kikuchi K, Takehara K. Demonstration of interleukin-2, interleukin-4 and interleukin-6 in sera from patients with localized scleroderma. Arch Dermatol Res. (1995) 287:193–7. doi: 10.1007/BF01262331
55. Querfeld C, Eckes B, Huerkamp C, Krieg T, Sollberg S. Expression of TGF-beta 1, -beta 2 and -beta 3 in localized and systemic scleroderma. J Dermatol Sci. (1999) 21:13–22. doi: 10.1016/S0923-1811(99)00008-0
56. Hasegawa M, Sato S, Nagaoka T, Fujimoto M, Takehara K. Serum levels of tumor necrosis factor and interleukin-13 are elevated in patients with localized scleroderma. Dermatology. (2003) 207:141–7. doi: 10.1159/000071783
57. Fuschiotti P, Larregina AT, Ho J, Feghali-Bostwick C, Medsger TA. Interleukin-13-producing CD8+ T cells mediate dermal fibrosis in patients with systemic sclerosis. Arthritis Rheumatol. (2013) 65:236–46. doi: 10.1002/art.37706
58. Fertin C, Nicolas JF, Gillery P, Kalis B, Banchereau J, Maquart FX. Interleukin-4 stimulates collagen synthesis by normal and scleroderma fibroblasts in dermal equivalents. Cell Mol Biol. (1991) 37:823–9.
59. Postlethwaite AE, Holness MA, Katai H, Raghow R. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4. J Clin Invest. (1992) 90:1479–85. doi: 10.1172/JCI116015
60. Wegrowski Y, Paltot V, Gillery P, Kalis B, Randoux A, Maquart FX. Stimulation of sulphated glycosaminoglycan and decorin production in adult dermal fibroblasts by recombinant human interleukin-4. Biochem J. (1995) 307:673–8. doi: 10.1042/bj3070673
61. Maeda D, Kubo T, Kiya K, Kawai K, Matsuzaki S, Kobayashi D, et al. Periostin is induced by IL-4/IL-13 in dermal fibroblasts and promotes RhoA/ROCK pathway-mediated TGF-β1 secretion in abnormal scar formation. J Plast Surg Handb Surg. (2019) 53:288–94. doi: 10.1080/2000656X.2019.1612752
62. Oriente A, Fedarko NS, Pacocha SE, Huang SK, Lichtenstein LM, Essayan DM. Interleukin-13 modulates collagen homeostasis in human skin and keloid fibroblasts. J Pharmacol Exp Ther. (2000) 292:988–94.
63. Leonardi A, Cortivo R, Fregona I, Plebani M, Secchi AG, Abatangelo G. Effects of Th2 cytokines on expression of collagen, MMP-1, and TIMP-1 in conjunctival fibroblasts. Invest Ophthalmol Vis Sci. (2003) 44:183–9. doi: 10.1167/iovs.02-0420
64. Gasparini G, Cozzani E, Parodi A. Interleukin-4 and interleukin-13 as possible therapeutic targets in systemic sclerosis. Cytokine. (2020) 125:154799. doi: 10.1016/j.cyto.2019.154799
65. McGaha T, Saito S, Phelps RG, Gordon R, Noben-Trauth N, Paul WE, et al. Lack of skin fibrosis in tight skin (TSK) mice with targeted mutation in the interleukin-4R alpha and transforming growth factor-beta genes. J Invest Dermatol. (2001) 116:136–43. doi: 10.1046/j.1523-1747.2001.00217.x
66. Ong C, Wong C, Roberts CR, Teh HS, Jirik FR. Anti-IL-4 treatment prevents dermal collagen deposition in the tight-skin mouse model of scleroderma. Eur J Immunol. (1998) 28:2619–29. doi: 10.1002/(ISSN)1521-4141
67. Kaviratne M, Hesse M, Leusink M, Cheever AW, Davies SJ, McKerrow JH, et al. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol. (2004) 173:4020–9. doi: 10.4049/jimmunol.173.6.4020
68. Fukuyama M, Ito T, Ohyama M. Alopecia areata: Current understanding of the pathophysiology and update on therapeutic approaches, featuring the Japanese Dermatological Association guidelines. J Dermatol. (2022) 49:19–36. doi: 10.1111/1346-8138.16207
69. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. (2014) 20:1043–9. doi: 10.1038/nm.3645
70. Dai Z, Xing L, Cerise J, Wang EHC, Jabbari A, de Jong A, et al. CXCR3 blockade inhibits T cell migration into the skin and prevents development of alopecia areata. J Immunol. (2016) 197:1089–99. doi: 10.4049/jimmunol.1501798
71. Barahmani N, Schabath MB, Duvic M. National Alopecia Areata Registry. History of atopy or autoimmunity increases risk of alopecia areata. J Am Acad Dermatol. (2009) 61:581–91. doi: 10.1016/j.jaad.2009.04.031
72. Ghaffari J, Rokni GR, Kazeminejad A, Abedi H. Association among thyroid dysfunction, asthma, allergic rhinitis and eczema in children with alopecia areata. Open Access Maced J Med Sci. (2017) 5:305–9. doi: 10.3889/oamjms.2017.050
73. Goh C, Finkel M, Christos PJ, Sinha AA. Profile of 513 patients with alopecia areata: associations of disease subtypes with atopy, autoimmune disease and positive family history. J Eur Acad Dermatol Venereol. (2006) 20:1055–60. doi: 10.1111/j.1468-3083.2006.01676.x
74. Holmes S, Harries M, Macbeth AE, Chiu WS, de Lusignan S, Messenger AG, et al. Alopecia areata and risk of atopic and autoimmune conditions: population-based cohort study. Clin Exp Dermatol. (2023) 48:325–31. doi: 10.1093/ced/llac104
75. Kridin K, Renert-Yuval Y, Guttman-Yassky E, Cohen AD. Alopecia areata is associated with atopic diathesis: results from a population-based study of 51,561 patients. J Allergy Clin Immunol Pract. (2020) 8:1323–8. doi: 10.1016/j.jaip.2020.01.052
76. Betz RC, Pforr J, Flaquer A, Redler S, Hanneken S, Eigelshoven S, et al. Loss-of-function mutations in the filaggrin gene and alopecia areata: strong risk factor for a severe course of disease in patients comorbid for atopic disease. J Invest Dermatol. (2007) 127:2539–43. doi: 10.1038/sj.jid.5700915
77. Kalkan G, Karakus N, Baş Y, Takçı Z, Ozuğuz P, Ateş O, et al. The association between Interleukin (IL)-4 gene intron 3 VNTR polymorphism and alopecia areata (AA) in Turkish population. Gene. (2013) 527:565–9. doi: 10.1016/j.gene.2013.05.086
78. Jagielska D, Redler S, Brockschmidt FF, Herold C, Pasternack SM, Garcia Bartels N, et al. Follow-up study of the first genome-wide association scan in alopecia areata: IL13 and KIAA0350 as susceptibility loci supported with genome-wide significance. J Invest Dermatol. (2012) 132:2192–7. doi: 10.1038/jid.2012.129
79. Bertolini M, Zilio F, Rossi A, Kleditzsch P, Emelianov VE, Gilhar A, et al. Abnormal interactions between perifollicular mast cells and CD8+ T-cells may contribute to the pathogenesis of alopecia areata. PloS One. (2014) 9:e94260. doi: 10.1371/journal.pone.0094260
80. McDiarmid AK, Swoboda PP, Erhayiem B, Ripley DP, Kidambi A, Broadbent DA, et al. Single bolus versus split dose gadolinium administration in extra-cellular volume calculation at 3 Tesla. J Cardiovasc Magn Reson. (2015) 17:6. doi: 10.1186/s12968-015-0112-6
81. Suárez-Fariñas M, Ungar B, Noda S, Shroff A, Mansouri Y, Fuentes-Duculan J, et al. Alopecia areata profiling shows TH1, TH2, and IL-23 cytokine activation without parallel TH17/TH22 skewing. J Allergy Clin Immunol. (2015) 136:1277–87. doi: 10.1016/j.jaci.2015.06.032
82. Fuentes-Duculan J, Gulati N, Bonifacio KM, Kunjravia N, Zheng X, Suárez-Fariñas M, et al. Biomarkers of alopecia areata disease activity and response to corticosteroid treatment. Exp Dermatol. (2016) 25:282–6. doi: 10.1111/exd.12918
83. Song T, Pavel AB, Wen HC, Malik K, Estrada Y, Gonzalez J, et al. An integrated model of alopecia areata biomarkers highlights both TH1 and TH2 upregulation. J Allergy Clin Immunol. (2018) 142:1631–1634.e13. doi: 10.1016/j.jaci.2018.06.029
84. Inui S, Noguchi F, Nakajima T, Itami S. Serum thymus and activation-regulated chemokine as disease activity and response biomarker in alopecia areata. J Dermatol. (2013) 40:881–5. doi: 10.1111/1346-8138.12273
85. Tembhre MK, Sharma VK. T-helper and regulatory T-cell cytokines in the peripheral blood of patients with active alopecia areata. Br J Dermatol. (2013) 169:543–8. doi: 10.1111/bjd.2013.169.issue-3
86. Shohat M, Mimouni D, Ben-Amitai D, Sredni B, Sredni D, Shohat B, et al. In vitro cytokine profile in childhood alopecia areata and the immunomodulatory effects of AS-101. Clin Exp Dermatol. (2005) 30:432–4. doi: 10.1111/j.1365-2230.2005.01817.x
87. Zhang X, Zhao Y, Ye Y, Li S, Qi S, Yang Y, et al. Lesional infiltration of mast cells, Langerhans cells, T cells and local cytokine profiles in alopecia areata. Arch Dermatol Res. (2015) 307:319–31. doi: 10.1007/s00403-015-1539-1
88. Bain KA, McDonald E, Moffat F, Tutino M, Castelino M, Barton A, et al. Alopecia areata is characterized by dysregulation in systemic type 17 and type 2 cytokines, which may contribute to disease-associated psychological morbidity. Br J Dermatol. (2020) 182:130–7. doi: 10.1111/bjd.18008
89. Czarnowicki T, He HY, Wen HC, Hashim PW, Nia JK, Malik K, et al. Alopecia areata is characterized by expansion of circulating Th2/Tc2/Th22, within the skin-homing and systemic T-cell populations. Allergy. (2018) 73:713–23. doi: 10.1111/all.13346
90. Patruno C, Napolitano M, Ferrillo M, Fabbrocini G. Dupilumab and alopecia: A Janus effect. Dermatol Ther. (2019) 32:e13023. doi: 10.1111/dth.13023
91. Ito T, Kageyama R, Nakazawa S, Honda T. Understanding the significance of cytokines and chemokines in the pathogenesis of alopecia areata. Exp Dermatol. (2020) 29:726–32. doi: 10.1111/exd.14129
92. Renert-Yuval Y, Guttman-Yassky E. The changing landscape of alopecia areata: the therapeutic paradigm. Adv Ther. (2017) 34:1594–609. doi: 10.1007/s12325-017-0542-7
93. Fukuyama M, Kinoshita-Ise M, Mizukawa Y, Ohyama M. Two-sided influence of dupilumab on alopecia areata co-existing with severe atopic dermatitis: A case series and literature review. J Cutaneous Imm Allergy. (2023) 6:13–7. doi: 10.1002/cia2.12289
94. Kulkarni M, Rohan CA, Travers JB, Serrao R. Long-term efficacy of dupilumab in alopecia areata. Am J Case Rep. (2022) 23:e936488. doi: 10.12659/AJCR.936488
95. Flanagan K, Sperling L, Lin J. Drug-induced alopecia after dupilumab therapy. JAAD Case Rep. (2019) 5:54–6. doi: 10.1016/j.jdcr.2018.10.010
96. Gallo R, Trave I, Parodi A. Massive acute alopecia of the scalp in a patient treated with dupilumab. Acta Derm Venereol. (2020) 100:adv00191. doi: 10.2340/00015555-3549
97. Marks DH, Mesinkovska N, Senna MM. Cause or cure? Review of dupilumab and alopecia areata. J Am Acad Dermatol. (2023) 88:651–3. doi: 10.1016/j.jaad.2019.06.010
98. McFeely O, Blasco MC, Doyle C, Beatty P, Andrawis M, Murphy L, et al. “I feel like a new woman”: atopic dermatitis and alopecia areata treated successfully by dupilumab. Clin Exp Dermatol. (2023) 48:266–7. doi: 10.1093/ced/llac088
99. Cai L, Wei Y, Zhao M, Zhuo J, Tao X, Lin M. Case report: Dupilumab therapy for alopecia areata in a 4-year-old patient resistant to baricitinib. Front Med (Lausanne). (2023) 10:1253795. doi: 10.3389/fmed.2023.1253795
100. Napolitano M, Fabbrocini G, Patruno C. Dupilumab-associated cutaneous adverse events among adult patients with atopic dermatitis: A retrospective study. J Dermatol. (2023) 50:880–7. doi: 10.1111/1346-8138.16764
101. Guttman-Yassky E, Renert-Yuval Y, Bares J, Chima M, Hawkes JE, Gilleaudeau P, et al. Phase 2a randomized clinical trial of dupilumab (anti-IL-4Rα) for alopecia areata patients. Allergy. (2022) 77:897–906. doi: 10.1111/all.15071
102. Kageyama R, Ito T, Hanai S, Morishita N, Nakazawa S, Fujiyama T, et al. Immunological properties of atopic dermatitis-associated alopecia areata. Int J Mol Sci. (2021) 22:2618. doi: 10.3390/ijms22052618
103. Boniface K, Seneschal J, Picardo M, Taïeb A. Vitiligo: focus on clinical aspects, immunopathogenesis, and therapy. Clin Rev Allergy Immunol. (2018) 54:52–67. doi: 10.1007/s12016-017-8622-7
104. Li S, Zhu G, Yang Y, Jian Z, Guo S, Dai W, et al. Oxidative stress drives CD8+ T-cell skin trafficking in patients with vitiligo through CXCL16 upregulation by activating the unfolded protein response in keratinocytes. J Allergy Clin Immunol. (2017) 140:177–189.e9. doi: 10.1016/j.jaci.2016.10.013
105. Martins C, Migayron L, Drullion C, Jacquemin C, Lucchese F, Rambert J, et al. Vitiligo skin T cells are prone to produce type 1 and type 2 cytokines to induce melanocyte dysfunction and epidermal inflammatory response through jak signaling. J Invest Dermatol. (2022) 142:1194–205. doi: 10.1016/j.jid.2021.09.015
106. Chen J, Li S, Li C. Mechanisms of melanocyte death in vitiligo. Med Res Rev. (2021) 41:1138–66. doi: 10.1002/med.21754
107. Boniface K, Jacquemin C, Darrigade AS, Dessarthe B, Martins C, Boukhedouni N, et al. Vitiligo skin is imprinted with resident memory CD8 T cells expressing CXCR3. J Invest Dermatol. (2018) 138:355–64. doi: 10.1016/j.jid.2017.08.038
108. Jacquemin C, Martins C, Lucchese F, Thiolat D, Taieb A, Seneschal J, et al. NKG2D defines a subset of skin effector memory CD8 T cells with proinflammatory functions in vitiligo. J Invest Dermatol. (2020) 140:1143–1153.e5. doi: 10.1016/j.jid.2019.11.013
109. Cheuk S, Schlums H, Gallais Sérézal I, Martini E, Chiang SC, Marquardt N, et al. CD49a expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity. (2017) 46:287–300. doi: 10.1016/j.immuni.2017.01.009
110. Richmond JM, Strassner JP, Rashighi M, Agarwal P, Garg M, Essien KI, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol. (2019) 139:769–78. doi: 10.1016/j.jid.2018.10.032
111. Wu J, Zhou M, Wan Y, Xu A. CD8+ T cells from vitiligo perilesional margins induce autologous melanocyte apoptosis. Mol Med Rep. (2013) 7:237–41. doi: 10.3892/mmr.2012.1117
112. van den Boorn JG, Konijnenberg D, Dellemijn TAM, van der Veen JPW, Bos JD, Melief CJM, et al. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol. (2009) 129:2220–32. doi: 10.1038/jid.2009.32
113. Yang L, Wei Y, Sun Y, Shi W, Yang J, Zhu L, et al. Interferon-gamma inhibits melanogenesis and induces apoptosis in melanocytes: A pivotal role of CD8+ Cytotoxic T lymphocytes in vitiligo. Acta Derm Venereol. (2015) 95:664–70. doi: 10.2340/00015555-2080
114. Webb KC, Tung R, Winterfield LS, Gottlieb AB, Eby JM, Henning SW, et al. Tumour necrosis factor-α inhibition can stabilize disease in progressive vitiligo. Br J Dermatol. (2015) 173:641–50. doi: 10.1111/bjd.14016
115. Boukhedouni N, Martins C, Darrigade AS, Drullion C, Rambert J, Barrault C, et al. Type-1 cytokines regulate MMP-9 production and E-cadherin disruption to promote melanocyte loss in vitiligo. JCI Insight. (2020) 5:e133772. doi: 10.1172/jci.insight.133772
116. Migayron L, Merhi R, Seneschal J, Boniface K. Resident memory T cells in nonlesional skin and healed lesions of patients with chronic inflammatory diseases: Appearances can be deceptive. J Allergy Clin Immunol. (2024) 153:606–14. doi: 10.1016/j.jaci.2023.11.017
117. Silverberg JI, Silverberg NB. Association between vitiligo and atopic disorders: a pilot study. JAMA Dermatol. (2013) 149:983–6. doi: 10.1001/jamadermatol.2013.4228
118. de Lusignan S, Alexander H, Broderick C, Dennis J, McGovern A, Feeney C, et al. Atopic dermatitis and risk of autoimmune conditions: Population-based cohort study. J Allergy Clin Immunol. (2022) 150:709–13. doi: 10.1016/j.jaci.2022.03.030
119. Acharya P, Mathur M. Association of atopic dermatitis with vitiligo: A systematic review and meta-analysis. J Cosmet Dermatol. (2020) 19:2016–20. doi: 10.1111/jocd.13263
120. Roh YS, Huang AH, Sutaria N, Choi U, Wongvibulsin S, Choi J, et al. Real-world comorbidities of atopic dermatitis in the US adult ambulatory population. J Am Acad Dermatol. (2022) 86:835–45. doi: 10.1016/j.jaad.2021.11.014
121. Birlea SA, Jin Y, Bennett DC, Herbstman DM, Wallace MR, McCormack WT, et al. Comprehensive association analysis of candidate genes for generalized vitiligo supports XBP1, FOXP3, and TSLP. J Invest Dermatol. (2011) 131:371–81. doi: 10.1038/jid.2010.337
122. Pehlivan S, Ozkinay F, Alper S, Onay H, Yuksel E, Pehlivan M, et al. Association between IL4 (-590), ACE (I)/(D), CCR5 (Delta32), CTLA4 (+49) and IL1-RN (VNTR in intron 2) gene polymorphisms and vitiligo. Eur J Dermatol. (2009) 19:126–8. doi: 10.1684/ejd.2008.0578
123. Imran M, Laddha NC, Dwivedi M, Mansuri MS, Singh J, Rani R, et al. Interleukin-4 genetic variants correlate with its transcript and protein levels in patients with vitiligo. Br J Dermatol. (2012) 167:314–23. doi: 10.1111/bjd.2012.167.issue-2
124. Al-Shobaili H, Settin A, Alzolibani A, Al Robaee A, Salem T, Al-Saif F, et al. Interleukin-4 (-590 C>T) and interleukin-4 receptor (Q551R A>G) gene polymorphisms in Saudi patients with vitiligo. Eur J Dermatol. (2013) 23:402–4. doi: 10.1684/ejd.2013.2009
125. Cheong KA, Chae SC, Kim YS, Kwon HB, Chung HT, Lee AY. Association of thymic stromal lymphopoietin gene -847C>T polymorphism in generalized vitiligo. Exp Dermatol. (2009) 18:1073–5. doi: 10.1111/j.1600-0625.2009.00897.x
126. Khan R, Gupta S, Sharma A. Circulatory levels of T-cell cytokines (interleukin [IL]-2, IL-4, IL-17, and transforming growth factor-β) in patients with vitiligo. J Am Acad Dermatol. (2012) 66:510–1. doi: 10.1016/j.jaad.2011.07.018
127. Vaccaro M, Cicero F, Mannucci C, Calapai G, Spatari G, Barbuzza O, et al. IL-33 circulating serum levels are increased in patients with non-segmental generalized vitiligo. Arch Dermatol Res. (2016) 308:527–30. doi: 10.1007/s00403-016-1675-2
128. Tembhre MK, Sharma VK, Sharma A, Chattopadhyay P, Gupta S. T helper and regulatory T cell cytokine profile in active, stable and narrow band ultraviolet B treated generalized vitiligo. Clin Chim Acta. (2013) 424:27–32. doi: 10.1016/j.cca.2013.05.005
129. Tawfik N, Abd Elhamid M, Omar H, Gomaa AHA. Assessment of serum interleukin-13 level in vitiligo patients and its correlation to disease severity. Egypt J Dermatol Venerol. (2023) 43:139. doi: 10.4103/ejdv.ejdv_46_22
130. Luo L, Zhu J, Guo Y, Li C. Mitophagy and immune infiltration in vitiligo: evidence from bioinformatics analysis. Front Immunol. (2023) 14:1164124. doi: 10.3389/fimmu.2023.1164124
131. Katayama I, Yang L, Takahashi A, Yang F, Wataya-Kaneda M. The two faces of mast cells in vitiligo pathogenesis. Explor Immunol. (2021) 1:269–84. doi: 10.37349/ei
132. Czarnowicki T, He H, Leonard A, Kim HJ, Kameyama N, Pavel AB, et al. Blood endotyping distinguishes the profile of vitiligo from that of other inflammatory and autoimmune skin diseases. J Allergy Clin Immunol. (2019) 143:2095–107. doi: 10.1016/j.jaci.2018.11.031
133. Rezk AF, Kemp DM, El-Domyati M, El-Din WH, Lee JB, Uitto J, et al. Misbalanced CXCL12 and CCL5 chemotactic signals in vitiligo onset and progression. J Invest Dermatol. (2017) 137:1126–34. doi: 10.1016/j.jid.2016.12.028
134. Jin R, Zhou M, Lin F, Xu W, Xu A. Pathogenic th2 cytokine profile skewing by IFN-γ-responding vitiligo fibroblasts via CCL2/CCL8. Cells. (2023) 12:217. doi: 10.3390/cells12020217
135. Gu L, Tseng S, Horner RM, Tam C, Loda M, Rollins BJ. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature. (2000) 404:407–11. doi: 10.1038/35006097
136. Islam SA, Chang DS, Colvin RA, Byrne MH, McCully ML, Moser B, et al. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat Immunol. (2011) 12:167–77. doi: 10.1038/ni.1984
137. Han J, Lee E, Kim E, Yeom MH, Kwon O, Yoon TH, et al. Role of epidermal γδ T-cell-derived interleukin 13 in the skin-whitening effect of Ginsenoside F1. Exp Dermatol. (2014) 23:860–2. doi: 10.1111/exd.12531
138. Choi H, Choi H, Han J, Jin SH, Park JY, Shin DW, et al. IL-4 inhibits the melanogenesis of normal human melanocytes through the JAK2-STAT6 signaling pathway. J Invest Dermatol. (2013) 133:528–36. doi: 10.1038/jid.2012.331
139. Purwar R, Kraus M, Werfel T, Wittmann M. Modulation of keratinocyte-derived MMP-9 by IL-13: a possible role for the pathogenesis of epidermal inflammation. J Invest Dermatol. (2008) 128:59–66. doi: 10.1038/sj.jid.5700940
140. Rani S, Bhardwaj S, Srivastava N, Sharma VL, Parsad D, Kumar R. Senescence in the lesional fibroblasts of non-segmental vitiligo patients. Arch Dermatol Res. (2017) 309:123–32. doi: 10.1007/s00403-016-1713-0
141. Kovacs D, Bastonini E, Ottaviani M, Cota C, Migliano E, Dell’Anna ML, et al. Vitiligo skin: exploring the dermal compartment. J Invest Dermatol. (2018) 138:394–404. doi: 10.1016/j.jid.2017.06.033
142. Lee JW, Kim TH, Park TJ, Kang HY. p16ink4a positivity of melanocytes in non-segmental vitiligo. Diagnostics (Basel). (2020) 10:878. doi: 10.3390/diagnostics10110878
143. Wang S, Zhou M, Lin F, Liu D, Hong W, Lu L, et al. Interferon-γ induces senescence in normal human melanocytes. PloS One. (2014) 9:e93232. doi: 10.1371/journal.pone.0093232
144. Dong BQ, Liao ZK, Le Y, Jiang S, Luo LF, Miao F, et al. Acceleration of melanocyte senescence by the proinflammatory cytokines IFNγ and TNFα impairs the repigmentation response of vitiligo patients to narrowband ultraviolet B (NBUVB) phototherapy. Mech Ageing Dev. (2023) 211:111779. doi: 10.1016/j.mad.2023.111779
145. Zhu M, Min S, Mao X, Zhou Y, Zhang Y, Li W, et al. Interleukin-13 promotes cellular senescence through inducing mitochondrial dysfunction in IgG4-related sialadenitis. Int J Oral Sci. (2022) 14:29. doi: 10.1038/s41368-022-00180-6
146. Ren H, Akabane AL, Kelleher K, Halverstam C, Hicks M, Schachter JR, et al. Vitiligo induced by dupilumab treatment: A case series. J Eur Acad Dermatol Venereol. (2023) 37(11):2259–61. doi: 10.1111/jdv.19132
147. Takeoka S, Kamata M, Yokoi I, Takehara A, Tada Y. Rapid enlargement of vitiligo vulgaris after initiation of dupilumab for atopic dermatitis: A case report. Acta Derm Venereol. (2021) 101:adv00581. doi: 10.2340/actadv.v101.545
Keywords: type-1 immunity, type-2 immunity, atopic dermatitis, localized scleroderma, alopecia areata, vitiligo
Citation: Migayron L, Bordes S, Closs B, Seneschal J and Boniface K (2024) Type-2 immunity associated with type-1 related skin inflammatory diseases: friend or foe? Front. Immunol. 15:1405215. doi: 10.3389/fimmu.2024.1405215
Received: 22 March 2024; Accepted: 14 May 2024;
Published: 29 May 2024.
Edited by:
Mark Mellett, University Hospital Zürich, SwitzerlandReviewed by:
Kazufumi Kunimura, Kyushu University, JapanRosalie Luiten, Academic Medical Center, Netherlands
Copyright © 2024 Migayron, Bordes, Closs, Seneschal and Boniface. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katia Boniface, a2F0aWEuYm9uaWZhY2VAdS1ib3JkZWF1eC5mcg==
†ORCID: Laure Migayron, orcid.org/0000-0003-2003-1923
Julien Seneschal, orcid.org/0000-0003-1139-0908
Katia Boniface, orcid.org/0000-0003-4428-1222