 Wenxia Qi1
Wenxia Qi1 Jiexiang Tian2Gang Wang2*Yanfeng Yan3Tao Wang2Yong Wei1,2Zhandong Wang1Guohua Zhang1Yuanyuan Zhang1,2Jia Wang1
Jiexiang Tian2Gang Wang2*Yanfeng Yan3Tao Wang2Yong Wei1,2Zhandong Wang1Guohua Zhang1Yuanyuan Zhang1,2Jia Wang1- 1Gansu University of Traditional Chinese Medicine, College of Integrative Medicine, Lanzhou, China
- 2Affiliated Hospital of Gansu University of Traditional Chinese Medicine, Department of Rheumatology and Orthopedics, Lanzhou, China
- 3Fourth Affiliated Hospital of Gansu University of Traditional Chinese Medicine, Department of Respiratory and Critical Care Medicine, Lanzhou, China
Sjögren’s Syndrome (SS) is an autoimmune disorder characterized by dysfunction of exocrine glands. Primarily affected are the salivary glands, which exhibit the most frequent pathological changes. The pathogenesis involves susceptibility genes, non-genetic factors such as infections, immune cells-including T and B cells, macrophage, dendritic cells, and salivary gland epithelial cells. Inflammatory mediators such as autoantibodies, cytokines, and chemokines also play a critical role. Key signaling pathways activated include IFN, TLR, BAFF/BAFF-R, PI3K/Akt/mTOR, among others. Comprehensive understanding of these mechanisms is crucial for developing targeted therapeutic interventions. Thus, this study explores the cellular and molecular mechanisms underlying SS-related salivary gland damage, aiming to propose novel targeted therapeutic approaches.
Introduction
SS is a prevalent autoimmune disorder primarily characterized by lymphocytic infiltration of exocrine glands, leading to dryness in the mouth and eyes (1). Additionally, about 30–40% of patients experience systemic complications affecting the kidneys, lungs, nervous system, and other organs (2–4). The primary target organs in SS are the exocrine glands, particularly the salivary glands (SG). The reduction in saliva production is closely linked to continuous immune cells infiltration around the salivary gland epithelial cells (SGECs) and progressive destruction of glandular structures. SG histopathological changes are predominantly marked by extensive lymphocytic infiltration into the glandular interstitium, significant SGECs destruction and atrophy, and alterations in the glandular ducts, including dilatation and narrowing, culminating in SG damage (5, 6). This chronic inflammation eventually leads to irreversible fibrosis in SGECs, characterized by an accumulation of excessive connective tissue and extracellular matrix, ultimately resulting in the loss of SG function (7). The pathogenesis of SS is intricately linked to immune system abnormalities, which involve the secretion of pro-inflammatory mediators through both innate and adaptive immune responses. Various pathological mechanisms include activation of the type I interferon (IFN) system, antigen presentation, and T and B lymphocyte activation, along with the formation of ectopic lymphoid structures (ELS) (8). Viral and genetic factors initiate the immune response, leading to lymphocytic infiltration in the SG and the release of numerous pro-inflammatory mediators, such as interleukins (IL), IFN, and members of the tumor necrosis factor (TNF) super family, which further damage the SG (9). Additionally, SGECs play a critical role in local immune responses and disease progression in SG, primarily through their secretion of cytokines and chemokines that activate and recruit T and B cells, as well as circulating peripheral immune cells (10). Inflammatory cytokines can activate specific pathways within SGECs by binding to lymphocytes and surface receptors on SGECs, further promoting the production of inflammatory factors and chemokines, thereby exacerbating the inflammatory response (11). A thorough understanding of the cellular and molecular mechanisms underlying SG damage in SS and strategies to maximize the preservation of SG secretory function are crucial for enhancing the quality of life for patients. Details can be found in Table 1 and Figure 1.
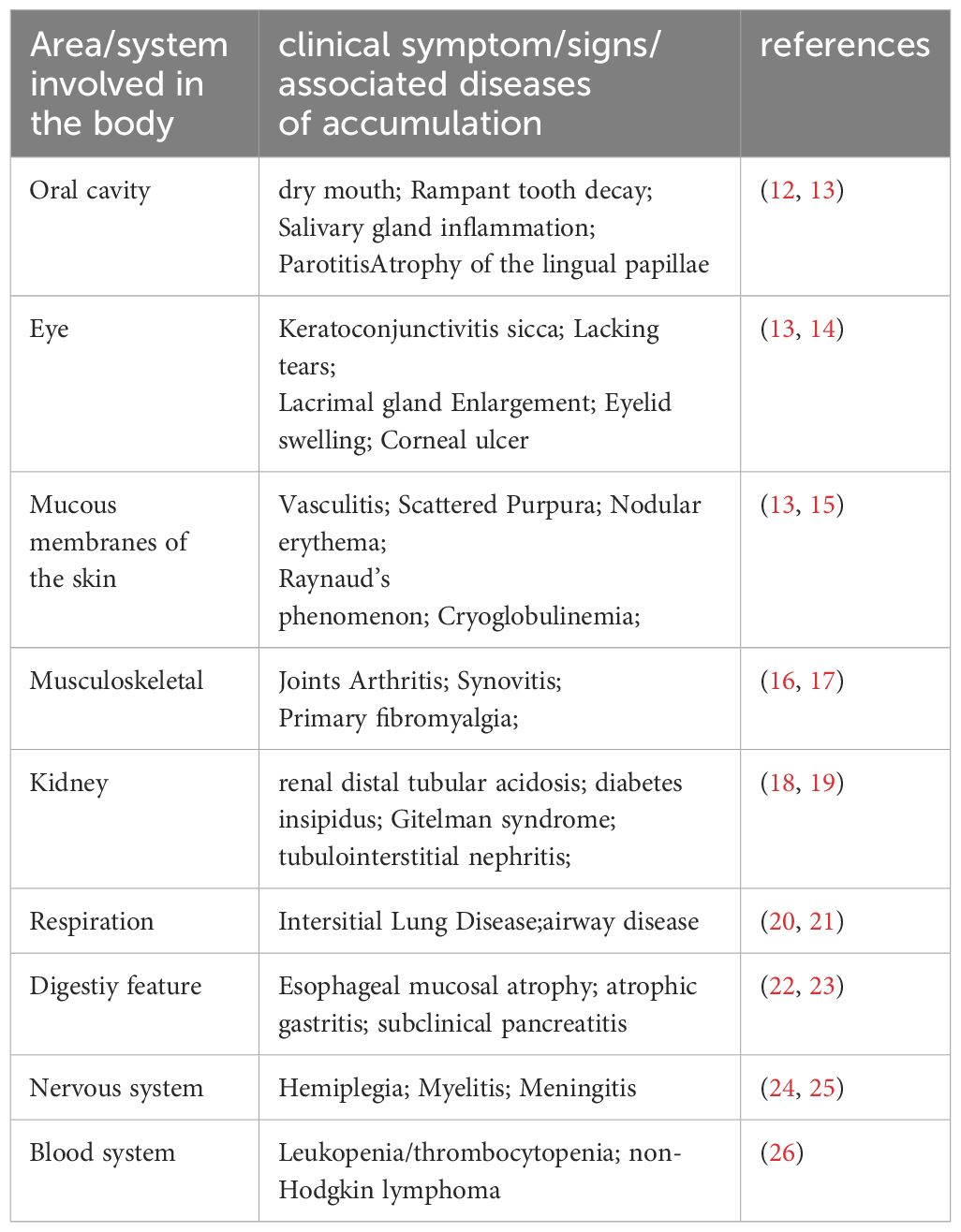
Table 1 Common clinical symptoms, signs, and related diseases involved in SS.
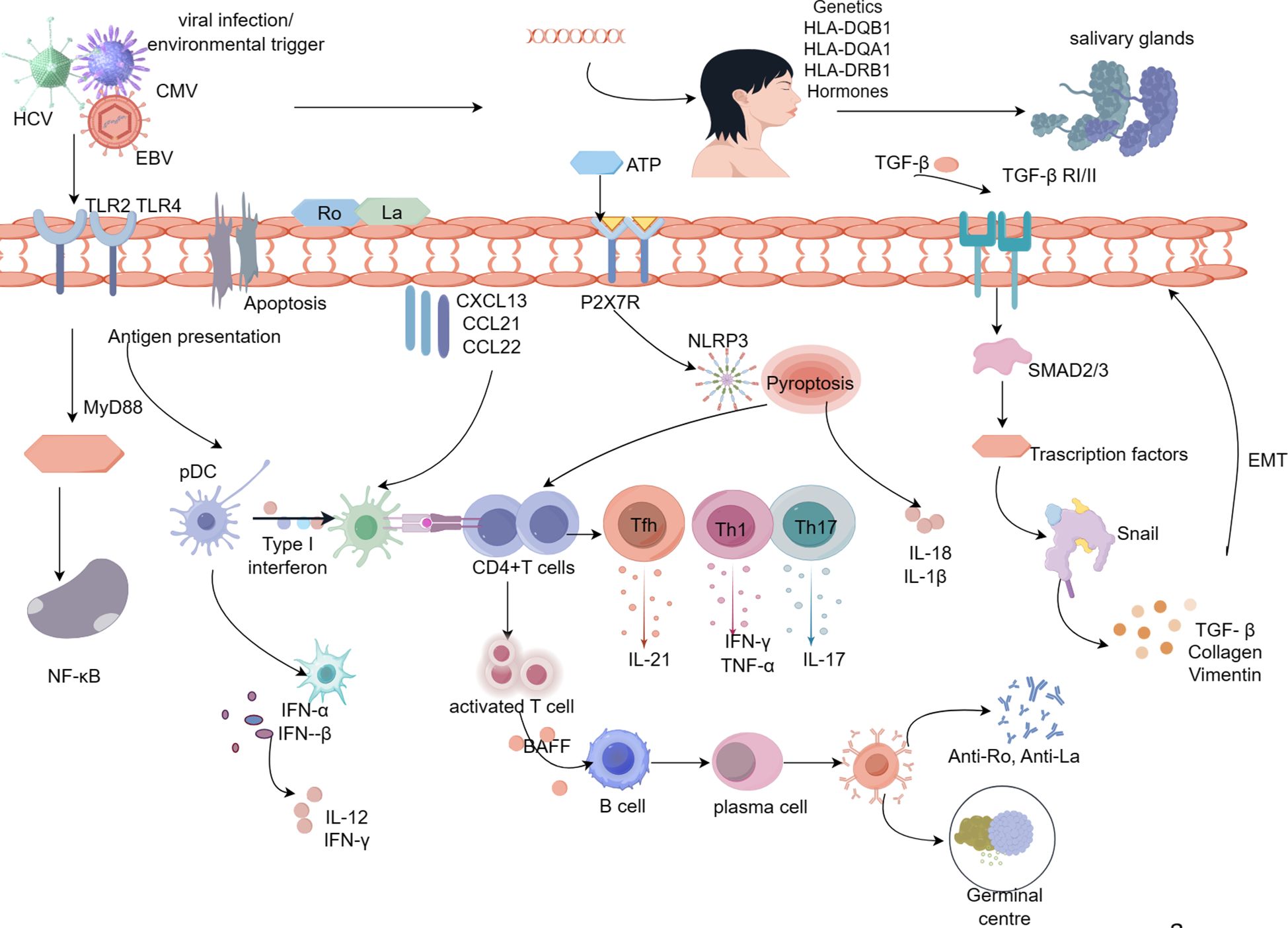
Figure 1 Pathologic mechanisms of salivary gland damage in Sjögren’s syndrome. Viral infection, hormonal imbalance, genetic susceptibility, and apoptosis lead to activation of SGECs, resulting in the release of the intracellular antigens Ro/La onto the surface of SGECs. Ro and La proteins activate immune cells on the extramembrane surface via TLR, and CD4+ T lymphocytes are activated via MHC-II molecules, which are expressed on the surface of SGECs.Activated CD4+T cells differentiate into Th17, Th1, and Tfh cells in response to inflammatory cytokines.SGECs interact with CD4+ T cells through inflammatory cytokines, repressors, leading to the production of pro-inflammatory cytokines (IL-21, IL-17, IFN-γ, TNF-α).With the involvement of BAFF, CD4+ T cells activate B lymphocytes, ultimately leading to the formation of autoantibodies and germinal centers. Abnormal upregulation of TGF-β1 in SGECs leads to morphological and functional mesenchymal changes in SGECs through activation of the TGF-β1/SMAD/Snail signaling pathway, which contributes to the process of SG fibrosis. Prolonged inflammatory stimulation results in a marked elevation of ATP, which activates P2X7R, and activated P2X7R allows NLRP3 to enter the cytoplasm thereby activating the NLRP3 inflammatory vesicles. activation of NLRP3 generates activated Caspase-1, which in turn cleaves the GSDMD, which punches holes in the cytosol at the N-terminal end, releasing mature IL-1β, IL-18, and thus activating the initial T cells, the differentiate into Th1 and Th17, which further secrete inflammatory cytokines.
Structural changes in salivary gland pathology in SS
In the human body, the SG encompasses the parotid, submandibular, and sublingual glands, along with numerous minor glands distributed throughout the oral mucosa. The structural composition of the SG primarily includes SGECs, immune cells, and stromal cells. The alveolar and ductal cells, which are key constituents of the SGECs, play vital roles in the gland. Salivary secretion is facilitated by the contraction of myoepithelial cells and is further regulated by parasympathetic neurotransmitters such as muscarinic agents. The secreted saliva initially travels through the intercalary ducts from smaller secretory ducts and ultimately reaches the oral cavity via larger excretory ducts (27, 28). Characteristic pathological features of SG in SS patients include extensive infiltration by T and B lymphocytes around the ducts, accompanied by atrophy and a decrease in vesicular cells (29). During the early and middle stages of the disease, there is an increase in foci infiltrated by immune cells, particularly B lymphocytes, around the ductal cells, leading to the formation of lymphoid tissue germinal centers (GC) within the SG and subsequent autoantibody production. Chronic inflammatory stimulation and injury result in a marked decline in follicular and ductal cells populations, which are gradually replaced by fibrous and adipose tissue (30, 31). Electron microscopy has revealed degeneration and atrophy of SGECs, fatty infiltration within the cytoplasm, and swelling of intracellular organelles—pathological alterations that significantly diminish the secretory function of the SG (32). SGECs fibrosis is now acknowledged as a significant pathological hallmark of SS (33). Moreover, lymphoepithelial lesions, characterized by ductal basal cells and infiltrating lymphocytes predominantly surrounded by B cells, are also notable. These lesions arise when basal cells, spared from lymphocyte attacks, proliferate, leading to lymphoepithelial hyperplasia and ultimately the development of lymphoepithelial lesion (34).
Genetic and environmental nuclear factors
Research has demonstrated that genetic factors are a significant risk factor for the development of SS. Two major genome-wide association studies (GWAS) conducted in 2013 identified risk genes in Han Chinese and European populations, revealing that many of these genes are involved in both innate and adaptive immune responses (35, 36). These studies highlighted a strong correlation between human leukocyte antigen (HLA) loci and SS, particularly noting a higher susceptibility associated with allelic variants such as HLA-DQB1*0201, HLA-DQA1*0501, and HLA-DQB1*0301 (DR3) (36). In the Chinese Han population, notable susceptibility loci include TNFAIP3 and GTF2I, which are largely related to HLA genes and the type I interferon (IFN) pathway (37, 38). While the number of GWAS focusing on SS is growing, the discovery of genetic variants with genome-wide significance remains limited compared to other autoimmune diseases (39). Epigenetics, which influences gene expression without altering the DNA sequence, primarily involves histone modification, non-coding RNA (ncRNA) activity, and DNA methylation. ncRNAs, which do not encode proteins, can form complex regulatory networks and play critical roles in bodily regulation. Abnormal expressions and functions of microRNA (miRNA), such as miR-146a, miR-155, and miR-21, can disrupt immune system functions and influence the initiation and progression of SS. Notably, miRNAs expressed in SS patients, like miR-146a/b and miR-30b-5p, may negatively regulate the B-cells activating factor (BAFF), crucial for B-cells maturation, survival, and proliferation, and pivotal in triggering SS (40). DNA methylation is also critical in SS; one study identified distinct methylation sites and regions in the whole blood of SS patients compared to healthy individuals, particularly within genes regulating type I IFN (41). Viral infections are central to the pathogenesis of SS and are considered potential triggers for initiating the autoimmune process. Viruses activate antigen-presenting cells (APCs) and autoreactive naïve T cells (42). SG serve as key sites for latent viral infections, making viral presence a major environmental factor in initiating innate immunity (43). The primary viruses implicated in SS include Epstein-Barr virus (EBV) (44), human herpesvirus (HHV) (45), human T-lymphotropic virus type 1 (HTLV), chronic hepatitis C virus (HCV) (46), and cytomegalovirus (CMV) (47). EBV is particularly significant in SS pathogenesis, capable of infecting SGECs by direct fusion with the plasma membrane or infecting B cells via the gP350/220 protein. It evades immune detection through strategies like molecular mimicry and can persist long-term within the host (48). Furthermore, EBV can exacerbate the autoimmune response by prompting the immune system to attack self-antigens through epitope spreading and bystander activation (49).
Common cells types and pathologic mechanisms in SS
T cells
Th1 and Th2 cells
Numerous studies have demonstrated that the immune response in SG affected by SS is predominantly mediated by Type 1 T helper cells (Th1) (50, 51). Elevated levels of Th1-related cytokines such as IFN-γ, TNF-α, and C-X-C motif chemokine receptor 3 (CXCR3) have been observed in the saliva of SS patients compared to those with normal SG function (52, 53), suggesting that increased lymphocyte infiltration is associated with an upregulation of Th1 cytokines. Specifically, IFN-γ plays a crucial role in SG dysfunction in the SS Ro60 humanized model (54). This cytokine is believed to contribute to SG dysfunction by disrupting tight junction structures within SS patients’ SG (55). Similarly, TNF-α, another pivotal Th1 cytokine, shows significantly higher expression in the saliva and SG of SS patients (56). Like IFN-γ, increased TNF-α levels compromise the integrity of tight junctions in salivary gland epithelial cells (SGECs) and diminish the population of vesicular cells, thereby impairing the SG’s secretory function (57). Amphiregulin (AREG), a critical growth factor, plays a vital role in the secretion of pro-inflammatory cytokines within the SG of SS patients. TNF-α has been shown to induce AREG secretion, which leads to damage in SG follicular and ductal cells (58). Additionally, TNF-α can trigger apoptosis and enhance the transcriptional expression of intercellular adhesion molecule-1 (ICAM-1) and macrophage inflammatory protein-3 (CCL20) in SGECs (59). Th2 cells, on the other hand, facilitate B cells responses through their cytokines (60). Th2 molecules have been identified in the intra-GC regions of SS patients, indicating that Th2 cells may play a role in regulating the initial B cells response (61).
Th17 and Treg cells
Th17 cells are a subtype of pro-inflammatory CD4+T cells that play a crucial role in initiating and advancing SS (62). Regulatory T cells (Treg), on the other hand, serve an inhibitory function by releasing soluble mediators or through direct cells-to-cells contact, thus maintaining the dynamic equilibrium of human immunity (63). Th17 cells produce a range of cytokines (IL-17, IL-17A, TNF-α), which collectively mediate the inflammatory response in the SG (64). Research has shown that the balance between Treg and TGF-β levels in the SG of early-stage SS model mice is significantly disrupted compared to normal controls due to the overactivation of Th17 and IL-17 cells (65). In SS patients, the expression of the transcriptional coactivator with PDZ-binding motif (TAZ) is markedly elevated (66). TAZ is known to promote Th17 differentiation and inhibit Treg development, leading to a disruption in immune homeostasis, which supports the involvement of Th17/Treg imbalance in SG damage. Clinical studies have confirmed that Th17 cells are significantly increased in the SG of SS patients compared to healthy individuals (67) and are positively correlated with the severity of the pathology (68). IL-17 induces the secretion of pro-inflammatory factors (TNF-α, IL-6, and IL-1β) and attracts immune cells to the SG by promoting the release of IL-8, C-X-C motif chemokine ligand 9, and C-C chemokine receptor type 3 (69). Furthermore, elevated IL-17A expression in the SG of SS patients has been linked to increased disease severity (70). Another Th17 cytokine, IL-22, also contributes to SG pathology. Firstly, IL-22 recruits B cells and lymphoid aggregates, facilitating the formation of ectopic lymphoid structures (ELS). Secondly, IL-22 enhances autoantibody production by stimulating the expression of cytokines such as CXCL12 and CXCL13 (71).
Tfh and Thr cells
Follicular helper T cells (Tfh) are a subset of CD4+T cells that enhance B cells differentiation and antibody production by expressing the chemokine receptor CXCR5, which interacts with the chemokine ligand CXCL13 on the surface of B lymphocytes in the germinal center (72). Elevated levels of CXCL13 have been detected in the SG of patients with SS (73). Some studies have linked increased serum concentrations of CXCL13 with abnormal B cells parameters and even lymphomagenesis (74). Tfh cells are abundantly present in the SG of SS patients and are positively correlated with disease activity (75). Moreover, Tfh cells are closely associated with the formation of ELS, and increased numbers of activated Tfh cells have been observed in the SG during the development of ectopic GC (61). IL-21 is a key effector cytokine of Tfh cells (76). Devangi and colleagues observed that IL-21 induces apoptosis in B cells and also inhibits growth and induces apoptosis in lymphoma cells, suggesting that IL-21 may also promote apoptosis in SGECs (77). Follicular regulatory T cells (Tfr), derived from FOXP3+ Treg cells, represent a distinct class of regulatory T cells. Tfr cells inhibit the function of Tfh and B cells within germinal centers, primarily by negatively regulating Tfh cells, thereby reducing or inhibiting autoantibody production (78). The balance between Tfh and Tfr cells is crucial for maintaining normal immune function. Research using a mouse model of Sjögren’s syndrome has shown that a reduction in Tfr cells leads to increased lymphocyte infiltration and antibody deposition in the SG (79).
CD8+T cells
CD8+T cells, also known as cytotoxic T lymphocytes (CTL), are a critical subpopulation of adaptive immune cells that play a pivotal role in the elimination of intracellular pathogens. CTL are activated, proliferate, differentiate, and become effective primarily through the recognition of antigenic peptide-MHC class I molecule complexes presented by target cells via T-cells receptors (80). These cells are characterized by their ability to use perforin and MHC class I molecule complexes. CTL can kill target cells by producing IFN-γ and exhibiting cytolytic activity through perforin/granzyme or Fas cells surface death receptor (Fas) signaling pathways (81). In autoimmune diseases, the destruction of target cells by autoreactive CTL can lead to the release of large amounts of autoantigens, which may trigger the overproduction of autoantibodies. Overactive or abnormal proliferation of CTL has been observed in the SG of patients with SS (82, 83). Immunofluorescence experiments have demonstrated an increase in tissue-resident CTL within the SGECs of SS patients. CTL contribute to SG injury through multiple biological pathways. Firstly, activated CTL produce high levels of IFN-γ and TNF-α, inducing inflammatory responses in the SG (84). Secondly, IFN-γ from CTL can compromise the integrity and functionality of SGECs tight junctions, resulting in SGECs damage (85).
B cells
In SS, the infiltrating cells in the SG are predominantly CD4+T cells and B cells. Initially, the infiltrating cells are mainly CD4+ T cells (86). Activated T cells stimulate B cells activation by producing pro-inflammatory cytokines, establishing a positive feedback loop where excessive B cells activation plays a central role in the pathogenesis of SS (87, 88). B cells contribute to the pathological process in SS by producing autoantibodies such as anti-antinuclear antibody (ANA), anti-Sjögren’s syndrome type A (anti-SSA), and anti-Sjögren’s syndrome type B (anti-SSB) antibodies (89, 90). BAFF, a member of the tumor necrosis factor superfamily, is crucial in influencing the proliferation, maturation, and survival of B cells. Produced by SG SGECs, APCs, and activated T and B cells, BAFF is a key pathogenic factor in SS. The interaction between BAFF and its receptor (BAFF-R) enhances B cells activation and proliferation, and its aberrant expression can lead to B cells dysfunction and disrupt immune homeostasis (91). Elevated BAFF levels in the SG of SS patients promote B cells maturation and proliferation, resulting in autoantibody production (88). Additionally, BAFF mediates the survival of B lymphocytes by activating the NF-κB pathway, which induces the production of B-cells lymphoma-2 (Bcl-2). Members of the Bcl-2 family can regulate SGECs apoptosis, thus supporting the survival of autoreactive B cells and the production of autoantibodies (92). Elevated BAFF levels in the serum and SG of SS patients have been found to correlate positively with autoantibody (anti-SSA/SSB) levels (93, 94). Tripartite Motif-containing 21 (TRIM21), a cytoplasmic Fc receptor, interacts with Ro60 to form Sjögren’s syndrome antigen A (SSA). TRIM21 expression enhances the binding of anti-SSA antibodies to SSA-reactive B cells, thereby driving B cells activation and anti-SSA antibody production.
Current studies have identified several novel B cells subpopulations that exert pathologic mechanisms in SS. Notable among these are abnormal B-cells subsets, such as circulating naïve B-cells and plasma cells, which are present in the SG of SS patients and positively correlate with anti-ANA antibody titers (95). T-box transcription factor TBX21(T-bet), predominantly expressed in CD4+ and CD8+T cells, regulates the development and function of Th1 cells by controlling the transcription of multiple genes. CD11c+ age-associated B cells (ABC) exhibit high levels of T-bet, which induces the release of IFN-γ and IL-12 (96, 97). Research has shown that an upregulation of the IL-21 signaling pathway in the SG of SS patients is associated with increased B-cells enrichment and disease activity (98), suggesting a role for CD11c+ ABC via IL-21 signaling (99). Fc receptor-like 4 (FcRL4) is part of the immunoglobulin superfamily and is typically expressed on human B cells, playing a regulatory role in immune responses including proliferation, differentiation, and antibody production. The proportion of FcRL4+ B cells in the SG of SS patients has been positively linked to the presence of lymphoepithelial lesions (100). FcRL4+ B cells are also closely associated with chemokine receptor CCR5 expression in the SG of SS patients, and their expression may enhance the migration of chemokines (CCL3 and CCL5) involved in the SG inflammatory response (101). Regulatory B cells subsets (Breg) areknown to mitigate autoimmune inflammatory responses. Bregs exert their regulatory functions through the production of regulatory cytokines and effector molecules such as IL-10, IL-35, and granzyme B (102). A negative correlation has been observed between IL-10-producing Breg cells and Tfh cells in both SS patients and SS model mice, indicating that Breg cells could represent a potential therapeutic target for SS (103).
Dendritic cells
Dendritic cells (DC) are specialized APCs that initiate and drive the differentiation of naïve T cells into effector T cells (104). DCs can be divided into myeloid dendritic cells (mDC) and plasmacytoid dendritic cells (pDCs). In the SG of patients with SS, early infiltrating cells primarily include CD4+CD45RO+T cells and CD20+B cells, subsequently joined by CD27+ and CD79a+ B cells, with CD38+ plasma cells located at the periphery of T- and B-cells infiltrates (105). pDCs are the most potent cellular producers of type I IFN. Activation of the type I IFN pathway in the SG of SS patients correlates positively with the titers of anti-Ro and anti-La autoantibodies (106, 107). Type I IFN promotes the inflammatory response through both autocrine and paracrine mechanisms and induces BAFF production by pDCs (108). Furthermore, pDCs express TLR7 and TLR9 on their surface, which can be triggered by self-antigens to produce substantial amounts of type I IFN (109). TLR-mediated activation of pDCs leads to increased secretion of pro-inflammatory cytokines such as TNF and IL-8. Additionally, type I IFN can prompt macrophage to produce CXCL13, which in turn drives the accumulation of CXCR5+CD19+B cells in the SG, exacerbating the inflammatory response (110). mDCs on the other hand, respond primarily to microbial pathogens and facilitate Th1-mediated adaptive immune responses predominantly through the production of IL-12. It serves as a pro-inflammatory cytokine that activates T-bet to promote the differentiation of naïve T cells into Th1 cells (111). Overexpression of IL-12 in the SG of SS model mice has been observed to induce SS-like symptoms, with an age-dependent increase in anti-SSB/La and anti-ANA antibodies (112).
Macrophage
Macrophage are among the most prevalent innate immune cells in the SG of SS patients. Studies have shown that macrophage are consistently present in the affected SG of SS patients and their abundance is positively correlated with the focality index. Activated macrophage produce inflammatory cytokines that result in damage to SGECs (113). macrophage exhibit high plasticity, differentiating into either classically activated (M1 type) or alternatively activated (M2 type) macrophage in response to various microenvironmental stimuli. M1-type macrophage exacerbate inflammation, accelerate extracellular matrix degradation and apoptosis, and enhance Th1-type immune responses. In contrast, M2 macrophage suppress T-cells proliferation and activation, modulate Th2-type immune responses, and contribute to tissue remodeling. The imbalance in M1/M2 polarization plays a critical role in the pathogenesis of SS (114, 115). M1 macrophage, prevalent in the early stages of SS, produce pro-inflammatory cytokines (TNF-α, IL-6, IL-12) that activate CD4+T cells, promoting their differentiation into the Th1 lineage and exacerbating SG inflammation (116). Conversely, M2 macrophage release anti-inflammatory mediators such as IL-10 and TGF-β), which help mitigate inflammation and facilitate tissue regeneration, thereby reducing autoimmune inflammation (117). As SS advances, chronic inflammation leads to irreversible SG fibrosis, primarily mediated by M2 macrophage. The TGF-β1 signaling pathway, which drives fibrosis, includes both SMAD-regulated and non-SMAD-regulated mechanisms. TGF-β1 encourages M2 macrophage polarization through activation of the SMAD2/3/4 trimeric complex, and this pathway also promotes the conversion of fibroblasts into myofibroblasts (117). Thus, the regulation of macrophage polarization homeostasis is essential in the pathogenesis of SS. In NOD/ShiLtJ mice, macrophage in the SG have been observed to produce high levels of the B-cells chemokine CXCL13, potentially facilitating the formation of ectopic GC within the SG (73).
Salivary gland epithelial cells
The primary target organs in SS are the exocrine glands, particularly the SG. The reduction of saliva in patients is closely linked to the infiltration of immune cells around SGECs and the destruction of glandular structures (118, 119). SGECs drive and regulate local inflammatory responses by facilitating the activation and differentiation of immune cells, creating a feedback loop where immune cells and the inflammatory microenvironment further activate SGECs (120, 121). Despite not being specialized APCs, SGECs express high levels of immunoreactive molecules that mediate lymphocyte homing, antigen presentation, and amplify interactions with immune cells. Innate immune responses such as TLR signaling, inflammatory vesicle signaling, and type I interferon signaling can be activated in SGECs (122, 123). In response to inflammatory stimuli, SGECs overexpress MHC-II and co-stimulatory molecules (CD80 and CD86), which activate CD4+T cells in the SG (43). Additionally, SGECs highly express intercellular adhesion molecule (ICAM-1) and vascular cells adhesion molecule (VCAM-1), facilitating the binding to T-lymphocyte function-associated antigen-1 (LFA1) and very late activation antigen-4 (VLA4). This interaction stabilizes synapses between SGECs and T-cells, further mediating inflammatory responses (124). Activated SGECs also express high levels of inflammatory cytokines and chemokines (CXCL12, CXCL13, IL-1, IL-6, TNF-α and BAFF). IL-1 and IL-6 enhance APCs and CD4+ T-cells activation, BAFF promotes B-cells proliferation and maturation, and CXCL12 and CXCL13 facilitate the migration of predominantly B-cells immune cells to the SG (125). Growth-associated oncogene-α (GRO-α) and its receptor CXCR2, members of the CXC chemokine family, are involved in the inflammatory response in SG (126). Higher expression levels of GRO-α and CXCR2 have been observed in the SG of SS patients compared to healthy controls. Induction of SGECs with IL-6/TNF-α revealed that increased CXCR2 expression correlated positively with IL-6/TNF-α levels, and GRO-α expression was proportionally linked to CXCR2 expression under pro-inflammatory conditions (127). A disintegrin and metalloproteinase 17 (ADAM17) plays a crucial role in cells signaling, adhesion, and cytokine release, impacting inflammatory and immune responses and cells proliferation (128). A significant reduction in CXCR2 expression was observed in SS patients’ SGECs when using an ADAM17 inhibitor. It is hypothesized that ADAM17 may regulate the SG inflammatory response by modulating the interaction between GRO-α and its CXCR2 receptor (129).
Uncontrolled apoptosis can lead to the release of large amounts of cellular contents into the extracellular space, resulting in autoantigen exposure and inflammatory lesion formation, thus inducing a strong immune response. Studies have demonstrated (130) that excessively apoptotic SGECs serve as endogenous antigens, prompting pDCs to highly express IFN-α, thereby activating IFN signaling in SS patients (131). TNF-α and IFN-γ cause structural and functional disruptions in SGECs tight junctions, leading to decreased salivary secretion. Further research has shown that apoptosis in SGECs leads to the cleavage and translocation of α-cytosolic lining proteins and SSA antigens into apoptotic particles, which were then activated by pDCs, resulting in a disturbed immune response and exacerbation of the inflammatory response in the SG (132). Genetic analysis of the SG has revealed a downregulation of anti-apoptosis-related genes and upregulation of pro-apoptosis genes in SGECs in SS samples, suggesting the presence of uncontrolled apoptosis of SGECs in SS (133). Apoptosis in SGECs also activates self-reactive lymphocytes, triggering the activation of T cells, which then induce SS-associated autoantibody production and redistribution of Ro/SSA and La/SSB to the cells surface (134).
Epithelial mesenchymal transfor mation (EMT) describes the process where epithelial cells differentiate into mesenchymal cells under specific physiological and pathological conditions. TGF-β1 is a principal driver of fibrosis in many chronic inflammatory diseases (135). The activation of EMT constitutes a significant pathological response of SGECs to chronic inflammation in SS. In SS patients, fibrosis of SGECs often results from tissue injury and inflammation (136), with fibrous mediators produced by inflammatory cells and epithelial cells, particularly TGF-β1, playing crucial roles in EMT (137). SMAD proteins facilitate TGF-β1 signaling through transcriptional regulation, impacting significantly on cells function and fate (138). The snail Family Transcriptional Repressor (Snail) emerges as a pivotal regulator of EMT, reducing the connectivity and polarity of epithelial cells and facilitating their transition to mesenchymal cells (139). Sisto et al. observed a strong positive expression of EMT-related proteins (waveform protein, collagen type I, and Snail) in SG from SS patients. Conversely, the expression level of the epithelial marker E-cadherin was diminished in diseased SG biopsies, suggesting that TGF-β1 initiates the EMT program in SGECs through the classical TGF-β1/SMAD/Snail signaling pathway (140). In conclusion, SGECs play a crucial role in SS lesions. Inhibiting SGECs activation and restoring their physiological functions may enhance salivary gland secretory function in SS patients.
AQPs and M3R are involved in regulating salivary gland secretion regulation
AquaporinS are a class of transmembrane proteins found in cells membranes, whose main role is to regulate the permeability of water molecules, thereby facilitating their passage through the cells membrane. Aquaporin 5 (AQP5) and muscarinic acetylcholine receptor subtype 3 (M3R) are integral to salivary gland secretion regulation (141, 142). AQP5, a transmembrane transporter protein, is primarily located in various secretory epithelial cells and glands such as the SG, lacrimal glands, and cornea. It enhances the cells membrane’s water permeability, facilitates water molecule transport, and participates in both secretion and absorption of water, as well as intra- and extracellular water balance (143). In coordination with AQP1 in capillary endothelial cells and myoepithelial cells, AQP5 transports saliva into the striated tubes and secretory ducts, playing a crucial role in regulating water transport rates and maintaining salivary secretion. Notably, AQP5 expression is either reduced or absent in SGECs of SS model mice, while being elevated in myoepithelial cells (144). The distribution of AQP5 is similarly diminished in the SGECs of SS patients, impeding trans-epithelial water transport within glandular vesicles and contributing to dry mouth symptoms (145). Research indicates that AQP5 expression is significantly downregulated in the SGECs of both SS model mice and SS patients (56, 146). AQP4 is found in myoepithelial cells that encircle the spinous lobules and intercalary ducts (147). In addition, it has been reported that in the SG of SS patients, there is increased AQP3 protein expression at the apical membrane of the adenohypophysis and decreased AQP1 and AQP4 protein expression in myoepithelial cellss. Therefore, as far as the current study is concerned, AQP1 and AQP3–5 may be involved in SG secretion (148).
M3R, a G-protein-coupled acetylcholine receptor, responds to acetylcholine stimulation by increasing calcium ion transport in SG alveolar cells, activating chloride channels, and mediating the translocation of AQP5 to the apical plasma membrane, thus enhancing salivary secretion (149). M3R is widely expressed in various tissues, including exocrine glands, indicating a significant role in both salivary and lacrimal secretion, particularly in the former (150). A research (151) reported that cholinergic signaling is compromised in M3R knockout mice, leading to SG hypoplasia. Furthermore, studies have shown that IL-17-producing M3R-reactive T cells may exacerbate SG inflammatory responses in SS (152). AQP5, as an effector protein of M3R, is essential for regulating salivary secretion. The anti-M3R antibody targets the M3R receptor, impairing its binding to muscarinic sites and thereby inhibiting intracellular signaling transduction, which affects AQP5 phosphorylation and its water transport capability, ultimately restricting glandular secretion (153).
Signaling pathway
IFN signaling pathway
Dysregulation of IFN signaling constitutes the primary pathophysiological basis of SS. Acting as a critical bridge between the innate and adaptive immune systems, IFN facilitates interactions with adaptive immune cells, perpetuating a cycle of immune activation. It modulates the immune responses of T and B cells and plays a role in SG injury (154). Enhanced IFN signaling has been observed in the SG of SS patients (155). IFNs are primarily categorized into IFN-α, IFN-β, and IFN-γ, and are activated predominantly through TLR and Retinoic Acid-Inducible Gene I-like receptor signaling (156). While most cells produce IFN-β, pDCss predominantly secrete IFN-α, linked to high expression levels of TLR7 and TLR9. IFN-α induces pDCs to express elevated levels of TLR7, resulting in a sustained increase of IFN-α in the SG of SS patients and maintaining the inflammatory milieu (157). Studies indicate that IFN-α enhances lymphocyte activation and migration to the SG via the JAK1/STAT1/2 signaling pathway in SGECs and by inducing the expression of CXCL13, BAFF, and CXCL10. Additionally, IFN-α stimulates the expression of TLR7 and the downstream signaling molecule Myeloid differentiation primary response 88 (MyD88) (158). Similarly, IFN-γ, although typically expressed at low levels in normal SGECs, is readily induced by TLR3, like IFN-α. IFN-γ also facilitates the production of autoimmune antibodies such as SSA/Ro and immunoglobulin G by stimulating BAFF expression in SGECs (159).
TLR signaling pathway
TLRs play an integral role in the pathogenesis of SS. Current research indicates that TLRs involved in the SG inflammatory response primarily include TLR2, TLR3, TLR4, TLR7, and TLR9 (160). Moreover, viral double-stranded RNA can induce apoptosis in SGECs by activating TLR3, which upregulates apoptotic proteins such as Bcl-2 modifier and ultra-long Bcl-2 interacting cells death mediator. Apoptotic SGECs then act as endogenous antigens that recruit immune cells to infiltrate the SG, thereby triggering an immune response (161, 162). TLR2 and TLR4 are significantly upregulated in the SGs of SS mice and activate the Mitogen-Activated Protein Kinase and NF-κB pathways via MyD88, leading to elevated expression of IL-6, monocyte chemotactic protein 1, and TNF-α in SGECs. These inflammatory factors are significantly reduced when MyD88 is knocked out (163). Conversely, TLR7 and TLR9 enhance B lymphocyte maturation and proliferation. TLR9 specifically induces B lymphocytes to express autoantibodies and inflammatory factors such as monocyte chemotactic protein 1, IL-8, and IL-6. TLR7 facilitates IFN-α synthesis by inducing STAT3^S727 and NF-κB phosphorylation in B lymphocytes (164). In SS patients, TLR7 not only increases B cells secretion of IFN-α but also promotes the differentiation of immature B cells into plasma cells, culminating in abnormal immune system activation (165). Therefore, TLR signaling is critical in regulating inflammatory factor expression, B-lymphocyte maturation, antibody synthesis, and SGECs apoptosis in SS.
BAFF/BAFF-R signaling pathway
BAFF, a member of the TNF family, induces B lymphocyte activation, maturation, migration, and autoantibody production, and is a principal cause of B lymphocyte over-activation in the SG of SS patients (166). BAFF expression is significantly higher in the SG of SS patients, correlating with serum IgG, ESR, anti-ANA levels, and disease activity. This excessive expression is a major contributor to SG damage. Studies indicate that elevated BAFF levels are present in the SG of all SS patients, while serum BAFF levels are comparatively lower in patients with GC formation than in those without (99). Consequently, the distribution of BAFF in SS patients is closely associated with lymphocyte migration to the SG and GC formation. SGECs are prompted by IFN-α to produce large amounts of BAFF, leading to the overactivation of B lymphocytes. BAFF-R, a specific receptor for BAFF, when bound to BAFF, activates the PI3K/AKT/mTOR signaling pathway in B lymphocytes. This activation leads to IKK phosphorylation and elevates the expression of NF-κB, triggering the production of inflammatory factors (IL-12, IFN-γ, IL-6, and IL-1β) and resulting in an inflammatory response in the SG (167, 168).
JAK-STAT signaling pathway
The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway is a crucial regulatory system for cells proliferation and differentiation. The JAK-STAT pathway comprises four JAK intracellular tyrosine kinases (JAK1, JAK2, JAK3, and TYK2) and seven transcription factors, STAT (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6), which mediate the transduction of various cytokines related to immune response, inflammation, and cellular activation and survival (169). Immunohistochemical analysis has shown that SGECs in the SG of patients with SS highly express JAK1 and JAK2 (170). Increased expression of STAT3 has been observed in SS SG (171, 172), which is stimulated by cytokines mediated by JAK1 and JAK2 (173, 174). Recent studies suggest that STAT3 is also involved in the process of episomal DNA methylation/hydroxymethylation in SS (175). In the cytokine signaling mediated by JAK/STAT, IL-6, IL-21, and IL-23 are implicated in the pathogenesis of SS. IL-21, a significant cytokine in the IFN signaling pathway, is notably elevated in SS patients, and SGRNA sequencing has revealed significantly higher levels of IL-21 and IL-21-inducible genes (IL-21R, JAK3, STAT1, and CXCL10) (176, 177). Additionally, recent studies have shown that baricitinib, a JAK inhibitor, can ameliorate the destruction of alveolar cells in the SG of SS patients by reducing IFN-γ-induced CXCL10 expression and CXCL10-dependent immune cells infiltration in the SGECs (170).
P2X7R/NLRP3 signaling pathway
Cellular pyroptosis, a recent discovery, is a novel form of inflammatory programmed cells death characterized by mild cellular swelling. Before this swelling, bubble-like protrusions form on the cells membrane, mediated by Gasdermin family proteins. These proteins create annular pores in the membrane, allowing cellular contents to flow out gradually and triggering an inflammatory response (178). The Purinergic 2X7 receptor (P2X7R), an Adenosine triphosphate (ATP)-gated ion channel, is expressed in various tissues including the central nervous system and different epithelial tissues (179). Research has indicated (180, 181) that P2X7R in the SG plays a crucial role in cholinergic receptor-mediated salivary secretion. P2X7R is a significant activator of the NOD-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasomes (182). Upon ATP stimulation, activated P2X7R facilitates the formation of large non-selective membrane pores that permit NLRP3 entry into the cytoplasm, thus activating the NLRP3 inflammasome (183). Activation of the NLRP3 inflammasome leads to the production of activated Caspase-1, which cleaves GSDMD, causing its N-terminus to perforate the cytosolic membrane. This process releases mature IL-1β and IL-18, which activate initial T-cells, prompting their differentiation into Th1 and Th17 cells and inducing the secretion of cytokines (IL-6, IL-17, IL-21, IL-22, IL-23) that provoke an inflammatory response (184). Baldini et al. demonstrated that P2X7R, Caspase-1, and IL-18 levels were elevated in the SG of SS patients at both RNA and protein levels (185). Khalafall et al. (186) showed through in vitro experimental studies on SS model mice that P2X7R activation prompted the assembly of NLRP3 inflammasomes and the maturation and release of IL-1β in mouse SGECs. IL-1β, a major cytokine involved in SS inflammation, primarily contributes to SGECs apoptosis and necrosis and facilitates the release of tissue-specific autoantigens. IL-18, a critical pro-inflammatory cytokine of the IL-1 family and a key downstream factor of inflammasomes, is upregulated in the SGECs of SS patients and closely linked to GC formation (187).
TGF-β1/SMAD/Snail signaling pathway
EMT plays a crucial role in activating the pathological fibrosis cascade response in chronic inflammatory diseases (188, 189). In the pathology of SS, various cytokines significantly alter the polarity and organization of the SGECs, impacting their secretory function and closely linking to EMT activation (190, 191). Chronic inflammation is a key factor in SG fibrosis, with CD4+T cells, macrophage, and epithelial cells contributing to the pathological accumulation of extracellular matrix (ECM) components typical of SG changes in SS (192). Increasing evidence indicates that TGF-β1 mediates its biological effects via the TGF-β/SMAD/Snail signaling pathway, playing a significant pathogenic role in various fibrotic diseases. In SS, aberrant up-regulation of TGF-β1 in SG exacerbates the fibrosis by activating the TGF-β1/SMAD/Snail signaling pathway, leading to morphological and functional mesenchymal transformations in SGECs (63).
Summary
The pathogenesis of SS-related salivary gland injury is complex, resulting from a multifaceted interplay of cellular and molecular factors. This process is still not fully understood, with ongoing research into its specific mechanisms. Although several potential therapeutic targets have been identified, and some targeted medications have shown promising efficacy in both ex vivo and in vivo studies, their translation into clinical practice remains limited, often requiring extended periods. Therefore, enhancing our understanding of SS salivary gland pathogenesis and utilizing preliminary findings to prevent or mitigate further glandular damage and preserve secretory function is essential for improving patient quality of life.
Author contributions
WQ: Writing – original draft. JT: Funding acquisition, Methodology, Writing – review & editing. YY: Writing – review & editing. TW: Software, Writing – review & editing. YW: Formal analysis, Software, Supervision, Visualization, Writing – review & editing. ZW: Software, Writing – review & editing. GZ: Conceptualization, Writing – review & editing. YZ: Formal analysis, Software, Visualization, Writing – review & editing. JW: Conceptualization, Writing – review & editing. GW: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research was supported by two National Natural Science Foundation of China (81960832, 81860858) and the Natural Science Foundation of Gansu Province for 2 projects (21JRI1RA158, 22JR11RA131).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bjordal O, Norheim KB, Rødahl E, Jonsson R, Omdal R. Primary sjögren’s syndrome. N Engl J Med. (2018) 379:96–7. doi: 10.1056/NEJMc1804598
2. Qin B, Wang J, Yang Z, Yang M, Ma N, Huang F, et al. Epidemiology of primary Sjögren’s syndrome: a systematic review and meta-analysis. Ann Rheum Dis. (2015) 74:1983–9. doi: 10.1136/annrheumdis-2014–205375
3. Zhang Q, Lv X, Wang Y, Wang B, He Y, Chen C, et al. Expression of CFTR, a hallmark gene of ionocytes, is downregulated in salivary glands of Sjögren’s syndrome patients. Arthritis Res Ther. (2022) 24:263. doi: 10.1186/s13075–022-02959–8
5. Horeth E, Oyelakin A, Song EAC, Che M, Bard J, Min S, et al. Transcriptomic and single-cells analysis reveals regulatory networks and cellsular heterogeneity in mouse primary sjögren’s syndrome salivary glands. Front Immunol. (2021) 12:729040. doi: 10.3389/fimmu.2021.729040
6. Kroese FGM, Haacke EA, Bombardieri M. The role of salivary gland histopathology in primary Sjögren’s syndrome: promises and pitfalls. Clin Exp Rheumatol. (2018) 36 Suppl 112:222–33.
7. Jonckheere S, Adams J, De Groote D, Campbell K, Berx G, Goossens S. Epithelial-mesenchymal transition (EMT) as a therapeutic target. cellss Tissues Organs. (2022) 211:157–82. doi: 10.1159/000512218
8. Mavragani CP, Moutsopoulos HM. Sjögren’s syndrome: Old and new therapeutic targets. J Autoimmun. (2020) 110:102364. doi: 10.1016/j.jaut.2019.102364
9. Mavragani CP. Mechanisms and new strategies for primary sjögren’s syndrome. Annu Rev Med. (2017) 68:331–43. doi: 10.1146/annurev-med-043015–123313
10. Katsiougiannis S, Stergiopoulos A, Moustaka K, Havaki S, Samiotaki M, Stamatakis G, et al. Salivary gland epithelial cells in Sjögren’s syndrome: Metabolic shift and altered mitochondrial morphology toward an innate immune cells function. J Autoimmun. (2023) 136:103014. doi: 10.1016/j.jaut.2023.103014
11. Tseng YC, Yang HY, Lin WT, Chang CB, Chien HC, Wang HP, et al. Salivary dysbiosis in Sjögren’s syndrome and a commensal-mediated immunomodulatory effect of salivary gland epithelial cellss. NPJ Biofilms Microbiomes. (2021) 7:21. doi: 10.1038/s41522-021-00192-w
12. Pedersen AM, Nauntofte B. Primary Sjögren’s syndrome: oral aspects on pathogenesis, diagnostic criteria, clinical features and approaches for therapy. Expert Opin Pharmacother. (2001) 2:1415–36. doi: 10.1517/14656566.2.9.1415
13. Generali E, Costanzo A, Mainetti C, Selmi C. Cutaneous and mucosal manifestations of sjögren’s syndrome. Clin Rev Allergy Immunol. (2017) 53:357–70. doi: 10.1007/s12016-017-8639-y
14. Bjordal O, Norheim KB, Rødahl E, Jonsson R, Omdal R. Primary Sjögren’s syndrome and the eye. Surv Ophthalmol. (2020) 65:119–32. doi: 10.1016/j.survophthal.2019.10.004
15. Llamas-Molina JM, Velasco-Amador JP, de la Torre-Gomar FJ, Carrero-Castaño A, Ruiz-Villaverde R. Localized cutaneous nodular amyloidosis in a patient with sjögren’s syndrome. Int J Mol Sci. (2023) 24:9409. doi: 10.3390/ijms24119409
16. Vitali C, Del Papa N. Pain in primary Sjögren’s syndrome. Best Pract Res Clin Rheumatol. (2015) 29:63–70. doi: 10.1016/j.berh.2015.05.002
17. ter Borg EJ, Kelder JC. Polyarthritis in primary Sjögren’s syndrome represents a distinct subset with less pronounced B cells proliferation a Dutch cohort with long-term follow-up. Clin Rheumatol. (2016) 35:649–55. doi: 10.1007/s10067–016-3175–3
18. François H, Mariette X. Renal involvement in primary Sjögren syndrome. Nat Rev Nephrol. (2016) 12:82–93. doi: 10.1038/nrneph.2015.174
19. Aiyegbusi O, McGregor L, McGeoch L, Kipgen D, Geddes CC, Stevens KI. Renal disease in primary sjögren’s syndrome. Rheumatol Ther. (2021) 8:63–80. doi: 10.1007/s40744-020-00264-x
20. Luppi F, Sebastiani M, Sverzellati N, Cavazza A, Salvarani C, Manfredi A. Lung complications of Sjogren syndrome. Eur Respir Rev. (2020) 29:200021. doi: 10.1183/16000617.0021–2020
21. Berardicurti O, Marino A, Genovali I, Navarini L, D'Andrea S, Currado D, et al. Interstitial lung disease and pulmonary damage in primary sjögren’s syndrome: A systematic review and meta-analysis. J Clin Med. (2023) 12:2586. doi: 10.3390/jcm12072586
22. Ebert EC. Gastrointestinal and hepatic manifestations of sjogren syndrome. J Clin Gastroenterol. (2012) 46:25–30. doi: 10.1097/MCG.0b013e3182329d9c
23. Popov Y, Salomon-Escoto K. Gastrointestinal and hepatic disease in sjogren syndrome. Rheumatic Dis Clinics North America. (2018) 44:143–51. doi: 10.1016/j.rdc.2017.09.010
24. Berkowitz AL, Samuels MA. The neurology of Sjögren’s syndrome and the rheumatology of peripheral neuropathy and myelitis. Pract Neurol. (2014) 14:14–22. doi: 10.1136/practneurol-2013–000651
25. Gwathmey KG, Satkowiak K. Peripheral nervous system manifestations of rheumatological diseases. J Neurological Sci. (2021) 424:117421. doi: 10.1016/j.jns.2021.117421
26. Rischmueller M, Tieu J, Lester S. Primary sjögren’s syndrome. Best Pract Res Clin Rheumatol. (2016) 30:189–220. doi: 10.1016/j.berh.2016.04.003
27. Porcheri C, Mitsiadis T. Physiology, pathology and regeneration of salivary glands. cellss. (2019) 8:976. doi: 10.3390/cellss8090976
28. Pedersen AML, Sørensen CE, Proctor GB, Carpenter GH, Ekström J. Salivary secretion in health and disease. J Oral Rehabilitation. (2018) 45:730–46. doi: 10.1111/joor.12664
29. Vitali C. Classification criteria for Sjogren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheumatic Diseases. (2002) 61:554–8. doi: 10.1136/ard.61.6.554
30. Erkılınç G, Doğru A, Arslan Y, Burak Öz R, Karahan N, Şahin M, et al. Evaluation of histopathological results of minor salivary gland biopsies in patients with the diagnosis of Sjögren’s syndrome. Arch Rheumatol. (2022) 37:49–58. doi: 10.46497/ArchRheumatol.2022.8607
31. Liao R, Yang HT, Li H, et al. Recent advances of salivary gland biopsy in sjögren’s syndrome. Front Med. (2022) 8:792593. doi: 10.3389/fmed.2021.792593
32. Li N, Li L, Wu M, Li Y, Yang Wu Y, et al. Integrated bioinformatics and validation reveal potential biomarkers associated with progression of primary sjögren’s syndrome. Front Immunol. (2021) 12:697157. doi: 10.3389/fimmu.2021.697157
33. Fragoulis GE, Fragkioudaki S, Reilly JH, Kerr SC, McInnes IB, Moutsopoulos HM. Analysis of the cells populations composing the mononuclear cells infiltrates in the labial minor salivary glands from patients with rheumatoid arthritis and sicca syndrome. J Autoimmunity. (2016) 73:85–91. doi: 10.1016/j.jaut.2016.06.008
34. van Ginkel MS, Haacke EA, Bootsma H, Arends S, van Nimwegen JF, Verstappen GM, et al. Presence of intraepithelial B-lymphocytes is associated with the formation of lymphoepithelial lesions in salivary glands of primary Sjögren’s syndrome patients. Clin Exp Rheumatol. (2019) 37 Suppl 118:42–8.
35. Li Y, Zhang K, Chen H, Sun F, Xu J, Wu Z, et al. A genome-wide association study in Han Chinese identifies a susceptibility locus for primary Sjögren’s syndrome at 7q11.23. Nat Genet. (2013) 45:1361–5. doi: 10.1038/ng.2779
36. for UK Primary Sjögren’s Syndrome Registry, Lessard CJ, Li H, Rasmussen A, Grundahl KM, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat Genet. (2013) 45:1284–92. doi: 10.1038/ng.2792
37. Imgenberg-Kreuz J, Rasmussen A, Sivils K, Nordmark G. Genetics and epigenetics in primary Sjögren’s syndrome. Rheumatology. (2021) 60:2085–98. doi: 10.1093/rheumatology/key330
38. Goules AV, Tzioufas AG. Primary Sjögren’s syndrome: Clinical phenotypes, outcome and the development of biomarkers. Autoimmun Rev. (2016) 15:695–703. doi: 10.1016/j.autrev.2016.03.004
39. Thorlacius GE, Björk A, Wahren-Herlenius M. Genetics and epigenetics of primary Sjögren syndrome: implications for future therapies. Nat Rev Rheumatol. (2023) 19:288–306. doi: 10.1038/s41584–023-00932–6
40. Wang-Renault SF, Boudaoud S, Nocturne G, Roche E, Sigrist N, Daviaud C, et al. Deregulation of microRNA expression in purified T and B lymphocytes from patients with primary Sjögren’s syndrome. Ann Rheum Dis. (2018) 77:133–40. doi: 10.1136/annrheumdis-2017–211417
41. Teruel M, Barturen G, Martínez-Bueno M, Castellini-Pérez O, Barroso-Gil M, Povedano E, et al. Integrative epigenomics in Sjögren´s syndrome reveals novel pathways and a strong interaction between the HLA, autoantibodies and the interferon signature. Sci Rep. (2021) 11:23292. doi: 10.1038/s41598–021-01324–0
42. Pacheco Y, Acosta-Ampudia Y, Monsalve DM, Chang C, Gershwin ME, Anaya JM. Bystander activation and autoimmunity. J Autoimmunity. (2019) 103:102301. doi: 10.1016/j.jaut.2019.06.012
43. Low H, Witte T. Aspects of innate immunity in Sjögren’s syndrome. Arthritis Res Ther. (2011) 13:218. doi: 10.1186/ar3318
44. Lucchesi D, Pitzalis C, Bombardieri M. EBV and other viruses as triggers of tertiary lymphoid structures in primary Sjögren’s syndrome. Expert Rev Clin Immunol. (2014) 10:445–55. doi: 10.1586/1744666X.2014.892417
45. Hou X, Hong X, Ou M, Meng S, Wang T, Liao S, et al. Analysis of Gene Expression and TCR/B cells Receptor Profiling of Immune cellss in Primary Sjögren’s Syndrome by Single-cells Sequencing. J Immunol. (2022) 209:238–49. doi: 10.4049/jimmunol.2100803
46. Brito-Zerón P, Gheitasi H, Retamozo S, Bové A, Londoño M, Sánchez-Tapias JM, et al. How hepatitis C virus modifies the immunological profile of Sjögren syndrome: analysis of 783 patients. Arthritis Res Ther. (2015) 17:250. doi: 10.1186/s13075–015-0766–3
47. Liu Z, Chu A. Sjögren’s syndrome and viral infections. Rheumatol Ther. (2021) 8:1051–9. doi: 10.1007/s40744–021-00334–8
48. Maslinska M, Kostyra-Grabczak K. The role of virus infections in Sjögren’s syndrome. Front Immunol. (2022) 13:823659. doi: 10.3389/fimmu.2022.823659
49. Möhl BS, Chen J, Park SJ, Jardetzky TS, Longnecker R. Epstein-Barr Virus Fusion with Epithelial cellss Triggered by gB Is Restricted by a gL Glycosylation Site. J Virol. (2017) 91:e01255–17. doi: 10.1128/JVI.01255–17
50. Psianou K, Panagoulias I, Papanastasiou AD, de Lastic AL, Rodi M, Spantidea PI, et al. Clinical and immunological parameters of Sjögren’s syndrome. Autoimmun Rev. (2018) 17:1053–64. doi: 10.1016/j.autrev.2018.05.005
51. Singh N, Cohen PL. The T cells in Sjogren’s syndrome: force majeure, not spectateur. J Autoimmun. (2012) 39:229–33. doi: 10.1016/j.jaut.2012.05.019
52. Zhao H, Li Q, Ye M, Yu J. Tear luminex analysis in dry eye patients. Med Sci Monit. (2018) 24:7595–602. doi: 10.12659/MSM.912010
53. Chen X, Aqrawi LA, Utheim TP, Tashbayev B, Utheim ØA, Reppe S, et al. Elevated cytokine levels in tears and saliva of patients with primary Sjögren’s syndrome correlate with clinical ocular and oral manifestations. Sci Rep. (2019) 9:7319. doi: 10.1038/s41598–019-43714–5
54. Yin H, Vosters JL, Roescher N, D'Souza A, Kurien BT, Tak PP, et al. Location of immunization and interferon-γ Are central to induction of salivary gland dysfunction in ro60 peptide immunized model of sjögren’s syndrome. PloS One. (2011) 6:e18003. doi: 10.1371/journal.pone.0018003
55. Torres-Aguilar H, Sosa-Luis SA, Aguilar-Ruiz SR. Infections as triggers of flares in systemic autoimmune diseases: novel innate immunity mechanisms. Curr Opin Rheumatol. (2019) 31:525–31. doi: 10.1097/BOR.0000000000000630
56. Yoshimura S, Nakamura H, Horai Y, Nakajima H, Shiraishi H, Hayashi T, et al. Abnormal distribution of AQP5 in labial salivary glands is associated with poor saliva secretion in patients with Sjögren’s syndrome including neuromyelitis optica complicated patients. Modern Rheumatol. (2016) 26:384–90. doi: 10.3109/14397595.2015.1083146
57. Sisto M, Lisi S, Lofrumento DD, Frassanito MA, Cucci L, D'Amore S, et al. Induction of TNF-alpha-converting enzyme-ectodomain shedding by pathogenic autoantibodies. Int Immunol. (2009) 21:1341–9. doi: 10.1093/intimm/dxp103
58. Sisto M, Lisi S, D’Amore M, Lofrumento DD. The metalloproteinase ADAM17 and the epidermal growth factor receptor (EGFR) signaling drive the inflammatory epithelial response in Sjögren’s syndrome. Clin Exp Med. (2015) 15:215–25. doi: 10.1007/s10238–014-0279–4
59. Wang Y, Shnyra A, Africa C, Warholic C, McArthur C. Activation of the extrinsic apoptotic pathway by TNF-alpha in human salivary gland (HSG) cellss in vitro, suggests a role for the TNF receptor (TNF-R) and intercellsular adhesion molecule-1 (ICAM-1) in Sjögren’s syndrome-associated autoimmune sialadenitis. Arch Oral Biol. (2009) 54:986–96. doi: 10.1016/j.archoralbio.2009.07.011
60. Crotty S. A brief history of T cells help to B cellss. Nat Rev Immunol. (2015) 15:185–9. doi: 10.1038/nri3803
61. Maehara T, Moriyama M, Hayashida JN, Tanaka A, Shinozaki S, Kubo Y, et al. Selective localization of T helper subsets in labial salivary glands from primary Sjögren’s syndrome patients. Clin Exp Immunol. (2012) 169:89–99. doi: 10.1111/j.1365-2249.2012.04606.x
62. Gan Y, Zhao X, He J, Liu X, Li Y, Sun X, et al. Increased interleukin-17F is associated with elevated autoantibody levels and more clinically relevant than interleukin-17A in primary sjögren’s syndrome. J Immunol Res. (2017) 2017:1–9. doi: 10.1155/2017/4768408
63. Sisto M, Lorusso L, Ingravallo G, Ribatti D, Lisi S. TGFβ1-Smad canonical and -Erk noncanonical pathways participate in interleukin-17-induced epithelial–mesenchymal transition in Sjögren’s syndrome. Lab Invest. (2020) 100:824–36. doi: 10.1038/s41374-020-0373-z
64. Alunno A, Carubbi F, Bartoloni E, Bistoni O, Caterbi S, Cipriani P, et al. Unmasking the pathogenic role of IL-17 axis in primary Sjögren’s syndrome: A new era for therapeutic targeting? Autoimmun Rev. (2014) 13:1167–73. doi: 10.1016/j.autrev.2014.08.022
65. Alunno A, Carubbi F, Bistoni O, Caterbi S, Bartoloni E, Mirabelli G, et al. T regulatory and T helper 17 cellss in primary sjögren’s syndrome: facts and perspectives. Mediators Inflammation. (2015) 2015:1–10. doi: 10.1155/2015/243723
66. Geng J, Yu S, Zhao H, Sun X, Li X, Wang P, et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cellss and Treg cellss. Nat Immunol. (2017) 18:800–12. doi: 10.1038/ni.3748
67. Voigt A, Bohn K, Sukumaran S, Stewart CM, Bhattacharya I, Nguyen CQ. Unique glandular ex-vivo Th1 and Th17 receptor motifs in Sjögren’s syndrome patients using single-cells analysis. Clin Immunol. (2018) 192:58–67. doi: 10.1016/j.clim.2018.04.009
68. Verstappen GM, Corneth OBJ, Bootsma H, Kroese FGM. Th17 cellss in primary Sjögren’s syndrome: Pathogenicity and plasticity. J Autoimmunity. (2018) 87:16–25. doi: 10.1016/j.jaut.2017.11.003
69. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol. (2017) 18:612–21. doi: 10.1038/ni.3742
70. Fei Y, Zhang W, Lin D, Wu C, Li M, Zhao Y, et al. Clinical parameter and Th17 related to lymphocytes infiltrating degree of labial salivary gland in primary Sjögren’s syndrome. Clin Rheumatol. (2014) 33:523–9. doi: 10.1007/s10067-013-2476-z
71. Barone F, Nayar S, Campos J, Cloake T, Withers DR, Toellner KM, et al. IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc Natl Acad Sci USA. (2015) 112:11024–9. doi: 10.1073/pnas.1503315112
72. Zhou DM, Xu YX, Zhang LY, Sun Y, Wang ZY, Yuan YQ, et al. The role of follicular T helper cellss in patients with Malignant lymphoid disease. Hematology. (2017) 22:412–8. doi: 10.1080/10245332.2017.1300623
73. Kramer JM, Klimatcheva E, Rothstein TL. CXCL13 is elevated in Sjögren’s syndrome in mice and humans and is implicated in disease pathogenesis. J Leukocyte Biol. (2013) 94:1079–89. doi: 10.1189/jlb.0113036
74. Traianos EY, Locke J, Lendrem D, Bowman S, Hargreaves B, Macrae V, et al. Serum CXCL13 levels are associated with lymphoma risk and lymphoma occurrence in primary Sjögren’s syndrome. Rheumatol Int. (2020) 40:541–8. doi: 10.1007/s00296-020-04524-5
75. Chen W, Yang F, Xu G, Ma J, Lin J. Follicular helper T cellss and follicular regulatory T cellss in the immunopathology of primary Sjögren’s syndrome. J Leukoc Biol. (2021) 109:437–47. doi: 10.1002/JLB.5MR1020-057RR
76. Zhang CJ, Wang C, Jiang M, Gu C, Xiao J, Chen X, et al. Act1 is a negative regulator in T and B cellss via direct inhibition of STAT3. Nat Commun. (2018) 9:2745. doi: 10.1038/s41467–018-04974–3
77. Mehta DS, Wurster AL, Whitters MJ, Young DA, Collins M, Grusby MJ. IL-21 induces the apoptosis of resting and activated primary B cellss. J Immunol. (2003) 170:4111–8. doi: 10.4049/jimmunol.170.8.4111
78. Sage PT, Sharpe AH. T follicular regulatory cellss in the regulation of B cells responses. Trends Immunol. (2015) 36:410–8. doi: 10.1016/j.it.2015.05.005
79. Fu W, Liu X, Lin X, Feng H, Sun L, Li S, et al. Deficiency in T follicular regulatory cellss promotes autoimmunity. J Exp Med. (2018) 215:815–25. doi: 10.1084/jem.20170901
80. Dijkgraaf FE, Kok L, Schumacher TNM. Formation of tissue-resident CD8 + T-cells memory. Cold Spring Harb Perspect Biol. (2021) 13:a038117. doi: 10.1101/cshperspect.a038117
81. De Araújo-Souza PS, Hanschke SCH, Viola JPB. Epigenetic control of interferon-gamma expression in CD8 T cellss. J Immunol Res. (2015) 2015:1–7. doi: 10.1155/2015/849573
82. Zhou H, Yang J, Tian J, Wang S. CD8+ T lymphocytes: crucial players in sjögren’s syndrome. Front Immunol. (2021) 11:602823. doi: 10.3389/fimmu.2020.602823
83. Mingueneau M, Boudaoud S, Haskett S, Reynolds TL, Nocturne G, Norton E, et al. Cytometry by time-of-flight immunophenotyping identifies a blood Sjögren’s signature correlating with disease activity and glandular inflammation. J Allergy Clin Immunol. (2016) 137:1809–1821.e12. doi: 10.1016/j.jaci.2016.01.024
84. Barr JY, Wang X, Meyerholz DK, Lieberman SM. CD8 T cellss contribute to lacrimal gland pathology in the nonobese diabetic mouse model of Sjögren syndrome. Immunol Cells Biol. (2017) 95:684–94. doi: 10.1038/icb.2017.38
85. Baker OJ, Camden JM, Redman RS, Jones JE, Seye CI, Erb L, et al. Proinflammatory cytokines tumor necrosis factor-α and interferon-γ alter tight junction structure and function in the rat parotid gland Par-C10 cells line. Am J Physiology-cells Physiol. (2008) 295:C1191–201. doi: 10.1152/ajpcells.00144.2008
86. Verstappen GM, Kroese FGM, Bootsma H. T cellss in primary Sjögren’s syndrome: targets for early intervention. Rheumatology. (2021) 60:3088–98. doi: 10.1093/rheumatology/kez004
87. Pontarini E, Lucchesi D, Bombardieri M. Current views on the pathogenesis of Sjögren’s syndrome. Curr Opin Rheumatol. (2018) 30:215–21. doi: 10.1097/BOR.0000000000000473
88. Nocturne G, Mariette X. B cellss in the pathogenesis of primary Sjögren syndrome. Nat Rev Rheumatol. (2018) 14:133–45. doi: 10.1038/nrrheum.2018.1
89. Theander E, Jonsson R, Sjöström B, Brokstad K, Olsson P, Henriksson G. Prediction of sjögren’s syndrome years before diagnosis and identification of patients with early onset and severe disease course by autoantibody profiling. Arthritis Rheumatol. (2015) 67:2427–36. doi: 10.1002/art.39214
90. Shen L, Gao C, Suresh L, et al. Central role for marginal zone B cellss in an animal model of Sjogren’s syndrome. Clin Immunol. (2016) 168:30–6. doi: 10.1016/j.clim.2016.04.008
91. Quartuccio L, Salvin S, Fabris M, Maset M, Pontarini E, Isola M, et al. BLyS upregulation in Sjogren’s syndrome associated with lymphoproliferative disorders, higher ESSDAI score and B-cells clonal expansion in the salivary glands. Rheumatology. (2013) 52:276–81. doi: 10.1093/rheumatology/kes180
92. Su T, Li J, Meng M, Zhao S, Xu Y, Ding X, et al. Bone marrow stromal cellss induced activation of nuclear factor κB signaling protects non-Hodgkin’s B lymphoma cellss from apoptosis. Tumor Biol. (2016) 37:10745–52. doi: 10.1007/s13277–016-4860–1
93. Varin MM, Le Pottier L, Youinou P, Saulep D, Mackay F, Pers JO. B-cells tolerance breakdown in Sjögren’s Syndrome: Focus on BAFF. Autoimmun Rev. (2010) 9:604–8. doi: 10.1016/j.autrev.2010.05.006
94. Mariette X. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjogren’s syndrome. Ann Rheumatic Diseases. (2003) 62:168–71. doi: 10.1136/ard.62.2.168
95. Jin L, Yu D, Li X, Yu N, Li X, Wang Y, et al. CD4+CXCR5+ follicular helper T cellss in salivary gland promote B cellss maturation in patients with primary Sjogren’s syndrome. Int J Clin Exp Pathol. (2014) 7:1988–96.
96. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)–driven accumulation of a novel CD11c+ B-cells population is important for the development of autoimmunity. Blood. (2011) 118:1305–15. doi: 10.1182/blood-2011–01-331462
97. Rubtsova K, Rubtsov AV, Van Dyk LF, Kappler JW. Marrack P. T-box transcription factor T-bet, a key player in a unique type of B-cells activation essential for effective viral clearance. Proc Natl Acad Sci USA. (2013) 110:E3216–24. doi: 10.1073/pnas.1312348110
98. Chen X, Jiang S, Zhou Z, Zhou Z, Xu X, Ying S, et al. Increased expression of interleukin-21-inducible genes in minor salivary glands are associated with primary Sjögren’s syndrome disease characteristics. Rheumatology. (2021) 60:2979–89. doi: 10.1093/rheumatology/keaa695
99. Carrillo-Ballesteros FJ, Palafox-Sánchez CA, Franco-Topete RA, Muñoz-Valle JF, Orozco-Barocio G, Martínez-Bonilla GE, et al. Expression of BAFF and BAFF receptors in primary Sjögren’s syndrome patients with ectopic germinal center-like structures. Clin Exp Med. (2020) 20:615–26. doi: 10.1007/s10238–020-00637–0
100. Haacke EA, Bootsma H, Spijkervet FKL, Visser A, Vissink A, Kluin PM, et al. FcRL4+ B-cellss in salivary glands of primary Sjögren’s syndrome patients. J Autoimmunity. (2017) 81:90–8. doi: 10.1016/j.jaut.2017.03.012
101. Verstappen GM, Ice JA, Bootsma H, Pringle S, Haacke EA, de Lange K, et al. Gene expression profiling of epithelium-associated FcRL4+ B cellss in primary Sjögren’s syndrome reveals a pathogenic signature. J Autoimmunity. (2020) 109:102439. doi: 10.1016/j.jaut.2020.102439
102. Lin X, Wang X, Xiao F, Ma K, Liu L, Wang X, et al. IL-10-producing regulatory B cellss restrain the T follicular helper cells response in primary Sjögren’s syndrome. Cells Mol Immunol. (2019) 16:921–31. doi: 10.1038/s41423-019-0227-z
103. Bombardieri M, Lewis M, Pitzalis C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat Rev Rheumatol. (2017) 13:141–54. doi: 10.1038/nrrheum.2016.217
104. Hillen MR, Ververs FA, Kruize AA, Van Roon JA. Dendritic cellss, T-cellss and epithelial cellss: a crucial interplay in immunopathology of primary Sjögren’s syndrome. Expert Rev Clin Immunol. (2014) 10:521–31. doi: 10.1586/1744666X.2014.878650
105. Cellsa M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cellss activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol. (2000) 1:305–10. doi: 10.1038/79747
106. Cao W. Pivotal functions of plasmacytoid dendritic cellss in systemic autoimmune pathogenesis. J Clin Cells Immunol. (2014) 5(2):212. doi: 10.4172/2155-9899.1000212
107. Bodewes ILA, Al-Ali S, Van Helden-Meeuwsen CG, Maria NI, Tarn J, Lendrem DW, et al. Systemic interferon type I and type II signatures in primary Sjögren’s syndrome reveal differences in biological disease activity. Rheumatology. (2018) 57:921–30. doi: 10.1093/rheumatology/kex490
108. Bodewes ILA, Björk A, Versnel MA, Wahren-Herlenius M. Innate immunity and interferons in the pathogenesis of Sjögren’s syndrome. Rheumatology. (2021) 60:2561–73. doi: 10.1093/rheumatology/key360
109. Alculumbre S, Raieli S, Hoffmann C, Chelbi R, Danlos FX, Soumelis V. Plasmacytoid pre-dendritic cellss (pDCs): from molecular pathways to function and disease association. Semin Cells Dev Biol. (2019) 86:24–35. doi: 10.1016/j.semcdb.2018.02.014
110. Zhao J, Kubo S, Nakayamada S, Shimajiri S, Zhang X, Yamaoka K, et al. Association of plasmacytoid dendritic cellss with B cells infiltration in minor salivary glands in patients with Sjögren’s syndrome. Mod Rheumatol. (2016) 26:716–24. doi: 10.3109/14397595.2015.1129694
111. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. (2003) 3:133–46. doi: 10.1038/nri1001
112. Vosters JL, Landek-Salgado MA, Yin H, Yin H, Swaim WD, Kimura H, Tak PP, et al. Interleukin-12 induces salivary gland dysfunction in transgenic mice, providing a new model of Sjögren’s syndrome. Arthritis Rheumatism. (2009) 60:3633–41. doi: 10.1002/art.24980
113. Zong Y, Yang Y, Zhao J, Li L, Luo D, Hu J, et al. Characterisation of macrophage infiltration and polarisation based on integrated transcriptomic and histological analyses in Primary Sjögren’s syndrome. Front Immunol. (2023) 14:1292146. doi: 10.3389/fimmu.2023.1292146
114. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
115. Cutolo M, Campitiello R, Gotelli E, Soldano S. The role of M1/M2 macrophage polarization in rheumatoid arthritis synovitis. Front Immunol. (2022) 13:867260. doi: 10.3389/fimmu.2022.867260
116. Baturone R, Soto M, Márquez M, Macías I, de Oca MM, Medina F, et al. Health-related quality of life in patients with primary Sjögren’s syndrome: relationship with serum levels of proinflammatory cytokines. Scandinavian J Rheumatol. (2009) 38:386–9. doi: 10.1080/03009740902973821
117. Lu X, Li N, Zhao L, Guo D, Yi H, Yang L, et al. Human umbilical cord mesenchymal stem cellss alleviate ongoing autoimmune dacryoadenitis in rabbits via polarizing macrophages into an anti-inflammatory phenotype. Exp Eye Res. (2020) 191:107905. doi: 10.1016/j.exer.2019.107905
118. Rivière E, Chivasso C, Pascaud J, Bechara R, Ly B, Delporte C, et al. Hyperosmolar environment and salivary gland epithelial cellss increase extra-cellsular matrix remodeling and lymphocytic infiltration in Sjögren’s syndrome. Clin Exp Immunol. (2023) 212:39–51. doi: 10.1093/cei/uxad020
119. Ren Y, Cui G, Gao Y. Research progress on inflammatory mechanism of primary Sjögren syndrome. Zhejiang Da Xue Xue Bao Yi Xue Ban. (2021) 50:783–94. doi: 10.3724/zdxbyxb-2021–0072
120. Tzioufas AG, Kapsogeorgou EK, Moutsopoulos HM. Pathogenesis of Sjögren’s syndrome: What we know and what we should learn. J Autoimmunity. (2012) 39:4–8. doi: 10.1016/j.jaut.2012.01.002
121. Manoussakis MN, Kapsogeorgou EK. The role of intrinsic epithelial activation in the pathogenesis of Sjögren’s syndrome. J Autoimmun. (2010) 35:219–24. doi: 10.1016/j.jaut.2010.06.011
122. Vakrakou AG, Svolaki IP, Evangelou K, Gorgoulis VG, Manoussakis MN. cells-autonomous epithelial activation of AIM2 (absent in melanoma-2) inflammasome by cytoplasmic DNA accumulations in primary Sjögren’s syndrome. J Autoimmunity. (2020) 108:102381. doi: 10.1016/j.jaut.2019.102381
123. Jiang J, Zhao M, Chang C, Wu H, Lu Q. Type I interferons in the pathogenesis and treatment of autoimmune diseases. Clin Rev Allergy Immunol. (2020) 59:248–72. doi: 10.1007/s12016–020-08798–2
124. Hsueh PY, Ju Y, Vega A, Edman MC, MacKay JA, Hamm-Alvarez SF. A multivalent ICAM-1 binding nanoparticle which inhibits ICAM-1 and LFA-1 interaction represents a new tool for the investigation of autoimmune-mediated dry eye. Int J Mol Sci. (2020) 21:2758. doi: 10.3390/ijms21082758
125. Fasano S, Mauro D, Macaluso F, Xiao F, Zhao Y, Lu L, et al. Pathogenesis of primary Sjögren’s syndrome beyond B lymphocytes. Clin Exp Rheumatol. (2020) 38 Suppl 126:315–23.
126. Sisto M, Lisi S. New insights into ADAMs regulation of the GRO-α/CXCR2 system: focus on sjögren’s syndrome. Int Rev Immunol. (2015) 34:486–99. doi: 10.3109/08830185.2014.975892
127. Lisi S, Sisto M, Lofrumento DD, D’Amore M, De Lucro R, Ribatti D. A potential role of the GRO-α/CXCR2 system in Sjögren’s syndrome: regulatory effects of pro-inflammatory cytokines. Histochem Cells Biol. (2013) 139:371–9. doi: 10.1007/s00418-012-1035-z
128. Lisi S, D’Amore M, Sisto M. ADAM17 at the interface between inflammation and autoimmunity. Immunol Lett. (2014) 162:159–69. doi: 10.1016/j.imlet.2014.08.008
129. Lisi S, Sisto M, Lofrumento DD, D’Amore M, De Lucro R, Ribatti D. GRO-α/CXCR2 system and ADAM17 correlated expression in Sjögren’s syndrome. Inflammation. (2013) 36:759–66. doi: 10.1007/s10753–013-9602–6
130. Witas R, Peck AB, Ambrus JL, Nguyen CQ. Sjogren’s syndrome and TAM receptors: A possible contribution to disease onset. J Immunol Res. (2019) 2019:4813795. doi: 10.1155/2019/4813795
131. Kyriakidis NC, Kapsogeorgou EK, Gourzi VC, Konsta OD, Baltatzis GE, Tzioufas AG. Toll-like receptor 3 stimulation promotes Ro52/TRIM21 synthesis and nuclear redistribution in salivary gland epithelial cellss, partially via type I interferon pathway. Clin Exp Immunol. (2014) 178:548–60. doi: 10.1111/cei.12432
132. Ainola M, Porola P, Takakubo Y, Przybyla B, Kouri VP, Tolvanen TA, et al. Activation of plasmacytoid dendritic cellss by apoptotic particles - mechanism for the loss of immunological tolerance in Sjögren’s syndrome. Clin Exp Immunol. (2018) 191:301–10. doi: 10.1111/cei.13077
133. Yang Y, Hou Y, Li J, Zhang F, Du Q. Characterization of antiapoptotic microRNAs in primary Sjögren’s syndrome. Cells Biochem Funct. (2020) 38:1111–8. doi: 10.1002/cbf.3569
134. Katsiougiannis S, Tenta R, Skopouli FN. Endoplasmic reticulum stress causes autophagy and apoptosis leading to cellsular redistribution of the autoantigens Ro/Sjögren’s syndrome-related antigen A (SSA) and La/SSB in salivary gland epithelial cellss. Clin Exp Immunol. (2015) 181:244–52. doi: 10.1111/cei.12638
135. Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cells state in development and disease. J Clin Invest. (2009) 119:1438–49. doi: 10.1172/JCI38019
136. Di Gregorio J, Robuffo I, Spalletta S, et al. The epithelial-to-mesenchymal transition as a possible therapeutic target in fibrotic disorders. Front Cells Dev Biol. (2020) 8:607483. doi: 10.3389/fcells.2020.607483
137. Inan S, Hayran M. cells signaling pathways related to epithelial mesenchymal transition in cancer metastasis. Crit Rev Oncog. (2019) 24:47–54. doi: 10.1615/CritRevOncog.2018029509
138. Yao Y, Chen R, Wang G, Zhang Y, Liu F. Exosomes derived from mesenchymal stem cellss reverse EMT via TGF-β1/Smad pathway and promote repair of damaged endometrium. Stem Cells Res Ther. (2019) 10:225. doi: 10.1186/s13287–019-1332–8
139. Hosseini K, Trus P, Frenzel A, Werner C, Fischer-Friedrich E. Skin epithelial cellss change their mechanics and proliferation upon snail-mediated EMT signalling. Soft Matter. (2022) 18:2585–96. doi: 10.1039/D2SM00159D
140. Sisto M, Lorusso L, Ingravallo G, Tamma R, Ribatti D, Lisi S. The TGF-β1 signaling pathway as an attractive target in the fibrosis pathogenesis of sjögren’s syndrome. Mediators Inflamm. (2018) 2018:1965935. doi: 10.1155/2018/1965935
141. Dela Cruz A, Kartha V, Tilston-Lunel A, Mi R, Reynolds TL, Mingueneau M, et al. Gene expression alterations in salivary gland epithelia of Sjögren’s syndrome patients are associated with clinical and histopathological manifestations. Sci Rep. (2021) 11:11154. doi: 10.1038/s41598-021-90569-w
142. Abe S, Tsuboi H, Kudo H, Asashima H, Ono Y, Honda F, et al. M3 muscarinic acetylcholine receptor-reactive Th17 cellss in primary Sjögren’s syndrome. JCI Insight. (2020) 5:e135982, 135982. doi: 10.1172/jci.insight.135982
143. Steinfeld S, Cogan E, King LS, Agre P, Kiss R, Delporte C. Abnormal distribution of aquaporin-5 water channel protein in salivary glands from Sjögren’s syndrome patients. Lab Invest. (2001) 81:143–8. doi: 10.1038/labinvest.3780221
144. Konttinen YT, Tensing EK, Laine M, Porola P, Törnwall J, Hukkanen M. Abnormal distribution of aquaporin-5 in salivary glands in the NOD mouse model for Sjögren’s syndrome. J Rheumatol. (2005) 32:1071–5.
145. Gresz V, Horvath A, Gera I, Nielsen S, Zelles T. Immunolocalization of AQP5 in resting and stimulated normal labial glands and in Sjögren’s syndrome. Oral Dis. (2015) 21:e114–120. doi: 10.1111/odi.12239
146. Lai Z, Yin H, Cabrera-Pérez J, Guimaro MC, Afione S, Michael DG, et al. Aquaporin gene therapy corrects Sjögren’s syndrome phenotype in mice. Proc Natl Acad Sci U S A. (2016) 113:5694–9. doi: 10.1073/pnas.1601992113
147. Sisto M, Lorusso L, Ingravallo G, Nico B, Ribatti D, Ruggieri S, et al. Abnormal distribution of AQP4 in minor salivary glands of primary Sjögren’s syndrome patients. Autoimmunity. (2017) 50:202–10. doi: 10.1080/08916934.2017.1341495
148. Tzartos JS, Stergiou C, Daoussis D, Zisimopoulou P, Andonopoulos AP, Zolota V, et al. Antibodies to aquaporins are frequent in patients with primary Sjögren’s syndrome. Rheumatology. (2017) 56:2114–22. doi: 10.1093/rheumatology/kex328
149. Delporte C, Soyfoo M. Aquaporins: Unexpected actors in autoimmune diseases. Autoimmun Rev. (2022) 21:103131. doi: 10.1016/j.autrev.2022.103131
150. Nguyen CQ, Peck AB. The interferon-signature of sjögren’s syndrome: how unique biomarkers can identify underlying inflammatory and immunopathological mechanisms of specific diseases. Front Immunol. (2013) 4:142. doi: 10.3389/fimmu.2013.00142
151. Wess J. Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu Rev Pharmacol Toxicol. (2004) 44:423–50. doi: 10.1146/annurev.pharmtox.44.101802.121622
152. Iizuka M, Tsuboi H, Asashima H, et al. M3 muscarinic acetylcholine receptor reactive IL-17 producing T cellss promotes development of Sjögren’s syndrome like sialadenitis. Modern Rheumatol. (2015) 25:158–60. doi: 10.3109/14397595.2014.884683
153. Mona M, Mondello S, Hyon JY, Hirota T, Kondo Y, Matsui M, et al. Clinical usefulness of anti-muscarinic type 3 receptor autoantibodies in patients with primary Sjögren’s syndrome. Clin Exp Rheumatol. (2021) 39:795–803. doi: 10.55563/clinexprheumatol/gy6udz
154. Mathian A, Felten R, Alarcon-Riquelme ME, Psarras A, Mertz P, Chasset F, et al. Type 1 interferons: A target for immune-mediated inflammatory diseases (IMIDs). Joint Bone Spine. (2024) 91:105627. doi: 10.1016/j.jbspin.2023.105627
155. Davies R, Hammenfors D, Bergum B, Vogelsang P, Gavasso S, Brun JG, et al. Aberrant cells signalling in PBMCs upon IFN-α stimulation in primary Sjögren’s syndrome patients associates with type I interferon signature. Eur J Immunol. (2018) 48:1217–27. doi: 10.1002/eji.201747213
156. Coccia EM, Battistini A. Early IFN type I response: Learning from microbial evasion strategies. Semin Immunol. (2015) 27:85–101. doi: 10.1016/j.smim.2015.03.005
157. Maria NI, Steenwijk EC, IJpma AS, van Helden-Meeuwsen CG, Vogelsang P, Beumer W, et al. Contrasting expression pattern of RNA-sensing receptors TLR7, RIG-I and MDA5 in interferon-positive and interferon-negative patients with primary Sjögren’s syndrome. Ann Rheum Dis. (2017) 76:721–30. doi: 10.1136/annrheumdis-2016–209589
158. Apostolou E, Kapsogeorgou EK, Konsta OD, Giotakis I, Saridaki MI, Andreakos E, et al. Expression of type III interferons (IFNλs) and their receptor in Sjögren’s syndrome. Clin Exp Immunol. (2016) 186:304–12. doi: 10.1111/cei.12865
159. Thompson N, Isenberg DA, Jury EC, Ciurtin C. Exploring BAFF: its expression, receptors and contribution to the immunopathogenesis of Sjögren’s syndrome. Rheumatol (Oxford). (2016) 55:1548–55. doi: 10.1093/rheumatology/kev420
160. Kiripolsky J, Kramer JM. Current and emerging evidence for toll-like receptor activation in sjögren’s syndrome. J Immunol Res. (2018) 2018:1246818. doi: 10.1155/2018/1246818
161. Nakamura H, Horai Y, Shimizu T, Kawakami A. Modulation of apoptosis by cytotoxic mediators and cells-survival molecules in sjögren’s syndrome. Int J Mol Sci. (2018) 19:2369. doi: 10.3390/ijms19082369
162. Manoussakis MN, Spachidou MP, Maratheftis CI. Salivary epithelial cellss from Sjogren’s syndrome patients are highly sensitive to anoikis induced by TLR-3 ligation. J Autoimmun. (2010) 35:212–8. doi: 10.1016/j.jaut.2010.06.010
163. Kiripolsky J, Romano RA, Kasperek EM, Yu G, Kramer JM. Activation of myd88-dependent TLRs mediates local and systemic inflammation in a mouse model of primary sjögren’s syndrome. Front Immunol. (2019) 10:2963. doi: 10.3389/fimmu.2019.02963
164. Karlsen M, Jonsson R, Brun JG, Appel S, Hansen T. TLR-7 and -9 stimulation of peripheral blood B cellss indicate altered TLR signalling in primary sjögren’s syndrome patients by increased secretion of cytokines. Scand J Immunol. (2015) 82:523–31. doi: 10.1111/sji.12368
165. Wang Y, Roussel-Queval A, Chasson L, Hanna Kazazian N, Marcadet L, Nezos A, et al. TLR7 signaling drives the development of sjögren’s syndrome. Front Immunol. (2021) 12:676010. doi: 10.3389/fimmu.2021.676010
166. Yoshimoto K, Suzuki K, Takei E, Ikeda Y, Takeuchi T. Elevated expression of BAFF receptor, BR3, on monocytes correlates with B cells activation and clinical features of patients with primary Sjögren’s syndrome. Arthritis Res Ther. (2020) 22:157. doi: 10.1186/s13075–020-02249–1
167. Salazar-Camarena DC, Ortíz-Lazareno P, Marín-Rosales M, Cruz A, Muñoz-Valle F, Tapia-Llanos R, et al. BAFF-R and TACI expression on CD3+ T cellss: Interplay among BAFF, APRIL and T helper cytokines profile in systemic lupus erythematosus. Cytokine. (2019) 114:115–27. doi: 10.1016/j.cyto.2018.11.008
168. Fu J, Shi H, Zhan T, Li H, Ye L, Xie L, et al. BST-2/Tetherin is involved in BAFF-enhanced proliferation and survival via canonical NF-κB signaling in neoplastic B-lymphoid cellss. Exp Cells Res. (2021) 398:112399. doi: 10.1016/j.yexcr.2020.112399
169. Xin P, Xu X, Deng C, Deng C, Liu S, Wang Y, et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol. (2020) 80:106210. doi: 10.1016/j.intimp.2020.106210
170. Aota K, Yamanoi T, Kani K, Ono S, Momota Y, Azuma M. Inhibition of JAK-STAT signaling by baricitinib reduces interferon-γ-induced CXCL10 production in human salivary gland ductal cellss. Inflammation. (2021) 44:206–16. doi: 10.1007/s10753-020-01322-w
171. Ciccia F, Guggino G, Rizzo A, Bombardieri M, Raimondo S, Carubbi F, et al. Interleukin (IL)-22 receptor 1 is over-expressed in primary Sjogren’s syndrome and Sjögren-associated non-Hodgkin lymphomas and is regulated by IL-18. Clin Exp Immunol. (2015) 181:219–29. doi: 10.1111/cei.12643
172. Vartoukian SR, Tilakaratne WM, Seoudi N, Bombardieri M, Bergmeier L, Tappuni AR, et al. Dysregulation of the suppressor of cytokine signalling 3-signal transducer and activator of transcription-3 pathway in the aetiopathogenesis of Sjögren’s syndrome. Clin Exp Immunol. (2014) 177:618–29. doi: 10.1111/cei.12377
173. Wakamatsu E, Matsumoto I, Yasukochi T, Naito Y, Goto D, Mamura M, et al. Overexpression of phosphorylated STAT-1alpha in the labial salivary glands of patients with Sjögren’s syndrome. Arthritis Rheumatol. (2006) 54:3476–84. doi: 10.1002/art.22176
174. Pertovaara M, Silvennoinen O, Isomäki P. Cytokine-induced STAT1 activation is increased in patients with primary Sjögren’s syndrome. Clin Immunol. (2016) 165:60–7. doi: 10.1016/j.clim.2016.03.010
175. Charras A, Arvaniti P, Le Dantec C, Arleevskaya MI, Zachou K, Dalekos GN, et al. JAK inhibitors suppress innate epigenetic reprogramming: a promise for patients with sjögren’s syndrome. Clin Rev Allergy Immunol. (2020) 58:182–93. doi: 10.1007/s12016-019-08743-y
176. Kang KY, Kim HO, Kwok SK, Ju JH, Park KS, Sun DI, et al. Impact of interleukin-21 in the pathogenesis of primary Sjögren’s syndrome: increased serum levels of interleukin-21 and its expression in the labial salivary glands. Arthritis Res Ther. (2011) 13:R179. doi: 10.1186/ar3504
177. Loureiro-Amigo J, Franco-Jarava C, Perurena-Prieto J, Palacio C, Martínez-Valle F, Soláns-Laqué R. Serum CXCL13, BAFF, IL-21 and IL-22 levels are related to disease activity and lymphocyte profile in primary Sjögren’s syndrome. Clin Exp Rheumatol. (2021) 39 Suppl 133:131–9. doi: 10.55563/clinexprheumatol/fp741f
178. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. (2020) 369:1633–7. doi: 10.1126/science.abb9818
179. Khakh BS, Alan North R. P2X receptors as cells-surface ATP sensors in health and disease. Nature. (2006) 442:527–32. doi: 10.1038/nature04886
180. Nakamoto T, Brown DA, Catalán MA, Gonzalez-Begne M, Romanenko VG, Melvin JE. Purinergic P2X7 receptors mediate ATP-induced saliva secretion by the mouse submandibular gland. J Biol Chem. (2009) 284:4815–22. doi: 10.1074/jbc.M808597200
181. Novak I, Jans IM, Wohlfahrt L. Effect of P2X 7 receptor knockout on exocrine secretion of pancreas, salivary glands and lacrimal glands: P2X 7 receptor in exocrine glands. J Physiol. (2010) 588:3615–27. doi: 10.1113/jphysiol.2010.190017
182. Jiang S, Zhang Y, Zheng JH, Li X, Yao YL, Wu YL, et al. Potentiation of hepatic stellate cells activation by extracellsular ATP is dependent on P2X7R-mediated NLRP3 inflammasome activation. Pharmacol Res. (2017) 117:82–93. doi: 10.1016/j.phrs.2016.11.040
183. Zhang GX, Wang MX, Nie W, Liu DW, Zhang Y, Liu HB. P2X7R blockade prevents NLRP3 inflammasome activation and pancreatic fibrosis in a mouse model of chronic pancreatitis. Pancreas. (2017) 46:1327–35. doi: 10.1097/MPA.0000000000000928
184. Shin JI, Lee KH, Joo YH, Lee JM, Jeon J, Jung HJ, et al. Inflammasomes and autoimmune and rheumatic diseases: A comprehensive review. J Autoimmunity. (2019) 103:102299. doi: 10.1016/j.jaut.2019.06.010
185. Baldini C, Rossi C, Ferro F, Santini E, Seccia V, Donati V, et al. The P2X 7 receptor–inflammasome complex has a role in modulating the inflammatory response in primary S jögren’s syndrome. J Intern Med. (2013) 274:480–9. doi: 10.1111/joim.12115
186. Khalafalla MG, Woods LT, Camden JM, Khan AA, Limesand KH, Petris MJ, et al. P2X7 receptor antagonism prevents IL-1β release from salivary epithelial cellss and reduces inflammation in a mouse model of autoimmune exocrinopathy. J Biol Chem. (2017) 292:16626–37. doi: 10.1074/jbc.M117.790741
187. Bombardieri M, Barone F, Pittoni V, Alessandri C, Conigliaro P, Blades MC, et al. Increased circulating levels and salivary gland expression of interleukin-18 in patients with Sjögren’s syndrome: relationship with autoantibody production and lymphoid organization of the periductal inflammatory infiltrate. Arthritis Res Ther. (2004) 6:R447. doi: 10.1186/ar1209
188. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. cells. (2009) 139:871–90. doi: 10.1016/j.cells.2009.11.007
189. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. (2012) 18:1028–40. doi: 10.1038/nm.2807
190. Ewert P, Aguilera S, Alliende C, Quest AF, Castro I, Cortés J, et al. Disruption of tight junction structure in salivary glands from Sjögren’s syndrome patients is linked to proinflammatory cytokine exposure. Arthritis Rheumatism. (2010) 62:1280–9. doi: 10.1002/art.27362
191. Barrera MJ, Bahamondes V, Sepúlveda D, Quest AF, Castro I, Cortés J, et al. Sjögren’s syndrome and the epithelial target: A comprehensive review. J Autoimmunity. (2013) 42:7–18. doi: 10.1016/j.jaut.2013.02.001
Keywords: Sjögren’s syndrome, salivary gland damage, pathomechanisms, cellular, molecular biology
Citation: Qi W, Tian J, Wang G, Yan Y, Wang T, Wei Y, Wang Z, Zhang G, Zhang Y and Wang J (2024) Advances in cellular and molecular pathways of salivary gland damage in Sjögren’s syndrome. Front. Immunol. 15:1405126. doi: 10.3389/fimmu.2024.1405126
Received: 22 March 2024; Accepted: 28 May 2024;
Published: 10 July 2024.
Edited by:
Efstathia K. Kapsogeorgou, National and Kapodistrian University of Athens, GreeceReviewed by:
Margherita Sisto, University of Bari Aldo Moro, ItalyUma Sriram, Temple University, United States
Copyright © 2024 Qi, Tian, Wang, Yan, Wang, Wei, Wang, Zhang, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Wang, MTI1NjkzNTg0NEBxcS5jb20=