 Jiao Xu
Jiao Xu Qingyue Zeng
Qingyue Zeng Shuangqing Li1
Shuangqing Li1 Qiaoli Su
Qiaoli Su Hong Fan
Hong Fan- 1General Practice Medical Center, West China Hospital, Sichuan University, Chengdu, China
- 2Department of Respiratory and Critical Care Medicine, West China Hospital, Sichuan University, Chengdu, China
Chronic obstructive pulmonary disease (COPD) is a common respiratory disease characterized by irreversible progressive airflow limitation, often manifested by persistent cough, sputum production and other respiratory symptoms that pose a serious threat to human health and affect the quality of life of patients. The disease is associated with chronic inflammation, which is associated with the onset and progression of COPD, but anti-inflammatory therapy is not first-line treatment. Inflammation has multiple manifestations and phenotypes, and this heterogeneity reveals different patterns of inflammation, making treatment difficult. This paper aims to explore the direction of more effective anti-inflammatory treatment by analyzing the nature of inflammation and the molecular mechanism of disease occurrence and development in COPD patients, and to provide new ideas for the treatment of COPD patients.
1 Introduction
Chronic obstructive pulmonary disease (COPD) is the third cause of death in the world, characterized by high morbidity and mortality, and has become a major disease burden worldwide (1, 2). Chronic inflammation plays an important role in the occurrence and development of COPD, mainly affecting the lung parenchyma and surrounding airway, leading to persistent respiratory symptoms and irreversible progressive airflow restriction (3). This chronic inflammation is characterized by an increase in the number of cells, including macrophages, lymphocytes, and neutrophils, which are mainly located in the pulmonary blood vessels, peripheral airways, and lung parenchyma. In some patients, there may also be an increase in the number of cells, such as eosinophils, T helper 2 (Th2) cells, or type 2 intrinsic lymphocytes (ILC2). These cells can release different inflammatory mediators together with other structural cells (4). Oxidative stress plays a key role in COPD related inflammation, as well as in smokers. COPD patients may also experience systemic inflammation, which exacerbates the severity of cardiovascular, endocrine and metabolic complications. Accelerated lung aging in COPD patients also causes senescent cells in the lung to release inflammatory proteins, including tumor necrosis factor alpha (TNF-α), Interleukin (IL)-1, IL-6, chemokine (C-X-C motif) ligand 8 (CXCL8), chemokine (C-C motif) ligand 2 (CCL2) and matrix metalloproteinases (MMPs). In the progress of diagnosis and treatment of diseases, it is very important to identify the disease phenotype with the best curative effect, and it is also important to explore biomarkers to identify this disease phenotype (4).
2 Pathology of COPD
The two main pathological types of COPD are small airway disease due to fibrosis around the bronchioles and emphysema due to destruction of the alveolar walls, which can be distinguished by computed tomography (CT) examination. Although some patients mainly present with small airway disease or emphysema, most patients have a mixed condition (5). These pathological changes are mainly caused by chronic inflammation. Inhalation of harmful gases or particles can cause inflammation of lung parenchyma and respiratory tract. Various inflammatory factors can lead to tissue destruction and then emphysema, damage the defense and repair function of respiratory tract, and eventually lead to small airway fibrosis and progressive irreversible airflow obstruction (6) (Figure 1). Usually, the more severe COPD symptoms are, the inflammation of the airway is also correspondingly increased and continues after smoking cessation (1). Even in mild patients, peripheral airway obstruction and loss can occur (7). The emphysema observed in smokers first appears close to the thickened and narrowed bronchioles that are the main obstructions in COPD, and the mechanism by which the small airways thicken so close to the lung tissue destroyed by emphysema remains unclear (6).
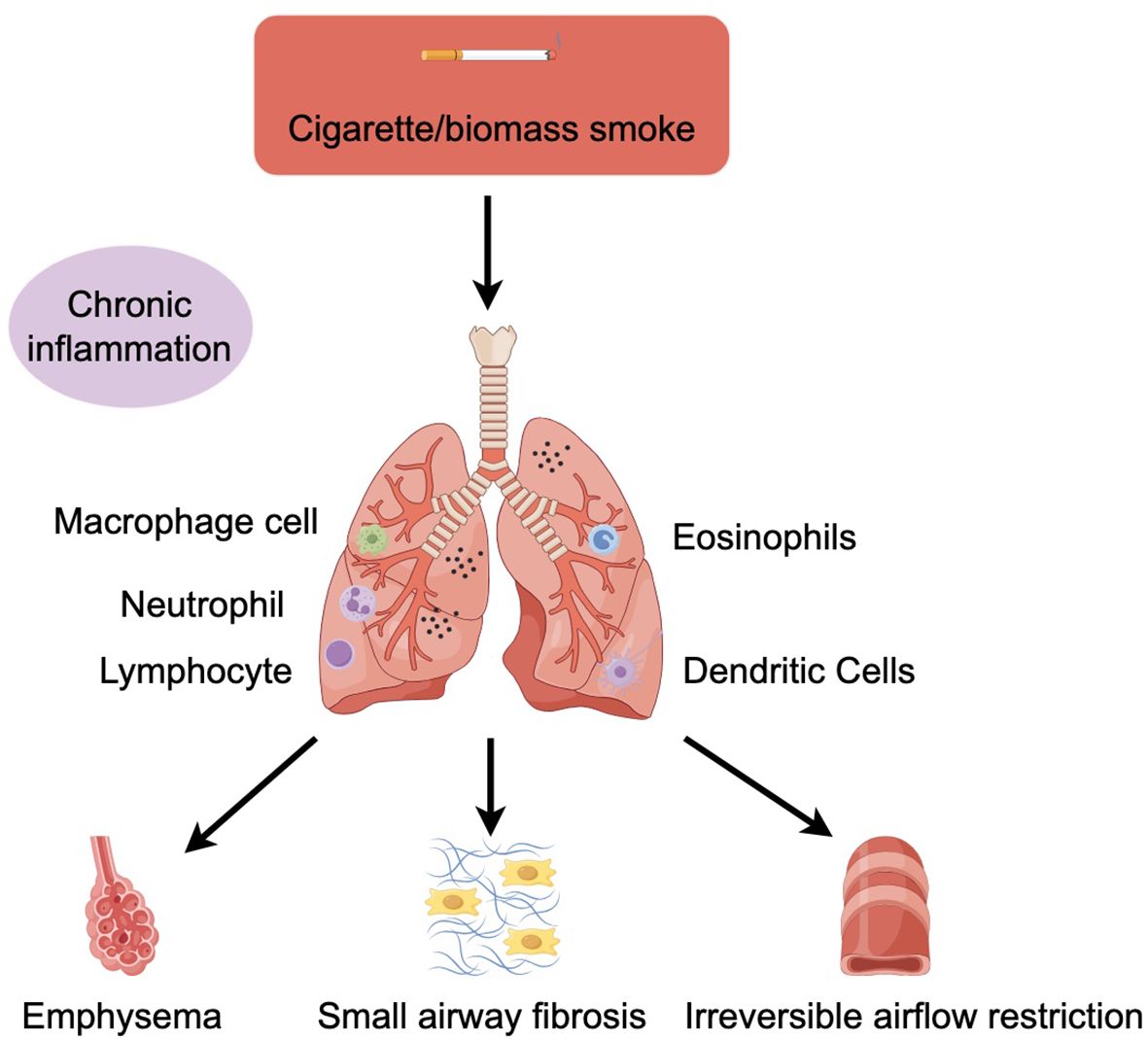
Figure 1. COPD is mainly associated with cigarette/biomass smoke. Cigarette/biomass smoke can cause inflammation of the lung parenchyma and airways, activating macrophages, neutrophil, lymphocyte, eosinophils, and dendritic cells in response to toxic particles in the smoke. Various inflammatory factors can lead to tissue destruction and then to emphysema, impairing the defence and repair function of the airways and ultimately leading to small airway fibrosis and progressive irreversible airflow limitation.
3 Characteristics of inflammation associated with COPD
Patients with COPD often have a characteristic inflammatory pattern, with a significant increase in the number of macrophages, neutrophils, T lymphocytes and B lymphocytes in airway secretions (8–10). This pattern of inflammation involves both innate and acquired immune responses (i.e. cellular and humoral immunity), which are linked by passive activation of dendritic cells. Barnes et al. (4) showed that COPD patients had a more severe inflammatory response than smokers without airway obstruction, and this inflammatory pattern, once established, persisted even after smoking cessation. Although smoking is a major environmental risk factor for COPD, only a proportion of smokers develop the disease (11). This may be due to differences in the response to smoking among individuals, including factors such as genetic susceptibility, epigenetic changes, and oxidative stress (10). These factors may amplify the inflammation caused by smoking. Studies have shown that inhalation of harmful gases or particles can stimulate macrophages and airway epithelial cells to release a variety of chemokines, and monocytes, neutrophils and lymphocytes can accumulate in the lungs under the influence of a variety of chemokines. This inflammation is still present in people who have stopped smoking. So far, the mechanism of this phenomenon is not clear (12). It has been suggested that this persistent inflammation may be related to abnormalities in the regulation of the immune system and individual differences (4).
3.1 Inflammatory cells
The formation of lung inflammation in patients with COPD is mainly associated with congenital and adaptive immunity, among them, the congenital immune contains neutrophils and macrophages, eosinophils and mast cells, natural killer cells, γδ T cells, such as inflammation, fine dominant immune response, adaptive immunity refers to T and B lymphocytes or dominant immune response. In this group of patients, the number of macrophages, neutrophils, and lymphocytes is increased, and these cells are mainly located in the pulmonary blood vessels, peripheral airway, and parenchyma of the lung. However, some patients may also show an increase in the number of eosinophils, ILC2, or Th2 cells. At the same time, airway and alveolar epithelial cells and endothelial cells and other structural cells are also activated. Inflammatory cells can release different inflammatory mediators together with other cells (such as epithelial cells and other structural cells) (4). Studies have confirmed that the lack of local IgA can indirectly lead to bacterial translocation and small airway inflammation, which develops into airway remodeling and eventually irreversible airflow restriction (13).
3.2 Macrophages
Macrophages play an important role in the chronic inflammation of COPD patients (Figure 2). The number of macrophages in the bronchoalveolar lavage fluid (BALF) and sputum of COPD patients can be significantly increased, typically reaching 5-10 times higher than normal values. Macrophages often accumulate at sites of alveolar wall destruction, a phenomenon we can observe in patients with emphysema and COPD, where the number of macrophages correlates with the severity of emphysema. Studies of this phenomenon have shown that macrophages are activated by factors such as cigarette smoke extracts and release inflammatory mediators such as TNF-α, CXCL1, CXCL8, CCL2, leukotriene (LT) B4 and reactive oxygen species (ROS), etc., which mediate the inflammatory response (14). Large increases in macrophages are seen in COPD patients and smokers and are mainly associated with increased circulating chemokines CCL2 and CXCL1, both of which are also found to be significantly elevated in the sputum and BALF of COPD patients (15).
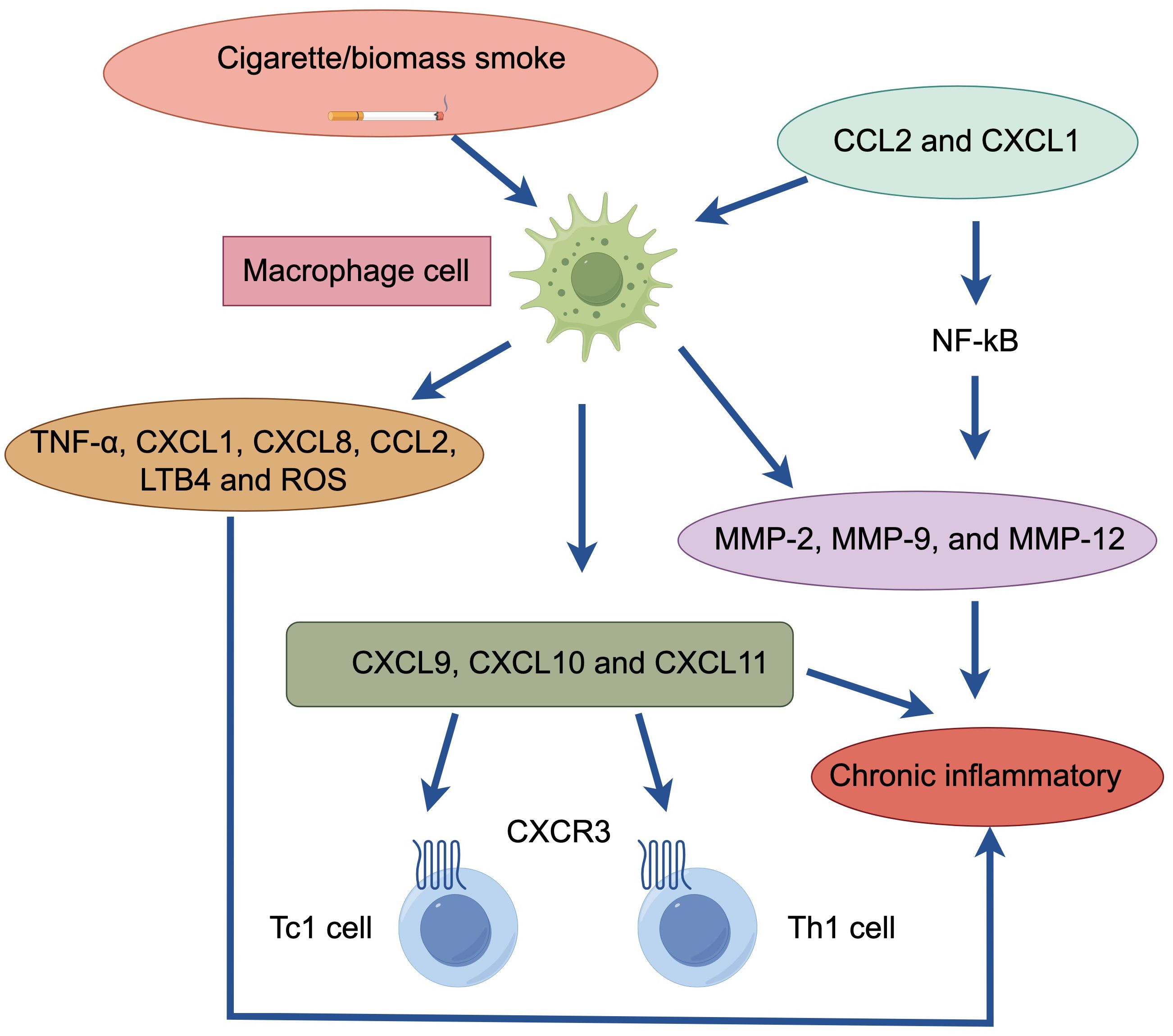
Figure 2. Macrophage cell in patients with COPD. Cigarette/biomass smoke activate macrophages. These macrophages release inflammatory mediators (TNF-α, CXCL1, CXCL8, CCL2, LTB4, ROS) and produce elastolytic enzymes (MMP-2, MMP-9, MMP-12), regulated by the transcription factor NF-κB. The chemokines (CCL2, CXCL1) produced by macrophages enhance the chemotactic response of monocytes, which respond strongly to CXCL1, further attracting macrophages. Additionally, macrophages generate chemotactic effects on CD8+ Tc1 and CD4+ Th1 cells (via CXCL9, CXCL10, CXCL11). This cascade of events ultimately leads to chronic inflammation.
Macrophages have different phenotypes with different activation pathways that lead to different organismal responses. Previous studies have shown that mouse M1-type macrophages are pro-inflammatory, whereas M2-type macrophages are anti-inflammatory, release IL-10, and exhibit significant phagocytic activity (16). However, these differences were not evident in human macrophages, probably because the macrophages in COPD patients are mainly M1-type, but further studies are needed to prove this (17).
Macrophages can secrete elastolytic enzymes, including MMP-2, MMP-9, and MMP-12 (18). MMP-9 is produced by alveolar macrophages and is one of the molecules that mediates the inflammatory response in COPD patients. Compared to smokers, alveolar macrophages in COPD patients secrete more inflammatory proteins and have higher elastolytic activity, suggesting increased activation of macrophages in COPD patients (19). Much of the upregulation of inflammatory proteins is regulated by the transcription factor NF-κB in macrophages, which is extensively activated when the disease is exacerbated, resulting in massive production of inflammatory proteins (20). This inflammatory protein can also be further secreted upon exposure to cigarette smoke, thereby accelerating the rate of inflammation. Macrophages mediate this mechanism even when kept continuously in culture for more than 3 days, so that macrophages in smokers and nonsmokers are fundamentally different (4).
Monocytes from COPD patients exhibit a stronger chemotactic response to CXCL1 than monocytes from smokers and non-smokers (21). In addition, macrophages can also exert chemotactic effects on CD8+ Tc1 and CD4+ Th1 cells by binding to CXCR3, a chemokine receptor expressed by CD8+ Tc1 and CD4+ Th1 cells, to release CXCL9, CXCL10 and CXCL11 chemokines (22).
It has been shown that in healthy individuals and smokers, corticosteroids inhibit the release of CXCL8, TNF-α and MMP-9 from macrophages, thereby alleviating the inflammatory response, but in patients with COPD, where the inflammatory response is mainly mediated by cytokines, chemokines and proteases, corticosteroid use is ineffective (23) and fails to work in macrophages of COPD patients, showing a relatively ineffective (19). Resistance to corticosteroids in COPD patients may be due to decreased histone deacetylase (HDAC) 2 activity due to increased secretion of cytokines (e.g., TNF-α and CXCL8) in macrophages (24).
Both alveolar-derived macrophages and monocyte-derived macrophages in patients with COPD exhibit reduced phagocytic uptake of bacteria, which may be the result of long-term colonization of the lower airways by bacteria such as Haemophilus influenzae or Streptococcus pneumoniae (25), which is present in at least 50% of patients with COPD and may increase the risk of acute exacerbation of COPD, in addition to causing chronic inflammation (26). In addition, macrophage phagocytosis, such as phagocytosis of apoptotic cells, is also defective in COPD patients, which may contribute to the persistence of inflammation in COPD patients (27). The cause of the defective phagocytosis of macrophages is still unclear, and some findings suggest that it may be related to the defective function of cellular microtubules, which are closely related to cytophagy (28).
3.3 Neutrophils
Increased sputum and BALF neutrophilia is a typical feature of COPD, and this neutrophil-induced inflammation can caused by regular exposure to cigarette smoke, infectious agent and oxidative stress (5). Neutrophil recruitment to the airways and parenchyma involves initial adhesion to endothelial cells through E-selectin, which is upregulated on endothelial cells in the airways of patients with COPD. Adherent neutrophils migrate into the respiratory tract under the direction of various neutrophil chemotactic factors, including LTB4, CXCL1, CXCL5, and CXCL8, levels of which are increased in airways of patients with COPD (4).
Neutrophils secrete several serine proteases including neutrophil elastase (NE), cathepsin G, proteinase-3, MMPs and myeloperoxidase (MPO) which may contribute to the alveolar damage (4, 29). Alpha‐1 antitrypsin (AAT) is a serum inhibitor of serine proteinases. AAT deficiency (AATD), which is due to a genetic abnormality in the antiproteases, is associated with neutrophilic inflammation in the lung and accounts for less than 1% of COPD patients (5). AATD that results in early onset emphysema, usually in smokers, due to inactivate NE and other serine proteases (30). Despite these theoretical mechanisms, however, only a few patients have AATD, and, thus, it has been difficult to explain why the lung is not normally protected in the majority of COPD patients.
It has been suggested that the size and extent of the neutrophil traffic may be related to the pathological changes that occur in individuals with COPD in case of emphysema (31). The NE is stored within neutrophil azurophil granules which diffuse away from the granule until the concentration has fallen sufficiently for the enzyme to be completely inactivated by the local concentration of AAT. AAT limits rather than prevents neutrophil protease-related damage. It binds to proteases via a one-to-one molar ratio. However, at the location where a degranulating neutrophil is located, the concentration of NE is much higher than that of AAT. This results in obligatory tissue damage around each degranulating cell until the concentration of protease is reduced by diffusion into the local tissue environment. Thus, the role of AAT is to slow down rather than completely stop the damage caused by these proteases (32). This theoretical relationship may explain not only the tissue destruction that occurs in the absence of AATD, but also the extensive destruction seen in deficient subjects. It can be predicted that in people who do not have AATD, there will always be some tissue damage in the immediate vicinity of a degranulating neutrophil. When the AAT concentrations are significantly reduced (as in patients with AATD), the area of damage that will occur can be predicted to be far greater. This would explain not only the increased susceptibility of patients with AATD to tissue damage, but it also provides an explanation for the development of similar but limited damage in non-AATD subjects.
The above findings have implications for the precise treatment of COPD. The World Health Organization (WHO) recommends that especially in areas with a high prevalence of AATD, all patients with COPD should be screened for AATD (1). AAT augmentation therapy with infusion of human plasma derived purified AAT has been shown to slow progression (33) (Figure 3).
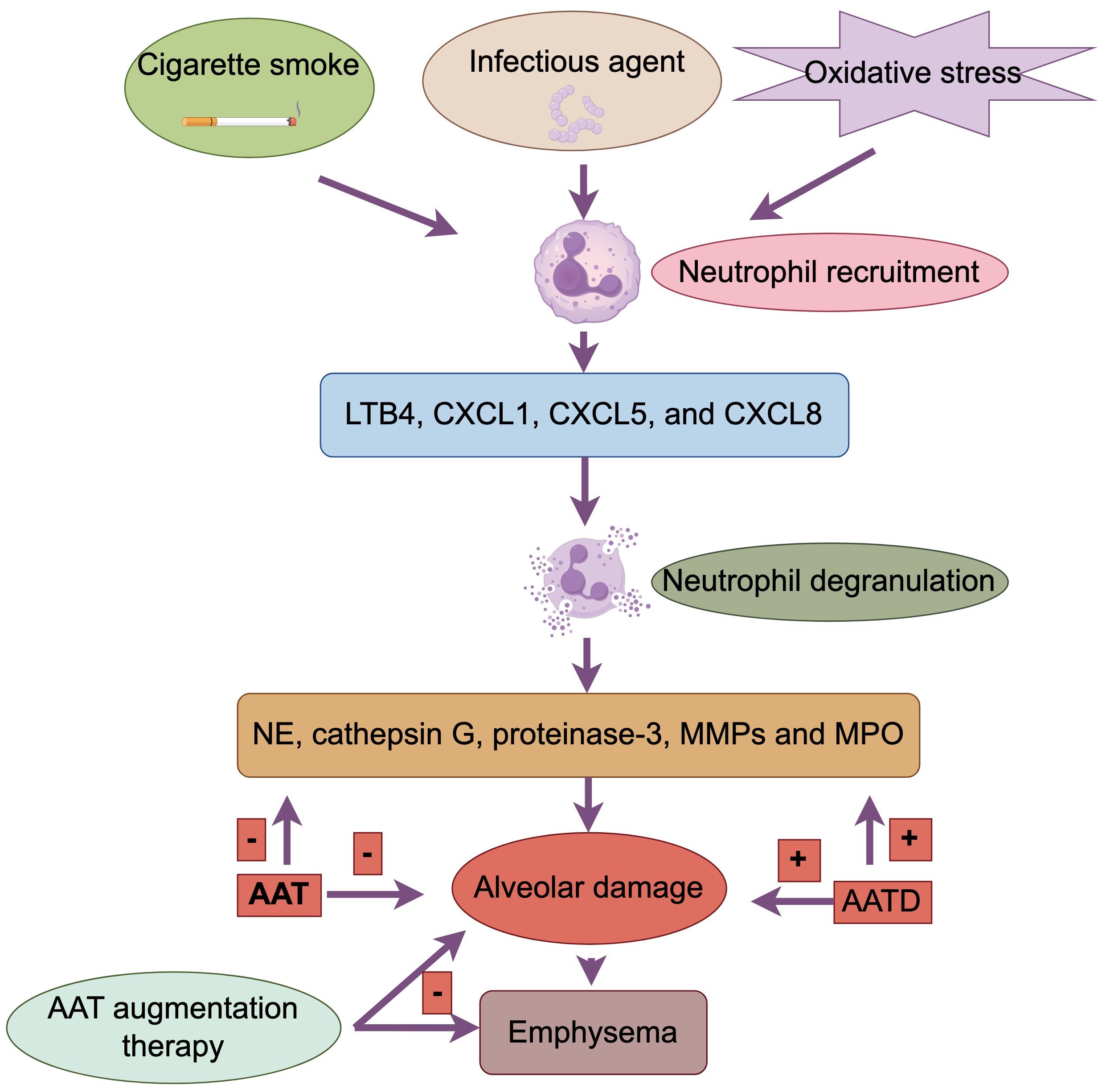
Figure 3. Neutrophil in patients with COPD. Cigarette smoke, infectious agents, and oxidative stress induce neutrophilia. Neutrophils migrate to the respiratory tract under the guidance of neutrophil chemotactic factors (LTB4, CXCL1, CXCL5, CXCL8). Neutrophils secrete serine proteases (NE, cathepsin G, proteinase-3, MMPs, MPO), leading to alveolar damage. Alpha-1 antitrypsin (AAT) inhibits serine proteases. AAT deficiency (AATD) results in insufficient inactivation of NE and other proteases, causing greater tissue damage. AAT, by binding to proteases, slows but does not completely prevent tissue damage. AAT augmentation therapy can slow the progression of COPD.
3.4 Eosinophils
As a heterogeneous lung lesion, COPD has multiple subtypes. With the deeper understanding of the inflammatory mechanisms of COPD, it has been found that neutrophilic inflammation is the most common type of inflammation, but 20% to 40% of patients also belong to the type 2 inflammatory endotype, characterized by elevated eosinophils (EOS) (34, 35). It is generally accepted that type 2 inflammation is characterized by upregulation of the expression of type 2 cytokines, such as IL-4, IL-5, and IL-13, produced by Th2 and ILC2 (36). Currently, the mechanism of airway type 2 inflammation formation in COPD is unclear. Some studies suggest that granulocyte-macrophage colony-stimulating factor (GM-CSF) and CCL5 secreted by the airway epithelium in patients with COPD may promote the survival and recruitment of EOS in the airways (37). Acute exacerbations are more frequent in patients with COPD characterized by type 2 inflammation compared with the predominantly neutrophil-driven, non-type 2 inflammatory endotype (35).
The differences in eosinophil counts may determine different responses to inhaled corticosteroid (ICS) therapy (38). By modeling eosinophil count as a continuous variable, a post hoc analysis showed that ICS-containing regimens had little to no effect when blood eosinophil count was less than 100 cells/μL, and thus this threshold could be used to identify patients with a low likelihood of benefit from treatment with ICS (39). In addition, decreased eosinophils in blood and sputum were associated with an increase in proteobacteria, primarily Haemophilus, as well as an increase in bacterial infections and pneumonia (40, 41). ICS are anti-inflammatory drugs used in combination with one or two long-acting bronchodilators (LABDs) for the treatment of COPD. Studies have shown that ICS reduce exacerbation rates, improve quality of life, and prevent mortality in COPD patients with a history of exacerbations (42, 43). Accordingly, GOLD recommends the use of eosinophil testing for COPD patients with a history of exacerbations in clinical practice, despite their appropriate use of LABDs, to identify patients who are best suited to receive ICS therapy (1). The threshold of a blood eosinophil count ≥ 300 cells/μL identifies the top of the continuous relationship between eosinophils and ICS, and can be used to identify patients with the greatest likelihood of treatment benefit with ICS. The thresholds of < 100 cells/μL and ≥ 300 cells/μL should be regarded as estimates, rather than precise cut-off values, that can predict different probabilities of treatment benefit (38, 44). There is no clear evidence that ICS treatment reduces blood eosinophil count, so it retains their predictive value independent of ICS treatment.
The pathogenesis of patients with COPD is complex, and although it is currently recommended that group E patients with blood eosinophil count ≥300/μL should be treated with the triple therapy of “ICS + LAMA + LABA”, some patients are still poorly controlled after the triple therapy [1]. The New England Journal recently published a new clinical study revealing the efficacy of using dupilumab in the treatment of patients with COPD with type 2 inflammatory features. Dupilumab, a fully human monoclonal antibody, blocks the shared receptor component for IL-4 and IL-13, key drivers of type 2 inflammation. The results showed that in COPD patients with type 2 inflammation as indicated by elevated blood eosinophil counts, those who received dupilumab had fewer exacerbations, better lung function and quality of life, and less severe respiratory symptoms than those who received placebo (45) (Figure 4). In summary, biologically targeted therapy for type 2 inflammation may be a new treatment option.
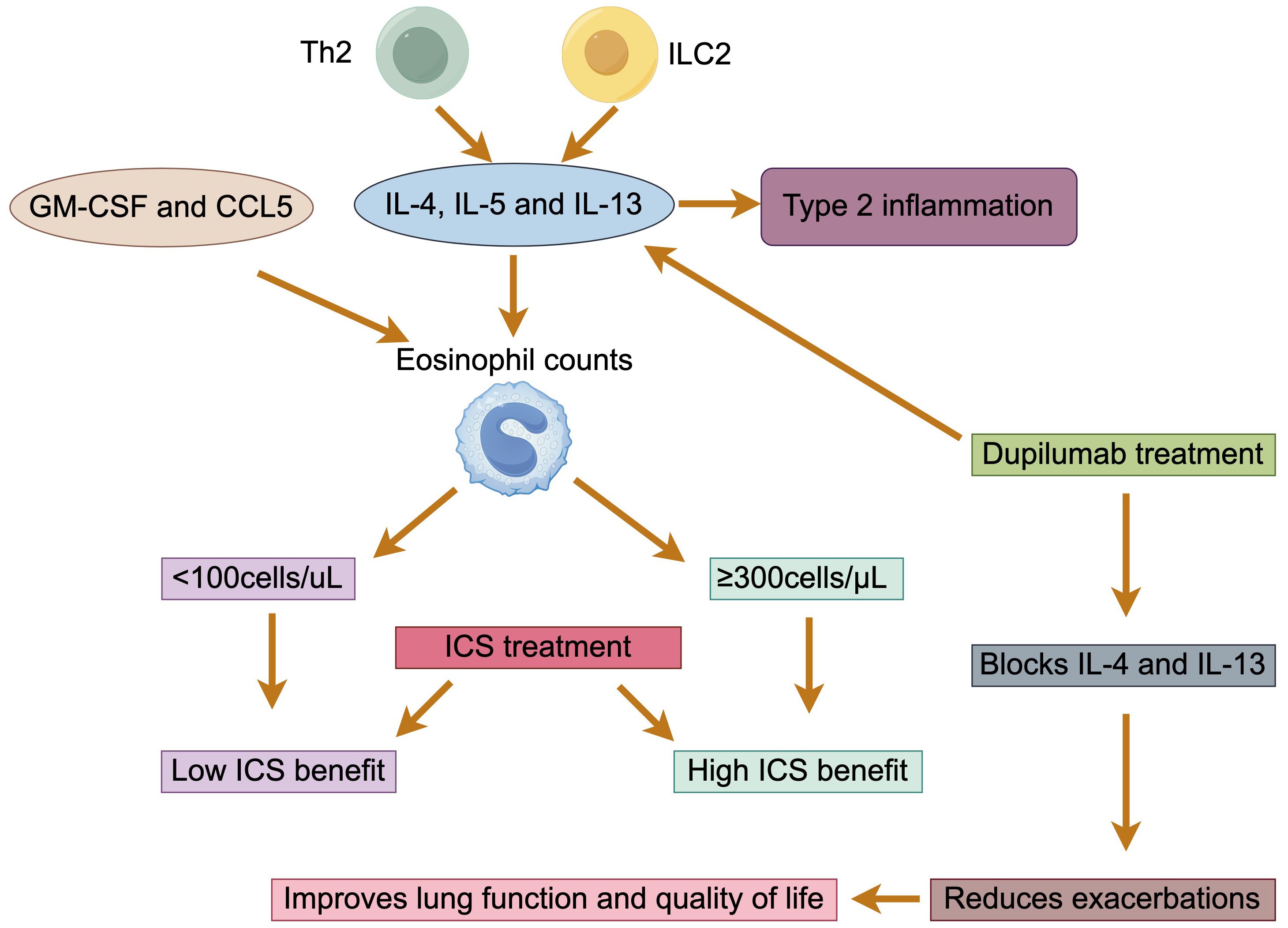
Figure 4. Eosinophils in patients with COPD. The type 2 inflammatory endotype is characterized by the production of IL-4, IL-5, and IL-13 by Th2 and ILC2 cells. GM-CSF and CCL5 facilitate the survival and recruitment of eosinophils. The effectiveness of ICS therapy is correlated with eosinophil counts: less than 100 cells/μL indicates low effectiveness, while counts of 300 cells/μL or higher indicate high effectiveness. Dupilumab, which blocks IL-4 and IL-13, can reduce exacerbations and improve both lung function and quality of life.
3.5 Lymphocytes
3.5.1 T lymphocytes
Lymphocytes play an important role in the mechanism of COPD. Smoking status, degree of airflow obstruction and emphysema were all associated with increased CD8+ cell counts and/or CD8+/CD4+ ratios (46). Numerous studies have found increased numbers of CD8+ T-lymphocytes in the blood and lower respiratory tract tissues of patients with COPD compared to controls, as well as in sputum and BAL, but the number of lymphocytes in these secretions is insignificant and very difficult to count (46, 47). CD4+ T cells differentiate into different functional T cell subsets upon activation, including Th1, Th2, Th9, Th17, Th22, Tfh, and regulatory T (Treg) cells, and CD8+ T cells also differentiate into diverse subsets, including Tc1, Tc2, Tc9, Tc17, Tc22 cells, Tfcs, and suppressive CD8 Tregs (48), of which Th1/Tc1, Th2/Tc2, Th17, Treg are closely associated with COPD (49).
Th1/Tc1 cells can be differentiated under IL-12 conditions and produce IFNγ through activation of the transcription factors, such as T-bet and STAT4. Th2/Tc2 cells can be differentiated under IL-4 conditions and produce IL-4, IL-5 and IL-13 through GATA-3 and GATA-3, STAT6, respectively (48). Data have shown that Th1 and Tc1 were significantly higher, while Th2 and Tc2 were significantly lower in the peripheral blood of acute exacerbation of COPD (AECOPD) patients. This is because Th1 cells secrete IFN-γ to inhibit the proliferation of Th2 cells, which aggravates the pathological damage caused by Th1-mediated protective immunity after imbalance and promotes the pulmonary inflammatory response in COPD patients. The increase in the number of Th1 cells is also able to induce the proliferation of Tc1 cells, which in turn inhibits the proliferation of Tc2 cells (47). In peripheral small airways, Th1/Tc1 cells can secrete and release IFN-γ to act on alveolar macrophages and attract the infiltration of neutrophils (47, 50). Since Th2/Tc2 cells secrete an abundance of type II cytokines that can promote IgE production and the recruitment of pathogenic cells, such as eosinophils, these cells impose significant influence on allergy reactions, particularly in the respiratory tract (49, 51).
Th17 cells are characterized by the release of proinflammatory cytokines, such as IL-17A, IL-17F, IL-21 and IL-22, which are associated with COPD progression and the exacerbation of alveolar destruction (52, 53). They are found mainly in the bronchial mucosa and express the transcription factor RORγt as a specific marker (54). The IL-17A and IL-17F cytokines are predominantly released by Th17 cells, which are linked to neutrophilic inflammation, as IL-17 induces airway epithelial cells to release neutrophil chemotactic chemokines such as CXC-chemokine ligand 1 (CXCL1) and CXCL8, attracting neutrophils into the airways (55). In contrast, Treg cells are responsible for the regulation of immune responses by suppressing inflammation and autoimmunity through the release of anti-inflammatory cytokines, such as IL-10 and transforming growth factor-β (TGF-β) (46). Intracellular expression of Foxp3 is currently considered as the most specific marker for Treg cells and these suppressive functions are dependent on the expression of the transcription factor Foxp3 (56). In the absence of IL-10, the IL-23 levels may increase, leading to their differentiation into Th17 cells (52, 57).
Studies over the past decade has emphasized the importance of maintaining a balance between Th17 cells and Treg cells in controlling the inflammatory response in COPD (58). An increased Th17 response and Th17/Treg ratio play critical roles in the progression of COPD (52, 59). Wang et al. (60) showed that patients with moderate and severe COPD had a higher frequency of Th17 cells, elevated levels of RORγt mRNA expression and increased serum levels of IL-17A, IL-6, IL-21, IL-22 and IL-23. The authors also showed a lower frequency of Treg cells, and decreased Foxp3 mRNA expression and serum level of IL-10 in those patients. Additionally, it was demonstrated that the increase in the Th17/Treg ratio was negatively correlated with the worsening of lung function in those patients. In accordance, flow cytometric analysis revealed a significantly higher Th17/Treg ratio in the COPD group compared to non-smoking patients (59).
T-lymphocyte-mediated dysfunction of cellular immunity may have an impact in the course of AECOPD, participating in and contributing to its deterioration (47). Blood immunophenotypic analysis showed that COPD patients with low circulating lymphocyte counts (especially reduced CD4+ T-cells) were more likely to experience acute exacerbations, including persistent exacerbation (61). Similarly, Freeman et al. (62) showed that both CD4+ and CD8+ T cells were significantly reduced in AECOPD, which may indicate extravasation of T cells to sites of inflammation or organized lymphoid tissue, and then increased to a constant level at steady state. They also concluded that a decrease in CD4+ and CD8+ T cells frequency could be a potential marker of AECOPD as it precedes the onset of symptoms. However, larger studies are needed to verify this possibility.
3.5.2 B lymphocytes
In the lungs of COPD patients, the number of B-lymphocytes is increased, especially in patients with severe disease (4). B lymphocytes may secrete autoantibodies against oxidized extracellular matrix proteins or against endothelial cells (46). Proteins that have been modified by carbonyl groups due to oxidative stress can stimulate the production of auto-antibodies (63). Serum anti-carbonyl-modified auto-protein antibody titers were significantly increased in patients with severe COPD compared to controls. Antibody levels were negatively correlated with disease severity and were predominantly of the IgG1 type (63). B-lymphocyte infiltration into the submucosal layer of the small airways of COPD patients has been reported, and lymphoid aggregates containing well-defined follicles and germinal centers increase with the severity of COPD disease (8, 64). In patients with very severe COPD, an increased number of B cells was found only in the connective tissue of the patients’ small airways compared to controls (65). Lymphoid follicles in the small airways of COPD patients consist of a large number of B-lymphocytes scattered with CD21+ and CD35+ follicular dendritic cells (66). B-cell activating factor plays a crucial role in regulating the function and proliferation of B-cells, and its levels are elevated in the lymphoid follicles of individuals with COPD (67). These changes may be related to the inflammatory response and disease progression in COPD.
3.6 Dendritic cells
Dendritic Cells (DCs) also play an important role in the pathogenesis of COPD. DCs are an important component of the immune system, mainly involved in initiating the immune response and regulating the innate immune response (68). In patients with COPD, the number of DCs in the airways is altered, and smoking causes DCs to release inflammatory chemokines, such as CCL20, which play an important role in the pathogenesis of COPD (69). Studies have shown that dendritic cell accumulation exists in the bronchial mucosa of patients who smoke and COPD, and that cigarette smoke alters dendritic cell maturation and function, thereby affecting the immune response in the lungs (70). Specifically, cigarette smoke activates epithelial cells and macrophages, causing them to release a variety of chemokines that attract and activate inflammatory cells in the lungs, including DCs (69, 71). These inflammatory cells, along with macrophages, polymorphonuclear leukocytes (PMN), and epithelial cells, release proteases such as MMP and NE, leading to elastin degradation and emphysema formation (69, 72). In addition, DCs, by releasing pro-inflammatory chemokines, may be involved in maintaining neutrophilic airway inflammation, which in turn leads to emphysematous lung tissue destruction (69). DCs are not only involved in the triggering of COPD pathogenesis, but may also play an important role in the pathologic process of COPD by modulating effector CD8+ T cell responses (69). Interventions targeting the function of DCs may provide new avenues for the treatment of COPD.
4 Oxidative stress
Oxidative stress is strongly associated with COPD. Smoking or dust irritation leads to lung cell damage, excessive mucus secretion and neutrophil accumulation generating large amounts of ROS, which oxidatively inactivate antiproteinases and destroy lung tissue structure. The accumulation of neutrophils also activates a large number of inflammatory factors, generating more ROS and exacerbating oxidative stress, especially during exacerbations (73, 74). Increased oxidative stress in the lungs of COPD patients has been demonstrated by measuring various markers of oxidative stress in breath. For example, ethane, a volatile product of lipid peroxidation, is increased in the exhaled breath of COPD patients and correlates with disease severity (75). COPD patients also had increased concentrations of H2O2, malondialdehyde, 4HNE (4-hydroxy-2-nonenal), and 8-isoprostane in exhaled breath condensate (76–79), and these markers increased further during exacerbations (80, 81). These increased markers of oxidative stress remained elevated even in ex-smokers, suggesting that they derive from endogenous oxidative stress and may be a reflection of ongoing lung inflammation (79). The transcription factors FOXO3a (Forkhead box O3a) and Nrf2 (nuclear factor erythroid 2-related factor 2) regulate multiple antioxidant genes, both of which are reduced in COPD lungs (82, 83).
There is growing evidence of mitochondrial dysfunction in COPD (84). This is manifested by increased mitochondrial mass, fusion, and leaky mitochondrial membranes, phenomena that result from an impaired autophagic mechanism that scavenges damaged mitochondria (i.e., mitochondrial autophagy) (85). Abnormally functioning leaky mitochondria may be a major source of ROS in COPD (86, 87).
A number of drugs have been developed that can selectively target mitochondria, but no clinical studies have been reported in patients with COPD. Given that mitochondrial dysfunction may be a major source of mitochondrial ROS in COPD, mitochondria-targeted antioxidants may have great therapeutic potential (88).
5 Systemic inflammation
Patients with COPD, especially during acute exacerbations, are often complicated by systemic inflammation, which often manifests itself in elevated levels of cytokines, chemokines, and inflammatory proteins or abnormal cell counts (89, 90). In addition to COPD patients, systemic inflammation can be found in patients who smoke, as evidenced by elevated systemic inflammatory markers such as C-reactive protein (CRP), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-alpha), activated leukocytes, and fibrinogen; however, the degree of systemic inflammation is more pronounced in COPD patients (4, 91, 92). In any case, systemic inflammation in patients with COPD may contribute to their systemic manifestations and may exacerbate comorbid diseases (4). The results of a large population-based study showed that an increase in systemic inflammatory markers was associated with an increased risk of developing diabetes, cardiovascular disease, and lung cancer (93). Patients with persistent systemic inflammation have a higher mortality rate and more frequent episodes of acute exacerbation (4).
Several studies have identified the presence of potentially pathogenic microorganisms (PPM) in the lower respiratory tract of patients with stable COPD, which may lead to systemic inflammation and increased serum C-reactive protein (CRP), interleukin-8 (IL-8) levels and plasma fibrinogen (FIB) (26, 94, 95). Long-term antibiotic therapy reduces the number of acute exacerbations of COPD and improves health-related quality of life, but it also increases the risk of bacterial resistance (96). Short-term antibiotic therapy reduces the risk of bacterial resistance and drug-related side effects, but its effectiveness is inconsistent. Some studies have shown that short-term oral antibiotics can reduce airway bacterial load, decrease airway inflammation, and even eliminate PPM for a short period of time, but the bacteria rapidly recolonize. However, most studies have found that short-term antibiotic therapy does not reduce airway inflammation or decrease acute exacerbations of COPD, and may even lead to increased bacterial resistance (97). In conclusion, the outlook for the treatment of systemic inflammation remains challenging and further research is needed to clarify the efficacy of antibiotic therapy and to explore new therapeutic approaches to reduce systemic inflammation and improve patients’ quality of life.
In addition to antibiotics, statin is an important intervention for addressing systemic inflammation. Simvastatin does not prevent exacerbations in patients with COPD who do not have metabolic or cardiovascular indications for statin therapy, according to a large, multicenter, randomized trial (98). However, in an observational study of patients with COPD treated with statins for cardiovascular and metabolic indications, an association between statin use and improved outcomes, including exacerbations and reduced mortality, was reported (99). In addition, a cohort study including 950 COPD outpatients found no association between statin use and risk of AECOPD or all-cause mortality (100). This result adds to the evidence suggesting that statin therapy should not be initiated at the outset in COPD solely for prevention, but should be prescribed in line with current cardiovascular disease guidelines, particularly to reduce mortality.
6 Conclusion
The inflammation pathogenesis of COPD is complex, mainly related to inflammatory cell, oxidative stress, and systemic inflammation. COPD encompasses several different clinical and pathophysiologic phenotypes, and it is essential to identify COPD phenotypes that are efficacious for specific therapies, which includes identifying disease types and biomarker testing. On the basis of the foregoing, AAT augmentation therapy and mitochondria-targeted antioxidants may have great therapeutic potential. Corticosteroids have shown more significant efficacy in COPD patients with eosinophilic inflammation. However, roflumilast, a phosphodiesterase-4 inhibitor, appears to be more effective in patients with a neutrophilic phenotype, which may become a therapeutic agent for the neutrophil-mediated inflammatory response (1, 101, 102). In patients prone to acute exacerbations, studies have shown that regular use of certain antibiotics such as azithromycin or erythromycin for one year reduces the risk of acute exacerbations (103). In addition to antibiotics, statin is an important intervention for addressing systemic inflammation in COPD.
Therefore, there is a greater need to use specific biomarkers to predict the effect of treatment, such as specific proteins like serine proteases. While much of the focus and difficulty is on patients with advanced disease, numerous studies have shown that patients in the early stages of the disease have a greater therapeutic benefit, and if effective, precise and safe anti-inflammatory therapies can be developed for different phenotypes of COPD, intervening in the early stages of the disease will show greater benefit, preventing disease progression and reducing the burden of disease.
Author contributions
JX: Conceptualization, Writing – original draft. QZ: Conceptualization, Writing – original draft. SL: Supervision, Writing – review & editing. QS: Investigation, Writing – review & editing. HF: Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Key Research and Development Program of China (2022YFC3601503) and the Key Research and Development Project of Sichuan Province (2022YFS0261).
Acknowledgments
Figures were made by Figdraw.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. GLOBAL STRATEGY FOR THE DIAGNOSIS, MANAGEMENT, AND PREVENTION OF CHRONIC OBSTRUCTIVE PULMONARY DISEASE (2023 REPORT) (2023). Available online at: https://goldcopd.org/2023-gold-report-2/.
2. Halpin DMG, Celli BR, Criner GJ, Frith P, López Varela MV, Salvi S, et al. The GOLD Summit on chronic obstructive pulmonary disease in low- and middle-income countries. Int J Tuberc Lung Dis. (2019) 23:1131–41. doi: 10.5588/ijtld.19.0397
3. Brightling C, Greening N. Airway inflammation in COPD: progress to precision medicine. Eur Respir J. (2019) 54. doi: 10.1183/13993003.00651-2019
4. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2016) 138:16–27. doi: 10.1016/j.jaci.2016.05.011
6. Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. (2009) 4:435–59. doi: 10.1146/annurev.pathol.4.110807.092145
7. McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. (2011) 365:1567–75. doi: 10.1056/NEJMoa1106955
8. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
9. Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. (2008) 8:183–92. doi: 10.1038/nri2254
10. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. (2011) 378:1015–26. doi: 10.1016/S0140-6736(11)60988-4
11. Yang IA, Jenkins CR, Salvi SS. Chronic obstructive pulmonary disease in never-smokers: risk factors, pathogenesis, and implications for prevention and treatment. Lancet Respir Med. (2022) 10:497–511. doi: 10.1016/S2213-2600(21)00506-3
12. Gamble E, Grootendorst DC, Hattotuwa K, O'Shaughnessy T, Ram FS, Qiu Y, et al. Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: a pooled analysis. Eur Respir J. (2007) 30:467–71. doi: 10.1183/09031936.00013006
13. Polosukhin VV, Richmond BW, Du RH, Cates JM, Wu P, Nian H, et al. Secretory IgA deficiency in individual small airways is associated with persistent inflammation and remodeling. Am J Respir Crit Care Med. (2017) 195:1010–21. doi: 10.1164/rccm.201604-0759OC
14. Barnes PJ. Alveolar macrophages as orchestrators of COPD. Copd. (2004) 1:59–70. doi: 10.1081/COPD-120028701
15. Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax. (2002) 57:590–5. doi: 10.1136/thorax.57.7.590
16. Gordon S, Plűddemann A. Tissue macrophage heterogeneity: issues and prospects. Semin Immunopathol. (2013) 35:533–40. doi: 10.1007/s00281-013-0386-4
17. Chana KK, Fenwick PS, Nicholson AG, Barnes PJ, Donnelly LE. Identification of a distinct glucocorticosteroid-insensitive pulmonary macrophage phenotype in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2014) 133:207–16.e1-11. doi: 10.1016/j.jaci.2013.08.044
18. Russell RE, Thorley A, Culpitt SV, Dodd S, Donnelly LE, Demattos C, et al. Alveolar macrophage-mediated elastolysis: roles of matrix metalloproteinases, cysteine, and serine proteases. Am J Physiol Lung Cell Mol Physiol. (2002) 283:L867–73. doi: 10.1152/ajplung.00020.2002
19. Culpitt SV, Rogers DF, Shah P, De Matos C, Russell RE, Donnelly LE, et al. Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2003) 167:24–31. doi: 10.1164/rccm.200204-298OC
20. Caramori G, Romagnoli M, Casolari P, Bellettato C, Casoni G, Boschetto P, et al. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax. (2003) 58:348–51. doi: 10.1136/thorax.58.4.348
21. Traves SL, Smith SJ, Barnes PJ, Donnelly LE. Specific CXC but not CC chemokines cause elevated monocyte migration in COPD: a role for CXCR2. J Leukoc Biol. (2004) 76:441–50. doi: 10.1189/jlb.1003495
22. Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. (2004) 1:e8. doi: 10.1371/journal.pmed.0010008
23. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2013) 131:636–45. doi: 10.1016/j.jaci.2012.12.1564
24. Barnes PJ. Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol. (2009) 71:451–64. doi: 10.1146/annurev.physiol.010908.163257
25. Taylor AE, Finney-Hayward TK, Quint JK, Thomas CM, Tudhope SJ, Wedzicha JA, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J. (2010) 35:1039–47. doi: 10.1183/09031936.00036709
26. Singh R, Mackay AJ, Patel AR, Garcha DS, Kowlessar BS, Brill SE, et al. Inflammatory thresholds and the species-specific effects of colonising bacteria in stable chronic obstructive pulmonary disease. Respir Res. (2014) 15:114. doi: 10.1186/s12931-014-0114-1
27. Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. (2003) 81:289–96. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x
28. Donnelly LE, Barnes PJ. Defective phagocytosis in airways disease. Chest. (2012) 141:1055–62. doi: 10.1378/chest.11-2348
29. DI Stefano A, Gnemmi I, Dossena F, Ricciardo FL, Maniscalco M, Lo Bello F, et al. Pathogenesis of COPD at the cellular and molecular level. Minerva Med. (2022) 113:405–23. doi: 10.23736/S0026-4806.22.07927-7. A, D.I.S.
30. Henao MP, Craig TJ. Understanding alpha-1 antitrypsin deficiency: A review with an allergist's outlook. Allergy Asthma Proc. (2017) 38:98–107. doi: 10.2500/aap.2017.38.4027
31. Stockley RA. Neutrophils and the pathogenesis of COPD. Chest. (2002) 121:151s–5s. doi: 10.1378/chest.121.5_suppl.151S
32. Sapey E. Neutrophil modulation in alpha-1 antitrypsin deficiency. Chronic Obstr Pulm Dis. (2020) 7:247–59. doi: 10.15326/jcopdf.7.3.2019.0164
33. Chapman KR, Chorostowska-Wynimko J, Koczulla AR, Ferrarotti I, McElvaney NG. Alpha 1 antitrypsin to treat lung disease in alpha 1 antitrypsin deficiency: recent developments and clinical implications. Int J Chron Obstruct Pulmon Dis. (2018) 13:419–32. doi: 10.2147/COPD
34. Yousuf A, Ibrahim W, Greening NJ, Brightling CE. T2 biologics for chronic obstructive pulmonary disease. J Allergy Clin Immunol Pract. (2019) 7:1405–16. doi: 10.1016/j.jaip.2019.01.036
35. David B, Bafadhel M, Koenderman L, De Soyza A. Eosinophilic inflammation in COPD: from an inflammatory marker to a treatable trait. Thorax. (2021) 76:188–95. doi: 10.1136/thoraxjnl-2020-215167
36. Gandhi NA, Bennett BL, Graham NM, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discovery. (2016) 15:35–50. doi: 10.1038/nrd4624
37. Costa C, Rufino R, Traves SL, Lapa Silva E JR, Barnes PJ, Donnelly LE. CXCR3 and CCR5 chemokines in induced sputum from patients with COPD. Chest. (2008) 133:26–33. doi: 10.1378/chest.07-0393
38. Singh D, Agusti A, Martinez FJ, Papi A, Pavord ID, Wedzicha JA, et al. Blood eosinophils and chronic obstructive pulmonary disease: A global initiative for chronic obstructive lung disease science committee 2022 review. Am J Respir Crit Care Med. (2022) 206:17–24. doi: 10.1164/rccm.202201-0209PP
39. Bafadhel M, Peterson S, De Blas MA, Calverley PM, Rennard SI, Richter K, et al. Predictors of exacerbation risk and response to budesonide in patients with chronic obstructive pulmonary disease: a post-hoc analysis of three randomised trials. Lancet Respir Med. (2018) 6:117–26. doi: 10.1016/S2213-2600(18)30006-7
40. Dicker AJ, Huang JTJ, Lonergan M, Keir HR, Fong CJ, Tan B, et al. The sputum microbiome, airway inflammation, and mortality in chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2021) 147:158–67. doi: 10.1016/j.jaci.2020.02.040
41. Martinez-Garcia MA, Faner R, Oscullo G, de la Rosa D, Soler-Cataluña JJ, Ballester M, et al. Inhaled steroids, circulating eosinophils, chronic airway infection, and pneumonia risk in chronic obstructive pulmonary disease. A network analysis. Am J Respir Crit Care Med. (2020) 201:1078–85. doi: 10.1164/rccm.201908-1550OC
42. Agusti A, Fabbri LM, Singh D, Vestbo J, Celli B, Franssen FME, et al. Inhaled corticosteroids in COPD: friend or foe? Eur Respir J. (2018) 52. doi: 10.1183/13993003.01219-2018
43. Lipson DA, Barnhart F, Brealey N, Brooks J, Criner GJ, Day NC, et al. Once-daily single-inhaler triple versus dual therapy in patients with COPD. N Engl J Med. (2018) 378:1671–80. doi: 10.1056/NEJMoa1713901
44. Lea S, Higham A, Beech A, Singh D. How inhaled corticosteroids target inflammation in COPD. Eur Respir Rev. (2023) 32. doi: 10.1183/16000617.0084-2023
45. Bhatt SP, Rabe KF, Hanania NA, Vogelmeier CF, Cole J, Bafadhel M, et al. Dupilumab for COPD with type 2 inflammation indicated by eosinophil counts. N Engl J Med. (2023) 389:205–14. doi: 10.1056/NEJMoa2303951
46. Caramori G, Casolari P, Barczyk A, Durham AL, Di Stefano A, Adcock I. COPD immunopathology. Semin Immunopathol. (2016) 38:497–515. doi: 10.1007/s00281-016-0561-5
47. Xue W, Ma J, Li Y, Xie C. Role of CD(4) (+) T and CD(8) (+) T lymphocytes-mediated cellular immunity in pathogenesis of chronic obstructive pulmonary disease. J Immunol Res. (2022) 2022:1429213. doi: 10.1155/2022/1429213
48. Koh CH, Lee S, Kwak M, Kim BS, Chung Y. CD8 T-cell subsets: heterogeneity, functions, and therapeutic potential. Exp Mol Med. (2023) 55:2287–99. doi: 10.1038/s12276-023-01105-x
49. Qin K, Xu B, Pang M, Wang H, Yu B. The functions of CD4 T-helper lymphocytes in chronic obstructive pulmonary disease. Acta Biochim Biophys Sin (Shanghai). (2022) 54:173–8. doi: 10.3724/abbs.2021009
50. Kemeny DM, Vyas B, Vukmanovic-Stejic M, Thomas MJ, Noble A, Loh LC, et al. CD8(+) T cell subsets and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (1999) 160:S33–7. doi: 10.1164/ajrccm.160.supplement_1.10
51. St Paul M, Ohashi PS. The roles of CD8(+) T cell subsets in antitumor immunity. Trends Cell Biol. (2020) 30:695–704. doi: 10.1016/j.tcb.2020.06.003
52. Lourenço JD, Ito JT, Martins MA, Tibério IFLC, Lopes FDTQDS. Th17/Treg imbalance in chronic obstructive pulmonary disease: clinical and experimental evidence. Front Immunol. (2021) 12:804919. doi: 10.3389/fimmu.2021.804919
53. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. (2008) 28:454–67. doi: 10.1016/j.immuni.2008.03.004
54. Di Stefano A, Caramori G, Gnemmi I, Contoli M, Vicari C, Capelli A, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. (2009) 157:316–24. doi: 10.1111/j.1365-2249.2009.03965.x
55. Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol. (2010) 72:495–516. doi: 10.1146/annurev-physiol-021909-135926
56. Isajevs S, Taivans I, Strazda G, Kopeika U, Bukovskis M, Gordjusina V, et al. Decreased FOXP3 expression in small airways of smokers with COPD. Eur Respir J. (2009) 33:61–7. doi: 10.1183/09031936.00145307
57. Saraiva M, Vieira P, O'Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med. (2020) 217. doi: 10.1084/jem.20190418
58. Thomas R, Qiao S, Yang X. Th17/Treg imbalance: implications in lung inflammatory diseases. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24054865
59. Zheng X, Zhang L, Chen J, Gu Y, Xu J, Ouyang Y. Dendritic cells and Th17/Treg ratio play critical roles in pathogenic process of chronic obstructive pulmonary disease. BioMed Pharmacother. (2018) 108:1141–51. doi: 10.1016/j.biopha.2018.09.113
60. Wang H, Ying H, Wang S, Gu X, Weng Y, Peng W, et al. Imbalance of peripheral blood Th17 and Treg responses in patients with chronic obstructive pulmonary disease. Clin Respir J. (2015) 9:330–41. doi: 10.1111/crj.12147
61. Ryu MH, Yun JH, Morrow JD, Saferali A, Castaldi P, Chase R, et al. Blood gene expression and immune cell subtypes associated with chronic obstructive pulmonary disease exacerbations. Am J Respir Crit Care Med. (2023) 208:247–55. doi: 10.1164/rccm.202301-0085OC
62. Freeman CM, Martinez CH, Todt JC, Martinez FJ, Han MK, Thompson DL, et al. Acute exacerbations of chronic obstructive pulmonary disease are associated with decreased CD4+ & CD8+ T cells and increased growth & differentiation factor-15 (GDF-15) in peripheral blood. Respir Res. (2015) 16:94. doi: 10.1186/s12931-015-0251-1
63. Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2011) 184:796–802. doi: 10.1164/rccm.201010-1605OC
64. Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. (2004) 364:709–21. doi: 10.1016/S0140-6736(04)16900-6
65. Olloquequi J, Ferrer J, Montes JF, Rodríguez E, Montero MA, García-Valero J. Differential lymphocyte infiltration in small airways and lung parenchyma in COPD patients. Respir Med. (2010) 104:1310–8. doi: 10.1016/j.rmed.2010.03.002
66. Brusselle GG, Demoor T, Bracke KR, Brandsma CA, Timens W. Lymphoid follicles in (very) severe COPD: beneficial or harmful? Eur Respir J. (2009) 34:219–30. doi: 10.1183/09031936.00150208
67. Seys LJ, Verhamme FM, Schinwald A, Hammad H, Cunoosamy DM, Bantsimba-Malanda C, et al. Role of B cell-activating factor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 192:706–18. doi: 10.1164/rccm.201501-0103OC
68. Peters M, Peters K, Bufe A. Regulation of lung immunity by dendritic cells: Implications for asthma, chronic obstructive pulmonary disease and infectious disease. Innate Immun. (2019) 25:326–36. doi: 10.1177/1753425918821732
69. Givi ME, Redegeld FA, Folkerts G, Mortaz E. Dendritic cells in pathogenesis of COPD. Curr Pharm Des. (2012) 18:2329–35. doi: 10.2174/138161212800166068
70. Vassallo R, Tamada K, Lau JS, Kroening PR, Chen L. Cigarette smoke extract suppresses human dendritic cell function leading to preferential induction of Th-2 priming. J Immunol. (2005) 175:2684–91. doi: 10.4049/jimmunol.175.4.2684
71. Kroening PR, Barnes TW, Pease L, Limper A, Kita H, Vassallo R. Cigarette smoke-induced oxidative stress suppresses generation of dendritic cell IL-12 and IL-23 through ERK-dependent pathways. J Immunol. (2008) 181:1536–47. doi: 10.4049/jimmunol.181.2.1536
72. Bracke KR, D'hulst AI, Maes T, Moerloose KB, Demedts IK, Lebecque S, et al. Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficient mice. J Immunol. (2006) 177:4350–9. doi: 10.4049/jimmunol.177.7.4350
73. Noguera A, Batle S, Miralles C, Iglesias J, Busquets X, MacNee W, et al. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax. (2001) 56:432–7. doi: 10.1136/thorax.56.6.432
74. Guo P, Li R, Piao TH, Wang CL, Wu XL, Cai HY. Pathological mechanism and targeted drugs of COPD. Int J Chron Obstruct Pulmon Dis. (2022) 17:1565–75. doi: 10.2147/COPD.S366126
75. Paredi P, Kharitonov SA, Leak D, Ward S, Cramer D, Barnes PJ. Exhaled ethane, a marker of lipid peroxidation, is elevated in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2000) 162:369–73. doi: 10.1164/ajrccm.162.2.9909025
76. Bartoli ML, Novelli F, Costa F, Malagrinò L, Melosini L, Bacci E, et al. Malondialdehyde in exhaled breath condensate as a marker of oxidative stress in different pulmonary diseases. Mediators Inflammation. (2011) 2011:891752. doi: 10.1155/2011/891752
77. Corradi M, Pignatti P, Manini P, Andreoli R, Goldoni M, Poppa M, et al. Comparison between exhaled and sputum oxidative stress biomarkers in chronic airway inflammation. Eur Respir J. (2004) 24:1011–7. doi: 10.1183/09031936.04.00002404
78. Dekhuijzen PN, Aben KK, Dekker I, Aarts LP, Wielders PL, van Herwaarden CL, et al. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (1996) 154:813–6. doi: 10.1164/ajrccm.154.3.8810624
79. Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, et al. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med. (2000) 162:1175–7. doi: 10.1164/ajrccm.162.3.2001063
80. Biernacki WA, Kharitonov SA, Barnes PJ. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax. (2003) 58:294–8. doi: 10.1136/thorax.58.4.294
81. Paredi P, Kharitonov SA, Barnes PJ. Analysis of expired air for oxidation products. Am J Respir Crit Care Med. (2002) 166:S31–7. doi: 10.1164/rccm.2206012
82. Barnes PJ, Baker J, Donnelly LE. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med. (2019) 200:556–64. doi: 10.1164/rccm.201810-1975TR
83. Hwang JW, Rajendrasozhan S, Yao H, Chung S, Sundar IK, Huyck HL, et al. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J Immunol. (2011) 187:987–98. doi: 10.4049/jimmunol.1001861
84. Birch J, Barnes PJ, Passos JF. Mitochondria, telomeres and cell senescence: Implications for lung ageing and disease. Pharmacol Ther. (2018) 183:34–49. doi: 10.1016/j.pharmthera.2017.10.005
85. Cloonan SM, Choi AM. Mitochondria in lung disease. J Clin Invest. (2016) 126:809–20. doi: 10.1172/JCI81113
86. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2015) 136:769–80. doi: 10.1016/j.jaci.2015.01.046
87. Belchamber KBR, Singh R, Batista CM, Whyte MK, Dockrell DH, Kilty I, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J. (2019) 54. doi: 10.1183/13993003.02244-2018
88. Barnes PJ. Oxidative stress-based therapeutics in COPD. Redox Biol. (2020) 33:101544. doi: 10.1016/j.redox.2020.101544
89. Gan WQ, Man SF, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. (2004) 59:574–80. doi: 10.1136/thx.2003.019588
90. Agustí A, Edwards LD, Rennard SI, MacNee W, Tal-Singer R, Miller BE, et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PloS One. (2012) 7:e37483. doi: 10.1371/journal.pone.0037483
91. Çolak Y, Afzal S, Lange P, Nordestgaard BG. Smoking, systemic inflammation, and airflow limitation: A Mendelian randomization analysis of 98 085 individuals from the general population. Nicotine Tob Res. (2019) 21:1036–44. doi: 10.1093/ntr/nty077
92. Strollo HC, Nouraie SM, Hoth KF, Riley CM, Karoleski C, Zhang Y, et al. Association of systemic inflammation with depressive symptoms in individuals with COPD. Int J Chron Obstruct Pulmon Dis. (2021) 16:2515–22. doi: 10.2147/COPD.S322144
93. Thomsen M, Dahl M, Lange P, Vestbo J, Nordestgaard BG. Inflammatory biomarkers and comorbidities in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2012) 186:982–8. doi: 10.1164/rccm.201206-1113OC
94. Marin A, Garcia-Aymerich J, Sauleda J, Belda J, Millares L, García-Núñez M, et al. Effect of bronchial colonisation on airway and systemic inflammation in stable COPD. Copd. (2012) 9:121–30. doi: 10.3109/15412555.2011.636407
95. Banerjee D, Khair OA, Honeybourne D. Impact of sputum bacteria on airway inflammation and health status in clinical stable COPD. Eur Respir J. (2004) 23:685–91. doi: 10.1183/09031936.04.00056804
96. Wang JX, Li HQ, Zhang F, Ning W. Systemic inflammation and the effects of short-term antibiotic treatment for PPM positive patients with stable COPD. Int J Chron Obstruct Pulmon Dis. (2019) 14:1923–32. doi: 10.2147/COPD
97. Brill SE, Law M, El-Emir E, Allinson JP, James P, Maddox V, et al. Effects of different antibiotic classes on airway bacteria in stable COPD using culture and molecular techniques: a randomised controlled trial. Thorax. (2015) 70:930–8. doi: 10.1136/thoraxjnl-2015-207194
98. Criner GJ, Connett JE, Aaron SD, Albert RK, Bailey WC, Casaburi R, et al. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N Engl J Med. (2014) 370:2201–10. doi: 10.1056/NEJMoa1403086
99. Ingebrigtsen TS, Marott JL, Nordestgaard BG, Lange P, Hallas J, Vestbo J. Statin use and exacerbations in individuals with chronic obstructive pulmonary disease. Thorax. (2015) 70:33–40. doi: 10.1136/thoraxjnl-2014-205795
100. Damkjær M, Håkansson K, Kallemose T, Ulrik CS, Godtfredsen N. Statins in high-risk chronic obstructive pulmonary disease outpatients: no impact on time to first exacerbation and all-cause mortality - the STATUETTE cohort study. Int J Chron Obstruct Pulmon Dis. (2021) 16:579–89. doi: 10.2147/copd.S296472
101. Magnussen H, Disse B, Rodriguez-Roisin R, Kirsten A, Watz H, Tetzlaff K, et al. Withdrawal of inhaled glucocorticoids and exacerbations of COPD. N Engl J Med. (2014) 371:1285–94. doi: 10.1056/NEJMoa1407154
102. Rennard SI, Calverley PM, Goehring UM, Bredenbröker D, Martinez FJ. Reduction of exacerbations by the PDE4 inhibitor roflumilast–the importance of defining different subsets of patients with COPD. Respir Res. (2011) 12:18. doi: 10.1186/1465-9921-12-18
103. Uzun S, Djamin RS, Kluytmans JA, Mulder PG, van't Veer NE, Ermens AA, et al. Azithromycin maintenance treatment in patients with frequent exacerbations of chronic obstructive pulmonary disease (COLUMBUS): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. (2014) 2:361–8. doi: 10.1016/S2213-2600(14)70019-0
Keywords: chronic obstructive pulmonary disease, chronic inflammation, inflammatory cells, oxidative stress, systemic inflammation
Citation: Xu J, Zeng Q, Li S, Su Q and Fan H (2024) Inflammation mechanism and research progress of COPD. Front. Immunol. 15:1404615. doi: 10.3389/fimmu.2024.1404615
Received: 21 March 2024; Accepted: 29 July 2024;
Published: 09 August 2024.
Edited by:
Garry M. Walsh, University of Aberdeen, United KingdomReviewed by:
Spyridon Fortis, The University of Iowa, United StatesStanley Chan, RMIT University, Australia
Copyright © 2024 Xu, Zeng, Li, Su and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Fan, ZmFuaG9uZ2ZhbkBxcS5jb20=; Qiaoli Su, MTg5ODA2MDEzNThAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship