 Hong Huang1
Hong Huang1 Xiong Cai
Xiong Cai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 11 June 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1404441
Succinate, traditionally viewed as a mere intermediate of the tricarboxylic acid (TCA) cycle, has emerged as a critical mediator in inflammation. Disruptions within the TCA cycle lead to an accumulation of succinate in the mitochondrial matrix. This excess succinate subsequently diffuses into the cytosol and is released into the extracellular space. Elevated cytosolic succinate levels stabilize hypoxia-inducible factor-1α by inhibiting prolyl hydroxylases, which enhances inflammatory responses. Notably, succinate also acts extracellularly as a signaling molecule by engaging succinate receptor 1 on immune cells, thus modulating their pro-inflammatory or anti-inflammatory activities. Alterations in succinate levels have been associated with various inflammatory disorders, including rheumatoid arthritis, inflammatory bowel disease, obesity, and atherosclerosis. These associations are primarily due to exaggerated immune cell responses. Given its central role in inflammation, targeting succinate pathways offers promising therapeutic avenues for these diseases. This paper provides an extensive review of succinate’s involvement in inflammatory processes and highlights potential targets for future research and therapeutic possibilities development.
Inflammation is fundamentally a protective response against chemical, mechanical, or microbial stimuli, typically manifesting as acute and beneficial to the host. However, when unresolved, this process can transition into chronic inflammation (1). A burgeoning area of research focuses on the intersection of metabolic changes and inflammation (2). Central to this discussion is succinate, a tricarboxylic acid (TCA) cycle intermediate primarily found in the mitochondrial matrix. It serves both as a substrate for succinate dehydrogenase (SDH) and as an electron donor in the electron transport chain (ETC) (3).
Interestingly, elevated succinate levels have been detected in the cytoplasm, suggesting its functions extend beyond those classically understood within the mitochondria (4, 5). Recent evidence underscores succinate’s role as a signaling molecule, particularly under hypoxic conditions common in inflammatory environments (6). Here, disruptions in the TCA cycle and respiratory chain lead to cytoplasmic succinate accumulation (7), contributing to several biological processes such as stabilization of hypoxia-inducible factor-1α (HIF-1α), generation of reactive oxygen species (ROS), and protein succinylation (8–10). Intracellular succinate is increasingly recognized as a pro-inflammatory mediator (10).
Moreover, extracellular succinate exerts significant physiological effects by activating succinate receptor 1 (SUCNR1, also known as GPR91), a G protein-coupled receptor on the plasma membrane (11, 12). Given its role as a GPCR, SUCNR1 is a promising target for pharmaceutical modulation using small molecules (13–15). While traditionally implicated in promoting pro-inflammatory responses (16–18), recent findings also suggest anti-inflammatory functions associated with SUCNR1 (19). The precise role of the succinate/SUCNR1 axis in either exacerbating or ameliorating disease remains an area of active investigation, but it is clear that this pathway is crucial in linking metabolism with immune regulation (20, 21). Despite significant advancements in this area, further research is warranted to comprehensively unravel both the intracellular and extracellular functions of succinate.
In this paper, we explore the multifaceted roles of succinate in inflammation, particularly its pivotal function as a signaling molecule within immune responses. We further discuss how these insights inform our understanding of specific diseases, provide opportunities for modulating succinate levels, and pave the way for novel therapeutic interventions.
Succinate, a central metabolite in the TCA cycle, is produced from α-ketoglutarate (α-KG) through a two-step process. First, α-KG is decarboxylated to succinyl-CoA by α-KG dehydrogenase, followed by conversion to succinate via succinyl-CoA synthetase. This reaction represents a substrate-level phosphorylation, generating GTP (or ATP in some organisms) directly (3). Under normal conditions, succinate is promptly oxidized to fumarate by SDH.
However, under conditions like immune cell activation or tumorigenesis where cells predominantly rely on anaerobic glycolysis, alternative pathways lead to succinate accumulation (7, 22). SDH, which consists of subunits SDHA, SDHB, SDHC, and SDHD, is integral to both the TCA cycle and the mitochondrial ETC as respiratory complex II. It is located within the inner mitochondrial membrane (23) and facilitates the conversion of succinate to fumarate while simultaneously reducing ubiquinone (UQ) to ubiquinol (UQH2) and flavin adenine dinucleotide (FAD) to FADH2—critical steps in ATP production (3, 24). Under hypoxic conditions, SDH activity diminishes, leading to the reversal of electron flow and enabling fumarate to serve as the terminal electron acceptor in the ETC, resulting in succinate accumulation (2, 25). Chouchani et al. reported that sources such as the purine nucleotide cycle and malate/aspartate shuttle contribute fumarate to SDH during ischemia (8), while anaerobic glycolysis primarily produces succinate via glutamine-dependent anaplerosis to α-KG (26). Additionally, succinate can be generated from the γ-aminobutyric acid (GABA) shunt, with the pathway’s activity linked to the expression levels of GABA transporter family members solute carrier 12 (SLC6A12) and SLC6A13 (9). Another significant source of intracellular succinate is the uptake of extracellular succinate by sodium-dependent transporters of the SLC13 family (27).
Given its charged nature at physiological pH, succinate’s membrane permeability is naturally restricted, necessitating specialized transporters for its movement from the mitochondrial matrix to the cytosol and across the plasma membrane into the extracellular space (28). Intracellularly, the dicarboxylate carrier (DIC), a member of the SLC25 transporter family, transports accumulated succinate across the mitochondrial inner membrane. The voltage-dependent anion channel (VDAC) facilitates this process at the outer membrane (29, 30). Once in the cytosol, succinate is exported to the extracellular environment via organic anion/dicarboxylate transporters (OATs) (27). Conditions of increased energy demand trigger anaerobic energy systems, leading to excessive lactate production and cellular acidification (31, 32). This acidification modifies succinate’s chemical structure, allowing it to cross cell membranes with assistance from plasma membrane monocarboxylate transporter 1 (MCT1) (33, 34). Additionally, Gudgeon et al. (35) observed that succinate uptake in CD4+ T cells is partially dependent on MCT1. In macrophages, enhancing transporter-mediated succinate uptake is crucial for augmenting and sustaining inflammatory responses (27). The key role of these succinate transporters lies in exporting succinate to different cellular compartments, where it serves as a signaling molecule (10). Targeting these transporters offers a potential strategy to regulate extracellular succinate levels, indirectly impacting SUCNR1 activation (Figure 1).
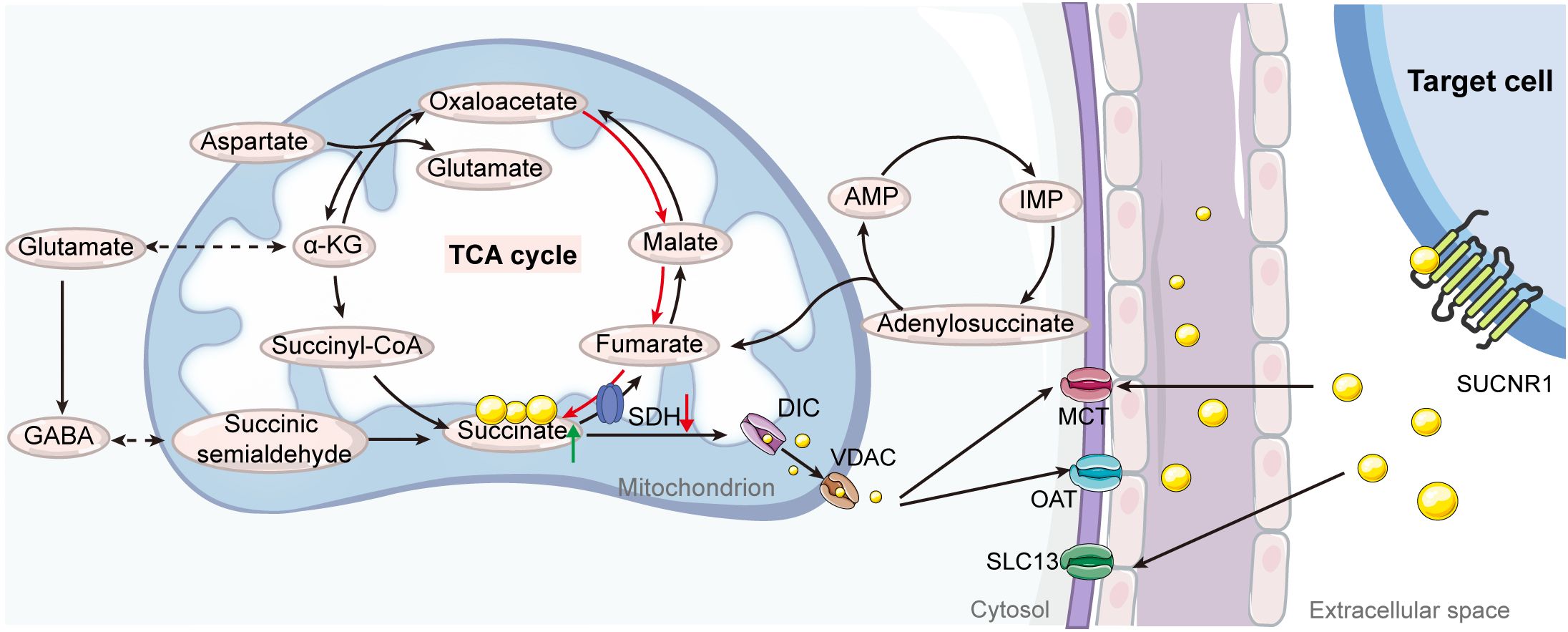
Figure 1 Illustration of the pathways of succinate production and transport. Under normal physiological conditions within the tricarboxylic acid (TCA) cycle, succinate is synthesized as an intermediate from the transformation of succinyl-CoA and is then converted into fumarate by succinate dehydrogenase (SDH). Under specific stress conditions or metabolic alterations, mitochondrial succinate can accumulate through several alternative pathways, including reverse activity of SDH, the purine nucleotide cycle, malate/aspartate shuttle, glutamine-dependent anaplerosis, and the γ-aminobutyric acid (GABA) shunt. When mitochondrial succinate levels exceed cellular requirements, it is transported into the cytosol through mitochondrial dicarboxylate carriers (DICs) and voltage-dependent anion channels (VDACs). The efflux of succinate from the cytosol to the extracellular space is mediated by organic anion transporters (OATs) and monocarboxylate transporters (MCTs). Additionally, extracellular succinate can be reabsorbed via the solute carrier family 13 (SLC13) and further facilitated by MCTs. AMP, adenosine monophosphate; IMP, inosine monophosphate.
Succinate, increasingly recognized as a signaling molecule, plays a pivotal role at the juncture of metabolism and inflammation (36). It serves as a crucial mediator linking intracellular and extracellular inflammatory signaling and regulates inflammation through various distinct mechanisms (37). This multifaceted relationship not only deepens our understanding of cellular responses during inflammation but also highlights potential therapeutic targets in inflammatory diseases.
HIF-1α, a key transcription factor, regulates cellular responses to hypoxia. Upon stabilization, HIF-1α translocates into the nucleus, binding to hypoxia-responsive elements to initiate transcription of genes (38), including those related to inflammation such as interleukin-1β (IL-1β) (9, 39). Prolyl hydroxylases (PHDs), part of the α-KG-dependent dioxygenase family, are integral to cellular oxygen sensing and regulate HIF-1α stability. An increase in cytoplasmic succinate, following PHD inhibition, activates HIF-1α, thereby amplifying the transcription of pro-inflammatory genes (40). Although this activation is beneficial for cellular adaptation to hypoxic conditions, excessive succinate can induce a “pseudohypoxia” state (41, 42), enhancing HIF-1α activity even in oxygen-rich environments, a condition frequently observed in tumors with mutated SDH (43). Consequently, inhibiting PHDs hinders HIF-1α degradation even in oxygen-rich environments, hence triggering the activation of a spectrum of target genes (41).
Additionally, succinate has been shown to indirectly stabilize HIF-1α by inducing the production of ROS (44, 45). ROS mediates the oxidation of Fe2+ to Fe3+, inhibiting PHD activity that relies on Fe2+. This inhibition leads to the activation of HIF-1α (46). In macrophages activated by lipopolysaccharide (LPS), elevated succinate levels enhance UQ reduction, leading to “reverse electron transport (RET)” which channels electrons back to respiratory complex I (47, 48). This process significantly increases ROS production, further stabilizing HIF-1α through PHD inhibition (40) (Figure 2). As a result, high levels of succinate not only disrupt ATP synthesis and oxidative phosphorylation but also enhance ROS generation, amplifying pro-inflammatory signals (49, 50).
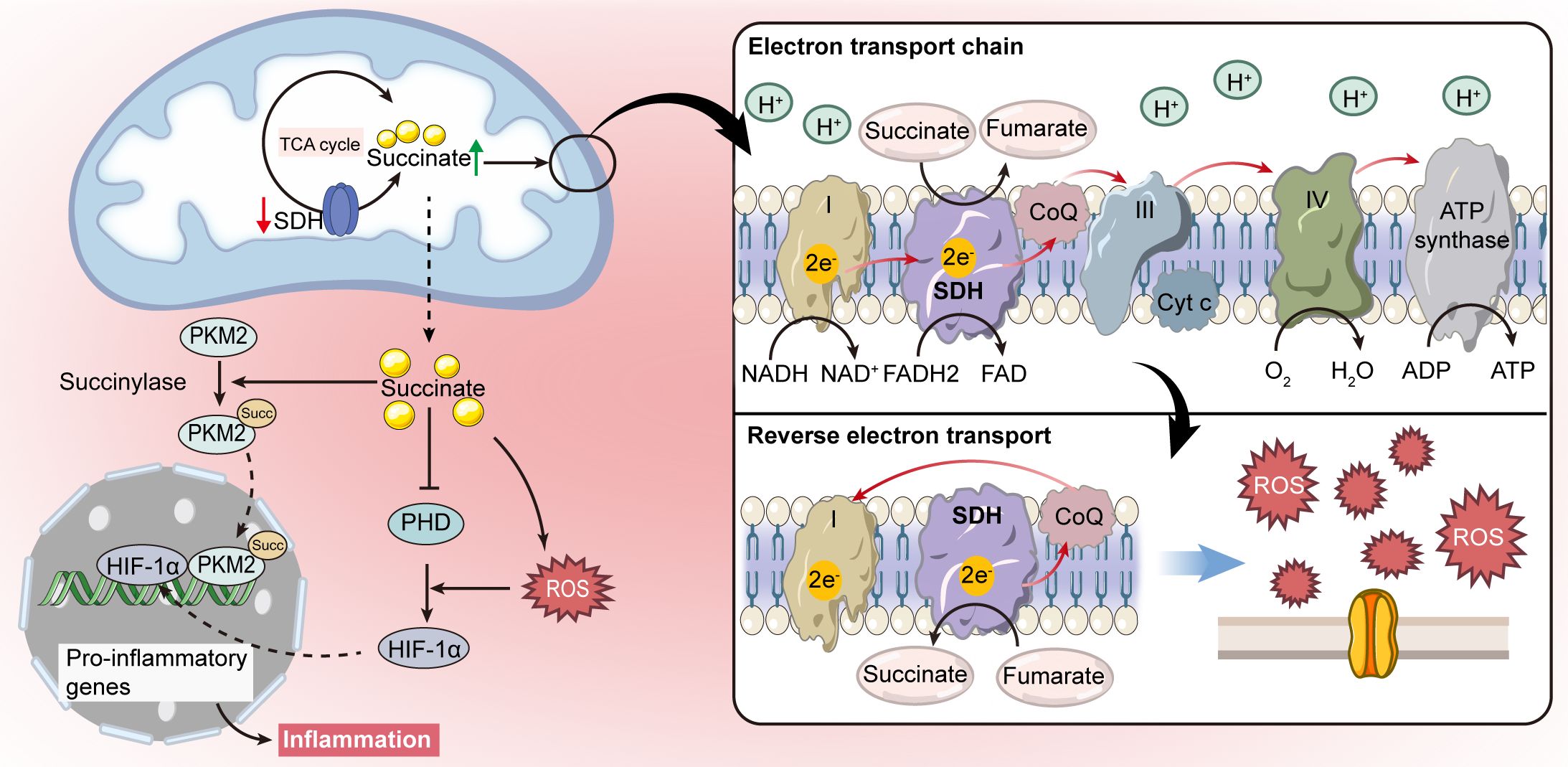
Figure 2 Intracellular signaling pathways of succinate. Succinate acts as a signal molecule that stabilizes hypoxia-inducible factor-1α (HIF-1α) by inhibiting prolyl hydroxylase (PHD), which leads to the production of pro-inflammatory cytokines. Succinate can also induce redox signaling by promoting mitochondrial reactive oxygen species (ROS) production through reverse electron transport (RET), further supporting HIF-1α stabilization. Moreover, succinate accumulation is linked to succinylation, which regulates metabolic enzyme activities.
Moreover, increased cytoplasmic succinate levels promote post-translational modifications, such as succinylation, which regulates the activity of metabolic enzymes in mitochondria (51–53). In LPS-activated macrophages, a metabolic shift occurs from reliance on mitochondrial ATP production to dependence on aerobic glycolysis, with pyruvate kinase M2 (PKM2) playing a critical role (54). Hypersuccinylation of PKM2 facilitates its nuclear translocation, promoting the formation of a PKM2-HIF-1α complex at the IL-1β gene promoter, as illustrated in Figure 2. This interaction intensifies the severity of dextran sulfate sodium (DSS)-induced colitis in mice (54, 55). Additionally, the sirtuin family enzyme SIRT5 is implicated in modulating PKM2 succinylation and its enzymatic activity, thus potentially attenuating pro-inflammatory responses in macrophages (54).
Upon release from cells, succinate interacts with its membrane receptor, SUCNR1, which is sensitively responsive to extracellular succinate levels (56). SUCNR1 is expressed across a variety of tissues, including the kidney, small intestine, liver, spleen, retina, and heart, as well as within specific immune cells such as macrophages, dendritic cells (DCs), and some T-cell subsets. This wide distribution underscores its significant role in modulating immune responses (17, 28, 57). The expression of SUCNR1 is influenced by the extracellular concentration of succinate, which under normal physiological conditions in plasma and urine remains below the activation threshold of the receptor (58). However, these concentrations are approximately twofold lower than the EC50 for SUCNR1, indicating that a substantial increase in succinate levels is necessary to fully engage the receptor (59). The signaling pathways activated by SUCNR1 involve two G proteins—Gq and Gi (11, 60). Activation of protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) cascades, along with calcium mobilization, are mediated through the Gq pathway, whereas the inhibition of cyclic adenosine monophosphate (cAMP) is facilitated by the Gi pathway (12). These mechanisms highlight the critical role of the succinate-SUCNR1 axis in the regulation of inflammatory processes.
While intracellular succinate is acknowledged as a necessary pro-inflammatory signal, its extracellular presence not only amplifies pro-inflammatory responses but, under certain conditions, can also exert anti-inflammatory effects (27). In induced neural stem cells (iNSCs) and somatic neural stem cells (NSCs) that utilized to mimic the inflammatory cascade, Peruzzotti-Jametti et al. showed that succinate induces an anti-inflammatory response through SUCNR1 signaling, leading to the release of prostaglandin E2 (PGE2), which modulates various cell types within the inflammatory microenvironment. Additionally, the absence of SUCNR1 in NSCs significantly reduces anti-inflammatory actions both in vitro and in vivo (61). Succinate has been proposed as a potential therapeutic for severe sepsis, primarily by enhancing ATP generation, which is crucial for rescuing tissues from the bioenergetic dysfunction associated with sepsis (62). Recent research suggests that activating SUCNR1 in macrophages within adipose tissue could initiate an anti-inflammatory response (63). Moreover, succinate may inhibit the release of inflammatory mediators like IL-6, IL-1β, nitric oxide (NO), and tumor necrosis factor (TNF) in inflammatory macrophages, independently of SUCNR1 signaling (64). However, succinate can also exacerbate inflammatory conditions by enhancing oxidative stress or driving inflammation, as observed in macrophages exposed to LPS, in mouse models of antigen-induced arthritis, and 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis (9, 16, 65). The role of succinate in inflammation and its broader physiological and pathological implications are increasingly focused on the dynamics of its receptor, SUCNR1 (66, 67). The dual pro- and anti-inflammatory capacities of succinate during inflammation appear to be highly context-dependent.
Macrophages are heterogeneous immune cells that play a crucial role in immune responses. Upon classical activation by the toll-like receptor 4 (TLR4) ligand LPS, macrophages transition from ATP production via oxidative phosphorylation to glycolysis, which is accompanied by an increase in succinate levels (68). This metabolic shift in macrophages is significant, as cancer cells have also been observed to release succinate into the extracellular space, thereby facilitating the polarization of macrophages into tumor-associated macrophages through SUCNR1 (67). Succinate serves not only as a marker of pro-inflammatory activity but also contributes to the pro-inflammatory capabilities of macrophages. In the inflammatory microenvironment, excessive pro-inflammatory activity can exacerbate conditions such as inflammatory bowel disease (IBD) (69, 70). Moreover, succinate acts as a chemokine, inducing cell migration in the U937 macrophage cell line via SUCNR1 activation (17). Gobelli et al. underscored the essential roles of SDHA and SDHB in LPS-mediated production of IL-10 and IL-1β, HIF-1α stabilization, and tyrosine phosphorylation of the transcription factor signal transducer and activator of transcription 3 (STAT3) in macrophages. However, they noted that these components are not necessary for the production of IL-6 and TNF-α (71).
The succinate-SUCNR1 axis, while recognized for inducing and exacerbating inflammation, also plays a crucial role in polarizing anti-inflammatory macrophages and regulating inflammation (72, 73). Intriguingly, although succinate is produced by pro-inflammatory macrophages, its receptor SUCNR1 is primarily found in anti-inflammatory macrophages (63, 74). Recent studies have highlighted its role in the inflammatory program, particularly in promoting anti-inflammatory activity through SUCNR1 (63). Park et al. demonstrated that macrophages treated with succinate could mitigate colitis (75). Specifically, adipose-derived mesenchymal stem cells (ADMSCs) were stimulated by intracellular succinate accumulation to secrete PGE2, a soluble molecule that reduced inflammation in macrophages and shifted their phenotype from pro-inflammtory to anti-inflammatory macrophages (76). Furthermore, extracellular succinate enhances this change by binding to SUCNR1 on anti-inflammatory macrophages via Gq and PLC pathway signaling (77). Notably, research indicates that a higher α-KG/succinate ratio favors anti-inflammatory macrophage activation, whereas a lower ratio promotes pro-inflammatory activation (78). This finding suggests that while succinate can promote anti-inflammatory macrophage polarization, the precise balance between its concentration and that of other metabolites is critical in determining macrophage polarization direction. Additional research is necessary to ascertain the specific concentration thresholds of succinate that influence this polarization process (Figure 3A).
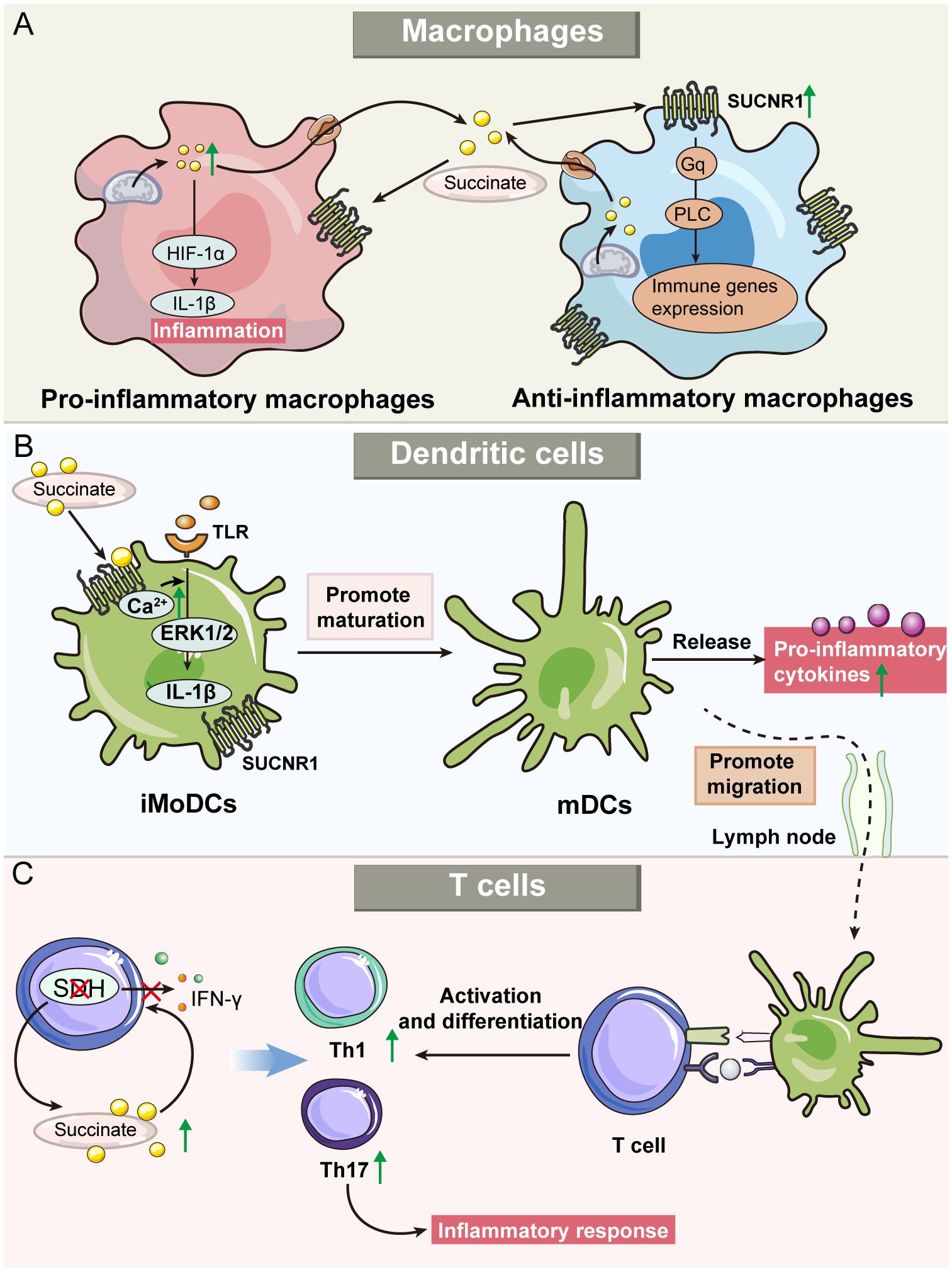
Figure 3 Roles of succinate and SUCNR1 in immune cells, including macrophages, dendritic cells (DCs), and T cells. (A) Succinate modulates the activities of both pro-and anti-inflammatory macrophages. Succinate serves as an indicator of pro-inflammatory activity, which promotes the production of IL-1β in macrophages and triggers inflammatory response. Furthermore, succinate also enhances the activity of anti-inflammatory macrophages through the SUCNR1-mediated Gq pathway. (B) Exogenous succinate regulates the function of DCs. Immature monocyte-derived DC (iMoDCs) express high levels of SUCNR1, and succinate promotes iMoDC maturation. In addition, succinate can upregulate the migration of mature DCs into the lymph nodes and enhance antigen presentation by DCs. (C) Succinate’s role in T cell inflammatory responses. Succinate dehydrogenase (SDH) deficiency leads to increased succinate, which regulates cytokine secretion, alters T-cell metabolism and promotes differentiation into Th1 and Th17, thereby regulating the inflammatory response.
DCs are crucial antigen-presenting cells that play a vital role in regulating the adaptive immune system. During inflammation, DCs capture antigens, mature, and migrate to lymphoid tissues to present these antigens to naive T cells. This process is accompanied by the secretion of a range of pro-inflammatory cytokines, further amplifying the immune response (79). In immature monocyte-derived DCs (iMoDCs), elevated expression of SUCNR1 was observed, which decreased as the cells matured (17). The binding of extracellular succinate to SUCNR1 on iMoDCs triggers the mobilization of intracellular calcium and phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2). This activity, in synergy with TLRs, promotes iMoDC maturation and secretion of IL-1β, thus enhancing the inflammatory response to pathogens. Additionally, succinate significantly increases the migratory capacity of DCs and their ability to activate T cells in an antigen-specific manner. Notably, these succinate-mediated effects are substantially reduced in the absence of SUCNR1, as demonstrated in knockout studies (17). Mice treated with succinate developed more severe arthritis, characterized by an increased presence of T helper cell 17 (Th17) cells and SUCNR1-expressing DCs in lymph nodes (80). Importantly, HIF-1α has been identified as a crucial mediator in the regulatory effects of DCs in inflammatory diseases. For example, in a colitis mouse model, the absence of HIF-1α in intestinal DCs exacerbated colitis symptoms (81). Similarly, in obesity models, HIF-1α deficiency in adipose tissue DCs led to enhanced inflammation (82) (Figure 3B).
Succinate not only indirectly modulates T-cell function through its effects on DCs but also exerts a direct regulatory impact within the inflammatory microenvironment. It has been observed that succinate can inhibit degranulation and cytokine secretion in CD4+ and CD8+ T cells, particularly affecting the production of interferon-γ (IFN-γ) (35). Additionally, inhibiting SDH during T-cell activation significantly impairs the expression of activation markers, production of inflammatory cytokines, and cell proliferation (83). Conversely, Chen et al. reported that SDH dysfunction, while hindering cell proliferation, actually enhances inflammatory responses in T cells. Specifically, T cells deficient in SDHB exhibited an elevated succinate/α-KG ratio, upregulation of pro-inflammatory gene markers, and a propensity to differentiate into pro-inflammatory Th1 and Th17 lineages (84). In experimental autoimmune uveitis in mice, succinate was shown to enhance the formation of neutrophil extracellular traps and the frequencies of Th1/Th17 cells, accompanied by increased production of IFN-γ and IL-17A through the succinate-SUCNR1 axis (85). These findings highlight the pivotal roles of SDH and succinate in the adaptive immune system (Figure 3C).
Succinate’s functions extend well beyond its role as a mere metabolic intermediate; it influences blood pressure, the renin-angiotensin system in the kidney, lipolysis in adipose tissue, skeletal muscle remodeling, and retinal vascularization, affecting the pathophysiology of a broad spectrum of diseases (11, 19). Dysregulation of succinate metabolism or its accumulation has been linked to various disorders ranging from metabolic syndromes to inflammatory diseases, including gastrointestinal disorders (58), diabetes mellitus (86), retinal complications (87), cardiovascular disease (88–90), fatty liver diseases (19), rheumatoid arthritis (91), sepsis (92), obesity (93), and some cancers (94) (Figure 4). Representative examples will be further discussed in the following section (Table 1).
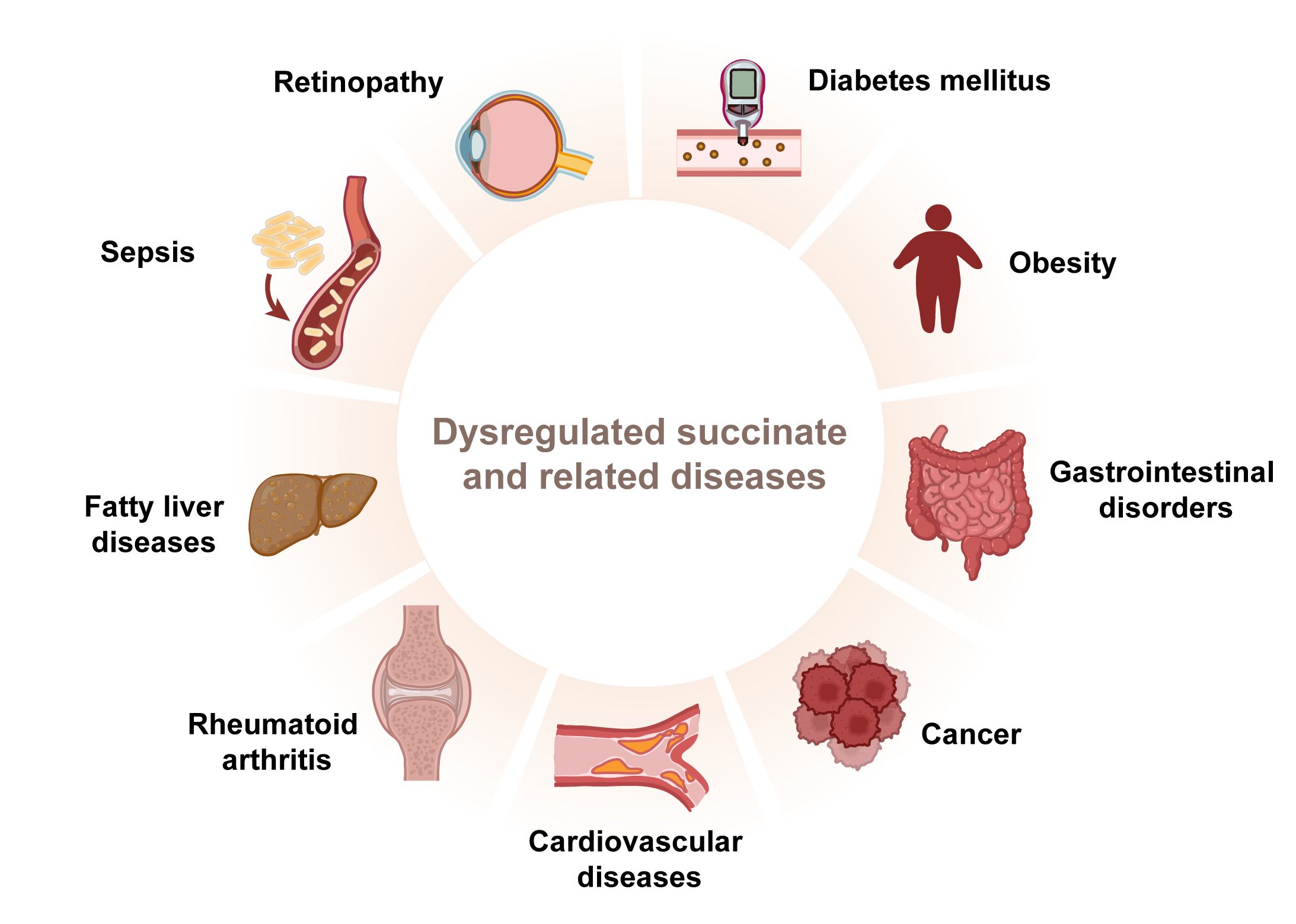
Figure 4 Dysregulated succinate contributes to various diseases. Succinate plays an important role in inflammatory diseases.
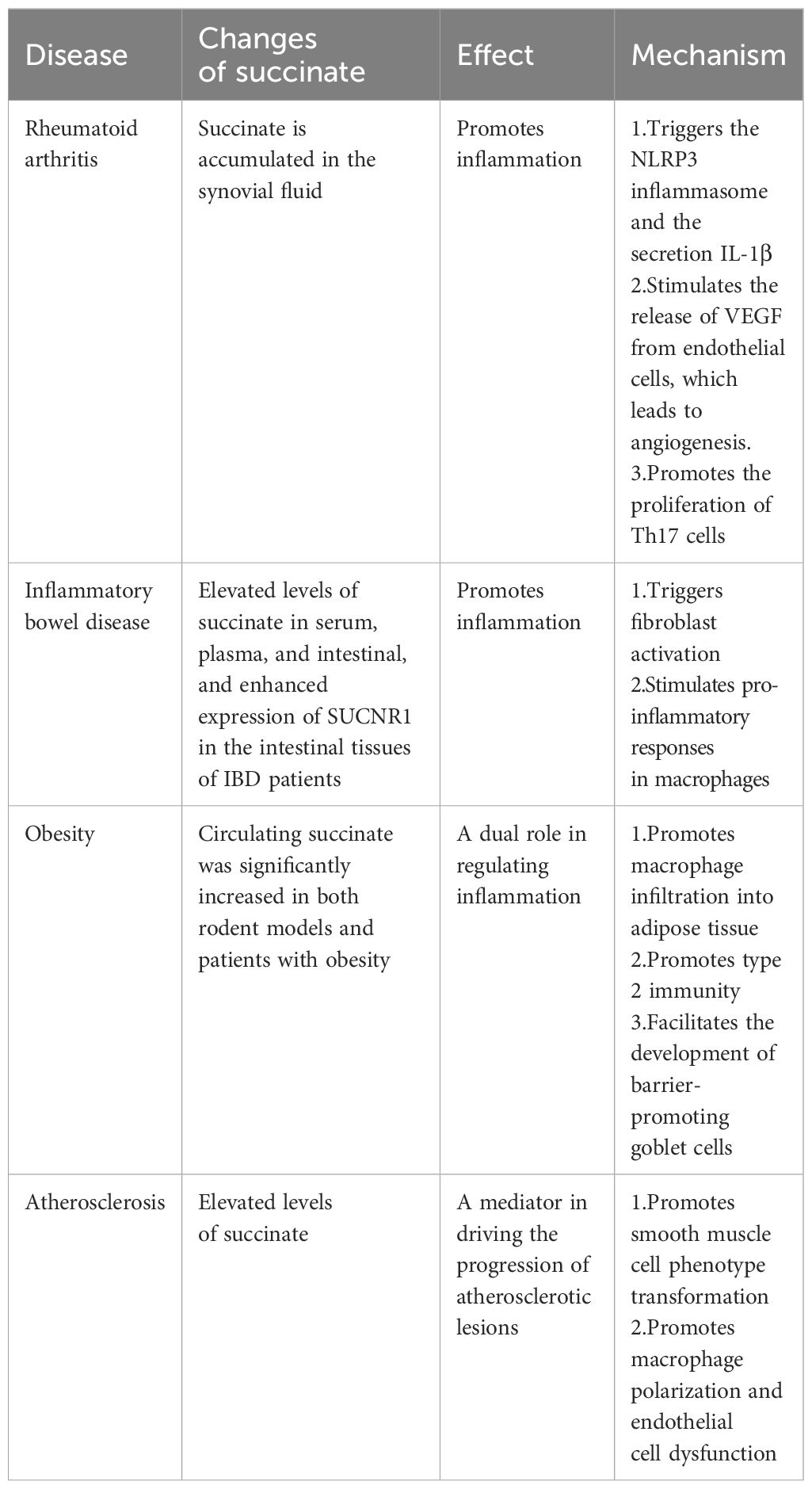
Table 1 Evidences of dysregulated succinate and its effects in diseases.
Rheumatoid arthritis, a chronic inflammatory disorder, is closely associated with metabolic dysregulation, which influences its progression and symptom severity (95, 96). Succinate levels are notably high in the synovial fluid of both antigen-induced arthritis mice and patients with rheumatoid arthritis (16, 97). Metabolic profiling has identified succinate as a distinctive metabolite differentiating rheumatoid arthritis from other arthropathies (98). This accumulation triggers macrophages in the synovial fluid to release IL-1β, mediated through SUCNR1 signaling (16). Additionally, succinate in synovial fibroblasts activates the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome, enhancing IL-1β secretion (99). Treatment of endothelial cells with LPS and exposure of these cells along with synovial fibroblasts to hypoxic conditions result in succinate accumulation, which induces angiogenesis through the release of vascular endothelial growth factor (VEGF), associated with HIF-1α stabilization and SUCNR1 activation by extracellular succinate (91). This endothelial activation and increased angiogenesis contribute to the pathogenesis of rheumatoid arthritis by facilitating leukocyte recruitment and migration and promoting hyperplasia in the synovial pannus (100). Moreover, SUCNR1 functions as a chemotactic gradient sensor, orchestrating DC migration toward lymph nodes, thereby leading to the proliferation of Th17 cells. These cells are closely associated with articular lesions and are known to intensify inflammation, neutrophil infiltration, mechanical hyperalgesia, and bone erosion within joints (80). These findings underscore the critical role of succinate in regulating inflammation in rheumatoid arthritis (101).
IBD, encompassing two primary phenotypes, Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic disorder characterized by recurrent inflammation of the gastrointestinal tract (102). Studies have shown increased levels of succinate in the sera, plasma, and intestinal tissues of IBD patients, along with enhanced expression of SUCNR1 in these tissues (65, 103, 104). This is supported by findings in mice, where those colonized with fecal samples from CD and UC patients displayed higher fecal succinate levels compared to those colonized from healthy individuals, indicating that succinate accumulation is a metabolic hallmark of IBD-associated dysbiosis (105). SUCNR1 not only triggers fibroblast activation and stimulates pro-inflammatory responses in macrophages but also contributes to colitis and intestinal fibrosis in murine models (65). Furthermore, a relationship has been reported between increased succinate levels in the gut lumen and microbiome dysbiosis in both IBD patients and animal models of intestinal inflammation (30, 106).
Recent studies indicate that circulating succinate levels are significantly elevated in both rodent models and patients with obesity (93, 107). The inflammation of adipose tissue, a hallmark of obesity, drives insulin resistance and subsequently, diabetes mellitus. Succinate and its receptor SUCNR1 exacerbate this obesity-induced inflammation, promoting the infiltration of macrophages into adipose tissue, which is a key contributor to the glucose intolerance observed in type 2 diabetes (21). Interestingly, the effects of SUCNR1 deficiency in macrophages vary depending on their location and the level of inflammation. In subcutaneous fat, SUCNR1-deficient macrophages tend to exhibit a pro-inflammatory phenotype, unlike their wild-type counterparts. Conversely, in visceral fat, which is typically pro-inflammatory, macrophages show reduced inflammation when SUCNR1 is deficient (63). Previous studies have demonstrated that the succinate-SUCNR1 axis plays a crucial role in promoting type 2 immunity in the gut, acting as an essential anti-inflammatory regulator (108). While obesity is associated with increased succinate levels (93), it often features impaired SUCNR1 signaling, leading to what is described as a “succinate-resistant state” (63). In SUCNR1-deficient mouse models, succinate-SUCNR1 signaling initiates type 2 immune responses in the intestine, facilitates the development of barrier-promoting goblet cells, mitigates high-fat diet (HFD)-induced mucosal barrier impairments and intestinal dysbiosis, and ultimately exhibits anti-obesity effects. This research highlights the protective function of the succinate-SUCNR1 axis against intestinal barrier dysfunction induced by HFD (109).
Atherosclerosis is a multifactorial pathological condition characterized by vascular inflammation and the accumulation of lipids and fibrous elements in the vessel walls (110). Elevated levels of succinate may act as a mediator driving the progression of atherosclerotic lesions by activating the SUCNR1 pathway, which results in phenotypical transformations of smooth muscle cells, macrophage polarization, and endothelial cell dysfunction (111). Studies have shown that patients with coronary heart disease exhibit notably higher levels of succinate and IL-1β compared with healthy individuals, with a positive correlation between succinate and IL-1β levels. The glycolytic metabolism triggered by the release of succinate is evident in atherosclerotic pathology. This, in turn, stimulates the succinate/IL-1β signaling pathway through SUCNR1, thereby exacerbating inflammatory responses (112).
Targeting succinate metabolism is emerging as a promising approach for the therapeutic management of various diseases. This section highlights pharmacological strategies that intervene in succinate metabolism and discusses their potential implications for therapeutic applications.
In the metabolism of pro-inflammatory macrophages, Mills et al. confirmed the critical role of mitochondrial SDH in regulating LPS-induced gene expression. Specifically, elevated succinate oxidation via active SDH increases the expression of inflammatory genes, while inhibition of SDH fosters an anti-inflammatory phenotype (113). Malonate, a competitive inhibitor at the carboxylate site of SDH, and its cell-permeable prodrug dimethyl malonate (DMM), which produces malonate intracellularly, lead to succinate accumulation. DMM has been shown to effectively suppress the expression of pro-inflammatory genes, such as IL-1β and HIF-1α, and simultaneously enhance the expression of anti-inflammatory genes like IL-1RA and type I IFN-inducible genes (113). Interestingly, DMM does not affect TNF-α levels, suggesting a specific pathway of action (113). In conditions like cellular hypoxia in synovial fibroblasts and ischemia, SDH is proposed to function in reverse, converting fumarate to succinate (8). DMM exhibits anti-inflammatory effects by inhibiting the HIF-1α/VEGF axis in hypoxia-treated synovial fibroblasts, preventing succinate accumulation, and inhibiting angiogenesis (91). Moreover, DMM reduces succinate accumulation during ischemia and controls oxidation during reperfusion, thereby decreasing mitochondrial ROS generation and attenuating ischemia-reperfusion injury (8). The malonate ester prodrug diacetoxymethyl malonate shows greater potency than DMM, owing to its accelerated esterase hydrolysis (114). Although succinate accumulates via different mechanisms in LPS-treated macrophages and other conditions, inhibition of SDH similarly reduces IL-1β production (91).
Itaconate, an endogenous small molecule synthesized by the enzyme cis-aconitate decarboxylase, acts as an immunomodulator and inhibits SDH, impacting metabolic and inflammatory processes in cells (115). Due to its structural similarity to succinate, itaconate was identified decades ago as a competitive inhibitor of SDH. This inhibition leads to elevated succinate accumulation and reduced mitochondrial ROS production, affecting mitochondrial respiratory chain complex I (116). Itaconate demonstrates anti-inflammatory properties in vitro and in vivo by modulating SDH levels, mitochondrial respiration, and cytokine production (IL-1β, IL-18, IL-6, IL-12, NO, and HIF-1α, but not TNF-α), particularly during macrophage activation and ischemia-reperfusion injury (117). Further studies show that SDH inhibition by itaconate also mitigates cerebral ischemia-reperfusion injury (118, 119). As an SDH inhibitor, itaconate manages oxidative stress and enhances physiological outcomes associated with ischemia-reperfusion injury (120). Research has revealed that both endogenous (117) and exogenous (121) itaconate-mediated SDH inhibition affects mitochondrial function. Importantly, while targeting succinate through this pathway offers valuable insights into its physiological roles, the therapeutic application must be approached cautiously, given the pivotal role of SDH in cellular energetics (Figure 5).
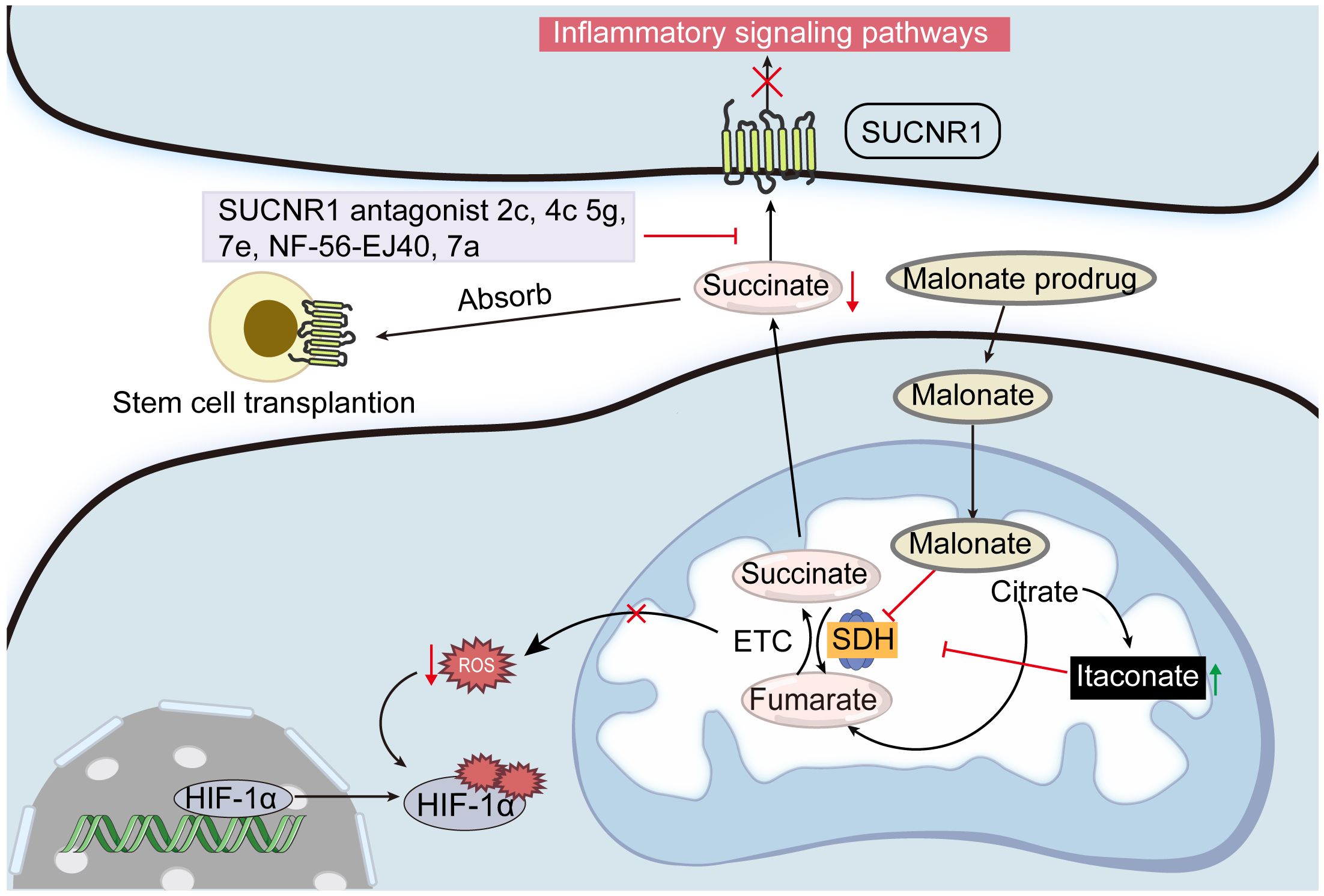
Figure 5 Potential strategies for controlling succinate concentrations. Inhibiting SDH activity with malonate and itaconate is one way to regulate the succinate production in mitochondria. Blocking SUCNR1 activity with its antagonists 2c, 4c, 5g, 7e, and NF-56-EJ40 may reduce the negative effects of excessive succinate accumulation. Furthermore, excessive succinate can be absorbed by transplanting stem cells that express SUCNR.
In recent years, there has been increasing attention on the role of SUCNR1 in various pathophysiological processes, significantly highlighting its potential as a therapeutic target (122). The accumulation of succinate in various inflammatory states further underscores the importance of targeting SUCNR1 to mitigate the detrimental effects of excess succinate (28). Advances in understanding the structure-function relationship of SUCNR1 have facilitated the development of structure-based antagonists (123). Systematic structure-activity relationship studies have identified compounds “2c” and “4c” as effective SUCNR1 antagonists both in vitro and in vivo. Moreover, compounds “5g” and “7e”, which are structurally distinct and orally bioavailable, have shown potential as promising leads in drug development (13). NF-56-EJ40, a high-affinity SUCNR1 antagonist, effectively reduces succinate/IL-1β signaling in macrophages and HUVECs, demonstrating a potent inhibitory effect (112, 124). Additionally, NF-56-EJ40 substantially attenuates succinate-mediated gene expression in primary human anti-inflammatory macrophages (77). A topically applied gel formulation of the SUCNR1 antagonist compound “7a” has been shown to reduce inflammatory events and osteoclastogenesis in vivo (125). The therapeutic efficacy of “7a” is directly attributed to the suppression of SUCNR1-mediated succinate signaling (126).
Furthermore, the transplantation of cells expressing SUCNR1 into an inflammatory environment is also a promising strategy to mitigate excess succinate. Mesenchymal stem cell (MSC) transplantation, known for its ability to differentiate into various cell types, is utilized to facilitate the repair and regeneration of damaged tissues (127). This method has been demonstrated to effectively decrease succinate levels in tissues, subsequently triggering beneficial effects in adjacent areas. Specifically, upon transplantation, adipose-derived MSCs absorb succinate, decreasing its accumulation and shifting macrophage to pro-inflammatory macrophages, which mitigates DSS-induced colitis in mice (76). Similarly, when transplanting NSCs into the cerebrospinal fluid of mice with experimental autoimmune encephalomyelitis, succinate secreted by type 1 mononuclear phagocytes binds to SUCNR1 on NSCs, thereby reducing the production of succinate in the cerebrospinal fluid and preventing the progression of neuroinflammation (61).
However, the critical involvement of SUCNR1 in essential physiological processes and its significance in regulating energy homeostasis must be carefully considered. The systemic application of SUCNR1 antagonists could lead to severe adverse effects due to the disruption of these essential functions. Additionally, while antagonists serve to inhibit receptor function, there might be scenarios where activating SUCNR1 could offer therapeutic benefits. The challenge lies in employing SUCNR1 antagonists effectively in the treatment of SUCNR1-mediated diseases without compromising its normal or advantageous functions (73).
The correlation between aberrant succinate changes and inflammatory diseases due to overreactive immune responses has been increasingly recognized. The significance of this research lies in its potential to advance the understanding and treatment of inflammatory diseases. Overall, the study of succinate presents a combination of challenges and opportunities, as the delicate balance between the beneficial and detrimental effects of succinate and its receptor is not yet fully understood. As research delves deeper into its complexities, one aspect remains clear: succinate will continue to be a pivotal focus in biomedical research.
HH: Conceptualization, Project administration, Writing – original draft. GL: Project administration, Writing – review & editing. YH: Project administration, Writing – review & editing. JC: Project administration, Writing – review & editing. JY: Formal analysis, Methodology, Writing – review & editing. QZ: Conceptualization, Project administration, Writing – review & editing. LL: Conceptualization, Project administration, Writing – review & editing. XC: Conceptualization, Project administration, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by grants from the National Natural Science Foundation of China General Program (82274506, 82104871), the Science and Technology Innovation Program of Hunan Province (2020SK1020, 2021RC4035), the Natural Science Foundation of Hunan (2024JJ5298, 2023JJ40477), the Natural Science Foundation of Guangxi (2020GXNSFAA297053), the Open-competing Disciple Construction Project of Hunan University of Chinese Medicine (HNUCM) (22JBZ003), and was financially supported by the Furong Distinguished Scholar Program of Hunan (XJT[2020]58), the 121 Training Project for Innovative Talents of Hunan (XRSH[2019]192), the Chinese Academy of Engineering Academician Liang Liu’s Workstation Project (KH[2023]3-23YS001), and the World First-class Discipline Incubation Project of HNUCM (XJF[2022]57).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Leuti A, Fazio D, Fava M, Piccoli A, Oddi S, Maccarrone M. Bioactive lipids, inflammation and chronic diseases. Adv Drug Delivery Rev. (2020) 159:133–69. doi: 10.1016/j.addr.2020.06.028
2. Atallah R, Olschewski A, Heinemann A. Succinate at the crossroad of metabolism and angiogenesis: Roles of SDH, HIF1alpha and SUCNR1. Biomedicines. (2022) 10:3089. doi: 10.3390/biomedicines10123089
3. Martínez-Reyes I, Chandel N. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. (2020) 11:102. doi: 10.1038/s41467-019-13668-3
4. Tuomas L, Christopher JM, Tuukka I, John ZC, Juho H, William G Jr K, et al. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J Biol Chem. (2015) 291:4256–65. doi: 10.1074/jbc.M115.688762
5. Justina K, Rasa B, Darius T, Arvydas S, Sonata T. Increased succinate accumulation induces ROS generation in in vivo ischemia/reperfusion-affected rat kidney mitochondria. BioMed Res Int. (2020) 2020:8855585. doi: 10.1155/2020/8855585
6. Zhang W, Lang R. Succinate metabolism: A promising therapeutic target for inflammation, ischemia/reperfusion injury and cancer. Front Cell Dev Biol. (2023) 11:1266973. doi: 10.3389/fcell.2023.1266973
7. Grimolizzi F, Arranz L. Multiple faces of succinate beyond metabolism in blood. Haematologica. (2018) 103:1586–92. doi: 10.3324/haematol.2018.196097
8. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. (2014) 515:431–5. doi: 10.1038/nature13909
9. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces il-1β through hif-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
10. Murphy M, O'Neill L. Krebs cycle reimagined: The emerging roles of succinate and itaconate as signal transducers. Cell. (2018) 174:780–4. doi: 10.1016/j.cell.2018.07.030
11. de Castro Fonseca M, Aguiar CJ, da Rocha Franco JA, Gingold RN, Leite MF. GPR91: Expanding the frontiers of krebs cycle intermediates. Cell Commun Signal. (2016) 14:3. doi: 10.1186/s12964-016-0126-1
12. Gilissen J, Jouret F, Pirotte B, Hanson J. Insight into sucnr1 (GPR91) structure and function. Pharmacol Therapeut. (2016) 159:56–65. doi: 10.1016/j.pharmthera.2016.01.008
13. Bhuniya D, Umrani D, Dave B, Salunke D, Kukreja G, Gundu J, et al. Discovery of a potent and selective small molecule hgpr91 antagonist. Bioorg Med Chem Lett. (2011) 21:3596–602. doi: 10.1016/j.bmcl.2011.04.091
14. Geubelle P, Gilissen J, Dilly S, Poma L, Dupuis N, Laschet C, et al. Identification and pharmacological characterization of succinate receptor agonists. Brit J Pharmacol. (2017) 174:796–808. doi: 10.1111/bph.13738
15. Rexen Ulven E, Trauelsen M, Brvar M, Lückmann M, Bielefeldt L, Jensen L, et al. Structure-activity investigations and optimisations of non-metabolite agonists for the succinate receptor 1. Sci Rep-UK. (2018) 8:10010. doi: 10.1038/s41598-018-28263-7
16. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med. (2016) 213:1655–62. doi: 10.1084/jem.20160061
17. Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, et al. Triggering the succinate receptor gpr91 on dendritic cells enhances immunity. Nat Immunol. (2008) 9:1261–9. doi: 10.1038/ni.1657
18. Rubić-Schneider T, Carballido-Perrig N, Regairaz C, Raad L, Jost S, Rauld C, et al. GPR91 deficiency exacerbates allergic contact dermatitis while reducing arthritic disease in mice. Allergy. (2017) 72:444–52. doi: 10.1111/all.13005
19. Marsal-Beltran A, Rodríguez-Castellano A, Astiarraga B, Calvo E, Rada P, Madeira A, et al. Protective effects of the succinate/SUCNR1 axis on damaged hepatocytes in NAFLD. Metabolism. (2023) 145:155630. doi: 10.1016/j.metabol.2023.155630
20. McCreath K, Espada S, Gálvez B, Benito M, de Molina A, Sepúlveda P, et al. Targeted disruption of the SUCNR1 metabolic receptor leads to dichotomous effects on obesity. Diabetes. (2015) 64:1154–67. doi: 10.2337/db14-0346
21. van Diepen JA, Robben JH, Hooiveld GJ, Carmone C, Alsady M, Boutens L, et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia. (2017) 60:1304–13. doi: 10.1007/s00125-017-4261-z
22. Fernández-Veledo S, Ceperuelo-Mallafré V, Vendrell J. Rethinking succinate: An unexpected hormone-like metabolite in energy homeostasis. Trends Endocrinol Metab. (2021) 32:680–92. doi: 10.1016/j.tem.2021.06.003
23. Bandara AB, Drake JC, Brown DA. Complex ii subunit sdhd is critical for cell growth and metabolism, which can be partially restored with a synthetic ubiquinone analog. BMC Mol Cell Biol. (2021) 22:35. doi: 10.1186/s12860-021-00370-w
24. Dalla Pozza E, Dando I, Pacchiana R, Liboi E, Scupoli MT, Donadelli M, et al. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin Cell Dev Biol. (2020) 98:4–14. doi: 10.1016/j.semcdb.2019.04.013
25. Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase – assembly, regulation and role in human disease. Mitochondrion. (2010) 10:393–401. doi: 10.1016/j.mito.2010.03.001
26. Wise D, Ward P, Shay J, Cross J, Gruber J, Sachdeva U, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U.S.A. (2011) 108:19611–6. doi: 10.1073/pnas.1117773108
27. Fremder M, Kim SW, Khamaysi A, Shimshilashvili L, Eini-Rider H, Park IS, et al. A transepithelial pathway delivers succinate to macrophages, thus perpetuating their pro-inflammatory metabolic state. Cell Rep. (2021) 36:109521. doi: 10.1016/j.celrep.2021.109521
28. Ariza A, Deen P, Robben J. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front Endocrinol. (2012) 3:22. doi: 10.3389/fendo.2012.00022
29. Palmieri F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflugers Arch. (2004) 447:689–709. doi: 10.1007/s00424-003-1099-7
30. Connors J, Dawe N, Van Limbergen J. The role of succinate in the regulation of intestinal inflammation. Nutrients. (2018) 11:25. doi: 10.3390/nu11010025
31. Reddy A, Bozi LHM, Yaghi OK, Mills EL, Xiao H, Nicholson HE, et al. pH-gated succinate secretion regulates muscle remodeling in response to exercise. Cell. (2020) 183:62–75.e17. doi: 10.1016/j.cell.2020.08.039
32. Prag HA, Gruszczyk AV, Huang MM, Beach TE, Young T, Tronci L, et al. Mechanism of succinate efflux upon reperfusion of the ischaemic heart. Cardiovasc Res. (2021) 117:1188–201. doi: 10.1093/cvr/cvaa148
33. Andrienko TN, Pasdois P, Pereira GC, Ovens MJ, Halestrap AP. The role of succinate and ros in reperfusion injury - a critical appraisal. J Mol Cell Cardiol. (2017) 110:1–14. doi: 10.1016/j.yjmcc.2017.06.016
34. Bisbach CM, Hass DT, Thomas ED, Cherry TJ, Hurley JB. Monocarboxylate transporter 1 (MCT1) mediates succinate export in the retina. Invest Ophthalmol Vis Sci. (2022) 63:1. doi: 10.1167/iovs.63.4.1
35. Gudgeon N, Munford H, Bishop EL, Hill J, Fulton-Ward T, Bending D, et al. Succinate uptake by t cells suppresses their effector function via inhibition of mitochondrial glucose oxidation. Cell Rep. (2022) 40:111193. doi: 10.1016/j.celrep.2022.111193
36. Ryan D, O'Neill L. Krebs cycle reborn in macrophage immunometabolism. Annu Rev Immunol. (2020) 38:289–313. doi: 10.1146/annurev-immunol-081619-104850
37. Frezza C. Mitochondrial metabolites: Undercover signalling molecules. Interface Focus. (2017) 7:20160100. doi: 10.1098/rsfs.2016.0100
38. Cowman S, Koh M. Revisiting the hif switch in the tumor and its immune microenvironment. Trends Cancer. (2022) 8:28–42. doi: 10.1016/j.trecan.2021.10.004
39. Taylor C, Scholz C. The effect of hif on metabolism and immunity. Nat Rev Nephrol. (2022) 18:573–87. doi: 10.1038/s41581-022-00587-8
40. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
41. Denko N. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. (2008) 8:705–13. doi: 10.1038/nrc2468
42. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. (2005) 7:77–85. doi: 10.1016/j.ccr.2004.11.022
43. Morin A, Letouzé E, Gimenez-Roqueplo A, Favier J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int J Cancer. (2014) 135:2237–48. doi: 10.1002/ijc.29080
44. Chandel N, McClintock D, Feliciano C, Wood T, Melendez J, Rodriguez A, et al. Reactive oxygen species generated at mitochondrial complex iii stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of o2 sensing. J Biol Chem. (2000) 275:25130–8. doi: 10.1074/jbc.M001914200
45. Guzy R, Sharma B, Bell E, Chandel N, Schumacker P. Loss of the sdhb, but not the sdha, subunit of complex ii triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. (2008) 28:718–31. doi: 10.1128/mcb.01338-07
46. Wu L-Y, He Y-L, Zhu L-L. Possible role of PHD inhibitors as hypoxia-mimicking agents in the maintenance of neural stem cells’ self-renewal properties. Front Cell Dev Biol. (2018) 6:169. doi: 10.3389/fcell.2018.00169
47. Scialo F, Fernandez-Ayala DJ, Sanz A. Role of mitochondrial reverse electron transport in ros signaling: Potential roles in health and disease. Front Physiol. (2017) 8:428. doi: 10.3389/fphys.2017.00428
48. Roca FJ, Whitworth LJ, Prag HA, Murphy MP, Ramakrishnan L. Tumor necrosis factor induces pathogenic mitochondrial ros in tuberculosis through reverse electron transport. Science. (2022) 376:eabh2841. doi: 10.1126/science.abh2841
49. Mateska I, Witt A, Hagag E, Sinha A, Yilmaz C, Thanou E, et al. Succinate mediates inflammation-induced adrenocortical dysfunction. Elife. (2023) 12:e83064. doi: 10.7554/eLife.83064
50. Kamarauskaite J, Baniene R, Trumbeckas D, Strazdauskas A, Trumbeckaite S. In vivo increased succinate accumulation induces ros generation in ischemia/reperfusion-affected rat kidney mitochondria. BioMed Res Int. (2020) 2020:8855585. doi: 10.1155/2020/8855585
51. Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. (2013) 50:919–30. doi: 10.1016/j.molcel.2013.06.001
52. Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, et al. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics. (2012) 11:100–7. doi: 10.1074/mcp.M111.015875
53. Lin H, Su X, He B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem Biol. (2012) 7:947–60. doi: 10.1021/cb3001793
54. Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, et al. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1β production and to prevent DSS-induced colitis in mice. Cell Rep. (2017) 19:2331–44. doi: 10.1016/j.celrep.2017.05.065
55. Palsson-McDermott E, Curtis A, Goel G, Lauterbach M, Sheedy F, Gleeson L, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:347. doi: 10.1016/j.cmet.2015.01.017
56. Krzak G, Willis CM, Smith JA, Pluchino S, Peruzzotti-Jametti L. Succinate receptor 1: An emerging regulator of myeloid cell function in inflammation. Trends Immunol. (2021) 42:45–58. doi: 10.1016/j.it.2020.11.004
57. Ryan D, Murphy M, Frezza C, Prag H, Chouchani E, O'Neill L, et al. Coupling krebs cycle metabolites to signalling in immunity and cancer. Nat Metab. (2019) 1:16–33. doi: 10.1038/s42255-018-0014-7
58. Yan XL, Liu XC, Zhang YN, Du TT, Ai Q, Gao X, et al. Succinate aggravates intestinal injury in mice with necrotizing enterocolitis. Front Cell Infect Microbiol. (2022) 12:1064462. doi: 10.3389/fcimb.2022.1064462
59. Okovityi SV, Rad’ko SV, Shustov EB. Succinate receptors (sucnr1) as a potential target for pharmacotherapy. Pharm Chem J. (2015) 49:573–7. doi: 10.1007/s11094-015-1331-8
60. Mills E, O'Neill L. Succinate: A metabolic signal in inflammation. Trends Cell Biol. (2014) 24:313–20. doi: 10.1016/j.tcb.2013.11.008
61. Peruzzotti-Jametti L, Bernstock JD, Vicario N, Costa ASH, Kwok CK, Leonardi T, et al. Macrophage-derived extracellular succinate licenses neural stem cells to suppress chronic neuroinflammation. Cell Stem Cell. (2018) 22:355–368.e313. doi: 10.1016/j.stem.2018.01.020
62. Protti A, Singer M. Bench-to-bedside review: Potential strategies to protect or reverse mitochondrial dysfunction in sepsis-induced organ failure. Crit Care. (2006) 10:228. doi: 10.1186/cc5014
63. Keiran N, Ceperuelo-Mallafre V, Calvo E, Hernandez-Alvarez MI, Ejarque M, Nunez-Roa C, et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat Immunol. (2019) 20:581–92. doi: 10.1038/s41590-019-0372-7
64. Harber KJ, de Goede KE, Verberk SGS, Meinster E, de Vries HE, van Weeghel M, et al. Succinate is an inflammation-induced immunoregulatory metabolite in macrophages. Metabolites. (2020) 10:372. doi: 10.3390/metabo10090372
65. Macias-Ceja DC, Ortiz-Masia D, Salvador P, Gisbert-Ferrandiz L, Hernandez C, Hausmann M, et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal Immunol. (2019) 12:178–87. doi: 10.1038/s41385-018-0087-3
66. Starling S. Succinate regulates muscle exercise adaptations. Nat Rev Endocrinol. (2020) 16:678–9. doi: 10.1038/s41574-020-00429-2
67. Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell. (2020) 77:213–227.e215. doi: 10.1016/j.molcel.2019.10.023
68. Kelly B, O'Neill L. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. (2015) 25:771–84. doi: 10.1038/cr.2015.68
69. Mizoguchi E, Low D, Ezaki Y, Okada T. Recent updates on the basic mechanisms and pathogenesis of inflammatory bowel diseases in experimental animal models. Intest Res. (2020) 18:151–67. doi: 10.5217/ir.2019.09154
70. Dharmasiri S, Garrido-Martin E, Harris R, Bateman A, Collins J, Cummings J, et al. Human intestinal macrophages are involved in the pathology of both ulcerative colitis and crohn disease. Inflammation Bowel Dis. (2021) 27:1641–52. doi: 10.1093/ibd/izab029
71. Gobelli D, Serrano-Lorenzo P, Esteban-Amo M, Serna J, Pérez-García M, Orduña A, et al. The mitochondrial succinate dehydrogenase complex controls the stat3-il-10 pathway in inflammatory macrophages. iScience. (2023) 26:107473. doi: 10.1016/j.isci.2023.107473
72. Na Y, Stakenborg M, Seok S, Matteoli G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol. (2019) 16:531–43. doi: 10.1038/s41575-019-0172-4
73. Wu KK. Extracellular succinate: A physiological messenger and a pathological trigger. Int J Mol Sci. (2023) 24:11165. doi: 10.3390/ijms241311165
74. Trauelsen M, Rexen Ulven E, Hjorth SA, Brvar M, Monaco C, Frimurer TM, et al. Receptor structure-based discovery of non-metabolite agonists for the succinate receptor GPR91. Mol Metab. (2017) 6:1585–96. doi: 10.1016/j.molmet.2017.09.005
75. Park I, Son M, Ma H, Kim J, Kim D, Kim S, et al. Succinate-treated macrophages attenuate dextran sodium sulfate colitis in mice. Intest Res. (2021) 19:349–53. doi: 10.5217/ir.2020.00075
76. Yuan Y, Ni S, Zhuge A, Li L, Li B. Adipose-derived mesenchymal stem cells reprogram m1 macrophage metabolism via phd2/hif-1alpha pathway in colitis mice. Front Immunol. (2022) 13:859806. doi: 10.3389/fimmu.2022.859806
77. Trauelsen M, Hiron TK, Lin D, Petersen JE, Breton B, Husted AS, et al. Extracellular succinate hyperpolarizes m2 macrophages through sucnr1/gpr91-mediated gq signaling. Cell Rep. (2021) 35:109246. doi: 10.1016/j.celrep.2021.109246
78. Liu P, Wang H, Li X, Chao T, Teav T, Christen S, et al. Α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. (2017) 18:985–94. doi: 10.1038/ni.3796
79. Gardner A, de Mingo Pulido Á, Ruffell B. Dendritic cells and their role in immunotherapy. Front Immunol. (2020) 11:924. doi: 10.3389/fimmu.2020.00924
80. Saraiva AL, Veras FP, Peres RS, Talbot J, Lima KA, Luiz JP, et al. Succinate receptor deficiency attenuates arthritis by reducing dendritic cell traffic and expansion of th17 cells in the lymph nodes. FASEB J. (2018) 32:6550–8. doi: 10.1096/fj.201800285
81. Flück K, Breves G, Fandrey J, Winning S. Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of protective regulatory t cells in murine colitis. Mucosal Immunol. (2016) 9:379–90. doi: 10.1038/mi.2015.67
82. Wood E, Macdougall C, Blythe H, Clément M, Colas R, Dalli J, et al. Hif1α activation in dendritic cells under sterile conditions promotes an anti-inflammatory phenotype through accumulation of intracellular lipids. Sci Rep. (2020) 10:20825. doi: 10.1038/s41598-020-77793-6
83. Nastasi C, Willerlev-Olsen A, Dalhoff K, Ford SL, Gadsboll ASO, Buus TB, et al. Inhibition of succinate dehydrogenase activity impairs human T cell activation and function. Sci Rep. (2021) 11:1458. doi: 10.1038/s41598-020-80933-7
84. Chen X, Sunkel B, Wang M, Kang S, Wang T, Gnanaprakasam J, et al. Succinate dehydrogenase/complex ii is critical for metabolic and epigenetic regulation of t cell proliferation and inflammation. Sci Immunol. (2022) 7:eabm8161. doi: 10.1126/sciimmunol.abm8161
85. Li H, Tan H, Liu Z, Pan S, Tan S, Zhu Y, et al. Succinic acid exacerbates experimental autoimmune uveitis by stimulating neutrophil extracellular traps formation via sucnr1 receptor. Br J Ophthalmol. (2023) 107:1744–9. doi: 10.1136/bjophthalmol-2021-320880
86. Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, Hanner F, et al. Succinate receptor gpr91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest. (2008) 118:2526–34. doi: 10.1172/JCI33293
87. Ozturk B. Understanding ischemic retinopathies: The role of succinate and its receptor in retinal pigment epithelium. Adv Exp Med Biol. (2023) 1415:527–31. doi: 10.1007/978-3-031-27681-1_77
88. Osuna-Prieto F, Martinez-Tellez B, Ortiz-Alvarez L, Di X, Jurado-Fasoli L, Xu H, et al. Elevated plasma succinate levels are linked to higher cardiovascular disease risk factors in young adults. Cardiovasc Diabetol. (2021) 20:151. doi: 10.1186/s12933-021-01333-3
89. Xu J, Zheng Y, Zhao Y, Zhang Y, Li H, Zhang A, et al. Succinate/il-1β signaling axis promotes the inflammatory progression of endothelial and exacerbates atherosclerosis. Front Immunol. (2022) 13:817572. doi: 10.3389/fimmu.2022.817572
90. Aguiar CJ, Rocha-Franco JA, Sousa PA, Santos AK, Ladeira M, Rocha-Resende C, et al. Succinate causes pathological cardiomyocyte hypertrophy through gpr91 activation. Cell Commun Signal. (2014) 12:78. doi: 10.1186/s12964-014-0078-2
91. Li Y, Liu Y, Wang C, Xia WR, Zheng JY, Yang J, et al. Succinate induces synovial angiogenesis in rheumatoid arthritis through metabolic remodeling and hif-1alpha/vegf axis. Free Radic Biol Med. (2018) 126:1–14. doi: 10.1016/j.freeradbiomed.2018.07.009
92. Liu H, Zhang H, Zhang X, Chen Q, Xia L. Role of succinic acid in the regulation of sepsis. Int Immunopharmacol. (2022) 110:109065. doi: 10.1016/j.intimp.2022.109065
93. Serena C, Ceperuelo-Mallafré V, Keiran N, Queipo-Ortuño M, Bernal R, Gomez-Huelgas R, et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. (2018) 12:1642–57. doi: 10.1038/s41396-018-0068-2
94. Kuo C, Wu J, Wu K. Cancer-derived extracellular succinate: A driver of cancer metastasis. J BioMed Sci. (2022) 29:93. doi: 10.1186/s12929-022-00878-z
95. McHugh J. Dual inhibitor targets fibroblast metabolism in ra. Nat Rev Rheumatol. (2023) 19:760. doi: 10.1038/s41584-023-01051-y
96. Masoumi M, Hashemi N, Moadab F, Didehdar M, Farahani R, Khorramdelazad H, et al. Immunometabolism dysfunction in the pathophysiology and treatment of rheumatoid arthritis. Curr Med Chem. (2023) 30:3119–36. doi: 10.2174/0929867329666220907151213
97. Borenstein D, Gibbs C, Jacobs R. Gas-liquid chromatographic analysis of synovial fluid. Succinic acid and lactic acid as markers for septic arthritis. Arthritis Rheum. (1982) 25:947–53. doi: 10.1002/art.1780250806
98. Bahn Y-S, Kim S, Hwang J, Xuan J, Jung YH, Cha H-S, et al. Global metabolite profiling of synovial fluid for the specific diagnosis of rheumatoid arthritis from other inflammatory arthritis. PloS One. (2014) 9:e97501. doi: 10.1371/journal.pone.0097501
99. Li Y, Zheng J-Y, Liu J-Q, Yang J, Liu Y, Wang C, et al. Succinate/NLRP3 inflammasome induces synovial fibroblast activation: Therapeutical effects of clematichinenoside ar on arthritis. Fronts Immunol. (2016) 7:532. doi: 10.3389/fimmu.2016.00532
100. Leblond A, Allanore Y, Avouac J. Targeting synovial neoangiogenesis in rheumatoid arthritis. Autoimmun Rev. (2017) 16:594–601. doi: 10.1016/j.autrev.2017.04.005
101. Clayton SA, MacDonald L, Kurowska-Stolarska M, Clark AR. Mitochondria as key players in the pathogenesis and treatment of rheumatoid arthritis. Front Immunol. (2021) 12:673916. doi: 10.3389/fimmu.2021.673916
102. Bretto E, Ribaldone D, Caviglia G, Saracco G, Bugianesi E, Frara S. Inflammatory bowel disease: Emerging therapies and future treatment strategies. Biomedicines. (2023) 11:2249. doi: 10.3390/biomedicines11082249
103. Ooi M, Nishiumi S, Yoshie T, Shiomi Y, Kohashi M, Fukunaga K, et al. Gc/ms-based profiling of amino acids and tca cycle-related molecules in ulcerative colitis. Inflammation Res. (2011) 60:831–40. doi: 10.1007/s00011-011-0340-7
104. Monfort-Ferré D, Caro A, Menacho M, Martí M, Espina B, Boronat-Toscano A, et al. The gut microbiota metabolite succinate promotes adipose tissue browning in crohn’s disease. J Crohns Colitis. (2022) 16:1571–83. doi: 10.1093/ecco-jcc/jjac069
105. Nagao-Kitamoto H, Shreiner AB, Gillilland MG, Kitamoto S, Ishii C, Hirayama A, et al. Functional characterization of inflammatory bowel disease–associated gut dysbiosis in gnotobiotic mice. Cell Mol Gastroenterol Hepatol. (2016) 2:468–81. doi: 10.1016/j.jcmgh.2016.02.003
106. Fernandez-Veledo S, Vendrell J. Gut microbiota-derived succinate: Friend or foe in human metabolic diseases? Rev Endocr Metab Disord. (2019) 20:439–47. doi: 10.1007/s11154-019-09513-z
107. Sadagopan N, Li W, Roberds SL, Major T, Preston GM, Yu Y, et al. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am J Hypertens. (2007) 20:1209–15. doi: 10.1016/j.amjhyper.2007.05.010
108. Nadjsombati MS, McGinty JW, Lyons-Cohen MR, Jaffe JB, DiPeso L, Schneider C, et al. Detection of succinate by intestinal tuft cells triggers a type 2 innate immune circuit. Immunity. (2018) 49:33–41.e37. doi: 10.1016/j.immuni.2018.06.016
109. Li X, Huang G, Zhang Y, Ren Y, Zhang R, Zhu W, et al. Succinate signaling attenuates high-fat diet-induced metabolic disturbance and intestinal barrier dysfunction. Pharmacol Res. (2023) 194:106865. doi: 10.1016/j.phrs.2023.106865
110. Grebe A, Hoss F, Latz E. Nlrp3 inflammasome and the il-1 pathway in atherosclerosis. Circ Res. (2018) 122:1722–40. doi: 10.1161/circresaha.118.311362
111. Zhang S, Liang Y, Li L, Chen Y, Wu P, Wei D. Succinate: A novel mediator to promote atherosclerotic lesion progression. DNA Cell Biol. (2022) 41:285–91. doi: 10.1089/dna.2021.0345
112. Xu J, Zheng Y, Zhao Y, Zhang Y, Li H, Zhang A, et al. Succinate/il-1beta signaling axis promotes the inflammatory progression of endothelial and exacerbates atherosclerosis. Front Immunol. (2022) 13:817572. doi: 10.3389/fimmu.2022.817572
113. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. (2016) 167:457–470.e413. doi: 10.1016/j.cell.2016.08.064
114. Beach TE, Prag HA, Pala L, Logan A, Huang MM, Gruszczyk AV, et al. Targeting succinate dehydrogenase with malonate ester prodrugs decreases renal ischemia reperfusion injury. Redox Biol. (2020) 36:101640. doi: 10.1016/j.redox.2020.101640
115. Shi X, Zhou H, Wei J, Mo W, Li Q, Lv X. The signaling pathways and therapeutic potential of itaconate to alleviate inflammation and oxidative stress in inflammatory diseases. Redox Biol. (2022) 58:102553. doi: 10.1016/j.redox.2022.102553
116. Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, et al. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J Biol Chem. (2016) 291:14274–84. doi: 10.1074/jbc.M115.685792
117. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
118. Cordes T, Lucas A, Divakaruni AS, Murphy AN, Cabrales P, Metallo CM. Itaconate modulates tricarboxylic acid and redox metabolism to mitigate reperfusion injury. Mol Metab. (2020) 32:122–35. doi: 10.1016/j.molmet.2019.11.019
119. Zhang D, Lu Z, Zhang Z, Man J, Guo R, Liu C, et al. A likely protective effect of dimethyl itaconate on cerebral ischemia/reperfusion injury. Int Immunopharmacol. (2019) 77:105924. doi: 10.1016/j.intimp.2019.105924
120. Li Z, Zheng W, Kong W, Zeng T. Itaconate: A potent macrophage immunomodulator. Inflammation. (2023) 46:1177–91. doi: 10.1007/s10753-023-01819-0
121. Németh B, Doczi J, Csete D, Kacso G, Ravasz D, Adams D, et al. Abolition of mitochondrial substrate-level phosphorylation by itaconic acid produced by lps-induced irg1 expression in cells of murine macrophage lineage. FASEB J. (2016) 30:286–300. doi: 10.1096/fj.15-279398
122. Li X, Xie L, Qu X, Zhao B, Fu W, Wu B, et al. Gpr91, a critical signaling mechanism in modulating pathophysiologic processes in chronic illnesses. FASEB J. (2020) 34:13091–105. doi: 10.1096/fj.202001037R
123. Velcicky J, Wilcken R, Cotesta S, Janser P, Schlapbach A, Wagner T, et al. Discovery and optimization of novel sucnr1 inhibitors: Design of zwitterionic derivatives with a salt bridge for the improvement of oral exposure. J Med Chem. (2020) 63:9856–75. doi: 10.1021/acs.jmedchem.0c01020
124. Haffke M, Fehlmann D, Rummel G, Boivineau J, Duckely M, Gommermann N, et al. Structural basis of species-selective antagonist binding to the succinate receptor. Nature. (2019) 574:581–5. doi: 10.1038/s41586-019-1663-8
125. Guo Y, Xu F, Thomas SC, Zhang Y, Paul B, Sakilam S, et al. Targeting the succinate receptor effectively inhibits periodontitis. Cell Rep. (2022) 40:111389. doi: 10.1016/j.celrep.2022.111389
126. Thomas SC, Guo Y, Xu F, Saxena D, Li X. A novel SUCNR1 inhibitor alleviates dysbiosis through inhibition of host responses without direct interaction with host microbiota. Mol Oral Microbiol. (2024). 39:80–90. doi: 10.1111/omi.12431
Keywords: succinate, inflammation, hypoxia-inducible factor-1α, succinate receptor 1, therapeutic intervention
Citation: Huang H, Li G, He Y, Chen J, Yan J, Zhang Q, Li L and Cai X (2024) Cellular succinate metabolism and signaling in inflammation: implications for therapeutic intervention. Front. Immunol. 15:1404441. doi: 10.3389/fimmu.2024.1404441
Received: 21 March 2024; Accepted: 27 May 2024;
Published: 11 June 2024.
Edited by:
Elsa Sanchez Lopez, University of California, San Diego, United StatesReviewed by:
Vasileia Ismini Alexaki, University Hospital Carl Gustav Carus, GermanyCopyright © 2024 Huang, Li, He, Chen, Yan, Zhang, Li and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiong Cai, Y2FpeGlvbmdAaG51Y20uZWR1LmNu; Liqing Li, bGlsaXFpbmc4N0BxcS5jb20=; Qin Zhang, ZGJveXpxMUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.