 Wen-Hua Qi1†
Wen-Hua Qi1† Li-Fan Hu2†Yu-Jiawei Gu1Xiao-Yan Zhang3Xue-Mei Jiang1
Li-Fan Hu2†Yu-Jiawei Gu1Xiao-Yan Zhang3Xue-Mei Jiang1 Wu-Jiao Li4Jun-Sheng Qi1Guo-Sheng Xiao1*
Wu-Jiao Li4Jun-Sheng Qi1Guo-Sheng Xiao1* Hang Jie5*
Hang Jie5*- 1College of Biological and Food Engineering, Chongqing Three Gorges University, Chongqing, China
- 2College of Environmental and Chemical Engineering, Chongqing Three Gorges University, Chongqing, China
- 3Chongqing Wanzhou NO. 3 Middle School, Chongqing, China
- 4Department of Laboratory Medicine, Shenzhen Children’s Hospital, Shenzhen, China
- 5Jinfo Mountain Forest Ecosystem Field Scientific Observation and Research Station of Chongqing, Chongqing Institute of Medicinal Plant Cultivation, Chongqing, China
Background: Forest musk deer (FMD, Moschus Berezovskii) is a critically endangered species world-widely, the death of which can be caused by pulmonary disease in the farm. Pulmonary fibrosis (PF) was a huge threat to the health and survival of captive FMD. MicroRNAs (miRNAs) and messenger RNAs (mRNAs) have been involved in the regulation of immune genes and disease development. However, the regulatory profiles of mRNAs and miRNAs involved in immune regulation of FMD are unclear.
Methods: In this study, mRNA-seq and miRNA-seq in blood were performed to constructed coexpression regulatory networks between PF and healthy groups of FMD. The hub immune- and apoptosis-related genes in the PF blood of FMD were explored through Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. Further, protein–protein interaction (PPI) network of immune-associated and apoptosis-associated key signaling pathways were constructed based on mRNA-miRNA in the PF blood of the FMD. Immune hub DEGs and immune hub DEmiRNAs were selected for experimental verification using RT-qPCR.
Results: A total of 2744 differentially expressed genes (DEGs) and 356 differentially expressed miRNAs (DEmiRNAs) were identified in the PF blood group compared to the healthy blood group. Among them, 42 DEmiRNAs were negatively correlated with 20 immune DEGs from a total of 57 correlations. The DEGs were significantly associated with pathways related to CD molecules, immune disease, immune system, cytokine receptors, T cell receptor signaling pathway, Th1 and Th2 cell differentiation, cytokine-cytokine receptor interaction, intestinal immune network for IgA production, and NOD-like receptor signaling pathway. There were 240 immune-related DEGs, in which 186 immune-related DEGs were up-regulated and 54 immune-related DEGs were down-regulated. In the protein-protein interaction (PPI) analysis of immune-related signaling pathway, TYK2, TLR2, TLR4, IL18, CSF1, CXCL13, LCK, ITGB2, PIK3CB, HCK, CD40, CD86, CCL3, CCR7, IL2RA, TLR3, and IL4R were identified as the hub immune genes. The mRNA-miRNA coregulation analysis showed that let-7d, miR-324-3p, miR-760, miR-185, miR-149, miR-149-5p, and miR-1842-5p are key miRNAs that target DEGs involved in immune disease, immune system and immunoregulation.
Conclusion: The development and occurrence of PF were significantly influenced by the immune-related and apoptosis-related genes present in PF blood. mRNAs and miRNAs associated with the development and occurrence of PF in the FMD.
1 Introduction
Musk deer (Moschus spp.), a rare and critically endangered species endemic to China, Vietnam, and other Asian countries, has attracted the attention of the government and the international organizations concerned. More than 80% of the musk deer are distributed in China, the musk production of which accounted for 90% of the world musk output before the 1950s. Due to habitat destruction and killing musk deer for musk harvesting, the population of musk deer has declined rapidly since the 1950s. For this reason, all musk deer have been classified as a Grade 1 protected animal in China since 2002 (1). Musk was secreted by the odor glands of male musk deer, which has multiple medicinal effects such as neuroleptic, anti-inflammatory, anti-thrombotic, and anti-tumor in the medicine industry (2, 3). Moreover, musk has been widely used in the high-end perfume industry (4, 5). Forest musk deer (FMD; Moschus Berezovskii) is a species of musk deer, which is the most farmed musk deer species, followed by the alpine musk deer (AMD; Moschus chrysogaster) in China. At present, the musk mainly came from the captive FMD and AMD in China.
In order to make sustainable use of musk deer resources and reduce the killing of wild musk deer, artificial musk deer farms have been set up in China since 1958 (6). Although China’s artificial breeding of musk deer has been experienced for more than 60 years, the population of captive musk deer is increasing slowly, which is closely related to the high mortality caused by multiple diseases. Although the captive breeding of FMD has made great progress, its long-term breeding is hampered by multiple diseases, including pulmonary disease (7). In the animal farms, the common diseases of musk deer industry included abscess, pulmonary disease, gastroenteritis, and parasitic diseases, which were important constraints to the growth of China’s musk industry (8). Pulmonary disease, a respiratory disease is closely associated with dozens of diseases including pneumonia, tuberculosis, and pulmonary fibrosis (PF) (9–12). Pulmonary disease is a primary life-threatening for captive FMD. PF is a fatal disease of the respiratory system that affects the health and survival of animals accompanied by an overproliferation of fibroblasts and an inflammatory reaction (13–15). Bacterial pneumonia, especially PF, severely affected the survival of FMD in captivity (16). There are many causes for the formation of PF, among which infectious bacterial are the important causes (17). It has been reported that Pseudomonas aeruginosa, Streptococcus pneumoniae, Staphylococcus aureus, and Streptococcus pneumoniae can induce the progression of PF (17, 18).
Blood is an essential part of the immune system and also the first line of defense against infectious disease (19, 20). The changes of gene expression in blood cells were influenced by pathological changes of animal body (21). Therefore, immune system status can be monitored by blood transcriptome. Multiple pathogenic infections and immune system dysfunction have been reported to be associated with the development of pneumonia, phthisis, and PF (9, 22, 23). Analysis of the blood transcriptome also helps to identify immune genes and their signaling pathways. MiRNAs modulated RNA silencing and post-transcriptional gene expression regulation, which took part in the immune system, immune diseases, and apoptosis. Consequently, the purpose of the present study was to establish a dependable blood miRNA biomarker for the diagnosis of PF. As far as we know, there is no current systematic and comprehensive analysis of miRNA–mRNA regulatory networks based on PF and healthy blood groups derived from FMD. The building of potential miRNA–mRNA regulatory networks is going to help identify the full range of molecular mechanisms by which miRNA affects PF and may be used in the diagnosis of the disease.
The present study screened for unigenes and miRNAs that were differentially expressed, as well as immune-related genes and apoptosis-related genes in the PF and healthy peripheral blood of FMD by high-throughput sequencing. The hub immune- and apoptosis-related genes in the PF blood of FMD were explored through Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. The mRNA–miRNA interaction network is important in regulating immune and apoptotic functions. The present study was aimed at constructing multiple protein–protein interaction (PPI) network of immune-associated and apoptosis-associated key signaling pathways in the PF blood of the FMD. DEmiRNAs may be involved in the regulation of hub immune- and apoptosis-related genes in the PF blood of FMD were identified by Spearman correlation analysis. Furthermore, the mRNA–miRNA regulatory network may help to understand the mechanisms involved in the development of PF blood in FMD.
2 Materials and methods
2.1 Sample collection
The sick FMDs were looked after by us all day long. We take blood as soon as they got worse. When the sick FMDs died, an autopsy was performed by veterinarian to determine the PF. The 17 FMD venous blood samples were taken at the Chongqing Institute of Medicinal Plant Cultivation in Chongqing, China (Table 1; Supplementary Figure S1). Fresh blood samples were promptly kept in RNAlater (Ambion Inc., Austin, TX, USA). The PF and healthy blood groups of FMD were taken between January 2021 and December 2022 in Chongqing, China. All animal experiments were approved by Chongqing Three Gorges University and Chongqing Institute of Medicinal Plant Cultivation. The collected blood samples are packaged, sealed, labeled, and immediately refrigerated at 4°C before being sent to the laboratory. For long-term storage, the blood should be rapidly frozen at −40°C immediately after collection and then stored at −80°C for no more than three months.
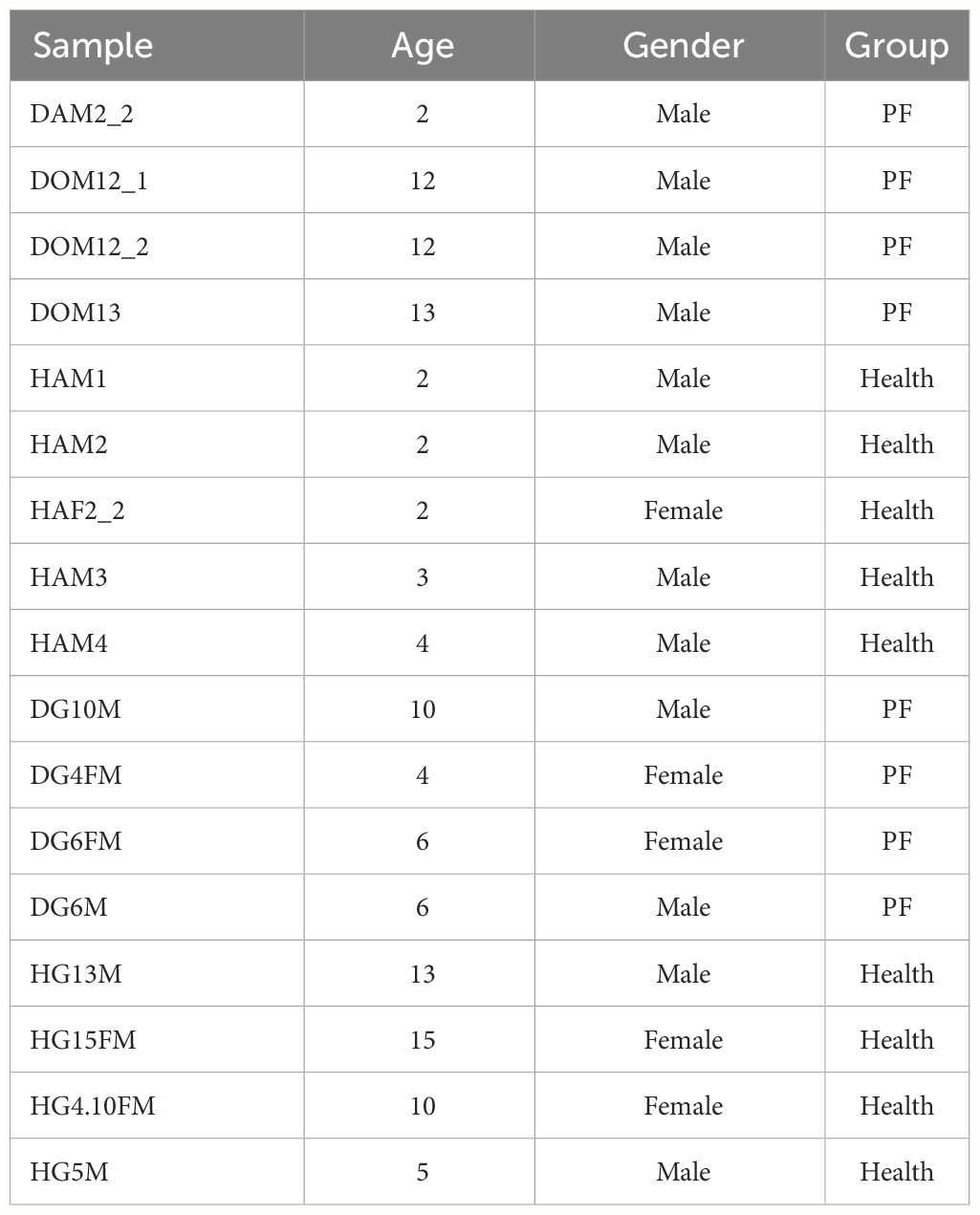
Table 1 Summary of sample information.
2.2 mRNA and miRNA sequencing and differential expression analysis
The total RNA was extracted using TRIzol reagent and then processed with RNase-free DNase I. A whole RNA pool was collected the venous blood of 17 FMD individuals to create an mRNA library or a small RNA library by using an Illumina HiSeq2500 platform (24, 25). Principal components analysis (PCA) of mRNAs and miRNA expression were performed in the nine samples and eight samples, respectively. The known miRNAs were identified by aligning them against the miRBase (26), and the novel miRNAs were predicted by using miranda (27). Expression levels of genes and miRNAs estimated as FPKM (fragments per kilobase million) and TPM (transcripts per million) indices, respectively, which were used to determine DEGs and DEmiRNAs between PF and healthy blood groups. The unigenes with Padj < 0.05 and |log2(fold change) | >1.0 were taken as the threshold for DEGs. DEmiRNAs were identified with the thresholds of p < 0.05 and | log2 (fold change) | >2.0.
2.3 miRNA target gene prediction and enrichment analysis
The target genes of DEmiRNAs in the two groups were predicted by using RNA hybridization (28), MiRanda (29), and PITA (Probability of Interaction by Target Accessibility, 30), and the results from the three algorithms were intersected. The potential target genes predicted by the above two softwares were combined, and the intersecting components were included as a set of candidate target genes. DEGs and DEmiRNA target mRNAs were further used to fulfil the GO and KEGG enrichment analyses. The p < 0.05 was designated as the threshold for significance.
2.4 miRNA–mRNA network integration
A set of 1,811 immune-related genes was obtained from ImmPort. Additionally, 306 apoptosis-related genes were recognized from the molecular signature database (MSigDB), which were intersected with the DEGs of the PF and healthy blood groups to regard as immune DEG set and apoptotic DEG set. In the typical immune pathway of this study, these immune DEGs were uploaded to STRING and obtain their interaction information (31, 32). The key potential regulatory networks of immune-related DEGs (or apoptosis-related DEGs) in the immune pathway (or apoptotic pathway) were pictured using the cytoscape software. It has been reported that DEGs and DEmiRNAs have potential negative regulatory relationships. On the basis of this idea, we researched the expression correlation of immune DEGs (or apoptotic DEGs) and DEmiRNAs by using the PCC (Pearson correlation coefficient). The negatively coexpressed DEmiRNA–DEG pairs with PCC < −0.7 and p < 0.05 were screened to construct miRNA-gene networks.
2.5 Real-time fluorescent quantitative PCR
Six immune hub DEGs (TLR2, TLR4, CXCL13, IL18, AKT1, and ITGB2) and six immune hub DEmiRNAs (let-7f-5p, let-7d, miR-30b-3p, miR-25-5p, miR-149-5p, and miR-760) were chosen for differential expression analysis. The primers for six immune-related DEGs and six DEmiRNAs were provided in Supplementary Table S1. Each sample’s mRNA and miRNA was reverse transcribed using PrimeScript™ RT reagent Kit (TaKaRa) and a miScript II RT Kit (Qiagen, Hilden, Germany). Quantitative PCR of the immune DEGs and immune DEmiRNAs were performed by using TB GreenR Premix Ex TaqTM II (Takara) and miScript SYBR Green PCR Kit (Qiagen), respectively. GAPDH and U6 snRNA were used as reference gene and miRNA, respectively. The relative expression of six DEGs and DEmiRNAs was computed using the 2−ΔΔCt method. The mean ± SE of three tests were presented in the data. Statistical significance was assessed by using t-tests as follows: *p ≤ 0.05 and **p ≤ 0.01.
3 Results
3.1 Overview of mRNA library of the two blood groups of healthy and BF
To investigate the changes of gene expression profiles in the PF pathological changes of FMD body and to compare their differences with healthy blood groups, nine cDNA libraries were built from the two blood groups of PF and healthy FMD. The unprocessed data were stored in the NCBI Sequence Read Archive (SRA) with the accession number PRJNA916839. After quality filtering, a total of 400,157,758 and 423,446,384 clean reads, representing a total of 62.12 and 80.09 Gb nucleotides, were generated for the PF and healthy blood groups, respectively (Supplementary Table S2). Approximately 86.33% of the clean reads was mapped to the FMD reference genome (33), with a match ratio ranging from 78.40 to 90.83% (Supplementary Table S3). After eliminating the low level of genes and transcripts, the reads were organized into 19,445 known genes and 4,907 annotated transcripts. A PCA was conducted, revealing that the samples from the PF and healthy blood groups formed two separate clusters based on the first principal component (PC1), which accounted for 66% of the variance (Supplementary Figure S2). This indicated that the sequencing data was suitable for further analysis. A mean of 11,884 (70.92%) and 12,229 (74.62%) unigenes with FPKM values greater than 0.5 were acquired from the PF and healthy blood group of FMD, respectively (Supplementary Figure S3). A detailed overview was described in Table 2.
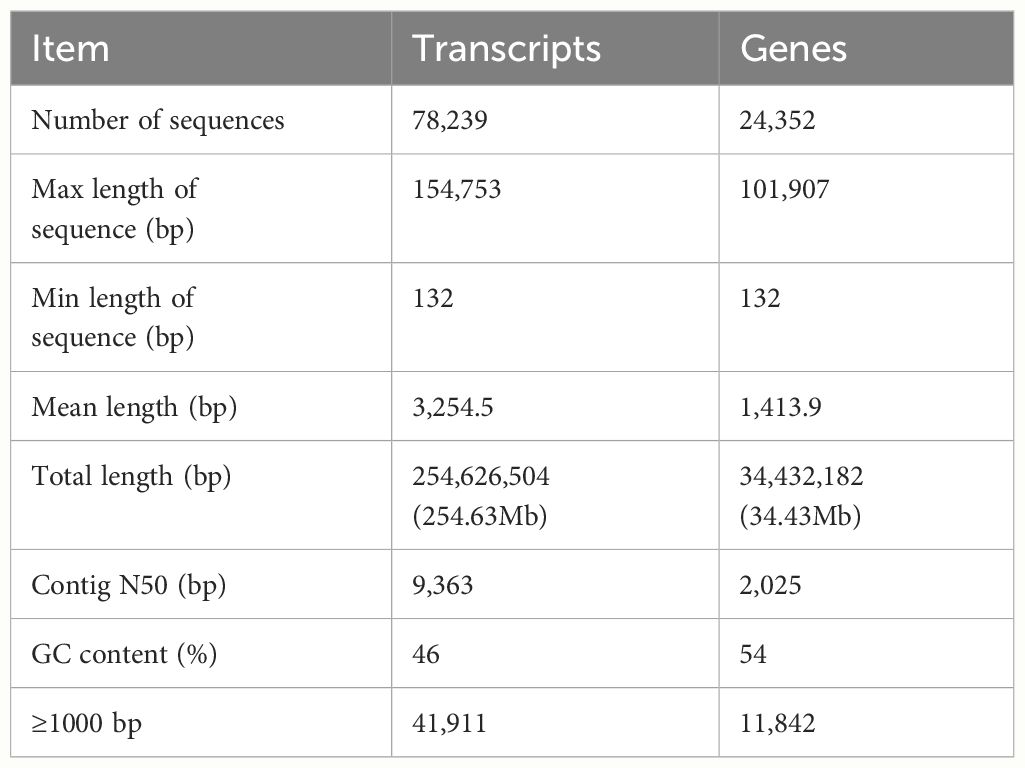
Table 2 Transcripts and genes of the merged assembly.
3.2 DEGs and functional annotation analysis
A total of 2,744 DEGs (1,657 upregulated and 1,087 downregulated genes) were identified in the PF blood groups compared to the healthy blood groups. In GO enrichment analysis, the DEGs in the PF blood group were found to be associated with various immune functions, including immune receptor activity (GO:0140375), adaptive immune response (GO:0002250), innate immune response (GO:0045087), regulation of immune response (GO:0050776), immune effector process (GO:0002252), immune system development (GO:0002520), regulation of immune system process (GO:0002682), activation of immune response (GO:0002253), cell activation involved in immune response (GO:0002263), immune response-regulating signaling pathway (GO:0002764), and myeloid cell activation involved in immune response (GO:0002275) (Supplementary Table S4). The 2744 DEGs were related to 116 KEGG pathways (Supplementary Table S5), in which the most common immune pathways were CD molecules, immune disease, the immune system, cytokine receptors, T-cell receptor signaling pathway, Th1 and Th2 cell differentiation, cytokine–cytokine receptor interaction, the intestinal immune network for IgA production, and NOD-like receptor signaling pathway.
3.3 Immune-related DEGs and apoptosis-related DEGs
Among 2744 DEGs, 240 immune DEGs were identified and verified in the PF blood groups compared to the healthy blood groups, including 186 upregulated and 54 downregulated genes (Supplementary Table S6). The significant expression patterns of immune-related DEGs between PF and healthy blood groups are shown in Supplementary Table S6. In addition, we conducted a hierarchical cluster analysis of top 60 immune DEGs across the nine samples, which indicated that immune-related DEG expression levels in the PF groups could be robustly separated from those in the healthy groups (Figure 1A). The upregulated immune DEGs gathered into one group, and the downregulated immune DEGs gathered into another group. Similarly, PF and healthy blood groups were gathered separately (Figure 1A), thus highlighting gene expression differences between PF and healthy blood groups. The GO analysis revealed that the 240 immune DEGs were significantly enriched in BP including regulation of immune response, adaptive immune response, cell activation involved in immune response, leukocyte activation involved in immune response, cytokine production involved in immune response, immune system process, regulation of immune effector process, innate immune response, immune effector process; CC including receptor complex, extracellular region, and plasma membrane signaling receptor complex, and MF including immune receptor activity, cytokine receptor activity, signaling receptor binding, and molecular transducer activity (Supplementary Table S7). Collectively, these terms were all strongly linked to immunological function. Furthermore, the analysis of KEGG showed that these 240 immune DEGs were significantly enriched in various pathways related to the immune function. These pathways included the NOD-like receptor signaling pathway, Toll-like receptor signaling pathway, TNF signaling pathway, cytokine–cytokine receptor interaction, Th17 cell differentiation, Th1 and Th2 cell differentiation, CD molecules, immune system and immune disease (Supplementary Table S8).

Figure 1 Analysis of immune-related DEGs in the PF and healthy blood of FMD. (A) Heatmap of immune-related DEGs. (B, C) The apoptosis-related DEGs interaction network in the key signaling pathway. (D, E) The immune-related DEGs interaction network in the key signaling pathway.
In the PF blood group, 56 apoptosis-related DEGs (42 upregulated and 14 downregulated genes) were discovered. The significant expression patterns and distribution of the apoptotic DEGs between PF and healthy blood groups are shown in Supplementary Table S9. Furthermore, the analysis of GO revealed that these 56 DEGs related to apoptosis were significantly enriched in BP including regulation of mitochondrial membrane permeability involved in apoptotic process, positive regulation of apoptotic process, negative regulation of extrinsic apoptotic signaling pathway, apoptotic signaling pathway, apoptotic mitochondrial changes, leukocyte apoptotic process, glial cell apoptotic process, and execution phase of apoptosis; CC including proteasome accessory complex, organelle lumen, and membrane-enclosed lumen; and MF including cysteine-type endopeptidase activity involved in apoptotic signaling pathway, cysteine-type endopeptidase activity involved in the execution phase of apoptosis, cysteine-type endopeptidase inhibitor activity involved in apoptotic process, and peptidase activator activity involved in apoptotic process (Supplementary Table S10). An interesting point was that these terms were also related to the process of apoptosis. Furthermore, the KEGG analysis demonstrated that these 56 apoptosis-related DEGs were obviously enriched in apoptosis, cell growth, and death, B-cell receptor signaling network, NOD-like receptor signaling pathway, and NF-kappa B-signaling pathway (as shown in Supplementary Table S11).
3.4 Interaction network analysis of DEGs
In our analysis, we projected a total of 7,658 mRNA-mRNA pairs in the BIPF and healthy blood groups. The apoptosis-related DEGs in the apoptosis, Toll-like receptor signaling pathway, MAPK signaling pathway, T-cell receptor signaling pathway, cell growth and death, and PI3K-Akt signaling pathway were piped to STRING to create PPI networks (Figures 1B, C); the hub genes identified in one of the MCODE models in the PPI network including TLR4, IKBKB, AKT1, MYD88, TP53, JUN, IRF3, IRF7, MAPK1, NGF, and IGF1R major belonged to apoptosis, Toll-like receptor signaling pathway, and MAPK signaling pathway (Figure 1B); the other one MCODE model including TP53, AKT1, IKBKB, PIK3CB, MAPK1, PIK3CG, IGF1R, TLR4, NGF, and CASP7, which were mainly involved in T-cell receptor signaling pathway, cell growth and death, and PI3K-Akt signaling pathway (Figure 1C).
Among the KEGG pathways of immune-related DEGs in the PF and healthy blood groups, we observed that the majority of interaction networks were associated with CD molecules, cytokine–cytokine receptor interaction, NOD-like receptor signaling pathway, TNF signaling pathway, NF-kappa B-signaling pathway, immune system, immune disease, Th17-cell differentiation, Th1 and Th2 cell differentiation (Figures 1D, E; Supplementary Figures S4, S5). The immune-related DEGs in the NF-kappa B-signaling pathway, CD molecules, NOD-like receptor signaling pathway, cytokine–cytokine receptor interaction, Th1 and Th2 cell differentiation, and TNF signaling pathway were inputted into the STRING database to create PPI networks (Figure 1D); the hub genes identified in one of the MCODE models in the PPI network including TLR4, TYK2, CD40, LCK, ZAP70, CD86, TLR2, CCR7, TLR3, IRF3, IRF7 major belonged to CD molecules, NF-kappa B-signaling pathway, and NOD-like receptor signaling pathway (Figure 1B); the other one MCODE model including TYK2, IL6R, CCR7, CSF3R, CXCL13, LCK, IL4R, PIK3CB, SOCS3, IL2RA, LTA, CD40, TNFSF11, and IL27RA were mainly in cytokine–cytokine receptor interaction, Th1 and Th2 cell differentiation, and TNF signaling pathway (Figure 1E). The most prevalent immune pathways associated with DEGs were involved in immune disease and immune system, in which interaction network of the related DEGs were built in Supplementary Figure S4; The hub genes, including TLR4, LCK, ZAP70, CD86, ITGB2, ICOS, CD28, CD3E, CD40, TLR2 major belonged to the immune disease, functioned as core nodes in the regulatory network (Supplementary Figure S4); The hub genes, containning PIK3CB, HCK, VAV1, ITK, TYK2, CCR7, FGR, IKBKB, IRF7, IRF3, PIK3CG, AKT1, and TLR3 were mainly in the regulatory network of immune system pathway (Supplementary Figure S4).
3.5 miRNA library construction and identification
Eight short RNA libraries were created using blood samples from two groups: PF and healthy individuals of FMD. The counts of raw reads and clean reads of high-throughput sRNA were listed in Supplementary Table S12. Out of all the clean reads, about 99.08% were successfully aligned to the FMD reference genome. The unique match ratio ranged from 72.45% to 77.89% (Supplementary Table S13). Initially, the sRNA data were adjusted to mitigate the impact of technical noise. A PCA was conducted, revealing that the samples from the PF and healthy blood groups formed two independent clusters. This distinct cluster was mainly based on the PC1, which accounted for 47% of the total variance (Supplementary Figure S6). The unprocessed data have been stored in the NCBI SRA with the accession number PRJNA667837. The clean reads were annotated using the FMD reference genome and categorized into miRNAs, tRNAs, rRNAs, and other categories by performing a blast analysis using Rfam and miRBase. The unannotated RNAs were used to recognize novel miRNAs. The majority of miRNAs had lengths ranging from 21 to 23 nt, and the majority of the sequencing reads had a length of 22 nucleotides in the eight miRNA libraries. The research discovered a total of 3,206 miRNAs, out of which 1,407 miRNAs (700 known and 707 novel) were shown to be co-expressed in both PF and healthy blood groups. An average of 707 (69.77%) and 546 (64.12%) miRNAs with TPM > 0.5 were obtained in the PF and healthy blood group of FMD, respectively (Supplementary Figure S7). A heatmap of DEmiRNAs in the eight samples was plotted, which showed an obvious separation of DEmiRNAs and group clusters (Figure 2A).
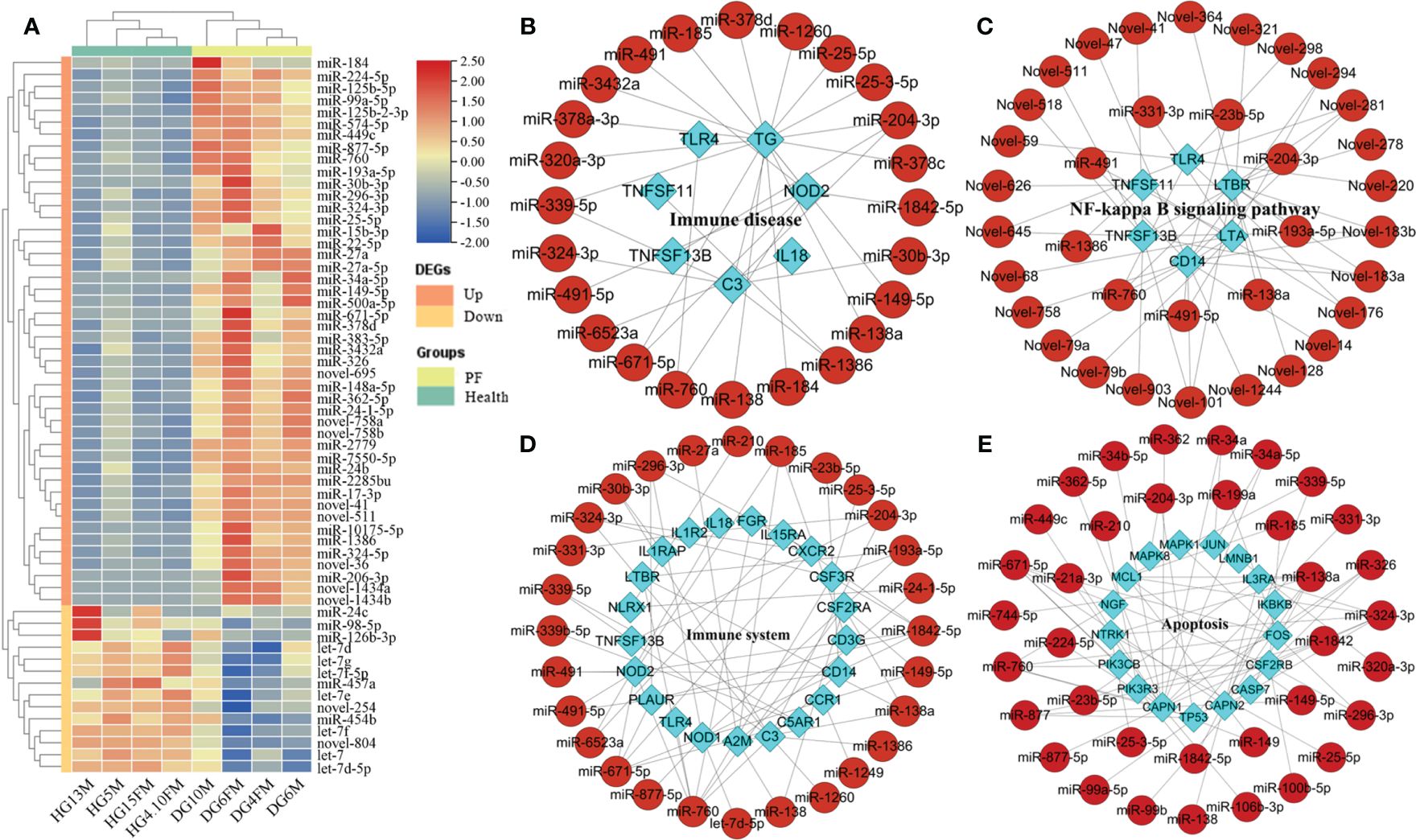
Figure 2 Analysis of DEmiRNAs in the PF and healthy blood of FMD. (A) Heatmap of DEmiRNAs. (B–D) DEmiRNA and target immune-related DEGs interaction network in the key signaling pathway. (E) DEmiRNA and apoptosis-related DEGs interaction network in the apoptosis pathway.
3.6 DEmiRNAs and their target genes enrichment analysis
Three hundred fifty-six DEmiRNAs (332 upregulated and 24 downregulated miRNAs) were identified from a comparison of the PF and healthy blood sRNA libraries in the M. berezovskii. In the DEmiRNAs, 123 known and 233 novel mature miRNAs were identified. Among 356 DEmiRNAs, 53 immune-associated known DEmiRNAs and 55 immune-associated novel DEmiRNAs were identified and verified (Supplementary Table S14). The GO analysis of the DEmiRNA target immune genes were significantly enriched in the immune term including adaptive immune response, negative/positive regulation of immune response, activation of the immune response, regulation of immune system process, cytokine receptor activity, ligand-activated transcription factor activity, G protein–coupled peptide receptor activity, and activation of the immune effector process (Supplementary Table S15). KEGG analysis of these DEmiRNA-target immune genes that they were involved in the Toll-like receptor signaling pathway, antigen processing and presentation, RIG-I-like receptor signaling pathway, HIF-1 signaling pathway, Th1 and Th2 cell differentiation, TGF-beta signaling pathway, immune system, immune disease, and NF-kappa B-signaling pathway (Supplementary Table S16).
3.7 Immune-related DEGs and DEmiRNAs (apoptosis-related DEGs and DEmiRNAs) interaction analysis
The interaction networks of immune-related DEGs (or apoptosis-related DEGs) and DEmiRNAs, which regulated the FMD immune and apoptosis response, were predicted by the STRING and pictured by cytoscape software. The interaction network of DEmiRNAs with their target immune-related DEGs (or apoptosis-related DEGs) has also been constructed and demonstrated in the critical signal pathway (Figure 2A). In the immune disease, the network contained 97 molecules and 173 interactions, including 88 DEmiRNAs and nine immune DEGs (Figure 2A), in which the known DEmiRNAs, such as miR-149-5p, miR-185, miR-204-3p, miR-324-3p, miR-671-5p, and miR-760, related to immune disease, functioned as hub miRNAs in the regulatory network, and the target immune DEGs of these miRNAs included TG, C3, NOD2, TLR4, TNFSF11, IL18, and TNFSF13B as the core node genes. In the immune system, the network contained 132 molecules and 320 interactions, including 104 DEmiRNAs and 28 immune DEGs (Figure 2B), in which the known DEmiRNAs, such as let-7d-5p, miR-324-3p, miR-185, miR-204-3p, miR-193a-5p, miR-671-5p, miR-760, and miR-1842-5p related to immune system, functioned as hub miRNAs in the regulatory network, and the target immune DEGs of these miRNAs included CXCR2, IL1R2, FGR, IL1RAP, IL15RA, C3, NOD1, NOD2, TLR2, TLR4, CCL3, IL18, and TNFSF13B as the core node genes. In the Toll-like receptor signaling pathway, the network contained 31 molecules and 34 interactions, including 27 DEmiRNAs and four immune DEGs (Supplementary Figure S8), in which the known DEmiRNAs, such as miR-23b-5p, miR-491, miR-491-5p, and miR-760 related to Toll-like receptor signaling pathway, functioned as hub miRNAs in the regulatory network, and the target immune DEGs of these miRNAs included TLR2, TLR4, CD14, and CCL3 as the core node genes.
In the NF-kappa B-signaling pathway, the network contained 42 molecules and 79 interactions, including 36 DEmiRNAs and 6 immune DEGs (Figure 2B), in which the known DEmiRNAs, such as miR-138a, miR-193a-5p, miR-204-3p, miR-23b-5p, miR-491, miR-491-5p, miR-331-3p, and miR-760 related to NF-kappa B-signaling pathway, functioned as hub miRNAs in the regulatory network, and the target immune DEGs of these miRNAs included LTA, TLR4, CD14, TNFSF11, TNFSF13B, and LTBR, as the core node genes. In the NOD-like receptor signaling pathway, the network contained 59 molecules and 72 interactions, including 54 DEmiRNAs and five immune DEGs (Supplementary Figure S9), in which the known DEmiRNAs, such as let-7d-5p, miR-149-5p, miR-185, miR-193a-5p, miR-204-3p, miR-23b-5p, miR-185, miR-210, miR-491, miR-491-5p, miR-671-5p, miR-877-5p, miR-760, and miR-1842-5p related to NOD-like receptor signaling pathway, functioned as hub miRNAs in the regulatory network, and the target immune DEGs of these miRNAs included IL18, NOD1, NOD2, TLR4, and NLRX1, as the core node genes. In the apoptosis signaling pathway, the network contained 136 molecules and 214 interactions, including 118 DEmiRNAs and 18 apoptosis-related DEGs (Figure 2B), in which the known DEmiRNAs, such as miR-138a, miR-877-5p, miR-149, miR-760, miR-138, miR-149-5p, miR-877, miR-671-5p,miR-339-5p, miR-326, miR-324-3p, miR-296-3p, miR-34a-5p, and miR-23b-5p, related to apoptosis signaling pathway, functioned as hub miRNAs in the regulatory network, and the target immune DEGs of these miRNAs included MCL1, MAPK8, MAPK1, FOS, IL3RA, TP53, PIK3R3, PIK3CB, CASP7, CSF2RB, NTRK1, IKBKB, and JUN, as the core node genes.
3.8 Validation of DEmRNAs and DEmiRNAs by qRT-PCR
In the study, six immune-related hub DEGs and six immune-related hub DEmiRNAs were chosen for qRT-PCR verification. The results showed that the FPKM of the six immune-related DEGs and the TPM of six DEmiRNAs from RNA-seq (p < 0.05; Figures 3A, 4A) were consistent with those from the RT-qPCR data (Figures 3B, 4B) based on the expression trends between the PF and healthy blood groups, which shown the high reliability of the RNA-seq results in the study.

Figure 3 Verification of immune-related hub DEGs by RT-qPCR. (A) Immune-related hub DEGs expression in terms of FPKM as assessed by mRNA sequencing. (B) qRT-PCR analysis of six immune-related hub DEGs. Data represent the means ± SE. * represents a significant difference and ** represents a very significant difference.

Figure 4 Verification of immune-related hub DEmiRNAs by RT-qPCR. (A) Immune-related hub DEmiRNAs expression in terms of TPM as assessed by RNA-seq. (B) qRT-PCR analysis of six immune-related hub miRNAs. Data represent the means ± SE. * represents a significant difference and ** represents a very significant difference.
4 Discussion
4.1 Innate immune response
Innate immunity is the first line of defense, which can mount resistance to reinfection. In the study, a differential expression analysis was conducted by utilizing mRNA-seq. 240 immune-related DEGs (54 downregulated and 186 upregulated) and 56 apoptosis-related DEGs (14 downregulated and 42 upregulated) were identified in the PF and healthy blood group of FMD. Genes rarely function alone, which form regulatory networks with other molecules to perform biological functions. At this point, immune-related DEGs and apoptosis-related DEGs were built to elucidate the regulatory relationships of DEGs. In the study, some immune-related DEGs (CXCL13, CCL3, ISG20, and IL1R2) were important to the innate immune response, which were upregulated in the PF blood groups. The interaction between CXCL13 and its receptor (CXCR5) has been implicated in the pathogenesis of numerous diseases and the immune responses of healthy organisms (34, 35). The downregulation of CXCL13 may recruit immune cells to the site of infection (34) and eliminate the proinflammatory response (36). CLL3 gene highly expressed in several autoimmune diseases (37–39), which attracted a variety of leukocytes in vitro (40, 41) and thus played protective roles in all kinds of infectious diseases (42–45). Furthermore, it has been documented that CCL3 can serve as a protective barrier against Chlamydia infection; conversely, CCL3 deficiency would render an individual more vulnerable to contracting infectious diseases (38, 39). ISG20, a protein induced by interferon, participated in the innate immune response and has been found to play a role in inflammatory responses and viral infections (46). In addition, the upregulation of ISG20, an exonuclease gene, could inhibit the DNA and RNA virus replication (47). NFKBIE, ISG20, and IL1R2 were crucial in regulating inflammation and host defense (47, 48). CCL23, IRF1, and IRF7 were identified for immune-related DEGs, which had high expression level. The chemokine CCL23 was secreted by different immune cells, which bound its receptor CCR1 and involved in immune response (49, 50). IRF7 could involve in regulation of type I interferon to against pathogens infections and the innate immune response (51). In our study, the upregulation of ISG20, CCL23, IRF7 suggested that the strong innate immune response in the FMD had been activated by the PF.
To further clarify the function of immune DEGs and apoptotic DEGs, KEGG pathway enrichment analysis was performed. In our study, the NOD-like receptor signaling pathway, Toll-like receptor signaling pathway, cytokine–cytokine receptor interactions, CD molecules, immune disease, and immune system were significantly immune-related pathways, which was conducted to elucidate further the function of immune-related DEGs. An examination of the topology of interaction networks involving immune-related DEGs (apoptosis-related DEGs) revealed that the majority of key DEGs were members of the chemokine, interleukin, and TLR families. In this study, five TLR genes (TLR1, TLR2, TLR3, TLR4, and TLR7) were identified for immune DEGs, which participated in pathogens reorganization. In TLRs, TLR1, TLR2, TLR4, and TLR7 were significantly upregulated in the PF, whereas TLR3 was significantly downregulated in the PF. While TLR5, TLR6 had no differences in the PF and healthy blood comparisons. TLRs, which stand for pattern recognition receptors, were identified as their role in recognizing pathogens and activating immune responses (52, 53). TLR4, TLR5, TLR6, TLR7, and TLR8 can identify viral proteins and extracellular bacterial and fungal cell wall components (54), which were vital to the immune responses of the host against a variety of invading pathogens (53). TLR2 and TLR3 were able to induce the immune response by recognizing glycoproteins from various viruses (55, 56). TLR4 participated the innate immunity of rodents against the respiratory syncytial virus (57). TLR7 and TLR8 have the capability to identify single-stranded RNA of viruses that are abundant in guanosine and uridine (58). The upregulation of TLR2 and TLR4, which function as recognition receptors for the immune disease, played a crucial role in the innate immune system of FMD. Interleukin-18 (IL-18) was a highly potent pro-inflammatory cytokine that regulated innate and acquired immune responses and involved in the host’s defense against infections (59). In our study, TLR2, TLR4, and IL18 were identified as hub gene, TLR2 and TLR4 were significantly upregulated, which promoted IL18 production. This research was consistent with previous reports (60). Chemokines were very important for the role of the innate immune system and control the migration and positioning of immune cells, which were a defense against infection and inflammation. CXCL13 and CX3CR1 are both members of the chemokine family. CXCL8/IL-8 was Proven to directly bind to Mycobacterium tuberculosis and potentially enhance the host resistance to infection (61). IL-1R2 was an endogenous inhibitor that prevented the transduction of the IL-1 signal (62). Prior research has demonstrated that IL1R2 exhibited efficacy as an in vivo anti-inflammation and can cure various diseases (63, 64). It was the assertion that IL1R2 secretion cannot eradicate the acute inflammatory response and restrict the inflammatory reaction to alleviate disease (65). Consequently, IL1R2 upregulation may play a significant role in maintaining the physiological equilibrium of FMD inflammatory.
4.2 Adaptive immune response
The regulatory mechanisms of the adaptive immune system was regulated by T cells, B cells, and their antigen-specific receptors (TCR and BCR) (66). Innate immune response can resist the pathogen, while adaptive immune response can finally clear the infection and achieve long-lasting and highly specific protection and sustained by memory T cells (67, 68). CD3E, CD3G, NFKBIE, TRBV4-1, TRBV12-3, TRAV26-1, IL4R, and IL2RA were found to be enriched in the Th1 and Th2 cell division pathways, respectively, in the study. TRAV14DV4, TRAV38-2DV8, and TRDC were also significantly downregulated in the PF blood groups in the study. The TCR family, comprising TRBV4-1, TRBV12-3, TRAV26-1, TRAV14DV4, TRAV38-2DV8, and TRDC, was essential for adaptive immune initiation and facilitated the recognition of an array of antigens (69). CD3E and CD3G were present on the surface of T lymphocytes, which performed important functions in the adaptive immune response (70). IL4R was expressed on eosinophils, macrophages, lung fibroblasts, T and B lymphocytes, which was over-expressed and played significant role in the immune system (71–74). IL2RA is mainly expressed on mature T cells and some other activated hematopoietic cell, which played essential roles in immune regulation of multiple diseases (75).
A variety of interleukins (IL2, IL4, IL5, IL6, IL7, IL10, IL13, IL14, and IL21) were found to participate in the proliferation and differentiation of B cells (76–80). In the study, only IL18, IL27, and IL36a had significantly different expression and were significantly upregulated in the PF blood groups. IL18 regulated both Th 1 and Th2 responses, which acted synergistically with IL12 in the Th1 paradigm, whereas IL18 with IL2 and without IL12 it can induce Th2 cytokine production from CD4+ T cells, natural killer (NK) cells, NKT cells, as well as from Th1 cells (81). IL-27, a member of the cytokine superfamily IL-6/IL-12, significantly regulated immune response (82). IL-27 was discovered to play a broad anti-inflammatory function in infectious and chronic immune-mediated diseases (83–85). IL36 is implicated in both the development and advancement of inflammatory and fibrotic disorders (1–7). Indeed, emerging evidence indicated that IL-36 could mediate the relationship between inflammation and fibrosis (86–88). IL-36a was a members of the IL36 cytokine family (89, 90). In addition to stimulating humoral immunity, IL27 could suppressed T-cell responses in autoimmune conditions and thus induced inflammation and cell death in certain situations (91). IL-18 is a crucial factor in facilitating the synthesis of specific cytokines (92, 93). The observation that the expression of IL18 and the receptor of IL18 (IL18RAP) was significantly increased in the PF blood group compared to the healthy blood group and the expressions of the two genes were negative. More research is required to determine whether the down expression of IL18 affected the effectiveness of the immune responses.
4.3 Integrated analysis of DEGSs–DEmiRNA
MiRNAs regulated many cellular processes and signaling pathways, including embryogenesis, cell proliferation and differentiation, apoptosis, and disease onset. In doing so, miRNAs functioned as potent inhibitors of protein translation via the degradation of mRNAs (94, 95). Nevertheless, the hub miRNAs accountable for the aberrant regulation of immune response and apoptosis in the PF blood group of FMD still need to be further study. In our research, total 282 DEmiRNAs were targeted by the 217 immune DEGs with immunity, and 39 apoptotic DEGs were identified as miRNAs associated with both immunity and apoptosis. A total of 25 genes shared by immune-related DEGs and apoptosis-related DEGs that were identified in the PF and healthy blood groups. DEmiRNA–DEGs interaction networks were constructed based on the principle that miRNAs restrained protein translation by binding to the 3′UTR of target mRNAs and degrading them. In the study, the immune DEGs-DEmiRNA (and apoptotic DEGs-DEmiRNA) interaction network was first constructed in the PF blood group of FMD. In the apoptosis-related DEG–DEmiRNA network of apoptosis pathway, elevated expression of 42 apoptosis-related DEmiRNAs could induce the down expression of MAPK1, MAPK8, TP53, PIK3R3, and NTRK1, while the above 42 apoptosis-related DEmiRNAs could induce the upregulation of IL3RA, CAPN1, CASP7, LMNB1, FOS, NGF, AKT1, JUN, PIK3CB, MCL1, CAPN2, CSF2RB, and IKBKB (Figure 2E). In the immune-related DEG–DEmiRNA network of Toll-like receptor signaling pathway, elevated expression of miR-760, miR-23b-5p, miR-491, and miR-491-5p could induce the down expression of CCL3; while the above 4 apoptosis-related DEmiRNA could induce the upregulation of CD14, TLR2, and IL18 (Figure 2D). In the immune-related DEG–DEmiRNA network of immune disease pathway, elevated expression of 24 immune-related DEmiRNAs could induce the down expression of CCL3 and TNFSF11, while the above 24 immune-related DEmiRNAs could induce the upregulation of TLR2, NOD2, TLR4, C3, TG, IL18, TNFSF13B, and TG (Figure 2B). In the immune-related DEG–DEmiRNA network of immune disease pathway, elevated expression of miR-331-3p, miR-491, miR-491-5p, miR-760, miR-23b-5p, miR-1386, miR-204-3p, miR-193a-5p, and miR-138a could induce the down expression of TNFSF11 and LTA; while the above nine immune-related DEmiRNAs could induce the upregulation of CD14, TLR4, TNFSF13B, and LTBR (Figure 2B). In the immune-related DEG–DEmiRNA network of immune system pathway, downregulated expression of let-7d-5p could induce the upregulated expression of TNFSF11 and LTA, while the above nine immune-related DEmiRNAs could induce the upregulation of CD14, TLR4, TNFSF13B, and LTBR (Figure 2C). The integrated analysis of DEmiRNA–DEG interaction networks results indicated that most hub mRNAs are members of the chemokine, interleukin, and TLR families. It was confirmed that upregulation of hub mRNAs, including CCL4, CXCL10, TLR2, TLR4, and TLR7, occurred in the PF blood groups.
Investigations into the role of miRNAs in the pathogenesis of PF are still rarely. The differential expression of let-7f-5p between the PF and healthy blood groups was identified in the study, suggesting that let-7f-5p may serve as a biomarker for the investigation of the PF mechanism. This result was accordance with previous report (96). It has been reported that the let-7 family miRNAs were demonstrated to participate in the regulation of PF (97). These studies further support the hypothesis that PI3K and the let-7 family may have significant functions in the PF. It has been reported that Let-7f-5p prevents the PF by modulating cellular reactive oxygen species, mitochondrial DNA damage, and cell apoptosis (98). A prior investigation has validated the notion that the target genes of let-7f could enhance the transcriptional program of PF in a model of lung fibrosis induced by bleomycin (99). Notably, the target gene PIK3CA of let-7f-5p was an essential element of the PI3K/Akt pathway, which was instrumental in the pathogenesis of PF (14). It has been demonstrated that vascular endothelial growth factor, reactive oxygen species, and COX2 are involved in the PF in the downstream of the PI3K/AKT signaling pathway (100, 101). It has been believed that COX-2 was shown to regulate the expression of Fas receptor in pulmonary fibroblasts (102). Notably, we found that the downregulated let-7f-5p might induce the upregulation of SLC6A4, POR, and ATP13A3. The results of the integrated analysis of DEmiRNA–DEGs indicated that 29 miRNAs (18 upregulated and 11 downregulated) and 267 genes) formed miRNA-target gene pairs (Figure 2), these complex network of which potentially regulate neuronal cell proliferation, immune cell death, epilepsy, neurodevelopmental disorders, and Wnt/β-catenin and PTEN signaling.
5 Conclusions
In conclusion, the development and occurrence of PF were significantly influenced by the immune-related and apoptosis-related genes present in PF blood. mRNAs and miRNAs associated with the development and occurrence of PF in the FMD were investigated using RNA-seq technology in this study. It was possible to establish the interaction network between immune-related DEGs and DEmiRNAs (apoptosis-related DEGs and DEmiRNAs) by contrasting the profiles and functional analyses of DEmiRNAs and DEGs in the PF and healthy blood of FMD. We obtained 240 immune-related DEGs by RNA-seq, with 186 upregulated and 54 downregulated immune-related DEGs in the PF blood group compared to the healthy blood group. According to functional enrichment analysis, several immune-related pathways and terms were enriched with immune-related DEGs. Fifty-six apoptosis-related DEGs were obtained in the PF and healthy blood groups of FMD. As determined by functional enrichment analysis, apoptosis-related DEGs were enriched in several immune-related terms and pathways. Based on our findings, a gene set consisting of TYK2, TLR2, TLR4, IL18, CSF1, CXCL13, LCK, ITGB2, PIK3CB, HCK, CD40, CD86, CCL3, CCR7, IL2RA, TLR3, and IL4R could potentially function as immunoassay markers for the purpose of monitoring and evaluating the immune status of FMD. By examining networks of immune-related DEGs and DEmiRNAs (apoptosis-related DEGs and DEmiRNAs), our research will offer fresh perspectives on the molecular mechanisms that regulate the progression of PF.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was approved by The Ethics Committee of Chongqing Three Gorges University and Chongqing Institute of Medicinal Plant Cultivation. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
W-HQ: Conceptualization, Investigation, Project administration, Supervision, Writing – original draft. L-FH: Data analysis, Investigation, Methodology, Software, Writing – original draft. Y-JG: Data analysis, Investigation, Visualization, Writing – review & editing. X-YZ: Investigation, Methodology, Software, Writing – review & editing. X-MJ: Data curation, Resources, Writing – review & editing. W-JL: Data curation, Formal analysis, Methodology, Software, Writing – review & editing. J-SQ: Conceptualization, Investigation, Project administration, Supervision, Writing – review & editing. G-SX: Conceptualization, Investigation, Project administration, Supervision, Writing – review & editing. HJ: Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Project administration.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Natural Science Foundation of Chongqing (No.2023NSCQ-MSX0404), National Natural Science Foundation (NSFC31702032, 32370560, 82274046) of P. R. China.
Acknowledgments
We thank Miaojie Yu and Ting Liu at Chongqing Three Gorges University for assisting the research. The authors thank Shanghai Origingene Bio-Pharm Technology Co., Ltd. for providing the sequencing and bioinformatics service for the project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1404108/full#supplementary-material
References
1. Yang Q, Meng X, Xia L, Feng Z. Conservation status and causes of decline of musk deer (Moschus spp.) in China. Biol Conserv. (2003) 109:333–42. doi: 10.1016/S0006-3207(02)00159-3
2. Wang J, Xing H, Qin X, Ren Q, Yang J, Li L. Pharmacological effects and mechanisms of muscone. J Ethnopharmacol. (2020) 262:113–20. doi: 10.1016/j.jep.2020.113120
3. Liu K, Xie L, Deng M, Zhang X, Luo J, Li X. Zoology, chemical composition, pharmacology, quality control and future perspective of Musk (Moschus): a review. Chin Med. (2021) 16:46. doi: 10.1186/s13020-021-00457-8
4. Shrestha M. Animal welfare in the musk deer. Appl Anim Behav Sci. (1998) 59:245–50. doi: 10.1016/S0168-1591(98)00139-7
5. Meng X, Gong B, Ma G, Xiang L. Quantified analyses of musk deer farming in China: a tool for sustainable musk production and ex situ conservation. Asian Australas J Anim Sci. (2011) 24:1473–82. doi: 10.5713/ajas.2011.11111
6. Fan J, Zheng X, Wang H, Qi H, Jiang B, Qiao M, et al. Analysis of genetic diversity and population structure in three forest musk deer captive populations with different origins. G3 (Bethesda). (2019) 9:1037–44. doi: 10.1534/g3.119.400001
7. Qiao J, Wu X, Su L. A review of mainly affected on musk-deer diseases: purulent, respiratory system and parasitic diseases. J Econ Anim. (2009) 13:104–7. doi: 10.13326/j.jea.2009.02.010
8. Lv XH, Qiao JY, Wu XM, Su LN. A review of mainly affected on musk-deer diseases: purulent, respiratory system and parasitic diseases. J Economic Anim. (2009) 13:104–7. doi: 10.3969/j.issn.1007-7448.2009.02.011
9. Umbrello M, Formenti P, Bolgiaghi L, Chiumello D. Current concepts of ARDS: a narrative review. Int J Mol Sci. (2016) 18:64. doi: 10.3390/ijms18010064
10. Huppert LA, Matthay MA, Ware LB. Pathogenesis of acute respiratory distress syndrome. Semin Respir Crit Care Med. (2019) 40:31–9. doi: 10.1055/s-0039-1683996
11. Banavasi H, Nguyen P, Osman H, Soubani AO. Management of ARDS -what works and what does not. Am J Med Sci. (2021) 362:13–23. doi: 10.1016/j.amjms.2020.12.019
12. Li Q, Deng MS, Wang RT, Luo H, Luo YY, Zhang DD, et al. PD-L1 upregulation promotes drug-induced pulmonary fibrosis by inhibiting vimentin degradation. Pharmacol Res. (2023) 187:106636. doi: 10.1016/j.phrs.2022.106636
13. Williams KJ. Gammaherpesviruses and pulmonary fibrosis: evidence from humans, horses, and rodents. Veterinary Pathol. (2014) 51:372–84. doi: 10.1177/0300985814521838
14. Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Su K, et al. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci Rep. (2017) 7:14272. doi: 10.1038/s41598-017-14612-5
15. Wang L, Li S, Yao Y, Yin W, Ye T. The role of natural products in the prevention and treatment of pulmonary fibrosis: a review. Food Funct. (2021) 12:990–1007. doi: 10.1039/D0FO03001E
16. Zhao W, Yu D, Cheng J, Wang Y, Yang Z, Yao X, et al. Identification and pathogenicity analysis of Streptococcus equinus FMD1, a beta-hemolytic strain isolated from forest musk deer lung. J Veterinary Med Sci. (2020) 82:172–6. doi: 10.1292/jvms.19-0566
17. Chioma OS, Drake WP. Role of microbial agents in pulmonary fibrosis. Yale J Biol Med. (2017) 90:219–27.
18. Knippenberg S, Ueberberg B, Maus R, Bohling J, Ding N, Tort Tarres M, et al. Streptococcus pneumoniae triggers progression of pulmonary fibrosis through pneumolysin. Thorax. (2015) 70:636–46. doi: 10.1136/thoraxjnl-2014-206420
19. Liew C, Ma J, Tang H, Zheng R, Dempsey A. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med. (2006) 147:126–32. doi: 10.1016/j.lab.2005.10.005
20. Chaussabel D, Pascual V, Banchereau J. Assessing the human immune system through blood transcriptomics. BMC Biol. (2010) 8:84. doi: 10.1186/1741-7007-8-84
21. Yang M, Huang Y, Wu H, Li C, Ling S, Sun J, et al. Blood transcriptome analysis revealed the immune changes and immunological adaptation of wildness training giant pandas. Mol Genet Genomics. (2022) 297:227–39. doi: 10.1007/s00438-021-01841-7
22. Hooven TA, Polin RA. Pneumonia. Semin Fetal Neonatal Med. (2017) 22:206–13. doi: 10.1016/j.siny.2017.03.002
23. Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol. (2020) 11:1722. doi: 10.3389/fimmu.2020.01722
24. Yan CC, Zhang XS, Zhou L, Yang Q, Zhou M, Zhang LW, et al. Effects of aging on gene expression in blood of captive Tibetan macaques ( Macaca thibetana) and comparisons with expression in humans. Zool Res. (2020) 41:557–63. doi: 10.24272/j.issn.2095-8137.2020.092
25. Yang Q, Yu J, Jiang L, Liu X, Liu F, Cai Y, et al. Identification and expression profile of microRNA in seven tissues of the Golden snub-nosed monkey (Rhinopithecus roxellanae). Mol Genet Genomics. (2020) 295:1547–58. doi: 10.1007/s00438-020-01720-7
26. Griffiths-Jones S, Saini HK, Van Dongen S, Enright AJ. miRBase: Tools for microRNA genomics. Nucleic Acids Res. (2007) 36:D154–8. doi: 10.1093/nar/gkm952
27. Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. (2012) 40:37–52. doi: 10.1093/nar/gkr688
28. Kruger J, Rehmsmeier M. Rnahybrid: microrna target prediction easy, fast and flexible. Nucleic Acids Res. (2006) 34:W451–4. doi: 10.1093/nar/gkl243
29. Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microrna targets predicts functional non-conserved and non-canonical sites. Genome Biol. (2010) 11:R90. doi: 10.1186/gb-2010-11-8-r90
30. Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. (2007) 39:1278–84. doi: 10.1038/ng2135
31. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. (2019) 47:D607–13. doi: 10.1093/nar/gky1131
32. Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. (2021) 49:D605–12. doi: 10.1093/nar/gkaa1074
33. Fan Z, Li W, Jin J, Cui K, Yan C, Peng C, et al. The draft genome sequence of forest musk deer (Moschus berezovskii). Gigascience. (2018) 7:giy038. doi: 10.1093/gigascience/giy038
34. Wang B, Wang M, Ao D, Wei X. CXCL13-CXCR5 axis: Regulation in inflammatory diseases and cancer. Biochim Biophys Acta Rev Cancer. (2022) 1877:188799. doi: 10.1016/j.bbcan.2022.188799
35. Pan Z, Zhu T, Liu Y, Zhang N. Role of the CXCL13/CXCR5 axis in autoimmune diseases. Front Immunol. (2022) 13:850998. doi: 10.3389/fimmu.2022.850998
36. Emu B, Luca D, Offutt C, Grogan JL, Rojkovich B, Williams MB, et al. Safety, pharmacokinetics, and biologic activity of pateclizumab, a novel monoclonal antibody targeting lymphotoxin α: results of a phase I randomized, placebo-controlled trial. Arthritis Res Ther. (2012) 14:R6. doi: 10.1186/ar3554
37. Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Miller SD. An important role for the chemokine macrophage inflammatory protein-1 alpha in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol. (1995) 155:5003–10. doi: 10.4049/jimmunol.155.10.5003
38. Cameron MJ, Arreaza GA, Grattan M, Meagher C, Meagher C, Burdick MD, et al. Differential expression of CC chemokines and the CCR5 receptor in the pancreas is associated with progression to type I diabetes. J Immunol. (2000) 165:1102–10. doi: 10.4049/jimmunol.165.2.1102
39. Crane IJ, Xu H, Manivannan A, McKillop-Smith S, Lamont G, Wallace C, et al. Effect of anti-macrophage inflammatory protein-1alpha on leukocyte trafficking and disease progression in experimental autoimmune uveoretinitis. Eur J Immunol. (2003) 33:402–10. doi: 10.1002/immu.200310014
40. Lee SC, Brummet ME, Shahabuddin S, Woodworth TG, Georas SN, Leiferman KM, et al. Cutaneous injection of human subjects with macrophage inflammatory protein-1 alpha induces significant recruitment of neutrophils and monocytes. J Immunol. (2000) 164:3392–401. doi: 10.4049/jimmunol.164.6.3392
41. DiPietro LA, Burdick M, Low QE, Kunkel SL, Strieter RM. MIP-1alpha as a critical macrophage chemoattractant in murine wound repair. J Clin Invest. (1998) 101:1693–8. doi: 10.1172/JCI1020
42. Olszewski MA, Huffnagle GB, Traynor TR, McDonald RA, Cook DN, Toews GB. Regulatory effects of macrophage inflammatory protein 1alpha/CCL3 on the development of immunity to Cryptococcus neoformans depend on expression of early inflammatory cytokines. Infect Immun. (2001) 69:6256–63. doi: 10.1128/IAI.69.10.6256-6263.2001
43. Lindell DM, Standiford TJ, Mancuso P, Leshen ZJ, Huffnagle GB. Macrophage inflammatory protein 1alpha/CCL3 is required for clearance of an acute Klebsiella pneumoniae pulmonary infection. Infect Immun. (2001) 69:6364–9. doi: 10.1128/IAI.69.10.6364-6369.2001
44. Takahashi H, Tashiro T, Miyazaki M, Kobayashi M, Pollard RB, Suzuki F. An essential role of macrophage inflammatory protein 1alpha/CCL3 on the expression of host’s innate immunities against infectious complications. J Leukoc Biol. (2002) 72:1190–7. doi: 10.1189/jlb.72.6.1190
45. Domachowske JB, Bonville CA, Gao JL, Murphy PM, Easton AJ, Rosenberg HF. The chemokine macrophage-inflammatory protein-1 alpha and its receptor CCR1 control pulmonary inflammation and antiviral host defense in paramyxovirus infection. J Immunol. (2000) 165:2677–82. doi: 10.4049/jimmunol.165.5.2677
46. Deymier S, Louvat C, Fiorini F, Cimarelli A. ISG20: an enigmatic antiviral RNase targeting multiple viruses. FEBS Open Bio. (2022) 12:1096–111. doi: 10.1002/2211-5463.13382
47. Weiss CM, Trobaugh DW, Sun C, Lucas TM, Diamond MS, Ryman KD, et al. The interferon-induced exonuclease ISG20 exerts antiviral activity through upregulation of type I interferon response proteins. mSphere. (2018) 3:e00209-18. doi: 10.1128/mSphere.00209-18
48. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. (2009) 1:a001651. doi: 10.1101/cshperspect.a001651
49. Arruda-Silva F, Bianchetto-Aguilera F, Gasperini S, Polletti S, Cosentino E, Tamassia N, et al. Human neutrophils produce CCL23 in response to various TLR-agonists and TNFα. Front Cell Infect Microbiol. (2017) 7:176. doi: 10.3389/fcimb.2017.00176
50. Novak H, Müller A, Harrer N, Günther C, Carballido JM, Woisetschläger M. CCL23 expression is induced by IL-4 in a STAT6-dependent fashion. J Immunol. (2007) 178:4335–41. doi: 10.4049/jimmunol.178.7.4335
51. Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. (2011) 12:399–414. doi: 10.1038/gene.2011.21
52. Loots AK, Cardoso-Vermaak E, Venter EH, Mitchell E, Kotzé A, Dalton DL. The role of toll-like receptor polymorphisms in susceptibility to canine distemper virus. Mamm Biol. (2018) 88:94–9. doi: 10.1016/j.mambio.2017.11.014
53. Duan T, Du Y, Xing C, Wang HY, Wang RF. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol. (2022) 13:812774. doi: 10.3389/fimmu.2022.812774
54. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
55. Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, et al. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol. (2002) 76:8729–36. doi: 10.1128/jvi.76.17.8729-8736.2002
56. Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. (2004) 10:1366–73. doi: 10.1038/nm1140
57. Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol. (2001) 75:10730–7. doi: 10.1128/JVI.75.22.10730-10737.2001
58. Melchjorsen J, Jensen SB, Malmgaard L, Rasmussen SB, Weber F, Bowie AG, et al. Activation of innate defense against a paramyxovirus is mediated by RIG-I and TLR7 and TLR8 in a cell-type-specific manner. J Virol. (2005) 79:12944–51. doi: 10.1128/JVI.79.20.12944-12951.2005
59. Ihim SA, Abubakar SD, Zian Z, Sasaki T, Saffarioun M, Maleknia S, et al. Interleukin-18 cytokine in immunity, inflammation, and autoimmunity: Biological role in induction, regulation, and treatment. Front Immunol. (2022) 13:919973. doi: 10.3389/fimmu.2022.919973
60. Estruch M, Bancells C, Beloki L, Sanchez-Quesada JL, Ordóñez-Llanos J, Benitez S. CD14 and TLR4 mediate cytokine release promoted by electronegative LDL in monocytes. Atherosclerosis. (2013) 229:356–62. doi: 10.1016/j.atherosclerosis.2013.05.011
61. Krupa A, Fol M, Dziadek BR, Kepka E, Wojciechowska D, Brzostek A, et al. Binding of CXCL8/IL-8 to mycobacterium tuberculosis modulates the innate immune response. Mediators Inflammation. (2015) 2015:124762. doi: 10.1155/2015/124762
62. Peters VA, Joesting JJ, Freund GG. IL-1 receptor 2 (IL-1R2) and its role in immune regulation. Brain Behav Immun. (2013) 32:1–8. doi: 10.1016/j.bbi.2012.11.006
63. Rauschmayr T, Groves RW, Kupper TS. Keratinocyte expression of the type 2 interleukin 1 receptor mediates local and specific inhibition of interleukin 1-mediated inflammation. Proc Natl Acad Sci USA. (1997) 94:5814–9. doi: 10.1073/pnas.94.11.5814
64. Bessis N, Guery L, Mantovani A, Vecchi A, Sims JE, Fradelizi D, et al. The type II decoy receptor of IL-1 inhibits murine collagen-induced arthritis. Eur J Immunol. (2000) 30:867–75. doi: 10.1002/1521-4141(200003)30:3<867::AID-IMMU867>3.3.CO;2-D
65. Mora-Buch R, Dotti I, Planell N, Calderon-Gomez E, Jung P, Masamunt MC, et al. Epithelial IL-1R2 acts as a homeostatic regulator during remission of ulcerative colitis. Mucosal Immunol. (2016) 9:950–9. doi: 10.1038/mi.2015.108
66. Martin SF. Adaptation in the innate immune system and heterologous innate immunity. Cell Mol Life Sci. (2014) 71:4115–30. doi: 10.1007/s00018-014-1676-2
67. Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. (2010) 125:S33–40. doi: 10.1016/j.jaci.2009.09.017
68. Stambas J, Lu C, Tripp RA. Innate and adaptive immune responses in respiratory virus infection: implications for the clinic. Expert Rev Respir Med. (2020) 14:1141–7. doi: 10.1080/17476348.2020.1807945
69. Connelley TK, Degnan K, Longhi CW, Morrison WI. Genomic analysis offers insights into the evolution of the bovine TRA/TRD locus. BMC Genomics. (2014) 15:994. doi: 10.1186/1471-2164-15-994
70. Zhang C, Dang D, Cong L, Sun H, Cong X. Pivotal factors associated with the immunosuppressive tumor microenvironment and melanoma metastasis. Cancer Med. (2021) 10:4710–20. doi: 10.1002/cam4.3963
71. Kotsimbos TC, Ghaffar O, Minshall EM, Humbert M, Durham SR, Pfister R, et al. Expression of the IL-4 receptor alpha-subunit is increased in bronchial biopsy specimens from atopic and nonatopic asthmatic subjects. J Allergy Clin Immunol. (1998) 102:859–66. doi: 10.1016/s0091-6749(98)70029-6
72. Andrews R, Rosa L, Daines M, Khurana Hershey G. Reconstitution of a functional human type II IL-4/IL-13 receptor in mouse B cells: demonstration of species specificity. J Immunol. (2001) 166:1716–22. doi: 10.4049/jimmunol.166.3.1716
73. Dubois GR, Schweizer RC, Versluis C, Bruijnzeel-Koomen CA, Bruijnzeel PL. Human eosinophils constitutively express a functional interleukin-4 receptor: interleukin-4 -induced priming of chemotactic responses and induction of PI-3 kinase activity. Am J Respir Cell Mol Biol. (1998) 19:691–9. doi: 10.1165/ajrcmb.19.4.3208
74. Bankaitis KV, Fingleton B. Targeting IL4/IL4R for the treatment of epithelial cancer metastasis. Clin Exp Metastasis. (2015) 32:847–56. doi: 10.1007/s10585-015-9747-9
75. Nguyen CH, Schlerka A, Grandits AM, Koller E, van der Kouwe E, Vassiliou GS, et al. IL2RA promotes aggressiveness and stem cell-related properties of acute myeloid leukemia. Cancer Res. (2020) 80:4527–39. doi: 10.1158/0008-5472.CAN-20-0531
76. Barker J, Verfaillie CM. A novel in vitro model of early human adult B lymphopoiesis that allows proliferation of pro-B cells and differentiation to mature B lymphocytes. Leukemia. (2000) 14:1614–20. doi: 10.1038/sj.leu.2401869
77. Ghalamfarsa G, Jadidi-Niaragh F, Hojjat-Farsangi M, Asgarian-Omran H, Yousefi M, Tahmasebi F, et al. Differential regulation of B-cell proliferation by IL21 in different subsets of chronic lymphocytic leukemia. Cytokine. (2013) 62:439–45. doi: 10.1016/j.cyto.2013.03.023
78. Magri M, Yatim A, Benne C, Balbo M, Henry A, Serraf A, et al. Notch ligands potentiate IL-7-driven proliferation and survival of human thymocyte precursors. Eur J Immunol. (2009) 39:1231–40. doi: 10.1002/eji.200838765
79. McKenzie AN, Culpepper JA, de Waal Malefyt R, Brière F, Punnonen J, Aversa G, et al. Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc Natl Acad Sci U.S.A. (1993) 90:3735–9. doi: 10.1073/pnas.90.8.3735
80. Meazza R, Gaggero A, Neglia F, Basso S, Sforzini S, Pereno R, et al. Expression of two interleukin-15 mRNA isoforms in human tumors does not correlate with secretion: role of different signal peptides. Eur J Immunol. (1997) 27:1049–54. doi: 10.1002/eji.1830270502
81. Vecchié A, Bonaventura A, Toldo S, Dagna L, Dinarello CA, Abbate A. IL-18 and infections: Is there a role for targeted therapies? J Cell Physiol. (2021) 236:1638–57. doi: 10.1002/jcp.30008
82. Yoshida H, Miyazaki Y. Regulation of immune responses by interleukin-27. Immunol Rev. (2008) 226:234–47. doi: 10.1111/j.1600-065X.2008.00710.x
83. Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. (2006) 7:937–45. doi: 10.1038/ni1376
84. Koltsova EK, Kim G, Lloyd KM, Saris CJ, von Vietinghoff S, Kronenberg M, et al. Interleukin-27 receptor limits atherosclerosis in Ldlr–/– mice. Circ Res. (2012) 111:1274–85. doi: 10.1161/CIRCRESAHA.112.277525
85. Hirase T, Hara H, Miyazaki Y, Ide N, Nishimoto-Hazuku A, Fujimoto H, et al. Interleukin 27 inhibits atherosclerosis via immunoregulation of macrophages in mice. Am J Physiol Heart Circulatory Physiol. (2013) 305:H420–9. doi: 10.1152/ajpheart.00198.2013
86. Artlett CM. The IL-1 family of cytokines. Do they have a role in scleroderma fibrosis? Immunol Lett. (2018) 195:30–7. doi: 10.1016/j.imlet.2017.11.012
87. Scheibe K, Kersten C, Schmied A, Vieth M, Primbs T, Carlé B, et al. Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology. (2019) 156:1082–97.e11. doi: 10.1053/j.gastro.2018.11.029
88. Nishida A, Inatomi O, Fujimoto T, Imaeda H, Tani M, Andoh A. Interleukin-36α induces inflammatory mediators from human pancreatic myofibroblasts via a MyD88 dependent pathway. Pancreas. (2017) 46:539–48. doi: 10.1097/MPA.0000000000000765
89. Dinarello C, Arend W, Sims J, Smith D, Blumberg H, O’Neill L, et al. IL-1 family nomenclature. Nat Immunol. (2010) 11:973. doi: 10.1038/ni1110-973
90. Tripodi D, Conti F, Rosati M, Maccauro G, Saggini A, Cianchetti E, et al. IL-36 a new member of the IL-1 family cytokines. J Biol Regul Homeost Agents. (2012) 26:7–14.
91. Nortey AN, Garces KN, Hackam AS. Exploring the role of interleukin-27 as a regulator of neuronal survival in central nervous system diseases. Neural Regener Res. (2022) 17:2149–52. doi: 10.4103/1673-5374.336134
92. Au WC, Raj NB, Pine R, Pitha PM. Distinct activation of murine interferon-alpha promoter region by IRF-1/ISFG-2 and virus infection. Nucleic Acids Res. (1992) 20:2877–84. doi: 10.1093/nar/20.11.2877
93. Uchida T, Kinoshita M, Fukasawa M, Habu Y, Shinomiya N, Seki S. IL-18 time-dependently modulates Th1/Th2 cytokine production by ligand-activated NKT cells. Eur J Immunol. (2007) 37:966–77. doi: 10.1002/eji.200636465
94. Hata A. Functions of microRNAs in cardiovascular biology and disease. Annu Rev Physiol. (2013) 75:69–93. doi: 10.1146/annurev-physiol-030212-183737
95. Wojciechowska A, Braniewska A, Kozar-Kamińska K. MicroRNA in cardiovascular biology and disease. Adv Clin Exp Med. (2017) 26:865–74. doi: 10.17219/acem/62915
96. Zhao W, Cheng J, Luo Y, Fu W, Zhou L, Wang X, et al. MicroRNA let-7f-5p regulates PI3K/AKT/COX2 signaling pathway in bacteria-induced pulmonary fibrosis via targeting of PIK3CA in forest musk deer. PeerJ. (2022) 10:e14097. doi: 10.7717/peerj.14097
97. Li D, Ji H, Zhao B, Xu C, Xia W, Han L, et al. Therapeutic effect of ulinastatin on pulmonary fibrosis via downregulation of TGF−β1, TNF−α and NF−κB. Mol Med Rep. (2018) 17:1717–23. doi: 10.3892/mmr.2017.8056
98. Sun L, Zhu M, Feng W, Lin Y, Yin J, Jin J, et al. Exosomal miRNA Let-7 from Menstrual Blood-Derived Endometrial Stem Cells Alleviates Pulmonary Fibrosis through Regulating Mitochondrial DNA Damage. Oxid Med Cell Longev. (2019) 2019:4506303. doi: 10.1155/2019/4506303
99. Xie T, Liang J, Guo R, Liu N, Noble PW, Jiang D. Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol Genomics. (2011) 43:479–87. doi: 10.1152/physiolgenomics.00222.2010
100. Laddha AP, Kulkarni YA. VEGF and FGF-2: Promising targets for the treatment of respiratory disorders. Respir Med. (2019) 156:33–46. doi: 10.1016/j.rmed.2019.08.003
101. Wu Q, Zhou Y, Feng FC, Jin YH, Wang ZC, Zhou XM. Probing into the mechanism of alkaline citrus extract promoted apoptosis in pulmonary fibroblasts of bleomycin-induced pulmonary fibrosis mice. Evid Based Complement Alternat Med. (2018) 2018:9658950. doi: 10.1155/2018/9658950
Keywords: blood transcriptome, immune response, miRNA-mRNA network, signal pathway analysis, RT-qPCR, forest musk deer
Citation: Qi W-H, Hu L-F, Gu Y-J, Zhang X-Y, Jiang X-M, Li W-J, Qi J-S, Xiao G-S and Jie H (2024) Integrated mRNA–miRNA transcriptome profiling of blood immune responses potentially related to pulmonary fibrosis in forest musk deer. Front. Immunol. 15:1404108. doi: 10.3389/fimmu.2024.1404108
Received: 20 March 2024; Accepted: 18 April 2024;
Published: 30 May 2024.
Edited by:
Gregory Todd Pharr, Mississippi State University, United StatesReviewed by:
Long Zhang, China West Normal University, ChinaXiuxiang Meng, Renmin University of China, China
Copyright © 2024 Qi, Hu, Gu, Zhang, Jiang, Li, Qi, Xiao and Jie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guo-Sheng Xiao, eGdzMDNAMTYzLmNvbQ==; Hang Jie, amllaGFuZ2lzZ29vZEAxMjYuY29t
†These authors have contributed equally to this work