 Zijian Chen
Zijian Chen Shizhou Cheng1†
Shizhou Cheng1† Zuhai Zhang
Zuhai Zhang- 1Department of Ophthalmology, The First Affiliated Hospital of Yangtze University, Jingzhou, Hubei, China
- 2Physical Examination Department, The First Affiliated Hospital of Yangtze University, Jingzhou, Hubei, China
Ocular toxoplasmosis (OT) is an intraocular infection caused by the parasite Toxoplasma gondii. OT is manifested as retinal choroiditis and is the most common infectious cause of posterior uveitis. Invasion of the retina by T. gondii leads to disruption of the blood-ocular barrier and promotes the migration of immune cells to the ocular tissues. Cytokines such as IFN-γ and IL-1β are effective for controlling parasite growth, but excessive inflammatory responses can cause damage to the host. In this review, we will discuss in detail the latest advances in the immunopathology and treatment of OT.
1 Introduction
Toxoplasma gondii is an intracellular apicomplexan protozoan parasite that attaches to the cell membrane via its apical complex and invades all nucleated cells of vertebrates by gliding motility (1). Toxoplasmosis is an infectious disease caused by T. gondii and is highly prevalent worldwide. Most primary T. gondii infections are asymptomatic (2), which makes early detection and treatment challenging in clinical practice. T. gondii comprise 3 major clonal lineages, namely type I, II and III (2, 3). Type I T. gondii is associated with severe OT, whereas type II strains are less virulent but are the most common cause of human infections (4). Type III strains are least virulent and are often found in domesticated and wild animals and less commonly in humans. Regardless of type, T. gondii can cause life-threatening disease in immunocompromised or immunosuppressed individuals, including those with HIV/AIDS (5). Additionally, these populations have a higher risk of recurrence due to reactivation of latent infection (6). Furthermore, infection during pregnancy can lead to severe neurological damage and even death in the fetus (7).
T. gondii invasion triggers a series of immune responses such as the release of various cytokines that are essential for defending the host against the parasite. The process by which T. gondii invades the eye is complex and involves the migration of the parasite across the blood-retina barrier into the retina, often resulting in infectious uveitis and other ocular complications. Therefore, early diagnosis of systemic toxoplasmosis is important for the treatment and prevention of OT and its complications. OT is typically self-limiting and frequently overlooked. However, its high recurrence rate underscores the importance of a safe and effective intervention. Current treatments (ethylaminopyrimidine and sulfadiazine) are ineffective for eradicating T. gondii and can cause multiple side effects, which may worsen the health of patients with compromised systemic conditions. In fact, adverse reactions to toxoplasmosis treatments have been reported to be the cause of treatment discontinuation in up to 40% of HIV patients (8).
In this review, we will discuss the systemic and ocular immune responses elicited by T. gondii infection, identify the limitations of current toxoplasmosis treatments, and provide insights into the development of novel therapeutic agents from an immunological standpoint.
2 T. gondii infection and dissemination
When an intermediate host consumes raw or undercooked meat containing T. gondii tissue cysts, the cysts rupture as they pass through the gastrointestinal tract, releasing bradyzoites. These bradyzoites infect the host intestinal epithelium and differentiate into tachyzoites that rapidly replicate and disseminate throughout the body (9). The most accepted hypothesis for the mode of T. gondii transmission within the host is the “Trojan horse” mechanism. In this mechanism, the parasite invades immune cells, especially dendritic cells (DC), and exploit their mobility to disseminate. It has been shown that T. gondii-infected DC cells synthesize and secrete the neurotransmitter γ-aminobutyric acid (GABA), which activates GABA-A receptors (10) and induces a hypermigratory phenotype in the cells, ultimately promoting parasite transmission (Figure 1). In addition, the parasite can spread through the bloodstream within monocytes (11, 12) and alter their phenotype to enhance migration (13).
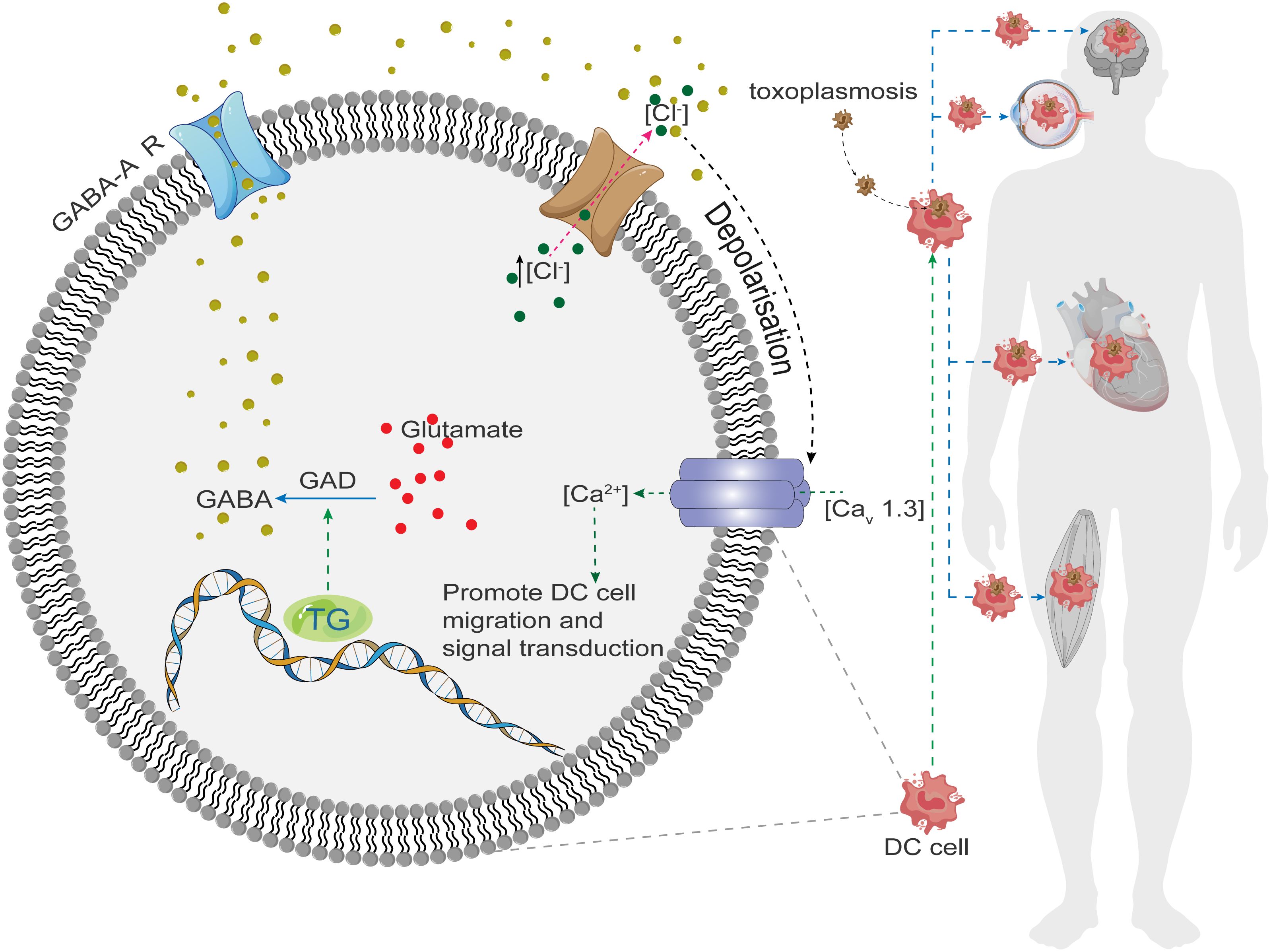
Figure 1 Toxoplasma gondii invades host cells and stimulates GABA synthase (GAD) to synthesize GABA, which binds to GABA-R on the membrane leading to the excretion of Cl- from the cell, generating a depolarization, which opens the downstream voltage-gated Ca2+ channel (Cav1.3), and Ca2+ inflows into the cell, where it plays the role of a second messenger to promote cell motility, migration, and signaling, and In turn, it spreads throughout the body, invading the brain, heart, muscles, and eyes.
3 Initial host immune responses
3.1 Cellular immunity
IFNγ-mediated macrophage activation and CD8+ T cell-mediated cytotoxicity play important roles in controlling T. gondii infections. CD8+ T cells are essential for mediating resistance against T. gondii in mice by stimulating the production of endogenous interferon-γ (IFN-γ) within the first few days of infection (14, 15). T. gondii rhoptry protein (ROP) activates the signal transducers and activators of transcription (STAT)3/6, which modulates intracellular signaling pathways and triggers the production of host IFN-γ and IL-12. In rodents, cellular immune responses mediated by DC cells, T cells, natural killer (NK) cells, macrophages, and cytokines (IL-12 and IFN-γ) are essential for overcoming primary infection and establishing control over latent chronic infection (16). Furthermore, T. gondii infection can induce cell pyroptosis and IL-1β and IL-18 release by activating NLRP1 inflammasomes (17–19).
3.2 IL-12-mediated immune responses
Mouse experiments have demonstrated that T. gondii induces IL-12 production via several pathways: (1) Recognition of T. gondii-derived profilin-like molecules by TLR11 induces IL-12 release in a myeloid differentiation primary response protein 88 (MyD88)-dependent manner (20); (2) Recognition of profilin-like proteins by TLR12 homodimers or TLR11/TLR12 heterodimer leads to IL-12 production; (3) Detection of T. gondii procyclin-18 (TgCyp18) by the chemokine receptor CCR5 stimulates IL-12 expression by DCs (21); (4) T. gondii-mediated TLR9 activation plays a central role in coordinating DC-mediated IL-12 production and subsequent IFN-γ secretion by CD4+ T cells (22).
It has been shown that injection of CD8(+) DCs with soluble T. gondii antigen leads to IL-12 production in mice (23, 24). IL-12 activates CD4+ T cells to secrete IFN-γ, which induces the expression of inducible nitric oxide synthase (iNOS) and the generation of nitric oxide (NO), leading to cytotoxic disruption of T. gondii vesicles. Khan et al. found that treatment of chronically infected hosts with antigen-specific CD4+ T cells restored CD8+ T cell function and prevented reactivation of latent infection (25). Furthermore, antigen-specific CD8+ T cells effectively eliminated T. gondii cysts from immunodeficient animals (26).
3.3 Indoleamine 2,3-dioxygenase
When an organism is infected with T. gondii, binding of IFN-γ, tumor necrosis factor-α (TNF-α), lipopolysaccharide (LPS), cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), CD80/CD86, and other pro-inflammatory cytokines to epithelial cells, DCs, and macrophages can induce the expression of indoleamine-2,3-dioxygenase (IDO) (27, 28). IDO is an intracellular enzyme that inhibits T. gondii growth (27). However, metabolites from the IDO-mediated kynurenine pathway (KP) have been shown to inhibit immune responses by suppressing CD8+ T cell, NK cell, DC, and macrophage functions, promoting Th2 cell and regulatory T cell (Treg) differentiation, as well as inducing the production of transforming growth factor-β (TGF-β) (27). Therefore, IDO expression must be finely regulated to enhance immune clearance of T. gondii while minimizing immunosuppression.
4 Migration to immune privileged regions of the eye
4.1 Migration of T. gondii to the retina
In most cases, T. gondii remains dormant and inactive in the host without eliciting an immune response (29). Upon entry into the human body, T. gondii are transformed into rapidly dividing tachyzoites in the small intestine. These tachyzoites infect most nucleated cells, triggering inflammatory responses that result in immune-mediated tissue damage (30). It has been reported that CD11+ DCs and CD11+ monocytes transport the parasite to brain tissues and the retina through the blood-brain barrier and blood-retina barrier, respectively (12, 31, 32) (Figure 2A). Alternatively, T. gondii may also reach the retina through direct migration of tachyzoites across the retinal vascular endothelium (33). This migration is partly dependent on the interaction between intercellular adhesion molecule-1 (ICAM-1) on retinal endothelial cells (34) and micronemal protein 2 (MIC2) on tachyzoites (35) (Figure 2B). T. gondii infection of brain endothelial cells has been shown to upregulate ICAM-1 expression and thereby facilitate leukocyte migration across the endothelial barrier (36). In addition to ICAM-1, vascular cell adhesion molecule-1 (VCAM-1), activated leukocyte adhesion molecule (ALCAM), and chemokines CXCL21 and CXCL10 are also involved in T. gondii migration across the retinal vascular endothelium (32).
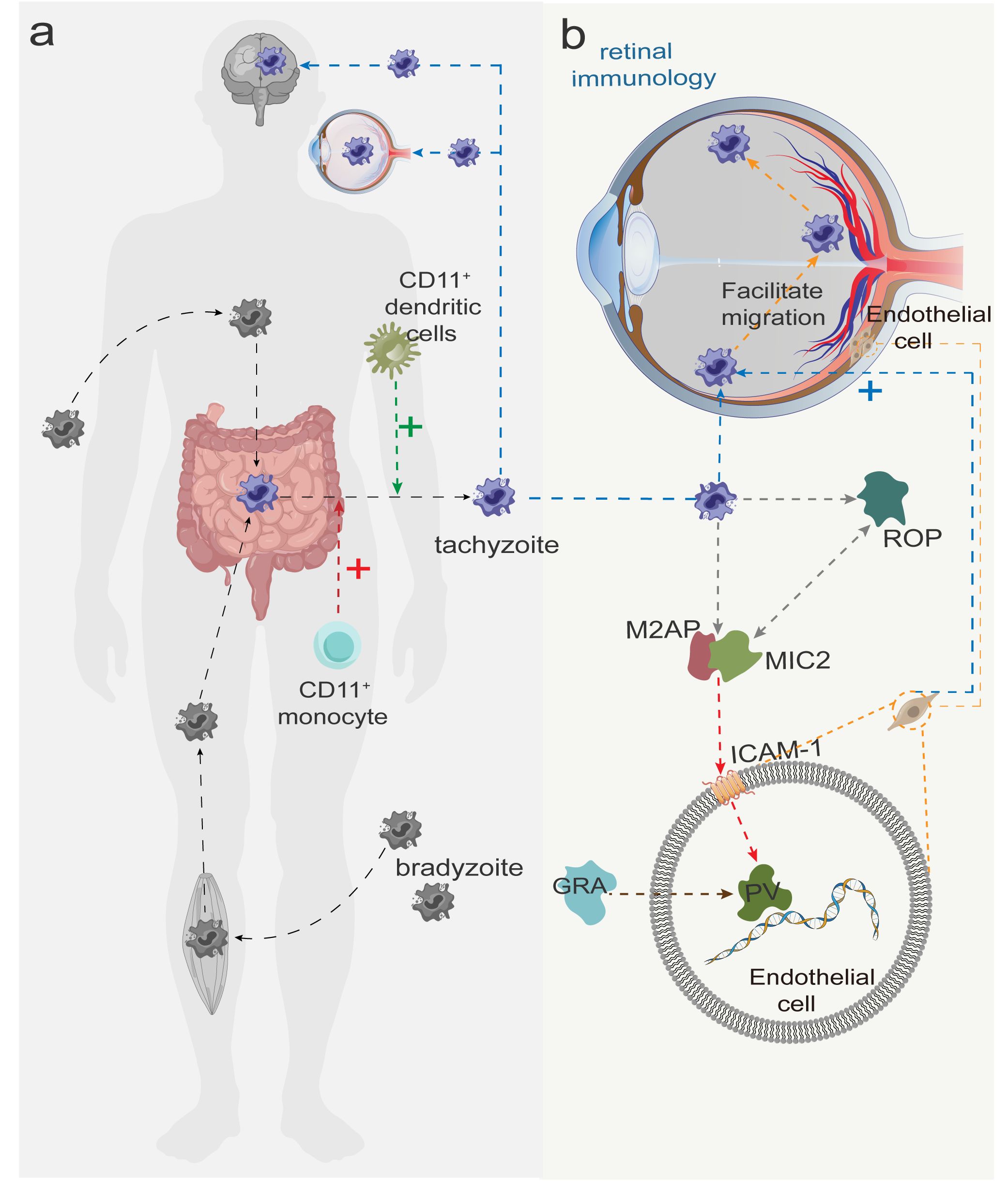
Figure 2 (A) Slow colonizers (dormant form of Toxoplasma gondii) are transmitted after invasion of the human body and are converted to tachyzoites in the small intestine. CD11+ dendritic cells and CD11+ monocytes facilitate the transport of tachyzoites to the brain and the retina (crossing the blood-brain and blood-eye barriers), with the red and green plus signs indicating facilitation. (B) After tachyzoites arrive at the retina with blood, MIC2 is first secreted from the tachyzoite tip and forms a MIC2-M2AP hexameric complex with MIC2-associated protein (M2AP) in a ratio of 1:1, recognizing the intercellular adhesion factor ICAM-2 on the host cell membrane. Subsequently, rod protein (rhoptry protein, ROP) is released and interacts with MIC2 to invade retinal endothelial cells and form parasitophorous vacuole (PV). Finally, dense granule protein (GRA) begin to be secreted, modifying the PV and facilitating the worm’s access to nutrients necessary for survival and replication.
4.2 Disruption of the blood-retina barrier
The inner blood-retina barrier (iBRB) is comprised of retinal endothelial cells, pericytes, Müller cells, and astrocytes. Choroidal vessels are separated from photoreceptor cells by the retinal pigment epithelium (RPE), which together with the Bruch’s membrane, form the outer BRB (oBRB). Entry into the retina through the choroidal layer requires the parasite to cross the oBRB. Song et al. found that human monocyte THP-1 cells infected with T. gondii can migrate across human RPE ARPE-19 cells. Infected monocytes disrupted the oBRB through focal adhesion kinase (FAK) signaling and partly through CXCL8 (37).
4.3 Retinal immune responses
Müller cells and astrocytes are two types of macroglial cells found in the retina of many vertebrates, including mammals, and are essential for maintaining the homeostasis and neural organization of the retina. Müller cells extend from their apical end at the outer limb membrane (OLM) to their basal end at the inner limiting membrane (ILM). Astrocytes are primarily located in the nerve fiber layer (NFL) and ganglion cell layer (GCL). These cells secrete prostaglandins (PGE), NO, and arachidonic acid (AA), as well as molecules that stabilize the tight junctions between vascular endothelial to protect neurons from potential inflammatory damage. Furtado et al. showed that tachyzoites travel to the eyes via the bloodstream and preferentially infect Müller cells and astrocytes in the retina (38). It is worth noting that bradyzoites are present as cysts in the inner layers of the retina and can infect both glial and neuronal cells (39). The form that T. gondii adopts can be influenced by variations in the host eye or the virulence of the strain. Therefore, further studies are warranted to examine how these factors impact the infectious stage of T. gondii.
Studies have shown that high extracellular levels of TNF-α exacerbate astrocyte-mediated inflammation and neurodegeneration. Molecules from inflammatory cells, platelets, and plasma can activate Müller cells to secrete a wide range of inflammatory mediators such as TNF-α, interleukins (IL), interferons, and ICAM-1. Müller cells can directly mediate cytotoxic effects by upregulating TNF-α or monocyte chemotactic protein 1 (MCP-1) expression (40, 41), causing further damage to the retina.
Lie et al. documented increased expression of various immune molecules in the infected RPE, including BIRC3, CCL2, CXL3, CXL8, ICAM-1, IL1RN, IL-6, IL-17RB, LRP8, and NF-κB1 (42). Retinal infection by T. gondii induces VEGF expression in the RPE via activin-like kinase 4 (ALK4) and hypoxia-inducible factor-1 (HIF-1). Upon injury, Müller cells release high levels of VEGF, which may disrupt the BRB and potentially facilitate increased T. gondii migration (40). Furthermore, VEGF production by reactive astrocytes can exacerbate disease progression by increasing vascular permeability, promoting neovascularization, and contributing to cytotoxicity and secondary damage to nearby neurons and glial cells (41).
4.4 Intricate balance between inflammatory and regulatory cytokines
IL-10 is a potent immunomodulatory cytokine and its deficiency increases the susceptibility to T. gondii infection in mice. IL-10 has been shown to inhibit T. gondii-induced inflammation (43) by suppressing Th1 cell differentiation (44), whereas IL-10 deficiency leads to 4- to 6-fold increase in IL-12 and IFN-γ (43, 45). Thus, IL-10 plays a key role in dampening inflammation and restricting excessive proinflammatory response. Previous studies have shown that both IL-27 and IL-33 expression is critical for IL-10 production by effector T cells in T. gondii infection models (46, 47).
Previous study has shown that IL-6-deficient mice infected with T. gondii have severe retinal inflammation and high parasite load, indicating that IL-6 plays a key role in protecting the retina from T. gondii infection (48). In contrast, a study by Rochet et al. demonstrated that topical application of IL-6 antibodies significantly improved retinal structure and reduced parasite load, suggesting that IL-6 can induce retinopathy in mice (49). Similarly, T. gondii infection upregulates IL-17 expression, and IL-17 neutralization is partially protective against fatal T. gondii-associated inflammation (43, 50). IL-17 is secreted by astrocytes during acute inflammation and is protective against neural cell apoptosis and tissue damage in active uveitis by maintaining homeostasis and inhibiting intracellular calcium increase (51). However, excess IL-17 can also lead to unwanted tissue damage (43). Therefore, inflammatory cytokines must be used with caution in the treatment of toxoplasmosis as they may hinder parasite eradication and exacerbate patient discomfort.
Neutrophils play an opposite role to CD8+ T cells during T. gondii infection. Studies have shown that RPE cells infected with T. gondii can activate neutrophils via GM-CSF, IL-6 and IL-18 to produce reactive oxygen species, TNF-α and IL-1β, which are highly damaging to the retinal tissues (52, 53).
In summary, the balance of various cytokines plays a crucial role in toxoplasmosis. Certain cytokines (e.g. IL-1β, IL-6, and IL-17) can act as double-edged swords that protect the host against T. gondii infection but also cause deleterious effects on host cells. Consequently, this delicate balance poses challenges in the development of anti-T. gondii drugs, as any disruption may negatively impact patient outcomes (Table 1).
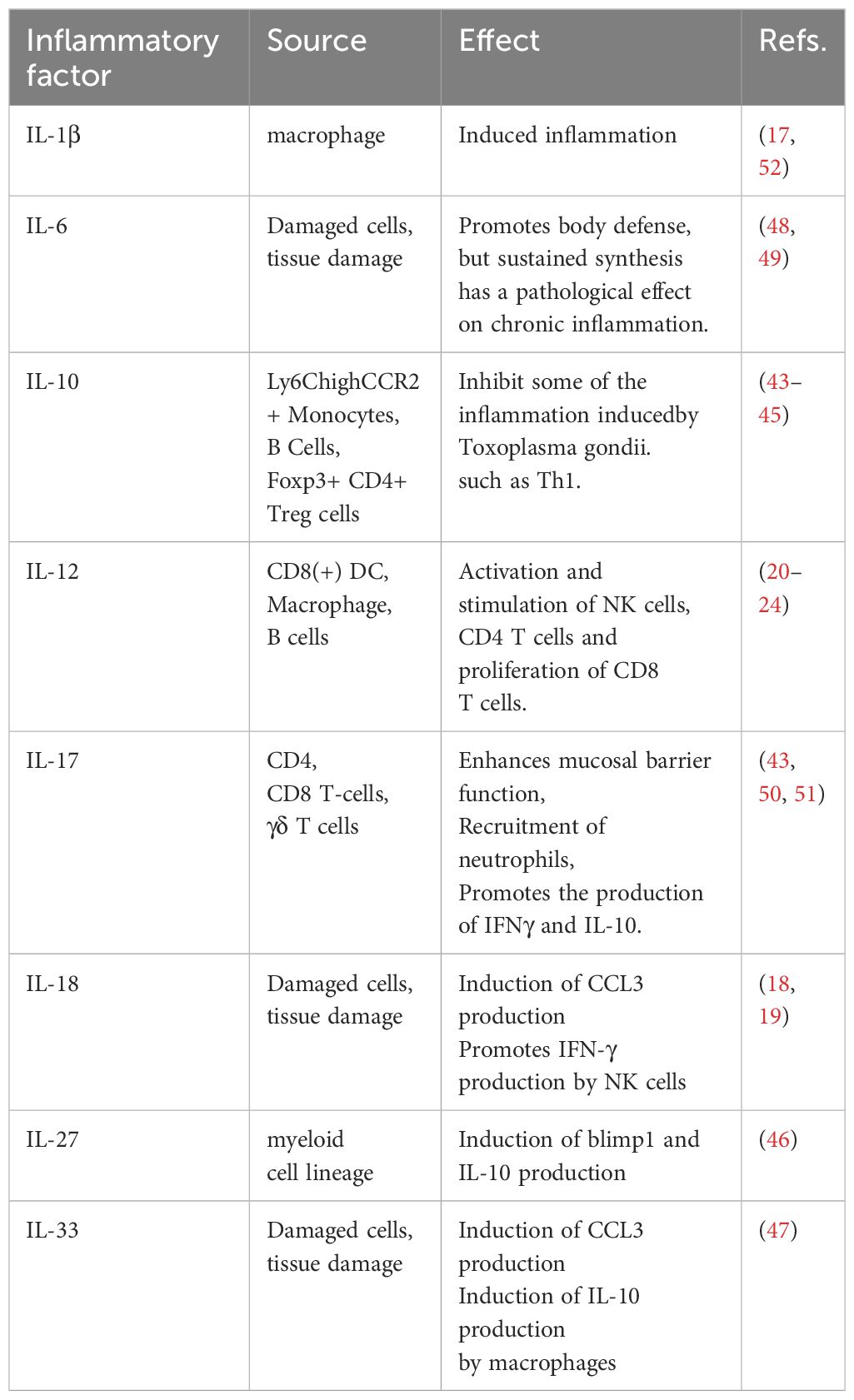
Table 1 The role of inflammatory factors in toxoplasmosis.
5 Ocular immune responses and clinical manifestations
5.1 T. gondii-induced eye damage
OT is a leading cause of infectious uveitis, which can lead to visual impairment and blindness (54). OT accounts for 30–50% of all cases of posterior uveitis and may recur in 40–79% of patients (55). T. gondii infection during pregnancy may lead to miscarriage or congenital toxoplasmosis. In contrast to congenital toxoplasmosis, toxoplasmosis infection acquired after birth is responsible for most OT cases (56). OT is typically asymptomatic in young children (57). However, patients over 45 years of age with active lesion have a 10-fold higher risk of visual impairment. The location of the lesion is critical as macular involvement can result in 8.95-fold higher risk of visual impairment than peripheral retinal damage (54). OT usually presents as posterior uveitis with unilateral choroidal retinopathy and vitritis. Common complications are increased intraocular pressure, cataract, posterior vitreous detachment, retinal detachment, retinal choroidal scarring, and retinal neovascularization (58, 59). Immunocompromised individuals are also more likely to develop OT, with HIV-positive patients having a 2.1 times higher risk of the disease than HIV-negative patients (60).
5.2 Clinical manifestations of OT
Retinal choroiditis is the most common feature of active intraocular inflammation in patients with OT. Early fundus fluorescein angiography (FFA) shows inflammation in areas of low fluorescence, followed by progressive leakage from surrounding retinal vessels and margins of the main lesion, resulting in areas of high fluorescence (61). Inflammation in the anterior chamber of the eye can block aqueous outflow channels leading to increased intraocular pressure (IOP), which is associated with increased anterior chamber cellularity and macular involvement (62). Severe vitritis is characterized by a bright white reflex observed by indirect fundoscopy that resembles “headlight in the fog” (59). Vitritis can cause anterior retinal membrane formation and subsequent vitreoretinal traction in the region of retinal choroiditis, leading to complications such as retinal detachment, vitreomacular traction syndrome, and vitreous hemorrhage. Acute OT presents with extensive retinal necrosis and is usually accompanied by vasculitis. It is predominantly characterized by multifocal segmental retinal arteritis (SRA), also known as Kyrieleis arteritis (63). Active lesions are white with blurred borders located near atrophic or hyperpigmented scars (29). During the relapsing phase, the lesions occur near previous scars with varying hyperpigmentation (61). Moreover, T. gondii infection also promotes the development of punctate extraretinal toxoplasmosis (PORT), neuroretinitis, retinal vascular occlusion, secondary Coats’ disease, Fuchs’ syndrome, and sclerochoroiditis (64), all of which are less commonly reported or present in particular individuals.
6 Diagnostic methods for OT
T. gondii-specific IgG antibodies are detected in most typical clinical cases, suggesting previous infection (congenital or acquired). Although a negative result for IgG antibodies almost excludes T. gondii infection, false-negative results can be observed in rare cases (65, 66). Laboratory diagnostic methods for systemic toxoplasmosis include polymerase chain reaction (PCR), serologic testing, immunohistochemical identification, in vitro culture, and animal inoculation. Rarely, detection of antigens in serum and body fluids, skin tests, and antigen-specific lymphocyte transformation have also been used (67). Detection of specific antibodies in the aqueous and vitreous humor as well as PCR-based assays can be effectively used to diagnose OT (68). In addition, the severity and prognosis of OT can be assessed by imaging tests such as fundus photography, optical coherence tomography (OCT), OCT angiography, ultrasound, confocal scanning laser ophthalmoscopy, FFA and indocyanine green angiography.
7 Breakthroughs in treatment modalities
7.1 Routine OT treatments
OT is a self-limiting disease that is incurable and prone to recurrence. Retinal choroiditis caused by T. gondii usually resolves within 1–2 months (57). The conventional treatment regimen for OT is a combination of pyrimethamine and sulfonamides (sulfadiazine) with or without systemic glucocorticoids (69), which act in different steps of the tetrahydrofolate synthesis pathway to effectively inhibit parasite growth. In particular, pyrimethamine blocks dihydrofolate reductase activity, and sulfadiazine is a competitive inhibitor of p-aminobenzoic acid (57) that negatively impacts nucleic acid synthesis in T. gondii. It has been reported that patients treated with glucocorticoids alone have unfavorable outcomes (70).
7.2 Alternative OT treatments
Trimethoprime-sulfamethoxazol is a common alternative medication for OT (71, 72) due to its good safety profile, accessibility and affordability. It has been found that trimethoprime-sulfamethoxazol in combination with prednisolone is safer and more effective than conventional OT treatment (73).
Alternative treatments with systemic or local (intravitreal) antibiotics (74, 75) have emerged in recent years. Intravitreal injection of clindamycin and dexamethasone has been shown to achieve effective control of retinal chorioretinitis (76, 77). In addition, antibiotic treatment may reduce the risk of recurrent T. gondii-related retinal choroiditis, but there was insufficient evidence to support its benefit in patient outcomes (75).
Two other antiparasitic drugs, atovaquone and azithromycin, have been found to be effective against OT in experimental studies (78) but not in preventing recurrence of retinal chorioretinitis in humans, which may be attributed to the drug resistance in T. gondii (79).
7.3 Treatment for OT complications
Verteporfin photodynamic therapy (V-PDT) and intravitreal injection of anti-VEGF antibody (bevacizumab) are safe and effective treatments for toxoplasmosis-associated choroidal neovascularization (CNV) in the macula, and the former has shown better results in children (80–82).
7.4 Exosome treatment for T. gondii-induced uveitis
Bai et al. verified the role of exosomes secreted by mesenchymal stem cells (MSCs) in an experimental rat model of autoimmune uveoretinitis (83). The authors found that periocular injection of hMSCs-derived exosomes reduced leukocyte infiltration in the eye and alleviated uveitis, attenuating harmful Th1 and Th17 cell-driven immune responses (84). Heightened Th1/Th17 immune responses have been reported in the eyes of OT patients (55), and high levels of Th1 cytokines such as IL-2, IFN-γ, IL-6, IL-17, and MCP-1 were detected in the aqueous humor of OT patients (85). IL-17A is highly expressed in the aqueous humor of OT patients (86) and in the retina of T. gondii-infected mice (87). The balance between Th17 and Th1 responses (especially IFN-γ) is critical for the outcome of infection. Sauer et al. reported a novel in vivo therapeutic approach for inhibiting intraocular inflammation through intravitreal injection of IL-17A monoclonal antibodies (86). Thus, MSCs may serve as a feasible treatment for ocular inflammation caused by T. gondii infection, and hMSCs-derived exosomes have a broad potential for the treatment of retinal chorioretinitis.
7.5 TgMyoA as a potential target for toxoplasmosis treatment
The MyoA motility complex of T. gondii has long been recognized as an attractive target for drug development (88–90). T. gondii motility is driven, at least in part, by this unusual XIVa-like myosin motor protein myosin A (MyoA), which is found only in apicomplexan parasites and a small number of ciliates. Depletion of T. gondii MyoA (TgMyoA) leads to significantly reduced parasite motility, host-cell invasion, and host-cell efflux (91). A drug screening study by Kelsen et al. identified KNX-002 as a compound that inhibits TgMyoA ATPase activity and parasite movement, supporting TgMyoA as a druggable target for toxoplasmosis. However, the risk of drug resistance warrants further investigation (92).
8 Discussion
Toxoplasmosis is a refractory infectious disease caused by the parasite T. gondii. Current treatments are unable to cure toxoplasmosis, and their side effects may exacerbate the health of some patients with poor systemic conditions. Therefore, the development of new, highly effective, and low-toxicity anti-T. gondii drugs and preventive vaccines is a key direction of future research. Furthermore, improving the diagnosis and surveillance of toxoplasmosis, interdisciplinary cooperation and public health education are also crucial for controlling the disease and ensuring timely and effective treatment.
T. gondii invasion in the eye is complicated by the presence of the BRB and the specialized functions of various retinal cells. This complexity makes it challenging to apply findings from non-ocular models to the pathophysiology of OT. New insights into the immune mechanisms of OT may provide clues for the development of topical medications that can minimize ocular complications and eradicate parasites from the eye to prevent recurrence. Therefore, further studies are warranted to clarify the mechanisms of retinal infiltration and inflammation in OT.
Author contributions
ZC: Writing – original draft, Writing – review & editing. SC: Writing – review & editing. XC: Writing – review & editing. ZZ: Writing – review & editing. YD: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dobrowolski JM, Sibley LD. Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell. (1996) 84:933–9. doi: 10.1016/S0092-8674(00)81071-5
2. Hill D, Dubey JP. Toxoplasma gondii: transmission, diagnosis and prevention. Clin Microbiol Infect. (2002) 8:634–40. doi: 10.1046/j.1469-0691.2002.00485.x
3. Howe DK, Sibley LD. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J Infect Dis. (1995) 172:1561–6. doi: 10.1093/infdis/172.6.1561
4. Grigg ME, Ganatra J, Boothroyd JC, Margolis TP. Unusual abundance of atypical strains associated with human ocular toxoplasmosis. J Infect Dis. (2001) 184:633–9. doi: 10.1086/322800
5. Belanger F, Derouin F, Grangeot-Keros L, Meyer L. Incidence and risk factors of toxoplasmosis in a cohort of human immunodeficiency virus-infected patients: 1988–1995. HEMOCO and SEROCO Study Groups. Clin Infect Dis. (1999) 28:575–81. doi: 10.1086/515147
6. Rostami A, Riahi SM, Abdollahzadeh Sagha S, Taghipour A, Sepidarkish M, Mohammadnia-Afrouzi M, et al. Seroprevalence estimates of latent and acute toxoplasma infections in HIV(+) people-call for action in underprivileged communities. Microorganisms. (2021) 9(10):2034. doi: 10.3390/microorganisms9102034
7. Attias M, Teixeira DE, Benchimol M, Vommaro RC, Crepaldi PH, De Souza W. The life-cycle of Toxoplasma gondii reviewed using animations. Parasites Vectors. (2020) 13:588. doi: 10.1186/s13071-020-04445-z
8. Porter SB, Sande MA. Toxoplasmosis of the central nervous system in the acquired immunodeficiency syndrome. N Engl J Med. (1992) 327:1643–8. doi: 10.1056/NEJM199212033272306
9. Robert-Gangneux F, Dardé ML. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin Microbiol Rev. (2012) 25:264–96. doi: 10.1128/CMR.05013-11
10. Bhandage AK, Barragan A. Calling in the Ca(V)alry-Toxoplasma gondii Hijacks GABAergic Signaling and Voltage-Dependent Calcium Channel Signaling for Trojan horse-Mediated Dissemination. Front Cell Infect Microbiol. (2019) 9:61. doi: 10.3389/fcimb.2019.00061
11. Unno A, Suzuki K, Xuan X, Nishikawa Y, Kitoh K, Takashima Y. Dissemination of extracellular and intracellular Toxoplasma gondii tachyzoites in the blood flow. Parasitol Int. (2008) 57:515–8. doi: 10.1016/j.parint.2008.06.004
12. Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gâtel D, Tardieux I. CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood. (2006) 107:309–16. doi: 10.1182/blood-2005-02-0666
13. Ueno N, Harker KS, Clarke EV, McWhorter FY, Liu WF, Tenner AJ, et al. Real-time imaging of Toxoplasma-infected human monocytes under fluidic shear stress reveals rapid translocation of intracellular parasites across endothelial barriers. Cell Microbiol. (2014) 16:580–95. doi: 10.1111/cmi.2014.16.issue-4
14. Shirahata T, Yamashita T, Ohta C, Goto H, Nakane A. CD8+ T lymphocytes are the major cell population involved in the early gamma interferon response and resistance to acute primary Toxoplasma gondii infection in mice. Microbiol Immunol. (1994) 38:789–96. doi: 10.1111/j.1348-0421.1994.tb01858.x
15. Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Sci (New York NY). (1988) 240:516–8. doi: 10.1126/science.3128869
16. Yap GS, Sher A. Effector cells of both nonhemopoietic and hemopoietic origin are required for interferon (IFN)-gamma- and tumor necrosis factor (TNF)-alpha-dependent host resistance to the intracellular pathogen, Toxoplasma gondii. J Exp Med. (1999) 189:1083–92. doi: 10.1084/jem.189.7.1083
17. Ewald SE, Chavarria-Smith J, Boothroyd JC. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect Immun. (2014) 82:460–8. doi: 10.1128/IAI.01170-13
18. Yasuda K, Nakanishi K, Tsutsui H. Interleukin-18 in health and disease. Int J Mol Sci. (2019) 20(3):649. doi: 10.3390/ijms20030649
19. Cai G, Kastelein R, Hunter CA. Interleukin-18 (IL-18) enhances innate IL-12-mediated resistance to Toxoplasma gondii. Infect Immun. (2000) 68:6932–8. doi: 10.1128/IAI.68.12.6932-6938.2000
20. Yarovinsky F. Innate immunity to Toxoplasma gondii infection. Nat Rev Immunol. (2014) 14:109–21. doi: 10.1038/nri3598
21. Sasai M, Yamamoto M. Innate, adaptive, and cell-autonomous immunity against Toxoplasma gondii infection. Exp Mol Med. (2019) 51:1–10. doi: 10.1038/s12276-019-0353-9
22. Benson A, Pifer R, Behrendt CL, Hooper LV, Yarovinsky F. Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe. (2009) 6:187–96. doi: 10.1016/j.chom.2009.06.005
23. Huang LY, Reis e Sousa C, Itoh Y, Inman J, Scott DE. IL-12 induction by a TH1-inducing adjuvant in vivo: dendritic cell subsets and regulation by IL-10. J Immunol. (2001) 167:1423–30. doi: 10.4049/jimmunol.167.3.1423
24. Liu J, Cao S, Kim S, Chung EY, Homma Y, Guan X, et al. Interleukin-12: an update on its immunological activities, signaling and regulation of gene expression. Curr Immunol Rev. (2005) 1:119–37. doi: 10.2174/1573395054065115
25. Khan IA, Hwang S, Moretto M. Toxoplasma gondii: CD8 T cells cry for CD4 help. Front Cell Infect Microbiol. (2019) 9:136. doi: 10.3389/fcimb.2019.00136
26. Sa Q, Ochiai E, Tiwari A, Mullins J, Shastri N, Mercier C, et al. Determination of a key antigen for immunological intervention to target the latent stage of Toxoplasma gondii. J Immunol (Baltimore Md: 1950). (2017) 198:4425–34. doi: 10.4049/jimmunol.1700062
27. Zhou Q, Shi Y, Chen C, Wu F, Chen Z. A narrative review of the roles of indoleamine 2,3-dioxygenase and tryptophan-2,3-dioxygenase in liver diseases. Ann Trans Med. (2021) 9:174. doi: 10.21037/atm
28. Ito H, Hoshi M, Ohtaki H, Taguchi A, Ando K, Ishikawa T, et al. Ability of IDO to attenuate liver injury in alpha-galactosylceramide-induced hepatitis model. J Immunol (Baltimore Md: 1950). (2010) 185:4554–60. doi: 10.4049/jimmunol.0904173
29. Kalogeropoulos D, Sakkas H, Mohammed B, Vartholomatos G, Malamos K, Sreekantam S, et al. Ocular toxoplasmosis: a review of the current diagnostic and therapeutic approaches. Int Ophthalmol. (2022) 42:295–321. doi: 10.1007/s10792-021-01994-9
30. Blader IJ, Saeij JP. Communication between Toxoplasma gondii and its host: impact on parasite growth, development, immune evasion, and virulence. APMIS: Acta Pathol Microbiol Immunol Scand. (2009) 117:458–76. doi: 10.1111/j.1600-0463.2009.02453.x
31. Lambert H, Hitziger N, Dellacasa I, Svensson M, Barragan A. Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell Microbiol. (2006) 8:1611–23. doi: 10.1111/j.1462-5822.2006.00735.x
32. Furtado JM, Bharadwaj AS, Ashander LM, Olivas A, Smith JR. Migration of Toxoplasma gondii-infected dendritic cells across human retinal vascular endothelium. Invest Ophthalmol Visual Sci. (2012) 53:6856–62. doi: 10.1167/iovs.12-10384
33. Tardieux I, Ménard R. Migration of Apicomplexa across biological barriers: the Toxoplasma and Plasmodium rides. Traffic (Copenhagen Denmark). (2008) 9:627–35. doi: 10.1111/j.1600-0854.2008.00703.x
34. Furtado JM, Bharadwaj AS, Chipps TJ, Pan Y, Ashander LM, Smith JR. Toxoplasma gondii tachyzoites cross retinal endothelium assisted by intercellular adhesion molecule-1 in vitro. Immunol Cell Biol. (2012) 90:912–5. doi: 10.1038/icb.2012.21
35. Barragan A, Brossier F, Sibley LD. Transepithelial migration of Toxoplasma gondii involves an interaction of intercellular adhesion molecule 1 (ICAM-1) with the parasite adhesin MIC2. Cell Microbiol. (2005) 7:561–8. doi: 10.1111/cmi.2005.7.issue-4
36. Lachenmaier SM, Deli MA, Meissner M, Liesenfeld O. Intracellular transport of Toxoplasma gondii through the blood-brain barrier. J Neuroimmunol. (2011) 232:119–30. doi: 10.1016/j.jneuroim.2010.10.029
37. Song HB, Jun HO, Kim JH, Lee YH, Choi MH, Kim JH. Disruption of outer blood-retinal barrier by Toxoplasma gondii-infected monocytes is mediated by paracrinely activated FAK signaling. PloS One. (2017) 12:e0175159. doi: 10.1371/journal.pone.0175159
38. Furtado JM, Ashander LM, Mohs K, Chipps TJ, Appukuttan B, Smith JR. Toxoplasma gondii migration within and infection of human retina. PloS One. (2013) 8:e54358. doi: 10.1371/journal.pone.0054358
39. Song HB, Jung BK, Kim JH, Lee YH, Choi MH, Kim JH. Investigation of tissue cysts in the retina in a mouse model of ocular toxoplasmosis: distribution and interaction with glial cells. Parasitol Res. (2018) 117:2597–605. doi: 10.1007/s00436-018-5950-3
40. Le YZ. VEGF production and signaling in Müller glia are critical to modulating vascular function and neuronal integrity in diabetic retinopathy and hypoxic retinal vascular diseases. Vision Res. (2017) 139:108–14. doi: 10.1016/j.visres.2017.05.005
41. de Hoz R, Rojas B, Ramírez AI, Salazar JJ, Gallego BI, Triviño A, et al. Retinal macroglial responses in health and disease. BioMed Res Int. (2016) 2016:2954721. doi: 10.1155/2016/2954721
42. Lie S, Rochet E, Segerdell E, Ma Y, Ashander LM, Shadforth AMA, et al. Immunological molecular responses of human retinal pigment epithelial cells to infection with Toxoplasma gondii. Front Immunol. (2019) 10:708. doi: 10.3389/fimmu.2019.00708
43. Dos Santos PV, de Toledo DNM, de Souza DMS, Menezes TP, Perucci LO, Silva ZM, et al. The imbalance in the relationship between inflammatory and regulatory cytokines during gestational toxoplasmosis can be harmful to fetuses: A systematic review. Front Immunol. (2023) 14:1074760. doi: 10.3389/fimmu.2023.1074760
44. Yap GS, Sher A. Cell-mediated immunity to Toxoplasma gondii: initiation, regulation and effector function. Immunobiology. (1999) 201:240–7. doi: 10.1016/S0171-2985(99)80064-3
45. Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kühn R, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol. (1996) 157:798–805. doi: 10.4049/jimmunol.157.2.798
46. Neumann C, Heinrich F, Neumann K, Junghans V, Mashreghi MF, Ahlers J, et al. Role of Blimp-1 in programing Th effector cells into IL-10 producers. J Exp Med. (2014) 211:1807–19. doi: 10.1084/jem.20131548
47. Nascimento DC, Melo PH, Piñeros AR, Ferreira RG, Colón DF, Donate PB, et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun. (2017) 8:14919. doi: 10.1038/ncomms14919
48. Lyons RE, Anthony JP, Ferguson DJ, Byrne N, Alexander J, Roberts F, et al. Immunological studies of chronic ocular toxoplasmosis: up-regulation of major histocompatibility complex class I and transforming growth factor beta and a protective role for interleukin-6. Infect Immun. (2001) 69:2589–95. doi: 10.1128/IAI.69.4.2589-2595.2001
49. Rochet É, Brunet J, Sabou M, Marcellin L, Bourcier T, Candolfi E, et al. Interleukin-6-driven inflammatory response induces retinal pathology in a model of ocular toxoplasmosis reactivation. Infect Immun. (2015) 83:2109–17. doi: 10.1128/IAI.02985-14
50. Guiton R, Vasseur V, Charron S, Arias MT, Van Langendonck N, Buzoni-Gatel D, et al. Interleukin 17 receptor signaling is deleterious during Toxoplasma gondii infection in susceptible BL6 mice. J Infect Dis. (2010) 202:427–35. doi: 10.1086/653738
51. Hu MH, Zheng QF, Jia XZ, Li Y, Dong YC, Wang CY, et al. Neuroprotection effect of interleukin (IL)-17 secreted by reactive astrocytes is emerged from a high-level IL-17-containing environment during acute neuroinflammation. Clin Exp Immunol. (2014) 175:268–84. doi: 10.1111/cei.12219
52. Philip R, Epstein LB. Tumour necrosis factor as immunomodulator and mediator of monocyte cytotoxicity induced by itself, gamma-interferon and interleukin-1. Nature. (1986) 323:86–9. doi: 10.1038/323086a0
53. Ashander LM, Lie S, Ma Y, Rochet E, Washington JM, Furtado JM, et al. Neutrophil activities in human ocular toxoplasmosis: an in vitro study with human cells. Invest Ophthalmol Vis Sci. (2019) 60:4652–60. doi: 10.1167/iovs.19-28306
54. Cifuentes-González C, Rojas-Carabali W, Pérez ÁO, Carvalho É, Valenzuela F, Miguel-Escuder L, et al. Risk factors for recurrences and visual impairment in patients with ocular toxoplasmosis: A systematic review and meta-analysis. PloS One. (2023) 18:e0283845. doi: 10.1371/journal.pone.0283845
55. Pfaff AW, de-la-Torre A, Rochet E, Brunet J, Sabou M, Sauer A, et al. New clinical and experimental insights into Old World and neotropical ocular toxoplasmosis. Int J Parasitol. (2014) 44:99–107. doi: 10.1016/j.ijpara.2013.09.007
56. Arantes TE, Silveira C, Holland GN, Muccioli C, Yu F, Jones JL, et al. Ocular involvement following postnatally acquired Toxoplasma gondii infection in Southern Brazil: A 28-year experience. Am J Ophthalmol. (2015) 159:1002–12.e2. doi: 10.1016/j.ajo.2015.02.015
57. Butler NJ, Furtado JM, Winthrop KL, Smith JR. Ocular toxoplasmosis II: clinical features, pathology and management. Clin Exp Ophthalmol. (2013) 41:95–108. doi: 10.1111/j.1442-9071.2012.02838.x
58. Balasundaram MB, Andavar R, Palaniswamy M, Venkatapathy N. Outbreak of acquired ocular toxoplasmosis involving 248 patients. Arch Ophthalmol (Chicago Ill: 1960). (2010) 128:28–32. doi: 10.1001/archophthalmol.2009.354
59. Delair E, Latkany P, Noble AG, Rabiah P, McLeod R, Brézin A. Clinical manifestations of ocular toxoplasmosis. Ocular Immunol Inflamm. (2011) 19:91–102. doi: 10.3109/09273948.2011.564068
60. Hodge WG, Seiff SR, Margolis TP. Ocular opportunistic infection incidences among patients who are HIV positive compared to patients who are HIV negative. Ophthalmology. (1998) 105:895–900. doi: 10.1016/S0161-6420(98)95033-3
61. Al-Dhibi HA, Al-Mahmood AM, Arevalo JF. A systematic approach to emergencies in uveitis. Middle East Afr J Ophthalmol. (2014) 21:251–8. doi: 10.4103/0974-9233.134687
62. Dodds EM, Holland GN, Stanford MR, Yu F, Siu WO, Shah KH, et al. Intraocular inflammation associated with ocular toxoplasmosis: relationships at initial examination. Am J Ophthalmol. (2008) 146:856–65.e2. doi: 10.1016/j.ajo.2008.09.006
63. Khadamy J. Atypical ocular toxoplasmosis: multifocal segmental retinal arteritis (Kyrieleis arteritis) and peripheral choroidal leision. Cureus. (2023) 15:e47060. doi: 10.7759/cureus.47060
64. Smith JR, Cunningham ET Jr. Atypical presentations of ocular toxoplasmosis. Curr Opin Ophthalmol. (2002) 13:387–92. doi: 10.1097/00055735-200212000-00008
65. Bidgoli S, Koch P, Caspers L. [Toxoplasmic chorioretinitis: positive PCR on vitreous with negative serology for Toxoplasma gondii]. J francais d’ophtalmologie. (2011) 34:384.e1–5. doi: 10.1016/j.jfo.2011.01.015
66. Rajput R, Denniston AK, Murray PI. False negative toxoplasma serology in an immunocompromised patient with PCR positive ocular toxoplasmosis. Ocular Immunol Inflamm. (2018) 26:1200–2. doi: 10.1080/09273948.2017.1332769
67. Saadatnia G, Golkar M. A review on human toxoplasmosis. Scand J Infect Dis. (2012) 44:805–14. doi: 10.3109/00365548.2012.693197
68. Montoya JG. Laboratory diagnosis of Toxoplasma gondii infection and toxoplasmosis. J Infect Dis. (2002) 185 Suppl 1:S73–82. doi: 10.1086/338827
69. Stanford MR, Gilbert RE. Treating ocular toxoplasmosis: current evidence. Memorias do Instituto Oswaldo Cruz. (2009) 104:312–5. doi: 10.1590/S0074-02762009000200027
70. Bosch-Driessen LE, Berendschot TT, Ongkosuwito JV, Rothova A. Ocular toxoplasmosis: clinical features and prognosis of 154 patients. Ophthalmology. (2002) 109:869–78. doi: 10.1016/S0161-6420(02)00990-9
71. Souza CE, Nascimento H, Lima A, Muccioli C, Belfort R Jr. Intravitreal injection of sulfamethoxazole and trimethoprim associated with dexamethasone as an alternative therapy for ocular toxoplasmosis. Ocular Immunol Inflamm. (2018) 26:1041–4. doi: 10.1080/09273948.2017.1307420
72. Choudhury H, Jindal A, Pathengay A, Bawdekar A, Albini T, Flynn HW Jr. The role of intravitreal trimethoprim/sulfamethoxazole in the treatment of toxoplasma retinochoroiditis. Ophthalmic Surg Lasers Imaging Retina. (2015) 46:137–40. doi: 10.3928/23258160-20150101-27
73. Rothova A, Meenken C, Buitenhuis HJ, Brinkman CJ, Baarsma GS, Boen-Tan TN, et al. Therapy for ocular toxoplasmosis. Am J Ophthalmol. (1993) 115:517–23. doi: 10.1016/S0002-9394(14)74456-3
74. Vasconcelos-Santos DV. Ocular manifestations of systemic disease: toxoplasmosis. Curr Opin Ophthalmol. (2012) 23:543–50. doi: 10.1097/ICU.0b013e328358bae5
75. Pradhan E, Bhandari S, Gilbert RE, Stanford M. Antibiotics versus no treatment for toxoplasma retinochoroiditis. Cochrane Database Syst Rev. (2016) 2016:Cd002218. doi: 10.1002/14651858
76. Zamora YF, Arantes T, Reis FA, Garcia CR, Saraceno JJ, Belfort R Jr., et al. Local treatment of toxoplasmic retinochoroiditis with intravitreal clindamycin and dexamethasone. Arquivos Brasileiros Oftalmol. (2015) 78:216–9. doi: 10.5935/0004-2749.20150056
77. Lasave AF, Díaz-Llopis M, Muccioli C, Belfort R Jr., Arevalo JF. Intravitreal clindamycin and dexamethasone for zone 1 toxoplasmic retinochoroiditis at twenty-four months. Ophthalmology. (2010) 117:1831–8. doi: 10.1016/j.ophtha.2010.01.028
78. Holland GN, Lewis KG. An update on current practices in the management of ocular toxoplasmosis. Am J Ophthalmol. (2002) 134:102–14. doi: 10.1016/S0002-9394(02)01526-X
79. Baatz H, Mirshahi A, Puchta J, Gümbel H, Hattenbach LO. Reactivation of toxoplasma retinochoroiditis under atovaquone therapy in an immunocompetent patient. Ocular Immunol Inflamm. (2006) 14:185–7. doi: 10.1080/09273940600659740
80. Mauget-Faÿsse M, Mimoun G, Ruiz-Moreno JM, Quaranta-El Maftouhi M, De Laey JJ, Postelmans L, et al. Verteporfin photodynamic therapy for choroidal neovascularization associated with toxoplasmic retinochoroiditis. Retina (Philadelphia Pa). (2006) 26:396–403. doi: 10.1097/00006982-200604000-00003
81. Oliveira LB, Reis PA. Photodynamic therapy-treated choroidal neovascular membrane secondary to toxoplasmic retinochoroiditis. Graefe’s Arch Clin Exp Ophthalmol = Albrecht von Graefes Archiv fur klinische und Experimentelle Ophthalmol. (2004) 242:1028–30. doi: 10.1007/s00417-004-0894-y
82. Kianersi F, Naderi Beni A, Naderi Beni Z, Ghanbari H. Intravitreal bevacizumab for treatment of choroidal neovascularization secondary to toxoplasmic retinochoroiditis: a case series. Semin Ophthalmol. (2015) 30:181–7. doi: 10.3109/08820538.2013.838278
83. Bai L, Shao H, Wang H, Zhang Z, Su C, Dong L, et al. Effects of mesenchymal stem cell-derived exosomes on experimental autoimmune uveitis. Sci Rep. (2017) 7:4323. doi: 10.1038/s41598-017-04559-y
84. Harrell CR, Simovic Markovic B, Fellabaum C, Arsenijevic A, Djonov V, Arsenijevic N, et al. Therapeutic potential of mesenchymal stem cell-derived exosomes in the treatment of eye diseases. Adv Exp Med Biol. (2018) 1089:47–57. doi: 10.1007/5584_2018_219
85. Lahmar I, Abou-Bacar A, Abdelrahman T, Guinard M, Babba H, Ben Yahia S, et al. Cytokine profiles in toxoplasmic and viral uveitis. J Infect Dis. (2009) 199:1239–49. doi: 10.1086/597478
86. Sauer A, Pfaff AW, Villard O, Creuzot-Garcher C, Dalle F, Chiquet C, et al. Interleukin 17A as an effective target for anti-inflammatory and antiparasitic treatment of toxoplasmic uveitis. J Infect Dis. (2012) 206:1319–29. doi: 10.1093/infdis/jis486
87. Kikumura A, Ishikawa T, Norose K. Kinetic analysis of cytokines, chemokines, chemokine receptors and adhesion molecules in murine ocular toxoplasmosis. Br J Ophthalmol. (2012) 96:1259–67. doi: 10.1136/bjophthalmol-2012-301490
88. Trivedi DV, Nag S, Spudich A, Ruppel KM, Spudich JA. The myosin family of mechanoenzymes: from mechanisms to therapeutic approaches. Annu Rev Biochem. (2020) 89:667–93. doi: 10.1146/annurev-biochem-011520-105234
89. Bookwalter CS, Kelsen A, Leung JM, Ward GE, Trybus KM. A Toxoplasma gondii class XIV myosin, expressed in Sf9 cells with a parasite co-chaperone, requires two light chains for fast motility. J Biol Chem. (2014) 289:30832–41. doi: 10.1074/jbc.M114.572453
90. Thomas JC, Green JL, Howson RI, Simpson P, Moss DK, Martin SR, et al. Interaction and dynamics of the Plasmodium falciparum MTIP-MyoA complex, a key component of the invasion motor in the malaria parasite. Mol bioSystems. (2010) 6:494–8. doi: 10.1039/b922093c
91. Egarter S, Andenmatten N, Jackson AJ, Whitelaw JA, Pall G, Black JA, et al. The toxoplasma Acto-MyoA motor complex is important but not essential for gliding motility and host cell invasion. PloS One. (2014) 9:e91819. doi: 10.1371/journal.pone.0091819
Keywords: toxoplasmosis, ocular toxoplasmosis, immune response, cytokines, therapy
Citation: Chen Z, Cheng S, Chen X, Zhang Z and Du Y (2024) New advances in immune mechanism and treatment during ocular toxoplasmosis. Front. Immunol. 15:1403025. doi: 10.3389/fimmu.2024.1403025
Received: 18 March 2024; Accepted: 30 April 2024;
Published: 10 May 2024.
Edited by:
Jing Yang, Yunnan Agricultural University, ChinaReviewed by:
Celio Geraldo Freire-de-Lima, Federal University of Rio de Janeiro, BrazilAulia Rahmi Pawestri, University of Brawijaya, Indonesia
Copyright © 2024 Chen, Cheng, Chen, Zhang and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zuhai Zhang, enVoYWlfemhhbmdAb3V0bG9vay5jb20=; Yanhua Du, ODQ3MTAwMTlAcXEuY29t
†These authors have contributed equally to this work