 Johannes Reusch1
Johannes Reusch1 Linda Elise Franken2
Linda Elise Franken2 Jakob Then1Philippe Ringler2,3
Jakob Then1Philippe Ringler2,3 Joachim Butzer1Thomas Juroschek1
Joachim Butzer1Thomas Juroschek1 Christian Klein4
Christian Klein4 Tilman Schlothauer1*†Laurent Larivière1*†
Tilman Schlothauer1*†Laurent Larivière1*†- 1Roche Pharma Research and Early Development, Therapeutic Modalities, Roche Innovation Center Munich, Roche Diagnostics GmbH, Penzberg, Germany
- 2Roche Pharma Research and Early Development, Therapeutic Modalities, Roche Innovation Center Basel, F.Hoffmann-La Roche Ltd, Basel, Switzerland
- 3Biozentrum, University of Basel, Basel, Switzerland
- 4Roche Pharma Research and Early Development, Discovery Oncology, Roche Innovation Center Zurich, Roche Glycart AG, Schlieren, Switzerland
TRIM21 is a pivotal effector in the immune system, orchestrating antibody-mediated responses and modulating immune signaling. In this comprehensive study, we focus on the interaction of TRIM21 with Fc engineered antibodies and subsequent implications for viral neutralization. Through a series of analytical techniques, including biosensor assays, mass photometry, and electron microscopy, along with structure predictions, we unravel the intricate mechanisms governing the interplay between TRIM21 and antibodies. Our investigations reveal that the TRIM21 capacity to recognize, bind, and facilitate the proteasomal degradation of antibody-coated viruses is critically dependent on the affinity and avidity interplay of its interactions with antibody Fc regions. We suggest a novel binding mechanism, where TRIM21 binding to one Fc site results in the detachment of PRYSPRY from the coiled-coil domain, enhancing mobility due to its flexible linker, thereby facilitating the engagement of the second site, resulting in avidity due to bivalent engagement. These findings shed light on the dual role of TRIM21 in antiviral immunity, both in recognizing and directing viruses for intracellular degradation, and demonstrate its potential for therapeutic exploitation. The study advances our understanding of intracellular immune responses and opens new avenues for the development of antiviral strategies and innovation in tailored effector functions designed to leverage TRIM21s unique binding mode.
1 Introduction
The tripartite motif-containing 21 (TRIM21) protein also known as Ro52 or RNF81, has emerged as a pivotal player in the dynamic interface between the adaptive and innate arms of the immune system (1). With its unique functions as effector and sensor occurring simultaneously, it triggers the antibody mediated degradation of viral capsids by the proteasome and activates innate immune signaling pathways (2–5). This unique intracellular receptor exhibits an ancient origin, and its importance in immune regulation and host defense has garnered significant attention and demands further exploration (6).
In healthy cells, a functional antibody is not capable of entering the cytosol since it neither has a mechanism to penetrate the cell membrane, nor can it exit the endosome during endocytosis (7, 8). However, it was reported that antibodies bound to incoming pathogens remain attached to viral capsids, even after endosomal escape, and are thus delivered into the cytosol (9). Here, TRIM21, an E3 ubiquitin ligase ubiquitously expressed in human cells, recognizes the Fc part of antibodies (associated to viruses) and directs the complex to the ubiquitin-proteasome system (UPS) for its degradation (10). This process is called antibody-dependent intracellular neutralization (ADIN). TRIM21 expression is drastically upregulated by interferon (IFN) stimulation thereby amplifying its neutralization activities (3). As an intracellular antibody Fc receptor, TRIM21 complements the traditional extracellular functions of antibodies by allowing them to function as immune regulators within cells and activate immune signaling pathways like the transcription factor NF-κB, AP-1 and IRF 3/5/7 (5, 11).
The Trim family of protein is characterized by a remarkably conserved N-terminal RBCC region (12). This region is composed of a really interesting new gene (RING) domain, one or two B-box domains, and a coiled-coil domain which represents the triumvirate giving the family its name (12–14). Trim proteins exhibit variations in their C-terminal regions, which are responsible for their specific functional attributes (15). For TRIM21, a C terminal PRYSPRY (PS) domain is responsible for target binding.
The RING domain functions as an E3 ubiquitin ligase, essential for recognizing and tagging proteins for degradation by attaching ubiquitin chains, signaling their breakdown by the proteasome (11, 16–18). The B-Box domain, specific to TRIM proteins, regulates TRIM21 by preventing auto-ubiquitination, thus regulating immune response activation (19–21). The coiled-coil domain (B-Box C-terminal; BBC) promotes dimerization of TRIM21 monomers (RBCC-PS), positioning RING domains at either end of an elongated antiparallel coiled coil domain, which is inactivated while the PRY-SPRY domain (B30.2) binds to IgG Fc, indicating its role in immune function (15).
In mammals, the TRIM21-IgG interaction is highly conserved, allowing the PRYSPRY domain to recognize IgGs from one species to various other species (22). Although TRIM21 has a lower affinity for the Fc region of IgA and IgM compared to IgG, targeting IgA still enables neutralization (23). As a homodimeric molecule, TRIM21 symmetrically binds to an IgG, engaging both Fc heavy chains simultaneously. This results in a 1:1 stoichiometry with the heterodimeric IgG (9). To date, only a single known apparent affinity/avidity dissociation constant (KD,AVIDITY) for dimeric TRIM21 binding to the IgG1 Fc wild type has been reported. This value is 600 pM, as measured by fluorescence anisotropy, while the kinetic rate parameters (kON and kOFF) remain undetermined. This interaction displays the strongest affinity observed in human IgG-Fc receptor interactions (9).
The binding affinity (KD,AFFINITY) between human IgG1 Fc WT and the recombinant human PRYSPRY domain has been measured to be in the range of 40 nM by isothermal titration calorimetry (ITC) to 200 nM applying surface plasmon resonance (SPR) technology, depending on technology and assay setup (9, 10, 24). The PRYSPRY domain (antibody Fc binding domain) interacts with both CH2 and CH3 domains, although the most substantial interaction is with the CH3 domain (24, 25). Like FcRn, the neonatal Fc receptor, TRIM21 interacts with the HNYH-motif where amino acids 429 – 436, especially H433, N434, H435, and Y436 of the CH3 Fc loop as well as I253 of CH2 are inserted into the PRYSPRY binding pocket (24, 26–30). FcRn prolongs the half-life of IgG by protecting it from lysosomal degradation and mediating its recycling back into the bloodstream. Unlike FcRn, TRIM21 – IgG interaction is reported to be largely pH-independent (from pH 5-8) but sensitive to salt. The ionic strength within cells is a critical determinant of biomolecular interactions, influencing protein activity, aggregation, and cellular processes essential for survival and proliferation (31, 32). Notably, the TRIM21-IgG interaction demonstrates a significant increase in affinity (5-fold) with reduced ionic strength, which is reflected by a slower off-rate when the salt concentration is decreased from 200 to 20 mM (22, 24). This sensitivity to ionic conditions demonstrates the importance of considering intracellular electrostatic environments when investigating the mechanism of action of TRIM21 and its role in immune complex degradation.
For the FcRn – IgG interaction there are reported mutations within the hotspot region, like YTE (M252Y/S254T/T256E), HH (T307H, N434H), which demonstrate increased FcRn binding, while mutating key residues into alanines (H310A, H433A and Y436A; AAA) completely abolishes it (33–41). In addition, there is a reported influence of the antibody variable domain (Fabs) on FcRn binding and pharmacokinetics, which has not been studied for TRIM21 (42, 43).
Recent findings highlight that TRIM21’s signaling requires higher activation than neutralization, which means that it is able to neutralize without initiating immune responses (2, 19). Engineered IgG1 variants with reduced TRIM21 binding still neutralize effectively but trigger less NF-κB signaling (10, 30). Mutations can significantly alter TRIM21 binding, with single mutations like H433A reducing activity, while others increase IgG1 Fc affinity up to 100-fold (10, 30). Slower dissociation rates from TRIM21 affect signaling more than neutralization, indicating possible low-level activity without an anti-viral state (10, 30, 44, 45). The distinction between effector and sensor activation thresholds allows for the effective clearance of minor viral threats without an extensive immune response.
Antiviral activity of TRIM21 has been demonstrated against a wide range of non-enveloped viruses, including adenovirus, Porcine reproductive and respiratory syndrome virus (PRRS), Japanese encephalitis virus (JEV), Hepatitis B virus (HBV) and rotavirus as well as certain bacteria (5, 9, 11, 46–51). It can interfere with infection by direct interaction with the viral proteins, as well as by regulating immune responses and by recognition of antibody decorated viral complexes (52). Upon the cytosolic entry of virus complexes opsonized by antibodies, the Fc portion of the antibodies is rapidly recognized by the catalytically inactive TRIM21 dimer. A proposed mechanism for the activation of viral degradation involves target-induced clustering (53). It is postulated that the intermolecular dimerization of the TRIM21 RING (between two or more TRIM21 dimers) domains triggers ubiquitination activity (54). This process potentially alleviates B-box inhibition of the RING domain, facilitates E2-Ubiquitin interaction, and constructs K63-linked ubiquitin chains on TRIM21 (19).
Previous research demonstrated that ADIN, which is triggered by catalytically active RING dimers, is remarkably efficient in IFN-stimulated mouse cells, requiring only 1.6 antibodies per virus to achieve effective neutralization (45). In contrast, IFN-stimulated human cells demand a higher antibody count, with 5 antibodies needed per virus for similar neutralization efficacy (45). It has been observed that the upregulation of TRIM21 can decrease the number of antibodies necessary for successful TRIM21-mediated neutralization (9, 45). In human cells that have not been stimulated by IFN, the ADIN process requires the recruitment of a greater number of TRIM21 molecules (53). Therefore, the nature of the target (oligomer) or the presence of multiple antibodies bound to the target could induce TRIM21 activation by promoting the clustering of multiple TRIM21 molecules in close proximity (53). TRIM21-antibody opsonization levels need to be above a certain threshold to enable RING dimerization, thus activating the innate neutralization pathway against the bound pathogen.
We sought to elucidate the detailed mechanism of action of the TRIM21-IgG interaction. To achieve this we generated several antibody Fc mutants, modifying TRIM21 binding. The used antibody variants express symmetrical, similar Fc heavy chains, or asymmetrical, varying heavy chains. We utilize a suite of complementary technologies to decipher TRIM21 binding. While it is established that single point mutations can modify PRYSPRY binding and that one TRIM21 homodimer interacts with one Fc heterodimer in a 1:1 stoichiometry with strong avidity, TRIM21 detailed mode of action remains unclear. This includes the precise nature of the natural dimer state, mutations that influence avidity, and determining how a synergistic interplay of TRIM21 mediated antibody variants bound to viruses leads to amplified avidity thereby influencing its mode of action.
In this study, we explore the mechanism by which the engagement of both heavy chains mediated via TRIM21 dimers takes place and examine the impact of Fc variants of antibodies on the balance between affinity and avidity, offering detailed insights into the mechanism of antibody binding. We demonstrate the occurrence of TRIM21 - anti-AAV antibody clustering on a biosensor, unveiling intricate binding dynamics. Gaining an in-depth knowledge of TRIM21 and its interactions with antibodies paves the way for innovative therapeutic approaches targeting infectious diseases, autoimmune conditions, and immune signaling pathways.
2 Results
2.1 Design of TRIM21 antibody biosensor assay configuration for dissecting its binding mode
To study the TRIM21 interactions in detail, there are two assay configurations possible on a (SPR) biosensor. Depending on the scientific question that should be addressed, either the antibody or TRIM21 is captured on the chip-surface (Figure 1). Furthermore, it is to be considered, that the bivalent antibody displays an Fc part with two binding moieties for TRIM21 and TRIM21 naturally forms a homodimer, which might cumulate in a more complex binding mode than a simple 1:1 interaction.
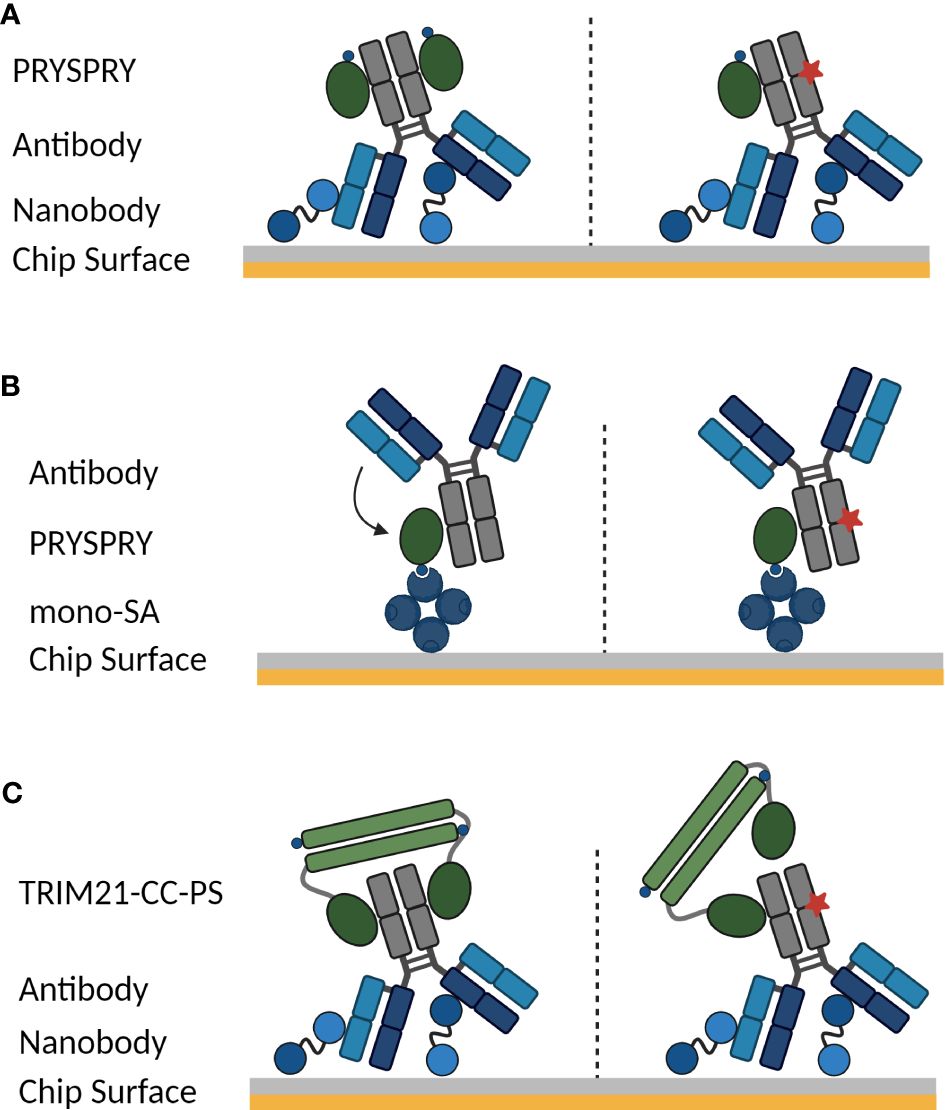
Figure 1 SPR assay orientations to characterize the interaction between TRIM21 and an antibody. Symmetrical and asymmetrical antibody Fc variants are investigated. In case of an asymmetrical Fc part, one Fc heavy chain contains a AAA mutation (schematically shown by red star), that completely abolishes TRIM21 binding. The used Fc variants and assay setups allow determining how Fc mutations influence the avidity-binding mode and dissecting avidity from affinity. (A) Antibody Fc variants are captured on the biosensor surface via an anti-Fab nanobody (vhh), Fc-only variants are coupled using standard amine coupling chemistry and cytokine Fc-Fusions are captured via anti-PGLALA F(ab’)2 fragment (55), while TRIM21 PRYSPRY domain is the analyte in solution (see Materials and Methods). Configuration (B) schematically shows the inverse to (A) while the PRYSPRY domain is captured via monovalent streptavidin. (C) To analyze the dimeric TRIM21 engagement of both IgG heavy chains, the antibody is captured via its Fab fragment, cytokine Fc-Fusions are captured via anti-PGLALA F(ab’)2 fragment (identical capture setup as in (A) and TRIM21-coiled-coil-PYRSPRY (TRIM21-CC-PS) is injected. Illustrations are created with BioRender.com.
To investigate the TRIM21-IgG binding dynamics, we employed a strategic approach using two recombinant TRIM21 constructs (Sequence in Materials and Methods). We initially focused on the production of the PRYSPRY domain. This was essential to quantify the affinities of TRIM21 for various Fc-engineered antibodies, providing a clear assessment of how specific Fc modifications impact the binding affinity. By examining the interaction with the PRYSPRY domain alone, we could discern the direct effects of Fc engineering on the affinity component of the binding without the influence of avidity. Subsequently, we produced the TRIM21-CC-PS construct, comprising the PRYSPRY and coiled-coil domains to emulate the natural dimeric state of TRIM21. This construct is pivotal for investigating bivalent interactions with IgG antibodies, as it closely represents the native dimer and maintains the 1:1 stoichiometry of TRIM21 binding to both heavy chains of an IgG, as confirmed by a previous study, making it an ideal tool for our biosensor assays (9).
If the antibody (ligand) is captured on the surface under physiological conditions and the PRYSPRY domain (antibody Fc binding domain) (analyte) is in solution as shown in Figure 1A, the monovalent PRYSPRY domain binds to the Fc part exclusively in a monovalent fashion. Here, the mode of binding is independent from the ligand surface density. For the interacting pair of PRYSPRY and an antibody, this assay configuration exclusively probes the affinity mode, as the bivalent nature of the antibody Fc part is in this set-up irrelevant. Notably, this assay orientation allows the affinity characterization of various antibody Fc variants modulating the affinity towards the PRYSPRY domain. It also enables to investigate the stoichiometry for PRYSPRY and the dimeric Fc part, which we address with symmetrical and asymmetrical Fc variants. The latter abolishes TRIM21 binding for one Fc heavy chain.
The alternative orientation with PRYSPRY domain on the surface and the antibody in solution (Figure 1B) allows to investigate further attributes that describe the TRIM21-IgG interaction in more detail. As an example is the potential Fab contribution to TRIM21 binding efficiency as observed for FcRn binding (42, 43, 56–58). The ligand density in this assay setup is crucial. The antibody may interact with one or two PRYSPRY domains (ligand) depending on ligand density, by interlinking two domains that are close enough for simultaneous engagement (affinity & avidity). To exclude avidity binding modes we measured at low ligand density.
To analyze the TRIM21-CC-PS - antibody interactions, the antibody is captured on the sensor surface while TRIM21-CC-PS is injected (Figure 1C), enabling the observation of avidity effects from bivalent binding. Kinetic rate parameters for avidity, specifically kON and kOFF, remain uncharacterized for TRIM21. These kinetic parameters are more informative than the equilibrium dissociation constant (KD), offering a comprehensive view of binding kinetics. By determining kON and kOFF, we enhance our understanding of the interaction, which is essential for assessing the impact of Fc engineering on the TRIM21-antibody interaction. This detailed approach is critical for unraveling the effects of Fc engineering on both affinity and avidity, providing key insights for the development of therapeutic antibodies with enhanced specificity and improved therapeutic efficacy.
2.2 Antibody Fc engineerings to dissect affinity towards the PRYSPRY domain
To resolve the influence of Fc engineerings on the affinity for the TRIM21 PRYSPRY domain we used the assay setup shown in Figure 1A. To determine the kinetic rate parameters of the PRYSPRY - antibody interaction and its stoichiometry, a human IgG1 Fc WT (mAb1 WT) was compared with an asymmetrical variant mAb1 WT-AAA (H310A, H433A, Y436A). SPR binding kinetic of both constructs (Figures 2A, B) result in the same kinetic rate parameters. To confirm loss of binding in our AAA mutant, a double AAA mutant (mAb1 AAA) was used as control (Figure 2C). MAb1 WT and WT-AAA demonstrate an affinity dissociation constant (KD,AFFINITY, Equation 1) of 43 nM and 40 nM, respectively (Figure 2D, detailed parameters in SI Supplementary Table S1).
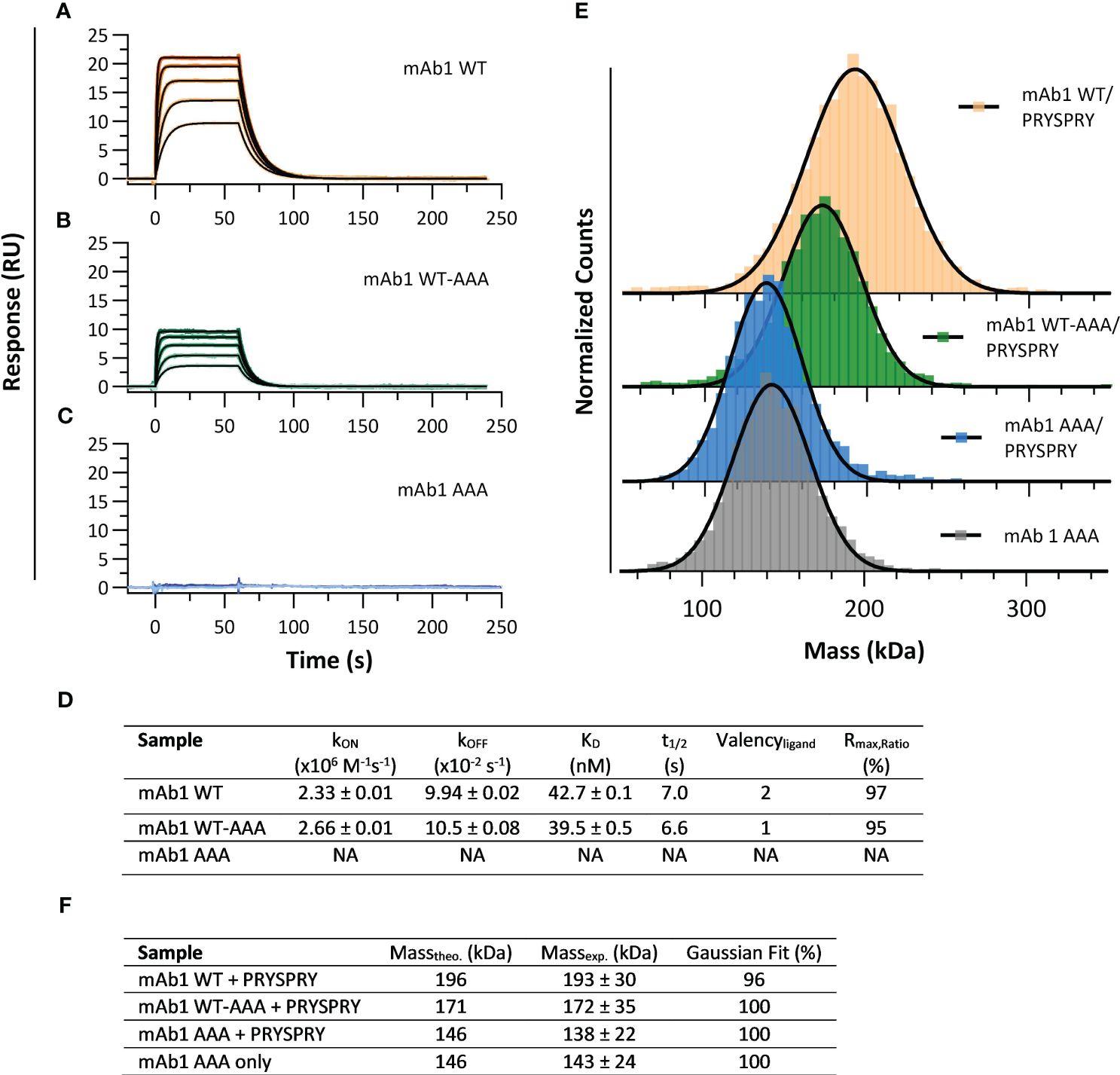
Figure 2 Interaction analysis of human IgG1 (mAb1) Fc variants and TRIM21 PRYSPRY domain. (A–C) showing sensorgrams (SPR data) where PRYSPRY was injected in five different concentrations as two-fold dilution series to immobilized mAb1 Fc variants (capture level approx. 60 RU). Each plot shows the measured raw data (colored gradient) and the global fit analysis as solid black lines. For immobilized mAb1 WT (A) and mAb1 WT-AAA (B) PRYSPRY was injected at 500 nM highest concentration and for mAb1 AAA (C) at 2000 nM. The sensorgrams show the affinity binding mode applying a mono-exponential fit model (Langmuir 1:1). The determined kinetic parameters are described in (D). The kON, kOFF and KD values are results from a global fit analysis ± fitting error. (E, F) show the complementary mass photometry (MP) data displaying a 2:1 binding stoichiometry confirming the SPR data. For the PRYSPRY - mAb1 WT complex, the data reveals a double bound state and for mAb1 WT-AAA a single bound state, while the control mAb1 AAA shows no binding at all. A Gaussian distribution model was used to analyze the MP data. For individual masses of the molecules, see SI Info Supplementary Figure S1.
The Rmax,Ratio (Equations 2, 3) represents the ratio of experimental and theoretical maximal feasible SPR signal between a ligand - analyte couple, and provides information about the stoichiometry of an interaction (59). The interaction between mAb1 WT and mAb1 WT-AAA - PRYSPRY, with assumed binding sites of two and one, respectively, approaches 100% efficiency, leading to stoichiometries of 2:1 and 1:1, as shown in Figure 2D. This confirms that the IgG Fc binds two PRYSPRY domains, one at each of the CH2-CH3 interfaces. Additionally, the data shows that there is no effect of the first PRYSPRY binding event on the binding efficiency of the second monomer.
To confirm the findings from the SPR analysis we applied mass photometry (MP) for mAb1 WT/WT-AAA/AAA as a complementary method (Figures 2E, F). Considering the fast kON and kOFF (Figure 2D) for the mAb1-PRYSPRY interaction, 200 nM mAb1 Fc variant were pre-incubated with 4000 nM PRYSPRY domain, and analyzed at a 1:250 dilution.
The non-binder variant mAb1 AAA with PRYSPRY (138 kDa) and without PRYSPRY domain (143 kDa), are in good agreement with each other and with the theoretical molecular weight (Figure 2F), demonstrating that PRYSPRY binding is abolished. An individual PRYSPRY domain appears at molecular weight of 32 kDa (Controls: Supplementary Figure S1). MAb1 WT + PRYSPRY gave a molecular weight of 193 kDa for the complex, which is in agreement with two PRYSPRY bound to one antibody (Gaussian Distribution Model), while mAb1 WT-AAA + PRYSPRY shows only the single bound state represented by 172 kDa.
To further examine how PRYSPRY binding can be altered by antibody constructs and Fc engineerings, we characterized additional variants, applying the assay setup shown in Figure 1A. All measured antibody formats share an identical IgG1 Fc framework, with or without specific mutations within TRIM21 binding interface. mAb1 WT and mAb2 WT exhibit different electrostatic and hydrophobic Fab patches. Two additionally investigated constructs are Briakinumab and Ustekinumab. Briakinumab shows a large positively charged Fab region, which is absent in Ustekinumab, and has been shown to have different FcRn affinities (42, 60). A cytokine-Fc Fusion and a Fc-only (CH2-CH3) WT were also included along with the mAb2 WT knob-into-hole (KiH) variant, which has specific mutations in the CH2-CH3 region (details in materials & methods). To compare the determined affinities, the kinetic rate parameters are displayed in a kinetic rate scale plot (Figure 3A). Detailed sensorgrams and kinetic parameters are shown in SI Info Supplementary Figure S2.
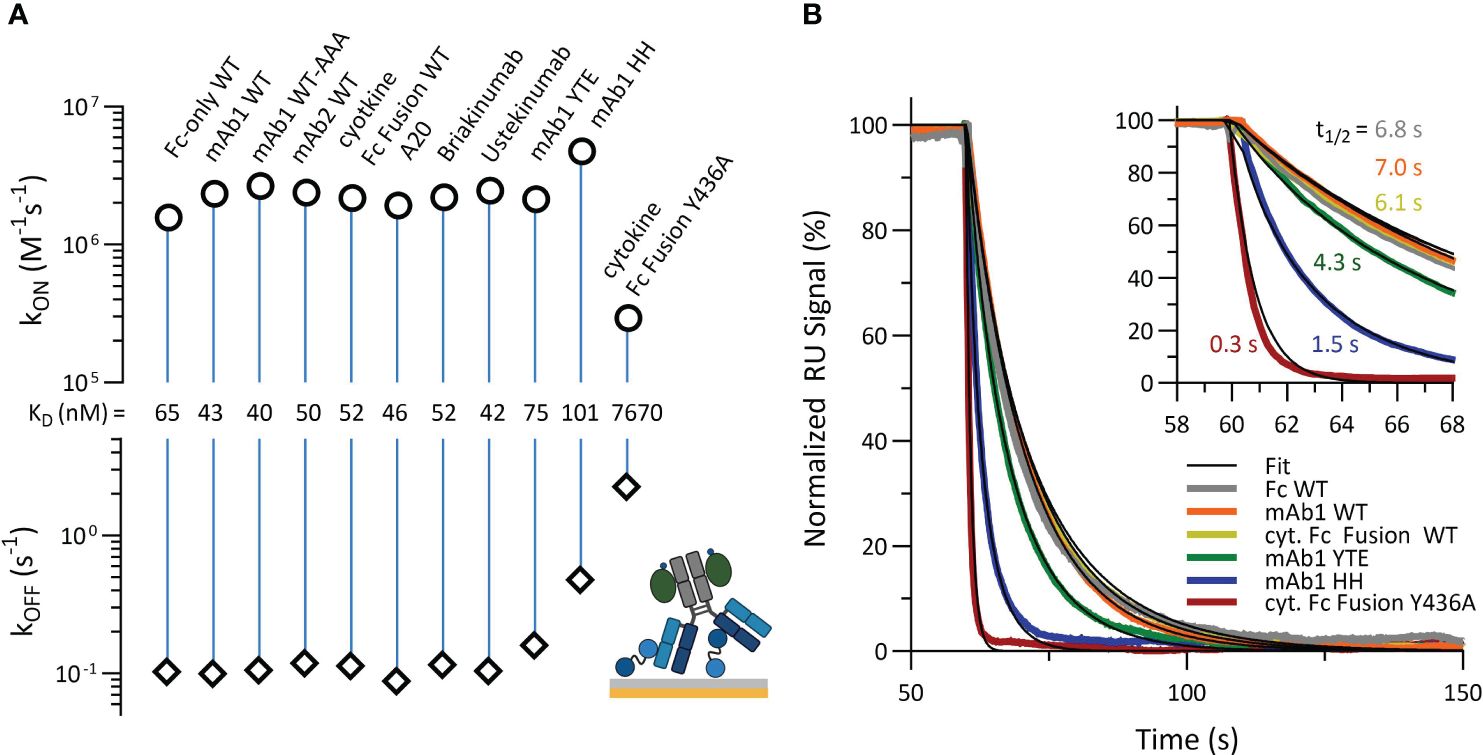
Figure 3 Kinetic characterization of TRIM21 PRYSPRY binding to immobilized antibody Fc variants and cytokine-Fc Fusion constructs, and Fc only variant (Raw data SI Info Supplementary Figure S2). Detailed SPR assay setup is described in materials and methods. (A) The Affinity Rate Scale Plot enables the kinetic comparison of several binding experiments at one glance. The association rate (kON) and corresponding dissociation rate (kOFF) are juxtaposed in opposition, connected via a vertical line, representing the binding strength (affinity). The further apart both parameters (kON and kOFF) the stronger the interaction is. Compared to mAb1 Fc WT, the Fc variants YTE (M252Y, S254T, T256E), HH (T307H, N434H) and Y436A show decreased PRYSPRY affinity. The YTE affinity is 1.7-fold, HH 2.4-fold and Y436A 180-fold decreased. As shown in (B) the altered binding strength is mostly off rate driven, which becomes apparent in the overlay of normalized dissociation. The start of dissociation is normalized to 100%.
In this assay orientation, the affinities of most antibody constructs for PRYSPRY are similar. Exempt are the Fc variants YTE (M252Y, S254T, T256E), HH (T307H, N434H) and Y436A, which contain Fc mutations at key residues in the binding interface for PRYSPRY. These variants have lower affinities for the PRYSPRY domain than WT, showing a sequence from highest to lowest strength of binding as follows: WT has the strongest affinity, followed by YTE, then HH, and Y436A has the weakest.
For YTE the effect is only off rate driven (1.6x faster compared to WT), while for HH and Y436A it is a combination of both on and off rates. In contrast to WT, HH shows a 5x faster off rate, while the on rate is 2x faster, compensating for a significantly weaker affinity (KD,AFFINITY) compared to the YTE variant. For the Y436A variant both rates are strongly decreased (8x on rate and 23x off rate compared to WT). Overlaying the normalized dissociation phases (Figure 3B) shows the significant effect of Fc engineering (t1/2: 7.0 s for WT vs 0.3 s for Y436A).
Here, we showed that our assay setup (Figure 1A) is suitable to determine the pure, undisturbed affinity of the PRYSPRY-IgG interaction. We found a 180-fold reduced affinity for Y436A compared to WT. The SPR and MP results reveal a PRYSPRY-IgG stoichiometry of 2:1 without any binding cooperativity. The chosen assay setup does not reveal an additional contribution of Fab binding, as such we apply the reversed assay orientation to address this topic in the following section.
2.3 Impact of antibody variable domain on TRIM21 PRYSPRY binding affinity
FcRn and TRIM21 share an overlapping CH2-CH3 binding interface. Antibodies with an identical Fc part but different Fab domains bind differently to FcRn, showing different affinities and pharmacokinetic properties (42). To uncover a potential PRYSPRY-IgG Fab contribution, we reversed the assay setup while allowing the Fab arms to move freely in solution (Figures 1B, 4, detailed data in SI Info Supplementary Figure S3). To prevent intermolecular interactions where a single antibody binds to two adjacent PRYSPRY domains, we used low PRYSPRY densities (detailed in Materials and Methods, Section 7.5). The surface density of the PRYSPRY domain was empirically optimized until the data could be accurately analyzed using a monophasic fit model. This model reflects the affinity-binding mode, ensuring that no avidity effects arise from interactions with two adjacent PRYSPRY domains.
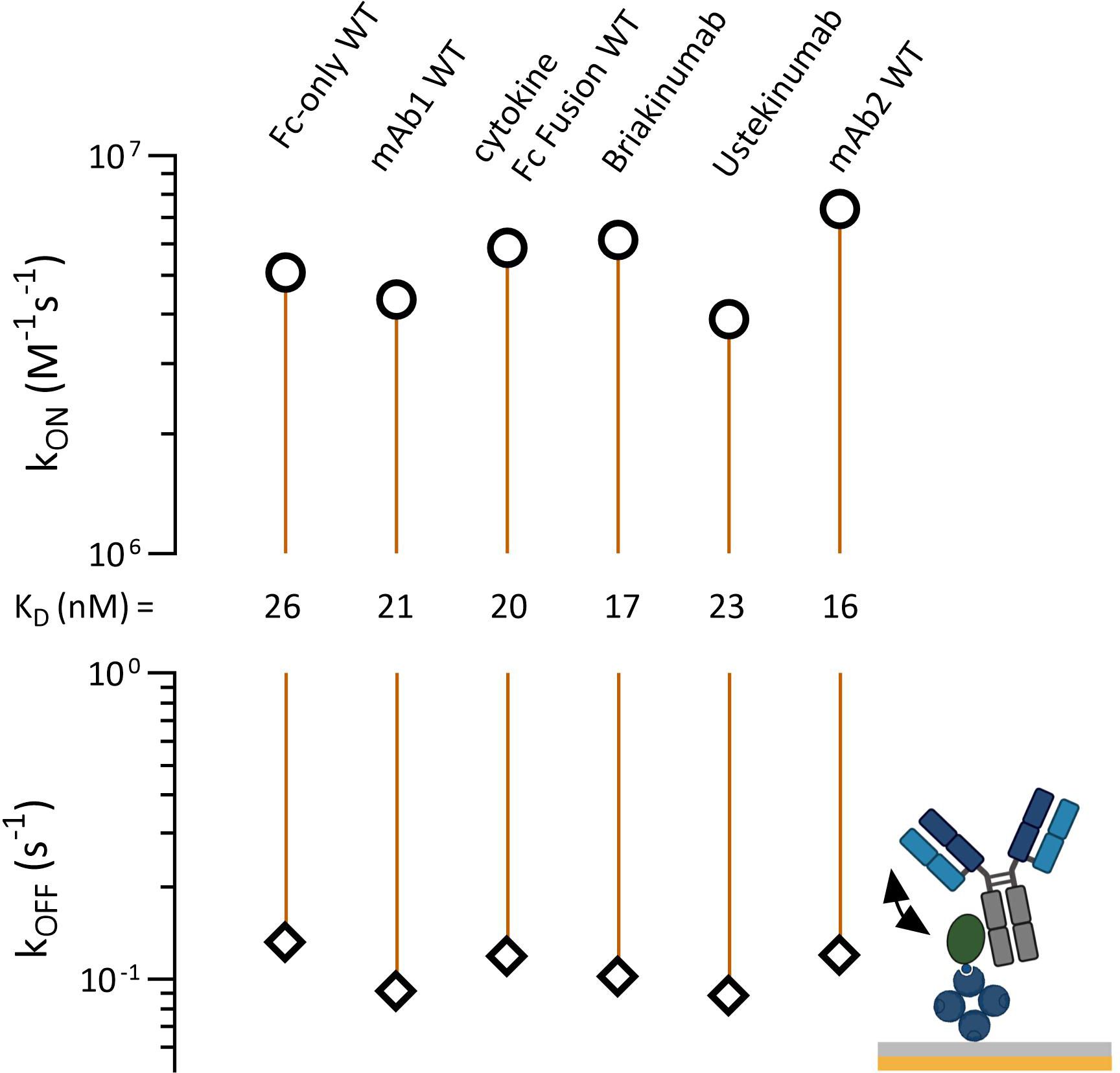
Figure 4 Affinity rate scale plot for captured TRIM21 PRYSPRY domain (ligand) and antibody (human IgG1) variable domain variants or antigen fusion constructs in solution (analyte). The injected constructs have different Fab regions but share the same Fc region. This allows the investigation of a potential Fab contribution to the PRYSPRY binding. All constructs were analyzed by applying a simple 1:1 Langmuir fit. The analyzed antibody variants do not show any Fab contribution. Notably, there is a faster on-rate (2x) for all constructs when compared to the reverse assay setup up (PRYSPRY as analyte). Raw data is shown in SI Info Supplementary Figure S3.
The reverse assay orientation reveals a two to three-fold increase in binding strength. This effect is primarily driven by on-rate changes, not influenced by Fab regions, as evidenced by same outcomes for the control (Fc-only WT), which lacks a Fab. The affinities across the characterized antibody panel remain consistent with each other, confirming the absence of any measurable Fab effect on the Fc-PRYSPRY interaction.
2.4 The dimeric state of TRIM21-CC-PS
To probe the dimeric engagement of both IgG heavy changes simultaneously, we first characterized the construct TRIM21-CC-PS (Figure 5). The expected theoretical molecular weight of this dimeric construct is 86 kDa. Applying SEC-MALS revealed a mass of approximately 90 kDa for 94% of the particles (4% showed 177kDa corresponding to a tetramer) (Figure 5A). MP gave 99% of particles at a weight of 82 ± 13 kDa (Figure 5B). Electron Microscopy analysis revealed that the PRYSPRY domains are facing away from each other with the coiled-coil domains in between mediating dimerization. Shown 2D classes indicate a slight variation in the position of the PRYSPRY domains with respect to each other (Figure 5C). Whether these variations are caused by the limitations of the applied technique or are biologically relevant has to be investigated.
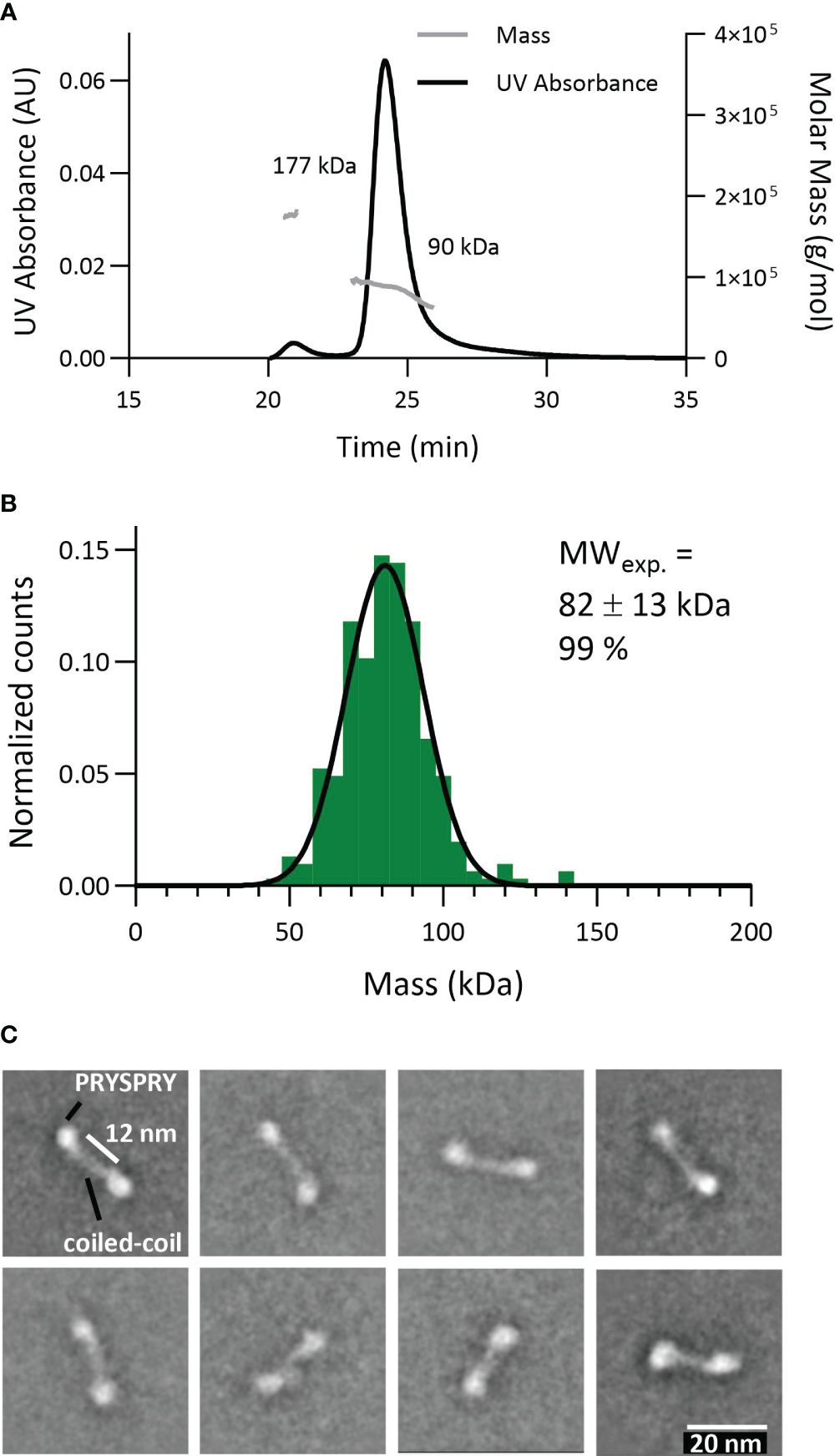
Figure 5 Characterization of the TRIM21 dimeric nature (TRIM21-CC-PS) applying different technologies. (A) SEC-MALS data reveals 94% TRIM21-CC-PS dimer (90 kDa). (B) Mass photometry technology shows 99% TRIM21-CC-PS with 82 kDa. (C) Selection of EM 2D classes confirming TRIM21-CC-PS dimers. The coiled coil domains facilitate dimerization whereas the C-terminal PRYSPRY domains are placed at the opposite end of each coiled-coil domain.
Here, we confirmed the dimeric nature of TRIM21-CC-PS with three techniques. Surprisingly, the EM data show that the coiled-coil domain is stretched and the distance between the two PRYSPRY domains is extended. The fact that we were able to resolve the shape of the coiled-coil is an indication that in its dimeric shape TRIM21 is relatively limited in its flexibility.
2.5 TRIM21-CC-PS – antibody Fc variant Interaction by mass photometry and electron microscopy
To investigate the TRIM21-CC-PS - Antibody Fc binding mode, we applied MP and electron microscopy (Figure 6). For the MP measurements we incubated various, increasing TRIM21-CC-PS concentrations with three antibody Fc variants (mAb1 WT, mAb1 WT-AAA and mAb1 AAA) each at different molar ratios.
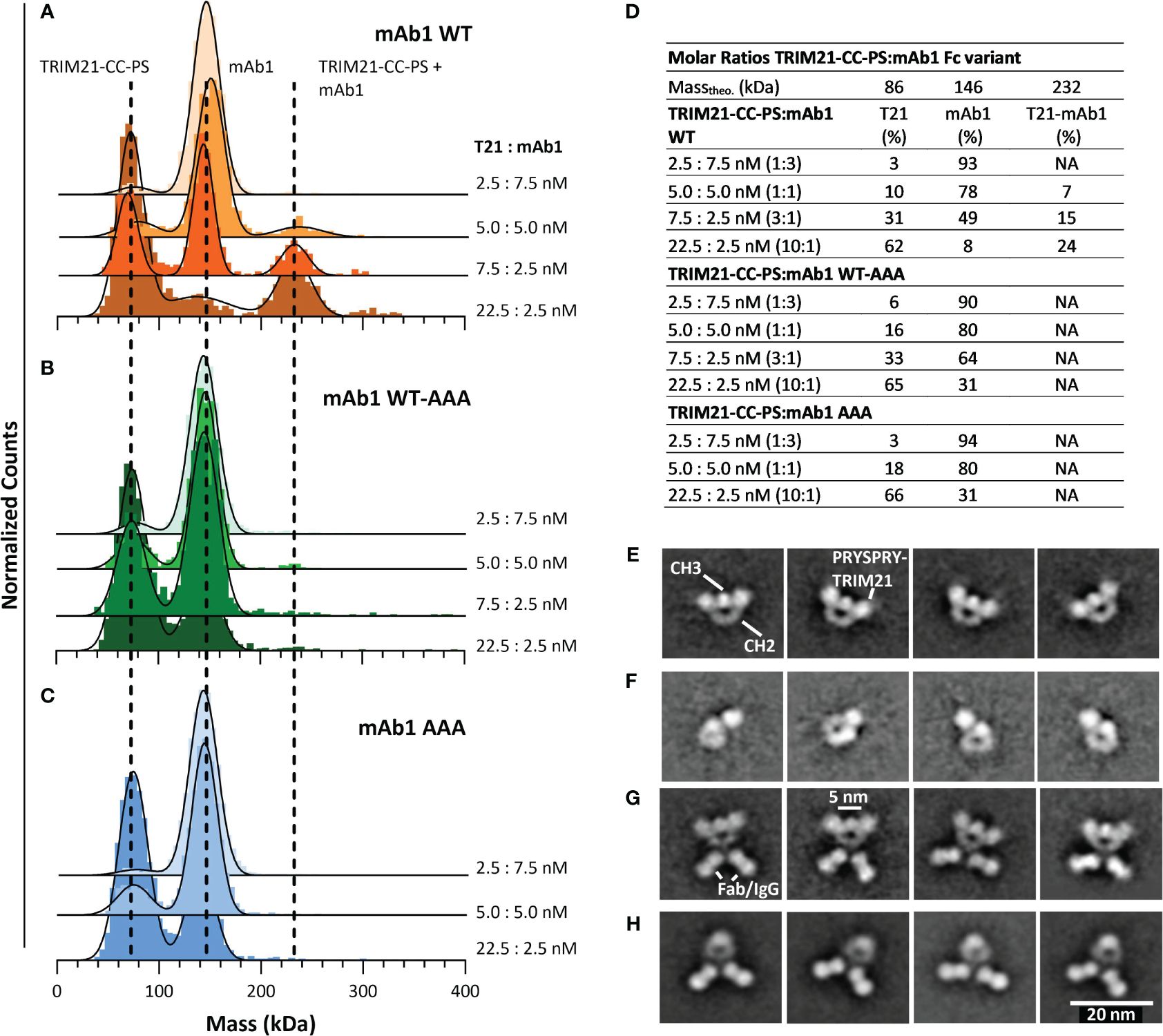
Figure 6 Characterizing the interaction of TRIM21-CC-PS with three different Antibody Fc variants. (A–D) Applying MP, dashed lines indicate the main peak of the respective species over all measurements. The applied 3-dimensional Gaussian Fit Distribution is shown in black lines. (A) MP of TRIM21-CC-PS with mAb 1 WT, (B) MP of TRIM21-CC-PS with mAb 1 WT-AAA (C) MP of TRIM21-CC-PS with mAb 1 AAA. Only mAb 1 WT shows binding to TRIM21-CC-PS at low nM concentration in accordance with its low nM binding strength. (D) The amount (%) of TRIM21-CC-PS - mAb1 WT complex increases with excess of TRIM21-CC-PS, while TRIM21-CC-PS - mAb1 WT-AAA shows single events of complexed species for the applied concentrations, that could not be fitted robustly. (E–H) selected 2D averages of EM data, resolving TRIM21-CC-PS with Fc WT (E), TRIM21-CC-PS with Fc WT-AAA (F), TRIM21-CC-PS with mAb1 WT (G) and mAb1 WT alone (H).
As shown in Figures 6A–C, incubating TRIM21-CC-PS with mAb 1 gave peaks at approximately 80 kDa and 140 kDa, representing uncomplexed TRIM21-CC-PS and mAb1, which is in agreement with their theoretical masses and individual mass measurements (Figure 5 and SI Info Supplementary Figure S1). At approximately 230 kDa appeared a third peak whose molecular weight corresponds to a complex consisting of one TRIM21-CC-PS with one mAb 1 WT (Figure 6A). With increasing TRIM21-CC-PS concentration, the equilibrium is shifted to the complexed species until nearly all antibodies are bound. At a 10-fold molar excess of TRIM21-CC-PS (Figure 6D), 24% of TRIM21-CC-PS and mAb1 WT are complexed, revealing a 1:1 stoichiometry where one dimer and engages both IgG heavy chains simultaneously (detailed data in SI Info Supplementary Figure S4). Detecting the TRIM21-CC-PS - mAb1 WT interaction at low nanomolar concentrations also allows insights into the strong interaction by the dimeric engagement, resulting in an avid binding mode.
The applied mAb1 WT-AAA concentrations showed a minor complexed fraction (approx. 2%), while the AAA variant shows no formed complexes (Figures 6B, C). For mAb1 WT-AAA variant, we expect binding, but only to one of the two Fc heavy chains resulting in weak affinity, which cannot be resolved at the concentrations used (similar affinities as for PRYSPRY). The symmetrical AAA mutation abolished TRIM21 binding completely.
To further elaborate the binding mode we analyzed TRIM21-CC-PS with mAb1 WT and the Fc-only variants WT and WT-AAA applying EM (Figures 6E–H) and MP (Supplementary Figure S6). The results demonstrate that the Fc-only WT-AAA variant binds one PRYSPRY domain, compared to WT (Figures 6E, F). The Mab1 WT - TRIM21-CC-PS complex confirms the binding of two PRYSPRY domains to the Fc portion compared to mAb1 WT alone (Figures 6G, H). Although the coiled-coil dimerization domain of TRIM21 is not resolved in our 2D class averages, and the analysis of raw images presents challenges due to the size and flexibility of the complexes, the clustering patterns observed in the antibody-TRIM21-CC-PS EM data suggest that a single TRIM21-CC-PS molecule has the capacity to bind to an Fc region through one or both of its PRYSPRY domains. Notably, our data demonstrates that TRIM21 can bind concurrently to two individual Fc regions. This multifaceted binding ability is further corroborated by our AAV binding experiments detailed in section 5.7. Here, we present evidence that TRIM21 can induce clustering of AAV particles through simultaneous interactions with two different Fc regions of one-armed A20 antibodies.
As depicted in Figure 5C, we do not observe a pre-bound/pre-formed positioning of the PRYSPRY domains that would already fit the spatial arrangement of antibodies Fc Part (CH2-CH3). The average distance between the two PRYSPRY domains measures ~10.1 ± 2.1 nm whereas the distance between two Fc-bound PRYSPRY domains is ~5.3 ± 0.5 nm. Therefore, for one TRIM21 to engage both heavy chains, the PRYSPRY domains will have to rearrange. As can be observed in the alphafold model (Uniprot ID P19474), the PRYSPRY domain attaches to its helix with a flexible linker. This linker could allow dimeric engagement by bringing the PRYSPRY domains closer to each other.
2.6 Binding kinetic of TRIM21-CC-PS with antibody Fc variants
Here, we examined the interaction between TRIM21-CC-PS and various antibody Fc variants through SPR experiments (assay setup Figure 1C), focusing on the avidity binding mode influenced by Fc engineering. Utilizing a low surface density setup to exclude intermolecular interactions, we explored both symmetrical and asymmetrical Fc variants, including YTE, HH, and Y436A, to discern the relationship between antibody affinity and avidity enhancement due to dimeric engagement (previously investigated for affinity binding, Figure 3).
Figure 7 shows the characterization of TRIM21-CC-PS with (antibody) Fc variants. When several antibody concentrations were injected over 180 seconds, a 1:1 fit model describes the data. The dissociation phase is characterized by a significantly reduced dissociation rate (kOFF,AVIDITY) in comparison to the affinity-only interactions (kOFF,AFFINITY). The sensorgram reveals an avidity only binding mode and a consistent 1:1 stoichiometry (exemplary mAb1 WT, Figure 7A and SI Info, Supplementary Figure S5).
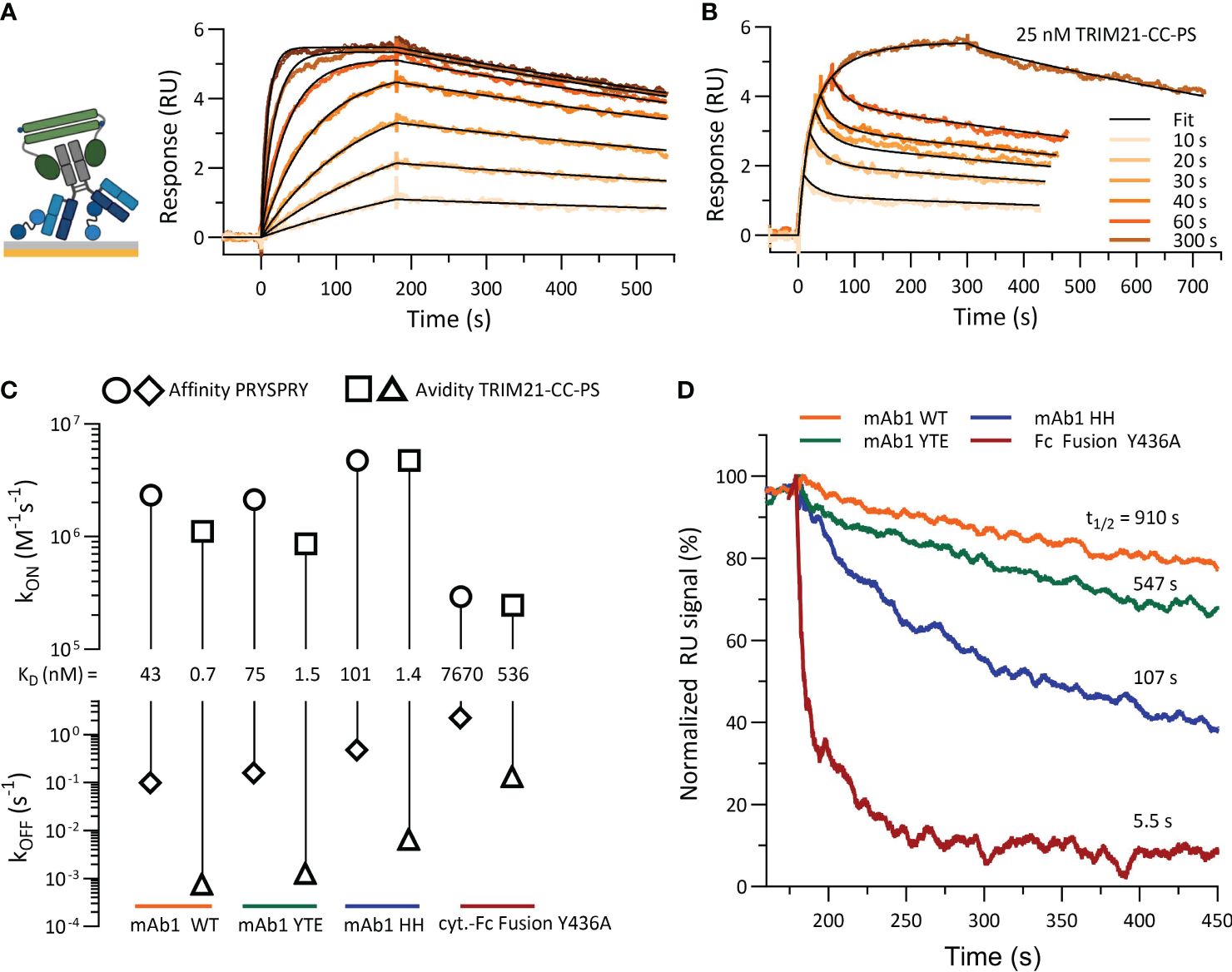
Figure 7 Characterization of TRIM21-CC-PS with Antibody Fc variants. (A) shows the sensorgram (SPR) of mAb1 WT (ligand, approx. 8–10 RU) and TRIM21-CC-PS (analyte) where TRIM21-CC-PS was injected in seven different concentration, each for 180 sec as two-fold dilution series with 100 nM as highest concentration. Applied fit model is a simple 1:1 interaction reflecting 100% avid bound, 1:1 antibody - TRIM21-CC-PS species. (B) Variation in association time (10 -300 sec) injecting a constant concentration of 25 nM TRIM21-CC-PS to captured mAb1 WT reveals a biphasic binding kinetic, which can be described by applying a two state model providing fast and slow kinetic rates. (C) Rate-scale-plot comparing affinity and avidity measurements of Fc variants towards PRYSPRY or TRIM21-CC-PS. (D) The altered binding strength from affinity to avidity is mostly off rate driven, which becomes apparent in the overlay of normalized dissociation phases (kOFF,AVIDITY) but can also occur as combination of both kinetic rate parameters, namely on and off rate.
Variation in the association time for a constant TRIM21-CC-PS concentration (25 nM) with mAb1 WT on the surface (ligand) uncovered a biphasic dissociation pattern, supporting a two-state binding model (Figure 7B) (Equations 4, 5). This model illustrates an initial rapid binding and dissociation phase (single bound state, affinity) followed by a slower, more stable avidity-driven complex formation (double bound state, avidity), each with its own association (kON) and dissociation (kOFF) rates (Supplementary Figures S5B, J). Applying both models results in identical dissociation constants (KD,AVIDITY = 0.7 nM), demonstrating a substantial avidity-enhanced binding strength compared to affinity alone (KD,AFFINITY = 43 nM) (Figures 7C, D).
Comparative analysis revealed that avidity enhancements (KD,AFFINITY/KD,AVIDITY) for Fc variants were significant, with the WT variant showing a 61-fold increase, YTE 50-fold, HH 72-fold, and Y436A 14-fold, when compared to their respective affinities (Figure 7C). However, when compared to the WT avidity component, YTE and HH were both about 2-fold less effective, whereas Y436A’s binding strength decreased dramatically by 766-fold. Despite YTE and HH having similar reductions in avidity compared to WT avidity, HH exhibited kinetic rates 5 times faster than YTE. The interaction of asymmetrical antibody Fc variants (WT-AAA, YTE-AAA, HH-AAA) with TRIM21-CC-PS showed only the affinity binding mode (SI Info, Supplementary Figure S5).
Complementary MP analysis (SI Info, Supplementary Figure S6 and Supplementary Table S2) corroborated our SPR findings, confirming a 1:1 stoichiometry for all symmetrical constructs and showing a trend where weaker bindings resulted in fewer avid complexes as the identical the concentration of antibody-TRIM21-CC-PS was used. Additional MP experiments with other antibody Fc WT constructs, such as mAb2 WT, Briakinumab, and Ustekinumab, revealed similar complexation levels to mAb1 WT, indicating that the variable domains of these antibodies do not affect their interaction with TRIM21-CC-PS. This further validates our SPR-derived conclusions.
2.7 TRIM21 within the context of viral neutralization
Determining whether TRIM21 compromises the efficacy of AAV-based gene therapies is critical, yet its role has not been established. To explore this potential involvement, our study focuses on the interactions between TRIM21 and recombinant adeno-associated virus vector (rAAVv) particles. We employed SPR to simulate the binding of TRIM21 to antibody-coated rAAVv particles (Figure 8A). Additionally, we have used electron microscopy (EM) to visually document these interactions, with findings depicted in Figure 8C. Our setup simulates the scenario after endosomal escape of antibody decorated AAV particles and their encounter with cytosolic TRIM21, proposing that TRIM21’s interaction with antibody-bound virus particles triggers ubiquitination through intermolecular RING dimerization, as suggested previously (20, 53). Our assay involved TRIM21-CC-PS, anti-AAV2 capsid antibody A20 variants (including a bivalent A20 with WT Fc and IgG variants with a single Fab arm against rAAVv-2, featuring either WT Fc or WT-AAA asymmetrical Fc), and rAAV serotype 2 (rAAV2), as depicted in (Figure 8A). Our research reveals complex interactions between TRIM21-CC-PS, A20 antibody variants, and rAAVv-2, showing that binding dynamics are more intricate than simple models suggest (Figure 8B). These interactions involve both affinity and avidity effects, with TRIM21-CC-PS binding to the antibodies Fc region and the Fab arm(s) binding to the rAAVv-2 capsid, contributing both to overall avidity.
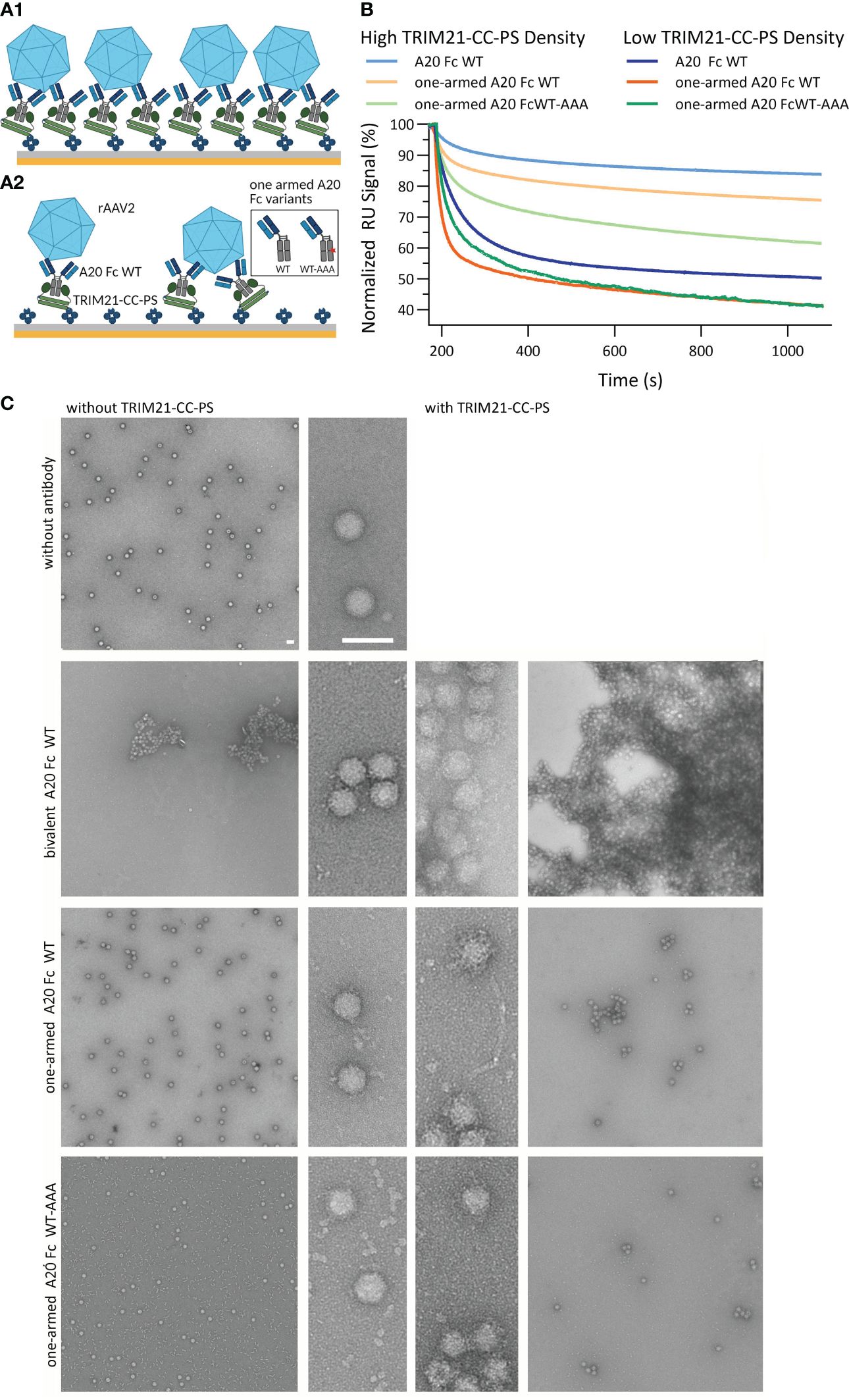
Figure 8 TRIM21-CC-PS-Antibody-AAV2 Characterization. SPR Assay data is shown in (A, B). (A1, A2) Schematic SPR assay configuration to analyze the affinity to avidity interplay of TRIM21-CC-PS, anti-capsid antibody variants A20 and rAAVv-2. Biotinylated TRIM21-CC-PS is captured via monovalent streptavidin achieving a captured level of 190 RU (A1, high density) and 35 RU (A2, low density). Subsequent, anti-AAV2 capsid antibody variants (bivalent A20 Fc WT, one-armed A20 Fc WT and Fc WT-AAA) are injected to saturate the TRIM21-CC-PS surface, followed by the injection of rAAV-2. (B) Overlay of the normalized dissociation phases (Start of Dissociation: 100%) after the injection of 3.32 nM rAAVv-2 over low and high TRIM21-CC-PS-A20 densities. At higher antibody densities, more avid complexation occurs and a higher degree of rAAVv-2 surface decoration is possible. This allows less complex to dissociate over time to due to simultaneous engagement of both, TRIM21-CC-PS and AAV2, mediated via the A20 antibody variants. (C) Electron microscopy images of rAAVv-2 interactions with antibodies alone (left column) and TRIM21 additionally (right column). The scale bars represent 50 nm.
All three anti-AAV2 antibody variants show affinities for the capsid in the same range (KD,AFFINITY 75 - 140 nM), whereas the bivalent A20 WT also demonstrated avid binding KD,AVIDITY = 5 nM SI Info, (Supplementary Figure S7). The full IgG A20 Fc WT shows strong bivalent engagement with TRIM21-CC-PS (KD,AVIDITY = 0.7 nM) unlike the asymmetrical AAA-WT variant, which lacks this avidity SI Info (Supplementary Figure S8).
The AAV2 viral capsid harbors n = 60 epitopes for A20, assuming intra- and intermolecular bivalent binding is possible, depending on antibody variant and concentration (61). By varying the A20 antibody levels (8-fold different capture level, Figure 8B) and analyzing EM data (Figure 8C), we observed that higher antibody densities on the virus surface enhance avidity. The bivalent A20 Fc WT variant promotes increased avidity, surpassing the monovalent A20 Fc WT and WT-AAA variants in binding efficiency, leading to faster dissociation.
Analysis of the dissociation phases indicate that all antibody variants demonstrate avidity, with slower and faster dissociating species suggesting complex formation between rAAVv-2 and antibodies. On a biosensor surface, ligands are randomly distributed by statistics. As such, two adjacent, single arm A20 constructs could still interact with one rAAVv, allowing avidity to occur. However, lower antibody densities result in a greater proportion of fast dissociating species, indicating a decrease in avidity (Figure 8B, low surface density).
EM images (Figure 8C) reveal that A20 antibodies as well as TRIM21-CC-PS facilitate AAV particle aggregation, owing to their ability to bind two separate molecules. While A20 causes severe aggregation, to the point that an additional effect of TRIM21 cannot be measured, the one-armed antibodies lose their ability to cause aggregation, allowing us to separate the effect of TRIM21 (Figure 8C, bottom two rows). Indeed, TRIM21 causes aggregation, albeit to a lesser extent. This confirms our SPR observation that reducing bivalent engagement modifies but does not negate avidity. Our findings emphasize the crucial role of the antibody as an immune regulator in mediating both, target binding (AAV) and effector functions via TRIM21 engagement achieving avidity, demonstrating the multifaceted nature of these interactions.
3 Discussion
This study systematically explores the interaction of TRIM21 with antibody Fc variants through SPR, MP, and EM to elucidate antibody binding to TRIM21 and investigate a TRIM21-mediated AAV neutralization mechanism. We meticulously optimized our SPR assays for each scientific hypothesis, facilitating low-density studies ideal for distinguishing between affinity and avidity. This approach yielded significant insights into the dynamics and mechanisms of TRIM21 interaction with human IgG Fc variants including a newly proposed binding mechanism.
TRIM21 is a highly conserved mammalian Fc receptor that is structurally and mechanistically distinct from previously identified Fc receptors (22). Our analysis confirmed the PRYSPRY–mAb1 WT interaction affinity at 43nM, aligning closely with prior ITC data (37 nM) (24) and differing from previous SPR results (212 nM) (62). This discrepancy originates from our refined assay conditions, which resulted in a 5-fold increase in binding rate (kON) without altering the dissociation rate (kOFF). Our methodology, mirroring ITC gold-standard accuracy, demonstrates the benefits of optimized assay setups for enhanced target interaction analysis, thereby addressing the discrepancies observed in prior SPR experiments. Additionally, our findings on the mAb1 WT-AAA Fc variant versus symmetrical Fc WT confirmed a 2:1 stoichiometry and identical affinities of two TRIM21 PRYSPRY domains for one IgG Fc homodimer (KD,AFFINITY,WT-AAA = 40 nM vs KD,AFFINITY,WT = 43 nM), supporting a non-cooperative binding model (24). This aligns with the symmetric TRIM21-IgG interaction in the previously reported crystal structures (24).
Despite TRIM21 and FcRn sharing the Fc CH2-CH3 binding site, the HNHY motif, mutations in the Fc region affect their interactions differently. We engineered mutations within this interface (YTE, HH, Y436A, AAA), previously identified to impact FcRn binding, to assess their effect on TRIM21 (33, 34, 38–41, 63). Our findings reveal a ranking of affinities for PRYSPRY, from strong to weak: WT, YTE, HH and Y436A. Notably, the Y436A mutation significantly weakens the binding to both FcRn (41) and TRIM21, with a 180-fold decrease in TRIM21 binding affinity, a previously unexplored aspect in the context of TRIM21 interaction. Through SPR analysis, we confirmed an avid equilibrium dissociation constant (KD,AVDIDITY) of 0.7 nM for mAb1 Fc WT (vs. 0.6 nM as determined by fluorescence anisotropy) and confirmed a 1:1 stoichiometry (9). In addition, we uncovered kinetic parameters alongside with an avidity enhancement (KD,AFFINITY/KD,AVIDITY) for the Fc variants of 14- to 72-fold. This suggests a substantial affinity to avidity transition due to bivalent engagement, particularly notable with the Y436A mutation, which demonstrates affinity in the µM range, while avidity is nM.
Interestingly, while the YTE and HH variants weaken TRIM21 binding compared to WT, they enhance FcRn binding (10-fold for YTE, HH is in similar range); in a pH-dependent manner, highlighting a divergent influence of these mutations on the two receptors (33, 34, 38). The AAA mutation, which eliminates FcRn binding (39, 40), also abolishes TRIM21 interaction. Remarkably, we found no Fab contribution that adds an additional complexity to TRIM21 as it does for FcRn (42, 43). A possible hypothesis to explain this phenomenon could be the pH independency known for TRIM21 (22). The divergence of TRIM21 and FcRn suggests potential implications for efficient antibody recycling and ADIN. A potential intracellular transition between FcRn and TRIM21 has to be explored. Our study emphasizes the importance of considering both affinity and avidity in TRIM21 interactions and challenges the direct extrapolation from one to the other, underlining the complexity of antibody engineering with respect to receptor binding.
Our configuration of unbound TRIM21 revealed that the C-terminal PRYSPRY domains within a TRIM21 dimer, where dimerization is mediated via the coiled coil domains, are positioned at opposite ends, away from each other (64). This finding is in contrast to previous studies on TRIM family proteins, such as TRIM25. It has been suggested, that unbound C-terminal domains, responsible for substrate binding, are centrally located within an elongated structure characterized by a coiled-coil domain (65, 66). A possible explanation for the changed TRIM21 configuration in the unbound state is the truncation of TRIM21 (TRIM21-CC-PS). Arguing against this is the fact, that the length of the coiled coil domain exceeds the distance measured between the two PRYSPRY domains by at least 3 nm. Also, the truncated sequence still maintains a stretch of 30 amino acids beyond the coiled-coil domain, suggesting a natural configuration at the observed binding location. Our analysis demonstrates that the PRYSPRY domains are spaced approximately 6–12 nm apart, diverging from the ~5 nm separation when bound to Fc, which challenges the notion of a pre-bound state compatible with the Fc structure and spatial arrangement (Figures 5, 6).
Zeng and colleagues (53) have proposed a TRIM21-IgG Fc-complex structural framework based on Small-angle X-ray scattering (SAXS) data, positioning the bound PRYSPRY domains centrally, which is in agreement with our findings. This configuration also accounts for avidity due to bivalent Fc engagement, observed in our SPR (Figure 7C) and MP data (Figure 6A), as well as the positioning of the bound PRYSPRY domain indicated by our negative staining techniques (Figures 6E–H). Beyond previous findings, our detailed examination underpins a novel conceptual framework for TRIM21 structural dynamics in both its bound and unbound states (Figure 9). In its unbound state, the dimer’s PRYSPRY domains converge at the coiled coil’s apex, a feature discernible through negative staining (Figure 5C). Binding to an Fc site results in the detachment of PRYSPRY from the coil, enhancing mobility due to its flexible linker, and thereby facilitating the engagement of the second site in a spatially favorable configuration. Our SPR data confirms, that binding occurs in a two-step process, initially rapid, followed by a slower event that forms the final TRIM21-IgG complex (Figure 7B). The slower phase occurs over a prolonged duration to accommodate the Fc geometry (Supplementary Figure S5J, kON2 = 0.02s-1). The transition markedly increases the complex flexibility, rendering the coiled-coil domain indistinct in negative stain averages of the TRIM21-CC-PS - Fc assembly.
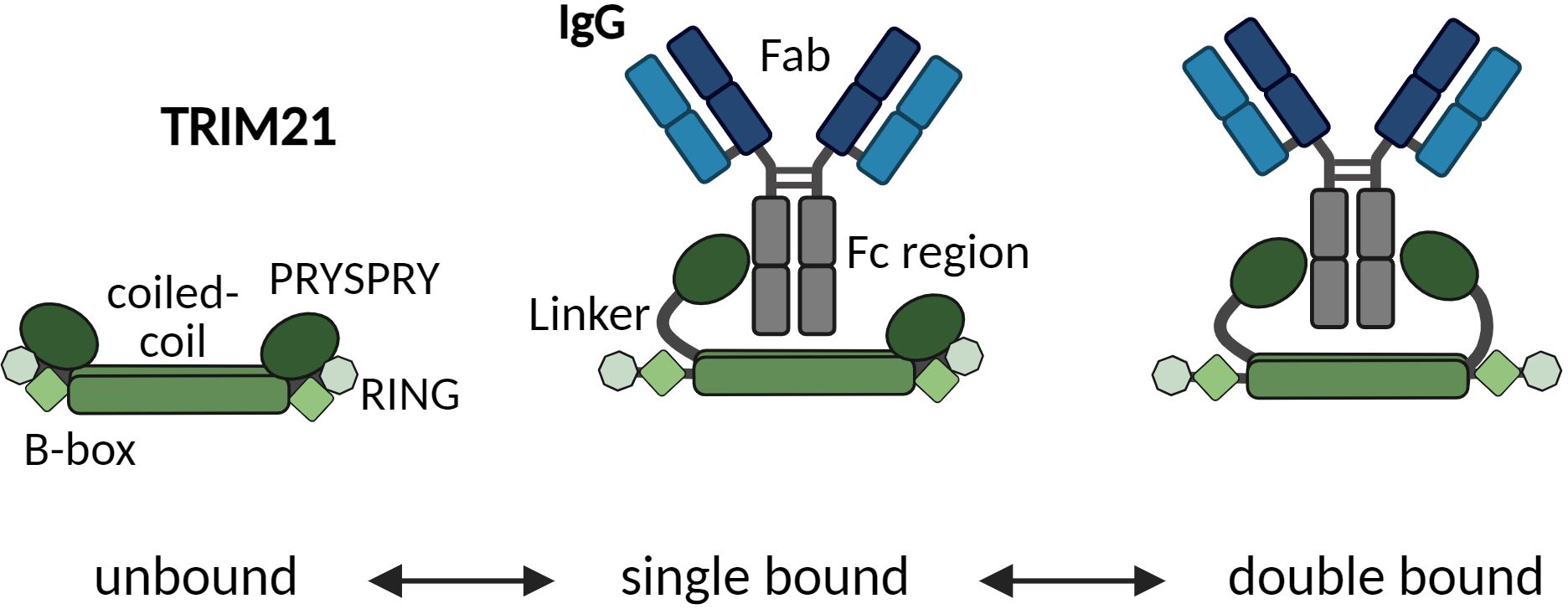
Figure 9 Schematic model suggesting how one TRIM21 dimer engages both sites of the Fc region in a two-step process. Upon Fc binding, the PRYSPRY detaches from the coiled-coil domain. The linker domain allows enough freedom of movement to allow engagement of the second PRYSPRY domain. Only after initial binding bivalent engagement of both Fc heavy chains is possible.
Integrating the structural insights from PRYSPRY domains in complex with Fc (PDB-ID: 2IWG) (24) with the predictive model of TRIM21 (Uniprot ID: P19474) and its dimeric form according to AlphaFold2 (for details see materials and methods) yields diverse models (67). While a minority retains the dimeric form of TRIM21 engaging a single Fc site (Supplementary Figure S10A1), most predictions indicate a separation of PRYSPRY domains from the coiled-coil (Supplementary Figures S10A2, B) agreeing with our model (Figure 9).
Combining our findings, we propose a tentative model in which the PRYSPRY engagement with the Fc domain may influence its interactions with other domains of TRIM21. This observation leads us to speculate that within a TRIM21 dimer, the PRYSPRY domain might have an additional, yet uncharacterized, role in modulating the interactions of the B-Box and the RING domains. We suggest that the binding of the PRYSPRY domain to the Fc domain could potentially release these intra-domain interactions, thereby affecting RING activation.
Lastly, our study delved into the TRIM21 antiviral mechanism, particularly its role in E3 ligase activation and neutralization of adeno-associated virus (AAV) using AAV as a model. We investigated how the anti-AAV2 capsid antibody A20 interacts with TRIM21 and rAAVv-2, noting A20 unique ability to neutralize AAV2 and AAV3 by targeting a specific conformational epitope (68, 69). Despite the lack of knowledge regarding the precise neutralization mode, subsequent to initial heparan sulfate proteoglycan (HSPG) primary receptor attachment, our data suggests A20 impedes viral entry by promoting large aggregate formation (Figure 8), potentially blocking endocytosis-mediated cell entry (61, 69, 70). We highlight the distinction between neutralizing and non-neutralizing antibodies in human blood, with the latter capable of cytosolic entry, as antibody-AAV complexes, post-endosomal escape (71, 72). However, the role of non-entry blocking antibodies in TRIM21-mediated AAV neutralization remains unexplored due to their unavailability for testing. Our findings indicate antibody clustering on AAV is feasible, aiding in the activation of TRIM21’s E3 ligase via RING dimerization, a critical step in neutralization (9, 20, 53). Comparing AAV with AdV, we noted the smaller size of AAV and fewer antibody binding sites could significantly influence its neutralization process. Unlike A20, which forms large aggregates, the anti-AdV5 antibody does not aggregate virions or hinder their attachment nor trafficking (3, 4, 44, 73, 74).
Our elucidated TRIM21 dimeric nature and its interaction with Fc-engineered antibodies revealed a new mechanism for bivalent engagement and demonstrated the impact of Fc mutations on both affinity and avidity. This knowledge is pivotal for advancing antibody engineering and understanding TRIM21 function in viral defense.
4 Materials and methods
4.1 Recombinant TRIM21 variants
Recombinant human TRIM21 proteins, including the PRYSPRY domain variant and the PRYSPRY-coiled-coil (TRIM21-CC-PS) variant, were produced and purified by Proteros Biostructures GmbH (Planegg, Germany). The production process involved the transformation of Escherichia coli strain BL21 DE3, the overexpression of TRIM21 under standard conditions (18°C, 1mM IPTG, 16h) and the purification by Ni-NTA resin (Qiagen, Hilden, Germany) and Superdex S-200 26/60 gel filtration (GE Healthcare, IL, US). Each plasmid used for the transformation encoded either the TRIM21 PRYSPRY domain or the TRIM21-CC-PS variant. Both variants were C-terminally fused to a His-Avi-Tag via a (4GS)1 linker. The gene used for this process is identified by NCBI Gene ID 6737, which corresponds to UniProtKB identifier P19474–1 (Isoform 1). The amino acid sequence for the TRIM21 PRYSPRY variant spans from amino acid 277 to 475, while the sequence for the TRIM21-CC-PS variant spans from amino acid 128 to 475. To verify the protein integrity SDS PAGE, SEC Analysis, Mass Photometry and Mass Spectrometry for mass identification were performed.
4.2 Antibody constructs/Fc variants
Recombinant human IgG1 Fc wildtype, Fc variants, Fc-only variants (core hinge-CH2-Ch3) with specific mutations (Fc WT, Fc WT-AAA, mAb1 WT, mAb1 WT-AAA, mAb1 AAA, mAb1 YTE, mAb1 YTE-AAA, mAb1 HH, mAb1 HH-AAA, mAb2 WT, mAb2 WT sym., anti-AAV2 capsid antibody A20 WT, A20 one arm WT, A20 one arm WT-AAA), and cytokine-Fc Fusion constructs (cytokine-Fc Fusion WT, cytokine-Fc Fusion WT-AAA, cytokine-Fc Fusion Y436A) as well as Ustekinumab and Briakinumab were produced and purified internally at Roche (Penzberg, Germany). Mutations (Kabat numbering scheme) are the following: WT: wildtype; AAA: H310A, H433A, Y436A; YTE: M252Y, S254T, T256E; HH: T307H, N434H and Y436A. The production process involved the transient transfection of HEK Expi293F cells, sourced from Thermo Fisher Scientific (MS, US), with plasmids encoding both the light and heavy chains. Asymmetrical variants were generated using the knob-into-hole technology (75). This technology refers to refers to mutations Y349C, T366S, L368A and Y407V (Hole) and S354C and T366W (Knob) both in the CH3-CH3 interface to promote heteromultimer formation (76, 77).
4.3 Virus supply
Replication-deficient, recombinant Adeno-associated virus vector particles serotype 2 (rAAVv-2) without carrying any transgene (empty rAAVv-2) were supplied by Virovek (CA, US), aliquoted up on arrival and frozen at -80°C until required. The electron microscopy experiments were performed using genome-filled rAAV2 vectors, supplied by Sirion Biotech (Germany).
4.4 SEC-MALS
The molecular weight of the sample was determined using size-exclusion chromatography combined with multi-angle light scattering (SEC-MALS). The SEC-MALS analysis was performed using a high-performance liquid chromatography (HPLC) instrument (Superdex200 Increase 10/300 GL, GE Healthcare). The HPLC instrument was coupled with a Wyatt Heleos II 18-angle light-scattering instrument (297-TS, Wyatt Technology) and a Wyatt Optilab rEX (766-rEX, Wyatt Technology) online refractive index detector. The sample was prepared at a concentration of 1 mg/mL, and 50 µg of the sample was injected into the system. The running buffer used was phosphate-buffered saline (PBS) at pH 7.4. The system was operated at a flow rate of 0.5 mL/min. To ensure the accuracy and reliability of the system, a system suitability test was performed by injecting bovine serum albumin (BSA) prior to the sample analysis. The results from the BSA injection were used to validate the performance of the system and to confirm that it was operating within the acceptable parameters for accurate molecular weight determination. The Zimm model has been used as a default data process procedure in ASTRA 8.2 (Wyatt Technology).
4.5 SPR experiments
4.5.1 General assay setup
The Biacore T200 system (GE Healthcare, Uppsala, Sweden) was used for all surface plasmon resonance (SPR) experiments. The C1 sensor chips (carboxymethylated matrix-free surface), amine coupling kit (containing 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), and ethanolamine-HCl), HBS-N (10 mM HEPES, 150 mM NaCl, pH 7.4), HBS-EP+ running buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05% (v/v) surfactant P20, pH 7.4), and regeneration solutions were also obtained from Cytiva.
The ligand was immobilized on the C1 sensor chip using standard amine coupling chemistry. Briefly, the sensor surface was conditioned with two one minute injections of conditioning solution (0.1 M glycine-NaOH, 0.3% (v/v) Triton X-100, pH 12) followed by “Extra wash” and then run “Prime” using HBS-N running buffer. The Chip was then activated by injecting a mixture of 0.4 M EDC and 0.1 M NHS at a flow rate of 10 µL/min for 7 minutes. The ligand was then injected at a concentration of 20 µg/mL in 10 mM sodium acetate buffer (pH 4.5) at a flow rate of 10 µL/min for 3 minutes unless stated otherwise. Unreacted groups were blocked by injecting 1 M ethanolamine-HCl (pH 8.5) at a flow rate of 10 µL/min for 7 minutes.
Next, a molecule of interest, diluted accordingly in HBS-EP+ running buffer was captured via the amine coupled ligand to a desired Response unit (RU) level at a flow rate of 5 µL/min.
Binding kinetics were measured at 25°C by injecting various concentrations of the analyte over the captured molecule of interest at a flow rate of 50 µL/min unless stated otherwise. Each injection was followed by a dissociation phase. The sensor surface was regenerated between each cycle by injecting 10 mM glycine-HCl (pH 2 or 3, see below) at a flow rate of 30 µL/min.
The sensorgrams were analyzed using the Biacore T200 evaluation software (v3.1) and GraphPad Prism (v8.4.2). The data were fitted to a 1:1 binding model to determine the association rate constant (kON), dissociation rate constant (kOFF), and equilibrium dissociation constant (KD). The KD was calculated as kOFF/kON. For analyzing complex binding kinetics the data were fitted by applying the Two State Reaction Model obtaining a KD value explained in more detail in the following section. The results are presented as mean ± standard deviation.
To ensure the reliability of the data, a blank run (buffer only) was performed before each experiment to check the baseline stability.
4.5.2 TRIM21 PRYSPRY injections to captured antibody (Fc) Variants, immobilized Fc only variants and captured cytokine-Fc fusion constructs
To investigate the interactions of TRIM21 PRYSPRY with various protein constructs, we utilized a consistent methodology across different experiments, applying standard amine coupling chemistry for immobilization on a C1 sensor chip. The experiments focused on three targets: antibody (Fc) variants, Fc only variants, and cytokine-Fc Fusion constructs.
For the antibody (Fc) variants and Fc only variants, we aimed for initial immobilization to achieve response unit (RU) signals of approximately 60 for antibody variants and between 60 and 80 for Fc only variants, using the CaptureSelect™ Human Fab-kappa Kinetics Biotin Conjugate ligand (7103302100, Thermo Fisher Scientific) for the former and direct immobilization for the latter. The cytokine-Fc Fusion constructs involved capturing using an in-house anti-PGLALA F(ab’)2 fragment (55) with a target RU signal between 35 and 40.
Subsequent steps involved injecting five different concentrations of TRIM21 PRYSPRY, prepared in a two-fold dilution series, across all experiments. Each concentration underwent an association phase of 60 seconds and a dissociation phase of 180 seconds. The integrity of the sensor surface for subsequent cycles was maintained through a regeneration process involving twice injections of 10 mM glycine-HCl (pH adjusted according to the specific experiment - pH 2.0 for antibody variants and cytokine-Fc fusions, pH 3.0 for Fc only variants) for 60 seconds each, followed by a 120-second injection of running buffer (HBS-EP+) at a flow rate of 30 µL/min. This ensured the complete removal of any bound molecules from the previous cycle and restoration of the sensor surface to its initial state, facilitating accurate and repeatable measurements across the experiments.
4.5.3 Antibody (Fc) variants/Fc only variants/cytokine-Fc fusion constructs injections to captured TRIM21 PRYSPRY
In the initial step, a monovalent Streptavidin (mono-SA, in house, Roche) was immobilized on a C1 sensor chip via standard amine coupling chemistry. The ligand was introduced at a concentration of 10 µg/mL in 10 mM sodium acetate buffer (pH 4.5) at a flow rate of 10 µL/min for 180 seconds. Subsequently, biotinylated TRIM21 PRYSPRY in running buffer (HBS-EP+) was captured via mono-SA, aiming for a response unit (RU) signal of 3 to prevent any inter-molecular interactions interlinking two TRIM21 PRYSPRY by one antibody. Following this, five different concentrations of TRIM21 PRYSPRY, as specified in the sensorgrams, were prepared in a two-fold dilution series and introduced over the sensor chip. Each concentration underwent a 60-second association phase, followed by a 180-second dissociation phase to monitor the rate of complex decay. To restore the sensor surface for subsequent cycles, a regeneration step was performed after each cycle. This involved the injection of 10 mM glycine-HCl (pH 3.0) twice, each for 15 seconds, followed by a 30-second injection of running buffer (HBS-EP+) at a flow rate of 20 µL/min. This process ensured the complete removal of any bound molecules from the previous cycle and the restoration of the sensor surface to its initial state.
4.5.4 TRIM21-CC-ps injections to immobilized antibody (Fc) variants or cytokine-Fc fusion constructs
First, CaptureSelect™ Human Fab-kappa Kinetics Biotin Conjugate ligand (7103302100, Thermo Fisher Scientific) to capture Antibody (Fc) Variants or anti-PGLALA F(ab’)2 fragment (in-house, Roche) to capture cytokine-Fc Fusion constructs was immobilized. The ligands were introduced at a concentration of 5 µg/mL in 10 mM sodium acetate buffer (pH 4.5) at a flow rate of 5 µL/min for 120 seconds. Second, TRIM21-CC-PS in running buffer (HBS-EP+) was captured via the ligand, aiming for a response unit (RU) signal of 8. Following this, several different concentrations of Trim2-CC, as specified in the sensorgrams, were prepared in a two-fold dilution series and introduced over the sensor chip. Each concentration was injected for 180-second association phase, followed by a 600-second dissociation phase. To restore the sensor surface for subsequent cycles, a regeneration step was performed after each cycle. This involved the injection of 10 mM glycine-HCl (pH 2.0) twice, each for 60 seconds, followed by a 120-second injection of running buffer (HBS-EP+) at a flow rate of 30 µL/min.
4.5.5 Mimicking an TRIM21-CC-PS – antibody decorated viral complex
The formation of an immune complex on a sensor chip, consisting of TRIM21-CC-PS, an antibody, and AAV2-wt as a viral model, was achieved using surface plasmon resonance (SPR) technology on a Biacore system. Initially, monovalent Streptavidin (mono-SA, in house, Roche) was immobilized on a C1 sensor chip (Flow cells 1–4) until saturation was reached. This was achieved by injecting mono-SA at a concentration of 10 µg/mL in 10 mM sodium acetate buffer (pH 4.5) at a flow rate of 5 µL/min for 120 seconds using standard amine coupling chemistry. The running buffer during this step was HBSN. Following a buffer change to HBS-EP, biotinylated TRIM21-CC-PS was captured on flow cell 2 to approximately 35 RU and on flow cell 3 to approximately 190 RU to generate different ligand densities. Flow cell 1 served as a reference surface. Subsequently, different anti-AAV2 capsid antibody constructs with TRIM21 relevant Fc mutations were injected at a concentration of 5 µg/mL at a flow rate of 20 µL/min for 180 seconds until TRIM21-CC-PS was saturated. Following this, three different concentrations of AAV2-wt-empty, as specified in the sensorgrams, were prepared in a three-fold dilution series and introduced over the sensor chip. Each concentration underwent a 180-second association phase, followed by a 900-second dissociation phase at a flow rate of 10 µL/min. To restore the sensor surface for subsequent cycles, a regeneration step was performed after each cycle. This involved the injection of 10 mM glycine-HCl (pH 3.0) for 20 seconds, followed by a 30-second injection of running buffer (HBS-EP+) at a flow rate of 20 µL/min.
4.5.6 1:1 Binding model
In a simple 1:1 interaction, an analyte (A) binds to a ligand (B) to form a complex (AB). The equilibrium dissociation constant (KD,AFFINITY) was calculated using the equation:
Rmax (maximum response) is a parameter that represents the maximum binding capacity of the ligand immobilized on the sensor chip surface. It is determined by the amount of ligand immobilized and the stoichiometry of the interaction. Rmax is measured in response units (RU). This equation assumes that all the immobilized ligand is active and capable of binding to the analyte. In practice, not all the immobilized ligand may be active, and the observed Rmax may be less than the calculated Rmax.
MWanalyte is the molecular weight of the analyte. MWligand is the molecular weight of the ligand. Rligand is the immobilization level of the ligand, measured in RU. Valencyligand is the number of analyte molecules that bind to each ligand molecule.
The Rmax,ratio, calculated as provides a measure of the proportion of active ligand and thereby the Stoichiometry.
4.5.7 Complex binding kinetics – two state reaction
The rationale behind the Two State Model lies in its ability to account for the additional kinetic steps that occur during the binding process. In contrast to a 1:1 binding model, in some interactions, the formation of the initial complex induces a conformational change, leading to a different, usually more stable, complex (AB*). This can be represented as follows:
In this model, the analyte first associates with the ligand with an association rate constant (kON1), forming an initial complex (AB). This complex then undergoes a conformational change with a rate constant (kON2) to form the final complex (AB*). Each of these steps is reversible, with the initial complex dissociating back to the free analyte and ligand with a rate constant (kOFF1), and the final complex reverting back to the initial complex with a rate constant (kOFF2).
The apparent or avidity equilibrium dissociation constant (KD) reflects the overall stability of the final AB* complex and is calculated using the formula:
4.6 Mass photometry technology
The mass photometry experiments were conducted using the OneMP system, TwoMP system and OneMP-MassFluidix HC system to analyze low-affinity interactions (Refeyn Ltd., Oxford, UK) at room temperature. The sample buffer used for all experiments was 1x PBS, pH 7.4 (Roche Diagnostics GmbH). The sample carrier slides (Refeyn Ltd., Oxford, UK) were prepared by cleaning them in an isopropanol ultrasonic bath for 10 minutes, followed by consecutive rinsing with H2O. The slides were then dried under a stream of clean nitrogen. A fourteen well, each well 3 mm in diameter, sample well gasket (Refeyn Ltd., Oxford, UK) providing four measurement spots, each capable of holding a volume of 20µL sample solution, was placed onto the carrier slide. The molecules of interest, TRIM21 PRYSPRY or TRIM21-CC-PS with antibody Fc variants, were prepared in the sample buffer at various concentrations, specifically four times higher than in the final droplet (for final concentrations and ratios, see histograms). The samples were thoroughly mixed and incubated at room temperature for 10 minutes to allow for complex formation. Approximately 5 µL of the sample was placed on a clean glass coverslip, containing 15µL of PBS, and loaded into the mass photometry system. The system was set to acquire data using the AquireMP 2.5 software (Refeyn Ltd.) for a period of 1 minute at a 1 kHz frame rate. The mass photometry data were analyzed using DiscoverMP software 2024 R1 (Refeyn Ltd.) and GraphPad Prism (Version 8.4.2, GraphPad Software, Inc., CA, USA). A 3-dimensional Gaussian distribution model was applied for the analysis. The ratio (in %) for each detected species, whether a single molecule or an interacting pair of molecules, was calculated by dividing the counts of events within a specific Gaussian distribution by the total number of detected events. The acquisition parameters were set according to the manufacturer’s instructions and optimized for the specific molecules of interest using the following parameters: number of averaged frames: 5, threshold 1: 1.5, and threshold 2: 0.25. The mass of the individual molecules and their complexes were determined from the intensity of the light scattered by the molecules. Results were reported as normalized counts, calculated by dividing events in each bin by the total number of events. To ensure the reliability of the data, a control experiment was performed with each molecule alone to confirm their individual masses (5 nM concentration in the final droplet). A mass calibration curve was generated by analyzing three different proteins with known molecular masses of approximately 66 kDa, 145.5 kDa, and 194 kDa, respectively (Roche Diagnostics GmbH). The calibration data was collected from the 2.9 μm × 10.8 μm instrument field of view for 100 seconds.
4.7 Electron microscopy experiments
Samples were diluted in D-PBS (Gibco Life Technologies) until a suitable concentration was reached for negative staining EM. Interaction mixtures (Figure 6) were mixed and incubated at RT for 1h prior to dilution and imaging. Recombinant AAVv-2-antibody interactions (Figure 8) were incubated for 30 minutes except for bivalent A20 Fc WT, which caused full precipitation at 30 minutes and was incubated for 5 minutes only. Images that additionally contain TRIM21-CC-PS were incubated for 12 minutes with antibody variants, followed by 18 minutes of incubation with TRIM21-CC-PS. Since bivalent A20 Fc WT alone was sufficient to fully precipitate rAAVv-2s, it was not possible to observe an additional effect of TRIM21-CC-PS. Electron microscopy grids (T600H-Cu 698 l/inch Hex. mesh Thin Bar; EMS) were coated with a home-made ~2nm carbon film by floating the carbon on H2O and letting the water level drop till the carbon covered the grids. After at least 2 days of drying the grids were used. Four ul of sample was incubated on a glow-discharged carbon coated grid for 30 seconds, followed by two steps of washing with H2O, a step of washing with UAc (2%) solution and incubation with UAc for 30s. As an internal standard and to improve the quality of the negative stain, 2ul of tobacco mosaic virus in solution was incubated for 10 seconds on the grid to each sample that did not include AAVs. This step was added after the 2 rounds of washing with H2O and followed by 2 extra H2O washing steps (TMV; kindly supplied by Ruben Diaz-Avalos, New York Structural Biology Center, USA).
Grids were loaded into a Jeol JEM-1400 Plus transmission electron microscope operating a Lab6 electron source at 120 kV. Electron micrographs were recorded on TVIPS XF416 4000 by 4000 pixel charge-coupled device camera (Tietz Video and Image Processing System, Gauting, Germany) at a nominal magnification of 100,000x yielding pictures with a pixel size corresponding to 0.1149 nm at the specimen level.
Images were gaussian blurred 4x in Fiji (78) before being imported into the EMAN2 software package (79). Reference-free alignment was performed on manually selected particles followed by classifications by multivariate statistical analysis. Images were used as indicated without CTF correction or further processing. Particles of interest were manually selected using interactive Particle Picking (e2boxer.py) and subsequently combined into a particle set. Particles were then sorted iteratively in multiple 2D classes using Reference Free Class Averaging (e2refine2d.py) allowing EMAN2 to discard 15% of the picked particles to come to a cleaner result. The center setting was varied, the classaverager was set to mean and default settings were kept for the other parameters. Scale Bars were added using Fiji (78).
4.8 Model predictions
Structural model predictions were generated using AlphaFold2 with standard settings (67). To generate the multimeric Fc bound model, two copies of the TRIM21 sequence (full and truncated) were entered as separate molecules and the Fc light and heavy chain were fused into one molecule by adding a long GGGS linker between light and heavy chain. Results were visualized with ChimeraX (80, 81).
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
JR: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. LF: Writing – original draft, Writing – review & editing, Formal analysis, Investigation, Visualization. JT: Writing – original draft, Investigation. PR: Writing – original draft, Investigation. JB: Writing – original draft, Investigation. TJ: Writing – original draft, Investigation. CK: Writing – review & editing, Conceptualization, Supervision. TS: Writing – review & editing, Conceptualization, Supervision. LL: Writing – review & editing, Conceptualization, Supervision.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We would like to acknowledge Matthias Lauer and Lubomir Kovacik for their contributions to the Nautilus at Roche Basel, the EM facility where the electron microscopy data was recorded. We highly appreciate Holger Kley, Roche Penzberg, providing SEC-MALS data for TRIM21.
Conflict of interest
CK declares employment, patents and stock ownership with Roche. Authors JR, JT, JB, TJ, TS, and LL were employed by the company Roche Diagnostics GmbH. Authors LF and PR were employed by the company F.Hoffmann-La Roche Ltd.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1401471/full#supplementary-material
Abbreviations
AAV, Adeno-Associated Virus; ADIN, antibody-dependent intracellular neutralization; AP-1, Activator Protein 1; EM, Electron Microscopy; Fc, Fragment crystallizable; FcRn, neonatal Fc receptor; HBV, Hepatitis B virus; hIgG1, human Immunoglobulin G isotype 1; HSPG, heparan sulfate proteoglycan; IFN, Interferon; IRF 3/5/7, Interferon Regulatory Factor 3, 5, and 7; ITC, Isothermal titration calorimetry; JAK-STAT, Janus Kinase-Signal Transducer and Activator of Transcription; JEV, Japanese encephalitis virus; KD, equilibrium dissociation constant; KiH, Knob-into-hole; kOFF, dissociation rate constant; kON, association rate constant; mAb, monoclonal Antibody; MP, Mass Photometry; MTL, mass transport limitation; NFkB, Nuclear Factor kappa-light-chain-enhancer of activated B cells; PK, Pharmacokinetics; PRRS, Porcine reproductive and respiratory syndrome virus; PS, PRYSPRY domain; rAAVv, recombinant Adeno-associated virus vector particles; RING, really interesting new gene; SAXS, Small-angle X-ray scattering; SPR, surface plasmon resonance; Trim, tripartite motif; TRIM21-CC-PS, tripartite motif-containing 21 coiled-coil-PRYSPRY; UPS, ubiquitin-proteasome system; VCP, valosin-containing protein; WT, wildtype.
References
1. Rajsbaum R, García-Sastre A, Versteeg GA. Trimmunity: the roles of the Trim E3-ubiquitin ligase family in innate antiviral immunity. J Mol Biol. (2014) 426:1265–84. doi: 10.1016/j.jmb.2013.12.005
2. Foss S, Bottermann M, Jonsson A, Sandlie I, James LC, Andersen JT. Trim21—from intracellular immunity to therapy. Front Immunol. (2019) 10:2049. doi: 10.3389/fimmu.2019.02049
3. McEwan WA, James LC. Trim21-dependent intracellular antibody neutralization of virus infection. Prog Mol Biol Transl. (2015) 129:167–87. doi: 10.1016/bs.pmbts.2014.10.006
4. Watkinson RE, McEwan WA, James LC. Intracellular antibody immunity. J Clin Immunol. (2014) 34:30–4. doi: 10.1007/s10875–014-0017–4
5. Watkinson RE, Tam JCH, Vaysburd MJ, James LC. Simultaneous neutralization and innate immune detection of a replicating virus by Trim21. J Virol. (2013) 87:7309–13. doi: 10.1128/jvi.00647–13
6. Boudinot P, Aa L, Jouneau L, Pasquier LD, Pontarotti P, Briolat V, et al. Origin and evolution of trim proteins: new insights from the complete Trim repertoire of zebrafish and pufferfish. PloS One. (2011) 6:e22022. doi: 10.1371/journal.pone.0022022
7. Randow F, MacMicking JD, James LC. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science. (2013) 340:701–6. doi: 10.1126/science.1233028
8. Rhodes DA, Isenberg DA. Trim21 and the function of antibodies inside cells. Trends Immunol. (2017) 38:916–26. doi: 10.1016/j.it.2017.07.005
9. Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (Trim21). Proc Natl Acad Sci. (2010) 107:19985–90. doi: 10.1073/pnas.1014074107
10. Foss S, Watkinson RE, Grevys A, McAdam MB, Bern M, Høydahl LS, et al. Trim21 immune signaling is more sensitive to antibody affinity than its neutralization activity. J Immunol. (2016) 196:3452–9. doi: 10.4049/jimmunol.1502601
11. McEwan WA, Tam JCH, Watkinson RE, Bidgood SR, Mallery DL, James LC. Intracellular antibody-bound pathogens stimulate immune signaling via the fc receptor Trim21. Nat Immunol. (2013) 14:327. doi: 10.1038/ni.2548
12. Ozato K, Shin DM, Chang TH, Morse HC 3rd. Trim family proteins and their emerging roles in innate immunity. Nat Rev Immunol. (2008) 8:849–60. doi: 10.1038/nri2413
13. Meroni G, Diez-Roux G. Trim/Rbcc, a novel class of ‘Single protein ring finger’ E3 ubiquitin ligases. BioEssays. (2005) 27:1147–57. doi: 10.1002/bies.20304
14. Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. The tripartite motif family identifies cell compartments. EMBO J. (2001) 20:2140–51. doi: 10.1093/emboj/20.9.2140
15. Wang H-T, Hur S. Substrate recognition by Trim and Trim-like proteins in innate immunity. Semin Cell Dev Biol. (2021) 111:76–85. doi: 10.1016/j.semcdb.2020.09.013
16. Fletcher AJ, Mallery DL, Watkinson RE, Dickson CF, James LC. Sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of Trim21. Proc Natl Acad Sci. (2015) 112:10014–9. doi: 10.1073/pnas.1507534112
17. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. (2002) 82:373–428. doi: 10.1152/physrev.00027.2001
18. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. (1998) 67:425–79. doi: 10.1146/annurev.biochem.67.1.425
19. Dickson C, Fletcher AJ, Vaysburd M, Yang J-C, Mallery DL, Zeng J, et al. Intracellular antibody signalling is regulated by phosphorylation of the Fc receptor Trim21. Elife. (2018) 7:e32660. doi: 10.7554/elife.32660
20. Kiss L, Clift D, Renner N, Neuhaus D, James LC. Ring domains act as both substrate and enzyme in a catalytic arrangement to drive self-anchored ubiquitination. Nat Commun. (2021) 12:1220. doi: 10.1038/s41467–021-21443–6
21. Kiss L, Zeng J, Dickson CF, Mallery DL, Yang J-C, McLaughlin SH, et al. A tri-ionic anchor mechanism drives Ube2n-specific recruitment and K63-chain ubiquitination in Trim ligases. Nat Commun. (2019) 10:4502. doi: 10.1038/s41467-019-12388-y
22. Keeble AH, Khan Z, Forster A, James LC. Trim21 is an Igg receptor that is structurally, thermodynamically, and kinetically conserved. Proc Natl Acad Sci. (2008) 105:6045–50. doi: 10.1073/pnas.0800159105
23. Bidgood SR, Tam JCH, McEwan WA, Mallery DL, James LC. Translocalized Iga mediates neutralization and stimulates innate immunity inside infected cells. Proc Natl Acad Sci. (2014) 111:13463–8. doi: 10.1073/pnas.1410980111
24. James LC, Keeble AH, Khan Z, Rhodes DA, Trowsdale J. Structural basis for Pryspry-mediated tripartite motif (Trim) protein function. Proc Natl Acad Sci. (2007) 104:6200–5. doi: 10.1073/pnas.0609174104
25. James LC. Intracellular antibody immunity and the cytosolic Fc receptor Trim21. Curr Top Microbiol Immunol. (2014) 382:51–66. doi: 10.1007/978–3-319–07911-0_3
26. Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ. Crystal structure at 2.2 a resolution of the Mhc-related neonatal Fc receptor. Nature. (1994) 372:336–43. doi: 10.1038/372336a0
27. Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, et al. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem. (2014) 289:7812–24. doi: 10.1074/jbc.M113.537563
28. Vaughn DE, Bjorkman PJ. Structural basis of Ph-dependent antibody binding by the neonatal Fc receptor. Structure. (1998) 6:63–73. doi: 10.1016/s0969–2126(98)00008–2
29. Bottermann M, James LC. Intracellular antiviral immunity. Adv Virus Res. (2018) 100:309–54. doi: 10.1016/bs.aivir.2018.01.002
30. Ng PML, Kaliaperumal N, Lee CY, Chin WJ, Tan HC, Au VB, et al. Enhancing antigen cross-presentation in human monocyte-derived dendritic cells by recruiting the intracellular Fc receptor Trim21. J Immunol. (2019) 202:2307–19. doi: 10.4049/jimmunol.1800462
31. Altamash T, Ahmed W, Rasool S, Biswas KH. Intracellular ionic strength sensing using nanoluc. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22020677
32. Liu B, Poolman B, Boersma AJ. Ionic strength sensing in living cells. ACS Chem Biol. (2017) 12:2510–4. doi: 10.1021/acschembio.7b00348
33. Dall’Acqua WF, Kiener PA, Wu H. Properties of human Igg1s engineered for enhanced binding to the neonatal Fc receptor (Fcrn). J Biol Chem. (2006) 281:23514–24. doi: 10.1074/jbc.M604292200
34. Booth BJ, Ramakrishnan B, Narayan K, Wollacott AM, Babcock GJ, Shriver Z, et al. Extending human Igg half-life using structure-guided design. MAbs. (2018) 10:1098–110. doi: 10.1080/19420862.2018.1490119
35. Mackness BC, Jaworski JA, Boudanova E, Park A, Valente D, Mauriac C, et al. Antibody Fc engineering for enhanced neonatal Fc receptor binding and prolonged circulation half-life. MAbs. (2019) 11:1276–88. doi: 10.1080/19420862.2019.1633883
36. Liu R, Oldham RJ, Teal E, Beers SA, Cragg MS. Fc-engineering for modulated effector functions-improving antibodies for cancer treatment. Antibodies. (2020) 9:64. doi: 10.3390/antib9040064
37. Qi T, Cao Y. In translation: Fcrn across the therapeutic spectrum. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22063048
38. Moessner Ekkehard, Tilman S, inventors; F HOFFMANN-LA ROCHE AG (HOFF-C), assignee. Immunoglobulin G Class Fragment Crystallizable (Fc)-Region Used for Treatment of Ocular Vascular Diseases, Comprises First Variant Fc-Region Polypeptide and Second Variant Fc-Region Polypeptide; Fc-Region Variants with Modified Fcrn-Binding and Methods of Use patent WO2016071376A2. INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT): World Intellectual Property Organization. (2015). Available at: https://worldwide.espacenet.com/publicationDetails/biblio?II=0&ND=3&adjacent=true&locale=de_EP&FT=D&date=20160512&CC=WO&NR=2016071376A2&KC=A2.
39. Schlothauer T, A S, inventors; HOFFMANN LA ROCHE & CO AG F (HOFF-C) assignee. New Polypeptide Used in Pharmaceutical Formulation for E.G. Treating Vascular Eye Diseases, and Transporting Soluble Receptor Ligand from Eye over Blood-Ocular-Barrier into Blood Circulation; Fc-Region Variants with Modified Fcrn- and Maintained Protein a-Binding Properties patent WO2015107026A1. INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT): World Intellectual Property Organization. (2015). Available at: https://worldwide.espacenet.com/publicationDetails/biblio?II=0&ND=3&adjacent=true&locale=de_EP&FT=D&date=20150723&CC=WO&NR=2015107026A1&KC=A1.
40. Joerg Thomas Regula WS, Tilman Schlothauer, inventor; F HOFFMANN-LA ROCHE assignee. New Immunoglobulin G (Igg) Class Fragment Crystallizable (Fc)-Region Useful in Antibody for Preparing Medicine Used for Treating Ocular Vascular Diseases, Comprises First and Second Variant Fc-Region Polypeptides; Fc-Receptor Binding Modified Asymmetric Antibodies and Methods of Use patent WO2014177459A2. INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT): World Intellectual Property Organization. (2014). Available at: https://worldwide.espacenet.com/publicationDetails/biblio?II=0&ND=3&adjacent=true&locale=de_EP&FT=D&date=20141106&CC=WO&NR=2014177459A2&KC=A2.
41. T S, inventor; HOFFMANN LA ROCHE, assignee. Fc-Region Variants with Improved Protein a-Binding; New Polypeptide Comprising First and Second Polypeptides Each Comprising Immunoglobulin (Ig) Hinge Region Comprising Cysteine Residue, Ig Constant Heavy Chain (Ch) 2 Domain and Ig Ch3-Domain, Used to Treat Vascular Eye Diseases patent WO2015107015A1. INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT): World Intellectual Property Organization. (2015). Available at: https://worldwide.espacenet.com/publicationDetails/biblio?II=0&ND=3&adjacent=true&locale=de_EP&FT=D&date=20150723&CC=WO&NR=2015107015A1&KC=A1.
42. Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, et al. Charge-mediated influence of the antibody variable domain on Fcrn-dependent pharmacokinetics. Proc Natl Acad Sci U.S.A. (2015) 112:5997–6002. doi: 10.1073/pnas.1408766112
43. Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, et al. Monoclonal antibodies with identical Fc sequences can bind to Fcrn differentially with pharmacokinetic consequences. Drug Metab Dispos. (2011) 39:1469–77. doi: 10.1124/dmd.111.039453
44. Bottermann M, Lode HE, Watkinson RE, Foss S, Sandlie I, Andersen JT, et al. Antibody-antigen kinetics constrain intracellular humoral immunity. Sci Rep-uk. (2016) 6:37457. doi: 10.1038/srep37457
45. McEwan WA, Hauler F, Williams CR, Bidgood SR, Mallery DL, Crowther RA, et al. Regulation of virus neutralization and the persistent fraction by Trim21. J Virol. (2012) 86:8482–91. doi: 10.1128/jvi.00728–12
46. Caddy SL, Vaysburd M, Wing M, Foss S, Andersen JT, O’Connell K, et al. Intracellular neutralisation of rotavirus by Vp6-specific Igg. PloS Pathog. (2020) 16:e1008732. doi: 10.1371/journal.ppat.1008732
47. Manocha GD, Mishra R, Sharma N, Kumawat KL, Basu A, Singh SK. Regulatory role of Trim21 in the type-I interferon pathway in Japanese encephalitis virus-infected human microglial cells. J Neuroinflamm. (2014) 11:24. doi: 10.1186/1742–2094-11–24
48. Mu T, Zhao X, Zhu Y, Fan H, Tang H. The E3 ubiquitin ligase Trim21 promotes Hbv DNA polymerase degradation. Viruses. (2020) 12. doi: 10.3390/v12030346
49. Rakebrandt N, Lentes S, Neumann H, James LC, Neumann-Staubitz P. Antibody- and Trim21-dependent intracellular restriction of Salmonella enterica. Pathog Dis. (2014) 72:131–7. doi: 10.1111/2049–632x.12192
50. Vaysburd M, Watkinson RE, Cooper H, Reed M, O’Connell K, Smith J, et al. Intracellular antibody receptor Trim21 prevents fatal viral infection. Proc Natl Acad Sci. (2013) 110:12397–401. doi: 10.1073/pnas.1301918110
51. Wei Y, Zeng S, Zou C, Zhang H, Peng O, Xue C, et al. Porcine Trim21 ring-finger E3 ubiquitin ligase is essential for anti-Prrsv activity. Vet Microbiol. (2021) 256:109043. doi: 10.1016/j.vetmic.2021.109043
52. Li X, Yang L, Chen S, Zheng J, Zhang H, Ren L. Multiple roles of Trim21 in virus infection. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24021683
53. Zeng J, Santos AF, Mukadam AS, Osswald M, Jacques DA, Dickson CF, et al. Target-induced clustering activates Trim-away of pathogens and proteins. Nat Struct Mol Biol. (2021) 28:278–89. doi: 10.1038/s41594-021-00560-2
54. Fiorentini F, Esposito D, Rittinger K. Does it take two to tango? Ring domain self-association and activity in Trim E3 ubiquitin ligases. Biochem Soc Trans. (2020) 48:2615–24. doi: 10.1042/bst20200383
55. Wessels U, Schick E, Ritter M, Kowalewsky F, Heinrich J, Stubenrauch K. Novel drug and soluble target tolerant antidrug antibody assay for therapeutic antibodies bearing the P329g mutation. Bioanalysis. (2017) 9:849–59. doi: 10.4155/bio-2017–0048
56. Jensen PF, Larraillet V, Schlothauer T, Kettenberger H, Hilger M, Rand KD. Investigating the interaction between the neonatal Fc receptor and monoclonal antibody variants by hydrogen/deuterium exchange mass spectrometry. Mol Cell Proteomics. (2015) 14:148–61. doi: 10.1074/mcp.M114.042044
57. Piche-Nicholas NM, Avery LB, King AC, Kavosi M, Wang M, O’Hara DM, et al. Changes in complementarity-determining regions significantly alter Igg binding to the neonatal Fc receptor (Fcrn) and pharmacokinetics. MAbs. (2018) 10:81–94. doi: 10.1080/19420862.2017.1389355
58. Schlothauer T, Rueger P, Stracke JO, Hertenberger H, Fingas F, Kling L, et al. Analytical Fcrn affinity chromatography for functional characterization of monoclonal antibodies. MAbs. (2013) 5:576–86. doi: 10.4161/mabs.24981
59. Day ES, Capili AD, Borysenko CW, Zafari M, Whitty A. Determining the affinity and stoichiometry of interactions between unmodified proteins in solution using biacore. Anal Biochem. (2013) 440:96–107. doi: 10.1016/j.ab.2013.05.012
60. Grevys A, Frick R, Mester S, Flem-Karlsen K, Nilsen J, Foss S, et al. Antibody variable sequences have a pronounced effect on cellular transport and plasma half-life. iScience. (2022) 25:103746. doi: 10.1016/j.isci.2022.103746
61. McCraw DM, O’Donnell JK, Taylor KA, Stagg SM, Chapman MS. Structure of adeno-associated virus-2 in complex with neutralizing monoclonal antibody A20. Virology. (2012) 431:40–9. doi: 10.1016/j.virol.2012.05.004
62. Foss S, Watkinson R, Sandlie I, James LC, Andersen JT. Trim21: A cytosolic Fc receptor with broad antibody isotype specificity. Immunol Rev. (2015) 268:328–39. doi: 10.1111/imr.12363
63. Burvenich IJ, Lee FT, O’Keefe GJ, Makris D, Cao D, Gong S, et al. Engineering anti-Lewis-Y Hu3s193 antibodies with improved therapeutic ratio for radioimmunotherapy of epithelial cancers. EJNMMI Res. (2016) 6:26. doi: 10.1186/s13550–016-0180–0
64. Fletcher AJ, James LC. Coordinated neutralization and immune activation by the cytosolic antibody receptor Trim21. J Virol. (2016) 90:4856–9. doi: 10.1128/JVI.00050–16
65. Sanchez JG, Chiang JJ, Sparrer KMJ, Alam SL, Chi M, Roganowicz MD, et al. Mechanism of Trim25 catalytic activation in the antiviral Rig-I pathway. Cell Rep. (2016) 16:1315–25. doi: 10.1016/j.celrep.2016.06.070
66. Sanchez JG, Okreglicka K, Chandrasekaran V, Welker JM, Sundquist WI, Pornillos O. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc Natl Acad Sci U.S.A. (2014) 111:2494–9. doi: 10.1073/pnas.1318962111
67. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with alphafold. Nature. (2021) 596:583–9. doi: 10.1038/s41586–021-03819–2
68. Moskalenko M, Chen L, van Roey M, Donahue BA, Snyder RO, McArthur JG, et al. Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: implications for gene therapy and virus structure. J Virol. (2000) 74:1761–6. doi: 10.1128/jvi.74.4.1761–1766.2000
69. Wobus CE, Hugle-Dorr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (Aav-2) capsid: epitope mapping and identification of capsid domains involved in Aav-2-cell interaction and neutralization of Aav-2 infection. J Virol. (2000) 74:9281–93. doi: 10.1128/jvi.74.19.9281–9293.2000
70. Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. (1998) 72:1438–45. doi: 10.1128/JVI.72.2.1438–1445.1998
71. Fitzpatrick Z, Leborgne C, Barbon E, Masat E, Ronzitti G, van Wittenberghe L, et al. Influence of pre-existing anti-capsid neutralizing and binding antibodies on Aav vector transduction. Mol Ther Methods Clin Dev. (2018) 9:119–29. doi: 10.1016/j.omtm.2018.02.003
72. Xiao PJ, Samulski RJ. Cytoplasmic trafficking, endosomal escape, and perinuclear accumulation of adeno-associated virus type 2 particles are facilitated by microtubule network. J Virol. (2012) 86:10462–73. doi: 10.1128/JVI.00935–12
73. Varghese R, Mikyas Y, Stewart PL, Ralston R. Postentry neutralization of adenovirus type 5 by an antihexon antibody. J Virol. (2004) 78:12320–32. doi: 10.1128/JVI.78.22.12320–12332.2004
74. Wohlfart CE, Svensson UK, Everitt E. Interaction between hela cells and adenovirus type 2 virions neutralized by different antisera. J Virol. (1985) 56:896–903. doi: 10.1128/JVI.56.3.896–903.1985
75. Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ Engineering of antibody Ch3 domains for heavy chain heterodimerization. Protein Eng. (1996) 9:617–21. doi: 10.1093/protein/9.7.617
76. Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. (2015) 67:95–106. doi: 10.1016/j.molimm.2015.01.003
77. Vaks L, Litvak-Greenfeld D, Dror S, Shefet-Carasso L, Matatov G, Nahary L, et al. Design principles for bispecific Iggs, opportunities and pitfalls of artificial disulfide bonds. Antibodies. (2018) 7. doi: 10.3390/antib7030027
78. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. (2012) 9:676–82. doi: 10.1038/nmeth.2019
79. Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, et al. Eman2: an extensible image processing suite for electron microscopy. J Struct Biol. (2007) 157:38–46. doi: 10.1016/j.jsb.2006.05.009
80. Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, et al. Ucsf chimerax: meeting modern challenges in visualization and analysis. Protein Sci. (2018) 27:14–25. doi: 10.1002/pro.3235
Keywords: TRIM21, therapeutic IgG, antibody Fc mutants, antibody mediated viral neutralization, binding kinetics, affinity, avidity, structure-function
Citation: Reusch J, Franken LE, Then J, Ringler P, Butzer J, Juroschek T, Klein C, Schlothauer T and Larivière L (2024) TRIM21 and Fc-engineered antibodies: decoding its complex antibody binding mode with implications for viral neutralization. Front. Immunol. 15:1401471. doi: 10.3389/fimmu.2024.1401471
Received: 15 March 2024; Accepted: 29 May 2024;
Published: 12 June 2024.
Edited by:
Carlos Rosales, National Autonomous University of Mexico, MexicoReviewed by:
William Alexander McEwan, University of Cambridge, United KingdomMark Chiu, Tavotek Biotherapeutics, United States
Copyright © 2024 Reusch, Franken, Then, Ringler, Butzer, Juroschek, Klein, Schlothauer and Larivière. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tilman Schlothauer, dGlsbWFuLnNjaGxvdGhhdWVyQHJvY2hlLmNvbQ==; Laurent Larivière, bGF1cmVudC5sYXJpdmllcmVAcm9jaGUuY29t
†These authors share last authorship