 Lin-Nan Shao
Lin-Nan Shao Wen-Qian Song
Wen-Qian Song- Blood Group Reference Laboratory, Dalian Blood Center, Dalian, China
Background: Mutations in the ABO gene, including base insertions, deletions, substitutions, and splicing errors, can result in blood group subgroups associated with the quantity and quality of blood group antigens. Here, we employed third-generation PacBio sequencing to uncover a novel AEL allele arising from an intron splice site mutation, which altered the expected A2 phenotype to manifest as an Ael phenotype. The study aimed to characterize the molecular mechanism underlying this phenotypic switch
Methods: A 53-year-old healthy male blood donor with an atypical agglutination pattern was investigated. PacBio sequencing was used to sequence the entire ABO gene of the proband. In silico analysis predicted aberrant splicing, which was experimentally verified using a minigene splicing assay.
Results: Based on serological characteristics, the proband was determined to have an Ael phenotype. Sequencing revealed heterozygosity for ABO*O.01.02 and a novel ABO*A2.01-like allele with an additional c.28 + 5G>A mutation in intron 1. In silico predictions also indicated that this mutation is likely to cause aberrant splicing. Minigene analysis suggested that this mutation disrupted the 5’-end canonical donor splice site in intron 1, activated a cryptic donor site, and resulted in a 167 bp insertion, producing a truncated glycosyltransferase (p.Lys11Glufs*66). Meanwhile, a small amount of the wild type transcript was also generated through normal splicing, contributing to the Ael phenotype.
Conclusion: A novel AEL allele was identified in a Chinese male blood donor on the ABO*A2.01 background, characterized by the c.28 + 5G>A variant. This study provides insights into the molecular basis of blood group antigen variation.
Introduction
The ABO blood group system, discovered by Karl Landsteiner more than a century ago (1), continues to be of paramount importance in blood transfusion, prenatal serological testing, and bone marrow as well as organ transplantation. The gene responsible for the ABO system is located on chromosome 9 and codes for the glycosyltransferases A and B enzymes, which are responsible for the production of A and B antigens on red blood cells (RBCs), respectively. The ABO gene encompasses over 18 kb and is composed of seven exons, each ranging in size from 28 to 688 bp. Exons 6 and 7, the final two exons, make up 823 bp of the transcribed 1062 bp mRNA and are responsible for encoding the catalytic domain of the ABO glycosyltransferases (2, 3). Numerous studies have demonstrated that base insertions, deletions, substitutions, and mutations within the coding exons of the ABO gene can give rise to subgroups (4–7), which directly influence the integrity and abundance of blood group antigens (8, 9). Additionally, certain ABO subgroups exhibit weak expression and can be attributed to variants within the introns at exon/intron boundaries (10) and in regulatory regions, including the CCAAT-binding factor/Nuclear Factor Y (CBF/NF-Y) binding site (11), the proximal promoter (4), and the +5.8-kb site (12). These subgroups are frequently misclassified as common ABO genotypes due to the predominant focus on coding regions. Third-generation long-read single-molecule real-time sequencing technology, as provided by Pacific BioSciences (PacBio), holds significant potential for the comprehensive assembly of haplotype sequences of blood group gene alleles (13).
The Ael phenotype represents a rare subgroup distinguished by its absence of agglutination in reaction to anti-A or anti-A,B antibodies. Nonetheless, Ael RBCs do interact with these antibodies, as shown by adsorption and elution tests (14). Flow cytometry (15) and immunogold electron microscopy (16, 17) have revealed very low levels of A antigen on Ael cells. Individuals with the Ael phenotype typically have serum containing anti-A1 antibodies and may also have antibodies that agglutinate A2 cells (18). Notably, glycosyltransferase A is undetectable in Ael serum or on RBC membranes, and the H-transferase activity in Ael serum is less than that in A1 or A2 serum (19). It is necessary to use serological and/or molecular biology techniques to accurately identify the blood type of such patients, in order to ensure that weak antigens are not missed and avoid blood transfusion in cases of blood type inconsistency.
In this study, we employ PacBio sequencing to describe a novel AEL allele where an intron splice site mutation, which would normally result in an A2 phenotype, anomalously leads to an Ael phenotype, and we aim to elucidate its underlying mechanism.
Materials and methods
Specimen collection
A 53-year-old healthy male with an anomalous agglutination pattern visited our blood center to donate blood. Upon providing informed consent, a specimen was collected using EDTA anticoagulant.
Serological tests
Serological tests were conducted, which included ABO blood typing using standard serologic methods, comprising forward and reverse typing. Forward typing employed anti-A, anti-B, anti-AB, and anti-H (anti-A and anti-B from Changchun Brother Biotech Co, Ltd., anti-AB from DIAGAST, and anti-H from Shanghai Hemo-pharmaceutical Biological Company) reagents to detect A, B, and H antigens, respectively, on red blood cells (RBCs). Reverse typing was carried out using A1, B, and O cells (from Shanghai Hemo-pharmaceutical Biological Company) with a tube test by qualified personnel, following the manufacturer’s guidelines. To confirm the presence of A antigens on the RBCs, adsorption and elution procedures were conducted using monoclonal anti-A antibody, following the standard protocol, with heat elution being the method employed.
PacBio third-generation long-read single-molecule real-time sequencing
Genomic DNA was extracted from peripheral whole blood samples using a commercially available HiPure Blood DNA Mini Kit, according to the manufacturer’s instructions. For long-range PCR, reaction mixtures contained 5 μL of 5× PrimeSTAR GXL buffer, 2 μL of dNTP mixture (2.5 mM each), 0.5 μL of PrimeSTAR GXL DNA polymerase, 0.26 μL of each PCR primer mix (100µM), 30 ng DNA templates, and DNase/RNase-free deionized water, resulting in a final reaction volume of 25 μL. The PCR cycling program comprised an initial denaturation at 94°C for 2 min, followed by 26 cycles of 98°C for 12 s, 68°C for 12 min (beginning from the 11th cycle, each subsequent cycle increased by 30 s), and a final extension step at 68°C for 10 min. The PCR products were used for preparing the library for PacBio sequencing. The entire ABO gene, including flanking regulatory regions in the three overlapping fragments, was amplified. The fragments overlapped more than 1 kb (Figure 1A).

Figure 1. Genetic structure of the ABO gene. (A) Three overlapping segments (blue and red arrows above) of the long-range PCR amplification cover the entire ABO gene. The vertical lines inside the blue and red arrows indicate mutation sites. Full-length gene haplotypes can be assembled based on the mutation sites within the overlapping region. NG_006669.1 was used as the reference allele sequence. (B) SD-Score prediction results of 5’ splice site sequences spanning three nucleotides at the 3’ end of exon 1 and six nucleotides at the 5’ end of intron 1 (from exon –3 to intron +6). The program returns differences in the SD-Score (ΔSD-Score), the information contents (ΔRi), and the position weight matrix (ΔCV) in the simulation of all possible single nucleotide variations.
In silico analysis
The effect of the splice site variation was predicted using the SD-Score web service program (https://www.med.nagoya-u.ac.jp/neurogenetics/SD_Score/sd_score.html). The frequency of a specific 5’ splice site sequence in the human genome can be used as an indicator of splicing signal intensity. The SD-Score represents a common logarithm of the frequency of a specific 5’ splice site in the human genome. For example, the SD-Score for CAG|GTGAGG, which was observed at 2562 sites in 189249 human 5’ GT splice sites, is log (2562/189249) =-1.868. The SD-Score of a splice site sequence that never appears in the human genome should be log (0/189249)= -∞ but is defined as log (0.25/189249)=-5.879 to simplify calculations. This algorithm predicts whether a mutation at the 5’ splice site will cause aberrant splicing, with a sensitivity of 97.1% and specificity of 94.7% (20). The ΔSD-Score is calculated by subtracting the wild-type SD-Score from the mutant SD-Score. Splicing patterns of potential splicing variants were predicted using the Rare Disease Data Center (RDDC) online RNA Splicer tool (https://rddc.tsinghua-gd.org/searchmiddle?to=SplitToolModel). The Berkeley Drosophila Genome Project (BDGP, http://www.fruitfly.org) splice site prediction program was employed to predict the generation and/or activation of new donor sites. 3D models of the wild type (WT) and novel mutant enzymes were generated via homology modeling with Phyre 2 (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index).
Minigene splicing assay
To investigate the potential splicing effects of the c.28 + 5G>A mutation, a minigene splicing assay was conducted in vitro. The minigene regions encompassing ABO exon 1, parts of intron 1, and exon 2 were amplified from the gDNA of the proband. Given the large size of intron 1 in the ABO gene, it was divided into two fragments—approximately 0.66 kb at the 5’-end and approximately 0.36 kb at the 3’-end—and these were subsequently constructed into a plasmid (Figure 2A).
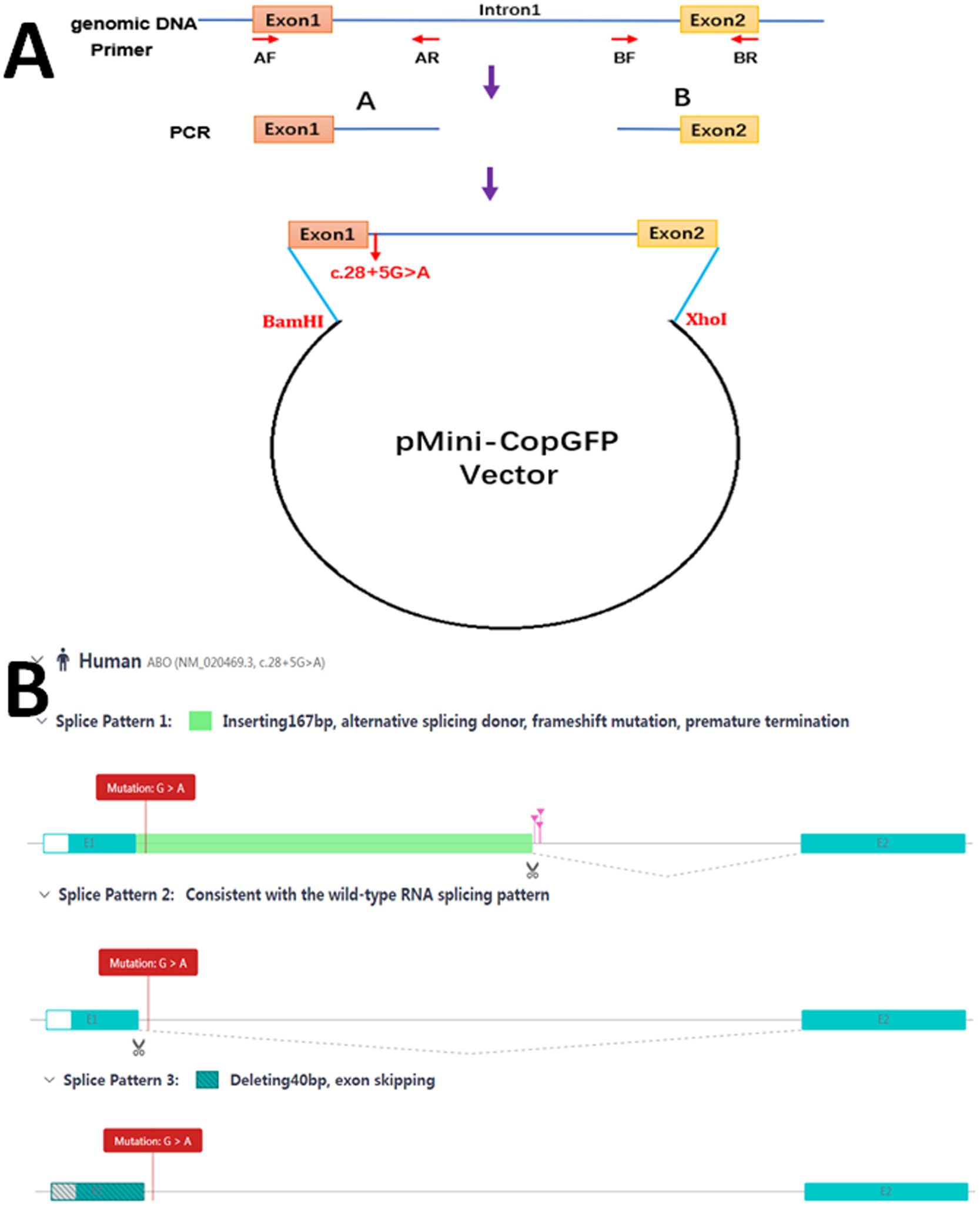
Figure 2. Schematic diagram for the mutation c.28 + 5G>A affecting splicing. (A) Plasmid construction flow chart; (B) The online RNA Splicer tool predicted three splice patterns: splice pattern 1 with 167 bp insertion, splice pattern 2 with the normal wild-type RNA splice, and splice pattern 3 with exon 40 bp deletion.
Two sets of primers, AF/AR and BF/BR (5’-AAGCTTGGTACCGAGCTCGGATCCAGACGCGGAGCCATGGCCGAGGTGTTGC-3’/5’-CACGTCGAGGGCGCCAAGAAAATTAAGGACAGGTGATCCAC-3’ and 5’-TTCTTGGCGCCCTCGACGTGCTCATTTCAGTGTGGTTC-3’/5’-TTAAACGGGCCCTCTAGACTCGAGCCAAACAAGACCAAGACAAGCATTATTA-3’), were developed to amplify the heterozygous c.28 + 5G>A mutation site from the gDNA fragment by seamless cloning (Vazyme Biotech Co., Ltd., Nanjing, China). The PCR products were then recombined and cloned into the two digestion sites (BamHI/XhoI) of the pMini-CopGFP vector (Hitrobio Biotechnology Co., Ltd., Beijing, China). The integrity of the WT and mutant plasmids was validated by Sanger sequencing. Subsequently, the plasmids were transfected into HEK293 cells. Total RNA was extracted from cells cultured for 48 h using a TRIzol reagent (Invitrogen, USA). RT-PCR was performed using the specific primer pair (5’-GGCTAACTAGAGAACCCACTGCTTA-3’ and 5’-CCAAACAAGACCAAGACAAGCATTA-3’). Afterward, the cDNA products were analyzed by 1% agarose gel electrophoresis and further validated through Sanger sequencing.
Results
Serological results
The RBCs of the proband showed no response to anti-A, anti-B, anti-A,B, or anti-A1, but exhibited strong agglutination (4+) when exposed to anti-H. The plasma of the proband demonstrated 1+ intensity agglutination with A1 cells, and 4+ intensity agglutination with B cells. The presence of A antigens on the proband’s RBCs was confirmed by the absorption-elution test (Table 1). The Ael phenotype is characterized RBCs that do not agglutinate with anti-A or anti-A,B antibodies, yet A antigens can be detected through absorption-elution testing. Additionally, the presence of anti-A1 antibodies in the serum further supports the classification of the proband as having an Ael phenotype.
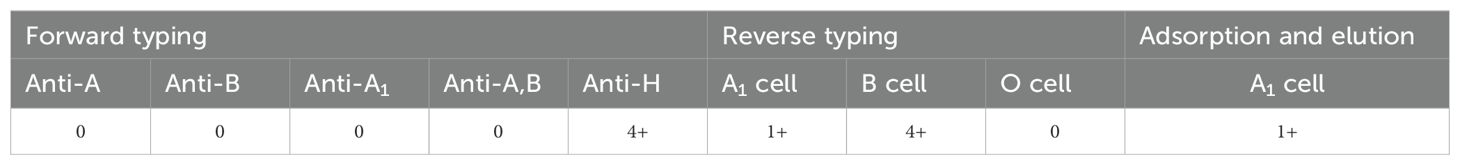
Table 1. Results of serologic grouping.
PacBio sequencing results
Through third-generation PacBio sequencing technology, two haplotypes of the full-length ABO gene were obtained. Table 2 summarizes the sequencing results performed in this study. The proband was found to be heterozygous for the ABO*O.01.02 and a novel ABO*A2.01-like allele. This new allele harbored an additional mutation, c.28 + 5G>A, in intron 1 (Figure 3). No variants were detected in the CBF/NF-Y binding site, proximal promoter, and +5.8-kb site when compared to the normal ABO*A2.01 allele.
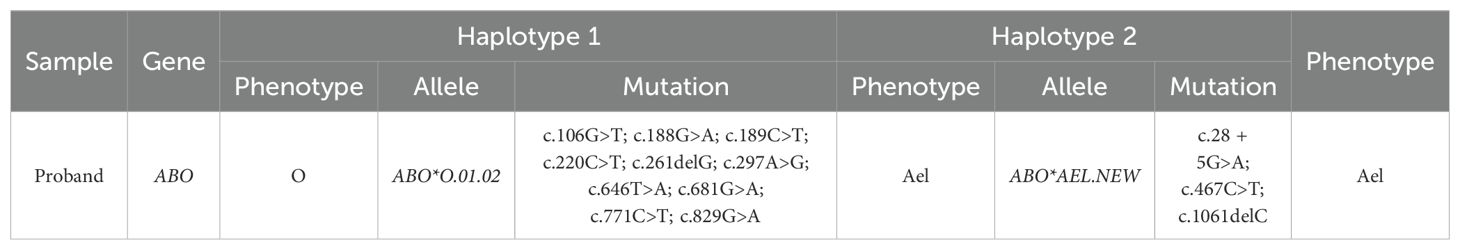
Table 2. PacBio single-molecule real-time sequencing results of ABO gene conducted in this study.
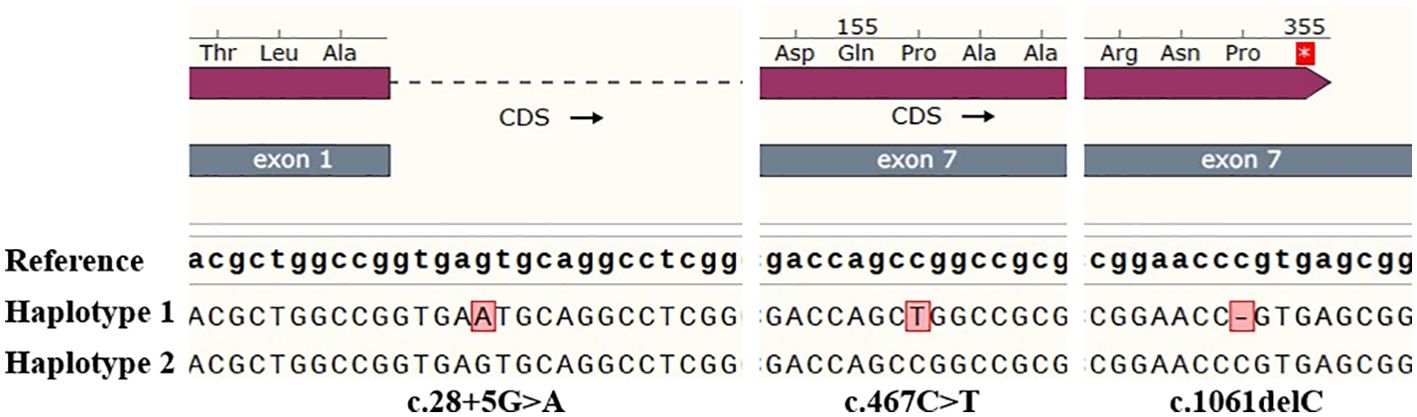
Figure 3. The nucleotide sequences around the mutations c.28 + 5G>A, c.467C>T and c.1061delC of the proband. Analyze through SnapGene v6.0.2 software.
Bioinformatics analysis
A 5’ splice site with a △SD-Score greater than −0.34 is considered not to affect pre-mRNA splicing. Conversely, a mutant site with a △SD-Score less than −0.34 and an SD-Score less than −2.9 indicates abnormal splicing. If the △SD-Score is less than −0.34 but the D-Score is above −2.9, the △Ri value is used. A △Ri value greater than −1.45 suggests normal splicing, whereas a value less than −1.45 indicates abnormal splicing. The splicing prediction consequences of the c.28 + 5G>A variant, as determined using the SD-Score algorithm, are shown in Figure 1B. The △SD-Score and △Ri value for this variant were −2.359 and −3.118, respectively, indicating that the c.28 + 5G>A variant, located at the exon/intron boundary, is likely to cause aberrant splicing. Three potential splice patterns were predicted using the online RNA splicer tool: splice pattern 1 with 167 bp insertion, splice pattern 2 with the normal WT RNA splice, and splice pattern 3 with 40 bp deletion and exon skipping (Figure 2B). Bioinformatics analysis with BDGP showed that the c.28 + 5G>A variant decreased the score of the canonical 5’ donor splice site from 0.99 to 0.00. The predicted new donor sites downstream are presented in Supplementary Figure S1.
Minigene splicing assay
We performed minigene analysis on both the WT and mutant type (MT) carrying the ABO c.28 + 5G>A mutation to further elucidate the nature of the abnormal splicing. Agarose gel electrophoresis of RT-PCR products demonstrated a single band for the WT, corresponding to a product of 181 bp, and two bands for the MT (MT-A: 181 bp and MT-B: 348 bp), with the MT-A band being significantly less intense than the MT-B band (Figure 4A). Sanger sequencing confirmed normal splicing for the WT and MT-A, whereas the MT-B exhibited abnormal splicing (Figure 4B), consistent with splice patterns 1 and 2 predicted by the online RNA Splicer tool and proximate donor site downstream predicted by the BDGP with a score of 0.40. The minigene analysis suggested that the c.28 + 5G>A variant can abolish the intron 1 5’-end canonical donor splice site and activate a cryptic donor splice site downstream of intron 1 in the ABO gene. This results in a 167 bp insertion in the mRNA leading to a truncated glycosyltransferase (p.Lys11Glufs*66) (Figure 4C). Amino acid sequence alignment results are provided in Supplementary Figure S2. Additionally, the crystallization of the WT and mutant glycosyltransferase A are presented (Figure 4D).
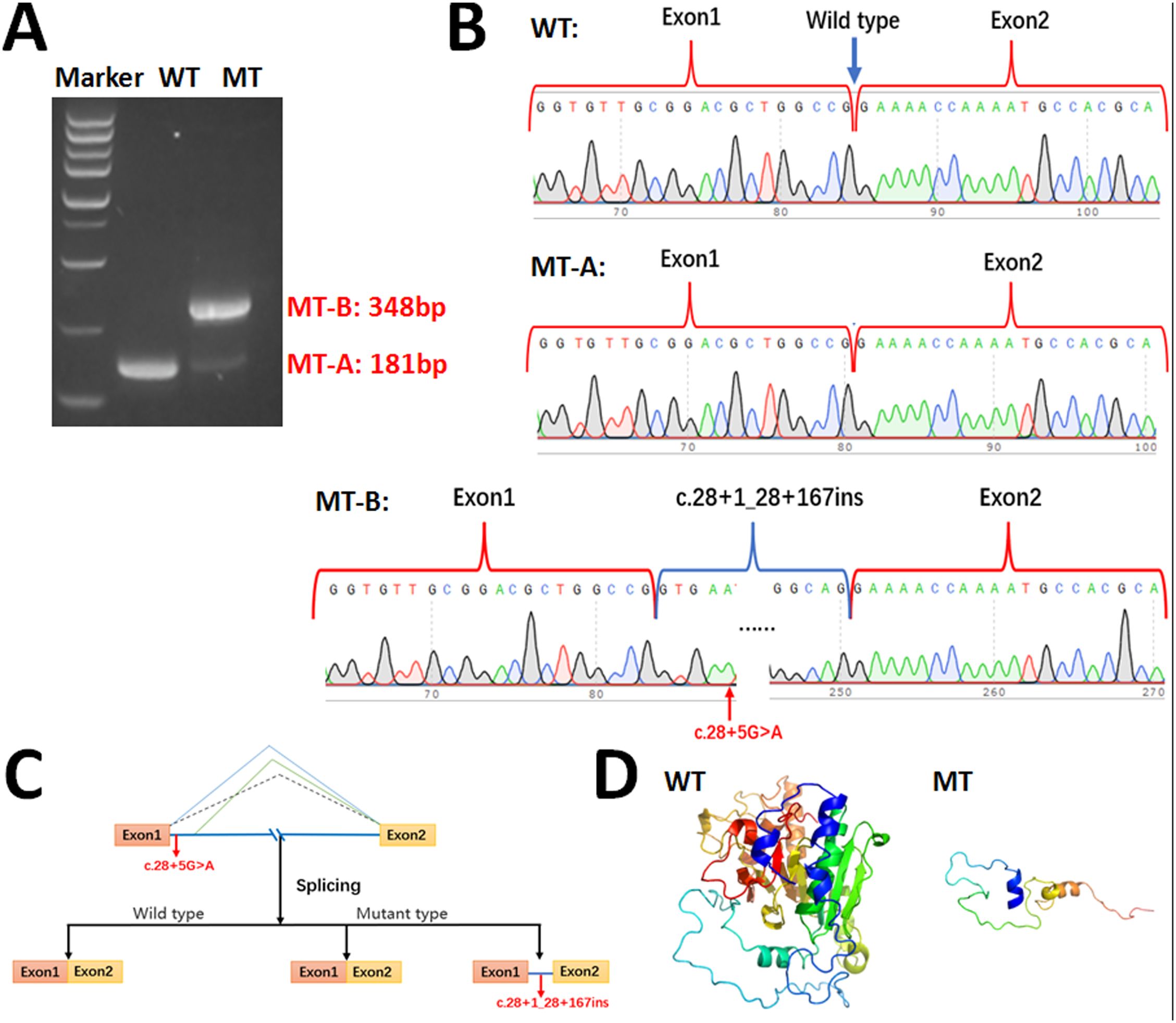
Figure 4. Minigene assay for ABO c.28 + 5G>A variant and schematic diagram of splicing pattern. (A) Gel electrophoresis of RT-PCR revealed a single band for wild type (181bp) and two bands for mutant type (181bp & 348bp). (B) Minigene product sequencing. The wild type minigene formed normal mRNA, but the c.28 + 5G>A variant of ABO caused normal splicing (MT-A) and abnormal splicing (MT-B), which abrogate the intron 1 canonical splice site and lead to activating a cryptic donor splice site downstream of intron 1 in the ABO gene and lead to a 167 bp insertion; (C) Schematic of splicing for ABO c.28 + 5G>A. (D) The wild type and truncated glycosyltransferase A.
Discussion
As of now, only eight AEL alleles, namely ABO*AEL.01–08, have been documented on the ISBT website (https://www.isbtweb.org/resource/001aboalleles.html, accessed Feb 26, 2024). All these alleles possess mutations within exon 7, except for ABO*AEL.04, similar to the novel allele described in this study, which also exhibits mutation at the intronic splicing site.
Pre-mRNA splicing is a fundamental process in eukaryotic gene expression, involving the accurate removal of introns and the linking of exons to produce mature mRNA. The successful completion and regulation of this process hinge on intricate biochemical interactions between cis- and trans-acting elements, including the 5’ and 3’ splice sites. Mutations at these sites can disrupt the authentic exon/intron boundary, modifying the spliceosome’s binding site and leading to abnormal splicing (21). The novel AEL allele identified in the proband, characterized by a mutation at the 5’ splice site of intron 1 (c.28 + 5G>A), was found to be associated with the Ael phenotype. In silico prediction analyses suggested that replacing the G at position c.28 + 5 with A, C, or T would result in altered splicing (Figure 1B).
To elucidate the mechanism underlying the c.28 + 5G>A mutation’s contribution to the Ael phenotype, we performed minigene assays. These experiments demonstrated that the mutation primarily abolishes the canonical donor site, causing a shift in the splice site towards the 3’ end and resulting in premature translation termination at position Leu75. This leads to the production of a truncated glycosyltransferase A lacking the catalytic domain, thereby affecting enzyme function relative to the WT glycosyltransferase A. Concurrently, the MT plasmid appeared to produce a minor amount of the WT transcript through normal splicing. Premature termination codons bearing transcript can be degraded by nonsense-mediated mRNA decay which is generally thought to be a eukaryotic mRNA surveillance pathway (22). However, WT transcript can generate trace amounts of functional glycosyltransferase A. This mechanism may account for the development of the Ael phenotype associated with c.28 + 5G>A. The results of the minigene assays are consistent with the aberrant splicing predicted by the SD-Score algorithm, the splice patterns 1 and 2 predicted by the online RNA Splicer tool, and the proximate donor site (score: 0.40) downstream predicted by the BDGP. Splicing site mutations can lead to exon skipping (23), however, minigene assays did not find evidence that the c.28 + 5G>A mutation causes the skipping of exon 1 (splice pattern 3) in this study.
Although the c.28 + 5G>A mutation has been previously reported (24), it differs from our findings, as it is cis-formed with ABO*B.01 rather than ABO*A2.01, leading to the Bel phenotype. However, their article did not conduct functional experiments to elucidate the mechanism. The ABO*AEL.04 allele has been reported to harbor a mutation at the 5’ splice site in intron 6 (c.374 + 5G>A) which consequently leads to the Ael phenotype (23, 25). This mutation is distinct from our finding of intron retention and instead causes exons skipping. Notably, our discovery of a mutation within the ABO*A2.01 backbone alters the expected A2 phenotype to an Ael phenotype, or more accurately, an A2el phenotype. Given the substantially reduced density of A antigen sites on A2 red blood cells compared to A1 RBCs (26), additional experimental validation is required to ascertain whether the A antigen sites on the A2el RBCs are indeed fewer than those on the Ael RBCs.
Our study offers several advantages. Firstly, we employed third-generation sequencing technology, which has not yet been widely popularized in the field of blood grouping, to capture two haplotypes containing the full-length and flanking regulatory regions of the ABO gene without ignoring mutations outside the coding region. Secondly, we identified a novel AEL allele and provided the first insight into the mechanism by which the c.28 + 5G>A mutation disrupts normal splicing, utilizing minigene technology. The nucleotide sequence of this new AEL allele has been submitted to the GenBank database (accession number: OR995727).
Conclusions
In conclusion, we identified a novel AEL allele in a Chinese male blood donor. This novel allele harbors the c.28 + 5G>A variant within the ABO*A2.01 allele background, which primarily causes abnormal splicing and 167 bp insertion in the mRNA, leading to the production of a truncated glycosyltransferase (p.Lys11Glufs*66).
Data availability statement
The data presented in the study are deposited in the GenBank database, accession number OR995727. The direct link is https://www.ncbi.nlm.nih.gov/nuccore/OR995727.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Dalian Blood Center. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LS: Formal analysis, Investigation, Resources, Writing – original draft. WS: Investigation, Writing – original draft. LZ: Resources, Writing – original draft. LP: Formal analysis, Writing – original draft. YD: Formal analysis, Writing – original draft. NX: Resources, Writing – original draft. SZ: Conceptualization, Writing – review & editing. XL: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors would like to thank the proband for his participation and cooperation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1396426/full#supplementary-material
Supplementary Figure 1 | Prediction results of donor sites using BDGP. (A) Splice site predictions for wild type with donor score cutoff 0.40 (exon/intron boundary shown in larger font). This server runs the NNSPLICE 0.9 version (January 1997) of the splice site predictor. The correlation coefficient (CC) for donor site prediction in an optimized network with one layer of hidden units is 0.855 versus 0.810 for a network with no hidden units; for the acceptor site prediction, the values are 0.824 and 0.750 respectively. At the 5% false positive level, 6% of the real donor sites and 9% of the real acceptor sites are missed. (B) The wild type sequence used to predict the results of donor sites. The nucleotides of exon 1 are shown in uppercase. The initiation codon ATG is underlined and the c.28 + 5G is marked in red. WT, wild type; MT, mutant type.
Supplementary Figure 2 | The amino acid sequence alignment results. The red part represents Helix, and the blue part represents Strand. WT, wild type; MT, mutant type. Images are generated by online web tool Novopro (https://www.novopro.cn/tools/secondary-structure-prediction.html).
References
1. Landsteiner K. Agglutination phenomena of normal human blood. Wien Klin Wochenschr. (1901) 113:768–9.
2. Yamamoto F. Review: ABO blood group system–ABH oligosaccharide antigens, anti-A and anti-B, A and B glycosyltransferases, and ABO genes. Immunohematology. (2004) 20:3–22. doi: 10.21307/immunohematology-2019-418
3. Liu HM, Chen YJ, Chen PS, Lyou JY, Hu HY, Ho YT, et al. A Novel Ael allele derived from a unique 816insG in Exon 7 of the ABO gene. J Formos Med Assoc. (2007) 106:969–74. doi: 10.1016/S0929-6646(08)60070-4
4. Cai X, Jin S, Liu X, Fan L, Lu Q, Wang J, et al. Molecular genetic analysis of ABO blood group variations reveals 29 novel ABO subgroup alleles. Transfusion. (2013) 53:2910–6. doi: 10.1111/trf.2013.53.issue-11pt2
5. Shao LN, Yang YC, Xia YX, Li CX, Zhou SH, Liang XH. Novel missense mutation c.797T>C (p.Met266Thr) gives rise to the rare B(A) phenotype in a Chinese family. Vox Sang. (2024) 119:383–7. doi: 10.1111/vox.v119.4
6. Liang S, Liang YL, Su YQ, Wu F, Peng L, Xu YP. Identification of a novel A allele with a c.36-39delA on the ABO*A1.02 allele. Transfusion. (2021) 61:E75–6. doi: 10.1111/trf.16642
7. He Y, Hong X, Zhang J, He J, Zhu F, Huang H. Analysis of the genomic sequence of ABO allele using next-generation sequencing method. Front Immunol. (2022) 13:814263. doi: 10.3389/fimmu.2022.814263
8. Seltsam A, Hallensleben M, Kollmann A, Blasczyk R. The nature of diversity and diversification at the ABO locus. Blood. (2003) 102:3035–42. doi: 10.1182/blood-2003-03-0955
9. Huang HB, Jin S, Liu X, Wang Z, Lu Q, Fan L, et al. Molecular genetic analysis of weak ABO subgroups in the Chinese population reveals ten novel ABO subgroup alleles. Blood Transfus. (2019) 17:217–22. doi: 10.2450/2018.0091-18
10. Wang C, Tang Y, Zhang P, Xiong L, Chen W, Lv X. Detection and phenotype analysis of a novel Ael blood group allele. Vox Sang. (2024) 119:74–8. doi: 10.1111/vox.v119.1
11. Kominato Y, Tsuchiya T, Hata N, Takizawa H, Yamamoto F. Transcription of human ABO histo-blood group genes is dependent upon binding of transcription factor CBF/NF-Y to minisatellite sequence. J Biol Chem. (1997) 272:25890–8. doi: 10.1074/jbc.272.41.25890
12. Sano R, Nakajima T, Takahashi K, Kubo R, Kominato Y, Tsukada J, et al. Expression of ABO blood-group genes is dependent upon an erythroid cell-specific regulatory element that is deleted in persons with the B(m) phenotype. Blood. (2012) 119:5301–10. doi: 10.1182/blood-2011-10-387167
13. Chen Z, He X. Application of third-generation sequencing in cancer research. Med Rev (Berl). (2021) 1:150–71. doi: 10.1515/mr-2021-0013
14. Sturgeon P, Moore BPL, Weiner W. Notations for two weak A variants: Aend and Ael. Vox Sang. (1964) 9:214–5. doi: 10.1111/j.1423-0410.1964.tb03685.x
15. Hult AK, Olsson ML. Many genetically defined ABO subgroups exhibit characteristic flow cytometric patterns. Transfusion. (2010) 50:308–23. doi: 10.1111/j.1537-2995.2009.02398.x
16. Hansen T, Namork E, Olsson ML, Chester MA, Heier HE. Different genotypes causing indiscernible patterns of A expression on Ael red blood cells as visualized by scanning immunogold electron microscopy. Vox Sang. (1998) 75:47–51. doi: 10.1046/j.1423-0410.1998.7510047.x
17. Heier HE, Namork E, Calkovsk á Z, Sandin R, Kornstad L. Expression of A antigens on erythrocytes of weak blood group A subgroups. Vox Sang. (1994) 66:231–6. doi: 10.1111/j.1423-0410.1994.tb00315.x
18. Denise MH ed. Mordern blood Banking and Transfusion Practices. 5th ed. Philadelphia: F.A.Davis Company (2005) p. 117–8.
19. Watkins WM. Blood group gene specified glycosyltransferases in rare ABO groups and in leukaemia. Rev Fr Transfus Immunohematol. (1978) 21:201–28. doi: 10.1016/S0338-4535(78)80043-9
20. Sahashi K, Masuda A, Matsuura T, Shinmi J, Zhang Z, Takeshima Y, et al. In vitro and in silico analysis reveals an efficient algorithm to predict the splicing consequences of mutations at the 5’ splice sites. Nucleic Acids Res. (2007) 35:5995–6003. doi: 10.1093/nar/gkm647
21. Jian X, Boerwinkle E, Liu X. In silico tools for splicing defect prediction: a survey from the viewpoint of end users. Genet Med. (2014) 16:497–503. doi: 10.1038/gim.2013.176
22. Maquat LE. When cells stop making sense: effects of nonsense codons on RNA metabolism in vertebrate cells. RNA. (1995) 1:453–65.
23. Chen DP, Sun CF, Ning HC, Peng CT, Wang WT, Tseng CP. Genetic and mechanistic evaluation for the weak A phenotype in Ael blood type with IVS6 + 5G>A ABO gene mutation. Vox Sang. (2015) 108:64–71. doi: 10.1111/vox.2014.108.issue-1
24. Hong X, Ying Y, Zhang J, Chen S, Xu X, He J, et al. Six splice site variations, three of them novel, in the ABO gene occurring in nine individuals with ABO subtypes. J Transl Med. (2021) 19:470. doi: 10.1186/s12967-021-03141-5
25. Sun CF, Yu LC, Chen IP, Chou DL, Twu YC, Wang WT, et al. Molecular genetic analysis for the Ael and A3 alleles. Transfusion. (2003) 43:1138–44. doi: 10.1046/j.1537-2995.2003.00500.x
Keywords: novel AEL allele, intron, splice site, minigene, PacBio, third-generation sequencing
Citation: Shao L-N, Song W-Q, Zhou L, Pan L-Z, Duan Y, Xiao N, Zhou S-H and Liang X-H (2024) Characterization of a novel AEL allele harboring a c.28 + 5G>A mutation on the ABO*A2.01 background: a study utilizing PacBio third-generation sequencing and functional assays. Front. Immunol. 15:1396426. doi: 10.3389/fimmu.2024.1396426
Received: 05 March 2024; Accepted: 05 December 2024;
Published: 23 December 2024.
Edited by:
Guirong Wang, Upstate Medical University, United StatesReviewed by:
Dario Balestra, University of Ferrara, ItalyVinzenz Lange, DKMS Life Science Lab GmbH, Germany
Copyright © 2024 Shao, Song, Zhou, Pan, Duan, Xiao, Zhou and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shi-Hang Zhou, enNoc2FpbEAxNjMuY29t; Xiao-Hua Liang, bGlhbmd4aWFvaHVhMTk2OEAxMjYuY29t