 Elan L’Estrange-Stranieri
Elan L’Estrange-Stranieri Timothy A. Gottschalk
Timothy A. Gottschalk Mark D. Wright1
Mark D. Wright1 Margaret L. Hibbs
Margaret L. Hibbs
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 28 June 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1395427
This article is part of the Research Topic Treat-to-target in systemic lupus erythematosus: Cytokine Transduction Pathways in SLE View all 7 articles
Systemic lupus erythematosus (SLE, lupus) is a debilitating, multisystem autoimmune disease that can affect any organ in the body. The disease is characterized by circulating autoantibodies that accumulate in organs and tissues, which triggers an inflammatory response that can cause permanent damage leading to significant morbidity and mortality. Lyn, a member of the Src family of non-receptor protein tyrosine kinases, is highly implicated in SLE as remarkably both mice lacking Lyn or expressing a gain-of-function mutation in Lyn develop spontaneous lupus-like disease due to altered signaling in B lymphocytes and myeloid cells, suggesting its expression or activation state plays a critical role in maintaining tolerance. The past 30 years of research has begun to elucidate the role of Lyn in a duplicitous signaling network of activating and inhibitory immunoreceptors and related targets, including interactions with the interferon regulatory factor family in the toll-like receptor pathway. Gain-of-function mutations in Lyn have now been identified in human cases and like mouse models, cause severe systemic autoinflammation. Studies of Lyn in SLE patients have presented mixed findings, which may reflect the heterogeneity of disease processes in SLE, with impairment or enhancement in Lyn function affecting subsets of SLE patients that may be a means of stratification. In this review, we present an overview of the phosphorylation and protein-binding targets of Lyn in B lymphocytes and myeloid cells, highlighting the structural domains of the protein that are involved in its function, and provide an update on studies of Lyn in SLE patients.
Systemic Lupus Erythematosus (SLE, lupus) is a chronic autoinflammatory disorder characterized by the production of autoantibodies, particularly towards nuclear antigen (anti-nuclear autoantibodies; ANAs) [reviewed by (1)]. ANAs can form immune complexes with self-antigen and deposit in any organ system, causing a diverse range of symptoms. SLE is known as a disease of a thousand faces and can be thought of as a prototypic autoimmune disorder, with underlying disease processes shared by many autoimmune conditions.
SLE reportedly affects one in every 1,000 to 5,000 individuals [reviewed by (2–4)]. Approximately nine times more women are impacted with the condition than men (2–4), with SLE having a higher prevalence, earlier onset, and greater severity in those with non-European ancestry (2, 4). The onset of SLE peaks in young to middle aged women (2, 3), with a later onset typically seen in men (3).
Milder forms of SLE are limited to arthritis and skin presentations, however, this can progress to life-threatening manifestations including renal disease (lupus nephritis; LN), serositis and neurological effects. Renal involvement is the most frequent serious manifestation of SLE, estimated to occur in 40-70% of SLE patients (5, 6), with one in ten LN patients progressing to end-stage renal failure within 5 years (7). Given the early onset of lupus in women, kidney disease is a significant driver of healthcare costs associated with the condition (4).
The first-line treatments for SLE include the long-term use of the antimalarial medication hydroxychloroquine to prevent future disease flares, and glucocorticoids to manage disease activity (8, 9). Other immunosuppressive agents can also be used as induction therapies or as alternatives to glucocorticoids/hydroxychloroquine when disease is refractory, including chemotherapeutics (methotrexate, cyclophosphamide) and anti-transplant-rejection medications (azathioprine, mycophenolate) (8). Achieving full remission without the need for ongoing immunosuppressive therapy is rare, and cumulative use carries significant risk of organ damage including bone disease, metabolic disorders, and retinopathy (8–11).
Advancements in immunosuppressive therapies, such as the introduction of corticosteroids and cyclophosphamide in the 1950s and 1960s, have seen an SLE patient’s prognosis greatly improve from a 10-year survival of 50-60% to 90% today (12, 13). However, there has been a lack of success in therapeutic advancement in the modern era; the 20-year overall survival of SLE patients is 75% (14, 15) and SLE is the tenth leading cause of death in young women in America, and the fifth within the African-American and Hispanic communities (16). Similarly in Australia, SLE has greater prevalence and severity amongst Asian Australians and First Nations people (17). Meta-analyses have shown that the highest increase in mortality risk is for kidney disease, followed by infection and cardiovascular disease (18, 19). An increased risk of certain cancers has also been observed in SLE (20), which is generally thought to be a result of chronic inflammation.
Research into superior therapies for SLE has been marred by a lack success (21–23). However, there have been a number of developments in recent years, including approval of two biologic therapies for SLE: belimumab to neutralize B-cell activating factor (BAFF) (24) and anifrolumab which antagonizes the type I interferon receptor (IFNARI) (25). Nonetheless, the response rate to biologics in conjunction with standard therapy is moderate, being 11% over placebo for belimumab in LN (24) and anifrolumab eliciting a 16.3% response rate in SLE patients without neuropsychiatric manifestations or active LN (25).
Given the modest overall response seen to biologics, there is a clear need to better understand the disease process in SLE to design more effective therapies. However, a remaining challenge in research is the diversity of SLE clinical presentations and underlying molecular causes. A key to superior therapies will therefore lie in our ability to stratify SLE patients for effective disease targeting in a precision medicine approach (26).
Lyn (Lck/yes-related novel tyrosine kinase) is a non-receptor protein tyrosine kinase that plays a unique role in immune cell signaling by propagating both activating and inhibitory pathways. Genetic manipulation studies in mice have illustrated this dichotomy, with reduction or enhancement of Lyn function giving rise to autoinflammatory diseases including lupus-like disease. Studies in SLE patients have also linked Lyn to SLE and related autoinflammatory conditions. Elucidating the mechanistic role of Lyn in SLE can therefore assist in the identification of new molecular targets for SLE therapy and shed light on whether perturbations in Lyn may serve as a biomarker for a precision medicine approach to treatment.
Lyn is a member of the Src family of non-receptor tyrosine kinases (SFKs) (27). SFKs are membrane anchored enzymes that phosphorylate receptors lacking intrinsic kinase activity to initiate signal transduction. Sequence homology has identified eight SFKs in addition to Lyn: Src, Lck, Hck, Fyn, Blk, Fgr, Yes, and Yrk (28). Lyn is most closely related to Hck (29) and is highly conserved across species (30–32). Src, Fyn, and Yes are ubiquitously expressed, whereas Hck, Fgr, Blk, and Lck are expressed only in hematopoietic lineages (33). Lyn is expressed broadly, detectable in hematopoietic stem cells (HSC), B cells, myeloid cells, NK cells, endothelial cells, epithelial cells, and neurons (34, 35). Lyn, however, is not expressed in conventional T cells, being downregulated at the thymocyte double negative (DN) stage of T cell development (Figure 1).
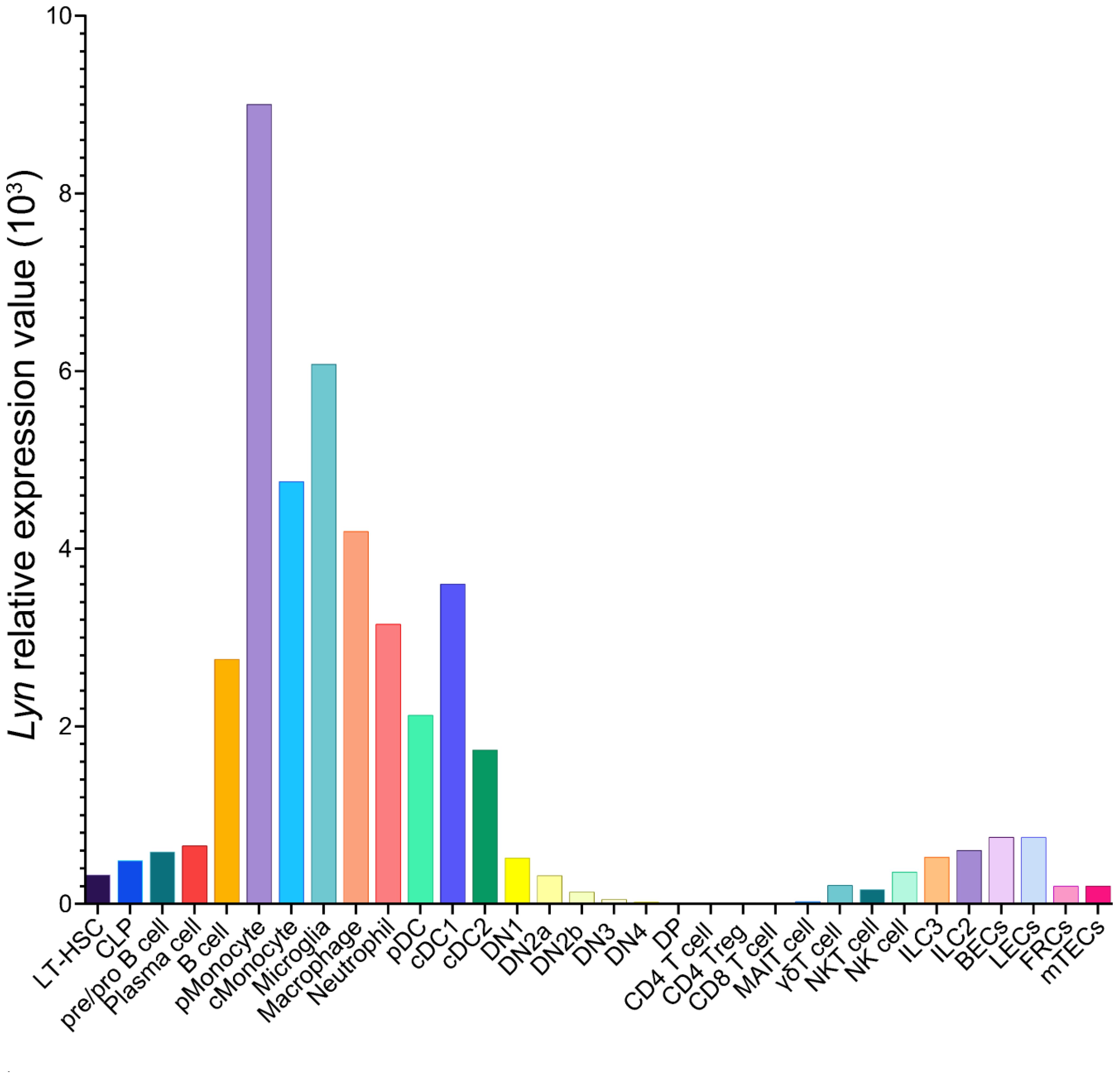
Figure 1 Lyn expression pattern in C57BL/6 mouse cells. Data from the Immunological Genome Project database of bulk RNA-sequencing on sorted cell populations (35). LT-HSC, long-lived hematopoietic stem cell; CLP, common lymphoid progenitor; pMonocyte, patrolling monocyte; cMonocyte, classical monocyte; pDC, plasmacytoid dendritic cell; cDC, conventional dendritic cell; DN, double negative thymocyte; DP, double positive thymocyte; MAIT, mucosal-associated invariant T cell; NK, natural killer; ILC, innate-like cell; BECs, blood endothelial cells; LECs, lymphatic endothelial cells; FRC, fibroblastic reticular cells; mTECs, medullary thymic epithelial cells.
Lyn, as with other SFKs, is a modular protein, containing four Src homology (SH1-4) domains and a unique domain (UD), comprehensively reviewed by others (28, 36) (Figure 2A). Two Lyn isoforms occur due to alternative splicing of Lyn pre-mRNA; a p56Lyn isoform (LynA) contains a 21-amino acid UD insert while the p53Lyn isoform (LynB) lacks this amino acid stretch (32, 39). Lyn is predominantly embedded in the intracellular side of the plasma membrane through the co-translational addition of saturated fatty acids to the N-terminus of Lyn (40, 41) (Figures 2A, B). The C-terminal end of Lyn comprises the protein tyrosine kinase (PTK) domain and a short tail containing the regulatory tyrosine residue (Y508) that determines Lyn’s activation status; when phosphorylated by the kinase Csk, pY508 places Lyn in an inactive closed conformation that blocks substrate access to the PTK (Figure 2B). Lyn is activated by dephosphorylating Y508 by phosphatases such as CD45 and CD148 (42), allowing Lyn to adopt its open conformation to allow for substrate phosphorylation to occur [reviewed by (37)] (Figure 2B).
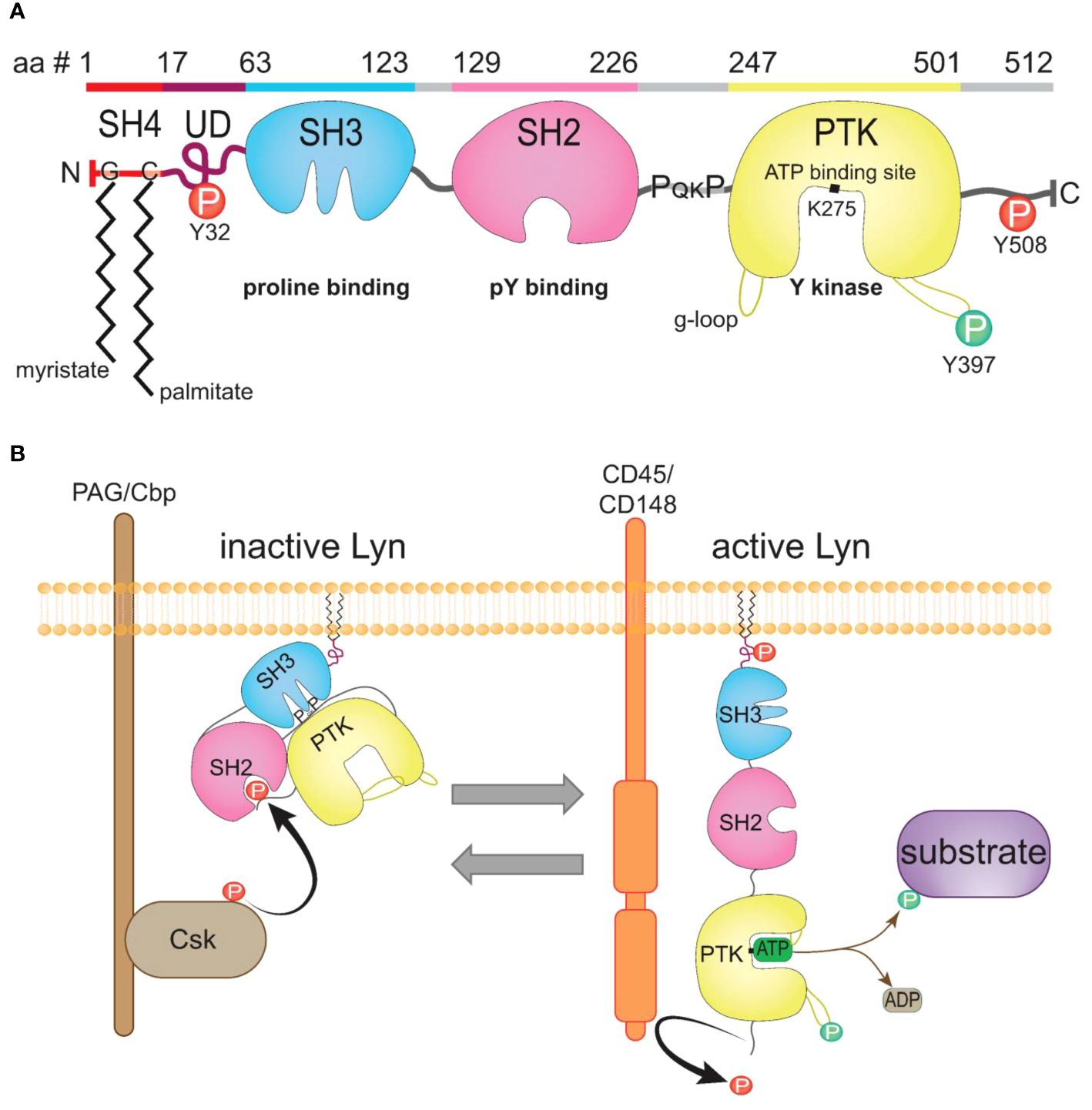
Figure 2 The structure of Lyn kinase and regulation of its activity. (A) The canonical LynA protein sequence; amino acid (aa) residues 1-512, with the domains highlighted and the corresponding schematic of structure depicted underneath (sequence from UniProt [31)]. Inhibitory pY residues (Y32, Y508, red), activating pY residue (Y397, green), the ATP-binding site (K275), myristate addition to residue G2 and palmitate to C3, and the SH3 proline-binding motif (PQKP) within the SH2-kinase linker sequence indicated. (B) Schematic of the active and inactive conformations of Lyn. Lyn is phosphorylated by Csk on Y508, placing Lyn in a closed inactive conformation. Lyn is activated by phosphatases such as CD45, which dephosphorylate Y508 on Lyn, removing the binding site for the SH2 domain, causing Lyn to unwind to an open and active conformation with ready access to its substrates. Lyn auto-phosphorylates Y397 to displace the activation loop from the active site, with the g-loop stabilizing ATP binding to K275 to enable phosphotransfer from ATP to substrate. Figure inspired by (37, 38).
Lyn plays a dual role in immune cell signaling by phosphorylating both activating and inhibitory immunoreceptors. In addition to Lyn’s known role in plasma membrane receptor phosphorylation, Lyn also phosphorylates substrates whilst in the Golgi apparatus during oxidative stress (43). A fraction of endogenous Lyn is also found in the nucleus (44, 45) where it can be activated by DNA damage, causing Lyn to phosphorylate inhibitory and activating nuclear targets (45–47). For activating receptors, the tyrosine motifs phosphorylated by Lyn and other SFKs are termed immunoreceptor tyrosine-based activation motifs (ITAMs) and have the conserved sequence YxxL6-10YxxL (48). Phosphorylated ITAMs act as binding sites for the recruitment and activation of downstream kinases such as Syk in B cells and myeloid cells and ZAP-70 in T cells (49, 50). Docking of Syk triggers its activation through autophosphorylation (51) and by direct phosphorylation via SFKs including Lyn (49). Active Syk initiates the activation of growth/inflammatory signaling pathways, including those regulated by mitogen-activated protein kinase (MAPK), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt).
Although other SFKs can phosphorylate inhibitory receptors (52, 53), Lyn plays a key role in inhibitory signaling whereupon it phosphorylates tyrosine residues in the conserved sequence I/V/LxYxxL/V, known as immunoreceptor tyrosine-based inhibitory motifs (ITIMs) (54). In contrast to ITAMs, ITIM phosphorylation recruits phosphatases to the plasma membrane and triggers their activation, which allows them to dephosphorylate and thereby turn off targets in the pathways activated by ITAM-recruited kinases (38, 54). ITIM-recruited phosphatases contain the SH2 phospho-tyrosine binding domain and include the lipid phosphatases SHIP-1/SHIP-2 and the protein tyrosine phosphatases SHP-1/SHP-2. As with Syk, Lyn also directly phosphorylates SHIP and SHP to cause their activation (55, 56), therefore phosphorylation of ITIM-containing inhibitory receptors typically occurs upon co-ligation with an activating receptor (57). Of note, SHP-1 and SHIP-1 can also be recruited directly to activating receptors to inhibit signaling; SHP-1 has been shown to bind IFNAR1, causing the dephosphorylation of associated Janus kinases and signal transducer and activator of transcription factors (STATs) (58, 59). Similarly, SHIP-1 is recruited to the M-CSF receptor (CSF-R1) to inhibit Akt phosphorylation (60, 61).
Interestingly, Lyn has also been shown to propagate inhibitory signaling from activating receptors, such as the FcαRI, FcγRIIa, and FcγRIIIa, with these signals termed inhibitory ITAM signals (ITAMi) (62–64). This occurs in the absence of receptor aggregation, or with low affinity binding to ligand (62, 64).
Lyn colocalizes with, and phosphorylates ITAMs of numerous activating receptors including Igα/BCR (65), CD19 (66), high affinity FcϵRl complex (67), high affinity FcγRI (CD64) (68, 69), and Fc Receptor Like 5 (FCRL5) (70) (Figure 3). Lyn also colocalizes with and propagates signaling from activating receptors that do not contain an ITAM, including the Epo receptor (74, 75), the G-CSF receptor (76–78), the β-chain of IL-3/5/GM-CSF receptors (79), CD14 (80) [the LPS co-receptor with TLR4 and MD-2 (81)], CD36 (a scavenger receptor implicated in both atherosclerosis and amyloid plaque accumulation) (82), c-Kit (83), IL-2R (84), CD40 (85), and FLT3 (86, 87).
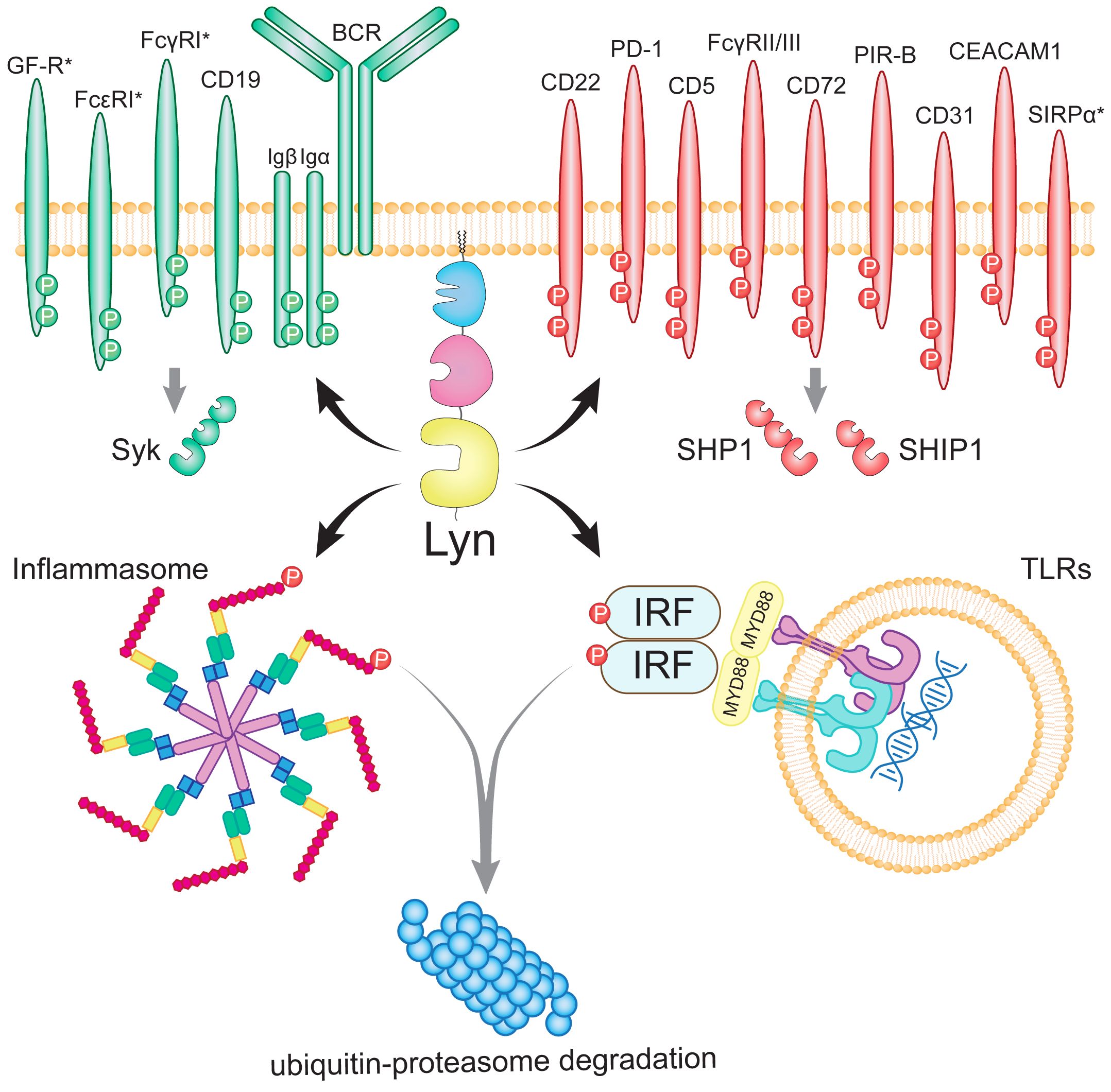
Figure 3 The network of Lyn’s phosphorylation targets in B lymphocytes and myeloid cells. Lyn drives activation by phosphorylating the ITAMs of activating receptors and Syk (indicated by green). Paradoxically, Lyn also promotes inhibition by phosphorylating the ITIMs of inhibitory receptors and SHP-1 and SHIP-1 (indicated by red). Lyn further promotes inhibition by phosphorylating the NLRP3 inflammasome (71) and the IRF family of transcription factors downstream of TLRs to target them for ubiquitin-proteasome degradation (72). Lyn has additionally been suggested to inhibit IRF5 through a protein-binding interaction (73). Myeloid-specific receptors (SIRPα, FcγRI, FcϵR and growth factor receptors; GF-R) are denoted with asterisks. Figure inspired by (37, 38).
The activating role of Lyn is exemplified by studies using mice expressing a constitutively active Lyn mutant, termed Lynup/up mice. The Lynup mutant was generated through a gain-of-function point mutation (p.Y508F), which removes the regulatory tyrosine residue in Lyn, resulting in Lyn being unable to ‘switch off’ and thereby having constitutive kinase activity (88). Lynup/up B cells exhibit chronic activation due to continuous ITAM phosphorylation by Lyn, seen in basal phosphorylation of Syk and PLCγ2, with B cells displaying a hyperactive phenotype and increased calcium influx upon stimulation (88, 89). Due to immune cell hyperactivation, Lynup/up mice develop lupus-like disease including glomerulonephritis (90), as well as severe inflammatory lung disease (91).
Highlighting the duplicitous role of Lyn in signaling, Lynup/up immune cells also show constitutive inhibitory signaling due to ITIM phosphorylation by Lyn, including phosphorylation of the inhibitory receptors CD22, FcγRIIB, SIRPα, PIR-B, as well as SHIP-1 and SHP-1 (90). However, the severe autoimmunity and autoinflammation exhibited by Lynup/up mice demonstrates that chronic Lyn-driven activating signaling overpowers Lyn’s inhibitory-driven role. Yet, how Lyn gain-of-function mutations result in immune hyperactivation despite enhanced inhibitory signaling remains unclear, with further comparative studies of Lyn loss-of-function and Lyn gain-of-function mice warranted to gain additional mechanistic insight.
De novo gain-of-function mutations in LYN have also been reported in four patients, including two missense mutations [p.Y508H (92) and p.Y508F (93)] and two nonsense mutations (p.Y508* and p.Y507*) (93) that lead to a truncated LYN protein, lacking the Y508 regulatory residue. Echoing the phenotype of Lynup/up mice, gain-of-function mutations in patients caused severe systemic inflammation, with disease onset occurring from birth (92, 93). Features of disease included fever, cutaneous neutrophilic vasculitis/atopic dermatitis, arthritis/arthralgia, colitis, and liver damage (92, 93). Patients exhibited elevated circulating proinflammatory cytokines and C-reactive protein (92, 93), as well as the presence of autoantibodies (93). Functional studies revealed enhanced activation of B cells, neutrophils, macrophages, and endothelial cells (93). Patients were treated with anti-IL-1β, anti-TNFα, as well as the SFK kinase inhibitor dasatinib, which improved systemic inflammation (92, 93).
Enhanced Lyn kinase activity has also been observed in a truncated N-terminal mutant of Lyn (LynΔN) that forms during apoptosis via direct cleavage by executioner caspases, relocating Lyn to the cytosol and nucleus (94–96). LynΔN is proinflammatory, with LynΔN/ΔN mice developing a severe psoriasis-like disease that is dependent on TNFα and inflammatory caspase induction of IL-1β and IL-18 activation (95, 97). Biopsies of human psoriatic skin lesions similarly show increased expression of caspases and LynΔN, implicating cleaved Lyn in human psoriasis (97).
Of the two Lyn isoforms, LynA has been implicated as having a greater role in activation. The UD insert in LynA contains a tyrosine residue (Y32) (Figure 2A) that is phosphorylated by active Lyn and Hck in trans (98), as well as by the epidermal growth factor receptor in epithelial cells (99). Phosphorylation of Y32 targets LynA for polyubiquitination by c-Cbl, causing rapid LynA degradation following activation (98), which is consistent with reports of reduced LynA levels after B cell activation (39). The rapid degradation of LynA has been shown to prevent Lyn-driven hyperactivating signaling in macrophages (100). This can be fine-tuned by increasing Lyn protein levels by inducing Lyn transcription with IFNγ or GM-CSF priming, which compensates for the reduction in LynA, leading to enhanced Lyn-driven macrophage activation to subsequent stimulation (100). The divergent function of the isoforms has been further explored in vivo by the Freedman group through the generation of mice expressing a single isoform; either LynA or LynB (101). This work demonstrated that the co-expression of both isoforms is required to prevent lupus-like disease; however, LynB-deficient mice developed more severe autoimmune pathology compared to LynA-deficient mice, with greater incidence of ANA detection and severe glomerulonephritis (101). This suggests that LynB may play a greater role in inhibitory signaling, and LynA may be more important for activating signaling (101). This is further supported by reports showing that the LynA isoform is overexpressed in cancerous cells, implying a dominant role for LynA in activation (99, 102).
Generally, the role of Lyn in activating signaling can be compensated for by other SFKs, whereas its function in inhibitory receptor signaling in B lymphocytes and myeloid cells is non-redundant, leading to a loss of immune cell inhibition and resultant lupus-like disease in Lyn-deficient mice (Lyn-/-) (103, 104). Lyn-/- mice are a well-studied model of lupus, exhibiting an age-dependent increase in pathogenic autoreactive antibodies including anti-dsDNA and anti-Sm IgG2b/c (105), IgA (106), and to a lesser extent IgE (107, 108). This promotes pathogenic immune complex deposition in the glomeruli, leading to recruitment of complement factor 3 deposition which drives inflammatory immune cell infiltration, culminating in glomerulonephritis. At the cellular level, Lyn deficiency results in hyperactive B cells to BCR crosslinking (109) and myeloid cells with enhanced TLR signaling (73, 110–112), integrin signaling (113, 114) and growth factor sensitivity (115).
B cells are necessary for autoimmune pathology in Lyn-/- mice by generating class-switched autoantibodies, as the disease is abrogated by a block in B cell development (Lyn-/-μMT-/-) (106, 116). Autoimmune pathology is also T cell-dependent, as Lyn-/- mice with an impairment in T cell development (Lyn-/-TCRβ-/-TCRδ-/-) or mice lacking the SAP signaling adaptor protein (Lyn-/-SAP-/-), which is critical for T cell-B cell interactions, both show attenuated production of anti-nuclear IgG autoantibodies (117). Given that T cells do not express Lyn, they are believed to be intrinsically unaffected in Lyn-/- mice, with their hyperactivation driven by other immune compartments and the inflammatory milieu. Supporting this, lethally irradiated Lyn+/+ mice reconstituted with mixed Lyn-/-Rag-/- and Lyn+/+ bone marrow exhibit T cell activation and autoimmune pathology, and adoptive transfer of Lyn+/+ T cells into aged Lyn-/- mice results in their effector memory differentiation and IFNγ production on par with host Lyn-/- T cells (111).
The autoimmune pathology seen in the global Lyn-deficient mice can be recapitulated by conditional deficiency of Lyn in B cells (Cd79a-cre x Lynflox/flox) (105), myeloid cells (Lyn−/−Rag−/−- Lyn+/+ chimera) (111), as well as CD11c+ cells (CD11c-cre x Lynflox/flox), which encompasses dendritic cells, age-associated B cells, type I innate lymphoid cells, macrophages, and patrolling monocytes, with the disease in fact being more severe in CD11c-cre x Lynflox/flox mice (112). Reflecting the inflammatory nature of the disease, pathology is dependent on inflammatory cytokines and is attenuated by IL-6 deficiency (106), IFNγ deficiency (111), IFNAR-1 deficiency (118), or treatment with neutralizing anti-BAFF antibody (111). Disease has repeatedly been shown to depend on toll-like receptor (TLR) signaling, as knockout of the MyD88 TLR adaptor protein abrogates disease (105, 112, 117, 119), as does disruption of the pattern recognition receptor adaptor protein CARD9 (120).
Lyn is also a haploinsufficient gene, with a loss-of-function in one allele (Lyn+/- mice) sufficient to cause autoimmune disease with age (121). Disease in Lyn+/- mice can also be accelerated to levels comparable to Lyn-/- mice by concordant loss-of-function of one allele of SHP-1 (Lyn+/−Mev+/−) or one allele of SHIP-1 (Lyn+/−SHIP-1+/−) (121), reflecting the polygenic additive model of SLE heritability.
A growing list of inhibitory targets of Lyn, including ITIM-containing receptors, have been identified and have themselves been implicated in SLE and autoimmune pathology (Figure 3). The collective loss of inhibitory signaling emanating from these receptors upon Lyn deficiency provides a mechanistic framework for the Lyn-/- autoimmune phenotype, which is largely driven by autoreactive B cells and hyperactive myeloid cells.
CD22 is expressed by B cells and is a member of the sialic-acid-binding immunoglobulin-type lectin (Siglec) family, which bind sialic acid on glycoproteins and lipids (122). Sialic acids are ubiquitous on vertebrate cells, and therefore act as a marker of self (123). Lyn is required for the ITIM phosphorylation of CD22 in order to recruit SHP-1 (124, 125) and to a lesser extent SHIP-1 (126). Co-ligation of the BCR and CD22 sends a ‘binding to self’ signal and therefore raises the activation threshold (127). Siglec-G is another B cell-specific Siglec that is believed to be similarly phosphorylated by Lyn (128, 129). CD22 deficiency alone causes an age-related expansion of autoreactive B cells without pathology (130), whilst double deficiency of both CD22 and Siglec-G results in glomerulonephritis (131). Sequence variation screening in SLE identified several substitutions in CD22 that were weakly associated with SLE (132), although this is not supported by genome-wide association studies (GWAS) (127). However, higher CD22 expression on circulating B cells has been associated with remission and low disease activity in SLE patients (133, 134).
Programmed cell death receptor-1 (PD-1) is an ITIM-bearing receptor that is expressed by B cells, activated T cells and T follicular helper cells (135). PD-1 binds to PD-L1 and PD-L2 on antigen-presenting cells (APCs), endothelial cells and epithelial cells (135) and upon co-ligation with the BCR, is phosphorylated by Lyn to induce the activation of SHP-2 (136). PD-1 inhibits B cell activation to CpG (TLR9 agonist) and anti-IgM stimulation (137), and T cells to TCR binding as well as modulating autoreactive T cell migration (135). PD-1-deficient mice develop SLE-like glomerulonephritis (138) with a number of studies identifying aberrant PD-1 and PD-L1/2 expression in SLE patients (139).
CD5 is a member of the Scavenger Receptor Cysteine-Rich family and is an ITIM-containing receptor constitutively expressed by mature T cells (140), anergic autoreactive B cells (141), a subset of IL-10-producing regulatory B cells (142, 143), and B1a cells. CD5 has been demonstrated to bind itself in cis and trans (144), as well as to the common inhibitory receptor, CD72 (145). B1 cells are an innate-like B cell population that have a degree of self-reactivity, producing the majority of baseline circulating IgM and IgA antibodies and, although they have protective functions, are implicated in autoimmune pathology (146, 147). CD5 is phosphorylated by Lyn in B1 cells, leading to the recruitment and activation of SHP-1 to negatively regulate BCR signaling and promote B1 cell apoptosis upon BCR cross-linking (148, 149). B1a cell numbers are expanded in Lynup/up mice (90) and Lyn-/- mice (105), likely reflecting perturbed CD5 signaling in Lyn-mutant mice.
FcγRIIb is the only inhibitory member of the FcγR family, which are receptors that bind the constant region of IgG (150). FcγRIIb contains a single ITIM motif that is phosphorylated by Lyn and recruits SHIP-1 (109, 151). B cells, plasma cells, plasmacytoid dendritic cells (pDCs) (152), neurons (153) and some memory CD8+ T cells (154, 155) express the FcγRIIb only, whereas myeloid cells also express the activating FcγRs (FcγRI, FcγRIII, FcγRIV) in addition to FcγRIIb (150, 156). FcγRIIb inhibits B cells upon co-ligation with the BCR, and prevents the hyperactivation of myeloid cells to immune complex binding (156). In pDCs, FcγRIIb prevents viral-immune complex uptake from triggering IFNα production (157), whilst in conventional dendritic cells (cDCs), FcγRIIb prevents spontaneous maturation in response to sterile immune complexes (158–160). Given its far reaching inhibitory functions, it is perhaps unsurprising that FcγRIIb-deficient mice develop severe lupus-like nephritis, with 60% mortality by 9-months-of-age (161). The overexpression of FcγRIIb has also been shown to ameliorate disease in other lupus models (162, 163), with FCGR2B being a strong susceptibility gene in SLE (164–166). Given the broad functions of FcγRIIb, loss of signaling emanating from this receptor in Lyn-deficient mice likely has wide-ranging effects contributing to the Lyn-/- phenotype.
Paired immunoglobulin Receptor-B (PIR-B) is an ITIM-containing receptor expressed in B cells, myeloid cells, and neurons, that binds to MHC-I in cis and trans (167). Lyn constitutively phosphorylates PIR-B to recruit and activate SHP-1 (88, 168) but additionally phosphorylates PIR-B in response to TLR9 stimulation with CpG-B (169). PIR-B has broad-ranging inhibitory functions, including negatively regulating TLR-induced B1 cell proliferation, B2 cell proliferation to BCR cross-linking (170), and myeloid cell integrin (171) and chemokine signaling (52), as well as proinflammatory macrophage polarization (172). PIR-B-deficient mice do not develop spontaneous autoimmune disease (169, 170); however, PIR-B deficiency on the C57BL/6-Faslpr background (an autoimmune susceptibility mutant), results in glomerulonephritis associated with enhanced production of rheumatoid factor autoantibodies by B1 cells (169). In DCs, PIR-B deficiency results in an immature phenotype (170, 173) and Th2-skewed responses (170); however, PIR-B-deficiency also enhances DC cross-presentation to cytotoxic T cells (173), and pDC production of IFNα to TLR9 stimulation with CpG-A (174). Therefore, in addition to promoting the hyperactivation of B lymphocytes and myeloid cells, diminished PIR-B signaling in Lyn-/- mice will also impact the DC compartment.
CD72 is a C-type lectin-like domain receptor expressed by B cells, pDCs, cDC2s, macrophages, and NK cells (35, 57, 175) and binds the lupus self-antigen ribonucleic protein Sm (176). The ITIM of CD72 is phosphorylated by Lyn and recruits SHP-1 to inhibit B cell activation upon co-ligation with the BCR (176, 177). CD72-deficient mice develop a spontaneous lupus-like disease with age (178). Further, a CD72 mutant with impaired ability to bind Sm/RNP exacerbates lupus-like disease on a susceptible background (176, 179). CD72 SNPs have also been associated with SLE (180), and B cells from SLE patients show reduced CD72 expression which correlates with disease activity (181, 182). CD72 also binds free CD100 (Semaphorin-4D), which is expressed ubiquitously, and is cleaved from the cell surface during activation, with soluble CD100 inhibiting myeloid cell activation through CD72 binding (175).
NLRP3 inflammasomes are multi-protein complexes that assemble in the cytoplasm in response to a wide range of stimuli and induce the maturation of caspase-1, leading to activation of the proinflammatory cytokines IL-18 and IL-1β in B cells, myeloid cells, and epithelial cells (183, 184). Lyn has been shown to phosphorylate NLRP3 causing its ubiquitination and degradation (Figure 3), with Lyn-deficient macrophages hyperproducing IL-1β (71). Further, Lyn activation downstream of BCR cross-linking also attenuates inflammasome formation (185). Impaired inhibition of NLRP3 in Lyn-/- mice is therefore likely a key contributor to the autoimmune pathology that develops.
SIRPα is a receptor of the signal regulatory protein (SIRP) family, being the only ITIM-bearing member (186), and is expressed on monocytes, macrophages, microglia, neutrophils, NK cells, neurons, pDCs and cDC2s (35, 186, 187). SIRPα is phosphorylated by Lyn to recruit SHP-1 and SHP-2 (88, 188, 189). Upon ligation with CD47, which is an integrin-associated protein expressed by all cells that acts as a marker of ‘self’, SIRPα sends a ‘don’t-eat-me’ signal to phagocytes (186), particularly in the context of inflammation (190, 191). SIRPα-deficient mice do not exhibit an overt phenotype at the steady-state (190). However, SIRPα suppresses severe systemic inflammation upon challenge with TLR-agonists and cytokines by attenuating macrophage activation (191). Whilst SIRPα has been shown to suppress inflammation, the role of SIRPα in autoimmunity is somewhat mixed, as SIRPα promotes DC priming of autoreactive T cells in collagen-induced arthritis (192), experimental autoimmune encephalomyelitis (193), and nonobese diabetic mice (194); however, SIRPα agonism conversely suppresses monocyte and neutrophil infiltration in experimental arthritis and colitis (195).
Given the role of Lyn in CD22 and Siglec-G ITIM phosphorylation, it is likely that Lyn is also implicated in the phosphorylation of other Siglec receptors. Siglec-F is an ITIM-containing receptor expressed by eosinophils, alveolar macrophages, microglia (196) as well as allergen-induced CD4+ T cells and lung stroma (197). Siglec-F is pro-apoptotic in eosinophils, with Siglec-F-deficient mice exhibiting eosinophilia upon allergen induction (197). Similarly, Lyn is also pro-apoptotic in eosinophils, with Lyn-/- mice exhibiting peritoneal eosinophilia (108) and lung eosinophilia upon ovalbumin challenge (198), and the treatment of eosinophils with antisense oligos to Lyn inhibits apoptosis (199). Furthermore, studies have implicated Lyn in microglial Siglec-F signaling, as upregulation of Siglec-F on microglia during neurodegenerative disease models is associated with increased activation of Lyn (196). However in eosinophils, Lyn deficiency does not perturb apoptosis to Siglec-F agonism compared to WT eosinophils (200). Therefore, whilst Lyn may be implicated in Siglec-F signaling, the details of Lyn’s involvement have not been elucidated.
Siglec-H is expressed by pDCs, microglia, DC progenitors, as well as intracellularly by marginal zone macrophages (201, 202) and triggers endocytosis upon ligation (201, 203). Siglec-H has been shown to inhibit pDC activation and IFNα production (204–206). Interestingly, Siglec-H deficiency outside of pDCs is sufficient to cause IFNα hyperproduction, as Siglec-H-deficient mice depleted of pDCs still hyperproduce IFNα during viral challenge (202). Siglec-H deficiency in mice results in mild autoimmune disease, and early infection by murine cytomegalovirus causes the development of severe lupus-like glomerulonephritis, which is dependent on IFNAR1 (206).
Unlike other Siglec receptors, Siglec-H does not contain an ITIM in its cytoplasmic tail (123). Instead, Siglec-H recruits DAP12, an ITAM-containing adaptor protein (203) that can be phosphorylated by SFKs (207). Given that Lyn-/- pDCs hyperproduce IFN-I in response to CpG (73, 112), it is possible that Lyn is involved in Siglec-H signaling, and that impaired Siglec-H signaling contributes to hyperproduction of IFN-I in Lyn-/- mice. However, whilst the SFK Hck has been ruled out as being involved in Siglec-H signaling (208), involvement of Lyn in Siglec-H signaling has not yet been assessed.
Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) is an Ig-like superfamily receptor which is highly expressed on endothelial cells towards the tight junctions, and to a lesser degree on most leukocytes and platelets (35, 209). PECAM-1 binds itself in trans, in addition to numerous other identified ligands, and is an ITIM-bearing receptor (209, 210). Lyn is required for the phosphorylation of PECAM-1 to recruit SHP-1/SHP-2 and phospholipase Cγ1 (210, 211). PECAM-1 is involved in leukocyte recruitment through homophilic binding between endothelial cells and leukocytes, initiating leukocyte diapedesis, which is largely independent of PECAM-1’s cytoplasmic signaling (210). PECAM-1 has also been shown to inhibit FcϵR signaling in mast cells (212), as well as FcγR (213) and TLR4 signaling (214) in human monocytes and macrophages. However, it is less clear whether the inhibitory function of PECAM-1 is recapitulated in mouse macrophages (215). PECAM-1 deficiency also causes hyperactivation of B cells to BCR cross-linking, with PECAM-1-deficient mice exhibiting reduced B2 cell numbers in the periphery and increased peritoneal B1 cells, as well autoantibody production, culminating in glomerulonephritis (216) – all features shared by Lyn-/- mice. Interestingly, Lyn has been implicated in strengthening epithelial barrier integrity (217), which may in part be through propagating PECAM-1 signaling, as PECAM-1 is required for maintaining epithelial barrier integrity (210, 218).
Carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) is the only ITIM-bearing member of the CEACAM family, and while it is constitutively expressed on epithelial cells, it is upregulated by activated leukocytes and endothelial cells (219). CEACAM-1 primarily binds itself in cis and trans, and acts as an inhibitory co-receptor, bringing phosphatases SHP-1/SHP-2 to the immune synapse to inhibit activation, and is important in preventing neutrophil, monocyte, B, T, and NK cell hyperactivation (219, 220). Lyn has been shown to phosphorylate the ITIM in CEACAM1 in myeloid cells (221, 222) and is important in myelopoiesis, with CEACAM1-/- mice having pronounced neutrophilia due to hyperresponsiveness to G-CSF (223). CEACAM1 deficiency also causes a reduction in Ly6C+ monocyte numbers during infection (224). Both neutrophilia and a reduction of Ly6C+ monocytes are features of Lyn-/- mice (225, 226), suggesting that perturbed CEACAM1 potentially contributes to the phenotype of Lyn-deficient mice.
In addition to Lyn’s tyrosine kinase function, there is also evidence that Lyn has a kinase-independent role through inhibitory protein-binding interactions.
Two Lyn mutants with reduced or no detectable kinase activity have been generated through ENU mutagenesis (227, 228). The p.T410K mutation destabilizes the activation loop, causing no detectable Lyn kinase activity, conceivably by preventing displacement of Lyn’s activation loop and thereby blocking substrate access to the active site (227). The p.E260G (228) mutation destabilizes the g-loop and the entire kinase domain, resulting in significantly reduced kinase activity (228, 229). LynT410K/T410K and LynE260G/E260G B cells exhibit hyper-responsiveness to BCR cross-linking on par with Lyn-/- B cells (227, 228). However, LynT410K/T410K mice were reported to have attenuated autoimmune pathology (227), while LynE260G/E260G mice have a delayed onset of pathology compared to Lyn-/- mice (228), indicating that Lyn has undefined inhibitory functions independent of its kinase activity in suppressing autoimmune pathology in vivo. However, the modifications in LynT410K and LynE260G are not true kinase-dead mutations as they occur outside of the catalytic site. These mutations have the potential to alter substrate-binding specificity (73) while retaining residual kinase functionality (229). Therefore, characterizing mice expressing an authentic kinase-dead mutation that affects the ATP-binding residue (p.K275X) is necessary to assess the true protein-binding functions of Lyn in vivo. The Lyn mutant p.K275X has been studied in vitro (44, 73, 98); however, mice expressing Lyn p.K275X have not yet been reported.
A mechanistic description of an inhibitory protein-binding interaction with Lyn and the interferon regulatory factor 5 (IRF5) transcription factor was provided by Ban et al. in 2016 (73). IRF5 induces the transcription of proinflammatory cytokines including IL-6, IL-12, and IFN-I and is strongly implicated in SLE pathology, being routinely identified in GWAS (230). The alleles of IRF5 associated with SLE induce the hyperactivation of IRF5 and cause spontaneous lupus-like disease in mice (231). Lyn was shown to bind IRF5 upon TLR7/9 stimulation downstream of the Myddosome oligomeric signaling complex (73). Lyn binding inhibited IRF5 activation by blocking access to IRF5 by the kinase IKKβ and the ubiquitin ligase TARF6 (73). This study showed that the kinase function of Lyn was dispensable for IRF5 inhibition, providing a potential kinase-independent mechanism that may explain the reduced pathology seen LynT410K/T410K and LynE260G/E260G mice. In further support of this, knockout of IRF5 was sufficient to attenuate TLR-induced cytokine production from DCs in vitro (Lyn-/-IRF5-/-), and monoallelic loss of IRF5 was sufficient to abrogate glomerulonephritis in vivo (Lyn-/-IRF5+/-) (73, 118), likely through suppressing transcription of proinflammatory cytokines, IFN-I, and oxidative phosphorylation pathway genes (118). This work also implicated a key role for this inhibitory function in the DC compartment, although analysis of DCs in vivo from Lyn-/- mice was not performed (73).
Later studies have, however, challenged the protein-binding inhibition of IRF5 by Lyn, and instead showed that Lyn inhibits the IRF family (IRF1, IRF5, IRF7, and IRF8) by phosphorylating a conserved tyrosine residue (p.Y118 in IRF5), which inhibits their activity by targeting IRFs for ubiquitination and proteasomal degradation (72) (Figure 3). Further conflicting findings have suggested that Lyn instead promotes IRF5 activation and nuclear translocation (232). Given the contradictory literature, further work is required to clarify the role of Lyn in relation to the IRF family, and whether this interaction is a protein-binding interaction or a kinase interaction.
Relative to the expansive number of experimental animal studies focused on Lyn, the role of Lyn in SLE patient disease has been under-researched; however, Lyn has similarly been implicated in human SLE disease.
Multiple LYN single nucleotide polymorphisms (SNPs) have been associated with SLE through GWAS. The SNPs rs7829816 and rs2667978 were first identified as being protective in SLE development in women of European descent in a 2008 GWAS, with an overall odds ratio of 0.77 and 0.81 respectively, although an association was not seen across all data sets analyzed (233). A replication study that analyzed 90 LYN SNPs in 2009 similarly detected associations for rs7829816 and rs2667978 in a pooled analysis of women of European ancestry, as well as a number of other LYN SNPs, though at a greatly reduced level of significance (234). Instead, this later study identified a stronger association with another SNP, rs6983130, which was associated with SLE with hematological presentations and the presence of autoantibodies including anti-dsDNA and anti-Sm (234). This study did not find an association for LYN in African-American and Korean populations, although these cohorts had a lower power of detection (234). A large-scale GWAS and meta-analysis conducted in 2015 on individuals of European descent similarly identified an association with the LYN SNP rs2667978 and SLE, although a strict genome-wide level of significance was not met (235). Similarly, a 2018 Spanish GWAS and meta-analysis of European descent individuals found an association between another LYN SNP rs17812659 and SLE, although again short of genome-wide level of significance (236). Collectively, GWAS have provided evidence for a genetic role of LYN in SLE, although this is weaker compared to routinely identified genes. However, all identified LYN SNPs reside in non-coding regions of the LYN gene without functional annotations, making implications of LYN SNP-associations with SLE unclear.
Studies have also investigated perturbations in LYN expression in SLE patients. Reduced LYN protein levels have been found in resting and activated circulating B cells in the majority of SLE patients, which was stable over time and independent of disease activity (237, 238). This was associated with reduced LYN mRNA expression (237, 239) as well as increased ubiquitination of the LYN protein (238), suggesting multiple mechanisms for reduced LYN levels in SLE B cells [reviewed by (240)]. These findings indicate that reduced Lyn activity through under-expression of Lyn may contribute to SLE pathogenesis in a manner similar to the disease process of Lyn+/- mice (121). In support of this, SNPs conferring elevated expression of CSK, the negative regulator of Lyn activity, have been identified in SLE patients and cause enhanced Lyn kinase inhibition and associated B cell hyperactivation (241). However, the aforementioned studies of LYN in SLE patients have focused on B lymphocytes, whilst myeloid cells were omitted from analyses (237–239). Therefore, it is unclear whether the status of LYN expression in SLE B lymphocytes is representative of LYN perturbations across the entire immune system in SLE; an important unanswered question given the critical role of Lyn in regulating activation of the myeloid compartment.
Recent RNA-sequencing and microarray studies in SLE patients have also contradicted earlier findings, instead reporting increased LYN mRNA expression in SLE patients. RNA-sequencing of antibody-secreting cells from SLE patients has revealed upregulated LYN expression compared to healthy controls (242). Similarly, a bulk RNA-sequencing analysis of SLE patient whole blood by Panousis et al. (2019) identified LYN as a differentially upregulated gene (243). Further, a longitudinal microarray analysis of pediatric SLE patient blood by Banchereau et al. (2016) linked increased LYN expression to disease activity in SLE patients, with expression quantitative trait loci analysis identifying seven SNPs in neighboring genes associated with the change in LYN expression (244). The baseline whole blood microarray analysis from SLE patients enrolled in the tabalumab (anti-BAFF) phase III trials also linked increased LYN expression to disease activity, IFN-I signature expression, and anti-dsDNA antibodies through weighted gene co-expression network analysis, identifying LYN in the ‘inflammatory response’ module (245). As previous work has shown that inflammatory cytokines induce the upregulation of Lyn in mouse macrophages to cause activation priming (100, 246), it is conceivable that SLE patients may exhibit augmented LYN expression, given the chronic inflammatory insult characteristic of SLE. In humans, LYN expression has also been found to be induced in autoimmune patient B cells by the use of TNFα blockers, resulting in enhanced Lyn activity and B cell hyperactivity, which was postulated to underlie the autoimmune syndrome that develops in a subset of TNFα-antagonist treated patients (247). However, SLE transcriptomic studies have not investigated LYN at the protein level and, therefore, LYN overexpression in SLE patients is yet to be substantiated.
Extensive studies in mice have illustrated the central role of Lyn in maintaining immune cell signaling balance by dualistically propagating both activating and inhibitory pathways. Beyond mouse models, there is a need to expand our understanding of LYN’s role in human SLE disease. Given Lyn can be pathogenic through both overexpression and under-expression, it is of benefit to further study the function of LYN in SLE disease. It is possible that perturbations in LYN may be cell type-specific and affect subsets of SLE patients. Potential therefore exists in repurposing drugs such as the Lyn inhibitor dasatinib (93) or the Lyn activator tolimidone (248) in a precision medicine approach to SLE treatment where LYN dysregulation is identified. Hence, additional studies are needed to consolidate differences in LYN expression in SLE patients as well as to discern the functional consequences of LYN SNPs and the veracity of Lyn as a biomarker for a precision medicine approach to SLE treatment.
EL’E-S: Writing – original draft, Visualization, Investigation, Formal analysis, Conceptualization. TG: Writing – review & editing, Supervision. MW: Writing – review & editing, Supervision. MH: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grant funding from the Lupus Research Alliance (to MLH) and the School of Translational Medicine, Monash University (to MW and MH). EL’E-S was supported by an Australian Government Research Training Program (RTP) Scholarship administered by Monash University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Moulton VR, Suarez-Fueyo A, Meidan E, Li H, Mizui M, Tsokos GC. Pathogenesis of human systemic lupus erythematosus: A cellular perspective. Trends Mol Med. (2017) 23:615–35. doi: 10.1016/j.molmed.2017.05.006
2. Pons-Estel GJ, Alarcon GS, Scofield L, Reinlib L, Cooper GS. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. (2010) 39:257–68. doi: 10.1016/j.semarthrit.2008.10.007
3. Rees F, Doherty M, Grainge MJ, Lanyon P, Zhang W. The worldwide incidence and prevalence of systemic lupus erythematosus: A systematic review of epidemiological studies. Rheumatol (Oxford). (2017) 56:1945–61. doi: 10.1093/rheumatology/kex260
4. Carter EE, Barr SG, Clarke AE. The global burden of sle: prevalence, health disparities and socioeconomic impact. Nat Rev Rheumatol. (2016) 12:605–20. doi: 10.1038/nrrheum.2016.137
5. Yu F, Haas M, Glassock R, Zhao MH. Redefining lupus nephritis: clinical implications of pathophysiologic subtypes. Nat Rev Nephrol. (2017) 13:483–95. doi: 10.1038/nrneph.2017.85
6. Hanly JG, O'Keeffe AG, Su L, Urowitz MB, Romero-Diaz J, Gordon C, et al. The frequency and outcome of lupus nephritis: results from an international inception cohort study. Rheumatol (Oxford). (2016) 55:252–62. doi: 10.1093/rheumatology/kev311
7. Tektonidou MG, Dasgupta A, Ward MM. Risk of end-stage renal disease in patients with lupus nephritis, 1971-2015: A systematic review and bayesian meta-analysis. Arthritis Rheumatol. (2016) 68:1432–41. doi: 10.1002/art.39594
8. Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, Boletis JN, et al. 2019 Update of the eular recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. (2019) 78:736–45. doi: 10.1136/annrheumdis-2019-215089
9. Ramamoorthy S, Cidlowski JA. Corticosteroids: mechanisms of action in health and disease. Rheum Dis Clin North Am. (2016) 42:15–31. doi: 10.1016/j.rdc.2015.08.002
10. Ponticelli C, Moroni G. Hydroxychloroquine in systemic lupus erythematosus (Sle). Expert Opin Drug Saf. (2017) 16:411–9. doi: 10.1080/14740338.2017.1269168
11. Chen HL, Shen LJ, Hsu PN, Shen CY, Hall SA, Hsiao FY. Cumulative burden of glucocorticoid-related adverse events in patients with systemic lupus erythematosus: findings from a 12-year longitudinal study. J Rheumatol. (2018) 45:83–9. doi: 10.3899/jrheum.160214
12. Tektonidou MG, Lewandowski LB, Hu J, Dasgupta A, Ward MM. Survival in adults and children with systemic lupus erythematosus: A systematic review and bayesian meta-analysis of studies from 1950 to 2016. Ann Rheum Dis. (2017) 76:2009–16. doi: 10.1136/annrheumdis-2017-211663
13. Mak A, Cheung MW, Chiew HJ, Liu Y, Ho RC. Global trend of survival and damage of systemic lupus erythematosus: meta-analysis and meta-regression of observational studies from the 1950s to 2000s. Semin Arthritis Rheum. (2012) 41:830–9. doi: 10.1016/j.semarthrit.2011.11.002
14. Rabbani MA, Habib HB, Islam M, Ahmad B, Majid S, Saeed W, et al. Survival analysis and prognostic indicators of systemic lupus erythematosus in Pakistani patients. Lupus. (2009) 18:848–55. doi: 10.1177/0961203309103410
15. Doria A, Iaccarino L, Ghirardello A, Zampieri S, Arienti S, Sarzi-Puttini P, et al. Long-term prognosis and causes of death in systemic lupus erythematosus. Am J Med. (2006) 119:700–6. doi: 10.1016/j.amjmed.2005.11.034
16. Yen EY, Singh RR. Brief report: lupus-an unrecognized leading cause of death in young females: A population-based study using nationwide death certificates, 2000-2015. Arthritis Rheumatol. (2018) 70:1251–5. doi: 10.1002/art.40512
17. Nikpour M, Bridge JA, Richter S. A systematic review of prevalence, disease characteristics and management of systemic lupus erythematosus in Australia: identifying areas of unmet need. Intern Med J. (2014) 44:1170–9. doi: 10.1111/imj.12568
18. Lee YH, Choi SJ, Ji JD, Song GG. Overall and cause-specific mortality in systemic lupus erythematosus: an updated meta-analysis. Lupus. (2016) 25:727–34. doi: 10.1177/0961203315627202
19. Yurkovich M, Vostretsova K, Chen W, Avina-Zubieta JA. Overall and cause-specific mortality in patients with systemic lupus erythematosus: A meta-analysis of observational studies. Arthritis Care Res (Hoboken). (2014) 66:608–16. doi: 10.1002/acr.22173
20. Goobie GC, Bernatsky S, Ramsey-Goldman R, Clarke AE. Malignancies in systemic lupus erythematosus: A 2015 update. Curr Opin Rheumatol. (2015) 27:454–60. doi: 10.1097/BOR.0000000000000202
21. Murphy G, Isenberg DA. New therapies for systemic lupus erythematosus - past imperfect, future tense. Nat Rev Rheumatol. (2019) 15:403–12. doi: 10.1038/s41584-019-0235-5
22. Dolgin E. Lupus in crisis: as failures pile up, clinicians call for new tools. Nat Biotechnol. (2019) 37:7–8. doi: 10.1038/nbt0119-7
23. Mahieu MA, Strand V, Simon LS, Lipsky PE, Ramsey-Goldman R. A critical review of clinical trials in systemic lupus erythematosus. Lupus. (2016) 25:1122–40. doi: 10.1177/0961203316652492
24. Furie R, Rovin BH, Houssiau F, Malvar A, Teng YKO, Contreras G, et al. Two-year, randomized, controlled trial of belimumab in lupus nephritis. N Engl J Med. (2020) 383:1117–28. doi: 10.1056/NEJMoa2001180
25. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. (2020) 382:211–21. doi: 10.1056/NEJMoa1912196
26. Fasano S, Milone A, Nicoletti GF, Isenberg DA, Ciccia F. Precision medicine in systemic lupus erythematosus. Nat Rev Rheumatol. (2023) 19:331–42. doi: 10.1038/s41584-023-00948-y
27. Yamanashi Y, Fukushige S, Semba K, Sukegawa J, Miyajima N, Matsubara K, et al. The yes-related cellular gene lyn encodes a possible tyrosine kinase similar to P56lck. Mol Cell Biol. (1987) 7:237–43. doi: 10.1128/mcb.7.1.237-243.1987
28. Boggon TJ, Eck MJ. Structure and regulation of src family kinases. Oncogene. (2004) 23:7918–27. doi: 10.1038/sj.onc.1208081
29. Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. (1988) 241:42–52. doi: 10.1126/science.3291115
30. Rider LG, Raben N, Miller L, Jelsema C. The cdnas encoding two forms of the lyn protein tyrosine kinase are expressed in rat mast cells and human myeloid cells. Gene. (1994) 138:219–22. doi: 10.1016/0378-1119(94)90811-7
31. UniProt C. Uniprot: A worldwide hub of protein knowledge. Nucleic Acids Res. (2019) 47:D506–D15. doi: 10.1093/nar/gky1049
32. Stanley E, Ralph S, McEwen S, Boulet I, Holtzman DA, Lock P, et al. Alternatively spliced murine lyn mrnas encode distinct proteins. Mol Cell Biol. (1991) 11:3399–406. doi: 101128/mcb.11.7.3399-3406.1991
33. Abram CL, Lowell CA. The diverse functions of src family kinases in macrophages. Front Biosci. (2008) 13:4426–50. doi: 10.2741/3015
34. Karlsson M, Zhang C, Mear L, Zhong W, Digre A, Katona B, et al. A single-cell type transcriptomics map of human tissues. Sci Adv. (2021) 7:eabh2169. doi: 10.1126/sciadv.abh2169
35. Heng TS, Painter MW, Immunological Genome Project C. The immunological genome project: networks of gene expression in immune cells. Nat Immunol. (2008) 9:1091–4. doi: 10.1038/ni1008-1091
36. Weerawarna PM, Richardson TI. Lyn kinase structure, regulation, and involvement in neurodegenerative diseases: A mini review. Kinases Phosphatases. (2023) 1:23–38. doi: 10.3390/kinasesphosphatases1010004
37. Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity. (2005) 22:9–18. doi: 10.1016/j.immuni.2004.12.004
38. Tsantikos E, Gottschalk TA, Maxwell MJ, Hibbs ML. Role of the lyn tyrosine kinase in the development of autoimmune disease. Int J Clin Rheumatol. (2014) 9:519. doi: 10.2217/ijr.14.44
39. Yamanashi Y, Miyasaka M, Takeuchi M, Ilic D, Mizuguchi J, Yamamoto T. Differential responses of P56lyn and P53lyn, products of alternatively spliced lyn mrna, on stimulation of B-cell antigen receptor. Cell Regul. (1991) 2:979–87. doi: 10.1091/mbc.2.12.979
40. Resh MD. Myristylation and palmitylation of src family members: the fats of the matter. Cell. (1994) 76:411–3. doi: 10.1016/0092-8674(94)90104-X
41. Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys Acta. (1999) 1451:1–16. doi: 10.1016/S0167-4889(99)00075-0
42. Skrzypczynska KM, Zhu JW, Weiss A. Positive regulation of lyn kinase by cd148 is required for B cell receptor signaling in B1 but not B2 B cells. Immunity. (2016) 45:1232–44. doi: 10.1016/j.immuni.2016.10.013
43. Matsuda D, Nakayama Y, Horimoto S, Kuga T, Ikeda K, Kasahara K, et al. Involvement of golgi-associated lyn tyrosine kinase in the translocation of annexin ii to the endoplasmic reticulum under oxidative stress. Exp Cell Res. (2006) 312:1205–17. doi: 10.1016/j.yexcr.2006.02.003
44. Ikeda K, Nakayama Y, Togashi Y, Obata Y, Kuga T, Kasahara K, et al. Nuclear localization of lyn tyrosine kinase mediated by inhibition of its kinase activity. Exp Cell Res. (2008) 314:3392–404. doi: 10.1016/j.yexcr.2008.08.019
45. Kharbanda S, Saleem A, Yuan ZM, Kraeft S, Weichselbaum R, Chen LB, et al. Nuclear signaling induced by ionizing radiation involves colocalization of the activated P56/P53lyn tyrosine kinase with P34cdc2. Cancer Res. (1996) 56:3617–21.
46. Yoshida K, Weichselbaum R, Kharbanda S, Kufe D. Role for lyn tyrosine kinase as a regulator of stress-activated protein kinase activity in response to DNA damage. Mol Cell Biol. (2000) 20:5370–80. doi: 10.1128/MCB.20.15.5370-5380.2000
47. Kumar S, Pandey P, Bharti A, Jin S, Weichselbaum R, Weaver D, et al. Regulation of DNA-dependent protein kinase by the lyn tyrosine kinase. J Biol Chem. (1998) 273:25654–8. doi: 10.1074/jbc.273.40.25654
48. Getahun A, Cambier JC. Of itims, itams, and itamis: revisiting immunoglobulin fc receptor signaling. Immunol Rev. (2015) 268:66–73. doi: 10.1111/imr.12336
49. Geahlen RL. Getting syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol Sci. (2014) 35:414–22. doi: 10.1016/j.tips.2014.05.007
50. Courtney AH, Lo WL, Weiss A. Tcr signaling: mechanisms of initiation and propagation. Trends Biochem Sci. (2018) 43:108–23. doi: 10.1016/j.tibs.2017.11.008
51. Mocsai A, Ruland J, Tybulewicz VL. The syk tyrosine kinase: A crucial player in diverse biological functions. Nat Rev Immunol. (2010) 10:387–402. doi: 10.1038/nri2765
52. Zhang H, Meng F, Chu CL, Takai T, Lowell CA. The src family kinases hck and fgr negatively regulate neutrophil and dendritic cell chemokine signaling via pir-B. Immunity. (2005) 22:235–46. doi: 10.1016/j.immuni.2005.01.004
53. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. Pd-1 inhibits T-cell receptor induced phosphorylation of the zap70/cd3zeta signalosome and downstream signaling to pkctheta. FEBS Lett. (2004) 574:37–41. doi: 10.1016/j.febslet.2004.07.083
54. Daeron M, Jaeger S, Du Pasquier L, Vivier E. Immunoreceptor tyrosine-based inhibition motifs: A quest in the past and future. Immunol Rev. (2008) 224:11–43. doi: 10.1111/j.1600-065X.2008.00666.x
55. Maxwell MJ, Yuan Y, Anderson KE, Hibbs ML, Salem HH, Jackson SP. Ship1 and lyn kinase negatively regulate integrin alpha iib beta 3 signaling in platelets. J Biol Chem. (2004) 279:32196–204. doi: 10.1074/jbc.M400746200
56. Yoshida K, Kharbanda S, Kufe D. Functional interaction between shptp1 and the lyn tyrosine kinase in the apoptotic response to DNA damage. J Biol Chem. (1999) 274:34663–8. doi: 10.1074/jbc.274.49.34663
57. Nitschke L, Tsubata T. Molecular interactions regulate bcr signal inhibition by cd22 and cd72. Trends Immunol. (2004) 25:543–50. doi: 10.1016/j.it.2004.08.002
58. Abram CL, Lowell CA. Shp1 function in myeloid cells. J Leukoc Biol. (2017) 102:657–75. doi: 10.1189/jlb.2MR0317-105R
59. David M, Chen HE, Goelz S, Larner AC, Neel BG. Differential regulation of the alpha/beta interferon-stimulated jak/stat pathway by the sh2 domain-containing tyrosine phosphatase shptp1. Mol Cell Biol. (1995) 15:7050–8. doi: 10.1128/MCB.15.12.7050
60. Wang Y, Keogh RJ, Hunter MG, Mitchell CA, Frey RS, Javaid K, et al. Ship2 is recruited to the cell membrane upon macrophage colony-stimulating factor (M-csf) stimulation and regulates M-csf-induced signaling. J Immunol. (2004) 173:6820–30. doi: 10.4049/jimmunol.173.11.6820
61. Baran CP, Tridandapani S, Helgason CD, Humphries RK, Krystal G, Marsh CB. The inositol 5'-phosphatase ship-1 and the src kinase lyn negatively regulate macrophage colony-stimulating factor-induced akt activity. J Biol Chem. (2003) 278:38628–36. doi: 10.1074/jbc.M305021200
62. Mkaddem SB, Murua A, Flament H, Titeca-Beauport D, Bounaix C, Danelli L, et al. Lyn and fyn function as molecular switches that control immunoreceptors to direct homeostasis or inflammation. Nat Commun. (2017) 8:246. doi: 10.1038/s41467-017-00294-0
63. Ben Mkaddem S, Hayem G, Jonsson F, Rossato E, Boedec E, Boussetta T, et al. Shifting fcgammariia-itam from activation to inhibitory configuration ameliorates arthritis. J Clin Invest. (2014) 124:3945–59. doi: 10.1172/JCI74572
64. Pasquier B, Launay P, Kanamaru Y, Moura IC, Pfirsch S, Ruffie C, et al. Identification of fcalphari as an inhibitory receptor that controls inflammation: dual role of fcrgamma itam. Immunity. (2005) 22:31–42. doi: 10.1016/j.immuni.2004.11.017
65. Yamanashi Y, Kakiuchi T, Mizuguchi J, Yamamoto T, Toyoshima K. Association of B cell antigen receptor with protein tyrosine kinase lyn. Science. (1991) 251:192–4. doi: 10.1126/science.1702903
66. Fujimoto M, Poe JC, Jansen PJ, Sato S, Tedder TF. Cd19 amplifies B lymphocyte signal transduction by regulating src-family protein tyrosine kinase activation. J Immunol. (1999) 162:7088–94. doi: 10.4049/jimmunol.162.12.7088
67. Eiseman E, Bolen JB. Engagement of the high-affinity ige receptor activates src protein-related tyrosine kinases. Nature. (1992) 355:78–80. doi: 10.1038/355078a0
68. Fitzer-Attas CJ, Lowry M, Crowley MT, Finn AJ, Meng F, DeFranco AL, et al. Fcgamma receptor-mediated phagocytosis in macrophages lacking the src family tyrosine kinases hck, fgr, and lyn. J Exp Med. (2000) 191:669–82. doi: 10.1084/jem.191.4.669
69. Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, et al. A critical role for syk in signal transduction and phagocytosis mediated by fcgamma receptors on macrophages. J Exp Med. (1997) 186:1027–39. doi: 10.1084/jem.186.7.1027
70. Zhu Z, Li R, Li H, Zhou T, Davis RS. Fcrl5 exerts binary and compartment-specific influence on innate-like B-cell receptor signaling. Proc Natl Acad Sci U.S.A. (2013) 110:E1282–90. doi: 10.1073/pnas.1215156110
71. Tang J, Xiao Y, Lin G, Guo H, Deng HX, Tu S, et al. Tyrosine phosphorylation of nlrp3 by the src family kinase lyn suppresses the activity of the nlrp3 inflammasome. Sci Signal. (2021) 14:eabe3410. doi: 10.1126/scisignal.abe3410
72. Tawaratsumida K, Redecke V, Wu R, Kuriakose J, Bouchard JJ, Mittag T, et al. A phospho-tyrosine-based signaling module using spop, csk, and lyn controls tlr-induced irf activity. Sci Adv. (2022) 8:eabq0084. doi: 10.1126/sciadv.abq0084
73. Ban T, Sato GR, Nishiyama A, Akiyama A, Takasuna M, Umehara M, et al. Lyn kinase suppresses the transcriptional activity of irf5 in the tlr-myd88 pathway to restrain the development of autoimmunity. Immunity. (2016) 45:319–32. doi: 10.1016/j.immuni.2016.07.015
74. Chin H, Arai A, Wakao H, Kamiyama R, Miyasaka N, Miura O. Lyn physically associates with the erythropoietin receptor and may play a role in activation of the stat5 pathway. Blood. (1998) 91:3734–45. doi: 10.1182/blood.V91.10.3734
75. Duprez V, Blank U, Chretien S, Gisselbrecht S, Mayeux P. Physical and functional interaction between P72(Syk) and erythropoietin receptor. J Biol Chem. (1998) 273:33985–90. doi: 10.1074/jbc.273.51.33985
76. Corey SJ, Burkhardt AL, Bolen JB, Geahlen RL, Tkatch LS, Tweardy DJ. Granulocyte colony-stimulating factor receptor signaling involves the formation of a three-component complex with lyn and syk protein-tyrosine kinases. Proc Natl Acad Sci U.S.A. (1994) 91:4683–7. doi: 10.1073/pnas.91.11.4683
77. Corey SJ, Dombrosky-Ferlan PM, Zuo S, Krohn E, Donnenberg AD, Zorich P, et al. Requirement of src kinase lyn for induction of DNA synthesis by granulocyte colony-stimulating factor. J Biol Chem. (1998) 273:3230–5. doi: 10.1074/jbc.273.6.3230
78. Zhu QS, Xia L, Mills GB, Lowell CA, Touw IP, Corey SJ. G-csf induced reactive oxygen species involves lyn-pi3-kinase-akt and contributes to myeloid cell growth. Blood. (2006) 107:1847–56. doi: 10.1182/blood-2005-04-1612
79. Yousefi S, Hoessli DC, Blaser K, Mills GB, Simon HU. Requirement of lyn and syk tyrosine kinases for the prevention of apoptosis by cytokines in human eosinophils. J Exp Med. (1996) 183:1407–14. doi: 10.1084/jem.183.4.1407
80. Stefanova I, Corcoran ML, Horak EM, Wahl LM, Bolen JB, Horak ID. Lipopolysaccharide induces activation of cd14-associated protein tyrosine kinase P53/56lyn. J Biol Chem. (1993) 268:20725–8. doi: 10.1016/S0021-9258(19)36840-1
81. Palsson-McDermott EM, O'Neill LA. Signal transduction by the lipopolysaccharide receptor, toll-like receptor-4. Immunology. (2004) 113:153–62. doi: 10.1111/j.1365-2567.2004.01976.x
82. Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, et al. A cd36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. (2002) 277:47373–9. doi: 10.1074/jbc.M208788200
83. Linnekin D, DeBerry CS, Mou S. Lyn associates with the juxtamembrane region of C-kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J Biol Chem. (1997) 272:27450–5. doi: 10.1074/jbc.272.43.27450
84. Torigoe T, Saragovi HU, Reed JC. Interleukin 2 regulates the activity of the lyn protein-tyrosine kinase in a B-cell line. Proc Natl Acad Sci U.S.A. (1992) 89:2674–8. doi: 10.1073/pnas.89.7.2674
85. Ren CL, Morio T, Fu SM, Geha RS. Signal transduction via cd40 involves activation of lyn kinase and phosphatidylinositol-3-kinase, and phosphorylation of phospholipase C gamma 2. J Exp Med. (1994) 179:673–80. doi: 10.1084/jem.179.2.673
86. Okamoto M, Hayakawa F, Miyata Y, Watamoto K, Emi N, Abe A, et al. Lyn is an important component of the signal transduction pathway specific to flt3/itd and can be a therapeutic target in the treatment of aml with flt3/itd. Leukemia. (2007) 21:403–10. doi: 10.1038/sj.leu.2404547
87. Robinson LJ, Xue J, Corey SJ. Src family tyrosine kinases are activated by flt3 and are involved in the proliferative effects of leukemia-associated flt3 mutations. Exp Hematol. (2005) 33:469–79. doi: 10.1016/j.exphem.2005.01.004
88. Harder KW, Parsons LM, Armes J, Evans N, Kountouri N, Clark R, et al. Gain- and loss-of-function lyn mutant mice define a critical inhibitory role for lyn in the myeloid lineage. Immunity. (2001) 15:603–15. doi: 10.1016/S1074-7613(01)00208-4
89. Kohlhas V, Hallek M, Nguyen PH. Constitutive activation of lyn kinase enhances bcr responsiveness, but not the development of cll in emicro-tcl1 mice. Blood Adv. (2020) 4:6106–16. doi: 10.1182/bloodadvances.2020002584
90. Hibbs ML, Harder KW, Armes J, Kountouri N, Quilici C, Casagranda F, et al. Sustained activation of lyn tyrosine kinase in vivo leads to autoimmunity. J Exp Med. (2002) 196:1593–604. doi: 10.1084/jem.20020515
91. Tsantikos E, Gottschalk TA, L'Estrange-Stranieri E, O'Brien CA, Raftery AL, Wickramasinghe LC, et al. Enhanced lyn activity causes severe, progressive emphysema and lung cancer. Am J Respir Cell Mol Biol. (2023) 69:99–112. doi: 10.1165/rcmb.2022-0463OC
92. Louvrier C, El Khouri E, Grall Lerosey M, Quartier P, Guerrot AM, Bader Meunier B, et al. De novo gain-of-function variations in lyn associated with an early-onset systemic autoinflammatory disorder. Arthritis Rheumatol. (2023) 75:468–74. doi: 10.1002/art.42354
93. de Jesus AA, Chen G, Yang D, Brdicka T, Ruth NM, Bennin D, et al. Constitutively active lyn kinase causes a cutaneous small vessel vasculitis and liver fibrosis syndrome. Nat Commun. (2023) 14:1502. doi: 10.1038/s41467-023-36941-y
94. Luciano F, Ricci JE, Auberger P. Cleavage of fyn and lyn in their N-terminal unique regions during induction of apoptosis: A new mechanism for src kinase regulation. Oncogene. (2001) 20:4935–41. doi: 10.1038/sj.onc.1204661
95. Marchetti S, Gamas P, Belhacene N, Grosso S, Pradelli LA, Colosetti P, et al. The caspase-cleaved form of lyn mediates a psoriasis-like inflammatory syndrome in mice. EMBO J. (2009) 28:2449–60. doi: 10.1038/emboj.2009.183
96. Luciano F, Herrant M, Jacquel A, Ricci JE, Auberger P. The P54 cleaved form of the tyrosine kinase lyn generated by caspases during bcr-induced cell death in B lymphoma acts as a negative regulator of apoptosis. FASEB J. (2003) 17:711–3. doi: 10.1096/fj.02-0716fje
97. Aira LE, Goncalves D, Bossowski JP, Rubio-Patino C, Chiche J, Paul-Bellon R, et al. Caspase 1/11 deficiency or pharmacological inhibition mitigates psoriasis-like phenotype in mice. J Invest Dermatol. (2019) 139:1306–17. doi: 10.1016/j.jid.2018.11.031
98. Brian BFT, Jolicoeur AS, Guerrero CR, Nunez MG, Sychev ZE, Hegre SA, et al. Unique-region phosphorylation targets lyna for rapid degradation, tuning its expression and signaling in myeloid cells. Elife. (2019) 8:e46043. doi: 10.7554/eLife.46043
99. Huang TH, Huo L, Wang YN, Xia W, Wei Y, Chang SS, et al. Epidermal growth factor receptor potentiates mcm7-mediated DNA replication through tyrosine phosphorylation of lyn kinase in human cancers. Cancer Cell. (2013) 23:796–810. doi: 10.1016/j.ccr.2013.04.027
100. Freedman TS, Tan YX, Skrzypczynska KM, Manz BN, Sjaastad FV, Goodridge HS, et al. Lyna regulates an inflammation-sensitive signaling checkpoint in macrophages. Elife. (2015) 4:e09183. doi: 10.7554/eLife.09183
101. Brian BFT, Sauer ML, Greene JT, Senevirathne SE, Lindstedt AJ, Funk OL, et al. A dominant function of lynb kinase in preventing autoimmunity. Sci Adv. (2022) 8:eabj5227. doi: 10.1126/sciadv.abj5227
102. Tornillo G, Knowlson C, Kendrick H, Cooke J, Mirza H, Aurrekoetxea-Rodriguez I, et al. Dual mechanisms of lyn kinase dysregulation drive aggressive behavior in breast cancer cells. Cell Rep. (2018) 25:3674–92 e10. doi: 10.1016/j.celrep.2018.11.103
103. Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, et al. Multiple defects in the immune system of lyn-deficient mice, culminating in autoimmune disease. Cell. (1995) 83:301–11. doi: 10.1016/0092-8674(95)90171-X
104. Nishizumi H, Taniuchi I, Yamanashi Y, Kitamura D, Ilic D, Mori S, et al. Impaired proliferation of peripheral B cells and indication of autoimmune disease in lyn-deficient mice. Immunity. (1995) 3:549–60. doi: 10.1016/1074-7613(95)90126-4
105. Lamagna C, Hu Y, DeFranco AL, Lowell CA. B cell-specific loss of lyn kinase leads to autoimmunity. J Immunol. (2014) 192:919–28. doi: 10.4049/jimmunol.1301979
106. Oracki SA, Tsantikos E, Quilici C, Light A, Schmidt T, Lew AM, et al. Ctla4ig alters the course of autoimmune disease development in lyn-/- mice. J Immunol. (2010) 184:757–63. doi: 10.4049/jimmunol.0804349
107. Charles N, Hardwick D, Daugas E, Illei GG, Rivera J. Basophils and the T helper 2 environment can promote the development of lupus nephritis. Nat Med. (2010) 16:701–7. doi: 10.1038/nm.2159
108. Lau M, Tsantikos E, Maxwell MJ, Tarlinton DM, Anderson GP, Hibbs ML. Loss of stat6 promotes autoimmune disease and atopy on a susceptible genetic background. J Autoimmun. (2012) 39:388–97. doi: 10.1016/j.jaut.2012.06.003
109. Chan VW, Meng F, Soriano P, DeFranco AL, Lowell CA. Characterization of the B lymphocyte populations in lyn-deficient mice and the role of lyn in signal initiation and down-regulation. Immunity. (1997) 7:69–81. doi: 10.1016/S1074-7613(00)80511-7
110. Keck S, Freudenberg M, Huber M. Activation of murine macrophages via tlr2 and tlr4 is negatively regulated by a lyn/pi3k module and promoted by ship1. J Immunol. (2010) 184:5809–18. doi: 10.4049/jimmunol.0901423
111. Scapini P, Hu Y, Chu CL, Migone TS, Defranco AL, Cassatella MA, et al. Myeloid cells, baff, and ifn-gamma establish an inflammatory loop that exacerbates autoimmunity in lyn-deficient mice. J Exp Med. (2010) 207:1757–73. doi: 10.1084/jem.20100086
112. Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA. Hyperactivated myd88 signaling in dendritic cells, through specific deletion of lyn kinase, causes severe autoimmunity and inflammation. Proc Natl Acad Sci U.S.A. (2013) 110:E3311–20. doi: 10.1073/pnas.1300617110
113. Malik M, Chen YY, Kienzle MF, Tomkowicz BE, Collman RG, Ptasznik A. Monocyte migration and lfa-1-mediated attachment to brain microvascular endothelia is regulated by sdf-1 alpha through lyn kinase. J Immunol. (2008) 181:4632–7. doi: 10.4049/jimmunol.181.7.4632
114. Pereira S, Lowell C. The lyn tyrosine kinase negatively regulates neutrophil integrin signaling. J Immunol. (2003) 171:1319–27. doi: 10.4049/jimmunol.171.3.1319
115. Hibbs ML, Harder KW. The duplicitous nature of the lyn tyrosine kinase in growth factor signaling. Growth Factors. (2006) 24:137–49. doi: 10.1080/08977190600581327
116. Harder KW, Quilici C, Naik E, Inglese M, Kountouri N, Turner A, et al. Perturbed myelo/erythropoiesis in lyn-deficient mice is similar to that in mice lacking the inhibitory phosphatases shp-1 and ship-1. Blood. (2004) 104:3901–10. doi: 10.1182/blood-2003-12-4396
117. Hua Z, Gross AJ, Lamagna C, Ramos-Hernandez N, Scapini P, Ji M, et al. Requirement for myd88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in lyn-deficient mice. J Immunol. (2014) 192:875–85. doi: 10.4049/jimmunol.1300683
118. Ban T, Kikuchi M, Sato GR, Manabe A, Tagata N, Harita K, et al. Genetic and chemical inhibition of irf5 suppresses pre-existing mouse lupus-like disease. Nat Commun. (2021) 12:4379. doi: 10.1038/s41467-021-24609-4
119. Silver KL, Crockford TL, Bouriez-Jones T, Milling S, Lambe T, Cornall RJ. Myd88-dependent autoimmune disease in lyn-deficient mice. Eur J Immunol. (2007) 37:2734–43. doi: 10.1002/eji.200737293
120. Ma J, Abram CL, Hu Y, Lowell CA. Card9 mediates dendritic cell–induced development of lyn deficiency–associated autoimmune and inflammatory diseases. Sci Signaling. (2019) 12:eaao3829. doi: 10.1126/scisignal.aao3829
121. Tsantikos E, Maxwell MJ, Kountouri N, Harder KW, Tarlinton DM, Hibbs ML. Genetic interdependence of lyn and negative regulators of B cell receptor signaling in autoimmune disease development. J Immunol. (2012) 189:1726–36. doi: 10.4049/jimmunol.1103427
122. Clark EA, Giltiay NV. Cd22: A regulator of innate and adaptive B cell responses and autoimmunity. Front Immunol. (2018) 9:2235. doi: 10.3389/fimmu.2018.02235
123. Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. (2007) 7:255–66. doi: 10.1038/nri2056
124. Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, et al. Polygenic autoimmune traits: lyn, cd22, and shp-1 are limiting elements of a biochemical pathway regulating bcr signaling and selection. Immunity. (1998) 8:497–508. doi: 10.1016/S1074-7613(00)80554-3
125. Smith KG, Tarlinton DM, Doody GM, Hibbs ML, Fearon DT. Inhibition of the B cell by cd22: A requirement for lyn. J Exp Med. (1998) 187:807–11. doi: 10.1084/jem.187.5.807
126. Poe JC, Fujimoto M, Jansen PJ, Miller AS, Tedder TF. Cd22 forms a quaternary complex with ship, grb2, and shc. A pathway for regulation of B lymphocyte antigen receptor-induced calcium flux. J Biol Chem. (2000) 275:17420–7. doi: 10.1074/jbc.M001892200
127. Muller J, Nitschke L. The role of cd22 and siglec-G in B-cell tolerance and autoimmune disease. Nat Rev Rheumatol. (2014) 10:422–8. doi: 10.1038/nrrheum.2014.54
128. Tsubata T. Ligand recognition determines the role of inhibitory B cell co-receptors in the regulation of B cell homeostasis and autoimmunity. Front Immunol. (2018) 9:2276. doi: 10.3389/fimmu.2018.02276
129. Rizzuto G, Brooks JF, Tuomivaara ST, McIntyre TI, Ma S, Rideaux D, et al. Establishment of fetomaternal tolerance through glycan-mediated B cell suppression. Nature. (2022) 603(7901):497–502. doi: 10.1038/s41586-022-04471-0
130. O'Keefe TL, Williams GT, Batista FD, Neuberger MS. Deficiency in cd22, a B cell-specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J Exp Med. (1999) 189:1307–13. doi: 10.1084/jem.189.8.1307
131. Jellusova J, Wellmann U, Amann K, Winkler TH, Nitschke L. Cd22 X siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. J Immunol. (2010) 184:3618–27. doi: 10.4049/jimmunol.0902711
132. Hatta Y, Tsuchiya N, Matsushita M, Shiota M, Hagiwara K, Tokunaga K. Identification of the gene variations in human cd22. Immunogenetics. (1999) 49:280–6. doi: 10.1007/s002510050494
133. El-Sayed ZA, Ragab SM, Khalifa KA, El Ashmawy RA. Altered cd19/cd22 balance in Egyptian children and adolescents with systemic lupus erythematosus. Egypt J Immunol. (2009) 16:27–38.
134. Suzuki J, Nakano S, Nakairi Y, Mitsuo A, Amano H, Morimoto S, et al. Cd19/22 balance relates to improvement of disease activity in systemic lupus erythematosus. Mod Rheumatol. (2006) 16:235–8. doi: 10.1007/s10165-006-0497-z
135. Sharpe AH, Pauken KE. The diverse functions of the pd1 inhibitory pathway. Nat Rev Immunol. (2018) 18:153–67. doi: 10.1038/nri.2017.108
136. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. Pd-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U.S.A. (2001) 98:13866–71. doi: 10.1073/pnas.231486598
137. Thibult ML, Mamessier E, Gertner-Dardenne J, Pastor S, Just-Landi S, Xerri L, et al. Pd-1 is a novel regulator of human B-cell activation. Int Immunol. (2013) 25:129–37. doi: 10.1093/intimm/dxs098
138. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the pd-1 gene encoding an itim motif-carrying immunoreceptor. Immunity. (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
139. Curran CS, Gupta S, Sanz I, Sharon E. Pd-1 immunobiology in systemic lupus erythematosus. J Autoimmun. (2019) 97:1–9. doi: 10.1016/j.jaut.2018.10.025
140. Voisinne G, Gonzalez de Peredo A, Roncagalli R. Cd5, an undercover regulator of tcr signaling. Front Immunol. (2018) 9:2900. doi: 10.3389/fimmu.2018.02900
141. Hippen KL, Tze LE, Behrens TW. Cd5 maintains tolerance in anergic B cells. J Exp Med. (2000) 191:883–90. doi: 10.1084/jem.191.5.883
142. Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique cd1dhicd5+ Phenotype controls T cell-dependent inflammatory responses. Immunity. (2008) 28:639–50. doi: 10.1016/j.immuni.2008.03.017
143. Yanaba K, Bouaziz JD, Matsushita T, Tsubata T, Tedder TF. The development and function of regulatory B cells expressing il-10 (B10 cells) requires antigen receptor diversity and tlr signals. J Immunol. (2009) 182:7459–72. doi: 10.4049/jimmunol.0900270
144. Brown MH, Lacey E. A ligand for cd5 is cd5. J Immunol. (2010) 185:6068–74. doi: 10.4049/jimmunol.0903823
145. Van de Velde H, von Hoegen I, Luo W, Parnes JR, Thielemans K. The B-cell surface protein cd72/lyb-2 is the ligand for cd5. Nature. (1991) 351:662–5. doi: 10.1038/351662a0
146. Morris G, Puri BK, Olive L, Carvalho AF, Berk M, Maes M. Emerging role of innate B1 cells in the pathophysiology of autoimmune and neuroimmune diseases: association with inflammation, oxidative and nitrosative stress and autoimmune responses. Pharmacol Res. (2019) 148:104408. doi: 10.1016/j.phrs.2019.104408
147. Baumgarth N. A hard(Y) look at B-1 cell development and function. J Immunol. (2017) 199:3387–94. doi: 10.4049/jimmunol.1700943
148. Ochi H, Watanabe T. Negative regulation of B cell receptor-mediated signaling in B-1 cells through cd5 and ly49 co-receptors via lyn kinase activity. Int Immunol. (2000) 12:1417–23. doi: 10.1093/intimm/12.10.1417
149. Bikah G, Carey J, Ciallella JR, Tarakhovsky A, Bondada S. Cd5-mediated negative regulation of antigen receptor-induced growth signals in B-1 B cells. Science. (1996) 274:1906–9. doi: 10.1126/science.274.5294.1906
150. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
151. Nishizumi H, Horikawa K, Mlinaric-Rascan I, Yamamoto T. A double-edged kinase lyn: A positive and negative regulator for antigen receptor-mediated signals. J Exp Med. (1998) 187:1343–8. doi: 10.1084/jem.187.8.1343
152. Flores M, Desai DD, Downie M, Liang B, Reilly MP, McKenzie SE, et al. Dominant expression of the inhibitory fcgammariib prevents antigen presentation by murine plasmacytoid dendritic cells. J Immunol. (2009) 183:7129–39. doi: 10.4049/jimmunol.0901169
153. Kam TI, Park H, Gwon Y, Song S, Kim SH, Moon SW, et al. Fcgammariib-ship2 axis links abeta to tau pathology by disrupting phosphoinositide metabolism in alzheimer's disease model. Elife. (2016) 5:e18691. doi: 10.7554/eLife.18691
154. Morris AB, Farley CR, Pinelli DF, Adams LE, Cragg MS, Boss JM, et al. Signaling through the inhibitory fc receptor fcgammariib induces cd8(+) T cell apoptosis to limit T cell immunity. Immunity. (2020) 52:136–50 e6. doi: 10.1016/j.immuni.2019.12.006
155. Starbeck-Miller GR, Badovinac VP, Barber DL, Harty JT. Cutting edge: expression of fcgammariib tempers memory cd8 T cell function in vivo. J Immunol. (2014) 192:35–9. doi: 10.4049/jimmunol.1302232
156. Verbeek JS, Hirose S, Nishimura H. The complex association of fcgammariib with autoimmune susceptibility. Front Immunol. (2019) 10:2061. doi: 10.3389/fimmu.2019.02061
157. Flores M, Chew C, Tyan K, Huang WQ, Salem A, Clynes R. Fcgammariib prevents inflammatory type I ifn production from plasmacytoid dendritic cells during a viral memory response. J Immunol. (2015) 194:4240–50. doi: 10.4049/jimmunol.1401296
158. Dhodapkar KM, Kaufman JL, Ehlers M, Banerjee DK, Bonvini E, Koenig S, et al. Selective blockade of inhibitory fcgamma receptor enables human dendritic cell maturation with il-12p70 production and immunity to antibody-coated tumor cells. Proc Natl Acad Sci U.S.A. (2005) 102:2910–5. doi: 10.1073/pnas.0500014102
159. Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory igg fc receptors on human dcs mediate opposing functions. J Clin Invest. (2005) 115:2914–23. doi: 10.1172/JCI24772
160. Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating fcgamma receptors on dendritic cells. J Exp Med. (2002) 195:1653–9. doi: 10.1084/jem.20020338
161. Bolland S, Ravetch JV. Spontaneous autoimmune disease in fc(Gamma)Riib-deficient mice results from strain-specific epistasis. Immunity. (2000) 13:277–85. doi: 10.1016/S1074-7613(00)00027-3
162. McGaha TL, Sorrentino B, Ravetch JV. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. (2005) 307:590–3. doi: 10.1126/science.1105160
163. Brownlie RJ, Lawlor KE, Niederer HA, Cutler AJ, Xiang Z, Clatworthy MR, et al. Distinct cell-specific control of autoimmunity and infection by fcgammariib. J Exp Med. (2008) 205:883–95. doi: 10.1084/jem.20072565
164. Kyogoku C, Dijstelbloem HM, Tsuchiya N, Hatta Y, Kato H, Yamaguchi A, et al. Fcgamma receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of fcgr2b to genetic susceptibility. Arthritis Rheum. (2002) 46:1242–54. doi: 10.1002/art.10257
165. Willcocks LC, Carr EJ, Niederer HA, Rayner TF, Williams TN, Yang W, et al. A Defunctioning Polymorphism in Fcgr2b Is Associated with Protection against Malaria but Susceptibility to Systemic Lupus Erythematosus. Proc Natl Acad Sci U.S.A. (2010) 107:7881–5. doi: 10.1073/pnas.0915133107
166. Chu ZT, Tsuchiya N, Kyogoku C, Ohashi J, Qian YP, Xu SB, et al. Association of fcgamma receptor iib polymorphism with susceptibility to systemic lupus erythematosus in chinese: A common susceptibility gene in the asian populations. Tissue Antigens. (2004) 63:21–7. doi: 10.1111/j.1399-0039.2004.00142.x
167. Takai T, Nakamura A, Endo S. Role of pir-B in autoimmune glomerulonephritis. J BioMed Biotechnol. (2011) 2011:275302. doi: 10.1155/2011/275302
168. Ho LH, Uehara T, Chen CC, Kubagawa H, Cooper MD. Constitutive tyrosine phosphorylation of the inhibitory paired ig-like receptor pir-B. Proc Natl Acad Sci U.S.A. (1999) 96:15086–90. doi: 10.1073/pnas.96.26.15086
169. Kubo T, Uchida Y, Watanabe Y, Abe M, Nakamura A, Ono M, et al. Augmented tlr9-induced btk activation in pir-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity. J Exp Med. (2009) 206:1971–82. doi: 10.1084/jem.20082392
170. Ujike A, Takeda K, Nakamura A, Ebihara S, Akiyama K, Takai T. Impaired dendritic cell maturation and increased T(H)2 responses in pir-B(-/-) mice. Nat Immunol. (2002) 3:542–8. doi: 10.1038/ni801
171. Pereira S, Zhang H, Takai T, Lowell CA. The inhibitory receptor pir-B negatively regulates neutrophil and macrophage integrin signaling. J Immunol. (2004) 173:5757–65. doi: 10.4049/jimmunol.173.9.5757
172. Ma G, Pan PY, Eisenstein S, Divino CM, Lowell CA, Takai T, et al. Paired immunoglobin-like receptor-B regulates the suppressive function and fate of myeloid-derived suppressor cells. Immunity. (2011) 34:385–95. doi: 10.1016/j.immuni.2011.02.004
173. Endo S, Sakamoto Y, Kobayashi E, Nakamura A, Takai T. Regulation of cytotoxic T lymphocyte triggering by pir-B on dendritic cells. Proc Natl Acad Sci U.S.A. (2008) 105:14515–20. doi: 10.1073/pnas.0804571105
174. Mitsuhashi Y, Nakamura A, Endo S, Takeda K, Yabe-Wada T, Nukiwa T, et al. Regulation of plasmacytoid dendritic cell responses by pir-B. Blood. (2012) 120:3256–9. doi: 10.1182/blood-2012-03-419093
175. Galuppo MK, de Rezende E, Forti FL, Cortez M, Cruz MC, Teixeira AA, et al. Cd100/sema4d increases macrophage infection by leishmania (Leishmania) amazonensis in a cd72 dependent manner. Front Microbiol. (2018) 9:1177. doi: 10.3389/fmicb.2018.01177
176. Akatsu C, Shinagawa K, Numoto N, Liu Z, Ucar AK, Aslam M, et al. Cd72 negatively regulates B lymphocyte responses to the lupus-related endogenous toll-like receptor 7 ligand sm/rnp. J Exp Med. (2016) 213:2691–706. doi: 10.1084/jem.20160560
177. Adachi T, Flaswinkel H, Yakura H, Reth M, Tsubata T. The B cell surface protein cd72 recruits the tyrosine phosphatase shp-1 upon tyrosine phosphorylation. J Immunol. (1998) 160:4662–5. doi: 10.4049/jimmunol.160.10.4662
178. Li DH, Winslow MM, Cao TM, Chen AH, Davis CR, Mellins ED, et al. Modulation of peripheral B cell tolerance by cd72 in a murine model. Arthritis Rheum. (2008) 58:3192–204. doi: 10.1002/art.23812
179. Xu M, Hou R, Sato-Hayashizaki A, Man R, Zhu C, Wakabayashi C, et al. Cd72(C) is a modifier gene that regulates fas(Lpr)-induced autoimmune disease. J Immunol. (2013) 190:5436–45. doi: 10.4049/jimmunol.1203576
180. Hitomi Y, Tsuchiya N, Kawasaki A, Ohashi J, Suzuki T, Kyogoku C, et al. Cd72 polymorphisms associated with alternative splicing modify susceptibility to human systemic lupus erythematosus through epistatic interaction with fcgr2b. Hum Mol Genet. (2004) 13:2907–17. doi: 10.1093/hmg/ddh318
181. Asmiyou A, Bakr AM, Shahin DA, Wahba Y. Cd40 and cd72 expression and prognostic values among children with systemic lupus erythematosus: A case-control study. Lupus. (2020) 29:1270–6. doi: 10.1177/0961203320941931
182. Vadasz Z, Haj T, Balbir A, Peri R, Rosner I, Slobodin G, et al. A regulatory role for cd72 expression on B cells in systemic lupus erythematosus. Semin Arthritis Rheum. (2014) 43:767–71. doi: 10.1016/j.semarthrit.2013.11.010
183. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery. (2020) 6:36. doi: 10.1038/s41421-020-0167-x
184. Spalinger MR, Schwarzfischer M, Scharl M. The role of protein tyrosine phosphatases in inflammasome activation. Int J Mol Sci. (2020) 21(15):5481. doi: 10.3390/ijms21155481
185. Lim KH, Chen LC, Hsu K, Chang CC, Chang CY, Kao CW, et al. Baff-driven nlrp3 inflammasome activation in B cells. Cell Death Dis. (2020) 11:820. doi: 10.1038/s41419-020-03035-2
186. Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (Sirpalpha) and cd47: structure, function, and therapeutic target. Annu Rev Immunol. (2014) 32:25–50. doi: 10.1146/annurev-immunol-032713-120142
187. Deuse T, Hu X, Agbor-Enoh S, Jang MK, Alawi M, Saygi C, et al. The sirpalpha-cd47 immune checkpoint in nk cells. J Exp Med. (2021) 218(3):e20200839. doi: 10.1084/jem.20200839
188. Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, Ullrich A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature. (1997) 386:181–6. doi: 10.1038/386181a0
189. Veillette A, Thibaudeau E, Latour S. High expression of inhibitory receptor shps-1 and its association with protein-tyrosine phosphatase shp-1 in macrophages. J Biol Chem. (1998) 273:22719–28. doi: 10.1074/jbc.273.35.22719
190. Bian Z, Shi L, Guo YL, Lv Z, Tang C, Niu S, et al. Cd47-sirpalpha interaction and il-10 constrain inflammation-induced macrophage phagocytosis of healthy self-cells. Proc Natl Acad Sci U.S.A. (2016) 113:E5434–43. doi: 10.1073/pnas.1521069113
191. Kidder K, Bian Z, Shi L, Liu Y. Inflammation unrestrained by sirpalpha induces secondary hemophagocytic lymphohistiocytosis independent of ifn-gamma. J Immunol. (2020) 205:2821–33. doi: 10.4049/jimmunol.2000652
192. Okuzawa C, Kaneko Y, Murata Y, Miyake A, Saito Y, Okajo J, et al. Resistance to collagen-induced arthritis in shps-1 mutant mice. Biochem Biophys Res Commun. (2008) 371:561–6. doi: 10.1016/j.bbrc.2008.04.124
193. Nishimura T, Saito Y, Washio K, Komori S, Respatika D, Kotani T, et al. Sirpalpha on cd11c(+) cells induces th17 cell differentiation and subsequent inflammation in the cns in experimental autoimmune encephalomyelitis. Eur J Immunol. (2020) 50:1560–70. doi: 10.1002/eji.201948410
194. Wong AS, Mortin-Toth S, Sung M, Canty AJ, Gulban O, Greaves DR, et al. Polymorphism in the innate immune receptor sirpalpha controls cd47 binding and autoimmunity in the nonobese diabetic mouse. J Immunol. (2014) 193:4833–44. doi: 10.4049/jimmunol.1401984
195. Xie MM, Dai B, Hackney JA, Sun T, Zhang J, Jackman JK, et al. An agonistic anti-signal regulatory protein alpha antibody for chronic inflammatory diseases. Cell Rep Med. (2023) 4:101130. doi: 10.1016/j.xcrm.2023.101130
196. Morshed N, Ralvenius WT, Nott A, Watson LA, Rodriguez FH, Akay LA, et al. Phosphoproteomics identifies microglial siglec-F inflammatory response during neurodegeneration. Mol Syst Biol. (2020) 16:e9819. doi: 10.15252/msb.20209819
197. Zhang M, Angata T, Cho JY, Miller M, Broide DH, Varki A. Defining the in vivo function of siglec-F, a cd33-related siglec expressed on mouse eosinophils. Blood. (2007) 109:4280–7. doi: 10.1182/blood-2006-08-039255
198. Beavitt SJ, Harder KW, Kemp JM, Jones J, Quilici C, Casagranda F, et al. Lyn-deficient mice develop severe, persistent asthma: lyn is a critical negative regulator of th2 immunity. J Immunol. (2005) 175:1867–75. doi: 10.4049/jimmunol.175.3.1867
199. Simon HU, Yousefi S, Dibbert B, Hebestreit H, Weber M, Branch DR, et al. Role for tyrosine phosphorylation and lyn tyrosine kinase in fas receptor-mediated apoptosis in eosinophils. Blood. (1998) 92:547–57. doi: 10.1182/blood.V92.2.547
200. Mao H, Kano G, Hudson SA, Brummet M, Zimmermann N, Zhu Z, et al. Mechanisms of siglec-F-induced eosinophil apoptosis: A role for caspases but not for shp-1, src kinases, nadph oxidase or reactive oxygen. PloS One. (2013) 8:e68143. doi: 10.1371/journal.pone.0068143
201. Zhang J, Raper A, Sugita N, Hingorani R, Salio M, Palmowski MJ, et al. Characterization of siglec-H as a novel endocytic receptor expressed on murine plasmacytoid dendritic cell precursors. Blood. (2006) 107:3600–8. doi: 10.1182/blood-2005-09-3842
202. Swiecki M, Wang Y, Riboldi E, Kim AH, Dzutsev A, Gilfillan S, et al. Cell depletion in mice that express diphtheria toxin receptor under the control of siglech encompasses more than plasmacytoid dendritic cells. J Immunol. (2014) 192:4409–16. doi: 10.4049/jimmunol.1303135
203. Blasius AL, Colonna M. Sampling and signaling in plasmacytoid dendritic cells: the potential roles of siglec-H. Trends Immunol. (2006) 27:255–60. doi: 10.1016/j.it.2006.04.005
204. Blasius AL, Cella M, Maldonado J, Takai T, Colonna M. Siglec-H is an ipc-specific receptor that modulates type I ifn secretion through dap12. Blood. (2006) 107:2474–6. doi: 10.1182/blood-2005-09-3746
205. Takagi H, Fukaya T, Eizumi K, Sato Y, Sato K, Shibazaki A, et al. Plasmacytoid dendritic cells are crucial for the initiation of inflammation and T cell immunity in vivo. Immunity. (2011) 35:958–71. doi: 10.1016/j.immuni.2011.10.014
206. Schmitt H, Sell S, Koch J, Seefried M, Sonnewald S, Daniel C, et al. Siglec-H protects from virus-triggered severe systemic autoimmunity. J Exp Med. (2016) 213:1627–44. doi: 10.1084/jem.20160189
207. Mason LH, Willette-Brown J, Taylor LS, McVicar DW. Regulation of ly49d/dap12 signal transduction by src-family kinases and cd45. J Immunol. (2006) 176:6615–23. doi: 10.4049/jimmunol.176.11.6615
208. Szumilas N, Corneth OBJ, Lehmann CHK, Schmitt H, Cunz S, Cullen JG, et al. Siglec-H-Deficient Mice Show Enhanced Type I Ifn Responses, but Do Not Develop Autoimmunity after Influenza or Lcmv Infections. Front Immunol. (2021) 12:698420. doi: 10.3389/fimmu.2021.698420
209. Woodfin A, Voisin MB, Nourshargh S. Pecam-1: A multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol. (2007) 27:2514–23. doi: 10.1161/ATVBAHA.107.151456
210. Privratsky JR, Newman DK, Newman PJ. Pecam-1: conflicts of interest in inflammation. Life Sci. (2010) 87:69–82. doi: 10.1016/j.lfs.2010.06.001
211. Ming Z, Hu Y, Xiang J, Polewski P, Newman PJ, Newman DK. Lyn and pecam-1 function as interdependent inhibitors of platelet aggregation. Blood. (2011) 117:3903–6. doi: 10.1182/blood-2010-09-304816
212. Wong MX, Roberts D, Bartley PA, Jackson DE. Absence of platelet endothelial cell adhesion molecule-1 (Cd31) leads to increased severity of local and systemic ige-mediated anaphylaxis and modulation of mast cell activation. J Immunol. (2002) 168:6455–62. doi: 10.4049/jimmunol.168.12.6455
213. Merchand-Reyes G, Robledo-Avila FH, Buteyn NJ, Gautam S, Santhanam R, Fatehchand K, et al. Cd31 acts as a checkpoint molecule and is modulated by fcgammar-mediated signaling in monocytes. J Immunol. (2019) 203:3216–24. doi: 10.4049/jimmunol.1900059
214. Rui Y, Liu X, Li N, Jiang Y, Chen G, Cao X, et al. Pecam-1 ligation negatively regulates tlr4 signaling in macrophages. J Immunol. (2007) 179:7344–51. doi: 10.4049/jimmunol.179.11.7344
215. Lovelace MD, Yap ML, Yip J, Muller W, Wijburg O, Jackson DE. Absence of platelet endothelial cell adhesion molecule 1, pecam-1/cd31, in vivo increases resistance to salmonella enterica serovar typhimurium in mice. Infect Immun. (2013) 81:1952–63. doi: 10.1128/IAI.01295-12
216. Wilkinson R, Lyons AB, Roberts D, Wong MX, Bartley PA, Jackson DE. Platelet endothelial cell adhesion molecule-1 (Pecam-1/cd31) acts as a regulator of B-cell development, B-cell antigen receptor (Bcr)-mediated activation, and autoimmune disease. Blood. (2002) 100:184–93. doi: 10.1182/blood-2002-01-0027
217. Han J, Zhang G, Welch EJ, Liang Y, Fu J, Vogel SM, et al. A critical role for lyn kinase in strengthening endothelial integrity and barrier function. Blood. (2013) 122:4140–9. doi: 10.1182/blood-2013-03-491423
218. Wimmer I, Tietz S, Nishihara H, Deutsch U, Sallusto F, Gosselet F, et al. Pecam-1 stabilizes blood-brain barrier integrity and favors paracellular T-cell diapedesis across the blood-brain barrier during neuroinflammation. Front Immunol. (2019) 10:711. doi: 10.3389/fimmu.2019.00711
219. Kim WM, Huang YH, Gandhi A, Blumberg RS. Ceacam1 structure and function in immunity and its therapeutic implications. Semin Immunol. (2019) 42:101296. doi: 10.1016/j.smim.2019.101296
220. Gray-Owen SD, Blumberg RS. Ceacam1: contact-dependent control of immunity. Nat Rev Immunol. (2006) 6:433–46. doi: 10.1038/nri1864
221. Skubitz KM, Campbell KD, Ahmed K, Skubitz AP. Cd66 family members are associated with tyrosine kinase activity in human neutrophils. J Immunol. (1995) 155:5382–90. doi: 10.4049/jimmunol.155.11.5382
222. Lu R, Pan H, Shively JE. Ceacam1 negatively regulates il-1beta production in lps activated neutrophils by recruiting shp-1 to a syk-tlr4-ceacam1 complex. PloS Pathog. (2012) 8:e1002597. doi: 10.1371/journal.ppat.1002597
223. Pan H, Shively JE. Carcinoembryonic antigen-related cell adhesion molecule-1 regulates granulopoiesis by inhibition of granulocyte colony-stimulating factor receptor. Immunity. (2010) 33:620–31. doi: 10.1016/j.immuni.2010.10.009
224. Horst AK, Bickert T, Brewig N, Ludewig P, van Rooijen N, Schumacher U, et al. Ceacam1+ Myeloid cells control angiogenesis in inflammation. Blood. (2009) 113:6726–36. doi: 10.1182/blood-2008-10-184556
225. Roberts ME, Barvalia M, Silva J, Cederberg RA, Chu W, Wong A, et al. Deep phenotyping by mass cytometry and single-cell rna-sequencing reveals lyn-regulated signaling profiles underlying monocyte subset heterogeneity and lifespan. Circ Res. (2020) 126:e61–79. doi: 10.1161/CIRCRESAHA.119.315708
226. Gottschalk TA, Vincent FB, Hoi AY, Hibbs ML. Granulocyte colony-stimulating factor is not pathogenic in lupus nephritis. Immun Inflammation Dis. (2021) 9(3):758–70. doi: 10.1002/iid3.430
227. Verhagen AM, Wallace ME, Goradia A, Jones SA, Croom HA, Metcalf D, et al. A kinase-dead allele of lyn attenuates autoimmune disease normally associated with lyn deficiency. J Immunol. (2009) 182:2020–9. doi: 10.4049/jimmunol.0803127
228. Barouch-Bentov R, Che J, Lee CC, Yang Y, Herman A, Jia Y, et al. A conserved salt bridge in the G loop of multiple protein kinases is important for catalysis and for in vivo lyn function. Mol Cell. (2009) 33:43–52. doi: 10.1016/j.molcel.2008.12.024
229. Poderycki M, Tomimori Y, Ando T, Xiao W, Maeda-Yamamoto M, Sauer K, et al. A minor catalytic activity of src family kinases is sufficient for maximal activation of mast cells via the high-affinity ige receptor. J Immunol. (2010) 184:84–93. doi: 10.4049/jimmunol.0901590
230. Almuttaqi H, Udalova IA. Advances and challenges in targeting irf5, a key regulator of inflammation. FEBS J. (2019) 286:1624–37. doi: 10.1111/febs.14654
231. Song S, De S, Nelson V, Chopra S, LaPan M, Kampta K, et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J Clin Invest. (2020) 130(12):6700–17. doi: 10.1172/JCI120288
232. Kong E, Li Y, Ma P, Zhang Y, Ding R, Hua T, et al. Lyn-mediated glycolysis enhancement of microglia contributes to neuropathic pain through facilitating irf5 nuclear translocation in spinal dorsal horn. J Cell Mol Med. (2023) 27:1664–81. doi: 10.1111/jcmm.17759
233. International Consortium for Systemic Lupus Erythematosus G, Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in itgam, pxk, kiaa1542 and other loci. Nat Genet. (2008) 40:204–10. doi: 10.1038/ng.81
234. Lu R, Vidal GS, Kelly JA, Delgado-Vega AM, Howard XK, Macwana SR, et al. Genetic associations of lyn with systemic lupus erythematosus. Genes Immun. (2009) 10:397–403. doi: 10.1038/gene.2009.19
235. Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. (2015) 47:1457–64. doi: 10.1038/ng.3434
236. Julia A, Lopez-Longo FJ, Perez Venegas JJ, Bonas-Guarch S, Olive A, Andreu JL, et al. Genome-wide association study meta-analysis identifies five new loci for systemic lupus erythematosus. Arthritis Res Ther. (2018) 20:100. doi: 10.1186/s13075-018-1604-1
237. Liossis SN, Solomou EE, Dimopoulos MA, Panayiotidis P, Mavrikakis MM, Sfikakis PP. B-cell kinase lyn deficiency in patients with systemic lupus erythematosus. J Investig Med. (2001) 49:157–65. doi: 10.2310/6650.2001.34042
238. Flores-Borja F, Kabouridis PS, Jury EC, Isenberg DA, Mageed RA. Decreased lyn expression and translocation to lipid raft signaling domains in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum. (2005) 52:3955–65. doi: 10.1002/art.21416
239. Liu Y, Dong J, Mu R, Gao Y, Tan X, Li Y, et al. Microrna-30a promotes B cell hyperactivity in patients with systemic lupus erythematosus by direct interaction with lyn. Arthritis Rheum. (2013) 65:1603–11. doi: 10.1002/art.37912
240. Liossis S-NC. The abnormal signaling of the B cell receptor and co-receptors of lupus B cells. Clin Immunol. (2024) 263:110222. doi: 10.1016/j.clim.2024.110222
241. Manjarrez-Orduno N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, et al. Csk regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat Genet. (2012) 44:1227–30. doi: 10.1038/ng.2439
242. Chen W, Hong SH, Jenks SA, Anam FA, Tipton CM, Woodruff MC, et al. Distinct transcriptomes and autocrine cytokines underpin maturation and survival of antibody-secreting cells in systemic lupus erythematosus. Nat Commun. (2024) 15:1899. doi: 10.1038/s41467-024-46053-w
243. Panousis NI, Bertsias GK, Ongen H, Gergianaki I, Tektonidou MG, Trachana M, et al. Combined genetic and transcriptome analysis of patients with sle: distinct, targetable signatures for susceptibility and severity. Ann Rheum Dis. (2019) 78:1079–89. doi: 10.1136/annrheumdis-2018-214379
244. Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell. (2016) 165:551–65. doi: 10.1016/j.cell.2016.03.008
245. Hoffman RW, Merrill JT, Alarcon-Riquelme MM, Petri M, Dow ER, Nantz E, et al. Gene expression and pharmacodynamic changes in 1,760 systemic lupus erythematosus patients from two phase iii trials of baff blockade with tabalumab. Arthritis Rheumatol. (2017) 69:643–54. doi: 10.1002/art.39950
246. Boulet I, Ralph S, Stanley E, Lock P, Dunn AR, Green SP, et al. Lipopolysaccharide- and interferon-gamma-induced expression of hck and lyn tyrosine kinases in murine bone marrow-derived macrophages. Oncogene. (1992) 7:703–10.
247. Karampetsou MP, Andonopoulos AP, Liossis SN. Treatment with tnfalpha blockers induces phenotypical and functional aberrations in peripheral B cells. Clin Immunol. (2011) 140:8–17. doi: 10.1016/j.clim.2011.01.012
Keywords: SLE, lupus, Lyn tyrosine kinase, Src family kinases, immunoreceptor signaling, inhibitory signaling, autoimmune disease, autoinflammation
Citation: L’Estrange-Stranieri E, Gottschalk TA, Wright MD and Hibbs ML (2024) The dualistic role of Lyn tyrosine kinase in immune cell signaling: implications for systemic lupus erythematosus. Front. Immunol. 15:1395427. doi: 10.3389/fimmu.2024.1395427
Received: 03 March 2024; Accepted: 17 June 2024;
Published: 28 June 2024.
Edited by:
Jennifer L. Barnas, University of Rochester, United StatesReviewed by:
Anne Satterthwaite, University of Texas Southwestern Medical Center, United StatesCopyright © 2024 L’Estrange-Stranieri, Gottschalk, Wright and Hibbs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margaret L. Hibbs, TWFyZ2FyZXQuSGliYnNAbW9uYXNoLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.