
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 April 2024
Sec. Molecular Innate Immunity
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1395035
 Stefania Bottardi1
Stefania Bottardi1 Taylorjade Layne1Ailyn C. Ramòn1,2Norreen Quansah1,2
Taylorjade Layne1Ailyn C. Ramòn1,2Norreen Quansah1,2 Hugo Wurtele1,2
Hugo Wurtele1,2 El Bachir Affar1,2
El Bachir Affar1,2 Eric Milot1,2*
Eric Milot1,2*Inflammation control is critical during the innate immune response. Such response is triggered by the detection of molecules originating from pathogens or damaged host cells by pattern-recognition receptors (PRRs). PRRs subsequently initiate intra-cellular signalling through different pathways, resulting in i) the production of inflammatory cytokines, including type I interferon (IFN), and ii) the initiation of a cascade of events that promote both immediate host responses as well as adaptive immune responses. All human PYRIN and HIN-200 domains (PYHIN) protein family members were initially proposed to be PRRs, although this view has been challenged by reports that revealed their impact on other cellular mechanisms. Of relevance here, the human PYHIN factor myeloid nuclear differentiation antigen (MNDA) has recently been shown to directly control the transcription of genes encoding factors that regulate programmed cell death and inflammation. While MNDA is mainly found in the nucleus of leukocytes of both myeloid (neutrophils and monocytes) and lymphoid (B-cell) origin, its subcellular localization has been shown to be modulated in response to genotoxic agents that induce apoptosis and by bacterial constituents, mediators of inflammation. Prior studies have noted the importance of MNDA as a marker for certain forms of lymphoma, and as a clinical prognostic factor for hematopoietic diseases characterized by defective regulation of apoptosis. Abnormal expression of MNDA has also been associated with altered levels of cytokines and other inflammatory mediators. Refining our comprehension of the regulatory mechanisms governing the expression of MNDA and other PYHIN proteins, as well as enhancing our definition of their molecular functions, could significantly influence the management and treatment strategies of numerous human diseases. Here, we review the current state of knowledge regarding PYHIN proteins and their role in innate and adaptive immune responses. Emphasis will be placed on the regulation, function, and relevance of MNDA expression in the control of gene transcription and RNA stability during cell death and inflammation.
Inflammation is a protective response produced by immune and non-immune cells in response to injury or infection, to restore tissue homeostasis through various repair processes (1). Inflammation can either be acute or chronic. Chronic inflammation, which is also referred to as long-term inflammation, is a disorganized form of inflammation sustained in time. It is common to various diseases, including cancers (reviewed in (2)). Acute inflammation is the rapid innate immune response to pathogen infection or tissue damage. Acute inflammation consists of two phases, initiation, and resolution. Initiation is characterized by the massive recruitment of neutrophils to the site of infection or tissue damage. Once the insult has been eliminated, neutrophils undergo cell death, and their recruitment diminishes. Monocytes involved in neutrophil clearance then accumulate at the site of infection or injured tissue. Both the initiation and resolution of acute inflammation are tightly regulated, and disruption of either phase of acute inflammation can have severe consequences (3, 4).
Type I and type II interferons (IFN) are critical for both phases of acute inflammation and for chronic inflammation. Their signalling leads to strong expression of IFN-stimulated genes (ISGs), which encode proteins that play a key role in innate and adaptive immune responses to pathogen infection (5, 6), and are also called upon during sterile inflammation induced in response to sterile cell death or tissue damage. IFN production and signalling are therefore strictly regulated to adequately respond to these insults (7). MNDA is a member of a large family of IFN inducible proteins, namely, the IFN inducible p200/hematopoietic IFN-inducible nuclear protein with a 200-amino-acid repeat (Ifi-200/HIN-200) domain (8–10). MNDA was first identified as a 55 kDa non-histone basic nuclear protein in the acute promyelocytic leukaemia cell line HL-60 (11, 12), and was subsequently found to be highly expressed in the human myeloid lineage, including primary myeloblasts, promyelocytes, peripheral monocytes, granulocytes and macrophages (13, 14). It is also expressed in several lymphoblastoid cell lines as well as in primary human leukaemia and lymphoma cells (14–18). MNDA expression has been shown to be strongly upregulated by type I IFN in myeloid and lymphoid cells (19).
Most Ifi-200/HIN-200 family factors are reported to act as pattern-recognition receptors (PRRs), which are critical for the sensing and binding of invading pathogen DNA or cytosolic self-DNA (20). The Ifi-200/HIN-200 protein family includes a sub-group of highly homologous murine [Ifi202a, Ifi202b, Ifi203, Ifi204, Ifi205, and Ifi206 (8)] and human (10, 14, 19, 21, 22) proteins. The latter includes IFN-γ inducible protein 16 (IFI16), absent in melanoma 2 (AIM2), IFN-inducible protein X/pyrin and HIN domain-containing protein 1 (IFIX/PYHIN1), and MNDA. In addition to the C-terminal HIN-200 domain(s), these proteins are characterised by an N-terminal PYRIN domain/domain in apoptosis and interferon response (PYD/DAPIN) (23, 24) (Figure 1). The organization of the PYD and HIN-200 domains is similar among these proteins, which are collectively referred to as the AIM2-like receptors (ALRs) or the PYHIN (contraction of PYRIN and HIN-200 domains) family (25).
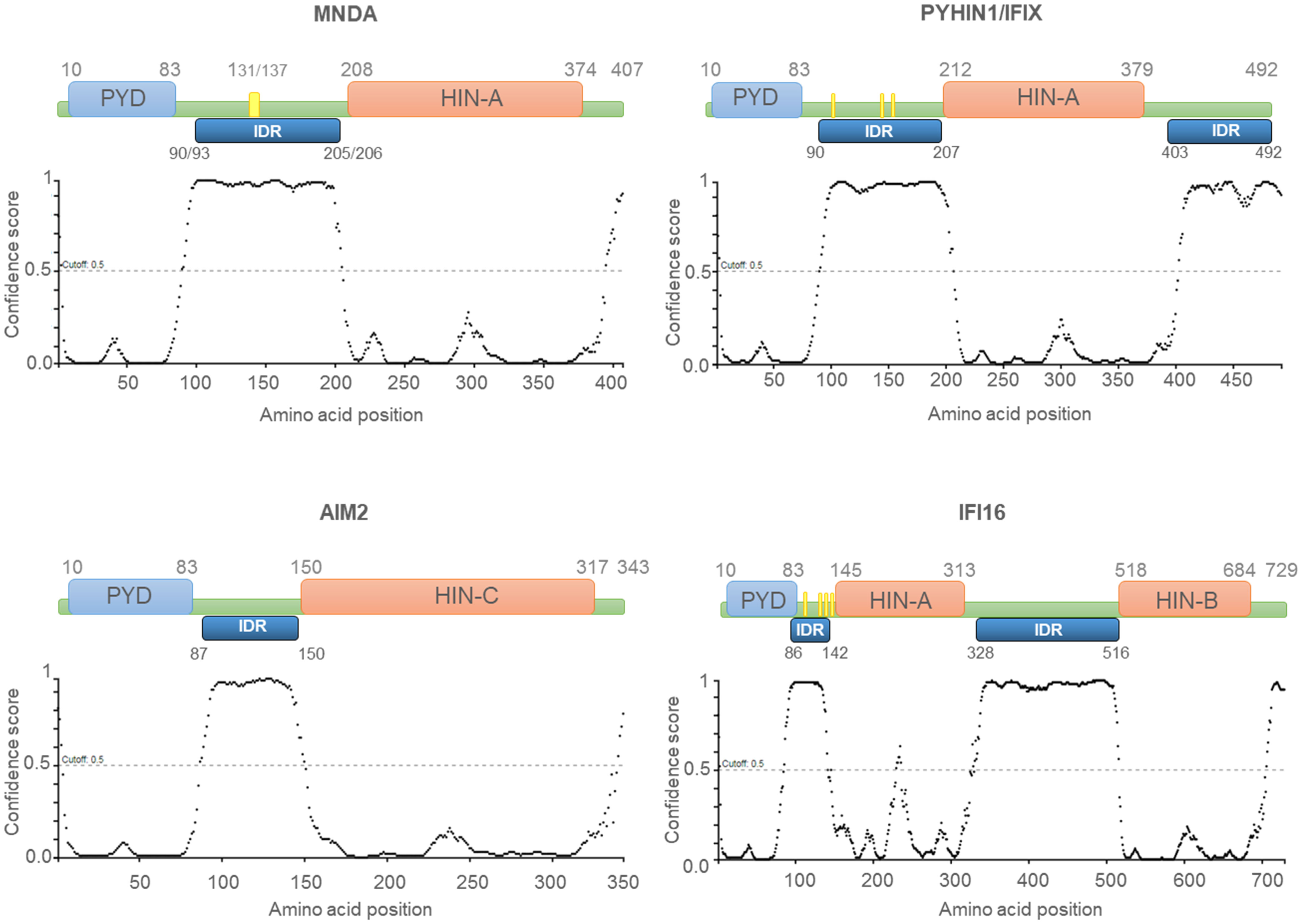
Figure 1 Human PYHIN proteins. Schematic diagram of the PYHIN protein structure with amino acid position. PYHIN proteins have a conserved domain architecture comprised of one PYRIN domain (PYD; light blue boxes), one or two hematopoietic IFN-inducible nuclear protein with a 200-amino-acid repeat (HIN-200; light orange boxes) domain/s, and at least one intrinsically disordered region (IDR; dark blue boxes). MNDA IDR consists of a stretch of 115/113 amino acids, spanning from residue 90/93 to residue 205/206. Except for AIM2, which has a predominant cytoplasmic distribution, the other PYHIN members contain either a multipartite nuclear localization signal (NLS) (PYHIN1/IFIX and IFI16) or a putative NLS (MNDA) (yellow boxes). IDR prediction was performed with PSIPRED (http://bioinf.cs.ucl.ac.uk/; 01/2024) and the results are plotted as scatter graphs. The canonical sequences of the PYHIN factors and the identification of their conserved domains were retrieved from the national library of medicine (NCBI) (https://www.ncbi.nlm.nih.gov/) and UniProt (https://www.uniprot.org/) databases. Reference sequences are as follows: MNDA: myeloid cell nuclear differentiation antigen [Homo sapiens]; NCBI Reference Sequence: NP_002423.1; UniProt ID: P41218. AIM2: Interferon-inducible protein AIM2 isoform 1 [Homo sapiens]; NCBI Reference Sequence: NP_004824.1; UniProt ID: O14862. IFI16: gamma-interferon-inducible protein 16 isoform 1 [Homo sapiens]; NCBI Reference Sequence: NP_001193496.1; UniProt ID: Q16666. PYHIN1/IFIX: Pyrin and HIN domain-containing protein 1 isoform alpha 1 [Homo sapiens]; NCBI Reference Sequence: NP_689714.2; UniProt ID: Q6K0P9. X-axis: amino acid number and position; Y-axis: confidence score.
Human PYHIN factors play important roles in the: (i) regulation of cell survival; (ii) innate and adaptive immune responses; and (iii) host resistance to tumours and viral infections (26–29). Some PYHIN factors were demonstrated to exert a critical function for exogenous DNA sensing, which can lead to the induction of IFNs and ISGs, or inflammasome regulation (30, 31). Sustained activation of IFN-mediated cellular responses, in particular those associated with inflammation-induced cell death, are drivers of autoimmune disorders such as systemic lupus erythematosus, systemic sclerosis, and Sjögren’s syndrome (32, 33). The mechanisms controlling IFN production and ISG induction are therefore of paramount importance to regulate tissue homeostasis, innate and adaptive immune responses, and to sustain immune surveillance of tumours.
The protein domains PYD and HIN-200 are critical for the biological functions of PYHIN factors. The PYD is a conserved motif composed of 80 amino acids, which can promote protein homo- and hetero-oligomerization (20, 24, 34). This domain is found in several proteins involved in signalling pathways associated with apoptosis, cell cycle and inflammation [reviewed in (8, 27, 29)]. Indeed, in response to sensing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), cytoplasmic PRRs containing PYDs can assemble to form a multimolecular platform known as the inflammasome (35). In turn, this leads to the activation of proinflammatory caspases and triggers the inflammatory cascade, which is essential to the innate immune response [reviewed in (36)].
The HIN-200 domain is highly conserved among mammals (37). Based on sequence similarities, three distinct classes of HIN-200 domains, HIN-A, HIN-B and HIN-C, have been described (Figure 1). The HIN-200 domains contain a tandem pair of β barrels or oligonucleotide/oligosaccharide binding (OB) formed by 70-80 amino acid residues (37–39), and is involved in protein interactions (39, 40). HIN-200 domains can also bind nucleic acids including single-stranded (ss) and double-stranded (ds) DNA via electrostatic interactions (20, 38, 41) between positively charged amino acids of the domain and phosphate groups of the DNA backbone (20). Despite their sequence similarity, the capacity of individual HIN-200 domain to bind specific DNA molecules is variable. Fluorescence polarization assays demonstrated that the interaction between the HIN-200 domain and the DNA is influenced by ionic strength but not by the DNA sequence (20). The affinity of these domains for nucleic acids can also be modulated by intra-molecular conformation of the protein. For instance, biomolecular interaction studies using alpha screen assays have established that while the HIN-B domain of IFI16 is sufficient for binding dsDNA, the presence of both HIN-A and HIN-B domains strengthen this association (42).
PYHIN factors contain other structural components that are likely to influence their affinity for nucleic acids. The in silico analysis of MNDA (43) as well as IFI16, AIM2, and IFIX revealed that they all possess at least one putative intrinsically disordered region (IDR) located between their PYD and HIN-200 domain (Figure 1). Most IDRs can interact with DNA and RNA molecules (44, 45), and can therefore influence the nucleic acid binding properties of PYHIN factors. In addition, consecutive aspartate (D) or glutamate (E) residues (referred to as D/E repeats) within the IDR domains can change the conformation from an open to a closed state through intramolecular electrostatic interactions with other functional domains, thereby causing auto-inhibition (46). This mechanism of auto-inhibition is of particular interest for PYHIN proteins, since a putative IDR located between their PYD and HIN-200 domains could facilitate conformational changes that occur after nucleic acid binding to the HIN-200 domain, as has been reported for AIM2 (Figure 2) [(20, 46) and see below]. Such a conformational change could ‘free’ the PYD and facilitate its interaction with other PYD-containing proteins.
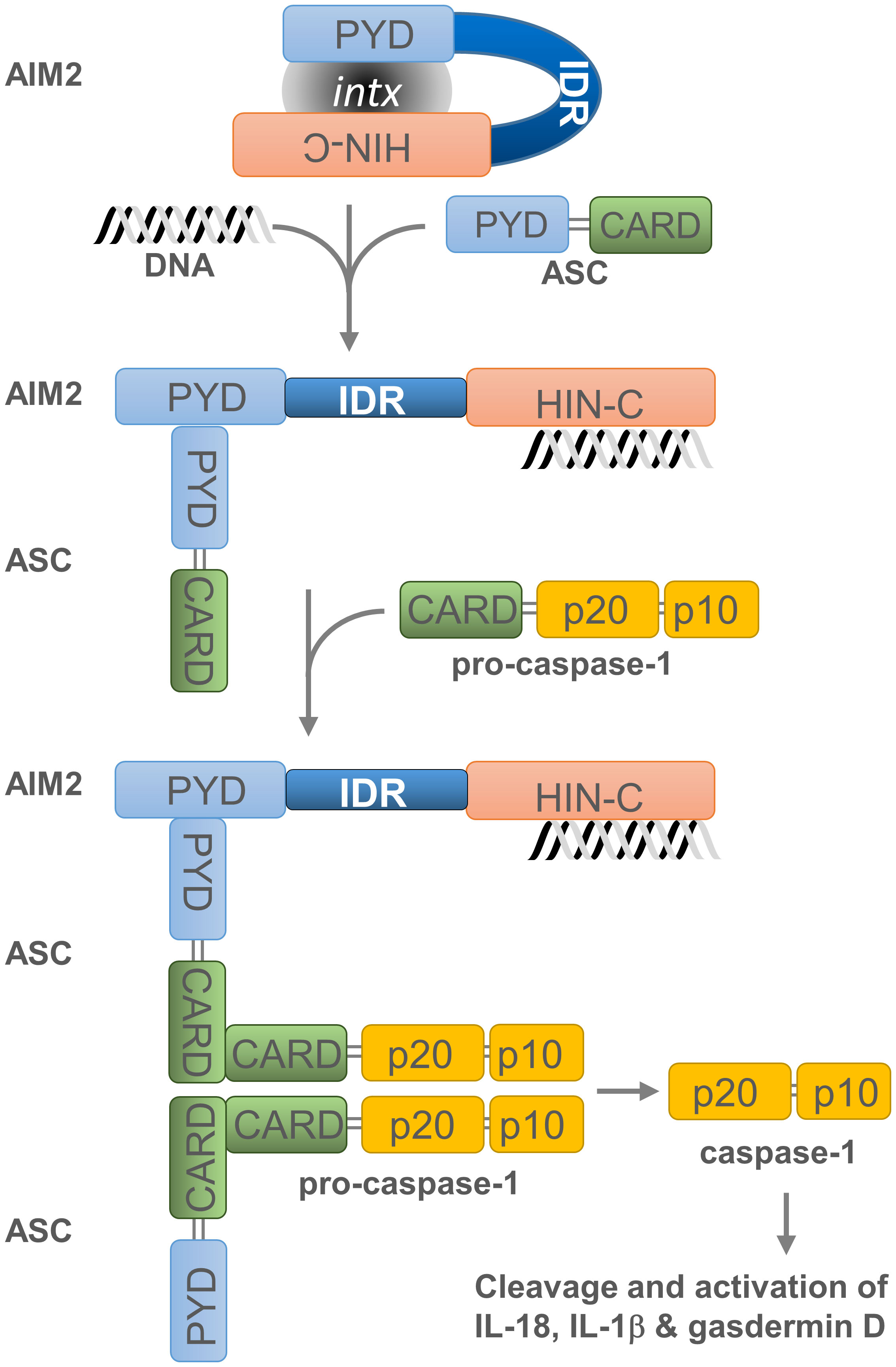
Figure 2 Assembly and activation of the AIM2 inflammasome. Overview of AIM2 inflammasome activation. Cytosolic dsDNA from invading pathogens or mislocated self-dsDNA (pathogen- or danger-associated molecular patterns: PAMPs or DAMPs) directly binds to the pattern recognition receptor (PRR) AIM2. This association, which relieves the intramolecular auto-inhibition imposed by the PYD-HIN-200 interaction, allows the recruitment of apoptosis-associated speck-like protein containing a CARD (ASC) as well as the binding and proximity-induced auto-proteolytic maturation of the pro-caspase-1. This inflammasome activation is likely to occur in the cytoplasmic compartment of the cell, where AIM2 is primarily located. Active caspase-1 can then cleave the pro-inflammatory cytokines IL-18 and IL-1β and the precursor of the pore-forming protein Gasdermin D (GSDMD). Cleaved GSDMD induces membrane pore formation, cell pyroptosis and the release of inflammatory cytokines. Grey oval indicated as ‘intx’ is a representation of the interaction between the PYD and HIN-200 (HIN-C) domains of AIM2.
Like AIM2, IFI16 can directly target and bind viral genomic DNA or RNA (47–51). Furthermore, IFI16, IFIX/PYHIN1, and MNDA play an anti-viral role by sequestering or interfering with Sp1-dependent gene expression, which is required for the replication of the human immunodeficiency virus 1 (HIV-1), the hepatitis B virus, and the simian virus 40 (SV40) as well as retro-transposition of long interspersed element-1 (LINE-1) sequences (13, 52). Mechanistically, the PYD region of these factors has been reported to compete with DNA to bind the transcription factor Sp1, thereby displacing the latter factor from viral gene promoters and interfering with Sp1-dependent viral gene expression (13). Interestingly, MNDA is involved in the regulation of IFN-α expression (53), which could also explain why it can complement the effect of viral genome detection by cytosolic sensors (13, 53).
AIM2 and IFI16 are the most thoroughly described PYHIN factors, and their biological importance has been well established. The characterization of their mechanisms of action has strongly influenced research into other PYHIN factors. The HIN-200 domains of AIM2 and IFI16 are essential for their interaction with pathogen dsDNA or dsRNA, and cytosolic mislocated self-DNA; because of this, the HIN-200 is critical for the activity of AIM2 and IFI16 as PRRs (29, 54), which is required to trigger inflammasome formation.
Untimely activation of AIM2-dependent inflammasome is prevented by several mechanisms. The auto-inhibition of AIM2 is due to the internal interaction between its PYD and HIN-200 domain (Figure 2). This inhibition is lifted when dsDNA binds to the HIN-200 domain which, in turn, enables dimerization of the AIM2 PYD with the PYD region of the inflammasome adaptor protein known as apoptosis-associated speck-like protein containing a CARD -or PYCARD- (ASC) (20). This PYD-PYD interaction favours the formation of a multimeric structure and permits the recruitment and activation of procaspase-1 through the CARD region of ASC. Subsequently, activated caspase-1 cleaves the pro-inflammatory cytokines interleukin 18 (IL-18) and 1β (IL-1β) (55–57) as well as the precursor of the pore-forming protein Gasdermin D (GSDMD) (58–61). This releases the N-terminal domain of GSDMD, which induces membrane pore formation, cell pyroptosis (or caspase-1-dependent and pro-inflammatory programmed cell death) (62), and the discharge of inflammatory cytokines (63).
While assembly and activation of the AIM2 inflammasome is dictated by the presence of cytosolic DNA, it is also dynamically influenced by interactions with other proteins. For example, PYD-only protein 3 (POP3) can inhibit AIM2 by interacting with its PYD region, thereby preventing the interaction of AIM2 with ASC and the consequent inflammasome formation (64). Similarly, the binding of murine Ifi202 to AIM2 interferes with AIM2-dependent signalling, preventing the assembly of the inflammasome that occurs after DNA recognition (65).
IFI16 has emerged as a component of an atypical, nuclear inflammasome. Upon detection of viral, microbial, or mislocated (cytoplasmic) self-DNA, IFI16 can form a functional AIM2-independent inflammasome that promotes caspase-1 activation and IL-1β cleavage (54). The PRR activity of IFI16 can also activate the stimulator of the interferon gene (STING) pathway in a cyclic GMP-AMP synthase (cGAS)-independent manner (30, 31, 66, 67). STING is associated with the endoplasmic reticulum (ER) and can detect cytosolic DNA or cyclic dinucleotides (68). Upon binding to its ligands, STING translocates from the ER to the Golgi (69, 70), where it recruits TANK binding kinase 1 (TBK1) also known as the nuclear factor kappa B (NF-κB) activating kinase. The TBK1 kinase activity elicits the nuclear relocalization and activation of NF-κB, as well as the phosphorylation, nuclear transfer and activation of interferon regulatory factor 3 (IRF3) (71–76). NF-κB then synergizes with IRF3 to activate the transcription of genes encoding type I IFNs and ISGs (77).
In addition to the above, a recent study showed that in response to viral RNA infection, IFI16 promotes retinoic acid-inducible gene l (RIG1) (also known as DEAD box protein 58 -DDX58-) gene expression and binds viral dsRNA through its HIN-A domain, which leads to a conformational change that facilitates the interaction between the PYD domains of IFI16 and RIG-1 (47). This interaction triggers RIG-1 polyubiquitination, activation, and association with mitochondrial antiviral signalling (MAVS) protein. This in turn triggers TBK1 activation, nuclear re-localization of IRF3, interferon regulatory factor 7 (IRF7), and NF-κB, and consequent transcriptional activation of ISGs and type I IFNs (53, 78, 79). Thoresen et al. (80) have shown that this RIG-1 signalling can occur rapidly during inflammation and is mechanistically distinct from RIG-1 oligomerization and aggregation that typically take place at the mitochondria membrane. The latter situation occurs subsequently, upon type I IFN induction, and leads to inflammasome activation as well as cell pyroptosis (62). Indeed, inflammasome activation and pyroptosis are usually observed at later stages of the acute phase of inflammation and during chronic inflammation [reviewed in (36, 81)].
The mechanism through which IFI16 coordinates inflammasome formation and the transcriptional activation of ISGs and type I IFN genes, is currently unclear. Interestingly, several IFI16 isoforms have been isolated (82). The short isoform IFI16-β, which does not include the PYD region, can interact with AIM2, thereby interfering with the formation of a functional AIM2-ASC complex (83). Thus, unlike the full-length protein, IFI16-β exerts a negative effect on inflammasome activation (54, 84, 85). IFI16-β contains both HIN-200 domains of the full-length protein and maintains the capacity to interact with and activate STING (83). Overall, the data suggests that IFI16-β synthesis during inflammation may dissociate the effect of IFI16 on ISG and type I IFN gene induction from its role in inflammasome activation.
Although IFIX and MNDA possess an HIN-200 domain, whether these proteins function as sensors of pathogen DNA is debated (23, 53, 86). It is also unknown whether they have the ability to interact with AIM2 and affect the AIM2 inflammasome activation, or if they can initiate inflammasome formation independently. Since IFIX and MNDA are located in the nucleus, they may contribute to the dynamics of the nuclear inflammasome or other aspects of the inflammatory process. Importantly, recent studies provide direct evidence that MNDA can control the expression of genes required for the cellular response to infection or sterile inflammation. This will be discussed more extensively below.
Proteomic and cellular localization studies have further defined the role of PYHIN factors. The protein interactome of individual overexpressed PYHIN factors in human HEK293 cells consists of over 300 proteins, with most interactors acting in intracellular signalling, cell cycle control, apoptosis, and transcriptional regulation. In addition, IFI16 and MNDA interactomes contain proteins involved in ribosome biogenesis and pre-rRNA processing, whereas IFIX and AIM2 strongly associate with proteins that modulate chromatin structure and DNA damage response (86). In the case of AIM2, these results are somewhat surprising: indeed, unlike IFI16 and IFIX, which contain a multipartite nuclear localization signal (NLS) (82, 87), or MNDA, which contains a putative NLS and shows a nuclear localization at steady state condition (88), AIM2 is mainly cytoplasmic. Nonetheless, AIM2 nuclear localization cannot be formally excluded (89), and the identified AIM2 interactome supports the idea that the cellular localization of PYHIN factors is relevant for their function.
Interestingly, while IFI16 and MNDA are predominantly nuclear, accumulation of these proteins in the cytosol has been detected in different cell types under certain conditions ( (31, 43, 90) and below). For instance, IFI16 has been detected in the cytoplasm of U2OS (91) and HeLa (92) cells, in UVB exposed keratinocytes (93), and in fibroblasts and endothelial cells upon infection with human herpesviruses, such as the Herpes simplex virus type 1 (HSV-1), the Kaposi’s sarcoma-associated herpesvirus (KSHV), and the Epstein-Barr virus (EBV) (54, 94–97). Once viral genomes are detected by IFI16 in the host cells, this PYHIN factor associates with the histone acetyltransferase p300, leading to the acetylation of several lysine residues within the IFI16 bipartite NLS (94, 98). These post-translational modifications impede the nuclear import of newly synthesized IFI16, thereby confining the protein to the cytoplasm.
MNDA is also relocated to the cytoplasm in response to signals related to the programmed cell death of neutrophils, and upon genotoxic stress or apoptosis induction in myeloid and lymphoid cells (43, 90). No post-translational modification has been associated with MNDA relocalization to the cytoplasm. Instead, cytoplasmic accumulation of MNDA has been linked to its cleavage by effector caspases (see following section).
MNDA has emerged as a stress-responsive factor whose localization undergoes dramatic changes during stress and following treatment with apoptosis-inducing agents. The predominant nucleolar localization of MNDA (43, 90, 99) was shown to be disrupted during programmed cell death and upon apoptosis induction or cellular stress induced by UVC radiation or cisplatin treatment (43, 90), with the protein being rapidly released into the nucleoplasm and subsequently relocated to the cytoplasm.
The ability of MNDA to bind dsDNA (100, 101) and its predominant localization at the nucleolus led authors to propose that it may act as a transcription factor (102). In silico analyses further suggested that it functions as a core transcription factor in monocytes (103, 104). Recent studies demonstrating that MNDA binds chromatin and modulates RNA polymerase II (Pol II) recruitment to myeloid cell leukaemia 1 (MCL1), B-cell lymphoma 2 (BCL2) and IRF7 genes (43, 53) provided direct evidence that MNDA participates to transcriptional regulation in leukocytes. While the HIN-200 domain and/or IDR are expected to be required for genomic DNA binding, no putative DNA binding site or consensus DNA sequence has been identified for MNDA. The assistance of other interacting partners, such as bona fide transcription factors (e.g., Yin Yang 1, or YY1) (105), may therefore be required for the targeting of MNDA to specific genes.
A critical step in the release of promoter-paused Pol II is the phosphorylation of negative elongation factor (NELF) and of the C-terminal domain (CTD) of Pol II by cyclin dependent kinase 9 (CDK9). This kinase is a component of the positive transcription elongation factor b (P-TEFb) complex, a key regulator of pre-mRNA synthesis of most genes in mammals (106–110). Bottardi et al. demonstrated that MNDA interacts with CDK9 and hexamethylene bis-acetamide-inducible protein 1 (HEXIM1) in myeloid and lymphoid cells, where it acts to control the expression of MCL1 and BCL2 genes (43). Indeed, in response to chemotherapeutic agents that induce genotoxic stress, MNDA binding to MCL1 and BCL2 pre-mRNAs is associated with lower levels of transcripts and consequently, MCL-1 and BCL-2 proteins. The effect of MNDA at these genes is not linked to variations in chromatin structure or to the compromised formation of the transcriptional pre-initiation complex. Instead, MNDA impairs optimal recruitment and distribution of Pol II and P-TEFb at the transcription start site and open reading frame of these genes. The peculiar chromatin distribution of Pol II and P-TEFb observed at specific genes in MNDA expressing cells (43), strongly suggests that MNDA hampers efficient transcriptional elongation by reducing Pol II processivity and affecting the reloading of P-TEFb and Pol II (43, 111). Overall, MNDA-dependent regulation of Pol II productive elongation and processing of RNA molecules contribute to fine-tuning the transcription of specific genes in both normal and stress-induced conditions.
Caspase-dependent cleavage of MNDA is associated with its relocalization and accumulation in the cytoplasm (90). The size of the MNDA protein fragments formed during caspase cleavage suggests that the PYD and HIN-200 domains are then separated into two different protein fragments (43, 90) (Figure 3). If, as in the case of AIM2, the PYD region of MNDA interacts with and is encumbered by its HIN-200 domain, this caspase-dependent cleavage may release the PYD region of MNDA, and promote its interaction with the PYD of ASC, which is required for formation and activation of the inflammasome (37) (Figures 2, 3). Further experiments will be necessary to define the precise effect of this cleavage and to identify the mechanism regulating this cleavage and, therefore, MNDA proteostasis.
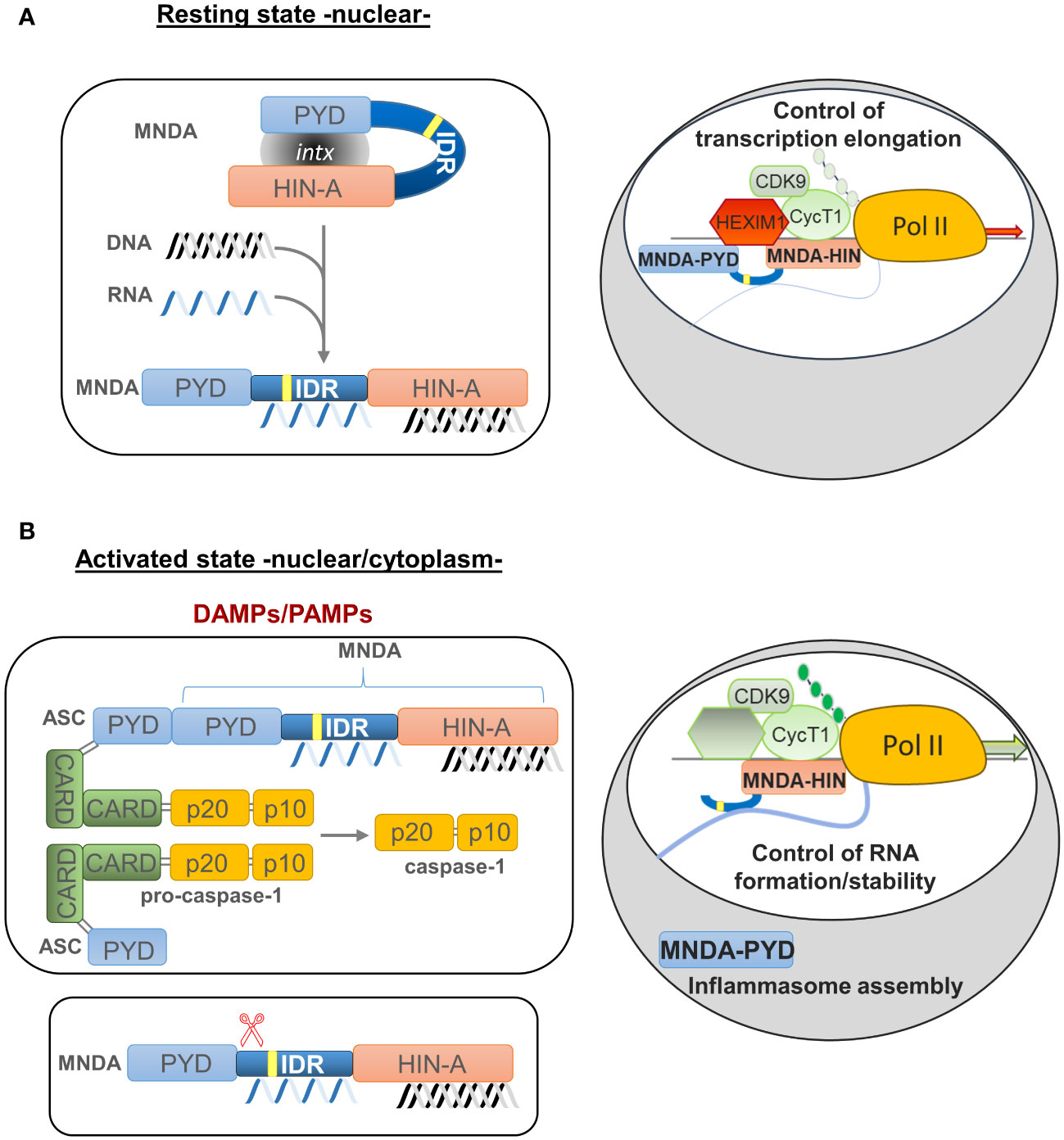
Figure 3 Model of MNDA nuclear function, cleavage and cytoplasmic relocalization. MNDA’s cellular localization and function vary in response to endogenous and exogenous stress signals. (A) Under normal conditions (resting state), MNDA’s PYD and HIN-200 (HIN-A) domains form an intramolecular association that results in a self-stabilized “resting” state, with the flexible link of the intrinsically disordered region (IDR) buckling. DNA binding to the HIN-200 domain and/or RNA association with the IDR or HIN-200 domain in the nucleoplasm weaken this intramolecular interaction and release constraints on the PYD signalling domain. The latter can then engage in homotypic or non-homotypic PYD-PYD interactions, for example with the nuclear protein ASC (apoptosis-associated speck-like protein containing a CARD) (not shown). MNDA can also associate with various nuclear proteins (86), including activating or repressive components of the positive transcription elongation factor b (P-TEFb) complex (43). In this “resting” state, MNDA could restrict the release of the promoter-proximal paused RNA polymerase II (Pol II) and, consequently, negatively affect transcriptional elongation of these genes. Grey oval indicated as ‘intx’ is a representation of the interaction between the PYD and HIN-200 (HIN-A) domains of MNDA. (B) In response to genotoxic or inflammation mediators such as danger- or pathogen-associated molecular patterns (DAMPs or PAMPs) (activated state), PYD-PYD interactions between MNDA and ASC proteins become possible, triggering inflammasome formation and, consequently, activation of pro-caspase-1. Among other substrates, catalytically active caspase-1 might induce proteolytic cleavage of MNDA. This would generate at least two characteristic MNDA fragments (43, 90). While the PYD-only fragment relocalizes to the cytoplasm where it may facilitate inflammasome assembly, the ‘IDR-HIN’ fragment would accumulate in the nucleus. Although it is not known whether the absence of PYD weakens the interaction of MNDA-HIN with repressive P-TEFb components such as HEXIM1, it has been shown that MNDA-dependent repression of transcription elongation is relieved following genotoxic stress (43). At the same time, MNDA may help stabilize newly transcribed RNA. This dual role of MNDA and the effect of MNDA on transcription elongation of MNDA targets, such as MCL1 and BCL2, could therefore be modulated by signals induced by genotoxic and inflammatory mediators.
The relocalization of MNDA from the nucleus to the cytoplasm correlates with the induction of effector caspases and cell death in circulating human neutrophils (90). Moreover, prolonged neutrophil survival, which results in the uncontrolled release of pro-inflammatory molecules and can lead to neutrophil-induced severe tissue injury propagating sepsis and possibly inducing septic shock (112–115), prevents MNDA cleavage and its cytoplasmic accumulation (90). Conversely, neutrophil cell death correlates with MNDA cleavage and accumulation in the cytoplasm. Likewise, in the myeloid cell line HL-60 exposed to genotoxic agents, MNDA expression promotes proteasomal degradation of the anti-apoptotic factor MCL-1 and favours apoptosis (90, 116). In agreement with these observations, MNDA also controls MCL-1 and BCL-2 protein and transcript levels in normal and leukemic B-cells, whereby higher MNDA expression levels are associated with markers of good clinical outcome (16, 43).
Consistent with their role as DNA sensors and immune activators, PYHIN factors are essential for rapid and powerful responses against endogenous and exogenous threats. Moreover, these factors must be tightly regulated to avoid inadequate innate immune response and dysregulated inflammasome activation. Accordingly, various genetic deletions of the human chromosomal 1q21-23 region, where MNDA and other PYHIN genes are clustered, have been linked to the development of diseases including breast and prostate cancers (21). Furthermore, the integrity of this chromosomal region is thought to be relevant to the regulation of acute and chronic inflammatory responses as well as autoimmune susceptibility [(117) and references therein]. Beside these large-scale rearrangements, point mutations and copy number variation of the MNDA gene have been identified in various hematopoietic cancers (Table 1). The association of abnormal MNDA expression levels and the onset of hematopoietic and inflammatory disorders are also consistent with the notion that MNDA dosage is critical during haematopoiesis (Table 2; Figure 4).
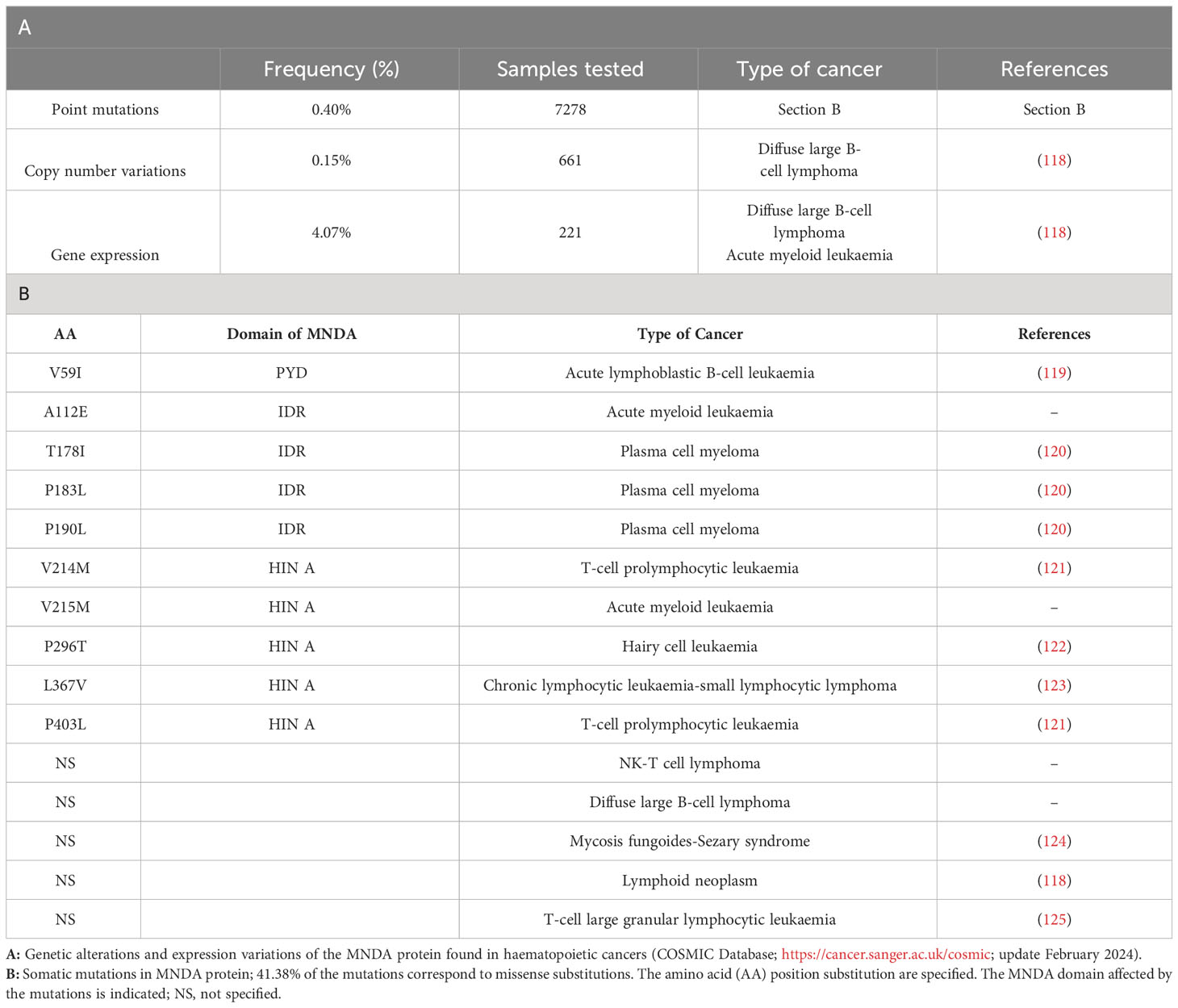
Table 1 MNDA mutations and expression level variations in hematopoietic malignant neoplasms.
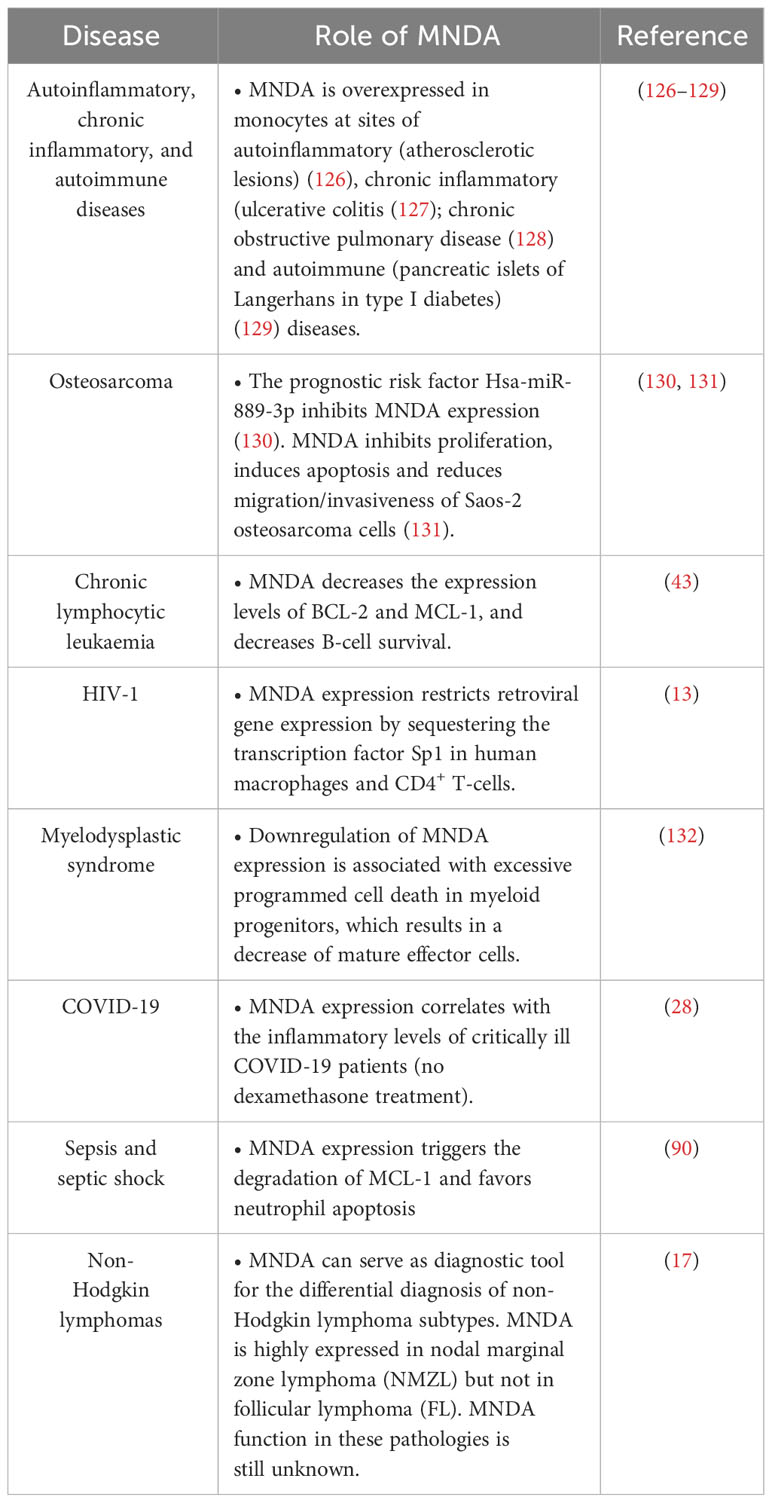
Table 2 MNDA expression levels and functions in various human diseases.
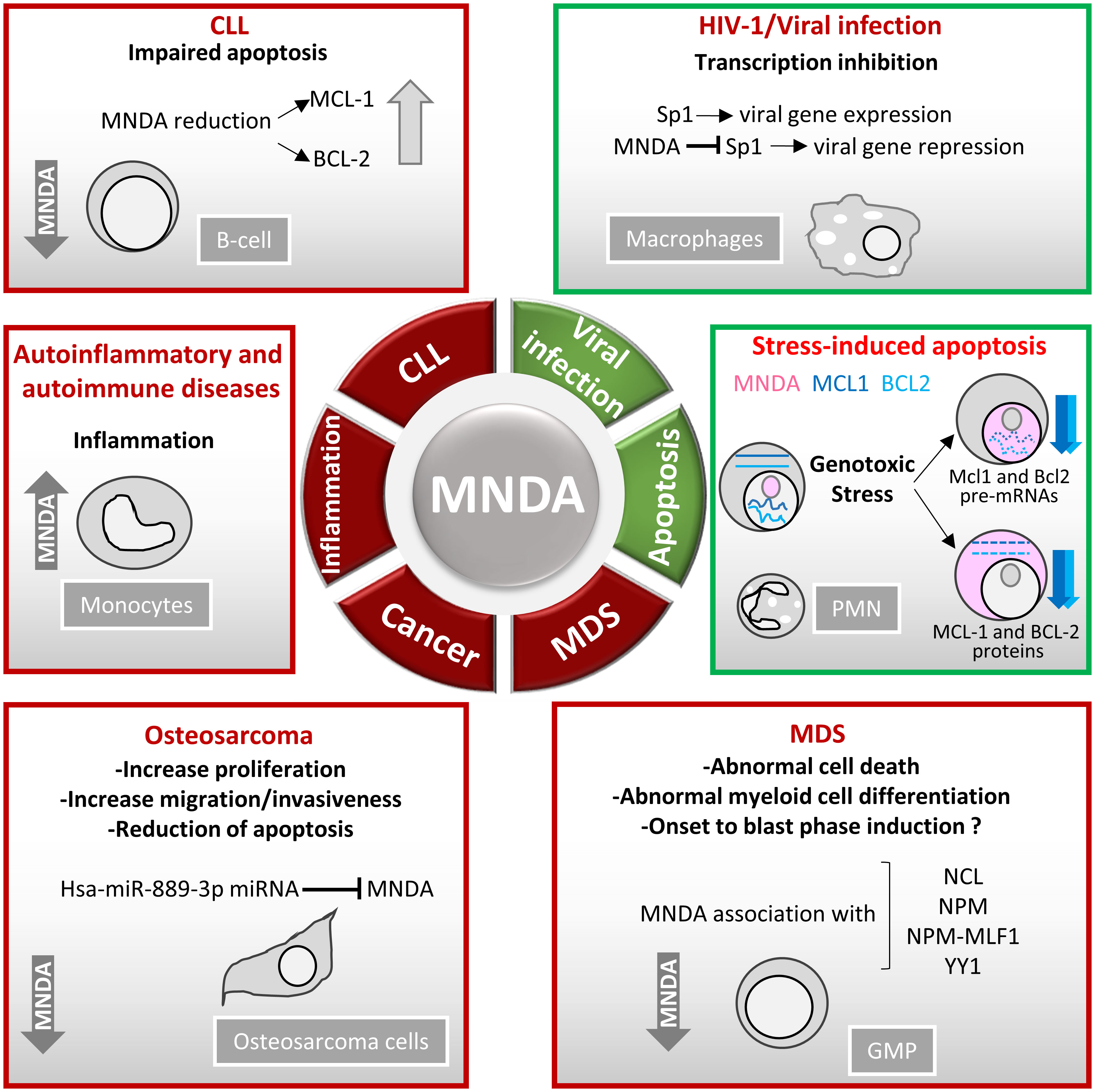
Figure 4 MNDA expression levels in human diseases. Schematic overview of the role of MNDA expression in human diseases and cellular functions. -In macrophages, MNDA can inhibit the DNA binding of the transcription factor Sp1 and thereby, blocks virus replication during human immunodeficiency virus 1 (HIV-1) infection (13). -Upon genotoxic stress in chronic lymphocytic leukaemia (CLL) cells or polymorphonuclear (PMN) leukocytes, MNDA represses MCL1 and BCL2 gene expression and enhances MCL-1 and BCL-2 protein degradation, promoting stress-induced apoptosis (90). -In granulocyte-monocyte progenitor (GMP) cells, reduced MNDA protein levels is associated with abnormal cell death and myeloid differentiation, and has been documented in familial (133) and sporadic (132–135) myelodysplastic syndromes (MDS). MNDA binds nucleophosmin 1 (NPM1) and the NPM-MLF1 (myeloid leukaemia factor 1) chimera product (99), which has been associated with MDS (136). Whether these associations alter trafficking signals and/or direct inappropriate cellular localization of the aforementioned proteins in MDS cells remains to be elucidated. MNDA also interacts with nucleolin (NCL) and Yin Yang 1 (YY1) proteins, which are involved in cell cycle regulation and genotoxic stress response (137, 138), suggesting a possible role of MNDA in the control of MDS cell survival. In accordance with this data and regardless the fact that the precise mechanism of action is still undefined, MNDA expression was proposed as an independent clinical marker for the evaluation of dyspoiesis (139). -In osteosarcoma cells, the Hsa-miR-889-3p microRNA, whose upregulation is a known prognostic risk factor (130), inhibits MNDA expression whereas MNDA overexpression counteracts proliferation, induces apoptosis and reduces migration and invasiveness in the SAOS-2 osteosarcoma model cell line (131). -MNDA overexpression has been documented in monocytes obtained from individuals diagnosed with autoinflammatory (atherosclerotic lesions) (126); autoimmune (pancreatic islets of Langerhans in type 1 diabetes) (129); chronic inflammatory diseases such as ulcerative colitis (127) and chronic obstructive pulmonary disease (128).
An association between PYHIN dysregulation and inflammatory disorders, e.g., autoimmune diseases and cancers, has been proposed to reflect PYHIN propensity to act as PRRs that sense and bind both exogenous and mislocated self-DNA (29, 140). Although there is no evidence that MNDA can act as a classical PRR (53), the regulation of transcription and apoptosis by this protein is likely to be important to control the inflammation response to infection and host cell damages. This view is supported by the strong correlation between the expression levels of MNDA and inflammation-related diseases and cancers. Indeed, MNDA is overexpressed in peripheral blood mononuclear cells of patients diagnosed with autoinflammatory diseases, such as inflammatory spondyloarthropathy (141); autoimmune diseases, like rheumatoid arthritis and psoriatic arthritis (141); and Sjögren patients (10, 142, 143). In line with the evidence that MNDA is overexpressed in macrophages at sites of inflammation (144), the number of MNDA positive monocytes in atherosclerotic aorta has been shown to increase during disease progression (126). The expression of MNDA is also significantly higher at inflammation sites in pancreatic islets of Langerhans isolated from type 1 diabetes patients, especially those developing complications like neuropathy [reviewed in (145)].
Cancer can be regarded as an inflammation-related disease. In particular, solid tumours are surrounded by stromal and inflammatory cells, which form the tumour microenvironment. In ‘hot’ tumours, this pro-inflammatory milieu can restraint tumour growth and enhance immunotherapeutic efficiency. However, in ‘cold’ tumours (146), the predominant myeloid-related innate immunity signature leads to the production of different mediators that can increase genomic DNA damage, and render the tumour cells refractory to immune checkpoint inhibitors and DNA repair mechanisms (147–149) while favouring immunotherapeutic resistance. It is therefore possible that factors modulating inflammation and hematopoietic cell death, such as MNDA, may act to limit cancer progression by controlling the extent of inflammation.
Mechanistically, cancer biology and inflammation rely on trained immunity, which is defined as the acquisition of memory by innate immune cells. It is a functional state that is associated with potent immune responses and can elicit robust adaptive immunity against infection and cancer. Initially considered to rely on monocytes and macrophages, trained immunity is also documented in neutrophils, natural killer (NK) cells, innate lymphoid cells and non-immune cells, like hematopoietic precursors, endothelial and epithelial cells (150). PYHIN proteins are expressed in epithelial cells (151, 152), which are nowadays considered as trained immunity-associated innate immune cells (153). Their possible roles as initial sensing of PAMPs and DAMPs and induction of ISG transcription in airway epithelial cells has been recently highlighted, in particular during respiratory viral infection such as caused by the single-stranded positive-sense RNA viruses known as severe acute respiratory syndrome coronavirus 2 (SARS‑CoV‑2) (154). Entry of SARS-CoV-2 into epithelial cells impairs the RIG-1 and melanoma differentiation-associated gene 5 (MDA-5) signalling and the localized first peak of IFN response. As a result, the required antiviral environment is not established and the ISG/IFN response, if any, must be assured by the underneath sentinel immune cells, such as macrophages. If these cells are not functionally intact and carry instead genetic defects (as described in young men with dysfunctional TLR7-variants), the IFN response by the host can be inadequate and dangerous [reviewed in (154)]. The specific example of SARS-CoV-2 infection emphasizes the role of PYHIN in cells others than sentinel immune cells, where they can add an extra layer of immunoprotection somehow specific to the tissue and type of perturbation. It is worth noting that it has been reported that the MNDA expression levels correlate with increased inflammatory levels during the acute phase of the disease (28).
The elevated expression of MNDA is a marker of various forms of myelodysplastic syndromes (MDS), which comprise erythro-myelopoiesis disorders. Indeed, MNDA expression has been proposed as an independent clinical marker for the evaluation of myeloid dyspoiesis (139). MDS are characterized by a loss of responsiveness of hematopoietic progenitor cells to environmental signals, as well as excessive apoptosis and/or susceptibility to programmed cell death (PCD) (155). PCD can be further classified as apoptotic or non-apoptotic cell death. The latter can take various forms, including pyroptosis, which is an immunoreactive form of cell death following recognition of intracellular pathogen sensors such as AIM2 or the nucleotide oligomerization domain (NOD)-like receptor (NLR) sensors. Unlike apoptosis, which is acknowledged as a form of non-inflammatory PCD, pyroptosis is mainly regulated by inflammation-related caspases, including caspase-1 that, upon activation, mediates membrane pore formation through the cleavage of GSDMD (see above) [reviewed in (156)]. It has been reported that a hallmark of MDS is the assembly of the NOD-like receptor protein 3 (NLRP3) inflammasome and the pyroptotic death of hematopoietic stem and progenitor cells, which can favour the cytopenia that marks the first phase of the disease (157). This is followed by a second or blastic phase, characterized by the accumulation of myeloid progenitors and reduced cell death (158). In approximately one third of the cases, MDS progresses to acute myeloid leukaemia (AML).
Granulocyte-macrophage progenitor cells isolated from familial and sporadic MDS patients present reduced MNDA expression (133–135). MDS cells are particularly sensitive to tumour necrosis factor (TNF)-related apoptosis inducing ligand (TRAIL)- apoptosis, a phenomenon that can be prevented in the K562 cell model by ectopic expression of MNDA (132). The effect of MNDA is probably specific to TRAIL-induced apoptosis during MDS since MNDA cannot protect K562 cells from caspase-dependent apoptotic inducers, e.g., H2O2 (132). In accordance with this, MNDA was demonstrated to act as a pro-apoptotic factor in several myeloid and lymphoid cell lines (90, 159).
Treatment regimens differ significantly based on the type of haematological disorder. Therefore, it is important to identify disease-specific biomarkers and characterize the molecular mechanisms behind these pathologies. Despite significant progress in biomarker classification (160, 161), several haematological diseases still lack specific biomarker profiles. For instance, the spreading of AML cells to different tissues and organs, which is referred to as extramedullary (EM) involvement (162), can be misdiagnosed and confused with large cell lymphoma or other lymphoproliferative disorders. In addition, EM AML are morphologically similar to blastic plasmacytoid dendritic cell neoplasm (BPDCN), which also arises at EM sites (163). Interestingly, MNDA can be detected by immunohistochemistry in the majority of EM AML tested, whereas in all cases of BPDCN, MNDA expression is undetectable (164).
MNDA can also be used as a diagnostic tool to distinguish between various forms of indolent low-grade lymphomas, which regroup slow growing mature B-cell non-Hodgkin lymphomas (NHL) such as marginal zone lymphoma (MZL) and follicular lymphoma (FL). MNDA is highly expressed in MZLs (nodal, extranodal, splenic) and in normal primary follicles (165), but is uncommon in FL (17, 166, 167).
Chronic lymphocytic leukaemia (CLL)/small lymphocytic lymphoma (SLL), which is a type of NHL, is a malignancy of monoclonal, mature, antigen-experienced, and apoptosis refractory CD19+/CD5+ B lymphoid cells, detected in peripheral blood at a concentration of ≥5000 cells/μL for a period of 3 months or more (168). This disease is called CLL when most of cancer cells are in the blood and bone marrow and SLL when they localize to the lymph nodes and spleen. However, these terminologies represent different forms of the same illness (which will be referred to as CLL hereafter). CLL is characterized by a heterogeneous clinical course. Numerous investigations indicate that CLL relapse is commonly associated with defects in pathways regulating the DNA damage response (in particular, mutations or deletion of the p53 tumour suppressor), inhibition of apoptosis (commonly caused by the overexpression of the BCL-2 family of anti-apoptotic proteins), and RNA metabolism (often affecting the splicing factor 3b subunit 3 -SF3B3-). Molecular and cytological defects have been regarded as direct consequences of the accumulation of somatic gene mutations acquired over time (169–171), as well as being the result of global and cumulative changes in the epigenetic landscape (172). Further characterization of known CLL biomarkers (173–176) and the identification of new biomarkers would be expected to improve prognostic accuracy, staging, and setting of novel therapeutic avenues. Unfortunately, with the notable exception of chromosome 13 focal deletion [del(13q)] (177), the functional impact of the somatic events that progressively transform normal B-cells towards a leukemic state have only been partially characterized, and because of this are by themselves inadequate to accurately define the disease outcome. Interestingly, the expression level of MNDA has been proposed as a predictive marker of the clinical progression of CLL (16, 43, 178). At the molecular level, an inverse correlation between the expression levels of MNDA and that of the anti-apoptotic proteins MCL-1 and BCL-2 has been observed in normal B- and CLL cells, confirming the importance of MNDA in the transcriptional control of BCL2 and MCL1 genes (43). CLL clones isolated from patients exhibiting reduced life expectancy and/or shorter disease-free period are indeed characterized by lower MNDA expression levels (16, 43). This observation is noteworthy since BCL-2 overexpression is found in most B-cell lymphoproliferative disorders, including CLL, and is responsible for the ‘apoptotic block’, which prevents cancer cells from initiating programmed cell death (179, 180). To counteract BCL-2 overexpression, BH3 mimetic drugs (like Venetoclax) and next generation BCL-2 inhibitors are suitable therapeutic choices (181, 182). However, their use in prolonged treatment frequently leads to resistance and clinical relapse. Among the best characterized mechanisms of resistance, are acquired BCL-2 mutations that reduce Venetoclax binding to BCL-2 (183), and overexpression of MCL-1 and BCL-XL proteins (184–186). Accordingly, MCL-1 overexpression is acknowledged as a negative prognostic marker in CLL and AML (43, 187–189). To counteract MCL-1 overexpression in refractory CLL, several MCL-1 inhibitors have been tested (190), either alone (191) or in combination with Venetoclax (192). However, MCL-1 inhibitors have been associated with significant side effects including cardiotoxicity (193, 194), limiting their therapeutic suitability. MCL1 transcriptional block has been assessed as clinical alternative, but this approach affects transcription broadly and cause toxic side effects [(149, 195) and references therein]. The finding that, in CLL cells, high MNDA expression is associated with downregulation of the anti-apoptotic factors MCL-1 and BCL-2 and with improved clinical course is promising. Further investigation is therefore justified to decipher how MNDA expression is controlled in CLL clones, and to fully define the molecular contribution of MNDA to the outcome of this disease.
In leukocytes, a precise coordination between apoptosis control and inflammation is required to regulate the response to PAMPs and DAMPs. The latter are primarily produced upon cell death resulting from infections or trauma, such as those caused by tissue lesions or genotoxins. DNA damage can be engendered by pathogens such as bacteria (reviewed in (196)), environmental pollution (like silica and asbestos particles, polycyclic aromatic hydrocarbons, pesticides, ionizing radiations, UV light, etc.), and anti-cancer drugs (such as alkylating agents) [(197) and references therein]. PAMPs and DAMPs trigger intracellular signalling leading to the activation of IFN genes and ISGs. IFN-regulated factors, including the PYHIN proteins, are therefore instrumental for innate and adaptive immune response to pathogen infection and also, to coordinate sterile inflammation (198). MNDA appears unlikely to act as a cytoplasmic DNA sensor, and does not activate the STING pathway or the ASC inflammasome cascade in HEK293 or THP-1 cells (53, 199). As discussed previously, the most important role of MNDA during inflammation is probably related to transcriptional control of genes required for the timely regulation of mechanisms governing the cellular response to infection or tissue damage. MNDA is not the unique PYHIN whose primary importance would be related to transcriptional regulation instead of PRR activity. Indeed, the mouse PYHIN protein Ifi207 is not involved in DNA sensing, but its association with active RNA polymerase II (RNA Pol II) and IRF7 in the nucleus is reported to enhance the transcriptional activation of IRF7-dependent cytokines, including the TNF gene in macrophages (200).
MNDA can induce IRF7 transcription, while repressing MCL1 and BCL2 transcription. The effect of MNDA on IRF7 results in increased expression of the type I interferon IFN-α (53). In addition to their critical role during inflammation, type I IFNs can synergize with apoptosis-promoting factors to increase caspases expression and create a molecular environment favourable for apoptosis (201). Thus, along with down-regulation of prosurvival MCL1 and BCL2 genes, IRF7 transcriptional induction may also be required for MNDA-mediated regulation of apoptosis in hematopoietic cells. Another important finding is that cytoplasmic MNDA favours proteasomal degradation of MCL-1, thereby promoting apoptosis (90). Interestingly, the beneficial proapoptotic effect of MNDA has been demonstrated in leukocytes during sepsis and in CLL cells, two haematological disorders characterized by dysregulated apoptosis.
Several important questions concerning the biology of MNDA and other PYHIN factors remain unanswered. Among them, their transcriptional regulation and the consequence of specific signalling events on PYHIN synthesis, stability and functional activation deserve further scrutiny. Investigation aimed at deciphering the intramolecular interactions between the PYD and HIN-200 domain, as well as the interactions between the various PYHIN factors, are expected to provide valuable information regarding the regulation of this family of factors. Further work is needed to explore how post-translational modifications can modulate the proteome of PYHIN factors and influence their cellular localization and function. Finally, since MNDA responds to different types of stress and controls the transcription of IRF7, it will be important to define whether this protein can influence cellular functions other than apoptosis, and to elucidate its importance in the context of innate and adaptive immune responses.
Whether acting as a PRR or stress-responsive transcriptional regulator, MNDA, together with the other PYHIN factors, are required for a timely response to pathogen infection or sterile inflammation. Further characterization of PYHIN factors is expected to lead to the development of novel therapies for haematological disorders characterized by abnormal apoptosis and inflammatory response.
SB: Conceptualization, Writing – original draft, Writing – review & editing, Visualization. TL: Writing – original draft, Writing – review & editing. AR: Writing – review & editing, Visualization. NQ: Visualization, Writing – review & editing. HW: Writing – review & editing, Funding acquisition. EA: Funding acquisition, Writing – review & editing. EM: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The author’s work is supported by grants from Natural Sciences and Engineering Research Council of Canada (NSERC) RGPIN-2019-05231 held by EM; by the NSERC RGPIN-2019-05082 held by HW; and by the NSERC, RGPIN-2021-04110 held by EBA. AR was supported by a scholarship award from the Fondation de l’Hôpital Maisonneuve-Rosemont.
The authors acknowledge the Centre de Recherche Hôpital Maisonneuve-Rosemont for providing facilities and other support, and their colleagues for useful discussions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Oronsky B, Caroen S, Reid T. What exactly is inflammation (and what is it not)? Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232314905
2. Fernandes Q, Inchakalody VP, Bedhiafi T, Mestiri S, Taib N, Uddin S, et al. Chronic inflammation and cancer; the two sides of a coin. Life Sci. (2024) 338:122390. doi: 10.1016/j.lfs.2023.122390
3. Alfaro S, Acuna V, Ceriani R, Cavieres MF, Weinstein-Oppenheimer CR, Campos-Estrada C. Involvement of inflammation and its resolution in disease and therapeutics. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms231810719
4. Milot E, Fotouhi-Ardakani N, Filep JG. Myeloid nuclear differentiation antigen, neutrophil apoptosis and sepsis. Front Immunol. (2012) 3:397. doi: 10.3389/fimmu.2012.00397
5. Kakiuchi T, Nariuchi H, Matsuhashi T. Fc receptors on IgM and IgG antibody-forming cells in an early immune response. Cell Immunol. (1981) 61:124–33. doi: 10.1016/0008-8749(81)90359-2
6. Kandalla PK, Subburayalu J, Cocita C, de Laval B, Tomasello E, Iacono J, et al. M-CSF directs myeloid and NK cell differentiation to protect from CMV after hematopoietic cell transplantation. EMBO Mol Med. (2023) 15:e17694. doi: 10.15252/emmm.202317694
7. Walter MR. The role of structure in the biology of interferon signalling. Front Immunol. (2020) 11:606489. doi: 10.3389/fimmu.2020.606489
8. Asefa B, Klarmann KD, Copeland NG, Gilbert DJ, Jenkins NA, Keller JR. The interferon-inducible p200 family of proteins: a perspective on their roles in cell cycle regulation and differentiation. Blood Cells Mol Dis. (2004) 32:155–67. doi: 10.1016/j.bcmd.2003.10.002
9. Fan X, Jiao L, Jin T. Activation and immune regulation mechanisms of PYHIN family during microbial infection. Front Microbiol. (2021) 12:809412. doi: 10.3389/fmicb.2021.809412
10. Mondini M, Costa S, Sponza S, Gugliesi F, Gariglio M, Landolfo S. The interferon-inducible HIN-200 gene family in apoptosis and inflammation: implication for autoimmunity. Autoimmunity. (2010) 43:226–31. doi: 10.3109/08916930903510922
11. Goldberger A, Hnilica LS, Casey SB, Briggs RC. Properties of a nuclear protein marker of human myeloid cell differentiation. J Biol Chem. (1986) 261:4726–31. doi: 10.1016/S0021-9258(17)38562-9
12. Hudson CR, Bellew T, Briggs JA, Casey SB, Briggs RC. Production and use of rat monoclonal antibodies to the human myeloid cell nuclear differentiation antigen. Hybridoma. (1988) 7:541–53. doi: 10.1089/hyb.1988.7.541
13. Bosso M, Prelli Bozzo C, Hotter D, Volcic M, Sturzel CM, Rammelt A, et al. Nuclear PYHIN proteins target the host transcription factor Sp1 thereby restricting HIV-1 in human macrophages and CD4+ T cells. PloS Pathog. (2020) 16:e1008752. doi: 10.1371/journal.ppat.1008752
14. Briggs RC, Briggs JA, Ozer J, Sealy L, Dworkin LL, Kingsmore SF, et al. The human myeloid cell nuclear differentiation antigen gene is one of at least two related interferon-inducible genes located on chromosome 1q that are expressed specifically in hematopoietic cells. Blood. (1994) 83:2153–62. doi: 10.1182/blood.V83.8.2153.2153
15. Geng Y, Choubey D. Differential induction of the 200-family proteins in Daudi Burkitt’s lymphoma cells by interferon-alpha. J Biol Regul Homeost Agents. (2000) 14:263–8.
16. Joshi AD, Hegde GV, Dickinson JD, Mittal AK, Lynch JC, Eudy JD, et al. ATM, CTLA4, MNDA, and HEM1 in high versus low CD38 expressing B-cell chronic lymphocytic leukemia. Clin Cancer Res. (2007) 13:5295–304. doi: 10.1158/1078-0432.CCR-07-0283
17. Kanellis G, Roncador G, Arribas A, Mollejo M, Montes-Moreno S, Maestre L, et al. Identification of MNDA as a new marker for nodal marginal zone lymphoma. Leukemia. (2009) 23:1847–57. doi: 10.1038/leu.2009.108
18. Miranda RN, Briggs RC, Shults K, Kinney MC, Jensen RA, Cousar JB. Immunocytochemical analysis of MNDA in tissue sections and sorted normal bone marrow cells documents expression only in maturing normal and neoplastic myelomonocytic cells and a subset of normal and neoplastic B lymphocytes. Hum Pathol. (1999) 30:1040–9. doi: 10.1016/S0046-8177(99)90221-6
19. Briggs JA, Burrus GR, Stickney BD, Briggs RC. Cloning and expression of the human myeloid cell nuclear differentiation antigen: regulation by interferon alpha. J Cell Biochem. (1992) 49:82–92. doi: 10.1002/jcb.240490114
20. Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. (2012) 36:561–71. doi: 10.1016/j.immuni.2012.02.014
21. Cridland JA, Curley EZ, Wykes MN, Schroder K, Sweet MJ, Roberts TL, et al. The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol Biol. (2012) 12:140. doi: 10.1186/1471-2148-12-140
22. Ludlow LE, Johnstone RW, Clarke CJ. The HIN-200 family: more than interferon-inducible genes? Exp Cell Res. (2005) 308:1–17. doi: 10.1016/j.yexcr.2005.03.032
23. Connolly DJ, Bowie AG. The emerging role of human PYHIN proteins in innate immunity: implications for health and disease. Biochem Pharmacol. (2014) 92:405–14. doi: 10.1016/j.bcp.2014.08.031
24. Fairbrother WJ, Gordon NC, Humke EW, O’Rourke KM, Starovasnik MA, Yin JP, et al. The PYRIN domain: a member of the death domain-fold superfamily. Protein Sci. (2001) 10:1911–8. doi: 10.1110/ps.13801
25. Caneparo V, Landolfo S, Gariglio M, De Andrea M. The absent in melanoma 2-like receptor IFN-inducible protein 16 as an inflammasome regulator in systemic lupus erythematosus: the dark side of sensing microbes. Front Immunol. (2018) 9:1180. doi: 10.3389/fimmu.2018.01180
26. Cerny J, Striz I. Adaptive innate immunity or innate adaptive immunity? Clin Sci (Lond). (2019) 133:1549–65. doi: 10.1042/CS20180548
27. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
28. Mairpady Shambat S, Gomez-Mejia A, Schweizer TA, Huemer M, Chang CC, Acevedo C, et al. Hyperinflammatory environment drives dysfunctional myeloid cell effector response to bacterial challenge in COVID-19. PloS Pathog. (2022) 18:e1010176. doi: 10.1371/journal.ppat.1010176
29. Schattgen SA, Fitzgerald KA. The PYHIN protein family as mediators of host defenses. Immunol Rev. (2011) 243:109–18. doi: 10.1111/j.1600-065X.2011.01053.x
30. Almine JF, O’Hare CA, Dunphy G, Haga IR, Naik RJ, Atrih A, et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun. (2017) 8:14392. doi: 10.1038/ncomms14392
31. Jonsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun. (2017) 8:14391. doi: 10.1038/ncomms14391
32. Mistry P, Kaplan MJ. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin Immunol. (2017) 185:59–73. doi: 10.1016/j.clim.2016.08.010
33. Sisto M, Ribatti D, Lisi S. Molecular mechanisms linking inflammation to autoimmunity in sjogren’s syndrome: identification of new targets. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232113229
34. Shen C, Lu A, Xie WJ, Ruan J, Negro R, Egelman EH, et al. Molecular mechanism for NLRP6 inflammasome assembly and activation. Proc Natl Acad Sci U S A. (2019) 116:2052–7. doi: 10.1073/pnas.1817221116
35. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
36. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery. (2020) 6:36. doi: 10.1038/s41421-020-0167-x
37. Shaw N, Liu ZJ. Role of the HIN domain in regulation of innate immune responses. Mol Cell Biol. (2014) 34:2–15. doi: 10.1128/MCB.00857-13
38. Albrecht M, Choubey D, Lengauer T. The HIN domain of IFI-200 proteins consists of two OB folds. Biochem Biophys Res Commun. (2005) 327:679–87. doi: 10.1016/j.bbrc.2004.12.056
39. Liao JC, Lam R, Brazda V, Duan S, Ravichandran M, Ma J, et al. Interferon-inducible protein 16: insight into the interaction with tumor suppressor p53. Structure. (2011) 19:418–29. doi: 10.1016/j.str.2010.12.015
40. Kumar S, Chera JS, Vats A, De S. Nature of selection varies on different domains of IFI16-like PYHIN genes in ruminants. BMC Evol Biol. (2019) 19:26. doi: 10.1186/s12862-018-1334-7
41. Ru H, Ni X, Zhao L, Crowley C, Ding W, Hung LW, et al. Structural basis for termination of AIM2-mediated signalling by p202. Cell Res. (2013) 23:855–8. doi: 10.1038/cr.2013.52
42. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. (2010) 11:997–1004. doi: 10.1038/ni.1932
43. Bottardi S, Guieze R, Bourgoin V, Fotouhi-Ardakani N, Douge A, Darracq A, et al. MNDA controls the expression of MCL-1 and BCL-2 in chronic lymphocytic leukemia cells. Exp Hematol. (2020) 88:68–82 e5. doi: 10.1016/j.exphem.2020.07.004
44. Srivastava A, Ahmad S, Gromiha MM. Deciphering RNA-recognition patterns of intrinsically disordered proteins. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19061595
45. van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. (2014) 114:6589–631. doi: 10.1021/cr400525m
46. Wang X, Bigman LS, Greenblatt HM, Yu B, Levy Y, Iwahara J. Negatively charged, intrinsically disordered regions can accelerate target search by DNA-binding proteins. Nucleic Acids Res. (2023) 51:4701–12. doi: 10.1093/nar/gkad045
47. Jiang Z, Wei F, Zhang Y, Wang T, Gao W, Yu S, et al. IFI16 directly senses viral RNA and enhances RIG-I transcription and activation to restrict influenza virus infection. Nat Microbiol. (2021) 6:932–45. doi: 10.1038/s41564-021-00907-x
48. Kim B, Arcos S, Rothamel K, Jian J, Rose KL, McDonald WH, et al. Discovery of widespread host protein interactions with the pre-replicated genome of CHIKV using VIR-CLASP. Mol Cell. (2020) 78:624–40 e7. doi: 10.1016/j.molcel.2020.04.013
49. Knipe DM. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology. (2015) 479-480:153–9. doi: 10.1016/j.virol.2015.02.009
50. Lo Cigno I, De Andrea M, Borgogna C, Albertini S, Landini MM, Peretti A, et al. The nuclear DNA sensor IFI16 acts as a restriction factor for human papillomavirus replication through epigenetic modifications of the viral promoters. J Virol. (2015) 89:7506–20. doi: 10.1128/JVI.00013-15
51. Merkl PE, Knipe DM. Role for a filamentous nuclear assembly of IFI16, DNA, and host factors in restriction of herpesviral infection. mBio. (2019) 10. doi: 10.1128/mBio.02621-18
52. Hotter D, Bosso M, Jonsson KL, Krapp C, Sturzel CM, Das A, et al. IFI16 targets the transcription factor sp1 to suppress HIV-1 transcription and latency reactivation. Cell Host Microbe. (2019) 25:858–72 e13. doi: 10.1016/j.chom.2019.05.002
53. Gu L, Casserly D, Brady G, Carpenter S, Bracken AP, Fitzgerald KA, et al. Myeloid cell nuclear differentiation antigen controls the pathogen-stimulated type I interferon cascade in human monocytes by transcriptional regulation of IRF7. Nat Commun. (2022) 13:14. doi: 10.1038/s41467-021-27701-x
54. Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe. (2011) 9:363–75. doi: 10.1016/j.chom.2011.04.008
55. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med. (2018) 215:827–40. doi: 10.1084/jem.20172222
56. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
57. Yabal M, Calleja DJ, Simpson DS, Lawlor KE. Stressing out the mitochondria: Mechanistic insights into NLRP3 inflammasome activation. J Leukoc Biol. (2019) 105:377–99. doi: 10.1002/JLB.MR0318-124R
58. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. (2016) 535:111–6. doi: 10.1038/nature18590
59. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
60. Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. (2016) 35:1766–78. doi: 10.15252/embj.201694696
61. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
62. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
63. Liu H, Chen Z, Weng X, Chen H, Du Y, Diao C, et al. Enhancer of zeste homolog 2 modulates oxidative stress-mediated pyroptosis in vitro and in a mouse kidney ischemia-reperfusion injury model. FASEB J. (2020) 34:835–52. doi: 10.1096/fj.201901816R
64. Khare S, Ratsimandresy RA, de Almeida L, Cuda CM, Rellick SL, Misharin AV, et al. The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat Immunol. (2014) 15:343–53. doi: 10.1038/ni.2829
65. Yin Q, Sester DP, Tian Y, Hsiao YS, Lu A, Cridland JA, et al. Molecular mechanism for p202-mediated specific inhibition of AIM2 inflammasome activation. Cell Rep. (2013) 4:327–39. doi: 10.1016/j.celrep.2013.06.024
66. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jonsson KL, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-kappaB signaling after nuclear DNA damage. Mol Cell. (2018) 71:745–60 e5. doi: 10.1016/j.molcel.2018.07.034
67. Kumar V. The trinity of cGAS, TLR9, and ALRs guardians of the cellular galaxy against host-derived self-DNA. Front Immunol. (2020) 11:624597. doi: 10.3389/fimmu.2020.624597
68. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. (2011) 478:515–8. doi: 10.1038/nature10429
69. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. (2009) 461:788–92. doi: 10.1038/nature08476
70. Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. (2009) 106:20842–6. doi: 10.1073/pnas.0911267106
71. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol. (2014) 88:5328–41. doi: 10.1128/JVI.00037-14
72. Balka KR, Louis C, Saunders TL, Smith AM, Calleja DJ, D’Silva DB, et al. TBK1 and IKKepsilon act redundantly to mediate STING-induced NF-kappaB responses in myeloid cells. Cell Rep. (2020) 31:107492. doi: 10.1016/j.celrep.2020.03.056
73. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
74. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. (2012) 5:ra20. doi: 10.1126/scisignal.2002521
75. Yum S, Li M, Fang Y, Chen ZJ. TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci U S A. (2021) 118. doi: 10.1073/pnas.2100225118
76. Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature. (2019) 567:394–8. doi: 10.1038/s41586-019-1000-2
77. Smale ST. Selective transcription in response to an inflammatory stimulus. Cell. (2010) 140:833–44. doi: 10.1016/j.cell.2010.01.037
78. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity. (2013) 38:855–69. doi: 10.1016/j.immuni.2013.05.007
79. Hartmann G. Nucleic acid immunity. Adv Immunol. (2017) 133:121–69. doi: 10.1016/bs.ai.2016.11.001
80. Thoresen DT, Galls D, Gotte B, Wang W, Pyle AM. A rapid RIG-I signaling relay mediates efficient antiviral response. Mol Cell. (2023) 83:90–104 e4. doi: 10.1016/j.molcel.2022.11.018
81. Zhang J, Wirtz S. Does pyroptosis play a role in inflammasome-related disorders? Int J Mol Sci. (2022) 23. doi: 10.3390/ijms231810453
82. Li D, Xie L, Qiao Z, Zhu J, Yao H, Qin Y, et al. IFI16 isoforms with cytoplasmic and nuclear locations play differential roles in recognizing invaded DNA viruses. J Immunol. (2021) 207:2699–709. doi: 10.4049/jimmunol.2100398
83. Wang PH, Ye ZW, Deng JJ, Siu KL, Gao WW, Chaudhary V, et al. Inhibition of AIM2 inflammasome activation by a novel transcript isoform of IFI16. EMBO Rep. (2018) 19. doi: 10.15252/embr.201845737
84. Dutta D, Dutta S, Veettil MV, Roy A, Ansari MA, Iqbal J, et al. BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-beta responses. PloS Pathog. (2015) 11:e1005030. doi: 10.1371/journal.ppat.1005030
85. Roy A, Dutta D, Iqbal J, Pisano G, Gjyshi O, Ansari MA, et al. Nuclear innate immune DNA sensor IFI16 is degraded during lytic reactivation of Kaposi’s sarcoma-associated herpesvirus (KSHV): role of IFI16 in maintenance of KSHV latency. J Virol. (2016) 90:8822–41. doi: 10.1128/JVI.01003-16
86. Diner BA, Li T, Greco TM, Crow MS, Fuesler JA, Wang J, et al. The functional interactome of PYHIN immune regulators reveals IFIX is a sensor of viral DNA. Mol Syst Biol. (2015) 11:787. doi: 10.15252/msb.20145808
87. Howard TR, Crow MS, Greco TM, Lum KK, Li T, Cristea IM. The DNA sensor IFIX drives proteome alterations to mobilize nuclear and cytoplasmic antiviral responses, with its acetylation acting as a localization toggle. mSystems. (2021) 6:e0039721. doi: 10.1128/mSystems.00397-21
88. Duhl DM, Gaczynski M, Olinski R, Briggs RC. Intranuclear distribution of the human myeloid cell nuclear differentiation antigen in HL-60 cells. J Cell Physiol. (1989) 141:148–53. doi: 10.1002/jcp.1041410122
89. Hu B, Jin C, Li HB, Tong J, Ouyang X, Cetinbas NM, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science. (2016) 354:765–8. doi: 10.1126/science.aaf7532
90. Fotouhi-Ardakani N, Kebir DE, Pierre-Charles N, Wang L, Ahern SP, Filep JG, et al. Role for myeloid nuclear differentiation antigen in the regulation of neutrophil apoptosis during sepsis. Am J Respir Crit Care Med. (2010) 182:341–50. doi: 10.1164/rccm.201001-0075OC
91. Barbe L, Lundberg E, Oksvold P, Stenius A, Lewin E, Bjorling E, et al. Toward a confocal subcellular atlas of the human proteome. Mol Cell Proteomics. (2008) 7:499–508. doi: 10.1074/mcp.M700325-MCP200
92. Berry A, Matthews L, Jangani M, Plumb J, Farrow S, Buchan N, et al. Interferon-inducible factor 16 is a novel modulator of glucocorticoid action. FASEB J. (2010) 24:1700–13. doi: 10.1096/fj.09-139998
93. Costa S, Borgogna C, Mondini M, De Andrea M, Meroni PL, Berti E, et al. Redistribution of the nuclear protein IFI16 into the cytoplasm of ultraviolet B-exposed keratinocytes as a mechanism of autoantigen processing. Br J Dermatol. (2011) 164:282–90. doi: 10.1111/j.1365-2133.2010.10097.x
94. Ansari MA, Dutta S, Veettil MV, Dutta D, Iqbal J, Kumar B, et al. Herpesvirus genome recognition induced acetylation of nuclear IFI16 is essential for its cytoplasmic translocation, inflammasome and IFN-beta responses. PloS Pathog. (2015) 11:e1005019. doi: 10.1371/journal.ppat.1005019
95. Ansari MA, Singh VV, Dutta S, Veettil MV, Dutta D, Chikoti L, et al. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J Virol. (2013) 87:8606–23. doi: 10.1128/JVI.00805-13
96. Johnson KE, Chikoti L, Chandran B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol. (2013) 87:5005–18. doi: 10.1128/JVI.00082-13
97. Singh VV, Kerur N, Bottero V, Dutta S, Chakraborty S, Ansari MA, et al. Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J Virol. (2013) 87:4417–31. doi: 10.1128/JVI.03282-12
98. Li T, Diner BA, Chen J, Cristea IM. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A. (2012) 109:10558–63. doi: 10.1073/pnas.1203447109
99. Xie J, Briggs JA, Morris SW, Olson MO, Kinney MC, Briggs RC. MNDA binds NPM/B23 and the NPM-MLF1 chimera generated by the t(3;5) associated with myelodysplastic syndrome and acute myeloid leukemia. Exp Hematol. (1997) 25:1111–7.
100. Li H, Wang ZX, Wu JW. Purification, characterization and docking studies of the HIN domain of human myeloid nuclear differentiation antigen (MNDA). Biotechnol Lett. (2014) 36:899–905. doi: 10.1007/s10529-013-1432-y
101. Li Y, Zhang C, Samad A, Zheng P, Li Y, Chen F, et al. Structural mechanism of dsDNA recognition by the hMNDA HIN domain: New insights into the DNA-binding model of a PYHIN protein. Int J Biol Macromol. (2023) 245:125461. doi: 10.1016/j.ijbiomac.2023.125461
102. Burrus GR, Briggs JA, Briggs RC. Characterization of the human myeloid cell nuclear differentiation antigen: relationship to interferon-inducible proteins. J Cell Biochem. (1992) 48:190–202. doi: 10.1002/jcb.240480210
103. Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. (2011) 144:296–309. doi: 10.1016/j.cell.2011.01.004
104. Suzuki T, Nakano-Ikegaya M, Yabukami-Okuda H, de Hoon M, Severin J, Saga-Hatano S, et al. Reconstruction of monocyte transcriptional regulatory network accompanies monocytic functions in human fibroblasts. PloS One. (2012) 7:e33474. doi: 10.1371/journal.pone.0033474
105. Xie J, Briggs JA, Briggs RC. Human hematopoietic cell specific nuclear protein MNDA interacts with the multifunctional transcription factor YY1 and stimulates YY1 DNA binding. J Cell Biochem. (1998) 70:489–506. doi: 10.1002/(ISSN)1097-4644
106. Booth GT, Parua PK, Sanso M, Fisher RP, Lis JT. Cdk9 regulates a promoter-proximal checkpoint to modulate RNA polymerase II elongation rate in fission yeast. Nat Commun. (2018) 9:543. doi: 10.1038/s41467-018-03006-4
107. Etchegaray JP, Zhong L, Li C, Henriques T, Ablondi E, Nakadai T, et al. The histone deacetylase SIRT6 restrains transcription elongation via promoter-proximal pausing. Mol Cell. (2019) 75:683–99 e7. doi: 10.1016/j.molcel.2019.06.034
108. Hunt G, Vaid R, Pirogov S, Pfab A, Ziegenhain C, Sandberg R, et al. Tissue-specific RNA Polymerase II promoter-proximal pause release and burst kinetics in a Drosophila embryonic patterning network. Genome Biol. (2024) 25:2. doi: 10.1186/s13059-023-03135-0
109. Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol. (2012) 13:543–7. doi: 10.1038/nrm3417
110. Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol. (2000) 20:2629–34. doi: 10.1128/MCB.20.8.2629-2634.2000
111. Egloff S. CDK9 keeps RNA polymerase II on track. Cell Mol Life Sci. (2021) 78:5543–67. doi: 10.1007/s00018-021-03878-8
112. Keel M, Ungethum U, Steckholzer U, Niederer E, Hartung T, Trentz O, et al. Interleukin-10 counterregulates proinflammatory cytokine-induced inhibition of neutrophil apoptosis during severe sepsis. Blood. (1997) 90:3356–63. doi: 10.1182/blood.V90.9.3356
113. Matute-Bello G, Liles WC, Radella F 2nd, Steinberg KP, Ruzinski JT, Jonas M, et al. Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med. (1997) 156:1969–77. doi: 10.1164/ajrccm.156.6.96-12081
114. Taneja R, Parodo J, Jia SH, Kapus A, Rotstein OD, Marshall JC. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med. (2004) 32:1460–9. doi: 10.1097/01.CCM.0000129975.26905.77
115. Vassallo A, Wood AJ, Subburayalu J, Summers C, Chilvers ER. The counter-intuitive role of the neutrophil in the acute respiratory distress syndrome. Br Med Bull. (2019) 131:43–55. doi: 10.1093/bmb/ldz024
116. Milot E, Filep JG. Regulation of neutrophil survival/apoptosis by Mcl-1. ScientificWorldJournal. (2011) 11:1948–62. doi: 10.1100/2011/131539
117. Choubey D, Deka R, Ho SM. Interferon-inducible IFI16 protein in human cancers and autoimmune diseases. Front Biosci. (2008) 13:598–608. doi: 10.2741/2705
118. Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, Burkhardt B, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. (2012) 44:1316–20. doi: 10.1038/ng.2469
119. Tran TH, Hunger SP. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Semin Cancer Biol. (2022) 84:144–52. doi: 10.1016/j.semcancer.2020.10.013
120. Tessoulin B, Moreau-Aubry A, Descamps G, Gomez-Bougie P, Maiga S, Gaignard A, et al. Whole-exon sequencing of human myeloma cell lines shows mutations related to myeloma patients at relapse with major hits in the DNA regulation and repair pathways. J Hematol Oncol. (2018) 11:137. doi: 10.1186/s13045-018-0679-0
121. Kiel MJ, Velusamy T, Rolland D, Sahasrabuddhe AA, Chung F, Bailey NG, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. (2014) 124:1460–72. doi: 10.1182/blood-2014-03-559542
122. Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. (2013) 73:4372–82. doi: 10.1158/0008-5472.CAN-12-3342
123. Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. (2011) 44:47–52. doi: 10.1038/ng.1032
124. McGirt LY, Jia P, Baerenwald DA, Duszynski RJ, Dahlman KB, Zic JA, et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood. (2015) 126:508–19. doi: 10.1182/blood-2014-11-611194
125. Cheon H, Xing JC, Moosic KB, Ung J, Chan VW, Chung DS, et al. Genomic landscape of TCRalphabeta and TCRgammadelta T-large granular lymphocyte leukemia. Blood. (2022) 139:3058–72. doi: 10.1182/blood.2021013164
126. Briggs RC, Atkinson JB, Miranda RN. Variable expression of human myeloid specific nuclear antigen MNDA in monocyte lineage cells in atherosclerosis. J Cell Biochem. (2005) 95:293–301. doi: 10.1002/jcb.20435
127. Flood P, Fanning A, Woznicki JA, Crowley T, Christopher A, Vaccaro A, et al. DNA sensor-associated type I interferon signaling is increased in ulcerative colitis and induces JAK-dependent inflammatory cell death in colonic organoids. Am J Physiol Gastrointest Liver Physiol. (2022) 323:G439–G60. doi: 10.1152/ajpgi.00104.2022
128. Esteve-Codina A, Hofer TP, Burggraf D, Heiss-Neumann MS, Gesierich W, Boland A, et al. Gender specific airway gene expression in COPD sub-phenotypes supports a role of mitochondria and of different types of leukocytes. Sci Rep. (2021) 11:12848. doi: 10.1038/s41598-021-91742-x
129. Jin Y, Sharma A, Carey C, Hopkins D, Wang X, Robertson DG, et al. The expression of inflammatory genes is upregulated in peripheral blood of patients with type 1 diabetes. Diabetes Care. (2013) 36:2794–802. doi: 10.2337/dc12-1986
130. Ge D, Chen H, Zheng S, Zhang B, Ge Y, Yang L, et al. Hsa-miR-889-3p promotes the proliferation of osteosarcoma through inhibiting myeloid cell nuclear differentiation antigen expression. BioMed Pharmacother. (2019) 114:108819. doi: 10.1016/j.biopha.2019.108819
131. Sun C, Liu C, Dong J, Li D, Li W. Effects of the myeloid cell nuclear differentiation antigen on the proliferation, apoptosis and migration of osteosarcoma cells. Oncol Lett. (2014) 7:815–9. doi: 10.3892/ol.2014.1811
132. Briggs RC, Shults KE, Flye LA, McClintock-Treep SA, Jagasia MH, Goodman SA, et al. Dysregulated human myeloid nuclear differentiation antigen expression in myelodysplastic syndromes: evidence for a role in apoptosis. Cancer Res. (2006) 66:4645–51. doi: 10.1158/0008-5472.CAN-06-0229
133. Pradhan A, Mijovic A, Mills K, Cumber P, Westwood N, Mufti GJ, et al. Differentially expressed genes in adult familial myelodysplastic syndromes. Leukemia. (2004) 18:449–59. doi: 10.1038/sj.leu.2403265
134. Hofmann WK, de Vos S, Komor M, Hoelzer D, Wachsman W, Koeffler HP. Characterization of gene expression of CD34+ cells from normal and myelodysplastic bone marrow. Blood. (2002) 100:3553–60. doi: 10.1182/blood.V100.10.3553
135. Kern W, Haferlach C, Schnittger S, Alpermann T, Haferlach T. Serial assessment of suspected myelodysplastic syndromes: significance of flow cytometric findings validated by cytomorphology, cytogenetics, and molecular genetics. Haematologica. (2013) 98:201–7. doi: 10.3324/haematol.2012.066787
136. Yoneda-Kato N, Look AT, Kirstein MN, Valentine MB, Raimondi SC, Cohen KJ, et al. The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene. (1996) 12:265–75.
137. Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. (2006) 6:493–505. doi: 10.1038/nrc1885
138. Nalabothula N, Indig FE, Carrier F. The nucleolus takes control of protein trafficking under cellular stress. Mol Cell Pharmacol. (2010) 2:203–12.
139. Somasundaram V, Soni S, Chopra A, Rai S, Mahapatra M, Kumar R, et al. Value of Quantitative assessment of Myeloid Nuclear Differentiation Antigen expression and other flow cytometric parameters in the diagnosis of Myelodysplastic syndrome. Int J Lab Hematol. (2016) 38:141–50. doi: 10.1111/ijlh.12458
140. Jakobsen MR, Paludan SR. IFI16: At the interphase between innate DNA sensing and genome regulation. Cytokine Growth Factor Rev. (2014) 25:649–55. doi: 10.1016/j.cytogfr.2014.06.004
141. Gu J, Marker-Hermann E, Baeten D, Tsai WC, Gladman D, Xiong M, et al. A 588-gene microarray analysis of the peripheral blood mononuclear cells of spondyloarthropathy patients. Rheumatol (Oxford). (2002) 41:759–66. doi: 10.1093/rheumatology/41.7.759
142. Chen L, Lu D, Yu K, He S, Liu L, Zhang X, et al. Bioinformatics analysis for identification of key genes in salivary gland and the potential of a combination of biomarkers for the diagnosis of SS. J Inflammation Res. (2021) 14:4143–53. doi: 10.2147/JIR.S322318
143. Hu S, Gao K, Pollard R, Arellano-Garcia M, Zhou H, Zhang L, et al. Preclinical validation of salivary biomarkers for primary Sjogren’s syndrome. Arthritis Care Res (Hoboken). (2010) 62:1633–8. doi: 10.1002/acr.20289
144. Lin JD, Nishi H, Poles J, Niu X, McCauley C, Rahman K, et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight. (2019) 4. doi: 10.1172/jci.insight.124574
145. Clark M, Kroger CJ, Tisch RM. Type 1 diabetes: A chronic anti-self-inflammatory response. Front Immunol. (2017) 8:1898. doi: 10.3389/fimmu.2017.01898
146. Gajewski TF. The next hurdle in cancer immunotherapy: overcoming the non-T-cell-inflamed tumor microenvironment. Semin Oncol. (2015) 42:663–71. doi: 10.1053/j.seminoncol.2015.05.011
147. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
148. Ritter B, Greten FR. Modulating inflammation for cancer therapy. J Exp Med. (2019) 216:1234–43. doi: 10.1084/jem.20181739
149. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. (2021) 6:263. doi: 10.1038/s41392-021-00658-5
150. Ochando J, Mulder WJM, Madsen JC, Netea MG, Duivenvoorden R. Trained immunity - basic concepts and contributions to immunopathology. Nat Rev Nephrol. (2023) 19:23–37. doi: 10.1038/s41581-022-00633-5
151. He Z, Dong H. The roles of short-chain fatty acids derived from colonic bacteria fermentation of non-digestible carbohydrates and exogenous forms in ameliorating intestinal mucosal immunity of young ruminants. Front Immunol. (2023) 14:1291846. doi: 10.3389/fimmu.2023.1291846
152. Massa D, Baran M, Bengoechea JA, Bowie AG. PYHIN1 regulates pro-inflammatory cytokine induction rather than innate immune DNA sensing in airway epithelial cells. J Biol Chem. (2020) 295:4438–50. doi: 10.1074/jbc.RA119.011400
153. Hartung F, Esser-von Bieren J. Trained immunity in type 2 immune responses. Mucosal Immunol. (2022) 15:1158–69. doi: 10.1038/s41385-022-00557-0
154. Englmeier L, Subburayalu J. What’s happening where when SARS-CoV-2 infects: are TLR7 and MAFB sufficient to explain patient vulnerability? Immun Ageing. (2022) 19:6. doi: 10.1186/s12979-022-00262-3
155. Parker JE, Mufti GJ. The myelodysplastic syndromes: a matter of life or death. Acta Haematol. (2004) 111:78–99. doi: 10.1159/000074488
156. Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. (2011) 243:206–14. doi: 10.1111/j.1600-065X.2011.01044.x
157. Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood. (2016) 128:2960–75. doi: 10.1182/blood-2016-07-730556
158. Dussiau C, Fontenay M. Mechanisms underlying the heterogeneity of myelodysplastic syndromes. Exp Hematol. (2018) 58:17–26. doi: 10.1016/j.exphem.2017.10.004
159. Bottardi S, Mavoungou L, Pak H, Daou S, Bourgoin V, Lakehal YA, et al. The IKAROS interaction with a complex including chromatin remodeling and transcription elongation activities is required for hematopoiesis. PloS Genet. (2014) 10:e1004827. doi: 10.1371/journal.pgen.1004827
160. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. (2022) 140:1200–28. doi: 10.1182/blood.2022015850
161. Tan B, Xin S, Hu Y, Feng C, Chen M. LBD: a manually curated database of experimentally validated lymphoma biomarkers. Database. (2022) 2022. doi: 10.1093/database/baac051
162. Eckardt JN, Stolzel F, Kunadt D, Rollig C, Stasik S, Wagenfuhr L, et al. Molecular profiling and clinical implications of patients with acute myeloid leukemia and extramedullary manifestations. J Hematol Oncol. (2022) 15:60. doi: 10.1186/s13045-022-01267-7
163. Pemmaraju N, Kantarjian H, Sweet K, Wang E, Senapati J, Wilson NR, et al. North American Blastic Plasmacytoid Dendritic Cell Neoplasm Consortium: position on standards of care and areas of need. Blood. (2023) 141:567–78. doi: 10.1182/blood.2022017865
164. Johnson RC, Kim J, Natkunam Y, Sundram U, Freud AG, Gammon B, et al. Myeloid cell nuclear differentiation antigen (MNDA) expression distinguishes extramedullary presentations of myeloid leukemia from blastic plasmacytoid dendritic cell neoplasm. Am J Surg Pathol. (2016) 40:502–9. doi: 10.1097/PAS.0000000000000595
165. Manohar V, Peerani R, Tan B, Gratzinger D, Natkunam Y. Myeloid cell nuclear differentiation antigen (MNDA) positivity in primary follicles: potential pitfall in the differential diagnosis with marginal zone lymphoma. Appl Immunohistochem Mol Morphol. (2020) 28:384–8. doi: 10.1097/PAI.0000000000000738
166. Metcalf RA, Monabati A, Vyas M, Roncador G, Gualco G, Bacchi CE, et al. Myeloid cell nuclear differentiation antigen is expressed in a subset of marginal zone lymphomas and is useful in the differential diagnosis with follicular lymphoma. Hum Pathol. (2014) 45:1730–6. doi: 10.1016/j.humpath.2014.04.004
167. Wang Z, Cook JR. IRTA1 and MNDA expression in marginal zone lymphoma: utility in differential diagnosis and implications for classification. Am J Clin Pathol. (2019) 151:337–43. doi: 10.1093/ajcp/aqy144
168. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. (2018) 131:2745–60. doi: 10.1182/blood-2017-09-806398
169. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. (2015) 526:525–30. doi: 10.1038/nature15395
170. Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. (2015) 526:519–24. doi: 10.1038/nature14666
171. Wang L, Fan J, Francis JM, Georghiou G, Hergert S, Li S, et al. Integrated single-cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemia. Genome Res. (2017) 27:1300–11. doi: 10.1101/gr.217331.116
172. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
173. Kang S, Ahn IE. Prognostic markers in the era of targeted therapies. Acta Haematol. (2023) 147(1):33–46. doi: 10.1159/000533704
174. Mansouri L, Thorvaldsdottir B, Sutton LA, Karakatsoulis G, Meggendorfer M, Parker H, et al. Different prognostic impact of recurrent gene mutations in chronic lymphocytic leukemia depending on IGHV gene somatic hypermutation status: a study by ERIC in HARMONY. Leukemia. (2023) 37:339–47. doi: 10.1038/s41375-022-01802-y
175. Mollstedt J, Mansouri L, Rosenquist R. Precision diagnostics in chronic lymphocytic leukemia: Past, present and future. Front Oncol. (2023) 13:1146486. doi: 10.3389/fonc.2023.1146486
176. Yun X, Zhang Y, Wang X. Recent progress of prognostic biomarkers and risk scoring systems in chronic lymphocytic leukemia. biomark Res. (2020) 8:40. doi: 10.1186/s40364-020-00222-3
177. Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. (2010) 17:28–40. doi: 10.1016/j.ccr.2009.11.019
178. Mittal AK, Hegde GV, Aoun P, Bociek RG, Dave BJ, Joshi AD, et al. Molecular basis of aggressive disease in chronic lymphocytic leukemia patients with 11q deletion and trisomy 12 chromosomal abnormalities. Int J Mol Med. (2007) 20:461–9. doi: 10.3892/ijmm
179. Cory S, Roberts AW, Colman PM, Adams JM. Targeting BCL-2-like proteins to kill cancer cells. Trends Cancer. (2016) 2:443–60. doi: 10.1016/j.trecan.2016.07.001
180. McConkey DJ, Chandra J, Wright S, Plunkett W, McDonnell TJ, Reed JC, et al. Apoptosis sensitivity in chronic lymphocytic leukemia is determined by endogenous endonuclease content and relative expression of BCL-2 and BAX. J Immunol. (1996) 156:2624–30. doi: 10.4049/jimmunol.156.7.2624
181. Iskierka-Jazdzewska E, Obracaj A, Urbaniak M, Robak T. New treatment options for newly-diagnosed and relapsed chronic lymphocytic leukemia. Curr Treat Opt Oncol. (2022) 23:775–95. doi: 10.1007/s11864-022-00974-0
182. Xu J, Dong X, Huang DCS, Xu P, Zhao Q, Chen B. Current advances and future strategies for BCL-2 inhibitors: potent weapons against cancers. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15204957
183. Tausch E, Close W, Dolnik A, Bloehdorn J, Chyla B, Bullinger L, et al. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica. (2019) 104:e434–e7. doi: 10.3324/haematol.2019.222588
184. Guieze R, Liu VM, Rosebrock D, Jourdain AA, Hernandez-Sanchez M, Martinez Zurita A, et al. Mitochondrial reprogramming underlies resistance to BCL-2 inhibition in lymphoid Malignancies. Cancer Cell. (2019) 36:369–84 e13. doi: 10.1016/j.ccell.2019.08.005
185. Tessoulin B, Papin A, Gomez-Bougie P, Bellanger C, Amiot M, Pellat-Deceunynck C, et al. BCL2-family dysregulation in B-cell Malignancies: from gene expression regulation to a targeted therapy biomarker. Front Oncol. (2018) 8:645. doi: 10.3389/fonc.2018.00645
186. Thijssen R, Roberts AW. Venetoclax in lymphoid Malignancies: new insights, more to learn. Cancer Cell. (2019) 36:341–3. doi: 10.1016/j.ccell.2019.09.008
187. Awan FT, Kay NE, Davis ME, Wu W, Geyer SM, Leung N, et al. Mcl-1 expression predicts progression-free survival in chronic lymphocytic leukemia patients treated with pentostatin, cyclophosphamide, and rituximab. Blood. (2009) 113:535–7. doi: 10.1182/blood-2008-08-173450
188. Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD, et al. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood. (1998) 91:991–1000. doi: 10.1182/blood.V91.3.991.991_991_1000
189. Pepper C, Lin TT, Pratt G, Hewamana S, Brennan P, Hiller L, et al. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood. (2008) 112:3807–17. doi: 10.1182/blood-2008-05-157131
190. Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S, et al. Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig Drugs. (2011) 20:1397–411. doi: 10.1517/13543784.2011.609167
191. Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. (2018) 9:5341. doi: 10.1038/s41467-018-07551-w
192. Yi X, Sarkar A, Kismali G, Aslan B, Ayres M, Iles LR, et al. AMG-176, an mcl-1 antagonist, shows preclinical efficacy in chronic lymphocytic leukemia. Clin Cancer Res. (2020) 26:3856–67. doi: 10.1158/1078-0432.CCR-19-1397
193. Rasmussen ML, Taneja N, Neininger AC, Wang L, Robertson GL, Riffle SN, et al. MCL-1 inhibition by selective BH3 mimetics disrupts mitochondrial dynamics causing loss of viability and functionality of human cardiomyocytes. iScience. (2020) 23:101015. doi: 10.1016/j.isci.2020.101015
194. Yuda J, Will C, Phillips DC, Abraham L, Alvey C, Avigdor A, et al. Selective MCL-1 inhibitor ABBV-467 is efficacious in tumor models but is associated with cardiac troponin increases in patients. Commun Med (Lond). (2023) 3:154. doi: 10.1038/s43856-023-00380-z
195. Tantawy SI, Timofeeva N, Sarkar A, Gandhi V. Targeting MCL-1 protein to treat cancer: opportunities and challenges. Front Oncol. (2023) 13:1226289. doi: 10.3389/fonc.2023.1226289
196. Martin OCB, Frisan T. Bacterial genotoxin-induced DNA damage and modulation of the host immune microenvironment. Toxins (Basel). (2020) 12. doi: 10.3390/toxins12020063
197. Tessmer I, Margison GP. The DNA alkyltransferase family of DNA repair proteins: common mechanisms, diverse functions. Int J Mol Sci. (2023) 25. doi: 10.3390/ijms25010463
198. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. (2010) 28:321–42. doi: 10.1146/annurev-immunol-030409-101311
199. Brunette RL, Young JM, Whitley DG, Brodsky IE, Malik HS, Stetson DB. Extensive evolutionary and functional diversity among mammalian AIM2-like receptors. J Exp Med. (2012) 209:1969–83. doi: 10.1084/jem.20121960
200. Baran M, Feriotti C, McGinley A, Carlile SR, Jiang Z, Calderon-Gonzalez R, et al. PYHIN protein IFI207 regulates cytokine transcription and IRF7 and contributes to the establishment of K. pneumoniae infection. Cell Rep. (2023) 42:112341. doi: 10.1016/j.celrep.2023.112341
Keywords: MNDA, PYHIN factors, innate immunity, transcription control, apoptosis, genotoxic stress
Citation: Bottardi S, Layne T, Ramòn AC, Quansah N, Wurtele H, Affar EB and Milot E (2024) MNDA, a PYHIN factor involved in transcriptional regulation and apoptosis control in leukocytes. Front. Immunol. 15:1395035. doi: 10.3389/fimmu.2024.1395035
Received: 02 March 2024; Accepted: 02 April 2024;
Published: 12 April 2024.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Julien Subburayalu, Technical University Dresden, GermanyCopyright © 2024 Bottardi, Layne, Ramòn, Quansah, Wurtele, Affar and Milot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Milot, ZS5taWxvdC4xQHVtb250cmVhbC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.