 Siena M. Mantooth
Siena M. Mantooth Yara Abdou
Yara Abdou Ana Rosa Saez-Ibañez3
Ana Rosa Saez-Ibañez3 David A. Zaharoff
David A. Zaharoff
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 13 May 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1385484
This article is part of the Research TopicVector-Based Gene Delivery in Cancer ImmunotherapyView all 6 articles
Breast cancer poses one of the largest threats to women’s health. Treatment continues to improve for all the subtypes of breast cancer, but some subtypes, such as triple negative breast cancer, still present a significant treatment challenge. Additionally, metastasis and local recurrence are two prevalent problems in breast cancer treatment. A newer type of therapy, immunotherapy, may offer alternatives to traditional treatments for difficult-to-treat subtypes. Immunotherapy engages the host’s immune system to eradicate disease, with the potential to induce long-lasting, durable responses. However, systemic immunotherapy is only approved in a limited number of indications, and it benefits only a minority of patients. Furthermore, immune related toxicities following systemic administration of potent immunomodulators limit dosing and, consequently, efficacy. To address these safety considerations and improve treatment efficacy, interest in local delivery at the site of the tumor has increased. Numerous intratumorally delivered immunotherapeutics have been and are being explored clinically and preclinically, including monoclonal antibodies, cellular therapies, viruses, nucleic acids, cytokines, innate immune agonists, and bacteria. This review summarizes the current and past intratumoral immunotherapy clinical landscape in breast cancer as well as current progress that has been made in preclinical studies, with a focus on delivery parameters and considerations.
Breast cancer (BC) is the most common cancer among women, with about 2.3 million new cases diagnosed globally in 2020 and 3 million new cases projected in the year 2040 (1). The BC five-year survival rate, according to the Surveillance, Epidemiology, and End Results (SEER) database, is 90% for all BC types and grades. Local and distant recurrence present one of the great problems in BC treatment. Metastases are responsible for >90% of BC deaths, and the 5-year relative survival rate for women with metastatic BC in the U.S. is 30%, according to SEER. However, only 6% of BC patients are initially diagnosed with stage IV disease (2–4). Thus, the majority of deaths are due to localized disease that progressed and metastasized after initial treatment. Despite great progress with diagnosis and treatment of BC, metastatic disease remains incurable. Some subtypes have higher likelihood of metastasis, with triple negative BC (TNBC) and human epidermal growth factor receptor 2 (HER2)-enriched tumors being more likely to recur within 5 years of treatment compared to the luminal subtypes (4, 5).
Immunotherapy, treatments to engage the immune system, has gained interest in the last decade for cancer treatment. The promise of immunotherapy is that it has the potential to generate durable cures, thus raising the tail of the survival curve (6, 7). The monoclonal antibody anti-programmed cell death protein 1 (aPD-1) checkpoint inhibitor pembrolizumab is an approved immunotherapy for unresectable locally advanced or metastatic TNBC (8) as well as a neoadjuvant and adjuvant for early-stage, high risk TNBC (9). Other immunotherapeutics, such as viruses, cells, cytokines, and innate immune agonists, are being or have been evaluated in clinical studies (10, 11). As of 2022, 778 total agents were actively being developed for use in breast cancer and TNBC, at various developmental stages (11). Rationally delivered immunotherapies can stimulate the immune system for lasting, durable responses. Furthermore, tumor infiltrating lymphocytes (TILs) have been demonstrated to be a positive biomarker for improved therapeutic efficacy in BC (12), and delivery of immunotherapeutics has the potential to increase TILs and other immune cells to generate robust anti-tumor activity.
However, immunotherapeutics delivered systemically can also lead to severe adverse events (AEs). For example, the use of checkpoint inhibitors in BC patients has led to serious immune related AEs, with the most frequently reported being rash (38%), infusion reaction (12%), and hypothyroidism (12%) (13–16). These toxicities limit the utilization of these drugs, and therefore, ways to optimize treatment delivery while minimizing toxicity are urgently needed.
One solution to address these challenges is to deliver immunotherapeutics locally at the site of the tumor as an intratumoral (i.t.) injection. I.t. delivery increases therapeutic concentration locally, which could improve treatment efficacy and reduce systemic AEs. Additionally, certain immunotherapeutics can locally stimulate an immune response at the site of the tumor as an in situ vaccine (17). The FDA-approval of i.t. oncolytic herpes virus talimogene laherparepvec (T-VEC) in metastatic melanoma demonstrates the potential for localized immunotherapy to induce systemic antitumor immunity (18).
Additionally, BC tumors typically present with an immunosuppressive tumor microenvironment, infiltrated by a host of immunosuppressive cells such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor associated macrophages (TAMs) (19). Thus, historically, BC has been considered an immunologically cold tumor, with few anti-tumor effector immune cells, rendering systemic immunotherapy ineffective for a majority of patients (20, 21). I.t. delivered immunotherapy could directly recruit effector immune cells into the tumor microenvironment while antagonizing and reducing immunosuppressive cells. When developing i.t. immunotherapies, numerous considerations should be taken into account. Delivery mediums, including but not limited to hydrogels, microparticles, and nanoparticles (NPs), can be engineered to improve retention within the tumor and prolong release locally. Timing of delivery relative to prior or concomitant treatments can also impact outcomes. For instance, neoadjuvant immunotherapy can reduce tumor burden prior to resection, and it may additionally be able to re-educate the immune system using tumor-specific, patient-specific antigens. Also, the frequency of treatment should be considered, as patients can have local pain and discomfort from i.t. injections (22).
In this review, we will present findings from clinical and preclinical BC studies that have evaluated an i.t. immunotherapy (Figure 1). Section II will introduce the different types of immunotherapies, providing a historical context and mechanisms of action. Section III will present completed and ongoing clinical trials of i.t. delivered immunotherapeutics for BC, highlighting the types, timing, and outcomes of treatment. Section IV will present preclinical strategies, with a focus on immunotherapy types, delivery timing and frequency, and delivery mediums. Section V will summarize key findings from the review. Section VI will reflect on future steps for the field of i.t. delivered BC immunotherapies.
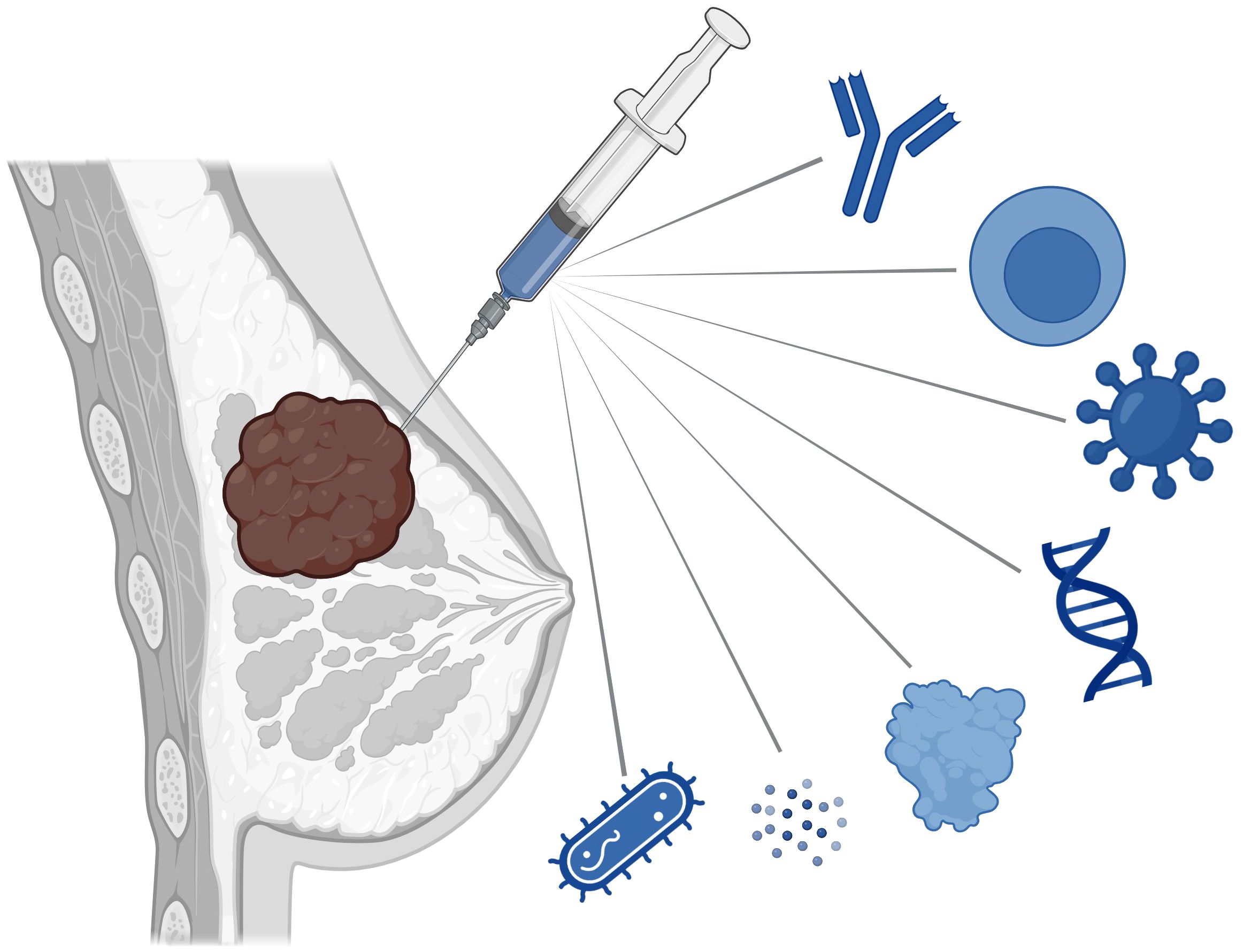
Figure 1 Intratumoral immunotherapies being explored in clinical and preclinical studies in breast cancer treatment include monoclonal antibodies, cellular therapies, viruses, nucleic acids, cytokines, innate immune agonists, and bacteria.
The following section will introduce the history and mechanism of seven technologies that have been studied in the context of BC, including checkpoint inhibitors and other monoclonal antibodies, cellular therapies, viruses, nucleic acids, cytokines/chemokines, innate immune agonists, and bacteria. Thus far, the only clinically approved immunotherapeutic for BC is the aPD-1 checkpoint inhibitor pembrolizumab (Keytruda) for TNBC. The remaining technologies are in clinical and/or preclinical development.
In the 1990’s, researchers Professor Tasuku Honjo and Professor James Allison discovered the cell surface receptors programmed death protein-1 (PD-1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), respectively, and their roles on the suppression of T cell activity. Honjo and Allison shared the Nobel Prize in Medicine in 2018 for their discovery that inhibition of PD-1 and CTLA-4 signaling can suppress negative immune regulation and provide a new form of cancer therapy (23). Today, aPD-(L)1 and aCTLA-4 have been approved for certain cancer indications in the clinic, and in particular, the aPD-1 checkpoint inhibitor pembrolizumab has been approved for certain BC indications (24). A next-generation checkpoint inhibitor antibody against lymphocyte activation gene-3 (LAG-3) was recently approved for a non-BC cancer indication (25). Other next-generation checkpoint inhibitors currently in development include antibodies against T cell immunoglobulin-3 (TIM-3), B7-H3 and B7-H4, A2aR and CD73, natural killer group protein 2A, and poliovirus receptor-related immunoglobulin domain containing (26, 27). Other monoclonal antibodies, which have not yet received regulatory approval, bind to costimulatory molecules, such as OX40, which enhances T cell receptor signaling (28) and CD40, which activates dendritic cells (DCs) (29).
Cellular therapies utilize either autologous or allogeneic cells, derived from non-stem and stem cells, which may or may not be genetically engineered or manipulated (30). The first recorded instance of cellular therapy was in the 19th century by Charles-Édouard Brown-Séquard who utilized extracts of animal testicles to prevent aging (31). In cancer therapy, chimeric antigen receptor (CAR) T cells, in which autologous T cells are genetically engineered to express a CAR directed against a particular target and kill tumor cells in a non-MHC-dependent manner, have demonstrated significant clinical success in blood tumors, and numerous efforts are being made to improve their efficacy in solid tumors. Other T cell immunotherapies utilize injections of activated, ex vivo differentiated and expanded allogenic T cells (AlloStim) to serve as an in situ cancer vaccine by evoking an immune response (32, 33). Another vaccination-based approach uses DCs to serve as a bridge between innate and adaptive immunity with the ability to present antigens to T cells. A variety of approaches have utilized DCs in cancer immunotherapy, the most common of which being ex vivo derived DCs loaded with tumor antigens (34). Finally, other less commonly utilized cellular products include other immune cells, such as natural killer (NK) cells and macrophages as well as fibroblasts, which can be engineered for enhanced anti-tumor immunity (35).
In the early 1900s, viruses were observed to improve the outcome of cancer treatment, and in the mid 1900’s, reports of certain wild type viruses provided variable benefit in cancer patients (36). Many of these viruses were not consistently effective, but adenoviruses demonstrated acceptable safety with potential to induce necrosis in cervical cancers (36). Oncolytic viruses, which specifically target and lyse cancer cells, have entered clinical trials, and T-VEC, a herpes virus also carrying the gene for cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF), is approved for the local treatment of unresectable metastatic melanoma (36, 37). Numerous oncolytic viruses are in preclinical and clinical development, including but not limited to adenoviruses, Newcastle disease viruses, herpes viruses, and measles viruses (38). Though some wild type viruses have clinical utility, oncolytic viruses can be engineered to enhance their anti-tumor action through confining viral replication only to cancer cells, targeting receptors that are more highly expressed on cancer cells, and incorporating transgenes for other anti-tumor biologic cargo (36). Other viral based techniques also utilize the ability of viruses to infect a cell as a gene delivery vehicle for intracellular delivery or to stimulate the immune system as an immune agonist.
In addition to viral mediated gene delivery, other nucleic acid-based deliveries include plasmid and mRNA-based delivery. Plasmids are small, circular structures of DNA, and for cancer immunotherapy, they can be engineered to encode tumor antigens and immunomodulatory or cytotoxic proteins. Plasmids can be delivered alone, with the assistance of physical forces, such as through sonication or electric pulses, or complexed with lipids, polymers, or other compounds for enhanced therapeutic efficacy (39). Plasmids are cost-effective, easy to manufacture, and demonstrate high stability. Poor delivery to the nucleus where plasmid-encoded genes are transcribed limits the efficacy of plasmid-based immunotherapies (39). On the other hand, mRNA therapeutics, which have recently gained public attention with SARS-CoV-2 vaccines, need only to be delivered into the cytoplasm of the cell for the transcription and translation of its encoded genetic material. Though less stable than DNA, mRNA can be encapsulated in polymer and/or lipid nano- and micro-particles for enhanced stability and transfection.
Cytokine and chemokine immunotherapies seek to modulate the tumor microenvironment (TME) from a tumor supporting, immune suppressive milieu to a tumor attacking, immune activated one. Cytokines are small proteins, often below 30kDa, which signal cell activation, growth, differentiation, and apoptosis (40). Cytokines serve as key modulators of the immune environment, and certain pro-inflammatory cytokines possess anti-tumor activity, which has led to the clinical approval of interleukin-2 (IL-2) and interferon-alpha (IFNα) for certain cancer indications (40). Granulocyte colony-stimulating factor (G-CSF), GM-CSF, vascular endothelial growth factor (VEGF), IL-12, and IFNγ have received the most attention in clinical trials, perhaps due to their relatively earlier discovery compared to the other cytokines, and many others are in clinical development (41). Notably, however, cytokines delivered systemically have a short half-life and can induce profound toxicities, including cytokine release syndrome, which can be fatal (42).
Chemokines are a class of cytokine that drive the chemotaxis or movement of immune cells to a particular site. Chemokines can serve pro- or anti-tumor functions, with the chemokines CXCL9 and CXCL10, ligands for CXCR3, being of the most interest in the anti-tumor category due to their ability to attract effector CD8+ and CD4+ T cells (43). No chemokines are currently clinically approved for therapeutic application.
Innate immune agonists offer another route to stimulate and recruit immune cells, thus altering the TME. Innate immune cells are equipped with pattern recognition receptors (PRRs), which recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Binding to these PRRs on innate immune cells can elicit an anti-tumor immune response through the induced production of cytokines and immune cell and environment repolarization. PRRs include the membrane-bound toll-like receptors (TLRs) and C-type lectin receptors (CLRs), as well as the cytosolic proteins retinoic acid-inducible gene- (RIG-I)-like receptors (RLRs), AIM2-like receptors (ALRs), NOD-like receptors (NLRs), and stimulator of interferon genes (STING) (44).
Bacteria also can be utilized as a cancer treatment, and the use of such dates back to 1863, in which a patient was intentionally infected with Streptococcus pyogenes. Though the tumor was reduced, the patient died from the infection (45). This and other trials with bacteria led to the physician William Coley injecting heat-inactivated Serratia marcescens and Streptococcus pyogenes into sarcoma, beginning a new field, now known as cancer immunotherapy (45). Localized bacteriotherapy can both serve to initiate an inflammatory response and to colonize the tumor itself, thereby leading to necrosis and cell lysis (46). In particular, the attenuated anaerobic bacterium Clostridium novyi-NT has locally initiated necrosis and cell lysis, leading to long-term cellular anti-tumor immunity (46).
As of January 2024, seventeen clinical trials successfully completed an evaluation of i.t. delivery of immunotherapeutics in BC. Clinical trials were sourced from clinicaltrials.gov and the Cancer Research Institute’s (CRI) Immuno-Oncology (IO) Intelligence database (Table 1, Figure 2). For clinical trial sourcing from clinicaltrials.gov, the key search terms used were “breast cancer” AND “intratumoral” OR “intralesional”. Trials were subsequently filtered to include immunotherapy results only. Additional clinical trials were sourced from the CRI’s IO Intelligence database, which is built from GlobalData’s Oncology Trial Database and subsequently curated by CRI based on CRI IO Analytics definition of different immunotherapy types and drug target information. For this publication, immunotherapy trials in which at least one form of BC was listed among the clinical trial indications were included and subsequently filtered by the appearance of the keywords “intratumoral” or “intralesional” in the trial title. Data cut date was January 26, 2024. Limitations to the trial sourcing methods included the impossibility of identifying trials involving treatments administered intratumorally if this was not clearly stated in the trial title.
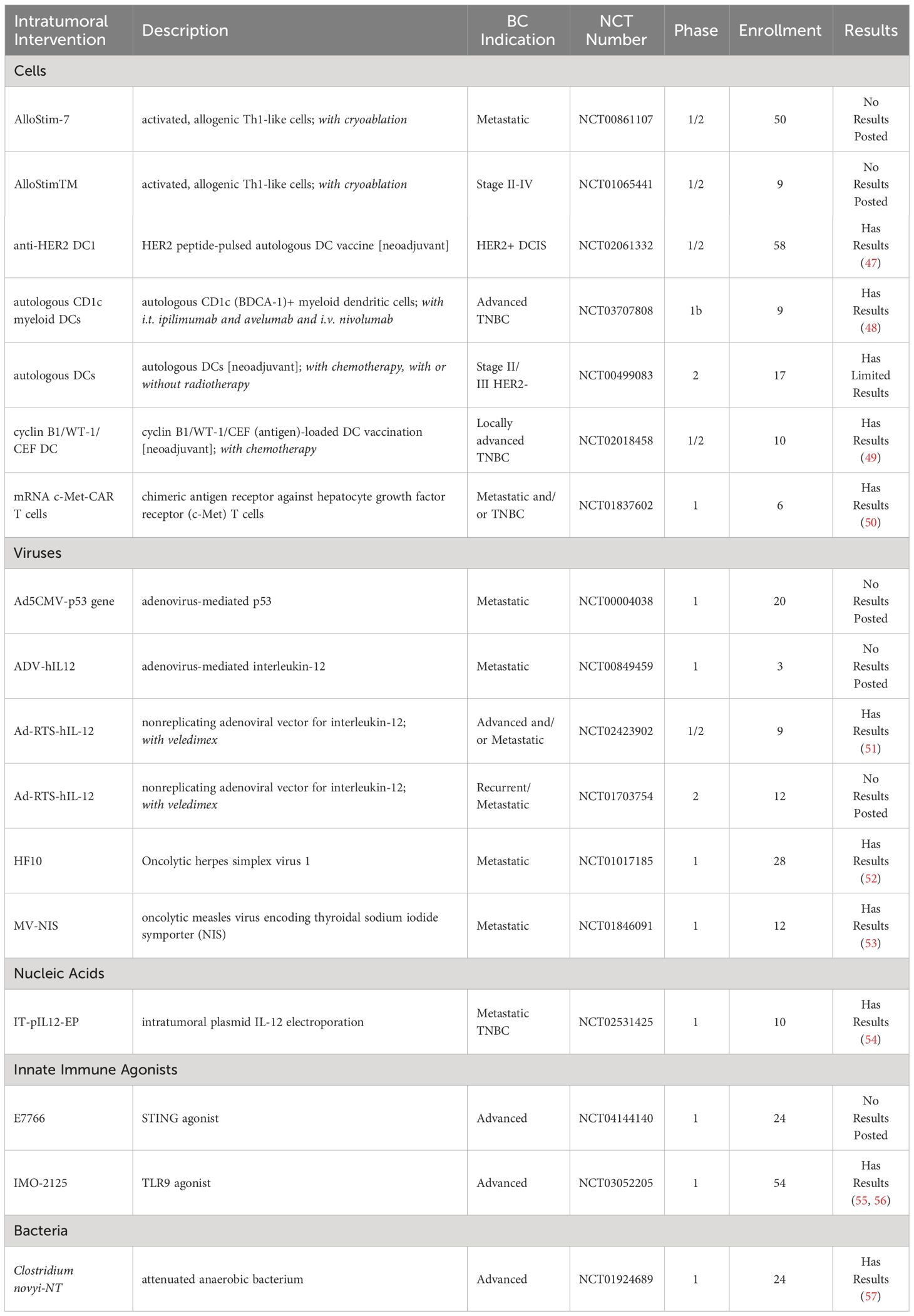
Table 1 Completed clinical trials of intratumoral therapies for breast cancer (clinicaltrials.gov, key search terms “breast cancer” AND “intratumoral” OR “intralesional”) as of January 2024.
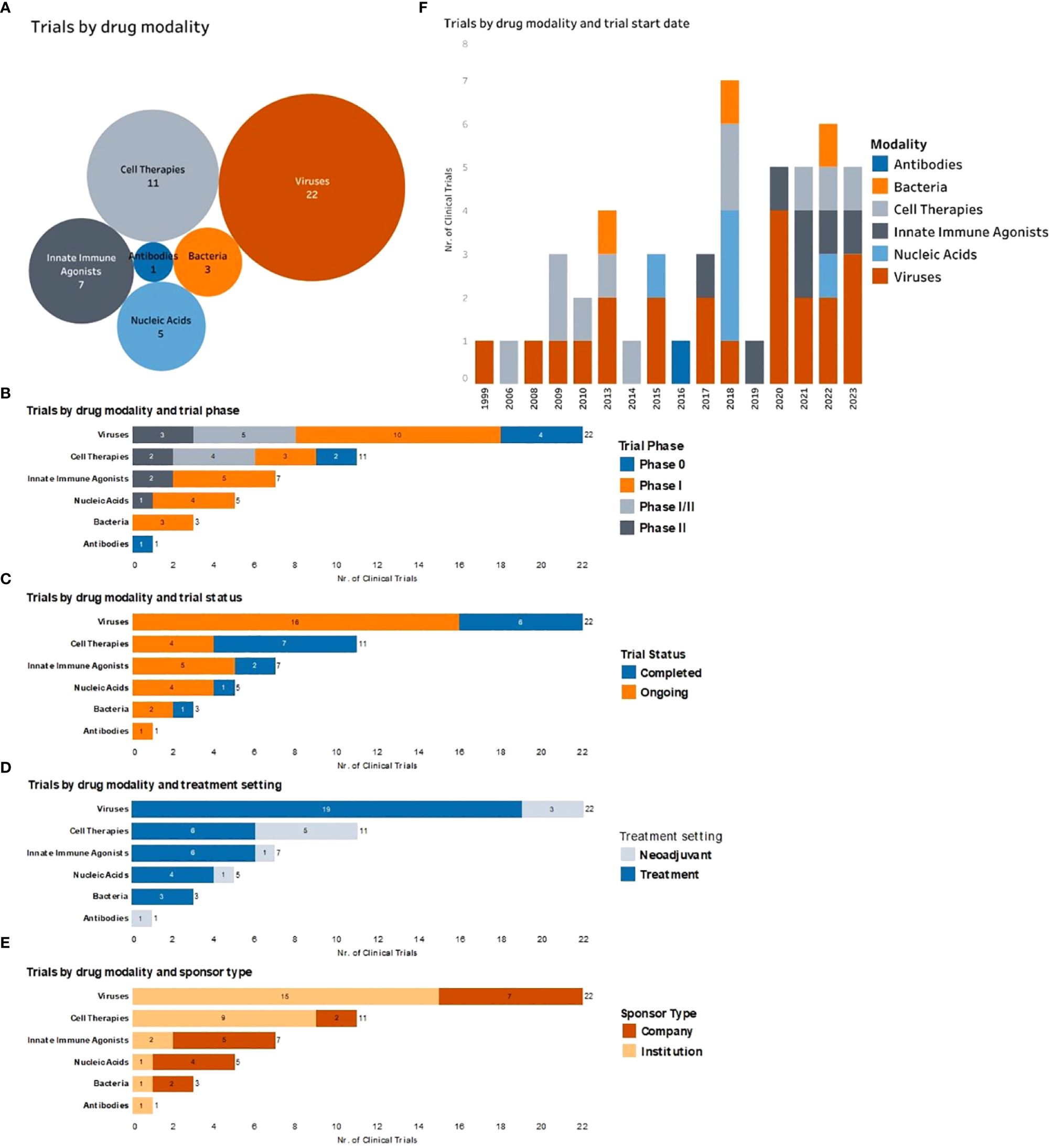
Figure 2 Complete and ongoing clinical studies of intratumorally delivered immunotherapies in breast cancer. (A) Bubble diagram representing trials by drug modality. Each bubble represents a different therapeutic modality. The size of each bubble corresponds to the number of trials within that group, also indicated by number. (B) Number of clinical trials by therapeutic modality being tested (y axe) and color-coded according to the trial phase. (C) Number of clinical trials by therapeutic modality being tested (y axe) and color-coded according to the trial status. (D) Number of trials by therapeutic modality being tested (y axe) and color-coded according to the treatment setting (equivalent to treatment timing). (E) Number of clinical trials by therapeutic modality being tested (y axe) and color-coded according to the type of trial sponsor. (F) Clinical trials organized by trial start year (x axe) and color-coded by type of therapeutic modality being tested.
Three clinical trials have evaluated DC-based vaccines in a neoadjuvant setting (NCT02061332, NCT02018458, NCT00499083). In a phase 1/2 trial, patients with HER2 overexpressing ductal carcinoma in situ (DCIS) or early invasive BC received a HER2-pulsed DC vaccine via intranodal and/or intralesional injection prior to surgery (NCT02061332). The treatment was determined to be safe with only grade 1 and grade 2 AEs reported. Tumor-specific T-cell responses as measured via systemic anti-HER2 CD4+ and CD8+ T cells pre- and post-treatment were increased with vaccination. The route of administration did not impact the immune or clinical response. Pathological complete response (pCR) was higher in patients with DCIS (28.6%) rather than invasive BC (8.3%). In patients with DCIS, the sentinel lymph node (LN) anti-HER2 CD4+ T cell response rather than peripheral blood anti-HER2 CD4+ or CD8+ T cell immune response was associated with pCR (47).
In a second phase 1/2 trial, patients with locally advanced TNBC received a neoadjuvant antigen-loaded DC vaccine concurrently with standard chemotherapy (doxorubicin/cyclophosphamide, then paclitaxel and carboplatin) (NCT02018458). The antigens selected for loading were overexpressed in TNBC, namely cyclin B1 and WT1, along with control viral antigens, CEF (cytomegalovirus, Epstein-Barr virus, and influenza virus) (49). Vaccines were administered i.t. four times pre- and s.c. thrice post- surgery. Patients had grade 1-2 AEs at the injection site (49). 5/10 patients had a pCR at surgery, while 3/10 still had macroscopic residual disease, and 2/10 had residual cancer burden scores of 1 (RCB1) (49). The RCB scoring system provides a ranking for the residual disease remaining following neoadjuvant therapy, with RCB0 equating to a pCR and RCB3 indicating extensive tumor burden. The vaccine was determined to be feasible and safe as a neoadjuvant to surgery for patients with TNBC (49).
The third clinical trial evaluated autologous DC vaccine following paclitaxel chemotherapy, prior to surgery and radiation, in patients with stage II/III HER2- BC in a phase 2 trial (NCT00499083). Study results are limited and forthcoming.
A phase 1 trial determined the feasibility and safety of i.t. delivery of CAR-T cells targeting the surface protein hepatocyte growth factor receptor (c-Met) for patients with metastatic BC patients (NCT01837602). c-Met is often over-expressed in several cancers, including BC. c-Met expression served as an eligibility requirement and was evaluated prior to patient enrollment (50). Low grade AEs were reported in some of the patients, including erythema and myalgia, but no CAR-T cell-related AEs above grade 3 were reported (50). CAR mRNA, albeit at low levels, was found in peripheral blood samples 20 minutes and 2 hours post-injection in two and one patients, respectively, but not at later time points (day 1 onward) (50). Necrosis, immune cell infiltration, and hemorrhage were observed within the injected tumor. Overall, the injection was determined to be well-tolerated, safe, and able to induce an i.t. inflammatory response (50).
A phase 1 clinical trial evaluated the safety and feasibility of i.t. delivery of autologous CD1c (BDCA-1)+ myeloid DCs in advanced solid tumors, including TNBC, along with i.t. ipilimumab and avelumab and intravenously (i.v.) delivered nivolumab (NCT03707808). 3/9 of the treated patients had TNBC (48). Only one patient (1/9) experienced a grade 3+ AE, and the most common AEs were local and generalized pruritus (3/9). All three BC patients had progressive disease post-treatment, though one of the patients initially had a mixed response with regression in a treated lesion but the occurrence of additional lesions. Overall, the treatment was demonstrated to be safe and feasible (48).
Other clinical trials delivering cells without posted study results include delivery of the activated, allogeneic Th1-like cells AlloStim sponsored by Immunovative Therapies, Ltd., with a phase 1/2 trial first posted in 2009 for patients with metastatic cancers including BC (NCT00861107) and a phase 1/2 trial first posted in 2010 for patients with stage II-IV cancers including BC (NCT01065441).
Six clinical trials utilized viruses either to directly lyse tumor cells or to deliver genes as a delivery vector. A non-replicating adenovirus encoding IL-12 (Ad-RTS-hIL-12) was evaluated in phase 1/2 (NCT02423902) and phase 2 (NCT01703754) clinical trials in patients with locally advanced or metastatic BC and with locally recurrent or metastatic BC, respectively. In the Ad-RTS-hIL-12 system, the expression of IL-12 was inducible via an oral activator ligand, veledimex (51). This platform increased IL-12, IFNγ, and CD8+ T cell infiltration and reduced Tregs within the injected tumor. A reduction in non-injected lesion size was also observed. Furthermore, using immune Response Evaluation Criteria in Solid Tumors (iRECIST) guidelines, 5/9 subjects demonstrated either partial response or stable disease, having a disease control rate of 44% and 22% at week 6 and 12, respectively, post-treatment. Because of the inducible production of IL-12 via veledimex, AEs were controllable, leading to a good safety profile (51).
In a phase 1 clinical trial, the oncolytic herpes simplex virus type 1, HF10, was delivered to patients with cutaneous and/or superficial cancers, including BC (NCT01017185). Initial data demonstrated that the injections were safe and well-tolerated, with mild AEs and rapid viral clearance from the saliva, urine, and blood (52).
In another phase 1 trial, the oncolytic measles virus encoding thyroidal sodium iodide symporter (NIS) (MV-NIS) was delivered once i.t. to evaluate safety and tolerability in patients with metastatic BC (NCT01846091). NIS is a plasma protein which assists in iodide transport. NIS uptake of systemically administrated Tc-99m pertechnetate or I-123 coupled with SPECT-CT imaging provided feedback on viral propagation. Six patients received increasing doses of MV-NIS, with no dose-limiting toxicities observed (53). AEs included grade 2 lymphopenia and fatigue and grade 1 flu-like symptoms. Clinically, at greater than six weeks post-treatment, 4/6 had stable disease, 1/6 had a clinical response, and 1/6 experienced disease progression. The treatment was determined to be safe, with evidence suggesting MV-NIS activity (53).
Other trials with unposted results included i.t. adenoviral-mediated delivery of unregulated IL-12 (NCT00849459) and p53 (NCT00004038) in patients with metastatic BC.
In a phase 1, non-randomized, open-label trial, metastatic TNBC patients received three doses of plasmid IL-12 intratumorally followed by electroporation on days 1, 5, and 8 (NCT02531425). Previously, in preclinical studies, plasmid IL-12 both expanded and activated CD8+ TILs and sensitized mice to checkpoint inhibitor therapy (54). These results were shown to be correlated with a higher chemokine CXCR3 gene signature (54). In the clinical trial, four of nine treated patients exhibited higher CD8+ TILs post-treatment, which were associated with increases in the expression of genes associated with T cell activation, cytokine signaling pathways, antigen presentation, and chemotaxis. Additionally, one patient previously unresponsive to aPD-1 therapy demonstrated clinical response to aPD-1 following plasmid IL-12 delivery and electroporation. Overall, the results suggested the treatment to be safe with an increased CXCR3 gene signature prognostic of a positive response (54).
To date, two clinical studies have evaluated an innate immune agonist in BC patients. A safety study was performed with the TLR9 agonist IMO-2125 in patients with refractory solid tumors, including BC (NCT03052205). I.t. injections were given on Days 1, 8, and 15. Fever, chills, and fatigue were the most common AEs. Immunologically, at 24 hours post-treatment, gene expression of IFN gamma, MHC I and II, and type 1 IFN pathways increased within the tumor. Overall, i.t. IMO-2125 was well-tolerated and induced favorable anti-tumor immune responses (55, 56).
The safety and tolerability of i.t. E7766, a STING agonist, was studied in patients with lymphoma and advanced solid tumors, including BC (NCT04144140). The study was completed in 2022, however, no results have been posted.
One completed phase 1 clinical trial tested the feasibility and safety of a single i.t. injection of Clostridium novyi-NT spores in patients with advanced solid tumors, including BC, in a 3 + 3 dose escalation study (NCT01924689). Two of the 24 patients had BC; one patient was not evaluable, and the other patient had stable disease (57). Overall, of the 22 evaluable patients, 9 had tumor regression and 19 had stable disease. Treatment promoted tumor-specific T-cell and systemic cytokine responses. However, toxicities were non-negligible, with grade ≥3 AEs comprised of gas gangrene (n=1), abscess limb (n=1), respiratory insufficiency (n=1), rash (n=1), pathologic fracture (n=1), sepsis (n=2), and soft-tissue infection (n=1) (57).
As of January 2024, 32 active and/or recruiting clinical trials are pursuing i.t. delivery of therapeutics for BC indications following the same search query as that from the completed clinical trials (Table 2, Figure 2). Reported updates, if provided, are as follows.
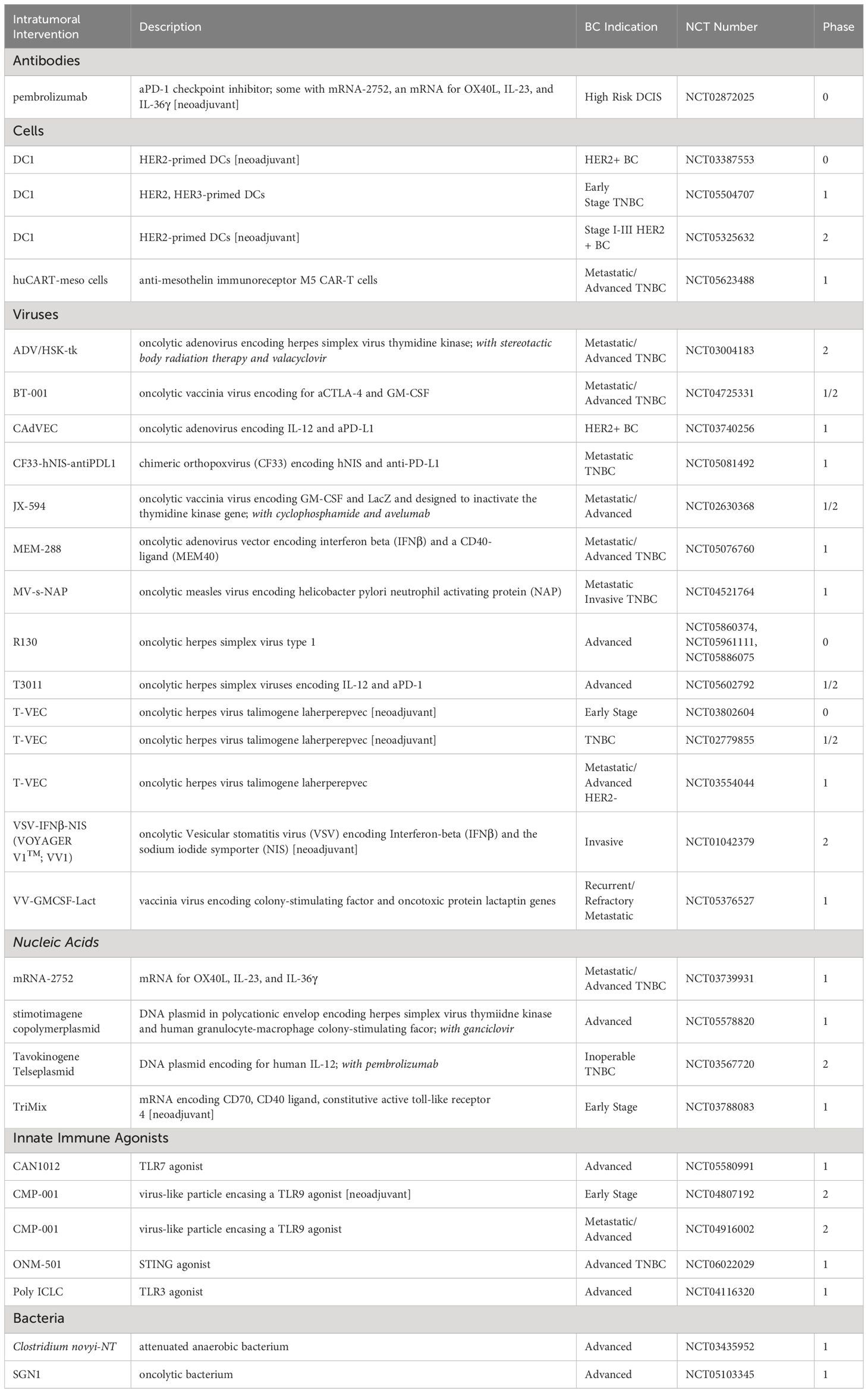
Table 2 Ongoing (recruiting and/or active) clinical trials of intratumoral immunotherapies for breast cancer (clinicaltrials.gov, key search terms “breast cancer” AND “intratumoral” OR “intralesional”) as of January 2024.
In a phase 1/2 clinical, DCs primed with HER2-antigens were i.t. delivered to patients with HER2+ BC (NCT05325632). Results from the phase 1 study indicated that of the 12 patients receiving treatment, three had complete responses, six had partial responses, and three had stable disease. Of these patients, nine had surgery, with six obtaining a pCR. Common AE’s included chills (50%), fatigue (42%), diarrhea (42%), headache (42%), and nausea (42%). Immunologically, the higher dose (100 million cells) had a higher HER2-specific T cell response than the lower dose (50 million cells) six weeks following DC treatment (58).
Intratumoral administration of pembrolizumab is being evaluated in patients with high risk DCIS in the neoadjuvant setting (NCT02872025). One cohort in this phase 1 trial is also receiving an mRNA construct encoding OX40L, IL-23, and IL-36γ (mRNA-2752). Higher immune cell infiltrates correlated with improved prognosis for patients with DCIS, providing the rationale for local pembrolizumab delivery (59). In an interim analysis, the injected doses, even at the smallest amount (2mg), led to increased CD8+ and overall T cell tumor infiltration (59). The overall DCIS volume was not dramatically altered with treatment, and additional anti-tumor responses were not observed, suggesting another agent may be required for tumor reduction (59).
The oncolytic adenovirus CAdVEC, which encodes for IL-12 and aPD-L1, was delivered in four metastatic BC patients as part of a phase 1 trial (NCT03740256) (60, 61). A single i.t. injection caused an influx of CD8+ T cells and TME repolarization without significant toxicity (60). Gene set enrichment analysis of tumors pre- and post- treatment indicated antigen processing and presentation enrichment (60). AE’s were either grade 1/2, with fatigue, fever, and injection site pain being the most frequently reported (61). Clinically, when combined with systemic checkpoint inhibitors, CAdVEC caused local and abscopal antitumor responses, with three partial responses observed (60, 61).
T-VEC delivered i.t. in the neoadjuvant setting with paclitaxel/doxorubicin/cyclophosphamide chemotherapy was evaluated in 37 patients with nonmetastatic TNBC in a phase 2 trial (NCT02779855) (62). The most common AEs included pain at the injection site, chills, fever, headache, and fatigue (62). Surgery was utilized for RCB assessment. The primary end point, RCB0 rate, was met (45.9%), and 2-year disease-free rate (overall 89%) had no recurrences in patients with RCB0-1 scores. Immunologically, i.t. immune activation was observed, with CD8+ effector T cell and CD3+CD45RO+ memory T cell i.t. density increased six weeks post-treatment (62).
T-VEC was also delivered i.t. in the adjuvant setting, in combination with chemotherapy or endocrine therapy in advanced/metastatic HER2- breast cancer (NCT03554044). Observed grade 3+ adverse events included neutropenia (5/19), thrombocytopenia (1/19), ulceration at the injection site (1/19), and anemia (1/19) (63). Response rates were obtained for 18/19 patients, with 11/18 obtaining a partial response, 4/18 had stable disease, with the remaining 3/18 progressing. Immunologically, the treatment altered the systemic immune profile (63).
I.t. administered oncolytic herpes simplex viruses encoding IL-12 and aPD-1 (T3011) in patients with advanced solid tumors (NCT05602792) reported an update of no dose limiting toxicities (DLTs) and no systemic viral transmission (64). The chimeric orthopoxvirus (CF33) encoding human NIS and anti-PD-L1 (CF33-hNIS-antiPDL1) was delivered in a phase 1 trial for metastatic TNBC (NCT05081492). Though studies are ongoing, an update at SABCS 2022 indicated no DLTs were reported in the six patients receiving dose levels 1 and 2 (1 and 3x105 pfu, respectively) (65).
mRNA-2752, which encodes OX40L, IL-23, and IL-36γ, was evaluated in advanced or metastatic solid cancers, including BC, and lymphoma (NCT03739931). Dose escalation studies revealed a DLT at the 8mg dose (66). Many inflammatory cytokines were upregulated in the tumor and in the plasma following treatment, which also led to T cell infiltration and activation (66).
Tavokinogene Telseplasmid (IT-tavo-EP), a DNA plasmid encoding for hIL-12 followed by electroporation, was delivered to sensitize patients with inoperable locally advanced or metastatic TNBC to pembrolizumab in a phase 2 trial (NCT03567720) (67). Of the 11 patients evaluated, three patients had a partial response, and some untreated tumors regressed (67). The objective response rate (ORR) was 27.3%. 68.8% of patients experienced an AE, with 37.5% being grade ≥3 (67). Findings suggested IT-tavo-EP could improve checkpoint inhibitor therapy in these heavily pretreated patients.
Clostridium novyi-NT, an attenuated anaerobic bacterium, was evaluated in advanced BCs and other solid malignancies in a phase 1 trial (NCT03435952) (68). No DLTs were observed, and the injection was determined to be feasible. Though neither of the two partial responses observed were in BC patients, the therapy exhibited anti-tumor potential (68).
Preclinically, i.t immunotherapies such as antibodies, cells, viruses, cytokines, and innate immune agonists have been utilized with varying degrees of success. Furthermore, in an effort to enhance intratumoral retention of immunotherapies after injection, a range of delivery systems have also been developed. The most frequently explored delivery systems include NPs, microspheres, and hydrogels. NPs are ~1-100 nm in diameter whereas microspheres/microparticles range from 1-1000 um in diameter. Hydrogels are three-dimensional polymeric networks encapsulating large amounts of water. The following subsections will highlight preclinical studies investigating each type of immunotherapy both without and with delivery strategies. Each therapeutic delivery highlights the timing of delivery, as well as its unique delivery system, should it contain one. Preclinical studies were obtained from a PubMed search using the terms: “intratumoral” AND “immunotherapy” AND “breast cancer”, as of January 2024.
Several preclinical studies have investigated the utility of i.t. delivery of monoclonal antibodies (mAbs) both with and without local retention strategies (Table 3).
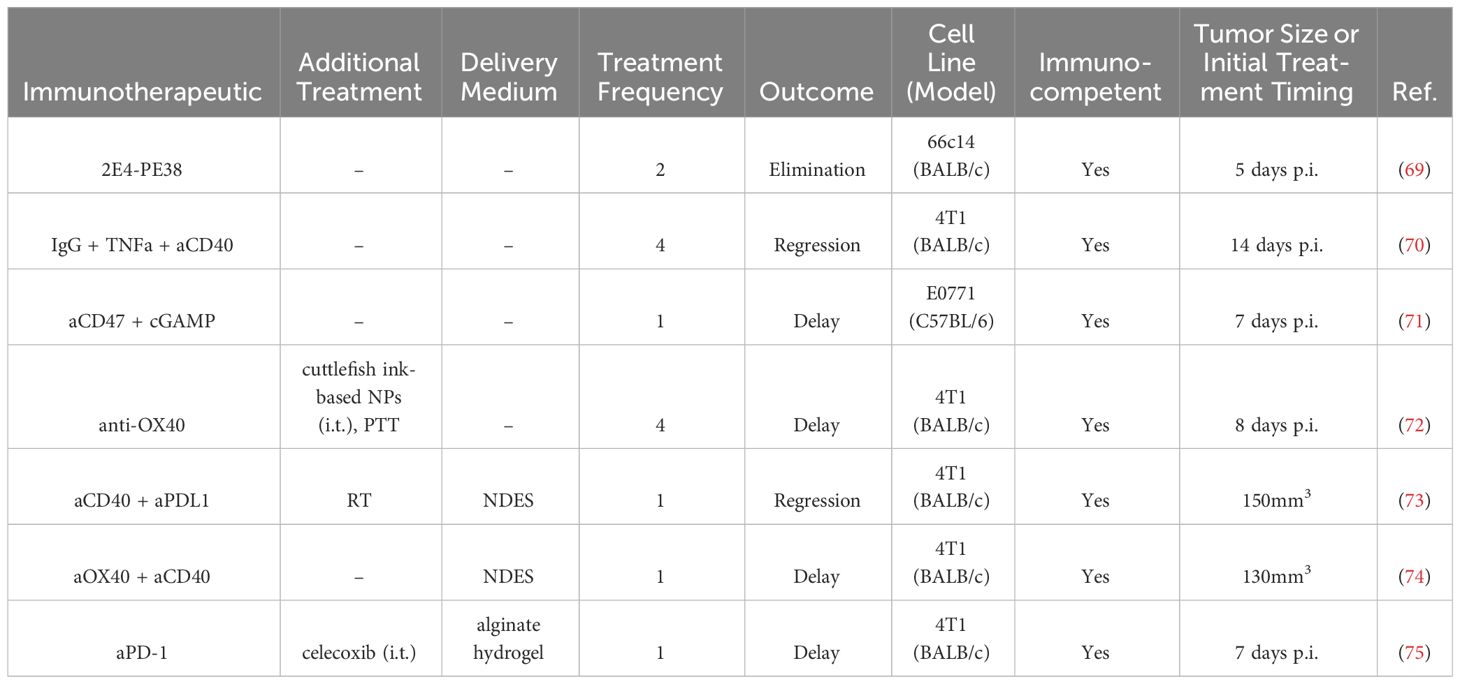
Table 3 Intratumoral antibody therapies in BC preclinical studies.
Anti-OX40 mAbs were i.t. delivered on day 10 and 11 post-4T1 tumor implantation in a murine model, which directly followed two consecutive days of administration of cuttlefish ink-based NPs, which are natural photothermal agents that have also immune activity (72). Treatment delayed tumor growth, while also inhibiting lung metastasis, reducing splenomegaly, increasing activated IFNγ+ CD4+ and CD8+ splenocytes, and increasing tumor infiltrating CD8+ T cells (72).
Anti-CD47 mAb delivered i.t. in combination with 2′3′-Cyclic GMP-AMP (cGAMP) seven days after tumor implantation led to a complete cure in 5/9 treated E0771 tumor-bearing mice after a single injection. Efficacy was dependent on STING- and type I interferon-responses. The combination treatment enhanced CD8+ T cell immunity and induced anti-tumor immunity (71).
In another study, allogenic immunoglobulin G (IgG) in combination with DC stimuli tumor necrosis factor alpha (TNFα) and aCD40 was delivered twice, two days apart, in two cycles 14-16 days post 4T1 tumor implantation in a mouse model. Treatment induced near complete regression of 4T1 tumors, with 100% survival at day 34 following implantation (70).
In a fourth study, the variable domain of anti-mCD25 was attached to a portion of Pseudomonas exotoxin A (2E4-PE38) and delivered on days 5 and 9 post-tumor implantation in a 66c14 murine BC tumor (69). The treatment increased functional IFNγ+ CD8+ T cells, cDCs, and macrophages, while decreasing Tregs. Cured mice were immune to rechallenge, and the effect was abrogated by CD8+ T cell depletion (69).
In an effort to improve therapeutic index and formulate synergistic drug combinations, aPD-1 and celecoxib, a nonsteroidal anti-inflammatory drug, were delivered within an alginate hydrogel injected subcutaneously adjacent to orthotopic 4T1 tumors seven days after tumor inoculation in a murine model (75). The combination within the gel delayed tumor growth and increased survival as well as decreased lung metastases, compared to the either single agent in the gel or the combination in a saline solution (75).
A nanofluidic drug-eluting seed (NDES) locally released the agonist mAbs aOX40 and aCD40 in an orthotopic 4T1 BC murine model to reduce delivery injections and localize treatment (74). The NDES was implanted using the trocar technique, which physicians regularly use to place drain tubes in patients. The NDES released payloads through physical and electrostatic interactions. Delivery of single agents (aOX40 or aCD40 alone) demonstrated slightly delayed tumor growth compared to untreated controls. Both single agents increased i.t. CD4+ and CD8+ T cells, as well as T cells expressing CD134/OX40, which is important for T cell expansion and survival. In the spleen, the percentage of CD8+ T cells secreting IFNγ and granzyme B increased. While the treatments only moderately slowed the growth of 4T1 tumors, the NDES device did demonstrate efficacy retaining the biologic activity in a sustained release system with reduced hepatocellular toxicity compared to systemically treated mice (74). In a follow-up study, aCD40 and aPD-L1 were delivered in the NDES device in larger tumor burdens of 150 mm3, with radiotherapy administered for three consecutive days prior to NDES implantation (73). The therapy increased survival and reduced tumor burden in a 4T1 murine model of BC. The antitumoral effects were associated with decreased liver inflammation, increased i.t. CD8+ T cells, higher proliferating Ki67+ CD4+ T cells, and reduced M2 macrophages. Though not directly shown, the dual delivery with RT was suggested to induce systemic antitumoral immunity (73).
Preclinical local cell delivery for BC treatment has included CAR-T cells, cytokine-induced killer (CIK) cells, fibroblasts, DCs, and xenogeneic mammary glandular cells. To date, no cellular therapy for BC treatment has incorporated a delivery system (Table 4).
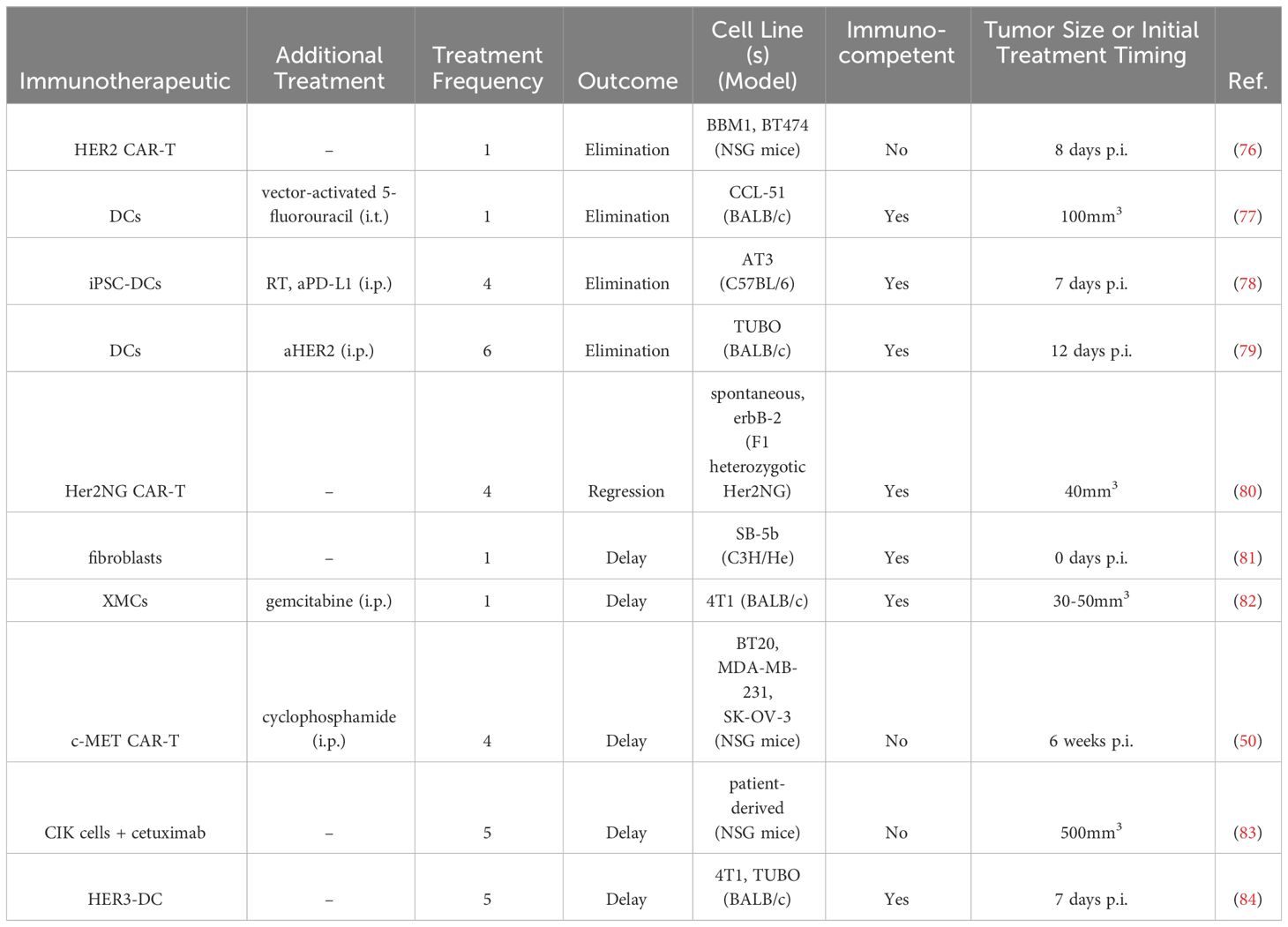
Table 4 Intratumoral cell therapies in BC preclinical studies.
HER2-targeted CAR-T cells were delivered intracranially to treat experimental HER2+ BC metastases in the brain. Experimental metastases were established via stereotactic injection of BBM1 and BT474 cell into the brain parenchymas of immunodeficient NOD/scid/γc(-/-) (NSG) mice (76). Local intracranial delivery of HER2 CAR T cells with either a 4-1BB or CD28 costimulatory domain eliminated brain metastases and extended survival past 100 days post tumor implantation, whereas control mice did not survive past 35 days of treatment. Intraventricular delivery of HER2 CAR-T cells with the 4-1BB co-stimulatory domain improved mouse survival compared to intracranial delivery (100% vs 50% at Day 100 post-tumor implantation), and furthermore it could eliminate leptomeningeal and multifocal disease (76).
Another CAR-T cell therapy targeted c-MET, which is expressed by 50% of all BCs (50). In vivo, 1.5x107 cells were delivered i.t. in the human xenograft SK-OV-3/luc model in NSG mice, concurrently with cyclophosphamide, on weeks 6, 7, 9, and 11 after tumor inoculation. Compared to controls, the c-MET CAR-T cells delayed tumor growth, which has led to a phase 0 clinical trial (50).
A third CAR-T cell therapy utilized F1 heterozygotic Her2NG offspring from Her2NG transgenic male mice and wild-type FVB/N strain female mice to evaluate CAR-T cells expressing scFvs that bind Her2NG (80). In the i.t. setting, 1.5x107 cells were delivered every two weeks for a total of four doses. Treatment led to regression of tumors, which eventually regrew. Though administered into one tumor locally, CAR-T cells were found in other mammary gland tumors as well as peripheral lymphoid organs.
CIK cells, which share functional characteristics of both T cells and NK cells, confer a unique benefit to treatment, as they do not require antigen for priming (83). CIK cells express FcγRIIIa (CD16a) and, in combination with monoclonal antibodies, can lyse tumor cells. In a patient-derived TNBC model in NSG mice, 1x107 CIK cells were delivered i.t. in combination with the anti-epidermal growth factor receptor (EGFR) mAb cetuximab once daily for five consecutive days when tumors had reached 500mm3. Treatment induced a delay in tumor growth as well as increased CD3+ T cell density within the tumor (83).
In another study, fibroblasts were utilized as vaccine carriers to treat BC that had metastasized to the brain (81). Fibroblasts were transfected with DNA from a breast tumor, SB-5b, which was spontaneously derived from a C3H/He mouse (H-2Kb). Fibroblasts additionally expressed MHC class I H-2Kb-alloantigens and were modified to secrete IL-2, IL-18, or GM-CSF. Mice treated intracranially with IL-2 expressing fibroblasts survived longer than mice in the control or other treatment groups. Splenocytes from these mice demonstrated higher cell lytic capacity as well as higher IFNγ production, demonstrating an increased anti-tumor activity (81).
DCs were also utilized as a vaccine after being transfected with an adenovirus encoding a secretory signal peptide with or without tumor associated antigen (Ad-sig or Ad-sig-TAA) linked to the extracellular domain of CD40 ligand (edcCD40L) (77). They were delivered i.t. in combination with the AdCDIRESE1A vector that encodes for bacterial cytosine deaminase, an enzyme that produces the chemotherapy 5-fluorouracil from 5-fluorocytosine. DCs activated with the Ad-sig-ecdCD40L vector and AdCDIRESE1A vector treatment eliminated tumors when injected i.t. in CCL-51 murine BC, enhancing survival compared to the other treatment groups. This combination treatment further reduced the development of lung metastasis compared to a single agent alone. When the gene for a TAA was incorporated into the adenovirus (rat H2N), and rH2N transgenic mice bearing rH2N+ NT2 breast tumors were treated i.t. with Ad-sig-rH2N/ecdCD40L DCs and the AdCDIRESE1A vector, tumor growth was delayed and survival was extended, as compared to the other treatment groups (77).
A more recent study with DCs evaluated their combination with aHER2 mAb after being pulsed with multiepitope MHC II HER2 peptides (79). In mice bearing HER2+ orthotopic TUBO tumors, DCs delivered i.t. together with intraperitoneal (i.p). aHER2 eliminated tumors and extended survival, compared to the single agents. Mice treated s.c. with this same combination only had a delay in tumor growth. The combination i.t. treatment led to increased IFNγ production from splenocytes and cells isolated from the tumor draining LN. Additionally, higher Th1 cytokines IFN-γ and TNF-α were found in the serum. Increased levels of CD4+ and CD8+ T cells, NK, NKT, and B cells were seen in the tumor. The treatment also led to the regression of untreated distant tumors (79).
A DC vaccine pulsed with HER3 antigen was i.t. delivered to subcutaneous 4T1 murine tumors seven days after tumor implantation (84). Treatments given twice a week for a total of six doses led to a delay in tumor growth and prolonged survival. Flow cytometry analysis revealed that greater numbers of CD4+ and CD8+ T cells per tumor mass had infiltrated the tumor compared to control treatments. Phenotypically, HER3-DC treatment increased the number of intratumoral CD4+ effector and memory cells. In culture, LN lymphocytes from the HER3-DC treated cohort produced higher amounts of IFNγ compared to control. Furthermore, these results were replicated in a HER2+ TUBO murine mammary carcinoma model, when DCs were delivered once weekly for a total of six doses (84).
DCs derived from induced pluripotent stem cells were i.t. delivered in combination with RT in the AT3 murine BC model thirteen days post tumor implantation (78). Three doses of DCs with two doses of RT led to slight tumor regression and prolonged survival, compared to DC treatment alone. Mechanistically, RT helped to direct the DCs to the tdLN and upregulate CD40 expression. The synergistic treatment increased intratumoral stem-like progenitor exhausted CD8+ T cells. In an abscopal tumor model, treatment of the primary tumor lead to tumor growth delay in the abscopal treatment, with a higher CD8+ TIL percentage of CD45+ cells in the distant tumor. Finally, RT and DC treatment sensitized AT-3 murine tumors to aPD-L1. Four DC i.t. injections, three RT treatments, and six aPD-L1 i.p. injections led to long-term, specific, durable cures in 30% of treated mice (78). Another cellular therapy investigated i.t. delivery of xenogeneic mammary glandular cells (XMCs) in combination with gemcitabine (82). Delivering xenogeneic tissue-specific cells was hypothesized both to induce an anti-inflammatory response and to promote cross-reactive antitumor T cells due to shared protein expression between XMCs and cancerous mammary tissue (85). In small 30-50mm3 4T1 mouse tumors, 1x106 XMCs isolated from porcine mammary glands were delivered i.t. once, along with three doses of gemcitabine delivered weekly i.p. The XMC treatment led to tumor growth delay, with higher percentages of necrotic areas and apoptotic cells and fewer Ki67+ proliferating cells. Additionally, i.t. CD4+ and CD8+ T cells and CD56+ NK cells increased, along with a decrease in CD11b+ MDSCs (82).
In summary, cellular therapies in preclinical murine models primarily evaluated efficacy of DCs (77–79, 84) and CAR-T cells (50, 76, 80). No treatment incorporated a local delivery system. No single cell therapy could eliminate tumors without an additional treatment in immunocompetent murine models (Table 4). However, with repeated injections and/or additional therapy, tumors could be eliminated in immunocompetent mice (77–79). I.t. delivery outperformed subcutaneous delivery (79), with indication of systemic anti-tumor effects (79, 80).
The largest amount of preclinical interest in BC i.t. immunotherapy has been in the area of virus delivery. Oncolytic virus and viral gene therapy were explored in multiple settings. All studies lacked delivery systems as encapsulation of viruses in polymeric particles or hydrogels impairs infectivity (Table 5).
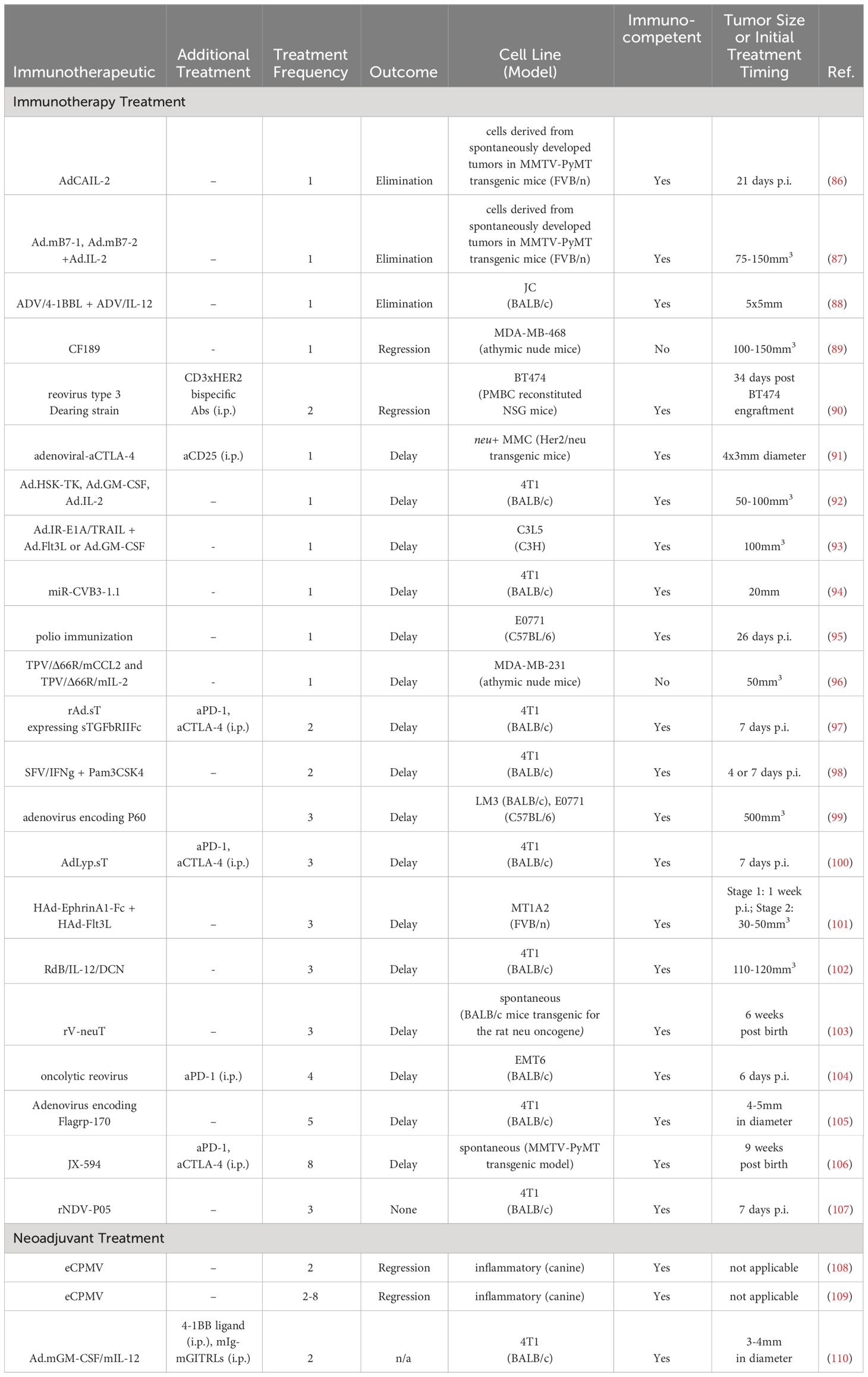
Table 5 Intratumoral viral therapies in BC preclinical studies.
Oncolytic adenoviruses encoding the LyP-1 peptide, which binds to the p32 receptor that is highly expressed on BC tumors and has a strong correlation with TNBC, were also engineered to express sTGFbRIIFc, which can inhibit transforming growth factor-β (TGFβ) signaling (100). I.t. delivery of these adenoviruses led to a delay in tumor growth and enhanced aPD-1 and aCTLA-4 checkpoint inhibitor therapy, with a reduction in lung metastases, in 4T1 murine tumors (100).
An oncolytic adenovirus expressing IL-12 and the proteoglycan decorin which can reduce TGFβ immunosuppression (RdB/IL12/DCN), was given every other day thrice to 110-120 mm3 4T1 mouse tumors (102). Treatment delayed tumor growth, increased proinflammatory cytokine expression in tumor tissue, increased i.t. CD8+ T cells, and reduced i.t. TGFβ which correlated with i.t. Treg reduction (102).
An oncolytic adenovirus which expressed a modified receptor for TGFβ (sTGFbRIIFc) to inhibit TGFβ signaling was delivered twice, on days 7 and 9/10 post implantation, in 4T1 murine tumors (97). Treatment downregulated TGFβ target genes and inhibited Th2 cytokine expression, while increasing Th1 cytokines. Additionally, peripheral blood memory T cells and splenic DCs increased, while MDSCs were reduced. Concurrent treatment with the adenovirus improved checkpoint inhibitor therapy (97).
An oncolytic adenovirus was constructed for use in BC cells that allow E1-deleted adenovirus viral replication that was restricted to BC cells via genome modification (inverse repeat adenovirus vectors) (93). Additionally, the E1A and TNF-related apoptosis inducing ligand (TRAIL) proteins were inserted into the vector to support viral replication and induce tumor apoptosis (Ad.IR-E1A/TRAIL). Researchers combined the Ad.IR-E1A/TRAIL oncolytic virus with an adenoviral vector carrying Flt3L or GM-CSF gene to induce a vaccination-type response. Different combinations of 1x109 pfu adenoviruses were delivered once to C3L5 murine tumors, and the most effective combination were Ad/IR-E1A/TRAIL + Ad.Flt3L, which outperformed combinations with Ad.GM-CSF and single therapies (93).
Another study attempted to enhance the efficacy of checkpoint inhibitors through the i.t. administration of an oncolytic vaccinia virus expressing GM-CSF (JX-594) in MMTV-PyMT transgenic mice which spontaneously develop tumors (106). JX-594 had previously been thought to serve as an in situ vaccine due to its ability to elicit an anti-tumor immune response and its synergy with aPD-1 in Renca tumors. JX-594 was delivered i.t. (1x107 pfu per tumor) eight times in the course of eight weeks, with systemic delivery of aPD-1 and aCTLA-4. Tumor growth was delayed and survival extended (106 days compared to 87 days), with an increase in i.t. CD4+, CD8+, CD8+ ICOS+, and CD8+ Granzyme B+ T cells (106).
The chimeric parapoxvirus CF189 was constructed as a therapeutic against TNBC (89). Different doses (103, 104, 105 PFU) were evaluated in MDA-MB-468 tumors in athymic nude mice, with regression observed in the lowest dose (103 PFU) when tumors 100-150mm3 in size receive one injection of the virus. The highest dose (105 PFU) in the injected tumor caused a slight tumor regression in the contralateral un-injected tumor, demonstrating the induction of systemic anti-tumor immunity from a local treatment (89).
A coxsackievirus B3 virus was modified with a targeting sequence miR145/143 to prevent normal tissue targeting and reduce toxicity (94). 4T1 murine tumors 20mm in size were treated with 1x106 PFU. Treatment delayed tumor growth and reduced metastasis in correlation with viral propagation and F4/80+, CD4+, and CD8+ immune cell infiltration, with reduced toxicity compared to wild type coxsackievirus B3 (94).
A tanapoxvirus (TPV) was engineered to express monocyte chemoattractant protein (CCL2) or IL-2 (TPV/D66R/mCCL2 and TPV/D66R/mIL-2) as a safer oncolytic virus with selectivity for tumor cells (96). One injection of 5x106 PFU in ~50mm3 MDA-MB-468 tumors in athymic nude mice led to tumor growth delay for both TPV/D66R/mCCL2 and TPV/D66R/mIL-2, with increased areas of necrosis and immune cell infiltration (96).
To increase efficacy of systemically delivered CD3-bispecific antibodies, an oncolytic, replication-competent reovirus type 3 Dearing strain was delivered twice 34-days after BT474 implantation in peripheral blood mononuclear cells (PBMC) reconstituted NSG mice (90). Three i.p. injections of CD3xHER2 bispecific antibodies were administered following the i.t. reoviral treatment. The reoviral treatment itself led to tumoral regression and, in combination with the bispecific Abs, led to additional regression (90).
Another oncolytic reovirus was used to treat palpable EMT6 murine tumors (104). Four injections of reovirus on days 6, 9, 12, and 14 post tumor inoculation at 5x108 PFU delayed tumor growth. Additional treatment with aPD-1 cured ~70% of treated mice at 110 days post tumor implantation. Immunologically, both reovirus and combination treatments increased splenic CD4+ and CD8+ T cells and TEM cells. Reoviral treatment upregulated i.t. Tregs, but aPD-1 reversed this phenomenon. The reoviral treatment and aPD-1 demonstrated systemic memory through re-challenge rejection as well as increased systemic anti-tumor IFNγ-producing CD8+ T cells (104).
Not all i.t. delivered oncolytic viruses are effective, especially as compared to systemic treatment. A recombinant Newcastle disease virus (NDV) virus strain 5 (rNDV-P05) was delivered to 4T1murine tumors thrice, spaced a week apart (107). No effect was observed, though the systemic administration of the virus did yield an anti-tumor effect (107).
Non-oncolytic viruses employed as gene vectors have also been well studied. An adenovirus vector carrying human IL-2 gene (AdCAIL-2) was delivered i.t. to tumor cells derived from tumors of MMTV-PyMT transgenic mice which, as noted above, spontaneously develop tumors (86). Direct injection with 5x108 pfu of AdCAIL-2 led to complete tumor regression in 8/9 mice within 3-4 weeks post-treatment. Antitumor activity was contingent on delivery location, as a subcutaneous injection of AdCAIL-2 on the opposite flank of the tumor did not lead to any delay in tumor growth. Tumor regression was associated with long-term anti-tumor immunity, as demonstrated by a lack of tumor growth in rechallenge studies (86).
A human adenovirus vector encoding EphrinA1-Fc, a ligand of tyrosine kinase receptor EphA2 overexpressed in BC which can inhibit tumor progression, and one expressing Flt3L were evaluated in small (30-50mm3) EphA2 overexpressing MT1A2 tumors in a murine model (101). Ad.EphrinA1-Fc led to tumor growth delay, which was further reduced by incorporating Ad.Flt3L (101).
To inflame the immune environment, immunostimulatory proteins and ligands αCTLA-4, αCD127, αCD3, IL-15, LIGHT, mda-7, and CD80 have been incorporated into adenoviruses (91). However, none displayed anti-tumor activity when delivered into 4x3mm HER2/neu positive mouse mammary carcinoma (MMC) tumors. Tumor delay was observed when the mAb aCD25 was delivered with i.t. adenoviral-aCTLA-4. Survival was extended ~6 days in the combination group compared to aCD25 mAb treatment alone (91).
An adenovirus encoding B7-1 (Ad.mB7-1) and B7-2 (Ad.mB7-2), ligands for T cell receptor CD28, were coupled with adenoviral-IL-2 delivery in PyMT and neu tumors to induce complete tumor regression after a single i.t. injection in a mouse model (87). In PyMT tumors, 7/15 tumors completely regressed following Ad.mB7-2/hIL-2 treatment, compared to 4/15 in the Ad.mB7-2 treatment group. PyMT-specific cytotoxic T cell activity was observed following these treatments via an in vitro cytotoxic T lymphocyte (CTL) assay. In neu tumors, 13/20 completely regressed in Ad.mB7-1/hIL-2, 9/15 in mB7-2/IL-2, demonstrating tumor dependent differences in response (87).
Three adenoviral constructs containing genes for herpes simplex virus thymidine kinase (HSV-TK), which confers cytotoxicity to the nucleoside analog ganciclovir by phosphorylating ganciclovir, GM-CSF, and IL-2, respectively, were engineered for immune stimulation as well as concurrent treatment with ganciclovir (92). In 50-100mm3 4T1 murine tumors, 1x1010 adenovirus particles injected at three different tumor sites. The greatest tumor delay involved the triple combination of GM-CSF, IL-2 and HSV-TK adenoviruses, which reduced lung metastases and correlated with an increase in lymphocyte infiltration. To evaluate how neoadjuvant treatment impacted metastasis, tumors were excised three days after adenoviral treatment, but no effect on metastasis was observed between surgery and no surgery groups 15 days after surgery (92).
Liver metastasis in the JC mouse mammary model were treated using an adenoviral-mediated delivery of the 4-1BB ligand (ADV/4-1BBL) co-delivered with an adenoviral delivery of IL-12 gene (ADV/IL-12) (88). 4-1BBL is a co-stimulatory receptor expressed on the surface of activated T cells. Liver metastases were treated once i.t. with ADV/4-1BBL and ADV/IL-12 in 5x5mm tumors. 78% of treated mice survived at least 150 days post tumor implantation, which was primarily mediated by CD4+ T cells. 5/7 of these mice rejected a tumor re-challenge. This ADV-mediated, i.t. treatment was compared to a systemically administered 4-1BBL mAb plus i.t. ADV/IL-12, which caused tumor rejection in 87% of mice. However, unlike the i.t. ADV/4-1BBL, the effect was mediated by CD8+ T cells, and only 3/8 of the mice rejected re-challenge (88).
An innate immune agonist, Flagrp-170, was delivered via adenoviral vector five times, given every other day, in 4-5mm diameter 4T1 murine tumors (105). Flagrp-170 contains an NF-κB-stimulating sequence from flagellin incorporated into the Grp-170 molecular chaperone and has demonstrated greater anti-tumor activity than flagellin alone. Treatment caused i.t. Th1-associated cytokine gene signature upregulation (IFNγ, IL-12, GM-CSF). Splenocytes and cells from the draining LN produced more IFNγ and IL-2 compared to untreated controls. The treatment delayed tumor growth, the effect of which was abrogated by inhibition of GM-CSF (105).
Expression of the transcription factor Foxp3 in both Tregs and in BC cells was targeted with a cell penetrating peptide P60 that binds to a region of Foxp3 encoded in an adenoviral vector (99). When HER2+ LM3 and TNBC E0771 mouse tumors reached a burden of 500 mm3, the vector was delivered once every three days for a total of three treatments, leading to tumor growth delay and decreased lung metastases. Treatment led to a reduction of Tregs in the tumor but not the spleen (99).
Recombinant vaccinia virus encoding for the neu oncogene was tested in neu transgenic BALB/c mice, and the stage of cancer, number of injections, and delivery route were all optimized (103). Mice survived longer when they received three injections (1x108 pfu/injection), were treated earlier, and were injected in the intramammary gland (intratumoral) compared to subcutaneously. Furthermore, intramammary gland injections led to higher anti-neu antibody titers and more cytotoxic splenocytes compared to subcutaneous vaccination (103).
A replication-deficient Semliki Forest virus (SFV) vector, expressing IFNγ (SFV/IFNγ) was utilized as a safer viral mediated delivery option and ideal for macrophage polarization, as SFVs do not infect human or murine macrophages but do infect other cells (98). When 4T1 murine tumors became palpable, SFV vectors were injected i.t., with a second dose administered six days later. Treatment delayed tumor growth, associated with an increase in CD4+ and CD8+ T cells and a decrease in Tregs, CD11b+ MDSCs and CD206+ M2 macrophages in the tumor (98).
Poliovirus delivery was utilized to leverage pre-existing anti-polio immunity induced from childhood vaccination to inflame the TME (95). Prior to E0771 tumor implantation, mice received prime and boost polio vaccination. 26 days post tumor implantation, 1x107 PFU of mRIPO, a version of a live-attenuated poliovirus type 1 vaccine adapted for mice, was delivered once. mRIPO treatment extended survival (50% at 40 days p.t. compared to less than 30 days for other treatment and control groups) and increased CD4+ T cells and eosinophils. CD4+ and CD8+ T cells displayed a higher functional status, indicated by reduced T cell exhaustion markers. UV-inactivated polio virus was also evaluated. 18 days post tumor implantation, the inactivated virus was delivered twice, six days apart, which also delayed tumor growth (95).
Two studies explicitly utilized vectors to induce an adaptive immune response prior to surgical resection. In a canine study of inflammatory BC, a rare type of the disease, empty cowpea mosaic virus (eCPMV) immunotherapy was delivered i.t. weekly (0.2–0.4 mg per dose) for a minimum of two injections to evaluate the safety and efficacy of treatment (108). eCPMV was not intended to infect cells but rather to serve as an innate immune agonist, similar to TLR and STING agonists. Five canine patients were treated, while five were provided firocoxib/cyclophosphamide/toceranib therapy as the control arm. After 14 days following the first injection, tumors had measurably decreased, allowing for surgery in two of the five patients who previously would not have been eligible. Phenotypically, the Treg+/CD8+ T cell ratio in peripheral blood decreased in the eCPMV treatment arm. eCPMV also altered the TME, with higher neutrophil counts and lower Ki-67 expression, as indicated by immunostaining. Combining eCPMV with the standard of care improved the mean survival compared to standard of care alone (134 vs. 67 days) (108).
In a follow-up study, a transcriptomal analysis of canine patients with inflammatory BC again treated with eCPMV revealed an increase in neutrophil recruitment and activation transcriptional profile (109). Tumors in this study also decreased in all six treated patients, which allowed three patients to undergo surgical treatment (109).
In another neoadjuvant study, an adenovirus encoding GM-CSF and IL-12 was delivered i.t. twice every three days alongside systemic 4-1BB ligand (two treatments, each three days apart) and mIg-mGITRLs (8 treatments, each three days apart) in 4T1 mouse tumors (110). mIg-mGITRLs are monoclonal antibody homodimers against glucocorticoid-induced tumor necrosis factor receptor family-related protein, which is primarily expressed on Tregs, and binding is thought to eliminate Tregs through ADCC. After 9 days following the first i.t. treatment, tumors were resected. The triple combination increased survival (>125 days in 65% of mice) and decreased tumor metastases. Mechanistically, within the spleen, the cytolytic activity of T cells increased and Treg function decreased (110).
Overall, viral delivery has been explored in both neoadjuvant and non-neoadjuvant settings, without delivery systems (Table 5). Most treatments did not have an additional systemic therapy, but those that did often incorporated checkpoint inhibitors (97, 100, 104, 106), reflecting the ability of certain viruses to sensitize the TME to these already FDA-approved therapies. Three therapies could eliminate tumors with a single injection (86–88), though most viral therapies caused only a delay in tumor growth even with repeated injections (Table 5). I.t. treatment could lead to a reduction in lung metastasis (92, 100) and growth delay in untreated, contralateral tumors (89). Route of delivery impacted treatment, as i.t. delivery outperformed subcutaneous delivery when tested in two studies (86, 103). In the neoadjuvant setting, treatment allowed patients in canine studies to undergo surgery when previously surgery had not been an option (108, 109), providing additional rationale to pursue i.t. viral therapy in the neoadjuvant setting.
Plasmid DNA encoding cytokines have been delivered i.t. to alter the TME of breast tumors in several preclinical studies. Because plasmids are negatively charged macromolecules, they require delivery assistance, through either electroporation or lipid nanoparticle formulation in order to enter BC cells (Table 6).
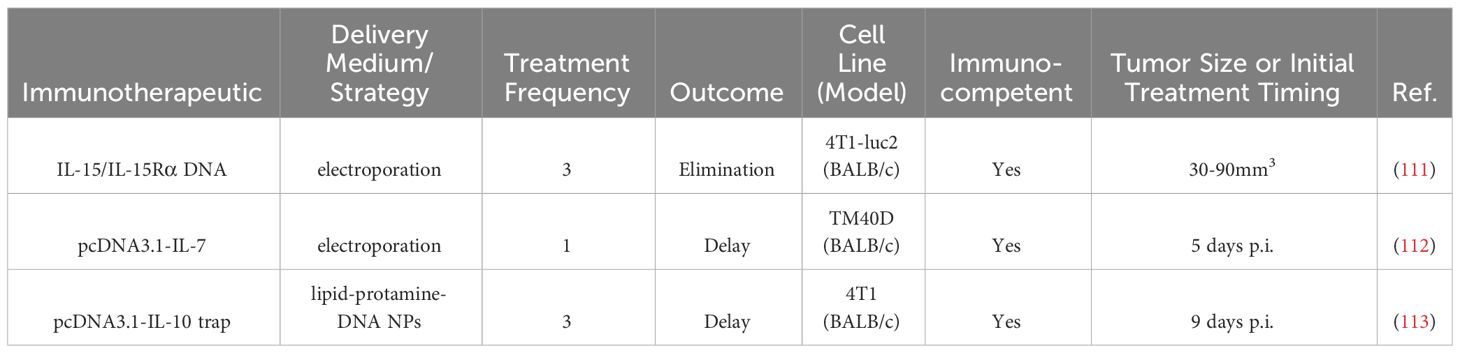
Table 6 Intratumoral nucleic acid therapies in BC preclinical studies.
Plasmid encoding for the inflammatory cytokine IL-15 and IL-15 receptor alpha (IL-15Rα), which stabilized and improves the bioactivity of IL-15, was delivered three times with electroporation within one week to 4T1-luc mammary mouse tumors (111). Tumors in 25% (2/8) of the mice were eliminated, which was associated with an increase in memory T cells, particularly CD8+ TEM and TCM and CD4+ TCM). Cured mice rejected tumor rechallenge (111).
The human cDNA of the cytokine IL-7, which can promote T cell survival, was encoded in a plasmid (pcDNA3.1-IL-7) and injected once i.t. followed by electric pulses in a TM40D mouse graft model of BC five days after tumor implantation (112). Treatment delayed tumor growth, the effect of which was dependent on CD8+ T cells. Serum IFNγ and splenocyte CTL cytotoxicity increased (112).
Rather than delivery of an immunostimulatory cytokine, another strategy to modulate the TME is to remove an immunosuppressive cytokine. A gene encoding an interleukin-10 (IL-10) trap protein was delivered i.t. in 4T1 murine tumors, as IL-10 had been correlated with reduced overall survival in patients with TNBC (113). The trap gene was loaded into lipid-protamine-DNA NPs and injected thrice i.t., leading to a delay in tumor growth and 80% survival at 26 days post-treatment as compared to 0% survival at this timepoint in the control groups (n=8-10 for groups). TNFα and IFNγ i.t. gene expression had increased post-treatment compared to controls (113).
As observed above, localized cytokines have the potential to modulate the TME from immunosuppressive to immunostimulatory. Several potent cytokine and chemokine therapies have been evaluated in preclinical BC studies with or without a supportive delivery vehicle (Table 7).
Table 7 Intratumoral cytokine and chemokine therapies in BC preclinical studies.
The cytokine GM-CSF was delivered at low and high doses i.t. in immunologically “cold” PyMT mouse tumors to evaluate the effects of dosing on tumor vasculature normalization and hypoxia (116). Delivered thrice per week over three weeks, low dose GM-CSF reduced tumor hypoxia while normalizing tumor vasculature. However, a five-week treatment with low dose GM-CSF did not affect tumor growth. When used as a pre- and concurrent treatment with aPD-1, low dose (5ng) but not high dose GM-CSF (100ng) led to tumor growth delay. Mechanistically, low dose GM-CSF reduced the inflammatory transcriptional profile of TAMs (116).
The proinflammatory cytokines IL-12 and TNFα were encapsulated in polylactic acid (PLA) microspheres and injected once i.t. in the weakly immunogenic MT-901 murine model of BC eleven days after tumor implantation (117). Following treatment, ~70% mice were disease-free, which was higher than IL-12 and GM-CSF-loaded microspheres (~40%) and either of the single cytokines alone-loaded microspheres (~2%). T cells derived from both the draining LN and spleen demonstrated higher activity, as measured through IFNγ production. The treatment also led to an increase in i.t. polymorphonuclear cells and CD8+ T cells, measurable five days post-treatment. When compared to treatment with surgery in rechallenge studies, all mice treated with the IL-12 and TNFα microspheres rejected the tumor, whereas only 20% of mice who had received surgery remained tumor-free. Local injection in a sustained delivery medium could eliminate tumors and establish immune memory (117).
A second study with PLA microspheres utilized a triple combination of cytokines, IL-12, IL-18, and TNFα (118). Mice bearing 4T1 tumors received a single i.t. injection of IL-12/IL-18/TNFα microspheres or dual-combination cytokines. The IL-12/TNFα combination led to the most pronounced delay in tumor growth, least metastatic lung nodules, and greatest survival. Surprisingly, the addition of IL-18 to the IL-12/TNFa combination reduced antitumor activity. Antitumor effects were contingent on both CD8+ T cells as well as NK cells. Local cytokine delivery enhanced CD8+ T cell numbers and reduced CD4+CD25+ Tregs in the tumor draining LN (118).
A sodium-alginate hydrogel was constructed to deliver lymphotactin, XCL-1, i.t. in combination with systemic delivery of doxorubicin-loaded poly(lactic acid-co-glycolic acid) (PLGA) NPs coated with cell membranes (119). The NPs would target the tumor, at which point the chemotherapy would induce immunogenic cell death (ICD) and the XCL-1 chemokines would recruit DCs into the tumor to enhance tumor antigen cross presentation. The sodium alginate was delivered as a solution and crosslinked in situ due to the presence of calcium ions in the tumor. The gel+XCL-1 was delivered once i.t. in 4T1-luc murine tumors the day after a series of three doxorubicin treatments and delayed tumor growth in the treated and untreated tumors, as well as increased survival at 30 days post-treatment. Furthermore, compared to doxorubicin-NP treatment alone, the XCL-1 treatment increased the homing of CD11c+ DCs to the tumor (119).
A neoadjuvant study pursued a combinatorial approach with i.t. and peritumoral immunotherapy and local radiotherapy prior to breast tumor resection in a mouse model (120). The cytokine Flt3L was i.t. injected daily over a period of five days, followed by 9 Gy radiotherapy the next day, and the TLR3 agonist poly(I:C) and aCD40 agonist delivered the following day peritumorally (termed “ISIM” treatment). Six days after poly(I:C)/aCD40 administration, tumors were resected. In 4T1-luc tumors, treatment delayed metastatic tumor growth and increased median survival (34 days vs. 26 days for the control). Furthermore, 6 days after poly(I:C)/aCD40, higher antigen-specific CD8+ were observed in the blood and lungs, along with higher percentages of effector T cells. As determined through depletion studies, prolonged survival was dependent on both CD4+ and CD8+ T cells. Batf3-/- mice with AT-3 tumors did not benefit from the triple combination, the improvement in survival seen in wild type mice was completely abrogated, though it could be moderately rescued by delivering bone-marrow–derived CD103+ DCs. The improvement in survival was demonstrated to be dependent on T cell egress from the LNs and immune activation in the tumor draining LN. Addition of systemic neoadjuvant aPD-L1 enhanced median survival (44 days vs. 35 days without aPD-L1) in 4T1-luc tumors. Additionally, increasing radiotherapy treatments to three and performing surgery only one day after poly(I:C)/aCD40 treatment both independently enhanced survival in mice bearing orthotopic 4T1-luc tumors (120). Prior studies with this ISIM treatment in the primary treatment setting demonstrated that it increased frequencies of activated, effector CD4+ and CD8+ T cells in AT-3 murine tumors (114) and decreased PMN-MSDCs in AT-3 and 4T1 BC murine tumors (115).
The chemokine CCL21, which recruits T cells, NK cells, and DCs, was formulated in Hydron, a hydrophilic polymeric hydrogel, and delivered to cl−66 mouse mammary tumors (122). Local delivery increased total tumor CD8+ T cell and NK cell numbers and enhanced i.t. DC recruitment. As a neoadjuvant, CCL21-Hydron was implanted four days prior to resection. CCL21-Hydron and resection improved overall survival (>70% at 100 days) compared to any other treatment group and delayed tumor growth in a re-challenge model (122).
In another local neoadjuvant study, our group co-formulated the cytokine IL-12 in the naturally occurring polysaccharide chitosan and delivered the co-formulation i.t. in 4T1 murine tumors three times, three days prior to tumor resection (121). Survival was extended (67% of mice were tumor-free >80 days post-resection), and lung metastases were reduced. Upon rechallenge, mice that had eliminated 4T1 tumors did not experience tumor recurrence. Splenocytes demonstrated a higher lytic capacity and enhanced tumor-specific responses, all without measurable systemic toxicity (121).
Research utilizing innate immune agonists to modulate the immune landscape intratumorally, has largely focused on TLR and STING agonists, with and without delivery systems (Table 8). Treatments with other agonists, such as RLR agonists, and neoadjuvant studies have also been performed. Innate immune agonist delivery represents the second largest group studied in preclinical studies.
Table 8 Intratumoral innate immune agonist therapies in BC preclinical studies.
In the metastatic 4T1 mouse model, the STING agonist cGAMP was delivered i.t. to increase the type I interferon response locally (125). After 16-24 hours, injection led to an i.t. influx of TILs and CD45+ CD11bmid Ly6C+ F4/80+ macrophages, which demonstrated phagocytic activity and produced the proinflammatory cytokine TNFα and chemokines Cxcl10 and Cxcl11. Two treatments of cGAMP at days 5 and 10 after tumor implantation yielded a modest but statistically significant delay in tumor growth, which was dependent on CD8+ T cells, as demonstrated through a CD8+ depletion study. The authors admitted a need for an appropriate delivery system for cGAMP, as cGAMP was likely not present for a sufficient amount of time to assess its full anti-tumor effects (125).
The STING agonist RR-CDA, a synthetic cyclic dinucleotide (CDN), was injected six times i.t. into spontaneously developing mammary tumor MMTV-PyMT mice 9 weeks post-birth (127). The i.t. treatment was combined with systemically delivered VEGF-R2 mAb (DC101) and aPD-1 mAb. The RR-CDA treatment alone delayed tumor growth, but both local and abscopal tumor control was enhanced with the triple combination treatment. The triple combination increased i.t. CD8+ T cell infiltration, decreased the density of the tumor vasculature, reduced lung metastases, and increased overall survival, which posed a strong rationale for STING therapy and antiangiogenic treatment in BC (127).
A novel STING agonist of vinylphosphonate-based CDNs was engineered to elicit a more potent immune response than other STING agonists evaluated in clinical trials, such as ADU-S100 (124). Additionally, masking the negative charge of the CDN by synthesizing it into a prodrug, enhanced potency, both in vivo and in vitro. When 4T1 murine tumors were ~65mm3, three i.t. deliveries, three days apart, of the highest concentration of prodrug led to tumor regression but not elimination, and this cohort had higher anti-tumoral CD8+ T cell levels compared to the vehicle treated group. Overall, prodrugs outperformed their parent compound, resulting in greater tumor growth delay (124).
Injection of the TLR3 agonist poly(I:C) was co-administered alongside a plasmid encoding the viral protein apoptin which induces apoptosis in tumor cells (128). Treatment of 4T1 murine tumors with the apoptin plasmid and poly(I:C) lead to significant growth delays at day 33 post-treatment, which was suggested to be due to induction of apoptosis, as analyzed through TUNEL assay. Locally, higher numbers of CD4+ and CD8+ infiltrated into the tumor, as analyzed via immunohistochemistry. The local treatment led to systemic increases of CD4+ and CD8+ T cell counts and the Th1 cytokines IFN-γ and IL-2, concurrent with a decrease in the Th2 cytokines IL-4 and IL-10 levels, as compared to other treatment and control groups (128).
Another novel agent was explored to modulate the local tumor immune environment, functioning like an innate immune agonist by non-specifically inflaming the TME. A tumor-targeted superantigen was constructed using the third loop of transforming growth factor α (TGFαL3) and staphylococcal enterotoxin type B (123). Seven days post 4T1 murine tumor implantation (20mm3), the superantigen was administered i.t. every other day for a total of four treatments. 5/14 (37.5%) of the treated tumors were eliminated and survived 6 months post-treatment, which outshone the systemically treated superantigen cohort (12.5% survival rate). The effect was correlated with an increase in IFNγ and TNFα production from splenocytes, a decrease in proliferation Ki-67 and micro vascularization CD31 levels, and an increase in tumor necrosis (123).
The influenza vaccine was delivered i.t. to induce local inflammation in a 4T1 mouse model (126). When tumors reached 10-25 mm2, the influenza vaccine was delivered thrice on consecutive days, which delayed tumor growth compared to a PBS control. Additional vaccinations (seven total) did not cause additional growth delay. Prior flu vaccination did not impair the anti-tumor response. Mechanistically, the 3x influenza vaccine reshaped the tumor immune microenvironment toward a Th1 profile, with increased IL-4 levels. Furthermore, the vaccine was demonstrated to work through its binding ability to sialic acid residues (126).
Local delivery systems have served to retain and release TLR agonists and RLR agonists preclinically. The innate immune agonist Poly(I:C), which binds to TLR3, MDA5, and RIG-I receptors, was complexed with the polymer polyethylenimine (PEI) and delivered i.t. in a 4T1 mouse model (141). The PEI-complexed Poly(I:C), called BO-112, was administered twice weekly, for a total of six doses, which led to 4T1 growth inhibition (141). In a follow-up study in TS/A tumors, the same treatment was combined with three doses of radiotherapy (RT) (134). The treatment synergized with RT, causing tumor regression in the primary tumor and tumor growth delay in an untreated abscopal tumor. In a bilateral tumor treatment scheme in which only one tumor was treated with both BO-112 and RT and the other was treated with RT alone, both tumors regressed, with over 20% survival at 150 days p.t. (134).
Several studies explored local delivery of the TLR7 agonist imiquimod. In the first study, imiquimod was encapsulated in a temperature responsive hydrogel composed of the naturally occurring polymers chitosan and hyaluronic acid, along with indocyanine green (IcG), for a combined immunostimulatory therapy with photothermal therapy (PTT) (130). Administering the imiquimod-IcG gel and applying laser therapy led to 4T1 murine tumor growth suppression in 8/10 mice. Greater suppression was induced with a low-dose systemic administration of the chemotherapeutic cyclophosphamide administered on day 0 and 7 post-laser treatment, with 10/12 mice experiencing growth suppression in this group. Additionally, the growth of untreated bilateral tumors was also delayed in both of these treatment groups, as compared to controls (130).
In a second study, imiquimod was delivered within an alginate hydrogel to localize treatment in a mouse model (132). Orthotopic 4T1-fLuc tumors were treated two weeks after tumor implantation with the alginate gel, co-formulated with doxorubicin, imiquimod, and aPD-L1. After 25 days post-treatment, no tumor nor metastases were observable, and all mice (10/10) survived at least 120 days post-treatment. Notably, when these therapeutics were delivered i.p., they had no effect on extending mouse survival, highlighting the importance of the delivery system and route (132).
In a third mouse study, imiquimod was loaded into acetylated chondroitin sulfateprotoporphyrin micelles (ACP-R837) and delivered five times at three-day intervals in 4T1 tumors (139). The treatment increased CD8+ T cell density and proinflammatory cytokines TNFα, IL-6, IL-1b, and IFNγ, while decreasing the anti-inflammatory cytokine IL-10. APC-R837 delayed tumor growth without displaying toxicity and extended survival. Mechanistically, it activated the death receptor signaling pathway, as indicated with an increase in caspase-8 expression. Furthermore, combination with systemically delivered micelles containing the chemotherapeutic doxorubicin caused the largest tumor apoptotic areas and lower Ki67 proliferation marker expression (139).
In a fourth imiquimod study, the agonist was formulated in a PLGA- poly(ethylene glycol)(PEG)-PLGA thermoresponsive hydrogel (131). In a bilateral EMT-6 murine tumor model, the imiquimod-gel was injected intratumorally in a single tumor eight days after tumor implantation. With a triple combination treatment of this imiquimod-gel, cryoablation, and systemically delivered aPD-1 and aCTLA-4, 1/18 mice had a 90-day, long-term survival with treated and untreated tumor elimination, demonstrating induction of an abscopal response (131).
Additionally, the TLR7/8 agonist resiquimod (R848) was encapsulated in platelet membrane-coated NPs (140). Five i.t. treatments with the NPs given five days after tumor implantation delayed the growth of 4T1 mouse tumors, decreased metastatic lung nodules, and enhanced survival with a median progression free survival of 23 days, which was 14 days longer than that of the control group (140).
Other studies pursued the TLR9 agonist unmethylated cytosine-phosphate-guanine (CpG) oligodeoxynucleotides, either alone or in combination with another therapy. In one study, CpG was loaded into 3-aminopropyltriethoxysilane-modified Fe3O4 NPs (FeNPs) (138). CpG-FeNPs were i.t. injected on days 7, 10, and 13 after tumor implantation in a mouse model, and delayed 4T1 tumor growth, with an inhibitory rate of 69%, and reduced lung metastases. Additionally, splenocytes displayed higher lytic activity against 4T1 cells than those from control groups (138).
CpG was combined with the chemotherapeutic doxorubicin and photothermal agent polydopamine (PDA) within a thiolated hyaluronic acid hydrogel (133). Orthotopic 4T1 mouse tumors were injected with the CpG/PDA/doxorubicin hydrogels and, 12-hours after injection, irradiated with a 780-nm NIR laser. One week following treatment, the tumors were irradiated again. Mice treated with the CpG/PDA and CpG/PDA/doxorubicin combinations had the highest levels of serum TNFα and IL-6, as well as splenic CD8+ T cells. At 12 days post-treatment, tumors from the mice treated with the CpG/PDA/doxorubicin and PTT had regressed the most. Finally, in a bilateral tumor model with one tumor treated with local CpG/PDA hydrogel plus PTT along with three treatments of systemic aPD-L1, the growth of the untreated bilateral tumor was delayed, as compared to control groups. This effect was only statistically significant with the addition of aPD-L1 as treatment with the local CpG/PDA hydrogel plus PTT alone did not lead to a statistical difference in anti-tumor rejection (133).
CpG and semiconducting polymer (SPII) nanorods were delivered i.t. in a responsive alginate hydrogel (135). Two hours post-injection, 4T1 murine tumors were treated with PTT. After 24 hours, TUNEL staining and Ki-67 staining of the treated tumor demonstrated higher levels of apoptosis and lower levels of cell proliferation, respectively. In a bilateral metastatic model, treatment with CpG/SPII/PTT therapy significantly reduced the growth of the untreated distant tumor and had no observable spontaneous metastatic lung nodules. Within the tumor, markers of immunogenic cell death [calreticulin and high-mobility group box 1 protein (HMGB1)] were increased, and the serum proinflammatory cytokines IL-6, TNFα, and IFNγ also were upregulated, as compared to other treatment groups. Finally, higher levels of mature DCs (CD11c+CD80+CD86+) in the tumor draining LNs and CD8+ cytotoxic T cells in untreated distant tumors were recorded (135).
Another in-situ forming alginate hydrogel locally delivered CpG and catalase labeled with 131I radioisotope to localize the effects of RT (129). When tumors had reached ~60 mm3, hydrogel treatment with concurrent RT in a subcutaneous 4T1 mouse model eliminated all tumors, with all mice (8/8) surviving 60 days post-treatment, compared to other controls which all succumbed to disease within less than a month post-treatment. In an orthotopic 4T1-fLuc model, 16 days-post-tumor inoculation, mice were treated once locally with the CpG/131I-Cat/alginate gel, followed by four systemic injections of aCTLA-4. After 90 days, 75% of mice were surviving without any sign of metastasis (129).
A final study of CpG also combined local brachytherapy, this time within thermoresponsive elastin-like polypeptides (ELPs) (136). When injected i.t. within the ELP delivery system in mice, CpG was still present 18 days later, and a high dose (100ug) led to a statistically significant delay in growth and reduction of lung metastases at 13 days post-treatment. Incorporating localized 131I brachytherapy led to further delay in tumor growth, extended survival, and reduced lung metastases (136).
Another innate immune pathway, RIG-I, leads to type I interferon production following 5’ triphosphorylated RNA activation. Because immunomodulatory 5’ triphosphorylated RNA (immRNA) are quickly degraded in vivo, they were loaded into extracellular vesicles (EVs) derived from red blood cells (RBCs) and delivered i.t. in 4T1 murine tumors (142). I.t. immRNA delivery in RBC-derived EVs six times every three days delayed tumor growth, while increasing the pro-inflammatory and decreasing the anti-inflammatory cytokine gene signatures. Furthermore, immRNA delivery led to an increase in apoptosis, as measured by TUNEL staining. A similar treatment with human CA1a breast tumors in NSG mice also led to a decrease in tumor growth, accompanied by an increase in i.t. neutrophils, macrophages, and DCs. A second RLR agonist, 3p-125b-ASO, also was engineered to silence the oncogene miR-125b, and i.t. delivery of 3p-125b-ASO in RBC-derived EVs following the same schedule as immRNA RBC-derived EVs also led to a reduction in tumor growth, increase in i.t. neutrophils, macrophages, DCs, NK cells, and T cells, increase in i.t. pro-inflammatory cytokines (TNFα, IL-6, IL-12p40), and increase in apoptosis (142).
A novel innate immune agonist was formulating using lipoaminoglycosides, with the polar headgroups being either tobramycin or kanamycin, which can stimulate cellular immunity (137). These lipoaminoglycosides self-assembled into vesicles. In the 4T1 murine model, vesicles were injected into 100-200mm3 tumors twice weekly. The treatment reduced tumor weight on day 19 post-treatment (137).
The inherent property of outer membrane vesicles, lipid-bilayer particles excreted from bacteria, to activate TLR ligands was leveraged as an immunotherapeutic and additionally engineered to display an anti-EGFR scFV on the surface (143). Delivered daily 11 times in 4T1 murine tumors once they reached an average diameter of 0.7 mm, the treatment had no effect on tumor growth at the 10ug dose level. However, an i.p. dose of 25ug led to almost complete tumor regression, which was correlated with an increase in necrosis, i.t. M1 macrophage polarization, and T and NK cell presence (143). This effect was a rare instance in which systemic injection outperformed a local delivery.
I.t. delivery of innate immune agonists was pursued in a neoadjuvant setting, without delivery systems. In a first study, the TLR7/8 agonist 3M-052 was delivered i.t. seven days prior to resection in a 4T1 murine model (145). Resected tumors had higher levels of immune cells (B cells, CD4+ and CD8+ T cells, NK cells, macrophages), and this neoadjuvant treatment extended survival, with a median of 20 days, as compared to the control of 10 days post-resection. Depletion of CD8+ T cells and inhibition of Type I IFN signaling reversed the metastasis-free survival benefit of the neoadjuvant treatment. Interestingly, when neoadjuvant aPD-1 was delivered systemically three times in combination with i.t. 3M-052 given once seven days prior to surgery, no improvement in survival or in i.t. immune cell infiltration was observed compared to the same treatment without aPD-1 (145).
I.t. CDN STING agonists were explored along with combinations of extended half-life IL-2 and aPD-1 delivered systemically (144). CDNs were delivered every three days for a total of three times, followed by surgery three days after the last administration, in 4T1-Luc murine tumors. Of the mice whose tumors were treated beginning at 100 mm3 (n=10), long-term survival was 60% at 90 days post-treatment. Of the mice whose tumors were treated beginning at 200 mm3 (n=10), long-term survival was slightly reduced to 30% at 90 days post-treatment. Additionally, neoadjuvant treatment (90% survival) was demonstrated to be superior to adjuvant treatment (10% survival) at 100 days post-tumor implantation. Immunologically, i.t. CDN and systemic IL-2 plus aPD-1 increased both i.t. and lung CD8+ T cells and effector CD4+ T cells. Depletion studies highlighted the importance of NK cells on therapeutic efficacy, which did not rely on CD8+ and CD4+ cells and neutrophils. NK cells were activated by the treatment, as shown by an increase in granzyme B, perforin, TNFα, IFNγ, IL-2, CD69, Ki67, and PD-1 expression. The systemic upregulation of IL-2 and type I interferons supported the activation of NK cells (144).
In summary, innate immune agonist immunotherapy was the second most pursued type of i.t. delivered immunotherapy within preclinical BC studies. Due to their small size, these agonists could greatly benefit from delivery systems for localization and long-term, sustained release combined with a reduction in treatment number. Hydrogels in particular necessitated only one i.t. injection (129–133, 135). Most innate immune agonists on their own did not induce a strong anti-tumor response as a monotherapy, and combination with chemotherapy, checkpoint inhibitors, PTT, and/or RT were required for robust anti-tumor responses (Table 8).
One preclinical study pursued i.t. delivery of a genetically modified Salmonella strain expressing a methioninase in nude mice bearing MDA-MB-231 xenografts (146). Salmonella had demonstrated preferential accumulation in the tumor in a previous study. Toxicity concerns were reduced by genetically engineering a strain that did not produce endotoxins, VNP20009, as determined by prior studies. As this strain alone could not eliminate tumors, it was additionally engineered to overexpress a methioninase to reduce tumor methionine levels, which can prevent mitosis, cause cell-cycle arrest, and induce apoptosis. I.t. delivery of this bacteria in 60-100 mm3 tumors led to tumor growth delay. Treated tumors underwent a liquefaction necrotic process. Furthermore, bacteria were predominately found in the tumor rather than the lung, liver, or kidney at ratios between 500–62,000:1. Finally, methionine was reduced in tumor cells but not systemically (146). The safety and feasibility of the treatment is being evaluated in a phase 1 clinical trial (NCT05103345).
The first major finding of this review is the increasing interest in i.t. delivery of BC immunotherapies. In the past two decades, the number of clinical trials increased steadily (Figure 2F). From 2004-2013, 11 clinical trials started, whereas a decade later from 2014-2023, 37 clinical trials began. Immunotherapy itself is strengthening its foothold as a primary therapeutic modality as well as the future of cancer treatment. Annually, a staggering 4 million patients worldwide are eligible for an immunotherapy, according to the CRI (147). Within BC, the impact of immunotherapies is just now being felt. Pembrolizumab was first FDA-approved for locally recurrent unresectable or metastatic PD-L1 positive TNBC in November 2020 and since then has also been approved for high-risk, early-stage TNBC (148). With the rising interest, more i.t. immunotherapies will likely be evaluated in BC.
The second major finding is the proven safety record of i.t. delivered immunotherapies in BC. Results from clinical studies conclude that i.t. immunotherapy is technically feasible and well-tolerated with a low toxicity profile (48–50, 54–56, 149). This feasibility and safety profile indicate that intratumoral injection is a relevant delivery method for BC treatment.
The third major finding is the evidence of both local and systemic efficacy, with induced immune changes, following i.t. immunotherapy. Clinical anti-tumor responses were noted in the following clinical trials: NCT02423902, NCT01703754 (51), NCT01846091 (53), NCT03740256 (60, 61), NCT02779855 (62), and NCT03567720 (67). Numerous preclinical studies across a range of immunotherapies have demonstrated elimination of treated tumors (69, 76–79, 86–88, 111, 117, 121, 123, 129–132, 144). Additionally, a major feature of i.t. immunotherapy is its potential to induce robust systemic immunity capable of controlling distant, untreated lesions. Anti-tumor effects in un-injected, abscopal tumors were observed with the adenovirus Ad-RTA-hIL-12 (51), and adenovirus CAdVEC (60, 61) in clinical studies. Preclinical treatments with antibodies, cells, viruses, cytokines, and innate immune agonists reduced or eliminated metastases (72, 75–77, 92, 94, 99, 100, 110, 118, 120, 121, 127, 129, 132, 135, 136, 138, 140, 145) and induced systemic anti-tumor effect in untreated tumors (79, 119, 130, 131, 133–135). Furthermore, i.t. therapies have both initiated T cell infiltration, as demonstrated in the following clinical trials: NCT02061332 (47), NCT01837602 (50), NCT02423902, NCT01703754 (51), NCT03740256 (60, 61), NCT02779855 (62), NCT02531425 (54), NCT03739931 (66), and NCT02872025 (59). Most, if not all, preclinical studies also demonstrated a skewing toward an anti-tumor immune response intratumorally. The potential of i.t. immunotherapy – treat locally, act systemically – promises great potential for BC patients.
First, a major trend and a significant opportunity is the movement of immunotherapies, including i.t. immunotherapies toward neoadjuvant and earlier stage treatments in BC patients. Immunotherapies are more likely to be effective in BC patients with lower disease burden and intact immune systems. With respect to neoadjuvant BC immunotherapy, currently, high risk patients receive chemotherapy prior to surgery. However, recurrence rates are still high, and side effects detrimentally affect patients’ quality of life. Immunotherapy promises to boost tumor specific immunity that eliminates residual cancer cells after surgery and prevents recurrence. Promising results from the clinical trials NCT02018458 (49), NCT02061332 (47), and NCT02779855 (62) and from the preclinical studies (108, 109, 120–122, 144, 145) encourage more consideration for neoadjuvant i.t. immunotherapy studies in further preclinical research and in developing clinical trials.
Second, the potential to find pCRs following neoadjuvant i.t. immunotherapy in BC is a major opportunity. pCRs may serve as a primary endpoint rather than recurrence which may take a decade or longer in BC patients for targets to be reached. It is expected that increasing numbers of BC trials will utilize neoadjuvant treatments, including i.t. immunotherapies, and measure pathological responses at the time of lumpectomy as the primary efficacy outcome.
Third, the type of immunotherapy delivered must be given additional consideration. For example, pembrolizumab given i.t. in high-risk DCIS did not decrease tumor volume in a phase 1 clinical trial (59). It is possible that inflaming the immune environment prior to or concurrent with checkpoint inhibitor therapy may synergize with checkpoint inhibitors to produce a better anti-tumor response. This combination treatment was noted in a metastatic TNBC patient receiving plasmid IL-12, whose tumor became sensitized to aPD-1 (54). Additionally, evaluation of preclinical studies did not note a clear trend for efficacy based on immunotherapeutic agent alone (Figure 3), which suggests that other factors (timing, frequency, delivery medium) also strongly impact treatment outcome. Currently, viral therapies dominate both clinical and preclinical research, but many of the competing platforms look promising.
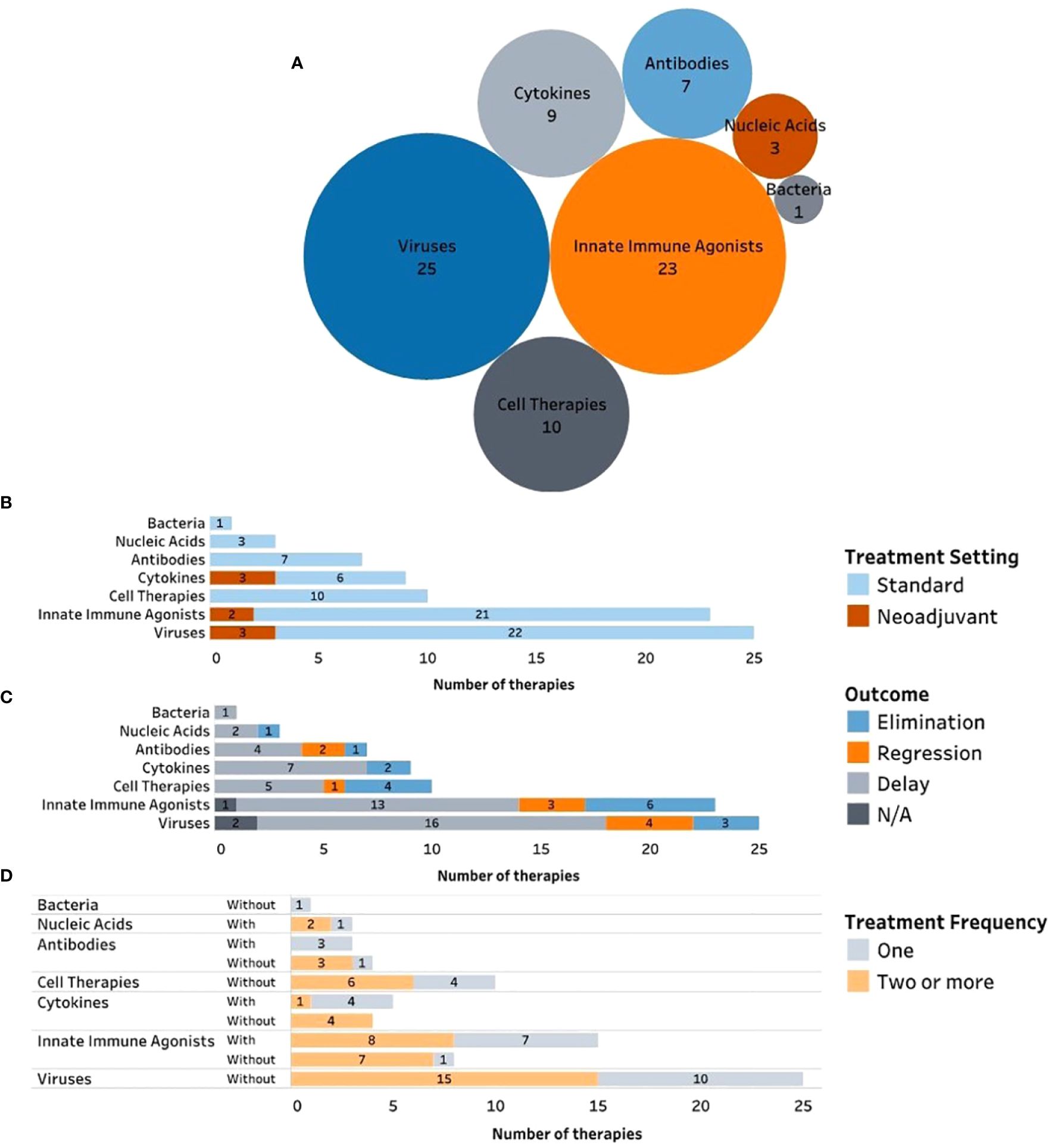
Figure 3 Preclinical intratumoral immunotherapies in breast cancer divided by (A) drug modality, (B) treatment setting, (C) outcome, and (D) treatment frequency.
Fourth, a delivery system for non-infectious agents should become a trend for i.t. therapy. Developing and incorporating delivery medium into intratumoral treatment can positively impact clinical treatment by decreasing the frequency of i.t. injections, as more numerous injections can decrease patient compliance, increase healthcare burden, and increase local pain associated with injections. Furthermore, delivery mediums can localize the therapeutic locally for extended periods of time, thereby increasing local therapeutic concentration and reducing systemic AEs. Currently evaluated delivery media primarily include hydrogels, microparticles, and NPs (Figure 3). Of these, hydrogels require fewer delivery injections than the others and potentially offer a long-term release profile. Furthermore, the tumor microenvironment is extremely dense, and saline-based delivery leaks from the injection site and returns via the needle track. Hydrogels can resist this higher-pressure environment and can be locally retained for extended periods of time, presenting an ideal delivery medium for i.t. therapies.
Overall, as the fields of immunotherapy and i.t. delivery continue to grow in BC, studies should continue to pursue treatment in earlier stages and carefully consider appropriate immunotherapeutics and delivery media to optimize patient treatment outcome.
SM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. YA: Conceptualization, Supervision, Writing – review & editing, Investigation. AS: Data curation, Formal analysis, Investigation, Validation, Visualization, Writing – review & editing. SU: Data curation, Formal analysis, Investigation, Validation, Visualization, Writing – review & editing. DZ: Conceptualization, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was funded in part by a Graduate Research Fellowship from the National Science Foundation (SM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Arnold M, Morgan E, Rumgay H, Mafra A, Singh D, Laversanne M, et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast. (2022) 66:15–23. doi: 10.1016/j.breast.2022.08.010
2. Abe O, Abe R, Enomoto K, Kikuchi K, Koyama H, Masuda H, et al. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet. (2005) 365:1687–717. doi: 10.1016/S0140-6736(05)66544-0
3. Harris JR, Lippman ME, Morrow M, Osborne CK. Diseases of the Breast, 2nd ed. (2000). Philadelphia, PA: Lippincott William & Wilkins.
4. Riggio AI, Varley KE, Welm AL. The lingering mysteries of metastatic recurrence in breast cancer. Br J Cancer. (2021) 124:13–26. doi: 10.1038/s41416-020-01161-4
5. Burguin A, Diorio C, Durocher F. Breast cancer treatments: Updates and new challenges. J Pers Med. (2021) 11:808. doi: 10.3390/jpm11080808
6. Sharma P, Allison JP. The future of immune checkpoint therapy . Available online at: https://www.science.org.
7. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell. (2015) 161:205–14. doi: 10.1016/j.cell.2015.03.030
8. Cortes J, Rugo HS, Cescon DW, Im S-A, Yusof MM, Gallardo C, et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. New Engl J Med. (2022) 387:217–26. doi: 10.1056/nejmoa2202809
9. Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, et al. Pembrolizumab for early triple-negative breast cancer. New Engl J Med. (2020) 382:810–21. doi: 10.1056/nejmoa1910549
10. Bhardwaj PV, Abdou YG. The evolving landscape of immune checkpoint inhibitors and antibody drug conjugates in the treatment of early-stage breast cancer. Oncologist. (2023) 28:832–44. doi: 10.1093/oncolo/oyad233
11. Abdou Y, Goudarzi A, Yu JX, Upadhaya S, Vincent B, Carey LA. Immunotherapy in triple negative breast cancer: beyond checkpoint inhibitors. NPJ Breast Cancer. (2022) 8:121. doi: 10.1038/s41523-022-00486-y
12. Zgura A, Gales L, Bratila E, Anghel R. Relationship between tumor infiltrating lymphocytes and progression in breast cancer. Maedica A J Clin Med. (2018) 13:317–20. doi: 10.26574/maedica.2018.13.4.317
13. Balibegloo M, Nejadghaderi SA, Sadeghalvad M, Soleymanitabar A, Salehi Nezamabadi S, Saghazadeh A, et al. Adverse events associated with immune checkpoint inhibitors in patients with breast cancer: A systematic review and meta-analysis. Int Immunopharmacol. (2021) 96:107796. doi: 10.1016/j.intimp.2021.107796
14. Valencia GA, Rioja P, Morante Z, Ruiz R, Fuentes H, Castaneda CA, et al. Immunotherapy in triple-negative breast cancer: A literature review and new advances. World J Clin Oncol. (2022) 13:219–36. doi: 10.5306/wjco.v13.i3.219
15. Criscitiello C, Corti C, Pravettoni G, Curigliano G. Managing side effects of immune checkpoint inhibitors in breast cancer. Crit Rev Oncol Hematol. (2021) 162:103354. doi: 10.1016/j.critrevonc.2021.103354
16. Conroy M, Naidoo J. Immune-related adverse events and the balancing act of immunotherapy. Nat Commun. (2022) 13:392. doi: 10.1038/s41467-022-27960-2
17. Hammerich L, Binder A, Brody JD. In situ vaccination: Cancer immunotherapy both personalized and off-the-shelf. Mol Oncol. (2015) 9:1966–81. doi: 10.1016/j.molonc.2015.10.016
18. Andtbacka RHI, Ross M, Puzanov I, Milhem M, Collichio F, Delman KA, et al. Patterns of clinical response with Talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM phase III clinical trial. Ann Surg Oncol. (2016) 23:4169–77. doi: 10.1245/s10434-016-5286-0
19. Kotsifaki A, Alevizopoulos N, Dimopoulou V, Armakolas A. Unveiling the immune microenvironment’s role in breast cancer: A glimpse into promising frontiers. Int J Mol Sci. (2023) 24:15332. doi: 10.3390/ijms242015332
20. Howard FM, Villamar D, He G, Pearson AT, Nanda R. The emerging role of immune checkpoint inhibitors for the treatment of breast cancer. Expert Opin Investig Drugs. (2022) 31:531–48. doi: 10.1080/13543784.2022.1986002
21. Zhao H, Yin X, Wang L, Liu K, Liu W, Bo L, et al. Identifying tumour microenvironment-related signature that correlates with prognosis and immunotherapy response in breast cancer. Sci Data. (2023) 10:119. doi: 10.1038/s41597-023-02032-2
22. Singh S, Cunningham C, Buchanan A, Jolly DJ, Nemunaitis J. Toxicity assessment of intratumoral injection of the herpes simplex type I thymidine kinase gene delivered by retrovirus in patients with refractory cancer. Mol Ther. (2001) 4:157–60. doi: 10.1006/mthe.2001.0430
23. Huang PW, Chang JWC. Immune checkpoint inhibitors win the 2018 Nobel Prize. BioMed J. (2019) 42:299–306. doi: 10.1016/j.bj.2019.09.002
24. Regulatory Approval Timeline of Active Immunotherapies . Available online at: https://www.cancerresearch.org/regulatory-approval-timeline-of-active-immunotherapies.
25. Chocarro L, Bocanegra A, Blanco E, Fernández-Rubio L, Arasanz H, Echaide M, et al. Cutting-edge: preclinical and clinical development of the first approved lag-3 inhibitor. Cells. (2022) 11:2351. doi: 10.3390/cells11152351
26. Marin-Acevedo JA, Kimbrough EMO, Lou Y. Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol. (2021) 14:1–29. doi: 10.1186/s13045-021-01056-8
27. Saez-Ibanez AR, Upadhaya S, Campbell J. Immuno-oncology clinical trials take a turn beyond PD1/PDL1 inhibitors. Nat Rev Drug Discovery. (2023) 22:442–3. doi: 10.1038/d41573-023-00066-0
28. Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. (2013) 73:7189–98. doi: 10.1158/0008-5472.CAN-12-4174
29. Bullock TNJ. CD40 stimulation as a molecular adjuvant for cancer vaccines and other immunotherapies. Cell Mol Immunol. (2022) 19:14–22. doi: 10.1038/s41423-021-00734-4
30. El-Kadiry AEH, Rafei M, Shammaa R. Cell therapy: types, regulation, and clinical benefits. Front Med (Lausanne). (2021) 8:756029. doi: 10.3389/fmed.2021.756029
31. Lefrère JJ, Berche P. La thérapeutique du docteur Brown-Séquard. Ann Endocrinol (Paris). (2010) 71:69–75. doi: 10.1016/j.ando.2010.01.003
32. Har-Noy M, Zeira M, Weiss L, Fingerut E, Or R, Slavin S. Allogeneic CD3/CD28 cross-linked Th1 memory cells provide potent adjuvant effects for active immunotherapy of leukemia/lymphoma. Leuk Res. (2009) 33:525–38. doi: 10.1016/j.leukres.2008.08.017
33. Har-Noy M, Zeira M, Weiss L, Slavin S. Completely mismatched allogeneic CD3/CD28 cross-linked Th1 memory cells elicit anti-leukemia effects in unconditioned hosts without GVHD toxicity. Leuk Res. (2008) 32:1903–13. doi: 10.1016/j.leukres.2008.05.007
34. Calmeiro J, Carrascal MA, Tavares AR, Ferreira DA, Gomes C, Falcão A, et al. Dendritic cell vaccines for cancer immunotherapy: The role of human conventional type 1 dendritic cells. Pharmaceutics. (2020) 12:1–20. doi: 10.3390/pharmaceutics12020158
35. Saez-Ibañez AR, Upadhaya S, Partridge T, Shah M, Correa D, Campbell J. Landscape of cancer cell therapies: trends and real-world data. Nat Rev Drug Discovery. (2022) 21:631–2. doi: 10.1038/d41573-022-00095-1
36. Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. (2020) 13:84. doi: 10.1186/s13045-020-00922-1
37. Ferrucci PF, Pala L, Conforti F, Cocorocchio E. Talimogene laherparepvec (T-VEC): an intralesional cancer immunotherapy for advanced melanoma. Cancers (Basel). (2021) 13:1383. doi: 10.3390/cancers13061383
38. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: A new class of immunotherapy drugs. Nat Rev Drug Discovery. (2015) 14:642–62. doi: 10.1038/nrd4663
39. Martínez-Puente DH, Pérez-Trujillo JJ, Zavala-Flores LM, García-García A, Villanueva-Olivo A, Rodríguez-Rocha H, et al. Plasmid DNA for therapeutic applications in cancer. Pharmaceutics. (2022) 14:1–27. doi: 10.3390/pharmaceutics14091861
40. Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Pérez-Gracia JL, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer. (2019) 120:6–15. doi: 10.1038/s41416-018-0328-y
41. Qiu Y, Su M, Liu L, Tang Y, Pan Y, Sun J. Clinical application of cytokines in cancer immunotherapy. Drug Des Devel Ther. (2021) 15:2269–87. doi: 10.2147/DDDT.S308578
42. Zhang Y, Guan XY, Jiang P. Cytokine and chemokine signals of T-cell exclusion in tumors. Front Immunol. (2020) 11:594609. doi: 10.3389/fimmu.2020.594609
43. Karin N. Chemokines in the landscape of cancer immunotherapy: How they and their receptors can be used to turn cold tumors into hot ones? Cancers (Basel). (2021) 13:1–23. doi: 10.3390/cancers13246317
44. Sameer AS, Nissar S. Toll-like receptors (TLRs): structure, functions, signaling, and role of their polymorphisms in colorectal cancer susceptibility. BioMed Res Int. (2021) 2021:1–14. doi: 10.1155/2021/1157023
45. Felgner S, Kocijancic D, Frahm M, Weiss S. Bacteria in cancer therapy: Renaissance of an old concept. Int J Microbiol. (2016) 2016:1–14. doi: 10.1155/2016/8451728
46. Staedtke V, Roberts NJ, Bai RY, Zhou S. Clostridium novyi-NT in cancer therapy. Genes Dis. (2016) 3:144–52. doi: 10.1016/j.gendis.2016.01.003
47. Lowenfeld L, Mick R, Datta J, Xu S, Fitzpatrick E, Fisher CS, et al. Dendritic cell vaccination enhances immune responses and induces regression of HER2pos DCIS independent of route: Results of randomized selection design trial. Clin Cancer Res. (2017) 23:2961–71. doi: 10.1158/1078-0432.CCR-16-1924
48. Schwarze JK, Awada G, Cras L, Tijtgat J, Forsyth R, Dufait I, et al. Intratumoral combinatorial administration of CD1c (BDCA-1)+ myeloid dendritic cells plus ipilimumab and avelumab in combination with intravenous low-dose nivolumab in patients with advanced solid tumors: A phase IB clinical trial. Vaccines (Basel). (2020) 8:1–16. doi: 10.3390/vaccines8040670
49. O’Shaughnessy J, Roberts LK, Smith JL, Levin MK, Timis R, Finholt JP, et al. Safety and initial clinical efficacy of a dendritic cell (DC) vaccine in locally advanced, triple-negative breast cancer (TNBC) patients (pts). J Clin Oncol. (2016) 34:1086. doi: 10.1200/JCO.2016.34.15_suppl.1086
50. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res. (2017) 5:1152–61. doi: 10.1158/2326-6066.CIR-17-0189
51. Lebel FM, Barrett JA, McArthur HL, Buck JY, Demars N, Mackenna R, et al. Demonstration of anti-tumor immunity via intratumoral regulated platform ad-RTS-hIL-12 in advanced breast cancer and recurrent glioblastoma patients. J Clin Oncol. (2018) 36:3038. doi: 10.1200/JCO.2018.36.15_suppl.3038
52. Ferris RL, Gross ND, Nemunaitis JJ, Andtbacka RHI, Argiris A, Ohr J, et al. Phase I trial of intratumoral therapy using HF10, an oncolytic HSV-1, demonstrates safety in HSV+/HSV- patients with refractory and superficial cancers. J Clin Oncol. (2014) 32:6082. doi: 10.1200/jco.2014.32.15_suppl.6082
53. Liu M, Peng K-W, Federspiel M, Russell S, Brunton B, Zhou Y, et al. Abstract P6-21-03: Phase I trial of intratumoral (IT) administration of a NIS-expressing derivative manufactured from a genetically engineered strain of measles virus (MV). Cancer Res. (2019) 79. doi: 10.1158/1538-7445.SABCS18-P6-21-03
54. Telli ML, Nagata H, Wapnir I, Acharya CR, Zablotsky K, Fox BA, et al. Intratumoral plasmid IL12 expands CD8þ T cells and induces a CXCR3 gene signature in triple-negative breast tumors that sensitizes patients to Anti–PD-1 therapy. Clin Cancer Res. (2021) 27:2481–93. doi: 10.1158/1078-0432.CCR-20-3944
55. Babiker H, Borazanci E, Subbiah V, Algazi AP, Schachter J, Lotem M, et al. 1031P Tilsotolimod engages the TLR9 pathway to promote antigen presentation and type I IFN signaling in solid tumours. Ann Oncol. (2020) 31:S711–2. doi: 10.1016/j.annonc.2020.08.1151
56. Babiker H, Borazanci E, Subbiah V, Maguire O, Rahimian S, Minderman H, et al. Safety, efficacy, and immune effects of intratumoral tilsotolimod in patients with refractory solid tumours: Updated results from ILLUMINATE-101. Ann Oncol. (2019) 30:v487. doi: 10.1093/annonc/mdz253.021
57. Janku F, Zhang HH, Pezeshki A, Goel S, Murthy R, Wang-Gillam A, et al. Intratumoral injection of clostridium novyi-NT spores in patients with treatment-refractory advanced solid tumors. Clin Cancer Res. (2021) 27:96–106. doi: 10.1158/1078-0432.CCR-20-2065
58. Han HS, Costa RL, Armaghani AJ, Hoover S, Kiluk J, Lee MC, et al. Neoadjuvant therapy of HER2 directed conventional dendritic cell (DC1) intratumoral (IT) therapy plus weekly paclitaxel, trastuzumab, and pertuzumab in patients with HER-2 positive breast cancer: NATASHA trial. J Clin Oncol. (2023) 41:596–6. doi: 10.1200/JCO.2023.41.16_suppl.596
59. Glencer AC, Wong JM, Hylton NM, Krings G, McCune E, Rothschild HT, et al. Modulation of the immune microenvironment of high-risk ductal carcinoma in situ by intralesional pembrolizumab injection. NPJ Breast Cancer. (2021) 7:59. doi: 10.1038/s41523-021-00267-z
60. Wang D, Porter CE, Lim B, Rosewell Shaw A, Robertson CS, Woods ML, et al. Ultralow-dose binary oncolytic/helper-dependent adenovirus promotes antitumor activity in preclinical and clinical studies. Sci Adv. (2023) 9:eade6790. doi: 10.1126/sciadv.ade6790
61. Chen N, Wang D, Porter CE, Shaw AR, Robertson CR, Woods ML, et al. Abstract P3-06-04: treatment of metastatic breast cancer with multipotent oncolytic/helper adenovirus CAdVEC. Cancer Res. (2023) 83. doi: 10.1158/1538-7445.sabcs22-p3-06-04
62. Soliman H, Hogue D, Han H, Mooney B, Costa R, Lee MC, et al. Oncolytic T-VEC virotherapy plus neoadjuvant chemotherapy in nonmetastatic triple-negative breast cancer: a phase 2 trial. Nat Med. (2023) 29:450–7. doi: 10.1038/s41591-023-02210-0
63. Huppert LA, Gliwa AS, Tait M, Quintal L, Starzinski S, Cheung A, et al. Phase Ib study of talimogene laherparepvec (T-VEC) in combination with chemotherapy (CT) or endocrine therapy (ET) in patients with metastatic, unresectable, or locoregionally recurrent HER2-negative breast cancer (BC). J Clin Oncol. (2023) 41:1091–1. doi: 10.1200/JCO.2023.41.16_suppl.1091
64. Ji D, Weitao Y, Tong X, Zhang C, Wang F, Chen Z, et al. A phase 1/2a study of T3011, an oncolytic HSV expressing IL-12 and PD-1 antibody, administered via intratumoral (IT) injection as monotherapy in advanced solid tumors. J Clin Oncol. (2023) 41:2520–0. doi: 10.1200/jco.2023.41.16_suppl.2520
65. Yuan Y, Rand JG, Zhang J, Kessler J, Modi B, Pillai R, et al. Abstract OT3-03-01: Phase I study of intratumoral administration of CF33-hNIS-antiPD-L1 (CHECKvacc) in patients with metastatic triple negative breast cancer. Cancer Res. (2023) 83. doi: 10.1158/1538-7445.sabcs22-ot3-03-01
66. Patel M, Jimeno A, Wang D, Stemmer S, Bauer T, Sweis R, et al. 539 Phase 1 study of mRNA-2752, a lipid nanoparticle encapsulating mRNAs encoding human OX40L/IL-23/IL-36γ, for intratumoral (ITu) injection +/- durvalumab in advanced solid tumors and lymphoma. J Immunother Cancer. (2021) 9:A569–9. doi: 10.1136/jitc-2021-sitc2021.539
67. Telli ML, Wapnir I, Devitt B, Cuff K, Soliman H, Vinayak S, et al. Abstract P3-09-04: Phase 2, open-label study of intratumoral tavokinogene telseplasmid (tavo) plus electroporation in combination with intravenous pembrolizumab therapy in patients with inoperable locally advanced or metastatic triple-negative breast cancer (mTNBC) (KEYNOTE- 890/OMS-I141). Cancer Res. (2020) 80. doi: 10.1158/1538-7445.sabcs19-p3-09-04
68. Janku F, Fu S, Murthy R, Karp D, Hong D, Tsimberidou A, et al. 383 First-in-man clinical trial of intratumoral injection of clostridium Novyi-NT spores in combination with pembrolizumab in patients with treatment-refractory advanced solid tumors. BMJ. (2020) 8(Suppl 3):A233.1–A233. doi: 10.1136/jitc-2020-sitc2020.0383
69. Onda M, Kobayashi K, Pastan I. Depletion of regulatory T cells in tumors with an anti-CD25 immunotoxin induces CD8 T cell-mediated systemic antitumor immunity. Proc Natl Acad Sci U.S.A. (2019) 116:4575–82. doi: 10.1073/pnas.1820388116
70. Carmi Y, Spitzer MH, Linde IL, Burt BM, Prestwood TR, Perlman N, et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature. (2015) 521:99–104. doi: 10.1038/nature14424
71. Kosaka A, Ishibashi K, Nagato T, Kitamura H, Fujiwara Y, Yasuda S, et al. CD47 blockade enhances the efficacy of intratumoral STING-targeting therapy by activating phagocytes. J Exp Med. (2021) 218:e20200792. doi: 10.1084/jem.20200792
72. Momenzadeh N, Hajian S, Shabankare A, Ghavimi R, Kabiri-Samani S, Kabiri H, et al. Photothermic therapy with cuttlefish ink-based nanoparticles in combination with anti-OX40 mAb achieve remission of triple-negative breast cancer. Int Immunopharmacol. (2023) 115:109622. doi: 10.1016/j.intimp.2022.109622
73. Liu HC, Viswanath DI, Pesaresi F, Xu Y, Zhang L, Di Trani N, et al. Potentiating antitumor efficacy through radiation and sustained intratumoral delivery of anti-CD40 and anti-PDL1. Int J Radiat Oncol Biol Phys. (2021) 110:492–506. doi: 10.1016/j.ijrobp.2020.07.2326
74. Chua CYX, Jain P, Susnjar A, Rhudy J, Folci M, Ballerini A, et al. Nanofluidic drug-eluting seed for sustained intratumoral immunotherapy in triple negative breast cancer. J Controlled Release. (2018) 285:23–34. doi: 10.1016/j.jconrel.2018.06.035
75. Li Y, Fang M, Zhang J, Wang J, Song Y, Shi J, et al. Hydrogel dual delivered celecoxib and anti-PD-1 synergistically improve antitumor immunity. Oncoimmunology. (2016) 5:1–12. doi: 10.1080/2162402X.2015.1074374
76. Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2 + breast cancer metastasis to the brain. Clin Cancer Res. (2018) 24:95–105. doi: 10.1158/1078-0432.CCR-17-2041
77. Akbulut H, Tang Y, Akbulut KG, Maynard J, Zhang L, Deisseroth A. Antitumor immune response induced by i.t. injection of vector-activated dendritic cells and chemotherapy suppresses metastatic breast cancer. Mol Cancer Ther. (2006) 5:1975–85. doi: 10.1158/1535-7163.MCT-06-0049
78. Oba T, Makino K, Kajihara R, Yokoi T, Araki R, Abe M, et al. In situ delivery of iPSC-derived dendritic cells with local radiotherapy generates systemic antitumor immunity and potentiates PD-L1 blockade in preclinical poorly immunogenic tumor models. J Immunother Cancer. (2021) 9:e002432. doi: 10.1136/jitc-2021-002432
79. Ramamoorthi G, Kodumudi K, Snyder C, Grover P, Zhang H, Greene MI, et al. Intratumoral delivery of dendritic cells plus anti-HER2 therapy triggers both robust systemic antitumor immunity and complete regression in HER2 mammary carcinoma. J Immunother Cancer. (2022) 10:e004841. doi: 10.1136/jitc-2022-004841
80. Globerson-Levin A, Waks T, Eshhar Z. Elimination of progressive mammary cancer by repeated administrations of chimeric antigen receptor-modified T cells. Mol Ther. (2014) 22:1029–38. doi: 10.1038/mt.2014.28
81. Lichtor T, Glick RP, Lin H, O-Sullivan IS, Cohen EP. Intratumoral injection of IL-secreting syngeneic/allogeneic fibroblasts transfected with DNA from breast cancer cells prolongs the survival of mice with intracerebral breast cancer. Cancer Gene Ther. (2005) 12:708–14. doi: 10.1038/sj.cgt.7700832
82. Huang CP, Liu LC, Chang CC, Wu CC, Shyr CR. Intratumoral xenogeneic tissue-specific cell immunotherapy inhibits tumor growth by increasing antitumor immunity in murine triple negative breast and pancreatic tumor models. Cancer Lett. (2022) 545:115478. doi: 10.1016/j.canlet.2021.10.044
83. Sommaggio R, Cappuzzello E, Dalla Pietà A, Tosi A, Palmerini P, Carpanese D, et al. Adoptive cell therapy of triple negative breast cancer with redirected cytokine-induced killer cells. Oncoimmunology. (2020) 9:1777046. doi: 10.1080/2162402X.2020.1777046
84. Basu A, Albert GK, Awshah S, Datta J, Kodumudi KN, Gallen C, et al. Identification of immunogenic MHC class II human HER3 peptides that mediate anti-HER3 CD4 th1 responses and potential use as a cancer vaccine. Cancer Immunol Res. (2022) 10:108–25. doi: 10.1158/2326-6066.CIR-21-0454
85. Huang CP, Yang CY, Shyr CR. Utilizing xenogeneic cells as a therapeutic agent for treating diseases. Cell Transplant. (2021) 30:9636897211011995. doi: 10.1177/09636897211011995
86. Addison CL, Braciak T, Ralston R, Muller WJ, Gauldie J, Graham FL. Intratumoral injection of an adenovirus expressing interleukin 2 induces regression and immunity in a murine breast cancer model. Proc Natl Acad Sci U.S.A. (1995) 92:8522–6. doi: 10.1073/pnas.92.18.8522
87. Emtage PCR, Wan Y, Muller W, Graham FL, Gauldie J. Enhanced interleukin-2 gene transfer immunotherapy of breast cancer by coexpression of B7-1 and B7-2. J Interferon Cytokine Res. (1998) 18:927–37. doi: 10.1089/jir.1998.18.927
88. Martinet O, Divino C, Zang Y, Gan Y, Mandeli J, Thung S, et al. T cell activation with systemic agonistic antibody versus local 4-1BB ligand gene delivery combined with interleukin-12 eradicate liver metastases of breast cancer. Gene Ther. (2002) 9:786–92. doi: 10.1038/sj.gt.3301687
89. Choi AH, O’Leary MP, Chaurasiya S, Lu J, Kim SI, Fong Y, et al. Novel chimeric parapoxvirus CF189 as an oncolytic immunotherapy in triple-negative breast cancer. Surg (United States). (2018) 163:336–42. doi: 10.1016/j.surg.2017.09.030
90. Groeneveldt C, Kinderman P, Van Den Wollenberg DJM, Van Den Oever RL, Middelburg J, Mustafa DAM, et al. Preconditioning of the tumor microenvironment with oncolytic reovirus converts CD3-bispecific antibody treatment into effective immunotherapy. J Immunother Cancer. (2020) 8:e001191. doi: 10.1136/jitc-2020-001191
91. Liu Y, Tuve S, Persson J, Beyer I, Yumul R, Li ZY, et al. Adenovirus-mediated intratumoral expression of immunostimulatory proteins in combination with systemic Treg inactivation induces tumor-destructive immune responses in mouse models. Cancer Gene Ther. (2011) 18:407–18. doi: 10.1038/cgt.2011.8
92. Majumdar AS, Zolotorev A, Samuel S, Tran K, Vertin B, Hall-Meier M, et al. Efficacy of herpes simplex virus thymidine kinase in combination with cytokine gene therapy in an experimental metastatic breast cancer model. Cancer Gene Ther. (2000) 7:1086–99. doi: 10.1038/sj.cgt.7700215
93. Bernt KM, Ni S, Tieu A-T, Lieber A. Assessment of a combined, adenovirus-mediated oncolytic and immunostimulatory tumor therapy. Cancer Res. (2005) 65:4343–52. doi: 10.1158/0008-5472.CAN-04-3527
94. Bahreyni A, Liu H, Mohamud Y, Xue YC, Zhang J, Luo H. A new miRNA-Modified coxsackievirus B3 inhibits triple negative breast cancer growth with improved safety profile in immunocompetent mice. Cancer Lett. (2022) 548:215849. doi: 10.1016/j.canlet.2022.215849
95. Brown MC, Beasley GM, McKay ZP, Yang Y, Desjardins A, Randazzo DM, et al. Intratumor childhood vaccine-specific CD4 + T-cell recall coordinates antitumor CD8 + T cells and eosinophils. J Immunother Cancer. (2023) 11:e006463. doi: 10.1136/jitc-2022-006463
96. Suryawanashi YR, Zhang T, Woyczesczyk HM, Christie J, Byers E, Kohler S, et al. T-independent response mediated by oncolytic tanapoxvirus recombinants expressing interleukin-2 and monocyte chemoattractant protein-1 suppresses human triple negative breast tumors. Med Oncol. (2017) 34:112. doi: 10.1007/s12032-017-0973-7
97. Yang Y, Xu W, Peng D, Wang H, Zhang X, Wang H, et al. An oncolytic adenovirus targeting transforming growth factor β Inhibits protumorigenic signals and produces immune activation: A novel approach to enhance anti-PD-1 and anti-CTLA-4 therapy. Hum Gene Ther. (2019) 30:1117–32. doi: 10.1089/hum.2019.059
98. Trofimova O, Korotkaja K, Skrastina D, Jansons J, Spunde K, Isaguliants M, et al. Alphavirus-driven interferon gamma (IFNG) expression inhibits tumor growth in orthotopic 4T1 breast cancer model. Vaccines (Basel). (2021) 9:1247. doi: 10.3390/vaccines9111247
99. Nicola Candia AJ, Garcia Fallit M, Peña Agudelo JA, Pérez Küper M, Gonzalez N, Moreno Ayala MA, et al. Targeting FOXP3 tumor-intrinsic effects using adenoviral vectors in experimental breast cancer. Viruses. (2023) 15:1813. doi: 10.3390/v15091813
100. Xu W, Yang Y, Hu Z, Head M, Mangold KA, Sullivan M, et al. LyP-1-modified oncolytic adenoviruses targeting transforming growth factor β Inhibit tumor growth and metastases and augment immune checkpoint inhibitor therapy in breast cancer mouse models. Hum Gene Ther. (2020) 31:863–80. doi: 10.1089/hum.2020.078
101. Tandon M, Vemula SV, Sharma A, Ahi YS, Mittal S, Bangari DS, et al. EphrinA1-EphA2 interaction-mediated apoptosis and FMS-like tyrosine kinase 3 receptor ligand-induced immunotherapy inhibit tumor growth in a breast cancer mouse model. J Gene Med. (2012) 14:77–89. doi: 10.1002/jgm.1649
102. Oh E, Choi I-K, Hong J, Yun C-O. Oncolytic adenovirus coexpressing interleukin-12 and decorin overcomes Treg-mediated immunosuppression inducing potent antitumor effects in a weakly immunogenic tumor model. Oncotarget. (2017) 8:4730–46. doi: 10.18632/oncotarget.13972
103. Masuelli L, Marzocchella L, Focaccetti C, Lista F, Nardi A, Scardino A, et al. Local delivery of recombinant vaccinia virus encoding for neu counteracts growth of mammary tumors more efficiently than systemic delivery in neu transgenic mice. Cancer Immunology Immunotherapy. (2010) 59:1247–58. doi: 10.1007/s00262-010-0850-0
104. Mostafa AA, Meyers DE, Thirukkumaran CM, Liu PJ, Gratton K, Spurrell J, et al. Oncolytic reovirus and immune checkpoint inhibition as a novel immunotherapeutic strategy for breast cancer. Cancers (Basel). (2018) 10:205. doi: 10.3390/cancers10060205
105. Yu X, Liu W, Chen S, Cheng X, Paez PA, Sun T, et al. Immunologically programming the tumor microenvironment induces the pattern recognition receptor NLRC4-dependent antitumor immunity. J Immunother Cancer. (2021) 9:e001595. doi: 10.1136/jitc-2020-001595
106. Chon HJ, Lee WS, Yang H, Kong SJ, Lee NK, Moon ES, et al. Tumor microenvironment remodeling by intratumoral oncolytic vaccinia virus enhances the efficacy of immune-checkpoint blockade. Clin Cancer Res. (2019) 25:1612–23. doi: 10.1158/1078-0432.CCR-18-1932
107. Ortega-Rivera OA, Gallegos-Alcalá P, Jiménez M, Luis Quintanar J, Torres-Juarez F, Rivas-Santiago B, et al. Inhibition of tumor growth and metastasis by Newcastle disease virus strain P05 in a breast cancer mouse model. J Breast Cancer. (2023) 26:186–200. doi: 10.4048/jbc.2023.26.e9
108. Alonso-Miguel D, Valdivia G, Guerrera D, Perez-Alenza MD, Pantelyushin S, Alonso-Diez A, et al. Neoadjuvant in situ vaccination with cowpea mosaic virus as a novel therapy against canine inflammatory mammary cancer. J Immunother Cancer. (2022) 10:1–13. doi: 10.1136/jitc-2021-004044
109. Barreno L, Sevane N, Valdivia G, Alonso-Miguel D, Suarez-Redondo M, Alonso-Diez A, et al. Transcriptomics of canine inflammatory mammary cancer treated with empty cowpea mosaic virus implicates neutrophils in anti-tumor immunity. Int J Mol Sci. (2023) 24:14034. doi: 10.3390/ijms241814034
110. Chen L, Huang TG, Meseck M, Mandeli J, Fallon J, Woo SLC. Rejection of metastatic 4T1 breast cancer by attenuation of treg cells in combination with immune stimulation. Mol Ther. (2007) 15:2194–202. doi: 10.1038/sj.mt.6300310
111. Guo S, Smeltz RB, Nanajian A, Heller R. IL-15/IL-15Rα Heterodimeric complex as cancer immunotherapy in murine breast cancer models. Front Immunol. (2021) 11:614667. doi: 10.3389/fimmu.2020.614667
112. Yuan CH, Yang XQ, Zhu CL, Liu SP, Wang BC, Wang FB. Interleukin-7 enhances the in vivo anti-tumor activity of tumor-reactive CD8+ T cells with induction of IFN-gamma in a murine breast cancer model. Asian Pacific J Cancer Prev. (2014) 15:265–71. doi: 10.7314/APJCP.2014.15.1.265
113. Shen L, Li J, Liu Q, Song W, Zhang X, Tiruthani K, et al. Local blockade of interleukin 10 and C-X-C motif chemokine ligand 12 with nano-delivery promotes antitumor response in murine cancers. ACS Nano. (2018) 12:9830–41. doi: 10.1021/acsnano.8b00967
114. Yokoi T, Oba T, Kajihara R, Abrams SI, Ito F. Local, multimodal intralesional therapy renders distant brain metastases susceptible to PD-L1 blockade in a preclinical model of triple-negative breast cancer. Sci Rep. (2021) 11:21992. doi: 10.1038/s41598-021-01455-4
115. Patel A, Oba T, Kajihara R, Yokoi T, Abrams SI, Ito F. Multimodal intralesional therapy for reshaping the myeloid compartment of tumors resistant to anti–PD-L1 therapy via IRF8 expression. J Immunol. (2021) 207:1298–309. doi: 10.4049/jimmunol.2100281
116. Mihalik NE, Steinberger KJ, Stevens AM, Bobko AA, Hoblitzell EH, Tseytlin O, et al. Dose-specific intratumoral GM-CSF modulates breast tumor oxygenation and antitumor immunity. J Immunol. (2023) 211:1589–604. doi: 10.4049/jimmunol.2300326
117. Sabel MS, Skitzki J, Stoolman L, Egilmez NK, Mathiowitz E, Bailey N, et al. Intratumoral IL-12 and TNF-α-loaded microspheres lead to regression of breast cancer and systemic antitumor immunity. Ann Surg Oncol. (2004) 11:147–56. doi: 10.1245/ASO.2004.03.022
118. Sabel MS, Su G, Griffith KA, Chang AE. Intratumoral delivery of encapsulated IL-12, IL-18 and TNF-α in a model of metastatic breast cancer. Breast Cancer Res Treat. (2010) 122:325–36. doi: 10.1007/s10549-009-0570-3
119. Xiong X, Zhao J, Su R, Liu C, Guo X, Zhou S. Double enhancement of immunogenic cell death and antigen presentation for cancer immunotherapy. Nano Today. (2021) 39:101225. doi: 10.1016/j.nantod.2021.101225
120. Oba T, Kajihara R, Yokoi T, Repasky EA, Ito F. Neoadjuvant in situ immunomodulation enhances systemic antitumor immunity against highly metastatic tumors. Cancer Res. (2021) 81:6183–95. doi: 10.1158/0008-5472.CAN-21-0939
121. Vo JLN, Yang L, Kurtz SL, Smith SG, prasanth KB, Ravindranathan S, et al. Neoadjuvant immunotherapy with chitosan and interleukin-12 to control breast cancer metastasis. Oncoimmunology. (2014) 3:1–10. doi: 10.4161/21624011.2014.968001
122. Ashour AE, Lin X, Wang X, Turnquist HR, Burns NM, Tuli A, et al. CCL21 is an effective surgical neoadjuvant for treatment of mammary tumors. Cancer Biol Ther. (2007) 6:1217–21. doi: 10.4161/cbt.6.8.4405
123. Yousefi F, Siadat SD, Saraji AA, Hesaraki S, Aslani MM, Mousavi SF, et al. Tagging staphylococcal enterotoxin B (SEB) with TGFaL3 for breast cancer therapy. Tumor Biol. (2016) 37:5305–16. doi: 10.1007/s13277-015-4334-x
124. Dejmek M, Brazdova A, Otava T, Polidarova MP, Klíma M, Smola M, et al. Vinylphosphonate-based cyclic dinucleotides enhance STING-mediated cancer immunotherapy. Eur J Med Chem. (2023) 259:115685. doi: 10.1016/j.ejmech.2023.115685
125. Ohkuri T, Kosaka A, Ishibashi K, Kumai T, Hirata Y, Ohara K, et al. Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunology Immunotherapy. (2017) 66:705–16. doi: 10.1007/s00262-017-1975-1
126. Daniels P, Cassoday S, Gupta K, Giurini E, Leifheit ME, Zloza A, et al. Intratumoral influenza vaccine administration attenuates breast cancer growth and restructures the tumor microenvironment through sialic acid binding of vaccine hemagglutinin. Int J Mol Sci. (2023) 25:225. doi: 10.3390/ijms25010225
127. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest. (2019) 129:4350–64. doi: 10.1172/JCI125413
128. Gupta SK, Tiwari AK, Gandham RK, Sahoo AP. Combined administration of the apoptin gene and poly (I:C) induces potent anti-tumor immune response and inhibits growth of mouse mammary tumors. Int Immunopharmacol. (2016) 35:163–73. doi: 10.1016/j.intimp.2016.03.034
129. Chao Y, Xu L, Liang C, Feng L, Xu J, Dong Z, et al. Combined local immunostimulatory radioisotope therapy and systemic immune checkpoint blockade imparts potent antitumour responses. Nat BioMed Eng. (2018) 2:611–21. doi: 10.1038/s41551-018-0262-6
130. Mei E, Chen C, Li C, Ding X, Chen J, Xi Q, et al. Injectable and biodegradable chitosan hydrogel-based drug depot contributes to synergistic treatment of tumors. Biomacromolecules. (2021) 22:5339–48. doi: 10.1021/acs.biomac.1c01279
131. Som A, Rosenboom J, Wehrenberg-Klee E, Chandler A, Ndakwah G, Chen E, et al. Percutaneous intratumoral immunoadjuvant gel increases the abscopal effect of cryoablation for checkpoint inhibitor resistant cancer. Adv Healthc Mater. (2023) 13:e2301848. doi: 10.1002/adhm.202301848
132. Chao Y, Liang C, Tao H, Du Y, Wu D, Dong Z, et al. Localized cocktail chemoimmunotherapy after in situ gelation to trigger robust systemic antitumor immune responses. Sci Adv. (2020) 6:1–14. doi: 10.1126/sciadv.aaz4204
133. Zhuang B, Chen T, Huang Y, Xiao Z, Jin Y. Chemo-photothermal immunotherapy for eradication of orthotopic tumors and inhibition of metastasis by intratumoral injection of polydopamine versatile hydrogels. Acta Pharm Sin B. (2022) 12:1447–59. doi: 10.1016/j.apsb.2021.09.001
134. Rodriguez-Ruiz ME, Serrano-Mendioroz I, Garate-Soraluze E, Sánchez-Mateos P, Barrio-Alonso C, Rodríguez López I, et al. Intratumoral BO-112 in combination with radiotherapy synergizes to achieve CD8 T-cell-mediated local tumor control. J Immunother Cancer. (2023) 11:e005011. doi: 10.1136/jitc-2022-005011
135. Shen J, Lin M, Ding M, Yu N, Yang C, Kong D, et al. Tumor immunosuppressive microenvironment modulating hydrogels for second near-infrared photothermal-immunotherapy of cancer. Mater Today Bio. (2022) 16:100416. doi: 10.1016/j.mtbio.2022.100416
136. Kelly G, Milligan JJ, Mastria EM, Kim S, Zelenetz SR, Dobbins J, et al. Intratumoral delivery of brachytherapy and immunotherapy by a thermally triggered polypeptide depot. J Controlled Release. (2022) 343:267–76. doi: 10.1016/j.jconrel.2022.01.024
137. Colombani T, Haudebourg T, Decossas M, Lambert O, Ada Da Silva G, Altare F, et al. Lipidic aminoglycoside derivatives: A new class of immunomodulators inducing a potent innate immune stimulation. Advanced Sci. (2019) 6:1900288. doi: 10.1002/advs.201900288
138. Zhang X, Wu F, Men K, Huang R, Zhou B, Zhang R, et al. Modified fe3O4 magnetic nanoparticle delivery of CpG inhibits tumor growth and spontaneous pulmonary metastases to enhance immunotherapy. Nanoscale Res Lett. (2018) 13:240. doi: 10.1186/s11671-018-2661-8
139. Wei X, Liu L, Li X, Wang Y, Guo X, Zhao J, et al. Selectively targeting tumor-associated macrophages and tumor cells with polymeric micelles for enhanced cancer chemo-immunotherapy. J Controlled Release. (2019) 313:42–53. doi: 10.1016/j.jconrel.2019.09.021
140. Bahmani B, Gong H, Luk BT, Haushalter KJ, DeTeresa E, Previti M, et al. Intratumoral immunotherapy using platelet-cloaked nanoparticles enhances antitumor immunity in solid tumors. Nat Commun. (2021) 12:1–12. doi: 10.1038/s41467-021-22311-z
141. Aznar MA, Planelles L, Perez-Olivares M, Molina C, Garasa S, Etxeberría I, et al. Immunotherapeutic effects of intratumoral nanoplexed poly I:C. J Immunother Cancer. (2019) 7:1–16. doi: 10.1186/s40425-019-0568-2
142. Peng B, Nguyen TM, Jayasinghe MK, Gao C, Pham TT, Vu LT, et al. Robust delivery of RIG-I agonists using extracellular vesicles for anti-cancer immunotherapy. J Extracell Vesicles. (2022) 11:e12187. doi: 10.1002/jev2.12187
143. Rezaei Adriani R, Mousavi Gargari SL, Bakherad H, Amani J. Anti-EGFR bioengineered bacterial outer membrane vesicles as targeted immunotherapy candidate in triple-negative breast tumor murine model. Sci Rep. (2023) 13:16403. doi: 10.1038/s41598-023-43762-y
144. Milling LE, Garafola D, Agarwal Y, Wu S, Thomas A, Donahue N, et al. Neoadjuvant STING activation, extended half-life IL2, and checkpoint blockade promote metastasis clearance via sustained NK-cell activation. Cancer Immunol Res. (2022) 10:26–39. doi: 10.1158/2326-6066.CIR-21-0247
145. Zanker DJ, Spurling AJ, Brockwell NK, Owen KL, Zakhour JM, Robinson T, et al. Intratumoral administration of the Toll-like receptor 7/8 agonist 3M-052 enhances interferon-driven tumor immunogenicity and suppresses metastatic spread in preclinical triple-negative breast cancer. Clin Transl Immunol. (2020) 9:e1177. doi: 10.1002/cti2.1177
146. Zhou S, Lin Y, Zhao Z, Lai Y, Lu M, Shao Z, et al. Targeted deprivation of methionine with engineered Salmonella leads to oncolysis and suppression of metastasis in broad types of animal tumor models. Cell Rep Med. (2023) 4:101070. doi: 10.1016/j.xcrm.2023.101070
147. Cancer Research Institute. Available online at: https://www.cancerresearch.org/our-impact.
148. Debien V, De Caluwé A, Wang X, Piccart-Gebhart M, Tuohy VK, Romano E, et al. Immunotherapy in breast cancer: an overview of current strategies and perspectives. NPJ Breast Cancer. (2023) 9:7. doi: 10.1038/s41523-023-00508-3
Keywords: breast cancer, cancer immunotherapy, intratumoral, localized delivery, neoadjuvant, hydrogel, delivery systems
Citation: Mantooth SM, Abdou Y, Saez-Ibañez AR, Upadhaya S and Zaharoff DA (2024) Intratumoral delivery of immunotherapy to treat breast cancer: current development in clinical and preclinical studies. Front. Immunol. 15:1385484. doi: 10.3389/fimmu.2024.1385484
Received: 12 February 2024; Accepted: 22 April 2024;
Published: 13 May 2024.
Edited by:
Steven Fiering, Dartmouth College, United StatesReviewed by:
Yanhua Gao, University of Pittsburgh, United StatesCopyright © 2024 Mantooth, Abdou, Saez-Ibañez, Upadhaya and Zaharoff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Zaharoff, ZGF6YWhhcm9AbmNzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.