 Lisa Feldman
Lisa Feldman- Division of Neurosurgery, City of Hope National Medical Center, Duarte, CA, United States
Glioblastoma (GBM) tumors are the most aggressive primary brain tumors in adults that, despite maximum treatment, carry a dismal prognosis. GBM tumors exhibit tissue hypoxia, which promotes tumor aggressiveness and maintenance of glioma stem cells and creates an overall immunosuppressive landscape. This article reviews how hypoxic conditions overlap with inflammatory responses, favoring the proliferation of immunosuppressive cells and inhibiting cytotoxic T cell development. Immunotherapies, including vaccines, immune checkpoint inhibitors, and CAR-T cell therapy, represent promising avenues for GBM treatment. However, challenges such as tumor heterogeneity, immunosuppressive TME, and BBB restrictiveness hinder their effectiveness. Strategies to address these challenges, including combination therapies and targeting hypoxia, are actively being explored to improve outcomes for GBM patients. Targeting hypoxia in combination with immunotherapy represents a potential strategy to enhance treatment efficacy.
Introduction
Glioblastoma (GBM) is an aggressive and devastating primary brain tumor characterized by a high proliferation rate, infiltrative growth, and resistance to traditional interventions. It is the most common primary brain tumor in adults, with an incidence ranging from two to five cases per 100,000 people (1). GBM accounts for approximately 48.3% of all primary malignant brain tumors (2) and affects individuals across various age groups. Its prevalence increases with age, peaking in the 75–84 years of age group, with a median age of 64 at diagnosis (3). GBM is more common in men than women, and more prevalent in Caucasians compared to Afro-Americans, Africans, American Indians, and Asians (3). This malignancy primarily affects the cerebral hemispheres, particularly in subcortical white matter and deep grey matter of the frontal and temporal lobes, although it can arise in any part of the brain (4).
GBM tumors are a type of diffuse astrocytic tumor classified as grade IV by the World Health Organization (WHO). According to the 2021 WHO classification of CNS tumors, they are now required to express isocitrate dehydrogenase (IDH-wild type) to be classified as GBM (5). GBM tumors are generally divided into primary and secondary tumors with the former arising from either progenitor or neural stem cells (90% of GBM), and the latter transforming from pre-existing, less aggressive tumors (10%) (6). Regardless, the pathogenesis of GBM involves wildly variable and complex genetic and molecular alterations. Amplification of the epidermal growth factor receptor (EGFR) gene, mutations in the IDH1 and IDH2 genes, and loss of tumor suppressor genes such as TP53 and PTEN serve as the most identified genetic abnormalities in GBM (7).
These molecular aberrations contribute to uncontrolled cell growth, angiogenesis, and invasion into the surrounding brain tissue (8). Therefore, current treatment modalities for GBM typically involve a combination of surgery, radiation therapy, and chemotherapy. The goal of surgery is to remove as much tumor as possible without compromising neurological function. Adjuvant radiation therapy follows surgical debulking by delivering targeted beams to the remaining cancer cells in the tumor bed to delay recurrence. Concurrently, chemotherapy, usually temozolomide (TMZ), is administered during and after radiation to enhance the treatment outcomes. Despite these aggressive treatment approaches, GBM inevitably recurs, underscoring the challenge of eradicating residual tumor cells and the need for innovative therapeutic interventions. Because these standard therapies only extend survival an average of 15 months (9), with less than 30% of patients living 2 years (10), neuro-oncologists are eager to develop new therapies.
Here, we review major challenges to developing successful treatments against GBM and focus on how hypoxia contributes to these obstacles.
The heterogeneity of GBM
Designing novel therapies against GBM has proven to be remarkably challenging due to the extensive variability of the GBM tumor microenvironment (TME) within brain tissue. GBM tumors metastasize outside the central nervous system (CNS) in less than 0.2% of cases (11), suggesting they require the unique milieu of brain parenchyma to survive. Together, the variety of unique resident brain cells, the favorable blood supply with high delivery of oxygen and nutrients to the brain, and the blood-brain barrier that creates a relatively immune-privileged state create an environment in which GBM tumor cells thrive. Once GBM cells develop and migrate into adjacent tissues, these tumors develop prototypical heterogeneity both within single tumors as well as across tumors in multifocal disease.
GBM is characterized by an overwhelming level of intratumoral heterogeneity, manifesting at various biological levels. At the genetic level, extensive genomic analyses have revealed diverse molecular alterations within individual tumors and across different lesions of multifocal GBM cases. The Cancer Genome Atlas (TCGA) research network has played a pivotal role in uncovering the genetic landscape of GBM, identifying mutations in genes such as EGFR, TP53, and PTEN that contribute to tumor heterogeneity (12, 13). This genetic diversity not only influences the aggressive behavior of GBM, but also poses challenges for developing targeted therapies.
In addition to genetic heterogeneity, GBM tumors exhibit profound cellular diversity. Amongst a background of mutated glial cells demonstrating rapid proliferation, migration, and infiltration throughout normal brain tissue, the GBM TME also includes cancer stem cells (CSCs) that demonstrate multipotent differentiation and true self-renewal properties (14, 15). Stromal cells, vascular endothelial cells, and both residing and infiltrating immune cells further contribute to specific niches within the TME (16). Traditionally, intratumoral heterogeneity is considered a result of distinct genetic alterations within various tumor cells (17). However, it is more likely the complex interaction of each of these cell types within the extracellular matrix (ECM) that contributes to heterogeneity. For example, within these specific niches, the proliferating or CSC-like tumor cells dynamically interact with resident parenchymal cells, such as microglia and macrophages, to orchestrate a unique milieu. The microscopic result of these various TMEs invariably leads to necrotic regions, endothelial proliferation around vessels, and hypoxic areas (16), all of which are classical pathological findings of GBMs.
The spatial heterogeneity observed within GBM tumors further adds to the complexity of the disease. Intra-tumoral variations in oxygen levels, nutrient availability, and blood flow create distinct microenvironments within the tumor mass. These variations lead to the formation of hypoxic regions, which are known to influence tumor progression, treatment resistance, and the emergence of more aggressive phenotypes (18).
To summarize, the heterogeneity of GBM is a complex phenomenon that covers various aspects like genetics, cells, microenvironment, and spatial diversity. The challenges posed by this heterogeneity highlight the intricate nature of GBM biology, which emphasizes the requirement for personalized and targeted therapeutic approach. The advancements in genomic profiling, single-cell analysis, and a deeper understanding of the TME pave the path for developing innovative strategies to address the diverse components that contribute to GBM heterogeneity.
The immunosuppressive GBM TME
GBM tumors are notorious for creating an immunosuppressive microenvironment that facilitates their growth and evades the host’s immune response. This immunosuppressive nature is orchestrated by a complex interplay of various cellular and molecular components within the TME (Figure 1). One prominent feature is the recruitment and activation of immunosuppressive cells, including regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), which inhibit the activity of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, crucial effectors of anti-tumor immunity (19, 20). The abundance of these immunosuppressive cells contributes to the establishment of an immune-tolerant milieu that shields GBM from immune-mediated destruction.

Figure 1 Factors contributing to immunosuppression in GBM.
VEGF is the master regulator of this immunosuppression. It downregulates intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1), adhesion molecules that reduce the infiltration of T-effector cells, activate antigen-specific regulator T cells (Tregs), and inhibit the maturation of dendritic cells (DCs) (21). Interestingly, a variety of brain tumors induce reduced expression of cell membrane sphingosine-1-phosphate receptor (S1P1), which causes trapping of T cells in bone marrow, and therefore actively induces an immunosuppressive state (22). Furthermore, Andaloussi et al. found a dramatic reduction in CD4+ and CD8+ cells, a reduction in the size of the thymus, and a decrease in thymic cellularity in the brains of mice implanted with murine glioma GL261 cells (23).
Upon reaching the GBM TME, T cells encounter a harshly immunosuppressive local state due to high concentrations of immunosuppressive modulators such as transforming growth factor β (TGF-β), prostaglandin E-2 (PGE2), and interleukin 10 (IL-10). This cytokine milieu shifts microglia and tumor-infiltrating myeloid cells to a quiescent phenotype that dampens immune responses and supports tumor growth (24). Additionally, GBM tumors exploit the expression of immune checkpoint molecules to dampen the anti-tumor immune response. Programmed cell death protein 1 (PD-1) and its ligand PD-L1, along with cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), serve as key immune checkpoints that, when engaged, downregulate T cell activity (25, 26). GBM cells and immune cells within the TME upregulate these checkpoint molecules, leading to T cell exhaustion and dysfunction (26). The upregulation of these immune checkpoints contributes to the immune escape mechanisms employed by GBM tumors, limiting the effectiveness of immunotherapeutic interventions.
Myeloid cells play a pivotal role in establishing an immunosuppressive microenvironment within GBM tumors. myeloid cells, including M2 macrophages, immature monocytes, and myeloid-derived suppressor cells (MDSCs), are the most common non-malignant cells identified in the GBM TME (27). Among these cells, MDSCs are frequently recruited to the GBM microenvironment, where they contribute to the inhibition of both innate and adaptive immune responses, dampening the activity of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, which are crucial effectors of anti-tumor immunity (20). These cells play a major role in tumor-mediated immunosuppression through further immune checkpoint molecule expression and anti-inflammatory cytokine release (28). Upregulation of programmed death-ligand 1 (PD-1) on tumor-infiltrating myeloid cells is a crucial GBM-derived method of immune evasion (29, 30). GBM cells themselves release interleukin 6 (IL-6), which is necessary for myeloid PD-L1 induction through a signal transducer and activator of transcription 3 (STAT3)-dependent fashion and disrupting IL6 signaling enhances immune-mediated antitumor effects against GBM (28).
L. Pang et al. recently identified Kunitz-type protease inhibitor TFPI2 as a crucial player in connecting various GBM cell populations, including self-renewing stem cells and immunosuppressive microglia. TFPI2 enhances stem cell renewal and tumor growth by activating the c-Jun N-terminal kinase–signal transducer and activator of transcription (STAT)3 pathway. Simultaneously, secreted TFPI2 triggers the polarization of immunosuppressive microglia via STAT6 signaling. In human GBM, TFPI2 is associated with stemness, immunosuppression, increased microglia, and poor prognosis. Inhibiting the TFPI2-CD51-STAT6 pathway both activates T cells and improves the efficacy of anti-PD1 immunotherapy against GBM in mice (31). Moreover, this same group also characterized legumain (LGMN) as a key protease that is transcriptionally regulated by HIF1α and enriched in tumor-associated macrophages. Increased LGMN activates the GSK-3β-STAT3 signaling pathway to promote TAM immunosuppression. Inhibiting HIF1α and LGMN enhances CD8+ T cell- anti-tumor immunity, impairs GBM tumor progression, and improves immunotherapy responses (anti-PD1 therapy) in mice (32). Altogether, these data suggest that targeting TFPI2, as well as HIF1α and LGMN, are exciting strategies to combat GBM.
Furthermore, myeloid cells within the GBM microenvironment actively promote the expansion and activation of regulatory T cells (Tregs). Tregs are a subset of T lymphocytes that play a crucial role in immune tolerance and homeostasis. The interaction between myeloid cells and Tregs creates a self-reinforcing loop of immunosuppression as Tregs, in turn, support the survival and function of MDSCs. This collaboration contributes to establishing an immunosuppressive network that shields GBM from effective immune surveillance and clearance (19).
Strategies to counteract the immunosuppressive effects of myeloid cells in GBM are actively being explored in the field of cancer immunotherapy. Targeting MDSCs or modulating their function to reduce their immunosuppressive capacity represents a potential avenue for enhancing the anti-tumor immune response. However, the remarkable plasticity and adaptability of myeloid cells in response to the GBM microenvironment pose significant challenges to the development of effective therapeutic interventions. A comprehensive understanding of the intricate interactions between myeloid cells and the immune system within the GBM TME is crucial for devising innovative and targeted immunotherapeutic strategies.
The blood–brain barrier (BBB) plays an active role in generating immunosuppression within the GBM TME. The BBB is a specialized endothelial barrier that separates the bloodstream from the brain tissue, tightly regulating the passage of toxins, drugs, and immune cells into the CNS. Altogether, there are at least five ways in which the BBB affects the neuroimmune status: through BBB permeability, modulation of the BBB transporters, removal of immunoreactive substances by cells of the BBB, transfer of immune cells through the BBB into the brain, and secretion of immunoreactive substances by cells of the BBB (33). In GBM, the stability of the BBB reflects the degree to which the BBB contributes to the establishment of an immunosuppressive milieu within the tumor. The selective permeability of the BBB limits the entry of circulating immune cells, including cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, into the tumor site. This restriction hampers the effectiveness of the anti-tumor immune response, as these cells are crucial effectors in recognizing and eliminating cancer cells (34). This restrictive behavior also prevents the delivery of many therapeutics into the tumor. Therefore, considerable efforts have been made to modulate or bypass the BBB, either invasively or non-invasively, for the purpose of drug delivery (35). However, the delicate balance between protecting the brain from potential immune-mediated damage and promoting an effective anti-tumor immune response poses a significant challenge in developing targeted and safe therapeutic approaches.
The BBB traditionally was thought to contribute to a relatively immune-privileged state of the brain, creating an environment where immune surveillance is inherently restricted. This immune-privileged status of the brain was thought to be related to the lack of a traditional lymphatic drainage system, limiting immune cells to efficiently traffic to and from the CNS and target tumor cells (36). More recently, however, the brain is no longer considered a truly immune-privileged organ, as microglia act as resident antigen-presenting cells and allow T cells to traffic in and out of the CNS (24). In GBM, this immune privilege is exploited by the tumor, enabling it to thrive in an environment that is less susceptible to immune-mediated destruction (36). The leaky BBB in GBM also leads to the influx of pro-tumoral immune cells into the TME, such as infiltrating MDSCs and Tregs. Once infiltrated in the tumor, these cells contribute to the immunosuppressive network by suppressing effector immune responses and promoting a tumor-favorable microenvironment (20, 37).
The BBB also regulates the transport of immune-modulating molecules into the brain. Endothelial cells within the BBB express various transporter systems on their surface, encompassing both active transport mechanisms and those functioning through facilitated diffusion. The roles of certain transporters undergo alterations in the context of neuroinflammation and are subject to modulation by signaling molecules, with the P-glycoprotein (Pgp) protein being a notable example. Pgp is a BBB efflux transporter that limits drug entry into the CNS through interactions with diverse ligands, including protease inhibitors, opiates, antiepileptics, cyclosporins, glucocorticoids, aldosterone, dexamethasone, and calcium channel blockers. Indeed, Pgp acts as a gatekeeper of the CNS as its functionality undergoes changes during inflammation, primarily resulting in the suppression of its transport activity in vivo. Consequently, induction of Pgp by proinflammatory cytokines such as TNFα, IL-1β, IL-6, IL-2, and IFNγ leads to a reduction in Pgp mRNA expression in CNS endothelial cells, affecting both synthesis and activity of the transporter (38, 39).
Understanding the intricate relationship between the BBB and immunosuppression in GBM is crucial for the development of therapeutic strategies. Strategies that aim to selectively breach the BBB or modulate its permeability are actively being explored to enhance immune cell infiltration into the tumor and improve the efficacy of immunotherapeutic interventions.
Aberrant neo-angiogenesis in GBMs
GBMs are notoriously vascular tumors in which endothelial cells support tumor growth by delivering nutrients to the tumor; however, blood vessels within GBM tumors are faulty because of their disorganized formation. GBM tumor cells, inflammatory, and stromal cells all contribute to an aggressive production of a disarrayed tangle of leaky, abnormal new blood vessels (40). Angiogenesis plays a major role in the development of these abnormal vessels; however, neovascularization via proliferation of existing endothelial cells and via hypoxia-inducible factor 1α (HIF-1α) recruitment of bone marrow-derived vascular cells also contributes to tumor angiogenesis (41). Glioma stem cells themselves also generate novel vasculature by transforming themselves into pericytes or endothelial cells (42, 43). This pro-angiogenesis drive is supported by the tumor’s inherent metabolic, inflammatory, and hypoxic conditions. Importantly, tumor cells that are often found in the periphery of the hypoxic necrotic core release unregulated levels of VEGF as well as additional paracrine factors that further lead to vascular bed inflammation and increased vascular permeability (44).
VEGF is a master regulator of the vascular aberrations in GBM, which in addition to driving faulty neo-angiogenesis, also result in the breakdown of the BBB. The BBB is comprised of astrocytes, endothelial cells, and tight junctions by pericyte feet, which together tightly regulate the transfer of molecules between the blood and brain parenchyma (45). The breakdown of the BBB results in increased vessel permeability with the influx of fluid into tumor tissue and surrounding brain tissue, resulting in cerebral edema and increased intracranial pressure. Simultaneously, the slowing of blood flow through these compromised blood vessels results in patchy oxygen delivery within the tumor bed. These focal areas of hypoxia develop from occluded vessels into pseudopallisading necrosis, a pathognomonic characteristic of GBM. These necrotic niches also recruit macrophages and other innate immune cells that further propagate angiogenesis and immunosuppression.
More recently, Pang et al., have shown that the circadian regulation of glioma stem cells alters the immunosuppressive TME in GBM tumors through paracrine and autocrine mechanisms (46, 47). One way the stem cells interact with the TME is via circadian regulation through CLOCKs, which include both transcriptional activators (such as CLOCK-BMAL1 complex) as well as inhibitors (such as cryptochrome 1 and 2, period 1,2,3 and REV-ERBα (48). CLOCK-BMAL1 complex in glioma stem cells leads to the upregulation of periostin (POSTN) that ultimately activates the TANK-binding kinase 1 (TBK1) in endothelial cells. The inhibition of POSTN and TBK1 in GBM mouse models, as well as pathological analysis in GBM patient samples, reveal that this pathway may be an interesting therapeutic strategy for targeting GBM angiogenesis (49).
Hypoxia within GBM
GBM tumors are characterized by areas of tissue hypoxia (50–53), which not only render them more aggressive (51) but are also critical for the maintenance of glioma stem cells, the tumoral cell population that is responsible for resistance to therapies (54, 55) and tumor recurrence (56). Hypoxic and inflammatory responses overlap powerfully in the GBM microenvironment (51), as hypoxic conditions restrict cytotoxic T lymphocyte development while promoting the proliferation and expression of inflammatory cytokines (57). Although not yet completely understood, glioma stem cells, which thrive in hypoxic conditions, preferentially inhibit the proliferation of activated T cells, as compared to differentiated tumor cells (58). Hypoxic conditions within GBM tumors activate the STAT3 signaling pathway, which mediates HIF-1α and leads to immunosuppression (59). Finally, the hypoxic TME recruits immunosuppressive cells such as tumor-associated macrophages, myeloid-derived suppressor cells, and Tregs (60), which all reduce the immune response through the expression of inhibitory molecules on the surface of effector immune cells (51).
New frontiers in treatment: immunotherapies
Immunotherapies are one of the newest frontiers in combating cancer (Figure 2). These therapies take advantage of a patient’s existing functional immune system by harvesting immune cells and retraining them to attack and kill tumor cells. These treatments have been FDA-approved for multiple blood-borne cancers (61) and are used investigationally for brain tumors. Immunotherapeutic strategies cover a wide range of treatments and include vaccines (62), adoptive cell therapy (63), monoclonal antibodies (64), oncolytic viruses (65, 66), and immune checkpoint blockers (67, 68).
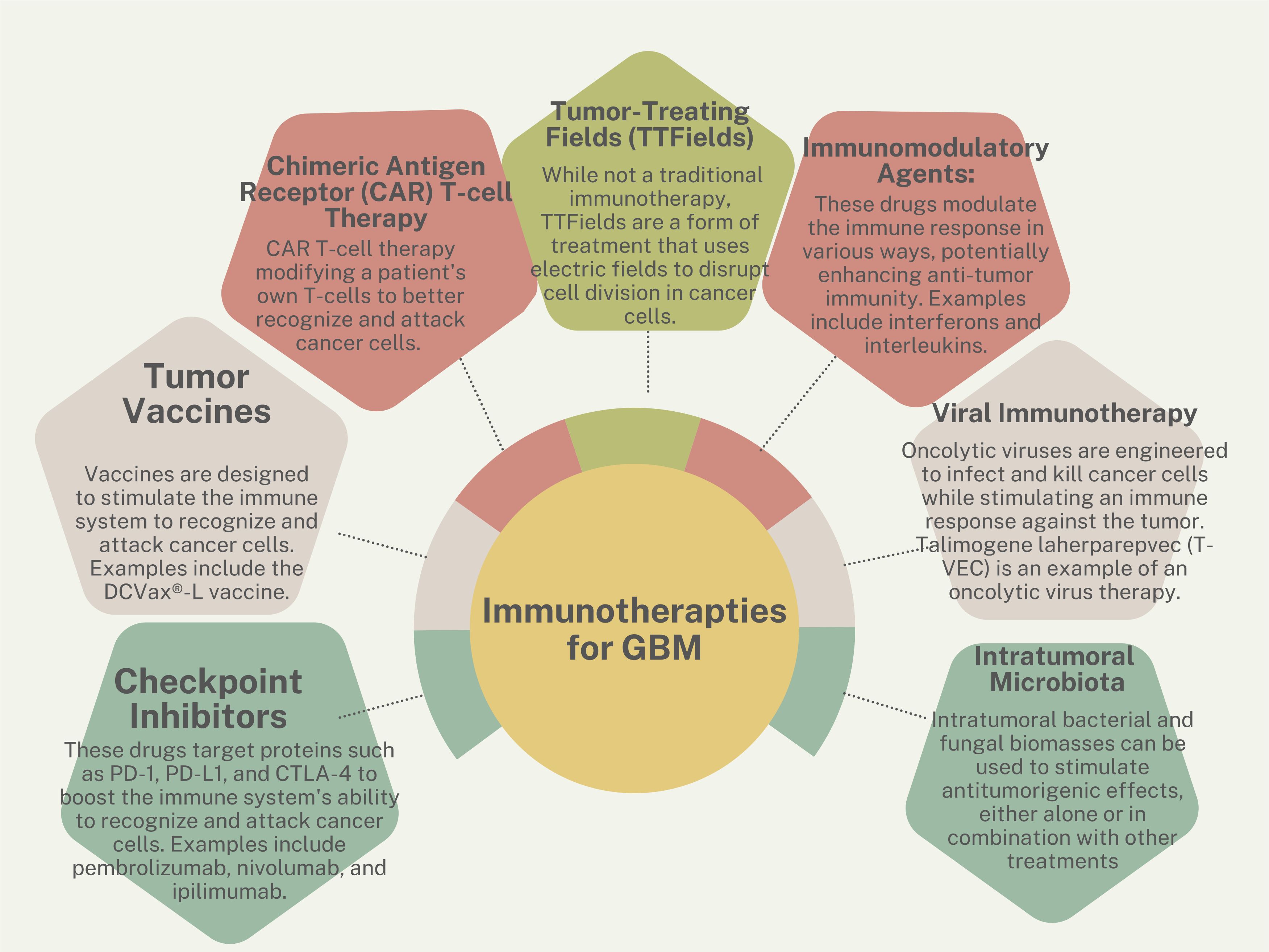
Figure 2 Immunotherapy strategies for GBM.
Vaccines
The field of GBM vaccine development has evolved with various approaches aimed at eliciting immune responses against tumor-specific antigens. Over two decades ago, one of the first antitumor vaccines was generated utilizing autologous dendritic cells (DCs) transduced with a replication-defective adenovirus encoding the full-length melanoma antigen MART-1/Melan-A (MART-1) to target melanoma (69). Since then, significant advancements in vaccine technology have been made to include class I-restricted peptide cocktails, multi-peptide vaccines, long peptide vaccines, peptide mimics, personalized peptide vaccines, and dendritic cell vaccines loaded with peptides, RNA, or tumor lysate.
Current clinical trials for glioma involve diverse vaccine formulations, targeting antigens like EGFRvIII, survivin, human cytomegalovirus proteins, and Wilm’s tumor protein-1 (70). Despite promising results in earlier phases, there have been challenges in translating efficacy from Phase II trials, exemplified by the EGFRvIII vaccine Rintega, which after completing the Phase III trial (ACT IV), showed that Rintega-treated patients had median overall survival of 20.4 months compared to 21.1 months in the control arm (71). Tumor progression was linked to reduced expression of EGFRvIII, indicating that Rintega triggered a distinct and potent immune response, leading to the effective elimination of GBM cells that expressed the targeted antigen. Subsequently, it was conjectured that tumors eluded immunologic regulation post-vaccination by discarding the targeted EGFRvIII antigen (72).
Heat-shock protein vaccines, particularly HSPPC-96, a dendritic cell vaccine derived from autologous tumor-derived HSP–peptide complex, have demonstrated positive immune responses in recurrent GBM patients. In a single-arm Phase II trial, HSPPC-96 was administered to patients after undergoing gross total resection of recurrent GBM. Treated patients enjoyed a median overall survival of 42.6 weeks (73). Additionally, ongoing trials explore the potential of personalized neoantigen vaccines and dendritic cell-based approaches in newly diagnosed GBM patients. The complex interplay between vaccine strategies, patient characteristics, and standard therapies necessitates careful evaluation in the quest for effective GBM immunotherapy.
Immune checkpoint inhibitors
Brain metastases occur in 8–10% of cancer patients (74, 75), with melanoma and lung cancer having the highest cumulative incidence (76). Like patients with GBM, the prognosis for patients with brain metastases is also poor, despite multidisciplinary treatments involving surgery, irradiation, and systemic therapies. Within the last decades, there has been excellent progress in understanding how solid cancer cells evade the immune system through immune checkpoints, including CTLA-4, PD-1, and PD-L1. Blockage of these immune checkpoints with antibodies such as ipilimumab (anti-CTLA-4), nivolumab (anti-PD-1), and pembrolizumab (also anti-PD-1) have shown efficacy in various solid tumors, particularly melanoma and non-small cell lung cancer, leading to prolonged survival in extracranial disease.
To systematically evaluate the use of checkpoint inhibitors in Neuro-Oncology, a review of relevant literature was conducted following PRISMA guidelines. The search identified 88 eligible publications, including studies on GBM and brain metastases. For brain metastases, 40 studies assessed ICIs without radiotherapy, and 40 studies explored the combination of ICIs with radiotherapy. The median intracranial progression-free survival for patients with brain metastases treated with ICIs was 2.7 months, and the overall progression-free survival was 3.0 months. The median survival for these patients was 8.0 months. The studies revealed varying efficacy across different tumor types and ICI agents. For example, ipilimumab monotherapy in melanoma brain metastases showed a pooled intracranial objective response rate of 9.0%, while pembrolizumab monotherapy in melanoma brain metastases demonstrated an intracranial objective response rate of 22.0–40.0%. Nivolumab monotherapy in NSCLC brain metastases had a pooled objective response rate of 14% (77).
For GBM, eight studies were included, encompassing phase I and II trials and retrospective analyses. The median survival of GBM patients treated with ICIs across these studies was 7.3 months, with a median progression-free survival of 2.1 months (77). As seen in multiple phase III clinical trials, there was no survival benefit in treating GBM patients with nivolumab. In the clinical trial CheckMate 143, median overall survival in patients treated with nivolumab was not different from those patients treated with bevacizumab (78). In the CheckMate 498 clinical trial, MGMT-methylated GBM patients treated with standard therapy of TMZ and radiotherapy survived longer than those patients treated PD-1 blockade and concomitant radiotherapy (79). Similarly, nivolumab with TMZ and radiotherapy were not superior to radiotherapy, TMZ or placebo in newly diagnosed MGMT-methylated GBM (80).
Chimeric antigen receptor engineered T cells
CAR T cells are an immunotherapy in which patients’ CD8 T cells are harvested and genetically modified to produce receptors that bind to antigens on the surface of cancer cells. After these CAR-T cells are expanded into millions of copies in the laboratory, they are re-introduced into the patient to recognize and kill cancer cells that carry the target antigen on the cell surface. Since 2017, six CAR T cell therapies have been approved by the FDA to treat lymphomas, leukemia, and multiple myeloma. Because CAR T cell therapy has been so successful in treating blood-borne cancers, neuro-oncologists have been hopeful CAR T cells might treat solid tumors such as GBM.
There are several target molecules expressed on GBM cells including EGFR/EGFRvIII, HER2, B7-H3, and CSPG4 that serve as targets for CAR T cells (81). Our team at City of Hope has designed interleukin 13 receptor alpha-2 (IL13Rα2)-targeting CAR T cells to treat GBM. IL-13 is a cytokine that T helper cells release to regulate inflammation. Binding to biological receptor, IL13Rα1, results in activation of the JAK/STAT pathway. However, when binding to its high-affinity decoy receptor IL13Rα2, a protein that is expressed in over 75% of GBMs and not expressed in normal brain, IL-13Rα2 leads to activation of the rapamycin pathway (82, 83), resulting in increased tumor invasiveness, and therefore poorer prognosis (84). Recent clinical trials utilizing CAR T cells to target CD19 led to extraordinary remission in relapsed or refractory B-cell lymphomas (85, 86), and the FDA has approved this treatment for pediatric (85) and refractory adult (87) acute lymphoblastic leukemia. Because hematological tumor cells can be accessed intravascularly, CAR T cell therapy is easily administered via an intravascular route, and upon delivery, can have direct contact with target tumor cells. In contrast, CAR T therapy for GBM has had marginal efficacy, mostly due to showing antitumor effects in only a subset of patients.
Many hindrances reduce CAR T efficacy for brain tumors. For example, GBM tumors themselves actively contribute to immune suppression through a host of well-orchestrated strategies within a harsh TME. Once CAR T cells reach target tumor cells, the microenvironment may suppress their activity and proliferation by expressing inhibitory cell-surface molecules (such as PD-L1, CD95) (88) or by releasing immunosuppressive tumor-derived soluble factors and cytokines (such as prostaglandin E2, IL6, IL10, and TGFβ) (89). The TME also preferentially promotes the trafficking of suppressive cell populations, such as regulatory T cells (Tregs), tumor-associated macrophages, microglia, and myeloid-derived suppressor cells (83, 90, 91).
Tumor treating fields
Tumor Treating Fields (TTFields) therapy is an innovative treatment modality for GBM, that is non-invasive and can be administered alongside standard treatments such as chemotherapy and radiation therapy. TTFields therapy involves the use of low intensity alternating electric fields delivered via transducer sticker arrays placed on the patient’s scalp. TTFields therapy is an antimitotic approach that disrupts the division of glioblastoma cells and their organelle assembly. This is achieved through the application of low intensity alternating electric fields directly to the tumor site. TTFields therapy is its favorable safety profile, as it primarily targets dividing cancer cells while sparing healthy brain tissue from damage.
Clinical studies evaluating the efficacy of TTFields therapy in GBM have shown relatively promising results. The landmark EF-14 was a randomized clinical trial involving 695 patients with GBM who had completed initial radiochemotherapy, were randomized to the addition of TTFields to maintenance temozolamide. The results demonstrated a significant improvement in both progression-free survival (6.7 months in the TTFields plus temozolomide group and 4.0 months in the temozolomide-alone group) and overall survival (median overall survival was 20.9 months in the TTFields-temozolomide group vs 16.0 months in the temozolomide-alone group (HR, 0.63; 95% CI, 0.53–0.76; P < .001)) in newly diagnosed glioblastoma patients who received TTFields therapy in addition to standard chemotherapy compared to those who received chemotherapy alone (92). Additionally, TTFields therapy has been associated with a favorable quality of life profile, with main risk.
While TTFields therapy represents a significant advancement in treating glioblastoma, ongoing research aims to optimize its efficacy and explore its potential in other cancer types. Studies are investigating various aspects of TTFields therapy, including optimal treatment schedules, combination with other therapeutic modalities, and mechanisms of action to enhance its anti-tumor effects further. As TTFields therapy continues to evolve, it holds promise as a valuable addition to the armamentarium of treatments for glioblastoma and potentially other malignancies, offering patients a novel and well-tolerated therapeutic option.
Immunomodulatory agents
Recently, cytokines such as IFN-α, TNF-α, and IL-12 have been used as novel therapeutic approaches to treat GBM treatment, aiming to alter the immunosuppressive TME to evoke an effective antitumor immune response (93). IFN-α, for instance, has been reported to enhance T cell and macrophage activity and inhibit tumor angiogenesis and immune-suppressive gene expression (94). Conversely, TNF-α fosters dendritic cell maturation and subsequent T cell activation, while IL-12 is associated with improved CAR-T cell functionality, increased infiltration of CD4+ T cells, and reduced T-regulatory cell frequency within the tumor microenvironment (95, 96).
However, IFN-α therapy is hindered by its significant systemic toxicity and limited efficacy at maximum tolerated doses, as evidenced by clinical trials reporting adverse effects such as hyperthermia, shivering, headaches, gastrointestinal symptoms, and orthostatic hypotension (97). This restricts its current use, highlighting the need for future strategies that combine IFN-α with other treatments to enhance both efficacy and tolerability (93).
IL-6 is a multifaceted cytokine that orchestrates several biological processes, such as the acute phase response, immune defense against infections, leukocyte infiltration at inflammation sites, leukocyte maturation, and endothelial cell characteristics (98). The IL-6 signaling pathway initiates with IL-6 binding to membrane-bound IL-6R (gp80) present on various cells like hepatocytes, neutrophils, monocytes, B cells, and T cells. Subsequently, the IL-6-IL-6R complex binds to membrane-bound gp130, expressed ubiquitously by all cells, forming an activated IL-6 receptor that triggers intracellular signaling. This classic IL-6 signaling pathway leads to anti-inflammatory responses. Alternatively, IL-6 can also act via the trans-signaling pathway, binding to soluble forms of IL-6R, and then binding to membrane-bound gp130, activating target cells lacking membrane-bound IL-6R and inducing pro-inflammatory responses (99, 100). Both pathways activate Janus kinases (JAKs) and signal transducer and activator of transcription (STATs) to modulate cellular responses (98).
Overexpression of IL-6 has been linked to poor survival in GBM patients based on data from The Cancer Genome Atlas (TCGA) and the Repository of Molecular Brain Neoplasia Data (REMBRANDT) (101). Within the GBM TME, IL-6 is secreted by tumor cells, glioma-associated microglia/macrophages (GAMs), and tumor-associated endothelial cells (102, 103), with its expression induced by treatments such as chemotherapy (104) and radiotherapy (105). IL-6 exerts its effects through IL-6R expressed on tumor cells and GAMs, promoting immunosuppressive GAMs and impairing T cell functions. Treatment with anti-IL-6 siltuximab, anti-IL-6R tocilizumab, or STAT3 inhibitor Stattic has been shown to mitigate PD-L1 expression and T cell apoptosis, underscoring the role of IL-6 signaling in regulating T cell responses via GAMs (28). Additionally, IL-6 sustains tumor progression by directly influencing glioblastoma cells to activate anti-apoptotic pathways, induce autophagy, and promote invasion through mechanisms involving MMPs and fascin-1 (106).
Viral immunotherapy
Oncolytic viruses (OVs) are weakly pathogenic viruses designed to selectively infect, replicate within, and kill tumor cells through apoptosis while avoiding infection of healthy cells (107). OVs stimulate the innate immune system through pathogen-associated molecular patterns and pattern recognition receptors that recruit immune cells such as neutrophils, macrophages, natural killer cells, Th1 cells, leading to cell lysis (108). This response also leads to an adaptive immune reaction to new cancer antigens, therefore possibly propagating long-term anti-tumor immunity (109). Moreover, OVs are able to convert the GBM TME from “cold” to “hot” by triggering inflammation, and improve the outcome of immune checkpoint inhibitors (110). OVs utilizing modified herpes simplex virus, adenovirus, measles virus, parvovirus, reovirus, and poliovirus, amongst others, are being studied in clinical trials against GBM worldwide (93). Because OVs are noted to have oncolytic activity but low specificity, various methods have been employed to improve tumor-specificity such as deleing virulence genes, adding tumor-specific promotors, or adding tumor suppressor gene miRNAs (111).
More than 20 oncolytic viruses have been evaluated in clinical trials for GBM. Key examples include HSV-1, adenovirus, reovirus, measles virus, Newcastle disease virus, and poliovirus (112). To circumvent the limitations imposed by the BBB, innovative delivery methods such as convection-enhanced delivery (CED) have been utilized. CED is particularly used for the recombinant nonpathogenic polio-rhinovirus chimera (PVSRIPO) and works by using a pressure gradient within a catheter to deliver therapeutic agents into the interstitial spaces of the brain parenchyma (113). Achieving efficient and safe delivery of oncolytic viruses is critical for successful virotherapy. The difficulty of delivering these viruses to the CNS and their potential elimination by the immune system has made intratumoral delivery the preferred approach. However, for optimal outcomes, the oncolytic virus should ideally be administered systemically to reach both primary and metastatic tumor sites, especially in the cases of multifocal GBM.
The predominant immune cells in the GBM TME are macrophages originating from peripheral monocytes, known as tumor-associated macrophages (TAMs) (114). TAMs primarily express the M2 surface marker and produce IL-10 and TGF-β with STAT3 expression, promoting an immunosuppressive “cold” TME (115). Additionally, T cell exhaustion, characterized by the upregulation of inhibitory molecules and increased Treg cells, is a hallmark of GBM (116). Moreover, expression levels of immunomodulatory chemokines CXCL2, CX3CL1, CCL5, and CCL2 are elevated in GBMs (117). Consequently, OVs could enhance alternative therapies by reversing these conditions to promote greater immune infiltration and improved tumor destruction (118). For example, delivering concomitant OVs with ICIs has shown promising outcomes. Infection with the measles virus in GBM models resulted in increased PD-L1 expression (119); and using both measles as well as anti-PD-1 antibodies resulted in longer survival in murine glioma models compared to survivals in animals treated with each therapy alone (120). Similarly, OVs can improve CAR T cell therapy against solid tumors (112). Care must be taken however, as application of a vesicular stomatitis virus encoding IFNβ resulted in the reverse outcome on CAR T cells targeting EGFRvIII in B16 murine glioma tumors (121). Further understanding of the complex interplay between OVs and other immunological treatments must be further characterized prior to use in larger-scale clinical trials.
Utilizing intratumoral bacteria
With ever-advancing technologies to study the TME, intratumoral bacterial and fungal biomasses have been characterized in multiple tumor types (122, 123). Intratumoral microbiota generates anti-tumorigenic effects by increasing antigen presentation, activating NK and T cells, improving immunosurveillance, and ultimately generating metabolites that suppress tumor progression (124). Intratumoral bacteria can induce potent immunogenic antitumor effects and significantly extend survival rates with effective immunological memory in various murine cancer models (125), such as against melanoma (126). Memory T cells pre-stimulated by bacteria-derived or tumor-derived antigens within GBM are found in peripheral blood and tumors (127). There is cross-reactivity between human tumor antigens and bacterial antigens due to molecular mimicry, allowing tumor antigen recognition by CD4+ T cells (127) and CD8+ cytotoxic T cells (128). Indeed, GBM contains distinct microbiota compared to adjacent normal tissues (129). Bacteria-specific peptides obtained from normal cell lines or GBM tumor samples are presented via MHC II molecules. After stimulation by these bacterial peptides, tumor-infiltrating lymphocytes secrete more pro-inflammatory cytokines and recruit CD8+ T cells to enhance tumor-killing (130). For patients where intratumoral bacterial nucleic acids or peptides are detected in very low quantities, subcutaneous injection of tumor antigen-engineered commensal bacteria to elicit distant anti-tumor immune responses is a novel and safe approach (126), and is an exciting option for GBM treatment (131). Through the subcutaneous delivery of lysed-engineered bacterial peptides, epidermal Langerhans cells further present tumor peptides in MHC molecules, allowing distant primed CD8+ T cells to enter the circulation and infiltrate tumors. This vaccination approach avoids HLA restriction (132) and is an exciting option to be used alone or in combination with other tumor vaccine strategies (131).
Strategies to improve immunotherapies
Various approaches have been suggested to address hypoxia in cancer, including inhibition of HIF signaling, the use of hypoxia-activated prodrugs (HAPs), targeting downstream pathways like the unfolded protein response (UPR) and mTOR, as well as metabolic interventions (133). Considering hypoxia’s central role in regulating tumor progression and immune suppression, it is plausible to explore hypoxia as a potential target in combined cancer immunotherapy. This section delves into the manipulation of hypoxia to enhance the effectiveness of cancer immunotherapy, drawing insights from both pre-clinical and clinical studies. Please refer to Box 1 for more details.
Hypoxia-activated prodrugs
HAPs, also referred to as bioreductive prodrugs, are inert compounds converted into active substances by enzymatic reduction, selectively targeting hypoxic TMEs (134). Despite promising pre-clinical hypoxic cytotoxicity, some HAPs demonstrated disappointing clinical efficacy, leading to their discontinuation. Tirapazamine (TRP) is the first HAP that showed clinical safety in 1994 (135), however, despite optimistic preclinical findings, phase III studies showed no therapeutic benefit compared to chemotherapy alone or chemoradiotherapy (136). Additionally, the mitomycin C derivative prodrug praziquantel (EO9) displayed efficacy in superficial bladder cancer patients (137), however, a phase III clinical trial evaluating the efficacy and safety of EO9 (NCT01469221) was unfortunately discontinued (134). A second generation TRP analogue, SN30000, demonstrated antitumor effects in xenograft models (138), and the New Zealand group working on this analogue hope to generate Phase I clinical data from this.
Notably, evofosfamide (TH-302), a TH-302, a prodrug of the cytotoxic alkylating agent bromo-isophosphoramide mustard, is favorably activated in hypoxic conditions. TH-302 has shown non-lymphotoxic properties and compatibility with immunotherapy (139), specifically with PD-1 and CTLA-4 blockade. Combining TH-302 with these immunotherapies achieved remarkable tumor cures and prolonged survival in prostate cancer mouse models (140). A recent phase I clinical trial (NCT03098160) evaluated TH-302 in combination with ipilimumab against various cancers, including melanoma, pancreatic, and prostate tumors, which showed no new safety issues and showed evidence of therapeutic activity (141). Encouraging results from a Phase II clinical trial involving TH-302 with doxorubicin for soft tissue sarcoma (142) led to an international, open-label phase III study using TH-302 in combination with doxorubicin as first-line therapy for locally advanced soft-tissue sarcoma (NCT01440088). Unfortunately, the combination did not show any overall survival benefit compared to those patients treated with single-drug doxorubicin alone (143). More recently, a Phase II clinical trial using TH302 for recurrent bevacizumab-refractory GBM showed that progression-free survival at 4 months following TH302-bevacizumab treatment was 31%, significantly higher than the historical rate of 3% (144).
Drugs targeting HIF signaling pathways
Drugs targeting hypoxia-inducible factors (HIFs) are under development, and are classified by their interference with HIF dimerization, DNA binding, mRNA or protein expression, or degradation (145). Combining classical chemotherapeutic agents, such as cisplatin, doxorubicin, and cyclophosphamide, with HIF pathway inhibition shows promise in enhancing antitumor immune responses (146). Studies indicate that downregulating HIF-1 expression by antisense HIF1-α enhances NK cell and CD8 T cell-mediated antitumor immunity, resulting in anti-tumor activity (147). PX-478 (S-2-amino-3-[4 V-N,N,-bis(2-chloroethyl) amino]-phenyl propionic acid N-oxide dihydrochloride) is a small-molecule that suppresses HIF-1α translation that, when given with a dendritic cell-based vaccine, dramatically reduces breast cancer in mice (148). Similarly, combining HIF-1-mediated ectoenzyme ENTPD2 inhibitors with anti-CTLA-4/PD-1 ICIs significantly increased T-cell infiltration in hepatocellular carcinoma tumors and extended survival in tumor-bearing mice (149).
Additional small molecules, including the Hsp90 inhibitor (17-AAG) (150), digoxin (151), and 2-methoxy estradiol (2ME2) (152), act non-specifically on HIF by inhibiting its synthesis or stabilization and have shown tumor inhibition. In contrast, PT2385 and its derivative, PT2399, uniquely bind to HIF2, disrupting its heterodimerization with HIF-β (153). The efficacy of PT2399 was validated in a preclinical model of clear-cell renal cancer cells with heightened HIF activation due to VHL mutation (154). Results from a recent Phase I clinical trial demonstrated the safety and efficacy of PT2385 as a direct HIF-2 inhibitor in locally advanced or metastatic clear cell renal carcinoma (155). More recently, a single-arm open-label phase II study of patients with recurrent GBM was treated with oral PT2385, which had limited anti-tumor effect, in part because of variable drug exposure (156). PT2977 (MK-6482), a more potent second-generation HIF-2 inhibitor, is currently undergoing a Phase III clinical trial in advanced renal cell carcinoma (NCT04195750) (157). It’s crucial to note that none of these studies evaluated tumor oxygenation status, emphasizing the need for further investigation to determine whether this treatment is more advantageous for specific patient populations exhibiting high HIF-2 expression or elevated tumor hypoxia.
Metabolic regulation
Several metabolic targets are being studied to reduce tumor hypoxia and improve treatment sensitivities to radiation as well as chemotherapies and immunotherapies. Oxygen is utilized by mitochondria through oxidative phosphorylation and the electron transport chain. Reducing cellular oxygen consumption rates by inhibiting oxidative phosphorylation increases oxygen in the TME and improves oxygen diffusion into hypoxic areas (157). One strategy to reduce tumor hypoxia in tumors is to lower the oxygen consumption rate in tissue. Reducing oxygen consumption by just 30% in poorly perfused tumor regions was more effective at reducing tumor hypoxia than increasing blood flow or increasing oxygen levels in the blood (158). Reducing oxygen consumption is an intriguing strategy because it also circumvents the need for drugs to diffuse into poorly oxygenated areas and can be used across multiple tumor types (157).
Metformin is an anti-diabetic biguanide that became an interesting metabolic targeting drug when it was noted that diabetic patients on metformin had a lower cancer incidence than diabetics on other medications, or non-diabetics (159). Metformin inhibits mitochondrial complex 1, which activates AMPK and inhibits mTOR, which overall reduces oxygen consumption rate (160). Although preclinical murine studies showed that metformin improved tumor oxygenation and increased radiotherapy sensitivity (161), metformin was not shown to benefit patients with non-small cell lung cancer in clinical trials (162, 163). There have been a number of clinical studies performed that had shown promising results in treating tumors, however due to insufficient sample sizes, the results have not been conclusive [for review see (164)].
Carbonic anhydrase isoform IX (CAIX), a cell-surface pH regulatory enzyme upregulated by HIF-1α and HIF-2α, is another exciting metabolic target. CAIX activates glycolysis, contributing to immune suppression in various solid malignancies (165). Targeting CAIX with monoclonal antibodies or small-molecule inhibitors, such as SLC-0111, decreases glycolytic metabolism and ultimately leads to increased immune activity (166). Since 1998, various clinical trials have been used to study anti-CAIX monoclonal antibodies against renal cell cancer, with various results such as complete responses, maintaining stable disease, and partial responses (see (167) for review). Blocking CAIX renders tumors more sensitive to ICIs due to enhanced Th1 responses and reduced tumor invasiveness. Therefore, inhibition of CAIX, in combination with immunotherapy, proves to be a potential strategy for improving clinical outcomes in hypoxic tumors (168).
Hypoxia-induced accumulation of extracellular adenosine leads to an immunosuppressive TME. Adenosine-A2A receptors (A2AR) is a G-protein-coupled receptor expressed in many immune cells such as cytotoxic T cells, macrophages, and regulatory T cells (169). In a pre-clinical study of squamous cell carcinoma, application of A2AR-blocker, SCH5826, decreased CD4+ T regs and promoted a CD8+ T cell-mediated response that reduced tumor growth (170). Indeed, inhibiting A2AR resulted in improved efficacy of chemotherapy and immunotherapy in multiple cancers such as lung adenocarcinoma, renal cell cancer, paraganglioma, and pheochromocytoma (171–174).
Given that metabolic reprogramming is so crucial to cancer cell growth and infiltration of immunosuppressive myeloid cells into the tumors, Khan et al. screened metabolic small-molecule compounds for impaired macrophage migration and determined that lactate dehydrogenase (LDH) inhibitor stiripentol, an anti-seizure medication was a top hit. They further characterized that LDHA-dependent ERK pathway leads to activation of YAP1/STAT3 and upregulation of chemokines CCL2 and CCL7. The team further determined that both genetic and pharmacological blockade of LDHA-mediated tumor-macrophage symbiosis both suppressed macrophage infiltration in murine GBMs as well as reduced tumor progression. LDHA-mediated glycolysis also results in chemo-radiotherapy resistance, therefore, targeting LDHA and its downstream pathways is a further attractive strategy to combat GBM (175, 176)
Supplemental oxygenation
Supplemental oxygen therapy, combined with existing immunotherapies, has the potential to decrease tumor hypoxia and extracellular adenosine accumulation. Respiratory hyperoxia with 60% oxygen enhances intra-tumoral infiltration of CTLs and improves antitumor responses when given with dual PD-1 and CTLA-4 therapy in mouse models (177). Indeed, adoptive immunotherapy combined with respiratory hyperoxia led to the complete obliteration of fibrosarcoma tumors in mice (177). The approach of using supplemental oxygen as an immunological co-adjuvant warrants further investigation in combination with existing cancer immunotherapies. Conversely, inhibiting both oxygen consumption and tumor hypoxia with metformin is correlated with improved efficacy of PD-1 blockade immunotherapy (178).
Hyperbaric oxygen (HBO) treatment delivers 100% oxygen gas at 2–4 times normal atmospheric pressure, which increases a patient’s saturated hemoglobin. In the 1970s and 80s, clinical trials showed that HBO treatment delivered before radiation treatment improved local control of squamous cell carcinoma and cervical cancer. However, HBO treatment had technical limitations related to decompression of the chamber, which led to tissue damage and therefore has since fell out of favor (179). Carbogen gas (composed of 95% oxygen and 5% carbon dioxide) breathing became an alternative to HBO as it could promote oxygen delivery via vasodilation and increased blood flow (180), but does not require an oxygen chamber, which allows the gas to be more easily delivered. Carbogen gas breathing showed inconsistent results, however, which might be related to the duration of time subjects breathed the gas prior to treatments (181).
Vessel normalization
Hypoxia, resulting from abnormal tumor vascularization, upregulates VEGF through HIF-1, creating a detrimental cycle of worsening hypoxia (139). VEGF/VEGFR-targeted therapies induce anti-angiogenic effects, reducing hypoxia and supporting the immune response. However, monotherapy with angiogenesis inhibitors can exacerbate tumor hypoxia, rendering both chemotherapies and radiation treatment less effective (182). Vascular normalization, achieved with low-dose anti-angiogenic agents, enhances immunotherapy efficacy and reduces toxicity. Clinical trials combining PD-1 blockade with anti-angiogenic agents, such as bevacizumab (183) or Lenvatinib (184), show promise in various cancers, including melanoma and hepatocellular carcinoma. In fact, ICIs induce interferon-γ production from CD4+/CD8+ T cells, play an important role in the normalization of blood vessels, and work as an anti-angiogenesis therapy themselves (185). Based on Phase III clinical trial results, the FDA has approved combinations of PD1/PD-L1 antibodies with anti-VEGF/VEGFR agents in lung and renal cancers (186). Additionally, there is much promise in utilizing anti-angiogenesis therapy with immunotherapy to augment radiotherapy (185). Nonetheless, the impact of hypoxic stress on tumor heterogeneity remains a question, emphasizing the need for tailored approaches based on tumor type, patient status, and reliable biomarkers.
In summary, the combination of hypoxia-targeting strategies with immunotherapy holds promise for enhancing therapeutic outcomes in cancer patients. However, the diverse responses to hypoxia-modifying therapy highlight the importance of personalized medicine, necessitating the identification of specific hypoxia markers for targeted therapies and robust anti-tumor immune responses.
Box 1: Advantages and disadvantages of methods to improve immunotherapies.
Inhibition of HIF signaling
Advantages: Combining classical chemotherapeutic agents, such as cisplatin, doxorubicin, and cyclophosphamide, with HIF pathway inhibition shows promise in enhancing antitumor immune responses.
Disadvantages: studies on the efficacy of PT2399, PT2385 and PT2977 HIF-2 inhibitors have not evaluated tumor oxygenation status, emphasizing the need for further investigation to determine whether this treatment is more advantageous for specific patient populations exhibiting high HIF-2 expression or elevated tumor hypoxia.
Hypoxia-activating prodrugs (HAPs)
Advantages: HAPs, also referred to as bioreductive prodrugs, are inert compounds converted into active substances by enzymatic reduction, selectively targeting hypoxic TMES Disadvantages: Despite promising pre-clinical hypoxic cytotoxicity, some HAPs demonstrated disappointing clinical efficacy, leading to their discontinuation.
Manipulation of downstream pathways:
Advantages: Supplemental oxygen therapy, combined with existing immunotherapies, has the potential to decrease tumor hypoxia and extracellular adenosine accumulation. Vascular normalization, achieved with low-dose anti-angiogenic agents, enhances immunotherapy efficacy and reduces toxicity and the FDA has approved combinations of PD1/PD-LI antibodies with anti-VEGF/VEGFR agents in lung and renal cancers.
Disadvantages: Carbogen gas, a supplemental oxygen therapy, can promote oxygen delivery, but has shown inconsistent results which might be related to the duration of time subjects breathed the gas prior to treatments. For vessel normalization, while there is much promise in utilizing anti-angiogenesis therapy with immunotherapy to augment radiotherapy, the impact of hypoxic stress on tumor heterogeneity remains a question, emphasizing the need for tailored approaches based on tumor type, patient status, and reliable biomarkers.
Interventions leading to metabolic changes:
Advantages: Reducing cellular oxygen consumption rates by inhibiting oxidative phosphorylation increases oxygen in the TME and improves oxygen diffusion into hypoxic areas.
Disadvantages: More clinical studies are needed to test efficacy of Metabolic targeting drugs, such as Metformin, inhibition of metabolic targets such as CAIX and A2AR, and metabolic small- molecule compounds such as LDH.
Future directions
Future advances leveraging RNA-seq and single-cell RNA sequencing techniques, along with bioinformatics and AI technology, hold immense potential in unraveling the intricacies of GBM TME. RNA-seq provides a high-throughput method to analyze gene expression profiles, allowing researchers to decipher the molecular patterns associated with different cell types within the TME. Moreover, sc-RNA-seq enables the dissection of individual cells, providing unprecedented insights into the heterogeneity and cellular interactions driving GBM progression and therapy resistance (53).
By harnessing the power of bioinformatics and AI, researchers can sift through vast amounts of genomic data generated from RNA-seq and sc-RNA-seq experiments to identify key molecular players and signaling pathways implicated in GBM pathogenesis. Advanced computational algorithms can integrate multi-omics data, such as genomics, transcriptomics, proteomics and epigenomics, to construct comprehensive models of the GBM TME. These models not only enhance our understanding of tumor biology but also facilitate the identification of novel therapeutic targets and the development of personalized treatment strategies. Furthermore, AI-driven approaches enable predicting patient outcomes and therapeutic responses, paving the way for precision medicine in GBM management.
Conclusion
It’s important to understand the dynamics of hypoxia in the GBM TME to develop targeted therapies that address the complexities of such an environment. The unique features of the TME, such as the presence of CSCs, interactions with stromal and immune cells, and the vascular and hypoxic niches, can be targeted to develop more effective and personalized treatments. However, developing clinically viable therapies requires overcoming the challenges posed by the remarkable heterogeneity inherent to GBM. Advances in precision medicine, immunotherapy, and innovative drug delivery systems are being explored to design therapies that account for the diverse elements of the GBM TME and to improve patient outcomes.
Author contributions
LF: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
I thank Dr. Erin Keebaugh and Ms. Bijal Shah for their assistance in manuscript preparation and editing.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014–2018. Neuro Oncol. (2021) 23:iii1–iii105. doi: 10.1093/neuonc/noab200
2. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol. (2020) 22:iv1–iv96. doi: 10.1093/neuonc/noaa200
3. Tamimi AF, Juweid M. "Glioblastoma: Epidemiology and Outcome of Glioblastoma," In: De Vleeschouwer S, editor. Glioblastoma, Brisbane (AU (2017).
4. Perry JR, Laperriere N, O’Callaghan CJ, Brandes AA, Menten J, Phillips C, et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N Engl J Med. (2017) 376:1027–37. doi: 10.1056/NEJMoa1611977
5. Fan X, Xiong Y, Wang Y. A reignited debate over the cell(s) of origin for glioblastoma and its clinical implications. Front Med. (2019) 13:531–9. doi: 10.1007/s11684-019-0700-1
6. Gaillard F GJ, Sharma R, et al. Glioblastoma, IDH-wildtype (2008). Available online at: https://radiopaedia.org/articles/glioblastoma-idh-wildtype?lang=us.
7. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
8. Farin A, Suzuki SO, Weiker M, Goldman JE, Bruce JN, Canoll P. Transplanted glioma cells migrate and proliferate on host brain vasculature: a dynamic analysis. Glia. (2006) 53:799–808. doi: 10.1002/glia.v53:8
9. Chang K, Zhang B, Guo X, Zong M, Rahman R, Sanchez D, et al. Multimodal imaging patterns predict survival in recurrent glioblastoma patients treated with bevacizumab. Neuro Oncol. (2016) 18:1680–7. doi: 10.1093/neuonc/now086
10. Stupp R, Brada M, van den Bent MJ, Tonn JC, Pentheroudakis G, Group EGW. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2014) 25 Suppl 3:iii93–101. doi: 10.1093/annonc/mdu050
11. Hsu E, Keene D, Ventureyra E, Matzinger MA, Jimenez C, Wang HS, et al. Bone marrow metastasis in astrocytic gliomata. J Neurooncol. (1998) 37:285–93. doi: 10.1023/A:1005909127196
12. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. (2013) 155:462–77.
13. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. (2016) 164:550–63.
14. Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. (2007) 7:733–6. doi: 10.1038/nrc2246
15. Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. (2006) 6:425–36. doi: 10.1038/nrc1889
16. Hambardzumyan D, Bergers G. Glioblastoma: defining tumor niches. Trends Cancer. (2015) 1:252–65. doi: 10.1016/j.trecan.2015.10.009
17. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science. (2013) 339:1546–58. doi: 10.1126/science.1235122
18. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
19. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with Malignant glioma. Cancer Res. (2006) 66:3294–302. doi: 10.1158/0008-5472.CAN-05-3773
20. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. (2018) 24:4175–86. doi: 10.1158/1078-0432.CCR-17-1846
21. De Vleeschouwer S, Bergers G. "Glioblastoma: To Target the Tumor Cell or the Microenvironment?," In: De Vleeschouwer S, editor. Glioblastoma, Brisbane (AU (2017).
22. Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. (2018) 24:1459–68. doi: 10.1038/s41591-018-0135-2
23. Andaloussi AE, Han Y, Lesniak MS. Progression of intracranial glioma disrupts thymic homeostasis and induces T-cell apoptosis in vivo. Cancer Immunol Immunother. (2008) 57:1807–16. doi: 10.1007/s00262-008-0508-3
24. Himes BT, Geiger PA, Ayasoufi K, Bhargav AG, Brown DA, Parney IF. Immunosuppression in glioblastoma: current understanding and therapeutic implications. Front Oncol. (2021) 11:770561. doi: 10.3389/fonc.2021.770561
25. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. (2015) 17:1064–75. doi: 10.1093/neuonc/nou307
26. Lombardo SD, Bramanti A, Ciurleo R, Basile MS, Pennisi M, Bella R, et al. Profiling of inhibitory immune checkpoints in glioblastoma: Potential pathogenetic players. Oncol Lett. (2020) 20:332. doi: 10.3892/ol
27. Parney IF, Waldron JS, Parsa AT. Flow cytometry and in vitro analysis of human glioma-associated macrophages. Laboratory investigation. J Neurosurg. (2009) 110:572–82. doi: 10.3171/2008.7.JNS08475
28. Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes tumor growth. Clin Cancer Res. (2019) 25:3643–57. doi: 10.1158/1078-0432.CCR-18-2402
29. Bloch O, Lim M, Sughrue ME, Komotar RJ, Abrahams JM, O’Rourke DM, et al. Autologous heat shock protein peptide vaccination for newly diagnosed glioblastoma: impact of peripheral PD-L1 expression on response to therapy. Clin Cancer Res. (2017) 23:3575–84. doi: 10.1158/1078-0432.CCR-16-1369
30. Filippone A, Lanza M, Mannino D, Raciti G, Colarossi C, Sciacca D, et al. PD1/PD-L1 immune checkpoint as a potential target for preventing brain tumor progression. Cancer Immunol Immunother. (2022) 71:2067–75. doi: 10.1007/s00262-021-03130-z
31. Pang L, Dunterman M, Guo S, Khan F, Liu Y, Taefi E, et al. Kunitz-type protease inhibitor TFPI2 remodels stemness and immunosuppressive tumor microenvironment in glioblastoma. Nat Immunol. (2023) 24:1654–70. doi: 10.1038/s41590-023-01605-y
32. Pang L, Guo S, Khan F, Dunterman M, Ali H, Liu Y, et al. Hypoxia-driven protease legumain promotes immunosuppression in glioblastoma. Cell Rep Med. (2023) 4:101238. doi: 10.1016/j.xcrm.2023.101238
33. Erickson MA, Banks WA. Neuroimmune axes of the blood-brain barriers and blood-brain interfaces: bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol Rev. (2018) 70:278–314. doi: 10.1124/pr.117.014647
34. Morimoto T, Nakazawa T, Maeoka R, Nakagawa I, Tsujimura T, Matsuda R. Natural killer cell-based immunotherapy against glioblastoma. Int J Mol Sci. (2023) 24(3):2111. doi: 10.3390/ijms24032111
35. Larsen JM, Martin DR, Byrne ME. Recent advances in delivery through the blood-brain barrier. Curr Top Med Chem. (2014) 14:1148–60. doi: 10.2174/1568026614666140329230311
36. Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. (2012) 33:579–89. doi: 10.1016/j.it.2012.07.004
37. Candolfi M, Curtin JF, Yagiz K, Assi H, Wibowo MK, Alzadeh GE, et al. B cells are critical to T-cell-mediated antitumor immunity induced by a combined immune-stimulatory/conditionally cytotoxic therapy for glioblastoma. Neoplasia. (2011) 13:947–60. doi: 10.1593/neo.11024
38. Fernandez C, Buyse M, German-Fattal M, Gimenez F. Influence of the pro-inflammatory cytokines on P-glycoprotein expression and functionality. J Pharm Pharm Sci. (2004) 7:359–71.
39. Roberts DJ, Goralski KB. A critical overview of the influence of inflammation and infection on P-glycoprotein expression and activity in the brain. Expert Opin Drug Metab Toxicol. (2008) 4:1245–64. doi: 10.1517/17425255.4.10.1245
40. Red-Horse K, Crawford Y, Shojaei F, Ferrara N. Endothelium-microenvironment interactions in the developing embryo and in the adult. Dev Cell. (2007) 12:181–94. doi: 10.1016/j.devcel.2007.01.013
41. Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. (2008) 13:206–20. doi: 10.1016/j.ccr.2008.01.034
42. Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. (2013) 153:139–52. doi: 10.1016/j.cell.2013.02.021
43. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. (2010) 468:829–33. doi: 10.1038/nature09624
44. Le Guelte A, Dwyer J, Gavard J. Jumping the barrier: VE-cadherin, VEGF and other angiogenic modifiers in cancer. Biol Cell. (2011) 103:593–605. doi: 10.1042/BC20110069
45. Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. (2015) 7:a020412. doi: 10.1101/cshperspect.a020412
46. Chen P, Hsu WH, Chang A, Tan Z, Lan Z, Zhou A, et al. Circadian regulator CLOCK recruits immune-suppressive microglia into the GBM tumor microenvironment. Cancer Discovery. (2020) 10:371–81. doi: 10.1158/2159-8290.CD-19-0400
47. Xuan W, Hsu WH, Khan F, Dunterman M, Pang L, Wainwright DA, et al. Circadian regulator CLOCK drives immunosuppression in glioblastoma. Cancer Immunol Res. (2022) 10:770–84. doi: 10.1158/2326-6066.CIR-21-0559
48. Xuan W, Khan F, James CD, Heimberger AB, Lesniak MS, Chen P. Circadian regulation of cancer cell and tumor microenvironment crosstalk. Trends Cell Biol. (2021) 31:940–50. doi: 10.1016/j.tcb.2021.06.008
49. Pang L, Dunterman M, Xuan W, Gonzalez A, Lin Y, Hsu WH, et al. Circadian regulator CLOCK promotes tumor angiogenesis in glioblastoma. Cell Rep. (2023) 42:112127. doi: 10.1016/j.celrep.2023.112127
50. Beppu T, Kamada K, Yoshida Y, Arai H, Ogasawara K, Ogawa A. Change of oxygen pressure in glioblastoma tissue under various conditions. J Neurooncol. (2002) 58:47–52. doi: 10.1023/A:1015832726054
51. Colwell N, Larion M, Giles AJ, Seldomridge AN, Sizdahkhani S, Gilbert MR, et al. Hypoxia in the glioblastoma microenvironment: shaping the phenotype of cancer stem-like cells. Neuro Oncol. (2017) 19:887–96. doi: 10.1093/neuonc/now258
52. Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A. (2015) 112:851–6. doi: 10.1073/pnas.1320611111
53. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. (2014) 344:1396–401. doi: 10.1126/science.1254257
54. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. (2006) 444:756–60. doi: 10.1038/nature05236
55. Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. (2009) 28:3949–59. doi: 10.1038/onc.2009.252
56. Park DM, Li J, Okamoto H, Akeju O, Kim SH, Lubensky I, et al. N-CoR pathway targeting induces glioblastoma derived cancer stem cell differentiation. Cell Cycle. (2007) 6:467–70. doi: 10.4161/cc.6.4.3856
57. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. (2013) 14:1173–82. doi: 10.1038/ni.2714
58. Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. (2010) 16:800–13. doi: 10.1158/1078-0432.CCR-09-2730
59. Gray GK, McFarland BC, Nozell SE, Benveniste EN. NF-kappaB and STAT3 in glioblastoma: therapeutic targets coming of age. Expert Rev Neurother. (2014) 14:1293–306. doi: 10.1586/14737175.2014.964211
60. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. (2009) 9:609–17. doi: 10.1038/nri2607
61. DeMaria PJ, Bilusic M. Cancer vaccines. Hematol Oncol Clin North Am. (2019) 33:199–214. doi: 10.1016/j.hoc.2018.12.001
62. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. (2019) 4:7. doi: 10.1038/s41541-019-0103-y
63. Lin YJ, Mashouf LA, Lim M. CAR T cell therapy in primary brain tumors: current investigations and the future. Front Immunol. (2022) 13:817296. doi: 10.3389/fimmu.2022.817296
64. Lampson LA. Monoclonal antibodies in neuro-oncology: Getting past the blood-brain barrier. MAbs. (2011) 3:153–60. doi: 10.4161/mabs.3.2.14239
65. Ghajar-Rahimi G, Kang KD, Totsch SK, Gary S, Rocco A, Blitz S, et al. Clinical advances in oncolytic virotherapy for pediatric brain tumors. Pharmacol Ther. (2022) 239:108193. doi: 10.1016/j.pharmthera.2022.108193
66. Li J, Meng Q, Zhou X, Zhao H, Wang K, Niu H, et al. Gospel of Malignant Glioma: Oncolytic virus therapy. Gene. (2022) 818:146217. doi: 10.1016/j.gene.2022.146217
67. Ghouzlani A, Kandoussi S, Tall M, Reddy KP, Rafii S, Badou A. Immune checkpoint inhibitors in human glioma microenvironment. Front Immunol. (2021) 12:679425. doi: 10.3389/fimmu.2021.679425
68. Guidon AC, Burton LB, Chwalisz BK, Hillis J, Schaller TH, Amato AA, et al. Consensus disease definitions for neurologic immune-related adverse events of immune checkpoint inhibitors. J Immunother Cancer. (2021) 9(7):e002890. doi: 10.1136/jitc-2021-002890corr1
69. Mukherji B, Chakraborty NG, Yamasaki S, Okino T, Yamase H, Sporn JR, et al. Induction of antigen-specific cytolytic T cells in situ in human melanoma by immunization with synthetic peptide-pulsed autologous antigen presenting cells. Proc Natl Acad Sci U S A. (1995) 92:8078–82. doi: 10.1073/pnas.92.17.8078
70. Winograd EK, Ciesielski MJ, Fenstermaker RA. Novel vaccines for glioblastoma: clinical update and perspective. Immunotherapy. (2016) 8:1293–308. doi: 10.2217/imt-2016-0059
71. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. (2017) 18:1373–85.
72. Binder DC, Ladomersky E, Lenzen A, Zhai L, Lauing KL, Otto-Meyer SD, et al. Lessons learned from rindopepimut treatment in patients with EGFRvIII-expressing glioblastoma. Transl Cancer Res. (2018) 7:S510–S3. doi: 10.21037/tcr
73. Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol. (2014) 16:274–9. doi: 10.1093/neuonc/not203
74. Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol. (2004) 22:2865–72. doi: 10.1200/JCO.2004.12.149
75. Schouten LJ, Rutten J, Huveneers HA, Twijnstra A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer. (2002) 94:2698–705. doi: 10.1002/cncr.10541
76. Cagney DN, Martin AM, Catalano PJ, Redig AJ, Lin NU, Lee EQ, et al. Incidence and prognosis of patients with brain metastases at diagnosis of systemic Malignancy: a population-based study. Neuro Oncol. (2017) 19:1511–21. doi: 10.1093/neuonc/nox077
77. Brahm CG, van Linde ME, Enting RH, Schuur M, Otten RHJ, Heymans MW, et al. The current status of immune checkpoint inhibitors in neuro-oncology: A systematic review. Cancers (Basel). (2020) 12(3):586. doi: 10.3390/cancers12030586
78. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkMate 143 phase 3 randomized clinical trial. JAMA Oncol. (2020) 6:1003–10. doi: 10.1001/jamaoncol.2020.1024
79. Omuro A, Reardon DA, Sampson JH, Baehring J, Sahebjam S, Cloughesy TF, et al. Nivolumab plus radiotherapy with or without temozolomide in newly diagnosed glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neurooncol Adv. (2022) 4:vdac025. doi: 10.1093/noajnl/vdac025
80. Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. (2022) 24:1935–49. doi: 10.1093/neuonc/noac116
81. Maggs L, Cattaneo G, Dal AE, Moghaddam AS, Ferrone S. CAR T cell-based immunotherapy for the treatment of glioblastoma. Front Neurosci. (2021) 15:662064. doi: 10.3389/fnins.2021.662064
82. Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. (2014) 16:1304–12. doi: 10.1093/neuonc/nou045
83. Dubinski D, Wolfer J, Hasselblatt M, Schneider-Hohendorf T, Bogdahn U, Stummer W, et al. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol. (2016) 18:807–18. doi: 10.1093/neuonc/nov280
84. Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC, et al. Glioma IL13Ralpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PloS One. (2013) 8:e77769. doi: 10.1371/journal.pone.0077769
85. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48.
86. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
87. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44.
88. Ricklefs FL, Alayo Q, Krenzlin H, Mahmoud AB, Speranza MC, Nakashima H, et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci Adv. (2018) 4:eaar2766. doi: 10.1126/sciadv.aar2766
89. Hao C, Parney IF, Roa WH, Turner J, Petruk KC, Ramsay DA. Cytokine and cytokine receptor mRNA expression in human glioblastomas: evidence of Th1, Th2 and Th3 cytokine dysregulation. Acta Neuropathol. (2002) 103:171–8. doi: 10.1007/s004010100448
90. Choi BD, Maus MV, June CH, Sampson JH. Immunotherapy for glioblastoma: adoptive T-cell strategies. Clin Cancer Res. (2019) 25:2042–8. doi: 10.1158/1078-0432.CCR-18-1625
91. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. (2006) 8:261–79. doi: 10.1215/15228517-2006-008
92. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA. (2017) 318:2306–16. doi: 10.1001/jama.2017.18718
93. Rocha Pinheiro SL, Lemos FFB, Marques HS, Silva Luz M, de Oliveira Silva LG, Faria Souza Mendes Dos Santos C, et al. Immunotherapy in glioblastoma treatment: Current state and future prospects. World J Clin Oncol. (2023) 14:138–59. doi: 10.5306/wjco.v14.i4.138
94. Weiss T, Puca E, Silginer M, Hemmerle T, Pazahr S, Bink A, et al. Immunocytokines are a promising immunotherapeutic approach against glioblastoma. Sci Transl Med. (2020) 12(564):eabb2311. doi: 10.1126/scitranslmed.abb2311
95. Agliardi G, Liuzzi AR, Hotblack A, De Feo D, Nunez N, Stowe CL, et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat Commun. (2021) 12:444. doi: 10.1038/s41467-020-20599-x
96. Enderlin M, Kleinmann EV, Struyf S, Buracchi C, Vecchi A, Kinscherf R, et al. TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther. (2009) 16:149–60. doi: 10.1038/cgt.2008.62
97. Feinberg B, Kurzrock R, Talpaz M, Blick M, Saks S, Gutterman JU. A phase I trial of intravenously-administered recombinant tumor necrosis factor-alpha in cancer patients. J Clin Oncol. (1988) 6:1328–34. doi: 10.1200/JCO.1988.6.8.1328
98. Kang S, Narazaki M, Metwally H, Kishimoto T. Historical overview of the interleukin-6 family cytokine. J Exp Med. (2020) 217(5):e20190347. doi: 10.1084/jem.20190347
99. Rose-John S. Interleukin-6 family cytokines. Cold Spring Harb Perspect Biol. (2018) 10(2):a028415. doi: 10.1101/cshperspect.a028415
100. Zaporowska-Stachowiak I, Springer M, Stachowiak K, Oduah M, Sopata M, Wieczorowska-Tobis K, et al. Interleukin-6 family of cytokines in cancers. J Interferon Cytokine Res. (2024) 44:45–59. doi: 10.1089/jir.2023.0103
101. Wang Q, He Z, Huang M, Liu T, Wang Y, Xu H, et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2alpha. Nat Commun. (2018) 9:559. doi: 10.1038/s41467-018-03050-0
102. Chang CY, Li MC, Liao SL, Huang YL, Shen CC, Pan HC. Prognostic and clinical implication of IL-6 expression in glioblastoma multiforme. J Clin Neurosci. (2005) 12:930–3. doi: 10.1016/j.jocn.2004.11.017
103. Hori T, Sasayama T, Tanaka K, Koma YI, Nishihara M, Tanaka H, et al. Tumor-associated macrophage related interleukin-6 in cerebrospinal fluid as a prognostic marker for glioblastoma. J Clin Neurosci. (2019) 68:281–9. doi: 10.1016/j.jocn.2019.07.020
104. Chen HY, Lin LT, Wang ML, Lee SH, Tsai ML, Tsai CC, et al. Musashi-1 regulates AKT-derived IL-6 autocrinal/paracrinal Malignancy and chemoresistance in glioblastoma. Oncotarget. (2016) 7:42485–501. doi: 10.18632/oncotarget.v7i27
105. Pasi F, Facoetti A, Nano R. IL-8 and IL-6 bystander signalling in human glioblastoma cells exposed to gamma radiation. Anticancer Res. (2010) 30:2769–72.
106. Li R, Li G, Deng L, Liu Q, Dai J, Shen J, et al. IL-6 augments the invasiveness of U87MG human glioblastoma multiforme cells via up-regulation of MMP-2 and fascin-1. Oncol Rep. (2010) 23:1553–9. doi: 10.3892/or
107. Mahmoud AB, Ajina R, Aref S, Darwish M, Alsayb M, Taher M, et al. Advances in immunotherapy for glioblastoma multiforme. Front Immunol. (2022) 13:944452. doi: 10.3389/fimmu.2022.944452
108. Foreman PM, Friedman GK, Cassady KA, Markert JM. Oncolytic virotherapy for the treatment of Malignant glioma. Neurotherapeutics. (2017) 14:333–44. doi: 10.1007/s13311-017-0516-0
109. Peruzzi P, Chiocca EA. Viruses in cancer therapy - from benchwarmers to quarterbacks. Nat Rev Clin Oncol. (2018) 15:657–8. doi: 10.1038/s41571-018-0077-0
110. Hu D, Tian Y, Xu J, Xie D, Wang Y, Liu M, et al. Oncolytic viral therapy as promising immunotherapy against glioma. MedComm – Future Med. (2023) 2:e61. doi: 10.1002/mef2.61
111. Lin D, Shen Y, Liang T. Oncolytic virotherapy: basic principles, recent advances and future directions. Signal Transduct Target Ther. (2023) 8:156. doi: 10.1038/s41392-023-01407-6
112. Hamad A, Yusubalieva GM, Baklaushev VP, Chumakov PM, Lipatova AV. Recent developments in glioblastoma therapy: oncolytic viruses and emerging future strategies. Viruses. (2023) 15(2):547. doi: 10.3390/v15020547
113. Mehta AM, Sonabend AM, Bruce JN. Convection-enhanced delivery. Neurotherapeutics. (2017) 14:358–71. doi: 10.1007/s13311-017-0520-4
114. Tseng JC, Levin B, Hirano T, Yee H, Pampeno C, Meruelo D. In vivo antitumor activity of Sindbis viral vectors. J Natl Cancer Inst. (2002) 94:1790–802. doi: 10.1093/jnci/94.23.1790
115. Zrachia A, Dobroslav M, Blass M, Kazimirsky G, Kronfeld I, Blumberg PM, et al. Infection of glioma cells with Sindbis virus induces selective activation and tyrosine phosphorylation of protein kinase C delta. Implications for Sindbis virus-induced apoptosis. J Biol Chem. (2002) 277:23693–701. doi: 10.1074/jbc.M111658200
116. Ramachandran M, Yu D, Dyczynski M, Baskaran S, Zhang L, Lulla A, et al. Safe and effective treatment of experimental neuroblastoma and glioblastoma using systemically delivered triple microRNA-detargeted oncolytic semliki forest virus. Clin Cancer Res. (2017) 23:1519–30. doi: 10.1158/1078-0432.CCR-16-0925
117. Liu Z, Zhao X, Mao H, Baxter PA, Huang Y, Yu L, et al. Intravenous injection of oncolytic picornavirus SVV-001 prolongs animal survival in a panel of primary tumor-based orthotopic xenograft mouse models of pediatric glioma. Neuro Oncol. (2013) 15:1173–85. doi: 10.1093/neuonc/not065
118. Lubin JA, Zhang RR, Kuo JS. Zika virus has oncolytic activity against glioblastoma stem cells. Neurosurgery. (2018) 82:E113–E4. doi: 10.1093/neuros/nyy047
119. Azambuja JH, Gelsleichter NE, Beckenkamp LR, Iser IC, Fernandes MC, Figueiro F, et al. CD73 downregulation decreases in vitro and in vivo glioblastoma growth. Mol Neurobiol. (2019) 56:3260–79. doi: 10.1007/s12035-018-1240-4
120. Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med. (2020) 26:39–46. doi: 10.1038/s41591-019-0694-x
121. Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. (2017) 32:253–67.e5. doi: 10.1016/j.ccell.2017.07.006
122. Dohlman AB, Klug J, Mesko M, Gao IH, Lipkin SM, Shen X, et al. A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors. Cell. (2022) 185:3807–22.e12.
123. Li X, Saxena D. The tumor mycobiome: A paradigm shift in cancer pathogenesis. Cell. (2022) 185:3648–51. doi: 10.1016/j.cell.2022.09.013
124. Wu J, Zhang P, Mei W, Zeng C. Intratumoral microbiota: implications for cancer onset, progression, and therapy. Front Immunol. (2023) 14:1301506. doi: 10.3389/fimmu.2023.1301506
125. Goto Y, Iwata S, Miyahara M, Miyako E. Discovery of intratumoral oncolytic bacteria toward targeted anticancer theranostics. Adv Sci (Weinh). (2023) 10:e2301679. doi: 10.1002/advs.202301679
126. Chen YE, Bousbaine D, Veinbachs A, Atabakhsh K, Dimas A, Yu VK, et al. Engineered skin bacteria induce antitumor T cell responses against melanoma. Science. (2023) 380:203–10. doi: 10.1126/science.abp9563
127. Naghavian R, Faigle W, Oldrati P, Wang J, Toussaint NC, Qiu Y, et al. Microbial peptides activate tumour-infiltrating lymphocytes in glioblastoma. Nature. (2023) 617:807–17. doi: 10.1038/s41586-023-06081-w
128. Zitvogel L, Kroemer G. Cross-reactivity between cancer and microbial antigens. Oncoimmunology. (2021) 10:1877416. doi: 10.1080/2162402X.2021.1877416
129. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. (2020) 368:973–80.
130. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
131. Xiong Z, Raphael I, Olin M, Okada H, Li X, Kohanbash G. Glioblastoma vaccines: past, present, and opportunities. EBioMedicine. (2024) 100:104963. doi: 10.1016/j.ebiom.2023.104963
132. Wang MM, Coupland SE, Aittokallio T, Figueiredo CR. Resistance to immune checkpoint therapies by tumour-induced T-cell desertification and exclusion: key mechanisms, prognostication and new therapeutic opportunities. Br J Cancer. (2023) 129:1212–24. doi: 10.1038/s41416-023-02361-4
133. Terry S, Faouzi Zaarour R, Hassan Venkatesh G, Francis A, El-Sayed W, Buart S, et al. Role of hypoxic stress in regulating tumor immunogenicity, resistance and plasticity. Int J Mol Sci. (2018) 19(10):3044. doi: 10.3390/ijms19103044
134. Hunter FW, Wouters BG, Wilson WR. Hypoxia-activated prodrugs: paths forward in the era of personalised medicine. Br J Cancer. (2016) 114:1071–7. doi: 10.1038/bjc.2016.79
135. Doherty N, Hancock SL, Kaye S, Coleman CN, Shulman L, Marquez C, et al. Muscle cramping in phase I clinical trials of tirapazamine (SR 4233) with and without radiation. Int J Radiat Oncol Biol Phys. (1994) 29:379–82. doi: 10.1016/0360-3016(94)90293-3
136. Reddy SB, Williamson SK. Tirapazamine: a novel agent targeting hypoxic tumor cells. Expert Opin Investig Drugs. (2009) 18:77–87. doi: 10.1517/13543780802567250
137. Hendricksen K, Cornel EB, de Reijke TM, Arentsen HC, Chawla S, Witjes JA. Phase 2 study of adjuvant intravesical instillations of apaziquone for high risk nonmuscle invasive bladder cancer. J Urol. (2012) 187:1195–9. doi: 10.1016/j.juro.2011.11.101
138. Hicks KO, Pruijn FB, Secomb TW, Hay MP, Hsu R, Brown JM, et al. Use of three-dimensional tissue cultures to model extravascular transport and predict in vivo activity of hypoxia-targeted anticancer drugs. J Natl Cancer Inst. (2006) 98:1118–28. doi: 10.1093/jnci/djj306
139. Noman MZ, Hasmim M, Lequeux A, Xiao M, Duhem C, Chouaib S, et al. Improving cancer immunotherapy by targeting the hypoxic tumor microenvironment: new opportunities and challenges. Cells. (2019) 8(9):1083. doi: 10.3390/cells8091083
140. Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest. (2018) 128:5137–49. doi: 10.1172/JCI96268
141. Hegde A, Jayaprakash P, Couillault CA, Piha-Paul S, Karp D, Rodon J, et al. A phase I dose-escalation study to evaluate the safety and tolerability of evofosfamide in combination with ipilimumab in advanced solid Malignancies. Clin Cancer Res. (2021) 27:3050–60. doi: 10.1158/1078-0432.CCR-20-4118
142. Chawla SP, Cranmer LD, Van Tine BA, Reed DR, Okuno SH, Butrynski JE, et al. Phase II study of the safety and antitumor activity of the hypoxia-activated prodrug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcoma. J Clin Oncol. (2014) 32:3299–306. doi: 10.1200/JCO.2013.54.3660
143. Tap WD, Papai Z, Van Tine BA, Attia S, Ganjoo KN, Jones RL, et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial. Lancet Oncol. (2017) 18:1089–103. doi: 10.1016/S1470-2045(17)30381-9
144. Brenner AJ, Floyd J, Fichtel L, Michalek J, Kanakia KP, Huang S, et al. Phase 2 trial of hypoxia activated evofosfamide (TH302) for treatment of recurrent bevacizumab-refractory glioblastoma. Sci Rep. (2021) 11:2306. doi: 10.1038/s41598-021-81841-0
145. Bhattarai D, Xu X, Lee K. Hypoxia-inducible factor-1 (HIF-1) inhibitors from the last decade (2007 to 2016): A “structure-activity relationship” perspective. Med Res Rev. (2018) 38:1404–42. doi: 10.1002/med.21477
146. Parayath N, Padmakumar S, Nair SV, Menon D, Amiji MM. Strategies for targeting cancer immunotherapy through modulation of the tumor microenvironment. Regenerative Eng Trans Med. (2020) 6:29–49. doi: 10.1007/s40883-019-00113-6
147. Sun X, Kanwar JR, Leung E, Lehnert K, Wang D, Krissansen GW. Gene transfer of antisense hypoxia inducible factor-1 alpha enhances the therapeutic efficacy of cancer immunotherapy. Gene Ther. (2001) 8:638–45. doi: 10.1038/sj.gt.3301388
148. Kheshtchin N, Arab S, Ajami M, Mirzaei R, Ashourpour M, Mousavi N, et al. Inhibition of HIF-1alpha enhances anti-tumor effects of dendritic cell-based vaccination in a mouse model of breast cancer. Cancer Immunol Immunother. (2016) 65:1159–67. doi: 10.1007/s00262-016-1879-5
149. Chiu DK, Xu IM, Lai RK, Tse AP, Wei LL, Koh HY, et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology. (2016) 64:797–813. doi: 10.1002/hep.28655
150. Mellatyar H, Asadi A, Akbarzadeh A, Sheervalilou R, Zarghami N. Spotlight on 17-AAG as an Hsp90 inhibitor for molecular targeted cancer treatment. Chem Biol Drug Des. (2019) 93:760–86.
151. Wang Y, Hou Y, Hou L, Wang W, Li K, Zhang Z, et al. Digoxin exerts anticancer activity on human nonsmall cell lung cancer cells by blocking PI3K/Akt pathway. Biosci Rep. (2021) 41(10):BSR20211056. doi: 10.1042/BSR20211056
152. Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. (2003) 3:363–75. doi: 10.1016/S1535-6108(03)00077-1
153. Semenza GL. Pharmacologic targeting of hypoxia-inducible factors. Annu Rev Pharmacol Toxicol. (2019) 59:379–403. doi: 10.1146/annurev-pharmtox-010818-021637
154. Wigerup C, Pahlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. (2016) 164:152–69. doi: 10.1016/j.pharmthera.2016.04.009
155. Courtney KD, Infante JR, Lam ET, Figlin RA, Rini BI, Brugarolas J, et al. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2alpha antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J Clin Oncol. (2018) 36:867–74. doi: 10.1200/JCO.2017.74.2627
156. Strowd R, Ellingson B, Raymond C, Yao J, Wen PY, Ahluwalia M, et al. Activity of a first-in-class oral HIF2-alpha inhibitor, PT2385, in patients with first recurrence of glioblastoma. J Neurooncol. (2023) 165:101–12. doi: 10.1007/s11060-023-04456-7
157. Vilaplana-Lopera N, Besh M, Moon EJ. Targeting hypoxia: revival of old remedies. Biomolecules. (2021) 11(11):1604. doi: 10.3390/biom11111604
158. Choueiri TK, Albiges L, Fan L, Perini RF, Zojwalla NJ, Powles T, et al. Phase III study of the hypoxia-inducible factor 2α (HIF-2α) inhibitor MK-6482 versus everolimus in previously treated patients with advanced clear cell renal cell carcinoma (ccRCC). J Clin Oncol. (2020) 38:TPS5094–TPS. doi: 10.1200/JCO.2020.38.15_suppl.TPS5094
159. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. (2005) 330:1304–5. doi: 10.1136/bmj.38415.708634.F7
160. Koritzinsky M. Metformin: A novel biological modifier of tumor response to radiation therapy. Int J Radiat Oncol Biol Phys. (2015) 93:454–64. doi: 10.1016/j.ijrobp.2015.06.003
161. Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res. (2013) 19:6741–50. doi: 10.1158/1078-0432.CCR-13-1787
162. Skinner H, Hu C, Tsakiridis T, Santana-Davila R, Lu B, Erasmus JJ, et al. Addition of metformin to concurrent chemoradiation in patients with locally advanced non-small cell lung cancer: the NRG-LU001 phase 2 randomized clinical trial. JAMA Oncol. (2021) 7:1324–32. doi: 10.1001/jamaoncol.2021.2318
163. Tsakiridis T, Pond GR, Wright J, Ellis PM, Ahmed N, Abdulkarim B, et al. Metformin in combination with chemoradiotherapy in locally advanced non-small cell lung cancer: the OCOG-ALMERA randomized clinical trial. JAMA Oncol. (2021) 7:1333–41. doi: 10.1001/jamaoncol.2021.2328
164. Lord SR, Harris AL. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic? Br J Cancer. (2023) 128:958–66. doi: 10.1038/s41416-023-02204-2
165. Chang WH, Lai AG. The hypoxic tumour microenvironment: A safe haven for immunosuppressive cells and a therapeutic barrier to overcome. Cancer Lett. (2020) 487:34–44. doi: 10.1016/j.canlet.2020.05.011
166. Chafe SC, McDonald PC, Saberi S, Nemirovsky O, Venkateswaran G, Burugu S, et al. Targeting hypoxia-induced carbonic anhydrase IX enhances immune-checkpoint blockade locally and systemically. Cancer Immunol Res. (2019) 7:1064–78. doi: 10.1158/2326-6066.CIR-18-0657
167. Campos NSP, Souza BS, Silva G, Porto VA, Chalbatani GM, Lagreca G, et al. Carbonic anhydrase IX: A renewed target for cancer immunotherapy. Cancers (Basel). (2022) 14(6):1392. doi: 10.3390/cancers14061392
168. Wang B, Zhao Q, Zhang Y, Liu Z, Zheng Z, Liu S, et al. Targeting hypoxia in the tumor microenvironment: a potential strategy to improve cancer immunotherapy. J Exp Clin Cancer Res. (2021) 40:24. doi: 10.1186/s13046-020-01820-7
169. Ye H, Zhao J, Xu X, Zhang D, Shen H, Wang S. Role of adenosine A2a receptor in cancers and autoimmune diseases. Immun Inflammation Dis. (2023) 11:e826. doi: 10.1002/iid3.826
170. Ma SR, Deng WW, Liu JF, Mao L, Yu GT, Bu LL, et al. Blockade of adenosine A2A receptor enhances CD8(+) T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol Cancer. (2017) 16:99. doi: 10.1186/s12943-017-0665-0
171. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. (2017) 127:929–41. doi: 10.1172/JCI89455
172. Giatromanolaki A, Kouroupi M, Pouliliou S, Mitrakas A, Hasan F, Pappa A, et al. Ectonucleotidase CD73 and CD39 expression in non-small cell lung cancer relates to hypoxia and immunosuppressive pathways. Life Sci. (2020) 259:118389. doi: 10.1016/j.lfs.2020.118389
173. Liu G, Zhang Q, Liu G, Li D, Zhang L, Gu Z, et al. Disruption of adenosine 2A receptor improves the anti-tumor function of anti-mesothelin CAR T cells both in vitro and in vivo. Exp Cell Res. (2021) 409:112886. doi: 10.1016/j.yexcr.2021.112886
174. Neo SY, Yang Y, Record J, Ma R, Chen X, Chen Z, et al. CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J Clin Invest. (2020) 130:1185–98. doi: 10.1172/JCI128895
175. Khan F, Lin Y, Ali H, Pang L, Dunterman M, Hsu WH, et al. Lactate dehydrogenase A regulates tumor-macrophage symbiosis to promote glioblastoma progression. Nat Commun. (2024) 15:1987. doi: 10.1038/s41467-024-46193-z
176. Khan F, Lin Y, Ali H, Pang L, Dunterman M, Hsu WH, et al. LDHA-regulated tumor-macrophage symbiosis promotes glioblastoma progression. Res Sq. (2023) 5:rs.3.rs-3401154. doi: 10.21203/rs.3.rs-3401154/v1
177. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. (2015) 7:277ra30. doi: 10.1126/scitranslmed.aaa1260
178. Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res. (2017) 5:9–16. doi: 10.1158/2326-6066.CIR-16-0103
179. Bennett MH, Feldmeier J, Smee R, Milross C. Hyperbaric oxygenation for tumour sensitisation to radiotherapy. Cochrane Database Syst Rev. (2018) 4:CD005007. doi: 10.1002/14651858.CD005007.pub4
180. Chaplin DJ, Horsman MR, Siemann DW. Further evaluation of nicotinamide and carbogen as a strategy to reoxygenate hypoxic cells in vivo: importance of nicotinamide dose and pre-irradiation breathing time. Br J Cancer. (1993) 68:269–73. doi: 10.1038/bjc.1993.326
181. Aquino-Parsons C, Hukin J, Green A. Concurrent carbogen and radiation therapy in children with high-risk brainstem gliomas. Pediatr Blood Cancer. (2008) 50:397–9.
182. Haibe Y, Kreidieh M, El Hajj H, Khalifeh I, Mukherji D, Temraz S, et al. Resistance mechanisms to anti-angiogenic therapies in cancer. Front Oncol. (2020) 10:221. doi: 10.3389/fonc.2020.00221
183. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. (2014) 2:632–42. doi: 10.1158/2326-6066.CIR-14-0053
184. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. (2016) 7:12624. doi: 10.1038/ncomms12624
185. Liu Z, Zhao Q, Zheng Z, Liu S, Meng L, Dong L, et al. Vascular normalization in immunotherapy: A promising mechanisms combined with radiotherapy. BioMed Pharmacother. (2021) 139:111607. doi: 10.1016/j.biopha.2021.111607
Keywords: glioblastoma, hypoxia, tumor microenvironment, novel treatments, immunotherapy
Citation: Feldman L (2024) Hypoxia within the glioblastoma tumor microenvironment: a master saboteur of novel treatments. Front. Immunol. 15:1384249. doi: 10.3389/fimmu.2024.1384249
Received: 09 February 2024; Accepted: 10 June 2024;
Published: 26 June 2024.
Edited by:
Bingdong Zhu, Lanzhou University, ChinaReviewed by:
Lipei Shao, National Institutes of Health (NIH), United StatesFatima Khan, Northwestern Medicine, United States
Copyright © 2024 Feldman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Feldman, bGZlbGRtYW5AY29oLm9yZw==