 Shuisen Wan1†
Shuisen Wan1† Hongkun Jiang
Hongkun Jiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol., 25 July 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1376258
Introduction: X-linked agammaglobulinemia (XLA) is a humoral immunodeficiency disorder characterized by recurrent infections, severe hypogammaglobulinemia, and a deficiency of circulating B cells. While the hallmark clinical manifestations of XLA typically include the respiratory, dermatological, and gastrointestinal systems, renal involvement is infrequent. In this article, we report two cases of XLA with concurrent renal disease, supplemented with a review of documented cases.
Case description: The two cases described involve twin brothers, both presenting with respiratory tract infections and renal manifestations. Subsequent genetic testing confirmed the diagnosis of XLA. The younger brother exhibited improvement following intravenous immunoglobulin (IVIG) therapy and anti-infection treatment. Due to financial constraints, the older brother received only anti-infection and symptomatic treatments. Seven months after discharge, the older brother developed nephritis. However, he showed improvement following IVIG treatment.
Conclusion: Immune profiling and genetic testing should be considered in male children with recurrent infections to facilitate the effective diagnosis of XLA. Regular monitoring is also imperative to detect and treat immune-mediated renal diseases in patients with XLA.
Inborn errors of immunity (IEI), previously referred to as primary immunodeficiency diseases (PID), constitute a spectrum of continuously evolving disorders primarily caused by single gene variants that induce alterations in the components and functions of immune cells and molecules. Clinical manifestations include heightened susceptibility to infections, autoimmune diseases, inflammatory conditions, allergies, bone marrow failure, and/or malignancies (1).
X-linked agammaglobulinemia (XLA) is a common primary antibody deficiency (PAD) within the spectrum of IEI, frequently diagnosed in pediatric practice (2). XLA is characterized by the most prevalent genetic anomaly resulting in impaired B cell development due to variants in the Bruton’s tyrosine kinase (BTK) gene, with an incidence estimated between 1:100,000 and 1:200,000 (3).
Key distinguishing features of XLA include increased vulnerability to infections accompanied by severe hypogammaglobulinemia and a deficiency of circulating peripheral blood B cells (B-cell percentage, < 2%). The BTK gene, located on the long arm of the X chromosome (Xq21.3–Xq22), spans a length of 37.5 kb and encodes a protein consisting of 659 amino acids. Comprising 19 exons, the BTK gene is susceptible to pathogenic variants in all structural domains and noncoding regions. The most common types include missense variants, followed by frame-shift variants, splice site variants, and nonsense variants, among others.
BTK, encoded by the BTK gene, belongs to the Tec family of non-receptor tyrosine kinases. BTK deficiency hinders the maturation of pro-B cells into later stages of pre-B cells in the B-cell differentiation pathway (4). Variants in the BTK gene disrupt B-cell development, leading to significantly reduced levels of mature B lymphocytes circulating in the peripheral blood of affected individuals. Consequently, this results in plasma cell generation disorders and a significant decrease across all subsets of B lymphocytes.
Approximately one-third of the cases of XLA demonstrate familial inheritance, while it is believed that new variants account for the remaining two-thirds (5). Although this condition predominantly affects men, women with extremely skewed X chromosome inactivation may exhibit similar clinical manifestations (6). Clinical onset is typically in early childhood, often after six months of age, coinciding with the weakening of maternal antibodies and the inability of affected individuals to produce their own immunoglobulins (7).
Patients with XLA may present with a diverse array of infections, encompassing both upper and lower respiratory tract infections, gastrointestinal infections, as well as invasive infections such as sepsis, meningitis, and osteomyelitis (7–12). While bacterial pathogens are primarily involved, viral and parasitic infections, including enteroviruses and Giardia lamblia, may also manifest (7–12).
Individuals diagnosed with XLA exhibit an elevated susceptibility to developing autoimmune diseases, with up to 15% of individuals experiencing conditions such as inflammatory bowel disease (IBD), autoimmune hemolytic anemia (AIHA), scleroderma, and immune complex-mediated glomerulonephritis (13–16). Additionally, reported cases document the co-occurrence of XLA with tumors (17, 18) and allergic rhinitis (19).
Once the diagnosis is established, immunoglobulin replacement therapy proves efficacious in managing XLA by significantly reducing infection rates and infection-associated complications, thereby improving patient survival rates (7–12).
Patients with XLA have deficiencies in humoral immunity, predisposing them to repeated infections, which in turn may lead to glomerular disease or tubulointerstitial disease in the context of infection and compromised immunity. However, detecting renal manifestations in patients with XLA can sometimes pose challenges as most relevant literature consists of case reports, and this makes it difficult to determine whether these renal findings represent sporadic occurrences or are linked to underlying pathophysiological mechanisms.
In this report, we have presented the clinical details of XLA in twin brothers presenting with renal manifestations, one of whom displayed a tubulointerstitial lesion on renal biopsy. Informed consent for the study was diligently obtained from the patients and their respective family members. Furthermore, we have incorporated a comprehensive review of the existing literature on renal biopsies conducted in patients with XLA to furnish a more detailed description of the clinical and pathological features as well as the prognostic implications associated with concurrent renal involvement.
At the age of 11, the patient presented at another healthcare facility with symptoms of fever, coughing, and headaches. During his hospitalization, he developed gross hematuria, persisting for 16 days, accompanied by a red skin rash on both lower limbs. In order to establish a definitive diagnosis and receive further treatment, the patient was transferred to our medical center.
At the time of admission to our facility, multiple scattered nonpruritic red rashes that did not blanch upon pressure were observed on both lower limbs. Laboratory investigations revealed the following findings: urine analysis revealed that the urine-specific gravity was within the normal range, suggestive of glomerular origin (80% abnormal morphology of red blood cells) and the presence of microscopic hematuria (red blood cell 4/4/HP) as well as significant proteinuria (+++, 1.702 g/24 h). All five parameters for urinary microalbumin were elevated compared to normal levels: β2-microglobulin (β2-MG) was 3.47 mg/L (0–0.23 mg/L), α1-microglobulin (α1-MG) was 58.9 mg/L (0–12 mg/L), microalbuminuria (MA) was 289 mg/L (0–30 mg/L), urinary IgG was 12.3 mg/L (0–9.6 mg/L), and transferrin receptor protein TRU was 13.7 mg/L (0–2.41 mg/L). Peripheral blood test results showed that the white blood cell (WBC) level was 12.16*109/L (4–10*109/L), neutrophil (NE) was 8.19*109/L (1.8–6.3*109/L), hemoglobin (Hb) was 116 g/L (120–140 g/L), platelet (PLT) was 369*109/L (100–300*109/L), serum albumin (ALB) was 38.9 g/L (40–55 g/L), the urea concentration was 8.79 mmol/L (2.85–7.14 mmol/L), creatinine (Cr) was 114 µmol/L (59–104 µmol/L), cystatin C (Cys-C) was 2.28 mg/L (0.53–0.95 mg/L), estimated glomerular filtration rate (eGFR) was 53.80 mL/min/1.73 m2, lactate dehydrogenase (LDH) was 238 U/L (135–225 U/L), C-reactive protein (CRP) at 8.10 mg/L (0–5 mg/L), and procalcitonin (PCT) at 0.09 ng/mL (0–0.05 ng/mL).
Results of the immunological examination revealed that the patient had significantly decreased levels of immunoglobulins (IgG 2.08 g/L, IgA < 0.07 g/L, IgM < 0.04 g/L), a deficiency in B cells (CD19 percentage: 0%, CD19 absolute value: 0/µL), a reduced NK cell count (CD16 + 56 percentage: 3% [5%–27%], CD16 + 56 absolute value: 30/mL [90–590/mL]), and a decrease in the proportion of CD4+ T cells among T cells (CD4/CD8 ratio: 0.80). Other immunological parameters showed no significant abnormalities.
The blood lipid analysis and coagulation profile yielded normal results. A chest CT scan indicated localized bronchial dilation with mild chronic inflammation in the lower lobe of the left lung. A renal ultrasound revealed normal morphology with enhanced cortical echoes. Renal emission computed tomography (ECT) indicated normal blood perfusion but reduced renal parenchymal function and delayed excretion.
Following admission, the child experienced recurrent fever with a maximum temperature of 39.2°C. Meropenem, combined with azithromycin, was intravenously administered for 3 days, after which the symptoms improved significantly. Renal biopsy findings were as follows (Figure 1): light microscopy revealed mild proliferation of mesangial cells and the mesangial matrix, with no obvious intraluminal proliferation. Occasional adhesion of glomerular tufts was observed, along with the presence of one fibrocellular crescent formation showing focal necrosis. Extensive granular and vacuolar degeneration was seen in the renal tubular epithelial cells, accompanied by desquamation and bare basement membrane formation. Multifocal atrophy and luminal narrowing of renal tubules were evident. Red blood cell casts were frequently observed within the tubular lumen. Interstitial edema, infiltration of individual nuclear cells in greater numbers between tubules, and mild fibrosis were also noted.
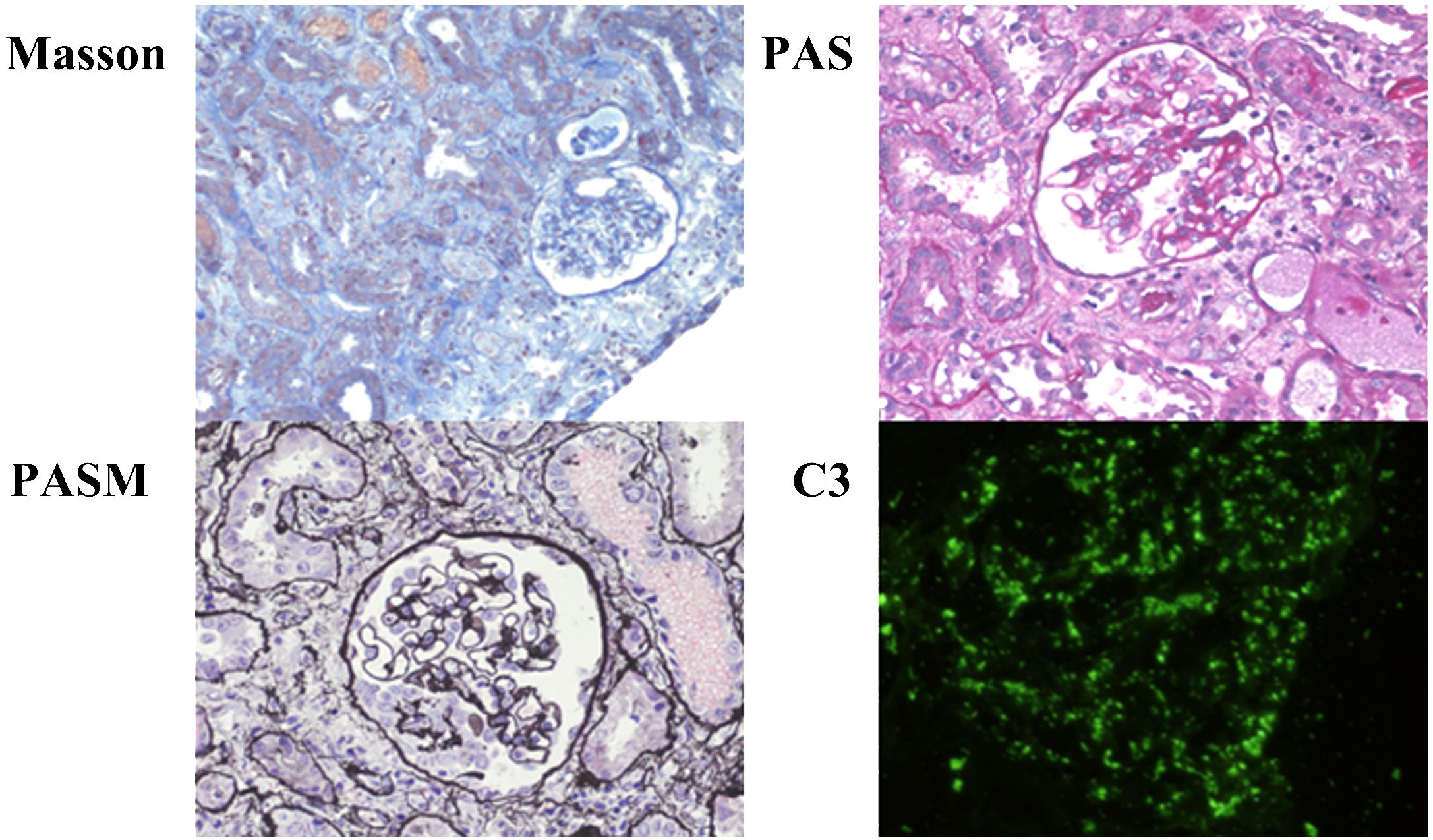
Figure 1 Renal biopsy of patient 1. Microscopic examination of renal biopsy reveals mild proliferation of mesangial cells and mesangial matrix in 16 glomeruli without obvious intraluminal proliferation. Occasional adhesions between glomerular capsules are observed, along with the formation of one-cell fibrous crescents showing focal necrosis. Extensive granular and vacuolar degeneration is observed in renal tubular epithelial cells, accompanied by detachment and exposure of basal membranes. Multi-focal tubular atrophy and luminal narrowing are present. Red blood cell casts frequently appear within the tubules. Interstitial edema is evident, along with the infiltration of numerous mononuclear cells in the intertubular spaces, accompanied by mild fibrosis. Immunofluorescence staining: C3 (+++) deposits appear arranged in a starry sky pattern within the glomeruli, IgA (−), IgM (−), Fib (−), IgG (−), and C1q (−).
Given the patient’s male sex and history of recurrent respiratory tract infections, two complete immunoglobulin tests were performed. The IgG, IgA, and IgM levels were all below 2 standard deviations from the mean values for children of the same age group. CD19+ B count was 0/µL. PID was suspected due to these findings as well as genetic testing results conducted on the patient, his mother, and his brother, revealing a variant in the BTK gene (p.Tyr40Cys) (Figure 2).
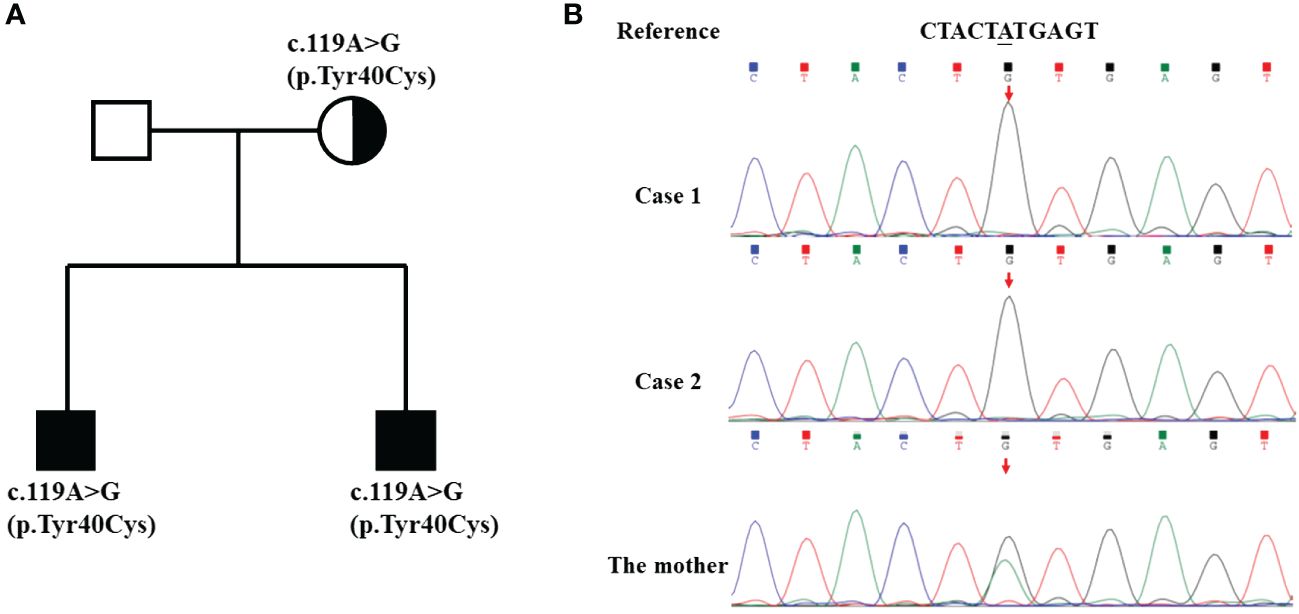
Figure 2 Pedigree and genomic sequence analysis of the BTK gene. (A) The pedigree of the family with XLA. The parents of the twins have no signs of XLA. (B) The c.119A>G mutation was detected in the twins and their mother.
Based on the patient’s medical history and clinical examination findings, the confirmed diagnosis was as follows: (1) X-linked agammaglobulinemia, (2) acute renal injury (pathological diagnosis: C3 starry sky deposition, tubulointerstitial lesion), (3) renal insufficiency (decompensated phase), (4) community-acquired pneumonia with bronchiectasis, and (5) ventricular dilation (sequelae of encephalitis).
Treatment involved the administration of a total intravenous immunoglobulin (IVIG) dosage of 15 g over three consecutive days. Subsequent urine tests revealed negative proteinuria, a red blood cell count of 2/4/HP with abnormal morphology in 70% of cells, and a white blood cell count of 2–8/HP. The patient was discharged from the hospital, and a monthly IVIG at a dosage of 10 g was initiated. During the course of the IVIG treatment, the patient intermittently experienced respiratory infections and exhibited mild proteinuria and microscopic hematuria, which improved following the administration of gamma globulin and anti-inflammatory therapy.
An 11-year-old male patient, the twin brother of case 1, presented with a history of recurrent respiratory infections occurring five to six times per year and consistently recovering after intravenous administration of antibiotics. He was admitted to our department with symptoms including a cough, a runny nose persisting for 3 days, and a fever lasting for 2 days.
Urine analysis revealed the presence of microscopic hematuria (26/HP) and proteinuria (2+). Peripheral blood tests showed elevated WBC at 15.51*109/L, neutrophil (NE#) at 12.14*109/L, lymphocyte (LY#) at 1.50*109/L, HBG at 130 g/L, PLT at 245*109/L, iron level (Fe) at 3.9 µmol/L, CRP at 82.60 mg/L, PCT at 0.11 ng/mL, and IgE at 5.87 IU/mL. Immunological examination revealed significantly decreased levels of immunoglobulins: IgG, 1.53 g/L; IgA, < 0.01 g/L; and IgM, 0.03 g/L. Liver function, renal function, and myocardial enzymes were normal. A lung CT scan indicated scattered inflammation in both lungs.
The patient was administered intravenous cefixime and azithromycin, as well as nebulized budesonide, salbutamol, and interferon therapy. Subsequent to the oral administration of fosinopril sodium, the fever subsided and the cough improved. A urine retest showed +1 proteinuria but no red blood cells under high magnification.
Genetic testing of the patient, his younger brother, and his mother confirmed a diagnosis of X-linked agammaglobulinemia. Based on the medical history and clinical laboratory test results, the following specific diagnoses were made: (1) XLA, (2) acute pneumonia, and (3) acute glomerulonephritis. However, due to financial constraints at that time, the child was unable to receive immunoglobulin treatment.
The child presented with foamy urine and sought further evaluation at another medical facility 7 months postdischarge. Subsequent investigations revealed proteinuria (1.365 g/24 h) and microscopic hematuria (34.05/HP). Renal function tests and the renal ultrasound scan showed no abnormalities. A renal biopsy indicated membranoproliferative glomerulonephritis (image unavailable), characterized by mesangial cell proliferation, increased matrix deposition, and a thickened basement membrane with a double contour appearance under light microscopy. Additionally, immunofluorescence staining revealed C3 and IgG deposits in the mesangium and capillary loops.
The child was treated with antibiotics administered intravenously or orally, along with fosinopril sodium and IVIG therapy, resulting in symptomatic improvement. Due to the family’s financial constraints, only 12.5 g of IVIG was administered during the child’s hospitalization. Upon reevaluation, a routine urine test showed reduced hematuria and proteinuria compared to pretreatment levels. After discharge from the hospital, a monthly IVIG at a dosage of 12.5 g was initiated for the child. He experienced respiratory tract infections one to two times a year.
A comprehensive summary of reported cases of XLA with concurrent renal disease that underwent renal biopsy, including the two cases that we examined, totaling 14 cases, is presented in Table 1 (20–30). This summary includes details such as the age at the time of diagnosis of XLA, age at onset of renal manifestations, initial clinical presentations of renal involvement, relevant laboratory tests, physical examinations, clinical diagnoses, treatments administered, and prognosis. A summary of histopathological findings from renal biopsies in these cases, including light microscopy observations, immunofluorescence study results, electron microscopy outcomes, as well as pathological diagnoses and genetic testing results, is presented in Table 2.
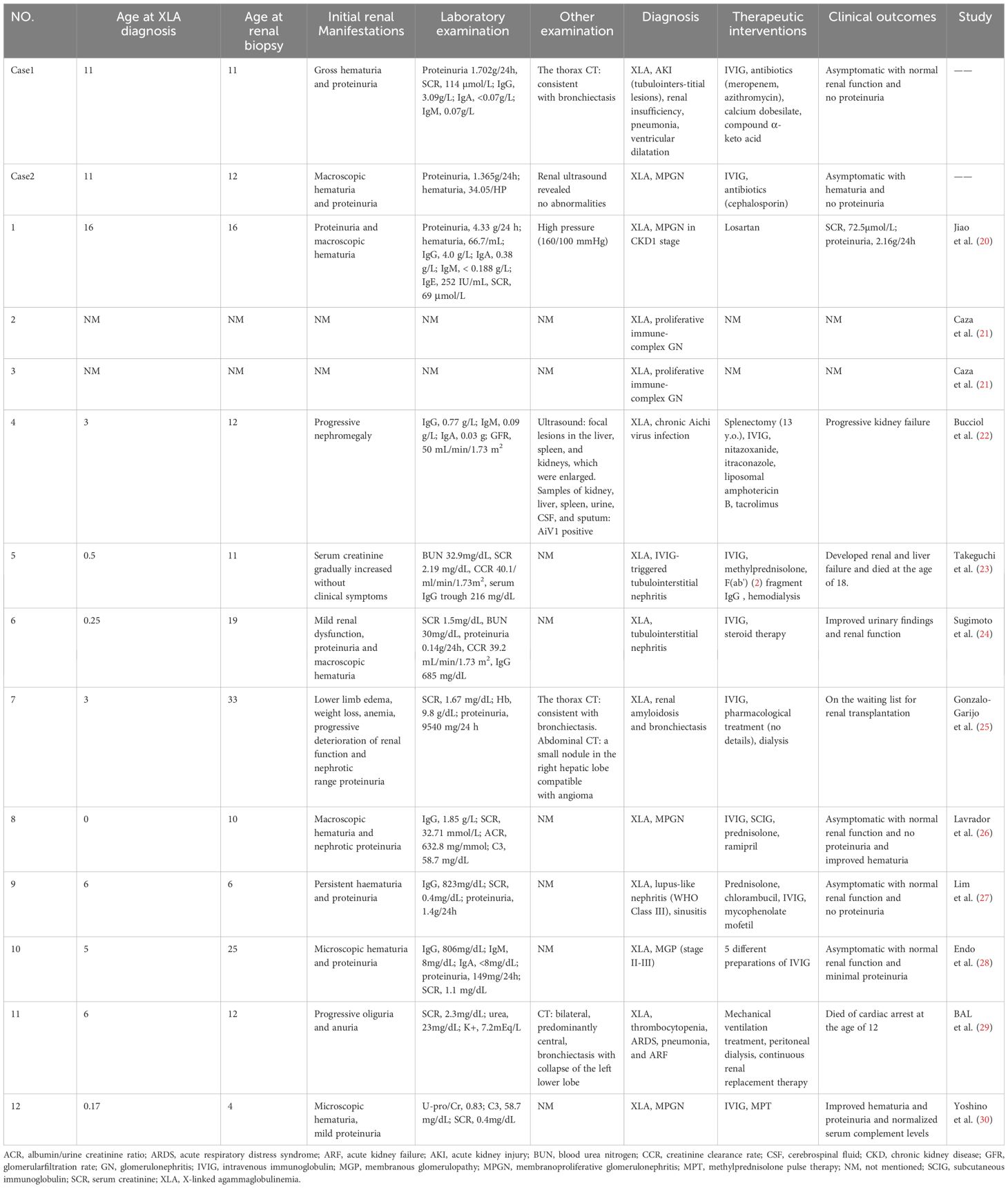
Table 1 Initial renal manifestations, relevant examinations, diagnosis, treatment, and prognosis of XLA patients with renal biopsy.
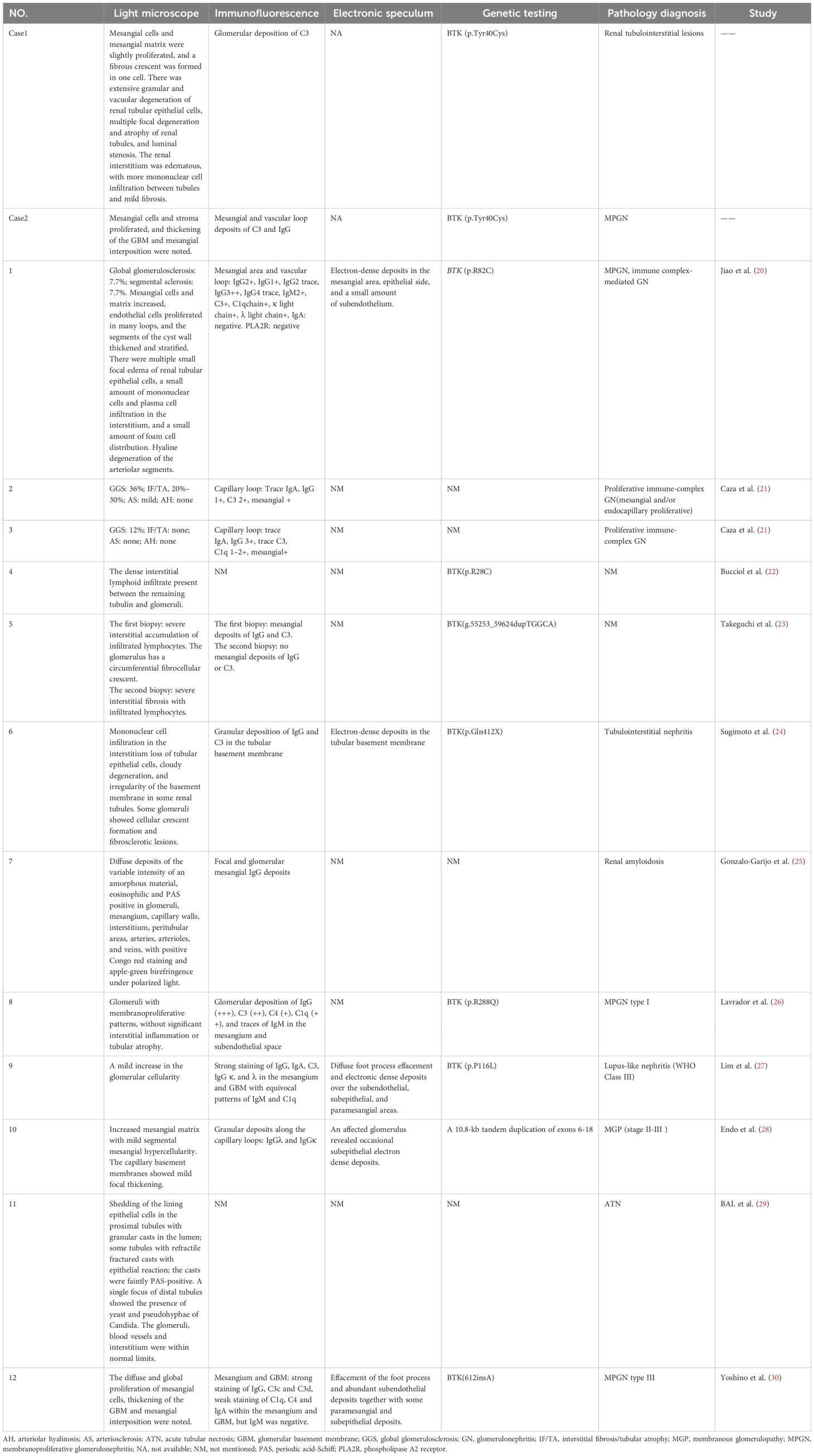
Table 2 Pathological findings of renal biopsy and genetic testing results.
The review involved 14 patients, including boys and adult men. The median age at diagnosis was 3 years (ranging from 0 to 16 years), while the median age at renal biopsy was 12 years (ranging from 4 to 33 years) (with some ages estimated based on information provided in the article). The common initial renal manifestations among all these cases comprised abnormal results in the urine analysis, such as proteinuria (nine of 14 cases; 64.3%) and hematuria (eight of 14 cases; 57.1%), along with decreased renal function (six of 14 cases; 42.9%). Out of the 14 patients, 13 underwent a renal biopsy, while a postmortem examination was done on one patient.
The most frequently observed pathological types detected in the examination of renal tissue were membranoproliferative glomerulonephritis (MPGN) and tubulointerstitial lesions. Other findings included immune complex-mediated glomerulonephritis, renal amyloidosis, membranous nephropathy, acute tubular necrosis, and lupus nephritis. Among the 14 cases, glomerulosclerosis was identified in four patients (28.6%), while mild interstitial fibrosis was present in three cases (21.4%), and crescent formation was noted in three cases (21.4%).
An immunofluorescence examination of renal tissues from 12 patients revealed the presence of immunoglobulin deposition in the majority of cases, including IgG (91.7%), IgA (33.3%), IgM (25.0%), C3 (83.3%), C1q (41.7%), and C4 (16.7%). The detection of immune complexes, immunoglobulins, and complement components in immunofluorescence suggested the involvement of humoral immunity in the observed renal manifestations. However, it remains unclear whether the deposited immunoglobulins originated from therapy or endogenous sources.
As shown in Table 1, patients with XLA and renal disease had low levels of immunoglobulins in their peripheral blood, and most of them received long-term IVIG treatment. In the cases reported by Takeguchi et al. (23), the initial renal biopsy confirmed the deposition of immunoglobulins and complement in glomeruli, while the subsequent renal biopsy after F(ab′)2 fragment IgG replacement therapy showed that these deposits had disappeared. Therefore, it was proposed that the deposition of immunoglobulins in glomeruli could originate from exogenous IVIG. Conversely, Jiao et al. (20) reported a case of MPGN occurring in a patient with XLA without prior immunoglobulin treatment, and they speculated that XLA itself could lead to MPGN, possibly due to dysregulation of peripheral B lymphocyte function and the production of abnormal endogenous antibodies. In our study, both twin patients had identical genotypes and experienced renal involvement before receiving immunoglobulin therapy, suggesting a plausible correlation between XLA and renal disease.
The standard initial dose of IVIG treatment for patients with XLA is typically 400–600 mg/kg per month, administered intravenously every three to four weeks or subcutaneously on a weekly or biweekly basis. The goal is to maintain serum IgG levels above 500 mg/dL in order to prevent potentially life-threatening recurrent bacterial infections (31, 32). However, long-term use of IVIG in patients with XLA necessitates careful monitoring for associated side effects. Apart from transient adverse reactions such as flushing, headache, fever, fatigue, and drowsiness, rare side effects such as renal damage, thrombosis formation, arrhythmias, aseptic meningitis, hemolytic anemia, and transfusion-related acute lung injury (TRALI) may also occur (33).
Renal impairment following IVIG may arise from several mechanisms: immune complex deposition in glomeruli, permeability nephritis due to immune-mediated hemolysis causing acute tubular obstruction, and transient vascular ischemia resulting from decreased renal perfusion (34–36). A minority of individuals with immunoglobulin-associated renal dysfunction progress to chronic renal failure or death (37, 38). These adverse reactions are contingent on the type of immunoglobulin preparation used and individual variations.
The current body of literature indicates that subcutaneous immunoglobulin (SCIG) demonstrates a reduced incidence of side effects, higher IgG trough levels, and lower infection rates in comparison to IVIG (39, 40). Further investigation is warranted to elucidate the source of immunoglobulin deposition in renal tissue and the associated immune mechanisms in XLA. In the management of patients with XLA receiving long-term IVIG therapy, timely evaluation of risk factors, administration of premedication, consideration of transitioning from IVIG to SCIG to minimize adverse reactions, and offering timely supportive treatment are crucial for optimizing the prognosis.
In conclusion, the following key points emerge:
1. Screening recommendations: Male children with recurrent infections should undergo thorough screenings for serum immunoglobulin, lymphocyte subsets, and other immune function evaluations. Additionally, genetic testing should be considered if warranted.
2. Genotype–phenotype correlation: The association between genotype and phenotype of XLA is unclear, as they may be influenced by factors such as gene modification, epigenetics, environment, or other unidentified variables.
3. Association with renal disease: XLA may be associated with immune-mediated renal diseases that often present with subtle or hidden phenotypes. Delayed diagnosis and delayed initiation of replacement therapy can lead to significant damage to renal function, precipitating various complications. For example, continued deterioration of renal function can result in the inability of the kidneys to excrete water, resulting in hypertension, pulmonary edema, and heart failure, which can be potentially fatal. In addition, susceptibility to infections, including urinary tract infections, lung infections, severe infections, and even sepsis, is heightened in affected individuals. Furthermore, in cases of renal failure, patients are prone to electrolyte imbalances such as hyperkalemia, posing a risk of cardiac arrest and death. Comprehensive renal-related examinations are crucial in patients with XLA, with prompt investigation of the immunopathogenesis for targeted therapeutic interventions.
4. Coexistence of autoimmune disease: XLA can also coexist with autoimmune diseases, potentially stemming from dysregulation of immune homeostasis triggered by genetic, immunological, and microbial factors, among others. B cells play a pivotal role in establishing long-term protective immunity.
Written informed consent was obtained from the minor(s)' legal guardian for the publication of any potentially identifiable images or data included in this article.
SW: Data curation, Formal analysis, Software, Writing – original draft. MC: Conceptualization, Formal analysis, Writing – original draft. JZ: Data curation, Software, Writing – review & editing. YB: Formal analysis, Software, Writing – review & editing. MS: Data curation, Software, Writing – review & editing. HJ: Conceptualization, Funding acquisition, Project administration, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study is funded by the National Natural Science Foundation of China (No.81300130), the Science and Technology Projects for People’s Livelihood of Liaoning Province (2021JH/10300008), Provincial Natural Science Foundation Joint Fund of Liaoning (2023-BSBA-363) and Basic Research Projects of Liaoning Province (JYTMS20230073).
We would like to acknowledge the hard and dedication of all the staff who implemented the intervention and evaluation components of the study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AIHA, autoimmune hemolytic anemia; BAFF, B-cell activating factor; BTK, Bruton’s tyrosine kinase; CRP, C-reactive protein; eGFR, estimated glomerular filtration rate; IBD, inflammatory bowel disease; IEI, inborn errors of immunity; IgAV, IgA vasculitis; IVIG, Iintravenous immune globulin; LPS, lipopolysaccharide; MPGN, membranoproliferative glomerulonephritis; MWS, Muckle–Wells syndrome; NLRP3, NACHT, LRR, and PYD domain-containing protein 3; PADs, primary antibody deficiencies; PID, primary immunodeficiency; RF, rheumatoid factor; SCIG, subcutaneous immunoglobulin; TLR, Toll-like receptor; TRALI, transfusion-related acute lung injury; XLA, X-linked agammaglobulinemia.
1. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2022) 42:1473–507. doi: 10.1007/s10875–022-01289–3
2. Suri D, Rawat A, Singh S. X-linked agammaglobulinemia. Indian J Pediatr. (2016) 83:331–7. doi: 10.1007/s12098–015-2024–8
3. Cardenas-Morales M, Hernandez-Trujillo VP. Agammaglobulinemia: from X-linked to autosomal forms of disease. Clin Rev Allergy Immunol. (2022) 63:22–35. doi: 10.1007/s12016–021-08870–5
4. Nomura K, Kanegane H, Karasuyama H, Tsukada S, Agematsu K, Murakami G, et al. Genetic defect in human X-linked agammaglobulinemia impedes a maturational evolution of pro-B cells into a later stage of pre-B cells in the B-cell differentiation pathway. Blood J Am Soc Hematol. (2000) 96:610–7.
5. Lougaris V, Soresina A, Baronio M, Montin D, Martino S, Signa S, et al. Long-term follow-up of 168 patients with X-linked agammaglobulinemia reveals increased morbidity and mortality. J Allergy Clin Immunol. (2020) 146:429–37. doi: 10.1016/j.jaci.2020.03.001
6. Takada H, Kanegane H, Nomura A, Yamamoto K, Ihara K, Takahashi Y, et al. Female agammaglobulinemia due to the bruton tyrosine kinase deficiency caused by extremely skewed X-chromosome inactivation. Blood. (2004) 103:185–7. doi: 10.1182/blood-2003–06-1964
7. Aadam Z, Kechout N, Barakat A, Chan KW, Ben-Ali M, Ben-Mustapha I, et al. X-linked agammagobulinemia in a large series of north african patients: frequency, clinical features and novel btk mutations. J Clin Immunol. (2016) 36:187–94. doi: 10.1007/s10875-016-0251-z
8. Abolhassani H, Vitali M, Lougaris V, Giliani S, Parvaneh N, Parvaneh L, et al. Cohort of Iranian patients with congenital agammaglobulinemia: mutation analysis and novel gene defects. Expert Rev Clin Immunol. (2016) 12:479–86. doi: 10.1586/1744666X.2016.1139451
9. Chen XF, Wang WF, Zhang YD, Zhao W, Wu J, Chen TX. Clinical characteristics and genetic profiles of 174 patients with X-linked agammaglobulinemia: report from shanghai, China (2000–2015). Med (Baltimore). (2016) 95:e4544. doi: 10.1097/MD.0000000000004544
10. Garcia-Garcia E, Staines-Boone AT, Vargas-Hernandez A, Gonzalez-Serrano ME, Carrillo-Tapia E, Mogica-Martinez D, et al. Clinical and mutational features of X-linked agammaglobulinemia in Mexico. Clin Immunol. (2016) 165:38–44. doi: 10.1016/j.clim.2016.02.010
11. Pac M, Bernatowska EA, Kierkus J, Ryzko JP, Cielecka-Kuszyk J, Jackowska T, et al. Gastrointestinal disorders next to respiratory infections as leading symptoms of X-linked agammaglobulinemia in children - 34-year experience of a single center. Arch Med Sci. (2017) 13:412–7. doi: 10.5114/aoms.2016.60338
12. Singh S, Rawat A, Suri D, Gupta A, Garg R, Saikia B, et al. X-linked agammaglobulinemia: twenty years of single-center experience from North West India. Ann Allergy Asthma Immunol. (2016) 117:405–11. doi: 10.1016/j.anai.2016.07.044
13. Behniafard N, Aghamohammadi A, Abolhassani H, Pourjabbar S, Sabouni F, Rezaei N. Autoimmunity in X-linked agammaglobulinemia: kawasaki disease and review of the literature. Expert Rev Clin Immunol. (2012) 8:155–9. doi: 10.1586/eci.11.94
14. Howard V, Greene JM, Pahwa S, Winkelstein JA, Boyle JM, Kocak M, et al. The health status and quality of life of adults with X-linked agammaglobulinemia. Clin Immunol. (2006) 118:201–8. doi: 10.1016/j.clim.2005.11.002
15. Ochs HD, Notarangelo LD. X-linked immunodeficiencies. Curr Allergy Asthma Rep. (2004) 4:339–48. doi: 10.1007/s11882–004-0082–5
16. Pessach IM. The relationship of X-linked primary immune deficiencies and autoimmunity. Curr Allergy Asthma Rep. (2010) 10:311–9. doi: 10.1007/s11882-010-0127-x
17. Simini G, Mackenzie S, Gohil S, Papanikolaou X, Manson J, Payne E. Successful use of allogeneic bone marrow transplantation in a patient with myelodysplastic syndrome presenting with autoimmune manifestations. Br J Haematol. (2021) 193:1275–7. doi: 10.1111/bjh.17437
18. Maffeis M, Notarangelo LD, Schumacher RF, Soncini E, Soresina A, Lanfranchi A, et al. Primary immunodeficiencies and oncological risk: the experience of the children's hospital of brescia. Front Pediatr. (2019) 7:232. doi: 10.3389/fped.2019.00232
19. Cinicola B, Uva A, Leonardi L, Moratto D, Giliani S, Carsetti R, et al. Case report: A case of X-linked agammaglobulinemia with high serum ige levels and allergic rhinitis. Front Immunol. (2020) 11:582376. doi: 10.3389/fimmu.2020.582376
20. Jiao CF, Zhao LL, Jiang L, Xu F, Cheng Z. X-linked agammaglobulinemia with membranoproliferative glomerulonephritis. Chin J Nephrol Dialysis Transplant. (2021) 30:394. doi: 10.3969/j.issn.1006–298X.2021.04.019
21. Caza TN, Hassen SI, Larsen CP. Renal manifestations of common variable immunodeficiency. Kidney360. (2020) 1:491–500. doi: 10.34067/KID.0000432020
22. Bucciol G, Moens L, Payne K, Wollants E, Mekahli D, Levtchenko E, et al. Chronic aichi virus infection in a patient with X-linked agammaglobulinemia. J Clin Immunol. (2018) 38:748–52. doi: 10.1007/s10875-018-0558-z
23. Takeguchi M, Korematsu S, Miyahara H, Kuga S, Izumi T. Ivig-triggered tubulointerstitial nephritis in X-linked agammaglobulinemia. Pediatr Int. (2017) 59:945–6. doi: 10.1111/ped.13329
24. Sugimoto K, Nishi H, Miyazawa T, Wada N, Izu A, Enya T, et al. Tubulointerstitial nephritis complicating ivig therapy for X-linked agammaglobulinemia. BMC Nephrol. (2014) 15:1–4. doi: 10.1186/1471-2369-15-109
25. Gonzalo-Garijo MA, Sanchez-Vega S, Perez-Calderon R, Perez-Rangel I, Corrales-Vargas S, Fernandez de Mera JJ, et al. Renal amyloidosis in a patient with X-linked agammaglobulinemia (Bruton's disease) and bronchiectasis. J Clin Immunol. (2014) 34:119–22. doi: 10.1007/s10875–013-9972–4
26. Lavrador V, Correia F, Sampaio R, Candido C, Sameiro-Faria M, Marques L, et al. Membranoproliferative glomerulonephritis and X-linked agammaglobulinemia: an uncommon association. Case Rep Pediatr. (2014) 2014:480947. doi: 10.1155/2014/480947
27. Lim L-M, Chang J-M, Wang I-F, Chang W-C, Hwang D-Y, Chen H-C. Atypical X-linked agammaglobulinaemia caused by a novel btk mutation in a selective immunoglobulin M deficiency patient. BMC Pediatr. (2013) 13:1–6. doi: 10.1186/1471-2431-13-150
28. Endo L, Giannobile J, Dobbs A, Foote J, Szymanska E, Warnock D, et al. Membranous glomerulopathy in an adult patient with X-linked agammaglobulinemia receiving intravenous gammaglobulin. J Investigat Allergol Clin Immunol. (2011) 21:405.
29. Bal A, Rawat A, Nada R, Singh S. A 12-year-old boy with X-linked agammaglobulinaemia who had breakthrough infection, thrombocytopenia and acute renal failure. Natl Med J India. (2009) 22:310–6.
30. Yoshino A, Honda M, Kanegane H, Obata K, Matsukura H, Sakazume S, et al. Membranoproliferative glomerulonephritis in a patient with X-linked agammaglobulinemia. Pediatr Nephrol. (2006) 21:36–8. doi: 10.1007/s00467-005-2029-z
31. Lee JH, Mohamed shanh N, Makmor-Bakry FHM, Islahudin FH, Alias H, Noh LM, et al. A systematic review and meta-regression analysis on the impact of increasing IgG trough level on infection rates in primary immunodeficiency patients on intravenous IgG therapy. J Clin Immunol. (2020) 40:682–98. doi: 10.1007/s10875-020-00788-5
32. Ballow M, Zachariah T. Practical aspects of immunoglobulin replacement. Ann Allergy Asthma Immunol. (2017) 119:299–303. doi: 10.1016/j.ekir.2017.12.002
33. Guo Y, Tian X, Wang X, Xiao Z. Adverse effects of immunoglobulin therapy. Front Immunol. (2018) 9:1299. doi: 10.3389/fimmu.2018.01299
34. Rault R, Piraino B, Johnston J, Oral A. Pulmonary and renal toxicity of intravenous immunoglobulin. Clin Nephrol. (1991) 36:83–6.
35. Hansen-Schmidt S, Silomon J, Keller F. Osmotic nephrosis due to high-dose immunoglobulin therapy containing sucrose (but not with glycine) in a patient with immunoglobulin a nephritis. Am J Kidney Dis. (1996) 28:451–3. doi: 10.1016/S0272-6386(96)90505-3
36. Cayco AV, Perazella MA, Hayslett JP. Renal insufficiency after intravenous immune globulin therapy: A report of two cases and an analysis of the literature. J Am Soc Nephrol. (1997) 8:1788–94. doi: 10.1681/ASN.V8111788
37. Orbach H, Tishler M, Shoenfeld Y. Intravenous immunoglobulin and the kidney—a two-edged sword. Semin Arthritis Rheumatism. (2004) 34:593–601. doi: 10.1016/j.semarthrit.2004.06.003
38. Orbach H, Katz U, Sherer Y, Shoenfeld Y. Intravenous immunoglobulin: adverse effects and safe administration. Clin Rev Allergy Immunol. (2005) 29:173–84. doi: 10.1385/CRIAI:29:3:173
39. Shrestha P, Karmacharya P, Wang Z, Donato A, Joshi AY. Impact of ivig vs. Scig on igg trough level and infection incidence in primary immunodeficiency diseases: A systematic review and meta-analysis of clinical studies. World Allergy Organ J. (2019) 12. doi: 10.1016/j.waojou.2019.100068
40. Shabaninejad H, Asgharzadeh A, Rezaei N, Rezapoor A. A comparative study of intravenous immunoglobulin and subcutaneous immunoglobulin in adult patients with primary immunodeficiency diseases: A systematic review and meta-analysis. Expert Rev Clin Immunol. (2016) 12:595–602. doi: 10.1586/1744666X.2016.1155452
Keywords: inborn errors of immunity, primary antibody deficiency, renal disease, tubulointerstitial lesions, X-linked Agammaglobulinemia
Citation: Wan S, Cao M, Zou J, Bai Y, Shi M and Jiang H (2024) Case report of renal manifestations in X-linked agammaglobulinemia. Front. Immunol. 15:1376258. doi: 10.3389/fimmu.2024.1376258
Received: 25 January 2024; Accepted: 14 May 2024;
Published: 25 July 2024.
Edited by:
Durga Prasanna Misra, Sanjay Gandhi Post Graduate Institute of Medical Sciences (SGPGI), IndiaReviewed by:
Giulia Costanzo, University of Cagliari, ItalyCopyright © 2024 Wan, Cao, Zou, Bai, Shi and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongkun Jiang, amlhbmdob25na3VuMDA3QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.