 Kenneth K. Y. Ting
Kenneth K. Y. Ting
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 22 April 2024
Sec. Nutritional Immunology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1375461
This article is part of the Research Topic Cross Talk Between Diet, Microbiome, and Cancer View all 5 articles
Excess dietary fructose consumption has been long proposed as a culprit for the world-wide increase of incidence in metabolic disorders and cancer within the past decades. Understanding that cancer cells can gradually accumulate metabolic mutations in the tumor microenvironment, where glucose is often depleted, this raises the possibility that fructose can be utilized by cancer cells as an alternative source of carbon. Indeed, recent research has increasingly identified various mechanisms that show how cancer cells can metabolize fructose to support their proliferating and migrating needs. In light of this growing interest, this review will summarize the recent advances in understanding how fructose can metabolically reprogram different types of cancer cells, as well as how these metabolic adaptations can positively support cancer cells development and malignancy.
Cancer cells undergo metabolic reprogramming, a process where they gradually acquire metabolic adaptations, to survive in a local tumor microenvironment (TME). These adaptations are critical in sustaining their high anabolic demands for uncontrolled proliferation, as well as conferring resistance against anti-tumor immunity. The first metabolic adaptation acquired by cancer cells was first observed by Otto Warburg in the 1920s (1, 2), where he found that cancer cells have adapted to oxidize glucose into lactate in greater amounts than other normal surrounding tissue despite residing in a normoxic environment. He later proposed that mitochondria dysfunction was the underlying cause behind the high aerobic glycolytic influx in cancer cells (3), which is now known as the Warburg effect. Since this discovery, many studies have now confirmed that cancer cells do undergo metabolic reprogramming, and further discovered that local cues in the TME, which is often influenced by lifestyle factors, can modulate the type of metabolic reprogramming undergone by cancer cells.
The daily consumption of fructose per person in United States has increased by 26% since the 1970s, highly correlating with the rise of incidence in obesity, metabolic diseases (ex. type 2 diabetes mellitus and non-alcoholic fatty liver disease (NAFLD)), neurological dysfunction and various types of cancer (4–7). This consequentially prompted the launch of many studies to determine if a TME enriched with high levels of fructose can reprogram cancer cells, enabling them to utilize fructose as a secondary source of fuel. As expected, an increasing number of studies have now shown that various types of cancer cells can indeed oxidize TME-derived fructose to support their proliferative and migrative needs, and these discoveries have been summarized in the following sections.
Although glucose and fructose share the same molecular formula (C6H12O6), their structural differences, such as the keto group in position 2 of the carbon chain in fructose, underlie their need to use different catabolic enzymes to facilitate their metabolism (Figure 1A). In general, the consumption of fructose (ex. in the form of sucrose or high fructose corn syrup) in hepatocytes is first phosphorylated to fructose-1-phosphate (F1P) by ketohexokinase (KHK) (8), which is a rate-limiting step of fructose metabolism. F1P is then converted into glyceraldehyde (GA) and dihydroxyacetone phosphate (DHAP) by Aldolase B (ALDOB). GA can then be phosphorylated into glyceraldehyde-3-phosphate (GA3P) by triose kinase and participate glycolysis as a substrate, or combine with DHAP to form fructose-1-6-bisphosphate (F1,6-BP) and enter the gluconeogenic pathway (9). Apart from this, F1P-derived DHAP can be converted to glycerol-3-phosphate (G3P) by Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), in which G3P can then combine with free fatty acids to synthesize triglycerides (10) (Figure 1B).
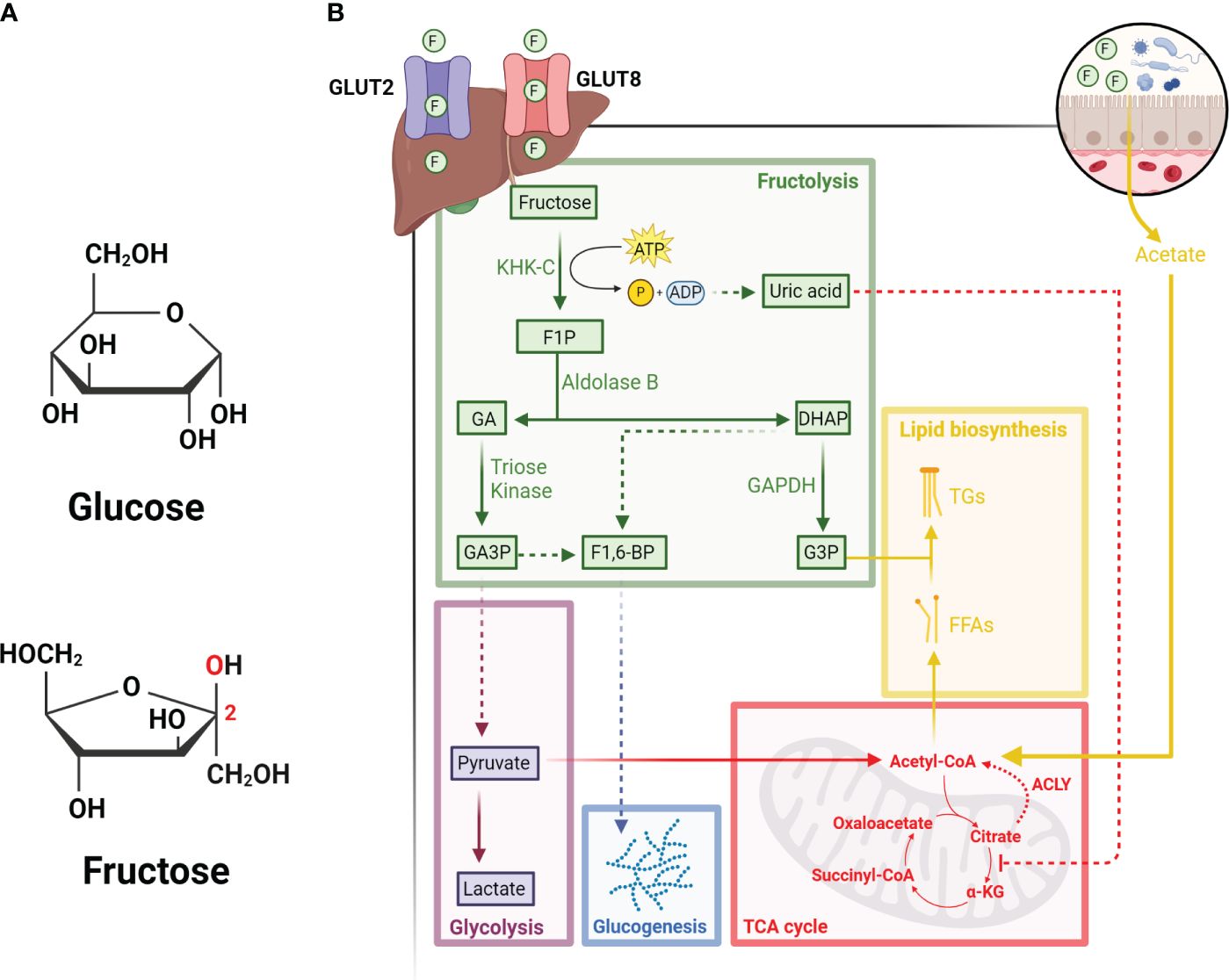
Figure 1 Hepatic metabolism of fructose. (A) Diagram illustrating the structural differences between glucose (top) and fructose (bottom). Red text indicates the carbonyl group located on carbon 2 of fructose. (B) Fructose enters hepatocytes from portal circulation through GLUT2 and GLUT8, followed by its phosphorylation by KHK-C into F1P. F1P is then converted into GA and DHAP by Aldolase B. GA can be phosphorylated by triose kinase into GA3P, which can either enter the glycolysis pathway or combine with DHAP to form F1-6-BP and enter the glucogenesis pathway. DHAP-derived from F1P can also be converted to G3P by GAPDH. The depletion of ATP due to rapid KHK-C activity leads to the accumulation of uric acid, which inhibits the activity of aconitase and increases the levels of citrate. Citrate can then be converted back to acetyl-CoA by ACLY and participate in the synthesis of free fatty acids (FFAs). Acetate derived from fructose metabolism in intestinal microbes also contributes to the lipogenic pools of acetyl-CoA. Finally, FFAs and G3P can be used to synthesize triglycerides (TGs), which can be stored as lipid droplets in the liver or secreted as very low-density lipoproteins into circulation. All figures are created with Biorender.com.
Unlike glycolysis, where multiple enzymes (i.e., phosphofructokinase and glucokinase) can be regulated through feedback inhibition, fructose metabolism bypasses these checkpoints. For instance, KHK-mediated phosphorylation of fructose to F1P is not feedback inhibited, therefore excess dietary fructose metabolism can lead to rapid F1P generation, ATP depletion and consequentially uric acid production (11). On the other hand, the generation of F1P can also stimulate the activity of glycolytic enzymes, such as liver-type pyruvate kinase, thus increasing the rate of glycolysis and subsequently the TCA cycle (11). Since F1P-derived production of uric acid can induce mitochondrial oxidative stress and inhibit aconitase activity, high fructose metabolism through the TCA cycle can lead to the accumulation of citrate. As the levels of citrate accumulates, it can then be converted to acetyl-CoA by ATP Citrate Lyase (ACLY) and support de novo lipogenesis (12). However, recent research has also shown that microbial-derived acetate post fructose metabolism by gut-microbiota is the primary source that supplies the lipogenic pools of acetyl-CoA, a process independent of ACLY activities (13).
Apart from the liver, recent advances in understanding fructose metabolism also demonstrate that the small intestine in fact metabolizes fructose prior to the liver (14). Specifically, fructose is first imported in the gut intracellularly by GLUT5, which is expressed at the apical side of enterocyte luminal membrane (14, 15), as well as GLUT2 or GLUT5, which is expressed at the basolateral pole of enterocyte (16, 17). Upon entry into the enterocyte, fructose is rapidly metabolized. Any excess fructose that cannot be metabolized by enterocyte will be delivered from the portal circulation to the liver (18, 19) through GLUT2 (20–22) or GLUT8 expressed on hepatocytes (23).
The metabolism of colon-rectal cancer (CRC) is highly dependent on glycolysis to sustain their uncontrolled proliferation (24–27). In fact, the hyperactivity of glycolysis in CRC significantly depletes glucose in local environment, thereby priming CRC to use fructose as an alternative source to further fuel its proliferation. For instance, Shen et al. have reported that multiple human CRC cell lines (ex. HCT116 and HT29) upregulated GLUT5 expression in response to glucose depletion, which led to the increased uptake of fructose and its metabolism to fuel central carbon metabolism (28). Furthermore, the study also found that fructose-induced levels of GLUT5 proteins could bind to KHK and block its lysosomal degradation, further sustaining its metabolism of fructose (28). Similar findings were also reported by Włodarczyk et al where they inhibited the function of GLUT5 with N-[4-(methylsulfonyl)-2-nitrophenyl]-1,3-benzodioxol-5-amine (MSBNA) in a CRC cell line (HT-29), and this significantly reduced their viability. Taken together, these studies have demonstrated that GLUT5 plays a critical role in supporting the growth of CRC through mediating fructose metabolism (29).
As described previously, cancer cells undergo metabolic reprogramming to adapt and survive in local environment. This is particularly important for cancer cells that undergo metastases as they are required to migrate and proliferate in a new environment. For instance, CRC could metastasize in the liver, which is the current leading cause of cancer-related deaths (30, 31). Given that fructose is predominantly metabolized in liver, this raises the possibility that dietary fructose overconsumption can provide a secondary energy source for CRC to utilize upon its metastasis to the liver. Indeed, Bu et al. performed meta-analysis of clinical CRC liver metastases samples, as well as in vivo metastatic models, and found that glycolysis, glucogenesis, fructose metabolism and PPP were upregulated (32). Mechanistically, CRC-derived liver metastases upregulated ALDOB in a GATA6-dependent manner (32). This upregulation is critical for metabolizing fructose to fuel central carbon metabolism, thereby supporting their proliferation (32). Knocking down ALDOB or fructose-restriction inhibited the growth of CRC-derived liver metastasis (32).
Apart from CRC, Goncalves et al. have used mouse models to demonstrate that chronic fructose consumption facilitates intestinal tumor growth, a process independent of the effects of obesity and metabolic syndrome (33). Specifically, the authors found that intestinal tumors upregulate fructose metabolism, which is critical for fueling glycolysis due to F1P-induced ATP depletion (33). The enhancement of glycolysis subsequently led to increased fatty acid synthesis, a process vital for cancer cell growth due to its need for cellular membrane formation, energy generation, and intracellular signaling (34, 35). Overall, these studies have demonstrated that cancer cells that reside in organs that directly metabolize fructose, such as the liver and intestine, can undergo reprogramming and utilize fructose to support its metabolic needs.
Although the effect of high fructose diet on CRC and intestinal cancer is well established, its effect on melanoma and lung cancer (LC) is conflicting, specifically in the context of anti-tumor immunity. Utilizing mice bearing B16 melanoma cells, Kuehm et al. found that the high levels of fructose in western diet led to the impairment of immune checkpoint blockade (ICB) therapeutic outcomes in these mice (36). Mechanistically, fructose activated cryoprotection in melanoma cells by inducing heme oxygenase-1 (HO-1) expression, allowing them to be resistant against immune-mediated killing during ICB therapy (36). Indeed, treating melanoma with inhibitor against HO-1 restored their susceptibility to ICB therapy both in vitro and in vivo (36). Similarly, in LC, GLUT5 expression was found to be induced, which led to the enhancement of fructose metabolism and increased synthesis of fatty acids (37). Notably, impairing GLUT5 expression in LC cell lines (ex. A549 and EKVX) significantly reduced its growth, while enforcing its expression exacerbated its malignancy (37). Similar findings were also reported by Weng et al, where they found that manipulating GLUT5 expression could modulate the malignancy of lung adenocarcinoma cells (ex. A549 and H1299), and that in comparison to glucose, these cells utilize fructose primarily for fatty acid accumulation and ATP production (38).
On the contrary, a recent study has shown that a high fructose diet is in fact beneficiary for anti-tumor immunity against melanoma and LC (39). Specifically, the authors in this study found that dietary fructose increased adipocyte-derived production of leptin, a hormone that is critical for T cell-mediated anti-tumor immunity (39). Indeed, B16-F10 melanoma-bearing mice with T cells that lack the expression of leptin receptor had increased tumor size post high fructose diet (39). Apart from this, the authors also selectively induced genetic deficiency of GLUT5 in adipocytes by utilizing Adipoq-Cre : Slc2a5fl/fl mice and found that upon their challenge with melanoma, these mice had lower serum leptin levels, lesser tumor-infiltrating CD8+ T cells and larger tumor size post high-fructose diet (39). Similar findings were reproduced in LLC murine lung cancer cells orthotopic tumor-bearing mice (39). Overall, these results have demonstrated that a high fructose diet may differentially affect the response of cancer cells against anti-tumor immunity.
In 1996, Zamora-León et al. first reported that breast cancer (BC) cell lines (ex. MCF-7 and MDA-468) selectively expressed GLUT5 (40), suggesting the possibility that BC can utilize fructose as a carbon fuel. Indeed, Fan et al. have later shown that BC cell lines (ex. MCF-7 and 4T1) incubated with fructose could proliferate at the same rate as incubating them with glucose, further demonstrating the metabolic plasticity of BC (41). Similar to other types of cancer cells, GLUT5 plays a critical role in supporting the growth of BC through mediating fructose metabolism as multiple studies have reported that impairing GLUT5 expression in BC cell lines (MCF-7 and 4T1) significantly reduced their proliferation rate (41, 42). Apart from proliferation, fructose metabolism has also been shown to enhance the migration ability of BC. For instance, Monzavi-Karbassi et al. reported that fructose could enhance the invasion of BC cell line (MDA-MB-468) (43). Specifically, the authors found that fructose altered cell-surface glycosylation of BC cells, which subsequently affected their binding to endothelial cells (43). Apart from this, fructose also changed the morphology and actin arrangement of these tumor cells, which correlated to an increase of their cell migration capacity (43). Similar findings were also reported by Jiang et al. where they found that fructose-induced 12-LOX signaling in 4T1 cells promoted their metastasis (44), as well as Fan et al. where they found that fructose increased metastasis of 4T1 cells in vivo (41). The detailed mechanism behind how fructose enhanced BC metastasis was then elucidated by Kim et al. where they found that fructose enhanced BC (MTV-TM-011) invasion in a KHK-A dependent manner (45). Mechanistically, KHK-A could bind to LRRC59 and KPNB1, transporting KHK-A into the nucleus where it could phosphorylate YWHAH, and promote SLUG recruitment to CDH1 promoter and reduced the expression of E-Cadherin (45). Taken together, these studies have collectively illustrated that fructolytic enzymes can support the growth and migration of BC through their classical role in mediating fructose metabolism, as well as performing moonlighting functions.
Abnormally high fructose levels have been previously reported in the bone marrow of acute myeloid leukemia (AML) patients (46), which raised the possibility that AML cells can also utilize fructose to fuel their growth. Indeed, Jeong et al. have reported that GLUT5 expression in a variety of AML cell lines (ex. MOLM13) is critical for regulating their fructose metabolism, which directly fuels the de novo serine synthesis pathway (SSP) (47). Specifically, the authors found that the increased activity of SSP in AML cells directly contributed to the increased production of α-ketoglutarate from glutamine, thus directly supporting TCA cycle anaplerosis (47). Knocking down Phosphoglycerate Dehydrogenase (PHGDH), a rate-limiting enzyme of SSP, reduced AML burden load in the presence of high fructose (47). Finally, the authors also found that AML cells unexpectedly utilized hexokinase (HK), rather than KHK, to metabolize fructose (47). Similar findings were also reported by Chen et al, where they have shown that various AML cell lines (ex. U937) upregulated GLUT5 and increased fructose metabolism under glucose-depleted condition to sustain their proliferation and survival (46). Metabolically, the enhanced fructose metabolism increased glycolytic flux in AML cells, as well as exacerbated their leukemic phenotypes (proliferation and migration) (46). Finally, inhibiting fructose metabolism in AML cells with in vitro and in vivo models with 2,5-anhydro-D-mannitol (2,5-AM), a fructose analog with high affinity to GLUT5, demonstrated therapeutic potential (46). Overall, these studies have shown that AML cells can oxidize fructose to support its growth through fueling its central carbon metabolism.
Like other types of cancer cells, fructose is also critical for supporting the growth of pancreatic cancer (PC). For instance, Carreño et al. showed that GLUT5 expression was upregulated in both human PC cell lines (ex. LNCaP and PC3) and malignant human prostate tissues, and that the enhancement of fructose metabolism is important for the growth of PC (48). Mechanistically, Liu et al. have shown that the metabolism of fructose in PC cell lines (ex. Panc-1 and MiaPaCa-2) was primarily used to support nucleic acid synthesis via the transketolase-regulated, non-oxidative branch of PPP (49). Specifically, the authors performed13C-labelled glucose and fructose tracing in PC cells and found that fructose was primarily oxidized to synthesize nucleic acids while glucose was oxidized to support glycolysis, TCA cycle and fatty acid synthesis. Overall, these studies have demonstrated that while fructose is important for the growth of PC, these cells have the ability to utilize glucose and fructose differentially to support its proliferation needs.
Similarly in glioma, Su et al. have reported that GLUT5 was overexpressed in both glioma cell lines (ex. LN22 and, U87) and glioma tissues, as well as demonstrating that glioma cell lines could utilize fructose to proliferate (50). In addition to this, the authors also found that inhibiting GLUT5 expression impaired glioma cells proliferation in vitro and the expression of GLUT5 was positively associated with poor survival of glioma patients (50). Apart from GLUT5, Gao et al. have also reported that KHK was overexpressed in glioma cells (ex. LN229 and U87), and that silencing its expression significantly reduced their proliferation and migration capacity (51). Furthermore, the authors have also discovered that fructose could enhance the stability of both mRNA and protein levels of KHK but the underlying mechanism behind its stabilization is not well understood (51). Taken together, both of these studies have confirmed that glioma can metabolize fructose to support its growth and migration by upregulating the expression of fructolytic enzymes, such as GLUT5 and KHK.
Finally, in contrast to other types of cancers, multiple studies have shown that fructose metabolism was in fact reduced in hepatocellular carcinoma (HCC) in comparison to normal hepatocytes (52, 53). For instance, Li et al. have reported that HCC utilized KHK-A, one of the two isoforms of KHK (with the other being KHK-C), to metabolize fructose. This finding was unexpected as KHK-C is primarily expressed in liver and it has a higher affinity for fructose than KHK-A (54). However, as illustrated by Li et al, HCC induced a c-Myc-dependent aberrant splicing change during the expression of KHK, which led to the production of KHK-A rather than KHK-C, and thus reducing the activity of fructose metabolism in HCC (52). Notably, the authors then found that this switch to KHK-A enabled it to phosphorylate phosphoribosyl pyrophosphate synthetase 1 and support nucleic acid synthesis through the PPP. Similar findings were also reported by Tee et al, where they have confirmed that KHK expression was downregulated in both HCC mouse models and human tissues, as well as demonstrating that the uptake of fructose in HCC cell lines (Huh7 and Hep3B cells) was reduced (53). More importantly, the authors have overexpressed KHK in HCC cell lines and found it significantly reduced their activity of glycolysis and TCA cycle, thus demonstrating that the suppression of KHK is a metabolic adaptation acquired by HCC to support their upregulation of glycolysis. The importance of glycolysis in facilitating HCC development under high fructose conditions has also been reported by Syamprasad et al, where they found that the expression of AKR1B1, the master regulator of polyol pathway that reroutes glucose to the generate sorbitol and fructose, was upregulated in HCC (55). Furthermore, the authors also found that inhibiting the function of AKR1B1 suppressed glycolytic reprogramming in HCC cell lines (PLC/PRF-5), while overexpressing it increased glycolysis. Finally, the authors discovered that inhibiting AKR1B1 reduced the pathologies induced by a high fructose diet and diethylnitrosamine administration, thus supporting the role that AKR1B1 plays in mediating the glycolytic reprogramming of HCC.
Apart from this, recent research has also shown that HCC can utilize the acetate secreted by gut microbes post high fructose metabolism (56). As previously described, microbial-derived acetate post fructose metabolism is critical for fueling the lipogenic pools of acetyl-CoA in hepatocytes (Figure 1B), and now a recent study performed from Zhou et al. have taken a step further and showed that microbial-derived acetate could also fuel de novo glutamine synthesis in HCC (56). This consequentially led to the enhancement of O-linked N-acetylglucosamine (O-GlcNAc) modification (O-GlcNAcylation), a post-translational modification of proteins that is critically dependent on the levels of glutamine. Specifically, the authors found that microbial-derived acetate enriched O-GlcNAcylation of CAPNS1 and eEF1A1, which are critical for cell proliferation and protein translational output, thus promoting the growth and metastasis of HCC. Taken together, this study has demonstrated that HCC can utilize microbial-derived acetate as a secondary source of carbon to meet its metabolic demands.
Based on the studies reviewed above, several conclusions can be drawn on how fructose contributes to the metabolic reprogramming of cancer cells (Figure 2). Firstly, fructose can be utilized as an alternative carbon source to support central carbon metabolism, which is critical for de novo lipid and nucleic acid synthesis, and thus sustaining cancer cell proliferation. However, this seemingly only happens in the TME where glucose is depleted and fructose is in abundance, thereby priming cancer cells to use fructose as an alternative carbon source. Secondly, different types of cancer cells can utilize fructose or its derived metabolites differentially to support their metabolic needs. For instance, fructose is primarily used in PC to support nucleic acid synthesis through the non-oxidative branch PPP rather than fueling fatty acid synthesis. On the other hand, HCC reduced its intrinsic fructose metabolism and utilized acetate derived from fructose metabolism of gut microbes to meet its metabolic demands. Thirdly, the upregulation of fructose metabolism in cancer cells is always dependent on an increased activity (i.e., increased expression) of fructose metabolic enzymes. Finally, inhibiting the metabolism of fructose in cancer cells that were fructose-adapted significantly reduced their malignancy. This demonstrates that cancer cells do respond to high fructose in the TME, and their adaptation to fructose is critical for their survival.
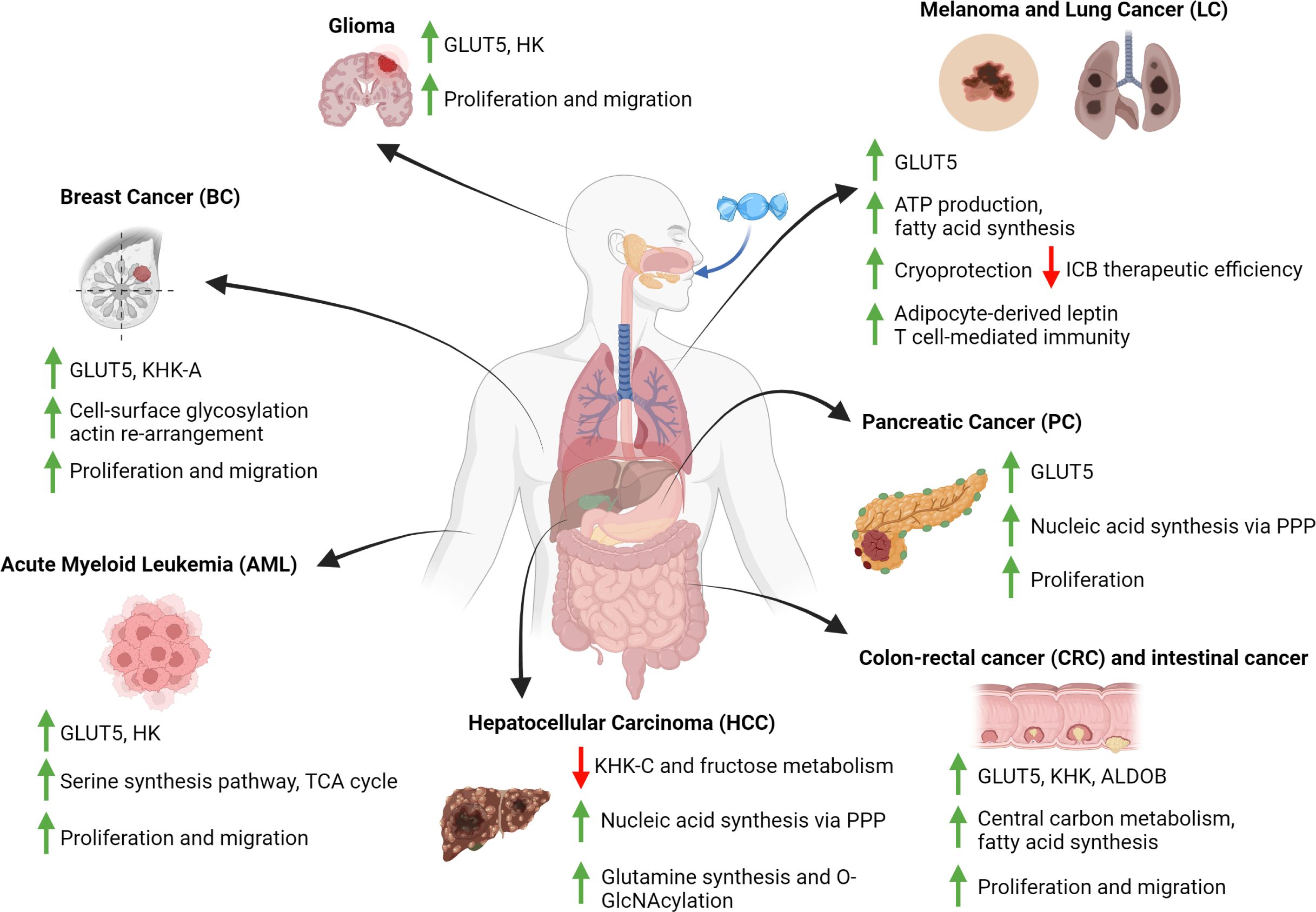
Figure 2 Fructose-induced metabolic reprogramming in cancer cells. Diagram that summarizes the metabolic consequences of fructose metabolism in various types of cancer cells. These consequences include changes to the expression of fructolytic enzymes, the fluxes of various metabolic pathways and the positive impacts to cancer cells development. All figures are created with Biorender.com.
Although the studies listed above elucidated how fructose supports the growth of various types of cancer cells, it remains unclear how the expression of fructose metabolic enzymes is regulated in response to fructose in the TME, including transcriptional and epigenetic mechanisms. A new study conducted by Huang et al. have shown that STAT6 is implicated in regulating the transcription in GLUT5 in a PC cell line (ex. DU145) (57), yet this remains to be characterized in other types of cancers. Secondly, how fructolytic enzymes can contribute to the pro-survival benefits of cancer cells, independent of their classical roles in metabolism, is only beginning to be appreciated, such as the secondary roles of KHK-A observed in BC (45) and HCC (38). Finally, it remains largely unclear how fructose-induced metabolic rewiring of cancer cells can modulate its interaction with immune cells. This is significant because a high fructose diet has always been linked to the induction of a chronic inflammatory environment (10), which is a critical factor that can modulate the development of cancer cells and anti-tumor responses (58). Therefore, future research should be warranted to investigate the consequences of excess dietary fructose metabolism, specifically how fructose diet-induced metabolites and inflammatory mediators can shape the TME and thus the crosstalk between tumor infiltrating immune cells and cancer cells.
KT: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. KT was supported by fellowships from Canadian Institutes of Health Research, Ontario Graduate Scholarship, fellowships from the University of Toronto and The Peterborough K. M. Hunter Charitable Foundation.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Warburg O. The metabolism of carcinoma cells1. J Cancer Res. (1925) 9:148–63. doi: 10.1158/jcr.1925.148
2. Warburg O, Wind F, Negelein E. Über den Stoffwechsel von Tumoren im Körper. Klinische Wochenschrift. (1926) 5:829–32. doi: 10.1007/BF01726240
3. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
4. Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weight gain, and the insulin resistance syndrome1,2,3. Am J Clin Nutr. (2002) 76:911–22. doi: 10.1093/ajcn/76.5.911
5. Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr. (2004) 79:537–43. doi: 10.1093/ajcn/79.4.537
6. Lustig RH, Schmidt LA, Brindis CD. The toxic truth about sugar. Nature. (2012) 482:27–9. doi: 10.1038/482027a
7. Ting KKY. Fructose overconsumption-induced reprogramming of microglia metabolism and function. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1375453
8. Geidl-Flueck B, Gerber PA. Insights into the Hexose Liver Metabolism-Glucose versus Fructose. Nutrients. (2017) 9. doi: 10.3390/nu9091026
9. Feinman RD, Fine EJ. Fructose in perspective. Nutr Metab (Lond). (2013) 10:45. doi: 10.1186/1743-7075-10-45
10. Febbraio MA, Karin M. “Sweet death”: fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. (2021) 33:2316–28. doi: 10.1016/j.cmet.2021.09.004
11. Keim NL, Havel PJ. Fructose: absorption and metabolism. In: Caballero B, editor. Encyclopedia of human nutrition, 3rd ed. Academic Press, Waltham (2013). p. 361–5.
12. Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem. (2012) 287:40732–44. doi: 10.1074/jbc.M112.399899
13. Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature. (2020) 579:586–91. doi: 10.1038/s41586-020-2101-7
14. Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, et al. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. (2018) 27:351–61.e3. doi: 10.1016/j.cmet.2017.12.016
15. Patel C, Douard V, Yu S, Gao N, Ferraris RP. Transport, metabolism, and endosomal trafficking-dependent regulation of intestinal fructose absorption. FASEB J. (2015) 29:4046–58. doi: 10.1096/fj.15-272195
16. Ferraris RP, Choe JY, Patel CR. Intestinal absorption of fructose. Annu Rev Nutr. (2018) 38:41–67. doi: 10.1146/annurev-nutr-082117-051707
17. Kellett GL, Helliwell PA. The diffusive component of intestinal glucose absorption is mediated by the glucose-induced recruitment of GLUT2 to the brush-border membrane. Biochem J. (2000) 350 Pt 1:155–62. doi: 10.1042/bj3500155
18. Lyssiotis CA, Cantley LC. F stands for fructose and fat. Nature. (2013) 502:181–2. doi: 10.1038/502181a
19. Vos MB, Lavine JE. Dietary fructose in nonalcoholic fatty liver disease. Hepatology. (2013) 57:2525–31. doi: 10.1002/hep.26299
20. Cheeseman CI. GLUT2 is the transporter for fructose across the rat intestinal basolateral membrane. Gastroenterology. (1993) 105:1050–6. doi: 10.1016/0016-5085(93)90948-C
21. Colville CA, Seatter MJ, Jess TJ, Gould GW, Thomas HM. Kinetic analysis of the liver-type (GLUT2) and brain-type (GLUT3) glucose transporters in Xenopus oocytes: substrate specificities and effects of transport inhibitors. Biochem J. (1993) 290:701–6. doi: 10.1042/bj2900701
22. Karim S, Adams DH, Lalor PF. Hepatic expression and cellular distribution of the glucose transporter family. World J Gastroenterol. (2012) 18:6771–81. doi: 10.3748/wjg.v18.i46.6771
23. DeBosch BJ, Chen Z, Saben JL, Finck BN, Moley KH. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J Biol Chem. (2014) 289:10989–98. doi: 10.1074/jbc.M113.527002
24. Bian Z, Zhang J, Li M, Feng Y, Wang X, Zhang J, et al. LncRNA-FEZF1-AS1 promotes tumor proliferation and metastasis in colorectal cancer by regulating PKM2 signaling. Clin Cancer Res. (2018) 24:4808–19. doi: 10.1158/1078-0432.CCR-17-2967
25. Kuo CC, Ling HH, Chiang MC, Chung CH, Lee WY, Chu CY, et al. Metastatic colorectal cancer rewrites metabolic program through a glut3-YAP-dependent signaling circuit. Theranostics. (2019) 9:2526–40. doi: 10.7150/thno.32915
26. Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X, et al. m6A-dependent glycolysis enhances colorectal cancer progression. Mol Cancer. (2020) 19:72. doi: 10.1186/s12943-020-01190-w
27. Ji Y, Yang C, Tang Z, Yang Y, Tian Y, Yao H, et al. Adenylate kinase hCINAP determines self-renewal of colorectal cancer stem cells by facilitating LDHA phosphorylation. Nat Commun. (2017) 8:15308. doi: 10.1038/ncomms15308
28. Shen Z, Li Z, Liu Y, Li Y, Feng X, Zhan Y, et al. GLUT5-KHK axis-mediated fructose metabolism drives proliferation and chemotherapy resistance of colorectal cancer. Cancer Lett. (2022) 534:215617. doi: 10.1016/j.canlet.2022.215617
29. Włodarczyk J, Włodarczyk M, Zielińska M, Jędrzejczak B, Dziki Ł, Fichna J. Blockade of fructose transporter protein GLUT5 inhibits proliferation of colon cancer cells: proof of concept for a new class of anti-tumor therapeutics. Pharmacol Rep. (2021) 73:939–45. doi: 10.1007/s43440-021-00281-9
30. André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. New Engl J Med. (2004) 350:2343–51. doi: 10.1056/NEJMoa032709
31. Wolpin BM, Mayer RJ. Systemic treatment of colorectal cancer. Gastroenterology. (2008) 134:1296–310. doi: 10.1053/j.gastro.2008.02.098
32. Bu P, Chen KY, Xiang K, Johnson C, Crown SB, Rakhilin N, et al. Aldolase B-mediated fructose metabolism drives metabolic reprogramming of colon cancer liver metastasis. Cell Metab. (2018) 27:1249–62.e4. doi: 10.1016/j.cmet.2018.04.003
33. Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang SK, Murphy CJ, et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science. (2019) 363:1345–9. doi: 10.1126/science.aat8515
34. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab. (2013) 18:153–61. doi: 10.1016/j.cmet.2013.05.017
35. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7:763–77. doi: 10.1038/nrc2222
36. Kuehm LM, Khojandi N, Piening A, Klevorn LE, Geraud SC, McLaughlin NR, et al. Fructose promotes cytoprotection in melanoma tumors and resistance to immunotherapy. Cancer Immunol Res. (2021) 9:227–38. doi: 10.1158/2326-6066.CIR-20-0396
37. Chen WL, Jin X, Wang M, Liu D, Luo Q, Tian H, et al. GLUT5-mediated fructose utilization drives lung cancer growth by stimulating fatty acid synthesis and AMPK/mTORC1 signaling. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.131596
38. Weng Y, Fan X, Bai Y, Wang S, Huang H, Yang H, et al. SLC2A5 promotes lung adenocarcinoma cell growth and metastasis by enhancing fructose utilization. Cell Death Discovery. (2018) 4:38. doi: 10.1038/s41420-018-0038-5
39. Zhang Y, Yu X, Bao R, Huang H, Gu C, Lv Q, et al. Dietary fructose-mediated adipocyte metabolism drives antitumor CD8(+) T cell responses. Cell Metab. (2023) 35(12):2107–18.e6.
40. Zamora-León SP, Golde DW, Concha II, CI R, Delgado-López F, Baselga J, et al. Expression of the fructose transporter GLUT5 in human breast cancer. Proc Natl Acad Sci U.S.A. (1996) 93:1847–52. doi: 10.1073/pnas.93.5.1847
41. Fan X, Liu H, Liu M, Wang Y, Qiu L, Cui Y. Increased utilization of fructose has a positive effect on the development of breast cancer. PeerJ. (2017) 5:e3804. doi: 10.7717/peerj.3804
42. Chan KK, Chan JYW, Chung KKW, Fung K-P. Inhibition of cell proliferation in human breast tumor cells by antisense oligonucleotides against facilitative glucose transporter 5. J Cell Biochem. (2004) 93:1134–42. doi: 10.1002/jcb.20270
43. Monzavi-Karbassi B, Hine RJ, Stanley JS, Ramani VP, Carcel-Trullols J, Whitehead TL, et al. Fructose as a carbon source induces an aggressive phenotype in MDA-MB-468 breast tumor cells. Int J Oncol. (2010) 37:615–22. doi: 10.3892/ijo
44. Jiang Y, Pan Y, Rhea PR, Tan L, Gagea M, Cohen L, et al. A sucrose-enriched diet promotes tumorigenesis in mammary gland in part through the 12-lipoxygenase pathway. Cancer Res. (2016) 76:24–9. doi: 10.1158/0008-5472.CAN-14-3432
45. Kim J, Kang J, Kang Y-L, Woo J, Kim Y, Huh J, et al. Ketohexokinase-A acts as a nuclear protein kinase that mediates fructose-induced metastasis in breast cancer. Nat Commun. (2020) 11:5436. doi: 10.1038/s41467-020-19263-1
46. Chen W-L, Wang Y-Y, Zhao A, Xia L, Xie G, Su M, et al. Enhanced fructose utilization mediated by SLC2A5 is a unique metabolic feature of acute myeloid leukemia with therapeutic potential. Cancer Cell. (2016) 30:779–91. doi: 10.1016/j.ccell.2016.09.006
47. Jeong S, Savino AM, Chirayil R, Barin E, Cheng Y, Park S-M, et al. High fructose drives the serine synthesis pathway in acute myeloid leukemic cells. Cell Metab. (2021) 33:145–59.e6. doi: 10.1016/j.cmet.2020.12.005
48. Carreño DV, Corro NB, Cerda-Infante JF, Echeverría CE, Asencio-Barría CA, Torres-Estay VA, et al. Dietary fructose promotes prostate cancer growth. Cancer Res. (2021) 81:2824–32. doi: 10.1158/0008-5472.CAN-19-0456
49. Liu H, Huang D, McArthur DL, Boros LG, Nissen N, Heaney AP. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. (2010) 70:6368–76. doi: 10.1158/0008-5472.CAN-09-4615
50. Su C, Li H, Gao W. GLUT5 increases fructose utilization and promotes tumor progression in glioma. Biochem Biophys Res Commun. (2018) 500:462–9. doi: 10.1016/j.bbrc.2018.04.103
51. Gao W, Li N, Li Z, Xu J, Su C. Ketohexokinase is involved in fructose utilization and promotes tumor progression in glioma. Biochem Biophys Res Commun. (2018) 503:1298–306. doi: 10.1016/j.bbrc.2018.07.040
52. Li X, Qian X, Peng LX, Jiang Y, Hawke DH, Zheng Y, et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat Cell Biol. (2016) 18:561–71. doi: 10.1038/ncb3338
53. Tee SS, Kim N, Cullen Q, Eskandari R, Mamakhanyan A, Srouji RM, et al. Ketohexokinase-mediated fructose metabolism is lost in hepatocellular carcinoma and can be leveraged for metabolic imaging. Sci Adv. (2022) 8:eabm7985. doi: 10.1126/sciadv.abm7985
54. Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, MacLean PS, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci. (2012) 109:4320–5. doi: 10.1073/pnas.1119908109
55. Syamprasad NP, Jain S, Rajdev B, Panda SR, Kumar GJ, Shaik KM, et al. AKR1B1 drives hyperglycemia-induced metabolic reprogramming in MASLD-associated hepatocellular carcinoma. JHEP Rep. (2024) 6:100974. doi: 10.1016/j.jhepr.2023.100974
56. Zhou P, Chang WY, Gong DA, Xia J, Chen W, Huang LY, et al. High dietary fructose promotes hepatocellular carcinoma progression by enhancing O-GlcNAcylation via microbiota-derived acetate. Cell Metab. (2023) 35:1961–75.e6. doi: 10.1016/j.cmet.2023.09.009
57. Huang X, Fang J, Lai W, Hu Y, Li L, Zhong Y, et al. IL-6/STAT3 axis activates glut5 to regulate fructose metabolism and tumorigenesis. Int J Biol Sci. (2022) 18:3668–75. doi: 10.7150/ijbs.68990
Keywords: fructose, cancer, metabolic reprogramming, tumor microenvironment (TME), ketohexokinase (KHK), GLUT5, metabolism, glycolysis
Citation: Ting KKY (2024) Fructose-induced metabolic reprogramming of cancer cells. Front. Immunol. 15:1375461. doi: 10.3389/fimmu.2024.1375461
Received: 23 January 2024; Accepted: 10 April 2024;
Published: 22 April 2024.
Edited by:
J. Mark Brown, Cleveland Clinic, United StatesReviewed by:
Robert N. Helsley, University of Kentucky, United StatesCopyright © 2024 Ting. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth K. Y. Ting, a2VubmV0aC50aW5nQG1haWwudXRvcm9udG8uY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.