
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 March 2024
Sec. Cytokines and Soluble Mediators in Immunity
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1374796
This article is part of the Research TopicThe immunological regulation of extracellular vesicles on chronic diseasesView all 12 articles
 Damian N. Di Florio1,2,3
Damian N. Di Florio1,2,3 Danielle J. Beetler1,2,3Elizabeth J. McCabe1
Danielle J. Beetler1,2,3Elizabeth J. McCabe1 Jon Sin4
Jon Sin4 Tsuneya Ikezu5
Tsuneya Ikezu5 DeLisa Fairweather1,2,6,7*
DeLisa Fairweather1,2,6,7*For many decades viral infections have been suspected as ‘triggers’ of autoimmune disease, but mechanisms for how this could occur have been difficult to establish. Recent studies have shown that viral infections that are commonly associated with viral myocarditis and other autoimmune diseases such as coxsackievirus B3 (CVB3) and SARS-CoV-2 target mitochondria and are released from cells in mitochondrial vesicles that are able to activate the innate immune response. Studies have shown that Toll-like receptor (TLR)4 and the inflammasome pathway are activated by mitochondrial components. Autoreactivity against cardiac myosin and heart-specific immune responses that occur after infection with viruses where the heart is not the primary site of infection (e.g., CVB3, SARS-CoV-2) may occur because the heart has the highest density of mitochondria in the body. Evidence exists for autoantibodies against mitochondrial antigens in patients with myocarditis and dilated cardiomyopathy. Defects in tolerance mechanisms like autoimmune regulator gene (AIRE) may further increase the likelihood of autoreactivity against mitochondrial antigens leading to autoimmune disease. The focus of this review is to summarize current literature regarding the role of viral infection in the production of extracellular vesicles containing mitochondria and virus and the development of myocarditis.
● Mitochondrial extracellular vesicles contain CVB3
● Extracellular vesicles containing mitochondrial components activate TLR4/NLRP3
● Autoantibodies against mitochondria are found in patients with myocarditis
● The autoimmune regulator AIRE may bind few mitochondrial genes
The immune system protects the host against infection by specifically recognizing and eliminating foreign pathogens, but in the process must avoid responding to host antigens. During maturation of the immune system, immune cells that react against self-antigens are eliminated providing an immune system that is ‘tolerant’ to self (1). T cells that escape central tolerance are additionally regulated with peripheral tolerance mechanisms that include the conversion of self-reactive T cells to regulatory T cells. Autoimmunity that progresses to autoimmune disease can occur if this process breaks down (2). Genetic and environmental factors contribute to the development of autoimmune diseases, but twin studies indicate that environmental factors are a significant contributor (3, 4). For many decades viral infections have been suspected as ‘triggers’ of autoimmune disease, but mechanisms for how this could occur have been difficult to establish (2, 5, 6). Recent findings suggest that subversion of host cellular extracellular vesicle (EV) processing by viral infections may lead not only to activation of the immune response against the virus but also against mitochondrial or other self-antigens thereby contributing to the development of autoimmune disease. In this review, we describe EVs with mitochondrial content, their relationship to viral infections such as coxsackievirus B3 (CVB3), and their potential role in driving autoimmune diseases with a focus on myocarditis.
In the last decade there has been a major increase in interest in EVs in their role in cell-to-cell communication, as biomarkers and as therapeutics (7, 8). Many terms and definitions are used to describe EVs, and in this review we use the term EVs to refer to all extracellular, lipid bilayer, sub-cellular particles and their functional contents with sizes ranging from several nm to several μm (9, 10). This umbrella term includes the widely recognized major subgroups termed exosomes, microvesicles, and apoptotic bodies, which are currently distinguishable only by their theorized origin and size but not by experimental means (11). EVs are engaged in cellular communication in both health and disease as transporters of molecular signals in the form of nucleic acids (e.g., DNA, mRNA, microRNA/miRs, long-coding RNA/lcRNA and circular RNA/circRNA), proteins and lipids (11).
When tissue environments are perturbed or cells become damaged as occurs during a viral infection, EV content changes based on cellular reprogramming in response to pathological stress (11, 12). EVs can either activate or inhibit innate and adaptive immune cell responses based on their content (13–15). EVs have been demonstrated to express major histocompatibility complex (MHC) class I or II and directly activate innate antigen presenting cells (APCs) or adaptive T and B cells in an antigen/self-antigen-specific manner (16, 17). Tetraspanins like CD9, CD63 and CD81, which are commonly used to characterize EVs, bind factors on innate immune cells like integrins (i.e., CD11b) that are important in activating and modulating immune responses (12, 18).
Importantly, many viruses use EV cellular machinery (i.e., exosome endosomal sorting complexes required for the transport/ESCRT pathway) for viral transmission such as cytomegalovirus, coxsackievirus, SARS-CoV-2, human immunodeficiency virus 1 (HIV-1), hepatitis viruses B, C and E (HBV, HCV, and HEV), and multiple members of the human herpesvirus (HHV) family (reviewed in (19–22). As a result, EVs can contain infectious virus, viral particles and/or viral proteins following infection that can subvert the immune response to promote viral replication. Important from an autoimmune disease context, a ‘mix’ of self and foreign antigens in/on EVs that may occur after viral infection may be presented to APCs and drive the immune response to target not only the infectious agent but also host antigens resulting in an autoimmune response.
The role of EVs in the development of autoimmune disease has been studied in patients and animal models. A review article by Tian et al. recently examined the role of EVs in a number of autoimmune diseases including thyroiditis, systemic lupus erythematosus (SLE), multiple sclerosis (MS), rheumatoid arthritis (RA), anti-phospholipid syndrome and type I diabetes (23). Many investigators have reported that the number of circulating EVs are elevated in patients with autoimmune disease compared to controls (24–26); however, the wide variety of methods and procedures for isolating EVs as well as differences in storage conditions makes it difficult to interpret these findings.
Studies examining changes in EV content and function may provide a clearer picture of their effects in patients with autoimmune disease. MicroRNA (miRs) content in blood EVs (exosomes) were identified as biomarkers that distinguished patients with relapsing-remitting MS (i.e., miR-15b-5p, miR-451a, miR-30b-5p, miR-342-3p) from those with progressive MS or healthy controls (27, 28). Eight out of the nine miRs that were identified in the study were confirmed in a separate group of patients indicating that the miRs/EVs could serve as biomarkers to predict MS type. Similar results have been found for other autoimmune diseases like type I diabetes (29). EVs have also been found to either promote inflammation/remodeling or to inhibit harmful immune responses for a number of autoimmune diseases including RA (30–33), Hashimoto’s thyroiditis (34), type I diabetes (35), SLE (36), and myocarditis (15). Additionally, EVs have been found with immunoglobulins (Ig) on their surface including IgG or internally in the form of self-antigen-complement-Ig immune complexes (ICs) (37, 38) suggesting that EVs may initiate and/or promote autoimmune damage and inflammation via ICs (39–42). Understanding the role of EVs in autoimmune disease is an emerging field with many questions still to be answered.
Another form of EVs that have received recent attention and may play a role in autoimmune disease are those that contain host cellular components such as mitochondria (e.g., primarily mitochondrial proteins or RNA) (43). The earliest evidence of mitochondria and mitochondrial components in vesicles comes from a description by Vishwa Nath in 1932 of work by Koltzoff in 1906 studying sperm cells from Paratelphusa spinigera (44). Koltzoff and Nath observed sub-cellular structures in crab spermatocytes undergoing a process that sounds similar to our current understanding of mitochondrial-derived vesicles (MDVs) (intracellular vesicles for mitochondrial transport) (44–46) or mitophagosomes (mitochondria fission products contained in autophagosomes for selective autophagy) (47, 48). Both MDVs and fragmented mitochondria fission products can be sent to autophagosomes for selective autophagy (46, 49) in a specific lysosomal degradation of mitochondria process referred to as ‘mitophagy’ (49). The formation of the endoplasmic reticulum barrier around fragmented mitochondrial pieces (i.e., the autophagosome), a process that occurs in receptor-mediated mitophagy, is what Nath suspected protected these structures (which he only knew as another membrane around a mitochondrial mass) from rupture and lysis when exposed to acetic acid (44). Another process that has been referred to as ‘mitoptosis’ involves selective removal of damaged mitochondria from the cell in vesicles (i.e., EVs) that are generally referred to as mitochondrial EVs (50) or mitovesicles (51) that contain whole or pieces of mitochondria (52, 53). See images of mitochondrial EVs budding from cardiac myocytes in Figure 1 (54). Importantly, this process can occur for healthy physiological removal or transfer of mitochondria as well as for damaged mitochondria (55).
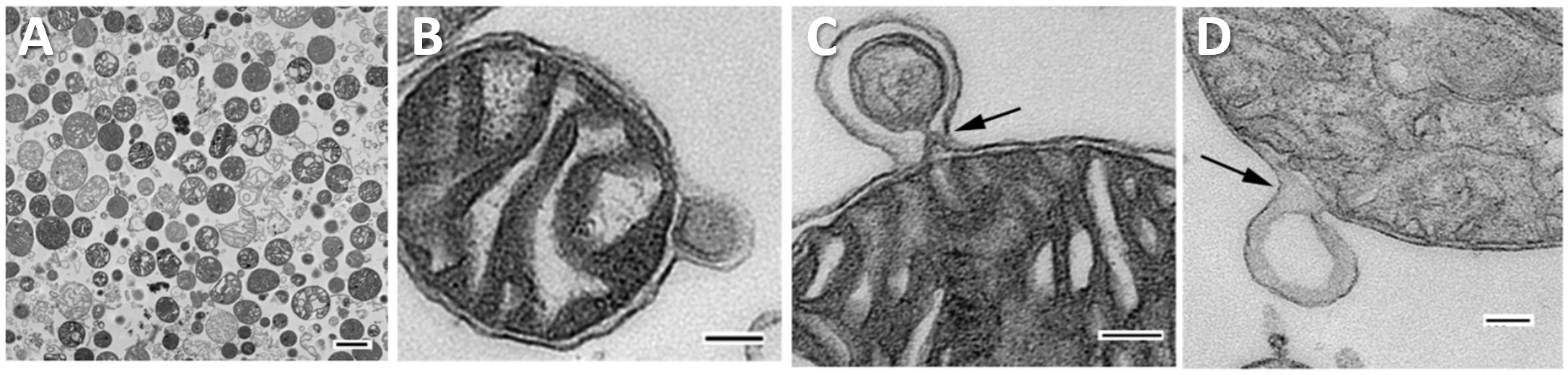
Figure 1 Mitochondrial derived vesicles (MDVs) from cardiac mitochondria. Widefield transmission electron microscopic images of mitochondria isolated from bovine heart. (A) 60-100 nm vesicles containing mitochondria. Scale bar, 500 nm, (B) MDV budding from mitochondria containing inner and outer mitochondrial membrane, (C) Protrusion of MDV from mitochondria showing constriction at its base, (D) MDV forming with only outer mitochondrial membrane. Panels (B–D), scale bars 100 nm. Reused with permission from (54).
EVs that contain mitochondria lack standardized definitions but are known to contain inner and outer mitochondrial membrane components, mitochondrial nucleic acid (i.e., DNA, RNA), and/or cardiolipin - a signature phospholipid that is more concentrated in mitochondrial membranes than cellular membranes (9). The two known major populations of mitochondrial EVs also differ in terms of their size: MDVs are smaller (30-100 nm) and EVs containing larger mitochondrial components or whole mitochondria have a larger size.
For decades, small non-enveloped RNA viruses like CVB3 were thought to cause host cell lysis as the primary method of viral dissemination, but recent evidence has demonstrated that infectious CVB3 and viral particles are released in mitochondrial EVs (47, 56). The first evidence that CVB3 infection disrupts cardiac mitochondria was published in 1964 using young Swiss white mice (Webster strain) inoculated with tissue culture-derived virus (57). Investigators utilized microscopy to assess subcellular changes to the myocardium during viral infection. Notably, they identified an increase in mitochondrial fission, disruption of mitochondrial cristae, and smaller mitochondria with additional membranes that enclosed them that likely depict mitophagosomes (57). A later study from 1997 identified CVB3 localization around and within cardiac mitochondria during myocarditis in mice (Figure 2) (58).
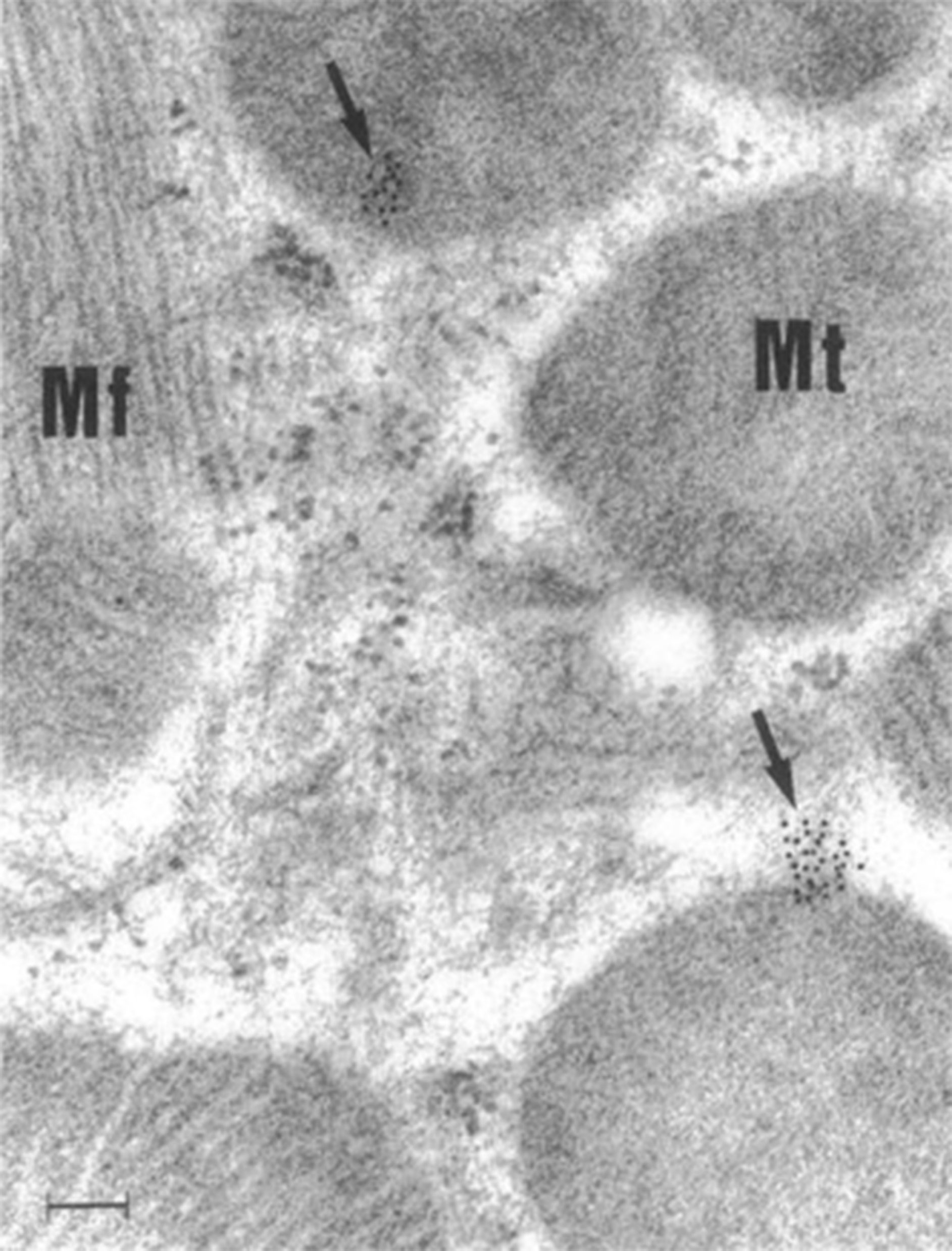
Figure 2 CVB3 localizes around and within murine cardiac mitochondria during myocarditis. Immunogold electron micrograph of mouse cardiomyocyte with CVB3 myocarditis on day 8 post infection. Black dots (arrows) are gold staining of CVB3 viral genome localizing around and in cardiomyocyte mitochondria. Scale bar, 100 nm. Mt, mitochondria; Mf, myofibril. Reused with permission from (58).
In 2017, Roberta Gottlieb’s laboratory published a study demonstrating CVB3 viral transmission via EVs containing mitochondrial components (47). Using an in vitro neural progenitor stem cell model of viral infection, Robinson et al. demonstrated that CVB3 localizes to the mitochondrial compartment of infected cells and is later ejected from cells in vesicles containing virus, inner and outer mitochondrial membrane components, and autophagy machinery (i.e., microtubule-associated protein light chain 3/LC3-II) (47). Using electron microscopy, they found these particles ranged in size from 100-200 nm in diameter and contained single or multiple virions (Figure 3) (47). They also observed that the ejected particles were infectious to adjacent uninfected host cells.

Figure 3 CVB3 identified in EVs using transmission electron microscopy. (A) Widefield transmission electron microscopic view of single virion (green arrow) in an extracellular EV or free virion (pink arrow) from culture of CVB3 in C2C12 cells. (B) Higher digital magnification (dashed purple box) of a virus-like particle revealed an icosahedral shape structure (dashed green polygon) slightly larger than 31 nm in diameter enclosed within a membrane structure. (C) Large EV containing multiple virions (green arrows). Scale bars = 100 nm. Reused with permission from (47).
The protein dynamin-related protein 1 (Drp1) is required for mitochondria to undergo fission. Drs. Gottlieb and Sin showed that CVB3 infection led to Drp1-induced mitochondrial fission resulting in damaged mitochondria being processed into mitophagosomes via mitophagy and released from host HL-1 cardiomyocytes in culture as mitochondrial EVs (48). The role of fission in the production of mitochondrial vesicles was confirmed by inhibition/blocking mitochondrial fission machinery using mitochondrial division inhibitor-1 (Mdivi-1) or direct inhibition of Drp1 with siRNA which resulted in less viral replication and fewer/no virus containing EVs in the culture supernatant (48). Dr. Sin’s group additionally showed that Tank binding kinase I (TBK1) increased phosphorylation of GABA type A receptor associated protein-like (GABARAPL) proteins leading to EVs that contain mitochondria being released from the cells (59). CVB3 infection has also been shown to perturb syntaxin-17 facilitated mitophagosome-lysosomal fusion, which may lead to build up and release of formed mitophagosomes from the cell (60). Thus, these studies confirm that CVB3 localizes to mitochondria and is released in mitochondrial vesicles. Further research is needed to better understand the molecular mechanisms of intracellular mitophagosome formation in the context of viral infections to determine how viruses take advantage of mitochondrial compartments and evade intracellular degradation by targeted autophagy. A summary of our current understanding of CVB3-mitochondria interaction and the development of EV populations containing mitochondria and virus is illustrated in Figure 4.
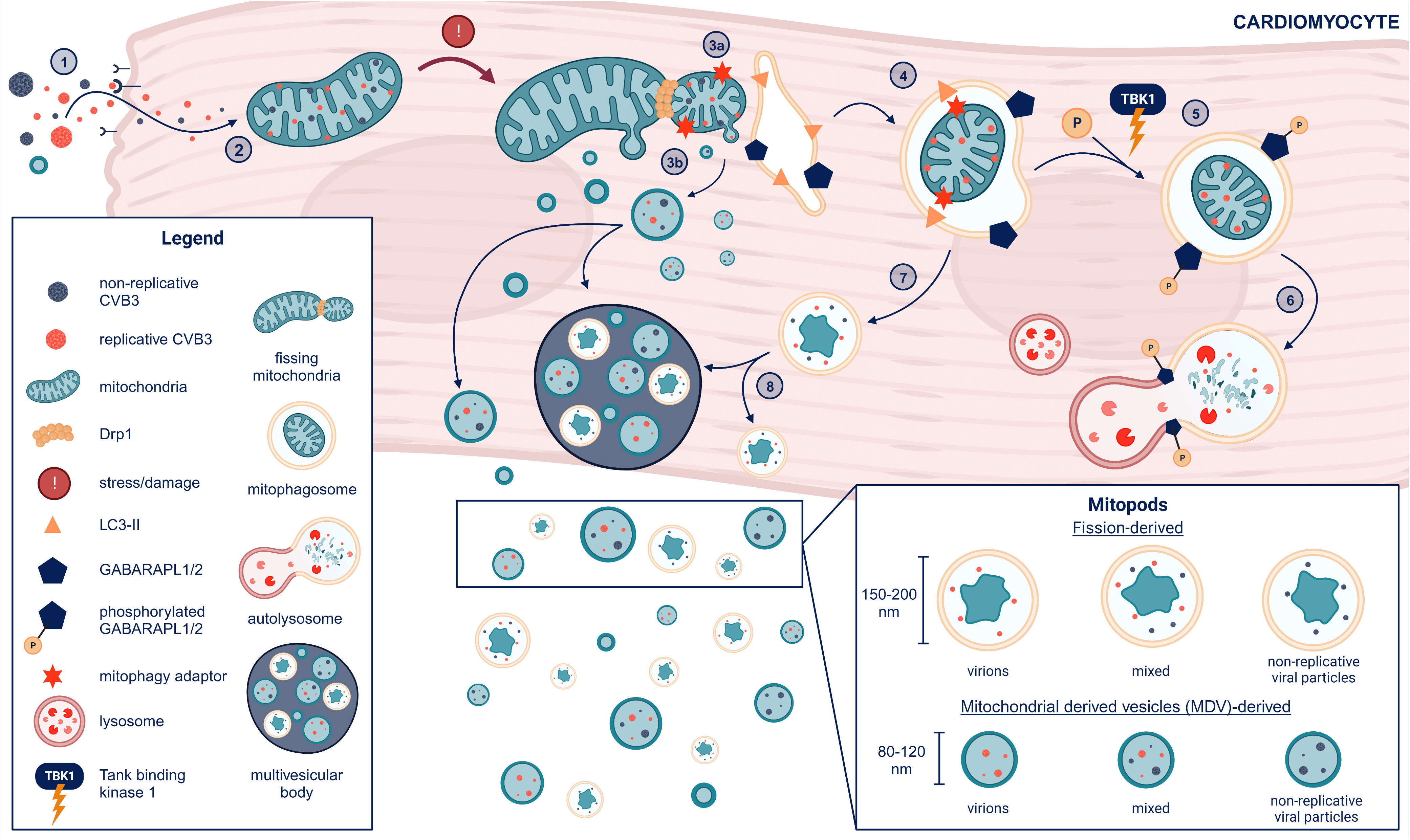
Figure 4 Formation of mitochondrial EVs from cardiomyocytes after CVB3 infection (1). CVB3 gains entry to cardiomyocytes via the coxsackievirus adrenoreceptor (CAR) or passive entry from previously formed mitochondrial EVs containing replicative virus (2). CVB3 mitochondrial localization induces mitochondrial stress and damage leading to (3a) mitochondrial-derived vesicle (MDV) formation and Drp1-mediated mitochondrial fission and recruitment of the endoplasmic reticulum (ER) for autophagosome formation alongside LC3 lipidation (LC3-II). (3b) MDVs containing replicative and or non-replicative viral particles may either eject from the cell or join multi-vesicular bodies before release from cardiomyocytes (MDVs can also be slated for receptor mediated mitophagy and potentially escape the cell without GABARAPL phosphorylation, which is not shown in this diagram) (4). LC3-II binds mitophagy adaptors situated on the outer mitochondrial membrane to form a mitophagosome with GABARAPL proteins on the endoplasmic reticulum (ER) facing the cytosol (5). Phosphorylation of the mitophagosome on GABARAPL proteins by tank-binding kinase 1 (TBK1) leads mitophagosomes to subsequent (6) lysosomal fusion and degradation (7). Non-phosphorylated mitophagosomes do not proceed to fusion with the lysosome but either (8) join the multivesicular body for cell release and dissemination or are ejected alone. The resulting EVs containing mitochondrial components and viral particles (replicative and/or non-replicative) we term as “mitopods” or mitochondrial escape-pods for CVB3. The two major sources of mitopods are MDV-derived or fission-derived. Another possible distinguishing feature of fission-derived versus MDV-derived mitopods would be an additional membrane derived from the ER. This figure was created using BioRender.
It turns out that many viruses are known to localize to mitochondria (61–64), utilize mitochondrial machinery for replication (48, 65), evade immune responses within EVs (66) and modify cellular processes (59, 60, 67). Importantly, most of the viruses that are associated with causing myocarditis [e.g., CVB, influenza, HIV, poliovirus, hepatitis C virus, SARS-CoV-22 (68, 69)] have been found to target mitochondria to gain a replicative advantage (61–64) and are ejected from cells/tissues in EVs (60, 65, 70, 71) suggesting that these mechanisms may provide an explanation for how viruses could cause autoimmune disease.
Dr. Peter Schultheiss, a major contributor to the fields of cardiology and myocarditis, began identifying and characterizing autoimmune antibodies in patients with myocarditis in the 1970s. In 1978, Bolte and Schultheiss reported that 76% of 17 patients with viral myocarditis had autoantibodies in sera and 41% of these were anti-nuclear antibodies (72). They went on to show that autoantibodies against the adenine nucleotide transporter (ANT), which is a component of the inner mitochondrial membrane, were elevated in patients with myocarditis and dilated cardiomyopathy (DCM) (73, 74). Myocarditis progresses to DCM in susceptible patients and animal models (75). Patients with suspected or confirmed viral myocarditis or cardiomyopathy/DCM had the highest reactivity to anti-mitochondrial antigen and highest expression of anti-mitochondrial antibodies (74). Further analysis found that the sera had uniquely specific reactivity toward cardiac mitochondrial antigen compared to liver or kidney mitochondrial antigen (76).
Another group independently reported that patients with various cardiomyopathies including myocarditis had autoantibodies reactive against mitochondrial proteins (77). They found that 13% with acute myocarditis, 31% of patients with DCM, and 33% with hypertrophic cardiomyopathy generated antibody responses specifically to the M7 antigen of the mitochondrial enzyme sarcosine dehydrogenase, and 25% of these reacted against the cardiac-specific form of the mitochondrial antigen (77). Another group observed autoantibodies against ANT in patients diagnosed with myocarditis or DCM (78). They also observed cardiac-specific reactivity and suggested mitochondrial autoimmune activity as a potential mechanism for the development of DCM following acute myocarditis (78). This agrees with our current understanding of the development of DCM following acute myocarditis (75, 79, 80).
Although viral-induced myocarditis is often categorized as a distinct condition from autoimmune myocarditis clinically and in animal models, the distinction between the two conditions is not clear-cut because patients with viral myocarditis and mouse models of viral myocarditis have been demonstrated to develop autoantibodies and autoreactive T and B cells against cardiac myosin and other self-antigens including mitochondria (2, 39, 79, 81–83).
Importantly, a study examining autoantibody levels that compared experimental autoimmune myocarditis (EAM) to CVB3-induced myocarditis in mice found that ANT was only produced after viral infection but not in EAM suggesting that viral infection was necessary for the production of mitochondrial autoantibodies whereas both models produced autoantibodies against cardiac myosin (84). Additionally, Lin et al. showed that depletion of Drp1 (required for fission) in mice using the mitochondrial fission inhibitor Mdivi-1 reduced CVB3 myocarditis and restored mitochondrial function in the heart (71) suggesting that mitochondrial EVs containing virus may increase myocarditis, although they did not examine this in the study. Overall, these findings suggest that viral infection may be an important mechanism to produce mitochondrial autoantibodies found in patients with autoimmune diseases.
Anti-mitochondrial antibodies (for example, antibodies that target cardiolipin, mitofusin 1, mitochondrial DNA or mitochondrial RNA) are commonly found in patients with rheumatic autoimmune diseases such as RA, SLE, and anti-phospholipid syndrome (85–87). Mobarrez et al. reports most larger EVs (0.7 - 3.0 μm) found in SLE patients contain functional mitochondrial components, as indicated by the presence of the translocase of outer mitochondrial membrane 20 (TOMM20) and hexokinase1 (25). Elevated levels of these type of EVs containing mitochondria are positively associated with increased SLE disease activity, proinflammatory cytokines, and anti-dsDNA antibodies, suggesting that these EVs may be involved in disease pathogenesis (25). Becker et al. recently reviewed the mechanism of immune activation leading to autoimmune disease by mitochondria in these rheumatic conditions but does not discuss the potential role of viral infections in the process or whether the mitochondrial EVs also contain virus or viral components (85). These findings suggest that damage to mitochondria resulting in autoimmune responses may be a common mechanism in the pathogenesis of many autoimmune diseases.
One possible mechanism where myocarditis and other autoimmune diseases could be induced and/or exacerbated by mitochondrial EVs is by activation of Toll-like receptor (TLR)4, interleukin (IL)-1β and leucin-rich repeat (LRR)-containing protein (NLRP)3, which is a pathway that has been demonstrated to increase myocarditis and viral replication in CVB3 models of myocarditis (88, 89). Mitochondria are known to be immunogenic and the TLR4/NLRP3 signaling pathway can be activated by mitochondrial components such as cytochrome c, mitochondrial transcription factor A (TFAM), ATP and cardiolipin, which can all be found in mitochondrial EVs, to initiate a proinflammatory and profibrotic immune response (90–94). Mitochondrial and viral antigens may be expressed on the cell surface or interior of EVs and activate APCs via MHC class II presentation, TLRs or be processed for presentation after the EV lipid membrane has ‘merged’ with an APC (23).
However, not all mitochondria found in EVs stimulate the innate immune response. In some cases, healthy mitochondria within EVs are found to fuse with recipient mitochondria in cultured cardiomyocytes and in a mouse model of myocardial infarction where they improve mitochondrial function and disease (95). These investigators showed a similar improvement in doxorubicin-induced toxicity in cultured cardiomyocytes (96). Thus, transfer of healthy intact mitochondria within EVs represents a novel and potentially viable therapy for patients with mitochondrial damage or dysfunction (97, 98).
As mentioned earlier, a key feature of the immune response that protects against the development of autoimmunity is the generation of tolerance to self-antigens that occurs in the thymus (1). Since the generation of T cell receptors (TCRs) in the thymus is a random process, negative selection of T cells that react too strongly to self-antigen is required to prevent autoimmunity. To determine whether there are too many self-reactive T cells, the thymus utilizes the autoimmune regulator gene (AIRE) and dendritic cells (99, 100).
AIRE is a transcriptional regulator that protects against self-reactivity by inducing the production of tissue-specific antigens normally not expressed in the thymus, a process that occurs in medullary thymic epithelial cells (mTECs) (99). Resident dendritic cells of the thymus take up self-proteins and present them to T cells. If reactivity to self-antigen is too strong, dendritic cells undergo cytokine signalling programs that destroy autoreactive T cells (99). Migratory and peripheral dendritic cell populations further contribute to negative selection of autoreactive T cells by selecting against cells reactive to peripheral antigen from other tissue microenvironments. Migratory dendritic cells take up antigen in their respective resident tissues and travel to the thymus whereas peripheral dendritic cells test autoreactive T cells in the periphery (i.e., their tissue of origin) thereby inducing, depending on the conditions, cell deletion, anergy or polarization toward a regulatory phenotype (100). It is estimated that there are 1,140 murine genes that interact with or localize to mitochondrial compartments (101), so AIRE should protect the host from developing autoimmunity against these mitochondrial antigens. One important question is whether mitochondrial genes represent a gap in the normal negative selection criteria in the thymus.
To our knowledge, no studies examining the function of AIRE describe its ability to produce mitochondrial self-antigen. Two major studies exist with publically available data of AIRE genomic binding and related expression (102, 103). A study by Bansal et al. reported murine transcriptomic data that examined AIRE binding using chromatin immunoprecipitation (ChIP) sequencing (ChIPseq). We examined whether any of the antigens that they reported for AIRE were directed against mitochondrial antigens using their published transcript-level data. They did not have protein/proteomic level data available to assess this question. They estimated AIREs coverage of genes by assessing transcription among AIRE knockout (KO) mice versus wild type (WT) controls in data derived from mouse mTECs (103). A negative log fold-change (LogFC) indicated downregulation of the transcript in the AIRE KO mice, suggesting that in WT conditions, AIRE may be responsible, in part, for regulating the transcription of a respective gene. To determine AIRE regulation of mitochondrial related genes, we performed a keyword search for “mitochondri” (which yielded results for mitochondrial, mitochondria, and mitochondrion) in the gene description column of their dataset, which yielded 315 genes. Among these, 20 were significantly downregulated at an FDR p ≤ 0.05 comparing AIRE WT to KO indicating that AIRE may regulate only 6.3% of the 315 mitochondrial related genes. We also assessed the potential role of AIRE to regulate 85 murine nuclear-encoded mitochondrial respiratory chain genes using keyword searches in the gene name column for “nduf,” “sdh,” “cox,” “uqcc,” and “atp,” which are the prefixes for gene names among components of respiratory complexes I-V, respectively. Their data showed a significant downregulation of 2 of 85 (2.3%) nuclear encoded mitochondrial respiratory chain genes in AIRE KO vs WT samples. Mitochondrial genes that were regulated by AIRE are listed in Table 1. Thus, only a small percentage of potential mitochondrial genes were regulated by AIRE using this method. More research is needed to better understand whether AIRE contributes to tolerance against mitochondrial antigens. Thus, a lack of mitochondrial tolerance may be one possible explanation for the development of mitochondrial targeted autoimmune responses in myocarditis and other autoimmune diseases.
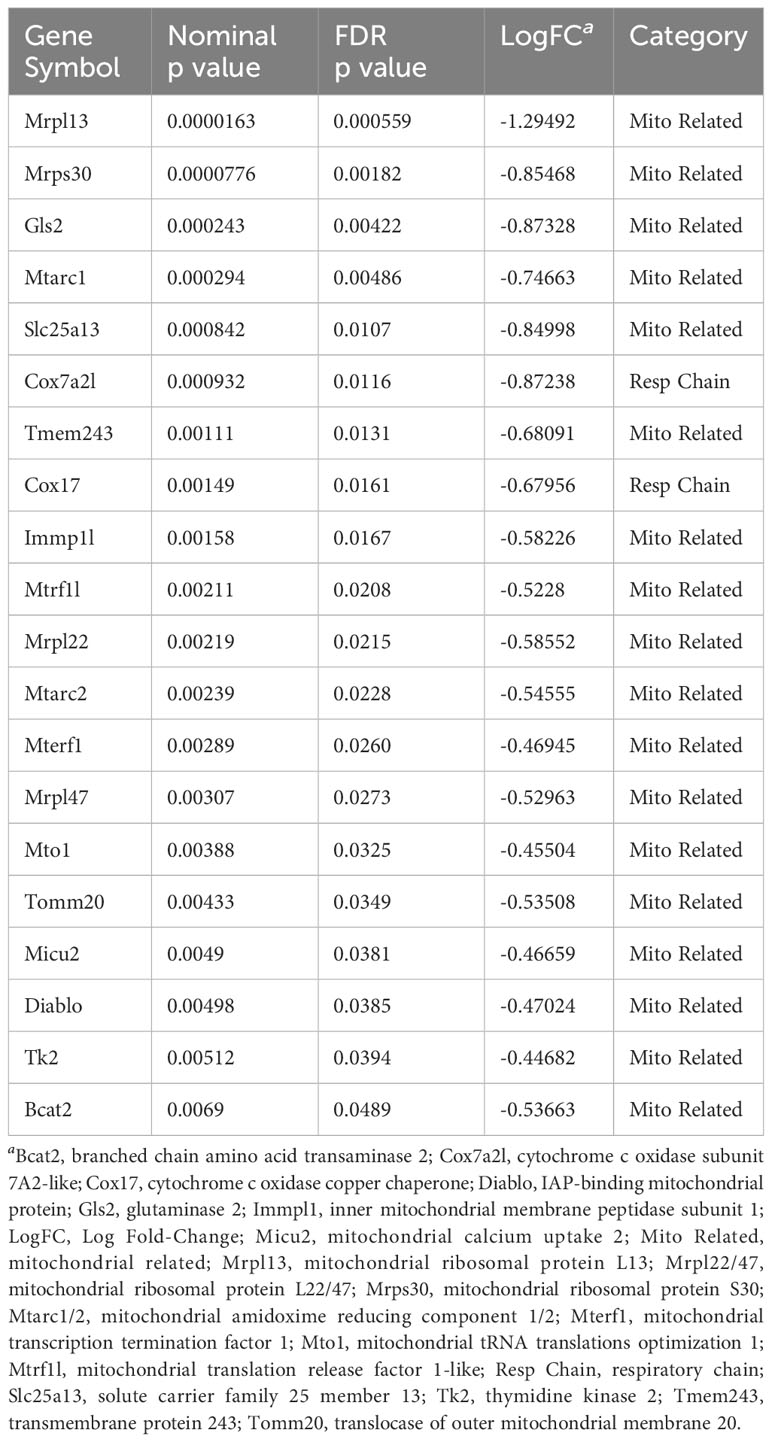
Table 1 Mitochondria related and respiratory complex genes expressed by AIRE in mice from Bansal et al. (103) in order of FDR p value.
In summary, we propose the following possible scenario for the role of mitochondrial EVs in the induction of autoimmune diseases like myocarditis. The initial infection with virus will activate antiviral TLRs like TLR3, 7, 8, 9 in the first few minutes/hours after infection. The virus will traffic to the mitochondria at the local site of infection and mitochondria within the virus’ favorite cell type/primary tropism to obtain a replicative advantage. The virus will be released from the cell in mitochondrial EVs. Mitochondrial components expressed within or on the surface of the EVs then activate TLR4 on APCs. The presence of virus/viral particles and mitochondrial components together may create a strong ‘adjuvant’ effect to activate the immune response. During the viremic stage of viral replication, which typically occurs in the first few days after viral infection, the virus within EVs can traffic through the bloodstream or lymphatics to the heart where infection of cardiac tissues can occur in a non-viral receptor specific manner via EVs or also with viral receptors if they are present in cardiac tissue. For example, CVB3 may enter cardiac cells via coxsackievirus-adenovirus receptor (CAR) which is expressed in the heart. Release of mitochondrial EVs from mitochondrially rich cardiomyocytes may drive a cardiac-specific autoimmune response because the mitochondrial content in/on EVs may contain heart specific mitochondrial antigens. TLR4 signaling has been found to be an important pathway in the pathogenesis of many autoimmune diseases including myocarditis. Autoantibodies against mitochondrial components are found in patients with many different autoimmune diseases including myocarditis and in viral animal models of myocarditis providing evidence of an autoimmune response against mitochondria. Whether mitochondrial EVs that originate from the heart occur at a sufficient level to activate a cardiac-specific autoimmune response may be one reason why myocarditis occurs only rarely. Defects in AIRE may also confer susceptibility to autoimmune responses against mitochondrial antigens in some patients.
For decades the question of whether viruses can cause autoimmune disease has lacked a plausible explanation. Evidence exists that viral infections cause myocarditis that is also associated with autoimmune responses against the heart in patients and animal models, yet how viruses could cause autoimmunity in myocarditis is not clear. Recent evidence substantiates that many viruses, and in particular the viruses that are associated with clinical cases of myocarditis, target mitochondria to promote viral replication and to evade the immune response they are ejected from cells within EVs. Often these EVs also contain mitochondrial components. It is known that EVs contain proteins, receptors and other components that identify them as originating from self-tissue. EVs that contain replicative virus and/or virus particles and mitochondrial components may form powerful danger signals to the immune system activating TLR4- a key pathway in the pathogenesis of myocarditis and DCM. Autoantibodies against mitochondrial components and specifically cardiac mitochondria are found in patients with myocarditis and DCM providing insight that viral infections may promote the release of mitochondrial antigens to activate an autoimmune response. Additionally, defects in AIRE may allow heightened self-reactivity against mitochondrial antigens. These mechanisms provide an explanation for how viral infections may initiate or promote autoimmune diseases like myocarditis.
DD: Conceptualization, Data curation, Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. DB: Conceptualization, Visualization, Writing – review & editing. EM: Writing – review & editing. JS: Conceptualization, Writing – review & editing. TI: Writing – review & editing. DF: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported in part by the NIH grant R01 HL164520 to DF and NIH TL1 TR002380 to DD, DB and DF.
We utilized previously published images from Robinson et al., 2014 (47) and Soubannier et al., 2012 (54) which are reprinted with copyright permissions via Creative Commons CC-BY-license. The image from Ukimura et al., 1997 (58) was reprinted after obtaining a license from Elsevier. We thank Dr. Heidi McBride for support and input on this review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sogkas G, Atschekzei F, Adriawan IR, Dubrowinskaja N, Witte T, Schmidt RE. Cellular and molecular mechanisms breaking immune tolerance in inborn errors of immunity. Cell Mol Immunol. (2021) 18:1122–40. doi: 10.1038/s41423-020-00626-z
2. Fairweather D, Kaya Z, Shellam GR, Lawson CM, Rose NR. From infection to autoimmunity. J Autoimmun. (2001) 16:175–86. doi: 10.1006/jaut.2000.0492
3. Shamim EA, Miller FW. Familial autoimmunity and the idiopathic inflammatory myopathies. Curr Rheumatol Rep. (2000) 2:201–11. doi: 10.1007/s11926-000-0080-0
4. Fairweather D, Root-Bernstein R. Autoimmune disease: Mechanism. In: Encyclopedia of life sciences. John Wiley & Sons, Ltd, Chichester, UK (2015). p. 1–7. doi: 10.1002/9780470015902.a0020193.pub2
5. Fairweather D, Rose NR. Type 1 diabetes: Virus infection or autoimmune disease? Nat Immunol. (2002) 3:338–40. doi: 10.1038/ni0402-338
6. Thomas OG, Olsson T. Mimicking the brain: Epstein-Barr virus and foreign agents as drivers of neuroimmune attack in multiple sclerosis. Front Immunol. (2023) 14:1304281. doi: 10.3389/fimmu.2023.1304281
7. Ciferri MC, Quarto R, Tasso R. Extracellular vesicles as biomarkers and therapeutic tools: from pre-clinical to clinical applications. Biol (Basel). (2021) 10:359. doi: 10.3390/biology10050359
8. Beetler DJ, Di Florio DN, Law EW, Groen CM, Windebank AJ, Peterson QP, et al. The evolving regulatory landscape in regenerative medicine. Mol Aspects Med. (2023) 91:101138. doi: 10.1016/j.mam.2022.101138
9. Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. (2018) 7:1535750. doi: 10.1080/20013078.2018.1535750
10. Witwer KW, Goberdhan DC, O'Driscoll L, Thery C, Welsh JA, Blenkiron C, et al. Updating MISEV: Evolving the minimal requirements for studies of extracellular vesicles. J Extracell Vesicles. (2021) 10:e12182. doi: 10.1002/jev2.12182
11. Yates AG, Pink RC, Erdbrugger U, Siljander PR, Dellar ER, Pantazi P, et al. In sickness and health: The functional role of extracellular vesicles in physiology and pathology in in vivo: Part II: Pathology. J Extracell Vesicles. (2022) 11:e12190. doi: 10.1002/jev2.12190
12. Rodrigues M, Fan J, Lyon C, Wan M, Hu Y. Role of extracellular vesicles in viral and bacterial infections: Pathogenesis, diagnostics, and therapeutics. Theranostics. (2018) 8:2709–21. doi: 10.7150/thno.20576
13. Admyre C, Bohle B, Johansson SM, Focke-Tejkl M, Valenta R, Scheynius A, et al. B cell-derived exosomes can present allergen peptides and activate allergen-specific T cells to proliferate and produce TH2-like cytokines. J Allergy Clin Immunol. (2007) 120:1418–24. doi: 10.1016/j.jaci.2007.06.040
14. Beetler DJ, Di Florio DN, Bruno KA, Ikezu T, March KL, Cooper LT Jr., et al. Extracellular vesicles as personalized medicine. Mol Aspects Med. (2023) 91:101155. doi: 10.1016/j.mam.2022.101155
15. Beetler DJ, Bruno KA, Watkins MM, Xu V, Chekuri I, Giresi P, et al. Reconstituted extracellular vesicles from human platelets decrease viral myocarditis in mice. Small. (2023) 19:e2303317. doi: 10.1002/smll.202303317
16. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. (1996) 183:1161–72. doi: 10.1084/jem.183.3.1161
17. Hwang I, Shen X, Sprent J. Direct stimulation of naive T cells by membrane vesicles from antigen-presenting cells: distinct roles for CD54 and B7 molecules. Proc Natl Acad Sci U S A. (2003) 100:6670–5. doi: 10.1073/pnas.1131852100
18. Thery C, Zitvogel L, Amigorena S. Exosomes: Composition, biogenesis and function. Nat Rev Immunol. (2002) 2:569–79. doi: 10.1038/nri855
19. Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, et al. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature. (2013) 496:367–71. doi: 10.1038/nature12029
20. Alenquer M, Amorim MJ. Exosome biogenesis, regulation, and function in viral infection. Viruses. (2015) 7:5066–83. doi: 10.3390/v7092862
21. Schorey JS, Harding CV. Extracellular vesicles and infectious diseases: New complexity to an old story. J Clin Invest. (2016) 126:1181–9. doi: 10.1172/JCI81132
22. Hernandez-Rodriguez J, Alba MA, Prieto-Gonzalez S, Cid MC. Diagnosis and classification of polyarteritis nodosa. J Autoimmun. (2014) 48–49:84–9. doi: 10.1016/j.jaut.2014.01.029
23. Tian J, Casella G, Zhang Y, Rostami A, Li X. Potential roles of extracellular vesicles in the pathophysiology, diagnosis, and treatment of autoimmune diseases. Int J Biol Sci. (2020) 16:620–32. doi: 10.7150/ijbs.39629
24. Sellam J, Proulle V, Jungel A, Ittah M, Miceli Richard C, Gottenberg JE, et al. Increased levels of circulating microparticles in primary Sjögren's syndrome, systemic lupus erythematosus and rheumatoid arthritis and relation with disease activity. Arthritis Res Ther. (2009) 11:R156. doi: 10.1186/ar2833
25. Mobarrez F, Fuzzi E, Gunnarsson I, Larsson A, Eketjall S, Pisetsky DS, et al. Microparticles in the blood of patients with SLE: Size, content of mitochondria and role in circulating immune complexes. J Autoimmun. (2019) 102:142–9. doi: 10.1016/j.jaut.2019.05.003
26. Saenz-Cuesta M, Irizar H, Castillo-Trivino T, Munoz-Culla M, Osorio-Querejeta I, Prada A, et al. Circulating microparticles reflect treatment effects and clinical status in multiple sclerosis. Biomark Med. (2014) 8:653–61. doi: 10.2217/bmm.14.9
27. Ebrahimkhani S, Vafaee F, Young PE, Hur SSJ, Hawke S, Devenney E, et al. Exosomal microRNA signatures in multiple sclerosis reflect disease status. Sci Rep. (2017) 7:14293. doi: 10.1038/s41598-017-14301-3
28. Selmaj I, Cichalewska M, Namiecinska M, Galazka G, Horzelski W, Selmaj KW, et al. Global exosome transcriptome profiling reveals biomarkers for multiple sclerosis. Ann Neurol. (2017) 81:703–17. doi: 10.1002/ana.24931
29. Guay C, Kruit JK, Rome S, Menoud V, Mulder NL, Jurdzinski A, et al. Lymphocyte-derived exosomal microRNAs promote pancreatic beta cell death and may contribute to type 1 diabetes development. Cell Metab. (2019) 29:348–361.e6. doi: 10.1016/j.cmet.2018.09.011
30. Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. (2010) 327:580–3. doi: 10.1126/science.1181928
31. Vinuela-Berni V, Doniz-Padilla L, Figueroa-Vega N, Portillo-Salazar H, Abud-Mendoza C, Baranda L, et al. Proportions of several types of plasma and urine microparticles are increased in patients with rheumatoid arthritis with active disease. Clin Exp Immunol. (2015) 180:442–51. doi: 10.1111/cei.12598
32. Lo Cicero A, Majkowska I, Nagase H, Di Liegro I, Troeberg L. Microvesicles shed by oligodendroglioma cells and rheumatoid synovial fibroblasts contain aggrecanase activity. Matrix Biol. (2012) 31:229–33. doi: 10.1016/j.matbio.2012.02.005
33. Headland SE, Jones HR, Norling LV, Kim A, Souza PR, Corsiero E, et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci Transl Med. (2015) 7:315ra190. doi: 10.1126/scitranslmed.aac5608
34. Cui X, Liu Y, Wang S, Zhao N, Qin J, Li Y, et al. Circulating exosomes activate dendritic cells and induce unbalanced CD4+ T cell differentiation in Hashimoto thyroiditis. J Clin Endocrinol Metab. (2019) 104:4607–18. doi: 10.1210/jc.2019-00273
35. Shigemoto-Kuroda T, Oh JY, Kim DK, Jeong HJ, Park SY, Lee HJ, et al. MSC-derived extracellular vesicles attenuate immune responses in two autoimmune murine models: Type 1 diabetes and uveoretinitis. Stem Cell Rep. (2017) 8:1214–25. doi: 10.1016/j.stemcr.2017.04.008
36. Dieker J, Hilbrands L, Thielen A, Dijkman H, Berden JH, van der Vlag J. Enhanced activation of dendritic cells by autologous apoptotic microvesicles in MRL/lpr mice. Arthritis Res Ther. (2015) 17:103. doi: 10.1186/s13075-015-0617-2
37. Cloutier N, Tan S, Boudreau LH, Cramb C, Subbaiah R, Lahey L, et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: The microparticle-associated immune complexes. EMBO Mol Med. (2013) 5:235–49. doi: 10.1002/emmm.201201846
38. Nielsen CT, Ostergaard O, Stener L, Iversen LV, Truedsson L, Gullstrand B, et al. Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheumatol. (2012) 64:1227–36. doi: 10.1002/art.34381
39. Fairweather D, Frisancho-Kiss S, Njoku DB, Nyland JF, Kaya Z, Yusung SA, et al. Complement receptor 1 and 2 deficiency increases coxsackievirus B3-induced myocarditis, dilated cardiomyopathy, and heart failure by increasing macrophages, IL-1beta, and immune complex deposition in the heart. J Immunol. (2006) 176:3516–24. doi: 10.4049/jimmunol.176.6.3516
40. Fairweather D, Petri MA, Coronado MJ, Cooper LT. Autoimmune heart disease: Role of sex hormones and autoantibodies in disease pathogenesis. Expert Rev Clin Immunol. (2012) 8:269–84. doi: 10.1586/eci.12.10
41. Ullal AJ, Reich CF 3rd, Clowse M, Criscione-Schreiber LG, Tochacek M, Monestier M, et al. Microparticles as antigenic targets of antibodies to DNA and nucleosomes in systemic lupus erythematosus. J Autoimmun. (2011) 36:173–80. doi: 10.1016/j.jaut.2011.02.001
42. Rother N, Yanginlar C, Pieterse E, Hilbrands L, van der Vlag J. Microparticles in autoimmunity: Cause or consequence of disease? Front Immunol. (2022) 13:822995. doi: 10.3389/fimmu.2022.822995
43. She Z, Xie M, Hun M, Abdirahman AS, Li C, Wu F, et al. Immunoregulatory effects of mitochondria transferred by extracellular vesicles. Front Immunol. (2020) 11:628576. doi: 10.3389/fimmu.2020.628576
44. Nath V. The spermatid and the sperm of the crab, Paratelphusa spinigera. J Cell Sci. (1932) 75:543–56. doi: 10.1242/jcs.s2-75.299.543
45. Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, et al. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol. (2008) 18:102–8. doi: 10.1016/j.cub.2007.12.038
46. Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. (2014) 33:2142–56. doi: 10.15252/embj.201488104
47. Robinson SM, Tsueng G, Sin J, Mangale V, Rahawi S, McIntyre LL, et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PloS Pathog. (2014) 10:e1004045. doi: 10.1371/journal.ppat.1004045
48. Sin J, McIntyre L, Stotland A, Feuer R, Gottlieb RA. Coxsackievirus B escapes the infected cell in ejected mitophagosomes. J Virol. (2017) 91:e01347-17. doi: 10.1128/JVI.01347-17
49. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. (2011) 12:9–14. doi: 10.1038/nrm3028
50. Jang SC, Crescitelli R, Cvjetkovic A, Belgrano V, Olofsson Bagge R, Sundfeldt K, et al. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J Extracell Vesicles. (2019) 8:1635420. doi: 10.1080/20013078.2019.1635420
51. D'Acunzo P, Perez-Gonzalez R, Kim Y, Hargash T, Miller C, Alldred MJ, et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci Adv. (2021) 7:eabe5085. doi: 10.1126/sciadv.abe5085
52. Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. (2012) 22:135–41. doi: 10.1016/j.cub.2011.11.057
53. Lyamzaev KG, Nepryakhina OK, Saprunova VB, Bakeeva LE, Pletjushkina OY, Chernyak BV, et al. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): Formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim Biophys Acta. (2008) 1777:817–25. doi: 10.1016/j.bbabio.2008.03.027
54. Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PloS One. (2012) 7:e52830. doi: 10.1371/journal.pone.0052830
55. Lyamzaev KG, Knorre DA, Chernyak BV. Mitoptosis, twenty years after. Biochem (Mosc). (2020) 85:1484–98. doi: 10.1134/S0006297920120020
56. Zhai X, Qin Y, Chen Y, Lin L, Wang T, Zhong X, et al. Coxsackievirus B3 induces the formation of autophagosomes in cardiac fibroblasts both in vitro and in vivo. Exp Cell Res. (2016) 349:255–63. doi: 10.1016/j.yexcr.2016.10.020
57. Rabin ER, Hassan SA, Jenson AB, Melnick JL. Coxsackie virus B3 myocarditis in mice. An electron microscopic, immunofluorescent and virus-assay study. Am J Pathol. (1964) 44:775–97.
58. Ukimura A, Deguchi H, Kitaura Y, Fujioka S, Hirasawa M, Kawamura K, et al. Intracellular viral localization in murine coxsackievirus-B3 myocarditis. Ultrastructural study by electron microscopic in situ hybridization. Am J Pathol. (1997) 150:2061–74.
59. Sawaged S, Mota T, Piplani H, Thakur R, Lall D, McCabe E, et al. TBK1 and GABARAP family members suppress coxsackievirus B infection by limiting viral production and promoting autophagic degradation of viral extracellular vesicles. PLoS Pathog. (2022) 18:e1010350. doi: 10.1371/journal.ppat.1010350
60. Tian L, Yang Y, Li C, Chen J, Li Z, Li X, et al. The cytotoxicity of coxsackievirus B3 is associated with a blockage of autophagic flux mediated by reduced syntaxin 17 expression. Cell Death Dis. (2018) 9:242. doi: 10.1038/s41419-018-0271-0
61. Taylor MP, Kirkegaard K. Modification of cellular autophagy protein LC3 by poliovirus. J Virol. (2007) 81:12543–53. doi: 10.1128/JVI.00755-07
62. Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. (2009) 186:255–68. doi: 10.1083/jcb.200903070
63. Gannage M, Dormann D, Albrecht R, Dengjel J, Torossi T, Ramer PC, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. (2009) 6:367–80. doi: 10.1016/j.chom.2009.09.005
64. Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG, et al. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci U S A. (2014) 111:6413–8. doi: 10.1073/pnas.1321114111
65. Oh SJ, Lim BK, Yun J, Shin OS. CVB3-mediated mitophagy plays an important role in viral replication via abrogation of interferon pathways. Front Cell Infect Microbiol. (2021) 11:704494. doi: 10.3389/fcimb.2021.704494
66. Blasius AL, Beutler B. Intracellular Toll-like receptors. Immunity. (2010) 32:305–15. doi: 10.1016/j.immuni.2010.03.012
67. Xin L, Xiao Z, Ma X, He F, Yao H, Liu Z. Coxsackievirus B3 induces crosstalk between autophagy and apoptosis to benefit its release after replicating in autophagosomes through a mechanism involving caspase cleavage of autophagy-related proteins. Infect Genet Evol. (2014) 26:95–102. doi: 10.1016/j.meegid.2014.05.005
68. Tschope C, Ammirati E, Bozkurt B, Caforio ALP, Cooper LT, Felix SB, et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat Rev Cardiol. (2021) 18:169–93. doi: 10.1038/s41569-020-00435-x
69. Fairweather D, Beetler DJ, Di Florio DN, Musigk N, Heidecker B, Cooper LT Jr. COVID-19, myocarditis and pericarditis. Circ Res. (2023) 132:1302–19. doi: 10.1161/CIRCRESAHA.123.321878
70. Chen YH, Du W, Hagemeijer MC, Takvorian PM, Pau C, Cali A, et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. (2015) 160:619–30. doi: 10.1016/j.cell.2015.01.032
71. Lin L, Zhang M, Yan R, Shan H, Diao J, Wei J. Inhibition of Drp1 attenuates mitochondrial damage and myocardial injury in coxsackievirus B3 induced myocarditis. Biochem Biophys Res Commun. (2017) 484:550–6. doi: 10.1016/j.bbrc.2017.01.116
72. Bolte HD, Schultheiss HP. Immunological results in myocardial diseases. Postgraduate Med J. (1978) 54:500–4. doi: 10.1136/pgmj.54.633.500
73. Schultheiss HP, Bolte HD. Immunological analysis of auto-antibodies against the adenine nucleotide translocator in dilated cardiomyopathy. J Mol Cell Cardiol. (1985) 17:603–17. doi: 10.1016/s0022-2828(85)80029-8
74. Schultheiss HP, Schwimmbeck P, Bolte HD, Klingenberg M. The antigenic characteristics and the significance of the adenine nucleotide translocator as a major autoantigen to antimitochondrial antibodies in dilated cardiomyopathy. Adv Myocardiol. (1985) 6:311–27.
75. Schultheiss HP, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE, et al. Dilated cardiomyopathy. Nat Rev Dis Primers. (2019) 5:32. doi: 10.1038/s41572-019-0084-1
76. Schultheiss HP, Klingenberg M. Immunochemical characterization of the adenine nucleotide translocator. Organ specificity and conformation specificity. Eur J Biochem. (1984) 143:599–605. doi: 10.1111/j.1432-1033.1984.tb08412.x
77. Klein R, Maisch B, Kochsiek K, Berg PA. Demonstration of organ specific antibodies against heart mitochondria (anti-M7) in sera from patients with some forms of heart diseases. Clin Exp Immunol. (1984) 58:283–92.
78. Ansari AA, Wang YC, Danner DJ, Gravanis MB, Mayne A, Neckelmann N, et al. Abnormal expression of histocompatibility and mitochondrial antigens by cardiac tissue from patients with myocarditis and dilated cardiomyopathy. Am J Pathol. (1991) 139:337–54.
79. Myers JM, Cooper LT, Kem DC, Stavrakis S, Kosanke SD, Shevach EM, et al. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight. (2016) 1:e85851. doi: 10.1172/jci.insight.85851
80. Fairweather D, Beetler DJ, Musigk N, Heidecker B, Lyle MA, Cooper LT Jr., et al. Sex and gender differences in myocarditis and dilated cardiomyopathy: An update. Front Cardiovasc Med. (2023) 10:1129348. doi: 10.3389/fcvm.2023.1129348
81. Caforio AL, Mahon NJ, Tona F, McKenna WJ. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: Pathogenetic and clinical significance. Eur J Heart Fail. (2002) 4:411–7. doi: 10.1016/s1388-9842(02)00010-7
82. Lv H, Havari E, Pinto S, Gottumukkala RV, Cornivelli L, Raddassi K, et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. (2011) 121:1561–73. doi: 10.1172/JCI44583
83. Gil-Cruz C, Perez-Shibayama C, De Martin A, Ronchi F, van der Borght K, Niederer R, et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science. (2019) 366:881–6. doi: 10.1126/science.aav3487
84. Neumann DA, Rose NR, Ansari AA, Herskowitz A. Induction of multiple heart autoantibodies in mice with coxsackievirus B3- and cardiac myosin-induced autoimmune myocarditis. J Immunol. (1994) 152:343–50. doi: 10.4049/jimmunol.152.1.343
85. Becker YLC, Duvvuri B, Fortin PR, Lood C, Boilard E. The role of mitochondria in rheumatic diseases. Nat Rev Rheumatol. (2022) 18:621–40. doi: 10.1038/s41584-022-00834-z
86. Burbano C, Villar-Vesga J, Orejuela J, Munoz C, Vanegas A, Vasquez G, et al. Potential involvement of platelet-derived microparticles and microparticles forming immune complexes during monocyte activation in patients with systemic lupus erythematosus. Front Immunol. (2018) 9:322. doi: 10.3389/fimmu.2018.00322
87. Chen PM, Tsokos GC. Mitochondria in the pathogenesis of systemic lupus erythematosus. Curr Rheumatol Rep. (2022) 24:88–95. doi: 10.1007/s11926-022-01063-9
88. Fairweather D, Yusung S, Frisancho S, Barrett M, Gatewood S, Steele R, et al. IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J Immunol. (2003) 170:4731–7. doi: 10.4049/jimmunol.170.9.4731
89. Eriksson U, Kurrer MO, Sonderegger I, Iezzi G, Tafuri A, Hunziker L, et al. Activation of dendritic cells through the interleukin 1 receptor 1 is critical for the induction of autoimmune myocarditis. J Exp Med. (2003) 197:323–31. doi: 10.1084/jem.20021788
90. Liu Q, Zhang D, Hu D, Zhou X, Zhou Y. The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol. (2018) 103:115–24. doi: 10.1016/j.molimm.2018.09.010
91. Schindler SM, Frank MG, Annis JL, Maier SF, Klegeris A. Pattern recognition receptors mediate pro-inflammatory effects of extracellular mitochondrial transcription factor A (TFAM). Mol Cell Neurosci. (2018) 89:71–9. doi: 10.1016/j.mcn.2018.04.005
92. Wenzel TJ, Bajwa E, Klegeris A. Cytochrome c can be released into extracellular space and modulate functions of human astrocytes in a Toll-like receptor 4-dependent manner. Biochim Biophys Acta Gen Subj. (2019) 1863:129400. doi: 10.1016/j.bbagen.2019.07.009
93. Bajwa E, Pointer CB, Klegeris A. The role of mitochondrial damage-associated molecular patterns in chronic neuroinflammation. Mediators Inflamm. (2019) 2019:4050796. doi: 10.1155/2019/4050796
94. Murray TE, Wenzel TJ, Simtchouk S, Greuel BK, Gibon J, Klegeris A. Extracellular cardiolipin modulates select immune functions of astrocytes in Toll-like receptor (TLR) 4-dependent manner. Mediators Inflamm. (2022) 2022:9946439. doi: 10.1155/2022/9946439
95. Ikeda G, Santoso MR, Tada Y, Li AM, Vaskova E, Jung JH, et al. Mitochondria-rich extracellular vesicles from autologous stem cell-derived cardiomyocytes restore energetics of ischemic myocardium. J Am Coll Cardiol. (2021) 77:1073–88. doi: 10.1016/j.jacc.2020.12.060
96. O'Brien CG, Ozen MO, Ikeda G, Vaskova E, Jung JH, Bayardo N, et al. Mitochondria-rich extracellular vesicles rescue patient-specific cardiomyocytes from doxorubicin injury: Insights into the SENECA Trial. JACC CardioOncol. (2021) 3:428–40. doi: 10.1016/j.jaccao.2021.05.006
97. Liu D, Gao Y, Liu J, Huang Y, Yin J, Feng Y, et al. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct Target Ther. (2021) 6:65. doi: 10.1038/s41392-020-00440-z
98. Clemente-Suarez VJ, Martin-Rodriguez A, Yanez-Sepulveda R, Tornero-Aguilera JF. Mitochondrial transfer as a novel therapeutic approach in disease diagnosis and treatment. Int J Mol Sci. (2023) 24:8848. doi: 10.3390/ijms24108848
99. Anderson MS, Su MA. AIRE expands: New roles in immune tolerance and beyond. Nat Rev Immunol. (2016) 16:247–58. doi: 10.1038/nri.2016.9
100. Hasegawa H, Matsumoto T. Mechanisms of tolerance induction by dendritic cells in vivo. Front Immunol. (2018) 9:350. doi: 10.3389/fimmu.2018.00350
101. Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. (2021) 49:D1541–D7. doi: 10.1093/nar/gkaa1011
102. LaFlam TN, Seumois G, Miller CN, Lwin W, Fasano KJ, Waterfield M, et al. Identification of a novel cis-regulatory element essential for immune tolerance. J Exp Med. (2015) 212:1993–2002. doi: 10.1084/jem.20151069
Keywords: autoimmune disease, extracellular vesicles, mitochondria, mitochondrial-derived vesicles, myocarditis, AIRE, coxsackievirus
Citation: Di Florio DN, Beetler DJ, McCabe EJ, Sin J, Ikezu T and Fairweather D (2024) Mitochondrial extracellular vesicles, autoimmunity and myocarditis. Front. Immunol. 15:1374796. doi: 10.3389/fimmu.2024.1374796
Received: 22 January 2024; Accepted: 28 February 2024;
Published: 14 March 2024.
Edited by:
Chun Wai Mai, UCSI University, MalaysiaReviewed by:
Giorgio Mangino, Sapienza University of Rome, ItalyCopyright © 2024 Di Florio, Beetler, McCabe, Sin, Ikezu and Fairweather. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: DeLisa Fairweather, RmFpcndlYXRoZXIuRGVMaXNhQG1heW8uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.