 Hui Zhao1†
Hui Zhao1† Jialin Song2†Xiujun Li3Zhaoyi Xia4,5Qian Wang1Jiaqi Fu1
Jialin Song2†Xiujun Li3Zhaoyi Xia4,5Qian Wang1Jiaqi Fu1 Yuqing Miao1*Dapeng Wang6*Xuguang Wang2*
Yuqing Miao1*Dapeng Wang6*Xuguang Wang2*- 1School of Materials and Chemistry, Institute of Bismuth and Rhenium, University of Shanghai for Science and Technology, Shanghai, China
- 2Department of Limb Trauma, Wendeng Orthopaedic Hospital of Shandong Province, Weihai, Shandong, China
- 3Department of Medicine, Chifeng University, Chifeng, China
- 4Department of Library, Children's Hospital Affiliated to Shandong University, Jinan, Shandong, China
- 5Department of Library, Jinan Children's Hospital, Shandong, Jinan, Shandong, China
- 6Department of Intensive Medicine, Wuxi People’s Hospital Affiliated to Nanjing Medical University, Wuxi, Jiangsu, China
Pulmonary hypertension (PH) is a malignant disease with progressive increase of pulmonary vascular pressure, which eventually leads to right heart failure. More and more evidences show that immune cells and inflammation play an important role in the occurrence and development of PH. In the context of pulmonary vascular diseases, immune cells migrate into the walls of the pulmonary vascular system. This leads to an increase in the levels of cytokines and chemokines in both the bloodstream and the surrounding tissues of the pulmonary vessels. As a result, new approaches such as immunotherapy and anti-inflammatory treatments are being considered as potential strategies to halt or potentially reverse the progression of PH. We reviewed the potential mechanisms of immune cells, cytokines and chemokines in PH development. The potential relationship of vascular cells or bone morphogenetic protein receptor 2 (BMPR2) in immune regulation was also expounded. The clinical application and future prospect of immunotherapy were further discussed.
1 Introduction
Pulmonary hypertension (PH) is a condition characterized by changes in the structure and function of the pulmonary vasculature, resulting in an increase in pulmonary vascular resistance and arterial pressure (1, 2). If left untreated, PH can progress to a severe form of right heart failure or even death. The underlying pathophysiology of PH is complex and not yet fully understood. However, it is widely accepted that the development and progression of PH are closely associated with vascular remodeling (3). In physiological state, the contraction and relaxation of blood vessels maintain a balanced state (4, 5). When immune inflammatory reaction occurs, various inflammatory factors and oxygen free radicals are produced to damage endothelium, leading to its dysfunction (6). The balance between vasodilating substances such as NO and vasoconstricting substances such as endothelin (ET) is broken, and the contractility of pulmonary blood vessels is abnormally increased (7–9). It is reported that the thickness of intima, media and adventitia and the average pulmonary artery pressure are correlated with the average perivascular inflammation score, which supports the role of perivascular inflammation in pulmonary vascular remodeling (10). In addition, the study also showed that the inflammatory pathology is more advanced when the bone morphogenetic protein type 2 receptor (BMPR2) is mutated. In the experimental PH, the fact that inflammation precedes vascular remodeling indicates that immune changes are the cause of vascular diseases rather than the result (11).
Further analysis of immune function in patients with PH proved that immune response exists in the process of PH occurrence and development (12). This can explain the accumulation of inflammatory cells around vessels and the excess of cytokines and chemokines (13, 14). In reality, maintaining a delicate balance between immunity and tolerance is crucial. Any disruption to this balance can result in chronic inflammation or even autoimmune disorders (15, 16). This review summarized the evidence and potential mechanism of immune cells and inflammation in PH in recent years. In addition, the potential relationship between vascular cells or BMPR2 in immune regulation was expounded. It is suggested that immunity and inflammation may be the key factors and promising therapeutic targets for PH development in Figure 1.
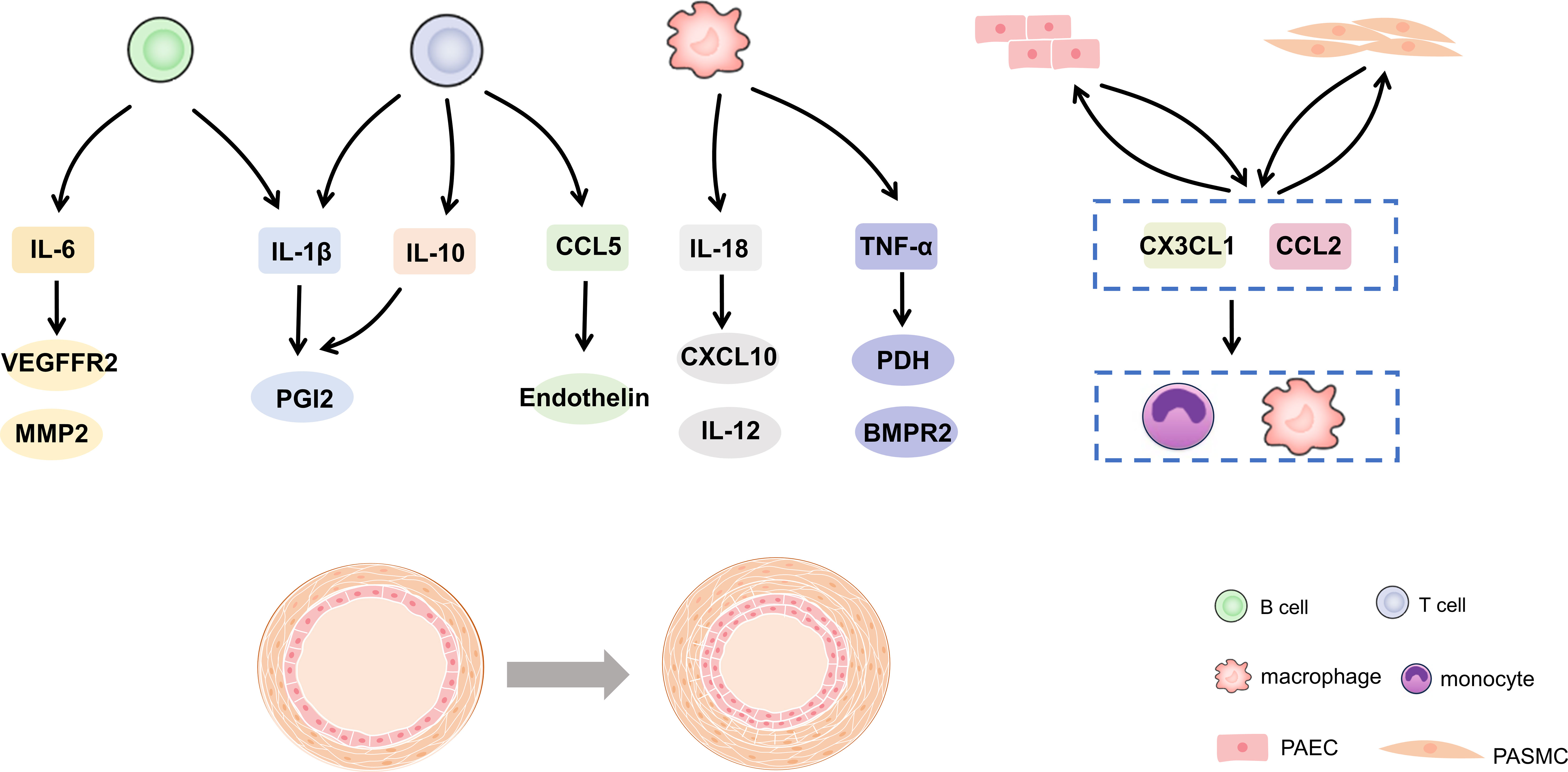
Figure 1 Cytokines and chemokines in pulmonary hypertension. IL, interleukin; CCL, C-C motif chemokine ligand; TNF, tumor necrosis factor-alpha; PGI2, prostacyclin; VEGFR2, vascular endothelial growth factor receptor 2; MMP, matrix metalloprotein; CXCL, C-X-C motif chemokine ligand; PDH, pyruvate dehydrogenase; BMPR2, bone morphogenetic protein type 2 receptor.
2 Immune cells and PH
Pulmonary vascular remodeling in pulmonary arterial hypertension (PAH) patients and PH animal models is often accompanied by different degrees of perivascular inflammatory infiltration, including T cells, B cells, macrophages, dendritic cells (DC), mast cells and neutrophils. This suggested that these immune cells may play an important role in the process of pulmonary vascular remodeling.
2.1 T cell
T cells are an important part of adaptive immune response and play an important role in the pathogenesis of PH, mainly helper T cells (Th cells) and regulatory T cells (Tregs) (17). Different types of T cells have specific functions and reactions in the inflammatory cascade reaction. Th cells produce pro-inflammatory response, while Tregs play a balanced response to achieve self-tolerance and prevent autoimmune (18). Studies have confirmed that the balance and homeostasis of T cells and their cytokines can prevent the loss of self-tolerance, and then affect the development of inflammation and PAH.
Based on the pro-inflammatory substances they release, Th cells can be categorized into different subsets, including Th1, Th2, and Th17 cells. Th1 and Th17 cells are capable of promoting inflammation through the production of various cytokines such as interleukin-6 (IL-6), IL-2, IL-21, interferon-gamma (IFN-γ), and tumor necrosis factor-alpha (TNF-α) (19, 20). The levels of peripheral Th17 cells, cytokines, and mRNA in patients with connective tissue diseases-associated PAH (CTD-PAH) were elevated (21). More importantly, the Th17/Tregs ratio was significantly related to the severity and prognosis of CTD-PAH (21). Moreover, the expression of T cell activation markers CD86 and CD40 was higher in idiopathic PAH (IPAH) patients following pretreatment with dexamethasone (22). Both with and without dexamethasone, PAH monocyte-derived DCs induced a higher activation and proliferation of CD4+ T cells, associated with a reduced expression of IL-4 (Th2 cells response) and a higher expression of IL-17 (Th17 cells response) (22). Animal studies have demonstrated that depleting CD4 cells or using SR1001, a Th17 cells development inhibitor, prevented the elevation of pressure and remodeling responses to chronic hypoxia (23). Furthermore, in a mice model of hypoxia-induced PH (HPH), the use of the receptor antibody MR16-1 to block IL-6 showed promising results. This treatment not only improved the condition of PH but also hindered the accumulation of macrophages and Th17 cells in the lungs (24).
And Th2 cells produce IL-4 and IL-13, which are described as antagonists of autoimmune pathological development. Researchers identified that Th2 CD4 T cells were necessary for Schistosoma-induced PH, given that deletion of CD4 T cells or inhibiting their Th2 cells function protected against type 2 inflammation and PH following Schistosoma exposure (25). They also observed that adoptive transfer of Schistosoma-sensitized CD4 Th2 cells was sufficient to drive type 2 inflammation and PH (25). Besides, it found that chemoattractant receptor homologous molecule expressed on Th2 cell (CRTH2) expression was up-regulated in circulating CD3CD4 T cells in patients with IPAH and in rodent PAH models (26). CRTH2 deficiency suppressed Th2 cells activation, including IL-4 and IL-13 secretion. CRTH2 disruption dramatically ameliorated pulmonary artery remodeling and PH in different PAH mice models (26).
Tregs play an important role in regulating the inflammatory response of Th cells to their own and foreign antigens. Tregs not only controls other T cells, but also regulates monocytes, macrophages, DCs, natural killer cells and B cells. In thymus-free rats with T cell immunodeficiency, pulmonary arterioles were blocked by proliferating endothelial cells. Blood vessels were surrounded by mast cells, B cells and macrophages, similar to human PAH (27). In addition, Tregs can regulate the proliferation of human pulmonary artery smooth muscle cells (HPASMCs). Tregs treatment significantly reduced the increase of right ventricular systolic pressure (RVSP) and Fulton index induced by hypoxia, decreased the expression of pro-inflammatory cytokines, and increased the level of IL-10 in vivo. This was attributed to the fact that Tregs treatment decreased the proliferation of HPASMCs and the expression of cyclin D1, cyclin-dependent kinase 4, p-Akt and p-ERK, and increased the expression of p27 in vitro (28). Furthermore, Tregs can inhibit the accumulation of collagen by inhibiting the secretion of transforming growth factor (TGF)-β1 and fibroblast growth factor 9 (29). Tregs also down-regulated cardiac fibroblasts by secreting IL-10, which helped to control the development of right ventricular hypertrophy (RVH) in PAH (30).
2.2 Macrophage
Macrophages play a crucial role as a key component of the innate immune system, and the produced antigen is presented to T cells to differentiate and activate adaptive immune system. Studies have shown that pulmonary inflammation mediated by peripulmonary macrophages is the key factor of pulmonary vascular remodeling. In PAH rat model and patients with left heart disease-related PH (LHD-PH), compared with the control group, macrophages increased, accompanied by an increase in lung IL-6 (31). At the same time, in the mice model of chronic thromboembolic PH (CTEPH), F4/80 positive monocytes/macrophages obviously accumulated in high-flow arteries (32). In human and animal models with PH, it has been reported that the level of monocyte recruitment chemokines in the lung is increased, and the number of peripheral blood monocytes is increased. Upon migration to the pulmonary vascular system, monocytes had the potential to differentiate into perivascular macrophages. This process was facilitated by the activation of chemokines chemokine ligand 2 (CCL2) and C-X3-C motif chemokine ligand 1 (CX3CL1) (33). Moreover, the recruitment and activation of macrophages around pulmonary vessels have an impact on the function of pulmonary vascular cells. SU5416 induced PH in athymic rats, and macrophages gathered around pulmonary arterioles and synthesize excessive leukotriene B4 (LTB4). LTB4 can damage endothelial cells (ECs) of nearby blood vessels, cause apoptosis, and cause abnormal proliferation of SMCs. Blocking macrophage-derived LTB4 biosynthesis or signal transduction can reverse the experimental PH, while depletion of CD68+ macrophages can prevent the occurrence of PH in thymus-free rats treated by sugen (34). Similarly, the development of HPH was related to the significant increase of levels of CX3CR1, CC chemokine receptor 2 (CCR2) and their respective ligands CX3CR1 and CCL2. CX3CR1 deficiency can prevent HPH by regulating monocyte recruitment, macrophage polarization and PASMCs proliferation (35).
2.3 B Cell, DC and neutrophil
B cells have the ability to differentiate into plasma cells, which are responsible for producing autoantibodies. In addition, B cells play a crucial role in immune responses by collaborating with antigen-presenting DCs and lymphoid organs. They achieved this through antigen presentation, the production of various cytokines, and the facilitation of T effector cell differentiation (31, 36). On the one hand, a functional role for B cells in PH was demonstrated in that either blocking B cells by an anti-CD20 antibody or B cells deficiency in JH-KO rats attenuated right ventricular systolic pressure and vascular remodeling in experimental PH (32). On the other hand, B cell depletion therapy was a potentially effective and safe adjuvant treatment for systemic sclerosis-PAH and systemic lupus erythematosus-PAH (37, 38).
In monocrotaline (MCT)-induced rats and hypoxic mice, the accumulation of pulmonary neutrophils increased (39, 40). Although neutrophils have received little attention in the pathogenesis of PAH, it has been found that neutrophil elastase can affect the pathogenesis. Neutrophil elastase isolated from PAH patients, PASMCs and PH rat models was enhanced (41–44). In MCT-induced and Sugen/hypoxic rat models, after treatment with elafin, an inhibitor of neutrophil elastase, the changes of pulmonary intima subsided and the lumen size increased (39).
The study revealed that the number of circulating myeloid DCs in IPAH patients was lower than that in the control group, and the immune deficiency of monocyte-derived DCs was obvious (45). However, the circulating plasma DCs (pDCs) value of IPAH patients did not change, and the pDCs value in lung tissue increased (46). pDCs secretes inflammatory factors and chemokines to promote the activation of immune cells. At the same time, these inflammatory factors and chemokines attracted monocytes to the lungs of patients with IPAH and CTD-PAH, and produced monocyte-derived DCs, which further aggravated inflammation (47).
3 Cytokine and PH
The increase of perivascular immune cell accumulation and intravascular infiltration, accompanied by the abnormal increase of some cytokines, leads to the increase of vascular cell inflammation and the formation of dysfunction. They played crucial roles as regulatory factors in PAH and are closely associated with the disease’s severity (48). At the same time, the circulating level of cytokines was an important marker for the diagnosis and treatment of PAH (49). Targeting specific cytokine responses and pathways is considered a promising therapeutic strategy.
Some cytokines and chemokines were related to the poor clinical outcome of PAH patients, and may be used as biomarkers of disease progression. Some, such as IL-1β and TNF-α, were related to the accumulation of extracellular matrix proteins (such as fibronectin), which were found in PAH lesions, while others, such as IL-6, are related to the proliferation of SMCs.
3.1 IL-1β
A study has provided evidence that the levels of IL-1β are significantly elevated in patients with PH. This increase in IL-1β had been found to be associated with a poorer prognosis for individuals with PH (50). The mice model of PH showed the same conclusion, and it has been proved that starting IL-1β receptor antagonist can reduce PH and RVH (51). Furthermore, there existed a correlation between IL-1β and prostacyclin (PGI2) (17, 52). IL-1β increased PGI2 production in a dose-dependent manner (52). Cyclooxygenase-2 (COX-2) was the key enzyme for PGI2 synthesis (52). After treating PASMCs with IL-1β, the level of COX-2 mRNA increased, which induced the expression of PGI2 (52, 53). However, IL-1β has been found to have a negative impact on adenosine cyclase, leading to a decrease in cyclic adenosine monophosphate (cAMP) production. This, in turn, weakened the role of PGI2 (54). Furthermore, IL-1β activated other cytokines and chemokines to play a role. The cleavage of IL-18 by IL-1β invertase resulted in the production of bioactive IL-18. This increase in bioactive IL-18 has been observed in patients with pulmonary vascular diseases. Additionally, the levels of the downstream chemokine CXCL10 were also elevated in these patients (55). The increased expression of IL-18 and CXCL10 may make the inflammatory environment permanent and eventually lead to the vascular occlusion characteristics of PAH (55).
3.2 IL-6
IL-6 is a cytokine secreted by lymphocytes, which has extensive pro-inflammatory properties and participates in the occurrence and development of PH. IL-6 has been found to have prognostic value in patients with PH. The serum level of IL-6 has been shown to be an independent predictor of survival in PH patients and can be used to predict the outcome of these patients (56). Animal model revealed that the RVH of rats injected with recombinant human IL-6 increased, and PH appeared under normal oxygen conditions (57). Similarly, lung-specific IL-6 overexpression mice spontaneously produced PH under chronic hypoxia, and showed muscular distal arterioles and proliferative lesions in vascular cells. The IL-6 receptor knockout rats were not affected by HPH, and the progressive accumulation of PASMCs was reduced (58).
IL-6 may promoted PH in the following ways: (1) IL-6 induced excessive proliferation of pulmonary artery ECs (PAECs) and PASMCs in the distal pulmonary vascular wall by up-regulating vascular endothelial growth factor receptor 2 (VEGFR2) and matrix metalloprotein -2 (20). (2) IL-6 affected many signal pathways, such as BMPR2/Smad, MAPK/P38 and MAPK/JNK (17). (3) IL-6 played a crucial role in regulating the balance between Th17 cells and Treg cells. It can stimulate the immune response of Th17 cells while inhibiting the suppressive function of Treg cells. This imbalance in the immune response, with an increased presence of Th17 cells and reduced Treg cell activity, has been associated with an increased risk of PAH (59).
3.3 IL-10, IL-18 and TNF-α
Abnormal levels of cytokines, including IL-10, IL-18, and TNF-a, have been observed in both human and animal models of PH.
IL-10 is released by T cells, which is negatively correlated with PGI2 therapy. And IL-10 was decreased in patients after cardiopulmonary bypass (60). At the same time, it has been proved that the decrease of IL-10 level is a risk factor for chronic obstructive pulmonary disease (COPD)-PH (61). However, exogenous IL-10 significantly reduced macrophage infiltration and vascular cell proliferation in the remodeled pulmonary artery. It also significantly reduced the lung levels of TGF-β1 and IL-6 (62). Therefore, the effect of IL-10 on the development of PH needs to be studied.
Under the influence of caspase-1, the inactive precursor form of IL-18 is converted into its active form, IL-18, which possesses biological activity (55). IL-18 acted on the cytokine-chemokine cascade reaction of type I immune response, affecting the expression of IL-12, IFN-γ and CXCL10. This promoted the recruitment of more lymphocytes to the vascular wall, which in turn exacerbated the pathological progression of PAH (55). Besides, IL-18 affected SMCs in an autocrine or paracrine way. IL-18 could thicken the smooth muscle layer by up-regulating the chemotaxis of metalloproteins and cells that constitute the vascular skeleton (63).
TNF-α is an important proinflammatory factor, which is significantly increased in the medial layer of pulmonary artery in COPD-PH patients (64). It is reported that transgenic mice that overexpress TNF-α will have severe PH value and right ventricular hypertrophy (65). Similarly, injection of TNF-a into the rat model led to increased vascular activity and remodeling (66). TNF-α may drove the proliferation of PAECs and PASMCs in HPH by inhibiting pyruvate dehydrogenase (PDH) activity, inhibiting BMPR2 and changing NOTCH signal transduction (63).
4 Chemokine and PH
4.1 CCL2
CCL2 is secreted by vascular ECs and SMCs, and its role is mediated by CCR2. CCL2 is an effective medium for the activation of monocytes and macrophages, which induces the secretion of cytokines and the expression of adhesion molecules (17, 67). It is reported that CCL2 induces the proliferation and migration of PAECs and SMCs in patients with PH, which leads to vascular remodeling and blood pressure increase (68). This may be because the increase of CCL2 produced by pulmonary vascular cells contributes to the increase of chemotactic activity of monocytes/macrophages. So that adventitial fibroblasts are activated (69, 70), the migration and proliferation of PASMCs increased, and pathological vascular remodeling was induced. In the presence of pulmonary ECs, the mobility of monocytes decreased significantly, while the blocking antibody of CCL2 increased significantly. The migration and proliferation of PASMCs in patients with PAH increased in response to CCL2 stimulation (71).
4.2 CCL5
CCL5 is expressed and secreted by T cells (17). Studies have confirmed that CCL5 can promote the proliferation of ECs. ET-converting enzyme-1 and ET promoted mitosis and vasoconstriction, and accelerated the pathological process of PH (72). In fact, compared with the control group, the expression of CCL5 mRNA in lung samples of PAH patients was increased, which may be derived from ECs (73). Moreover, the crosstalk between CCR5 and CCR2 mediated the synergy between macrophages and PASMCs, thus promoting inflammatory cell infiltration and PASMCs migration and proliferation during the development of PAH (74).
4.3 CX3CL1
CX3CL1 exists as a cell adhesion molecule on ECs or as a chemotactic protein, and its function is mediated by CX3CR1 (67). Under the pathological condition of PH, CX3CL1 led to the adhesion of macrophages expressing CX3CR1 to PAECs, which led to PAECs dysfunction and trigger perivascular inflammatory reaction (75). In addition, CX3CL1 promoted the proliferation of pericytes and PASMCs, which further aggravated the remodeling of pulmonary small vessels, leading to further aggravation of PH (76). CX3CR1 deficiency protected against HPH by modulating monocyte recruitment, macrophage polarization, and PAMCs proliferation (35).
5 Vascular cells and BMPR2
5.1 Vascular cells
ECs play a key role in maintaining vascular homeostasis under various stimuli, and regulate inflammation through mediators such as NO, ET, cell adhesion molecules, cytokines and chemokines. Under pathological conditions such as inflammation and hypoxia, PAECs can reduce the production of vasodilators (such as NO) and vascular growth factors, which is beneficial to the vasoconstriction of the distal pulmonary artery (77). Circulating ECs may be involved in the process of vascular injury, tumorigenesis or interaction with immune cells. In addition, endothelial progenitor cells were bone marrow-derived cells involved in homeostasis, and they were also physiological and pathological angiogenesis cells. The increase of proinflammatory cytokines was also beneficial to the activation of platelet adhesion and coagulation cascade reaction, which led to further occlusion of arterioles (78).
It has been suggested that inflammation can recruit SMCs population and enhance their contribution to pulmonary vascular remodeling. Because excessive proliferation of SMCs was observed in locally occluded blood vessels. Some studies also showed that continuous hypoxia can induce the recruitment of mesenchymal progenitor cells around blood vessels. The recruitment of these cells was very important for the occurrence of PAH (79) [142,143]. The changes of inflammatory vascular system may be caused by these migrating cells and resident SMCs, which restore the ability needed for vascular remodeling.
5.2 BMPR2 and immune
On the one hand, BMPR2 gene mutation has been identified as the main genetic cause of PAH. On the other hand, BMPR2 plays an important role in maintaining the immune system (80, 81). The disruption of BMPR2 signaling pathway causes an increased degree of inflammation and decreases the ability of the immune system to resolve it. Inhibition of BMPR2 gene expression led to unregulated proliferation and survival of endothelial cells through disordered TGF-β signaling, thus promoting vascular remodeling (82). In order to cope with the decrease of BMPR2 function in ECs, it is assumed that the integrity of ECs dysfunction may be damaged, which may lead to apoptosis, the release of TGF-β and the development of anti-apoptotic clones (83). On the contrary, SMCs proliferate due to TGF-β signaling, and undergo excessive growth reaction, leading to vascular remodeling (84).
Loss of BMPR2 expression enhanced ECs inflammatory response through various mechanisms mediated by ROS, nicotinamide adenine dinucleotide phosphate oxidase and NF-κB activity (85, 86). In innate immune response, BMPR2 deficiency increased the recruitment of macrophages to the perivascular area of pulmonary vascular. Mutated BMPR2 can also increase the production of granulocyte macrophage colony-stimulating factor, thus activating macrophages (87, 88). Activated macrophages secreted proinflammatory cytokines, such as IL-6 and IL-8 (86). In addition, the imbalance of BMPR2 in adaptive cellular immunity will destroy the development of T cells and lead to the increase of ECs apoptosis. The decrease of BMPR2 expression was also related to CD4 T cells depletion. Lack of T cells population led to inflammation around the artery, which was dominated by macrophage and B cells activity, thus aggravating PAH (11, 46).
6 Immunosuppression therapy in PH
At present, it is generally believed that there is a connection between inflammation and PH, but there are still several problems. Firstly, it is not clear whether inflammation is enough to promote the development of PH. Secondly, what are the causes of immune and inflammatory changes in PH lung tissues? Finally, does this have pathological or clinical significance? It is necessary to better understand the mechanism of immune cells, cytokines and chemokines in PH affecting abnormal angiogenesis and pulmonary artery remodeling. Although it is necessary to understand the immune/inflammatory components of PH more clearly, recent studies have shown promising results in animal models regarding the efficacy of anti-inflammatory therapies in reducing or even reversing the effects of PH. Currently, therapeutic drugs targeting the three pathological pathways of PH primarily focus on vasodilation. However, it is worth noting that these drugs also exhibit immunomodulatory properties (89). At present, there is no approved treatment specifically targeting the inflammatory process associated with pulmonary vascular diseases.
Tacrolimus is a calcineurin inhibitor, which can regulate immunity and has anti-inflammatory activity. Low-dose tacrolimus has been proved to reverse the disease progression of PAH rats induced by mc and hypoxia, restore the normal function of pulmonary artery endothelial cells, and activate other functions of BMPR2 receptor signal transduction by removing FKBP12 from BMPR 1 type co-receptor (90).Based on B cell depletion therapy, patients who received rituximab (an anti-CD20 monoclonal antibody) showed a decrease in rheumatoid factor, IL-12 and IL-17 (37). Hydroxymethylglutaryl coenzyme A (statins) have been proved to have anti-inflammatory and immunomodulatory functions (91, 92). Patients with IPAH, HPAH and CTD-PAH were treated with simvastatin, which inhibited lymphocyte function and improved right ventricular remodeling (93, 94). In addition, compared with the control group, the level of circulating inflammatory markers in patients with chronic embolic PH and PAH increased (95). After treatment with corticosteroids or immunosuppressants, the clinical symptoms of some patients improved (96). The above evidence reveals that suppressing immunity and inflammation can be used as a potential strategy for PH treatment. However, immunosuppressants are a kind of drugs that can effectively inhibit immune cell function, reduce immune response and inhibit inflammation. So far, no immunosuppressants have been approved for the treatment of multiple PH. The therapeutic effect of most immunomodulators on PH is still in the clinical trial stage.
7 Conclusion
It is an obvious fact that immune cells and inflammation play an important role in the pathophysiology of PH. There is increasing evidence that PH is not only caused by dynamic vasoconstriction. Inflammatory cells and their chemokines and cytokines affect pulmonary vascular system. However, many molecular and cellular mechanisms remain unsolved. It is significant for immunotherapy and anti-inflammation to better understand how inflammation and immunity participate in the development of polycyclic aromatic hydrocarbons.
Author contributions
HZ: Conceptualization, Writing – original draft. JS: Investigation, Writing – original draft. XL: Investigation, Visualization, Writing – review & editing. ZX: Visualization, Writing – review & editing. QW: Writing – review & editing, Formal Analysis. JF: Writing – review & editing, Methodology. YM: Project administration, Writing – review & editing. DW: Conceptualization, Project administration, Writing – review & editing. XW: Conceptualization, Project administration, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Program of Taihu-Talent Project of Wuxi government (2021THRC-TD-ZZYXK-2021).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. (2022) 43:3618–731. doi: 10.1183/13993003.00879-2022
2. Johnson S, Sommer N, Cox-Flaherty K, Weissmann N, Ventetuolo CE, Maron BA. Pulmonary hypertension: A contemporary review. Am J Respir Crit Care Med. (2023) 208:528–48. doi: 10.1164/rccm.202302-0327SO
3. Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. (2019) 53:1801887. doi: 10.1183/13993003.01887-2018
4. Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. Bmj. (2018) 360:j5492. doi: 10.1136/bmj.j5492
5. Hu Y, Chi L, Kuebler WM, Goldenberg NM. Perivascular inflammation in pulmonary arterial hypertension. Cells. (2020) 9:2338. doi: 10.3390/cells9112338
6. Zheng D, Liu J, Piao H, Zhu Z, Wei R, Liu K. ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front Immunol. (2022) 13:1039241. doi: 10.3389/fimmu.2022.1039241
7. Krüger-Genge A, Blocki A, Franke RP, Jung F. Vascular endothelial cell biology: an update. Int J Mol Sci. (2019) 20:4411. doi: 10.3390/ijms20184411
8. Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol (Oxf). (2017) 219:22–96. doi: 10.1111/apha.2017.219.issue-1
9. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. (2011) 146:873–87. doi: 10.1016/j.cell.2011.08.039
10. Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 186:261–72. doi: 10.1164/rccm.201201-0164OC
11. Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res. (2011) 109:867–79. doi: 10.1161/CIRCRESAHA.110.236927
12. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. (2023) 61:2200879. doi: 10.1183/13993003.00879-2022
13. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. (2022) 22:251–65. doi: 10.1038/s41577-021-00584-1
14. Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. (2008) 111:5271–81. doi: 10.1182/blood-2008-01-078204
15. Amaya-Uribe L, Rojas M, Azizi G, Anaya JM, Gershwin ME. Primary immunodeficiency and autoimmunity: A comprehensive review. J Autoimmun. (2019) 99:52–72. doi: 10.1016/j.jaut.2019.01.011
16. Romagnani S. Immunologic influences on allergy and the TH1/TH2 balance. J Allergy Clin Immunol. (2004) 113:395–400. doi: 10.1016/j.jaci.2003.11.025
17. Wang RR, Yuan TY, Wang JM, Chen YC, Zhao JL, Li MT, et al. Immunity and inflammation in pulmonary arterial hypertension: From pathophysiology mechanisms to treatment perspective. Pharmacol Res. (2022) 180:106238. doi: 10.1016/j.phrs.2022.106238
18. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. (2014) 115:165–75. doi: 10.1161/CIRCRESAHA.113.301141
19. Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. (2008) 205:799–810. doi: 10.1084/jem.20071258
20. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. (2009) 104:236–44, 28p following 244. doi: 10.1161/CIRCRESAHA.108.182014
21. Gaowa S, Zhou W, Yu L, Zhou X, Liao K, Yang K, et al. Effect of Th17 and Treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediators Inflammation. (2014) 2014:247372. doi: 10.1155/2014/247372
22. Hautefort A, Girerd B, Montani D, Cohen-Kaminsky S, Price L, Lambrecht BN, et al. T-helper 17 cell polarization in pulmonary arterial hypertension. Chest. (2015) 147:1610–20. doi: 10.1378/chest.14-1678
23. Maston LD, Jones DT, Giermakowska W, Howard TA, Cannon JL, Wang W, et al. Central role of T helper 17 cells in chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L609–l624. doi: 10.1152/ajplung.00531.2016
24. Hashimoto-Kataoka T, Hosen N, Sonobe T, Arita Y, Yasui T, Masaki T, et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci U.S.A. (2015) 112:E2677–86. doi: 10.1073/pnas.1424774112
25. Kumar R, Mickael C, Kassa B, Sanders L, Koyanagi D, Hernandez-Saavedra D, et al. Th2 CD4(+) T cells are necessary and sufficient for schistosoma-pulmonary hypertension. J Am Heart Assoc. (2019) 8:e013111. doi: 10.1161/JAHA.119.013111
26. Chen G, Zuo S, Tang J, Zuo C, Jia D, Liu Q, et al. Inhibition of CRTH2-mediated Th2 activation attenuates pulmonary hypertension in mice. J Exp Med. (2018) 215:2175–95. doi: 10.1084/jem.20171767
27. Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, et al. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med. (2007) 175:1280–9. doi: 10.1164/rccm.200608-1189OC
28. Chu Y, Xiangli X, Xiao W. Regulatory T cells protect against hypoxia-induced pulmonary arterial hypertension in mice. Mol Med Rep. (2015) 11:3181–7. doi: 10.3892/mmr.2014.3106
29. Peng X, Moore MW, Peng H, Sun H, Gan Y, Homer RJ, et al. CD4+CD25+FoxP3+ Regulatory Tregs inhibit fibrocyte recruitment and fibrosis via suppression of FGF-9 production in the TGF-β1 exposed murine lung. Front Pharmacol. (2014) 5:80. doi: 10.3389/fphar.2014.00080
30. Cao Y, Xu W, Xiong S. Adoptive transfer of regulatory T cells protects against Coxsackievirus B3-induced cardiac fibrosis. PLoS One. (2013) 8:e74955. doi: 10.1371/journal.pone.0074955
31. Kherbeck N, Tamby MC, Bussone G, Dib H, Perros F, Humbert M, et al. The role of inflammation and autoimmunity in the pathophysiology of pulmonary arterial hypertension. Clin Rev Allergy Immunol. (2013) 44:31–8. doi: 10.1007/s12016-011-8265-z
32. Breitling S, Hui Z, Zabini D, Hu Y, Hoffmann J, Goldenberg NM, et al. The mast cell-B cell axis in lung vascular remodeling and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L710–l721. doi: 10.1152/ajplung.00311.2016
33. Florentin J, Coppin E, Vasamsetti SB, Zhao J, Tai YY, Tang Y, et al. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J Immunol. (2018) 200:3612–25. doi: 10.4049/jimmunol.1701287
34. Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, et al. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med. (2013) 5:200ra117. doi: 10.1126/scitranslmed.3006674
35. Amsellem V, Abid S, Poupel L, Parpaleix A, Rodero M, Gary-Bobo G, et al. Roles for the CX3CL1/CX3CR1 and CCL2/CCR2 chemokine systems in hypoxic pulmonary hypertension. Am J Respir Cell Mol Biol. (2017) 56:597–608. doi: 10.1165/rcmb.2016-0201OC
36. Ulrich S, Taraseviciene-Stewart L, Huber LC, Speich R, Voelkel N. Peripheral blood B lymphocytes derived from patients with idiopathic pulmonary arterial hypertension express a different RNA pattern compared with healthy controls: a cross sectional study. Respir Res. (2008) 9:20. doi: 10.1186/1465-9921-9-20
37. Zamanian RT, Badesch D, Chung L, Domsic RT, Medsger T, Pinckney A, et al. Safety and efficacy of B-cell depletion with rituximab for the treatment of systemic sclerosis-associated pulmonary arterial hypertension: A multicenter, double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. (2021) 204:209–21. doi: 10.1164/rccm.202009-3481OC
38. Hennigan S, Channick RN, Silverman GJ. Rituximab treatment of pulmonary arterial hypertension associated with systemic lupus erythematosus: a case report. Lupus. (2008) 17:754–6. doi: 10.1177/0961203307087610
39. Taylor S, Dirir O, Zamanian RT, Rabinovitch M, Thompson AAR. The role of neutrophils and neutrophil elastase in pulmonary arterial hypertension. Front Med (Lausanne). (2018) 5:217. doi: 10.3389/fmed.2018.00217
40. Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. (2006) 168:659–69. doi: 10.2353/ajpath.2006.050599
41. Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. (2000) 6:698–702. doi: 10.1038/76282
42. Spiekerkoetter E, Alvira CM, Kim YM, Bruneau A, Pricola KL, Wang L, et al. Reactivation of gammaHV68 induces neointimal lesions in pulmonary arteries of S100A4/Mts1-overexpressing mice in association with degradation of elastin. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L276–89. doi: 10.1152/ajplung.00414.2007
43. Kim YM, Haghighat L, Spiekerkoetter E, Sawada H, Alvira CM, Wang L, et al. Neutrophil elastase is produced by pulmonary artery smooth muscle cells and is linked to neointimal lesions. Am J Pathol. (2011) 179:1560–72. doi: 10.1016/j.ajpath.2011.05.051
44. Rose F, Hattar K, Gakisch S, Grimminger F, Olschewski H, Seeger W, et al. Increased neutrophil mediator release in patients with pulmonary hypertension–suppression by inhaled iloprost. Thromb Haemost. (2003) 90:1141–9. doi: 10.1160/TH03-03-0173
45. Wang W, Yan H, Zhu W, Cui Y, Chen J, Wang X, et al. Impairment of monocyte-derived dendritic cells in idiopathic pulmonary arterial hypertension. J Clin Immunol. (2009) 29:705–13. doi: 10.1007/s10875-009-9322-8
46. Marsh LM, Jandl K, Grünig G, Foris V, Bashir M, Ghanim B, et al. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur Respir J. (2018) 51:1701214. doi: 10.1183/13993003.01214-2017
47. Itoh T, Nagaya N, Ishibashi-Ueda H, Kyotani S, Oya H, Sakamaki F, et al. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology. (2006) 11:158–63. doi: 10.1111/j.1440-1843.2006.00821.x
48. Groth A, Vrugt B, Brock M, Speich R, Ulrich S, Huber LC. Inflammatory cytokines in pulmonary hypertension. Respir Res. (2014) 15:47. doi: 10.1186/1465-9921-15-47
49. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, MaChado RD, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. (2010) 122:920–7. doi: 10.1161/CIRCULATIONAHA.109.933762
50. Zhong DX, Zhang Y, Jin Q, Zhang XC, Zhang F, Chen DD, et al. Increased serum PCSK9 in patients with idiopathic pulmonary arterial hypertension: insights from inflammatory cytokines. Pulm Circ. (2021) 11:20458940211051292. doi: 10.1177/20458940211051292
51. Voelkel NF, Tuder RM, Bridges J, Arend WP. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol. (1994) 11:664–75. doi: 10.1165/ajrcmb.11.6.7946395
52. Itoh A, Nishihira J, Makita H, Miyamoto K, Yamaguchi E, Nishimura M. Effects of IL-1beta, TNF-alpha, and macrophage migration inhibitory factor on prostacyclin synthesis in rat pulmonary artery smooth muscle cells. Respirology. (2003) 8:467–72. doi: 10.1046/j.1440-1843.2003.00491.x
53. Wen FQ, Watanabe K, Tanaka H, Yoshida M. Cytokines and lipopolysaccharide enhance basal and thrombin-stimulated production of PGI2 by cultured human pulmonary artery smooth muscle cells. Prostaglandins Leukot Essent Fatty Acids. (1997) 56:185–92. doi: 10.1016/S0952-3278(97)90532-9
54. El-Haroun H, Clarke DL, Deacon K, Bradbury D, Clayton A, Sutcliffe A, et al. IL-1beta, BK, and TGF-beta1 attenuate PGI2-mediated cAMP formation in human pulmonary artery smooth muscle cells by multiple mechanisms involving p38 MAP kinase and PKA. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L553–62. doi: 10.1152/ajplung.00044.2006
55. Ross DJ, Strieter RM, Fishbein MC, Ardehali A, Belperio JA. Type I immune response cytokine-chemokine cascade is associated with pulmonary arterial hypertension. J Heart Lung Transplant. (2012) 31:865–73. doi: 10.1016/j.healun.2012.04.008
56. Heresi GA, Aytekin M, Hammel JP, Wang S, Chatterjee S, Dweik RA. Plasma interleukin-6 adds prognostic information in pulmonary arterial hypertension. Eur Respir J. (2014) 43:912–4. doi: 10.1183/09031936.00164713
57. Golembeski SM, West J, Tada Y, Fagan KA. Interleukin-6 causes mild pulmonary hypertension and augments hypoxia-induced pulmonary hypertension in mice. Chest. (2005) 128:572s–3s. doi: 10.1378/chest.128.6_suppl.572S-a
58. Tamura Y, Phan C, Tu L, Le Hiress M, Thuillet R, Jutant EM, et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest. (2018) 128:1956–70. doi: 10.1172/JCI96462
59. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
60. Lei Y, Zhen J, Ming XL, Jian HK. Induction of higher expression of IL-beta and TNF-alpha, lower expression of IL-10 and cyclic guanosine monophosphate by pulmonary arterial hypertension following cardiopulmonary bypass. Asian J Surg. (2002) 25:203–8. doi: 10.1016/S1015-9584(09)60176-7
61. Yang D, Wang L, Jiang P, Kang R, Xie Y. Correlation between hs-CRP, IL-6, IL-10, ET-1, and chronic obstructive pulmonary disease combined with pulmonary hypertension. J Healthc Eng. (2022) 2022:3247807. doi: 10.1155/2022/3247807
62. Ito T, Okada T, Miyashita H, Nomoto T, Nonaka-Sarukawa M, Uchibori R, et al. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial hypertension in rats. Circ Res. (2007) 101:734–41. doi: 10.1161/CIRCRESAHA.107.153023
63. Sutendra G, Dromparis P, Bonnet S, Haromy A, McMurtry MS, Bleackley RC, et al. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFα contributes to the pathogenesis of pulmonary arterial hypertension. J Mol Med (Berl). (2011) 89:771–83. doi: 10.1007/s00109-011-0762-2
64. Joppa P, Petrasova D, Stancak B, Tkacova R. Systemic inflammation in patients with COPD and pulmonary hypertension. Chest. (2006) 130:326–33. doi: 10.1378/chest.130.2.326
65. Fujita M, Mason RJ, Cool C, Shannon JM, Hara N, Fagan KA. Pulmonary hypertension in TNF-alpha-overexpressing mice is associated with decreased VEGF gene expression. J Appl Physiol (1985). (2002) 93:2162–70. doi: 10.1152/japplphysiol.00083.2002
66. Stevens T, Janssen PL, Tucker A. Acute and long-term TNF-alpha administration increases pulmonary vascular reactivity in isolated rat lungs. J Appl Physiol (1985). (1992) 73:708–12. doi: 10.1152/jappl.1992.73.2.708
67. Pullamsetti SS, Savai R, Janssen W, Dahal BK, Seeger W, Grimminger F, et al. Inflammation, immunological reaction and role of infection in pulmonary hypertension. Clin Microbiol Infect. (2011) 17:7–14. doi: 10.1111/j.1469-0691.2010.03285.x
68. El Chami H, Hassoun PM. Immune and inflammatory mechanisms in pulmonary arterial hypertension. Prog Cardiovasc Dis. (2012) 55:218–28. doi: 10.1016/j.pcad.2012.07.006
69. Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, Sokol RJ, et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol. (2011) 187:2711–22. doi: 10.4049/jimmunol.1100479
70. Ye Y, Xu Q, Wuren T. Inflammation and immunity in the pathogenesis of hypoxic pulmonary hypertension. Front Immunol. (2023) 14:1162556. doi: 10.3389/fimmu.2023.1162556
71. Sanchez O, Marcos E, Perros F, Fadel E, Tu L, Humbert M, et al. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. (2007) 176:1041–7. doi: 10.1164/rccm.200610-1559OC
72. Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. (2009) 54:S10–s19. doi: 10.1016/j.jacc.2009.04.006
73. Dorfmüller P, Zarka V, Durand-Gasselin I, Monti G, Balabanian K, Garcia G, et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med. (2002) 165:534–9. doi: 10.1164/ajrccm.165.4.2012112
74. Abid S, Marcos E, Parpaleix A, Amsellem V, Breau M, Houssaini A, et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur Respir J. (2019) 54:1802308. doi: 10.1183/13993003.02308-2018
75. Zhang J, Hu H, Palma NL, Harrison JK, Mubarak KK, Carrie RD, et al. Hypoxia-induced endothelial CX3CL1 triggers lung smooth muscle cell phenotypic switching and proliferative expansion. Am J Physiol Lung Cell Mol Physiol. (2012) 303:L912–22. doi: 10.1152/ajplung.00014.2012
76. Xiang Y, Zheng F, Zhang Q, Zhang R, Pan H, Pang Z, et al. Tanreqing injection regulates cell function of hypoxia-induced human pulmonary artery smooth muscle cells (HPASMCs) through TRPC1/CX3CL1 signaling pathway. Oxid Med Cell Longev. (2022) 2022:3235102. doi: 10.1155/2022/3235102
77. Perros F, Ranchoux B, Izikki M, Bentebbal S, Happé C, Antigny F, et al. Nebivolol for improving endothelial dysfunction, pulmonary vascular remodeling, and right heart function in pulmonary hypertension. J Am Coll Cardiol. (2015) 65:668–80. doi: 10.1016/j.jacc.2014.11.050
78. Diller GP, Thum T, Wilkins MR, Wharton J. Endothelial progenitor cells in pulmonary arterial hypertension. Trends Cardiovasc Med. (2010) 20:22–9. doi: 10.1016/j.tcm.2010.03.003
79. Burke DL, Frid MG, Kunrath CL, Karoor V, Anwar A, Wagner BD, et al. Sustained hypoxia promotes the development of a pulmonary artery-specific chronic inflammatory microenvironment. Am J Physiol Lung Cell Mol Physiol. (2009) 297:L238–50. doi: 10.1152/ajplung.90591.2008
80. Tatius B, Wasityastuti W, Astarini FD, Nugrahaningsih DAA. Significance of BMPR2 mutations in pulmonary arterial hypertension. Respir Investig. (2021) 59:397–407. doi: 10.1016/j.resinv.2021.03.011
81. Farha S, Asosingh K, Xu W, Sharp J, George D, Comhair S, et al. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood. (2011) 117:3485–93. doi: 10.1182/blood-2010-09-306357
82. Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. (2006) 98:209–17. doi: 10.1161/01.RES.0000200180.01710.e6
83. McDonald PP, Fadok VA, Bratton D, Henson PM. Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF-beta in macrophages that have ingested apoptotic cells. J Immunol. (1999) 163:6164–72. doi: 10.4049/jimmunol.163.11.6164
84. Morrell NW. Pulmonary hypertension due to BMPR2 mutation: a new paradigm for tissue remodeling? Proc Am Thorac Soc. (2006) 3:680–6. doi: 10.1513/pats.200605-118SF
85. Kim CW, Song H, Kumar S, Nam D, Kwon HS, Chang KH, et al. Anti-inflammatory and antiatherogenic role of BMP receptor II in endothelial cells. Arterioscler Thromb Vasc Biol. (2013) 33:1350–9. doi: 10.1161/ATVBAHA.112.300287
86. Soon E, Crosby A, Southwood M, Yang P, Tajsic T, Toshner M, et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production. A gateway to pulmonary arterial hypertension. Am J Respir Crit Care Med. (2015) 192:859–72. doi: 10.1164/rccm.201408-1509OC
87. Andruska A, Spiekerkoetter E. Consequences of BMPR2 deficiency in the pulmonary vasculature and beyond: contributions to pulmonary arterial hypertension. Int J Mol Sci. (2018) 19:2499. doi: 10.3390/ijms19092499
88. Sawada H, Saito T, Nickel NP, Alastalo TP, Glotzbach JP, Chan R, et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J Exp Med. (2014) 211:263–80. doi: 10.1084/jem.20111741
89. Cohen-Kaminsky S, Hautefort A, Price L, Humbert M, Perros F. Inflammation in pulmonary hypertension: what we know and what we could logically and safely target first. Drug Discovery Today. (2014) 19:1251–6. doi: 10.1016/j.drudis.2014.04.007
90. Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. (2013) 123:3600–13. doi: 10.1172/JCI65592
91. Gomberg-Maitland M, Olschewski H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur Respir J. (2008) 31:891–901. doi: 10.1183/09031936.00097107
92. Wilkins MR, Ali O, Bradlow W, Wharton J, Taegtmeyer A, Rhodes CJ, et al. Simvastatin as a treatment for pulmonary hypertension trial. Am J Respir Crit Care Med. (2010) 181:1106–13. doi: 10.1164/rccm.200911-1699OC
93. Hillyard DZ, Cameron AJ, McDonald KJ, Thomson J, MacIntyre A, Shiels PG, et al. Simvastatin inhibits lymphocyte function in normal subjects and patients with cardiovascular disease. Atherosclerosis. (2004) 175:305–13. doi: 10.1016/j.atherosclerosis.2004.03.018
94. Kao PN. Simvastatin treatment of pulmonary hypertension: an observational case series. Chest. (2005) 127:1446–52. doi: 10.1016/S0012-3692(15)34501-3
95. Quarck R, Nawrot T, Meyns B, Delcroix M. C-reactive protein: a new predictor of adverse outcome in pulmonary arterial hypertension. J Am Coll Cardiol. (2009) 53:1211–8. doi: 10.1016/j.jacc.2008.12.038
Keywords: inflammation, immunity, cytokines, chemokines, pulmonary hypertension, immunosuppressive therapy
Citation: Zhao H, Song J, Li X, Xia Z, Wang Q, Fu J, Miao Y, Wang D and Wang X (2024) The role of immune cells and inflammation in pulmonary hypertension: mechanisms and implications. Front. Immunol. 15:1374506. doi: 10.3389/fimmu.2024.1374506
Received: 22 January 2024; Accepted: 26 February 2024;
Published: 11 March 2024.
Edited by:
Danchen Wu, Queen’s University, CanadaReviewed by:
Yonglei Wu, First Hospital of Jiaxing, ChinaDong Liu, Shanghai Jiao Tong University, China
Xiaojian Liu, The First Hospital of Tongxiang, China
Copyright © 2024 Zhao, Song, Li, Xia, Wang, Fu, Miao, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuguang Wang, d3hnODg4NjYyMDI0QDE2My5jb20=; Dapeng Wang, d2FuZ2RhcGVuZzEwNTNAMTYzLmNvbQ==; Yuqing Miao, eXFtaWFvQHVzc3QuZWR1LmNu
†These authors have contributed equally to this work and share first authorship