 Zeev Elkoshi
Zeev Elkoshi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol. , 02 April 2024
Sec. Cytokines and Soluble Mediators in Immunity
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1371753
This article is part of the Research Topic The Role of TGF-beta Superfamily Members in Immune Homeostasis and Disease View all 6 articles
The risk of cancer is higher in patients with asthma compared to those with allergic rhinitis for many types of cancer, except for certain cancers where a contrasting pattern is observed. This study offers a potential explanation for these observations, proposing that the premalignant levels of circulating transforming growth factor-β (TGF-β), IL-1β, and IL-6 as well as the reactivity of the TGF-β/Smad signaling pathway at the specific cancer site, are crucial factors contributing to the observed disparities. Circulating TGF-β, IL- β and IL-6 levels also help clarify why asthma is positively associated with obesity, Type 2 diabetes, hypertension, and insulin resistance, whereas allergic rhinitis is negatively linked to these conditions. Furthermore, TGF-β/Smad pathway reactivity explains the dual impact of obesity, increasing the risk of certain types of cancer while offering protection against other types of cancer. It is suggested that the association of asthma with cancer and metabolic dysregulations is primarily linked to the subtype of neutrophilic asthma. A binary classification of TGF-β activity as either high (in the presence of IL-1β and IL-6) or low (in the presence or absence of IL-1β and IL-6) is proposed to differentiate between allergy patients prone to cancer and metabolic dysregulations and those less prone. Glycolysis and oxidative phosphorylation, the two major metabolic pathways utilized by cells for energy exploitation, potentially underlie this dichotomous classification by reprogramming metabolic pathways in immune cells.
Asthma and allergic rhinitis (AR) are two allergic conditions classified as Type I Hypersensitivity which is characterized by high IgE antibody release from plasma cells. Allergens are the triggers of allergic reactions. Antigen-presenting cells process allergens and present them to naïve CD4+T cells. In the presence of IL-4, naïve CD4+T cells differentiate into allergen-specific Th2 cells. Simultaneously, B cells process allergens and present them to these allergen-specific Th2 cells through MHC-II. Consequently, these Th2 cells secrete IL-4 and IL-13, initiating the production of antigen-specific IgE antibodies by B cells, a process which leads to the differentiation of B cells into plasma cells that generate substantial amounts of specific IgE antibodies. These IgEs subsequently bind to high-affinity IgE receptors found on the surfaces of mast cells and basophils. Upon re-exposure to the antigen, it can bind to the IgE antibodies already attached to mast cells and basophils, triggering the release of allergy mediators such as histamine, tryptase, proteoglycans, and chymase (1).
Two epidemiological studies examined the risk of cancer in the Swedish population among individuals with asthma or AR. In one study, standardized incidence ratios (SIRs) were assessed for various types of cancer occurrences in a group of 138,723 individuals diagnosed with hay fever or AR. This cohort was identified from three different Swedish healthcare databases. Data from the Swedish Cancer Registry was utilized for the analysis of cancer risk (2). Another study followed a similar methodology, evaluating SIRs for different cancer types in a group of 439,738 Swedish patients with asthma (3). Remarkably, in 16 out of 23 cancer types evaluated:
The overall risk of cancer in asthmatic patients is 1.19 (95% CI: 1.17-1.21) versus 1.03(95% CI: 0.99 -1.07) in allergic rhinitis patients. The data is presented in Table 1.
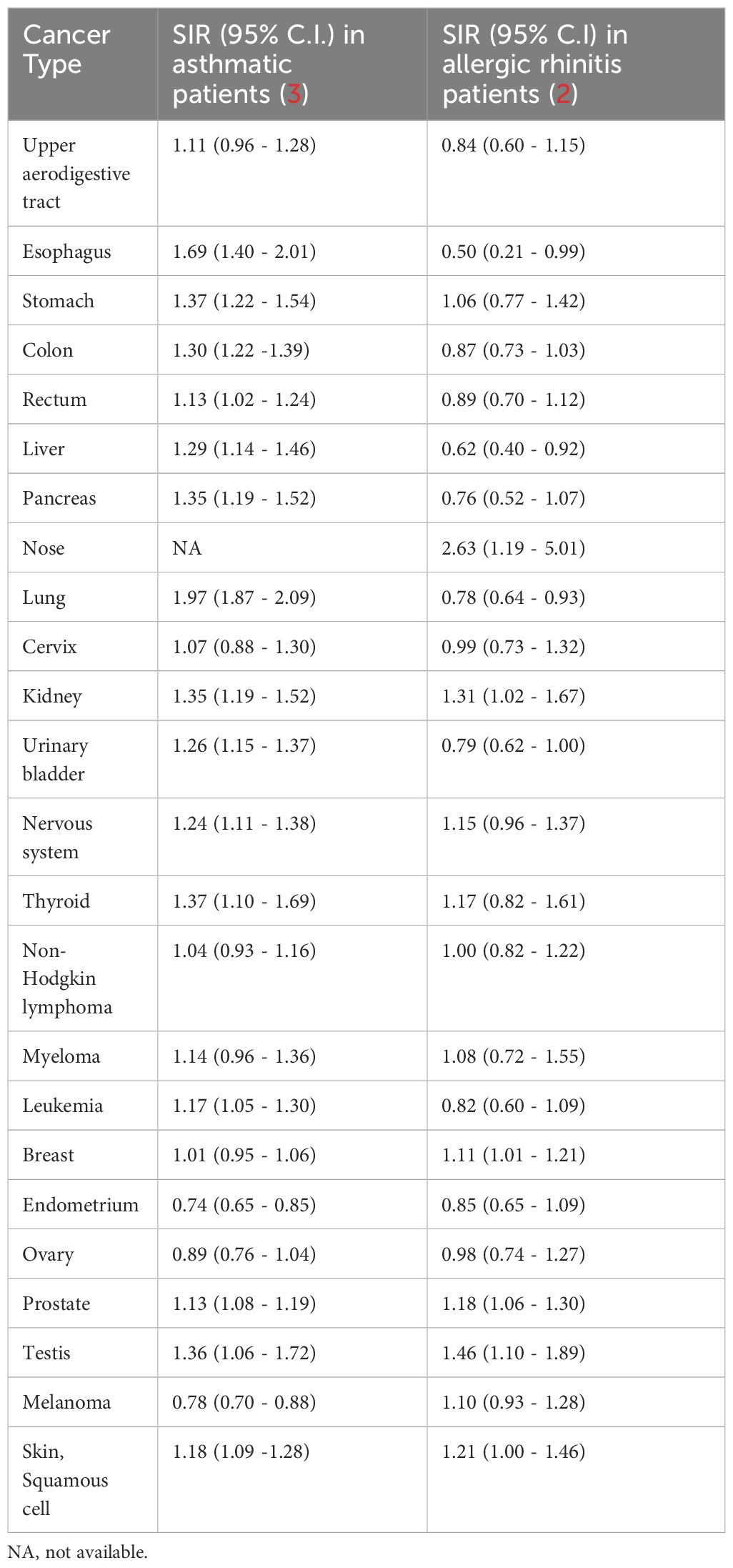
Table 1 SIRs for specific cancer types in asthmatic patients and in allergic rhinitis patients.
Endocrine glands related cancer, which appears in the studies by Hemminki and Liu, was omitted in Table 1, since it includes pancreatic, prostate, and breast cancers which are separately referred to in the list.
Metabolic dysregulations refer to disruptions or abnormalities in the normal metabolic processes of the body, including energy production, nutrient utilization, and waste elimination. A dysfunction in these metabolic processes can lead to various health issues such as obesity, Type 2 diabetes (DM2), insulin resistance, glucose intolerance dyslipidemia, and hypertension (4). Several studies investigated the association between metabolic dysregulations and allergic diseases. A positive association was found between asthma and DM2 in a recent literature review (5), and between asthma and hypertension (6). A positive association was also found between abdominal obesity and asthma in a meta-analysis published in 2019 (7). The evidence for these findings is substantiated by experimental models. Various murine models of obesity, including ob/ob, db/db, and Cpefat mice, manifest inherent airway hyperresponsiveness, a prominent characteristic of asthma (8). Additionally, other obese mouse models, such as TALLYHO/Jng mice, and mice subjected to a high-fat diet, exhibit asthma-like symptoms upon allergen sensitization and challenge (9). It is important to highlight that ob/ob and db/db mice experience insulin resistance and a gradual deterioration in glycemic control starting from 4 weeks of age (10). Consequently, these mouse models illustrate a direct association between DM2 and asthma. In support of this correlation, a study showed that metformin, an anti-glycemic drug, decreased both serum insulin levels and airway hyperresponsiveness in 12-week-old C57BL/6J mice that had been fed a high-fat diet for 8 weeks (11). In contrast to the positive association observed between metabolic disorders and asthma, a study by Lee et al. (12) reported an inverse association between AR and diabetes mellitus (the study data acquisition was biased towards DM2). Another study reported a lower prevalence of AR in patients with higher levels of fasting plasma glucose (13). A large population-based study in 30,590 Korean patients found that AR prevalence was significantly lower in subjects with metabolic syndrome (OR 0.84; 95% CI 0.76–0.93), high blood pressure (OR 0.85; 95% CI 0.77–0.94), or impaired fasting glucose (OR 0.81; 95% CI 0.73–0.89) (14). Likewise, a reverse association was observed between central obesity in children and AR (15).
In the next sections, a potential explanation for these observations will be proposed.
A notable distinction in the pathophysiology of asthma and allergic rhinitis becomes evident when comparing the circulating levels of TGF-β in both conditions. In one study, mean serum TGF-β levels evaluated in asthmatic patients were two orders of magnitude higher than the values observed in normal controls (39.59 ng/ml vs. 0.26 ng/ml). In steroid naϊve mild asthma patients TGF-β levels were even higher (47.44 ng/ml) (16). In another study, the mean value over 70 asthmatic patients (who did not use antihistaminic, oral, or parenteral steroid within the previous 4 weeks before blood sampling) was 41.7 ng/ml compared to a mean of 27.7 ng/ml over 15 healthy controls (p < 0.05) (17). Seven-fold increase in mean TGF-β serum levels was reported in 15 patients with moderate asthma (who did not use corticosteroids) compared to 14 healthy controls (36.78 ng/ml vs. 5.49 ng/ml) (18). In another study, TGF-β levels in the serum and bronchoalveolar lavage fluid (BALF) of asthmatic patients were reported as significantly higher compared to controls (serum: 0.186 ng/ml vs 0.119 ng/ml; BALF: 0.215 ng/ml vs 0.121 ng/ml) (for both: p < 0.05) (19). On the other hand, a study by Rosas et al. (20) reports significantly lower mean plasma values of TGF-β in AR patients compared to values observed in a control group (0.276 ng/ml vs. 0.932 ng/ml) (p < 0.005). Chai et al. (21) also reported a lower mean value for TGF-β blood level (following mitogen stimulation) in AR patients compared to healthy controls (19.1 ng/ml vs. 23.8 ng/ml). In unstimulated blood samples, the mean values were 17.9 ng/ml for AR compared to 23.8 ng/ml in healthy controls. While these differences were not statistically significant, a tendency toward lower TGF-β levels in AR is observed (21). These last results are confounded by the co-existence of asthma in some patients, which likely induces an increase in TGF-β levels. Additionally, some patients received anti-allergic treatment, which is also known to induce an increase in TGF-β levels (22). It is reasonable to assume that the levels of TGF-β in AR would have been lower without these confounding factors. Nevertheless, the values reported by Chai et al. in AR patients are comparable to those reported by Ciprandi et al. (23) in AR patients outside the pollen season. In addition, Qiu et al. (24) demonstrated statistically significant lower TGF-β serum levels in AR patients compared to healthy controls. Another study that evaluated TGFβ mRNA levels in nasal mucosa of AR patients found a statistically significant lower value in AR patents compared to healthy controls (25). It must be noted that TGF-β levels are sensitive to allergen exposure (23, 24), and the levels following exposure to the allergen or allergen-specific immunotherapy are higher compared to the values observed in patients not exposed to the allergen (23, 24). However, even after an exposure to the allergens, TGF-β levels are not higher in AR than those observed in healthy subjects (24).
Despite variations in reported serum TGF-β values among different studies, a consistent observation is that circulating TGF-β levels are significantly higher in individuals with asthma compared to healthy controls. Conversely, in patients with AR who have not been exposed to allergens, TGF-β levels are lower compared to controls. An outstanding result was reported by Bayrak Degirmenci et al. (26) who found that TGFβ levels in AR patients were higher compared to healthy controls. However, the patients in this study were evaluated in the symptomatic period (i.e. after exposure to the allergen).
The asthma patient population comprises several phenotypes. Broadly, asthma may be divided into two main classes: “type 2-high” and “type 2-low” classes. In “type 2-high” asthma, which encompasses about half of the patients, Th2 cytokines, namely IL-4, IL-5, and IL-13, are the primary drivers of the disease, and patients are characterized by high frequencies of allergen-specific immunoglobulin E (sIgE) antibodies in blood. In “type 2-low” asthma, airway inflammation is mediated by IL-1β, IL-6, and neutrophils, with a lack of Th2 response and low frequencies of blood sIgE antibodies (27, 28). This type of asthma, also termed “non-eosinophilic asthma” or “neutrophilic asthma,” involves IL-6 produced by airway epithelial cells and neutrophils, IL-1β produced following inflammasome activation, and TGF-β produced by neutrophils and eosinophils. The ORMDL3 and GSDMB genes are strongly associated with asthma. Elevated levels of ORMDL3 expression in RAW264.7 macrophages result in increased production of IL-6, IL-1β, and TGF-β1. Additionally, it has been observed that GSDMB induces TGF-β1 expression in primary human bronchial epithelial cells (29). It is of note Serum mean IL-23 levels were reported to be significantly higher in untreated severe asthmatics than in a control group of healthy volunteers (111.38 pg/ml vs. 30.63 pg/ml; p< 0.001) (30). In the presence of IL-21 and IL-6, TGF-β induces the differentiation of Th17 cells from naïve CD4+ T cells in mice (31), fostering a pro-inflammatory environment. The differentiation of naive human CD4+ T cells into Th17 cells requires TGF-β, IL-1β, and either IL-6, IL-21, or IL-23 (32). Neutrophilic asthma is therefore associated with Th17 differentiation. Th17 cells release IL-17, IL-21, and IL-22, which are dominant cytokines in neutrophilic asthma. It has also been reported that the association of asthma with 17q21 polymorphisms in ORMDL3 and GSDMA is linked to early-life secretion of IL-17 (33). IL-17 is responsible for the recruitment of neutrophils into the lungs (34) while neutrophils further contribute to the pro-inflammatory characteristics of the environment by releasing TGF-β and inducing the cleavage of pro-IL-1β to IL-1β (35) (a positive-feedback mechanism). During infection, neutrophils polarize macrophages toward the pro-inflammatory M1 phenotype (36). Similarly, M1 polarization is associated with airway neutrophilia in asthma (37). The rate of Th17 cell differentiation increases with TGF-β levels (up to a point where TGF-β blocks the IL-23 receptor) (31). Therefore, high TGF-β levels (below the turndown point) observed in neutrophilic asthma accelerate Th17 cell differentiation. In addition, the phenotype of neutrophilic patients is characterized by significantly elevated sputum IL-1β gene expression and sputum IL-1β protein levels compared to eosinophilic phenotype (38). Elevated IL-1β levels must also expedite Th17 differentiation.
In summary, high TGF-β levels in asthma compared to AR, and high IL-1β levels in neutrophilic asthma compared to eosinophilic asthma promote a Th17 related pro-inflammatory environment in asthmatic patients especially in patients with neutrophilia.
Along the course of tumor development, the tumor microenvironment (TME) develops from basically anti-tumor at early tumor stage to a pro-tumor environment, at late tumor stages. Neutrophils transform from the pro-inflammatory N1 phenotype to the anti-inflammatory N2 phenotype. Similarly, macrophages transform from the M1 pro-inflammatory phenotype to the M2 anti-inflammatory phenotype. A correlation between regulatory T cells (Treg cells) frequency (in both the TME and circulation) and cancer progression was observed in several types of cancer [ref. (39) and references therein]. Tregs suppress the pro-inflammatory activities of macrophages, neutrophils, NK cells, T cells and B cells (40). In addition, myeloid-derived suppressor cells (MDSC), defined as pathologically activated neutrophils and monocytes with potent immunosuppressive activity (41), accumulate in the circulation and in the TME during cancer progression (42). TGF-β contributes to an immunosuppressive TME. Studies have shown that TGF-β can induce the transition of N1 to N2 (43) and M1 to M2 (44) phenotypes. In the presence of IL-2, TGF-β promotes the differentiation of naïve T cells into suppressive inducible regulatory T cells (iTregs) (45, 46). Moreover, TGF-β enhances the immunosuppressive function of MDSC (47).
Drawing from these observations, the present report proposes that the difference in circulating TGF-β, could be accountable for the increased risk of various cancer types among individuals with neutrophilic asthma in comparison to those with AR, while the interference of the TGF-β/Smad signaling pathway leads to an opposite outcome. There are two mechanisms that could contribute to an increased incidence of cancer when TGF-β levels are elevated:
(a) Elevated TGF-β levels in the context of neutrophilic asthma create a pro-inflammatory environment that contributes to the initiation and early development of cancer. This effect parallels the impact of pre-existing autoimmune diseases on cancer risk (39). Inflammation has the potential to induce mutations that upregulate oncogenes or mutations that downregulate tumor-suppressor genes, thereby enhancing tumor initiation. Additionally, inflammation fosters cancer cell proliferation, as exemplified by the inactivation of nuclear factor-kappa B (NFκB) in immune cells (48).
(b) TGF-β is implicated in inducing and promoting the self-renewal of cancer stem cells. Cancer stem cell renewal or expansion was reported in glioma (49, 50), triple-negative breast cancer (51), and mammary cancer colonies induced in bone microenvironment (52). In pancreatic cancer, TGF-β enhanced the invasiveness of cancer stem cells (53).
The next two sections present lines of evidence to support the proposed hypothesis.
(a) 5α-dihydroxytestosterone (DHT) has been reported to regulate TGF-β signaling in prostate epithelial cells by transcriptionally suppressing TGF-β receptor II (TGF-βRII) (54). Notably, Equation 1 does not apply to prostate cancer (Table 1). It does not hold for testicular cancer as well (Table 1), as the testicles are a significant site of androgen synthesis.
(b) Estrogens and progesterone have been reported to inhibit TGF-β signaling by reducing the phosphorylation of Smad2/3 (55–57). Consequently, Equation 1 is not applicable to ovarian and endometrial cancers (Table 1). Ovarian tissue estrogen levels are at least 100-fold higher than circulating levels (58). Many of the endometrial cancer risk factors involve excess estrogens or estrogen signaling, and endometrial cancer cases usually express high levels of estrogen receptor α (59).
(c) Similar to the sex organs, the skin locally synthesizes considerable amounts of sex hormones (60, 61). No wonder that melanoma and squamous cell skin carcinoma do not obey Equation 1 (Table 1).
(d) Benign breast disease is associated with increased estrogen levels in breast tissue (62) and in the circulation (63). Nearly 30% of all breast cancers develop in women with prior benign breast disease (64). This explains why Equation 1 is not obeyed for breast cancer (Table 1).
Nasal cancer shows the highest cancer risk among all cancer types observed in patients with a history of AR (Table 1). Consistent with this finding, significantly increased immunoreactivity for TGF-β and TGF-β receptors was reported in the nasal mucosa of AR patients compared to the nasal mucosa of normal controls (65). Increased IL-1β and IL-6 levels (needed for TH17 differentiation) in nasal lavage was reported in AR patients following allergen challenge (66).
Yadav et al. (67) have illustrated the importance of the TGF-β/Smad3 signaling pathway in regulating glucose and energy homeostasis. These authors demonstrated that Smad3 deficient mice are protected from diet-induced obesity and diabetes. Moreover, a strong correlation between TGF-β1 levels and adiposity in rodents and humans was reported in this study. It also became evident that TGF-β1, along with other members of the TGF-β superfamily, disrupts adipogenicity. Adipogenicity involves the “healthy” expansion of adipose tissue through the proliferation of adipose progenitor cells as a means of storing excess energy. In fact, TGF-β promotes the proliferation of preadipocytes while inhibiting their differentiation into adipocytes. Accumulation of dysfunctional adipose tissue in obesity increases risk for cardiometabolic diseases (68).
Pancreas beta (β) cell dysfunction or loss is the common pathological feature in all types of diabetes mellitus. The TGF-β/Smad signaling pathway has been shown to play a central role in the pathophysiology of pancreatic β cells. More specifically, deletion of Smad3 simultaneously improves pancreatic β cell function, apoptosis, and systemic insulin resistance with the consequence of eliminated overt diabetes in diabetic mouse models (69–71), revealing Smad3 as a key mediator and ideal therapeutic target for DM2. Importantly, IL-6 is constitutively expressed by β and α pancreatic cells from healthy or DM2 patients (72), and IL-1β is produced by α pancreatic cells at comparable levels in both non-diabetic and diabetic (DM1 and DM2) donors (73).
There is evidence of elevated TGF-β production and gene expression by peripheral blood monocytes in hypertensive patients (74, 75). Additionally, hypertensive individuals have been found to exhibit heightened circulating levels of IL-1β and IL-6 (76).
Due to the association of asthma in general, and neutrophilic asthma in particular, with elevated circulating TGF-β levels, the positive connection between neutrophilic asthma and obesity, DM2, and hypertension becomes evident. Similarly, the inverse association between allergic rhinitis, obesity, hypertension and DM2 is also clarified, as allergic rhinitis is linked to reduced circulating TGF-β. It appears that the elevated levels of TGF-β and IL-1β observed in the subset of neutrophilic asthma patients [comprising 20%-30% of asthmatic patients (77)] may be responsible for the differences in the association of asthma and AR with metabolic dysregulations.
It should be noted that elevated levels of IL-1β and IL-6 were reported in AR. Multiple studies, demonstrated an elevation in IL-1β levels in nasal secretion and nasal mucosa of AR patients following allergen exposure (78). An increase in blood levels of IL-1β was reported in children with moderate to severe persistent AR (79). In addition, studies by Mirzai (25) and Gao (80) revealed increased expression of IL-6 in the nasal mucosa and serum, respectively, among patients with AR compared to controls. Nevertheless, despite the elevated levels of IL-1β and IL-6 in AR, the comparatively low levels of TGF-β in AR likely constrain Th17 differentiation.
By simple deduction, it turns out that for cancer types with impaired TGF-β signaling (cancer types where Equation 1 is not obeyed), obesity is expected to be associated with a reduced risk of cancer (see Figure 1). Five out of seven cancer types with impaired TGF-β signaling indeed demonstrate reduced risk of cancer in obese individuals compared to controls with normal body mass index (BMI): skin melanoma, prostate cancer, breast cancer (premenopausal) (81), skin squamous cell carcinoma (82), and testicular cancer (83, 84). As an exception, BMI is a consistent and leading risk factor for endometrial cancer in premenopausal women (85), another type of cancer where Equation 1 in not obeyed. The link between BMI and ovarian cancer varies among studies (86).
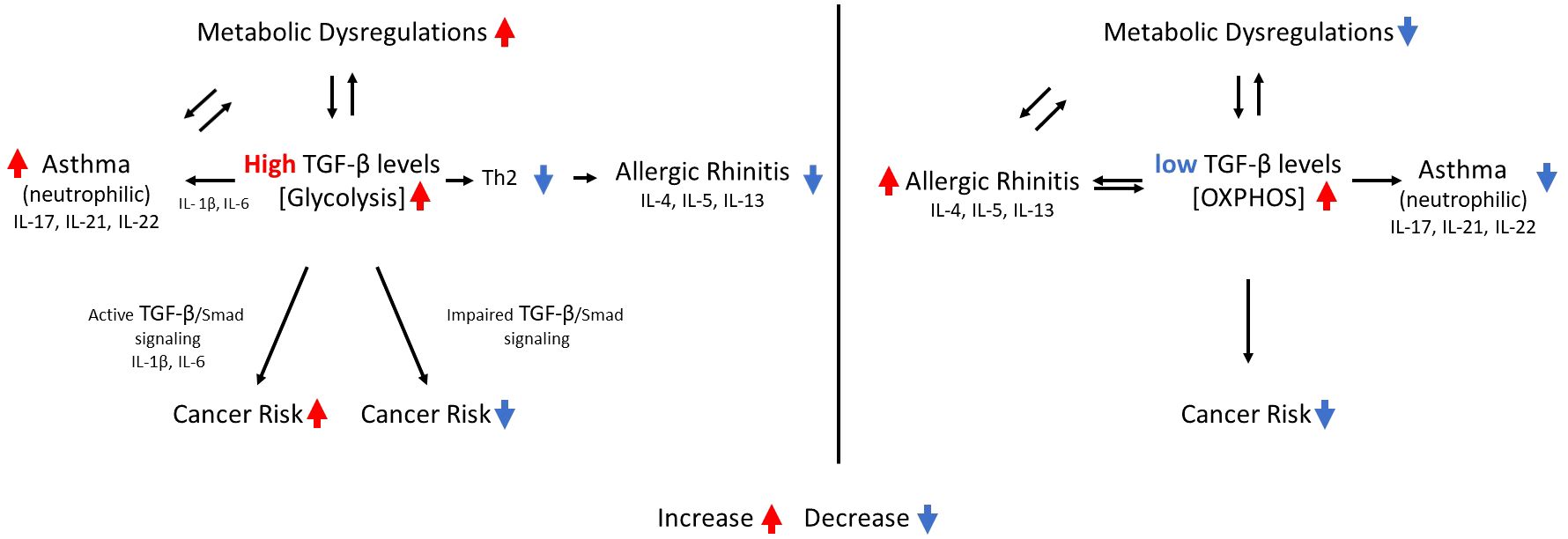
Figure 1 the interrelations between circulating TGF-β levels, asthma, allergic rhinitis, metabolic dysregulations, cancer risk and metabolic pathways. A single arrow indicates a causative effect. Two arrows pointing in opposite directions indicate an association between the two events.
A study involving 423 asthma patients found that obese patients demonstrated more neutrophils in sputum and blood than nonobese patients (87). This result is consistent with two other studies (88, 89). In a large study that included 727 adult asthma patients, Todd et al. (90) found no statistically significant difference in sputum neutrophil count among five groups with increasing median BMI. However, in this study, the mean neutrophil count consistently increased with increasing BMI. A more recent study reports elevated blood neutrophils with a higher BMI in 166 adolescent asthma patients (91). In another recent study, the serum leptin level was higher in obese than in lean female mice. This study demonstrated that leptin/obR signaling (obR is the leptin receptor), plays an important role in the pathogenesis of obesity-related neutrophilic airway inflammation in females, by promoting M1 macrophage polarization (92). A recent review discusses the contribution of neutrophils to obesity-associated inflammation (93). It is also noted that obesity in non-asthmatic adolescents is associated with increased sputum mRNA expression of IL6, IL8, IL13, IL17, IL23, and IFN-γ (94).
In contrast to neutrophilic asthma, allergic rhinitis inflammation is mainly mediated by Th2 cytokines. Although allergic rhinitis patients demonstrate increased levels of Th17 cytokines as well (95), blood frequencies of Th17 cells in asthmatic children were reported to be statistically significantly higher compared to those in children with allergic rhinitis (96). This difference would likely increase if only children with neutrophilic asthma were included in this study. As a Th2 disease, the presence of IL-4 and IL-13 is needed for AR development. Allergic rhinitis is associated with IL-4 and IL-13 polymorphism (97), and dual blockade of IL-4 and IL-13 with dupilumab, an IL-4Rα antibody, is required to inhibit type 2 inflammation such as AR (98, 99). At the same time, high TGF-β levels which are essential for the development of neutrophilic asthma, inhibits the development of Th2 reactions (100), and hence suppress allergic rhinitis (see Figure 1). It must be noted that TGFβ is also produced by eosinophils (as well as epithelial cells, macrophages, and fibroblasts) (101). Yet, the increased levels of IL-1β in neutrophilic asthma (as indicated by Simpson et al. (38)) likely contribute to a more pronounced differentiation of Th17 cells with this phenotype. It was also reported that Th17/Treg homeostasis, but not Th1/Th2 homeostasis, is implicated in the exacerbation of human bronchial asthma (102), underscoring the role of neutrophils in airway inflammation. Collectively, the distinct association of asthma with long-term comorbidities can be primarily attributed to neutrophilic patients, due to the elevated circulatory levels of TGF-β, IL-1β and IL-6 in this type of asthma.
For simplicity, this study adopts a dichotomous model where high TGF-β activity in conjunction with IL-1β and IL-6 is linked to asthma, metabolic dysregulations, and an elevated risk of cancer, while it is inversely associated with the occurrence of AR. In contrast, low TGF-β activity (with or without the presence of IL-1β and IL-6 levels), are associated with the inverse effects. In previous publications, the binary classification of immune response as “high Treg” or “low Treg”, proved useful in elucidating the association of certain pathogens with cancer and the association of others with autoimmune diseases. It also explained why certain specific pathogens are involved in coinfections. The effectiveness or ineffectiveness of specific immune-modulating drugs in the treatment of autoimmunity and cancer was also clarified by this binary model (103). The binary Tregs classification was also found to be useful in predicting the efficacy or inefficacy of several immune-modulating drugs in the treatment of severe coronavirus disease 2019 (COVID-19) (104, 105). Since regulatory T cells are a major source of TGF-β, the “high TGF-β” or “low TGF-β” classification is closely related to the “high Treg” and “low Treg” classification.
A binary classification of immune responses aligns with the two pathways of nutrients metabolism used by eukaryotic living cells to utilize and store energy: oxidative phosphorylation (OXPHOS) and glycolysis. OXPHOS consists of two closely interconnected components: the electron transport chain, which in eukaryotes occurs in the mitochondria, and chemiosmosis. NADH and succinate, produced in the citric acid cycle, undergo oxidation, releasing energy used by ATP synthase to facilitate the formation of ATP (adenosine triphosphate). The process of glycolysis, on the other hand, occurs in the cytoplasm and produces 2 ATP molecules per glucose molecule. Pyruvate, the end-product of glycolysis, serves as fuel for OXPHOS. Under aerobic conditions, pyruvate enters the mitochondria for oxidation to acetyl CoA, initiating the tricarboxylic acid (TCA) cycle and OXPHOS, which can yield 36 ATPs per glucose molecule. Conversely, under anaerobic conditions, pyruvate is reduced to lactate, by lactate dehydrogenase A (LDH-A) in the cytoplasm. The resulting lactate is then excreted into the extracellular space through monocarboxylate transporters. Normal cells usually release energy for ATP synthesis through OXPHOS (with glucose and fatty acids as fuels). However, most cancer cells preferentially utilize glycolysis, even in the presence of oxygen, a phenomenon known as the Warburg effect (106). While the glycolytic pathway from glucose to pyruvate is less efficient in energy exploitation than OXPHOS, its rate is faster. Inflammatory cells, including Th1, Th17, CD8 T cells, and M1 macrophages, primarily use glycolysis for energy storage. On the other hand, immune cells associated with the anti-inflammatory arm of the immune system, such as Tregs, M2 macrophages, memory T cells, and resting microglia, rely predominantly on OXPHOS and fatty acid oxidation for ATP production (106, 107).
TGF-β has been associated with increased glycolysis in cancers (108–111) and osteoarthritis (112). In addition, as aforementioned, in the absence of IL-1β and/or IL-6, it induces immunosuppression. In fact, Gauthier et al. (113) recently demonstrated that TGF-β uncouples glycolysis and inflammation in macrophages. By enhancing the expression and activity of a glycolytic enzyme, TGF-β induced glycolysis in macrophages, but, at the same time, suppressed their pro-inflammatory cytokines.
Rates of glycolysis in the liver, in pancreas β cells, and in adipose tissue are increased in DM2 due to compensatory hyperinsulinemia (114). It was also reported that aerobic glycolysis is increased in CD4 T cells in asthma (115). Lactate content was significantly higher in sputum supernatants from asthmatic patients, notably those with greater than 61% neutrophils, relative to healthy controls. One source of lactate production in asthmatic patients were primary nasal epithelial cells (116). Taken together, elevated TGF-β levels and increased glycolysis are associated with asthma (especially neutrophilic asthma), with an increased risk of cancer, with obesity, with DM2, hypertension, and with insulin resistance.
No studies describing OXPHOS activity in AR could be found. However, atopic dermatitis, which is associated with a low-producer TGF-β1 cytokine genotype (117), exhibits high OXPHOS activity in nonlesional skin (118). On the other hand, TGF-β1 expression was found to be markedly enhanced in atopic dermatitis lesions (119). In agreement, a mouse model of lesional atopic dermatitis demonstrated metabolic shift toward enhanced anaerobic glycolysis (120). Urticaria shows lower serum TGF-β levels compared to normal levels, but the difference was not statistically significant (121). Mast cells, the primary cells involved in producing urticaria wheals (122), utilize the OXPHOS process as a crucial step for mast cell degranulation and cytokine synthesis (123, 124). The connection between TGF-β levels and cell metabolism extends beyond allergic diseases. Sepsis, for instance, is linked to elevated circulating levels of TGF-β (125, 126). In line with this, glycolysis is the primary pathway determining the allocation of energy to organ cells during sepsis (127). The levels of TGF-β are elevated in joint synovial fluid of rheumatoid arthritis (RA) patients (128, 129). Glycolysis is increased in fibroblast-like synoviocytes in RA (130, 131).
In the cancer context, elevated TGF-β levels reprogram macrophage metabolism toward the glycolytic pathway while simultaneously suppressing their pro-inflammatory machinery (113) and diminishing their anti-cancer efficacy. Within adipose tissue, TGF-β-associated glycolysis in macrophages contributes to low-grade inflammation (132), a characteristic feature of obesity (133). Thus, the effect of TGF-β on both cancer cells and adipose tissue is, at least partially, mediated by macrophages and potentially other immune cells.
This data is presented in Table 2.
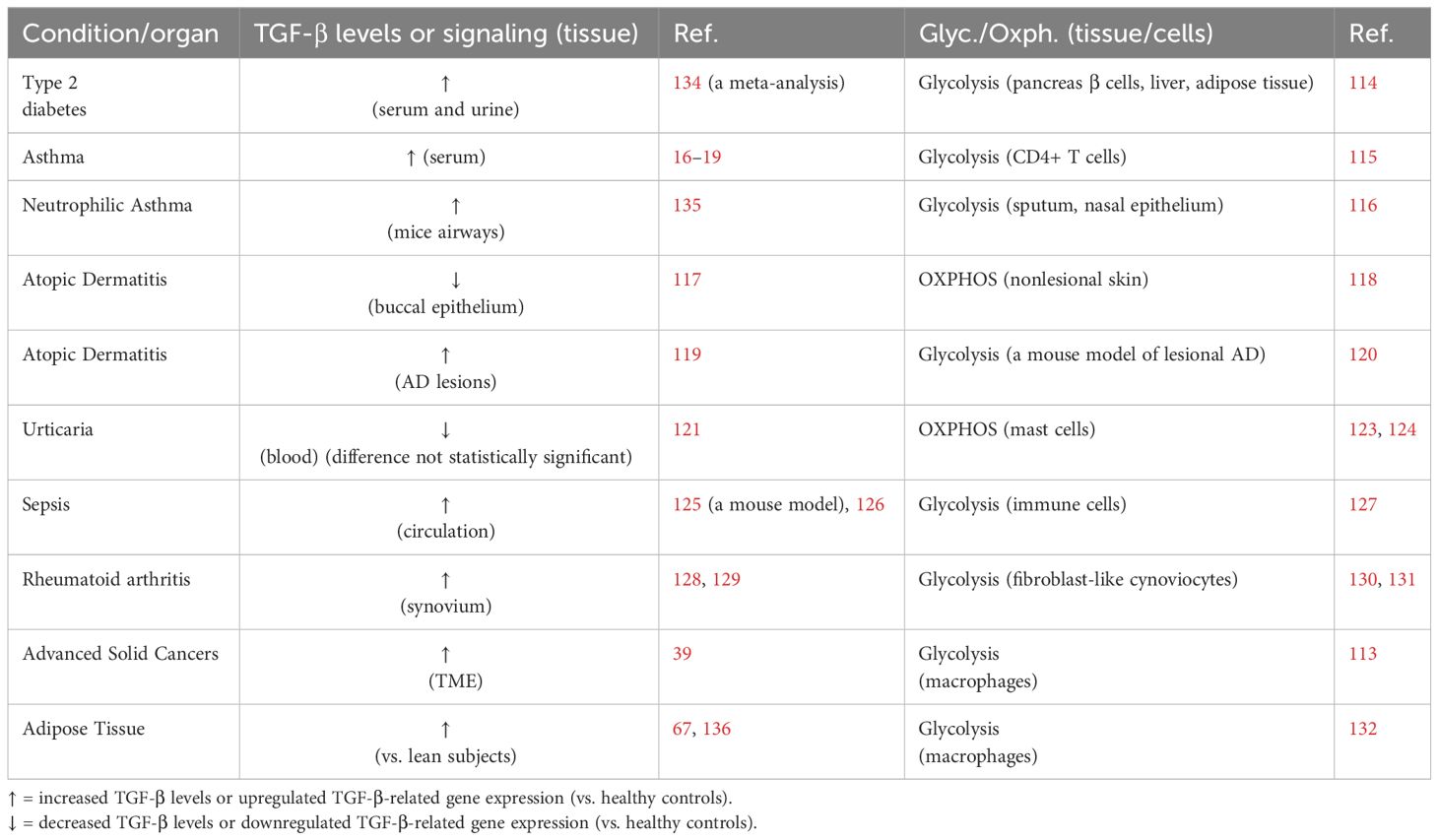
Table 2 The association between TGF-β levels or TGF-β/Smad signaling and metabolic bias (glycolysis vs. OXPHOS) in various diseases and in adipose tissue.
Regarding cancer initiation, high levels of glucose uptake observed in poorly differentiated different types of cancer reflect persistence of the glycolytic metabolism of stem cells in malignant cells that fail to fully differentiate (137). Biasing cancer stem cells’ metabolism toward the aerobic glycolytic pathway (the Warburg effect) drives their persistence in certain cancer types (breast cancer, nasopharyngeal carcinoma, hepatocellular carcinoma, uterine endometrial) (138, 139). As discussed above, TGF-β induces glycolysis and, therefore, promotes self-renewal in these cancers. However, stem cells of other cancer types (glioma, glioblastoma. lung cancer, pancreatic ductal adenocarcinoma, and leukemia) have demonstrated OXPHOS as their preferred energy production process (138, 139).
A clear distinction must be drawn between the association and causality of events. To establish causality, it is necessary for a change introduced in one event to result in some observable change in the second event. Reverting to the previous discussions, efforts have been undertaken to examine the causal relationship between TGF-β signaling and asthma. This has been pursued by studying the impact of anti-TGF-β/Smad agents on airway inflammation. There are indications that the role of TGF-β changes along the course of asthma development. TGF-β seems to inhibit the development of asthma in the initial stage of the disease, however it supports and maintains established inflammatory asthma. This temporal change in the role of TGF-β was reflected in the work by Fredriksson et al. (140) who demonstrated a contrasting effect of rapamycin on asthma which was depended on the stage of the disease. Rapamycin is known to activate the TGF-β signaling (141–143). Airway inflammation in a murine model was attenuated when rapamycin was administered during the induction stage of asthma, while inflammation was induced when rapamycin was administered in established asthma. In addition, rapamycin induced airway hyperactivity (i.e., exacerbation of the disease) in moderate lymphangioleiomyomatosis (a rare and progressive lung disease leading to the blockage of airways and blood vessels) (144). Here rapamycin was administered to patients with established lung inflammation. When CD4(+) T helper cells engineered to produce latent TGF-β1 were transferred to OVA-sensitized SCID mice 1-2 days before the mice were re-challenged with ovalbumin (OVA), airway inflammation was attenuated (145). Similarly, when TGF-β-secreting bone marrow stroma cells were injected intravenously to mice with ragweed induced asthma at the time of the antigen challenge, they protected the mice from most asthma-specific pathological changes, including Th2 related inflammatory cytokines and immunoglobulins (146).
Conversely, when nintedanib, a TGF-β/Smad pathway inhibitor (147), was given orally once a day (started day 35) to OVA-sensitized mice, after the starting of repeated OVA challenge (started day 31), airway inflammation was ameliorated (148). Similarly, when anti-TGF-β antibodies were injected to OVA-sensitized mice 30 minutes before each OVA-challenge for seven days, airway inflammation improved (149). Here again, the anti-TGF-β agent was administered (for 6 days) to mice after repeated OVA challenges. Pirfenidone is an antifibrotic agent that inhibits the TGF-β/Smad pathway (150). When OVA-sensitized mice were put on pirfenidone diet started before OVA-rechallenge and extended into a period of OVA-rechallenges, the results were mixed: pirfenidone had no effect on airway hyper-responsiveness, however pirfenidone significantly reduced distal bronchiolar cellularity and proximal and distal bronchiolar mucin content (151).
The pro-inflammatory role of TGF-β in established asthma may be related to asthma highly inflammatory environment that includes high concentrations of IL-6 and IL-21 (152), which in mice induce Th17 differentiation in the presence of low levels of TGF-β (31, 153) (in human, the presence of IL-1β is also required (32)). Th17 cytokines, including IL-17, play a role in neutrophilic inflammation in severe asthma (154) and IL-21is increased in human asthmatic airways (155). In the early stage of asthma, the concentrations of IL-6 and IL-21 are likely insufficient to induce Th17 differentiation. This inadequacy may explain the failure to reduce airway inflammation through TGF-β inhibition at this stage. Moreover, at this stage TGF-β suppresses the activity of the immune system with the result of reducing the inflammatory symptoms of asthma, observed in animal models of asthma.
It should be added that two studies demonstrate an opposite picture. Alcorn et al. (156) report that anti-TGF-β antibodies applied prior to each QVA challenge following a period of OVA sensitization, induced airway hypersensitivity with no significant effect on airway inflammation in a mouse model. Similarly, McMillan et al. (157) have shown that anti-TGF-beta antibodies administered therapeutically, with dosing starting after the onset of established eosinophilic airway inflammation in mice, significantly reduced peribronchiolar extracellular matrix deposition, airway smooth muscle cell proliferation, and mucus production in the lung without affecting established airway inflammation and Th2 cytokine production. The contrasting results between different studies may be related to varying levels of TGF-β in each asthma model. As Zhou et al. (31) have shown, in mice, TGF-β orchestrates Th17 cell differentiation in a concentration-dependent manner. At low concentrations, TGF-β synergizes with IL-6 and IL-21 to promote IL-23R expression, favoring Th17 cell differentiation. This effect increases with TGF-β level and reaches an apex at a specific TGF-β level. TGF-β concentrations higher than this apex point repress IL-23R expression and favor Foxp3+ Treg cells. It should be remembered that at the same time, TGF-β suppresses Th2 (and Th1) cells (46) and inhibits mast cells activation (158), both involved in airway inflammation.
It is noted that a protective effect of TGF-β in early stage and a detrimental effect in late state on disease development is also typical to cancer (39, 159).
Overall, inhibiting TGF-β/Smad signaling seems to provide short-term benefits for patients with neutrophilic asthma, considering the established state of the disease. The question of potential serious adverse events associated with such treatment remains. However, the causative relationship between TGF-β/Smad signaling and long-term comorbidities is still unclear.
The TGF-β superfamily member activin-A serves various biological functions and shares signaling pathways with TGF-β1. Ex vivo stimulations of nasal tissue from patients with nasal polyps have suggested a direct induction of each other’s expression at the mRNA level by activin-A and TGF-β1 (160). Additionally, recent findings indicate that Activin-A induces apoptosis of human lung adenocarcinoma A549 cells through the endoplasmic reticulum stress pathway (161). If this mechanism extends to other cancer types, it could elucidate the negative correlation between AR and the risk of these cancers. In contrast to the two mechanisms proposed earlier, this mechanism also predicts a beneficial effect of AR on cancer prognosis.
It is suggested here, that irrespective of the specific disease, allergy patients should individually be classified into two distinct groups: patients with high level of circulating TGF-β and Th17 cytokines and those with levels lower than or close to normal levels. The author hypothesizes that allergy patients belonging to the first group will display cell metabolism biased toward glycolysis. They will be more prone to develop obesity, insulin resistance, hypertension and DM2. These patients will also be subject to a higher risk of cancer compared with normal subjects. Patients belonging to the second group will display OXPHOS-biased cell metabolism, a lower association with metabolic dysregulations, and a reduced risk of cancer compared with normal subjects. Subjects belonging to the first group should be more closely watched for metabolic syndrome and cancer.
It is notable that asthma and AR often coexist. Estimates suggest that 19-38% of patients with AR also have concurrent asthma (162), while 85–95% of patients with asthma also experience AR (163). Drawing from the earlier discussions, the author posits that, in most cases, AR will tend to coexist more frequently with eosinophilic asthma and less so with neutrophilic asthma.
The distinct inflammatory pathways of AR and neutrophilic asthma likely contribute to their contrasting associations with metabolic dysregulations and cancer risk. Given that TGF-β, IL-1β and IL-6 are essential for Th17 cell differentiation, the connection between neutrophilic asthma (characterized by high levels of these cytokines) and metabolic dysregulation and the risk of cancer becomes apparent. Specifically, in neutrophilic asthma, levels of IL-1β surpass those observed in eosinophilic asthma (notably, IL-6 levels in asthmatic patients do not correlate with the presence of neutrophils in the airways (164)). In comparison to AR, neutrophilic asthma displays higher values of both TGF-β and IL-1β circulating levels. The difference in cytokine levels between these two allergic conditions accounts for the opposing associations of neutrophilic asthma and allergic rhinitis with most types of cancer and metabolic dysregulations.
It is noteworthy that the involvement of TGF-β in metabolic dysregulations has been previously documented (165). Nevertheless, to the author’s knowledge, this is the first piece of work that establishes a connection between TGF-β/Smad pathway, IL-1β and IL-6 levels, metabolic pathways, the type of allergy, metabolic dysregulations, and the risk of cancer within a unified framework.
The schematic relationship between TGF-β levels, TGF-β signaling, metabolic pathways, metabolic dysregulations, allergic rhinitis and cancer risk, is depicted in Figure 1.
The current study proposes a binary classification of TGF-β/Smad pathway activity as either high or low, aiming to elucidate the divergent connections between various allergic diseases and the risk of cancer, as well as their contrasting association with metabolic dysregulations. High levels of TGF-β and Th17 inflammatory cytokines in neutrophilic asthma, and low levels of TGF-β in allergic rhinitis may provide an explanation for the differences in long-term comorbidities between neutrophilic asthma and allergic rhinitis. The binary nature of these relationships is suggested to be governed by the two metabolic pathways of eukaryotic cells: glycolysis and oxidative phosphorylation.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
ZE: Writing – review & editing, Writing – original draft, Validation, Supervision, Methodology, Investigation, Data curation, Conceptualization.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The author expresses gratitude to Prof. Shaul Hestrin from the Department of Comparative Medicine, Stanford University School of Medicine, and Dr. Shivang Sunil Parikh from The Ragon Institute of MGH, MIT, and Harvard, for their valuable discussions and advice.
ZE was employed by the company Taro Pharmaceutical Industries Ltd.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The views and opinions expressed, and/or conclusions drawn, in this article are those of the author and do not necessarily reflect those of Taro Pharmaceutical Industries Ltd., its affiliates, directors or employees.
1. Buelow B, Routes JM. Immediate hypersensitivity reaction. Medscape. (2020). Available at: https://emedicine.medscape.com/article/136217-overview?form=fpf#a5.
2. Hemminki K, Försti A, Fallah M, Sundquist J, Sundquist K, Ji J. Risk of cancer in patients with medically diagnosed hay fever or allergic rhinitis. Int J Cancer. (2014) 135:2397–403. doi: 10.1002/ijc.28873
3. Liu X, Hemminki K, Försti A, Sundquist J, Sundquist K, Ji J. Cancer risk and mortality in asthma patients: A Swedish national cohort study. Acta Oncol. (2015) 54:1120–7. doi: 10.3109/0284186X.2014.1001497
4. Wondmkun YT. Obesity, insulin resistance, and type 2 diabetes: associations and therapeutic implications. Diabetes Metab Syndr Obes. (2020) 13:3611–6. doi: 10.2147/DMSO.S275898
5. Torres RM, Souza MDS, Coelho ACC, de Mello LM, Souza-MaChado C. Association between asthma and type 2 diabetes mellitus: mechanisms and impact on asthma control-A literature review. Can Respir J. (2021) 2021:8830439. doi: 10.1155/2021/8830439
6. Zolotareva O, Saik OV, Königs C, Bragina EY, Goncharova IA, Freidin MB, et al. Comorbidity of asthma and hypertension may be mediated by shared genetic dysregulation and drug side effects. Sci Rep. (2019) 9:16302. doi: 10.1038/s41598-019-52762-w
7. Jiang D, Wang L, Bai C, Chen O. Association between abdominal obesity and asthma: a meta-analysis. Allergy Asthma Clin Immunol. (2019) 15:16. doi: 10.1186/s13223-019-0333-6
8. Shore SA. Obesity and asthma: lessons from animal models. J Appl Physiol (1985). (2007) 102:516–28. doi: 10.1152/japplphysiol.00847.2006
9. Kong J, Yang F, Bai M, Zong Y, Li Z, Meng X, et al. Airway immune response in the mouse models of obesity-related asthma. Front Physiol. (2022) 13:909209. doi: 10.3389/fphys.2022.909209
10. Daniels Gatward LF, Kennard MR, Smith LIF, King AJF. The use of mice in diabetes research: The impact of physiological characteristics, choice of model and husbandry practices. Diabetes Med. (2021) 38:e14711. doi: 10.1111/dme.14711
11. Gu C, Loube J, Lee R, Bevans-Fonti S, Wu TD, Barmine JH, et al. Metformin alleviates airway hyperresponsiveness in a mouse model of diet-induced obesity. Front Physiol. (2022) 13:883275. doi: 10.3389/fphys.2022.883275
12. Lee TK, Jeon YJ, Jung SJ. Bi-directional association between allergic rhinitis and diabetes mellitus from the national representative data of South Korea. Sci Rep. (2021) 11:4344. doi: 10.1038/s41598-021-83787-9
13. Hashimoto Y, Futamura A. Prevalence of allergic rhinitis is lower in subjects with higher levels of fasting plasma glucose. Diabetes Care. (2010) 33:e143. doi: 10.2337/dc10-1338
14. Hwang IC, Lee YJ, Ahn HY, Lee SM. Association between allergic rhinitis and metabolic conditions: a nationwide survey in Korea. Allergy Asthma Clin Immunol. (2016) 12:5. doi: 10.1186/s13223-015-0108-7
15. Han YY, Forno E, Gogna M, Celedón JC. Obesity and rhinitis in a nationwide study of children and adults in the United States. J Allergy Clin Immunol. (2016) 137:1460–5. doi: 10.1016/j.jaci.2015.12.1307
16. Manuyakorn W, Kamchaisatian W, Atamasirikul K, Sasisakulporn C, Direkwattanachai C, Benjaponpitak S. Serum TGF-beta1 in atopic asthma. Asian Pac J Allergy Immunol. (2008) 26:185–9.
17. Ozyilmaz E, Canbakan S, Capan N, Erturk A, Gulhan M. Correlation of plasma transforming growth factor beta 1 with asthma control test. Allergy Asthma Proc. (2009) 30:35–40. doi: 10.2500/aap.2009.30.3192
18. Karagiannidis C, Hense G, Martin C, Epstein M, Rückert B, Mantel PY, et al. Activin A is an acute allergen-responsive cytokine and provides a link to TGF-beta-mediated airway remodeling in asthma. J Allergy Clin Immunol. (2006) 117:111–8. doi: 10.1016/j.jaci.2005.09.017
19. Jiang K, Chen HB, Wang Y, Lin JH, Hu Y, Fang YR. Changes in interleukin-17 and transforming growth factor beta 1 levels in serum and bronchoalveolar lavage fluid and their clinical significance among children with asthma. Transl Pediatr. (2013) 2:154–9. doi: 10.3978/j.issn.2224-4336.2013.10.03
20. Rosas A, Valencia MP, Sánchez M, Bautista M, Rico G, Vega GB. Transforming growth factor beta and platelets in allergic rhinitis and sinusitis. Rev Alerg Mex. (2011) 58:93–8.
21. Chai SK, Altman GM, Yazdanbakhsh M, Tsuji J, Godat L, Takaro TK. Production of interleukin 10 and transforming growth factor beta in concomitant allergy and autoimmunity. Ann Allergy Asthma Immunol. (2005) 94:279–85. doi: 10.1016/S1081-1206(10)61309-9
22. Lee SS, Won TB, Kim JW, Rhee CS, Lee CH, Hong SC, et al. Effects of dexamethasone on the expression of transforming growth factor-beta in the mouse model of allergic rhinitis. Laryngoscope. (2007) 117:1323–8. doi: 10.1097/MLG.0b013e318064e84d
23. Ciprandi G, De Amici M, Tosca M, Marseglia G. Serum transforming growth factor-beta levels depend on allergen exposure in allergic rhinitis. Int Arch Allergy Immunol. (2010) 152:66–70. doi: 10.1159/000260085
24. Qiu Q, Lu H, Lu C, Chen S, Han H. Variations in TGF-beta, IL-10, and IL-17 after specific immunotherapy and correlations with symptoms in patients with allergic rhinitis. J Investig Allergol Clin Immunol. (2012) 22:311–2.
25. Mirzaei Y, Savari Z, Yazdani-Nafchi F, Salehi-Vanani N, Fallahi E, Pirayesh A, et al. The expression analysis of IL-6, IL-18, IL-21, IL-23, and TGF-β mRNA in the nasal mucosa of patients with Allergic rhinitis. Afr Health Sci. (2022) 22:630–40. doi: 10.4314/ahs.v22i1.73
26. Bayrak Degirmenci P, Aksun S, Altin Z, Bilgir F, Arslan IB, Colak H, et al. Allergic rhinitis and its relationship with IL-10, IL-17, TGF-β, IFN-γ, IL 22, and IL-35. Dis Markers. (2018) 2018:9131432. doi: 10.1155/2018/9131432
27. Hammad H, Lambrecht BN. The basic immunology of asthma. Cell. (2021) 184:1469–85. doi: 10.1016/j.cell.2021.02.016
28. Maison N, Omony J, Illi S, Thiele D, Skevaki C, Dittrich AM, et al. T2-high asthma phenotypes across lifespan. Eur Respir J. (2022) 60:2102288. doi: 10.1183/13993003.02288-2021
29. Das S, Miller M, Broide DH. Chromosome 17q21 genes ORMDL3 and GSDMB in asthma and immune diseases. Adv Immunol. (2017) 135:1–52. doi: 10.1016/bs.ai.2017.06.001
30. Ciprandi G, Cuppari C, Salpietro A, Tosca M, Grasso L, Rigoli L, et al. Serum IL-23 in asthmatic children. J Biol Regul Homeost Agents. (2012) 26:S53–61.
31. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. (2008) 453:236–40. doi: 10.1038/nature06878
32. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. (2008) 9:641–9. doi: 10.1038/ni.1610
33. Lluis A, Schedel M, Liu J, Illi S, Depner M, von Mutius E, et al. Asthma-associated polymorphisms in 17q21 influence cord blood ORMDL3 and GSDMA gene expression and IL-17 secretion. J Allergy Clin Immunol. (2011) 127:1587–94.e6. doi: 10.1016/j.jaci.2011.03.015
34. Bantulà M, Roca-Ferrer J, Arismendi E, Picado C. Asthma and obesity: two diseases on the rise and bridged by inflammation. J Clin Med. (2021) 10:169. doi: 10.3390/jcm10020169
35. Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheumatol. (2009) 60:3642–50. doi: 10.1002/art.24959
36. Prame Kumar K, Nicholls AJ, Wong CHY. Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. (2018) 371:551–65. doi: 10.1007/s00441-017-2753-2
37. Chang C, Chen G, Wu W, Chen D, Chen S, Gao J, et al. Exogenous IL-25 ameliorates airway neutrophilia via suppressing macrophage M1 polarization and the expression of IL-12 and IL-23 in asthma. Respir Res. (2023) 24:260. doi: 10.1186/s12931-023-02557-5
38. Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J. (2014) 43:1067–76. doi: 10.1183/09031936.00105013
39. Elkoshi Z. Cancer and autoimmune diseases: A tale of two immunological opposites? Front Immunol. (2022) 25:821598. doi: 10.3389/fimmu.2022.821598
40. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol. (2020) 10:3100. doi: 10.3389/fimmu.2019.03100
41. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
42. Kalathil SG, Thanavala Y. Importance of myeloid derived suppressor cells in cancer from a biomarker perspective. Cell Immunol. (2021) 361:104280. doi: 10.1016/j.cellimm.2020.104280
43. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
44. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget. (2016) 7:52294–306. doi: 10.18632/oncotarget.10561
45. Schmidt A, Éliás S, Joshi RN, Tegnér J. In vitro differentiation of human CD4+FOXP3+ Induced regulatory T cells (iTregs) from naïve CD4+ T cells using a TGF-β-containing protocol. J Vis Exp. (2016) 118):55015. doi: 10.3791/55015
46. Chen W. TGF-β Regulation of T cells. Annu Rev Immunol. (2023) 41:483–512. doi: 10.1146/annurev-immunol-101921-045939
47. Cao P, Sun Z, Zhang F, Zhang J, Zheng X, Yu B, et al. TGF-β Enhances immunosuppression of myeloid-derived suppressor cells to induce transplant immune tolerance through affecting arg-1 expression. Front Immunol. (2022) 13:919674. doi: 10.3389/fimmu.2022.919674
48. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
49. Peñuelas S, Anido J, Prieto-Sánchez RM, Folch G, Barba I, Cuartas I, et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. (2009) 15:315–27. doi: 10.1016/j.ccr.2009.02.011
50. Nana AW, Yang PM, Lin HY. Overview of transforming growth factor β Superfamily involvement in glioblastoma initiation and progression. Asian Pac J Cancer Prev. (2015) 16:6813–23. doi: 10.7314/apjcp.2015.16.16.6813
51. Nie Z, Wang C, Zhou Z, Chen C, Liu R, Wang D. Transforming growth factor-beta increases breast cancer stem cell population partially through upregulating PMEPA1 expression. Acta Biochim Biophys Sin (Shanghai). (2016) 48:194–201. doi: 10.1093/abbs/gmv130
52. Futakuchi M, Lami K, Tachibana Y, Yamamoto Y, Furukawa M, Fukuoka J. The effects of TGF-β Signaling on cancer cells and cancer stem cells in the bone microenvironment. Int J Mol Sci. (2019) 20:5117. doi: 10.3390/ijms20205117
53. Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. (2011) 9:433–46. doi: 10.1016/j.stem.2011.10.001
54. Song K, Wang H, Krebs TL, Kim SJ, Danielpour D. Androgenic control of transforming growth factor-beta signaling in prostate epithelial cells through transcriptional suppression of transforming growth factor-beta receptor II. Cancer Res. (2008) 68:8173–82. doi: 10.1158/0008-5472.CAN-08-2290
55. Ito I, Hanyu A, Wayama M, Goto N, Katsuno Y, Kawasaki S, et al. Estrogen inhibits transforming growth factor beta signaling by promoting Smad2/3 degradation. J Biol Chem. (2010) 285:14747–55. doi: 10.1074/jbc.M109.093039
56. Hernández-Vega AM, Camacho-Arroyo I. Crosstalk between 17β-estradiol and TGF-β Signaling modulates glioblastoma progression. Brain Sci. (2021) 11:564. doi: 10.3390/brainsci11050564
57. Kunzmann S, Ottensmeier B, Speer CP, Fehrholz M. Effect of progesterone on Smad signaling and TGF-β/Smad-regulated genes in lung epithelial cells. PloS One. (2018) 13:e0200661. doi: 10.1371/journal.pone.0200661
58. Lindgren PR, Bäckström T, Cajander S, Damber MG, Mählck CG, Zhu D, et al. The pattern of estradiol and progesterone differs in serum and tissue of benign and Malignant ovarian tumors. Int J Oncol. (2002) 21(3):583–9. doi: 10.3892/ijo
59. Rodriguez AC, Blanchard Z, Maurer KA, Gertz J. Estrogen signaling in endometrial cancer: a key oncogenic pathway with several open questions. Horm Cancer. (2019) 10:51–63. doi: 10.1007/s12672-019-0358-9
60. Zouboulis CC, Chen WC, Thornton MJ, Qin K, Rosenfield R. Sexual hormones in human skin. Horm Metab Res. (2007) 39:85–95. doi: 10.1055/s-2007-961807
61. Parikh R, Sorek E, Parikh S, Michael K, Bikovski L, Tshori S, et al. Skin exposure to UVB light induces a skin-brain-gonad axis and sexual behavior. Cell Rep. (2021) 36:109579. doi: 10.1016/j.celrep.2021.109579
62. Ernster VL, Wrensch MR, Petrakis NL, King EB, Miike R, Murai J, et al. Benign and Malignant breast disease: initial study results of serum and breast fluid analyses of endogenous estrogens. J Natl Cancer Inst. (1987) 79:949–60.
63. Samoli E, Trichopoulos D, Lagiou A, Zourna P, Georgila C, Minaki P, et al. The hormonal profile of benign breast disease. Br J Cancer. (2013) 108:199–204. doi: 10.1038/bjc.2012.493
64. Visscher DW, Frost MH, Hartmann LC, Frank RD, Vierkant RA, McCullough AE, et al. Clinicopathologic features of breast cancers that develop in women with previous benign breast disease. Cancer. (2016) 122:378–85. doi: 10.1002/cncr.29766
65. Salib RJ, Kumar S, Wilson SJ, Howarth PH. Nasal mucosal immunoexpression of the mast cell chemoattractants TGF-beta, eotaxin, and stem cell factor and their receptors in allergic rhinitis. J Allergy Clin Immunol. (2004) 114:799–806. doi: 10.1016/j.jaci.2004.07.010
66. Gosset P, Malaquin F, Delneste Y, Wallaert B, Capron A, Mählck CG, et al. Interleukin-6 and interleukin-1 alpha production is associated with antigen-induced late nasal response. J Allergy Clin Immunol. (1993) 92:878–90. doi: 10.1016/0091-6749(93)90066-O
67. Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. (2011) 14:67–79. doi: 10.1016/j.cmet.2011.04.013
68. Lee MJ. Transforming growth factor beta superfamily regulation of adipose tissue biology in obesity. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1160–71. doi: 10.1016/j.bbadis.2018.01.025
69. El-Gohary Y, Tulachan S, Wiersch J, Guo P, Welsh C, Prasadan K, et al. A smad signaling network regulates islet cell proliferation. Diabetes. (2014) 63:224–36. doi: 10.2337/db13-0432
70. Sheng J, Wang L, Tang PM, Wang HL, Li JC, Xu BH, et al. Smad3 deficiency promotes beta cell proliferation and function in db/db mice via restoring Pax6 expression. Theranostics. (2021) 11:2845–59. doi: 10.7150/thno.51857
71. Wang HL, Wang L, Zhao CY, Lan HY. Role of TGF-beta signaling in beta cell proliferation and function in diabetes. Biomolecules. (2022) 12:373. doi: 10.3390/biom12030373
72. Rajendran S, Anquetil F, Quesada-Masachs E, Graef M, Gonzalez N, McArdle S, et al. IL-6 is present in beta and alpha cells in human pancreatic islets: Expression is reduced in subjects with type 1 diabetes. Clin Immunol. (2020) 211:108320. doi: 10.1016/j.clim.2019.108320
73. Anquetil F, Sabouri S, Thivolet C, Rodriguez-Calvo T, Zapardiel-Gonzalo J, Amirian N, et al. Alpha cells, the main source of IL-1β in human pancreas. J Autoimmun. (2017) 81:68–73. doi: 10.1016/j.jaut.2017.03.006
74. Porreca E, Di Febbo C, Mincione G, Reale M, Baccante G, Guglielmi MD, et al. Increased transforming growth factor-beta production and gene expression by peripheral blood monocytes of hypertensive patients. Hypertension. (1997) 30:134–9. doi: 10.1161/01.hyp.30.1.134
75. Lijnen PJ, Petrov VV, Fagard RH. Association between transforming growth factor-beta and hypertension. Am J Hypertens. (2003) 16:604–11. doi: 10.1016/s0895-7061(03)00847-1
76. Tanase DM, Gosav EM, Radu S, Ouatu A, Rezus C, Ciocoiu M, et al. Arterial hypertension and interleukins: potential therapeutic target or future diagnostic marker? Int J Hypertens. (2019) 2019:3159283. doi: 10.1155/2019/3159283
77. Kang N, Song WJ. Discovering biomarkers of neutrophilic asthma: A clinician's perspective. Allergy Asthma Immunol Res. (2022) 14:1–4. doi: 10.4168/aair.2022.14.1.1
78. Wang HR, Wei SZ, Song XY, Wang Y, Zhang WB, Ren C, et al. IL-1β and allergy: focusing on its role in allergic rhinitis. Mediators Inflamm. (2023) 2023:1265449. doi: 10.1155/2023/1265449
79. Han MW, Kim SH, Oh I, Kim YH, Lee J. Serum IL-1β can be a biomarker in children with severe persistent allergic rhinitis. Allergy Asthma Clin Immunol. (2019) 15:58. doi: 10.1186/s13223-019-0368-8
80. Gao S, Yu L, Zhang J, Li X, Zhou J, Zeng P, et al. Expression and clinical significance of VCAM-1, IL-6, and IL-17A in patients with allergic rhinitis. Ann Palliat Med. (2021) 10:4516–22. doi: 10.21037/apm-21-546
81. Recalde M, Pistillo A, Davila-Batista V, Leitzmann M, Romieu I, Viallon V, et al. Longitudinal body mass index and cancer risk: a cohort study of 2.6 million Catalan adults. Nat Commun. (2023) 14:3816. doi: 10.1038/s41467-023-39282-y
82. Pothiawala S, Qureshi AA, Li Y, Han J. Obesity and the incidence of skin cancer in US Caucasians. Cancer Causes Control. (2012) 23:717–26. doi: 10.1007/s10552-012-9941-x
83. Bjørge T, Lie AK, Engeland A. The impact of height and body mass index on the risk of testicular cancer in 600,000 Norwegian men. Cancer Causes Control. (2006) 17:983–7. doi: 10.1007/s10552-006-0032-8
84. Davies TW, Prener A, Engholm G. Body size and cancer of the testis. Acta Oncol. (1990) 29:287–90. doi: 10.3109/02841869009089999
85. Wise MR, Jordan V, Lagas A, Showell M, Wong N, Lensen S, et al. Obesity and endometrial hyperplasia and cancer in premenopausal women: A systematic review. Am J Obstet Gynecol. (2016) 214:689.e1–689.e17. doi: 10.1016/j.ajog.2016.01.175
86. Foong KW, Bolton H. Obesity and ovarian cancer risk: A systematic review. Post Reprod Health. (2017) 23:183–98. doi: 10.1177/2053369117709225
87. Telenga ED, Tideman SW, Kerstjens HA, Hacken NH, Timens W, Postma DS, et al. Obesity in asthma: more neutrophilic inflammation as a possible explanation for a reduced treatment response. Allergy. (2012) 67:1060–8. doi: 10.1111/j.1398-9995.2012.02855.x
88. Scott HA, Gibson PG, Garg ML, Wood LG. Airway inflammation is augmented by obesity and fatty acids in asthma. Eur Respir J. (2011) 38:594–602. doi: 10.1183/09031936.00139810
89. Haldar P, Pavord ID, Shaw DE, Berry MA, Thomas M, Brightling CE, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. (2008) 178:218–24. doi: 10.1164/rccm.200711-1754OC
90. Todd DC, Armstrong S, D'Silva L, Allen CJ, Hargreave FE, Parameswaran K. Effect of obesity on airway inflammation: a cross-sectional analysis of body mass index and sputum cell counts. Clin Exp Allergy. (2007) 37:1049–54. doi: 10.1111/j.1365-2222.2007.02748.x
91. Rhee H, Love T, Harrington D. Blood neutrophil count is associated with body mass index in adolescents with asthma. JSM Allergy Asthma. (2018) 3:1019.
92. Wang Y, Wan R, Hu C. Leptin/obR signaling exacerbates obesity-related neutrophilic airway inflammation through inflammatory M1 macrophages. Mol Med. (2023) 29:100. doi: 10.1186/s10020-023-00702-w
93. Uribe-Querol E, Rosales C. Neutrophils actively contribute to obesity-associated inflammation and pathological complications. Cells. (2022) 11:1883. doi: 10.3390/cells11121883
94. Gutmann D, Dressler M, Eickmeier O, Herrmann E, Kirwil M, Schubert R, et al. Proinflammatory pattern in the lower airways of non-asthmatic obese adolescents. Cytokine. (2024) 173:156452. doi: 10.1016/j.cyto.2023.156452
95. Amin K, Issa SM, Ali KM, Aziz MI, Hama Amieen HM, Bystrom J, et al. Evidence for eosinophil and IL-17 mediated inflammation in allergic rhinitis. Clin Mol Allergy. (2020) 18:6. doi: 10.1186/s12948-020-00117-6
96. Qing M, Yongge L, Wei X, Yan W, Zhen L, Yixin R, et al. Comparison of Th17 cells mediated immunological response among asthmatic children with or without allergic rhinitis. Asian Pac J Allergy Immunol. (2019) 37:65–72. doi: 10.12932/AP-140317-0047
97. Shirkani A, Mansouri A, Farid Hosseini R, Jabbari Azad F, Alsadat Mahmoudian R, Montazer M, et al. The role of interleukin-4 and 13 gene polymorphisms in allergic rhinitis: A case control study. Rep Biochem Mol Biol. (2019) 8:111–8.
98. Le Floc'h A, Allinne J, Nagashima K, Scott G, Birchard D, Asrat S, et al. Dual blockade of IL-4 and IL-13 with dupilumab, an IL-4Rα antibody, is required to broadly inhibit type 2 inflammation. Allergy. (2020) 75:1188–204. doi: 10.1111/all.14151
99. Campion NJ, Doralt A, Lupinek C, Berger M, Poglitsch K, Brugger J, et al. Dupilumab reduces symptom burden in allergic rhinitis and suppresses allergen-specific IgE production. Allergy. (2023) 78:1687–91. doi: 10.1111/all.15653
100. Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol. (2000) 165:4773–7. doi: 10.4049/jimmunol.165.9.4773
101. Al-Alawi M, Hassan T, Chotirmall SH. Transforming growth factor β and severe asthma: a perfect storm. Respir Med. (2014) 108:1409–23. doi: 10.1016/j.rmed.2014.08.008
102. Zou XL, Chen ZG, Zhang TT, Feng DY, Li HT, Yang HL. Th17/Treg homeostasis, but not Th1/Th2 homeostasis, is implicated in exacerbation of human bronchial asthma. Ther Clin Risk Manage. (2018) 14:1627–36. doi: 10.2147/TCRM.S172262
103. Elkoshi Z. The binary classification of chronic diseases. J Inflammation Res. (2019) 12:319–33. doi: 10.2147/JIR.S227279
104. Elkoshi Z. The binary model of chronic diseases applied to COVID-19. Front Immunol. (2021) 12:716084. doi: 10.3389/fimmu.2021.716084
105. Elkoshi Z. SARS-coV-2 omicron (B.1.1.529) variant: corticosteroids treatment/respiratory coinfection. Front Immunol. (2022) 13:856072. doi: 10.3389/fimmu.2022.856072
106. Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. (2017) 8:289. doi: 10.3389/fimmu.2017.00289
107. Soto-Heredero G, Gómez de Las Heras MM, Gabandé-Rodríguez E, Oller J, Mittelbrunn M. Glycolysis - a key player in the inflammatory response. FEBS J. (2020) 287:3350–69. doi: 10.1111/febs.15327
108. Lee SY, Jeon HM, Ju MK, Jeong EK, Kim CH, Yoo MA, et al. Dlx-2 is implicated in TGF-β- and Wnt-induced epithelial-mesenchymal, glycolytic switch, and mitochondrial repression by Snail activation. Int J Oncol. (2015) 46:1768–80. doi: 10.3892/ijo.2015.2874
109. Zhang D, Wang Y, Shi Z, Liu J, Sun P, Hou X, et al. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. (2015) 10:1335–48. doi: 10.1016/j.celrep.2015.02.006
110. Rodríguez-García A, Samsó P, Fontova P, Simon-Molas H, Manzano A, Castaño E, et al. TGF-β1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. (2017) 284:3437–54. doi: 10.1111/febs.14201
111. Hu ML, Wang XY, Chen WM. TGF-β1 upregulates the expression of lncRNA UCA1 and its downstream HXK2 to promote the growth of hepatocellular carcinoma. Eur Rev Med Pharmacol Sci. (2018) 22:4846–54. doi: 10.26355/eurrev_201808_15620
112. Wang C, Silverman RM, Shen J, O'Keefe RJ. Distinct metabolic programs induced by TGF-β1 and BMP2 in human articular chondrocytes with osteoarthritis. J Orthop Translat. (2018) 12:66–73. doi: 10.1016/j.jot.2017.12.004
113. Gauthier T, Yao C, Dowdy T, Jin W, Lim YJ, Patiño LC, et al. TGF-β uncouples glycolysis and inflammation in macrophages and controls survival during sepsis. Sci Signal. (2023) 16:eade0385. doi: 10.1126/scisignal.ade0385
114. Guo X, Li H, Xu H, Woo S, Dong H, Lu F, et al. Glycolysis in the control of blood glucose homeostasis. Acta Pharm Sin B. (2012) 2:358–67. doi: 10.1016/j.apsb.2012.06.002
115. Ostroukhova M, Goplen N, Karim MZ, Michalec L, Guo L, Liang Q, et al. The role of low-level lactate production in airway inflammation in asthma. Am J Physiol Lung Cell Mol Physiol. (2012) 302:L300–7. doi: 10.1152/ajplung.00221.2011
116. Qian X, Aboushousha R, van de Wetering C, Chia SB, Amiel E, Schneider RW, et al. IL-1/inhibitory κB kinase ϵ-induced glycolysis augment epithelial effector function and promote allergic airways disease. J Allergy Clin Immunol. (2018) 142:435–450.e10. doi: 10.1016/j.jaci.2017.08.043
117. Arkwright PD, Chase JM, Babbage S, Pravica V, David TJ, Hutchinson IV. Atopic dermatitis is associated with a low-producer transforming growth factor beta(1) cytokine genotype. J Allergy Clin Immunol. (2001) 108:281–4. doi: 10.1067/mai.2001.117259
118. Leman G, Pavel P, Hermann M, Crumrine D, Elias PM, Minzaghi D, et al. Mitochondrial activity is upregulated in nonlesional atopic dermatitis and amenable to therapeutic intervention. J Invest Dermatol. (2022) 142(10):2623–2634.e12. doi: 10.1016/j.jid.2022.01.035
119. Toda M, Leung DY, Molet S, Boguniewicz M, Taha R, Christodoulopoulos P, et al. Polarized in vivo expression of IL-11 and IL-17 between acute and chronic skin lesions. J Allergy Clin Immunol. (2003) 111:875–81. doi: 10.1067/mai.2003.1414
120. Pavel P, Leman G, Hermann M, Ploner C, Eichmann TO, Minzaghi D, et al. Peroxisomal fatty acid oxidation and glycolysis are triggered in mouse models of lesional atopic dermatitis. JID Innov. (2021) 1:100033. doi: 10.1016/j.xjidi.2021.100033
121. Degirmenci PB, Kırmaz C, Vatansever S, Onur E, Nal E, Erdin S, et al. Analysis of the association of chronic spontaneous urticaria with interlekin-4, -10, transforming growth factor-β1, interferon-γ, interleukin-17A and -23 by autologous serum skin test. Postepy Dermatol Alergol. (2017) 34:70–6. doi: 10.5114/pdia.2016.57679
122. Church MK, Kolkhir P, Metz M, Maurer M. The role and relevance of mast cells in urticaria. Immunol Rev. (2018) 282:232–47. doi: 10.1111/imr.12632
123. Erlich TH, Yagil Z, Kay G, Peretz A, Migalovich-Sheikhet H, Tshori S, et al. Mitochondrial STAT3 plays a major role in IgE-antigen-mediated mast cell exocytosis. J Allergy Clin Immunol. (2014) 134:460–9. doi: 10.1016/j.jaci.2013.12.1075
124. Chelombitko MA, Chernyak BV, Fedorov AV, Zinovkin RA, Razin E, Paruchuru LB. The role played by mitochondria in fcϵRI-dependent mast cell activation. Front Immunol. (2020) 16:584210. doi: 10.3389/fimmu.2020.584210
125. Ahmad S, Choudhry MA, Shankar R, Sayeed MM. Transforming growth factor-beta negatively modulates T-cell responses in sepsis. FEBS Lett. (1997) 402:213–8. doi: 10.1016/s0014-5793(96)01535-9
126. Miller-Graziano CL, Szabo G, Griffey K, Mehta B, Kodys K, Catalano D. Role of elevated monocyte transforming growth factor beta (TGF beta) production in posttrauma immunosuppression. J Clin Immunol. (1991) 11:95–102. doi: 10.1007/BF00917745
127. Liu J, Zhou G, Wang X, Liu D. Metabolic reprogramming consequences of sepsis: adaptations and contradictions. Cell Mol Life Sci. (2022) 79:456. doi: 10.1007/s00018-022-04490-0
128. Cheon H, Yu SJ, Yoo DH, Chae IJ, Song GG, Sohn J. Increased expression of pro-inflammatory cytokines and metalloproteinase-1 by TGF-beta1 in synovial fibroblasts from rheumatoid arthritis and normal individuals. Clin Exp Immunol. (2002) 127:547–52. doi: 10.1046/j.1365-2249.2002.01785.x
129. Gonzalo-Gil E, Criado G, Santiago B, Dotor J, Pablos JL, Galindo M. Transforming growth factor (TGF)-β signalling is increased in rheumatoid synovium but TGF-β blockade does not modify experimental arthritis. Clin Exp Immunol. (2013) 174:245–55. doi: 10.1111/cei.12179
130. de Oliveira PG, Farinon M, Sanchez-Lopez E, Miyamoto S, Guma M. Fibroblast-like synoviocytes glucose metabolism as a therapeutic target in rheumatoid arthritis. Front Immunol. (2019) 10:1743. doi: 10.3389/fimmu.2019.01743
131. Garcia-Carbonell R, Divakaruni AS, Lodi A, Vicente-Suarez I, Saha A, Cheroutre H, et al. Critical role of glucose metabolism in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. (2016) 68:1614–26. doi: 10.1002/art.39608
132. Sharma M, Boytard L, Hadi T, Koelwyn G, Simon R, Ouimet M, et al. Enhanced glycolysis and HIF-1α activation in adipose tissue macrophages sustains local and systemic interleukin-1β production in obesity. Sci Rep. (2020) 10:5555. doi: 10.1038/s41598-020-62272-9
133. Hildebrandt X, Ibrahim M, Peltzer N. Cell death and inflammation during obesity: "Know my methods, WAT(son)". Cell Death Differ. (2023) 30:279–92. doi: 10.1038/s41418-022-01062-4
134. Qiao YC, Chen YL, Pan YH, Ling W, Tian F, Zhang XX, et al. Changes of transforming growth factor beta 1 in patients with type 2 diabetes and diabetic nephropathy: A PRISMA-compliant systematic review and meta-analysis. Med (Baltimore). (2017) 96:e6583. doi: 10.1097/MD.0000000000006583
135. Whitehead GS, Thomas SY, Nakano K, Royer DJ, Burke CG, Nakano H, et al. A neutrophil/TGF-β axis limits the pathogenicity of allergen-specific CD4+ T cells. JCI Insight. (2022) 7:e150251. doi: 10.1172/jci.insight.150251
136. Fain JN, Tichansky DS, Madan AK. Transforming growth factor beta1 release by human adipose tissue is enhanced in obesity. Metabolism. (2005) 54:1546–51. doi: 10.1016/j.metabol.2005.05.024
137. Riester M, Xu Q, Moreira A, Zheng J, Michor F, Downey RJ. The Warburg effect: persistence of stem-cell metabolism in cancers as a failure of differentiation. Ann Oncol. (2018) 29:264–70. doi: 10.1093/annonc/mdx645
138. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. (2016) 114:1305–12. doi: 10.1038/bjc.2016.152
139. Yasuda T, Ishimoto T, Baba H. Conflicting metabolic alterations in cancer stem cells and regulation by the stromal niche. Regener Ther. (2021) 17:8–12. doi: 10.1016/j.reth.2021.01.005
140. Fredriksson K, Fielhaber JA, Lam JK, Yao X, Meyer KS, Keeran KJ, et al. Paradoxical effects of rapamycin on experimental house dust mite-induced asthma. PloS One. (2012) 7:e33984. doi: 10.1371/journal.pone.0033984
141. Wu Y, Wang W, Peng XM, He Y, Xiong YX, Liang HF, et al. Rapamycin upregulates connective tissue growth factor expression in hepatic progenitor cells through TGF-β-smad2 dependent signaling. Front Pharmacol. (2018) 9:877. doi: 10.3389/fphar.2018.00877
142. van der Poel HG. Mammalian target of rapamycin and 3-phosphatidylinositol 3-kinase pathway inhibition enhances growth inhibition of transforming growth factor-beta1 in prostate cancer cells. J Urol. (2004) 172:1333–7. doi: 10.1097/01.ju.0000138829.97838.19
143. Osman B, Doller A, Akool e-S, Holdener M, Hintermann E, Pfeilschifter J, et al. Rapamycin induces the TGFbeta1/Smad signaling cascade in renal mesangial cells upstream of mTOR. Cell Signal. (2009) 21:1806–17. doi: 10.1016/j.cellsig.2009.07.016
144. Steagall WK, Stylianou M, Pacheco-Rodriguez G, Yu ZX, Moss J. Unexpected sirolimus-stimulated airway hyperreactivity in lymphangioleiomyomatosis. ERJ Open Res. (2023) 9:00305–2023. doi: 10.1183/23120541.00305-2023
145. Hansen G, McIntire JJ, Yeung VP, Berry G, Thorbecke GJ, Chen L, et al. CD4(+) T helper cells engineered to produce latent TGF-beta1 reverse allergen-induced airway hyperreactivity and inflammation. J Clin Invest. (2000) 105:61–70. doi: 10.1172/JCI7589
146. Nemeth K, Keane-Myers A, Brown JM, Metcalfe DD, Gorham JD, Bundoc VG, et al. Bone marrow stromal cells use TGF-beta to suppress allergic responses in a mouse model of ragweed-induced asthma. Proc Natl Acad Sci U S A. (2010) 107:5652–7. Gorham, Jared D [corrected to Gorham, James D]; Bundoc, Victor G [corrected to Bundoc, Virgilio G]. doi: 10.1073/pnas.0910720107
147. Rangarajan S, Kurundkar A, Kurundkar D, Bernard K, Sanders YY, Ding Q, et al. Novel mechanisms for the antifibrotic action of nintedanib. Am J Respir Cell Mol Biol. (2016) 54:51–9. doi: 10.1165/rcmb.2014-0445OC
148. Lee HY, Hur J, Kim IK, Kang JY, Yoon HK, Lee SY, et al. Effect of nintedanib on airway inflammation and remodeling in a murine chronic asthma model. Exp Lung Res. (2017) 43:187–96. doi: 10.1080/01902148.2017.1339141
149. Wei Y, Zhang Z, Wang F, Zhou S. Assessment of tumor growth factor-β1 neutralizing antibody in the treatment of allergic rhinitis and asthma. Exp Ther Med. (2018) 15:649–56. doi: 10.3892/etm.2017.5501
150. Hamidi SH, Kadamboor Veethil S, Hamidi SH. Role of pirfenidone in TGF-β pathways and other inflammatory pathways in acute respiratory syndrome coronavirus 2 (SARS-Cov-2) infection: a theoretical perspective. Pharmacol Rep. (2021) 73:712–27. doi: 10.1007/s43440-021-00255-x
151. Mansoor JK, Decile KC, Giri SN, Pinkerton KE, Walby WF, Bratt JM, et al. Influence of pirfenidone on airway hyperresponsiveness and inflammation in a Brown-Norway rat model of asthma. Pulm Pharmacol Ther. (2007) 20:660–8. doi: 10.1016/j.pupt.2006.07.005
152. Halwani R, Sultana A, Vazquez-Tello A, Jamhawi A, Al-Masri AA, Al-Muhsen S. Th-17 regulatory cytokines IL-21, IL-23, and IL-6 enhance neutrophil production of IL-17 cytokines during asthma. J Asthma. (2017) 54:893–904. doi: 10.1080/02770903.2017.1283696
153. Wang J, Zhao X, Wan YY. Intricacies of TGF-β signaling in Treg and Th17 cell biology. Cell Mol Immunol. (2023) 20:1002–22. doi: 10.1038/s41423-023-01036-7
154. Margelidon-Cozzolino V, Tsicopoulos A, Chenivesse C, de Nadai P. Role of th17 cytokines in airway remodeling in asthma and therapy perspectives. Front Allergy. (2022) 3:806391. doi: 10.3389/falgy.2022.806391
155. Lajoie S, Lewkowich I, Herman NS, Sproles A, Pesce JT, Wynn TA, et al. IL-21 receptor signalling partially mediates Th2-mediated allergic airway responses. Clin Exp Allergy. (2014) 44:976–85. doi: 10.1111/cea.12341
156. Alcorn JF, Rinaldi LM, Jaffe EF, van Loon M, Bates JH, Janssen-Heininger YM, et al. Transforming growth factor-beta1 suppresses airway hyperresponsiveness in allergic airway disease. Am J Respir Crit Care Med. (2007) 176:974–82. doi: 10.1164/rccm.200702-334OC
157. McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. J Immunol. (2005) 174:5774–80. doi: 10.4049/jimmunol.174.9.5774
158. Gomez G, Ramirez CD, Rivera J, Patel M, Norozian F, Wright HV, et al. TGF-beta 1 inhibits mast cell Fc epsilon RI expression. J Immunol. (2005) 174:5987–93. doi: 10.4049/jimmunol.174.10.5987
159. Fabregat I, Moreno-Càceres J, Sánchez A, Dooley S, Dewidar B, Giannelli G, et al. IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. (2016) 283:2219–32. doi: 10.1111/febs.13665
160. Chaker AM, Zissler UM, Poulos N, Wagenmann M, Bas M, Gürth F, et al. Activin-A is a pro-inflammatory regulator in type-2-driven upper airway disease. Int Arch Allergy Immunol. (2018) 176:15–25. doi: 10.1159/000487930
161. Zhang F, Qi Y, Li J, Liu B, Liu Z, Cui X. Activin A induces apoptosis of human lung adenocarcinoma A549 cells through endoplasmic reticulum stress pathway. Oncol Rep. (2024) 51:29. doi: 10.3892/or.2023.8688
162. Compalati E, Ridolo E, Passalacqua G, Braido F, Villa E, Canonica GW. The link between allergic rhinitis and asthma: the united airways disease. Expert Rev Clin Immunol. (2010) 6:413–23. doi: 10.1586/eci.10.15
163. Togias A. Unique mechanistic features of allergic rhinitis. J Allergy Clin Immunol. (2000) 105:S599–604. doi: 10.1067/mai.2000.106885
164. Neveu WA, Allard JL, Raymond DM, Bourassa LM, Burns SM, Bunn JY, et al. Elevation of IL-6 in the allergic asthmatic airway is independent of inflammation but associates with loss of central airway function. Respir Res. (2010) 11:28. doi: 10.1186/1465-9921-11-28
Keywords: TGF-β, IL-1β, asthma, allergic rhinitis, cancer risk, obesity, type 2 diabetes, cell metabolism
Citation: Elkoshi Z (2024) TGF-β, IL-1β, IL-6 levels and TGF-β/Smad pathway reactivity regulate the link between allergic diseases, cancer risk, and metabolic dysregulations. Front. Immunol. 15:1371753. doi: 10.3389/fimmu.2024.1371753
Received: 16 January 2024; Accepted: 15 March 2024;
Published: 02 April 2024.
Edited by:
Piotr Jan Kraj, Old Dominion University, United StatesReviewed by:
Hridesh Banerjee, University of Pittsburgh, United StatesCopyright © 2024 Elkoshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zeev Elkoshi, emVldi5lbGtvc2hpQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.