 Julia Philippova
Julia Philippova Julia Shevchenko
Julia Shevchenko Sergey Sennikov
Sergey Sennikov- Laboratory of Molecular Immunology, Federal State Budgetary Scientific Institution Research Institute of Fundamental and Clinical Immunology, Novosibirsk, Russia
Disialoganglioside GD2 is a promising target for immunotherapy with expression primarily restricted to neuroectodermal and epithelial tumor cells. Although its role in the maintenance and repair of neural tissue is well-established, its functions during normal organism development remain understudied. Meanwhile, studies have shown that GD2 plays an important role in tumorigenesis. Its functions include proliferation, invasion, motility, and metastasis, and its high expression and ability to transform the tumor microenvironment may be associated with a malignant phenotype. Structurally, GD2 is a glycosphingolipid that is stably expressed on the surface of tumor cells, making it a suitable candidate for targeting by antibodies or chimeric antigen receptors. Based on mouse monoclonal antibodies, chimeric and humanized antibodies and their combinations with cytokines, toxins, drugs, radionuclides, nanoparticles as well as chimeric antigen receptor have been developed. Furthermore, vaccines and photoimmunotherapy are being used to treat GD2-positive tumors, and GD2 aptamers can be used for targeting. In the field of cell therapy, allogeneic immunocompetent cells are also being utilized to enhance GD2 therapy. Efforts are currently being made to optimize the chimeric antigen receptor by modifying its design or by transducing not only αβ T cells, but also γδ T cells, NK cells, NKT cells, and macrophages. In addition, immunotherapy can combine both diagnostic and therapeutic methods, allowing for early detection of disease and minimal residual disease. This review discusses each immunotherapy method and strategy, its advantages and disadvantages, and highlights future directions for GD2 therapy.
1 Introduction
Tumor immunotherapy targeting tumor-associated antigen (TAA) using monoclonal antibodies (mAbs) or immunocompetent cells can improve standard therapeutic methods, including surgery, chemotherapy, and radiation. The immunologic approach aims to stimulate and train the body’s own immune system to cope with malignant cells, which is a safer approach (1). In addition, immunotherapy can combine both diagnostic and therapeutic methods, allowing for early detection of the disease and minimal residual disease. Immunotherapy is also an effective method for combating metastasis and chemoresistant diseases, and treatment with mAbs shows encouraging results in long-term and overall relapse-free survival (2, 3).
Disialoganglioside GD2 is a surface TAA that is expressed by a wide range of tumors of neuroectodermal and epithelial origin, such as neuroblastoma (4), melanoma (5), glioma (6), retinoblastoma (7), medulloblastoma (8), small-cell lung cancer (9), various types of breast cancer (10, 11) and sarcoma (12–14) bladder cancer (15), colorectal cancer (16), and prostate cancer (17). GD2 can be detected on normal central and peripheral nervous system cells, melanocytes (18), lymphocytes, dendritic cells (19), and mesenchymal stem cells (20). Nevertheless, GD2 expression is significantly higher in tumor cells, making this target suitable not only for therapy but also for diagnosis and assessment of disease prognosis (21). GD2 also possesses genetic stability, i.e., the expression level does not decrease during treatment, and most of the antigen remains on the cell surface after binding by antibodies and recognition by immune cells (22). At the same time, GD2 immunotherapy has some limitations, mainly related to the occurrence of side effects and low efficacy in the treatment of extensive solid masses. In this review, different approaches to GD2 immunotherapy, their advantages and disadvantages, and the search for new strategies to improve current developments are presented.
2 Structure and synthesis of disialoganglioside GD2, its role and function
2.1 Structure and synthesis of disialoganglioside GD2
Ganglioside GD2 is a carbohydrate-containing sphingolipid (glycosphingolipid) consisting of a ceramide (sphingosine linked by an amide group to a fatty acid) with two sialic acid residues attached via three monosaccharide links (23, 24). The intracellular synthesis of GD2 occurs in the Golgi apparatus, which starts with the formation of ceramide (lipid domain) (25), followed by the addition of monosaccharide links by means of glycosyltransferases – GM3 synthetase (ST3Gal V) and GD3 synthetase (ST8Sia I, GD3S) (23, 25). The lipid domain is then incorporated into the plasma membrane, whereas the carbohydrate residues are located on the cell surface. GD2 is synthesized from the ganglioside precursors GD3 or GM3 by β1,4-N-acetylgalactosaminyltransferase (GalNAcT, GD2S).
2.2 Function of disialoganglioside GD2 and its role in oncogenesis
The functions of GD2 during normal development of the organism are understudied; it is assigned a role in the maintenance and repair of neural tissue through the regulation of complement activation and inflammation (26). At the same time, numerous studies demonstrate the importance of GD2 in oncogenesis; and its function, high expression, and ability to exert remodeling effects on the tumor microenvironment (TME) may be associated with malignant phenotypes. GD2 can promote proliferation, invasion, motility, and metastasis of various tumor cell types (27, 28), by inducing phosphorylation through the hepatocyte growth factor (HGF) receptor and c-Met pathway of breast cancer (29) or tyrosine kinase receptors and FAK pathways of osteosarcoma (30). ASC amino acid transporter 2 (ASCT2) promotes the malignant phenotype of small-cell lung cancer by enhancing cellular uptake of glutamine, leading to enhanced cell proliferation and migration through phosphorylation of the mTOR1 pathway (31). GD2 also plays a key role in melanoma cell adhesion, growth, proliferation, and invasion by interacting with integrin β1 (32). The ST8SIA1 (GD3 synthetase) gene was shown to regulate GD2 biosynthesis; and when it is knocked out, the inhibition of the FAK/AKT/mTOR signaling pathway and suppression of growth and metastasis in breast cancer is observed (33). It was also reported that increased GD2 expression in cancer cells is associated with NF-κB, and treatment with IKK (inhibition of NF-κB signaling) inhibitors in an experimental model reducing breast cancer metastasis to the lung by more than 5-fold (34), which also suggests the influence of GD2 on metastasis and cell migration. In addition, high GD2 expression is characteristic of diffuse mediastinal glioma cells with the H3K27M mutation, a rare but quite aggressive malignancy (35). GD2 expression was shown to be elevated in oral malignant osteosarcoma samples (30) and neuroblastoma with MYCN amplification (36), which also negatively affects the forecast. Recently, sialic acid-binding Ig-like lectins Siglec-7 were discovered to be expressed on NK cells (37). GD2 is able to suppress NK cell function through binding to Siglec-7, thereby maintaining immunosuppressive TME (38). In addition, GD2 also inhibits the functional activity of T cells and dendritic cells (39), while promoting the recruitment of MDSCs (myeloid-derived suppressor cells) (40) and Tregs (regulatory T cells) (41) to TME. Anti-GD2 mAb treatment inhibits the mTOR/MAPK signaling pathway in breast cancer cells (42), which results in inhibition of tumor migration and growth, and competes with Siglec-7 for binding to GD2 (38).
3 Monoclonal antibodies
Since the 1980s, anti-GD2 mAbs have been actively investigated as theranostic agents in cancer immunotherapy. The unconjugated antibodies recognize TAAs and bind to surface receptors of tumor and immunocompetent TME cells, and depending on the type of the receptor, exert antitumor effects through various mechanisms, including antibody-dependent cell-mediated cytotoxicity/antibody-dependent cellular phagocytosis (ADCC/ADCP), complement-dependent cytotoxicity (CDC), and direct cytotoxicity (Figure 1). During ADCC/ADCP, mAbs bind to Fcγ receptors and promote destruction (NK cells (43, 44), neutrophils (45), γδ T-cells (46)) or phagocytosis (macrophages (47)) of tumor cells. In CDC, the classical complement pathway is activated with the formation of the membrane attack complex (MAC) and recruitment of NK-, T-, NKT-cells, neutrophils, macrophages, and dendritic cells (48). Direct cytotoxicity is realized by the blockade of growth factor receptors, with mAbs binding to receptors on the membrane surface or soluble forms, or inducing apoptosis or necrosis axes (49). mAbs against GD2 can exert a direct cytotoxic action on gangliosides, likely leading to mitochondrial damage by translocation of GD2 from the cell membrane to intracellular compartments (50).
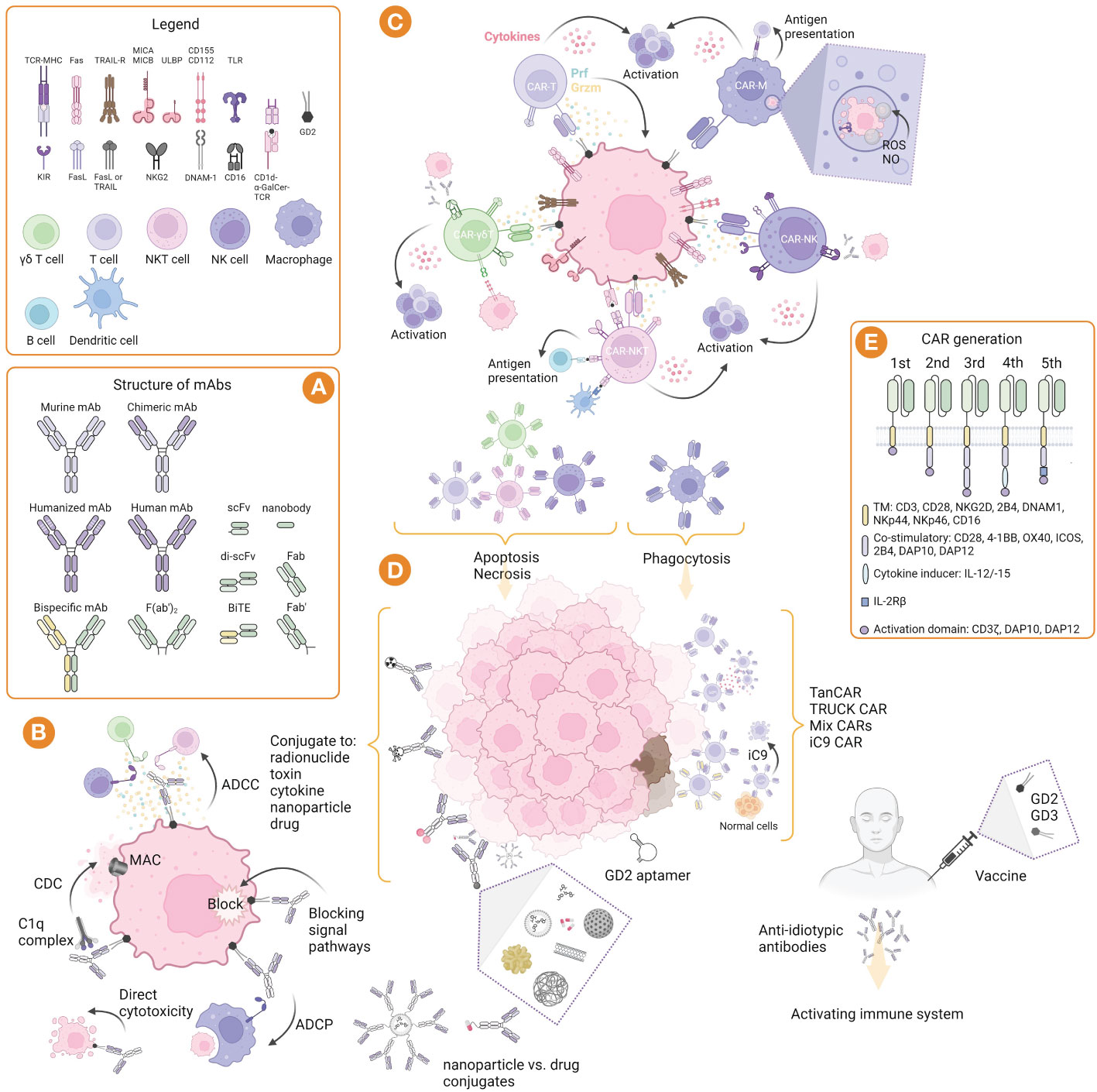
Figure 1 GD2-targeted immunotherapy: strategies, structure, and mechanisms of action. (A) Structure of murine, chimeric, humanized and bispecific mAbs, and their derivatives such as nanobodies and bispecific T cell engager (BiTE). (B) Mechanism of mAbs action: induction of CDC involving complement component 1q complex, followed by the complement cascade and formation of the membrane attack complex (MAC); induction of ADCC mediated by γδ T cells, NK cells, and NKT cells, as well as ADCP mediated by macrophages; blocking of signal pathways and direct cytotoxicity by induction of apoptosis. (C) Mechanisms of immune effector cell cytotoxicity that allow their properties to be exploited in adoptive and CAR therapies. (D) Strategies of immunotherapy include nacked mAbs, as well as conjugated mAbs with radionuclides, toxins, cytokines, nanoparticles, and drugs; CAR cells can be used alone or directed to two or more targets, as well as their modifications, such as TanCAR (bispecific CAR), TRUCK (T cells redirected for antigen-unrestricted cytokine-initiated killing) CAR, and iC9 (inducible caspase 9) CAR; GD2 aptamer can be conjugated to other molecules and toxins for drug delivery or imaging; GD2 vaccines can be used alone or in combination with GD3 vaccines to form anti-idiotypic antibodies and activate the immune system. (E) Structure of CARs generation including domains for αβ and γδ T cells, NK cells, NKT cells, and macrophages. Created with BioRender.com.
Three anti-GD2 drugs dinutuximab (Unituxin®), dinutuximab beta (Qarziba®), and naxitamab (Danyelza®) were formally approved in clinical practice for the treatment of patients with high-risk neuroblastoma. Despite clinical successes, there are several therapy-limiting challenges, including sensitization-related side effects, immunosuppressive TME, loss of antigen expression, production of neutralizing human anti-murine/-chimeric/-human antibodies (HAMA, HACA, and HAHA), extensive masses, etc. Therefore, new strategies are required to modify antibodies and conjugate/combine with other drugs for successful treatment (Table 1). Table 2 shows comparative characteristics of murine, chimeric, and humanized mAbs.
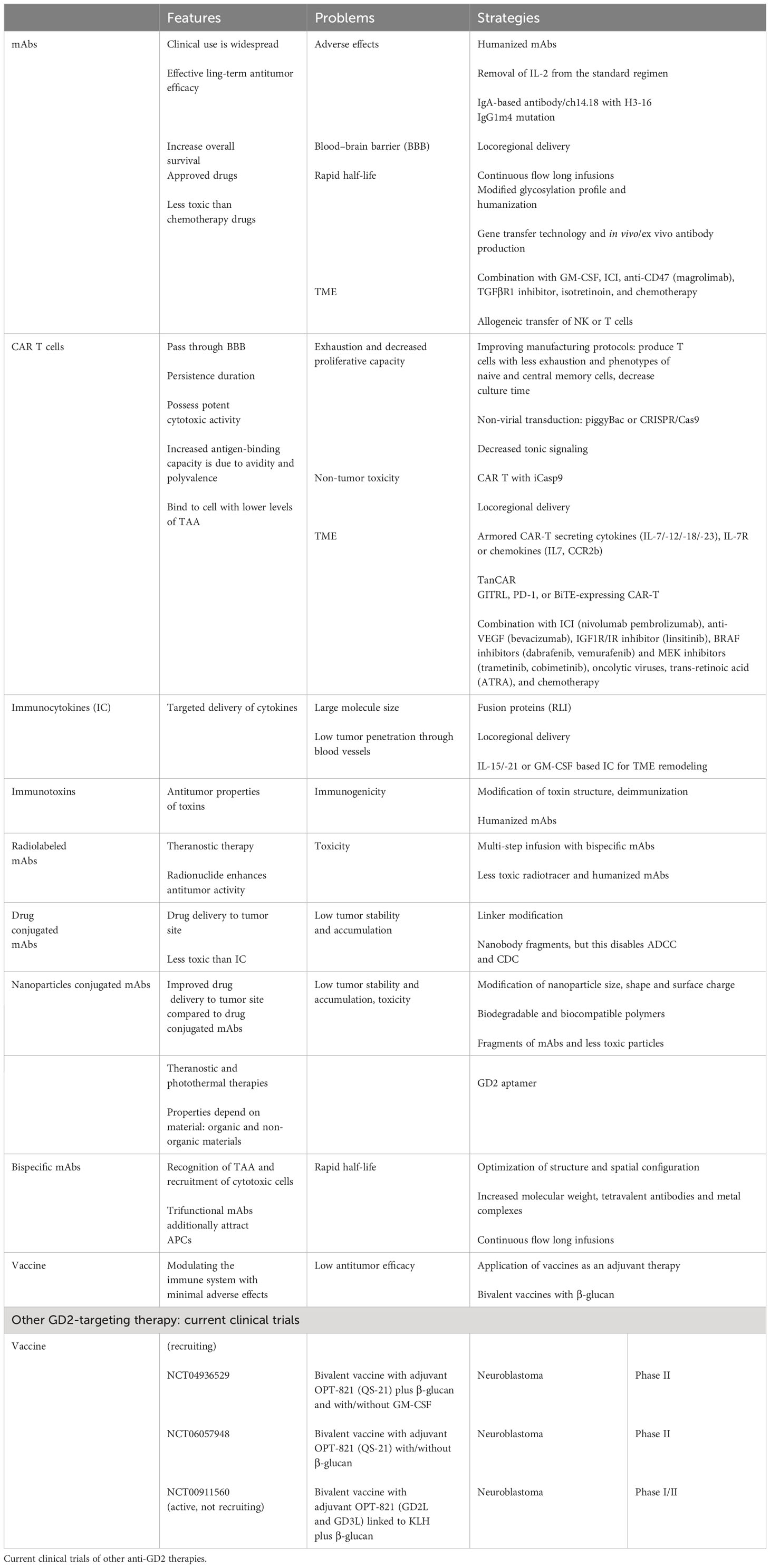
Table 1 Approaches to anti-GD2 therapies: features, problems, and strategies.
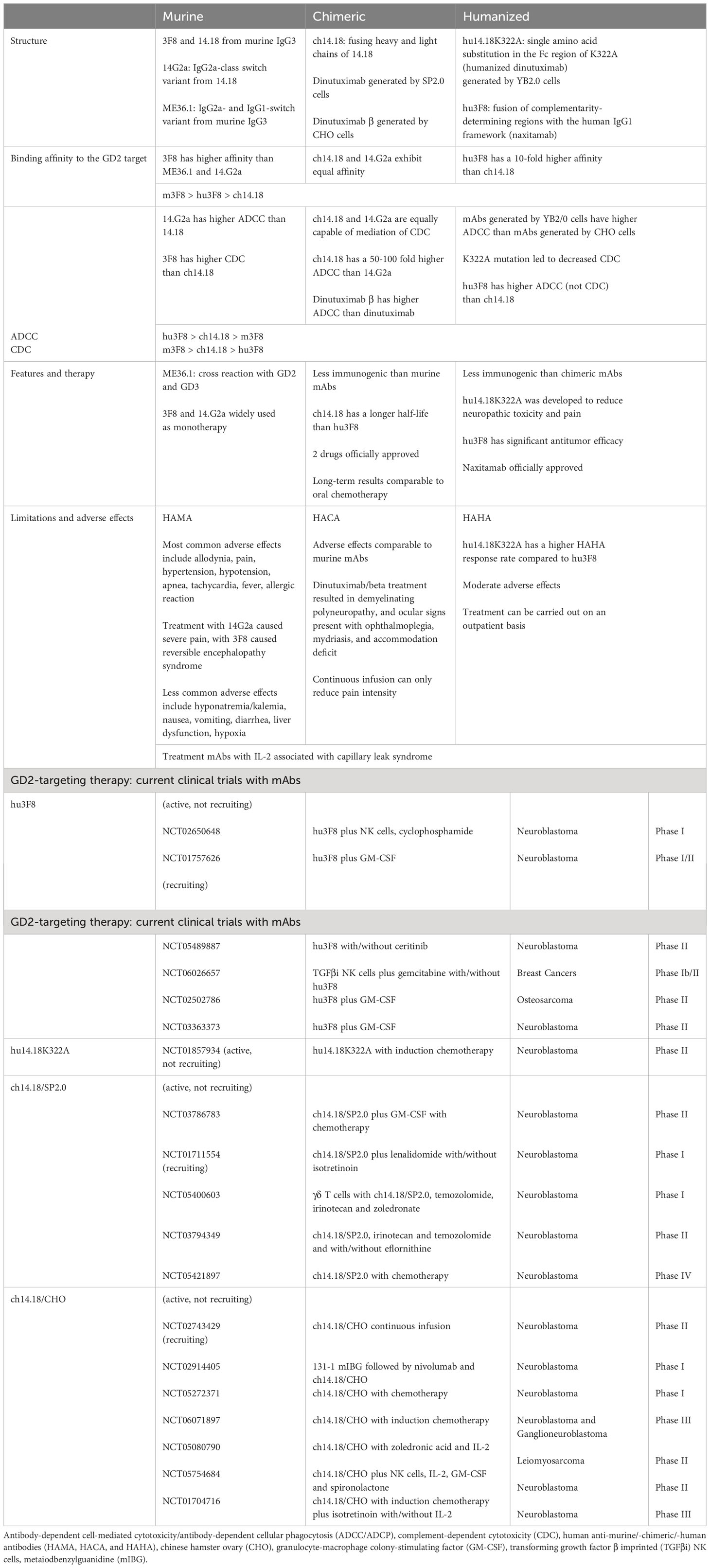
Table 2 Comparison of murine, chimeric, and humanized mAbs. Current clinical trials of anti-GD2 mAbs.
3.1 Murine mAbs: 3F8, 14G2a and ME36.1
Hybridoma technology was used to develop the first murine mAbs 3F8 and 14.18 of the IgG3 subclass (51, 52). Mouse mAb showed not only stable binding to GD2 antigen (53), but also the ability to mediate CDC (51) and ADCC (52, 54). Later, mAb 14G2a was developed based on the IgG2a-class switch variant of 14.18, which showed higher ADCC than 14.18 in vitro and in vivo (55). mAb ME36.1, derived from murine IgG3 and being IgG2a- and IgG1-class switch variants, can cross-link to GD2 and GD3 (56). In clinical practice, 3F8 (57–59) and 14G2a (60–62) were widely used as monotherapy. However, a high level of HAMA and several side effects were reported among patients. In particular, in a rat model, the development of severe pain requiring high doses of morphine was observed after the administration of 14G2a (63). In order to enhance the therapeutic potential of mAbs, including overcoming prolonged severe lymphopenia (64), GM-CSF (64–66), isotretinoin (13-cis-retinoic acid, a vitamin A derivative) (67), oral β-glucan (68, 69), and adoptive transfer of NK cells were added to 3F8 (70). mAb 14G2a was also tested in combination with IL2 (71). The results reported difficulty in treating bulky masses or progressive disease (64, 65) and the development of severe side effects (71). In addition, the development of posterior reversible encephalopathy syndrome (PRES) was observed with 3F8 treatment (72), which calls into question further testing of murine mAb.
3.2 Chimeric mAbs: ch14.18/SP2.0 (dinutuximab)
In order to reduce immunogenicity and neutralizing antibody levels, chimeric murine-human ch14.18 antibodies were developed by combining murine IgG3 mAb 14.18 (IgG2a switch variant 14G2a) chimeric fragments with Fc fragments of human IgG1 produced by the SP2.0 cell line (73). It was shown that ch14.18/SP2.0 and 14.G2a equally exhibited antitumor activity, antigen affinity, and ability to mediate CDC. However, ch14.18-mediated ADCC in vitro was 50-100-fold more effective compared to 14.G2a (74). Pharmacokinetic analysis showed that ch14.18/SP2.0 had a longer half-life compared to 14G2a (75), but its clearance was accelerated after repeated administration, probably, due to HACA formation (76). At the same time, the CDC is higher in mouse antibody 3F8 than in ch14.18, which is due to the difference between human and mouse IgG1 and IgG3 immunoglobulins (77).
Studies of monotherapy with dinutuximab (76, 78–81) did not show any treatment benefit except for reduced immunogenicity. However, in the long term, the antitumor effect was comparable to the use of oral chemotherapy (82). Combination therapy of dinutuximab with IL-2 and/or GM-CSF was also evaluated in several studies (83–85), and in combination with the murine antibody R24 (86). Administration of cytokines enhances ADCC (83, 84, 86), but HACA titers get increased in response to chimeric antibody administration (85). The Children’s Oncology Group reported improved survival with the combination of dinutuximab with GM-CSF, IL-2, and isotretinoin (ANBL0032) (87) compared to standard therapy with isotretinoin (88), prompting the FDA and EMA to approve this combination for maintenance therapy of high-risk neuroblastoma in pediatric patients after ASCT (89). Subsequent ANBL0032 studies of the same patient cohort questioned the use of IL2 as a therapeutic agent, as no benefit was found and GM-CSF may induce an endogenous IL2 response (90).
Although immunotherapy with mAb showed encouraging results, the problem of delayed relapses remains relevant and requires the development of new methods and drugs. One approach may be aimed at modulating TME. Thus, it was shown that the addition of irinotecan and temozolomide chemopreparations to dinutuximab with GM-CSF would enhance the antitumor effect at minimal doses of mAb (ANBL1221) (91). It was also shown that γδ T cells can provide better antitumor activity in combination with dinutuximab and temozolomide, while being superior to αβ T cells due to their functional properties (92). Magrolimab (anti-CD47 mAb) (38), galunisertib (TGFβR1 inhibitor) (93), and anti-CD105 (94) may be added to dinutuximab to enhance its efficacy. The addition of magrolimab can provide potent synergism with dinutuximab and enhance the antitumor response toward phagocytosis, while anti-CD105 induces ADCC by cells expressing the Fc receptor. In mouse models, it was shown that immunotherapy with dinutuximab in combination with NK cells, initiated prior to tumor resection, can reduce disease severity and increase survival (95). Another approach is to modify mAb and improve the delivery method. Silk fibroin was proposed as a delivery platform for bioactive dinutuximab, which can provide a higher concentration of mAb in the tumor (96).
3.3 Chimeric mAb: ch14.18/CHO (dinutuximab beta)
The technology to produce ch14.18 according to GMP standards was based on antibody production by SP2.0 and NS0 cell lines, which are non-secreting murine melanoma cells that carry murine xenotropic retrovirus, making it much more difficult to purify antibodies for the use in clinical trials (97). The antibodies produced by the chinese hamster ovary (CHO) cell line are similar in structure to human serum antibodies and have a glycosylation type involving small amounts of sialic N-glycolylneuroamic acid, which provides a prolonged half-life and a reduced immunogenicity profile (98). In addition, CHO does not carry murine retrovirus, so in order to improve production, the CHO cell line was used to produce dinutuximab beta (ch14.18/CHO). Comparative analysis of ch14.18/CHO and ch14.18/SP2.0 showed similar CDC for the antibodies in vitro, while ADCC was higher for dinutuximab beta even at low antibody concentrations. In vivo evaluation revealed suppression of metastasis in the animal model, which was probably due to the enhancement of NK-depended ADCC (99). The SIOPEN (The International Society of Pediatric Oncology Europe Neuroblastoma group) clinical trial confirmed the feasibility of dinutuximab beta because the toxicity and pharmacokinetics profile were similar to dinutuximab with objective responses (100). Subsequent SIOPEN clinical trials of a combination of dinutuximab beta with/without subcutaneous administration of IL2, isotretinoin, and standard chemotherapy regimens showed improved 5-year survival. However, due to side effects and lack of benefit, IL2 is not recommended for further use (101–103).
The use of different regimens and combinations of dinutuximab beta with different therapeutic approaches was also actively explored in recent studies. The clinical use of dinutuximab beta and haploidentical stem cell transplantation (haplo SCT) can improve survival with an acceptable toxicity profile (104) and a low risk of graft versus host reaction (GvHD) induction (105). In addition, dinutuximab beta stimulates haplo SCT towards NK cell differentiation with enhanced ADCC and potent secretion of pro-inflammatory cytokines (sIL2R, TNFα, and IL6), which emphasizes combinational functionality (106). Application of immunocytokine FAP-IL-2v related to fibroblast activation protein stimulates NK-mediated ADCC without induction of Treg compared to IL2 (107). The addition of γδ T cells and dinutuximab beta also promotes ADCC-mediated tumor cell lysis, and systemic administration of zoledronic acid is safe and leads to T cell expansion (108). Prolonged infusion (109) or the use of dinutuximab beta immediately after induction therapy (110) demonstrate an acceptable toxicity profile and objective responses. In particular, the use of at least one cycle of dinutuximab beta before surgery can lead not only to remission but also to tumor necrosis and normalization of oncomarkers (110). In addition, prolonged infusion not only results in effective immunomodulation, but also allows for reduced pain toxicity (111). It was also shown that dinutuximab beta, despite its antitumor activity, leads to MDSC induction (112). Therefore, the addition of chemical agents such as 5-FU or vorinostat (113), can suppress MDSC differentiation (112), and the use of nivolumab (a PD-1 inhibitor) eliminates their immunosuppressive effects (114). Clinical use of dinutuximab beta and nivolumab in two patients with relapsed/refractory neuroblastoma resulted in complete and good partial remission (115). The combination of dinutuximab beta with dual blockade of immune checkpoints PD-1 and TIGIT more effectively inhibits tumor growth compared to a single blocker (116).
3.4 Humanized mAbsAbs: hu14.18K322A and hu3F8 (naxitamab)
Antibody humanization involves optimization of the antibody variant region, which subsequently affects the frequency of immune response (HAMA and HACA) to the murine fragment, in particular, the elevation of complement component C3a and activation of the cascade (76). MAb hu14.18K322A has identical C-regions of IgG1-κ as ch14.18, except for a point mutation of the amino acid sequence replacing alanine with lysine 322 in the C(H)2 domains of the Fc fragment (117), which prevents complement activation (118). In addition, antibodies produced by the cell line of the rat hybridoma YB2/0 cell line strongly mediate ADCC as a result of reduced fucosylation compared to CHO-derived antibodies (119), which was confirmed in preclinical studies (120). However, hu14.18K322A has a reduced ability to mediate CDC and is less likely to induce mechanical allodynia in animal models compared to dinutuximab (120). Retrospective analysis also confirmed a difference in pain side effects between hu14.18K322A and dinutuximab, with the use of humanized antibodies requiring less opioids (121). Preclinical studies showed that hu14.18K322A in combination with αCD40/CpG enhanced NK-dependent antitumor response (122), and was also nonspecifically taken up by tumor cells (123). The combination of hu14.18K322A, IL15Rα/IL15, and GM-CSF was also shown to induce greater tumor regression in vivo compared to therapy with hu14.18K322A and GM-CSF with/without IL2 (124). Clinical studies showed that HAMA production was observed in 40% of patients (125), and the concentration of hu14.18K322A required for cell lysis was 3.5-4 times lower than that of dinutuximab (126). The combination of hu14.18K322A with NK cells, cytokines, or chemopreventive agents can lead to clinically significant responses (126). The addition of hu14.18K322A to induction therapy resulted in an early antitumor response (127), and subsequent efficacy evaluation showed significant tumor shrinkage and an encouraging 3-year survival rate (128). A study of the pharmacokinetic profile showed no differences between daily and weekly regimens (129), demonstrating the advantage of hu14.18K322A over the long-term administration of dinutuximab.
Mouse antibody 3F8 was also humanized by transferring the complementarity determining region (CDR) of heavy and light chains to the human IgG1-κ framework based on their homology (130). It was shown that hu3F8 was 200-fold more effective in enhancing ADCC in vitro but mediated CDC less compared to m3F8, and was superior to other antibodies in its ability to bind to GD2 antigen and antitumor activity in vivo. Clinical use of hu3F8 in combination with GM-CSF revealed clear advantages in achieving significant antitumor results, durable response and safety (131), which prompted the FDA to formally approve naxitamab for the treatment of high-risk neuroblastoma (132). The HAMA response rate for hu3F8 was comparatively lower than for hu14.18K322A (131). Thus, naxitamab had low immunogenicity and required several cycles of treatment to provide comparable efficacy (133). The safety profile allows naxitamab to be used in an outpatient setting compared to dinutuximab, which requires an inpatient regimen (2, 3). The clinical benefit and long-term survival prospectively raise the question of replacing chemotherapy with autologous stem cell transplantation with naxitamab in combination with GM-CSF in patients with first complete remission (1). The advantages of utilization and distinctive properties over other mAbs make hu3F8 promising for use in various GD2 therapy strategies, including CAR-T cells and conjugated antibodies. It was also reported on the improved in silico affinity of hu3F8 with a single D32H mutation in CDR1-VL by altering the electrostatic surface potential, which enhanced in vitro and in vivo cytotoxicity while maintaining tissue specificity (134).
3.5 Anti-GD2 mAbs and neurotoxicity
Dose-limiting neurotoxicity induced by mAbs, which requires patient care and analgesic therapy, is one of the key issues to be addressed. Severe pain is believed to be caused by the binding of mAb to GD2 on nerve fibers (135), which locally activates CDC through the C1q binding domain, generating anaphylatoxins such as C5 or C3 (136). Hence, most studies have focused on reducing complement activation. Various approaches have been taken to modify monoclonal antibodies. Therefore, a modified version of murine 3F8 called heat-modified murine 3F8 (HM3F8) was created (137). This modified version lacks effector functions, specifically ADCC and CDC, and can target GD2 or cross-reactive epitopes on nerves, resulting in the prevention of neuropathic pain after subsequent administration of unmodified antibodies. A novel IgA-based version of ch14.18 has been developed to reduce neuropathic pain (138). Unlike the IgG-based version, IgA-based ch14.18 does not cause neurotoxicity due to the absence of a C1q binding site. A new form of ch14.18, derived from the IgA2 isotype and based on IgA3.0, has an extended elimination period, high stability, and does not cause neurotoxicity (139). A comparative analysis showed that humanized mAbs have lower CDC compared to mouse mAbs. As described above, hu14.18K322A, with a point mutation in the Fc-fragment of the C1q domain (117) was designed to reduce CDC and, therefore, neurotoxicity. However, a recent study showed that the K322A mutation has inconsistent complement activity and may not be effective for therapeutic purposes (140). Kulanthaivadivel et al. also suggested that FcγR-dependent cytotoxicity may cause neurotoxicity. Therefore, a proposed alternative mutation format for IgG2a does not bind to FcγR and C1q. It has also been reported that a humanized H3-16 IgG1m4 antibody with an Fc mutation based on ch14.18 can reduce CDC (141). In a rat pain model, H3-16 IgG1m4 demonstrated decreased allodynia compared to dinutuximab. Naxitamab is a potential candidate for outpatient use among the presented antibodies, but its therapy can be complicated by painful side effects. Therefore, reducing neurotoxicity remains an important issue.
O-acetyl-GD2 (OAcGD2) is a derivative of GD2 that is expressed by cancer tissues but not by peripheral nerves (142). This property allows to avoid neurotoxicity. Preclinical studies have shown that the murine antibody 8B6 targeting OAcGD2 inhibits tumor growth even in the absence of ADCC and CDC (143), and its chimeric form does not cause allodynic pain (144). Additional studies are required to evaluate the benefits of using antibodies that target OAcGD2 in reducing neurotoxicity compared to anti-GD2 antibodies.
3.6 Immunocytokines
Immunocytokines (ICs) were developed in order to provide targeted delivery directly to the target, and thus, achieve high concentrations in the TME and reduce systemic side effects. The first anti-GD2 IC was obtained by fusing the C-terminal CH3 domain of mAb ch14.18 to IL2, which showed more efficient antigen-binding activity compared to mAb (145). Preclinical studies showed that ch14.18-IL2 exerted commensurate activity with systemic administration of the cytokine (146) and provided a prolonged effect of IL2 by increasing the half-life (147). The ability of ch14.18-IL2 to induce T cells directly into the TME (148, 149), induce NK-depended ADCC and exert more effective antitumor activity compared to ch14.18 and/or IL2 (150, 151) was tested in animal models.
To reduce immunogenicity, hu14.18-IL2 was developed, which demonstrated similar antitumor mechanisms in vivo (152–154). Clinical use of hu14.18-IL2 showed activation/modulation of the immune system by increasing lymphocyte counts or sIL2R levels. However, no clinically significant effect was achieved against massive disease (155–159). Probable reasons for the low antitumor efficacy may be the large size of the IC molecule, which degrades as it passes through the liver (160) or has low permeability into the tumor from the bloodstream (161). Intratumoral administration of IC can provide a more effective antitumor effect than intravenous administration (162), and enhance migration of NK cells into the tumor focus (163). To reduce IL2-dependent side effects, IC was produced by fusing IL2 to the C-terminal of mAb hu14.18 light chains (164). This construct is thought to impede the binding of IL2 to IL2Rs of intermediate affinity, which are associated with the manifestation of side effects, allowing the targeting of high-affinity receptors responsible for antitumor effects. Separately, ICs based on IL15 and IL21, similar in structure and function to IL2, were developed, that were safer and capable of exerting a remodeling effect on TME (165). A study of ICs hu14.18-IL2/IL15/IL21 in combination with chemotherapy showed that hu14.18-IL15 and hu14.18-IL21 could induce complete tumor regression and improved survival compared with hu14.18-IL2, and their application contributed to an increase in CD8+ T cells and M1 and a decrease in Treg and MSDC in the tumor (166). IC based on hu14.18 and GM-CSF may serve as an alternative, with hu14.18-GM-CSF showing enhanced ADCC in vitro compared to hu14.18 and/or GM-CSF (167).
Since the dominant mechanism of effector cell activation by IL15 in vivo is trans-presentation of the IL15Rα/IL15 complex (168), Burkett et al. developed RLI fusion proteins (sushi-IL15Rα and IL15 are connected using a flexible linker). RLIs are functionally more active than IL15 or IL15 plus IL15 plus IL15Rα/IL15 (169), in particular by enhancing cytokine recognition by receptors (170). Development of an IC based on RLI coupled to the C-terminal of the heavy chain of the c.60C3 chimeric antibody against GD2 may increase the half-life due to the small molecular weight of IL15 (169–171). c.60C3-RLI retains the cytokine potential of the fusion protein and the effector functions of the antibody (ADCC and CDC), and its in vitro and in vivo antitumor therapeutic activity is higher than that of RLI and mAb alone or in combination (172, 173). For example, the combination of dinutuximab, RLI N-803, and NK cells significantly increases antitumor activity (174).
3.7 Immunotoxins
In the classical sense, immunotoxins are bifunctional chimeric molecules consisting of an antibody fragment bound to a toxin of plant or bacterial origin (175). Thus, immunotoxins have the antigen-specific properties of an antibody and the activity of a toxin capable of penetrating and destroying a tumor cell by endocytosis (175). In the first studies, full-length mAb 14G2a was combined with plant toxins that inactivated ribosomes, ricin A (176) and gelonin (177). Preclinical studies showed that immunotoxin 14G2a-ricin A can effectively inhibit tumor growth in vivo (178, 179). Additionally, immunotoxin 14G2a-gelonin has been shown to be significantly more effective than native gelonin (177). Other immunotoxins, such as those based on scFv mAb 5F11 and diphtheria toxin (180), as well as mAb 14.18 and pseudomonad exotoxin A have also been developed (181). Immunotoxins using the Fv fragment lack the function to mediate ADCC or CDC (180, 181), however, the use of a small antibody fragment promotes better penetration into tumor cells (175). There were no further attempts to develop anti-GD2 immunotoxins, which may be associated with their immunogenicity and major problems in solid tumors. However, the implementation of new approaches aimed at reducing immunogenicity by modifying the structure of toxins or humanizing antibodies, as well as the use of immunomodulatory drugs, may add to the arsenal of strategies (182).
3.8 Radiolabeled mAbs and infrared photoimmunotherapy for cancer
Radiolabeled mAbs 131I-3F8 were first tested for imaging GD2-positive tumors in mouse models, proving their antigen-specific properties (183). Further clinical application of 131I-3F8 demonstrated a significant accumulation of labeled antibodies in high-dose tumors. Scintigraphy with 131I-3F8 compared with biopsy, 131I metaiodbenzylguanidine (MIBG), and standard diagnostic methods revealed more abnormal sites, including metastases, primarily due to increased sensitivity to neuroblastoma (184). The 131I-14G2a antibody was also used for imaging in clinical practice (62), and 99mTc-ch14.18 was more effective in detecting early metastases compared to MIBG (185). On the other hand, mAbs can promote tumor regression, which fits well into the concept of theranostic approach, where labeled antibodies have both diagnostic and therapeutic potential, making radioimmunotherapy (RIT) a feasible approach for the treatment of GD2-positive tumors. The main principles guiding the choice of labeled antibody are high antigen expression and antibody affinity, as well as the biodistribution, pharmacokinetic, and dynamic properties of mAbs (186). This is primarily associated with side effects that particularly affect hematopoiesis and excretory organs. Direct injection of antibodies, e.g. directly into the brain ventricular cavity, is preferred. In particular, this allows anatomical barriers (GEB) to be crossed and the liquor is devoid of leukocytes and proteins that can neutralize mAbs. Clinical trials with intraventricular administration via intrathecal or intraventricular catheter of 131I-3F8 (127, 187, 188) showed that the therapy was well tolerated (headache, fever, and vomiting, with no delayed side effects) and can be an adjunct to the main treatment, also in metastatic disease. However, intravenous administration showed no difference in progression-free survival and overall survival between patients receiving 3F8 + GM-CSF + CRA) and 131I-3F8 (67). However, this may be explained by stage 4 neuroblastoma complicated by MYCN, which requires further investigation.
Further attempts are made to improve labeled mAbs using different approaches and agents. Thus, multi-step targeting was proposed using the anti-GD2 antibody 5F11 (5F11-scFv-streptavidin) fused to streptavidin and its biotinylated radioactive ligand 111In with a DOTA chelating complex that binds mAb and radiolabeled mAb (189). Antigen pre-targeting showed an improved tumor-to-nontumor ratio, but accelerated clearance was observed due to the high immunogenicity of streptavidin. The development of high-affinity scFv to biotinylated DOTA chelator may improve the pre-targeting imaging and therapy strategy (190). Multistep radioimmunotherapy with BiAb, consisting of GD2-targeted hu3F8 and the mouse hapten antibody C825 with high affinity to chelating DOTA in complex with the metals 177Lu and 99Y, showed a complete antitumor response in a mouse model with minimal toxicity (191). Current imaging techniques rely on positron emission tomography (PET), which has advantages over SPECT in the highly accurate detection of tumors and metastases (192). The antibodies ch14.18/SP2.0 (193), ch14.18/CHO (194), and hu14.18K322 (123, 195) were adapted for PET using the radioactive isotope 64Cu in complex with the chelators DOTA, NOTA, SarAr, and their derivatives. The selection of radiopharmaceutical is determined by its safety, stability of the complex, rate of excretion and absorption by tumors and other tissues. For instance, NOTA chelator compared to DOTA binds more stably to 64Cu, which can accumulate in various organs and tissues (194, 196). The biodistribution of the 64Cu-SarAr complex after 48 h in the spleen and kidney was shown to be higher than that of other chelator complexes (196), while the safety data are lacking, making clinical application difficult (194). In addition, a decrease in the positive charge of chelators affects biodistribution, in particular, it reduces renal uptake of labeled antibodies (197). There were also no differences in biodistribution and antigen binding between 64Cu-p-NH2-Bn-DOTA in complex with ch14.18 and hu14.18K322 (123, 196). However, their radioimmunologic potential is directly dependent on clinical characteristics and requires further comparative analysis. Subsequent development of labeled anti-GD2 antibodies may focus on the selection of radiolabeled antibodies, chelators, and different antibody platforms (198).
Photoimmunotherapy (NIR-PIT) is a new approach in tumor treatment. It was shown that the GD2 antigen was suitable for this therapy. The essence of NIR-PIT is targeted delivery of anti-GD2 antibody conjugate with photoactivating chemical substance (water-soluble silicon-phthalocyanine derivative near-infrared derivative (IRdye700DX)) followed by exposure to NIR light with a wavelength of 690 nm, which leads to selective cell death (199, 200).
3.9 Delivery: mAbs with nanoparticles and drug conjugates
Antigen-specific targeting of anti-GD2 mAbs allows antibodies to be used as transporters of toxic agents and drugs directly into the TME, which may enhance the therapy of solid neoplasms. Conjugated antibodies or their Fab fragments with nanoparticles like radiolabeled mAbs can be used in combination with therapeutic and diagnostic approaches or separately (201). The properties and functions of nanoparticles depend on the material (viruses, lipids, polymers, metals and their oxides, hydrocarbon derivatives, etc.) as well as the antitumor agents loaded in them. Liposomes are spherical phospholipid vesicles capable of penetrating through the tumor vasculature and consolidating at the target site (202). Full-length anti-GD2 mAb and their Fab fragments were conjugated to liposomes loaded with the 13-cis-retinoic acid derivative phenretidine (203), the proto-oncogene suppressing antisense oligonucleotides c-myb (204) and c-myc (205), the chemopreventive agent doxyrubicin (206), siRNAs against vascular endothelial growth factor-A (VEGF-A) (207) and the anaplastic lymphoma caspase (ALK) gene (208, 209), the topoisomerase I inhibitor irinotecan (210) and the sepantronium bromide survivin YM155 (211). Porous silica-based nanoparticles have a homogeneous, inert, and stable structure and a non-toxic safety profile compared to liposomes (212, 213). MAbs ch14.18 bound to porous silica were used to deliver siRNA-34a targeting a wide range of pro-apoptotic genes (213). Iron oxide can be used as a potential binding molecule between the conjugate and mAbs based on catecholamine reactions (214). Non-covalent polymeric carcinostatics (scFv-polymer-carcinostatics) were also shown to be superior in antigen-binding properties and cytotoxic effect compared to covalent ones (215). Carbon nanotube nanoparticles (216) and gold nanorods (217) further enhance mAbs by photothermal degradation when exposed to an infrared laser. Another approach involves the use of compounds of graphene quantum tubes (218) or iron oxide (219) with polyethylene glycol and polyethylenimine, hollow gold particles (220) for tumor diagnosis.
Antibody-drug conjugated (ADC) antibodies consisting of an antibody-linker-drug composition are widely used in cancer immunotherapy (221). Over 80 ADCs are under clinical development, and recent developments are aimed at improving activity, specificity, safety, increasing serum half-life, and decreasing immunogenicity. Compared to immunotoxins, ADCs are less immunogenic, and therefore, less toxic (222). Initial development of anti-GD2 ADC using 14G2a and a synthetic analog of calicheamicin showed significant suppression of liver metastases in a mouse model (223). It is noteworthy that until recently, there were no conducted studies, although ADC-based therapies showed good antitumor responses. However, after 20 years, an ADC based on ch14.18 and monomethylauristatin E (MMAE) and F (MMAF) was developed that showed potent antitumor activity with the antibodies retaining stability, antigen-binding properties, and in vivo biodistribution profile (224), making this a promising area for further study. It has been shown that higher antigen density leads to a stronger internalization of mAbs (225). Therefore, MMAF-conjugated mAbs will be more effective in killing tumor cells with high GD2 density, as MMAE penetrates tumor cells better than MMAF (224). The development of antibody fragments, so-called minibodies, based on ch14.18 (two scFv linked by a linker to the CH3 domain of IgG1) conjugated to MMAE and MMAF (FDC), is also reported (226). The results show the therapeutic potential of FDC compared to ADC, including improved pharmacokinetic characteristics, reduced side effects associated with the absence of Fc-fragments, and pronounced cytotoxic properties.
Internalization of anti-GD2 antibodies can provide a means to deliver drugs or toxins directly into the tumor cell. However, it can also be a mechanism for tumors to evade immunotherapy with naked mAbs. Conjugating mAbs with endocytosis inhibitors, such as EIPA (5-(N-ethyl-N-isopropyl) amiloride), chlorpromazine, MBCD (methyl beta-cyclodextrin), and cytochalasin-D, has shown potential to inhibit antibody internalization (225). In addition, MBCD-conjugated mAb can enchance ADCC that may improve the efficacy antitumor therapy.
3.10 GD2 aptamers
In addition to mAb, “chemical antibody” aptamers were developed, which are single-stranded DNA or RNA molecules selected by an iterative selection process called systematic ligand evolution by exponential enrichment (SELEX) (227). High affinity aptamers recognize the GD2 antigen, so they can be conjugated to other molecules and toxins for drug delivery or imaging (228, 229). The main advantages of aptamers over mAbs include small molecular size and high permeability through blood vessels and GEB, high affinity, non-immunogenicity, safety, and low cost. At the same time, the structure of the molecules can be easily synthesized and modified for various therapeutic purposes due to geometric conformational flexibility and synthetic dynamics (229, 230). To date, two GD2 aptamers with doxirubicin incorporated into the structure were developed, one containing a pH-sensitive motif to reduce side effects and the ability to be activated in an anaerobic environment by TME (DB67) (229), and the other – by MYCN-siRNA (DB99) (230).
3.11 Bispecific antibodies
Bispecific antibodies (BiAbs), compared to classical antigen-specific antibodies, are able to recognize TAAs and additionally attract cytotoxic cells by targeting costimulatory molecules or receptors (231). Bispecific T-cell activators (BiTE), compared to BiAb, typically consist of two scFv as a polypeptide chain, with the light and heavy chains connected to a flexible linker (232). Various BiAb constructs targeting GD2 and CD3 were tested in preclinical models. In particular, BiAb were obtained by fusing IgG anti-GD2 antibody with scFv anti-CD3 antibody (233), chemical heteroconjugation of mAbs anti-GD2 and anti-CD3 (178, 234), scFv anti-GD2 with scFv anti-CD3 (BiTE) (22), which demonstrate binding of GD2-positive tumors and activated T cells in an MHC-independent manner, exhibiting cytotoxic properties through the perforin/granzyme axis. The hu3F8-based BiAb was shown to induce rapid T cell infiltration and expansion, mediating potent T cell-dependent cytotoxicity (TDCC) (235). Adoptive transfer of ex vivo proliferated T cells armed with GD2-BiAb leads to rapid tumor infiltration and induces a potent antitumor response (GD2-EAT) with significantly lower production of cytokines that induce CRS (236). At the same time, over-activation of T cells by BiAb may be resolved by aglycosylation of IgG-scFv (233). Combination treatment with the checkpoint inhibitors pembrolizumab (PD-1) or atezolizumab (PD-L1) enhanced armed BiAbs T-cell function and tumor control when administered sequentially and continuously (237). BiAb-directed T cells demonstrate superior cytotoxic properties and are less depleted than GD2.CAR-T cells (238). The present studies are aimed at optimizing the structure of BiAbs, taking into account the size of the constructs and affinity to tumor antigens, which affects biodistribution and cytotoxicity in vitro and in vivo. Thus, it was shown that for anti-GD2 BiAbs, the optimal option was to place the antigen and T-cell binding domains in a cis-configuration with a two-wall IgG-[L]-scFv platform and the use of two cis-modules additionally increased cytotoxicity (239). The rapid half-life of BiAbs requires continuous administration, which can be solved by increasing the molecular weight of the antibodies, in particular, by using tetravalent antibodies with two binding sites (240) or in complex with metals (191).
Trifunctionalized BiAbs (TrAbs) consist of heterodimeric isotopes of murine IgG2a and rat IgG2b. Their function is enhanced by the presence of an Fc region, which provides high affinity binding via FcγR to antigen-presenting cells (APCs) in addition to T cells; in particular, dendritic cells, monocytes, macrophages (241, 242), and lower affinity to NK cells (243). In mouse models, TrAbs SUREK-based vaccines were shown to promote T-cell recognition of TAAs (244), as well as the development of humoral response, in addition to a given GD2 antigen (242). In addition, treatment with anti-GD2 TrAbs SUREK was superior to dinutuximab beta against neuroblastoma (245), and when combined with an antitumor vaccine and immune checkpoint inhibitors, it stimulated the endogenous response and enhanced the antitumor effect (243). Due to low binding to the GD2 antigen (242), TrAb can be further utilized as an additional boost to the main therapy.
4 Anti-GD2 and idiotopic vaccine
The basic idea behind antitumor vaccines is to create a specific immune response in response to TAA administration. By nature, GD2 is a carbohydrate antigen. Thus, to enhance immunogenicity, strong protein-framed adjuvants such as keyhole limpet hemocyanin (KLH) (246) or non-toxic diphtheria toxin CRM197 (247) followed by subcutaneous injection of Quillaja saponaria (QS) (248) or monophosphoryl lipid A (249) are needed to enhance the cellular response (250). Active immunization of patients with GD2-KLH/MPL-A did not induce antibody formation against GD2 (249), and despite the serologic response from GD2-KLH/OPT-821 (equivalent to QS-21), there was no significant difference in progression-free survival between the control and subject groups (248). However, the right approach to vaccine development could potentially improve therapy. Subsequent trials of a bivalent GD2/GD3-KLH/OPT-821 vaccine combined with oral administration of β-glucan (a C-type lectin receptor activator) showed encouraging results with no serious toxicity (251); and subsequent immunization of an expanded cohort demonstrated a strong humoral response, with a high anti-GD2-IgG1 titer associated with better survival (252).
There were also attempts to develop idiotypic vaccines, also knows as anti-Id vaccines. The fundamental concept behind these vaccines is to prolong a humoral or cellular response by using anti-Id vaccines against already developed anti-idiotypic antibodies after previous therapy with anti-GD2 mAbs (253). Since TAAs are autoantigens, especially carbohydrate antigens, there is a tolerance of immune response to them, so the use of anti-Id vaccines would be able to overcome this barrier (254). Anti-Id mAbs murine 1A7 against ch14.18 (255) and rat A1G4 against 3F8 were developed (256). Clinical use of 1A7 showed no toxic effects, but objective responses were minimal (255). However, a ganglidiomab antibody against anti-GD2 antibody family 14.18 was later developed, which induced a humoral response in murine models (257) and among patients after therapy with anti-GD2 mAbs, demonstrating good tolerability without significant side effects (258). The development of anti-Id antibodies mimicking human and mouse GD2 ganglidiximab, which is capable of mediating ADCC and CDC, and which may be useful for tailoring humoral responses to paratopic regions mimicking GD2, was also reported (259).
5 Cell therapy
Cell-based immunotherapy involves the adoptive transfer of GD2-targeted genetically modified, virally vector-mediated (retroviral or lentiviral), or non-viral approaches (sleeping beauty transposition), or ex vivo stimulated NK-, NKT-, and T-cells in combination with anti-GD2 mAbs and other drugs for chemotherapy. Chimeric antigen receptor (CAR) cells are suitable for GD2-targeted therapy because they have unique properties to recognize targets of different classes, including glycolipids and carbohydrates, which have lower mutation rates (260). CAR recognizes the target in an MHC-independent manner using a single-chain variable fragment (scFv) derived from mAb. Since the construct includes costimulatory domains (CD27, CD28, 4-1BB, ICOS, OX40, and etc.), cells activated after encountering the CAR antigen do not need additional stimulation. This chapter presents different approaches and strategies to improve CAR therapy (Table 1), in particular through combination therapy and gene modification of different effector immune cell populations (Table 3).
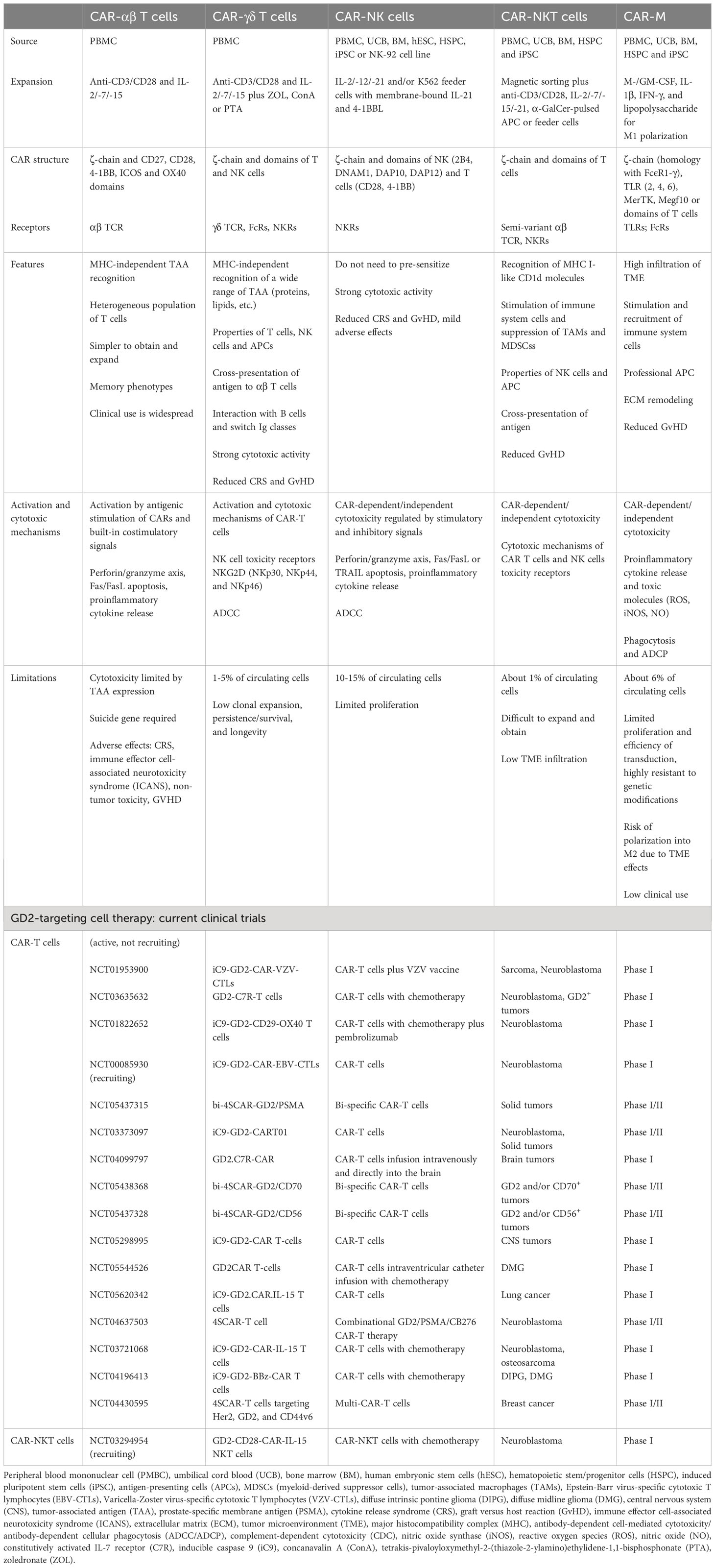
Table 3 Comparison of αβ T, γδ T, NK, NKT cells and macrophages with CAR. Current clinical trials of anti-GD2 CAR therapy.
5.1 CAR-T cells
The cytotoxic potential of CAR-T cells is widely used in clinical practice, and, unlike TCR, it is realized by the formation of a non-classical immune synapse that has an advantage in kinetics and enhanced signal transduction with comparable amounts of lytic molecule release (261). The main antitumor effects of CAR-T cells are realized through the major cytotoxic axis of perforins and granzymes (targeting antigen-positive fraction), as well as through the Fas/FasL axis (targeting antigen-negative fraction) and the release of cytokines (stromal cell sensitization) such as IL2, IL15, IFNγ, TNFα, etc. (262) (Figure 1). The expression profile of surface markers affects clinical responses. At the same time, CAR-T cells with a memory-like phenotype (CD62L, CCR7, CD45RA and CD45RO) provide high antitumor efficacy, whereas acquisition of a depleted cell phenotype (PD-1, LAG-3 and TIM-3) is associated with limited efficacy (263). It was also found that targeting GD2 with CAR-T compared to mAb had several advantages: 1) CAR polyvalency on the surface of T cells may have a higher overall avidity than a soluble antibody in divalent form, thereby increasing the probability of binding to tumor cells with lower GD2 expression; 2) additional cytotoxic mechanisms of T cells allow for more efficient destruction of tumor cells; 3) the duration of T cell persistence in the circulation may provide relapse control (264); CAR-T cells have the ability to cross the blood-brain barrier (265) compared to mAbs (58). However, despite their clear therapeutic potential, CAR-T may be limited by their rapid loss of functional properties and development of a depletion stage, as well as by their low in vivo proliferation and ability to infiltrate the tumor, which may be associated with immunosuppressive TME and its extracellular matrix, and lack of co-stimulatory stimulus when interacting with tumor cells. AICD (activation-induced cell death), which may be mediated by antigen re-stimulation (266) or Fas-FasL interaction, is suggested to be another limitation of CAR-T cell function (267). Nevertheless, excessive functional CAR-T activity is often associated with the manifestation of side effects including neurotoxicity, cytokine release syndrome (CRS), and GvHD (268). In addition, CAR-T cells are limited in large-scale individualized preparation. Given this series of challenges, strategies to improve CAR-T therapy focus on various modifications of the CAR structure as well as the route of delivery and targeted delivery.
5.2 Design of GD2.CAR cells
Functional properties and stability in the body depend on CAR design, including scFv, spacer and costimulatory domains, as well as additional components that enhance cell performance. First-generation GD2 CAR studies have demonstrated the importance of CD28 costimulatory signaling in specific antigen recognition for T cell survival and expansion, as well as for enhancing the immune response, including through IL2 secretion (269). However, the presence of the antigen-binding domain and CD3 ξ-chain alone does not provide sufficient stimulus to ensure the functional properties of CAR-T cells (270). An alternative mode of activation was based on the physiological stimulation of native TCR through interaction with APCs, which was achieved by transduction of virus-specific T cells (271, 272). The persistence of EBV-specific GD2.CAR-T cells was shown to be longer compared to autologous activated GD2.CAR-T cells (273). However, in the long term, despite the presence of antitumor efficacy with simultaneous CAR and TCR stimulation, virus-specific CAR-T cells was less retained in the bloodstream (264).
The costimulatory domains CD28 and 4-1BB (CD137) are the most commonly incorporated into CAR constructs, with T cell functions depending on domain selection. The CD28 domain was shown to enhance proliferation and IL2 release (274), and is a more potent driver of antitumor response compared to 4-1BB (275). In addition, the CD28 domain also enhances cytokine production and cytotoxicity for CARs with low avidity by lowering the threshold of antigen affinity (276). In contrast, the inclusion of the 4-1BB domain is associated with increased persistence, proliferation (277), and potent therapeutic activity in vivo (278). Antigen-independent signaling can induce earlier depletion of CAR T cells, with the CD28 domain increasing key aspects of depletion, while in contrast, the 4-1BB domain can improve antitumor effects by producing higher levels of cytokines, reduced expression of depletion markers, and increased resistance in vivo regardless of antigen-dependent or independent effects (277). However, it was shown that 4-1BB-based tonic CAR signaling could induce T cell apoptosis through continuous TRAF-dependent activation of the NFκB pathway and Fas-dependent cell death (279). Transcriptional analysis showed that GD2.28z.CAR T cells exhibited higher expression of genes encoding inhibitory receptors such as LAG3, HAVCR2 (TIM-3), CTLA4, BTLA, and CD244 (2B4), and the depletion-related transcription factors TBX21 (T-bet), EOMES, PRDM1 (Blimp-1), and IKZF2 (Helios) compared to GD2. BBz.CAR T cells that express memory-related transcription factors such as KLF6, JUN, and JUNB (277).
Tandem use of costimulatory domains can provide improved signal transduction as well as compensate for the deficiencies of a single domain. Thus, it was shown that the use of the CD28 domain alone could not sustain prolonged growth, activity, and survival of T cells (269). The OX40 domain (CD134), which is expressed after T cell activation following antigen and CD28 stimulation, is required for ongoing proliferation and cytokine production (280, 281). It is also noted that the replacement of OX40 with the 4-1BB domain or ligand can reduce or prevent AICD and/or PD-1-mediated suppression (266). It was shown that GD2.CD28.OX40z T cells resulted in maximal NF-κB activation associated with increased and prolonged proliferation and enhanced cytokine release compared with the inclusion of CD28 or OX40 alone (282). However, in another study, GD2.CD28.OX40z secreted less INFγ and IL2 after 30+ days, and their phenotype correlated with exhaustion status compared to GD2.4-1BB.CD28z (283). Phosphoproteomic analysis also showed that GD2.CD28.OX40z had the highest number of phosphorylation sites, suggesting that the cells were overstimulated. The ICOS domain (a member of the CD28 family) can also be added to the CAR construct to enhance the antitumor activity of CD8+ T cells by differentiating CD4+ T cells into the Th1/Th17 phenotype (284). In addition, the ICOS domain has a better effect on CAR-T survival compared to CD28 and also complements the function of the 4-1BB domain, including reducing tonic signaling (284), which likely contributed to the better persistence and antitumor activity of GD2 CAR-T cells in vivo (285). At the same time, combined stimulation of CD28 and 4-1BB may contribute to cytokine storm due to forced stimulation of CD28 (286). However, for third-generation GD2 CAR-T cells, the combination of CD28 and 4-1BB domains is the most optimal. 4-1BB signaling promotes the restoration of CD28-induced depletion and the most homogeneous distribution of CARs on the cell surface, with GD2.4-1BB.CD28z exhibiting effective antitumor activity in vivo (283). For virus-specific CAR-T cells, it was shown that the most optimal domain is CD28, as GD2.CD28z better supports the TCR function (287).
Chimeric TCR signaling is more efficient when mediated by the ξ-chain compared to the γ-chain FcϵRI (270). The choice of the variable fragment is dictated by the conditions of optimal affinity and high specificity of antigen recognition; therefore, ch14.18 is often used as scFv. However, scFv derived from 14g2a causes rapid depletion of GD2.CAR-T cells due to tonic signaling during ex vivo expansion (277). A humanized antibody can be used as a substitute for ch14.18. Thus, it was shown that scFv derived from the humanized antibody KM8138 did not cause anti-idiotypic rejection of CAR-T cells with preservation of their functional activity (288). In addition, scFv derived from hu3F8 allowed CAR-T cells to better target the tumor and promoted increased cytolytic activity compared to scFv based on mAbs CE7 and 14g2a (289, 290). It was also reported that the inclusion of a mutation in the spacer Fc domain avoided off-target toxicity by reducing high-affinity FcγR binding (280), as it can prevent binding to γ receptors of immune cells (291). In a preclinical model, this strategy provokes high neurotoxicity despite high in vivo efficacy (265, 292), making it not feasible.
Altering tonic signaling by reducing positively charged CAR sites through mutations or increasing ionic strength (increasing pH in the culture medium due to carnosine during ex vivo propagation) improves efficiency and reduces the fatigability of GD2.CAR-T cells (293). Application of PI3K (294) or Akt-pathway (295) inhibitors can block tonic CAR signaling at the initial stage of preparation, while additionally reducing terminal T cell differentiation. A strategy to reduce GD2.CAR-T depletion can be aimed at temporarily halting CAR signaling by turning on the C-terminal destabilizing domain of FK506 binding protein 12 (FKBP) using a drug-regulated system or the multikinase inhibitor dasatinib (296). Tonic signal transduction can be reduced by altering the TRAC locus, and cells have a delayed ability to differentiate in vitro and in vivo (297).
5.3 CAR-T cells and TME
Overcoming immunosuppressive and heterogeneous TME of solid tumors is one of the leading tasks to achieve the efficiency of GD2.CAR-T cells. Several approaches were used to realize this goal, including those aimed at increasing cell migration and enhancing cell cytotoxic properties. TRUCK CARs (“T cells redirected for antigen-unrestricted cytokine-initiated killing”) have the ability to produce transgenic pro-inflammatory cytokines IL7, IL12, IL15, IL18, IL23, and their combinations by CAR signaling induced by NFAT (nuclear factor of activated cells) (298, 299). TRUCKs GD2.CAR T cells secreting IL18 (300) or IL12/18 (301), were shown to have enhanced effector properties and also promote monocyte recruitment to the tumor. Co-expression of transgenic IL15 significantly increased GD2.CAR-T engraftment and also promoted additional sustained tumor control (292). In addition, IL15 also promotes differentiation into memory and stem cell-like phenotypes, with GD2.CAR-T exhibiting reduced PD-1 expression and increased survival in both peripheral blood and tissues (302). GD2.CAR T cells co-expressing chemokines IL7 and CCR2b were also developed, which had chemotaxis ability, improved proliferation and survival in vivo in addition to strong antitumor activity (303).
The combined use of CAR-T cells and oncolytic viruses (OVs) also aims to immunomodulate TME. OVs can be delivered by CAR-T cells to tumors systemically and provide direct lysis, or generate in vivo expansion of T cells with native TCR specificity to viral or virus-encoded antigens and enhance antitumor activity by inducing phenotypic changes in T cells with dual specificity (304). In addition, OV armed with various chemokines and cytokines can be used for CAR-T therapy. It was shown that the use of GD2.CAR-T cells and OVs armed with the chemokine RANTES and IL15 directly accelerated caspase pathways in tumor-exposed T cells, with RANTES and IL15 promoting CAR-T recruitment to the tumor and ensuring their local survival (305). In order to overcome the lack of immunogenicity of solid tumors, modification of GD2.CAR-T cells (NCT01953900) specific to varicella-zoster virus (VZV CAR-T) can restore cell function by preserving sensitivity to stimulation via TCR either by VZV vaccine or after co-culture of VZV CAR-T and APC treated with VZV peptides (306).
Tregs, MDSCs, tumor-associated macrophages (TAMs) M2, immune checkpoint molecules (PD-1 and CTLA-4), and growth factors and anti-inflammatory cytokines and chemokines are the main components of TME (307), targeted by CAR-T therapy in combination with drugs. It was shown that when tumors were re-expressed with GD2.CAR-T cells, there was increased expression of PD-1 and PD-L1 inhibitory molecules, requiring therapy with immune checkpoint inhibitors (290). Preclinical trials of GD2.CAR-T cells with nivolumab (a PD-1 inhibitor) (308) and bevacizumab (a vascular endothelial growth factor VEGF inhibitor) (309) demonstrated the efficacy of the combinations, which could be used in clinical practice. However, the combination of GD2.CAR-T cells with pembrolizumab (PD-1 inhibitor) did not show to have an expressed effect in patients with neuroblastoma, which may be related to the timing and duration of PD-1 inhibition and tumor type (310). In particular, pembrolizumab and nivolumab were shown to be particularly effective against melanoma and lung cancer (311). In addition, the positive antitumor effect of PD-1 blockade is directed to the inhibition of AICD (266). Inhibitory MDSCs are an obstacle in the antitumor response and may worsen the prognosis for patients with cancer (310). In preclinical models, GD2.CAR-T cells did not exert antitumor effects, which was likely to be associated with the inhibition of human cells by murine MDSCs (312). Patients’ initial PBMCs may also be a barrier to CAR-T cell expansion at the initial stage (313), including because of MDSCs that suppress the expression of genes involved in cell activation, signal transduction, inflammation, and secretion of cytokines and chemokines (314). Combination with trans-retinoic acid (ATRA) may improve the antitumor activity of GD2.CAR-T cells by reducing the suppressor effect of MDSCs (314). IGF1R/IR inhibitors (linsitinib) show synergism with GD2.CAR-T cells (315), while its use can inhibit Treg (316) and M2 macrophage differentiation (317). Supplying GD2.CAR-T with additional GITRL expression may also enhance T cell efficiency in TME (318). The development of CAR-T cells that produce antibodies to PD-L1 was shown to reduce tumor growth in a mouse model (319).
The FDA-approved BRAF (dabrafenib, vemurafenib) and MEK (trametinib, cobimetinib) inhibitors aim to stop MAPK signaling leading to unregulated cell growth and differentiation, and their benefits for the treatment of solid tumors were shown in clinical practice (320). Combining CAR-T cells with BRAF/MEK inhibitors has the potential to be a new effective treatment option, but there is a question about the effect of inhibitors on T cell function. The dabrafenib/trametinib combination was shown to have no effect on the cytotoxic functions of CAR-T cells compared to vemurafenib (321) or the vemurafenib/cobimetinib combination (322). In addition, the combination of GD2.CAR-T cells and trametinib improves in vivo and in vitro antitumor efficacy compared to cell monotherapy, in particular by suppressing T cell depletion as well as reducing PD-L1 expression on neuroblastoma cells (323). The PD-1/PD-L1 axis can be blocked by doxorubicin, which also enhances the cytotoxic effect of GD2.CAR-T cells (324). Conditioning with cyclophosphamide/fludarabine (Cy/Flu) (lymphodepletion) also shows antitumor efficacy, including by an increase in CAR-T cells proliferation (310, 325).
The problem of the heterogeneous structure of solid tumors can be solved by targeting multiple target antigens. In particular, sequential administration of CD171- and GD2-specific CAR-T cells enhanced antitumor response and helped to prevent antigen loss in preclinical trials (289). Targeting GD2 and HER2 can be combined in a single bispecific CAR (TanCAR) consisting of two separate linked scFv domains for TAA recognition, which also compensates for antigen escape (326). The addition of tazemetostat to the treatment regimen may increase the expression of GD2 antigen, and therefore, increase susceptibility to targeting (327). In the future, BiTE-secreting CAR-T cells (328, 329) may be developed for GD2.CAR-T therapy to effectively kill heterogeneous tumors.
5.4 Production of CAR-T cells, safety, and delivery
Another promising direction for CAR-T technology is to produce CARs without viral transduction, which may have advantages in production, facilitating monitoring of vector replication ability, and eliminating accidental integration of viral elements into the human genome (297). Viral transduction of T lymphocytes also results in the proliferation of not only CAR-T cells but also CAR-NK cells (330). GD2.CAR-T cells were successfully generated using the piggyBac (323) and CRISPR/Cas9 systems (297). CAR-T cells derived from CRISPR/Cas9 had a less depleted phenotype compared to retrovirus-transduced cells (297). All-in-one lentiviral constructs with a single vector utilize a single vector integration event, which also reduces the potential risk of a potential mutagenesis side effect (301). In addition, lentiviral vectors can transduce cells regardless of their division status, whereas retroviral vectors can do it only during mitosis (331). The use of the CliniMACS Prodigy device allows for the large-scale production of finished GD2.CAR-T cells (300, 332), which greatly simplifies the production of the final product.
Prolonged culturing during the production stage can cause earlier depletion of CAR-T cells. Initial stimulation leads to potent production of INFγ and TNF, and loss of IL2 secretion is identified as the first stage leading to depletion (266). The use of the GDAIN protocol improves the survival of GD2.CAR-T cells and promotes differentiation of central memory or naive/stem-like T cells (effector memory phenotype is associated with a poor antitumor response). GD2.CAR-T production by apheresis and elutriation (washed lymphocytes) is better than by magnetic sorting of anti-CD3/CD28 or adhesion to anti-CD3/CD28 plastic, which significantly affects CAR-T quantity and quality (333). The combination of CD3 and CD28 with IL7 and IL15 gives the best balance of CAR-T expansion and potent effector cells while maintaining the stem/memory phenotype (stem/memory subset – CD45RA, CCR7, and CD95) (334).
Integration of an inducible “safety switch” (iCasp9) into the CAR construct allows the removal of mis-activated cells to avoid excessive off-tumor toxicity as well as CRS and MAS (335). GD2.CAR-T cells with iCasp9 were tested in various clinical trials (302, 324, 336, 337), noting that the treatment was safe with minimal side effects. The UniCAR platform ensures safety by adding a specific on/off module, thus avoiding off-target toxicity in the periphery (338). Local administration of CAR-T cells is not only effective but also safe compared to systemic administration (339–341). The delivery of GD2.CAR-T cells encapsulated in chitosan-PEG in situ injectable hydrogel is an excellent solution for the treatment of retinoblastoma to reduce inflammation and prevent retinal detachment (341).
5.5 GD2-targeting CAR and adoptive therapy: NK, NKT, γδ T cells, and macrophages
NK cells are part of the innate immune system responsible for protecting the body from malignancy. Unlike T cells, which are MHC-restricted and require sensitization and the presence of a tumor target, NK cells are able to rapidly activate and destroy tumor cells through direct cytotoxicity, formation of proinflammatory cytokines and chemokines, as well as by manifesting ADCC through the membrane receptor FcγRIII (CD16) or the apoptosis axis through TRAIL or Fas/FasL (43, 44). NK cell activity is regulated by both activating signals (DNAM-1, NKG2D, CD226, NKp30, NKp44, NKp46, etc.) and inhibitory signals (KIR, CD94/NKG2A, TIGIT, etc.) through the interaction of cell membrane receptors with ligands on target cells (342, 343). It was shown that the effect mAbs exert on NK cells was not limited to ADCC, with FcγRIII-mediated signaling being able to block KIR inhibition (344). NK cell therapy with NK cells achieved considerable success in tumor regression and disease stabilization, and one of its major advantages is the absence of GvHD, making it attractive for allogeneic transfer (345). Preclinical studies showed that the antitumor activity and functional properties of NK cells could be further enhanced when combined with ch14.18 (93, 346, 347), hu14.18-IL2 (348), and galunisertib (TGFβR1 inhibitor) (93), IL2 and IL15 (349), IL21 (346, 347). The addition of IL21 enhances ADCC, activating receptor expression and granzyme release, while galunisertib has a remodeling effect on TME. In addition, cytokine-induced killer cells using IL2 and IL7 in combination with anti-GD2 can significantly increase the rate of cell death compared to the treatment with each of them separately (350). Clinical trials with adoptive transfer of haploidentical NK cells in combination with hu14.18K322A, GM-CSF and IL2 (126, 351), m3F8 (70) show promising results. In addition, the toxicity profile associated with mAbs was not altered by the administration of NK cells indicating their safety.
A large share of studies in GD2.CAR therapy focused on T cells, but its efficacy was hampered by TME, side effects, and the associated cost of treatment. From this point of view, it was hypothesized that NK cells had several advantages and might become better CAR drivers than T cells (352). NK cells are safer, do not cause GvHD and other side effects, produce mainly INFγ and GM-CSF (unlike T cells that induce CRS by TNFα, IL1, and IL6), can be activated by a variety of receptors, and are able to mediate ADCC. The NK-92 cell line is used to develop CARs, including GD2.CAR-NK cells. NK-92 is believed to be an ideal CAR host because it has natural antitumor properties and is easy to scale and modify (353). However, NK-92 cells cannot mediate ADCC because they lack CD16, carry an abnormal genome, and are irradiated before use, which may reduce their potential. Therefore, other sources of NK cells, (e.g. pluripotent stem cells) are required to test the hypothesis for GD2. Nevertheless, preclinical studies showed that GD2.CAR-NK cells could effectively kill tumor cells in vitro (354, 355) and in vivo (356, 357), as well as enhanced INFγ production (357). In addition, armored GD2.CAR-NK cells expressing IL12 show tendencies to recruit monocytes (355).
T cells with natural killer cell properties (NKT cells) and γδ T cells combine the innate and adaptive properties of the immune response and represent a subset of T cells that express different receptors, including those characteristic of NK cells (358). γδ T cells are characterized by expression of heterodimeric TCRγδ, whereas NKT cells express semi-invariant TCRαβ. The direct mechanism of NKT-cell and γδ T-cell cytotoxicity includes perforin/granzyme B-mediated cytolysis, TNF and TRAIL production, and Fas/FasL-dependent apoptosis (359, 360). NKT cells are characterized by reactivity to glycolipids of their own and microbial origin via the MHC I-like molecule CD1d and the α-GalCer glycolipid antigen presented by it (361). In addition, NKT cells exert potent antitumor potential by stimulating NK- and dendritic cells and priming αβ T cells (362), rapid and efficient migration to TME (363) and suppressing TAM and MDSC immunosuppression (364). In turn, γδ T cells can act as APCs for T cells at the tumor site (365), kill the tumor via ADCC and FcγRIII (CD16), and interact with B cells and switch Ig classes (46).
Their antitumor potential and ability to recognize a wide range of antigens, exert direct and indirect cytotoxicity, and influence immunosuppressive TME make NKT- and γδ T cells potential candidates for GD2-specific CAR therapy. Unlike CAR-T cells, their activation does not depend on CAR signaling because they can recognize antigens in an MHC-independent manner, and therefore, do not induce GvHD responses. Despite few studies, GD2.CAR-γδ T cells showed to be capable of antigen cross-presentation, leading to clonal expansion of αβ T cells, with cytotoxicity equivalent to GD2.CAR-αβ T cells (366). In addition, GD2.CAR-γδ T cells can target tumors with low antigen density compared to CAR-αβ T cells (367). Replacing the ξ-chain with DAP10 (chimeric costimulatory receptors) downregulates tonic signaling with preserved activity and cytotoxicity, but GD2.CAR-γδ T cells rapidly acquire depletion status (368), requiring further modification. GD2.CAR-NKT cells have low persistence (369), but the inclusion of IL15 in the construct may address this problem and enhance tumor infiltration and antitumor activity in vivo (370). It was also observed that GD2.CAR-NKT cells did not contribute to the development of GvHD, whereas GD2.CAR T cells were lethal (369). In the GD2.CAR clinical trial, IL15-enhanced NKT cells showed safety and objective responses (371). CAR-NKT cells, compared to CAR-NK cells, were also shown to better regulate the immune system, infiltrate tissues, are resistant, and differ by memory phenotype (372).
Since TME is a major obstacle for CAR-T, arming M1 macrophages with CAR (CAR-M) was proposed as an alternative approach. Macrophages can penetrate the tumor much more easily, while having high phagocytic capacity, secreting proinflammatory cytokines and lytic molecules (ROS, iNOS, NO), presenting antigens, and interacting with immune cells (373). However, several challenges were encountered to realize this approach, particularly, in vitro and in vivo propagation and gene transfer into primary macrophages. GD2.CAR-M were derived from pluripotent stem cells using the CRISPR/Cas9 method and have potent cytotoxic activity in vitro and in vivo (374), which may serve as a platform for further testing.
6 Conclusion
Current research is aimed at improving the safety and efficacy of treatment. The success of antitumor therapy largely depends on a properly selected target antigen. GD2 expression is detected on the cell surface of a wide range of solid tumors at high levels. It is restricted to neoplasms and is not lost after treatment. Suitable agents for targeting GD2 are those capable of recognizing antigens of glycolipid origin, such as monoclonal antibodies or a chimeric antigen receptor. Preclinical and clinical studies showed that combination therapy was the most promising treatment compared to monotherapy, in particular, targeting not only tumor cells but also the microenvironment. In addition, the treatment should be safe, scalable, and cost-effective. From this point of view, the most promising area of genetic engineering is humanized monoclonal antibodies, which showed clinical efficacy and were officially approved. At the same time, cell therapy shows promising results. CAR cells have a direct cytotoxic effect on tumor cells, and various CAR modifications also make it possible to influence both TME and immunocompetent cells. In the future, optimization of new generations of CAR design and protocols for obtaining genetically modified cells should be aimed at improving safety and overcoming early cellular depletion. Such modifications may include replacing chimeric scFv with humanized scFv, reducing tonic signaling from the CAR receptor, using NK, NKT cells, or macrophages that do not induce GvHD reactions and pronounced toxic side effects, and optimizing protocols to produce scaled ready-to-use cells without functional signs of depletion.
Author contributions
JP: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. JS: Writing – review & editing. SS: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was carried out with the support of the Russian Science Foundation, project number 21-65-00004 (https://rscf.ru/project/21-65-00004/, accessed on 20 April 2021).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mora J, Castañeda A, Gorostegui M, Varo A, Perez-Jaume S, Simao M, et al. Naxitamab combined with granulocyte-macrophage colony-stimulating factor as consolidation for high-risk neuroblastoma patients in first complete remission under compassionate use-updated outcome report. Cancers. (2023) 15:2535. doi: 10.3390/cancers15092535
2. Cabral J, Fernandez EI, Toy B, Secola R. Multidisciplinary clinical care in the management of patients receiving anti-GD2 immunotherapy for high-risk neuroblastoma. Paediatric Drugs. (2023) 25:13–25. doi: 10.1007/s40272-022-00544-9
3. Mora J, Chan GC, Morgenstern DA, Nysom K, Bear MK, Tornøe K, et al. Outpatient administration of naxitamab in combination with granulocyte-macrophage colony-stimulating factor in patients with refractory and/or relapsed high-risk neuroblastoma: Management of adverse events. Cancer Rep (Hoboken N.J.). (2023) 6:e1627. doi: 10.1002/cnr2.1627
4. Schulz G, Cheresh DA, Varki NM, Yu A, Staffileno LK, Reisfeld RA. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res. (1984) 44:5914–20.
5. Hersey P, Jamal O, Henderson C, Zardawi I, D'Alessandro G. Expression of the gangliosides GM3, GD3 and GD2 in tissue sections of normal skin, naevi, primary and metastatic melanoma. Int J Cancer. (1988) 41:336–43. doi: 10.1002/ijc.2910410303
6. Fredman P, von Holst H, Collins VP, Ammar A, Dellheden B, Wahren B, et al. Potential ganglioside antigens associated with human gliomas. Neurological Res. (1986) 8:123–6. doi: 10.1080/01616412.1986.11739744
7. Portoukalian J, David MJ, Gain P, Richard M. Shedding of GD2 ganglioside in patients with retinoblastoma. Int J Cancer. (1993) 53:948–51. doi: 10.1002/ijc.2910530614
8. Longee DC, Wikstrand CJ, Månsson JE, He X, Fuller GN, Bigner SH, et al. Disialoganglioside GD2 in human neuroectodermal tumor cell lines and gliomas. Acta neuropathologica. (1991) 82:45–54. doi: 10.1007/BF00310922
9. Grant SC, Kostakoglu L, Kris MG, Yeh SD, Larson SM, Finn RD, et al. Targeting of small-cell lung cancer using the anti-GD2 ganglioside monoclonal antibody 3F8: a pilot trial. Eur J Nucl Med. (1996) 23:145–9. doi: 10.1007/BF01731837
10. Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest. (2012) 122:2066–78. doi: 10.1172/JCI59735
11. Orsi G, Barbolini M, Ficarra G, Tazzioli G, Manni P, Petrachi T, et al. GD2 expression in breast cancer. Oncotarget. (2017) 8:31592–600. doi: 10.18632/oncotarget.16363
12. Heiner JP, Miraldi F, Kallick S, Makley J, Neely J, Smith-Mensah WH, et al. Localization of GD2-specific monoclonal antibody 3F8 in human osteosarcoma. Cancer Res. (1987) 47:5377–81.
13. Chang HR, Cordon-Cardo C, Houghton AN, Cheung NK, Brennan MF. Expression of disialogangliosides GD2 and GD3 on human soft tissue sarcomas. Cancer. (1992) 70:633–8. doi: 10.1002/1097-0142(19920801)70:3<633::aid-cncr2820700315>3.0.co;2-f
14. Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Luecke A, Dierkes C, et al. The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br J Cancer. (2012) 106:1123–33. doi: 10.1038/bjc.2012.57
15. Vantaku V, Donepudi SR, Ambati CR, Jin F, Putluri V, Nguyen K, et al. Correction: Expression of ganglioside GD2, reprogram the lipid metabolism and EMT phenotype in bladder cancer. Oncotarget. (2019) 10:6843–4. doi: 10.18632/oncotarget.27311
16. Lupatov AY, Gisina AM, Kim YS, Bykasov SA, Volchenko NN, Sidorov DV, et al. Ékspressiia gangliozida GD2 na kletkakh kolorektal'noĭ adenokartsinomy [Expression of ganglioside GD2 on colorectal adenocarcinoma cells]. Biomeditsinskaia khimiia. (2020) 66:95–9. doi: 10.18097/PBMC20206601095
17. Bhat AM, Mohapatra BC, Luan H, Mushtaq I, Chakraborty S, Dutta S, et al. Role of GD2 and its biosynthetic enzyme GD3 synthase in prostate cancer tumorigenesis. bioRxiv. (2023). doi: 10.1101/2023.03.18.533299
18. Lammie G, Cheung N, Gerald W, Rosenblum M, Cordoncardo C. Ganglioside gd(2) expression in the human nervous-system and in neuroblastomas - an immunohistochemical study. Int J Oncol. (1993) 3:909–15. doi: 10.3892/ijo.3.5.909
19. Hersey P, Jamal O. Expression of the gangliosides GD3 and GD2 on lymphocytes in tissue sections of melanoma. Pathology. (1989) 21:51–8. doi: 10.3109/00313028909059531
20. Jin HJ, Nam HY, Bae YK, Kim SY, Im IR, Oh W, et al. GD2 expression is closely associated with neuronal differentiation of human umbilical cord blood-derived mesenchymal stem cells. Cell Mol Life Sci. (2010) 67:1845–58. doi: 10.1007/s00018-010-0292-z
21. Sorokin M, Kholodenko I, Kalinovsky D, Shamanskaya T, Doronin I, Konovalov D, et al. RNA sequencing-based identification of ganglioside GD2-positive cancer phenotype. Biomedicines. (2020) 8:142. doi: 10.3390/biomedicines8060142
22. Cheng M, Ahmed M, Xu H, Cheung NK. Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to redirect T cells for tumor therapy. Int J Cancer. (2015) 136:476–86. doi: 10.1002/ijc.29007
23. Yu RK, Tsai YT, Ariga T, Yanagisawa M. Structures, biosynthesis, and functions of gangliosides–an overview. J oleo Sci. (2011) 60:537–44. doi: 10.5650/jos.60.537
24. National Center for Biotechnology Information. PubChem Compound Summary for CID 53481124, Ganglioside GD2 (d18:0/24:1(15Z) (2023). Available online at: https://pubchem.ncbi.nlm.nih.gov/compound/53481124.
25. Berois N, Osinaga E. Glycobiology of neuroblastoma: impact on tumor behavior, prognosis, and therapeutic strategies. Front Oncol. (2014) 4:114. doi: 10.3389/fonc.2014.00114
26. Ohmi Y, Ohkawa Y, Yamauchi Y, Tajima O, Furukawa K, Furukawa K. Essential roles of gangliosides in the formation and maintenance of membrane microdomains in brain tissues. Neurochemical Res. (2012) 37:1185–91. doi: 10.1007/s11064-012-0764-7
27. Cheresh DA, Pierschbacher MD, Herzig MA, Mujoo K. Disialogangliosides GD2 and GD3 are involved in the attachment of human melanoma and neuroblastoma cells to extracellular matrix proteins. J Cell Biol. (1986) 102:688–96. doi: 10.1083/jcb.102.3.688
28. Liang YJ, Ding Y, Levery SB, Lobaton M, Handa K, Hakomori SI. Differential expression profiles of glycosphingolipids in human breast cancer stem cells vs. cancer non-stem cells. Proc Natl Acad Sci United States America. (2013) 110:4968–73. doi: 10.1073/pnas.1302825110
29. Julien S, Bobowski M, Steenackers A, Le Bourhis X, Delannoy P. How do gangliosides regulate RTKs signaling? Cells. (2013) 2:751–67. doi: 10.3390/cells2040751
30. Shibuya H, Hamamura K, Hotta H, Matsumoto Y, Nishida Y, Hattori H, et al. Enhancement of Malignant properties of human osteosarcoma cells with disialyl gangliosides GD2/GD3. Cancer Sci. (2012) 103:1656–64. doi: 10.1111/j.1349-7006.2012.02344.x
31. Esaki N, Ohkawa Y, Hashimoto N, Tsuda Y, Ohmi Y, Bhuiyan RH, et al. ASC amino acid transporter 2, defined by enzyme-mediated activation of radical sources, enhances Malignancy of GD2-positive small-cell lung cancer. Cancer Sci. (2018) 109:141–53. doi: 10.1111/cas.13448
32. Yesmin F, Bhuiyan RH, Ohmi Y, Yamamoto S, Kaneko K, Ohkawa Y, et al. Ganglioside GD2 enhances the Malignant phenotypes of melanoma cells by cooperating with integrins. Int J Mol Sci. (2021) 23:423. doi: 10.3390/ijms23010423
33. Nguyen K, Yan Y, Yuan B, Dasgupta A, Sun J, Mu H, et al. ST8SIA1 Regulates Tumor Growth and Metastasis in TNBC by Activating the FAK-AKT-mTOR Signaling Pathway. Mol Cancer Ther. (2018) 17:2689–2701. doi: 10.1158/1535-7163.MCT-18-0399
34. Battula VL, Nguyen K, Sun J, Pitner MK, Yuan B, Bartholomeusz C, et al. IKK inhibition by BMS-345541 suppresses breast tumorigenesis and metastases by targeting GD2+ cancer stem cells. Oncotarget. (2017) 8:36936–49. doi: 10.18632/oncotarget.16294
35. Wingerter A, El Malki K, Sandhoff R, Seidmann L, Wagner DC, Lehmann N, et al. Exploiting gangliosides for the therapy of ewing's sarcoma and H3K27M-mutant diffuse midline glioma. Cancers. (2021) 13:520. doi: 10.3390/cancers13030520
36. Sha Y, Han L, Sun B, Zhao Q. Identification of a glycosyltransferase signature for predicting prognosis and immune microenvironment in neuroblastoma. Front Cell Dev Biol. (2022) 9:769580. doi: 10.3389/fcell.2021.769580
37. Jandus C, Boligan KF, Chijioke O, Liu H, Dahlhaus M, Démoulins T, et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J Clin Invest. (2014) 124:1810–20. doi: 10.1172/JCI65899
38. Theruvath J, Menard M, Smith BAH, Linde MH, Coles GL, Dalton GN, et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat Med. (2022) 28:333–44. doi: 10.1038/s41591-021-01625-x
39. Shurin GV, Shurin MR, Bykovskaia S, Shogan J, Lotze MT, Barksdale EM. Neuroblastoma-derived gangliosides inhibit dendritic cell generation and function. Cancer Res. (2001) 61:363–9.
40. Wondimu A, Liu Y, Su Y, Bobb D, Ma JS, Chakrabarti L, et al. Gangliosides drive the tumor infiltration and function of myeloid-derived suppressor cells. Cancer Res. (2014) 74:5449–57. doi: 10.1158/0008-5472.CAN-14-0927
41. Jales A, Falahati R, Mari E, Stemmy EJ, Shen W, Southammakosane C, et al. Ganglioside-exposed dendritic cells inhibit T-cell effector function by promoting regulatory cell activity. Immunology. (2011) 132:134–43. doi: 10.1111/j.1365-2567.2010.03348.x
42. Ly S, Anand V, El-Dana F, Nguyen K, Cai Y, Cai S, et al. Anti-GD2 antibody dinutuximab inhibits triple-negative breast tumor growth by targeting GD2+ breast cancer stem-like cells. J immunotherapy Cancer. (2021) 9:e001197. doi: 10.1136/jitc-2020-001197
43. Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SE, Yagita H, et al. Activation of NK cell cytotoxicity. Mol Immunol. (2005) 42:501–10. doi: 10.1016/j.molimm.2004.07.034
44. Bald T, Krummel MF, Smyth MJ, Barry KC. The NK cell-cancer cycle: advances and new challenges in NK cell-based immunotherapies. Nat Immunol. (2020) 21:835–47. doi: 10.1038/s41590-020-0728-z
45. Heemskerk N, van Egmond M. Monoclonal antibody-mediated killing of tumour cells by neutrophils. Eur J Clin Invest. (2018) 48 Suppl 2:e12962. doi: 10.1111/eci.12962
46. Yazdanifar M, Barbarito G, Bertaina A, Airoldi I. γδ T cells: the ideal tool for cancer immunotherapy. Cells. (2020) 9:1305. doi: 10.3390/cells9051305
47. Gül N, van Egmond M. Antibody-dependent phagocytosis of tumor cells by macrophages: A potent effector mechanism of monoclonal antibody therapy of cancer. Cancer Res. (2015) 75:5008–13. doi: 10.1158/0008-5472.CAN-15-1330
48. Golay J, Taylor RP. The role of complement in the mechanism of action of therapeutic anti-cancer mAbs. Antibodies (Basel Switzerland). (2020) 9:58. doi: 10.3390/antib9040058
49. Glassman PM, Balthasar JP. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol Med. (2014) 11:20–33. doi: 10.7497/j.issn.2095-3941.2014.01.002
50. Doronin II, Vishnyakova PA, Kholodenko IV, Ponomarev ED, Ryazantsev DY, Molotkovskaya IM, et al. Ganglioside GD2 in reception and transduction of cell death signal in tumor cells. BMC Cancer. (2014) 14:295. doi: 10.1186/1471-2407-14-295
51. Cheung NK, Saarinen UM, Neely JE, Landmeier B, Donovan D, Coccia PF. Monoclonal antibodies to a glycolipid antigen on human neuroblastoma cells. Cancer Res. (1985) 45:2642–9.
52. Cheresh DA, Rosenberg J, Mujoo K, Hirschowitz L, Reisfeld RA. Biosynthesis and expression of the disialoganglioside GD2, a relevant target antigen on small cell lung carcinoma for monoclonal antibody-mediated cytolysis. Cancer Res. (1986) 46:5112–8.
53. Saito M, Yu RK, Cheung NK. Ganglioside GD2 specificity of monoclonal antibodies to human neuroblastoma cell. Biochem Biophys Res Commun. (1985) 127:1–7. doi: 10.1016/s0006-291x(85)80117-0
54. Kushner BH, Cheung NK. Clinically effective monoclonal antibody 3F8 mediates nonoxidative lysis of human neuroectodermal tumor cells by polymorphonuclear leukocytes. Cancer Res. (1991) 51:4865–70.
55. Mujoo K, Kipps TJ, Yang HM, Cheresh DA, Wargalla U, Sander DJ, et al. Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res. (1989) 49:2857–61.
56. Thurin J, Thurin M, Kimoto Y, Herlyn M, Lubeck MD, Elder DE, et al. Monoclonal antibody-defined correlations in melanoma between levels of GD2 and GD3 antigens and antibody-mediated cytotoxicity. Cancer Res. (1987) 47:1229–33.
57. Cheung NK, Lazarus H, Miraldi FD, Abramowsky CR, Kallick S, Saarinen UM, et al. Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and Malignant melanoma. J Clin Oncol. (1987) 5:1430–40. doi: 10.1200/JCO.1987.5.9.1430
58. Cheung NK, Kushner BH, Yeh SD, Larson SM. 3F8 monoclonal antibody treatment of patients with stage 4 neuroblastoma: a phase II study. Int J Oncol. (1998) 12:1299–306. doi: 10.3892/ijo.12.6.1299
59. Cheung NK, Kushner BH, Cheung IY, Kramer K, Canete A, Gerald W, et al. Anti-G(D2) antibody treatment of minimal residual stage 4 neuroblastoma diagnosed at more than 1 year of age. J Clin Oncol. (1998) 16:3053–60. doi: 10.1200/JCO.1998.16.9.3053
60. Saleh MN, Khazaeli MB, Wheeler RH, Dropcho E, Liu T, Urist M, et al. Phase I trial of the murine monoclonal anti-GD2 antibody 14G2a in metastatic melanoma. Cancer Res. (1992) 52:4342–7.
61. Handgretinger R, Baader P, Dopfer R, Klingebiel T, Reuland P, Treuner J, et al. A phase I study of neuroblastoma with the anti-ganglioside GD2 antibody 14.G2a. Cancer immunology immunotherapy CII. (1992) 35:199–204. doi: 10.1007/BF01756188
62. Murray JL, Cunningham JE, Brewer H, Mujoo K, Zukiwski AA, Podoloff DA, et al. Phase I trial of murine monoclonal antibody 14G2a administered by prolonged intravenous infusion in patients with neuroectodermal tumors. J Clin Oncol. (1994) 12:184–93. doi: 10.1200/JCO.1994.12.1.184
63. Slart R, Yu AL, Yaksh TL, Sorkin LS. An animal model of pain produced by systemic administration of an immunotherapeutic anti-ganglioside antibody. Pain. (1997) 69:119–25. doi: 10.1016/s0304-3959(96)03247-2
64. Kushner BH, Kramer K, Cheung NK. Phase II trial of the anti-G(D2) monoclonal antibody 3F8 and granulocyte-macrophage colony-stimulating factor for neuroblastoma. J Clin Oncol. (2001) 19:4189–94. doi: 10.1200/JCO.2001.19.22.4189
65. Cheung NK, Sowers R, Vickers AJ, Cheung IY, Kushner BH, Gorlick R. FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J Clin Oncol. (2006) 24:2885–90. doi: 10.1200/JCO.2005.04.6011
66. Cheung NK, Cheung IY, Kramer K, Modak S, Kuk D, Pandit-Taskar N, et al. Key role for myeloid cells: phase II results of anti-G(D2) antibody 3F8 plus granulocyte-macrophage colony-stimulating factor for chemoresistant osteomedullary neuroblastoma. Int J Cancer. (2014) 135:2199–205. doi: 10.1002/ijc.28851
67. Cheung NK, Cheung IY, Kushner BH, Ostrovnaya I, Chamberlain E, Kramer K, et al. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol. (2012) 30:3264–70. doi: 10.1200/JCO.2011.41.3807
68. Sait S, Modak S. Anti-GD2 immunotherapy for neuroblastoma. Expert Rev Anticancer Ther. (2017) 17:889–904. doi: 10.1080/14737140.2017.1364995
69. Cardenas FI, Mauguen A, Cheung IY, Kramer K, Kushner BH, Ragupathi G, et al. Phase I trial of oral yeast-derived β-glucan to enhance anti-GD2 immunotherapy of resistant high-risk neuroblastoma. Cancers. (2021) 13:6265. doi: 10.3390/cancers13246265
70. Modak S, Le Luduec JB, Cheung IY, Goldman DA, Ostrovnaya I, Doubrovina E, et al. Adoptive immunotherapy with haploidentical natural killer cells and Anti-GD2 monoclonal antibody m3F8 for resistant neuroblastoma: Results of a phase I study. Oncoimmunology. (2018) 7:e1461305. doi: 10.1080/2162402X.2018.1461305
71. Frost JD, Hank JA, Reaman GH, Frierdich S, Seeger RC, Gan J, et al. A phase I/IB trial of murine monoclonal anti-GD2 antibody 14.G2a plus interleukin-2 in children with refractory neuroblastoma: a report of the Children's Cancer Group. Cancer. (1997) 80:317–33. doi: 10.1002/(sici)1097-0142(19970715)80:2<317::aid-cncr21>3.0.co;2-w
72. Garg RK, Kumar N, Malhotra HS. Posterior reversible encephalopathy syndrome in eclampsia. Neurol India. (2018) 66:1316–23. doi: 10.4103/0028-3886.241364
73. Gillies SD, Lo KM, Wesolowski J. High-level expression of chimeric antibodies using adapted cDNA variable region cassettes. J Immunol Methods. (1989) 125:191–202. doi: 10.1016/0022-1759(89)90093-8
74. Mueller BM, Romerdahl CA, Gillies SD, Reisfeld RA. Enhancement of antibody-dependent cytotoxicity with a chimeric anti-GD2 antibody. J Immunol (Baltimore Md. 1950). (1990) 144:1382–6. doi: 10.4049/jimmunol.144.4.1382
75. Uttenreuther-Fischer MM, Huang CS, Yu AL. Pharmacokinetics of human-mouse chimeric anti-GD2 mAb ch14.18 in a phase I trial in neuroblastoma patients. Cancer immunology immunotherapy CII. (1995) 41:331–8. doi: 10.1007/BF01526552
76. Handgretinger R, Anderson K, Lang P, Dopfer R, Klingebiel T, Schrappe M, et al. A phase I study of human/mouse chimeric antiganglioside GD2 antibody ch14.18 in patients with neuroblastoma. Eur J Cancer (Oxford Engl 1990). (1995) 31A:261–7. doi: 10.1016/0959-8049(94)00413-Y
77. Cheung NK, Walter EI, Smith-Mensah WH, Ratnoff WD, Tykocinski ML, Medof ME. Decay-accelerating factor protects human tumor cells from complement-mediated cytotoxicity in vitro. J Clin Invest. (1988) 81:1122–8. doi: 10.1172/JCI113426
78. Saleh MN, Khazaeli MB, Wheeler RH, Allen L, Tilden AB, Grizzle W, et al. Phase I trial of the chimeric anti-GD2 monoclonal antibody ch14.18 in patients with Malignant melanoma. Hum antibodies hybridomas. (1992) 3:19–24. doi: 10.3233/HAB-1992-3104
79. Yu AL, Uttenreuther-Fischer MM, Huang CS, Tsui CC, Gillies SD, Reisfeld RA, et al. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J Clin Oncol. (1998) 16:2169–80. doi: 10.1200/JCO.1998.16.6.2169
80. Simon T, Hero B, Faldum A, Handgretinger R, Schrappe M, Niethammer D, et al. Consolidation treatment with chimeric anti-GD2-antibody ch14.18 in children older than 1 year with metastatic neuroblastoma. J Clin Oncol. (2004) 22:3549–57. doi: 10.1200/JCO.2004.08.143
81. Simon T, Hero B, Faldum A, Handgretinger R, Schrappe M, Niethammer D, et al. Infants with stage 4 neuroblastoma: the impact of the chimeric anti-GD2-antibody ch14.18 consolidation therapy. Klinische Padiatrie. (2005) 217:147–52. doi: 10.1055/s-2005-836518
82. Simon T, Hero B, Faldum A, Handgretinger R, Schrappe M, Klingebiel T, et al. Long term outcome of high-risk neuroblastoma patients after immunotherapy with antibody ch14.18 or oral metronomic chemotherapy. BMC Cancer. (2011) 11:21. doi: 10.1186/1471-2407-11-21
83. Albertini MR, Hank JA, Schiller JH, Khorsand M, Borchert AA, Gan J, et al. Phase IB trial of chimeric antidisialoganglioside antibody plus interleukin 2 for melanoma patients. Clin Cancer Res. (1997) 3:1277–88.
84. Ozkaynak MF, Sondel PM, Krailo MD, Gan J, Javorsky B, Reisfeld RA, et al. Phase I study of chimeric human/murine anti-ganglioside G(D2) monoclonal antibody (ch14.18) with granulocyte-macrophage colony-stimulating factor in children with neuroblastoma immediately after hematopoietic stem-cell transplantation: a Children's Cancer Group Study. J Clin Oncol. (2000) 18:4077–85. doi: 10.1200/JCO.2000.18.24.4077
85. Murray JL, Kleinerman ES, Jia SF, Rosenblum MG, Eton O, Buzaid A, et al. Phase Ia/Ib trial of anti-GD2 chimeric monoclonal antibody 14.18 (ch14.18) and recombinant human granulocyte-macrophage colony-stimulating factor (rhGM-CSF) in metastatic melanoma. J immunotherapy emphasis tumor Immunol. (1996) 19:206–17. doi: 10.1097/00002371-199605000-00005
86. Choi BS, Sondel PM, Hank JA, Schalch H, Gan J, King DM, et al. Phase I trial of combined treatment with ch14.18 and R24 monoclonal antibodies and interleukin-2 for patients with melanoma or sarcoma. Cancer immunology immunotherapy. (2006) 55:761–74. doi: 10.1007/s00262-005-0069-7
87. Gilman AL, Ozkaynak MF, Matthay KK, Krailo M, Yu AL, Gan J, et al. Phase I study of ch14.18 with granulocyte-macrophage colony-stimulating factor and interleukin-2 in children with neuroblastoma after autologous bone marrow transplantation or stem-cell rescue: a report from the Children's Oncology Group. J Clin Oncol. (2009) 27:85–91. doi: 10.1200/JCO.2006.10.3564
88. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. New Engl J Med. (2010) 363:1324–34. doi: 10.1056/NEJMoa0911123
89. FDA. UNITUXINTM (dinutuximab) injection, for intravenous use Initial U.S. Approval: 2015 (2015). Available online at: https://www.accessdata.fda.gov.
90. Yu AL, Gilman AL, Ozkaynak MF, Naranjo A, Diccianni MB, Gan J, et al. Long-term follow-up of a phase III study of ch14.18 (Dinutuximab) + Cytokine immunotherapy in children with high-risk neuroblastoma: COG study ANBL0032. Clin Cancer Res. (2021) 27:2179–89. doi: 10.1158/1078-0432.CCR-20-3909
91. Mody R, Yu AL, Naranjo A, Zhang FF, London WB, Shulkin BL, et al. Irinotecan, temozolomide, and dinutuximab with GM-CSF in children with refractory or relapsed neuroblastoma: A report from the children's oncology group. J Clin Oncol. (2020) 38:2160–9. doi: 10.1200/JCO.20.00203
92. Zoine JT, Knight KA, Fleischer LC, Sutton KS, Goldsmith KC, Doering CB, et al. Ex vivo expanded patient-derived γδ T-cell immunotherapy enhances neuroblastoma tumor regression in a murine model. Oncoimmunology. (2019) 8:1593804. doi: 10.1080/2162402X.2019.1593804
93. Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, et al. TGFβR1 blockade with galunisertib (LY2157299) enhances anti-neuroblastoma activity of the anti-GD2 antibody dinutuximab (ch14.18) with natural killer cells. Clin Cancer Res. (2017) 23:804–13. doi: 10.1158/1078-0432.CCR-16-1743
94. Wu HW, Sheard MA, Malvar J, Fernandez GE, DeClerck YA, Blavier L, et al. Anti-CD105 antibody eliminates tumor microenvironment cells and enhances anti-GD2 antibody immunotherapy of neuroblastoma with activated natural killer cells. Clin Cancer Res. (2019) 25:4761–74. doi: 10.1158/1078-0432.CCR-18-3358
95. Zobel MJ, Zamora AK, Wu HW, Sun J, Lascano D, Malvar J, et al. Initiation of immunotherapy with activated natural killer cells and anti-GD2 antibody dinutuximab prior to resection of primary neuroblastoma prolongs survival in mice. J immunotherapy Cancer. (2020) 8:e001560. doi: 10.1136/jitc-2020-001560
96. Ornell KJ, Chiu B, Coburn JM. Development of a dinutuximab delivery system using silk foams for GD2 targeted neuroblastoma cell death. J Biomed materials Res Part A. (2021) 109:1393–405. doi: 10.1002/jbm.a.37131
97. Shepherd AJ, Wilson NJ, Smith KT. Characterisation of endogenous retrovirus in rodent cell lines used for production of biologicals. Biologicals. (2003) 31:251–60. doi: 10.1016/s1045-1056(03)00065-4
98. Zhang JH, Shan LL, Liang F, Du CY, Li JJ. Strategies and considerations for improving recombinant antibody production and quality in chinese hamster ovary cells. Front bioengineering Biotechnol. (2022) 10:856049. doi: 10.3389/fbioe.2022.856049
99. Zeng Y, Fest S, Kunert R, Katinger H, Pistoia V, Michon J, et al. Anti-neuroblastoma effect of ch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Mol Immunol. (2005) 42:1311–9. doi: 10.1016/j.molimm.2004.12.018
100. Ladenstein R, Weixler S, Baykan B, Bleeke M, Kunert R, Katinger D, et al. Ch14.18 antibody produced in CHO cells in relapsed or refractory Stage 4 neuroblastoma patients: a SIOPEN Phase 1 study. mAbs. (2013) 5:801–9. doi: 10.4161/mabs.25215
101. Ladenstein R, Pötschger U, Valteau-Couanet D, Luksch R, Castel V, Yaniv I, YLode H. N, et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol. (2018) 19:1617–29. doi: 10.1016/S1470-2045(18)30578-3
102. Ladenstein R, Pötschger U, Valteau-Couanet D, Luksch R, Castel V, Ash S, et al. Investigation of the role of dinutuximab beta-based immunotherapy in the SIOPEN high-risk neuroblastoma 1 trial (HR-NBL1). Cancers. (2020) 12:309. doi: 10.3390/cancers12020309
103. Wieczorek A, Manzitti C, Garaventa A, Gray J, Papadakis V, Valteau-Couanet D, et al. Clinical phenotype and management of severe neurotoxicity observed in patients with neuroblastoma treated with dinutuximab beta in clinical trials. Cancers. (2022) 14:1919. doi: 10.3390/cancers14081919
104. Lang P, Flaadt T, Ebinger M, Schlegel P, Lode H, Ladenstein R, et al. Abstract A013: Haploidentical stem cell transplantation and subsequent immunotherapy with antiGD2 antibody for patients with relapsed metastatic neuroblastoma. Cancer Immunol Res. (2019) 7:A013–3. doi: 10.1158/2326-6074.CRICIMTEATIAACR18-A013
105. Flaadt T, Ladenstein RL, Ebinger M, Lode HN, Arnardóttir HB, Poetschger U, et al. Anti-GD2 antibody dinutuximab beta and low-Dose interleukin 2 after haploidentical stem-Cell transplantation in patients with relapsed neuroblastoma: A multicenter, phase I/II trial. J Clin Oncol. (2023) 41:3135–48. doi: 10.1200/JCO.22.01630
106. Seitz CM, Flaadt T, Mezger M, Lang AM, Michaelis S, Katz M, et al. Immunomonitoring of stage IV relapsed neuroblastoma patients undergoing haploidentical hematopoietic stem cell transplantation and subsequent GD2 (ch14.18/CHO) antibody treatment. Front Immunol. (2021) 12:690467. doi: 10.3389/fimmu.2021.690467
107. Siebert N, Leopold J, Zumpe M, Troschke-Meurer S, Biskupski S, Zikoridse A, et al. The immunocytokine FAP-IL-2v enhances anti-neuroblastoma efficacy of the anti-GD2 antibody dinutuximab beta. Cancers. (2022) 14:4842. doi: 10.3390/cancers14194842
108. Fisher JP, Flutter B, Wesemann F, Frosch J, Rossig C, Gustafsson K, et al. Effective combination treatment of GD2-expressing neuroblastoma and Ewing's sarcoma using anti-GD2 ch14.18/CHO antibody with Vγ9Vδ2+ γδT cells. Oncoimmunology. (2015) 5:e1025194. doi: 10.1080/2162402X.2015.1025194
109. Mueller I, Ehlert K, Endres S, Pill L, Siebert N, Kietz S, et al. Tolerability, response and outcome of high-risk neuroblastoma patients treated with long-term infusion of anti-GD2 antibody ch14.18/CHO. mAbs. (2018) 10:55–61. doi: 10.1080/19420862.2017.1402997
110. Spasov N, Spasova M. Early use of dinutuximab beta in patients with high-risk neuroblastoma. Case Rep Pediatr. (2021) 2021:6610955. doi: 10.1155/2021/6610955
111. Siebert N, Troschke-Meurer S, Marx M, Zumpe M, Ehlert K, Gray J, et al. Impact of HACA on immunomodulation and treatment toxicity following ch14.18/CHO long-term infusion with interleukin-2: results from a SIOPEN phase 2 trial. Cancers. (2018) 10:387. doi: 10.3390/cancers10100387
112. Siebert N, Zumpe M, von Lojewski L, Troschke-Meurer S, Marx M, Lode HN. Reduction of CD11b+ myeloid suppressive cells augments anti-neuroblastoma immune response induced by the anti-GD2 antibody ch14.18/CHO. Oncoimmunology. (2020) 9:1836768. doi: 10.1080/2162402X.2020.1836768
113. Kroesen M, Büll C, Gielen PR, Brok IC, Armandari I, Wassink M, et al. Anti-GD2 mAb and Vorinostat synergize in the treatment of neuroblastoma. Oncoimmunology. (2016) 5:e1164919. doi: 10.1080/2162402X.2016.1164919
114. Siebert N, Zumpe M, Jüttner M, Troschke-Meurer S, Lode HN. PD-1 blockade augments anti-neuroblastoma immune response induced by anti-GD2 antibody ch14.18/CHO. Oncoimmunology. (2017) 6:e1343775. doi: 10.1080/2162402X.2017.1343775
115. Ehlert K, Hansjuergens I, Zinke A, Otto S, Siebert N, Henze G, et al. Nivolumab and dinutuximab beta in two patients with refractory neuroblastoma. J immunotherapy Cancer. (2020) 8:e000540. doi: 10.1136/jitc-2020-000540
116. Siebert N, Zumpe M, Schwencke CH, Biskupski S, Troschke-Meurer S, Leopold J, et al. Combined blockade of TIGIT and PD-L1 enhances anti-neuroblastoma efficacy of GD2-directed immunotherapy with dinutuximab beta. Cancers. (2023) 15:3317. doi: 10.3390/cancers15133317
117. Levine JD, Gooding J, Donatoni P, Borden L, Goetzl EJ. The role of the polymorphonuclear leukocyte in hyperalgesia. J Neurosci. (1985) 5:3025–9. doi: 10.1523/JNEUROSCI.05-11-03025.1985
118. Thommesen JE, Michaelsen TE, Løset GÅ., Sandlie I, Brekke OH. Lysine 322 in the human IgG3 C(H)2 domain is crucial for antibody dependent complement activation. Mol Immunol. (2000) 37:995–1004. doi: 10.1016/s0161-5890(01)00010-4
119. Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. (2003) 278:3466–73. doi: 10.1074/jbc.M210665200
120. Sorkin LS, Otto M, Baldwin WM 3rd, Vail E, Gillies SD, Handgretinger R, et al. Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain. (2010) 149:135–42. doi: 10.1016/j.pain.2010.01.024
121. Anghelescu DL, Goldberg JL, Faughnan LG, Wu J, Mao S, Furman WL, et al. Comparison of pain outcomes between two anti-GD2 antibodies in patients with neuroblastoma. Pediatr Blood Cancer. (2015) 62:224–8. doi: 10.1002/pbc.25280
122. Alderson KL, Luangrath M, Elsenheimer MM, Gillies SD, Navid F, Rakhmilevich AL, et al. Enhancement of the anti-melanoma response of Hu14.18K322A by αCD40 + CpG. Cancer immunology immunotherapy. (2013) 62:665–75. doi: 10.1007/s00262-012-1372-8
123. Vavere AL, Butch ER, Dearling JL, Packard AB, Navid F, Shulkin BL, et al. 64Cu-p-NH2-Bn-DOTA-hu14.18K322A, a PET radiotracer targeting neuroblastoma and melanoma. J Nucl Med. (2012) 53:1772–8. doi: 10.2967/jnumed.112.104208
124. Nguyen R, Moustaki A, Norrie JL, Brown S, Akers WJ, Shirinifard A, et al. Interleukin-15 enhances anti-GD2 antibody-mediated cytotoxicity in an orthotopic PDX model of neuroblastoma. Clin Cancer Res. (2019) 25:7554–64. doi: 10.1158/1078-0432.CCR-19-1045
125. Navid F, Sondel PM, Barfield R, Shulkin BL, Kaufman RA, Allay JA, et al. Phase I trial of a novel anti-GD2 monoclonal antibody, Hu14.18K322A, designed to decrease toxicity in children with refractory or recurrent neuroblastoma. J Clin Oncol. (2014) 32:1445–52. doi: 10.1200/JCO.2013.50.4423
126. Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P, et al. A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin Cancer Res. (2017) 23:6441–9. doi: 10.1158/1078-0432.CCR-17-0379
127. Furman WL, Federico SM, McCarville MB, Shulkin BL, Davidoff AM, Krasin MJ, et al. A phase II rrial of Hu14.18K322A in combination with induction chemotherapy in children with newly diagnosed high-risk neuroblastoma. Clin Cancer Res. (2019) 25:6320–8. doi: 10.1158/1078-0432.CCR-19-1452
128. Furman WL, McCarville B, Shulkin BL, Davidoff A, Krasin M, Hsu CW, et al. Improved outcome in children with newly diagnosed high-risk neuroblastoma treated with chemoimmunotherapy: updated results of a phase II study using hu14.18K322A. J Clin Oncol. (2022) 40:335–44. doi: 10.1200/JCO.21.01375
129. Bishop MW, Hutson PR, Hank JA, Sondel PM, Furman WL, Meagher MM, et al. A Phase 1 and pharmacokinetic study evaluating daily or weekly schedules of the humanized anti-GD2 antibody hu14.18K322A in recurrent/refractory solid tumors. mAbs. (2020) 12:1773751. doi: 10.1080/19420862.2020.1773751
130. Cheung NK, Guo H, Hu J, Tassev DV, Cheung IY. Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. Oncoimmunology. (2012) 1:477–86. doi: 10.4161/onci.19864
131. Kushner BH, Cheung IY, Modak S, Basu EM, Roberts SS, Cheung NK. Humanized 3F8 anti-GD2 monoclonal antibody dosing with granulocyte-macrophage colony-stimulating factor in patients with resistant neuroblastoma: A phase 1 clinical trial. JAMA Oncol. (2018) 4:1729–35. doi: 10.1001/jamaoncol.2018.4005
132. FDA. Danyelza (naxitamab-gqgk) injection, for intravenous use prescribing information. (2020). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761171lbl.pdf.
133. Cheung IY, Kushner BH, Modak S, Basu EM, Roberts SS, Cheung NV. Phase I trial of anti-GD2 monoclonal antibody hu3F8 plus GM-CSF: Impact of body weight, immunogenicity and anti-GD2 response on pharmacokinetics and survival. Oncoimmunology. (2017) 6:e1358331. doi: 10.1080/2162402X.2017.1358331
134. Zhao Q, Ahmed M, Guo HF, Cheung IY, Cheung NK. Alteration of electrostatic surface potential enhances affinity and tumor killing properties of anti-ganglioside GD2 monoclonal antibody hu3F8. J Biol Chem. (2015) 290:13017–27. doi: 10.1074/jbc.M115.650903
135. Yuki N, Yamada M, Tagawa Y, Takahashi H, Handa S. Pathogenesis of the neurotoxicity caused by anti-GD2 antibody therapy. J neurological Sci. (1997) 149:127–30. doi: 10.1016/s0022-510x(97)05390-2
136. Xiao WH, Yu AL, Sorkin LS. Electrophysiological characteristics of primary afferent fibers after systemic administration of anti-GD2 ganglioside antibody. Pain. (1997) 69:145–51. doi: 10.1016/s0304-3959(96)03280-0
137. Kushner BH, Kramer K, Modak S, Cheung NK. Successful multifold dose escalation of anti-GD2 monoclonal antibody 3F8 in patients with neuroblastoma: a phase I study. J Clin Oncol. (2011) 29:1168–74. doi: 10.1200/JCO.2010.28.3317
138. Evers M, Stip M, Keller K, Willemen H, Nederend M, Jansen M, et al. Anti-GD2 IgA kills tumors by neutrophils without antibody-associated pain in the preclinical treatment of high-risk neuroblastoma. J immunotherapy Cancer. (2021) 9:e003163. doi: 10.1136/jitc-2021-003163
139. Stip MC, Evers M, Nederend M, Chan C, Reiding KR, Damen MJ, et al. IgA antibody immunotherapy targeting GD2 is effective in preclinical neuroblastoma models. J immunotherapy Cancer. (2023) 11:e006948. doi: 10.1136/jitc-2023-006948
140. Kulanthaivadivel, Sathurthika. Dataset in support of the University of Southampton Doctoral Thesis 'Understanding and improving the mechanisms of action of anti-GD2 monoclonal antibody therapy in neuroblastoma'. University of Southampton (2024). doi: 10.5258/SOTON/D2936
141. Liu XY, Chen YL, Liu GJ, Deng XN, Cui Y, Tan J, et al. Development of a variant of dinutuximab with enhanced antitumor efficacy and reduced induction of neuropathic pain. FEBS Open Bio. (2022) 12:1644–56. doi: 10.1002/2211-5463.13464
142. Alvarez-Rueda N, Desselle A, Cochonneau D, Chaumette T, Clemenceau B, Leprieur S, et al. A monoclonal antibody to O-acetyl-GD2 ganglioside and not to GD2 shows potent anti-tumor activity without peripheral nervous system cross-reactivity. PloS One. (2011) 6:e25220. doi: 10.1371/journal.pone.0025220
143. Cochonneau D, Terme M, Michaud A, Dorvillius M, Gautier N, Frikeche J, et al. Cell cycle arrest and apoptosis induced by O-acetyl-GD2-specific monoclonal antibody 8B6 inhibits tumor growth in vitro and in vivo. Cancer Lett. (2013) 333:194–204. doi: 10.1016/j.canlet.2013.01.032
144. Terme M, Dorvillius M, Cochonneau D, Chaumette T, Xiao W, Diccianni MB, et al. Chimeric antibody c.8B6 to O-acetyl-GD2 mediates the same efficient anti-neuroblastoma effects as therapeutic ch14.18 antibody to GD2 without antibody induced allodynia. PloS One. (2014) 9:e87210. doi: 10.1371/journal.pone.0087210
145. Gillies SD, Reilly EB, Lo KM, Reisfeld RA. Antibody-targeted interleukin 2 stimulates T-cell killing of autologous tumor cells. Proc Natl Acad Sci United States America. (1992) 89:1428–32. doi: 10.1073/pnas.89.4.1428
146. Hank JA, Surfus JE, Gan J, Jaeger P, Gillies SD, Reisfeld RA, et al. Activation of human effector cells by a tumor reactive recombinant anti-ganglioside GD2 interleukin-2 fusion protein (ch14.18-IL2). Clin Cancer Res. (1996) 2:1951–9. doi: 10.1097/00002371-199611000-00039
147. Kendra K, Gan J, Ricci M, Surfus J, Shaker A, Super M, et al. Pharmacokinetics and stability of the ch14.18-interleukin-2 fusion protein in mice. Cancer immunology immunotherapy. (1999) 48:219–29. doi: 10.1007/s002620050569
148. Becker JC, Varki N, Gillies SD, Furukawa K, Reisfeld RA. An antibody-interleukin 2 fusion protein overcomes tumor heterogeneity by induction of a cellular immune response. Proc Natl Acad Sci United States America. (1996) 93:7826–31. doi: 10.1073/pnas.93.15.7826
149. Becker JC, Pancook JD, Gillies SD, Furukawa K, Reisfeld RA. T cell-mediated eradication of murine metastatic melanoma induced by targeted interleukin 2 therapy. J Exp Med. (1996) 183:2361–6. doi: 10.1084/jem.183.5.2361
150. Lode HN, Xiang R, Varki NM, Dolman CS, Gillies SD, Reisfeld RA. Targeted interleukin-2 therapy for spontaneous neuroblastoma metastases to bone marrow. J Natl Cancer Institute. (1997) 89:1586–94. doi: 10.1093/jnci/89.21.1586
151. Lode HN, Xiang R, Dreier T, Varki NM, Gillies SD, Reisfeld RA. Natural killer cell-mediated eradication of neuroblastoma metastases to bone marrow by targeted interleukin-2 therapy. Blood. (1998) 91:1706–15. doi: 10.1182/blood.V91.5.1706.1706_1706_1715
152. Lode HN, Xiang R, Pertl U, Förster E, Schoenberger SP, Gillies SD, et al. Melanoma immunotherapy by targeted IL-2 depends on CD4(+) T-cell help mediated by CD40/CD40L interaction. J Clin Invest. (2000) 105:1623–30. doi: 10.1172/JCI9177
153. Neal ZC, Imboden M, Rakhmilevich AL, Kim KM, Hank JA, Surfus J, et al. NXS2 murine neuroblastomas express increased levels of MHC class I antigens upon recurrence following NK-dependent immunotherapy. Cancer immunology immunotherapy. (2004) 53:41–52. doi: 10.1007/s00262-003-0435-2
154. Neal ZC, Yang JC, Rakhmilevich AL, Buhtoiarov IN, Lum HE, Imboden M, et al. Enhanced activity of hu14.18-IL2 immunocytokine against murine NXS2 neuroblastoma when combined with interleukin 2 therapy. Clin Cancer Res. (2004) 10:4839–47. doi: 10.1158/1078-0432.CCR-03-0799
155. King DM, Albertini MR, Schalch H, Hank JA, Gan J, Surfus J, et al. Phase I clinical trial of the immunocytokine EMD 273063 in melanoma patients. J Clin Oncol. (2004) 22:4463–73. doi: 10.1200/JCO.2004.11.035
156. Osenga KL, Hank JA, Albertini MR, Gan J, Sternberg AG, Eickhoff J, et al. A phase I clinical trial of the hu14.18-IL2 (EMD 273063) as a treatment for children with refractory or recurrent neuroblastoma and melanoma: a study of the Children's Oncology Group. Clin Cancer Res. (2006) 12:1750–9. doi: 10.1158/1078-0432.CCR-05-2000
157. Ribas A, Kirkwood JM, Atkins MB, Whiteside TL, Gooding W, Kovar A, et al. Phase I/II open-label study of the biologic effects of the interleukin-2 immunocytokine EMD 273063 (hu14.18-IL2) in patients with metastatic Malignant melanoma. J Trans Med. (2009) 7:68. doi: 10.1186/1479-5876-7-68
158. Shusterman S, London WB, Gillies SD, Hank JA, Voss SD, Seeger RC, et al. Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children's Oncology Group (COG) phase II study. J Clin Oncol. (2010) 28:4969–75. doi: 10.1200/JCO.2009.27.8861
159. Albertini MR, Hank JA, Gadbaw B, Kostlevy J, Haldeman J, Schalch H, et al. Phase II trial of hu14.18-IL2 for patients with metastatic melanoma. Cancer immunology immunotherapy CII. (2012) 61:2261–71. doi: 10.1007/s00262-012-1286-5
160. Gillies SD, Young D, Lo KM, Roberts S. Biological activity and in vivo clearance of antitumor antibody/cytokine fusion proteins. Bioconjugate Chem. (1993) 4:230–5. doi: 10.1021/bc00021a008
161. Griffon-Etienne G, Boucher Y, Brekken C, Suit HD, Jain RK. Taxane-induced apoptosis decompresses blood vessels and lowers interstitial fluid pressure in solid tumors: clinical implications. Cancer Res. (1999) 59:3776–82.
162. Johnson EE, Lum HD, Rakhmilevich AL, Schmidt BE, Furlong M, Buhtoiarov IN, et al. Intratumoral immunocytokine treatment results in enhanced antitumor effects. Cancer immunology immunotherapy. (2008) 57:1891–902. doi: 10.1007/s00262-008-0519-0
163. Buhtoiarov IN, Neal ZC, Gan J, Buhtoiarova TN, Patankar MS, Gubbels JA, et al. Differential internalization of hu14.18-IL2 immunocytokine by NK and tumor cell: impact on conjugation, cytotoxicity, and targeting. J leukocyte Biol. (2011) 89:625–38. doi: 10.1189/jlb.0710422
164. Gillies SD. A new platform for constructing antibody-cytokine fusion proteins (immunocytokines) with improved biological properties and adaptable cytokine activity. Protein engineering design selection PEDS. (2013) 26:561–9. doi: 10.1093/protein/gzt045
165. Conlon KC, Miljkovic MD, Waldmann TA. Cytokines in the treatment of cancer. J Interferon Cytokine Res. (2019) 39:6–21. doi: 10.1089/jir.2018.0019
166. Nguyen R, Zhang X, Sun M, Abbas S, Seibert C, Kelly MC, et al. Anti-GD2 antibodies conjugated to IL15 and IL21 mediate potent antitumor cytotoxicity against neuroblastoma. Clin Cancer Res. (2022) 28:3785–96. doi: 10.1158/1078-0432.CCR-22-0717
167. Metelitsa LS, Gillies SD, Super M, Shimada H, Reynolds CP, Seeger RC. Antidisialoganglioside/granulocyte macrophage-colony-stimulating factor fusion protein facilitates neutrophil antibody-dependent cellular cytotoxicity and depends on FcgammaRII (CD32) and Mac-1 (CD11b/CD18) for enhanced effector cell adhesion and azurophil granule exocytosis. Blood. (2002) 99:4166–73. doi: 10.1182/blood.v99.11.4166
168. Burkett PR, Koka R, Chien M, Chai S, Chan F, Ma A, et al. IL-15R alpha expression on CD8+ T cells is dispensable for T cell memory. Proc Natl Acad Sci United States America. (2003) 100:4724–9. doi: 10.1073/pnas.0737048100
169. Mortier E, Quéméner A, Vusio P, Lorenzen I, Boublik Y, Grötzinger J, et al. Soluble interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective and potent agonist of IL-15 action through IL-15R beta/gamma. Hyperagonist IL-15 x IL-15R alpha fusion proteins. J Biol Chem. (2006) 281:1612–9. doi: 10.1074/jbc.M508624200
170. Zhou Y, Quan G, Liu Y, Shi N, Wu Y, Zhang R, et al. The application of Interleukin-2 family cytokines in tumor immunotherapy research. Front Immunol. (2023) 14:1090311. doi: 10.3389/fimmu.2023.1090311
171. Yang Y, Lundqvist A. Immunomodulatory effects of IL-2 and IL-15; implications for cancer immunotherapy. Cancers. (2020) 12:3586. doi: 10.3390/cancers12123586
172. Vincent M, Bessard A, Cochonneau D, Teppaz G, Solé V, Maillasson M, et al. Tumor targeting of the IL-15 superagonist RLI by an anti-GD2 antibody strongly enhances its antitumor potency. Int J Cancer. (2013) 133:757–65. doi: 10.1002/ijc.28059
173. Vincent M, Quéméner A, Jacques Y. Antitumor activity of an immunocytokine composed of an anti-GD2 antibody and the IL-15 superagonist RLI. Oncoimmunology. (2013) 2:e26441. doi: 10.4161/onci.26441
174. Chu Y, Nayyar G, Jiang S, Rosenblum JM, Soon-Shiong P, Safrit JT, et al. Combinatorial immunotherapy of N-803 (IL-15 superagonist) and dinutuximab with ex vivo expanded natural killer cells significantly enhances in vitro cytotoxicity against GD2+ pediatric solid tumors and in vivo survival of xenografted immunodeficient NSG mice. J immunotherapy Cancer. (2021) 9:e002267. doi: 10.1136/jitc-2020-002267
175. Allahyari H, Heidari S, Ghamgosha M, Saffarian P, Amani J. Immunotoxin: A new tool for cancer therapy. Tumour Biol. (2017) 39:1010428317692226. doi: 10.1177/1010428317692226
176. Wargalla UC, Reisfeld RA. Rate of internalization of an immunotoxin correlates with cytotoxic activity against human tumor cells. Proc Natl Acad Sci United States America. (1989) 86:5146–50. doi: 10.1073/pnas.86.13.5146
177. Mujoo K, Reisfeld RA, Cheung L, Rosenblum MG. A potent and specific immunotoxin for tumor cells expressing disialoganglioside GD2. Cancer immunology immunotherapy. (1991) 34:198–204. doi: 10.1007/BF01742313
178. Manzke O, Russello O, Leenen C, Diehl V, Bohlen H, Berthold F. Immunotherapeutic strategies in neuroblastoma: antitumoral activity of deglycosylated Ricin A conjugated anti-GD2 antibodies and anti-CD3xanti-GD2 bispecific antibodies. Med Pediatr Oncol. (2001) 36:185–9. doi: 10.1002/1096-911X(20010101)36:1<185::AID-MPO1044>3.0.CO;2-J
179. Gottstein C, Schön G, Tawadros S, Kube D, Wargalla-Plate UC, Hansmann ML, et al. Antidisialoganglioside ricin A-chain immunotoxins show potent antitumor effects in vitro and in a disseminated human neuroblastoma severe combined immunodeficiency mouse model. Cancer Res. (1994) 54:6186–93.
180. Thomas PB, Delatte SJ, Sutphin A, Frankel AE, Tagge EP. Effective targeted cytotoxicity of neuroblastoma cells. J Pediatr Surg. (2002) 37:539–44. doi: 10.1053/jpsu.2002.30856
181. Tur MK, Sasse S, Stöcker M, Djabelkhir K, Huhn M, Matthey B, et al. An anti-GD2 single chain Fv selected by phage display and fused to Pseudomonas exotoxin A develops specific cytotoxic activity against neuroblastoma derived cell lines. Int J Mol Med. (2001) 8:579–84. doi: 10.3892/ijmm.8.5.579
182. Mazor R, Pastan I. Immunogenicity of immunotoxins containing Pseudomonas exotoxin A: causes, consequences, and mitigation. Front Immunol. (2020) 11:1261. doi: 10.3389/fimmu.2020.01261
183. Miraldi FD, Nelson AD, Kraly C, Ellery S, Landmeier B, Coccia PF, et al. Diagnostic imaging of human neuroblastoma with radiolabeled antibody. Radiology. (1986) 161:413–8. doi: 10.1148/radiology.161.2.3763911
184. Yeh SD, Larson SM, Burch L, Kushner BH, Laquaglia M, Finn R, et al. Radioimmunodetection of neuroblastoma with iodine-131-3F8: correlation with biopsy, iodine-131-metaiodobenzylguanidine and standard diagnostic modalities. J Nucl Med. (1991) 32:769–76.
185. Reuland P, Geiger L, Thelen MH, Handgretinger R, Haase B, Müller-Schauenburg W, et al. Follow-up in neuroblastoma: comparison of metaiodobenzylguanidine and a chimeric anti-GD2 antibody for detection of tumor relapse and therapy response. J Pediatr hematology/oncology. (2001) 23:437–42. doi: 10.1097/00043426-200110000-00009
186. Larson SM, Carrasquillo JA, Cheung NK, Press OW. Radioimmunotherapy of human tumours. Nat Rev Cancer. (2015) 15:347–60. doi: 10.1038/nrc3925
187. Kramer K, Humm JL, Souweidane MM, Zanzonico PB, Dunkel IJ, Gerald WL, et al. Phase I study of targeted radioimmunotherapy for leptomeningeal cancers using intra-Ommaya 131-I-3F8. J Clin Oncol. (2007) 25:5465–70. doi: 10.1200/JCO.2007.11.1807
188. Kramer K, Kushner BH, Modak S, Pandit-Taskar N, Smith-Jones P, Zanzonico P, et al. Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. J neuro-oncology. (2010) 97:409–18. doi: 10.1007/s11060-009-0038-7
189. Cheung NK, Modak S, Lin Y, Guo H, Zanzonico P, Chung J, et al. Single-chain Fv-streptavidin substantially improved therapeutic index in multistep targeting directed at disialoganglioside GD2. J Nucl Med. (2004) 45:867–77.
190. Orcutt KD, Slusarczyk AL, Cieslewicz M, Ruiz-Yi B, Bhushan KR, Frangioni JV, et al. Engineering an antibody with picomolar affinity to DOTA chelates of multiple radionuclides for pretargeted radioimmunotherapy and imaging. Nucl Med Biol. (2011) 38:223–33. doi: 10.1016/j.nucmedbio.2010.08.013
191. Cheal SM, Xu H, Guo HF, Zanzonico PB, Larson SM, Cheung NK. Preclinical evaluation of multistep targeting of diasialoganglioside GD2 using an IgG-scFv bispecific antibody with high affinity for GD2 and DOTA metal complex. Mol Cancer Ther. (2014) 13:1803–12. doi: 10.1158/1535-7163.MCT-13-0933
192. Palumbo B, Buresta T, Nuvoli S, Spanu A, Schillaci O, Fravolini ML, et al. SPECT and PET serve as molecular imaging techniques and in vivo biomarkers for brain metastases. Int J Mol Sci. (2014) 15:9878–93. doi: 10.3390/ijms15069878
193. Voss SD, Smith SV, DiBartolo N, McIntosh LJ, Cyr EM, Bonab AA, et al. Positron emission tomography (PET) imaging of neuroblastoma and melanoma with 64Cu-SarAr immunoconjugates. Proc Natl Acad Sci United States America. (2007) 104:17489–93. doi: 10.1073/pnas.0708436104
194. Schmitt J, Schwenck J, Maurer A, Przybille M, Sonanini D, Reischl G, et al. Translational immunoPET imaging using a radiolabeled GD2-specific antibody in neuroblastoma. Theranostics. (2022) 12:5615–30. doi: 10.7150/thno.56736
195. Butch ER, Mead PE, Amador Diaz V, Tillman H, Stewart E, Mishra JK, et al. Positron emission tomography detects in vivo expression of disialoganglioside GD2 in mouse models of primary and metastatic osteosarcoma. Cancer Res. (2019) 79:3112–24. doi: 10.1158/0008-5472.CAN-18-3340
196. Dearling JL, Voss SD, Dunning P, Snay E, Fahey F, Smith SV, et al. Imaging cancer using PET–the effect of the bifunctional chelator on the biodistribution of a (64)Cu-labeled antibody. Nucl Med Biol. (2011) 38:29–38. doi: 10.1016/j.nucmedbio.2010.07.003
197. Dearling JL, Paterson BM, Akurathi V, Betanzos-Lara S, Treves ST, Voss SD, et al. The ionic charge of copper-64 complexes conjugated to an engineered antibody affects biodistribution. Bioconjugate Chem. (2015) 26:707–17. doi: 10.1021/acs.bioconjchem.5b00049
198. White JM, Escorcia FE, Viola NT. Perspectives on metals-based radioimmunotherapy (RIT): moving forward. Theranostics. (2021) 11:6293–314. doi: 10.7150/thno.57177
199. Inagaki FF, Kato T, Furusawa A, Okada R, Wakiyama H, Furumoto H, et al. Disialoganglioside GD2-targeted near-infrared photoimmunotherapy (NIR-PIT) in tumors of neuroectodermal origin. Pharmaceutics. (2022) 14:2037. doi: 10.3390/pharmaceutics14102037
200. Kobayashi H, Choyke PL. Near-infrared photoimmunotherapy of cancer. Accounts Chem Res. (2019) 52:2332–9. doi: 10.1021/acs.accounts.9b00273
201. Farahavar G, Abolmaali SS, Gholijani N, Nejatollahi F. Antibody-guided nanomedicines as novel breakthrough therapeutic, diagnostic and theranostic tools. Biomaterials Sci. (2019) 7:4000–16. doi: 10.1039/C9BM00931K
202. Wang S, Chen Y, Guo J, Huang Q. Liposomes for tumor targeted therapy: A review. Int J Mol Sci. (2023) 24:2643. doi: 10.3390/ijms24032643
203. Raffaghello L, Pagnan G, Pastorino F, Cosimo E, Brignole C, Marimpietri D, et al. In vitro and in vivo antitumor activity of liposomal Fenretinide targeted to human neuroblastoma. Int J Cancer. (2003) 104:559–67. doi: 10.1002/ijc.10991
204. Brignole C, Pastorino F, Marimpietri D, Pagnan G, Pistorio A, Allen TM, et al. Immune cell-mediated antitumor activities of GD2-targeted liposomal c-myb antisense oligonucleotides containing CpG motifs. J Natl Cancer Institute. (2004) 96:1171–80. doi: 10.1093/jnci/djh221
205. Pastorino F, Brignole C, Marimpietri D, Pagnan G, Morando A, Ribatti D, et al. Targeted liposomal c-myc antisense oligodeoxynucleotides induce apoptosis and inhibit tumor growth and metastases in human melanoma models. Clin Cancer Res. (2003) 9:4595–605.
206. Pastorino F, Brignole C, Marimpietri D, Sapra P, Moase EH, Allen TM, et al. Doxorubicin-loaded Fab' fragments of anti-disialoganglioside immunoliposomes selectively inhibit the growth and dissemination of human neuroblastoma in nude mice. Cancer Res. (2003) 63:86–92.
207. Adrian JE, Wolf A, Steinbach A, Rössler J, Süss R. Targeted delivery to neuroblastoma of novel siRNA-anti-GD2-liposomes prepared by dual asymmetric centrifugation and sterol-based post-insertion method. Pharm Res. (2011) 28:2261–72. doi: 10.1007/s11095-011-0457-y
208. Di Paolo D, Brignole C, Pastorino F, Carosio R, Zorzoli A, Rossi M, et al. Neuroblastoma-targeted nanoparticles entrapping siRNA specifically knockdown ALK. Mol Ther. (2011) 19:1131–40. doi: 10.1038/mt.2011.54
209. Di Paolo D, Ambrogio C, Pastorino F, Brignole C, Martinengo C, Carosio R, et al. Selective therapeutic targeting of the anaplastic lymphoma kinase with liposomal siRNA induces apoptosis and inhibits angiogenesis in neuroblastoma. Mol Ther. (2011) 19:2201–12. doi: 10.1038/mt.2011.142
210. Monterrubio C, Paco S, Olaciregui NG, Pascual-Pasto G, Vila-Ubach M, Cuadrado-Vilanova M, et al. Targeted drug distribution in tumor extracellular fluid of GD2-expressing neuroblastoma patient-derived xenografts using SN-38-loaded nanoparticles conjugated to the monoclonal antibody 3F8. J Controlled release. (2017) 255:108–19. doi: 10.1016/j.jconrel.2017.04.016
211. Gholizadeh S, Dolman EM, Wieriks R, Sparidans RW, Hennink WE, Kok RJ. Anti-GD2 immunoliposomes for targeted delivery of the survivin inhibitor sepantronium bromide (YM155) to neuroblastoma tumor cells. Pharm Res. (2018) 35:85. doi: 10.1007/s11095-018-2373-x
212. Park JH, Gu L, von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ. Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat materials. (2009) 8:331–6. doi: 10.1038/nmat2398
213. Tivnan A, Orr WS, Gubala V, Nooney R, Williams DE, McDonagh C, et al. Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles. PloS One. (2012) 7:e38129. doi: 10.1371/journal.pone.0038129
214. Xu Y, Baiu DC, Sherwood JA, McElreath MR, Qin Y, Lackey KH, et al. Linker-free conjugation and specific cell targeting of antibody functionalized iron-oxide nanoparticles. J Mater Chem B. (2014) 2:6198–6206. doi: 10.1039/C4TB00840E
215. Pola R, Král V., Filippov SK, Kaberov L, Etrych T, Sieglová I, et al. Polymer cancerostatics targeted by recombinant antibody fragments to GD2-positive tumor cells. Biomacromolecules (2019) 20(1):412–21. doi: 10.1021/acs.biomac.8b01616
216. Wang CH, Huang YJ, Chang CW, Hsu WM, Peng CA. In vitro photothermal destruction of neuroblastoma cells using carbon nanotubes conjugated with GD2 monoclonal antibody. Nanotechnology. (2009) 20:315101. doi: 10.1088/0957-4484/20/31/315101
217. Peng CA, Wang CH. Anti-neuroblastoma activity of gold nanorods bound with GD2 monoclonal antibody under near-infrared laser irradiation. Cancers. (2011) 3:227–40. doi: 10.3390/cancers3010227
218. Lin YS, Chen Y, Tsai YH, Tseng SH, Lin KS. In vivo imaging of neuroblastomas using GD2-targeting graphene quantum dots. J Pediatr Surg. (2021) 56:1227–32. doi: 10.1016/j.jpedsurg.2021.03.035
219. Baiu DC, Artz NS, McElreath MR, Menapace BD, Hernando D, Reeder SB, et al. High specificity targeting and detection of human neuroblastoma using multifunctional anti-GD2 iron-oxide nanoparticles. Nanomedicine (London England). (2015) 10:2973–88. doi: 10.2217/nnm.15.138
220. Jiao P, Otto M, Geng Q, Li C, Li F, Butch ER, et al. Enhancing both CT imaging and natural killer cell-mediated cancer cell killing by a GD2-targeting nanoconstruct. J materials Chem B. (2016) 4:513–20. doi: 10.1039/C5TB02243F
221. Dean AQ, Luo S, Twomey JD, Zhang B. Targeting cancer with antibody-drug conjugates: Promises and challenges. mAbs. (2021) 13:1951427. doi: 10.1080/19420862.2021.1951427
222. Janthur WD, Cantoni N, Mamot C. Drug conjugates such as Antibody Drug Conjugates (ADCs), immunotoxins and immunoliposomes challenge daily clinical practice. Int J Mol Sci. (2012) 13:16020–45. doi: 10.3390/ijms131216020
223. Lode HN, Reisfeld RA, Handgretinger R, Nicolaou KC, Gaedicke G, Wrasidlo W. Targeted therapy with a novel enediyene antibiotic calicheamicin theta(I)1 effectively suppresses growth and dissemination of liver metastases in a syngeneic model of murine neuroblastoma. Cancer Res. (1998) 58:2925–8.
224. Kalinovsky DV, Kibardin AV, Kholodenko IV, Svirshchevskaya EV, Doronin II, Konovalova MV, et al. Therapeutic efficacy of antibody-drug conjugates targeting GD2-positive tumors. J immunotherapy Cancer. (2022) 10:e004646. doi: 10.1136/jitc-2022-004646
225. Tibbetts R, Yeo KK, Muthugounder S, Lee MH, Jung C, Porras-Corredor T, et al. Anti-disialoganglioside antibody internalization by neuroblastoma cells as a mechanism of immunotherapy resistance. Cancer immunology immunotherapy. (2022) 71:153–64. doi: 10.1007/s00262-021-02963-y
226. Kalinovsky DV, Kholodenko IV, Kibardin AV, Doronin II, Svirshchevskaya EV, Ryazantsev DY, et al. Minibody-based and scFv-based antibody fragment-drug conjugates selectively eliminate GD2-positive tumor cells. Int J Mol Sci. (2023) 24:1239. doi: 10.3390/ijms24021239
227. Mirian M, Kouhpayeh S, Shariati L, Boshtam M, Rahimmanesh I, Darzi L, et al. Generation of HBsAg DNA aptamer using modified cell-based SELEX strategy. Mol Biol Rep. (2021) 48:139–46. doi: 10.1007/s11033-020-05995-2
228. Sabbih GO, Danquah MK. Neuroblastoma GD2 expression and computational analysis of aptamer-based bioaffinity targeting. Int J Mol Sci. (2021) 22:9101. doi: 10.3390/ijms22169101
229. Zhang L, Wang M, Zhu Z, Ding C, Chen S, Wu H, et al. A novel pH-sensitive multifunctional DNA nanomedicine: an enhanced and harmless GD2 aptamer-mediated strategy for guiding neuroblastoma antitumor therapy. Int J nanomedicine. (2021) 16:3217–40. doi: 10.2147/IJN.S302450
230. Zhang L, Wang M, Zhu Z, Chen S, Wu H, Yang Y, et al. A GD2-aptamer-mediated, self-assembling nanomedicine for targeted multiple treatments in neuroblastoma theranostics. Mol Ther Nucleic Acids. (2021) 26:732–48. doi: 10.1016/j.omtn.2021.08.021
231. Thakur A, Lum LG. Cancer therapy with bispecific antibodies: Clinical experience. Curr Opin Mol Ther. (2010) 12:340–9.
232. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. (2015) 93:290–6. doi: 10.1038/icb.2014.93
233. Xu H, Cheng M, Guo H, Chen Y, Huse M, Cheung NK. Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer Immunol Res. (2015) 3:266–77. doi: 10.1158/2326-6066.CIR-14-0230-T
234. Yankelevich M, Kondadasula SV, Thakur A, Buck S, Cheung NK, Lum LG. Anti-CD3 × anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatr Blood Cancer. (2012) 59(7):1198–205. doi: 10.1002/pbc.24237
235. Park JA, Xu H, Cheung I, Cheung N-KV. Abstract B38: Tetravalent bispecific antibodies specific for HER2 and disialoganglioside GD2 to engage polyclonal T cells for osteosarcoma therapy. Cancer Res. (2018) 78:B38–8. doi: 10.1158/1538-7445.PEDCA17-B38
236. Park JA, Santich BH, Xu H, Lum LG, Cheung NV. Potent ex vivo armed T cells using recombinant bispecific antibodies for adoptive immunotherapy with reduced cytokine release. J immunotherapy Cancer. (2021) 9(5):e002222. doi: 10.1136/jitc-2020-002222
237. Park JA, Cheung NV. GD2 or HER2 targeting T cell engaging bispecific antibodies to treat osteosarcoma. J Hematol Oncol. (2020) 13(1):172. doi: 10.1186/s13045-020-01012-y
238. Hoseini SS, Dobrenkov K, Pankov D, Xu XL, Cheung NK. Bispecific antibody does not induce T-cell death mediated by chimeric antigen receptor against disialoganglioside GD2. Oncoimmunology. (2017) 6:e1320625. doi: 10.1080/2162402X.2017.1320625
239. Santich BH, Park JA, Tran H, Guo HF, Huse M, Cheung NV. Interdomain spacing and spatial configuration drive the potency of IgG-[L]-scFv T cell bispecific antibodies. Sci Trans Med. (2020) 12:eaax1315. doi: 10.1126/scitranslmed.aax1315
240. Reusch U, Harrington KH, Gudgeon CJ, Fucek I, Ellwanger K, Weichel M, et al. Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin Cancer Res. (2016) 22:5829–38. doi: 10.1158/1078-0432.CCR-16-0350
241. Deppisch N, Ruf P, Eissler N, Neff F, Buhmann R, Lindhofer H, et al. Efficacy and tolerability of a GD2-directed trifunctional bispecific antibody in a preclinical model: subcutaneous administration is superior to intravenous delivery. Mol Cancer Ther. (2015) 14:1877–83. doi: 10.1158/1535-7163.MCT-15-0156
242. Ruf P, Schäfer B, Eissler N, Mocikat R, Hess J, Plöscher M, et al. Ganglioside GD2-specific trifunctional surrogate antibody Surek demonstrates therapeutic activity in a mouse melanoma model. J Trans Med. (2012) 10:219. doi: 10.1186/1479-5876-10-219
243. Ivasko SM, Anders K, Grunewald L, Launspach M, Klaus A, Schwiebert S, et al. Combination of GD2-directed bispecific trifunctional antibody therapy with Pd-1 immune checkpoint blockade induces anti-neuroblastoma immunity in a syngeneic mouse model. Front Immunol. (2023) 13:1023206. doi: 10.3389/fimmu.2022.1023206
244. Eissler N, Ruf P, Mysliwietz J, Lindhofer H, Mocikat R. Trifunctional bispecific antibodies induce tumor-specific T cells and elicit a vaccination effect. Cancer Res. (2012) 72:3958–66. doi: 10.1158/0008-5472.CAN-12-0146
245. Zirngibl F, Ivasko SM, Grunewald L, Klaus A, Schwiebert S, Ruf P, et al. GD2-directed bispecific trifunctional antibody outperforms dinutuximab beta in a murine model for aggressive metastasized neuroblastoma. J immunotherapy Cancer. (2021) 9:e002923. doi: 10.1136/jitc-2021-002923
246. Livingston PO, Ragupathi G. Cancer vaccines targeting carbohydrate antigens. Hum Vaccines. (2006) 2:137–43. doi: 10.4161/hv.2941
247. Jaffe J, Wucherer K, Sperry J, Zou Q, Chang Q, Massa MA, et al. Effects of conformational changes in peptide-CRM197 conjugate vaccines. Bioconjugate Chem. (2019) 30:47–53. doi: 10.1021/acs.bioconjchem.8b00661
248. Rosenbaum E, Chugh R, Ryan CW, Agulnik M, Milhem MM, George S, et al. A randomised phase II trial of a trivalent ganglioside vaccine targeting GM2, GD2 and GD3 combined with immunological adjuvant OPT-821 versus OPT-821 alone in metastatic sarcoma patients rendered disease-free by surgery. Eur J Cancer (Oxford Engl 1990). (2022) 176:155–63. doi: 10.1016/j.ejca.2022.09.003
249. Becker R, Eichler MK, Jennemann R, Bertalanffy H. Phase I clinical trial on adjuvant active immunotherapy of human gliomas with GD2-conjugate. Br J Neurosurg. (2002) 16:269–75. doi: 10.1080/02688690220148860
250. Pirahmadi S, Zakeri S, Mehrizi AA, Djadid ND, Raz AA, Sani JJ. Combining Monophosphoryl Lipid A (MPL), CpG Oligodeoxynucleotide (ODN), and QS-21 Adjuvants Induces Strong and Persistent Functional Antibodies and T Cell Responses against Cell-Traversal Protein for Ookinetes and Sporozoites (CelTOS) of Plasmodium falciparum in BALB/c Mice. Infection Immun. (2019) 87:e00911–18. doi: 10.1128/IAI.00911-18
251. Kushner BH, Cheung IY, Modak S, Kramer K, Ragupathi G, Cheung NK. Phase I trial of a bivalent gangliosides vaccine in combination with β-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res. (2014) 20:1375–82. doi: 10.1158/1078-0432.CCR-13-1012
252. Cheung IY, Cheung NV, Modak S, Mauguen A, Feng Y, Basu E, et al. Survival impact of anti-GD2 antibody response in a phase II ganglioside vaccine trial among patients with high-risk neuroblastoma with prior disease progression. J Clin Oncol. (2021) 39:215–26. doi: 10.1200/JCO.20.01892
253. Kieber-Emmons T, Monzavi-Karbassi B, Pashov A, Saha S, Murali R, Kohler H. The promise of the anti-idiotype concept. Front Oncol. (2012) 2:196. doi: 10.3389/fonc.2012.00196
254. Ladjemi MZ. Anti-idiotypic antibodies as cancer vaccines: achievements and future improvements. Front Oncol. (2012) 2:158. doi: 10.3389/fonc.2012.00158
255. Foon KA, Sen G, Hutchins L, Kashala OL, Baral R, Banerjee M, et al. Antibody responses in melanoma patients immunized with an anti-idiotype antibody mimicking disialoganglioside GD2. Clin Cancer Res. (1998) 4:1117–24.
256. Cheung NK, Canete A, Cheung IY, Ye JN, Liu C. Disialoganglioside GD2 anti-idiotypic monoclonal antibodies. Int J Cancer. (1993) 54:499–505. doi: 10.1002/ijc.2910540324
257. Lode HN, Schmidt M, Seidel D, Huebener N, Brackrock D, Bleeke M, et al. Vaccination with anti-idiotype antibody ganglidiomab mediates a GD(2)-specific anti-neuroblastoma immune response. Cancer immunology immunotherapy. (2013) 62:999–1010. doi: 10.1007/s00262-013-1413-y
258. Klingel L, Siebert N, Troschke-Meurer S, Zumpe M, Ehlert K, Huber S, et al. Immune response and outcome of high-risk neuroblastoma patients immunized with anti-idiotypic antibody ganglidiomab: results from compassionate-use treatments. Cancers. (2022) 14:5802. doi: 10.3390/cancers14235802
259. Eger C, Siebert N, Seidel D, Zumpe M, Jüttner M, Brandt S, et al. Generation and characterization of a human/mouse chimeric GD2-mimicking anti-idiotype antibody ganglidiximab for active immunotherapy against neuroblastoma. PloS One. (2016) 11:e0150479. doi: 10.1371/journal.pone.0150479
260. Berry LJ, Moeller M, Darcy PK. Adoptive immunotherapy for cancer: the next generation of gene-engineered immune cells. Tissue Antigens. (2009) 74:277–89. doi: 10.1111/j.1399-0039.2009.01336.x
261. Davenport AJ, Cross RS, Watson KA, Liao Y, Shi W, Prince HM, et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Natl Acad Sci United States America. (2018) 115:E2068–76. doi: 10.1073/pnas.1716266115
262. Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. (2019) 20:1283. doi: 10.3390/ijms20061283
263. Si X, Xiao L, Brown CE, Wang D. Preclinical evaluation of CAR T cell function: in vitro and in vivo models. Int J Mol Sci. (2022) 23:3154. doi: 10.3390/ijms23063154
264. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
265. Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. (2018) 6:36–46. doi: 10.1158/2326-6066.CIR-17-0211
266. Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol Ther. (2016) 24:1135–49. doi: 10.1038/mt.2016.63
267. Künkele A, Johnson AJ, Rolczynski LS, Chang CA, Hoglund V, Kelly-Spratt KS, et al. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol Res. (2015) 3:368–79. doi: 10.1158/2326-6066.CIR-14-0200
268. Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. (2017) 9:1183–97. doi: 10.15252/emmm.201607485
269. Krause A, Guo HF, Latouche JB, Tan C, Cheung NK, Sadelain M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med. (1998) 188:619–26. doi: 10.1084/jem.188.4.619
270. Rossig C, Bollard CM, Nuchtern JG, Merchant DA, Brenner MK. Targeting of G(D2)-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int J Cancer. (2001) 94:228–36. doi: 10.1002/ijc.1457
271. Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. (2002) 99:2009–16. doi: 10.1182/blood.v99.6.2009
272. Savoldo B, Rooney CM, Di Stasi A, Abken H, Hombach A, Foster AE, et al. Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood. (2007) 110:2620–30. doi: 10.1182/blood-2006-11-059139
273. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. (2008) 14:1264–70. doi: 10.1038/nm.1882
274. Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer. (2011) 129:2935–44. doi: 10.1002/ijc.25960
275. Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. (2015) 28:415–28. doi: 10.1016/j.ccell.2015.09.004
276. Drent E, Poels R, Ruiter R, van de Donk NWCJ, Zweegman S, Yuan H, et al. Combined CD28 and 4-1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered T cells. Clin Cancer Res. (2019) 25:4014–25. doi: 10.1158/1078-0432.CCR-18-2559
277. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
278. Li W, Guo L, Rathi P, Marinova E, Gao X, Wu MF, et al. Redirecting T cells to glypican-3 with 4-1BB zeta chimeric antigen receptors results in th1 polarization and potent antitumor activity. Hum Gene Ther. (2017) 28:437–48. doi: 10.1089/hum.2016.025
279. Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, et al. Tonic 4-1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector-dependent. Cell Rep. (2017) 21:17–26. doi: 10.1016/j.celrep.2017.09.015
280. Mallett S, Fossum S, Barclay AN. Characterization of the MRC OX40 antigen of activated CD4 positive T lymphocytes–a molecule related to nerve growth factor receptor. EMBO J. (1990) 9:1063–8. doi: 10.1002/embj.1990.9.issue-4
281. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev. (2009) 229:173–91. doi: 10.1111/j.1600-065X.2009.00766.x
282. Pulè MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther J Am Soc Gene Ther. (2005) 12:933–41. doi: 10.1016/j.ymthe.2005.04.016
283. Quintarelli C, Orlando D, Boffa I, Guercio M, Polito VA, Petretto A, et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology. (2018) 7:e1433518. doi: 10.1080/2162402X.2018.1433518
284. Guedan S, Posey AD, Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. (2018) 3:e96976. doi: 10.1172/jci.insight.96976
285. Ji C, You F, Zhang T, Fan S, Han Z, Xiang S, et al. Novel anti-GD2 CAR-T cells exhibit superior cytotoxicity against neuroblastoma. Eur J Inflammation. (2020) 18. doi: 10.1177/2058739220961193
286. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
287. Omer B, Castillo PA, Tashiro H, Shum T, Huynh MTA, Cardenas M, et al. Chimeric antigen receptor signaling domains differentially regulate proliferation and native T cell receptor function in virus-specific T cells. Front Med. (2018) 5:343. doi: 10.3389/fmed.2018.00343
288. Thomas S, Straathof K, Himoudi N, Anderson J, Pule M. An optimized GD2-targeting retroviral cassette for more potent and safer cellular therapy of neuroblastoma and other cancers. PloS One. (2016) 11:e0152196. doi: 10.1371/journal.pone.0152196
289. Andersch L, Radke J, Klaus A, Schwiebert S, Winkler A, Schumann E, et al. CD171- and GD2-specific CAR-T cells potently target retinoblastoma cells in preclinical in vitro testing. BMC Cancer. (2019) 19:895. doi: 10.1186/s12885-019-6131-1
290. Sujjitjoon J, Sayour E, Tsao ST, Uiprasertkul M, Sanpakit K, Buaboonnam J, et al. GD2-specific chimeric antigen receptor-modified T cells targeting retinoblastoma - assessing tumor and T cell interaction. Trans Oncol. (2021) 14:100971. doi: 10.1016/j.tranon.2020.100971
291. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. (2015) 3:125–35. doi: 10.1158/2326-6066.CIR-14-0127
292. Gargett T, Ebert LM, Truong NTH, Kollis PM, Sedivakova K, Yu W, et al. GD2-targeting CAR-T cells enhanced by transgenic IL-15 expression are an effective and clinically feasible therapy for glioblastoma. J immunotherapy Cancer. (2022) 10:e005187. doi: 10.1136/jitc-2022-005187
293. Chen J, Qiu S, Li W, Wang K, Zhang Y, Yang H, et al. Tuning charge density of chimeric antigen receptor optimizes tonic signaling and CAR-T cell fitness. Cell Res. (2023) 33:341–54. doi: 10.1038/s41422-023-00789-0
294. Trier AM, Mack MR, Kim BS. The neuroimmune axis in skin sensation, inflammation, and immunity. J Immunol (Baltimore Md. 1950). (2019) 202:2829–35. doi: 10.4049/jimmunol.1801473
295. Zhang Q, Ding J, Sun S, Liu H, Lu M, Wei X, et al. Akt inhibition at the initial stage of CAR-T preparation enhances the CAR-positive expression rate, memory phenotype and in vivo efficacy. Am J Cancer Res. (2019) 9:2379–96.
296. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Sci (New York N.Y.). (2021) 372:eaba1786. doi: 10.1126/science.aba1786
297. Mueller KP, Piscopo NJ, Forsberg MH, Saraspe LA, Das A, Russell B, et al. Production and characterization of virus-free, CRISPR-CAR T cells capable of inducing solid tumor regression. J immunotherapy Cancer. (2022) 10:e004446. doi: 10.1136/jitc-2021-004446
298. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
299. Chmielewski M, Abken H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv Cell Gene Ther. (2020) 3:e84. doi: 10.1002/acg2.84
300. Glienke W, Dragon AC, Zimmermann K, Martyniszyn-Eiben A, Mertens M, Abken H, et al. GMP-compliant manufacturing of TRUCKs: CAR T cells targeting GD2 and releasing inducible IL-18. Front Immunol. (2022) 13:839783. doi: 10.3389/fimmu.2022.839783
301. Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, et al. Design and characterization of an "All-in-one" Lentiviral vector system combining constitutive anti-GD2 CAR expression and inducible cytokines. Cancers. (2020) 12:375. doi: 10.3390/cancers12020375
302. Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin Cancer Res. (2019) 25:2915–24. doi: 10.1158/1078-0432.CCR-18-1811
303. Li G, Zhang Q, Han Z, Zhu Y, Shen H, Liu Z, et al. IL-7 and CCR2b co-expression-mediated enhanced CAR-T survival and infiltration in solid tumors. Front Oncol. (2021) 11:734593. doi: 10.3389/fonc.2021.734593
304. Evgin L, Kottke T, Tonne J, Thompson J, Huff AL, van Vloten J, et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci Trans Med. (2022) 14:eabn2231. doi: 10.1126/scitranslmed.abn2231
305. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. (2014) 74:5195–205. doi: 10.1158/0008-5472.CAN-14-0697
306. Tanaka M, Tashiro H, Omer B, Lapteva N, Ando J, Ngo M, et al. Vaccination targeting native receptors to enhance the function and proliferation of chimeric antigen receptor (CAR)-modified T cells. Clin Cancer Res. (2017) 23:3499–509. doi: 10.1158/1078-0432.CCR-16-2138
307. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. (2021) 12:81. doi: 10.1186/s13287-020-02128-1
308. Zhang G, Zhao Y, Liu Z, Liu W, Wu H, Wang X, et al. GD2 CAR-T cells in combination with Nivolumab exhibit enhanced antitumor efficacy. Trans Oncol. (2023) 32:101663. doi: 10.1016/j.tranon.2023.101663
309. Bocca P, Di Carlo E, Caruana I, Emionite L, Cilli M, De Angelis B, et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology. (2017) 7:e1378843. doi: 10.1080/2162402X.2017.1378843
310. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. (2017) 25:2214–24. doi: 10.1016/j.ymthe.2017.05.012
311. Daskivich TJ, Belldegrun A. Words of wisdom. Re: Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. Eur Urol. (2015) 67:816–7. doi: 10.1016/j.eururo.2014.12.052
312. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res. (2016) 4:869–80. doi: 10.1158/2326-6066.CIR-15-0230
313. Stroncek DF, Ren J, Lee DW, Tran M, Frodigh SE, Sabatino M, et al. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy. (2016) 18:893–901. doi: 10.1016/j.jcyt.2016.04.003
314. Tumino N, Weber G, Besi F, Del Bufalo F, Bertaina V, Paci P, et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J Hematol Oncol. (2021) 14:191. doi: 10.1186/s13045-021-01193-0
315. de Billy E, Pellegrino M, Orlando D, Pericoli G, Ferretti R, Businaro P, et al. Dual IGF1R/IR inhibitors in combination with GD2-CAR T-cells display a potent anti-tumor activity in diffuse midline glioma H3K27M-mutant. Neuro-oncology. (2022) 24:1150–63. doi: 10.1093/neuonc/noab300
316. Miyagawa I, Nakayamada S, Nakano K, Yamagata K, Sakata K, Yamaoka K, et al. Induction of regulatory T cells and its regulation with insulin-like growth factor/insulin-like growth factor binding protein-4 by human mesenchymal stem cells. J Immunol (Baltimore Md. 1950). (2017) 199:1616–25. doi: 10.4049/jimmunol.1600230
317. Spadaro O, Camell CD, Bosurgi L, Nguyen KY, Youm YH, Rothlin CV, et al. IGF1 shapes macrophage activation in response to immunometabolic challenge. Cell Rep. (2017) 19:225–34. doi: 10.1016/j.celrep.2017.03.046
318. Philippova JG, Kuznetsova MS, Shevchenko JA, Tereshchenko VP, Fisher MS, Kurilin VV, et al. Phenotype and effector functions of GD2-specific CAR-T lyphocytes in vitro. Immunologiya. (2022) 43:525–35. doi: 10.33029/0206-4952-2022-43-5-525-535
319. Suarez ER, Chang d, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
320. Gouda MA, Subbiah V. Expanding the benefit: dabrafenib/trametinib as tissue-agnostic therapy for BRAF V600E-positive adult and pediatric solid tumors. Am Soc Clin Oncol Educ book. Am Soc Clin Oncol Annu Meeting. (2023) 43:e404770. doi: 10.1200/EDBK_404770
321. Gargett T, Fraser CK, Dotti G, Yvon ES, Brown MP. BRAF and MEK inhibition variably affect GD2-specific chimeric antigen receptor (CAR) T-cell function in vitro. J immunotherapy (Hagerstown Md. 1997). (2015) 38:12–23. doi: 10.1097/CJI.0000000000000061
322. Dörrie J, Babalija L, Hoyer S, Gerer KF, Schuler G, Heinzerling L, et al. BRAF and MEK inhibitors influence the function of reprogrammed T cells: consequences for adoptive T-cell therapy. Int J Mol Sci. (2018) 19:289. doi: 10.3390/ijms19010289
323. Tomida A, Yagyu S, Nakamura K, Kubo H, Yamashima K, Nakazawa Y, et al. Inhibition of MEK pathway enhances the antitumor efficacy of chimeric antigen receptor T cells against neuroblastoma. Cancer Sci. (2021) 112:4026–36. doi: 10.1111/cas.15074
324. Chulanetra M, Morchang A, Sayour E, Eldjerou L, Milner R, Lagmay J, et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am J Cancer Res. (2020) 10:674–87.
325. Yu L, Huang L, Lin D, Lai X, Wu L, Liao X, et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J Cancer Res Clin Oncol. (2022) 148:2643–52. doi: 10.1007/s00432-021-03839-5
326. Hegde MG, Corder A, Byrd TT, Fousek K, Heslop HE, Gottschalk S, et al. Abstract LB-199: A rationally designed bispecific chimeric antigen receptor molecule that simultaneously redirects T cells to target HER2 and GD2 in osteosarcoma. Cancer Res. (2014) 74:LB–199-LB-199. doi: 10.1158/1538-7445.AM2014-LB-199
327. Reppel L, Tsahouridis O, Akulian J, Davis IJ, Lee H, Fucà G, et al. Targeting disialoganglioside GD2 with chimeric antigen receptor-redirected T cells in lung cancer. J immunotherapy Cancer. (2022) 10:e003897. doi: 10.1136/jitc-2021-003897
328. Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. (2020) 17:418–34. doi: 10.1038/s41571-020-0347-5
329. Allen C, Zeidan AM, Bewersdorf JP. BiTEs, DARTS, biKEs and triKEs-are antibody based therapies changing the future treatment of AML? Life (Basel Switzerland). (2021) 11:465. doi: 10.3390/life11060465
330. Prapa M, Caldrer S, Spano C, Bestagno M, Golinelli G, Grisendi G, et al. A novel anti-GD2/4-1BB chimeric antigen receptor triggers neuroblastoma cell killing. Oncotarget. (2015) 6:24884–94. doi: 10.18632/oncotarget.4670
331. Dufait I, Liechtenstein T, Lanna A, Bricogne C, Laranga R, Padella A, et al. Retroviral and lentiviral vectors for the induction of immunological tolerance. Scientifica. (2012) 2012:694137. doi: 10.6064/2012/694137
332. Kaiser AD, Assenmacher M, Schröder B, Meyer M, Orentas R, Bethke U, et al. Towards a commercial process for the manufacture of genetically modified T cells for therapy. Cancer Gene Ther. (2015) 22:72–8. doi: 10.1038/cgt.2014.78
333. Stroncek DF, Lee DW, Ren J, Sabatino M, Highfill S, Khuu H, et al. Elutriated lymphocytes for manufacturing chimeric antigen receptor T cells. J Trans Med. (2017) 15:59. doi: 10.1186/s12967-017-1160-5
334. Gargett T, Brown MP. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy. (2015) 17:487–95. doi: 10.1016/j.jcyt.2014.12.002
335. Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a "safety switch" to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. (2014) 5:235. doi: 10.3389/fphar.2014.00235
336. Liu Z, Zhou J, Yang X, Liu Y, Zou C, Lv W, et al. Safety and antitumor activity of GD2-Specific 4SCAR-T cells in patients with glioblastoma. Mol Cancer. (2023) 22:3. doi: 10.1186/s12943-022-01711-9
337. Yang L, Ma X, Liu Y-C, Zhao W, Yu L, Qin M, et al. Chimeric antigen receptor 4SCAR-GD2-modified T cells targeting high-risk and recurrent neuroblastoma: A phase II multi-center trial in China. Blood. (2017) 130:3335. doi: 10.1182/blood.V130.Suppl_1.3335.3335
338. Mitwasi N, Feldmann A, Bergmann R, Berndt N, Arndt C, Koristka S, et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget. (2017) 8:108584–603. doi: 10.18632/oncotarget.21017
339. Wong HY, Schwarz H. CD137 / CD137 ligand signalling regulates the immune balance: A potential target for novel immunotherapy of autoimmune diseases. J Autoimmun. (2020) 112:102499. doi: 10.1016/j.jaut.2020.102499
340. Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. (2022) 603:934–41. doi: 10.1038/s41586-022-04489-4
341. Wang K, Chen Y, Ahn S, Zheng M, Landoni E, Dotti G, et al. GD2-specific CAR T cells encapsulated in an injectable hydrogel control retinoblastoma and preserve vision. Nat Cancer. (2020) 1:990–7. doi: 10.1038/s43018-020-00119-y
342. Zhang C, Liu Y. Targeting NK cell checkpoint receptors or molecules for cancer immunotherapy. Front Immunol. (2020) 11:1295. doi: 10.3389/fimmu.2020.01295
343. Habets DHJ, Schlütter A, van Kuijk SMJ, Spaanderman MEA, Al-Nasiry S, Wieten L. Natural killer cell profiles in recurrent pregnancy loss: Increased expression and positive associations with TACTILE and LILRB1. Am J Reprod Immunol (New York N.Y. 1989). (2022) 88:e13612. doi: 10.1111/aji.13612
344. Leung W. Use of NK cell activity in cure by transplant. Br J haematology. (2011) 155:14–29. doi: 10.1111/j.1365-2141.2011.08823.x
345. Ruggeri L, Aversa F, Martelli MF, Velardi A. Allogeneic hematopoietic transplantation and natural killer cell recognition of missing self. Immunol Rev. (2006) 214:202–18. doi: 10.1111/j.1600-065X.2006.00455.x
346. Liu Y, Wu HW, Sheard MA, Sposto R, Somanchi SS, Cooper LJ, et al. Growth and activation of natural killer cells ex vivo from children with neuroblastoma for adoptive cell therapy. Clin Cancer Res. (2013) 19:2132–43. doi: 10.1158/1078-0432.CCR-12-1243
347. Barry WE, Jackson JR, Asuelime GE, Wu HW, Sun J, Wan Z, et al. Activated natural killer cells in combination with anti-GD2 antibody dinutuximab improve survival of mice after surgical resection of primary neuroblastoma. Clin Cancer Res. (2019) 25:325–33. doi: 10.1158/1078-0432.CCR-18-1317
348. Bates PD, Rakhmilevich AL, Cho MM, Bouchlaka MN, Rao SL, Hales JM, et al. Combining immunocytokine and ex vivo activated NK cells as a platform for enhancing graft-versus-tumor effects against GD2+ Murine neuroblastoma. Front Immunol. (2021) 12:668307. doi: 10.3389/fimmu.2021.668307
349. Castriconi R, Dondero A, Cilli M, Ognio E, Pezzolo A, De Giovanni B, et al. Human NK cell infusions prolong survival of metastatic human neuroblastoma-bearing NOD/scid mice. Cancer immunology immunotherapy. (2007) 56:1733–42. doi: 10.1007/s00262-007-0317-0
350. Zhang C, Xiong X, Li Y, Huang K, Liu L, Peng X, et al. Cytokine-induced killer cells/natural killer cells combined with anti-GD2 monoclonal antibody increase cell death rate in neuroblastoma SK-N-SH cells. Oncol Lett. (2019) 18:6525–35. doi: 10.3892/ol.2019.11020
351. Talleur AC, Triplett BM, Federico S, Mamcarz E, Janssen W, Wu J, et al. Consolidation therapy for newly diagnosed pediatric patients with high-risk neuroblastoma using busulfan/melphalan, autologous hematopoietic cell transplantation, anti-GD2 antibody, granulocyte-macrophage colony-stimulating factor, interleukin-2, and haploidentical natural killer cells. Biol Blood marrow Transplant. (2017) 23:1910–7. doi: 10.1016/j.bbmt.2017.07.011
352. Marofi F, Al-Awad AS, Sulaiman Rahman H, Markov A, Abdelbasset WK, Ivanovna Enina Y, et al. CAR-NK cell: A new paradigm in tumor immunotherapy. Front Oncol. (2021) 11:673276. doi: 10.3389/fonc.2021.673276
353. Zhang J, Zheng H, Diao Y. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. Int J Mol Sci. (2019) 20:317. doi: 10.3390/ijms20020317
354. Esser R, Müller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. (2012) 16:569–81. doi: 10.1111/j.1582-4934.2011.01343.x
355. Rudek LS, Zimmermann K, Galla M, Meyer J, Kuehle J, Stamopoulou A, et al. Generation of an NFκB-driven alpharetroviral "All-in-one" Vector construct as a potent tool for CAR NK cell therapy. Front Immunol. (2021) 12:751138. doi: 10.3389/fimmu.2021.751138
356. Zuo P, Li Y, He C, Wang T, Zheng X, Liu H, et al. Anti-tumor efficacy of anti-GD2 CAR NK-92 cells in diffuse intrinsic pontine gliomas. Front Immunol. (2023) 14:1145706. doi: 10.3389/fimmu.2023.1145706
357. Mitwasi N, Feldmann A, Arndt C, Koristka S, Berndt N, Jureczek J, et al. "UniCAR"-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci Rep. (2020) 10 2141. doi: 10.1038/s41598-020-59082-4
358. Mak TW, Saunders ME, Jett BD eds. Chapter 11 - NK, γδ T and NKT cells. In: Primer to the Immune Response, 2nd ed. Elsevier Inc. 2014, 247–68. doi: 10.1016/B978-0-12-385245-8.00011-X
359. Delfanti G, Dellabona P, Casorati G, Fedeli M. Adoptive immunotherapy with engineered iNKT cells to target cancer cells and the suppressive microenvironment. Front Med. (2022) 9:897750. doi: 10.3389/fmed.2022.897750
360. Li Y, Li G, Zhang J, Wu X, Chen X. The dual roles of human γδ T cells: anti-tumor or tumor-promoting. Front Immunol. (2021) 11:619954. doi: 10.3389/fimmu.2020.619954
361. Metelitsa LS. Anti-tumor potential of type-I NKT cells against CD1d-positive and CD1d-negative tumors in humans. Clin Immunol (Orlando Fla.). (2011) 140:119–29. doi: 10.1016/j.clim.2010.10.005
362. Simonetta F, Lohmeyer JK, Hirai T, Maas-Bauer K, Alvarez M, Wenokur AS, et al. Allogeneic CAR invariant natural killer T cells exert potent antitumor effects through host CD8 T-cell cross-priming. Clin Cancer Res. (2021) 27:6054–64. doi: 10.1158/1078-0432.CCR-21-1329
363. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. (2009) 119:1524–36. doi: 10.1172/JCI37869
364. Ko HJ, Lee JM, Kim YJ, Kim YS, Lee KA, Kang CY. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: an alternative cell-based antitumor vaccine. J Immunol (Baltimore Md. 1950). (2009) 182:1818–28. doi: 10.4049/jimmunol.0802430
365. Khan MW, Curbishley SM, Chen HC, Thomas AD, Pircher H, Mavilio D, et al. Expanded human blood-derived γδT cells display potent antigen-presentation functions. Front Immunol. (2014) 5:344. doi: 10.3389/fimmu.2014.00344
366. Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther. (2018) 26:354–65. doi: 10.1016/j.ymthe.2017.12.001
367. Rozenbaum M, Meir A, Aharony Y, Itzhaki O, Schachter J, Bank I, et al. Gamma-delta CAR-T cells show CAR-directed and independent activity against leukemia. Front Immunol. (2020) 11:1347. doi: 10.3389/fimmu.2020.01347
368. Fisher J, Sharma R, Don DW, Barisa M, Hurtado MO, Abramowski P, et al. Engineering γδT cells limits tonic signaling associated with chimeric antigen receptors. Sci Signaling. (2019) 12:eaax1872. doi: 10.1126/scisignal.aax1872
369. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood. (2014) 124:2824–33. doi: 10.1182/blood-2013-11-541235
370. Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. NKT cells coexpressing a GD2-specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin Cancer Res. (2019) 25:7126–38. doi: 10.1158/1078-0432.CCR-19-0421
371. Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med. (2020) 26:1686–90. doi: 10.1038/s41591-020-1074-2
372. Theoharis S. Current state of the art of allogeneic CAR approaches - pile 'Em high and sell 'Em cheap. J Pharm Sci. (2021) 110:1909–14. doi: 10.1016/j.xphs.2021.02.006
373. Chen Y, Yu Z, Tan X, Jiang H, Xu Z, Fang Y, et al. CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2021) 139:111605. doi: 10.1016/j.biopha.2021.,27111605
Keywords: disialoganglioside GD2, cancer immunotherapy, monoclonal antibody, cell therapy, CAR cells, vaccine, neuroblastoma, clinical trials
Citation: Philippova J, Shevchenko J and Sennikov S (2024) GD2-targeting therapy: a comparative analysis of approaches and promising directions. Front. Immunol. 15:1371345. doi: 10.3389/fimmu.2024.1371345
Received: 16 January 2024; Accepted: 26 February 2024;
Published: 15 March 2024.
Edited by:
Zohreh Amoozgar, Harvard Medical School, United StatesReviewed by:
Amy Erbe, University of Wisconsin-Madison, United StatesAhad Khalilnezhad, Harvard Medical School, United States
Copyright © 2024 Philippova, Shevchenko and Sennikov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergey Sennikov, c2Vubmlrb3ZzdkBnbWFpbC5jb20=