
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 14 March 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1368586
This article is part of the Research TopicCell Death in Cancer ImmunologyView all 15 articles
 Elisha R. Verhaar1,2
Elisha R. Verhaar1,2 Anouk Knoflook1
Anouk Knoflook1 Novalia Pishesha3,4
Novalia Pishesha3,4 Xin Liu1
Xin Liu1 Willemijn J. C. van Keizerswaard1
Willemijn J. C. van Keizerswaard1 Kai W. Wucherpfennig5
Kai W. Wucherpfennig5 Hidde L. Ploegh1,2*
Hidde L. Ploegh1,2*MICA and MICB are Class I MHC-related glycoproteins that are upregulated on the surface of cells in response to stress, for instance due to infection or malignant transformation. MICA/B are ligands for NKG2D, an activating receptor on NK cells, CD8+ T cells, and γδ T cells. Upon engagement of MICA/B with NKG2D, these cytotoxic cells eradicate MICA/B-positive targets. MICA is frequently overexpressed on the surface of cancer cells of epithelial and hematopoietic origin. Here, we created nanobodies that recognize MICA. Nanobodies, or VHHs, are the recombinantly expressed variable regions of camelid heavy chain-only immunoglobulins. They retain the capacity of antigen recognition but are characterized by their stability and ease of production. The nanobodies described here detect surface-disposed MICA on cancer cells in vitro by flow cytometry and can be used therapeutically as nanobody-drug conjugates when fused to the Maytansine derivative DM1. The nanobody-DM1 conjugate selectively kills MICA positive tumor cells in vitro.
The Class I MHC-like molecules MICA and MICB are stress-induced surface glycoproteins, absent from healthy cells but upregulated on virus-infected or malignantly transformed human cells (1). MICA/B are ligands for NKG2D, an activating receptor on NK cells, CD8+ T cells, and γδ T cells (2). Upon engagement of NKG2D, these cytotoxic cells can eradicate MICA-positive targets, assisted by secretion of cytokines (3–5). Elevated levels of MICA/B occur in hematopoietic malignancies, as well as in epithelial solid tumors such as colorectal cancer (6), ovarian cancer (7), cervical cancer (8), breast cancer (9), pancreatic cancer (10), melanoma (11) and cholangiocarcinoma (12). MICA/B are thus considered possible targets for immunotherapy.
Nanobodies, a registered trademark, are also referred to as VHHs. They are the smallest immunoglobulin fragments that retain the capacity of antigen binding. They are the recombinantly expressed variable regions of camelid heavy chain-only immunoglobulins (13). Nanobodies have a short circulatory half-life, are poorly immunogenic, and show excellent tissue penetration compared to conventional full-sized immunoglobulins (14, 15). Many nanobodies do not require disulfide bonds for their stability, nor do they depend on glycosylation for expression. They are therefore easily and affordably produced in prokaryotic cells (16–18). Nanobodies have proven valuable as the point of departure for the construction of PET imaging agents (19–24), nanobody-drug conjugates (25–27), and chimeric antigen receptors in cell-based therapies (28–38).
Because MICA is expressed on stressed and cancerous cells, the ability to detect such aberrations in vivo would be an important diagnostic tool to detect premalignant and malignant lesions. Here, we report the generation of nanobodies that recognize MICA, and apply these nanobodies to detect surface-bound MICA in vitro by flow cytometry. Fused to the microtubule inhibitor Maytansine (DM1), these nanobodies can be used therapeutically as nanobody-drug conjugates.
We immunized an alpaca with 250 ug of the purified extracellular portion of MICA*009 (obtained by baculovirus expression in the lab of K.W. Wucherpfennig (39)) comprising the α1, α2, and α3 domains in alum adjuvant, followed by 3 booster injections at 2-week intervals. Immunizations were carried out by Camelid Immunogenics. The immune response of the animal was checked by immunoblot (Supplementary Figure 1). Briefly, 1 μg of antigen was resolved by SDS PAGE and transferred to a PVDF membrane. The membrane was incubated with at 1:5000 dilution of alpaca serum collected 2 weeks after the last boost. HRP-linked goat-anti-llama (0.05 μg/mL; Bethyl, NC9656984) was used as the secondary antibody. Membranes were developed with ECL Western Lightning Plus. Mononuclear cells from peripheral blood of the immunized alpaca were isolated by Ficoll gradient separation. The VHH library was generated according to an established protocol (Maas et al., 2007). Briefly, RNA was extracted (RNeasy RNA purification kit, Qiagen) and cDNA was prepared (Superscript III first-strand synthesis system, Invitrogen). The DNA sequences from conventional and heavy-chain only Ig genes are not distinguishable based on the use of specific primers, but two distinct hinge regions are generated between the VHH domain and the CH2 region. We amplified the VHH repertoire from the alpaca using VHH-specific primers that target these hinge sequences (Supplementary Table 1). We pooled the VHH PCR products and ligated them into a phagemid vector in-frame with the pIII gene of the M13 phagemid to construct a phagemid library display. We performed two rounds of panning against MICA*009 immobilized on an ELISA plate, following previously described protocols (40).
DNA from positive clones was sequenced and 9 clones were selected for further characterization. The relevant VHH sequences were subcloned into a pHEN6 expression vector with C-terminal modifications, so that each nanobody sequence included an LPETG motif recognized by sortase A, followed by a (His)6-tag to facilitate recovery and purification. Briefly, VHH sequences were amplified from the phagemid vector by PCR (primers in Supplementary Table 1) and the pHEN6 vector was linearized using the NcoI and BstEII restriction enzymes. Gibson assembly was performed following manufacturer’s directions (Gibson Assembly® Master Mix, NEB). Positive VHH clones were expressed in WK6 E.Coli in terrific broth and periplasmic protein expression was activated by induction with isopropyl β-D-thiogalactopyranoside (1 mM) at an OD600 of 0.6. VHHs were harvested from the periplasm by osmotic shock. The C-terminal (His)6-tag allows purification of the recombinant proteins using Ni-NTA agarose beads (Qiagen), followed by FPLC purification on an S75 column by FPLC (ÄKTA, Cytiva Life Sciences). Sortase reactions were performed by incubating each nanobody with a 10-fold molar excess of GGG-nucleophile in the presence of 25 µM Sortase 7M (41) overnight at 4°C. Because the LPETG sequence is cleaved during transpeptidation, the (His)6-tag immediately C-terminal of the LPETG motif is lost. This allows enrichment of the desired modified product by depletion of His-tagged sortase and unreacted nanobody on a NiNTA matrix, while the unbound fraction contains the modified nanobody.
An ELISA was performed to determine the concentration at which each biotinylated nanobody showed ~80% binding to recombinant MICA*009 (5 mg/mL) immobilized on an ELISA plate. Biotinylated nanobody at a concentration that yielded 80% of the maximum attainable binding value was then mixed with a 500-fold excess of unlabeled competitor nanobody and allowed to compete for binding to 5 μg/mL MICA*009 coated on an ELISA plate. Plates were incubated with streptavidin-HRP (0.00025 μg/mL) for 45-60 minutes at room temperature. After addition of TMB substrate, absorbance was read out at 450 nm on a Spectramax iD5 plate reader (Molecular Devices). If the unlabeled nanobody binds to an epitope distinct from that recognized by the biotinylated nanobody, no diminution of the signal at 450 nm is expected. Nanobodies that recognize the same epitope as that seen by the biotinylated nanobody will show a reduction in the signal at 450nm.
We estimated the binding affinity of VHH-A1 and VHH-H3 by performing an ELISA as previously described (42). Briefly, we incubated plates coated with 100μL PBS containing 2.5 μg/mL recombinant MICA*009 or GFP as negative control with biotinylated VHH-A1 and VHH-H3 in various concentrations (10-fold serial dilutions; 0.000001 nM – 1000 nM). Streptavidin-HRP at 0.00025 μg/mL was used as detection agent. After addition of TMB substrate, absorbance was read at 450 nm on a Spectramax iD5 plate reader (Molecular Devices). Binding affinity was estimated by calculating the IC50 obtained from three experimental replicates with each sample added in duplicates. Recombinant MICA*009 was produced by transfection of EXPI-293 cells with pcDNA3.1(+) vector encoding for extracellular MICA*009 containing a C-terminal LPETG sortase motif followed by a His (6)-tag to facilitate recovery and purification on a NiNTA matrix (Supplementary Figure 2). EXPI-293 cells were transfected using the ExpiFectamine™ 293 Transfection Kit, according to manufacturer’s directions (Gibco).
B16F10 and EL-4 cells and their MICA+ transfectants were a gift from the lab of Kai Wucherpfennig. B16F10 cells were cultured in complete DMEM (DMEM with 4.5 g/L glucose, substituted with 10% Fetal Bovine Serum (FBS) and 100 U/mL penicillin/streptomycin). EL-4 cells were cultured in complete RPMI 1640 (RPMI 1640, substituted with 10% FBS and 100 U/mL penicillin/streptomycin). Cells were maintained at optimal densities in a humidified 5% CO2 incubator at 37°C.
EL-4 WT and MICA+ cells, or B16F10 WT and MICA+ cells, were stained with biotinylated VHH-A1 and VHH-H3 for 30 minutes on ice, washed, and incubated with a cocktail of Streptavidin-conjugated PE at 0.0025 μg/mL (Invitrogen) and 2 μg/mL propidium iodide (Life technologies) for EL-4 or LIVE/DEAD™ Fixable Violet Dead Cell Stain Kit (Invitrogen) for B16F10, both according to manufacturer’s directions for 30 minutes on ice. Cells were analyzed on an LSR Fortessa flow cytometer (BD Biosciences). Gating strategies were based on cell lines stained with the appropriate controls, where single cells and live cells were appropriately selected.
VHH-DM1 was produced in a sortase-mediated transpeptidation reaction. Briefly, 500-1000 μg of VHH containing a C-terminal LPETG-motif was mixed with a 10-fold molar excess of GGG-DM1 and incubated with 25 μM Sortase for 16 hours at 4°C. GGG-DM1 was produced in-house by modifying a GGG-peptide linker to contain a maleimide group and allowing it to react with the thiol group on DM1 (Broadpharm) (Supplementary Figure 3A). Unreacted VHH and Sortase, both containing a (His)6-tag, were depleted by incubation with NiNTA agarose (Qiagen or Prometheus). Excess free GGG-DM1 was removed by desalting on a PD-10 desalting column (Cytiva). We plated 4000 cells/well in a 96-well plate and incubated cells with serial 3-fold dilutions of VHH-drug adduct or free DM4 (Broadpharm), a structural analog of DM1 (Supplementary Figure 3B) at 37°C in a humidified 5% CO2 atmosphere. After 72 hours, we measured cell viability by CellTiter GloTM assay according to the manufacturer’s directions (Promega). For co-culture experiments, MICA expression was determined after a 72-hour incubation. Each treatment was performed in duplicate. For flow cytometry, the duplicate wells of each condition were combined, and the cell mixture was stained with 0.0006 μg/mL biotinylated anti-human MICA/B antibody (Clone 6D4, Biolegend) for 30 minutes on ice. Cells were washed and incubated with Streptavidin-conjugated PE at 0.0025 μg/mL (Invitrogen) and LIVE/DEAD™ Fixable Violet Dead Cell Stain Kit according to manufacturer’s directions (Invitrogen) for 30 minutes on ice. Cells were washed and viability and MICA positivity were determined by flow cytometry on an LSR Fortessa flow cytometer (BD Biosciences).
All statistical analysis was performed with GraphPad Prism 8. Flow cytometry data was analyzed with FlowJo (v10.8.1 and v10.9.0).
We immunized an alpaca with purified recombinant MICA*009 in alum adjuvant, followed by 3 booster injections at 2-week intervals. We checked the immune response of the animal by immunoblot using serum samples collected prior to each boost. Having recorded a positive response after the 3rd boost, construction of a phage display library, followed by screening for MICA-reactive hits, yielded positive clones. DNA from positive clones was sequenced and 9 clones were selected for further characterization. Because nanobodies interact with their antigen mainly via their CDR3 region, and to a lesser extent via the germline-encoded CDR1 and CDR2 (43), we chose clones that were unique in their CDR3. A detailed comparison of the nanobody clones based on sequence similarity in the framework and CDR regions is described in the caption of Figure 1.
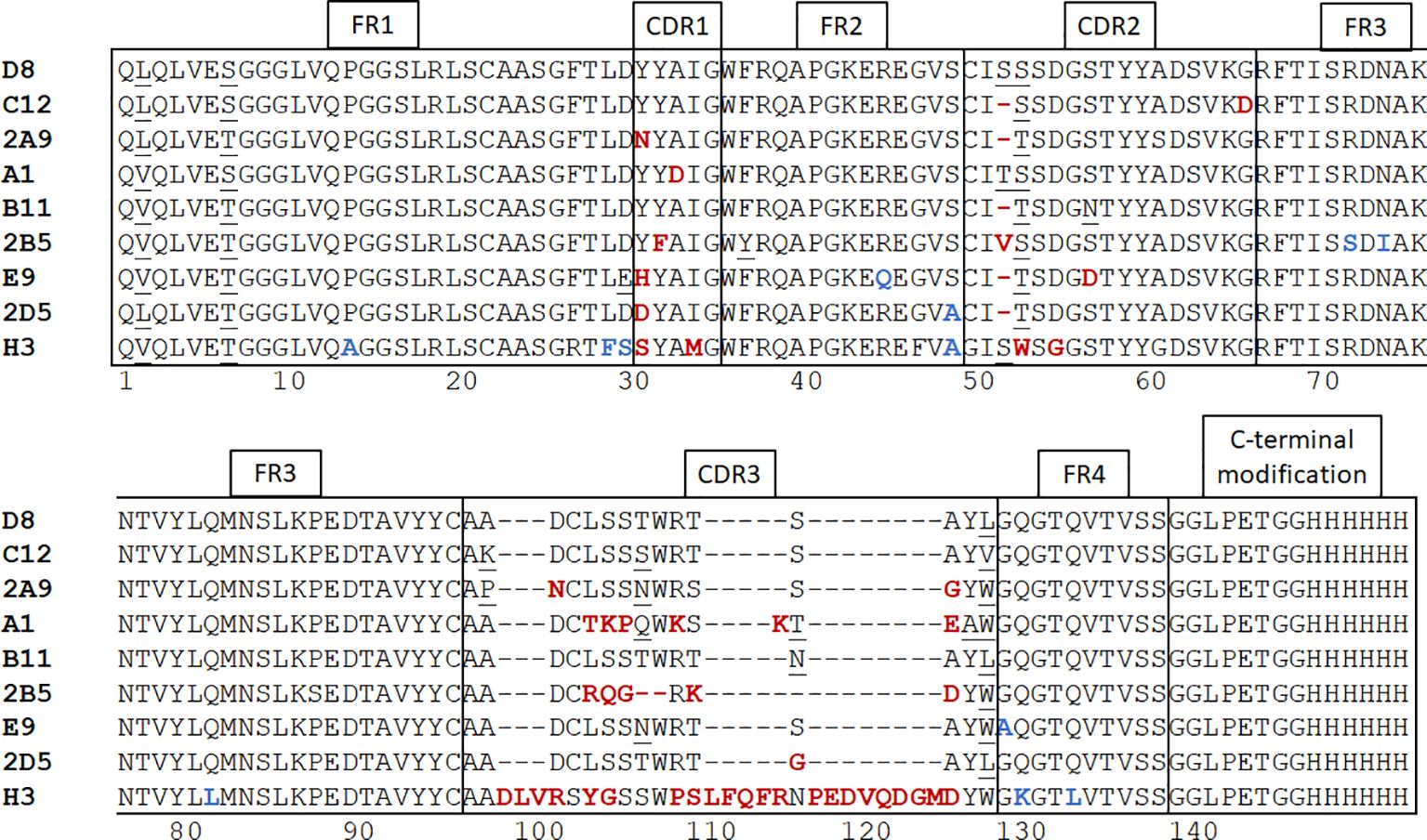
Figure 1 Alpaca immunization and nanobody panning. After construction of a phage display library and screening for positive clones with plate-based panning, nanobody sequences were determined and 9 unique clones were selected. Neutral amino acid substitutions attributable to somatic hypermutations are underscored. Unique substitutions in framework regions are highlighted in blue and in CDR’s are highlighted in red. Nanobodies harboring such mutations are more likely derived from different germline V regions rather than somatic hypermutation. The framework regions of nanobodies D8 and C12 are identical. The alpaca IGHHV-3-3*01 gene is the possible germline version of these nanobodies (44). The single difference of VHH A1 with D8 and C12 in its framework regions is an L2V substitution. A1 may thus be derived from the same germline V gene as D8 and C12 by a single (somatic) point mutation. The framework regions of nanobodies 2A9 and 2D5 are mostly identical to each other, with a single S49A substitution between them. Nanobody E9 has both a D29E and a R45Q substitution, indicating that E9 may be derived from a different V gene. In comparison with the other MICA-specific nanobodies, H3 has the largest number of differences in its framework regions and is clearly derived from a different germline V gene, likely the alpaca IGHHV3-1*01 (44). The CDR1 and CDR2 regions are mostly conserved. The most obvious deviation is a deletion at position 53 in VHH C12, B11, 2A9, 2D5, and E9. The MICA-specific nanobodies have CDR3 regions of 13-16 amino acids, but H3 has a 31-residue CDR3. Except for VHH H3, A1 and 2B5, the remaining CDR3 regions are enriched for the sequence “AxDCLSSxWRx”. The VHH sequences were subcloned into the pHen6 expression vector and modified at the C-terminus to contain an LPETG motif and (His)6 tag.
Relevant VHH sequences were subcloned into a pHEN6 expression vector to encode a VHH product with C-terminal modifications, so that each VHH sequence included an LPETG motif at its C-terminus, recognized by sortase A, and a (His)6-tag to facilitate recovery and purification (Figure 1). This arrangement enables the installation of fluorophores, biotin, and other substituents by a site-specific and efficient sortase-catalyzed transpeptidation reaction (41). Because the LPETG sequence is cleaved during transpeptidation, the (His)6-tag immediately C-terminal of the LPETG motif is lost. This allows enrichment of the desired modified product by depletion of His-tagged sortase and unreacted nanobody on a NiNTA matrix, while the unbound fraction contains the modified nanobody.
To determine whether the isolated MICA-specific nanobodies recognized similar or distinct epitopes on MICA, we performed cross-competition experiments by ELISA. Competition of unlabeled nanobodies with a biotinylated nanobody for binding to MICA showed that this set of nanobodies recognizes two distinct epitopes, one defined by the H3 nanobody and the second by all the other nanobodies. None of the nanobodies compete for binding with the 7C6 monoclonal antibody, an agent that inhibits shedding of MICA (45) (Figure 2A). Typically, not all nanobodies are suitable for use in immunoblotting experiments, but the biotinylated versions of A1 and H3 yielded a strong and specific signal in immunoblots on recombinant MICA (Figure 2B). The binding affinities of VHH-A1 and VHH-H3 are both in the nanomolar range, at ~0.2 and ~0.4 nM for A1 and H3 respectively (Figure 2C), as estimated by ELISA assay. By examining the binding of the A1 and H3 nanobodies to a subset of MICA/B allelic products, available in purified form, we conclude that the A1 and H3 nanobodies recognize the MICA*008 and MICA*009 alleles (Figure 2D) which, combined, cover 51.1% of the Caucasian population (46). To verify that A1 and H3 also recognize surface-disposed MICA, we used B16F10 transfectants that express MICA*009, and EL-4 transfectants that express MICA*008, with B16F10 and EL-4 wild type cells serving as negative controls. Both A1 and H3 showed excellent staining of the MICA transfectants by flow cytometry and yielded no signal for the untransfected parental cell lines (Figure 2E) with a significant difference determined by mean fluorescence intensity (MFI) (Figure 2F). Gating strategies are shown in Supplementary Figure 4.
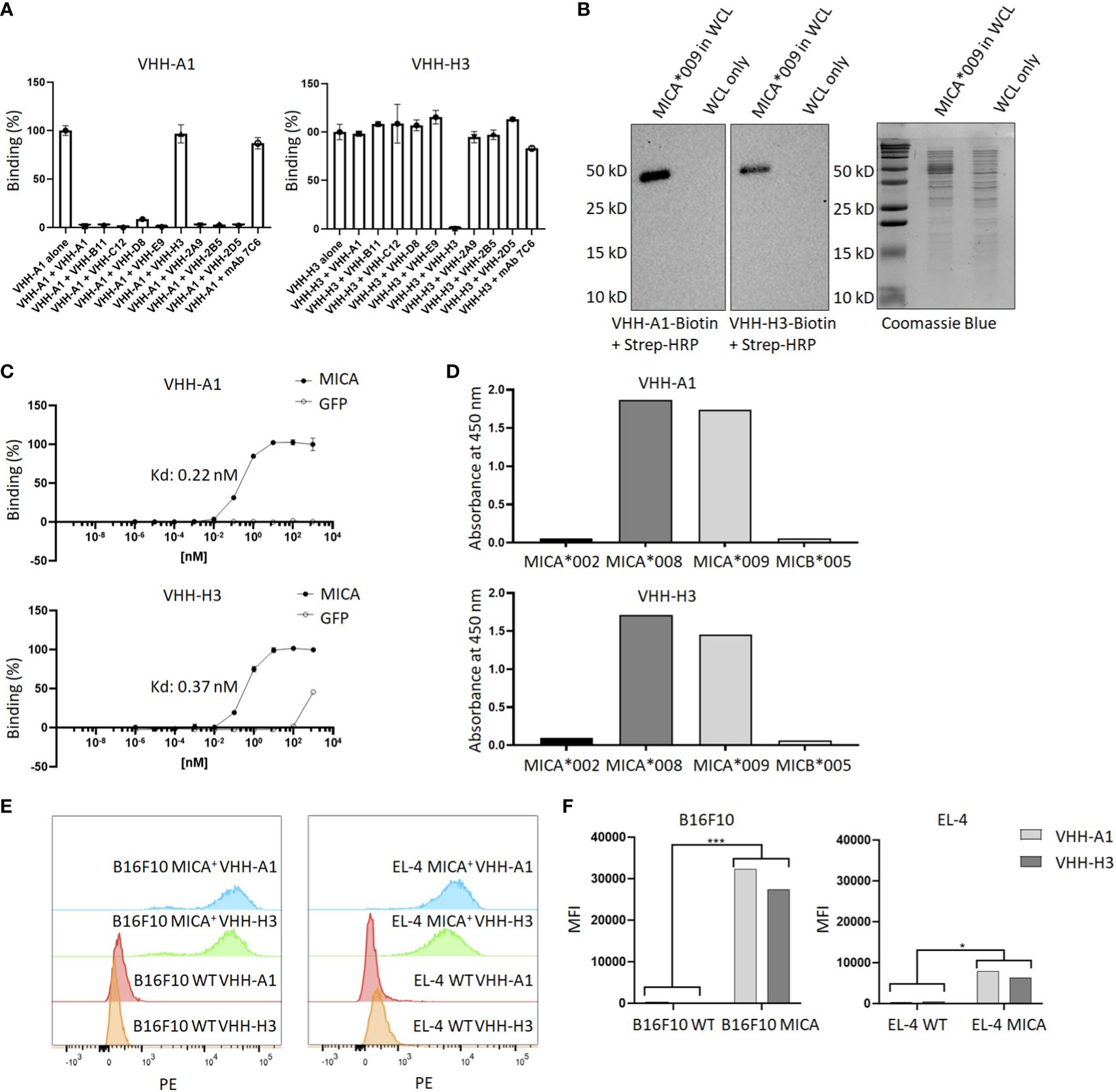
Figure 2 Characterization of MICA-specific VHHs. (A) Cross-competition ELISA shows that VHH-A1 and VHH-H3 recognize distinct epitopes on MICA. Neither VHH cross-competes for binding with the monoclonal antibody 7C6. (B) VHH-A1 and VHH-H3 recognize MICA in immunoblot. 500 ng recombinant MICA*009 in non-specific E. coli whole cell lysate (WCL) was separated by SDS-PAGE and transferred to a PVDF membrane. Blots were stained with 1 μg/mL biotinylated VHH-A1 or VHH-H3 respectively. Detection with strep-HRP (0.3 ng/mL) shows a clear signal for both VHHs. (C) Binding affinity as estimated by ELISA coated with 2.5 μg/mL recombinant MICA*009, or GFP as the negative control. Estimated Kd values are 0.22 nM and 0.37 nM for VHH-A1 and VHH-H3 respectively. (D) ELISA coated with different recombinant MICA alleles shows that VHH-A1 and VHH-H3 both recognize MICA*008 and MICA*009. (E) Flow cytometry with biotinylated VHH-A1 and VHH-H3, using streptavidin-conjugated PE as secondary agent, shows a clear signal in the PE channel for MICA+ EL-4 and B16F10 cells, but not for the WT cells, indicating recognition of membrane-disposed MICA on the surface of cells by both nanobodies. Gating strategies for flow cytometry are shown in Supplementary Figure 4. (F) We calculated the MFI after flow cytometry. The MFI of B16F10 WT cells was 394 for VHH-A1 and 299 for VHH-H3. The MFI of B16F10 MICA+ cells was 23430 for VHH-A1 and 27411 for VHH-H3. The MFI of EL-4 WT was 310 for VHH-A1 and 511 for VHH-H3. MFI of EL-4 MICA+ cells was 7955 for VHH-A1 and 6417 for VHH-H3. We averaged the MFI from the WT or MICA+ cells and determined a significant difference in nanobody staining of WT versus MICA+ cells (p = 0.00713 for B16F10; p = 0.0128 for EL-4, calculated by multiple T-test).
The reactivity of VHH-A1 and VHH-H3 make them appealing candidates for the construction of nanobody-drug conjugates. To test this, we ligated the Maytansine derivative DM1, a microtubule disrupting agent, to VHH-A1 or to a VHH that targets mouse MHC-II (VHHMHC-II) (47) as a negative control via a sortase-mediated transpeptidation reaction (Figure 3A) and confirmed successful ligation with SDS-PAGE (Figure 3B). We performed an in vitro cytotoxicity assay by titration of VHHMHC-II-DM1, VHHA1-DM1, or free DM4 (a functional analog of DM1) on EL-4 WT and MICA+ cells. EL-4 MICA+ cells were sensitive to VHHA1-DM1, with a stronger cytotoxic effect at lower doses of the VHH-drug conjugate compared to VHHMHC-II-DM1, as estimated by IC50. The IC50 of VHHA1-DM1 treated EL-4 MICA+ cells was comparable to that of cells treated with free DM4. Similarly treated WT cells showed no obvious reduction in viability with either nanobody-drug conjugate (Figure 3C).
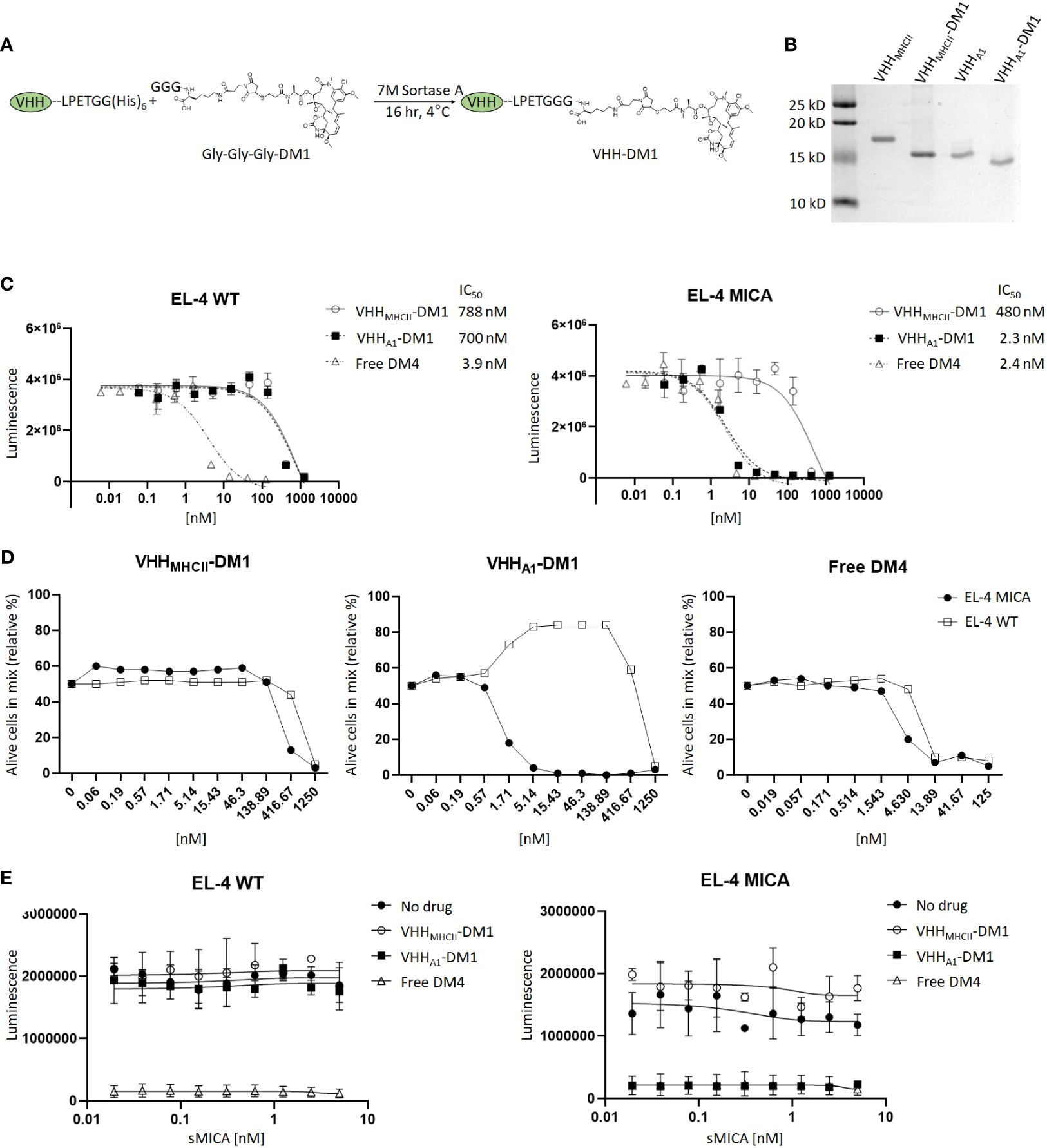
Figure 3 Anti-MICA VHHs as nanobody-drug conjugate with the Maytansine derivative DM1. (A) We ligated the microtubule inhibitor Maytansine GGG-DM1 to VHH-A1 or VHHMHC-II as non-targeting control through sortase-mediated transpeptidase reaction. (B) Because GGG-DM1 has a slight positive charge, the modified VHHs will migrate slightly lower on the SDS-PAGE gel compared to the unmodified VHHs. (C) The in vitro cytotoxicity assay was performed with limited dilutions of VHHMHC-II-DM1, VHHA1-DM1, or free DM4 on EL-4 WT cells and their MICA+ counterparts. After incubation for 72 hours, we measured cell viability by CellTiter Glo™ assay. MICA+ cells treated with VHHA1-DM1 showed a significant reduction in IC50, and thus a reduction in viability with smaller amounts of drug added, compared to similarly treated WT cells, or cells treated with the non-targeting VHHMHCII-DM1. (D) We co-cultured EL-4 WT and EL-4 MICA+ cells at a 1:1 ratio and added VHHMHCII-DM1, VHHA1-DM1, or free DM4 at different concentrations. Viability of EL-4 WT and MICA+ cells was determined using a live/dead cell stain. MICA+ cells were stained with a biotinylated anti-MICA mAb, using streptavidin-PE as secondary agent. Gating on live cells and PE showed elimination of MICA+ cells at VHH-drug adduct concentrations between 1.71 nM and 416 nM for VHHA1-DM1. A difference in [WT : MICA] was not observed in cells treated with VHHMHCII-DM1 or free DM4. Gating strategies for flow cytometry are shown in Supplementary Figure 5. (E) We incubated EL-4 WT and MICA+ cells with 2.5 nM of VHHMHCII-DM1, VHHA1-DM1, or free DM4 in the presence of sMICA (two-fold dilutions; 0-5 nM/0-170 ng/mL) for 72 hours. We measured viability by CellTiter Glo™ assay. We did not observe a decreased effect on cytotoxicity of VHHA1-DM1 on MICA+ cells with addition of sMICA in the medium.
To further validate selectivity of VHHA1-DM1 for MICA+ cells, we co-cultured EL-4 WT and EL-4 MICA+ cells at a 1:1 ratio, and added VHHMHCII-DM1, VHHA1-DM1, or free DM4 at different concentrations. We determined the ratio of viable EL-4 WT and EL-4 MICA+ cells after 72 hours by flow cytometry using a live/dead cell stain. We stained the MICA+ cells in the co-culture with a biotinylated αMICA mAb, using streptavidin-conjugated PE as secondary reagent. Gating on live cells and MICA+ cells showed specific elimination of MICA+ cells at adduct concentrations between 1.71 nM and 416 nM for VHHA1-DM1. A difference in ratio between WT and MICA+ cells was not observed in cells treated with VHHMHCII-DM1 or free DM4. Because WT cells proliferate slightly faster than MICA+ cells in culture, the distribution shifted to ~65% WT and 35% MICA+ cells after 72 hours in culture. Thus, numbers were normalized according to the percentage of cells of either line in the untreated (“0 nM”) group (Figure 3D). Gating strategies are shown in Supplementary Figure 5.
Tumor cells can downregulate surface expression of MICA through shedding, mediated by proteolytic cleavage at the α3 domain. Increased levels of soluble MICA (sMICA) in the serum of patients are associated with poor prognosis and worse disease progression (10, 48–50). To address the possible competition of sMICA for binding with the anti-MICA nanobody, we performed an in vitro cytotoxicity assay. EL-4 WT and MICA+ cells were incubated with VHHMHCII-DM1, VHHA1-DM1, or free DM4 at a fixed concentration of 2.5 nM, in the presence of sMICA at various concentrations (serial 2-fold dilutions; 0-5 nM/0-170 ng/mL). We observed no reduction in cytotoxicity of VHHA1-DM1 on MICA+ cells upon addition of sMICA to the medium (Figure 3E). Publications report concentrations of sMICA in the serum of MICA+ patients in the range of 0.1-15 ng/mL (51–53) which is at least 10-fold lower than the sMICA concentration in our competition assay. We thus expect little to no impact of sMICA in patients’ serum on the ability of these nanobodies to target membrane-bound MICA in vivo.
MICA and MICB are Class I MHC-related proteins expressed on stressed and cancerous cells. Their presence can serve not only as a diagnostic marker but may also be exploited as a target for therapy. While the typical immunoglobulins exert their functional properties through Fc effector functions, their size compromises efficient tissue penetration. Nanobodies offer an appealing alternative to immunoglobulins for the purpose of launching an immune attack on MICA-positive tumors. Nanobodies are characterized by their small size, showing superior tissue penetration compared to intact immunoglobulins, and ease of production and modification (14, 15, 17, 18). Lastly, nanobodies are poorly immunogenic, presumably because of their considerable sequence homology with human VH regions (44). Because nanobodies lack an Fc portion, for them to exert cytotoxic activity they require functionalization, for example with a cytotoxic drug creating a nanobody-drug conjugate, as done here for the VHH-DM1 adducts. Compared to antibody-drug conjugates using conventional immunoglobulins, the small size of the nanobody allows superior penetration into tumor tissue. Furthermore, owing to the relatively short circulatory half-life, the nanobody-drug conjugate that is not bound to its target will be eliminated more quickly from the circulation, resulting in less systemic cytotoxicity by slow release of the drug attached to the antibody-drug conjugate.
We produced and characterized in further detail two nanobodies, A1 and H3, that recognize the MICA alleles *008 and *009 with nM affinities. An analysis of the MICA-specific nanobodies shows that they are unique sequences, thus the isolated nanobodies were likely derived from a few different germline V genes (see Figure 1 and legend). The germline sequences of the V genes of the (outbred) alpaca used for immunization are not known. We can only compare the sequences of the MICA-specific nanobodies with each other, and with reference germline sequences from unrelated alpacas.
The alpaca IGHHV-3-3*01 gene is the possible germline version of the D8 and C12 nanobodies (44). The single difference of VHH A1 with D8 and C12 in its framework regions is an L2V substitution, thus A1 may be derived from the same germline V gene as D8 and C12 by somatic mutation. Nanobody E9 has a D29E and an R45Q substitution, indicating that E9 may be derived from a different V gene. In comparison with the other MICA-specific nanobodies, H3 has the largest number of differences in its framework regions and is clearly derived from a different germline V gene, likely the alpaca IGHHV3-1*01 (44).
Highly similar CDR regions, specifically CDR3, imply recognition of related antigens (54–57). For the MICA-specific nanobodies, the CDR1 and CDR2 regions are mostly conserved. The most obvious deviation in the CDR2 region is a deletion at position 53 in VHH C12, B11, 2A9, 2D5, and E9. Somatic hypermutation can produce deletions and insertions in V genes (58–60) but given the overall similarity in framework regions, the use of a distinct V gene that lacks residue 53 is the more plausible explanation. Except for H3, A1 and 2B5, the remaining CDR3 regions are enriched for the sequence “AxDCLSSxWRx”.
We show that these nanobodies bind to surface-disposed MICA on cells and can thus be used for diagnostic and therapeutic purposes. The specific targeting of MICA+ cells make them suitable candidates as diagnostic markers, as building blocks for nanobody-drug conjugate, or for the construction of chimeric antigen receptors (29, 30, 37, 61). MICA and MICB are highly polymorphic in the human population, with hundreds of alleles for MICA and MICB identified so far (46, 62). The isolated nanobodies were tested for recognition of the MICA alleles *002, *008 and *009, and MICB allele *005. Of the tested alleles, the nanobodies recognize MICA*008 and MICA*009, which together cover over 50% of the investigated German population (46). Expanding the nanobody pool to cover a larger portion of the alleles of MICA and MICB should be considered. We recognize the limitations of using a MICA+ cell line obtained by transfection. The availability of a suitable patient-derived cell line that expresses the correct alleles of MICA is a limiting factor, an issue worth exploring in future research.
We created a nanobody-drug conjugate by conjugating the microtubule inhibitor DM1 to VHH-A1. We show increased cytotoxicity of MICA+ tumor cells compared to WT tumor cells in vitro, with efficacy comparable to that of free drug but with much higher specificity for MICA+ cells. The production of these nanobody adducts should be scaled up for testing on in vivo tumor models. The creation of different VHH-drug combinations, for example by inclusion of DNA damaging agents or other cytotoxic drugs (63, 64), or even radiopharmaceuticals for targeted radiotherapy (65, 66), deserves consideration as well.
Cleavage of the α3 domain involving the disulphide isomerase ERp5 and ADAM-type proteases such as ADAM10 and ADAM17 (48–50, 67, 68), and thus shedding of the MICA/B from the cancer cell surface, may lead to immune evasion and failure to be recognized by NKG2D-positive cytotoxic cells. The monoclonal antibody 7C6 inhibits the shedding of MICA/B, and thus increases the density of MICA/B proteins on the surface of tumor cells (45) Although we saw no reduction in efficacy of VHHA1-DM1 on MICA+ cells upon addition of sMICA to the medium, the combination of anti-MICA nanobody adducts with the 7C6 antibody might therefore be therapeutically more attractive than either treatment alone.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: 10.6084/m9.figshare.25289806.
The animal study was approved by IACUC University of Massachusetts Amherst. The study was conducted in accordance with the local legislation and institutional requirements.
EV: Writing – original draft, Visualization, Validation, Supervision, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. AK: Writing – review & editing, Investigation, Data curation. NP: Writing – review & editing, Resources, Investigation. XL: Writing – review & editing, Resources. WvK: Writing – review & editing, Investigation. KW: Writing – review & editing, Resources. HP: Writing – review & editing, Writing – original draft, Supervision, Project administration, Funding acquisition, Conceptualization.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the NIH Pioneer Grant (DP1AI150593-05) to HP and an R01 from NCI (CA238039) to KW.
We gratefully acknowledge Dr. Thomas Balligand for helpful discussions.
Author KW serves on the scientific advisory boards of DEM BioPharma, Solu Therapeutics, D2M Biotherapeutics and Nextechinvest. He is a co-founder of Immunitas Therapeutics and receives sponsored research funding from Novartis and Fate Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1368586/full#supplementary-material
1. Agaugue S, Hargreaves A, De Sousa P, De Waele P, Gilham D. The high expression of NKG2D ligands on tumor and the lack of surface expression on healthy tissues provides a strong rationale to support NKG2D-based therapeutic approaches for cancer. Ann Oncol. (2018) 29:viii420. doi: 10.1093/annonc/mdy288.052
2. Bauer S, Groh V, Wu J, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Sci (1979). (1999) 285:727–9. doi: 10.1126/science.285.5428.727
3. Fuertes MB, Domaica CI, Zwirner NW. Leveraging NKG2D ligands in immuno-oncology. Front Immunol. (2021) 12:713158. doi: 10.3389/fimmu.2021.713158
4. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and its ligands: “One for all, all for one.” Front Immunol. (2018) 9:476. doi: 10.3389/fimmu.2018.00476
5. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. (2013) 31:413–41. doi: 10.1146/annurev-immunol-032712-095951
6. McGilvray RW, Eagle RA, Watson NFS, Al-Attar A, Ball G, Jafferji I, et al. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res. (2009) 15:6993–7002. doi: 10.1158/1078-0432.CCR-09-0991
7. Li K, Mandai M, Hamanishi J, Matsumura N, Suzuki A, Yagi H, et al. Clinical significance of the NKG2D ligands, MICA/B and ULBP2 in ovarian cancer: high expression of ULBP2 is an indicator of poor prognosis. Cancer Immunology Immunotherapy. (2009) 58:641–52. doi: 10.1007/s00262-008-0585-3
8. Cho H, Chung JY, Kim S, Braunschweig T, Kang TH, Kim J, et al. MICA/B and ULBP1 NKG2D ligands are independent predictors of good prognosis in cervical cancer. BMC Cancer. (2014) 14:1–11. doi: 10.1186/1471-2407-14-957
9. de Kruijf EM, Sajet A, van Nes JG, Putter H, THBM Smit V, Eagle RA, et al. NKG2D ligand tumor expression and association with clinical outcome in early breast cancer patients: an observational study (2012). Available online at: http://www.biomedcentral.com/1471-2407/12/24.
10. Chen J, Xu H, Zhu XX. Abnormal expression levels of sMICA and NKG2D are correlated with poor prognosis in pancreatic cancer. Ther Clin Risk Manag. (2015) 12:11–8. doi: 10.2147/TCRM.S96869
11. Vetter CS, Groh V, thor Straten P, Spies T, Brocker E-B, Becker JC. Expression of stress-induced MHC class I related chain molecules on human melanoma. J Invest Dermatol. (2002) 118:600–5. doi: 10.1046/j.1523-1747.2002.01700.x
12. Tsukagoshi M, Wada S, Yokobori T, Altan B, Ishii N, Watanabe A, et al. Overexpression of natural killer group 2 member D ligands predicts favorable prognosis in cholangiocarcinoma. Cancer Sci. (2016) 107:116–22. doi: 10.1111/cas.12853
13. Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hammers C, Bajyana Songa E, et al. Naturally occurring antibodies devoid of light chains. Nature1. (1993) 363:446–8.
14. Fang T, Lu X, Berger D, Gmeiner C, Cho J, Schalek R, et al. Nanobody immunostaining for correlated light and electron microscopy with preservation of ultrastructure. Nat Methods. (2018) 15:1029–32. doi: 10.1016/j.physbeh.2017.03.040
15. Tijink BM, Laeremans T, Budde M, Stigter-Van Walsum M, Dreier T, De Haard HJ, et al. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: Taking advantage of modular Nanobody technology. Mol Cancer Ther. (2008) 7:2288–97. doi: 10.1158/1535-7163.MCT-07-2384
16. Kijanka M, Dorresteijn B, Oliveira S, Van Bergen En Henegouwen PMP. Nanobody-based cancer therapy of solid tumors. Nanomedicine. (2015) 10:161–74. doi: 10.2217/nnm.14.178
17. Van Der Linden RHJ, Frenken LGJ, De Geus B, Harmsen MM, Ruuls RC, Stok W, et al. Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim Biophys Acta. (1999) 1431:37–46. doi: 10.1016/S0167-4838(99)00030-8
18. Tanha J, Xu P, Chen Z, Ni F, Kaplan H, Narang SA, et al. Optimal design features of camelized human single-domain antibody libraries. J Biol Chem. (2001) 276:24774–80. doi: 10.1074/jbc.M100770200
19. Rashidian M, Keliher EJ, Dougan M, Juras PK, Cavallari M, Wojtkiewicz GR, et al. Use of 18F-2-fluorodeoxyglucose to label antibody fragments for immuno-positron emission tomography of pancreatic cancer. ACS Cent Sci. (2015) 1:142–7. doi: 10.1021/acscentsci.5b00121
20. Ingram JR, Dougan M, Rashidian M, Knoll M, Keliher EJ, Garrett S, et al. PD-L1 is an activation-independent marker of brown adipocytes. Nat Commun. (2017) 8:1–15. doi: 10.1038/s41467-017-00799-8
21. Rashidian M, LaFleur MW, Verschoor VL, Dongre A, Zhang Y, Nguyen TH, et al. Immuno-PET identifies the myeloid compartment as a key contributor to the outcome of the antitumor response under PD-1 blockade. PNAS. (2019) 116:16971–80. doi: 10.1073/pnas.1905005116
22. Van Elssen CHMJ, Rashidian M, Vrbanac V, Wucherpfennig KW, El Habre Z, Sticht J, et al. Noninvasive imaging of human immune responses in a human xenograft model of graft-versus-host disease. J Nucl Med. (2017) 58:1003–8. doi: 10.2967/jnumed.116.186007
23. Jailkhani N, Ingram JR, Rashidian M, Rickelt S, Tian C, Mak H, et al. Noninvasive imaging of tumor progression, metastasis, and fibrosis using a nanobody targeting the extracellular matrix. PNAS. (2019) 116:14181–90. doi: 10.1073/pnas.1817442116
24. Fang T, Van Elssen CHMJ, Duarte JN, Guzman JS, Chahal JS, Ling J, et al. Targeted antigen delivery by an anti-class II MHC VHH elicits focused αmUC1(Tn) immunity. Chem Sci. (2017) 8:5591–7. doi: 10.1039/c7sc00446j
25. Altintas I, Heukers R, van der Meel R, Lacombe M, Amidi M, Van Bergen En Henegouwen PMP, et al. Nanobody-albumin nanoparticles (NANAPs) for the delivery of a multikinase inhibitor 17864 to EGFR overexpressing tumor cells. J Controlled Release. (2013) 165:110–8. doi: 10.1016/j.jconrel.2012.11.007
26. Fang T, Duarte JN, Ling J, Li Z, Guzman JS, Ploegh HL. Structurally-defined αMHC-II nanobody-drug conjugates: Therapeutic and imaging platforms for B-cell lymphoma. Angewandte Chemie Int Edition. (2016) 55:2416–20. doi: 10.1016/j.pep.2015.11.007.Simple
27. Bachran C, Schröder M, Conrad L, Cragnolini JJ, Tafesse FG, Helming L, et al. The activity of myeloid cell-specific VHH immunotoxins is target-, epitope-, subset- and organ dependent. Sci Rep. (2017) 7:2–11. doi: 10.1038/s41598-017-17948-0
28. Hajari Taheri F, Hassani M, Sharifzadeh Z, Behdani M, Arashkia A, Abolhassani M. T cell engineered with a novel nanobody-based chimeric antigen receptor against VEGFR2 as a candidate for tumor immunotherapy. IUBMB Life. (2019) 71:1259–67. doi: 10.1002/iub.2019
29. De Munter S, Ingels J, Goetgeluk G, Bonte S, Pille M, Weening K, et al. Nanobody based dual specific CARs. Int J Mol Sci. (2018) 19:1–11. doi: 10.3390/ijms19020403
30. Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. PNAS. (2019) 116:7624–31. doi: 10.1073/pnas.1817147116
31. Xie YJ, Dougan M, Ingram JR, Pishesha N, Fang T, Momin N, et al. Improved antitumor efficacy of chimeric antigen receptor T cells that secrete single-domain antibody fragments. Cancer Immunol Res. (2020) 8:518–30. doi: 10.1158/2326-6066.CIR-19-0734
32. You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. (2019) 9:64–78.
33. Hambach J, Riecken K, Cichutek S, Schütze K, Albrecht B, Petry K, et al. Targeting CD38-expressing multiple myeloma and burkitt lymphoma cells in vitro with nanobody-based chimeric antigen receptors (Nb-CARs). Cells. (2020) 9:1–14. doi: 10.3390/cells9020321
34. Rajabzadeh A, Ahmadvand D, Salmani MK, Rahbarizadeh F, Hamidieh AA. A VHH-based anti-MUC1 chimeric antigen receptor for specific retargeting of human primary T cells to MUC1-positive cancer cells. Cell J. (2021) 22:502–13. doi: 10.22074/cellj.2021.6917
35. Jamnani FR, Rahbarizadeh F, Shokrgozar MA, Mahboudi F, Ahmadvand D, Sharifzadeh Z, et al. T cells expressing VHH-directed oligoclonal chimeric HER2 antigen receptors: Towards tumor-directed oligoclonal T cell therapy. Biochim Biophys Acta (BBA) - Gen Subj. (2014) 1840:378–86. doi: 10.1016/j.bbagen.2013.09.029
36. Rahbarizadeh F, Ahmadvand D, Moghimi S. CAR T-cell bioengineering: Single variable domain of heavy chain antibody targeted CARs. Adv Drug Delivery Rev. (2019) 141:41–6. doi: 10.1016/j.addr.2019.04.006
37. Bao C, Gao Q, Li LL, Han L, Zhang B, Ding Y, et al. The application of nanobody in CAR-T therapy. Biomolecules. (2021) 11:1–18. doi: 10.3390/biom11020238
38. Zajc CU, Salzer B, Taft JM, Reddy ST, Lehner M, Traxlmayr MW. Driving CARs with alternative navigation tools – the potential of engineered binding scaffolds. FEBS J. (2021) 288:2103–18. doi: 10.1111/febs.15523
39. Badrinath S, Dellacherie MO, Li A, Zheng S, Zhang X, Sobral M, et al. A vaccine targeting resistant tumours by dual T cell plus NK cell attack. Nature. (2022) 606:992–8. doi: 10.1038/s41586-022-04772-4
40. Pardon E, Laeremans T, Triest S, Rasmussen SGF, Wohlkönig A, Ruf A, et al. A general protocol for the generation of Nanobodies for structural biology. Nat Protoc. (2014) 9:674–93. doi: 10.1038/nprot.2014.039
41. Jeong HJ, Abhiraman GC, Story CM, Ingram JR, Dougan SK. Generation of Ca2+-independent sortase A mutants with enhanced activity for protein and cell surface labeling. PloS One. (2017) 12:1–10. doi: 10.1371/journal.pone.0189068
42. Beatty JD, Beatty BG, Vlahos WG. Measurement of monoclonal antibody affinity by non-competitive enzyme immunoassay. J Immunol Methods. (1987) 100:173–9. doi: 10.1016/0022-1759(87)90187-6
43. Truong TTT, Huynh VQ, Vo NT, Nguyen HD. Studying the characteristics of nanobody CDR regions based on sequence analysis in combination with 3D structures. J Genet Eng Biotechnol. (2022) 20:1–13. doi: 10.1186/s43141-022-00439-9
44. Klarenbeek A, El Mazouari K, Desmyter A, Blanchetot C, Hultberg A, de Jonge N, et al. Camelid Ig V genes reveal significant human homology not seen in therapeutic target genes, providing for a powerful therapeutic antibody platform. MAbs. (2015) 7:693–706. doi: 10.1080/19420862.2015.1046648
45. De Andrade LF, En Tay R, Pan D, Luoma AM, Ito Y, Badrinath S, et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Sci (1979). (2018) 359:1537–42. doi: 10.1126/science.aao0505
46. Klussmeier A, Massalski C, Putke K, Schäfer G, Sauter J, Schefzyk D, et al. High-throughput MICA/B genotyping of over two million samples: workflow and allele frequencies. Front Immunol. (2020) 11:314. doi: 10.3389/fimmu.2020.00314
47. Fang T, Duarte JN, Ling J, Li Z, Guzman JS, Ploegh HL. Structurally defined αMHC-II nanobody-drug conjugates: A therapeutic and imaging system for B-cell lymphoma. Angewandte Chemie - Int Edition. (2016) 55:2416–20. doi: 10.1002/anie.201509432
48. Kaiser BK, Yim D, Chow I-T, Gonzales S, Dai Z, Mann HH, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature. (2007) 447:482–6. doi: 10.1038/nature05768
49. Salih HR, Rammensee H-G, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol. (2002) 169:4098–102. doi: 10.4049/jimmunol.169.8.4098
50. Xing S, Ferrari de Andrade L. NKG2D and MICA/B shedding: a ‘tag game’ between NK cells and Malignant cells. Clin Transl Immunol. (2020) 9:1–10. doi: 10.1002/cti2.1230
51. Hervier B, Ribon M, Tarantino N, Mussard J, Breckler M, Vieillard V, et al. Increased concentrations of circulating soluble MHC class I-related chain A (sMICA) and sMICB and modulation of plasma membrane MICA expression: potential mechanisms and correlation with natural killer cell activity in systemic lupus erythematosus. Front Immunol. (2021) 12:633658. doi: 10.3389/fimmu.2021.633658
52. Li JJ, Pan K, Gu MF, Chen MS, Zhao JJ, Wang H, et al. Prognostic value of soluble MICA levels in the serum of patients with advanced hepatocellular carcinoma. Chin J Cancer. (2013) 32:141–8. doi: 10.5732/cjc.012.10025
53. Arai J, Otoyama Y, Fujita K, Goto K, Tojo M, Katagiri A, et al. Baseline soluble MICA levels act as a predictive biomarker for the efficacy of regorafenib treatment in colorectal cancer. . BMC Cancer. (2022) 22:1–10. doi: 10.1186/s12885-022-09512-5
54. Henry Dunand CJ, Wilson PC. Restricted, canonical, stereotyped and convergent immunoglobulin responses. Philos Trans R Soc Lond B Biol Sci. (2015) 370:1–8. doi: 10.1098/rstb.2014.0238
55. Tian C, Hromatka BS, Kiefer AK, Eriksson N, Noble SM, Tung JY, et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat Commun. (2017) 8:1–13. doi: 10.1038/s41467-017-00257-5
56. Tsuji I, Vang F, Dominguez D, Karwal L, Sanjali A, Livengood JA, et al. Somatic hypermutation and framework mutations of variable region contribute to anti-zika virus-specific monoclonal antibody binding and function. J Virol. (2022) 96:1–18. doi: 10.1128/jvi.00071-22
57. Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell. (2013) 153:126–38. doi: 10.1016/j.cell.2013.03.018
58. Briney BS, Willis JR, Crowe JE. Location and length distribution of somatic hypermutation-associated DNA insertions and deletions reveals regions of antibody structural plasticity. Genes Immun. (2012) 13:523–9. doi: 10.1038/gene.2012.28
59. Wilson PC, De Bouteiller O, Liu Y-J, Potter K, Banchereau J, Capra JD, et al. Somatic Hypermutation Introduces Insertions and Deletions into Immunoglobulin V Genes (1998). Available online at: http://www.jem.org.
60. Bemark M, Neuberger MS. By-products of immunoglobulin somatic hypermutation. Genes Chromosomes Cancer. (2003) 38:32–9. doi: 10.1002/gcc.10241
61. Albert S, Arndt C, Feldmann A, Bergmann R, Bachmann D, Koristka S, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology. (2017) 6:1–17. doi: 10.1080/2162402X.2017.1287246
62. Koskela S, Tammi S, Clancy J, Lucas JAM, Turner TR, Hyvarinen K, et al. MICA and MICB allele assortment in Finland. HLA Immune Response Genet. (2023) 102:52–61. doi: 10.1111/tan.15023
63. Fuentes-Antrás J, Genta S, Vijenthira A, Siu LL. Antibody–drug conjugates: in search of partners of choice. Trends Cancer. (2023) 9:339–54. doi: 10.1016/j.trecan.2023.01.003
64. Fu Y, Ho M. DNA damaging agent-based antibody-drug conjugates for cancer therapy. Antib Ther. (2018) 1:43–53. doi: 10.1093/abt/tby007
65. Milenic DE, Brady ED, Brechbiel MW. Antibody-targeted radiation cancer therapy. Nat Rev Drug Discovery. (2004) 3:488–98. doi: 10.1038/nrd1413
66. Lin M, Paolillo V, Le DB, Macapinlac H, Ravizzini GC. Monoclonal antibody based radiopharmaceuticals for imaging and therapy. Curr Probl Cancer. (2021) 45. doi: 10.1016/j.currproblcancer.2021.100796
67. Liu G, Atteridge CL, Wang X, Lundgren AD, Wu JD. Cutting edge: the membrane type matrix metalloproteinase MMP14 mediates constitutive shedding of MHC class I chain-related molecule A independent of A disintegrin and metalloproteinases. J Immunol. (2010) 184:3346–50. doi: 10.4049/jimmunol.0903789
Keywords: MICA, NKG2D, NKG2D ligands, cancer, nanobodies, VHHs, immuno-oncology, nanobody drug conjugate
Citation: Verhaar ER, Knoflook A, Pishesha N, Liu X, van Keizerswaard WJC, Wucherpfennig KW and Ploegh HL (2024) MICA-specific nanobodies for diagnosis and immunotherapy of MICA+ tumors. Front. Immunol. 15:1368586. doi: 10.3389/fimmu.2024.1368586
Received: 10 January 2024; Accepted: 29 February 2024;
Published: 14 March 2024.
Edited by:
Laura Senovilla, Spanish National Research Council (CSIC), SpainReviewed by:
Eva M. Galvez, Spanish National Research Council (CSIC), SpainCopyright © 2024 Verhaar, Knoflook, Pishesha, Liu, van Keizerswaard, Wucherpfennig and Ploegh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hidde L. Ploegh, aGlkZGUucGxvZWdoQGNoaWxkcmVucy5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.