
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 February 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1367875
 Patrizia Leone1
Patrizia Leone1 Eleonora Malerba2
Eleonora Malerba2 Nicola Susca1
Nicola Susca1 Elvira Favoino3
Elvira Favoino3 Federico Perosa3Giuliano Brunori4
Federico Perosa3Giuliano Brunori4 Marcella Prete1*Vito Racanelli5
Marcella Prete1*Vito Racanelli5The tumor microenvironment is a highly complex and dynamic mixture of cell types, including tumor, immune and endothelial cells (ECs), soluble factors (cytokines, chemokines, and growth factors), blood vessels and extracellular matrix. Within this complex network, ECs are not only relevant for controlling blood fluidity and permeability, and orchestrating tumor angiogenesis but also for regulating the antitumor immune response. Lining the luminal side of vessels, ECs check the passage of molecules into the tumor compartment, regulate cellular transmigration, and interact with both circulating pathogens and innate and adaptive immune cells. Thus, they represent a first-line defense system that participates in immune responses. Tumor-associated ECs are involved in T cell priming, activation, and proliferation by acting as semi-professional antigen presenting cells. Thus, targeting ECs may assist in improving antitumor immune cell functions. Moreover, tumor-associated ECs contribute to the development at the tumor site of tertiary lymphoid structures, which have recently been associated with enhanced response to immune checkpoint inhibitors (ICI). When compared to normal ECs, tumor-associated ECs are abnormal in terms of phenotype, genetic expression profile, and functions. They are characterized by high proliferative potential and the ability to activate immunosuppressive mechanisms that support tumor progression and metastatic dissemination. A complete phenotypic and functional characterization of tumor-associated ECs could be helpful to clarify their complex role within the tumor microenvironment and to identify EC specific drug targets to improve cancer therapy. The emerging therapeutic strategies based on the combination of anti-angiogenic treatments with immunotherapy strategies, including ICI, CAR T cells and bispecific antibodies aim to impact both ECs and immune cells to block angiogenesis and at the same time to increase recruitment and activation of effector cells within the tumor.
The vascular endothelium is a thin heterogeneous monolayer of very specialized cells, the endothelial cells (ECs), that line the luminal side of blood and lymphatic vessels, and form a barrier regulating the passage of substances, cells and pathogens from the blood to the tissues (1).
ECs differ in various organs and along the vascular tree in functions, cellular morphology (size, thickness and position of the nucleus), gene expression profile, cell surface properties, production of extracellular matrix, and expression of intercellular junctions (2, 3). The expression of the most common EC markers, such as CD31, CD34, and von Willebrand Factor (vWF) changes for distinct vessel types and different anatomic compartments of the same organ (4).
ECs are involved in numerous processes including the regulation of vascular hemodynamics, vascular permeability, coagulation, cell extravasation, fibrinolysis, inflammation and angiogenesis, and metabolic homeostasis (1, 3). As ECs are in direct contact with the blood flow, they can sense its hemodynamic changes and respond by the secretion of vasoactive substances. Moreover, ECs are characterized by great plasticity to adjust the vascular system to hemodynamic forces regulating the vascular tone. They instantaneously respond to flow stimuli with changes in cell morphology, electrochemical activities, and gene expression (5, 6). In addition, together with smooth muscle cells of the vessel wall, ECs regulate the blood flow to tissues by adjusting the degree of vascular relaxation/constriction. Blood flow regulation is essential also for the delivery and exchange of oxygen, fluids, nutrients and hormones that occur in the capillaries. The presence or absence of gaps on the endothelial monolayer, and the formation of extensive tight junctions among ECs and pinocytotic vesicles can regulate endothelium permeability and make the endothelium ‘continuous’ (e.g. gut-vascular barrier), ‘fenestrated’ (e.g. glomeruli in the kidney) or ‘discontinuous’ (e.g. liver sinusoids) (1).
Endothelial heterogeneity can arise also in response to stress or pathological conditions. The vascular endothelium is emerging as a dynamic organ able to modulate its environment and respond to external stresses, and as an important paracrine/endocrine organ that releases a wide variety of anti-inflammatory and pro-inflammatory vasoactive molecules, such as nitric oxide (NO), prostacyclin (PGI2), reactive oxygen species, endothelin-1 and tumor necrosis factor-alpha (TNF-α), growth factors, cytokines, metabolites of arachidonic acid and many peptides like endothelin, urotensin, adenosine, purines, and others (7). An imbalance in the synthesis and/or release of such mediators can be caused by stress events, inflammation, hypoxia, and cancer resulting in EC activation and dysfunctional disease-promoting endothelial phenotype (3). Once the activated stressor is removed, activated ECs can return to quiescent status through mechanisms that have not been clarified so far, but that involve EC intrinsic autophagy (8). When compared to normal ECs, tumor-associated ECs are abnormal in terms of phenotype, genetic expression profile, and functions. Tumor blood vessels appear irregular, fragile, and leaky, and the endothelial network seems chaotic with an anomalous blood flow (9–11). Evidence suggests that this abnormal vascular architecture contributes to tumor growth and metastasis (12). Tumor-associated ECs are characterized by great plasticity and the ability to trans-differentiate into mesenchymal cells through a process termed endothelial-to-mesenchymal transition (EndMT). In the context of cancer, EndMT is an important adaptive process of the tumor microenvironment that gives rise to cells with multipotent potential and promotes tumor proliferation, spreading, and resistance to chemotherapy (13).
Tumor-associated ECs exhibit stem cell-like origin thus being crucial for tumor neo-angiogenesis (14–16). Moreover, tumor development results in a hypoxic milieu, which stimulates the expression of hypoxia-inducible factor (HIF), pro-angiogenic chemokines and factors leading to a strong angiogenesis stimulation (17).
Tumor-associated ECs act also as gatekeepers for tumor-infiltrating immune cells and actively participate in the priming, activation, or downregulation of effector immune cells, thereby directly influencing antitumor immunity by mechanisms that remain partially understood. Furthermore, tumor-associated ECs can establish a link with immune checkpoint molecules and influence the response to antitumor therapies (18). They are involved in the formation of tertiary lymphoid structures (TLS) which contribute to boosting immune responses (19).This study aims to examine the critical role of ECs within the tumor microenvironment, with a particular focus on their relationship with immune cells and their impact on immunotherapy. A better understanding of tumor-associated EC functions could help identify tumor-associated EC specific drug targets to improve cancer therapy.
Compared with ECs under physiological conditions (normal ECs), ECs in tumors display a markedly altered phenotype at the structural, molecular, and functional levels (Table 1). In general, disease promotes a loss of specialized capillary function and the acquisition of specific functions to support cancer cell survival. High tumor cell proliferation, tumor development and growth require the rapid formation of a complex vascular network which ensures sufficient oxygen and energy supply, and facilitates the removal of metabolic waste and carbon dioxide. Tumor growth results in a hypoxic microenvironment that cooperates with oncogenic processes to activate angiogenesis (20, 21). During tumor angiogenesis, ECs actively communicate with other angiogenesis driver cells, including pericytes, vascular smooth muscle cells, macrophages, skeletal muscle cells, and tumor cells through multiple mechanisms, such as cell-cell adhesion, formation of junctional complexes, secretion of paracrine cytokines and metabolites. Together these intercellular communications and the related cellular pathway activation form a very complex integrative signal network that drives angiogenesis and shapes endothelial phenotype (22). The coordinated execution and integration of such complex signaling programs is essential for physiological angiogenesis, while its dysregulation is critically linked to many major human diseases, including cancer. Angiogenesis is regulated by a large number of pro- and anti-angiogenic factors, whose levels dictate whether ECs will be in a quiescent or an angiogenic/activated state. When activators and inhibitors are in balance, the vasculature is quiescent and ECs are non-proliferative. During tumor development, pro-angiogenic factors and pro-angiogenic signaling pathways become dominant. Increased levels of HIF, vascular endothelial growth factor A (VEGFA), platelet-derived growth factor (PDGF), fibroblast growth factors (FGFs) or ANGPT2, as well as pro-angiogenic chemokines and receptors, lead to the “angiogenic switch”, the perpetual activation of ECs and the formation of new vessels from pre-existing vascular beds (23, 24). Through the binding to their receptors (VEGF receptor 2 (VEGFR2), PDGFR, TIE2), greatly upregulate on activated ECs, pro-angiogenic factors increase vascular permeability and promote EC proliferation, migration and assembly (23, 25).
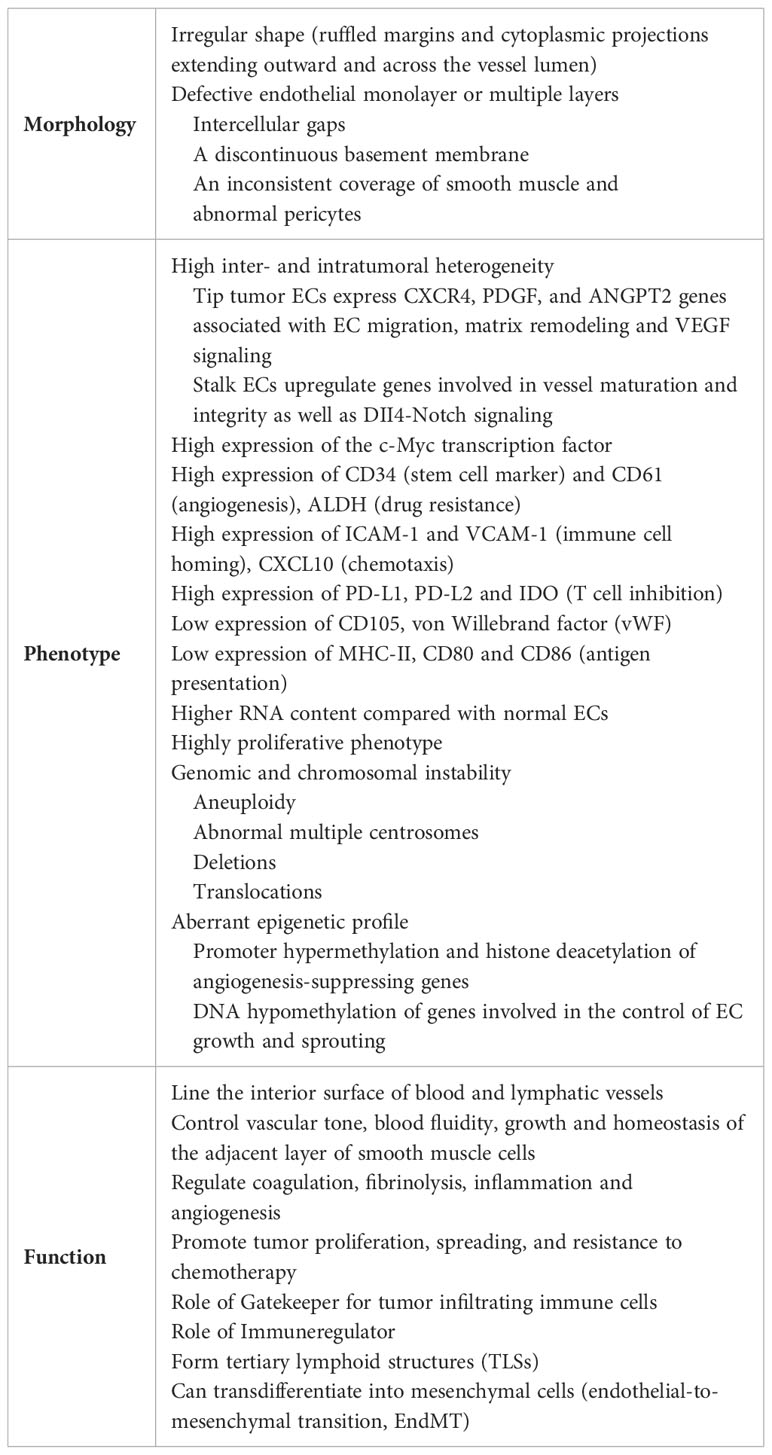
Table 1 Main features of tumor endothelial cells (ECs).
Moreover, pro-angiogenic factors can activate the genetic reprogramming of tumor-associated ECs supporting a highly proliferative phenotype with a great propensity for migration (26, 27). Hypoxia and reactive oxygen species (ROS) induce genetic and chromosomal instability through an elevated mutational frequency (28, 29). Fluorescence in situ hybridization (FISH) analysis showed that tumor-associated ECs are aneuploidy and have multiple karyotypic aberrations including abnormal multiple centrosomes, deletions and translocations (30). Furthermore, tumor-associated ECs display an aberrant epigenetic profile with silencing occurring in combination with promoter hypermethylation and histone deacetylation of angiogenesis-suppressing genes, and upregulation of genes involved in the control of EC growth and sprouting through DNA hypomethylation (31).
Morphologically tumor-associated ECs are characterized by irregular shape with ruffled margins and many cytoplasmic projections extending outward and across the vessel lumen. The intercellular connections are lost resulting in a defective endothelial monolayer or multiple layers with small and large intercellular gaps or holes, a discontinuous basement membrane, an inconsistent coverage of smooth muscle and abnormal pericytes (9, 10). This irregular and leaky architecture of tumor vessels facilitates the passage of fluid, blood, fibrin and tumor cells into the surrounding tissue promoting tumor metastasis (12). Furthermore, vessel leakiness increases the intratumoral interstitial pressure and reduces perfusion and blood flow causing tumor hypoxia and a metabolic EC switch to anaerobic glycolysis and acidosis (32).
Recent technological advances like single cell RNA sequencing (scRNAseq) were able to elucidate the wide inter- and intratumoral heterogeneity defining cellular subpopulations of tumor-associated ECs and providing functional information of the detected gene expression profiles. Tumor-associated ECs have a higher RNA content (two- to four-fold increase compared with normal EC) due to increased rates of transcription and high metabolic demands of nucleotide biosynthesis (purine and pyrimidine) or oxidative phosphorylation and glycolysis (15, 33). They exhibit elevated expression of the c-Myc transcription factor and its targets involved in tumor angiogenesis (15, 34). Compared with normal ECs, tumor-associated ECs display higher expression of the stem cell marker CD34 and the angiogenesis promoting markers CD61 (Integrin b3) and CD105 (Endoglin) (35), and lower or absent expression of von Willebrand factor (vWF) (36). Moreover, tip tumor-associated ECs express genes associated with EC migration, matrix remodeling and VEGF signaling such as CXCR4, PDGF, and ANGPT2, whereas stalk ECs upregulate genes involved in vessel maturation and integrity as well as DII4-Notch signaling (14, 37). Using scRNAseq in combination with orthogonal bulk multi-omics approaches, Goveia et al. have distinguished different subgroups of arterial, capillary and tip ECs expressing gene signatures related to basement-membrane breaching, vascular integrity, homeostasis, immune cell recruitment, and antigen presentation (14). In addition, they observed that specific EC phenotypes were differentially sensitive to anti-VEGF drugs and VEGFR tyrosine kinase inhibitors; tip and breach tumor-associated ECs were more sensitive to VEGF blockade than postcapillary vein and proliferating ECs, and capillary ECs were less sensitive to VEGFR tyrosine kinase inhibitors (14). Furthermore, the resistance to anti-VEGF therapy is due to the upregulation of alternative pro-angiogenic signals in tumor-associated ECs, such as genes involved in posttranscriptional collagen modification that could represent possible alternative angiogenic candidates (14).
However, selective targeting of different EC subpopulations in vivo is very difficult because of the limited number of truly unique surface molecules. Most of the reported EC targeting approaches are successfully restricted to organ-specific EC and not to a single EC subset (1).
Computational multi-pathway models that integrate multiple cell types and simultaneously simulate cell-cell communication have elucidated the individual molecular and cellular signaling components that function in concert to regulate angiogenesis. These models could also guide experimental studies to uncover new multilevel features of pathological angiogenesis and support the development of therapeutic strategies that target multiple pathways (22).
The finding that tumor-associated ECs are highly proliferative and display abnormal gene expression profiles and chromosomal instability has suggested a stem cell-like origin. Stem cell-like ECs, also called endothelial progenitor cells (EPCs) with the capacity to self-renew and form blood vessels have been identified at the inner surface of preexisting blood vessels (38, 39). Recently, by scRNAseq several studies have identified ECs with a resident endothelial stem cell signature (14–16). However, the definition, cell lineage, and the specific mechanism by which EPCs differentiate into ECs are not yet known (40). A specific marker to identify EPCs is lacking given that potential typical markers such as CD34, CD31, VEGFR2 and CD133 are shared by vascular ECs and hematopoietic cells (myeloid cells and mesenchymal stem cells) (41). In glioblastoma has been found a CD133+ cancer stem-cell-like population which included a fraction of EPCs (42). Patel et al. have isolated mouse CD34+CD45- vascular endothelial (VE)-cadherin+ EPCs with different expressions of CD31 and VEGFR2 based on the differentiation status which can develop entire blood vessels in wounds, inflamed skin, and tumors (43). Along with CD34 and CD133, tumor-associated ECs exhibit other stem cell markers including CD90, Sca-1, MDR1 ALP, and Oct-4, and form stem cell-like clusters with increased proliferative and invasive capacity and aldehyde dehydrogenase (ALDH) activity involved in resistance to therapy (26, 27, 44, 45).
Moreover, tumor cells can undergo vasculogenic mimicry in which they trans-differentiate into endothelial-like cells with the ability to develop matrix-rich vessel-like networks formed by vessels that do not arise from preexisting vessels (46). Interestingly, it was found that in glioblastoma and lymphoma, tumor cells can give rise to ECs. Indeed, they share the same somatic and cytogenetic mutations (42, 47).
The tumor can also produce molecules such as pleiotrophin and macrophage colony-stimulating factor (M-CSF) which guide the differentiation of monocytes and dendritic cells into endothelial-like cells (48, 49).
Being endothelium a thin layer of cells that line the interior surface of blood vessels, ECs are in close contact with circulating innate and adaptive immune cells and mediate their interactions with tumor stroma. Tumor-associated ECs play a highly tuned role of sentinels and immune regulators by performing tissue-specific and vessel type-specific immunomodulatory functions including immune cell recruitment, activation and antigen presentation (Figure 1) (10, 50).
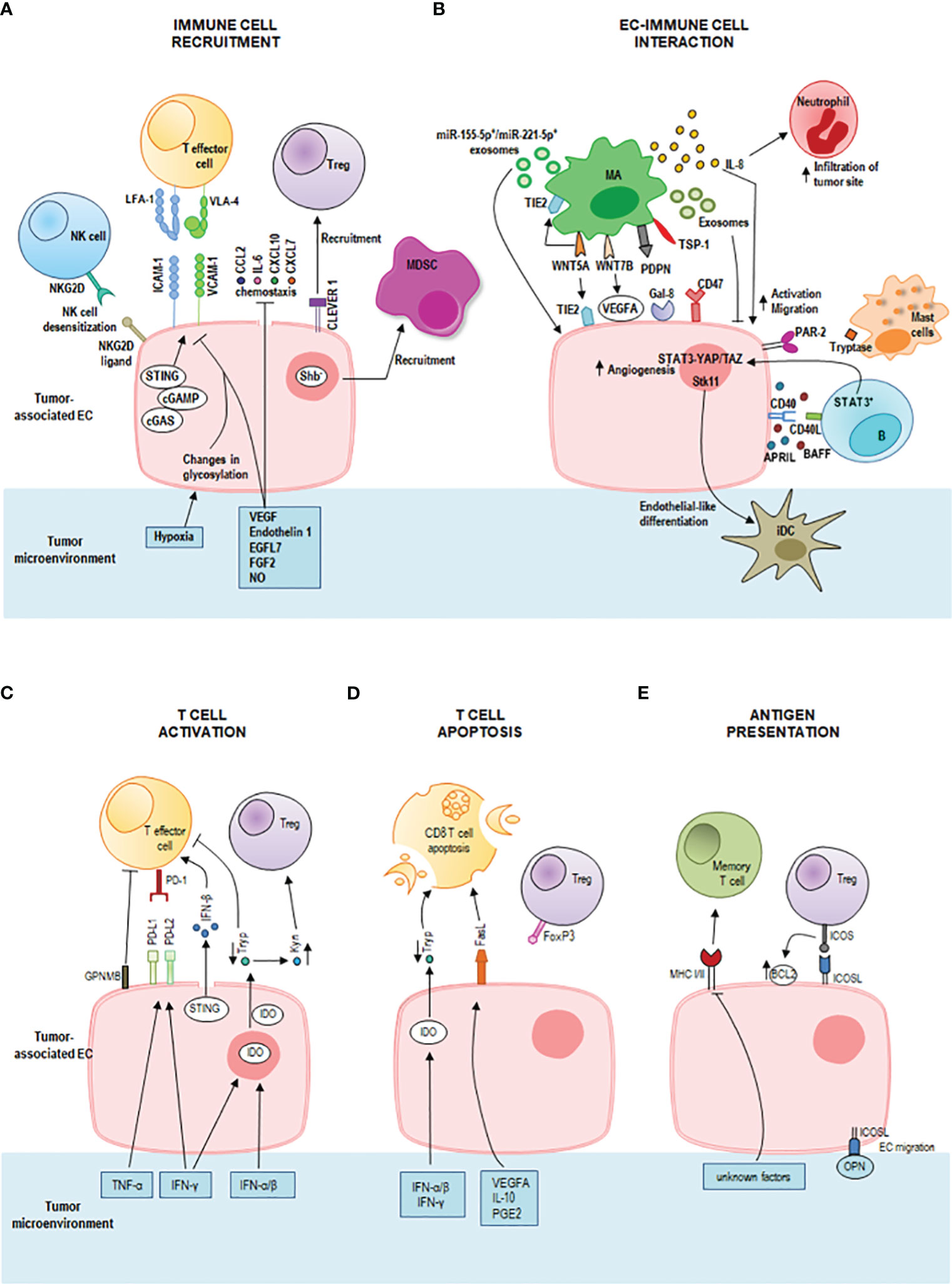
Figure 1 Immunoregulatory properties of tumor endothelial cells (ECs) in the tumor microenvironment. (A) Activated ECs recruit effector immune cells guiding their infiltration into the tumor microenvironment through a multi-staged adhesion process which includes binding of the integrins lymphocyte function-associated antigen-1 (LFA-1) and very late antigen-4 (VLA-4) on T cells to the respective ligands intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion protein-1 (VCAM-1) on tumor-associated ECs. The tumor-deriving cytokines vascular endothelial growth factor (VEGF), endothelin-1 (ET1), EGF-like domain-containing protein 7 (EGFL7), and fibroblast growth factor 2 (FGF2) downregulate gene expression of adhesion molecules and chemoattractants (e.g., CCL2, IL6, CXCL10, and CXCL7) to inhibit immune cell infiltration. Also, nitric oxide (NO) in the tumor microenvironment inhibits immune cell recruitment. Tumor hypoxia results in changes in EC glycosylation which impair adhesion and cell-matrix interactions. Tumor-associated ECs represent a selective barrier that favors regulatory T (Treg) cell traffic by the common lymphatic endothelial and vascular endothelial receptor 1 (CLEVER 1), but represses effector T cells, dendritic cells, natural killer (NK) cells and neutrophil granulocytes promoting immunological tolerance. Tumor-associated ECs can cause desensitization of NK cells through the expression of NKG2D ligands that interact with the receptor NKG2D on NK cells. The intracellular cGAS-cGAMP-STING pathway can enhance adhesion molecules and promote T cell infiltration. Shb deprivation in tumor-associated ECs led to the recruitment of myeloid-derived suppressor cells (MDSCs). (B) IL-8 signaling promotes EC activation and the migration of tumor cells, ECs, and infiltrating neutrophils at the tumor site. Macrophage-derived WNT7B increases VEGFA expression on tumor-associated ECs. Macrophage-derived WNT5A stimulates EC proliferation and migration and upregulates TIE2 in macrophages (MAs) and ECs. Tumor-associated macrophages-derived exosomes suppress EC migration by the miR-146b-5p/TRAF6/NF-kB/MMP2 pathway, whereas M2 macrophage-derived exosomal miR-155-5p and miR-221-5p mediate macrophage-EC interactions. Podoplanin (PDPN) on tumor-associated MAs mediates the attachment of MAs to lymphatic ECs by interaction with Galectin-8 (Gal-8) and stimulates new lymph vessel formation and the migration of tumor cells through the lymphatic system. Tumor-associated MAs express thrombospondin-1 which interacts with CD47 receptor expressed by tumor cells and tumor-associated ECs. Downregulation of CD47 in tumor-associated ECs increases angiogenesis, vascular integrity and stability, VEGF-A and VEGFR2 expression. A bidirectional crosstalk between ECs and B cells is realized by CD40 on microvascular ECs and CD40L on tumor-associated B cells. This interaction increases the EC release of BAFF and APRIL which further enhance the expression of CD40L on B cells. Moreover, tumor-associated B cells stimulate ECs via the signal transducer and activator of transcription 3 (STAT3)-YAP/TAZ signaling. Tumor-associated ECs induce endothelial-like differentiation of immature dendritic cells (iDC) within the tumor microenvironment by the expression of serine/threonine kinase 11 (Stk11). Mast cells release tryptase which acts on the proteinase-activated receptor-2 (PAR-2) stimulating both vascular endothelial and tumor cell proliferation. (C) STING activates also T cells by inducing a strong IFN-β production. The pro-inflammatory tumor-derived cytokines IFN-γ and TNF-α increase the expression of the immune checkpoint ligands programmed death-ligand 1 (PD-L1) and programmed death-ligand 2 (PD-L2) on the tumor-associated EC surface. These ligands bind to the receptor programmed death 1 (PD-1) on T cells inhibiting T cell functions. ECs also induce tumor-infiltrating T cell exhaustion through the glycoprotein nonmetastatic melanoma protein B (GPNMB). Interferon (IFN)-γ, IFN-α and IFN-β upregulate indoleamine 2,3-dioxygenase (IDO) production by tumor-associated ECs promoting the metabolism of tryptophan (Tryp) to kynurenine (Kyn). Tryp depletion inhibits T cell proliferation, whereas Kyn accumulation promotes Treg differentiation and activation. (D) T cell apoptosis is triggered by IDO-mediated Tryp degradation and by upregulation of Fas ligand (FasL) on tumor-associated EC by tumor-derived VEGFA, interleukin (IL)-10 and prostaglandin E2 (PGE2). Tumor-associated ECs acquired the ability to kill effector CD8+ T cells but not Treg cells (due to expression of FoxP3). (E) Tumor-associated ECs can act as semi-professional antigen presenting cells. They can present processed antigens to memory T cells via class I and class II major histocompatibility complex (MHC) molecules, yet they do not express the co-stimulatory molecules CD80 and CD86 required for naïve T cell activation. They express the inducible co-stimulator ligand (ICOSL) which interacts with the receptor ICOS on Treg cells sustaining their activation and proliferation. ICOS/ICOSL interaction also increases the expression of the antiapoptotic protein Bcl-2. ICOSL can bind osteopontin (OPN) promoting EC migration.
Whereas during immune homeostasis ECs allow immune cells to patrol by providing immune surveillance to the surrounding tissues, during inflammation, activated ECs recruit effector immune cells guiding their infiltration into the tumor microenvironment (51). The immune cell extravasation consists of three sequential steps, rolling, adhesion and transmigration (diapedesis) which are mediated by interactions between receptors and ligands on the endothelium and leukocytes, and involve vascular adhesion molecules and chemokines (Figure 1A). Initially, leukocytes establish transient selectin-mediated interactions with ECs, roll along the vessel wall, and are activated by chemokines presented on the endothelial surface. These chemokines by binding G protein-coupled receptor (GPCR) on leukocytes trigger an intracellular signaling cascade that induces the integrin-mediated firm adhesion of leukocytes by upregulation of integrin affinity for their respective ligands on ECs. As a last step, leukocytes pass across the vascular basement membrane and migrate along a chemotactic gradient toward the site of tissue damage (52, 53). Among the interactions between adhesion molecules and adhesion receptors, the binding of the integrins leukocyte function-associated antigen-1 (LFA-1) and very late activated antigen-4 (VLA-4) on T cells to the respective ligands intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) on ECs critically regulates leukocyte adhesion and spreading (54).
During pathological processes, such as chronic inflammation and cancer, the decrease of expression of leukocyte adhesion molecules along with endothelium dysfunction and endothelial anergy (the lack of response to inflammatory activation) can strongly impair immune cell migration and tissue infiltration. Specifically, tumors secrete cytokines such as VEGF, endothelin 1, EGF-like domain-containing protein 7 (EGFL7), and fibroblast growth factor 2 (FGF2) which induce downregulation of endothelial expression of selectins, adhesion molecules, chemokines to inhibit leukocytes migration and generate a tumor immune-privileged microenvironment (10). Also, NO in the tumor microenvironment inhibits immune cell recruitment (55). Shb deprivation in tumor-associated ECs enhances the recruitment of myeloid-derived suppressor cells (MDSCs) that confer a suppressive immune response, thus promoting tumor metastasis (56) (Figure 1A).
Tumor-associated ECs can establish a selective barrier allowing specific T cell subsets to infiltrate the tumor site more effectively. They favor Treg cell traffic (57), but repress effector T cells, dendritic cells, NK cells and neutrophil granulocytes promoting immunological tolerance (52).
Upon inflammation, tumor-associated ECs can also enhance the expression of specific adhesion markers, such as the common lymphatic endothelial and vascular endothelial receptor 1 (CLEVER 1), to direct immune cells. CLEVER 1 is constitutively expressed on lymphatic EC, on sinusoidal EC in the liver and spleen, and on high endothelial venules (HEVs), and its expression can be induced on tumor-associated EC in blood vessels, where it favors selective transmigration of Treg cells and type II macrophages from the blood into the tumor (58–61).
Changes in EC glycosylation promoted by the tumor hypoxic milieu contribute to EC dysfunction impairing endothelial barrier function, adhesion, cell-matrix interactions and cell signaling (62) (Figure 1A). Moreover, tumor hypoxia induces the chronic release of pro-angiogenic factors resulting in EC anergy (63). Of note, EC anergy is a reversible process and may be used as a therapeutic strategy. Preclinical data show that blocking the aforementioned mechanisms in tumor ECs increases the antitumor immune response. For instance, anti Clever 1 antibody treatment reduces the recruitment of Treg into the tumor and decreases tumor progression in vivo (59). Anti-VEGF therapies may contribute to the reprogramming of the tumor microenvironment supporting EC activation and effector T cell recruitment (64).
Through the expression of NKG2D ligands, tumor-associated ECs can interact with NK cells causing desensitization of antitumor NK responses (65) (Figure 1A). Moreover, cytokines such as TNF-α and IL-1β activate ECs and induce the release of CCL2 and CCL7 that recruit NK cells and the expression of ICAM-1 and VCAM-1 that enable a stable contact between ECs and NK cells. These findings suggest that these molecules might be potential targets to enhance NK cell infiltration into solid tumors and to increase the antitumor efficacy of NK cell therapy (66).
Tumor-associated ECs can interact with tumor-associated macrophages (Figure 1B) enhancing the metastatic potential of tumor cells by increasing endothelial permeability which promotes circulating tumor cell adhesion and transmigration into the tissue (67). In epithelial ovarian cancer, tumor-associated macrophages stimulate EC function by IL-8 production (Figure 1B) (68). IL-8 signaling triggers angiogenic responses in ECs, promotes proliferation and survival of endothelial and tumor cells, and increases the migration of tumor cells, ECs, and infiltrating neutrophils at the tumor site. In addition, stress and drug-induced IL-8 signaling has been shown to confer chemotherapeutic resistance in cancer cells (69).
Moreover, tumor-associated macrophages express high levels of WNT family proteins, especially of WNT7B and WNT5A (Figure 1B). WNT7B promotes the angiogenic switch and vascular remodeling by increasing VEGFA expression on tumor-associated ECs (70). WNT5A stimulates EC proliferation and migration and upregulates TIE2 in macrophages and ECs (71). TIE2-expressing macrophages can mimic vascular structure through the expression of EC markers and the formation of capillary-like structures in response to VEGF, paving the way for vessel maturation with replacement by true ECs (71). TIE2 is also involved in the regulation of endothelial permeability (72). Tumor-associated macrophage-derived exosomes can suppress EC migration by the miR-146b-5p/TRAF6/NF-kB/MMP2 pathway, and epithelial ovarian cancer-derived exosomes can reverse this process (73). In addition, M2 macrophage-derived exosomal miR-155-5p and miR-221-5p are involved in macrophage-EC interactions. By suppressing E2F2 expression in ECs, they promote angiogenesis and support the development of pancreatic ductal adenocarcinoma (74, 75). In breast cancer, a tumor-associated macrophage subset expressing podoplanin (PDPN) is involved in the attachment of this macrophage subset to lymphatic ECs. PDPN interacts with Gal-8 on lymphatic ECs to promote new lymph vessel formation and the migration of tumor cells through the lymphatic system. Experimental removal of PDPN-expressing macrophages or inhibition of Gal-8 significantly reduces tumor metastasis in mouse breast cancer models (76). Tumor-associated macrophages express the signal-regulatory protein α (SIRPα) and the thrombospondin-1 which interact with the CD47 receptor, a don’t eat me signal, expressed by tumor cells and tumor-associated ECs. Interestingly, the downregulation of CD47 in tumor-associated ECs increases angiogenesis, vascular integrity and stability, VEGF-A and VEGFR2 expression and promotes tumor growth (77). Thus, although the treatment with antibodies against CD47 can induce antitumor immune responses by blocking the inhibitory CD47-SIRPα signaling in tumor cells, it may also potentially promote tumor progression by blocking CD47 signaling in ECs. Furthermore, blocking CD47 confers radioresistance to ECs in vitro and protects soft tissue, bone marrow, and tumor-associated leukocytes in irradiated mice (78).
In chronic lymphocytic leukemia, it has been observed that the constitutive release of soluble BAFF and APRIL increased upon engagement of CD40 on microvascular ECs by CD40L aberrantly expressed on tumor cells. Endothelial BAFF and APRIL further amplified chronic lymphocytic leukemia cell survival by enhancing the expression of leukemic CD40L suggesting a bidirectional crosstalk between ECs and B cells (Figure 1B) (79). Moreover, tumor-associated B cells stimulate ECs via the signal transducer and activator of transcription 3 (STAT3)-YAP/TAZ signaling promoting tumor angiogenesis (Figure 1B) (80, 81).
In esophageal squamous cell carcinoma, the concomitant presence of tumor cells expressing high levels of high-mobility group box 1 (HMGB1) and peritumoral regions with high density of proliferating B cells is an unfavorable prognostic factor. These B cells activate pro-angiogenic phenotypes and promote the growth of both ECs and tumor cells (82)).
Altogether these findings identify (STAT3)-YAP/TAZ signaling, proliferating B cells as well as HMGB1 signals as potential therapeutic targets for anti-angiogenesis therapy.
ECs can regulate dendritic cell differentiation by the expression of serine/threonine kinase 11 (Stk11) (Figure 1B). Deletion of Stk11 in mouse ECs strongly reduces mature DC numbers and increases spontaneous tumorigenesis (83). The activation of MAPK/ERK1/2 signaling stimulates immature DCs to undergo endothelial-like differentiation within the tumor microenvironment derived from the human esophageal squamous cell carcinoma cells and represents an alternative pathway of tumor angiogenesis (84, 85).
Mast cells can also modulate angiogenesis through the release of classical pro-angiogenic factors and nonclassical pro-angiogenic granule-associated mediators. One of the latter is the tryptase which is released by mast cells after c-Kit receptor activation. Tryptase acts on the proteinase-activated receptor-2 (PAR-2) stimulating both vascular endothelial and tumor cell proliferation (Figure 1B) (86).
ECs are also the main source of spontaneous IFN-β production in growing tumors, a molecule involved in the generation of spontaneous antitumor immune responses (87). The release of IFN-β upon recognition of intracellular foreign nucleic acids is driven by the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway, which has emerged as a critical link between innate and adaptive immunity (88) (Figure 1C). It has been demonstrated that intratumoral administration of exogenous cGAMP or STING agonist (cyclic diguanylate monophosphate; c-di-GMP) increases endothelial STING expression and IFN-β release and strongly boosts antitumor immunity leading to control of tumor growth in a mouse model of melanoma, breast cancer and glioma (87, 89–92). In murine glioblastoma models, treatment with STING agonists associated with biodegradable intracranial implants has demonstrated a profound shift in the tumor immune landscape, with massive intratumoral infiltration of innate immune cells, such as NK cells, and an increase in survival (89). This finding warrants further examination of STING agonists alone or in combination with other immunotherapies. The combination of cytotoxic cationic silica nanoparticles (CSiNPs) with STING agonists can activate tumor-infiltrating antigen-presenting cells resulting in increased expansion of antigen specific CD8+ T cells, and potent tumor growth inhibition in murine melanoma (93). Despite the very encouraging preclinical results, limitations to the use of CSiNPs-STING agonists in anticancer therapy lie in their chemical features.
Endothelial STING expression is also associated with normalizing of tumor blood vessels, increased adhesion molecule expression, enhanced T cell infiltration, and prolonged survival in human colon and breast cancer (94).
In addition, STING activation synergizes with VEGFR2 blockade and/or immune checkpoint inhibitors (ICI) leading to normalization of the tumor vasculature and the tumor microenvironment, significant upregulation of adhesion molecules (ICAM-1 and VCAM-1) on ECs, and complete regression of immunotherapy-resistant tumors (90, 94). Further examination of this combination strategy of STING-based immunotherapy is ongoing in clinical trials.
The pro-inflammatory cytokines IFN-γ and TNF-α enhance the expression of the immune checkpoint ligands programmed death-ligand 1 (PD-L1) and programmed death-ligand 2 (PD-L2) on the tumor-associated EC surface. These ligands bind to the receptor programmed death 1 (PD-1) on T cells inhibiting T cell functions (Figure 1C). Recently, Sakano et al. have demonstrated that hepatocellular carcinoma-associated ECs can induce tumor-infiltrating T cell exhaustion through the expression of the glycoprotein nonmetastatic melanoma protein B (GPNMB), suggesting that GPNMB might be a novel therapeutic target in hepatocellular carcinoma (95) (Figure 1C).
Upon type I and type II IFN stimulation, tumor-associated ECs can express high levels of the immunosuppressive enzyme indoleamine 2,3‐dioxygenase (IDO) which is associated with T cell apoptosis, inhibition of T cell proliferation and activation of Treg cells (96, 97) (Figures 1C, D). IDO inhibition has been shown to enhance the efficacy of ICI and represents a promising strategy for cancer therapy. The combination of ICI with IDO inhibitors yields a synergistic effect in the activation of antitumor immune responses, and clinical trials to evaluate their efficacy are ongoing (98, 99).
Tumor-derived VEGFA, IL-10 and prostaglandin E2 cooperatively induce upregulation of the death mediator Fas ligand (FasL) in ECs which gain the ability to kill effector CD8+ T cells but not Treg cells (due to expression of FoxP3) (100) (Figure 1D). Pharmacological inhibition of these factors results in lower FasL expression on ECs and higher CD8+ T cell infiltration by preventing effector T cell apoptosis (100).
In addition, tumor-associated ECs can act as semi-professional antigen presenting cells and can activate memory T cells. They express both class I and class II MHC molecules and low levels of the co-stimulatory molecules CD40, CD80 and CD86 required for naïve T cell activation. Tumor-associated ECs express the inducible co-stimulator ligand (ICOSL) which sustains activation and proliferation of the Treg population, and increases its suppressor function (101, 102) (Figure 1E).
Moreover, the crosstalk between Treg cells and ECs via ICOS/ICOSL interaction increases the expression of the antiapoptotic protein Bcl-2 on the endothelial surface and improves the sensitivity of B-lymphoma cells towards ABT-199, a potent Bcl-2 inhibitor (103). In addition, a recent study has demonstrated that ICOSL can bind osteopontin, and their interaction induces EC and tumor cell migration (104) (Figure 1E).
If immune responses fail to eradicate the triggering stimulus, the formation of tertiary lymphoid structures (TLS) at sites with persistent inflammatory stimulation takes place. TLS are highly organized cellular aggregates resembling secondary lymphoid organs that develop in non-lymphoid tissues during chronic infectious diseases, autoimmune and inflammatory disorders, and cancers (19, 105). Like secondary lymphoid organs, their organization includes T and B cell compartmentalization, antigen presenting cells such as dendritic cells, stromal cells, conduits, and a highly organized vascular system of HEVs and lymphoid vessels (106–108). Although still partly unclear, TLS generation (Figure 2) is thought to be similar to that of secondary lymphoid organs considering the anatomical resemblance (109). Local production of CXCL13 and IL-7 by lymphocytes or stromal cells recruits lymphoid tissue inducer (LTi) cells (110) which interact with local stromal cells via lymphotoxin-α1β2 (LTα1β2) (111). Th17 cells, B cells, or M1-polarized macrophages can substitute LTi cells in the initiation of TLS genesis in various pathological conditions (112–115). Stromal cells release vascular endothelial growth factor C (VEGFC) which promotes the formation of HEVs and secrete adhesion molecules, such as VCAM1, ICAM1 and mucosal addressin cell adhesion molecule 1 (MADCAM1), and various chemokines, notably CXCL12, CXCL13, CC-chemokine ligand 19 (CCL19) and CCL21 (116). Together, these molecules work in concert to regulate the subsequent recruitment of immune cells and vascularization (117, 118). HEVs are blood vessels located in the TLS periphery and involved in immune cell recruitment and transmigration (119). Similarly to HEVs in lymph nodes, HEVs in TLS allow naïve and memory lymphocytes to pass from the bloodstream into the parenchyma of the tissue where they can interact with their cognate antigen. Moreover, HEV ECs can form pockets, a kind of “waiting areas”, in which lymphocytes reside for several minutes before entering the lymph node (120). Acting as lymphocyte portals, ECs lining HEVs express at their luminal surface high levels of ICAM-1, and depending on the organ and state of maturation, peripheral node addressin (PNAd) and/or mucosal addressin cell adhesion molecule (MAdCAM-1), ligands for the lymphocyte-homing receptor l-selectin (CD62L) that mediate the initial capture and rolling interactions of lymphocytes along the vessel wall (121, 122). Lymphocytes pass through the EC layer of the HEVs, crawl inside the perivascular channel and finally arrive at the lymph node parenchyma (123). Lymphocytes organize into T cell and B cell zones which compose the inner zone of TLS with a central B cell zone surrounded by a peripheral T cell-rich area. B cells undergo antibody class switching and somatic hypermutation becoming antibody-producing plasma cells (124). CD4+ and CD8+ T cells recognize peptide tumor antigens presented by antigen presenting cells and undergo activation, proliferation, and differentiation into CD4+ T follicular helper (TFH) cells and CD8+ effector memory cytotoxic cells which often represent the dominant subsets (124–126). CD4+ T helper 1 (TH1) cells and Treg cells can also be present (127), as well as CD68+ macrophages for clearance of apoptotic cells (128, 129). TFH cells express CXCR5 and migrate at the interface between B and T cell zones, where they support the activation of antigen specific B cells, and the generation of memory B cells and antibody-producing long-lived plasma cells (130–132). A subset of regulatory IL-10+ B cells that coexists with Treg cells has also been described in human breast and ovarian cancers (133, 134).
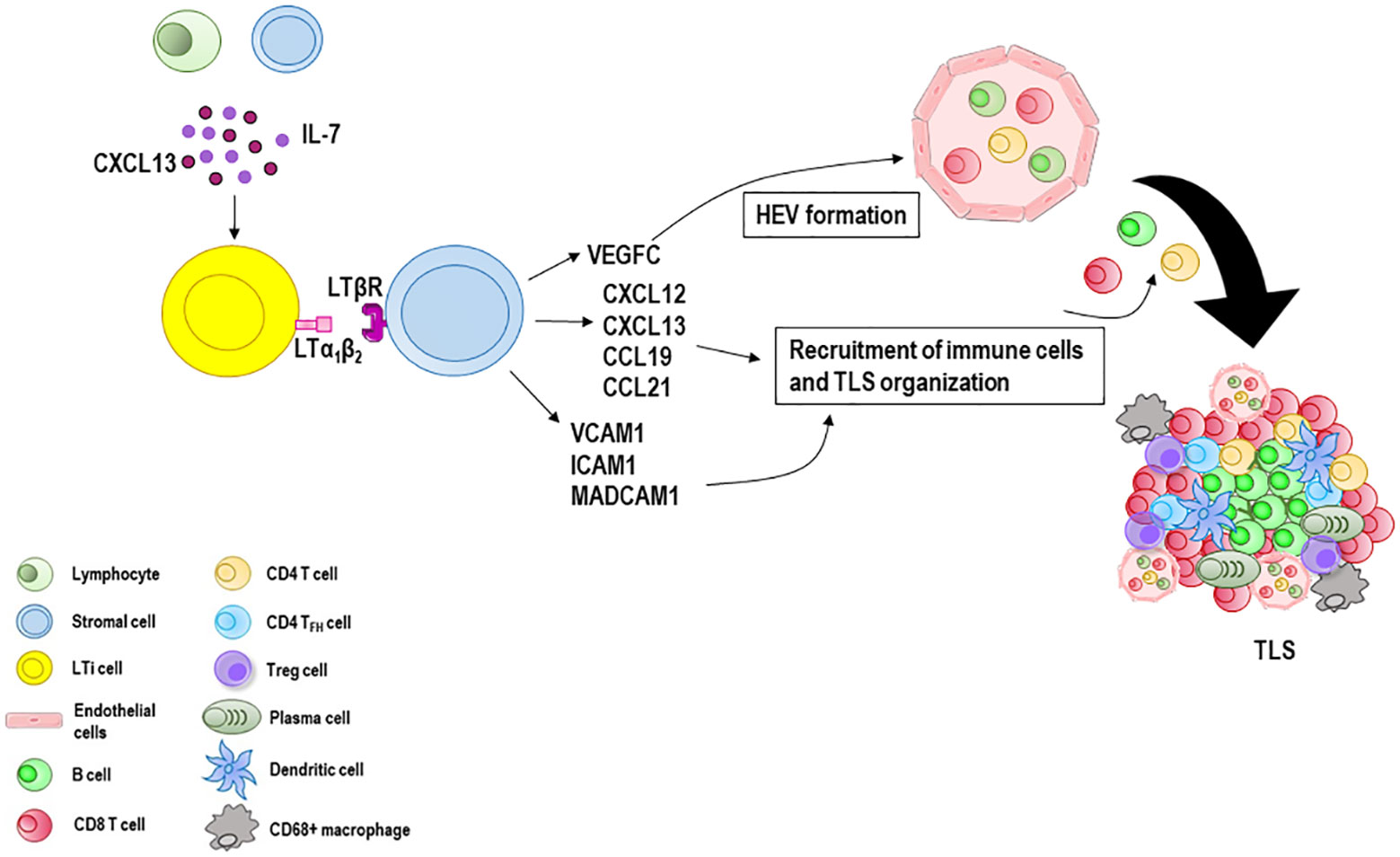
Figure 2 Tertiary lymphoid structures (TLS) formation and maturation. When immune responses fail to eradicate the triggering stimulus, the formation of TLS at sites with persistent inflammatory stimulation takes place. Lymphocytes and stromal cells release CXCL13 and IL-7 involved in the recruitment of lymphoid tissue inducer (LTi) cells which interact with local stromal cells via lymphotoxin-α1β2 (LTα1β2). Many immune cells such as B cells, M1 macrophages and Th17 cells can substitute LTi cells in the initiation of TLS genesis in various pathological conditions. Stromal cells secrete vascular endothelial growth factor C (VEGFC) which promotes the formation of the high endothelial venules (HEVs) and release adhesion molecules, such as vascular cell adhesion molecule 1 (VCAM1), intercellular adhesion molecule 1 (ICAM1) and mucosal addressin cell adhesion molecule 1 (MADCAM1), and various chemokines including CXCL12, CXCL13, CCL19 and CCL21. Together, these molecules recruit lymphocytes from HEVs and form a central B cell zone and a peripheral T cell zone enriched with CD8 T cells. CD4 T cells, CD4 T follicular helper (TFH) cells and Treg cells can also be present, as well as CD68+ macrophages and antibody-producing long-lived plasma cells.
Unlike secondary lymphoid organs, TLS lack a capsule and are exposed to local antigens, danger-associated molecular patterns and cytokines. They represent a niche in which immune cells interact with each other and with the surrounding microenvironment to generate or reactivate or potentiate the immune response (135). The presence of TLS has been related to robust immune reactions to clear infections (136), or to severe tissue destruction in autoimmune diseases (137). In tumors, TLS may exert either pro- or anti-tumor activities and their presence has been associated with variable outcomes. Important parameters for the impact of TLS on tumor control are the number, composition, and location. A high number of TLS within tumor microenvironment correlates with increased infiltration of adaptive immune cells and a better prognosis in ovarian carcinoma (138), non-small-cell lung carcinoma (NSCLC) (139), breast (140), renal cell, pancreatic (141), and hepatocellular cancers (142), gastrointestinal tumors (143) and melanoma (125, 144).
The proportion of mature dendritic cells in TLS able to activate effector T cells is strongly associated with better outcome and long survival in NSCLC and rectal cancers (145, 146). High proportions of Treg cells within TLS have been linked to worse outcomes in several types of cancer, including ovarian, breast and hepatocellular cancer (138, 147, 148), and in a mouse lung adenocarcinoma model (149).
The location of TLS is also crucial. The presence of TLS close to tumor beds is associated with favorable patient prognosis. In contrast, their location at a distance from tumor beds, outside the invasive margin, correlates with poor outcome (150).
Emerging evidence associates the presence of preexisting TLS with better response to immune checkpoint-blocking therapies in melanoma (144, 151), sarcoma (152, 153), NSCLC (153–155), renal cell carcinoma (151, 153, 156) and urothelial cancer (153, 157). The mechanism remains still unclear, but it could involve B cells and their interaction with T cells (151). TLS are sites for maturation, selection, clonal amplification, isotype switching of B cells, and generation of antibody-producing plasma cells which contribute to the antitumor response. Moreover, B cells can release an array of cytokines, through which they activate and recruit other immune effector cells, and they may act as antigen presenting cells, driving tumor specific T cell activation and expansion (151). However, pro-antitumor activities have also been described for B cells due to the existence of a heterogeneous population including regulatory B cells with immunosuppressive phenotype (133, 134, 158–160). A better understanding of these specific TLS mediators could optimize treatments aimed at enhancing TLS-dependent antitumor immunity safely and effectively. Recently, antitumor drugs, chemokines, cytokines, agonistic antibodies, and engineered dendritic cells expressing the TH1 cell transcription factor T-bet or CCL21 have been used in combination with traditional antitumor treatments to promote TLS formation, with good results. These agents, in combination, can turn cold immune-suppressed tumors into hot immunogenic tumors, promoting tumor infiltration by effector T cells and increasing the recruitment of immune cells that participate in TLS development (135). In a mouse model of breast and pancreatic tumors, the combined anti-VEGFR2 and anti-PDL1 therapy overcame T cell exhaustion phenotype and increased antitumor immune response stimulating TLS neogenesis (161). In a cohort of 59 patients with pancreatic ductal adenocarcinoma, an intradermal vaccine with an irradiated allogeneic granulocyte colony-stimulating factor (GM-CSF) in combination with cyclophosphamide increased TLS development (162). Furthermore, in patients with cervical neoplasia, enhanced TLS formation and maturation could be observed in regressing lesions after human papillomavirus vaccination (163).
The development of lymphatic vessels and their function are regulated by the VEGFC/VEGFR3 signaling (164). Activation of VEGFC/VEGFR3 signaling in lymphatic endothelial cells (LECs) increases the proliferation of LECs and the formation of lymphatic vessels, leading to the increase of lymphatic metastasis of tumor cells (165). Overexpression of VEGFR3 and its ligand VEGFC has been found in several tumors including colorectal cancer, endometrial, prostate cancer, ovarian carcinoma and breast cancer, and is associated with increased formation of lymphatic vessels, lymph node and distant metastases, as well as a poor prognosis (166–170).
Additionally, the VEGFC/VEGFR3 signaling may also modulate antitumor immune response promoting immune tolerance. VEGFC was found to protect tumors against preexisting antitumor immunity in a mouse melanoma model. In these mice, VEGFC activated LECs were able to cross-present tumor antigens leading to dysfunctional activation of tumor-specific CD8+ T cells (171). Moreover, in patients with acute myeloid leukemia, low-functioning NK cells exhibited great levels of VEGFR3 which promotes chemotherapy resistance (172). In lung adenocarcinoma, tumor-associated macrophages induced the expression of VEGFC and VEGFR3 in tumor cells inducing lymphangiogenesis and metastasis, and VEGFR3 inhibition enhanced lung adenocarcinoma cell chemosensitivity through upregulation of proteins p53 and PTEN (173).
Accordingly, the Food and Drug Administration (FDA) approved several drugs targeting VEGFC/VEGFR3, such as sorafenib, sunitinib, pazopanib, and axitinib, which have given good results in the treatment of renal cell carcinoma, hepatocellular carcinoma, thyroid cancer, gastrointestinal stromal tumors and soft tissue sarcoma (174–184).
Endothelial-to-mesenchymal transition (EndMT) is the phenotypic and functional transformation of endothelial to mesenchymal cells (fibroblast-like cells). During this process, ECs progressively lose their characteristic markers such as CD31, VE-cadherin, VEGFR2, and gain mesenchymal markers including fibroblast-specific protein-1 (FSP1), alpha 2 smooth muscle actin (α-SMA), vimentin and N-cadherin (185). Changes include also loss of the ability to form functional vessels, loss of cell-cell junctions and polarity, and the acquisition of cellular motility, and invasive and contractile properties (186). It occurs during embryonic development and in many pathological conditions including inflammation and cancer. Furthermore, cells in transition show a pro-inflammatory secretory phenotype related to the synthesis of extracellular matrix proteins such as fibronectin and collagens (186).
Numerous studies have demonstrated that TGF-β is the main inducer of EndMT (187–191). Other modulators of EndMT are hypoxia, EC metabolic alterations such as endothelial fatty acid oxidation and epigenetic regulators including microRNAs and long non-coding RNAs (13, 192).
EndMT has now been discovered in several pathologies, and especially in many tumors including colorectal carcinoma (193), pancreatic ductal adenocarcinoma (194), lung cancer (195), or glioblastoma (GBM) (196, 197), esophageal cancer (198), oral squamous carcinoma (199) and breast cancer (200).
During tumor development, ECs predominantly differentiate via EndMT into cancer-associated fibroblasts (CAFs) which contribute to the tumor microenvironmental plasticity and profoundly affect tumor growth and metastases (201). Moreover, EndMT participates in angiogenesis sprouting and vascular remodeling to support tumor cell dissemination, and it is involved in resistance to cancer therapy, such as chemotherapy, anti-angiogenic therapy, and radiation therapy (202). CAFs are known to produce soluble factors (IL-6 and IL-8) associated with chemoresistance and to regulate chemotherapy uptake by capturing active drugs and decreasing the expression of drug transporters. In addition, CAFs protect tumor cells from the oxidative stress induced by chemotherapy (203). Tumor vasculature EndMT-related phenotypic alterations due to fibrotic changes and specific gene loss may result in tumor resistance to and relapse after anti-VEGF therapy. Vascular damage after radiation therapy induces EndMT that reactivates dormant cancer stem cells and supports the shift of tumor associated macrophages toward an M2 phenotype, conferring tumor radioresistance (195).
Among the different cell types involved in tumor progression and metastasis, tumor-associated ECs stand out for their remarkable contribution at different steps of the metastatic process including cell-cell and cell-extracellular matrix adhesions, cell invasion and/or migration and angiogenesis. Tumor-associated ECs participate in blood vessel formation which supports tumor cell growth and dissemination by providing the exit routes for metastasis in distant organs. The abnormal and leaky architecture of tumor blood vessels along with the weakened EC junctions facilitate the detachment of tumor cells from the surrounding matrix, the migration into tumor vessels via binding to the chemokine receptor CXCR7, and the metastatic spread. This metastatic process is also supported by tumor microenvironment deriving pro-inflammatory cytokines (IL-3, IL-6, and IL-8) and growth factors (bFGF, G/GM-CSF, IGF1, PDGF, and TGF-β), as well as other factors like Notch, calcineurin, biglycan, Jag1, and Slit2 (204).
TGF-β induces upregulation of the T cell inhibitory receptor T cell immunoglobulin and mucin domain-3 (TIM-3) on the EC surface and favors tumor cell proliferation, survival and migration by activating NF-κB in melanoma cells in a galectin-9-independent manner (205, 206). TIM-3 expression on lymphoma-derived ECs sustains tumor onset, growth, and dissemination by inhibiting activation of CD4+ T cells and Th1 polarization, and correlates with poor patient outcome (74). Moreover, TIM-3 promotes tube formation and decreases tight junction formation in vascular ECs indicating that TIM-3 expression favors tumor invasion and metastasis by inducing angiogenesis and increasing capillary permeability (207, 208).
Sustained activation of Notch1 signaling in tumor-associated ECs induces VCAM1 expression, neutrophil recruitment and a senescent, pro-inflammatory endothelium which promotes tumor cell adhesion, intravasation and metastasis. In a mouse model of ovarian carcinoma, treatment with VCAM1 and Notch 1 receptor blocking antibodies blocked Notch-driven metastasis (209). Downregulation of the tumor suppressive angiocrine factor Slit2 and upregulation of its inhibitor receptor EphA2 on tumor-associated ECs promote tumor proliferation and motility (210).
PGI2 in the tumor microenvironment can bind to and activate the peroxisome proliferator-activated receptor β/δ (PPAR β/δ) on ECs inducing EC proliferation and angiogenesis (211, 212). PPAR β/δ acts as a critical “hub node” transcription factor that shifts the angiogenic balance towards the pro-angiogenic phenotype favoring tumor development, progression, and metastasis (213–216).
Circulating tumor-associated ECs can also attach to metastasizing tumor cells protecting them from anoikis-mediated apoptosis (apoptosis in cells upon loss of attachment to the extracellular matrix) (217).
Tumor-associated ECs display high expression of B7-H3 (CD276), a critical regulator of the adaptive immune response that can distinguish between physiological and pathological angiogenesis (218, 219). B7-H3 upregulates VEGFA expression and angiogenesis by activating the NF-κB pathway in colorectal cancer and it may be a promising target for colorectal cancer treatment (220). Moreover, tumor-associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas (221) and renal cell carcinomas (222). B7-H3 expression in Merkel cell carcinoma-associated ECs correlates with locally aggressive primary tumor features and increasing vascular density (223).
Tumor-associated ECs can also express CD137 (4-1BB), a member of the TNF receptor superfamily acting as a costimulatory immune receptor. Treatment of tumor-bearing immunocompromised Rag(-/-) mice with agonist CD137 monoclonal antibody not only enhances T cell activation but also stimulates tumor-associated ECs augmenting cell surface expression of ICAM-1, VCAM-1, and E-selectin with consequent promotion of CD8+ T cell recruitment into the malignant tissue (224).
In addition, tumor creates a local milieu favorable for tumor growth and metastasis. In high metastatic tumors, the tumor microenvironment influences EC epigenome promising upregulation of biglycan expression by DNA demethylation and enabling tumor intravasation and metastasis (225).
The tumor-associated EC altered glycosylation of surface adhesion molecules including ICAM-1, VCAM-1, and PECAM, and glycan-binding proteins (lectins) also promote tumor progression and metastasis by modifying the adhesive properties of ECs (62).
Many in vitro and in vivo studies using animal models have demonstrated the pivotal roles of galectins, a family of glycan-binding proteins, in tumor invasiveness, metastasis and angiogenesis (Reviewed in (226):. The blockade of galactin (Gal)-3 with an anti-gal-3 antibody has been shown to inhibit liver metastases of human adenocarcinoma xenotransplants in SCID mice (227). In a metastatic model, breast tumor cells expressing high levels of Gal-3 were more resistant to apoptosis induced by reactive nitrogen and oxygen species, suggesting that Gal-3 can also sustain the survival of metastasizing tumor cells (228).
Gal-1, Gal-3, and Gal-8 have been demonstrated to engage integrins or other cell surface proteins to mediate adhesion of tumor cells to extracellular matrix proteins and homotypic cell adhesion, or to inhibit adhesion favoring tumor cell detachment, dissemination through blood or lymph vessels and the attachment to ECs or basement membrane proteins at distal sites. Gal-3 produced by ECs can also foster angiogenesis by promoting EC migration and vessel formation (229–234), and by enhancing the VEGF- and basic fibroblast growth factor (bFGF)-mediated angiogenic response (235). Moreover, Gal-3 and Gal-1 directly interact with VEGFR2 increasing its pro-angiogenic function (236) and HIF1a favoring tumor progression and metastasis (237, 238).
Accordingly, it has been observed that altered galectin expression in human tumors correlates with the aggressiveness of the tumor, greater extent of vascularization and poor disease outcome (226, 239). Vaccination against Gal-1 promotes cytotoxic T cell infiltration in melanoma and reduces tumor burden in the immunized mice compared to the control group (240).
Gal-8 is markedly upregulated in inflamed human and mouse corneas and promotes VEGF-C-mediated lymphatic EC migration and sprouting. Treatment with Gal-8 inhibitors strongly reduces inflammatory lymphangiogenesis in vivo, suggesting Gal-8 as a promising therapeutic target for pathological lymphangiogenesis (241).
Finally, besides the impact of tumor-associated ECs on tumor growth and the metastatic process, they may affect tumor therapy resistance. The heterogeneity of tumor-associated ECs plays a central role in resistance to anti-angiogenic drugs. Treatment with anti-VEGFA antibodies and tyrosine-kinase inhibitors of VEGF receptors stimulates the production of different growth factors as alternative pro-angiogenic molecules, including angiopoietins (ANGs), epidermal growth factor (EGFs), FGFs, hepatocyte growth factor (HGF), TGFs, stromal cell-derived factor 1 (SDF1) and simultaneously, the upregulation of the respective receptors on ECs (242). EC-expressed Jag1 contributes to the development of a malignant vascular niche that is associated with an aggressive course and chemotherapy resistance (243). In addition, tumor-associated ECs produce non-conventional growth factors such as biglycan, lysyl oxidase (LOX) and pentraxin, sustaining angiogenesis processes (204). Tumor-associated ECs display cytogenetic and epigenetic abnormalities, expression of stemness genes, metabolic adaptation and sequestration of drugs in autophagic lysosomes leading to drug resistance (30, 204). Anti-VEGFA treatment and hypoxia induce glycosylation mediated resistance increasing Gal-1 production. Gal-1 binds to VEGFR2 leading to unresponsiveness to anti-VEGF therapy, and disruption of the Gal-1-VEGFR2 axis promotes vessel normalization, immune cell recruitment, tumor growth inhibition and restores sensitivity to anti-angiogenic therapy (229).
The efficacy of cancer therapy depends mainly on the ability of T cells to infiltrate tumors. The endothelium works as a barrier between the blood and the tumor. As such, it directly interacts with immune cells and plays a crucial role in recruiting and activating T cells (18).
Targeting ECs is thereby a potential strategy to optimize T cell-mediated antitumor immune responses. One strategy involves using anti-angiogenic agents to inhibit angiogenesis, overcome EC anergy, and enhance tumor T cell infiltration (244). Moreover, it has been observed that anti-angiogenic therapeutics can also normalize the dysfunctional tumor vasculature resulting in increased tumor blood perfusion and reduced tumor hypoxia, which in turn enhances drug accessibility and decreases hypoxia-mediated treatment resistance (245, 246). A single infusion of the VEGF specific antibody bevacizumab is sufficient to promote vessel normalization in rectal carcinoma patients (247). Similarly, in glioblastoma patients, treatment with cediranib, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes the structure and function of the tumor vasculature increasing tumor perfusion and improving patient survival (248–251).
Preclinical studies have demonstrated that vascular normalizing doses of anti-angiogenic treatments reprogram tumor microenvironment from immunosuppressive to immunosupportive and enhance immunotherapy efficacy. For instance, low doses of an anti-VEGFR2 antibody could normalize breast tumor vasculature, redirect tumor-associated macrophages to an immune stimulatory M1-like phenotype and increase T cell tumor infiltration (252).
Immunologic approaches targeting tumor vasculature such as a model based on T cells engineered to express a chimeric antigen receptor (CAR) targeted VEGFR2 also increase tumor T cell infiltration (253). Likewise, ICI efficacy may be achieved through Th1-mediated vessel normalization indicating a mutual regulatory loop in which ICI activate IFN-γ+ Th1 cells that induce vessel normalization which, in turn, promotes T cell recruitment (254).
Based on these findings, combined therapeutic strategies could synergistically potentiate antitumor treatments. Combination of anti-angiogenic drugs with ICI may revert both immune and EC anergy promoting the access of cytotoxic T cells to tumors and enhancing their antitumor effects.
A dose-dependent synergism exists between anti-angiogenic therapy and ICI blockade. Low-dose anti-VEGFR2 antibody treatments can sensitize breast cancer to PD-1 blockade via upregulation of PD-1 on immune cells by stimulating the secretion of osteopontin and TGF-β (255). Treatment with a combination of anti-VEGFR2 and anti-PD-L1 antibodies induces HEV formation in pancreatic and breast tumor mouse models promoting simultaneously T cell infiltration and activation (161).
The concurrent neutralization of VEGFA and ANGPT2 by a bispecific antibody promotes vascular regression, tumor necrosis, blood vessel normalization, and increases cytotoxic T cell infiltration in both genetically engineered and transplant tumor models, including metastatic breast cancer, pancreatic neuroendocrine tumor, and melanoma. Moreover, the anti-angiogenic therapy enhances the sensitivity of mouse tumors to PD-1 blockade via upregulation of the PD-L1 expression in tumor-associated EC (256). In patients with metastatic melanoma, the combination of bevacizumab with the anti-CTLA-4 monoclonal antibody ipilimumab reverses tumor-associated EC anergy increasing the expression of the adhesion molecules E-selectin, ICAM1, and VCAM1 which enhances the recruitment of T cells in the tumor bed and improves the clinical outcome (257). The anti-PD-L1 monoclonal antibody atezolizumab in combination with bevacizumab is found to have antitumor activity with good tolerability in patients with metastatic renal cell carcinoma (258, 259), hepatocellular carcinoma (260), metastatic NSCLC without targetable mutations in association with chemotherapy (261) and metastatic NSCLC which fails to respond to atezolizumab monotherapy (262).
A novel bispecific antibody, HB0025, which concurrently blocks both the PD-L1 and VEGF pathways, has demonstrated anti-cancer activities both in vitro and in vivo and is currently under clinical trial (NCT04678908) (263, 264). An active clinical trial (NCT05116007) is ongoing to evaluate the efficacy and safety of the anti-PD-1 and -VEGF bispecific antibody AK112 combined with chemotherapy in patients with extensive stage small cell lung cancer. Furthermore, there is a multitude of ongoing clinical trials that are investigating the synergistic effect achieved through the combination of anti-angiogenic treatments with immunotherapy (Table 2). Encouraging preliminary results are described in various cancer types with a meager response to therapy. For instance, the combination of the anti-PD-1 monoclonal antibody pembrolizumab with the tyrosine kinase inhibitor cabozantinib improves progression free survival and overall survival compared with single-agent pembrolizumab in patients with recurrent metastatic head and neck squamous cell carcinoma. The overall response rate correlates positively with baseline CD8+ T cell infiltration (265).
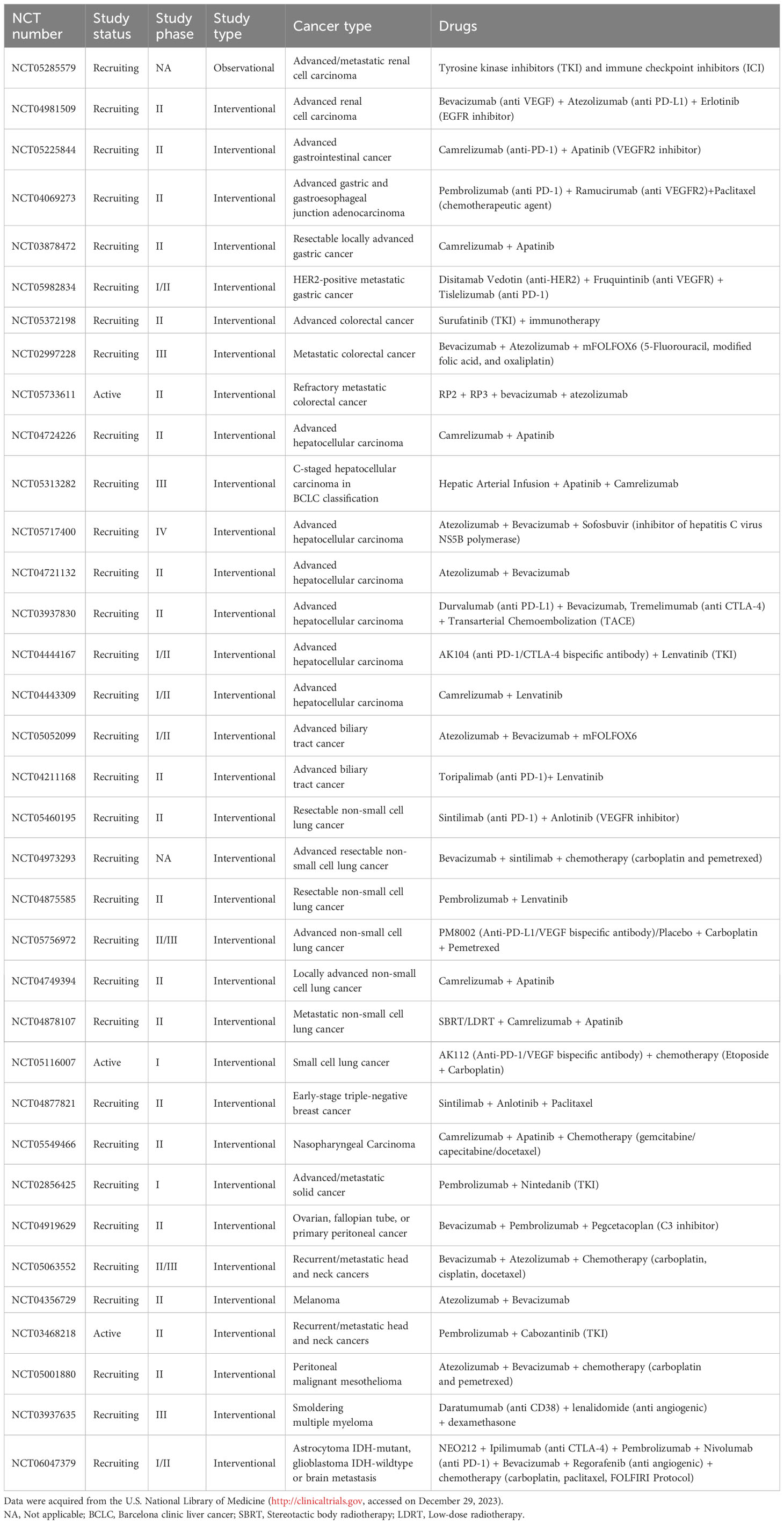
Table 2 Combination of anti-angiogenic treatments with immunotherapy in ongoing clinical trials.
The phase II BREAKPOINT trial (NCT03463681) met its primary endpoint showing prospective activity and safety of cabozantinib after an adjuvant or first-line ICI-based immunotherapy in patients with metastatic renal cell carcinoma (266).
Like all treatments, the combination of anti-angiogenic drugs with ICI has limitations and faces important challenges. Firstly, it is necessary to establish the appropriate drug dosage, and optimize the schedule of tumor immunotherapy and anti-angiogenesis therapy given that the dose of each drug, the sequence (simultaneous or sequential treatment), and the time of the combination can significantly impact the efficacy of the combination therapy.
Secondly, the combination of anti-angiogenic drugs with ICI has adverse effects, some of them are dose-dependent. High doses of anti-angiogenic drugs can damage tumor blood vessels resulting in hypoxia, immunosuppression and tumor treatment resistance (246). Other side effects include high-grade hypertension, decreased platelet count, proteinuria, the nonspecific activation of the immune system and hepatic toxicity (267).
Thirdly, combination therapy may not always be the best option for all patients, and there is no method to identify patients that can benefit from it. It is necessary to consider specific patient characteristics, such as the presence of autoimmune diseases or a previous organ transplantation history to avoid the nonspecific activation of the immune system that ICI treatment can cause. Specific attention should also be paid to elderly patients, patients at risk of hemorrhages, and patients suffering from severe cardiovascular diseases, hepatopathies or gastrointestinal problems as they may be exposed to a higher risk of exacerbations (268).
Furthermore, there is a lack of validated predictive biomarkers that can allow patient selection and stratification, and guide treatment decisions before and during therapy. Recent studies have suggested PD-L1 expression as a potential biomarker; they have reported that ICI combined with angiogenesis inhibitors improved the outcome in patients with positive PD-L1 expression affected by renal cell carcinoma, NSCLC, advanced HER2-positive breast cancer and untreated, unresectable hepatocellular carcinoma (260, 269–273). Moreover, pre-existing immunity (high expression of CD274, T cell effector signature and intratumoral CD8+ T cell density), high expression of Treg, MDSC and VEGFR2 in tumor tissue could also be potential candidate biomarkers for prediction of response and resistance to ICI and anti-angiogenic combination therapy (274). In addition to biomarkers derived from analysis of tumor tissue, circulating biomarkers, such as levels of VEGFA, soluble VEGFR2 and circulating ECs could be considered to monitor treatment responses. Moreover, non-invasive imaging biomarkers, including computed tomography (CT)-based perfusion scan, dynamic contrast-enhanced ultrasonography, dynamic contrast-enhanced magnetic resonance imaging and magnetic resonance imaging-based diffusion-weighted imaging and perfusion could be helpful to examine tumor vessel perfusion and vascularity during treatment (275). Increasing evidence suggests a role of the composition and function of the gut microbiome as a predictive biomarker (276). Based on these findings, a new approach in cancer management should aim to discover a composite biomarker of response to therapy that uses data from imaging, tumor tissue-derived, circulating and gut microbiome-derived biomarkers. The identification of a composite biomarker, that can be easily and continuously monitored, may support the assessment of treatment efficacy and the development of a personalized treatment based on patient-customized adjustment of therapeutic regimens.
Alternative strategies target tumor-derived extracellular vesicles (EVs) carrying pro-angiogenic factors such as VEGF, IL-6, microRNAs which stimulate EC functions or mediate drug resistance (277, 278). Recent studies have tried to improve the efficacy of anti‐angiogenic therapies by blocking tumor-derived EVs. For instance, patients with elevated EV‐VEGF levels can be treated with VEGF‐binding protein inhibitors or VEGFR2‐neutralizing antibodies to block EV-VEGF which are resistant to bevacizumab treatment (278).
Finally, a study has investigated the use of nanotechnology-based approaches to target ECs. The nanoformulated STING activator ZnCDA activates ECs resulting in disruption of tumor vasculature, increase of tumor-targeted drug accumulation and improvement of tumor-associated macrophage functions (279).
ECs play a critical role in tumor development which has gone beyond angiogenesis. During tumor progression, multiple cellular pathways can interact and regulate each other through signal competition, redundancy, shared downstream signaling network, and many crosstalk and cross-regulation mechanisms. Understanding the effects of ECs on tumor progression and their interactions with the other components of the tumor microenvironment requires computational models that integrate multiple cell types.
ECs are now considered active participants in the tumor microenvironment, secreting angiogenic factors such as cytokines, growth factors, and chemokines, and interacting with immune cells. Crosstalk between tumor endothelium and immune cells strongly impacts the immune response to tumors and contributes to immunosuppression by downregulation of antigen presentation and recruitment of immune effector cells. Moreover, tumor-associated ECs support the expansion of immunosuppressive cell populations, such as Treg cells, contributing to tumor immune escape. In addition, tumor-associated ECs of blood and lymphatic vessels are directly involved in the formation of distant metastasis. Emerging therapeutic strategies aim at modulating both ECs and immune cells, not only to block angiogenesis but also to enhance the recruitment and activation of effector cells within the tumor. A combination of anti-angiogenic treatments with immunotherapy strategies, including ICI, CAR T cells, and bispecific antibodies has emerged. Many pivotal clinical trials have proven the validity of these combinations which are now successfully used in patients with metastatic melanoma, renal cell carcinoma, hepatocellular carcinoma and metastatic NSCLC. Moreover, a multitude of clinical trials focusing on these combined therapeutic strategies are currently ongoing and new studies aimed at evaluating the use of nanotechnology-based approaches to target ECs have emerged.
Understanding the interplay between ECs, immune cells and tumor cells can provide important insights into the mechanisms of tumor progression and help overcome the limitations of current therapeutic interventions, including the emergence of treatment resistance and the mechanisms of immune escape.
PL: Conceptualization, Data curation, Writing – original draft. EM: Data curation, Writing – review & editing. NS: Data curation, Writing – review & editing. EF: Data curation, Writing – review & editing. FP: Data curation, Writing – review & editing. GB: Data curation, Writing – review & editing. MP: Data curation, Writing – review & editing. VR: Conceptualization, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Italian Association for Cancer Research (AIRC) through an Investigator Grant no. 20441 to VR. The sponsors of this study are non-profit organizations that support science in general; they had no role in gathering, analyzing, or interpreting the data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hennigs JK, Matuszcak C, Trepel M, Korbelin J. Vascular endothelial cells: heterogeneity and targeting approaches. Cells (2021) 10:2712–51. doi: 10.3390/cells10102712
2. Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med (2012) 2:a006429. doi: 10.1101/cshperspect.a006429
3. Kruger-Genge A, Blocki A, Franke RP, Jung F. Vascular endothelial cell biology: an update. Int J Mol Sci (2019) 20:4411–33. doi: 10.3390/ijms20184411
4. Pusztaszeri MP, Seelentag W, Bosman FT. Immunohistochemical expression of endothelial markers cd31, cd34, von willebrand factor, and fli-1 in normal human tissues. J Histochem Cytochem (2006) 54:385–95. doi: 10.1369/jhc.4A6514.2005
5. Reinhart WH. Shear-dependence of endothelial functions. Experientia (1994) 50:87–93. doi: 10.1007/BF01984940
6. Resnick N, Gimbrone MA Jr. Hemodynamic forces are complex regulators of endothelial gene expression. FASEB J (1995) 9:874–82. doi: 10.1096/fasebj.9.10.7615157
7. Shao Y, Saredy J, Yang WY, Sun Y, Lu Y, Saaoud F, et al. Vascular endothelial cells and innate immunity. Arterioscler Thromb Vasc Biol (2020) 40:e138–e52. doi: 10.1161/ATVBAHA.120.314330
8. Schaaf MB, Houbaert D, Mece O, Agostinis P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ (2019) 26:665–79. doi: 10.1038/s41418-019-0287-8
9. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol (2002) 160:985–1000. doi: 10.1016/S0002-9440(10)64920-6
10. Nagl L, Horvath L, Pircher A, Wolf D. Tumor endothelial cells (Tecs) as potential immune directors of the tumor microenvironment - new findings and future perspectives. Front Cell Dev Biol (2020) 8:766. doi: 10.3389/fcell.2020.00766
11. St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, et al. Genes expressed in human tumor endothelium. Science (2000) 289:1197–202. doi: 10.1126/science.289.5482.1197
12. Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med (2012) 2:a006536. doi: 10.1101/cshperspect.a006536
13. Clere N, Renault S, Corre I. Endothelial-to-mesenchymal transition in cancer. Front Cell Dev Biol (2020) 8:747. doi: 10.3389/fcell.2020.00747
14. Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, et al. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell (2020) 37:21–36 e13. doi: 10.1016/j.ccell.2019.12.001
15. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med (2018) 24:1277–89. doi: 10.1038/s41591-018-0096-5
16. Rohlenova K, Goveia J, Garcia-Caballero M, Subramanian A, Kalucka J, Treps L, et al. Single-cell rna sequencing maps endothelial metabolic plasticity in pathological angiogenesis. Cell Metab (2020) 31:862–77 e14. doi: 10.1016/j.cmet.2020.03.009
17. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer (2019) 18:157. doi: 10.1186/s12943-019-1089-9
18. Fang J, Lu Y, Zheng J, Jiang X, Shen H, Shang X, et al. Exploring the crosstalk between endothelial cells, immune cells, and immune checkpoints in the tumor microenvironment: new insights and therapeutic implications. Cell Death Dis (2023) 14:586. doi: 10.1038/s41419-023-06119-x
19. Dieu-Nosjean MC, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautes-Fridman C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev (2016) 271:260–75. doi: 10.1111/imr.12405
20. Dayan F, Mazure NM, Brahimi-Horn MC, Pouyssegur J. A dialogue between the hypoxia-inducible factor and the tumor microenvironment. Cancer Microenviron (2008) 1:53–68. doi: 10.1007/s12307-008-0006-3
21. Leone P, Buonavoglia A, Fasano R, Solimando AG, De Re V, Cicco S, et al. Insights into the regulation of tumor angiogenesis by micro-rnas. J Clin Med (2019) 8:2030–50. doi: 10.3390/jcm8122030
22. Zhang Y, Wang H, Oliveira RHM, Zhao C, Popel AS. Systems biology of angiogenesis signaling: computational models and omics. WIREs Mech Dis (2022) 14:e1550. doi: 10.1002/wsbm.1550
23. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature (2000) 407:249–57. doi: 10.1038/35025220
24. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell (2011) 146:873–87. doi: 10.1016/j.cell.2011.08.039
25. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer (2003) 3:401–10. doi: 10.1038/nrc1093
26. Matsuda K, Ohga N, Hida Y, Muraki C, Tsuchiya K, Kurosu T, et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem Biophys Res Commun (2010) 394:947–54. doi: 10.1016/j.bbrc.2010.03.089
27. Ohga N, Ishikawa S, Maishi N, Akiyama K, Hida Y, Kawamoto T, et al. Heterogeneity of tumor endothelial cells: comparison between tumor endothelial cells isolated from high- and low-metastatic tumors. Am J Pathol (2012) 180:1294–307. doi: 10.1016/j.ajpath.2011.11.035
28. Hojo T, Maishi N, Towfik AM, Akiyama K, Ohga N, Shindoh M, et al. Ros enhance angiogenic properties via regulation of nrf2 in tumor endothelial cells. Oncotarget (2017) 8:45484–95. doi: 10.18632/oncotarget.17567
29. Kondoh M, Ohga N, Akiyama K, Hida Y, Maishi N, Towfik AM, et al. Hypoxia-induced reactive oxygen species cause chromosomal abnormalities in endothelial cells in the tumor microenvironment. PloS One (2013) 8:e80349. doi: 10.1371/journal.pone.0080349
30. Hida K, Hida Y, Amin DN, Flint AF, Panigrahy D, Morton CC, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res (2004) 64:8249–55. doi: 10.1158/0008-5472.CAN-04-1567
31. Ciesielski O, Biesiekierska M, Panthu B, Vialichka V, Pirola L, Balcerczyk A. The epigenetic profile of tumor endothelial cells. Effects of combined therapy with antiangiogenic and epigenetic drugs on cancer progression. Int J Mol Sci (2020) 21:2606–28. doi: 10.3390/ijms21072606
32. Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, et al. Inhibition of the glycolytic activator pfkfb3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell (2016) 30:968–85. doi: 10.1016/j.ccell.2016.10.006
33. Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated C-myc. Cell (2012) 151:56–67. doi: 10.1016/j.cell.2012.08.026
34. Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, et al. C-myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev (2002) 16:2530–43. doi: 10.1101/gad.1024602
35. Sievert W, Tapio S, Breuninger S, Gaipl U, Andratschke N, Trott KR, et al. Adhesion molecule expression and function of primary endothelial cells in benign and Malignant tissues correlates with proliferation. PloS One (2014) 9:e91808. doi: 10.1371/journal.pone.0091808
36. Naschberger E, Schellerer VS, Rau TT, Croner RS, Sturzl M. Isolation of endothelial cells from human tumors. Methods Mol Biol (2011) 731:209–18. doi: 10.1007/978-1-61779-080-5_18
37. Blanco R, Gerhardt H. Vegf and notch in tip and stalk cell selection. Cold Spring Harb Perspect Med (2013) 3:a006569. doi: 10.1101/cshperspect.a006569
38. Naito H, Kidoya H, Sakimoto S, Wakabayashi T, Takakura N. Identification and characterization of a resident vascular stem/progenitor cell population in preexisting blood vessels. EMBO J (2012) 31:842–55. doi: 10.1038/emboj.2011.465
39. Wakabayashi T, Naito H, Suehiro JI, Lin Y, Kawaji H, Iba T, et al. Cd157 marks tissue-resident endothelial stem cells with homeostatic and regenerative properties. Cell Stem Cell (2018) 22:384–97 e6. doi: 10.1016/j.stem.2018.01.010
40. Yao X, Zeng Y. Tumour associated endothelial cells: origin, characteristics and role in metastasis and anti-angiogenic resistance. Front Physiol (2023) 14:1199225. doi: 10.3389/fphys.2023.1199225
41. Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol (2008) 28:1584–95. doi: 10.1161/ATVBAHA.107.155960
42. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature (2010) 468:829–33. doi: 10.1038/nature09624
43. Patel J, Seppanen EJ, Rodero MP, Wong HY, Donovan P, Neufeld Z, et al. Functional definition of progenitors versus mature endothelial cells reveals key soxf-dependent differentiation process. Circulation (2017) 135:786–805. doi: 10.1161/CIRCULATIONAHA.116.024754
44. Hida K, Maishi N, Akiyama K, Ohmura-Kakutani H, Torii C, Ohga N, et al. Tumor endothelial cells with high aldehyde dehydrogenase activity show drug resistance. Cancer Sci (2017) 108:2195–203. doi: 10.1111/cas.13388
45. Ohmura-Kakutani H, Akiyama K, Maishi N, Ohga N, Hida Y, Kawamoto T, et al. Identification of tumor endothelial cells with high aldehyde dehydrogenase activity and a highly angiogenic phenotype. PloS One (2014) 9:e113910. doi: 10.1371/journal.pone.0113910
46. Ge H, Luo H. Overview of advances in vasculogenic mimicry - a potential target for tumor therapy. Cancer Manag Res (2018) 10:2429–37. doi: 10.2147/CMAR.S164675
47. Streubel B, Chott A, Huber D, Exner M, Jager U, Wagner O, et al. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. N Engl J Med (2004) 351:250–9. doi: 10.1056/NEJMoa033153
48. Chen H, Campbell RA, Chang Y, Li M, Wang CS, Li J, et al. Pleiotrophin produced by multiple myeloma induces transdifferentiation of monocytes into vascular endothelial cells: A novel mechanism of tumor-induced vasculogenesis. Blood (2009) 113:1992–2002. doi: 10.1182/blood-2008-02-133751
49. Fernandez Pujol B, Lucibello FC, Zuzarte M, Lutjens P, Muller R, Havemann K. Dendritic cells derived from peripheral monocytes express endothelial markers and in the presence of angiogenic growth factors differentiate into endothelial-like cells. Eur J Cell Biol (2001) 80:99–110. doi: 10.1078/0171-9335-00136
50. Amersfoort J, Eelen G, Carmeliet P. Immunomodulation by endothelial cells - partnering up with the immune system? Nat Rev Immunol (2022) 22:576–88. doi: 10.1038/s41577-022-00694-4
51. Carman CV, Martinelli R. T lymphocyte-endothelial interactions: emerging understanding of trafficking and antigen-specific immunity. Front Immunol (2015) 6:603. doi: 10.3389/fimmu.2015.00603
52. De Sanctis F, Ugel S, Facciponte J, Facciabene A. The dark side of tumor-associated endothelial cells. Semin Immunol (2018) 35:35–47. doi: 10.1016/j.smim.2018.02.002
53. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7:678–89. doi: 10.1038/nri2156
54. Harjunpaa H, Llort Asens M, Guenther C, Fagerholm SC. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front Immunol (2019) 10:1078. doi: 10.3389/fimmu.2019.01078
55. De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr., et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest (1995) 96:60–8. doi: 10.1172/JCI118074
56. He Q, Jamalpour M, Bergquist E, Anderson RL, Gustafsson K, Welsh M. Mouse breast carcinoma monocytic/macrophagic myeloid-derived suppressor cell infiltration as a consequence of endothelial dysfunction in shb-deficient endothelial cells increases tumor lung metastasis. Int J Mol Sci (2021) 22:11478–92. doi: 10.3390/ijms222111478
57. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via ccl28 and T(Reg) cells. Nature (2011) 475:226–30. doi: 10.1038/nature10169
58. Irjala H, Elima K, Johansson EL, Merinen M, Kontula K, Alanen K, et al. The same endothelial receptor controls lymphocyte traffic both in vascular and lymphatic vessels. Eur J Immunol (2003) 33:815–24. doi: 10.1002/eji.200323859
59. Karikoski M, Marttila-Ichihara F, Elima K, Rantakari P, Hollmen M, Kelkka T, et al. Clever-1/stabilin-1 controls cancer growth and metastasis. Clin Cancer Res (2014) 20:6452–64. doi: 10.1158/1078-0432.CCR-14-1236
60. Martens JH, Kzhyshkowska J, Falkowski-Hansen M, Schledzewski K, Gratchev A, Mansmann U, et al. Differential expression of a gene signature for scavenger/lectin receptors by endothelial cells and macrophages in human lymph node sinuses, the primary sites of regional metastasis. J Pathol (2006) 208:574–89. doi: 10.1002/path.1921
61. Prevo R, Banerji S, Ni J, Jackson DG. Rapid plasma membrane-endosomal trafficking of the lymph node sinus and high endothelial venule scavenger receptor/homing receptor stabilin-1 (Feel-1/clever-1). J Biol Chem (2004) 279:52580–92. doi: 10.1074/jbc.M406897200
62. Chandler KB, Costello CE, Rahimi N. Glycosylation in the tumor microenvironment: implications for tumor angiogenesis and metastasis. Cells (2019) 8:544–65. doi: 10.3390/cells8060544
63. Griffioen AW, Damen CA, Blijham GH, Groenewegen G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood (1996) 88:667–73.
64. Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, et al. Vegf suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of nf-kappab-induced endothelial activation. FASEB J (2015) 29:227–38. doi: 10.1096/fj.14-250985
65. Thompson TW, Kim AB, Li PJ, Wang J, Jackson BT, Huang KTH, et al. Endothelial cells express nkg2d ligands and desensitize antitumor nk responses. Elife (2017) 6:e30881–902. doi: 10.7554/eLife.30881
66. Won Jun H, Kyung Lee H, Ho Na I, Jeong Lee S, Kim K, Park G, et al. The role of ccl2, ccl7, icam-1, and vcam-1 in interaction of endothelial cells and natural killer cells. Int Immunopharmacol (2022) 113:109332. doi: 10.1016/j.intimp.2022.109332
67. Doak GR, Schwertfeger KL, Wood DK. Distant relations: macrophage functions in the metastatic niche. Trends Cancer (2018) 4:445–59. doi: 10.1016/j.trecan.2018.03.011
68. Wang X, Zhao X, Wang K, Wu L, Duan T. Interaction of monocytes/macrophages with ovarian cancer cells promotes angiogenesis in vitro. Cancer Sci (2013) 104:516–23. doi: 10.1111/cas.12110
69. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res (2008) 14:6735–41. doi: 10.1158/1078-0432.CCR-07-4843
70. Yeo EJ, Cassetta L, Qian BZ, Lewkowich I, Li JF, Stefater JA 3rd, et al. Myeloid wnt7b mediates the angiogenic switch and metastasis in breast cancer. Cancer Res (2014) 74:2962–73. doi: 10.1158/0008-5472.CAN-13-2421
71. Dalton HJ, Armaiz-Pena GN, Gonzalez-Villasana V, Lopez-Berestein G, Bar-Eli M, Sood AK. Monocyte subpopulations in angiogenesis. Cancer Res (2014) 74:1287–93. doi: 10.1158/0008-5472.CAN-13-2825
72. Hellenthal KEM, Brabenec L, Wagner NM. Regulation and dysregulation of endothelial permeability during systemic inflammation. Cells (2022) 11:1935–56. doi: 10.3390/cells11121935
73. Wu Q, Wu X, Ying X, Zhu Q, Wang X, Jiang L, et al. Suppression of endothelial cell migration by tumor associated macrophage-derived exosomes is reversed by epithelial ovarian cancer exosomal lncrna. Cancer Cell Int (2017) 17:62. doi: 10.1186/s12935-017-0430-x
74. Huang X, Bai X, Cao Y, Wu J, Huang M, Tang D, et al. Lymphoma endothelium preferentially expresses tim-3 and facilitates the progression of lymphoma by mediating immune evasion. J Exp Med (2010) 207:505–20. doi: 10.1084/jem.20090397
75. Yang Y, Guo Z, Chen W, Wang X, Cao M, Han X, et al. M2 macrophage-derived exosomes promote angiogenesis and growth of pancreatic ductal adenocarcinoma by targeting E2f2. Mol Ther (2021) 29:1226–38. doi: 10.1016/j.ymthe.2020.11.024
76. Bieniasz-Krzywiec P, Martin-Perez R, Ehling M, Garcia-Caballero M, Pinioti S, Pretto S, et al. Podoplanin-expressing macrophages promote lymphangiogenesis and lymphoinvasion in breast cancer. Cell Metab (2019) 30:917–36 e10. doi: 10.1016/j.cmet.2019.07.015
77. Gao L, Chen K, Gao Q, Wang X, Sun J, Yang YG. Cd47 deficiency in tumor stroma promotes tumor progression by enhancing angiogenesis. Oncotarget (2017) 8:22406–13. doi: 10.18632/oncotarget.9899
78. Maxhimer JB, Soto-Pantoja DR, Ridnour LA, Shih HB, Degraff WG, Tsokos M, et al. Radioprotection in normal tissue and delayed tumor growth by blockade of cd47 signaling. Sci Transl Med (2009) 1:3ra7. doi: 10.1126/scitranslmed.3000139
79. Cols M, Barra CM, He B, Puga I, Xu W, Chiu A, et al. Stromal endothelial cells establish a bidirectional crosstalk with chronic lymphocytic leukemia cells through the tnf-related factors baff, april, and cd40l. J Immunol (2012) 188:6071–83. doi: 10.4049/jimmunol.1102066
80. Shen Y, Wang X, Liu Y, Singhal M, Gurkaslar C, Valls AF, et al. Stat3-yap/taz signaling in endothelial cells promotes tumor angiogenesis. Sci Signal (2021) 14:eabj8393. doi: 10.1126/scisignal.abj8393
81. Yang C, Lee H, Pal S, Jove V, Deng J, Zhang W, et al. B cells promote tumor progression via stat3 regulated-angiogenesis. PloS One (2013) 8:e64159. doi: 10.1371/journal.pone.0064159
82. Kam NW, Wu KC, Dai W, Wang Y, Yan LYC, Shakya R, et al. Peritumoral B cells drive proangiogenic responses in hmgb1-enriched esophageal squamous cell carcinoma. Angiogenesis (2022) 25:181–203. doi: 10.1007/s10456-021-09819-0
83. Zhao Q, Han YM, Song P, Liu Z, Yuan Z, Zou MH. Endothelial cell-specific expression of serine/threonine kinase 11 modulates dendritic cell differentiation. Nat Commun (2022) 13:648. doi: 10.1038/s41467-022-28316-6
84. Gottfried E, Kreutz M, Haffner S, Holler E, Iacobelli M, Andreesen R, et al. Differentiation of human tumour-associated dendritic cells into endothelial-like cells: an alternative pathway of tumour angiogenesis. Scand J Immunol (2007) 65:329–35. doi: 10.1111/j.1365-3083.2007.01903.x
85. Lu J, Zhao J, Liu K, Zhao J, Yang H, Huang Y, et al. Mapk/erk1/2 signaling mediates endothelial-like differentiation of immature dcs in the microenvironment of esophageal squamous cell carcinoma. Cell Mol Life Sci (2010) 67:2091–106. doi: 10.1007/s00018-010-0316-8
86. Ammendola M, Leporini C, Marech I, Gadaleta CD, Scognamillo G, Sacco R, et al. Targeting mast cells tryptase in tumor microenvironment: A potential antiangiogenetic strategy. BioMed Res Int (2014) 2014:154702. doi: 10.1155/2014/154702
87. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. Sting activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci USA (2015) 112:15408–13. doi: 10.1073/pnas.1512832112
88. Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, et al. Cyclic gmp-amp synthase is activated by double-stranded DNA-induced oligomerization. Immunity (2013) 39:1019–31. doi: 10.1016/j.immuni.2013.10.019
89. Berger G, Knelson EH, Jimenez-Macias JL, Nowicki MO, Han S, Panagioti E, et al. Sting activation promotes robust immune response and nk cell-mediated tumor regression in glioblastoma models. Proc Natl Acad Sci USA (2022) 119:e2111003119. doi: 10.1073/pnas.2111003119
90. Berger G, Marloye M, Lawler SE. Pharmacological modulation of the sting pathway for cancer immunotherapy. Trends Mol Med (2019) 25:412–27. doi: 10.1016/j.molmed.2019.02.007
91. Chen YP, Xu L, Tang TW, Chen CH, Zheng QH, Liu TP, et al. Sting activator C-di-gmp-loaded mesoporous silica nanoparticles enhance immunotherapy against breast cancer. ACS Appl Mater Interfaces (2020) 12:56741–52. doi: 10.1021/acsami.0c16728
92. Ohkuri T, Ghosh A, Kosaka A, Zhu J, Ikeura M, David M, et al. Sting contributes to antiglioma immunity via triggering type I ifn signals in the tumor microenvironment. Cancer Immunol Res (2014) 2:1199–208. doi: 10.1158/2326-6066.CIR-14-0099
93. An M, Yu C, Xi J, Reyes J, Mao G, Wei WZ, et al. Induction of necrotic cell death and activation of sting in the tumor microenvironment via cationic silica nanoparticles leading to enhanced antitumor immunity. Nanoscale (2018) 10:9311–9. doi: 10.1039/c8nr01376d
94. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. Sting activation reprograms tumor vasculatures and synergizes with vegfr2 blockade. J Clin Invest (2019) 129:4350–64. doi: 10.1172/JCI125413
95. Sakano Y, Noda T, Kobayashi S, Sasaki K, Iwagami Y, Yamada D, et al. Tumor endothelial cell-induced cd8(+) T-cell exhaustion via gpnmb in hepatocellular carcinoma. Cancer Sci (2022) 113:1625–38. doi: 10.1111/cas.15331
96. Georganaki M, Ramachandran M, Tuit S, Nunez NG, Karampatzakis A, Fotaki G, et al. Tumor endothelial cell up-regulation of ido1 is an immunosuppressive feed-back mechanism that reduces the response to cd40-stimulating immunotherapy. Oncoimmunology (2020) 9:1730538. doi: 10.1080/2162402X.2020.1730538
97. Munn DH, Mellor AL. Ido in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
98. Fujiwara Y, Kato S, Nesline MK, Conroy JM, DePietro P, Pabla S, et al. Indoleamine 2,3-dioxygenase (Ido) inhibitors and cancer immunotherapy. Cancer Treat Rev (2022) 110:102461. doi: 10.1016/j.ctrv.2022.102461
99. Le Naour J, Galluzzi L, Zitvogel L, Kroemer G, Vacchelli E. Trial watch: ido inhibitors in cancer therapy. Oncoimmunology (2020) 9:1777625. doi: 10.1080/2162402X.2020.1777625
100. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium fasl establishes a selective immune barrier promoting tolerance in tumors. Nat Med (2014) 20:607–15. doi: 10.1038/nm.3541
101. Leone P, Di Lernia G, Solimando AG, Cicco S, Saltarella I, Lamanuzzi A, et al. Bone marrow endothelial cells sustain a tumor-specific cd8(+) T cell subset with suppressive function in myeloma patients. Oncoimmunology (2019) 8:e1486949. doi: 10.1080/2162402X.2018.1486949
102. Lim WC, Olding M, Healy E, Millar TM. Human endothelial cells modulate cd4(+) T cell populations and enhance regulatory T cell suppressive capacity. Front Immunol (2018) 9:565. doi: 10.3389/fimmu.2018.00565
103. Zheng Z, Xu PP, Wang L, Zhao HJ, Weng XQ, Zhong HJ, et al. Mir21 sensitized B-lymphoma cells to abt-199 via icos/icosl-mediated interaction of treg cells with endothelial cells. J Exp Clin Cancer Res (2017) 36:82. doi: 10.1186/s13046-017-0551-z
104. Raineri D, Dianzani C, Cappellano G, Maione F, Baldanzi G, Iacobucci I, et al. Osteopontin binds icosl promoting tumor metastasis. Commun Biol (2020) 3:615. doi: 10.1038/s42003-020-01333-1
105. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol (2014) 35:571–80. doi: 10.1016/j.it.2014.09.006
106. Link A, Hardie DL, Favre S, Britschgi MR, Adams DH, Sixt M, et al. Association of T-zone reticular networks and conduits with ectopic lymphoid tissues in mice and humans. Am J Pathol (2011) 178:1662–75. doi: 10.1016/j.ajpath.2010.12.039
107. Penaranda C, Tang Q, Ruddle NH, Bluestone JA. Prevention of diabetes by fty720-mediated stabilization of peri-islet tertiary lymphoid organs. Diabetes (2010) 59:1461–8. doi: 10.2337/db09-1129
108. Stranford S, Ruddle NH. Follicular dendritic cells, conduits, lymphatic vessels, and high endothelial venules in tertiary lymphoid organs: parallels with lymph node stroma. Front Immunol (2012) 3:350. doi: 10.3389/fimmu.2012.00350
109. Ruddle NH. Lymphatic vessels and tertiary lymphoid organs. J Clin Invest (2014) 124:953–9. doi: 10.1172/JCI71611
110. Meier D, Bornmann C, Chappaz S, Schmutz S, Otten LA, Ceredig R, et al. Ectopic lymphoid-organ development occurs through interleukin 7-mediated enhanced survival of lymphoid-tissue-inducer cells. Immunity (2007) 26:643–54. doi: 10.1016/j.immuni.2007.04.009
111. Colbeck EJ, Ager A, Gallimore A, Jones GW. Tertiary lymphoid structures in cancer: drivers of antitumor immunity, immunosuppression, or bystander sentinels in disease? Front Immunol (2017) 8:1830. doi: 10.3389/fimmu.2017.01830
112. Deteix C, Attuil-Audenis V, Duthey A, Patey N, McGregor B, Dubois V, et al. Intragraft th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol (2010) 184:5344–51. doi: 10.4049/jimmunol.0902999
113. Guedj K, Khallou-Laschet J, Clement M, Morvan M, Gaston AT, Fornasa G, et al. M1 macrophages act as ltbetar-independent lymphoid tissue inducer cells during atherosclerosis-related lymphoid neogenesis. Cardiovasc Res (2014) 101:434–43. doi: 10.1093/cvr/cvt263
114. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of rorgamma T and lti cells. J Exp Med (2011) 208:125–34. doi: 10.1084/jem.20100052
115. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity (2011) 35:986–96. doi: 10.1016/j.immuni.2011.10.015
116. Furtado GC, Marinkovic T, Martin AP, Garin A, Hoch B, Hubner W, et al. Lymphotoxin beta receptor signaling is required for inflammatory lymphangiogenesis in the thyroid. Proc Natl Acad Sci USA (2007) 104:5026–31. doi: 10.1073/pnas.0606697104
117. Luther SA, Ansel KM, Cyster JG. Overlapping roles of cxcl13, interleukin 7 receptor alpha, and ccr7 ligands in lymph node development. J Exp Med (2003) 197:1191–8. doi: 10.1084/jem.20021294
118. Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol (2003) 3:292–303. doi: 10.1038/nri1054
119. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res (2011) 71:5678–87. doi: 10.1158/0008-5472.CAN-11-0431
120. Mionnet C, Sanos SL, Mondor I, Jorquera A, Laugier JP, Germain RN, et al. High endothelial venules as traffic control points maintaining lymphocyte population homeostasis in lymph nodes. Blood (2011) 118:6115–22. doi: 10.1182/blood-2011-07-367409
121. Ruddle NH. High endothelial venules and lymphatic vessels in tertiary lymphoid organs: characteristics, functions, and regulation. Front Immunol (2016) 7:491. doi: 10.3389/fimmu.2016.00491
122. Vella G, Guelfi S, Bergers G. High endothelial venules: A vascular perspective on tertiary lymphoid structures in cancer. Front Immunol (2021) 12:736670. doi: 10.3389/fimmu.2021.736670
123. Choe K, Moon J, Lee SY, Song E, Back JH, Song JH, et al. Stepwise transmigration of T- and B cells through a perivascular channel in high endothelial venules. Life Sci Alliance (2021) 4:736670–685. doi: 10.26508/lsa.202101086
124. Fridman WH, Meylan M, Petitprez F, Sun CM, Italiano A, Sautes-Fridman C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol (2022) 19:441–57. doi: 10.1038/s41571-022-00619-z
125. Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating cd8+ T cells. Cancer Res (2014) 74:705–15. doi: 10.1158/0008-5472.CAN-13-1342
126. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. Cd4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest (2013) 123:2873–92. doi: 10.1172/JCI67428
127. Hennequin A, Derangere V, Boidot R, Apetoh L, Vincent J, Orry D, et al. Tumor infiltration by tbet+ Effector T cells and cd20+ B cells is associated with survival in gastric cancer patients. Oncoimmunology (2016) 5:e1054598. doi: 10.1080/2162402X.2015.1054598
128. Baratin M, Simon L, Jorquera A, Ghigo C, Dembele D, Nowak J, et al. T cell zone resident macrophages silently dispose of apoptotic cells in the lymph node. Immunity (2017) 47:349–62.e5. doi: 10.1016/j.immuni.2017.07.019
129. Schumacher TN, Thommen DS. Tertiary lymphoid structures in cancer. Science (2022) 375:eabf9419. doi: 10.1126/science.abf9419
130. Di Caro G, Bergomas F, Grizzi F, Doni A, Bianchi P, Malesci A, et al. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin Cancer Res (2014) 20:2147–58. doi: 10.1158/1078-0432.CCR-13-2590
131. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med (2014) 189:832–44. doi: 10.1164/rccm.201309-1611OC
132. Kroeger DR, Milne K, Nelson BH. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin Cancer Res (2016) 22:3005–15. doi: 10.1158/1078-0432.CCR-15-2762
133. Ishigami E, Sakakibara M, Sakakibara J, Masuda T, Fujimoto H, Hayama S, et al. Coexistence of regulatory B cells and regulatory T cells in tumor-infiltrating lymphocyte aggregates is a prognostic factor in patients with breast cancer. Breast Cancer (2019) 26:180–9. doi: 10.1007/s12282-018-0910-4
134. Wei X, Jin Y, Tian Y, Zhang H, Wu J, Lu W, et al. Regulatory B cells contribute to the impaired antitumor immunity in ovarian cancer patients. Tumour Biol (2016) 37:6581–8. doi: 10.1007/s13277-015-4538-0
135. Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer (2019) 19:307–25. doi: 10.1038/s41568-019-0144-6
136. Moyron-Quiroz JE, Rangel-Moreno J, Hartson L, Kusser K, Tighe MP, Klonowski KD, et al. Persistence and responsiveness of immunologic memory in the absence of secondary lymphoid organs. Immunity (2006) 25:643–54. doi: 10.1016/j.immuni.2006.08.022
137. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol (2014) 14:447–62. doi: 10.1038/nri3700
138. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10:942–9. doi: 10.1038/nm1093
139. Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol (2008) 26:4410–7. doi: 10.1200/JCO.2007.15.0284
140. Wang B, Liu J, Han Y, Deng Y, Li J, Jiang Y. The presence of tertiary lymphoid structures provides new insight into the clinicopathological features and prognosis of patients with breast cancer. Front Immunol (2022) 13:868155. doi: 10.3389/fimmu.2022.868155
141. Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer (2015) 112:1782–90. doi: 10.1038/bjc.2015.145
142. Calderaro J, Petitprez F, Becht E, Laurent A, Hirsch TZ, Rousseau B, et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol (2019) 70:58–65. doi: 10.1016/j.jhep.2018.09.003
143. Lin Q, Tao P, Wang J, Ma L, Jiang Q, Li J, et al. Tumor-associated tertiary lymphoid structure predicts postoperative outcomes in patients with primary gastrointestinal stromal tumors. Oncoimmunology (2020) 9:1747339. doi: 10.1080/2162402X.2020.1747339
144. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature (2020) 577:561–5. doi: 10.1038/s41586-019-1914-8
145. Asam S, Nayar S, Gardner D, Barone F. Stromal cells in tertiary lymphoid structures: architects of autoimmunity. Immunol Rev (2021) 302:184–95. doi: 10.1111/imr.12987
146. McMullen TP, Lai R, Dabbagh L, Wallace TM, de Gara CJ. Survival in rectal cancer is predicted by T cell infiltration of tumour-associated lymphoid nodules. Clin Exp Immunol (2010) 161:81–8. doi: 10.1111/j.1365-2249.2010.04147.x
147. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol (2006) 24:5373–80. doi: 10.1200/JCO.2006.05.9584
148. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol (2007) 25:2586–93. doi: 10.1200/JCO.2006.09.4565
149. Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, et al. Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity (2015) 43:579–90. doi: 10.1016/j.immuni.2015.08.006
150. Fridman WH, Meylan M, Pupier G, Calvez A, Hernandez I, Sautes-Fridman C. Tertiary lymphoid structures and B cells: an intratumoral immunity cycle. Immunity (2023) 56:2254–69. doi: 10.1016/j.immuni.2023.08.009
151. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature (2020) 577:549–55. doi: 10.1038/s41586-019-1922-8
152. Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature (2020) 577:556–60. doi: 10.1038/s41586-019-1906-8
153. Vanhersecke L, Brunet M, Guegan JP, Rey C, Bougouin A, Cousin S, et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of pd-L1 expression. Nat Cancer (2021) 2:794–802. doi: 10.1038/s43018-021-00232-6
154. Cottrell TR, Thompson ED, Forde PM, Stein JE, Duffield AS, Anagnostou V, et al. Pathologic features of response to neoadjuvant anti-pd-1 in resected non-small-cell lung carcinoma: A proposal for quantitative immune-related pathologic response criteria (Irprc). Ann Oncol (2018) 29:1853–60. doi: 10.1093/annonc/mdy218
155. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct pd-1(+) cd8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with pd-1 blockade. Nat Med (2018) 24:994–1004. doi: 10.1038/s41591-018-0057-z
156. Meylan M, Petitprez F, Becht E, Bougouin A, Pupier G, Calvez A, et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity (2022) 55:527–41 e5. doi: 10.1016/j.immuni.2022.02.001
157. Gao J, Navai N, Alhalabi O, Siefker-Radtke A, Campbell MT, Tidwell RS, et al. Neoadjuvant pd-L1 plus ctla-4 blockade in patients with cisplatin-ineligible operable high-risk urothelial carcinoma. Nat Med (2020) 26:1845–51. doi: 10.1038/s41591-020-1086-y
158. Lee-Chang C, Bodogai M, Martin-Montalvo A, Wejksza K, Sanghvi M, Moaddel R, et al. Inhibition of breast cancer metastasis by resveratrol-mediated inactivation of tumor-evoked regulatory B cells. J Immunol (2013) 191:4141–51. doi: 10.4049/jimmunol.1300606
159. Roya N, Fatemeh T, Faramarz MA, Milad SG, Mohammad-Javad S, Najmeh SV, et al. Frequency of il-10+Cd19+ B cells in patients with prostate cancer compared to patients with benign prostatic hyperplasia. Afr Health Sci (2020) 20:1264–72. doi: 10.4314/ahs.v20i3.31
160. Wang WW, Yuan XL, Chen H, Xie GH, Ma YH, Zheng YX, et al. Cd19+Cd24hicd38hibregs involved in downregulate helper T cells and upregulate regulatory T cells in gastric cancer. Oncotarget (2015) 6:33486–99. doi: 10.18632/oncotarget.5588
161. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-pd-L1 therapy stimulates tumor immunity through hev formation. Sci Transl Med (2017) 9:eaak9679–707. doi: 10.1126/scitranslmed.aak9679
162. Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res (2014) 2:616–31. doi: 10.1158/2326-6066.CIR-14-0027
163. Maldonado L, Teague JE, Morrow MP, Jotova I, Wu TC, Wang C, et al. Intramuscular therapeutic vaccination targeting hpv16 induces T cell responses that localize in mucosal lesions. Sci Transl Med (2014) 6:221ra13. doi: 10.1126/scitranslmed.3007323
164. Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol (2004) 5:74–80. doi: 10.1038/ni1013
165. Hsu MC, Pan MR, Hung WC. Two birds, one stone: double hits on tumor growth and lymphangiogenesis by targeting vascular endothelial growth factor receptor 3. Cells (2019) 8:270–90. doi: 10.3390/cells8030270
166. Decio A, Taraboletti G, Patton V, Alzani R, Perego P, Fruscio R, et al. Vascular endothelial growth factor C promotes ovarian carcinoma progression through paracrine and autocrine mechanisms. Am J Pathol (2014) 184:1050–61. doi: 10.1016/j.ajpath.2013.12.030
167. Goussia A, Simou N, Zagouri F, Manousou K, Lazaridis G, Gogas H, et al. Associations of angiogenesis-related proteins with specific prognostic factors, breast cancer subtypes and survival outcome in early-stage breast cancer patients. A hellenic cooperative oncology group (Hecog) trial. . PloS One (2018) 13:e0200302. doi: 10.1371/journal.pone.0200302
168. Wang J, Taylor A, Showeil R, Trivedi P, Horimoto Y, Bagwan I, et al. Expression profiling and significance of vegf-a, vegfr2, vegfr3 and related proteins in endometrial carcinoma. Cytokine (2014) 68:94–100. doi: 10.1016/j.cyto.2014.04.005
169. Yang ZS, Xu YF, Huang FF, Ding GF. Associations of nm23h1, vegf-C, and vegf-3 receptor in human prostate cancer. Molecules (2014) 19:6851–62. doi: 10.3390/molecules19056851
170. Zhu G, Huang Q, Zheng W, Huang Y, Hua J, Yang S, et al. Lps upregulated vegfr-3 expression promote migration and invasion in colorectal cancer via a mechanism of increased nf-kappab binding to the promoter of vegfr-3. Cell Physiol Biochem (2016) 39:1665–78. doi: 10.1159/000447868
171. Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, et al. Vegf-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep (2012) 1:191–9. doi: 10.1016/j.celrep.2012.01.005
172. Lee JY, Park S, Kim DC, Yoon JH, Shin SH, Min WS, et al. A vegfr-3 antagonist increases ifn-gamma expression on low functioning nk cells in acute myeloid leukemia. J Clin Immunol (2013) 33:826–37. doi: 10.1007/s10875-013-9877-2
173. Li Y, Weng Y, Zhong L, Chong H, Chen S, Sun Y, et al. Vegfr3 inhibition chemosensitizes lung adenocarcinoma A549 cells in the tumor-associated macrophage microenvironment through upregulation of P53 and pten. Oncol Rep (2017) 38:2761–73. doi: 10.3892/or.2017.5969
174. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet (2014) 384:319–28. doi: 10.1016/S0140-6736(14)60421-9
175. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the asia-pacific region with advanced hepatocellular carcinoma: A phase iii randomised, double-blind, placebo-controlled trial. Lancet Oncol (2009) 10:25–34. doi: 10.1016/S1470-2045(08)70285-7
176. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet (2006) 368:1329–38. doi: 10.1016/S0140-6736(06)69446-4
177. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med (2007) 356:125–34. doi: 10.1056/NEJMoa060655
178. Haas NB, Manola J, Uzzo RG, Flaherty KT, Wood CG, Kane C, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (Ecog-acrin E2805): A double-blind, placebo-controlled, randomised, phase 3 trial. Lancet (2016) 387:2008–16. doi: 10.1016/S0140-6736(16)00559-6
179. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med (2008) 359:378–90. doi: 10.1056/NEJMoa0708857
180. Motzer RJ, Haas NB, Donskov F, Gross-Goupil M, Varlamov S, Kopyltsov E, et al. Randomized phase iii trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma. J Clin Oncol (2017) 35:3916–23. doi: 10.1200/JCO.2017.73.5324
181. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med (2007) 356:115–24. doi: 10.1056/NEJMoa065044
182. Ravaud A, Motzer RJ, Pandha HS, George DJ, Pantuck AJ, Patel A, et al. Adjuvant sunitinib in high-risk renal-cell carcinoma after nephrectomy. N Engl J Med (2016) 375:2246–54. doi: 10.1056/NEJMoa1611406
183. Sleijfer S, Ray-Coquard I, Papai Z, Le Cesne A, Scurr M, Schoffski P, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: A phase ii study from the european organisation for research and treatment of cancer-soft tissue and bone sarcoma group (Eortc study 62043). J Clin Oncol (2009) 27:3126–32. doi: 10.1200/JCO.2008.21.3223
184. Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase iii trial. J Clin Oncol (2010) 28:1061–8. doi: 10.1200/JCO.2009.23.9764
185. Yoshimatsu Y, Watabe T. Emerging roles of inflammation-mediated endothelial-mesenchymal transition in health and disease. Inflammation Regener (2022) 42:9. doi: 10.1186/s41232-021-00186-3
186. Piera-Velazquez S, Jimenez SA. Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol Rev (2019) 99:1281–324. doi: 10.1152/physrev.00021.2018
187. Arciniegas E, Sutton AB, Allen TD, Schor AM. Transforming growth factor beta 1 promotes the differentiation of endothelial cells into smooth muscle-like cells in vitro. J Cell Sci (1992) 103:521–9. doi: 10.1242/jcs.103.2.521
188. Goumans MJ, van Zonneveld AJ, ten Dijke P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: A switch to cardiac fibrosis? Trends Cardiovasc Med (2008) 18:293–8. doi: 10.1016/j.tcm.2009.01.001
189. Mihira H, Suzuki HI, Akatsu Y, Yoshimatsu Y, Igarashi T, Miyazono K, et al. Tgf-beta-induced mesenchymal transition of ms-1 endothelial cells requires smad-dependent cooperative activation of rho signals and mrtf-A. J Biochem (2012) 151:145–56. doi: 10.1093/jb/mvr121
190. van Meeteren LA, ten Dijke P. Regulation of endothelial cell plasticity by tgf-beta. Cell Tissue Res (2012) 347:177–86. doi: 10.1007/s00441-011-1222-6
191. Xiao L, Dudley AC. Fine-tuning vascular fate during endothelial-mesenchymal transition. J Pathol (2017) 241:25–35. doi: 10.1002/path.4814
192. Xiong J, Kawagishi H, Yan Y, Liu J, Wells QS, Edmunds LR, et al. A metabolic basis for endothelial-to-mesenchymal transition. Mol Cell (2018) 69:689–98 e7. doi: 10.1016/j.molcel.2018.01.010
193. Fan CS, Chen WS, Chen LL, Chen CC, Hsu YT, Chua KV, et al. Osteopontin-integrin engagement induces hif-1alpha-tcf12-mediated endothelial-mesenchymal transition to exacerbate colorectal cancer. Oncotarget (2018) 9:4998–5015. doi: 10.18632/oncotarget.23578
194. Fan CS, Chen LL, Hsu TA, Chen CC, Chua KV, Li CP, et al. Endothelial-mesenchymal transition harnesses hsp90alpha-secreting M2-macrophages to exacerbate pancreatic ductal adenocarcinoma. J Hematol Oncol (2019) 12:138. doi: 10.1186/s13045-019-0826-2
195. Choi SH, Kim AR, Nam JK, Kim JM, Kim JY, Seo HR, et al. Tumour-vasculature development via endothelial-to-mesenchymal transition after radiotherapy controls cd44v6(+) cancer cell and macrophage polarization. Nat Commun (2018) 9:5108. doi: 10.1038/s41467-018-07470-w
196. Huang M, Liu T, Ma P, Mitteer RA Jr., Zhang Z, Kim HJ, et al. C-met-mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J Clin Invest (2016) 126:1801–14. doi: 10.1172/JCI84876
197. Huang M, Zhang D, Wu JY, Xing K, Yeo E, Li C, et al. Wnt-mediated endothelial transformation into mesenchymal stem cell-like cells induces chemoresistance in glioblastoma. Sci Transl Med (2020) 12:eaay7522–551. doi: 10.1126/scitranslmed.aay7522
198. Nie L, Lyros O, Medda R, Jovanovic N, Schmidt JL, Otterson MF, et al. Endothelial-mesenchymal transition in normal human esophageal endothelial cells cocultured with esophageal adenocarcinoma cells: role of il-1beta and tgf-beta2. Am J Physiol Cell Physiol (2014) 307:C859–77. doi: 10.1152/ajpcell.00081.2014
199. Wang SH, Chang JS, Hsiao JR, Yen YC, Jiang SS, Liu SH, et al. Tumour cell-derived wnt5b modulates in vitro lymphangiogenesis via induction of partial endothelial-mesenchymal transition of lymphatic endothelial cells. Oncogene (2017) 36:1503–15. doi: 10.1038/onc.2016.317
200. Wilkus-Adamczyk K, Brodaczewska K, Majewska A, Kieda C. Microenvironment commits breast tumor ecs to dedifferentiation by micro-rna-200-B-3p regulation and extracellular matrix remodeling. Front Cell Dev Biol (2023) 11:1125077. doi: 10.3389/fcell.2023.1125077
201. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res (2007) 67:10123–8. doi: 10.1158/0008-5472.CAN-07-3127
202. Choi KJ, Nam JK, Kim JH, Choi SH, Lee YJ. Endothelial-to-mesenchymal transition in anticancer therapy and normal tissue damage. Exp Mol Med (2020) 52:781–92. doi: 10.1038/s12276-020-0439-4
203. Platel V, Corre I, Clere N. Endothelial-to-mesenchymal transition (Endomt): roles in tumorigenesis, metastatic extravasation and therapy resistance. J Oncol (2019) 2019:8361945. doi: 10.1155/2019/8361945
204. Maishi N, Annan DA, Kikuchi H, Hida Y, Hida K. Tumor endothelial heterogeneity in cancer progression. Cancers (Basel) (2019) 11:1511–27. doi: 10.3390/cancers11101511
205. Rogava M, Braun AD, van der Sluis TC, Shridhar N, Tuting T, Gaffal E. Tumor cell intrinsic toll-like receptor 4 signaling promotes melanoma progression and metastatic dissemination. Int J Cancer (2022) 150:142–51. doi: 10.1002/ijc.33804
206. Wu FH, Yuan Y, Li D, Lei Z, Song CW, Liu YY, et al. Endothelial cell-expressed tim-3 facilitates metastasis of melanoma cells by activating the nf-kappab pathway. Oncol Rep (2010) 24:693–9.
207. Cong Y, Cui Y, Zhu S, Cao J, Zou H, Martin TA, et al. Tim-3 promotes cell aggressiveness and paclitaxel resistance through nf-kappab/stat3 signalling pathway in breast cancer cells. Chin J Cancer Res (2020) 32:564–79. doi: 10.21147/j.issn.1000-9604.2020.05.02
208. Cong Y, Wang X, Wang S, Qiao G, Li Y, Cao J, et al. Tim-3 promotes tube formation and decreases tight junction formation in vascular endothelial cells. Biosci Rep (2020) 40:BSR20202130–43. doi: 10.1042/BSR20202130
209. Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, et al. Endothelial notch1 activity facilitates metastasis. Cancer Cell (2017) 31:355–67. doi: 10.1016/j.ccell.2017.01.007
210. Brantley-Sieders DM, Dunaway CM, Rao M, Short S, Hwang Y, Gao Y, et al. Angiocrine factors modulate tumor proliferation and motility through epha2 repression of slit2 tumor suppressor function in endothelium. Cancer Res (2011) 71:976–87. doi: 10.1158/0008-5472.CAN-10-3396
211. Liu Y, Colby JK, Zuo X, Jaoude J, Wei D, Shureiqi I. The role of ppar-delta in metabolism, inflammation, and cancer: many characters of a critical transcription factor. Int J Mol Sci (2018) 19:3339–53. doi: 10.3390/ijms19113339
212. Piqueras L, Reynolds AR, Hodivala-Dilke KM, Alfranca A, Redondo JM, Hatae T, et al. Activation of pparbeta/delta induces endothelial cell proliferation and angiogenesis. Arterioscler Thromb Vasc Biol (2007) 27:63–9. doi: 10.1161/01.ATV.0000250972.83623.61
213. Abdollahi A, Schwager C, Kleeff J, Esposito I, Domhan S, Peschke P, et al. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc Natl Acad Sci USA (2007) 104:12890–5. doi: 10.1073/pnas.0705505104
214. Du S, Wagner N, Wagner KD. The emerging role of ppar beta/delta in tumor angiogenesis. PPAR Res (2020) 2020:3608315. doi: 10.1155/2020/3608315
215. La Paglia L, Listi A, Caruso S, Amodeo V, Passiglia F, Bazan V, et al. Potential role of angptl4 in the cross talk between metabolism and cancer through ppar signaling pathway. PPAR Res (2017) 2017:8187235. doi: 10.1155/2017/8187235
216. Leone P, Solimando AG, Prete M, Malerba E, Susca N, Derakhshani A, et al. Unraveling the role of peroxisome proliferator-activated receptor beta/delta (Ppar beta/delta) in angiogenesis associated with multiple myeloma. Cells (2023) 12:1011–28. doi: 10.3390/cells12071011
217. Yadav A, Kumar B, Yu JG, Old M, Teknos TN, Kumar P. Tumor-associated endothelial cells promote tumor metastasis by chaperoning circulating tumor cells and protecting them from anoikis. PloS One (2015) 10:e0141602. doi: 10.1371/journal.pone.0141602
218. Kontos F, Michelakos T, Kurokawa T, Sadagopan A, Schwab JH, Ferrone CR, et al. B7-H3: an attractive target for antibody-based immunotherapy. Clin Cancer Res (2021) 27:1227–35. doi: 10.1158/1078-0432.CCR-20-2584
219. Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B. Genes that distinguish physiological and pathological angiogenesis. Cancer Cell (2007) 11:539–54. doi: 10.1016/j.ccr.2007.04.017
220. Wang R, Ma Y, Zhan S, Zhang G, Cao L, Zhang X, et al. B7-H3 promotes colorectal cancer angiogenesis through activating the nf-kappab pathway to induce vegfa expression. Cell Death Dis (2020) 11:55. doi: 10.1038/s41419-020-2252-3
221. Zang X, Sullivan PS, Soslow RA, Waitz R, Reuter VE, Wilton A, et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Mod Pathol (2010) 23:1104–12. doi: 10.1038/modpathol.2010.95
222. Crispen PL, Sheinin Y, Roth TJ, Lohse CM, Kuntz SM, Frigola X, et al. Tumor cell and tumor vasculature expression of B7-H3 predict survival in clear cell renal cell carcinoma. Clin Cancer Res (2008) 14:5150–7. doi: 10.1158/1078-0432.CCR-08-0536
223. Aung PP, Parra ER, Barua S, Sui D, Ning J, Mino B, et al. B7-H3 expression in merkel cell carcinoma-associated endothelial cells correlates with locally aggressive primary tumor features and increased vascular density. Clin Cancer Res (2019) 25:3455–67. doi: 10.1158/1078-0432.CCR-18-2355
224. Palazon A, Teijeira A, Martinez-Forero I, Hervas-Stubbs S, Roncal C, Penuelas I, et al. Agonist anti-cd137 mab act on tumor endothelial cells to enhance recruitment of activated T lymphocytes. Cancer Res (2011) 71:801–11. doi: 10.1158/0008-5472.CAN-10-1733
225. Maishi N, Ohba Y, Akiyama K, Ohga N, Hamada J, Nagao-Kitamoto H, et al. Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Sci Rep (2016) 6:28039. doi: 10.1038/srep28039
226. Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer (2005) 5:29–41. doi: 10.1038/nrc1527
227. Inufusa H, Nakamura M, Adachi T, Aga M, Kurimoto M, Nakatani Y, et al. Role of galectin-3 in adenocarcinoma liver metastasis. Int J Oncol (2001) 19:913–9. doi: 10.3892/ijo.19.5.913
228. Song YK, Billiar TR, Lee YJ. Role of galectin-3 in breast cancer metastasis: involvement of nitric oxide. Am J Pathol (2002) 160:1069–75. doi: 10.1016/S0002-9440(10)64927-9
229. Croci DO, Cerliani JP, Dalotto-Moreno T, Mendez-Huergo SP, Mascanfroni ID, Dergan-Dylon S, et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-vegf refractory tumors. Cell (2014) 156:744–58. doi: 10.1016/j.cell.2014.01.043
230. Delgado VM, Nugnes LG, Colombo LL, Troncoso MF, Fernandez MM, Malchiodi EL, et al. Modulation of endothelial cell migration and angiogenesis: A novel function for the "Tandem-repeat" Lectin galectin-8. FASEB J (2011) 25:242–54. doi: 10.1096/fj.09-144907
231. He J, Baum LG. Endothelial cell expression of galectin-1 induced by prostate cancer cells inhibits T-cell transendothelial migration. Lab Invest (2006) 86:578–90. doi: 10.1038/labinvest.3700420
232. Hsieh SH, Ying NW, Wu MH, Chiang WF, Hsu CL, Wong TY, et al. Galectin-1, a novel ligand of neuropilin-1, activates vegfr-2 signaling and modulates the migration of vascular endothelial cells. Oncogene (2008) 27:3746–53. doi: 10.1038/sj.onc.1211029
233. Nangia-Makker P, Honjo Y, Sarvis R, Akahani S, Hogan V, Pienta KJ, et al. Galectin-3 induces endothelial cell morphogenesis and angiogenesis. Am J Pathol (2000) 156:899–909. doi: 10.1016/S0002-9440(10)64959-0
234. Thijssen VL, Postel R, Brandwijk RJ, Dings RP, Nesmelova I, Satijn S, et al. Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc Natl Acad Sci USA (2006) 103:15975–80. doi: 10.1073/pnas.0603883103
235. Markowska AI, Liu FT, Panjwani N. Galectin-3 is an important mediator of vegf- and bfgf-mediated angiogenic response. J Exp Med (2010) 207:1981–93. doi: 10.1084/jem.20090121
236. Markowska AI, Jefferies KC, Panjwani N. Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells. J Biol Chem (2011) 286:29913–21. doi: 10.1074/jbc.M111.226423
237. White NM, Masui O, Newsted D, Scorilas A, Romaschin AD, Bjarnason GA, et al. Galectin-1 has potential prognostic significance and is implicated in clear cell renal cell carcinoma progression through the hif/mtor signaling axis. Br J Cancer (2014) 110:1250–9. doi: 10.1038/bjc.2013.828
238. Zhao XY, Zhao KW, Jiang Y, Zhao M, Chen GQ. Synergistic induction of galectin-1 by ccaat/enhancer binding protein alpha and hypoxia-inducible factor 1alpha and its role in differentiation of acute myeloid leukemic cells. J Biol Chem (2011) 286:36808–19. doi: 10.1074/jbc.M111.247262
239. Laderach DJ, Gentilini LD, Giribaldi L, Delgado VC, Nugnes L, Croci DO, et al. A unique galectin signature in human prostate cancer progression suggests galectin-1 as a key target for treatment of advanced disease. Cancer Res (2013) 73:86–96. doi: 10.1158/0008-5472.CAN-12-1260
240. Femel J, van Hooren L, Herre M, Cedervall J, Saupe F, Huijbers EJM, et al. Vaccination against galectin-1 promotes cytotoxic T-cell infiltration in melanoma and reduces tumor burden. Cancer Immunol Immunother (2022) 71:2029–40. doi: 10.1007/s00262-021-03139-4
241. Chen WS, Cao Z, Sugaya S, Lopez MJ, Sendra VG, Laver N, et al. Pathological lymphangiogenesis is modulated by galectin-8-dependent crosstalk between podoplanin and integrin-associated vegfr-3. Nat Commun (2016) 7:11302. doi: 10.1038/ncomms11302
242. Haibe Y, Kreidieh M, El Hajj H, Khalifeh I, Mukherji D, Temraz S, et al. Resistance mechanisms to anti-angiogenic therapies in cancer. Front Oncol (2020) 10:221. doi: 10.3389/fonc.2020.00221
243. Cao Z, Ding BS, Guo P, Lee SB, Butler JM, Casey SC, et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell (2014) 25:350–65. doi: 10.1016/j.ccr.2014.02.005
244. Huinen ZR, Huijbers EJM, van Beijnum JR, Nowak-Sliwinska P, Griffioen AW. Anti-angiogenic agents - overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol (2021) 18:527–40. doi: 10.1038/s41571-021-00496-y
245. Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat Med (2001) 7:987–9. doi: 10.1038/nm0901-987
246. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell (2014) 26:605–22. doi: 10.1016/j.ccell.2014.10.006
247. Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, et al. Direct evidence that the vegf-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med (2004) 10:145–7. doi: 10.1038/nm988
248. Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, et al. Azd2171, a pan-vegf receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell (2007) 11:83–95. doi: 10.1016/j.ccr.2006.11.021
249. Gerstner ER, Ye X, Duda DG, Levine MA, Mikkelsen T, Kaley TJ, et al. A phase I study of cediranib in combination with cilengitide in patients with recurrent glioblastoma. Neuro Oncol (2015) 17:1386–92. doi: 10.1093/neuonc/nov085
250. Sorensen AG, Batchelor TT, Zhang WT, Chen PJ, Yeo P, Wang M, et al. A "Vascular normalization index" as potential mechanistic biomarker to predict survival after a single dose of cediranib in recurrent glioblastoma patients. Cancer Res (2009) 69:5296–300. doi: 10.1158/0008-5472.CAN-09-0814
251. Sorensen AG, Emblem KE, Polaskova P, Jennings D, Kim H, Ancukiewicz M, et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res (2012) 72:402–7. doi: 10.1158/0008-5472.CAN-11-2464
252. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA (2012) 109:17561–6. doi: 10.1073/pnas.1215397109
253. Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, et al. Gene therapy using genetically modified lymphocytes targeting vegfr-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest (2010) 120:3953–68. doi: 10.1172/JCI43490
254. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature (2017) 544:250–4. doi: 10.1038/nature21724
255. Li Q, Wang Y, Jia W, Deng H, Li G, Deng W, et al. Low-dose anti-angiogenic therapy sensitizes breast cancer to pd-1 blockade. Clin Cancer Res (2020) 26:1712–24. doi: 10.1158/1078-0432.CCR-19-2179
256. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and vegfa inhibition elicits antitumor immunity that is enhanced by pd-1 checkpoint blockade. Sci Transl Med (2017) 9:eaak9670–84. doi: 10.1126/scitranslmed.aak9670
257. Wu X, Giobbie-Hurder A, Liao X, Lawrence D, McDermott D, Zhou J, et al. Vegf neutralization plus ctla-4 blockade alters soluble and cellular factors associated with enhancing lymphocyte infiltration and humoral recognition in melanoma. Cancer Immunol Res (2016) 4:858–68. doi: 10.1158/2326-6066.CIR-16-0084
258. Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (Immotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet (2019) 393:2404–15. doi: 10.1016/S0140-6736(19)30723-8
259. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun (2016) 7:12624. doi: 10.1038/ncomms12624
260. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med (2020) 382:1894–905. doi: 10.1056/NEJMoa1915745
261. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous nsclc. N Engl J Med (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
262. Lee J, Koh J, Kim HK, Hong S, Kim K, Park S, et al. Bevacizumab plus atezolizumab after progression on atezolizumab monotherapy in pretreated patients with nsclc: an open-label, two-stage, phase 2 trial. J Thorac Oncol (2022) 17:900–8. doi: 10.1016/j.jtho.2022.04.001
263. Cui X, Jia H, Xin H, Zhang L, Chen S, Xia S, et al. A novel bispecific antibody targeting pd-L1 and vegf with combined anti-tumor activities. Front Immunol (2021) 12:778978. doi: 10.3389/fimmu.2021.778978
264. Du YQ, Zhang J, Sun ML, Wen Q, Zheng YW, Tolcher AW, et al. Safety and efficacy of hb0025, an anti-pd-1/vegf bispecific antibody fusion protein, in patients with advanced solid tumors: preliminary results from an fih trial. J Clin Oncol (2023) 41. doi: 10.1200/JCO.2023.41.16_suppl.2589
265. Saba NF, Steuer CE, Ekpenyong A, McCook-Veal A, Magliocca K, Patel M, et al. Pembrolizumab and cabozantinib in recurrent metastatic head and neck squamous cell carcinoma: A phase 2 trial. Nat Med (2023) 29:880–7. doi: 10.1038/s41591-023-02275-x
266. Procopio G, Claps M, Pircher C, Porcu L, Sepe P, Guadalupi V, et al. A multicenter phase 2 single arm study of cabozantinib in patients with advanced or unresectable renal cell carcinoma pre-treated with one immune-checkpoint inhibitor: the breakpoint trial (Meet-uro trial 03). Tumori (2023) 109:129–37. doi: 10.1177/03008916221138881
267. Li B, Jin J, Guo D, Tao Z, Hu X. Immune checkpoint inhibitors combined with targeted therapy: the recent advances and future potentials. Cancers (Basel) (2023) 15:2858–78. doi: 10.3390/cancers15102858
268. Rossi E, Bersanelli M, Gelibter AJ, Borsellino N, Caserta C, Doni L, et al. Combination therapy in renal cell carcinoma: the best choice for every patient? Curr Oncol Rep (2021) 23:147. doi: 10.1007/s11912-021-01140-9
269. Cheng AL, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Updated efficacy and safety data from imbrave150: atezolizumab plus bevacizumab vs. Sorafenib for unresectable hepatocellular carcinoma. J Hepatol (2022) 76:862–73. doi: 10.1016/j.jhep.2021.11.030
270. Choueiri TK, Motzer RJ, Rini BI, Haanen J, Campbell MT, Venugopal B, et al. Updated efficacy results from the javelin renal 101 trial: first-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann Oncol (2020) 31:1030–9. doi: 10.1016/j.annonc.2020.04.010
271. Emens LA, Esteva FJ, Beresford M, Saura C, De Laurentiis M, Kim SB, et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, her2-positive advanced breast cancer (Kate2): A phase 2, multicentre, randomised, double-blind trial. Lancet Oncol (2020) 21:1283–95. doi: 10.1016/S1470-2045(20)30465-4
272. Lee MS, Ryoo BY, Hsu CH, Numata K, Stein S, Verret W, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (Go30140): an open-label, multicentre, phase 1b study. Lancet Oncol (2020) 21:808–20. doi: 10.1016/S1470-2045(20)30156-X
273. Socinski MA, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, et al. Impower150 final overall survival analyses for atezolizumab plus bevacizumab and chemotherapy in first-line metastatic nonsquamous nsclc. J Thorac Oncol (2021) 16:1909–24. doi: 10.1016/j.jtho.2021.07.009
274. Zhu AX, Abbas AR, de Galarreta MR, Guan Y, Lu S, Koeppen H, et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat Med (2022) 28:1599–611. doi: 10.1038/s41591-022-01868-2
275. Lu L, Zhan M, Li XY, Zhang H, Dauphars DJ, Jiang J, et al. Clinically approved combination immunotherapy: current status, limitations, and future perspective. Curr Res Immunol (2022) 3:118–27. doi: 10.1016/j.crimmu.2022.05.003
276. Chen J, Siliceo SL, Ni Y, Nielsen HB, Xu A, Panagiotou G. Identification of robust and generalizable biomarkers for microbiome-based stratification in lifestyle interventions. Microbiome (2023) 11:178. doi: 10.1186/s40168-023-01604-z
277. Gao J, Zhang X, Jiang L, Li Y, Zheng Q. Tumor endothelial cell-derived extracellular vesicles contribute to tumor microenvironment remodeling. Cell Commun Signal (2022) 20:97. doi: 10.1186/s12964-022-00904-5
278. Zhang S, Yang J, Shen L. Extracellular vesicle-mediated regulation of tumor angiogenesis- implications for anti-angiogenesis therapy. J Cell Mol Med (2021) 25:2776–85. doi: 10.1111/jcmm.16359
Keywords: endothelial cells, tumor, microenvironment, T cells, tumor immune evasion
Citation: Leone P, Malerba E, Susca N, Favoino E, Perosa F, Brunori G, Prete M and Racanelli V (2024) Endothelial cells in tumor microenvironment: insights and perspectives. Front. Immunol. 15:1367875. doi: 10.3389/fimmu.2024.1367875
Received: 09 January 2024; Accepted: 05 February 2024;
Published: 15 February 2024.
Edited by:
Peter Hamar, Semmelweis University, HungaryReviewed by:
Chen Zhao, Nanjing Medical University, ChinaCopyright © 2024 Leone, Malerba, Susca, Favoino, Perosa, Brunori, Prete and Racanelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcella Prete, bWFyY2VsbGEucHJldGVAdW5pYmEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.