 Yuchu Xiang1†
Yuchu Xiang1† Xudong Liu2,3†
Xudong Liu2,3† Yifan Wang4†
Yifan Wang4† Dawei Zheng5
Dawei Zheng5 Qiuxing Meng6,7Lingling Jiang8Sha Yang9
Qiuxing Meng6,7Lingling Jiang8Sha Yang9 Sijia Zhang10Xin Zhang11Yan Liu12*
Sijia Zhang10Xin Zhang11Yan Liu12* Bo Wang2,13*
Bo Wang2,13*- 1West China Hospital of Sichuan University, Sichuan University, Chengdu, China
- 2Institute of Medical Microbiology and Hygiene, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 3College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
- 4State Key Laboratory for Oncogenes and Related Genes, Division of Cardiology, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai Cancer Institute, Shanghai, China
- 5The College of Life Science, Sichuan University, Chengdu, China
- 6Department of Laboratory Medicine, Liuzhou People’s Hospital, Liuzhou, China
- 7Guangxi Health Commission Key Laboratory of Clinical Biotechnology (Liuzhou People’s Hospital), Liuzhou, China
- 8Guangxi Medical University Cancer Hospital, Nanning, China
- 9Institute of Pharmaceutical Science, China Pharmaceutical University, Nanjing, China
- 10Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 11Zhongshan Hospital of Fudan University, Xiamen, Fujian, China
- 12Department of Organ Transplantation, Guizhou Provincial People’s Hospital, Guiyang, Guizhou, China
- 13Department of Urology, Guizhou Provincial People’s Hospital, Guiyang, Guizhou, China
Resistance to targeted therapy and immunotherapy in non-small cell lung cancer (NSCLC) is a significant challenge in the treatment of this disease. The mechanisms of resistance are multifactorial and include molecular target alterations and activation of alternative pathways, tumor heterogeneity and tumor microenvironment change, immune evasion, and immunosuppression. Promising strategies for overcoming resistance include the development of combination therapies, understanding the resistance mechanisms to better use novel drug targets, the identification of biomarkers, the modulation of the tumor microenvironment and so on. Ongoing research into the mechanisms of resistance and the development of new therapeutic approaches hold great promise for improving outcomes for patients with NSCLC. Here, we summarize diverse mechanisms driving resistance to targeted therapy and immunotherapy in NSCLC and the latest potential and promising strategies to overcome the resistance to help patients who suffer from NSCLC.
1 Introduction
Lung cancer is a common cancer and one of the leading causes of cancer deaths (1–4). It can be classified into two main groups based on pathologic features: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC accounts for 80-85% of all lung cancer cases (5). It can be further divided into three main subtypes: adenocarcinoma (40%), squamous cell carcinoma (25-30%), and large cell carcinoma (5-10%) (6). According to the World Health Organization (WHO), the 5-year survival rate of NSCLC is only 5-10% (7).
Common therapies for NSCLC include surgical intervention, radiation, chemotherapy, targeted therapy, and immunotherapy. Surgical resection is the most trustworthy and efficient approach for managing patients with NSCLC in terms of diagnosis, staging, treatment, and palliative care. For this method to be feasible, the cancer must be fully resectable (8). In addition, it is worth noting that around 70% of individuals diagnosed with NSCLC have either locally progressed or metastatic disease. Chemotherapy induces remission in people with locally advanced and metastatic illnesses (9). While chemotherapy is suitable for numerous NSCLC patients, the utilization of traditional chemotherapeutic drugs has reached a therapeutic plateau. Radiotherapy is a conventional therapeutic method that includes stereotactic RT and hadron therapy as the primary approaches. Hadrons are subatomic particles composed of quarks, such as protons, neutrons, or heavy ions, which are influenced by powerful nuclear forces.
Although conventional treatments have been the standard of treatment, the increasing clinical use of targeted therapies and immunotherapy has provided new therapeutic ideas for a wide range of healthcare professionals. However, the emergence of drug resistance during treatment remains a thorny issue (10).
Targeted therapy is drugs that specifically target genetic alterations or signaling pathways involved in tumor growth and survival. These therapies have revolutionized the treatment of lung cancer, particularly NSCLC (11). Nevertheless, tumor cells can develop resistance to these medications via several mechanisms, including targeted modifications and activation of alternate pathways. The former refers to the ability of cancer cells to generate novel genetic modifications or mutations that provide resistance to specific therapies, such as epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), or proto-oncogene tyrosine protein kinase-1 (ROS1) mutations. The latter one refers to the observation that tumor cells can circumvent certain pathways, like mesenchymal to epithelial transition factor (MET) amplification, kirsten rat sarcoma viral oncogene (KRAS) mutations (12), that are targeted for treatment by activating alternative signaling pathways that promote the survival and proliferation of the cells. This phenomenon is commonly referred to as “adaptive resistance.” NSCLC, similar to other forms of cancer, is distinguished by its genetic and molecular diversity. Lung tumors might comprise diverse groups of cancer cells with varying genetic modifications. Although targeted medicines can effectively combat the main clone, they may not have the capability to eradicate secondary subclones that possess drug-resistant mutations (13).
Immunotherapeutic resistance refers to the ability of cancer cells to evade or overcome the effects of immunotherapy, which is a treatment approach that harnesses the body’s immune system to fight cancer (14). While immunotherapy has shown significant promise in treating NSCLC, some patients may experience resistance to these therapies (15). In the context of NSCLC, immunotherapeutic resistance can occur through several mechanisms (16–20): 1. Loss or downregulation of immune cell recognition; 2. Tumor microenvironment changes; 3. Immune checkpoint activation; 4. Tumor heterogeneity.
In this review, we review the mechanisms of resistance to targeted therapy and immunotherapy in NSCLC, and list the promising strategies for overcoming the resistance from different perspectives.
2 Targeted therapy resistance in NSCLC
Herein, we summarized the mechanisms of the main targets that generate resistance in targeted therapy for NSCLC (Figure 1). In the clinical work, the targets can be divided into two groups: common clinical gene targets and uncommon clinical gene targets.
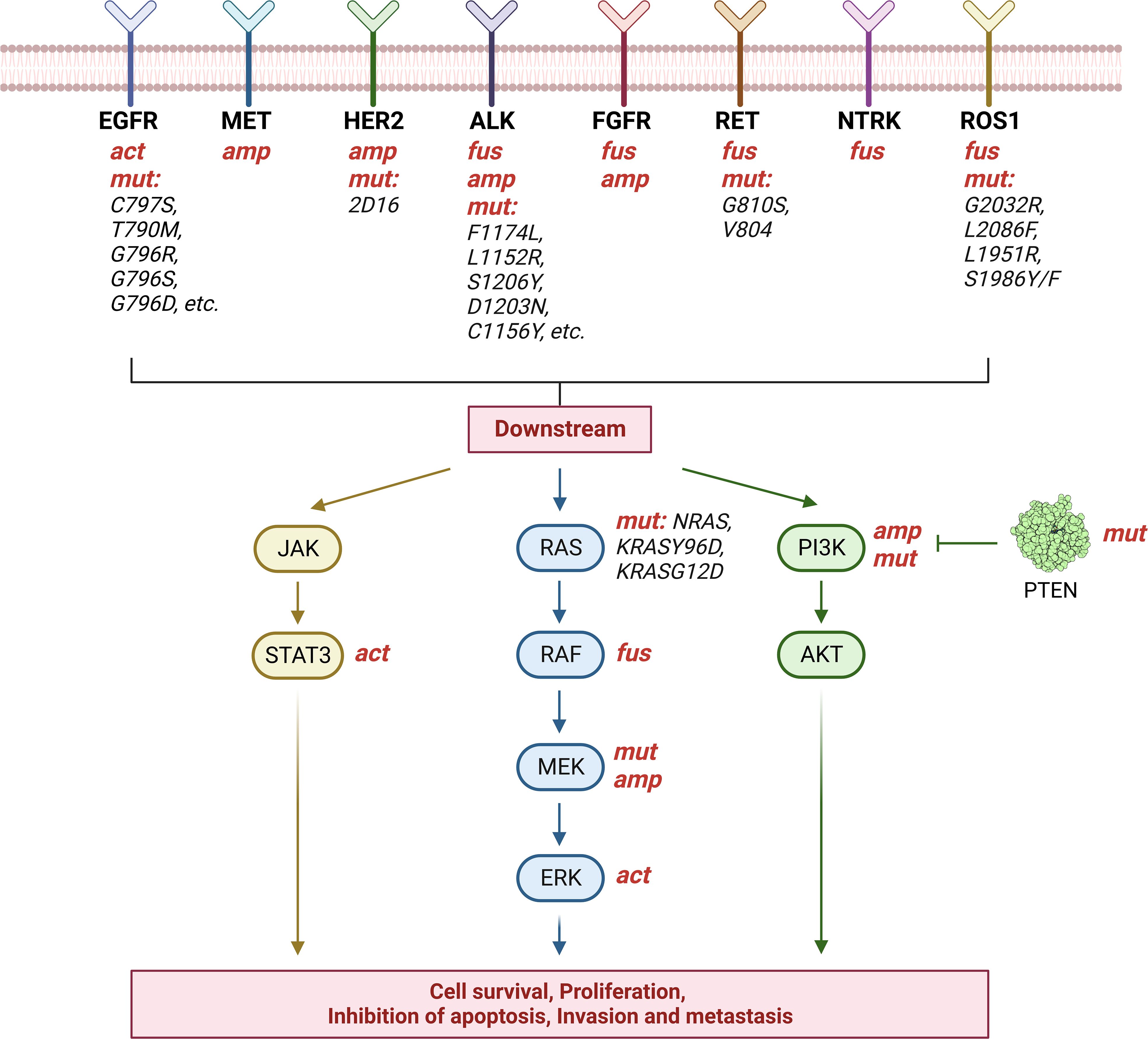
Figure 1 Targeted therapy resistance in NSCLC. The image shows the mechanisms of the main targets that generate resistance in targeted therapy for NSCLC. The above eight targets generate drug resistance through gene mutation(mut), activation(act), amplification(amp), or fusion(fus). Mutated targets have been marked in black. In particular, mutations in PTEN, an inhibitor of PI3K, also lead to tumor progression. Created with BioRender.com.
2.1 Common clinical gene targets
2.1.1 EGFR: the mechanisms of resistance
The EGFR gene is the most prevalent oncogenic driver in NSCLC, occurring in 10–15% of all NSCLC cases (21). TKIs targeting EGFR mutations have emerged as the preferred first-line treatment option for patients with EGFR-mutant NSCLC. Currently, three generations of EGFR-TKI drugs have been developed. The first generation comprises gefitinib, erlotinib, and icotinib, among which the acquisition of the T790M resistance mutation is a common occurrence (22). The second generation includes afatinib and dacomitinib; however, these drugs encounter significant challenges in overcoming resistance, thereby limiting their efficacy in patients with acquired resistance to first-generation TKIs. The third generation encompasses osimertinib, rociletinib, and avitinib, and it targets resistance mutations such as EGFR L718Q, L844V, and C797S mutations.
We categorize the mechanisms of resistance generation into target-dependent and target-independent. Taking EGFR as an example, the subsequent targets will not be repeated, EGFR-dependent refers to those mediated by acquired resistance mutations in the structural domain of the EGFR kinase, whereas EGFR-independent refers to those mediated by alterations in non-target kinases, such as bypass signaling activation or phenotypic transformation (23).
1) EGFR target-dependent resistance mechanisms:
Patients receiving first- or second-generation EGFR-TKIs primarily develop EGFR target-dependent resistance, whereas only approximately 20% of patients receiving the third-generation TKI osimertinib as second-line therapy exhibit target-dependent resistance mechanisms (24).
The most common resistance mutations include the C797S mutation in exon 20 of EGFR (25) and the T790M mutation (22), which often co-occur. In cases where only the C797S mutation is present without T790M, NSCLC with drug resistance may still maintain sensitivity to quinazoline-based EGFR inhibitors such as gefitinib, erlotinib, and afatinib (26, 27). In trans-C797S/T790M cases, cells are resistant to third-generation EGFR-TKIs but remain sensitive to combined treatment with first- and third-generation TKIs (28). In cis-C797S/T790M cases, EGFR-TKI treatment alone or in combination is ineffective (25), indicating resistance to all EGFR-TKIs (29).
Various EGFR mutations have been identified as mechanisms of resistance to third-generation EGFR-TKIs (24, 30–32). These include EGFR G796R, G796S, and G796D mutations (33, 34), S768I mutations (35), L718X and L792X mutations (31), L798I mutation (36), and L858R/T790M/L792H and L858R/T790M/G796R mutations (37), which have been associated with resistance to osimertinib. Among these, the L792X mutation interferes with the spatial binding of the EGFR kinase structural domain and can coexist with trans-G796/C97X mutations (31, 37). Other rare EGFR-TKI resistance mutations include residues L718 and G719 in the Adenosine triphosphate (ATP) binding site (30, 31), as well as the G724S mutation in the P-loop structural domain of the kinase (38–40). It has been suggested that G724S is an allele-specific drug resistance mutation that occurs in the context of Ex19Del (41) and limits the activity of third-generation EGFR-TKIs both in vitro and in vivo by inducing conformational changes in the glycine-rich loop (38), but further investigation is needed (39). In addition, amplification of EGFR wild-type alleles, rather than mutant alleles, has been shown to be sufficient to generate acquired resistance (36, 42).
2) EGFR target-independent resistance mechanisms:
For EGFR target-independent resistance, MET amplification has been identified as the most common mechanism of resistance observed during treatment with osimertinib (30), followed by human epidermal growth factor receptor 2 (HER2) amplification (43).
MET-mediated resistance primarily occurs through MET gene amplification (24, 30, 36), which leads to the activation of downstream signaling pathways, including STAT, mitogen-activated protein kinase (MAPK), and phosphatidylinositol 3-kinase (PI3K), thereby bypassing the EGFR pathway. HER2, an ErbB2 receptor tyrosine kinase (RTK), also contributes to EGFR-TKI resistance by activating the MAPK and PI3K pathways. HER2 amplification has been detected in 12% of tumor samples from patients without coexisting T790M mutations treated with first-generation EGFR-TKIs (44). Moreover, HER2 exon 16 skipping deletion (HER2D16) mediates resistance to osimertinib through an Src-independent mechanism. Combining osimertinib with the pan-HER small molecule inhibitor afatinib has shown synergistic potential in overcoming HER2D16 resistance (45). Additionally, fusion events involving various oncogenes, including fibroblast growth factor receptor (FGFR) (24), B-Raf proto-oncogene (BRAF) (46, 47), ROS1 (48), ret proto-oncogene (RET) (24, 46, 47), neurotrophic tropomyosin kinase receptors (NTRK) (47), and ALK (30), have been implicated in EGFR-TKI resistance.
In addition to the resistance mechanisms mentioned previously, several other factors contribute to EGFR-induced NSCLC drug resistance. Firstly, RAS mutations, encompassing NRAS, KRAS, and BRAFV600E mutations, have been prominently associated with resistance to EGFR-TKIs (24, 30, 46). Furthermore, phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) amplification or mutation is detected in approximately 3-5% of first-generation EGFR TKI resistance cases (30, 49) and 5-12% of third-generation TKI resistance cases (24, 30, 36). Additionally, FGFR amplification, including Src family kinase (SFK) and focal adhesion kinase (FAK), has been identified as a notable mechanism of resistance (50, 51).
Furthermore, alterations in cell cycle-related genes, such as amplification or mutation of CDK4 and CDK6, as well as CDKN2A, play a significant role in resistance development (24, 30). Moreover, histological phenotypic transformations, such as the transition to small cell lung carcinoma (SCLC), epithelial-to-mesenchymal transition (EMT), and squamous cell carcinoma transition (SCCT), also contribute significantly to resistance development. Lastly, epigenetic modifications constitute another dimension of this intricate resistance landscape.
These resistance mechanisms highlight the complexity of EGFR-TKI resistance in NSCLC, necessitating further research to improve treatment strategies and develop novel therapeutic approaches.
2.1.2 EGFR: the strategies to overcome resistance
To overcome resistance to third-generation EGFR-TKIs, various strategies have been explored, including the use of first- or second-generation TKIs for EGFR target-dependent resistance. For the classical C797S/T790M mutation, clinical evidence supporting the efficacy of combination therapy with first- and third-generation EGFR TKIs has been reported (29), such as nazatinib in combination with gefitinib and osimertinib in combination with gefitinib. Studies have shown that the combination of osimertinib and gefitinib has consistent efficacy as a first-line treatment for EGFR-mutated NSCLC, which provides insights into the use of dual EGFR-TKIs (52). First- and second-generation TKIs are not affected by the C797S resistance mutation, making them potential treatment options for patients with this mutation (25). Additionally, the second-generation TKI afatinib retains its binding ability to the ATP pocket even in the presence of the G724S mutation, making it a candidate for combination therapy with osimertinib to overcome acquired G724S resistance mutations (40). Afatinib has also shown efficacy against L718Q resistance mutations (53).
Targeting non-dependent resistance to third-generation EGFR-TKIs is more challenging and often requires combination therapies.
1) EGFR-TKIs + Chemotherapy.
Combining EGFR-TKIs with chemotherapy is a common approach to delay resistance development. In the NEJ005 phase II trial, the combination of gefitinib with carboplatin plus pemetrexed demonstrated prolonged progression-free survival (PFS) and overall survival (54, 55). The NEJ009 phase III trial compared chemotherapy plus gefitinib to gefitinib alone, and the combination group showed significantly longer PFS (56). Similar conclusions were reached in other phase III trials (57). The ongoing FLAURA 2 phase III trial is evaluating the combination of carboplatin (or cisplatin) pemetrexed with osimertinib, aiming to investigate whether the observed benefits of combination therapy apply to osimertinib treatment (58).
2) EGFR-TKIs + Specific small molecule inhibitors
The combination of specific inhibitors with EGFR-TKIs, selected based on the underlying mechanism of secondary resistance, has demonstrated improved therapeutic outcomes. For instance, pralsetinib (BLU667) for RET rearrangements, brigatinib for ALK fusions, trametinib and selumetinib for MEK inhibition, entrectinib for NTRK mutations, sotorasib as a KRAS inhibitor, cabozantinib as a multi-target inhibitor, and inhibitors targeting AXL, JAK1, and BRAFV600E are potential treatment options.
MET amplification is a well-studied mechanism of drug resistance, and treatment options for MET-amplified patients have been investigated. Studies have shown that the combination of octreotide and MET-TKI-savolitinib exhibits enhanced antitumor activity, suggesting its potential as a treatment option for MET-driven EGFR TKI-resistant patients (59). Combination therapy with third-generation EGFR-TKIs and MET inhibitors is emerging as a common and promising approach for first-line treatment of NSCLC patients with MET-amplified EGFR mutations. A phase III clinical trial evaluating first-line amivantamab plus galazetinib in combination with osimertinib in EGFR-mutated NSCLC is currently underway (60). In patients with MET amplification pretreated with EGFR-TKIs, the first-generation EGFR-TKI gefitinib in combination with the MET inhibitor capmatinib has demonstrated effectiveness (61). Tepotinib, another MET TKI, showed a higher objective response rate (ORR) in combination with gefitinib in phase Ib/II trials (62). Moreover, the natural product berberine has shown MET-inhibiting ability and synergistic induction of apoptosis with osimertinib, thus overcoming MET-enhanced induction of osimertinib-resistant cancers (63). Preclinical trials with HQP8361 (64) and dictamnine (65), two MET receptor inhibitors, have demonstrated anti-osimertinib resistance properties. Increased MET copy number has also been identified as a common mechanism of resistance to rosatinib, which can be overcome by the MET inhibitor crizotinib (36).
AXL RTK activation is another resistance mechanism. Combining AXL inhibitors with axitinib has shown efficacy in overcoming resistance to axitinib in EGFR-mutant NSCLC (66). Novel AXL inhibitors, including AXL/MET dual inhibitors and AXL-specific antibody-drug conjugates, have exhibited potent effects in overcoming osimertinib resistance in preclinical trials (67, 68).
Additionally, many EGFR-TKI resistance mechanisms involving bypass pathway activation can be addressed through bypass inhibition. In vitro studies have shown that the addition of PI3K inhibitors to EGFR-TKIs can overcome resistance, and combining MEK and PI3K inhibitors has proven to be an effective therapeutic strategy for treating NSCLC with acquired resistance to EGFR-TKIs (69). In cases of drug-resistant NSCLC resulting from FGFR gene family amplification, combined inhibition of FGFR and AKT has shown efficacy in FGFR1-overexpressing osimertinib-resistant NSCLC cells (70). Simultaneous inhibition of SFK/FAK and EGFR may also hold promise as a therapeutic strategy (50). RET fusion activates a bypass pathway leading to resistance to EGFR-TKIs, and this pathway can be effectively targeted in the clinic using a selective RET inhibitor (BLU-667) (47). Activation of the insulin-like growth factor 1 receptor (IGF1R) can be addressed through the use of IGF1R inhibitors in combination with octreotide, and clinical trials have been conducted to evaluate their safety and efficacy (71, 72). Furthermore, exon 16 skipping HER2 deletion (HER2D16) leads to osimertinib resistance, and unlike in breast cancer, HER2D16 mediates resistance in NSCLC through a Src-independent mechanism, making it insensitive to Src inhibitors (45). Combining osimertinib with the pan-HER small molecule inhibitor afatinib has shown synergistic potential in overcoming HER2D16 resistance (45). EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC Inhibition (73). ROS1 rearrangement caused by EGFR-TKIs can be overcome by combining crizotinib (74, 75).
3) Fourth-generation EGFR-TKIs
Currently, fourth-generation EGFR-TKIs are undergoing clinical investigation, with a primary focus on targeting T790M/C797S drug-resistant mutations. EAI045, the first selective small-molecule variant inhibitor, has shown efficacy in mouse NSCLC models when used in combination with cetuximab. However, its clinical introduction has been hindered by significant adverse effects (76). JBJ-04-125-02 has demonstrated greater durability compared to EAI001 and has shown the ability to slow down C797S resistance either as a monotherapy or in combination with other agents (77). Another promising fourth-generation EGFR-TKI, tQB3804, has shown the ability to overcome resistance mediated by multiple mutations and is currently undergoing phase I clinical trials (78).
4) Antibody-drug conjugates (ADCs)
In addition to the approaches mentioned above, antibody-drug conjugates (ADCs) have emerged as an effective treatment strategy for overcoming drug resistance. Although most of the them target HER family, an increasing number of trials have shown that this ADC has therapeutic effects in patients who are resistant to EGFR-TKIs. Clinical activity of HER3-DXd, an ADC targeting HER3, has been observed independent of the resistance mechanism in a phase I clinical trial involving patients resistant to EGFR-TKIs, suggesting a novel approach for treating EGFR-TKI-resistant NSCLC patients (79).
2.1.3 KRAS: the mechanisms of resistance
KRAS is a small GTPase that exhibits reduced ability to hydrolyze GTP or interact with GTPase-activating protein (GAP) when mutated. This results in KRAS being locked into an active GTP-binding state, promoting cancer cell growth and anti-apoptosis (80). KRAS mutation represents the most prevalent genetic alteration observed in NSCLC (81). The most common KRAS mutations in NSCLC is KRAS(G12C) accounting for 45%, followed by KRAS(G12V) and KRAS(G12D) (82, 83).
Currently, the clinical agents targeting KRAS mutation in NSCLC include AMG 510 (sotorasib) (84) and MRTX849 (adagrasib) (85). These inhibitors covalently bind to cysteine residues in the switch-II pocket, which is generated by the G12C mutation. This binding favors GDP over GTP binding, reducing interactions between mutant KRAS and effector or regulatory molecules.
However, monotherapy with KRAS inhibitors is prone to adaptive resistance for a number of reasons, including the following:
1) RAS target-dependent resistance mechanisms:
The reactivation of the RAS pathway in an adaptive manner is crucial in the emergence of resistance to inhibitors targeting KRAS G12C (86). For instance, increased HER2 copy number leads to the persistence of MAPK pathway signaling and resistance to inhibitors. This resistance can be overcome by targeting SHP2 (87). RTKs such as the ErbB family or FGFR can also maintain KRAS in an activated GTP-bound state via SHP2, attenuating the effects of KRAS(G12C) inhibitors (88, 89). Additionally, amplification of MET can lead to resistance of NSCLC to KRAS(G12C) inhibitors (86). A patient treated with MRTX849 developed polyclonal acquired resistance and reactivation of RAS-MAPK signaling. Some cancer cells develop non-G12C KRAS mutations, resulting in resistance to KRAS inhibitors. For example, a study identified a novel KRAS Y96D mutation that interferes with protein-drug interactions in the switch-II pocket and confers resistance to inhibitors in a KRAS G12C cancer model (90). Acquired oncogenic mutations in KRAS, NRAS, or BRAF have also been observed during G12C inhibitor treatment (91).
2) KRAS target-idependent resistance mechanisms:
Certain cancer cells become independent of KRAS dependence by activating alternative pathways. For instance, BET-regulated YAP1 upregulation was found to contribute to resistance acquisition in KRAS; LKB1 and KRAS; TP53 mutant NSCLC cells (92).
2.1.4 KRAS: the strategies to overcome resistance
Currently, combination therapy is the primary approach to overcome KRAS inhibitor resistance. Dual blockade of MET and KRASG12C has shown significant efficacy in MET-amplified, KRASG12C-mutant NSCLC (86). The MET inhibitor crizotinib restored sensitivity to sotorasib by inhibiting RAS-MEK-ERK and AKT signaling (86). Inhibition of SHP2 reduced the conversion of GDP to GTP-bound KRAS and overcame adaptive resistance to MAPK pathway-targeting drugs, including KRAS(G12C) inhibitors (93). Additionally, the combination of TBK1 and MEK inhibition, along with intermittent BET inhibition, has been shown to overcome resistance to KRAS inhibitors by Kitajima et al (92). Furthermore, in a xenograft mouse model, the combination of the clinical KRAS inhibitors AMG-510 and MRTX849 with carboplatin and palbociclib resulted in significant regression of lung tumors (94, 95).
Feedback reactivation of the adaptive RAS pathway, non-G12C KRAS mutations, and bypass activation are the three major mechanisms underlying KRAS inhibitor resistance. Given these challenges, the use of combination therapy has arisen as a successful approach to treatment. The combined therapy of crizotinib + sotolacib has been employed to address the issue of resistance to sotolacib caused by MET amplification (86). Similarly, for resistance arising from HER2 amplification, the combination of TNO155 + sotorasib has demonstrated promising results (87). Given that feedback reactivation of the adaptive RAS pathway represents a key mechanism driving resistance to KRAS G12C inhibitors, targeted inhibition of SHP2 assumes paramount importance in overcoming KRAS resistance.
2.1.5 ALK: the mechanisms of resistance
ALK is a transmembrane RTK, which belongs to a subfamily within the insulin receptor superfamily (96). ALK is encoded by ALK gene, which is located on the short arm of chromosome 2 (2p23) (97). In the process of oncogenesis, the ALK gene translocates with another partner gene, leading to a fusion oncogene that overexpresses in cancers (98). A minority (3–13%) of NSCLC cases have been demonstrated to exhibit rearrangements in the ALK gene (99).
A series of ALK-TKIs, including crizotinib, ceritinib, alectinib, brigatinib, ensartinib, and lorlatinib, have been approved for ALK-positive patients worldwide. More specifically, crizotinib, ceritinib, and lorlatinib work as ATP-competitive inhibitors (100–102); alectinib and brigatinib can inhibit ALK protein by preventing phosphorylation (103); ensartinib acts by inhibiting ALK fusions engineered to have point mutations (104).
The first-generation ALK inhibitor is crizotinib. Its resistance mechanisms can be categorized into ALK-dependent mechanisms and ALK-independent mechanisms (105). The former is constructed by point mutations, including F1174L, L1152R, S1206Y, 1151Tins, I1171T, D1203N, V1180L, C1156Y, F1164V, G1269A, G1202R, and G1269S (106, 107), as well as gene amplification, which implicated in 9% of the secondary mutations of the ALK gene cases (108). While ALK-independent mechanisms refer to the activation of bypass signaling through the activation of other oncogenes, like EGFR, extracellular signal regulated kinase (ERK), Crk-like adaptor protein etc. Also, the primary resistance of crizotinib, whose RR is about 60%, should not be ignored. The primary resistance is the different variants of the EML4–ALK fusion protein or the false-positive diagnoses of ALK translocation (109).
The second-generation ALK inhibitors, ceritinib, alectinib, and brigatinib, were introduced due to the drug-resistance of the first-generation ALK inhibitor, which show predominant effects on crizotinib-resistant patients (110). Nevertheless, the wide use of second-generation ALK inhibitors can inevitably lead to drug resistance and tumor relapse (111), which can also be divided into ALK-dependent mechanisms and ALK-independent. More than half of the ceritinib-resistant specimens showed secondary mutations, such as G1123S, F1174C/L/V, and G1202R mutations (112). And ALK-independent mechanisms included activating mutation of MEK, the overexpression of p-glycoprotein ABCB1, SRC activation and overexpression of SRC homology 2 domain (SHP2), and activation of IGF-1 R, KIT, and EGFR (112). G1202R, G1206C, G1206Y, E1210K, F1245C, G1269A, G1269S, V1180L, and I1171T/N/S mutations can be relevant to the alectinib-resistance by affecting the binding between the drug and the ALK fusion protein or other ways (112, 113). Some bypass tracks, like C-MET activation, Coactivation of MET and Proto-oncogene tyrosine-protein kinase Src (SRC), Yes-associated protein 1 (YAP 1), Amphiregulin, the overexpression of ATP-binding cassette subfamily C member 11 (ABCC11), Transformation to SCLC, and EMT, can cause alectinib-resistance as well (112, 114–118). It is also reported that the brigatinib-resistance may be associated with the mutation of G1202R, I1171T, I1171N, V1180L, G1202R, T1151M, L1196M, E1408V, and ALK amplification (119–121), and some compound mutations including E1210K+S1206C and E1210K+D1203N (110). We can only make a conclusion that the ALK-dependent mechanisms of brigatinib are still unclear. The brigatinib-resistance is also caused by MTOR T1834-T1837del, JAK3R948C mutation, CDKN2A/B loss, and NFE2L2E79Q mutation, and MET amplification, mutation of BRAF-V600E, and KRAS -G12D (112).
Lorlatinib, a third-generation ALK/ROS1-TKI, acts as an ATP-competitive macrocyclic. However, just like the first- and second-generation ALK inhibitors, acquired lorlatinib-resistance still existed in nearly all patients (112). It has been reported that compound mutations can be identified among more than one-third of lorlatinib-resistance cases. And the ALK-independent mechanisms included activation of EGFR, TP53, NRAS-G12D, and MAP3K1 mutations, neurofibromatosis type 2 (NF2) loss-of-function mutations, EMT, and transformation to neuroendocrine carcinoma (105, 110, 114, 122, 123).
2.1.6 ALK: the strategies to overcome resistance
By activating bypass signaling to deal with ALK TKI resistance, the combination with other inhibitors targeting different kinases can be a beneficial addition as well. For example, the joint utilization of the MEK inhibitor selumetinib and ceritinib has been proven to show significant therapy effect on MAP2K1K57N activation mutation of MEK NSCLC cell line, and dual blockage of ALK/MEK can overcome even delay the ALK-TKL resistance (105). Gilteritinib effectively counteracts the development of resistance to lorlatinib in cancer cases with ALK rearrangements (124).
In addition, a research has demonstrated good responses to immunotherapy with immune checkpoint inhibitors, namely nivolumab, pembrolizumab, and atezolizumab, in advanced NSCLC (125). In addition, ALK-TKIs and immune checkpoint inhibitors are also used in combination therapy, including crizotinib with nivolumab, or ipilimumab, or pembrolizumab; alectinib with atezolizumab; ceritinib with nivolumab and lorlatinib with avelumab (105).
2.1.7 ROS1: the mechanisms of resistance
The ROS1 gene, encoding a RTKRTK of the insulin receptor superfamily, shares significant homology with ALK (126). When the ROS1 gene undergoes a genetic alteration, such as a fusion with another gene, it can lead to the formation of a fusion protein with abnormal signaling properties. This fusion protein is constitutively activated, resulting in uncontrolled cell growth and division, which is a hallmark of cancer (127). ROS1-dependent cancers are a specific group of tumors that rely on the abnormal activity of the ROS1 fusion protein for their growth and survival. These cancers include certain types of NSCLC (126, 128), glioblastoma, cholangiocarcinoma, and others. In recent years, targeted therapeutic agents like lorlatinib and crizotinib, have emerged as promising treatment options for ROS1-positive NSCLC patients. Despite the initial success of ROS1-targeted therapies, the development of resistance remains a challenge. Several resistance mechanisms have been identified, including:
1) ROS1 target-dependent resistance mechanisms:
ROS1 kinase domain mutations may include gene fusions or mutations that result in constitutive activation of ROS1 kinase activity. For example, ROS1 gene fusions with other genes, such as CD74, SLC34A2, and EZR (129, 130), have been identified in NSCLC. These fusions lead to the overexpression of ROS1 and the constitutive activation of its kinase domain, promoting uncontrolled cell growth and tumor formation. ROS1 kinase domain mutations are detected in more than 30% of crizotinib-resistant and nearly 50% of lorlatinib-resistant cases (131). Lorlatinib does not possess sufficient efficacy against the ROS1G2032R mutation, which is known to be resistant to crizotinib. Furthermore, lorlatinib also lacks potency against the ROS1L2086F mutation (131). Consequences of several ROS1 mutations include both functional and steric effects. ROS1G2032R causes a steric clash with the piperidine ring of crizotinib, thereby impeding its effective binding as a TKI (132). Similarly to the ALKL1196M resistance substitution, ROS1L1951R also engenders steric hindrance against crizotinib binding (133). In a comparable manner, the αC helix undergoes a positional change induced by ROS1S1986Y/F, which further obstructs crizotinib binding (134). NSCLC induced by a mutation in CD74–ROS1 develops resistance to crizotinib (135). Moreover, experimental evidence demonstrates that ROS1G2032R induces epithelial-mesenchymal transition and amplifies the migratory and invasive capacities of ROS1 fusion-driven cancer cells through the upregulation of Twist1 (136).
2) ROS1 target-dependent resistance mechanisms:
However, in addition to the genetic alterations within the ROS1 gene itself, there are extrinsic mechanisms that can affect the activity and response of ROS1-dependent cancers. Activation of bypass signaling pathways is involved in this process, such as KRAS, NRAS, EGFR, HER2, KIT, BRAF, and MEK (134, 137–139), have been implicated. In the clinical setting, mutations in KRASG12D and BRAFV600E have emerged during crizotinib treatment (140).
2.1.8 ROS1: the strategies to overcome resistance
Researchers have made significant progress in addressing ROS1-targeted therapy resistance. Several strategies and treatment options have been explored.
Next-generation ROS1 inhibitors are being developing. Currently undergoing clinical evaluation in ALK and ROS1 fusion-positive NSCLC cancers, PF-06463922, a next-generation ROS1/ALK small-molecule inhibitor, exhibits potent inhibition of various oncogenic ROS1 fusion variants and selective activity against a wide range of kinases (133). Repotrectinib (TPX-0005) shows effectiveness against acquired solvent-front mutations in ROS1, NTRK1–3, and ALK, making it a promising treatment for patients with ROS1-, NTRK1–3-, or ALK-rearranged malignancies who have progressed on prior TKIs (141).
Combination therapy is also one of the potentially effective treatment approaches. Combining ROS1-targeted agents with other targeted therapies or immunotherapies has been investigated to enhance treatment efficacy and overcome resistance. For example, combining ROS1 inhibitors with MEK inhibitors or immune checkpoint inhibitors has shown potential in preclinical and clinical studies (142, 143).
Furthermore, ongoing research focuses on identifying novel targets and resistance mechanisms to inform the development of new treatment strategies. For instance, targeting bypass or compensatory signaling pathways, such as MAPK, MET, or AXL, may provide additional avenues to combat resistance (144–146).
2.1.9 BRAF: the mechanisms of resistance
BRAF, a member of the Raf kinase family, plays a crucial role in cell growth, proliferation, and differentiation through the MAPK pathway (147). BRAF mutations can be classified into V600 and non-V600 mutations. V600E mutations are the most common type of BRAF mutation in NSCLC (55%), followed by G469A (35%), D594G (10%), and others (148, 149). The V600E mutation leads to an overload of the MAPK pathway and reduces the activation of apoptotic mechanisms regulated by BAD and cysteine cascade events (150).
In monotherapy studies of NSCLC patients, darafenib demonstrated activity in a prospective study of NSCLC with BRAF mutations (151). Targeted agents for advanced BRAF-mutated NSCLC include vemurafenib, dabrafenib, or sorafenib (152). In vitro studies have confirmed the effectiveness of the MEK-TKI trametinib alone in BRAF V600E-mutated NSCLC cell lines (153). However, targeted therapy is currently not applicable to patients with non-V600 BRAF mutations (154). BRAF inhibitors effectively inhibit V600 mutation monomers, but in non-V600 mutation dimers, the drug can only bind to one of the sites, significantly reducing affinity for the second site and downstream ERK inhibition. Consequently, BRAF non-V600 mutant tumors are theoretically insensitive to BRAF inhibitors, as supported by in vitro experiments (155). Certain EGFR-TKIs, such as gefitinib, afatinib, and osimertinib, have been shown to directly inhibit the G469V BRAF mutant, suppressing NSCLC cell growth in vitro (154).
The mechanisms of BRAF inhibitor resistance in NSCLC have not been fully elucidated. For BRAF V600E mutant NSCLC, acquired resistance after dabrafenib monotherapy is caused by oncogenic KRAS mutations and subsequent sustained BRAF non-dependent MEK activation (156). Kim et al. (157) found that treatment of a dalafenib-resistant (GSR) cell line (V600E NSCLC) with dalafenib resulted in upregulation of EGFR, activation of the EGFR-RAS-CRAF pathway, and sustained activation of ERK1/2, enhancing EGFR-mediated RAS activity. In BRAF non-V600E mutant NSCLC, a study demonstrated that single-agent trametinib upregulates the AKT signaling pathway in this cell line, indicating resistance to single-agent MEK inhibitors (158). Additionally, BRAF non-V600E mutations often coexist with RAS mutations, which may contribute to drug resistance (159). Acquired BRAF mutations can also occur in tumors with EGFR mutations and are suspected to be a mechanism of resistance to the EGFR-TKI osimertinib (160).
2.1.10 BRAF: the strategies to overcome resistance
Although BRAF inhibitors have shown efficacy in various cancers, most patients eventually develop resistance. Combination therapies have been found to be more effective than monotherapy and can delay or overcome resistance. In BRAF V600E-mutated NSCLC, the combination of dabrafenib and trametinib targeting BRAF and MEK demonstrated promising response rates compared to dabrafenib monotherapy (161). However, this therapy is not effective in patients with non-V600 mutated tumors. Limited studies exist on drug resistance strategies for NSCLC with BRAF non-V600E mutations. When trametinib was combined with vemurafenib to treat cell lines, the AKT pathway was not upregulated, suggesting that combination therapy may be beneficial in overcoming drug resistance (158). Chen et al. (162) found that atypical BRAF mutant cells were resistant to the BRAF inhibitor vemurafenib but sensitive to the RAF dimer inhibitor LY3009120, presenting a potential therapeutic option. For acquired BRAF mutations caused by EGFR mutations, drug combinations effectively alleviate BRAF mutation-induced osimertinib resistance. An in vitro study revealed that osimertinib resistance caused by acquired BRAF G469A mutation could be restored with selumetinib and trametinib, rather than dabrafenib treatment (163). Furthermore, several cases of acquired BRAF V600E mutations have shown that patients respond well to the concurrent combination of dabrafenib, trametinib, and osimertinib (164–167). Li et al. (168) provided clinical evidence that trametinib plus osimertinib is effective in patients with EGFR-mutated NSCLC carrying acquired BRAF p.D594N mutation. Sun et al. (169) demonstrated that concurrent treatment with vemurafenib and osimertinib, targeting both EGFR and BRAF, resulted in regression of BRAF V600E mutation-induced osimertinib resistance.
BRAF V600E NSCLC is a prevalent subtype known for its resistance to BRAF inhibitors, mainly due to the existence of KRAS mutations leading to continuous activation of the MEK pathway regardless of BRAF (156). However, the combination of dabrafenib and trametinib has demonstrated efficacy in alleviating this resistance (161). Notably, the BRAF inhibitor (BRAFi) and MEK inhibitor (MEKi) combination has received approval from the US Food and Drug Administration (FDA) for various cancer types (170, 171). Conversely, resistance mechanisms in NSCLC with BRAF non-V600E mutations are not well studied and require more research. Additionally, BRAF mutation serves as a mechanism of drug resistance in EGFR mutation-induced NSCLC. In this context, the combination of BRAF inhibitors has shown promise in overcoming resistance to osimertinib induced by BRAF mutations (163–169).
2.2 Uncommon clinical gene targets
2.2.1 RET
The RET gene encodes a RTKRTK that exerts its oncogenic effects through chromosomal rearrangements generating hybrid proteins (172) as well as mutational activation (173). The proportion of patients with NSCLC who have RET rearrangements is approximately 1%-2% (174–176).
Resistance mechanisms to RET multikinase inhibitors and selective inhibitors can be classified into three main categories:
secondary RET mutations (e.g. RET Gatekeeper mutation V804L/M), acquisition of mutations in genes other than RET, and bypass activation (177). The majority of NSCLC patients with RET fusion have not yet revealed a clear mechanism of drug resistance, preclinical studies have demonstrated that both RET G810S (178) and RET V804 (179, 180) mutations can confer resistance to RET inhibitors in NSCLC. Another avenue of resistance is through MET-mediated bypass activation, which can be overcome by co-inhibition strategies (181, 182). Moreover, RET fusions have been identified as a mechanism of acquired resistance to osimertinib in EGFR mutant NSCLC cases (47). Continued research in this area will provide valuable insights into overcoming resistance and developing effective treatment approaches.
2.2.2 NRAS
The neuroblastoma rat sarcoma virus oncogene homolog (NRAS) is a member of the RAS family, discovered subsequent to the identification of KRAS and HRAS (183). NRAS encodes small GTPases that play a crucial role in regulating cell cycle, proliferation, maturation, and differentiation by transducing signals from membrane-bound RTKs to the nucleus. Mutations in NRAS lead to persistent activation of Ras-GTP, thereby promoting tumorigenesis and metastasis. NRAS mutations are infrequent in NSCLC patients, and there are currently no approved therapies specifically targeting NRAS-mutated NSCLC.
LXH254, a type II pan-RAF inhibitor with high selectivity for BRAF and CRAF, has exhibited antitumor activity in preclinical models of NRAS mutations (184). Park et al. reported that human NRAS-mutant NSCLC cells display moderate sensitivity to PAN-RAF inhibitors, and dual-targeted inhibition of PLK1 and PAN-RAF using Volasertib in combination with LXH254 is more effective in inhibiting long-term cell survival compared to LXH254 alone. This combination therapy may serve as a valuable source of inspiration and guidance for clinical treatment (185).
Nonetheless, substantial gaps remain in the research concerning resistance to NRAS-targeted therapy in NSCLC treatment. It has been demonstrated that miR-145-5p enhances the sensitivity of acquired gefitinib-resistant cells to gefitinib by suppressing NRAS and MEST expression (186). To advance our understanding of NRAS-associated resistance mechanisms in NSCLC treatment, further rigorous investigations are warranted.
2.2.3 PIK3CA
Alterations in the phosphatidylinositol 3-kinase (PI3K) pathway are prevalent in cancer (187). Downstream signaling pathways of PI3K involve AKT and mTOR, while phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), encoding the catalytic subunit p110α of the PI3K complex, is frequently mutated and amplified in various cancer types. Genetic alterations in PIK3CA contribute to oncogenesis in NSCLC and activate mutations in both EGFR and KRAS (188). The incidence of PIK3CA mutations in NSCLC patients is approximately 3.7% (189).
Currently, there is no approved targeted therapy specifically for PIK3CA. The most promising candidate is Alpelisib (BYL719), a potent and selective PI3Kα inhibitor currently undergoing phase II clinical studies for the treatment of NSCLC. Regarding the resistance of PIK3CA-targeted therapy in NSCLC, an in vitro study involving a panel of cancer cell lines revealed that BRAF and phosphatase and tensin homolog (PTEN) mutations, along with concurrent PIK3CA and KRAS mutations, have the potential to render cells insensitive to BYL719 treatment (190). A clinical case report on a NSCLC patient with resistance to alpelisib identified a MET exon 14 skipping mutation, possibly serving as a resistance mechanism to alpelisib (191). Previous studies have also implicated MET exon 14 skipping mutations as mechanisms of resistance to TKI inhibitors (192). Additionally, miR-10a enhances the resistance of circulating tumor cells (CTCs) to cisplatin and inhibits the PI3K/Akt pathway by targeting PIK3CA, providing a novel therapeutic target for NSCLC treatment (193). Inhibiting EZH2 enhances the sensitivity of PIK3CA-driven NSCLC to PI3K inhibition (194). Limited research exists on specific therapeutic strategies for overcoming treatment resistance induced by PIK3CA mutations in NSCLC. However, studies on breast cancer offer insights that could be applicable to NSCLC. PDK-1 signaling is known to activate the mTOR complex without activating AKT (195), and IGF-1 and other growth factors can also activate mTOR signaling, leading to resistance to PI3K inhibitors (196). Therefore, combining PI3K, AKT, mTOR, or CDK 4/6 inhibitors can enhance therapeutic efficacy. Furthermore, while PI3K inhibitors reduce AKT signaling, AKT inhibitors can slow down the development of resistance to PI3K inhibitors in breast cancer cells (197). Synergistic inhibition of CDK4/6 and PI3K inhibitors may overcome resistance to PI3K inhibitor monotherapy (198).
It is noteworthy that the majority of studies investigating PIK3CA resistance have primarily focused on breast cancer cells, while there remains a paucity of research specifically examining resistance mechanisms in NSCLC. Therefore, further investigations into the molecular mechanisms underlying PIK3CA resistance in NSCLC are warranted to enhance our understanding and inform the development of effective therapeutic strategies.
2.2.4 HER2
HER2, a member of the HER family along with HER1 (also known as EGFR), HER3, and HER4, forms heterodimers with other HER family receptors or homodimers when highly expressed (199). These interactions subsequently lead to tyrosine kinase phosphorylation and activation of downstream tumorigenic signaling pathways, including RAS-RAF-MEK-ERK and PI3K-AKT. HER2 alterations, which are widely acknowledged as a key factor in the development of various types of solid tumors, primarily consist of HER2 mutation, HER2 amplification, and HER2 overexpression. The incidence rates of these alterations in NSCLC are approximately 1%-6.7%, 2%-22%, and 7.7%-23%, respectively (200–204).
Early targeted therapy for HER2 mutations in NSCLC patients primarily involved pan-HER-TKIs such as afatinib (205–208), daclatinib (209), neratinib (210), poziotinib (211), and pyrotinib. However, the results of these prospective studies with small sample sizes were unsatisfactory (206, 208, 210, 212). The antibody-drug conjugate T-DM1 has demonstrated favorable short-term efficacy in advanced NSCLC with HER2 mutations, but response duration is limited (213, 214). A relatively new anti-HER2 ADC, T-DXd, comprising the topoisomerase I inhibitor deruxtecan, trastuzumab, and cleavable linkers, has shown promising results. The FDA recently approved a 5.4 mg/kg dose of T-DXd based on data from the clinical studies DESTINY-Lung01 (215) and DESTINY-Lung02 (216) for patients with advanced and metastatic HER2-mutated NSCLC who have received prior systemic therapy. A retrospective analysis of 101 patients with advanced NSCLC harboring HER2 mutations and treated with trastuzumab showed higher overall efficacy when combined with chemotherapy compared to chemotherapy alone (217). In vitro experiments have demonstrated that T-DM1 can overcome resistance to gefitinib and osimertinib (218). Another study involving 9 patients with advanced NSCLC and HER2 mutations reported higher efficacy with trastuzumab in combination with chemotherapy compared to afatinib treatment (219). T-DM1 monotherapy has also shown activity in patients with HER2 amplification and concurrent EGFR mutations (213). Furthermore, preclinical models with HER2 amplification have exhibited significant tumor shrinkage when treated with a combination of T-DM1 and the pan-HER inhibitor neratinib, similar to trastuzumab-duocarmycin, another HER2-targeted ADC (213).
The role of HER2 amplification or overexpression as a driver in primary NSCLC remains unclear and requires further thorough investigation. Current research suggests that HER2 amplification is a mechanism by which various types of NSCLC develop drug resistance. HER2 amplification leads to resistance to EGFR-TKIs in NSCLC (30), and HER2 mutations exhibit similar effects (220), with HER2D16 leading to osimertinib resistance via the Src non-dependent pathway (221). In addition, HER2 amplification also contributes to afatinib resistance in HER2-mutated NSCLC (222) and ALK-TKI resistance in ALK-rearranged NSCLC (223). Furthermore, HER2 amplification can cause acquired resistance to MET inhibitors during crizotinib treatment (221) and resistance in KRASG12C NSCLC, which can be overcome by co-targeting SHP2 (87).Current research indicates that HER2 amplification serves as a prominent mechanism underlying the development of drug resistance in various lung cancer types. This resistance extends to EGFR-TKI (30), HER2 self-mutation inhibitors (222), ALK-TKI (223), MET inhibitors (221), and KRAS G12C inhibitors (87). Notably, afatinib, a HER2-targeted inhibitor, has demonstrated limited efficacy in NSCLC patients, failing to meet the anticipated disease control outcomes (206). This lack of response could be attributed to the inherent resistance of HER2 mutants to pan HER TKIs. Moreover, the activation of YES1 leads to the emergence of resistance to neratinib in breast and lung cancers that have HER2 amplification (224).
While studies investigating the mechanisms of resistance to ADCs have primarily focused on breast cancer cells (225, 226), there remains a significant gap in our understanding of these mechanisms within the context of NSCLC cells. Further investigation is required to elucidate the specific resistance mechanisms operating in NSCLC cells, thus enabling the development of effective therapeutic strategies.
2.2.5 c-MET
c-MET is a member of the tyrosine kinase receptor superfamily and is encoded by the MET proto-oncogene. Structurally, it is a transmembrane tyrosine kinase receptor protein comprised of a heterodimer consisting of a 45 kDa extracellular α subunit and a 145 kDa transmembrane β subunit (227). The extracellular regions of both subunits serve as the site for ligand recognition, while the intracellular region possesses tyrosine kinase activity. This activity is responsible for activating tyrosine kinase upon ligand binding and subsequently initiating downstream cascade signals (228). In patients with NSCLC, MET exon 14 jump mutations are present in around 5.6% of patients (229). On the other hand, MET amplification has been observed in a broader range of 2% to 20% in various studies. This variation may be attributed to disparities in the definition of amplification levels and the methods used for detection (230).
MET inhibitors can be categorized into three main classes: small molecule MET receptor inhibitors (e.g., crizotinib, tivantinib, savolitinib, tepotinib, cabozantinib, and foretinib), MET receptor monoclonal antibodies (e.g., onartuzumab, emibetuzumab), and antibodies targeting their ligands HGF (e.g., ficlatuzumab and rilotumumab) (231). Of these, Crizotinib has gained FDA approval for treating ALK-positive patients with advanced NSCLC. It is an ATP-competitive, non-selective inhibitor with multiple targets, including c-Met and ALK (232). Additionally, Merck’s TepMetko (tepotinib) has obtained approval in Japan for treating patients with advanced NSCLC harboring MET exon 14 skipping mutations. This represents the world’s first approved single-target inhibitor for c-MET.
Resistance mechanisms to inhibitors targeting MET exon 14 skipping mutations can be categorized into primary and secondary resistance. Primary resistance primarily arises from mutations and alterations in exon splice sites (233, 234), whereas secondary resistance is often associated with HER2 amplification (221). MET amplification commonly contributes to resistance development in various other NSCLC treatments, particularly in the context of acquired resistance to EGFR-TKI (235, 236). Moreover, modifications in the signaling molecules’ functionality have also been identified as contributing factors in the emergence of drug resistance.
Overcoming resistance to targeted therapies in NSCLC is a significant challenge, but there are several promising strategies that researchers are exploring. Clinical trials and ongoing research efforts are essential to further explore and optimize these approaches for overcoming resistance to targeted therapies in NSCLC. Above all, In the recent 5 years, the promising strategies for overcoming the resistance to targeted therapies in NSCLC are summarized in Table 1.
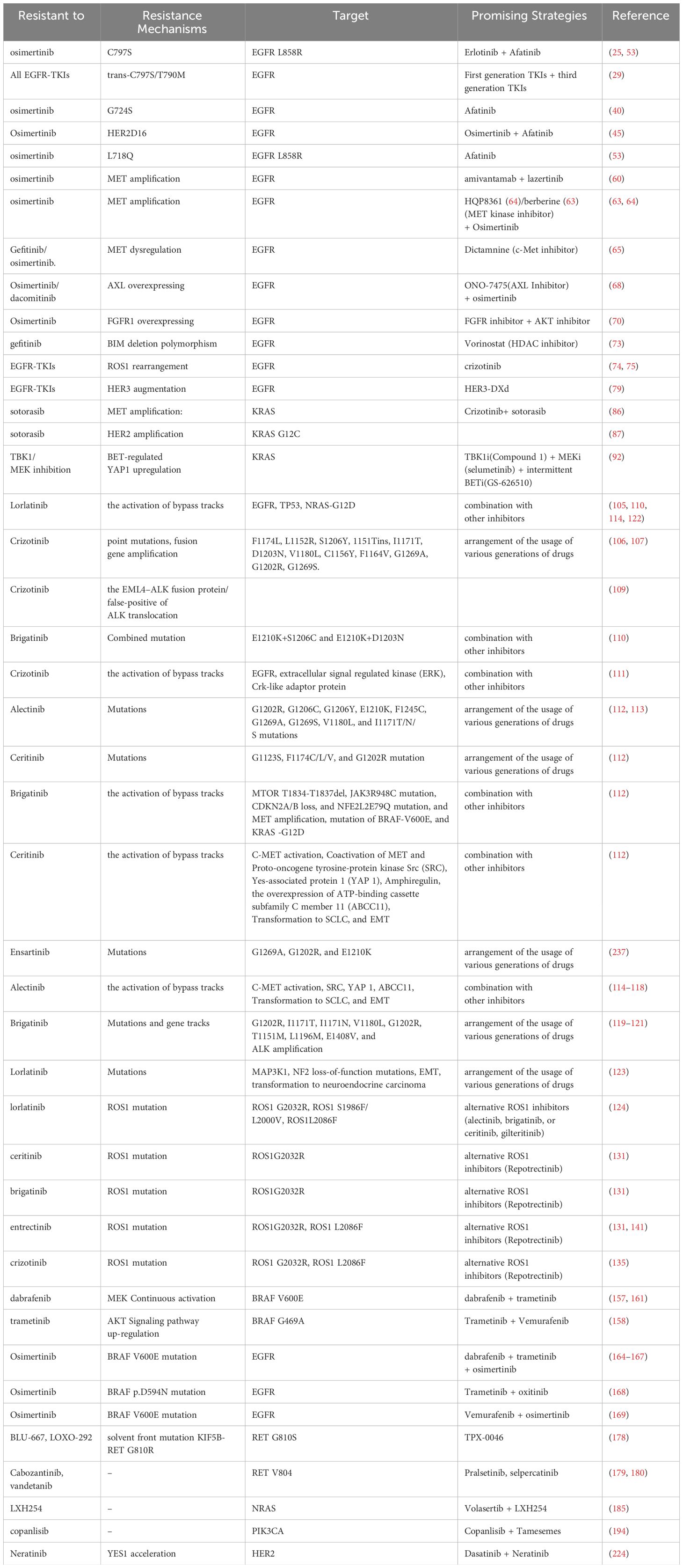
Table 1 The promising strategies for overcoming the resistance to targeted therapies in NSCLC in recent 5 years.
3 The mechanisms of immunotherapeutic resistance in NSCLC
3.1 Immune checkpoint inhibitors
3.1.1 The definition of immune checkpoint inhibitors resistance
Targeting the PD-L1/PD-1 axis and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) with ICIs has shown significant improvements in survival outcomes for patients with NSCLC (238). Several studies have demonstrated that both single-agent and combination ICI therapies have significantly enhanced clinical efficacy endpoints in locally advanced, metastatic NSCLC and extensive-stage small-cell lung cancer patients without EGFR or ALK gene mutations (239). Specifically, for advanced NSCLC patients, ICI treatment can provide long-term benefits, with median response durations ranging from 12 to 25 months, particularly in those with PD-L1 expression ≥50% (240).
Despite the durable benefits indicating the establishment of long-term immune memory, some patients who initially responded to ICI therapy eventually experienced relapse and acquired resistance, leading to clinical and biological immune treatment failure (241, 242). Primary and acquired resistance have emerged as a significant challenge in further improving the prognosis of patients with advanced or metastatic lung cancer (243). Primary resistance is characterized by disease progression within a period of at least 6 weeks (two cycles) but not exceeding 6 months of ICI treatment (244). Acquired resistance, on the other hand, refers to disease progression occurring after at least 6 months of clinical benefit, including objective response or stable disease lasting for at least 6 months (245). Definitions of post-ICI treatment resistance encompass both toxicity-related and unrelated resistance, including resistance occurring after completion of the planned treatment regimen (246). This definition is also applicable to the investigation of adjuvant/neoadjuvant immunotherapy in NSCLC (247). Studies have indicated that the binding of anti-PD-1 receptors decreases after the last dose of treatment for a period of 2-3 months. Therefore, if a patient who previously benefited from ICI therapy experiences disease progression within 12 weeks of the last dose, it is classified as acquired resistance. In such cases, restarting ICI treatment may offer potential benefits (248). On the other hand, patients who received ICI treatment but showed no initial benefit and experience disease progression after stopping treatment for any reason are classified as having primary resistance. However, a recent expert group has suggested that all relapses occurring after initial objective response (excluding stable disease) should be considered acquired resistance, regardless of its timing (249). The concepts of primary and acquired resistance to ICI treatment have evolved from the understanding of resistance in other anti-tumor therapies, such as chemotherapy and tyrosine kinase inhibitors. However, unlike the clear definition of resistance in those therapies, a unified definition for ICI immune resistance patterns has not been established (250). Currently, the most influential concept is proposed by Society for Immunotherapy of Cancer (SITC), which identifies three distinct resistance patterns to anti-PD-1/PD-L1 therapy: primary resistance, acquired resistance, and progression on or after therapy discontinuation (251).
The tumor microenvironment (TME) plays a crucial role in the immune response to ICI treatment (243). Furthemore, the TME evolves based on different embryonic cell genetic backgrounds and tumor genetic compositions (249). In addition, therapeutic interventions such as radiotherapy, surgery, chemotherapy, and immunotherapy, along with clinical and disease characteristics, can influence the TME, potentially affecting the efficacy of single- or combined ICI treatment. Considering different disease progression times and patterns, immune resistance characteristics in NSCLC patients can be categorized into two groups: 1) early progression, including fast progression (FP) or hyper-progressive disease (HPD); 2) late progression after initial treatment benefit (249). These distinct progression times and patterns underscore the immune resistance process observed in diverse patient populations receiving ICI treatment. However, due to the complex nature of biology and the rapid integration of immunotherapy in clinical practice, a unified approach to prevent, define, and manage immune resistance has not yet been established, necessitating further research.
3.1.2 The mechanisms underlying ICI resistance
Resistance to ICI is caused by immune system antigen presentation impairment and the TME. The former prevents T cell priming, activation, trafficking, and migration, and the latter can increase resistance by overexpressing T cell co-inhibitory receptors and immunosuppressive cells (244), which can be divided into endogenous and exogenous mechanisms (Figure 2).
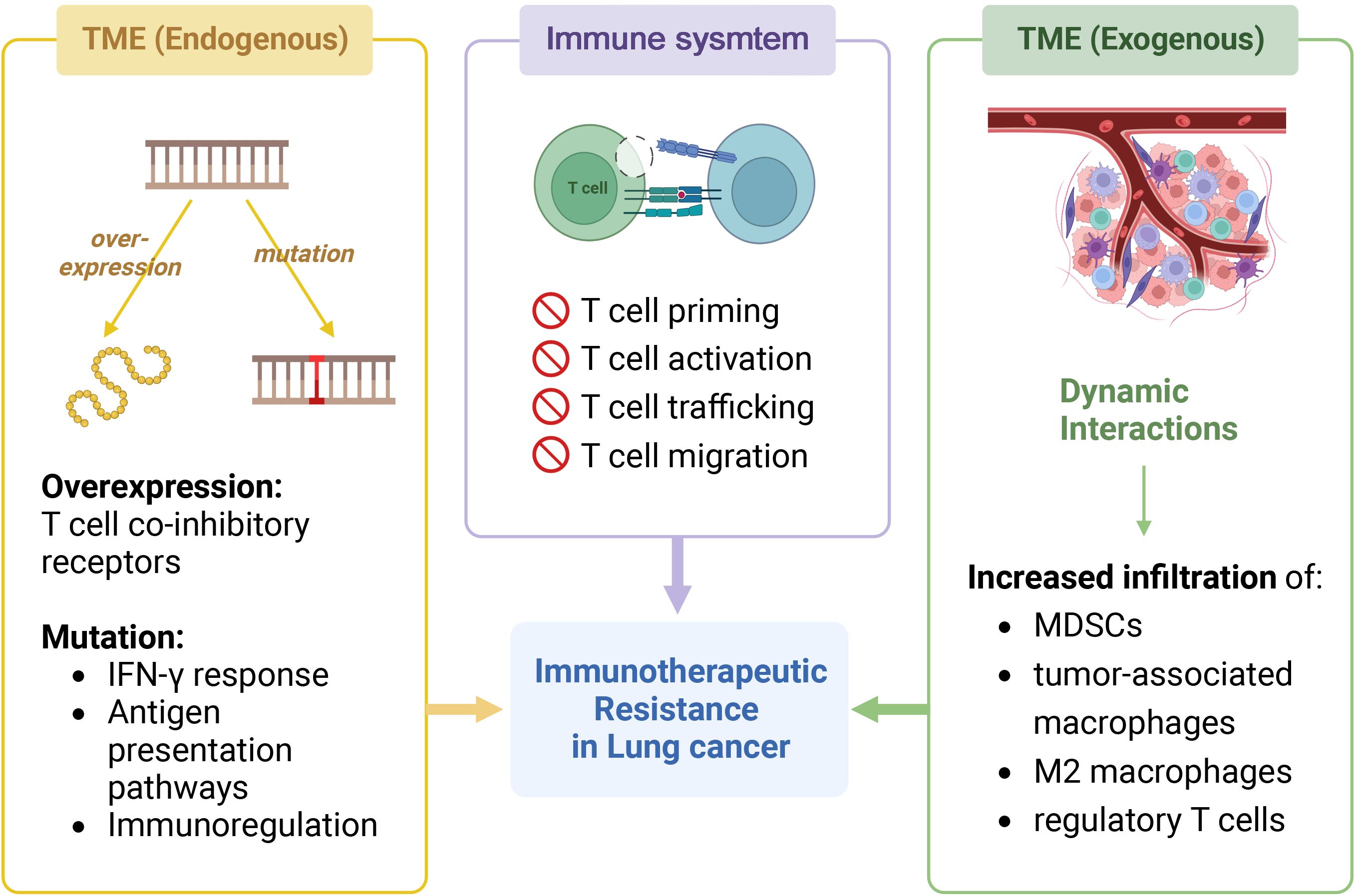
Figure 2 Immunotherapeutic resistance in NSCLC. The image shows the mechanisms that generate resistance in immunotherapy for NSCLC. It can be classified into two parts: immune system and tumor microenvironment (TME), the latter can be further divided into endogenous and exogenous. In immune system, impaired antigen presentation leads to the failure to activate normal immune responses such as T cell priming, activation, trafficking, and migration. In addition, endogenous alterations in the tumor microenvironment leads to the development of drug resistance, including gene overexpression and gene mutation. The former includes T cell co-inhibitory receptors on immunosuppressive cells, and the latter includes genes involved in IFqN-γ response, antigen presentation pathways, and immunoregulation. Exogenous factors such as dynamic interactions within the TME lead to increased infiltration of myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages, M2 macrophages, and tumor-associated macrophages. Created with BioRender.com.
The endogenous mechanisms for TME resistance can be categorized as follows:
1) Constitutive or acquired mutations in TME genes reduce antigen expression and recognition, preventing T cell activation (252).
2) Mutations in genes relevant to IFN-γ response and antigen presentation, such as antigen processing machinery, signaling transducers, and transcription activators, can disrupt the expression of co-inhibitory signaling and major histocompatibility complex (MHC) molecules (253). Defects in IFN signaling, regardless of MHC-I levels, are the main cause of impaired anti-tumor responses (254).
3) Immunoregulation gene mutations can also affect carcinogenic pathways and the immunosuppressive TME (255). Deficit of β-2 microglobulin can affect MHC-I molecule expression, affecting T cell antigen recognition potential and reducing cytotoxic activity (254). Thus, IL-2 agonists or other non-MHC-I strategies are needed to activate CD8 and NK cells (254).
Exogenous TME factors also matter (254). Dynamic interactions in the TME disrupt immunosuppression, pro-inflammatory cytokines, and mediators like co-stimulatory and co-inhibitory signals, increasing myeloid-derived suppressor cells (MDSCs), tumor-associated, M2 macrophage, and regulatory T cell infiltration (249). Hypoxia increases VEGF expression, which mobilizes and infiltrates MDSCs into tumor sites (256).
However, acquired resistance mechanisms are linked to cancer cell and TME adaptive changes during immune therapy. These changes can be caused by epigenetic regulation or protein translation modification (257). Clonal evolution of cancer cells can also cause acquired resistance by impairing anti-tumor immune responses through gene mutations (258). Furthermore, resistance patterns affect treatment strategy selection (256). Enhanced adjuvant therapy can restore immune sensitivity in acquired resistance patients, allowing immunotherapy (259). Additionally, patient selection criteria and immune resistance patterns may affect research results (259).
3.2 Cancer vaccines
Cancer vaccines have emerged as a vital tool in NSCLC treatment, offering potential solutions to challenges associated with conventional therapies and improved patient outcomes. Firstly, cancer vaccines specifically target tumor antigens, focusing the immune response on cancer cells while minimizing harm to healthy tissues, potentially reducing side effects compared to non-specific treatments (260). Secondly, by activating the immune system, cancer vaccines stimulate innate and adaptive immune responses, leading to systemic immune memory and long-term protection against cancer recurrence (261). Personalization is another advantage, as vaccines can be tailored to individual tumor characteristics, enhancing treatment efficacy based on specific antigens or mutations (262). Additionally, the versatility of cancer vaccines allows for combination strategies with other therapies, such as immune checkpoint inhibitors or targeted therapies, to enhance overall immune responses and overcome resistance mechanisms (263). Lastly, cancer vaccines can be applied throughout the various stages of NSCLC, serving as adjuvant therapy post-surgery or as a therapeutic option for metastatic disease (264).
All current investigations into lung cancer vaccines are focused on NSCLC, primarily in participants with advanced disease. Notable vaccines under study for NSCLC include Belagenpumatucel-L (Lucanix), which stimulates the production of TGF-β2 to induce cancer cell death. Phase 2 and 3 trial results indicated that belagenpumatucel-L improved survival rates of NSCLC patients (265, 266). Another vaccine, Stimuvax or Tecemotide, triggers an immune response against mucin 1, a protein overexpressed in NSCLC (267). While a 2011 study showed some positive results, they were not particularly strong (268–270). In contrast, MAGE, the initial cancer/testis antigen (CTA) discovered, exhibits discernible expression in approximately 30%-50% of NSCLC patients, particularly in cases of squamous cell carcinoma (LUSC) (271). The safety of a recombinant adjuvanted MAGE-A3 was assessed in patients with resected MAGE-A3 positive NSCLC through the phase III MAGRIT study. However, the final findings of the study did not demonstrate a superior disease-free survival (DFS) in the MAGE-A3 treated group compared to the placebo control group (60.5 months vs. 57.9 months, respectively) (272). CIMAvax-EGF, a protein-specific vaccine intended to inhibit the EGFR, a protein excessively expressed on NSCLC cells, has exhibited both safety and efficacy in several clinical trials conducted within Cuba (273). In a randomized controlled phase II study encompassing 80 patients with stage IIIB/IV NSCLC who had previously undergone first-line chemotherapy, CIMAvax-EGF demonstrated both safety and immunogenicity (274). Racotumomab prompts antigen development against NeuGcGM3, a type of lipid, and a phase 2 and 3 study demonstrated significantly prolonged overall survival rates compared to the placebo (275). The TG4010, a viral vector vaccine, consists of a modified Vaccinia Virus Ankara (MVA) that carries the human MUC1 gene and interleukin 2 (276). In a phase I clinical trial, Rochlitz et al. observed a promising safety profile for TG4010, with one lung cancer patient experiencing significant reduction in metastasis over a 14-month period (277). A phase II randomized clinical trial suggests that combining TG4010 with chemotherapy in first-line advanced or metastatic NSCLC patients (stages IIIB and IV) may improve the efficacy of chemotherapy (278).
4 New strategies for prevention and combating immunotherapy resistance
4.1 Enhance T cell priming
New approaches to enhance T cell priming for T cell engineering and vaccination have emerged. These include Tumor-Infiltrating Lymphocyte (TIL) therapy, adoptive cell therapy utilizing Chimeric Antigen Receptor T cells (CAR-T cells), and personalized vaccines (279).
TIL therapy involves isolating TIL from tumor tissue, expanding and screening them to identify tumor antigens, and reintroducing them into the patient (279). Clinical trials are currently underway to compare the efficacy of TIL adoptive cell therapy alone or in combination with ICI in first-line and resistant NSCLC patients, with positive results demonstrating tumor regression and disease control in previously unresponsive patients (280). In contrast, CAR-T cells therapy involves genetically engineering T lymphocytes to express synthetic tumor antigen receptors, enabling them to recognize antigens independently of MHC presentation (255). Although adoptive cell therapy has shown promise in hematologic malignancies, its efficacy in solid tumors, including NSCLC, remains limited, highlighting the need for improved target antigens (255). Personalized vaccines based on tumor-specific neoantigens present another avenue for augmenting T cell-mediated anti-tumor responses. An ongoing study is evaluating the NEO-PV-01 personalized vaccine in combination with pembrolizumab and chemotherapy as a first-line (1L) treatment for NSCLC (NCT03380871) (). However, only a small fraction of mutations encode neoantigens recognizable by T cells. Other potential neoantigens include peptides resulting from single-nucleotide variations, insertions and deletions causing frameshifts, structural variations, and even post-translational events resulting from abnormal RNA splicing mediated by viral genome integration (281).
4.2 Overcoming the resistance to ICI
4.2.1 Targeting VEGF
Due to the mechanical damage caused by VEGF-mediated angiogenesis and vessel remodeling, lymphocytes encounter impairments in their transendothelial migration process (282). VEGF, acting as an extracellular mediator, exerts significant influence on tumor immunity through diverse mechanisms. These include the inhibition of dendritic cell maturation and antigen presentation, upregulation of immune suppressive mediators such as PD-L1, PD-L2, IDO-1, IL-6, and IL-10, induction of regulatory T cells, and the accumulation of MDSCs (282). Several studies have investigated monoclonal antibodies and small molecules targeting the VEGF axis, aiming to restore VEGF-mediated MDSC infiltration and subsequent immune suppression (283). In the Impower 150 study, which focused on non-squamous NSCLC patients, the combination of anti-VEGF monoclonal antibody bevacizumab, atezolizumab, and chemotherapy as first-line treatment resulted in a significant prolongation of overall survival (OS) compared to chemotherapy alone (284). Additional drugs, including monoclonal antibodies such as ramucirumab and multitargeted kinase inhibitors like lenvatinib, sitravatinib, nintedanib, and axitinib, are currently undergoing evaluation for their efficacy in first-line and ICI-resistant NSCLC patients (244). In a phase Ib/II study involving 22 NSCLC patients (a majority of whom had received prior treatment, with 52% having received prior ICI therapy, the combination of lenvatinib and pembrolizumab showed promising preliminary results, with an objective response rate (ORR) of 33% and a median progression-free survival (PFS) of 5.9 months (285). Based on these encouraging findings, the LEAP series III clinical trials are currently underway to validate the effectiveness of lenvatinib and pembrolizumab as both first-line and later-line treatments (286). Moreover, a phase III study is investigating the efficacy of sitravatinib in combination with nivolumab for ICI-resistant patients (287).
4.2.2 Cytokines
Research is underway to evaluate cytokines that can be specifically targeted. Notably, the use of the anti-IL-1β monoclonal antibody canakinumab has shown promising results in reducing NSCLC incidence and mortality in patients with atherosclerosis (288). This has sparked significant interest in IL-1β as a potential therapeutic target. However, the combination of canakinumab and docetaxel chemotherapy did not provide any benefits to ICI-resistant NSCLC patients, as observed in the phase III CANOPY-2 study (289). Currently, canakinumab is undergoing validation in phase III clinical trials as a first-line treatment in combination with immunotherapy and chemotherapy for adjuvant therapy, aiming to reduce immune resistance (290). On the other hand, studies targeting IL-10 and TGF-β have yielded disappointing results. In the phase II CYPRESS1 and CYPRESS2 studies, the use of pegilodecakin, a PEGylated recombinant human IL-10, in combination with ICI did not show benefits in terms of ORR and survival in NSCLC patients. Moreover, it led to significant adverse reactions (291). Similarly, despite the initial promising efficacy and safety of bintrafusp alfa, a bifunctional protein targeting TGF-β and PD-L1, it failed to provide benefits to ICI-naïve NSCLC patients with high PD-L1 expression compared to pembrolizumab in the phase III Lung-037 study. The trial was terminated recently due to futility (292). In addition to these findings, there is a growing body of research focusing on the gut microbiome as a potential target for immune anti-tumor therapy. Evidence suggests that the enrichment of different microbial communities and the use of broad-spectrum antibiotics can serve as predictors of ICI efficacy in NSCLC patients. This indicates that altering the composition of the gut microbiome, such as through fecal microbiota transplantation, could be a treatment strategy to enhance immunotherapy efficacy or reverse ICI resistance (293).
In summary, the degree and duration of immune anti-tumor response in cancer patients are dependent on the complexity of the immune system and its dynamic interaction with the TME, as well as the evolutionary process of tumor clone selection. However, resistance often emerges in patients at different stages, such as early in treatment, after a prolonged period of clinical benefit, or even after treatment discontinuation for any reason. These resistance mechanisms can take various forms. In addition, the factors influencing immune response and the underlying mechanisms of specific immune resistance are multifaceted. Currently, research focused on reducing resistance to ICI is still in its early stages. Most strategies primarily rely on biomarkers, which may limit the efficacy of newly developed therapies.
In fact, personalized therapies based on biomarkers and specific resistance patterns may offer the most promising approach for predicting, preventing, and mitigating ICI resistance. In addition to PD-L1, numerous other biomarkers have been evaluated for predicting ICI efficacy or resistance. These include molecular and genetic characteristics such as tumor mutation burden, neoantigen load, and selective gene mutations, as well as specific immune cell subtypes like TILs and MDSCs. Furthermore, immune signaling pathways have also been explored as potential biomarkers.
Advanced analytical tools can enable the development of highly complex multiparameter evaluation methods for the TME and biomarkers. By integrating genomic, transcriptomic, and spatial positioning data, these tools can facilitate the identification of patients who are more likely to benefit from immunotherapy. Here, we summarized the Phase III Clinical Studies of Anti-ICI Resistance Strategies In Advanced NSCLC in recent 5 years in Table 2.
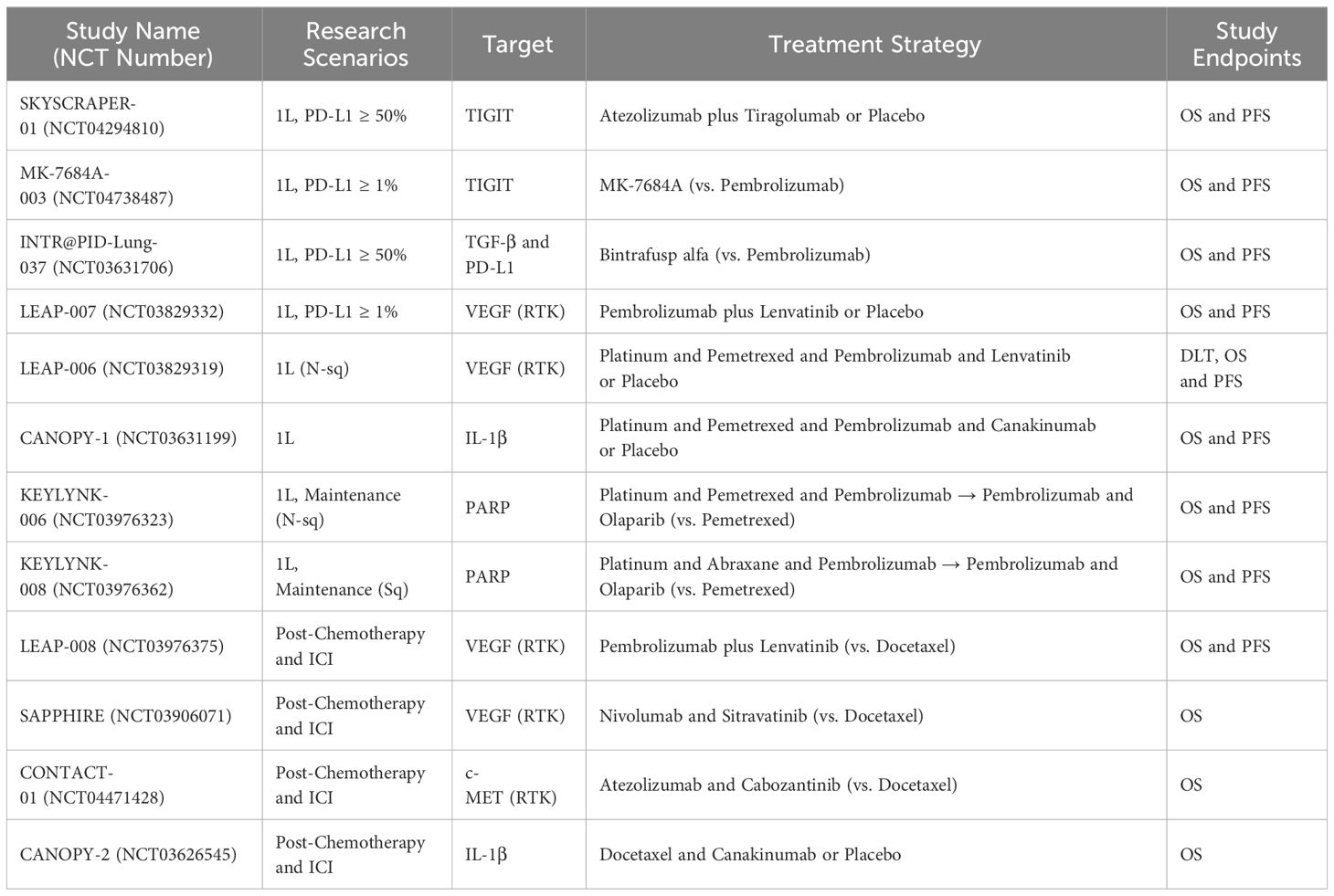
Table 2 Phase III clinical studies of Anti-ICI resistance strategies in advanced NSCLC.
4.3 Overcoming the resistance to CTLA-4 immunotherapy
CTLA-4, also known as CD152, is a transmembrane protein expressed in activated CD4+ and CD8+ T cells (294–298). Under physiological conditions, CTLA-4 and CD80/CD86 binding can inhibit T-cell activation signals and prevent autoimmune disease (299, 300). Blocking CTLA-4 can directly target inhibitory signals on effector T cells and reduce the inhibitory effect of regulatory T cells (Tregs) (301–305) thus effectively enhancing the antitumor effect of T cells.
However, while immune checkpoint blocking therapies (ICBs) are revolutionizing therapeutic algorithms for cancers, the frequently observed innate, adaptive or acquired drug resistance remains an inevitable obstacle to a durable antitumor activity, thus leading to non-response or tumor relapse.
Increased Tregs and dendritic cells (DCs) in the tumor environment may be responsible for acquired resistance and provide another therapeutic target to prevent or overcome resistance. Tregs can be identified with the cell surface markers forkhead box protein 3 (Foxp3), CD25, CD357, lymphocyte-activation gene 3 (LAG3), CTLA-4, and low CD127. Foxp3 are crucial in the immunosuppressive activity of suppressor T cells or Tregs within the lung cancer TME. Foxp3 is a transcription factor that is upregulated in TILs and tumor cells and coveys a negative prognostic factor in the NSCLC and maybe a future target for resistant tumors. LAG3 is also a co-inhibitory molecule on TILs, Tregs, DCs, and NK cells that dampens T cell activation via its binding to MHC II receptors, making it another possible therapeutic target after resistance. T cell immunoglobulin and mucin domain-3-containing molecule 3 (TIM3) is a cell surface protein typically seen on DCs that interacts with Galectin-9 on T cells leading to inhibition of the T cell response. TIM-3 expression can be seen on TILs, and its interaction with galectin-9 on Tregs or tumor cells can lead to T cell inhibition (306). Increased TIM-3 expression has been seen as a marker of poor prognosis but may also provide an alternative checkpoint target for therapy after PD-1 failure (306, 307).
In order to overcome resistance, potential therapeutic strategies include enhancing antigen procession and presentation, reinforcing the activity and infiltration of T cells, and destroying immunosuppression microenvironment. In future, determining the driving factors behind ICB resistance by tools of precision medicine may maximize clinical benefits from ICBs. Moreover, efforts in individualized dosing, intermittent administration and/or combinatory regimens have opened new directions for overcoming ICB resistance.
Above all, in recent 5 years, the promising strategies for overcoming the resistance to CTLA-4 immunotherapy in NSCLC can be listed in Table 3.
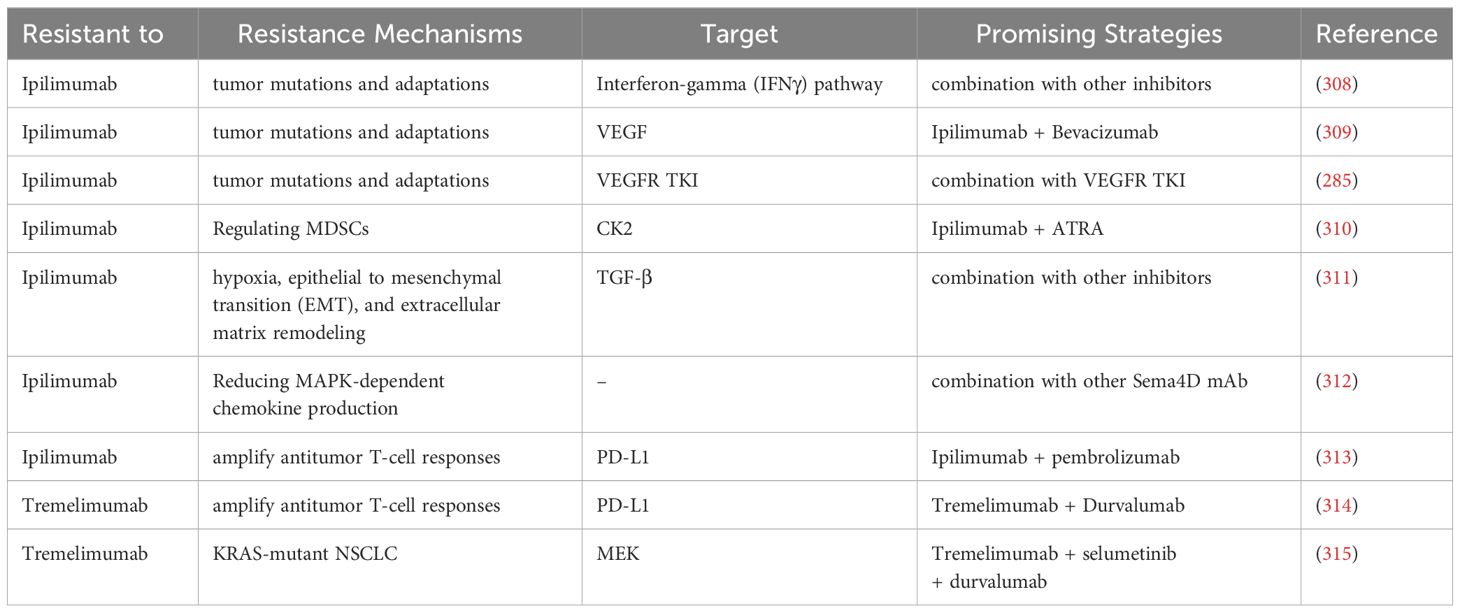
Table 3 The promising strategies for overcoming the resistance to CTLA-4 immunotherapy of NSCLC in recent 5 years.
4.4 Overcoming the resistance to cancer vaccines
Despite the potential benefits observed in NSCLC treatment, resistance to cancer vaccines remains a significant challenge. Resistance to cancer vaccines can arise through tumor intrinsic and extrinsic resistance. Tumor intrinsic resistance encompasses six aspects: mutations in signaling pathways supporting tumor-immune control, loss of tumor antigen expression, changes in antigen processing pathways, loss of HLA expression, epigenetic changes, and increased expression of immunosuppressive ligands (316–318). Extrinsic resistance to cancer vaccines may be caused by immunosuppressive cells (such as CAFs, MDSCs, Tregs, and M2 macrophages) and immunosuppressive cytokines, which can directly or indirectly inhibit the activation of effector T cells and DC-mediated T cells in TME (319–323). Lung cancer cells have been shown to produce a variety of immunosuppressive molecules, including TGF-β, prostaglandin E2, IL-10, and cyclooxygenase-2, which can affect DC processing and presentation, as well as the acquisition and expression of CTL effector cell function (324–326). Strategies for developing effective immunotherapy for NSCLC have previously faced challenges. However, recent successes in identifying T cell responses to tumor-specific antigens in patients with NSCLC suggest that this goal may now be achievable (327). The identification of lung tumor-associated antigens and presenting them in the optimal context may enable the immune system to generate anti-lung tumor effector cells (328).
Researchers are currently exploring various strategies to overcome resistance. Some representative strategies include:
1) Combination therapies: Clinical trials have been initiated to explore the combination of ICBs with various established cancer vaccines in the hope of providing more effective tumor-specific T-cell responses in patients with advanced-stage NSCLC. These include the combination of TG4010, nivolumab and anti-PD1 antibody (329); the CIMAvax-EGF vaccine with anti-PD1 antibodies (273); and the melanoma antigen family A, 3 (MAGE-A3) vaccine with ICBs (330).
2) Personalized vaccines: The vaccine Vx-001, targeting TERT, a tumor antigen derived from universal tumor antigens and recognized as neoantigens, showed strong immunogenicity. However, a phase II clinical trial in NSCLC patients did not demonstrate any significant improvements in overall survival, disease control rate, or time to treatment failure (331). Linette et al. present the development of a clinical grade neoantigen vaccine formulation (FRAME-001) with stability for up to 32 weeks, intended for immunotherapy in advanced NSCLC in combination with pembrolizumab, showcasing the potential of personalized therapeutic cancer vaccines targeting tumor-specific neoantigens (332).
3) Modulating the TME: Cancer vaccines that target tumor-associated antigens (TAAs) may face challenges due to cancer cell evasion mechanisms, making the stable components of the cancer microenvironment an alternative vaccination target to avoid immune anergy caused by genetic mutations (333). The utilization of combined cancer vaccines alongside diverse immunotherapies or standardized therapies has emerged as an effective approach in recent years to counteract the immunosuppressive TME and enhance clinical outcomes (334). The field of NSCLC treatment is currently divided into three main categories, which are DC vaccines (NCT05195619, NCT03546361), DNA-based vaccine(NCT05242965), peptide-based cancer vaccines (NCT05254184, NCT03879694, NCT03633110).
4) Adjuvants and immune stimulants: The combination of a DNA vaccine containing a fusion gene of MUC1 and VEGFR2, along with the use of GM-CSF as an adjuvant showed an increased inhibition in the growth of MUC1-expressing tumours and prolonged mouse survival (335). A combination vaccine involving irradiated lung adenocarcinoma cells and transfected K562 cells with hCD40L and hGM-CSF induced tumor regression in metastatic lung adenocarcinoma by activating dendritic cells, resulting in a median overall survival of 7.9 months and median progression-free survival of 1.7 months, with 5 out of 14 patients showing CD8+ T-cell activation after vaccination (336). In a phase I study, intratumoral administration of autologous DC transduced with an adenoviral vector expressing the CCL21 gene (Ad-CCL21-DC) in patients with advanced NSCLC resulted in increased tumor CD8+ T-cell infiltration, induction of systemic tumor antigen-specific immune responses, and enhanced tumor PD-L1 expression (337).
5) mRNA-based vaccines: CV9201 and CV9202 have been investigated in NSCLC (338). In particular, CV9202 encoding six tumor-associated antigens(NY-ESO-1, MAGE-C1, MAGE-C2, Survivin, 5T4 and MUC) demonstrated good tolerance and elicited antigen-specific immune responses in a phase Ib study of stage IV NSCLC patients (339).
Despite these limitations, lung cancer vaccines continue to be a promising and actively investigated area. Recent phase III trials targeting specific subpopulations with L-BLP25, MAGE-A3, and belagenpumatucel-L vaccines have demonstrated potential benefits, highlighting the importance of combining vaccines with chemotherapy and radiation as part of a multimodal approach to target lung cancer. Consulting with a healthcare professional or an oncologist can provide the most up-to-date information regarding the treatment options available for lung cancer and drug resistance. Here, we summarized the promising strategies to overcoming the resistance of cancer vaccines to NSCLC in the past 5 years in Table 4.
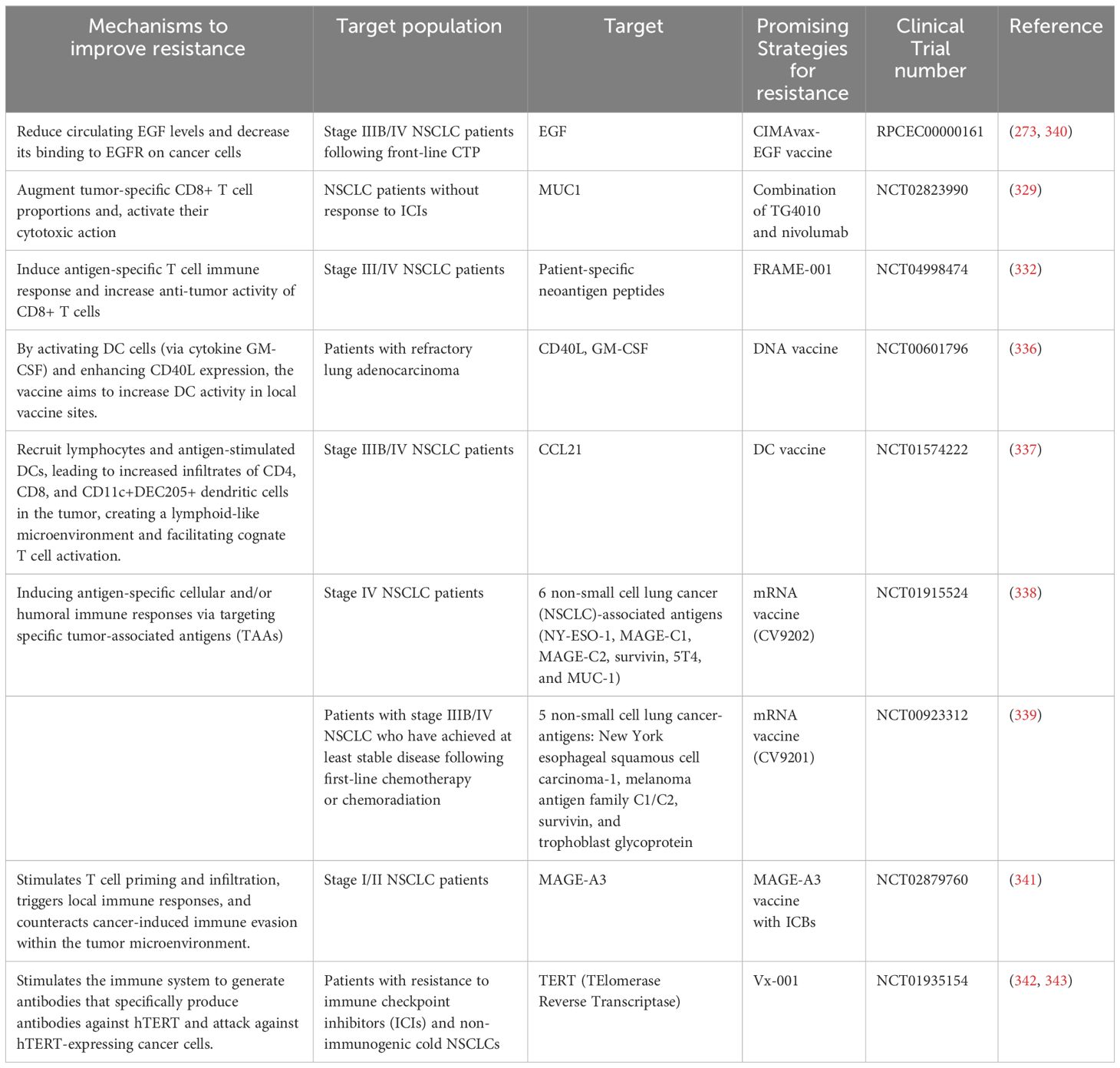
Table 4 The promising strategies to overcome drug resistance with cancer vaccines to NSCLC.
4.5 New strategy: immune system-modulators
Immune system modulators play a crucial role in cancer treatment, encompassing drugs and proteins. These components function by either augmenting the immune system’s capacity to identify and combat cancer cells or inhibiting signals that enable cancer cells to elude immune detection. Examples of such modulators include cytokines, BCG, and various other drugs (344).
In the context of lung cancer, immunomodulators have been employed to enhance the immune response against cancer cells, potentially leading to improved patient outcomes. Apart from checkpoint inhibitors, another class of immunomodulator utilized in lung cancer treatment is cytokines. Numerous cytokine therapies have been investigated for their efficacy in treating NSCLC. Among these, interleukin-2 (IL-2) is one of the most extensively studied cytokines. IL-2 has been demonstrated to stimulate the immune system, specifically the activation and proliferation of T cells. Its therapeutic application has been explored in both NSCLC and SCLC (345–348). For instance, IL-2 therapy, such as ALKS 4230, has shown promise in improving survival rates among patients with advanced NSCLC (349). In the case of NSCLC, IL-2 has been administered in combination with other treatments, such as chemotherapy and radiation therapy, yielding mixed results (350). Some studies have reported enhanced response rates and overall survival, while others have not observed significant benefits (351). Additionally, patients with IL-2-expressing tumors have exhibited significantly improved 5-year overall survival rates in cases of radically resected NSCLC (352). Other cytokines, including IFN-α and IFN-γ, have also been investigated for their potential in NSCLC treatment. These cytokines have demonstrated antitumor effects and the ability to stimulate the immune system, yet their application in lung cancer remains investigational (345, 353, 354). Furthermore, Bacille Calmette-Guérin (BCG), a live attenuated vaccine traditionally used for tuberculosis prevention, has found application in cancer treatment. BCG can be injected directly into the lungs to stimulate an immune response against cancer cells. It can be administered alone or in conjunction with other therapies, such as chemotherapy or radiation therapy (355).
Currently, the FDA has approved 16 different immunomodulators for the treatment of various cancer types. These immunomodulators include nine checkpoint inhibitors, four cytokines, two adjuvants, and a small molecule with immunomodulatory properties. Most of these approvals are for advanced or resistant cancers, but recently some have also been approved as first-line systemic treatments for certain metastatic cancers. Among the approved cytokine drugs are Aldesleukin (Proleukin®), Granulocyte-macrophage colony-stimulating factor (GM-CSF), Interferon alfa-2a, Interferon alfa-2b (Intron A®), and Peginterferon alfa-2b (Sylatron®/PEG-Intron®) (356). In addition, adjunct agents such as Imiquimod and Poly ICLC (Hiltonol®) have been used in cancer treatment (357, 358).
5 Conclusion
In this review, we discuss the mechanisms of resistance to targeted therapy and immunotherapy in NSCLC and outline promising strategies for overcoming resistance from different perspectives.
For targeted therapy, we categorized numerous NSCLC-related targets into common and uncommon clinical gene targets. EGFR is the predominant oncogene in NSCLC, with resistance mechanisms involving EGFR-dependent and EGFR-independent pathways. Clinical evidence supports that resistance to the classical C797S/T790M mutation can be overcome by combining first- and third-generation EGFR TKIs. Various mutation types can be targeted using a combination of EGFR-TKIs and chemotherapy, or specific small molecule inhibitors. Furthermore, fourth-generation EGFR-TKIs and ADCs have been identified as successful treatment approaches to address resistance. Resistance to KRAS inhibitors develops due to the adaptive reactivation of the RAS pathway, the presence of non-G12C KRAS mutations, and the activation of alternative pathways. Employing combination therapy is the primary method to address resistance. Several ALK-TKIs have been authorized for treating global ALK-positive patients with the ALK gene, but they may lead to ALK-TKI resistance by activating the bypass signaling pathway. Combining an ALK-TKI with an immune checkpoint inhibitor can help overcome resistance. ROS1 resistance may develop from intrinsic factors like gene fusions or mutations leading to continuous activation of ROS1 kinase, and extrinsic factors such as activation of alternative signaling pathways. Clinical trials are assessing new ROS1 inhibitors for ALK and ROS1 fusion-positive NSCLC cancers. Research is also exploring the combination of ROS1-targeted drugs with other treatments to enhance effectiveness. BRAF is also a commonly targeted gene, with the V600E mutation being the most prevalent type in NSCLC. However, the mechanisms of resistance to BRAF inhibitors in NSCLC are not completely understood. The more uncommon gene targets include RET, NRAS, PIK3CA, HER2, and c-MET. PIK3CA is commonly mutated and amplified in various cancer types. There are no approved targeted treatments for PIK3CA. Research on PIK3CA resistance has mainly concentrated on breast cancer cells, with limited focus on understanding resistance mechanisms in NSCLC. The FDA has approved T-DXd, an ADC, for treating patients with HER2-mutated NSCLC. The first c-MET single-target inhibitor in the world was authorized for treating patients with advanced NSCLC harboring a MET exon 14 skipping mutation.
Immunotherapy employing ICIs that target the PD-L1/PD-1 axis and CTLA-4 has shown significant improvements in the survival of lung cancer patients. However, drug resistance remains a significant issue, with the TME playing a crucial role. Various strategies are employed to combat resistance to ICI therapy. For instance, enhancing T cell priming through strategies including TIL therapy, CAR-T cell therapy, and personalized vaccines, targeting VEGF, or using cytokines. ICBs are revolutionizing cancer treatment. Potential strategies to overcome resistance involve improving antigen processing and delivery, enhancing T-cell activity and infiltration, and disrupting the immunosuppressive microenvironment. Cancer vaccines are a significant treatment method for NSCLC. They can target tumor antigens, minimize side effects, and can be combined with other treatments like immune checkpoint inhibitors or targeted therapies. Such vaccinations can be used at all stages of NSCLC, either as an additional therapy after surgery or as a treatment for metastatic disease. Immune system modulators have been utilized to enhance the immune response against lung cancer cells. The FDA has approved 16 immunomodulators for treating different types of cancer, with IL-2 being an extensively researched cytokine.
In conclusion, while many patients with NSCLC still benefit from target therapy and immunotherapy, the resistance to targeted therapy and immunotherapy is a significant challenge and still poses challenges in the treatment of NSCLC. However, combination therapies that target multiple pathways or mechanisms and biomarker identification-based approaches show promise for overcoming resistance. Comprehending the underlying mechanisms of resistance and the development of novel therapies are essential for devising successful approaches. Ongoing research in these fields will help overcome resistance and hold promise for improving patient outcomes in the future.
Author contributions
YX: Writing – original draft, Writing – review & editing. XL: Writing – original draft, Writing – review & editing. YW: Writing – original draft, Writing – review & editing. DZ: Writing – original draft. QM: Writing – original draft. LJ: Writing – original draft. SY: Writing – original draft. SZ: Writing – original draft. XZ: Writing – original draft. YL: Writing – original draft, Writing – review & editing. BW: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. BW was supported by China Scholarship Council (CSC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. (2016) 66:115–32. doi: 10.3322/caac.21338
2. Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. LANCET. (2021) 398:535–54. doi: 10.1016/s0140-6736(21)00312-3
3. Cheng TY, Cramb SM, Baade PD, Youlden DR, Nwogu C, Reid ME. The International Epidemiology of Lung Cancer: Latest Trends, Disparities, and Tumor Characteristics. J Thorac Oncol. (2016) 11:1653–71. doi: 10.1016/j.jtho.2016.05.021
4. Romaszko AM, Doboszyńska A. Multiple primary lung cancer: A literature review. Adv Clin Exp Med. (2018) 27:725–30. doi: 10.17219/acem/68631
5. Qin Q, Ren Y, Zhong D. [Research Progress of CDK4/6 Inhibitors in Non-small Cell Lung Cancer]. Zhongguo Fei Ai Za Zhi. (2020) 23:176–81. doi: 10.3779/j.issn.1009-3419.2020.03.07
6. Nicholson AG, Tsao MS, Beasley MB, Borczuk AC, Brambilla E, Cooper WA, et al. The 2021 WHO Classification of Lung Tumors: Impact of Advances Since 2015. J Thorac Oncol. (2022) 17:362–87. doi: 10.1016/j.jtho.2021.11.003
7. Qi H, Fan L. [PI3K/Akt/mTOR signaling pathway and non-small cell lung cancer]. Zhongguo Fei Ai Za Zhi. (2010) 13:1149–54. doi: 10.3779/j.issn.1009-3419.2010.12.13
8. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. (2008) 83:584–94. doi: 10.4065/83.5.584
9. Pfister DG, Johnson DH, Azzoli CG, Sause W, Smith TJ, Jr SB, et al. American Society of Clinical Oncology Treatment of Unresectable Non–Small-Cell Lung Cancer Guideline: Update 2003. J OF Clin Oncol. (2004) 22:330–53. doi: 10.1200/jco.2004.09.053
10. Ashrafi A, Akter Z, Modareszadeh P, Modareszadeh P, Berisha E, Alemi PS, et al. Current Landscape of Therapeutic Resistance in Lung Cancer and Promising Strategies to Overcome Resistance. Cancers. (2022) 14:4562. doi: 10.3390/cancers14194562
11. Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. (2015) 4:36–54. doi: 10.3978/j.issn.2218-6751.2014.05.01
12. Facchinetti F, Proto C, Minari R, Garassino M, Tiseo M. Mechanisms of Resistance to Target Therapies in Non-small Cell Lung Cancer. Handb Exp Pharmacol. (2018) 249:63–89. doi: 10.1007/164_2017_16
13. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. (2018) 15:81–94. doi: 10.1038/nrclinonc.2017.166
14. Sambi M, Bagheri L, Szewczuk MR. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. J Oncol. (2019) 2019:4508794. doi: 10.1155/2019/4508794
15. Boyero L, Sánchez-Gastaldo A, Alonso M, Noguera-Uclés JF, Molina-Pinelo S, Bernabé-Caro R. Primary and Acquired Resistance to Immunotherapy in Lung Cancer: Unveiling the Mechanisms Underlying of Immune Checkpoint Blockade Therapy. Cancers. (2020) 12:3729. doi: 10.3390/cancers12123729
16. Anichini A, Perotti VE, Sgambelluri F, Mortarini R. Immune Escape Mechanisms in Non Small Cell Lung Cancer. Cancers. (2020) 12:3605. doi: 10.3390/cancers12123605
17. Bai R, Chen N, Li L, Du N, Bai L, Lv Z, et al. Mechanisms of Cancer Resistance to Immunotherapy. Front Oncol. (2020) 10:1290. doi: 10.3389/fonc.2020.01290
18. Said SS, Ibrahim WN. Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives. Pharmaceutics. (2023) 15(4):1143. doi: 10.3390/pharmaceutics15041143
19. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
20. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. (2014) 14:535–46. doi: 10.1038/nrc3775
21. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. (2010) 10:760–74. doi: 10.1038/nrc2947
22. Yu HA, Arcila ME, Hellmann MD, Kris MG, Ladanyi M, Riely GJ. Poor response to erlotinib in patients with tumors containing baseline EGFR T790M mutations found by routine clinical molecular testing. Ann Oncol. (2014) 25:423–8. doi: 10.1093/annonc/mdt573
23. Cooper AJ, Sequist LV, Lin JJ. Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management. Nat Rev Clin Oncol. (2022) 19:499–514. doi: 10.1038/s41571-022-00639-9
24. Papadimitrakopoulou VA, Wu YL, Han JY, Ahn MJ, Ramalingam SS, John T, et al. LBA51 - Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. (2018) 29:viii741. doi: 10.1093/annonc/mdy424.064
25. Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, et al. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin Cancer Res. (2015) 21:3924–33. doi: 10.1158/1078-0432.Ccr-15-0560
26. Rangachari D, To C, Shpilsky JE, VanderLaan PA, Kobayashi SS, Mushajiang M, et al. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J Thorac Oncol. (2019) 14:1995–2002. doi: 10.1016/j.jtho.2019.07.016
27. Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, et al. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res. (2015) 21:3913–23. doi: 10.1158/1078-0432.Ccr-14-2789
28. Arulananda S, Do H, Musafer A, Mitchell P, Dobrovic A, John T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non-Small Cell Lung Cancer. J Thorac Oncol. (2017) 12:1728–32. doi: 10.1016/j.jtho.2017.08.006
29. Wang Z, Yang JJ, Huang J, Ye JY, Zhang XC, Tu HY, et al. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J Thorac Oncol. (2017) 12:1723–7. doi: 10.1016/j.jtho.2017.06.017
30. Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. LBA50 - Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann Oncol. (2018) 29:viii740. doi: 10.1093/annonc/mdy424.063
31. Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin Cancer Res. (2018) 24:3097–107. doi: 10.1158/1078-0432.Ccr-17-2310
32. Passaro A, Malapelle U, Attili I, de Marinis F. Overcoming resistance to osimertinib in non-small cell lung cancer: Hopes, doubts, and in-between. Cancer. (2020) 126:2594–6. doi: 10.1002/cncr.32810
33. Ou SI, Cui J, Schrock AB, Goldberg ME, Zhu VW, Albacker L, et al. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer. (2017) 108:228–31. doi: 10.1016/j.lungcan.2017.04.003
34. Zheng D, Hu M, Bai Y, Zhu X, Lu X, Wu C, et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget. (2017) 8:49671–9. doi: 10.18632/oncotarget.17913
35. Leventakos K, Kipp BR, Rumilla KM, Winters JL, Yi ES, Mansfield AS. S768I Mutation in EGFR in Patients with Lung Cancer. J Thorac Oncol. (2016) 11:1798–801. doi: 10.1016/j.jtho.2016.05.007
36. Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. (2016) 7:11815. doi: 10.1038/ncomms11815
37. Zhang Q, Zhang XC, Yang JJ, Yang ZF, Bai Y, Su J, et al. EGFR L792H and G796R: Two Novel Mutations Mediating Resistance to the Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib. J Thorac Oncol. (2018) 13:1415–21. doi: 10.1016/j.jtho.2018.05.024
38. Fassunke J, Müller F, Keul M, Michels S, Dammert MA, Schmitt A, et al. Overcoming EGFR(G724S)-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat Commun. (2018) 9:4655. doi: 10.1038/s41467-018-07078-0
39. Zhang Y, He B, Zhou D, Li M, Hu C. Newly emergent acquired EGFR exon 18 G724S mutation after resistance of a T790M specific EGFR inhibitor osimertinib in non-small-cell lung cancer: a case report. Onco Targets Ther. (2019) 12:51–6. doi: 10.2147/ott.S188612
40. Peled N, Roisman LC, Miron B, Pfeffer R, Lanman RB, Ilouze M, et al. Subclonal Therapy by Two EGFR TKIs Guided by Sequential Plasma Cell-free DNA in EGFR-Mutated Lung Cancer. J Thorac Oncol. (2017) 12:e81–4. doi: 10.1016/j.jtho.2017.02.023
41. Brown BP, Zhang YK, Westover D, Yan Y, Qiao H, Huang V, et al. On-target Resistance to the Mutant-Selective EGFR Inhibitor Osimertinib Can Develop in an Allele-Specific Manner Dependent on the Original EGFR-Activating Mutation. Clin Cancer Res. (2019) 25:3341–51. doi: 10.1158/1078-0432.Ccr-18-3829
42. Nukaga S, Yasuda H, Tsuchihara K, Hamamoto J, Masuzawa K, Kawada I, et al. Amplification of EGFR Wild-Type Alleles in Non-Small Cell Lung Cancer Cells Confers Acquired Resistance to Mutation-Selective EGFR Tyrosine Kinase Inhibitors. Cancer Res. (2017) 77:2078–89. doi: 10.1158/0008-5472.Can-16-2359
43. Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann Oncol. (2018) 29:viii740. doi: 10.1093/annonc/mdy424.063
44. Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discovery. (2012) 2:922–33. doi: 10.1158/2159-8290.Cd-12-0108
45. Hsu CC, Liao BC, Liao WY, Markovets A, Stetson D, Thress K, et al. Exon 16-Skipping HER2 as a Novel Mechanism of Osimertinib Resistance in EGFR L858R/T790M-Positive Non-Small Cell Lung Cancer. J Thorac Oncol. (2020) 15:50–61. doi: 10.1016/j.jtho.2019.09.006
46. Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. (2018) 4:1527–34. doi: 10.1001/jamaoncol.2018.2969
47. Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discovery. (2018) 8:1529–39. doi: 10.1158/2159-8290.Cd-18-1022
48. Zeng L, Yang N, Zhang Y. GOPC-ROS1 Rearrangement as an Acquired Resistance Mechanism to Osimertinib and Responding to Crizotinib Combined Treatments in Lung Adenocarcinoma. J Thorac Oncol. (2018) 13:e114–6. doi: 10.1016/j.jtho.2018.02.005
49. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. (2011) 3:75ra26. doi: 10.1126/scitranslmed.3002003
50. Ichihara E, Westover D, Meador CB, Yan Y, Bauer JA, Lu P, et al. SFK/FAK Signaling Attenuates Osimertinib Efficacy in Both Drug-Sensitive and Drug-Resistant Models of EGFR-Mutant Lung Cancer. Cancer Res. (2017) 77:2990–3000. doi: 10.1158/0008-5472.Can-16-2300
51. Karachaliou N, Chaib I, Cardona AF, Berenguer J, Bracht JWP, Yang J, et al. Common Co-activation of AXL and CDCP1 in EGFR-mutation-positive Non-smallcell Lung Cancer Associated With Poor Prognosis. EBioMedicine. (2018) 29:112–27. doi: 10.1016/j.ebiom.2018.02.001
52. Rotow JK, Costa DB, Paweletz CP, Awad MM, Marcoux P, Rangachari D, et al. Concurrent osimertinib plus gefitinib for first-line treatment of EGFR-mutated non-small cell lung cancer (NSCLC). J Clin Oncol. (2020) 38:9507–7. doi: 10.1200/JCO.2020.38.15_suppl.9507
53. Starrett JH, Guernet AA, Cuomo ME, Poels KE, van Alderwerelt van Rosenburgh IK, Nagelberg A, et al. Drug Sensitivity and Allele Specificity of First-Line Osimertinib Resistance EGFR Mutations. Cancer Res. (2020) 80:2017–30. doi: 10.1158/0008-5472.Can-19-3819
54. Sugawara S, Oizumi S, Minato K, Harada T, Inoue A, Fujita Y, et al. Randomized phase II study of concurrent versus sequential alternating gefitinib and chemotherapy in previously untreated non-small cell lung cancer with sensitive EGFR mutations: NEJ005/TCOG0902. Ann Oncol. (2015) 26:888–94. doi: 10.1093/annonc/mdv063
55. Oizumi S, Sugawara S, Minato K, Harada T, Inoue A, Fujita Y, et al. Updated survival outcomes of NEJ005/TCOG0902: a randomised phase II study of concurrent versus sequential alternating gefitinib and chemotherapy in previously untreated non-small cell lung cancer with sensitive EGFR mutations. ESMO Open. (2018) 3:e000313. doi: 10.1136/esmoopen-2017-000313
56. Hosomi Y, Morita S, Sugawara S, Kato T, Fukuhara T, Gemma A, et al. Gefitinib Alone Versus Gefitinib Plus Chemotherapy for Non-Small-Cell Lung Cancer With Mutated Epidermal Growth Factor Receptor: NEJ009 Study. J Clin Oncol. (2020) 38:115–23. doi: 10.1200/jco.19.01488
57. Noronha V, Patil VM, Joshi A, Menon N, Chougule A, Mahajan A, et al. Gefitinib Versus Gefitinib Plus Pemetrexed and Carboplatin Chemotherapy in EGFR-Mutated Lung Cancer. J Clin Oncol. (2020) 38:124–36. doi: 10.1200/jco.19.01154
58. Jänne P, Planchard D, Howarth P, Todd A, Kobayashi K. OA07.01 Osimertinib Plus Platinum/Pemetrexed in Newly-Diagnosed Advanced EGFRm-Positive NSCLC; The Phase 3 FLAURA2 Study. J Thorac Oncol. (2019) 14:S222–3. doi: 10.1016/j.jtho.2019.08.441
59. Sequist LV, Han JY, Ahn MJ, Cho BC, Yu H, Kim SW, et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. (2020) 21:373–86. doi: 10.1016/s1470-2045(19)30785-5
60. Cho BC, Felip E, Hayashi H, Thomas M, Lu S, Besse B, et al. MARIPOSA: phase 3 study of first-line amivantamab + lazertinib versus osimertinib in EGFR-mutant non-small-cell lung cancer. Future Oncol. (2022) 18:639–47. doi: 10.2217/fon-2021-0923
61. Wu YL, Zhang L, Kim DW, Liu X, Lee DH, Yang JC, et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J Clin Oncol. (2018) 36:3101–9. doi: 10.1200/jco.2018.77.7326
62. Wu YL, Cheng Y, Zhou J, Lu S, Zhang Y, Zhao J, et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): an open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir Med. (2020) 8:1132–43. doi: 10.1016/s2213-2600(20)30154-5
63. Chen Z, Vallega KA, Chen H, Zhou J, Ramalingam SS, Sun S-Y. The natural product berberine synergizes with osimertinib preferentially against MET-amplified osimertinib-resistant lung cancer via direct MET inhibition. Pharmacol Res. (2022) 175:105998. doi: 10.1016/j.phrs.2021.105998
64. Yu D, Li Y, Sun KD, Gu J, Chen Z, Owonikoko TK, et al. The novel MET inhibitor, HQP8361, possesses single agent activity and enhances therapeutic efficacy of AZD9291 (osimertinib) against AZD9291-resistant NSCLC cells with activated MET. Am J Cancer Res. (2020) 10:3316–27. doi: 10.21203/rs.3.rs-25317/v1
65. Yu J, Zhang L, Peng J, Ward R, Hao P, Wang J, et al. Dictamnine, a novel c-Met inhibitor, suppresses the proliferation of lung cancer cells by downregulating the PI3K/AKT/mTOR and MAPK signaling pathways. Biochem Pharmacol. (2022) 195:114864. doi: 10.1016/j.bcp.2021.114864
66. Taniguchi H, Yamada T, Wang R, Tanimura K, Adachi Y, Nishiyama A, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Article. Nat Commun. (2019) 10:259. doi: 10.1038/s41467-018-08074-0
67. Tanaka H, Sakagami H, Kaneko N, Konagai S, Yamamoto H, Matsuya T, et al. Mutant-Selective Irreversible EGFR Inhibitor, Naquotinib, Inhibits Tumor Growth in NSCLC Models with EGFR-Activating Mutations, T790M Mutation, and AXL Overexpression. Mol Cancer Ther. (2019) 18:1366–73. doi: 10.1158/1535-7163.Mct-18-0976
68. Okura N, Nishioka N, Yamada T, Taniguchi H, Tanimura K, Katayama Y, et al. ONO-7475, a Novel AXL Inhibitor, Suppresses the Adaptive Resistance to Initial EGFR-TKI Treatment in EGFR-Mutated Non–Small Cell Lung Cancer. Clin Cancer Res. (2020) 26:2244–56. doi: 10.1158/1078-0432.Ccr-19-2321
69. Sato H, Yamamoto H, Sakaguchi M, Shien K, Tomida S, Shien T, et al. Combined inhibition of MEK and PI3K pathways overcomes acquired resistance to EGFR-TKIs in non-small cell lung cancer. Cancer Sci. (2018) 109:3183–96. doi: 10.1111/cas.13763
70. Terp MG, Jacobsen K, Molina MA, Karachaliou N, Beck HC, Bertran-Alamillo J, et al. Combined FGFR and Akt pathway inhibition abrogates growth of FGFR1 overexpressing EGFR-TKI-resistant NSCLC cells. NPJ Precis Oncol. (2021) 5:65. doi: 10.1038/s41698-021-00208-w
71. Hayakawa D, Takahashi F, Mitsuishi Y, Tajima K, Hidayat M, Winardi W, et al. Activation of insulin-like growth factor-1 receptor confers acquired resistance to osimertinib in non-small cell lung cancer with EGFR T790M mutation. Thorac Cancer. (2020) 11:140–9. doi: 10.1111/1759-7714.13255
72. Yang X, Xia Y, Xu L, Liang L, Zhuo M, Wu M, et al. Efficacy and Safety of Combination Treatment With Apatinib and Osimertinib After Osimertinib Resistance in Epidermal Growth Factor Receptor-Mutant Non-small Cell Lung Carcinoma—A Retrospective Analysis of a Multicenter Clinical Study. Article. Front Mol Biosci. (2021) 8:639892. doi: 10.3389/fmolb.2021.639892
73. Nakagawa T, Takeuchi S, Yamada T, Ebi H, Sano T, Nanjo S, et al. EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res. (2013) 73:2428–34. doi: 10.1158/0008-5472.Can-12-3479
74. Shaw AT, Riely GJ, Bang YJ, Kim DW, Camidge DR, Solomon BJ, et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann Oncol. (2019) 30:1121–6. doi: 10.1093/annonc/mdz131
75. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. (2014) 371:1963–71. doi: 10.1056/NEJMoa1406766
76. Wang S, Song Y, Liu D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. (2017) 385:51–4. doi: 10.1016/j.canlet.2016.11.008
77. To C, Jang J, Chen T, Park E, Mushajiang M, De Clercq DJH, et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discovery. (2019) 9:926–43. doi: 10.1158/2159-8290.Cd-18-0903
78. Liu X, Zhang X, Yang L, Tian X, Dong T, Ding CZ, et al. Abstract 1320: Preclinical evaluation of TQB3804, a potent EGFR C797S inhibitor. Cancer Res. (2019) 79:1320–0. doi: 10.1158/1538-7445.Am2019-1320
79. Jänne PA, Baik C, Su WC, Johnson ML, Hayashi H, Nishio M, et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated Non-Small Cell Lung Cancer. Cancer Discovery. (2022) 12:74–89. doi: 10.1158/2159-8290.Cd-21-0715
80. Köhler J, Jänne PA. If Virchow and Ehrlich Had Dreamt Together: What the Future Holds for KRAS-Mutant Lung Cancer. Int J Mol Sci. (2021) 22(6):3025. doi: 10.3390/ijms22063025
81. Cai C, Yao S, Zou Y, Lu H, Chen X, Wang Y, et al. KRAS(G12C) mutation-induced TOPK overexpression contributes to tumour progression in non-small cell lung cancer. J Cell Mol Med. (2023) 27(12):1637–52. doi: 10.1111/jcmm.17640
82. Prior IA, Hood FE, Hartley JL. The Frequency of Ras Mutations in Cancer. Cancer Res. (2020) 80:2969–74. doi: 10.1158/0008-5472.Can-19-3682
83. Haigis KM. KRAS Alleles: The Devil Is in the Detail. Trends Cancer. (2017)3:686–97. doi: 10.1016/j.trecan.2017.08.006
84. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med. (2021) 384:2371–81. doi: 10.1056/NEJMoa2103695
85. Riely GJ, Ou SHI, Rybkin I, Spira A, Papadopoulos K, Sabari JK, et al. 99O_PR KRYSTAL-1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. J Thorac Oncol. (2021) 16:S751–2. doi: 10.1016/S1556-0864(21)01941-9
86. Suzuki S, Yonesaka K, Teramura T, Takehara T, Kato R, Sakai H, et al. KRAS Inhibitor Resistance in MET-Amplified KRAS (G12C) Non-Small Cell Lung Cancer Induced By RAS- and Non-RAS-Mediated Cell Signaling Mechanisms. Clin Cancer Res. (2021) 27:5697–707. doi: 10.1158/1078-0432.Ccr-21-0856
87. Ho CSL, Tüns AI, Schildhaus HU, Wiesweg M, Grüner BM, Hegedus B, et al. HER2 mediates clinical resistance to the KRAS(G12C) inhibitor sotorasib, which is overcome by co-targeting SHP2. Eur J Cancer. (2021) 159:16–23. doi: 10.1016/j.ejca.2021.10.003
88. Moll HP, Pranz K, Musteanu M, Grabner B, Hruschka N, Mohrherr J, et al. Afatinib restrains K-RAS–driven lung tumorigenesis. Sci Trans Med. (2018) 10:eaao2301. doi: 10.1126/scitranslmed.aao2301
89. Kruspig B, Monteverde T, Neidler S, Hock A, Kerr E, Nixon C, et al. The ERBB network facilitates KRAS-driven lung tumorigenesis. Sci Trans Med. (2018) 10:eaao2565. doi: 10.1126/scitranslmed.aao2565
90. Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discovery. (2021) 11:1913–22. doi: 10.1158/2159-8290.Cd-21-0365
91. Zhao Y, Murciano-Goroff YR, Xue JY, Ang A, Lucas J, Mai TT, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. (2021) 599:679–83. doi: 10.1038/s41586-021-04065-2
92. Kitajima S, Asahina H, Chen T, Guo S, Quiceno LG, Cavanaugh JD, et al. Overcoming Resistance to Dual Innate Immune and MEK Inhibition Downstream of KRAS. Cancer Cell. (2018) 34:439–452.e6. doi: 10.1016/j.ccell.2018.08.009
93. Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med. (2020) 218(1):e20201414. doi: 10.1084/jem.20201414
94. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discovery. (2020) 10:54–71. doi: 10.1158/2159-8290.Cd-19-1167
95. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019/11/01. (2019) 575:217–23. doi: 10.1038/s41586-019-1694-1
96. Hallberg B, Palmer RH. The role of the ALK receptor in cancer biology. Ann Oncol. (2016) 27 Suppl 3:iii4–iii15. doi: 10.1093/annonc/mdw301
97. Golding B, Luu A, Jones R, Viloria-Petit AM. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol Cancer. (2018) 17(1):52. doi: 10.1186/s12943-018-0810-4
98. Du X, Shao Y, Qin H-F, Tai Y-H, Gao H-J. ALK- rearrangement in non-small-cell lung cancer (NSCLC). Thorac Cancer. (2018) 9:423–30. doi: 10.1111/1759-7714.12613
99. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. (2010) 363:1693–703. doi: 10.1056/NEJMoa1006448
100. Rothschild SI. Ceritinib-a second-generation ALK inhibitor overcoming resistance in ALK-rearranged non-small cell lung cancer. Trans Lung Cancer Res. (2014) 3:379–81. doi: 10.3978/j.issn.2218-6751.2014.11.09
101. Dhillon S, Clark M. Ceritinib: first global approval. Drugs. (2014) 74:1285–91. doi: 10.1007/s40265-014-0251-3
102. Baba K, Goto Y. Lorlatinib as a treatment for ALK-positive lung cancer. Future Oncol (London England). (2022) 18:2745–66. doi: 10.2217/fon-2022-0184
103. Beardslee T, Lawson J. Alectinib and Brigatinib: New Second-Generation ALK Inhibitors for the Treatment of Non-Small Cell Lung Cancer. J advanced practitioner Oncol. (2018) 9:94–101.
104. Horn L, Infante JR, Reckamp KL, Blumenschein GR, Leal TA, Waqar SN, et al. Ensartinib (X-396) in ALK-Positive Non-Small Cell Lung Cancer: Results from a First-in-Human Phase I/II, Multicenter Study. Clin Cancer research: an Off J Am Assoc Cancer Res. (2018) 24:2771–9. doi: 10.1158/1078-0432.Ccr-17-2398
105. Sharma GG, Mota I, Mologni L, Patrucco E, Gambacorti-Passerini C, Chiarle R. Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes. Cancers (Basel). (2018) 10(3):62. doi: 10.3390/cancers10030062
106. Davare MA, Vellore NA, Wagner JP, Eide CA, Goodman JR, Drilon A, et al. Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proc Natl Acad Sci. (2015) 112:E5381–90. doi: 10.1073/pnas.1515281112
107. Wu J, Savooji J, Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol. (2016) 9:19. doi: 10.1186/s13045-016-0251-8
108. Awad MM, Shaw AT. ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin Adv Hematol Oncol. (2014) 12:429–39.
109. Toyokawa G, Seto T. Updated Evidence on the Mechanisms of Resistance to ALK Inhibitors and Strategies to Overcome Such Resistance: Clinical and Preclinical Data. Oncol Res Treat. (2015) 38:291–8. doi: 10.1159/000430852
110. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discovery. (2016) 6:1118–33. doi: 10.1158/2159-8290.Cd-16-0596
111. Qian M, Zhu B, Wang X, Liebman M. Drug resistance in ALK-positiveNon-small cell lungcancer patients. Semin Cell Dev Biol. (2017) 64:150–7. doi: 10.1016/j.semcdb.2016.09.016
112. Haratake N, Toyokawa G, Seto T, Tagawa T, Okamoto T, Yamazaki K, et al. The mechanisms of resistance to second- and third-generation ALK inhibitors and strategies to overcome such resistance. Expert Rev Anticancer Ther. (2021) 21:975–88. doi: 10.1080/14737140.2021.1940964
113. Drizou M, Kotteas EA, Syrigos N. Treating patients with ALK-rearranged non-small-cell lung cancer: mechanisms of resistance and strategies to overcome it. Clin Transl Oncol. (2017) 19:658–66. doi: 10.1007/s12094-016-1605-y
114. Petrilli AM, Fernández-Valle C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene. (2016) 35:537–48. doi: 10.1038/onc.2015.125
115. Gouji T, Takashi S, Mitsuhiro T, Yukito I. Crizotinib can overcome acquired resistance to CH5424802: is amplification of the MET gene a key factor? J Thorac Oncol. (2014) 9:e27–8. doi: 10.1097/jto.0000000000000113
116. Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. (2015) 15:73–9. doi: 10.1038/nrc3876
117. Arai S, Takeuchi S, Fukuda K, Taniguchi H, Nishiyama A, Tanimoto A, et al. Osimertinib Overcomes Alectinib Resistance Caused by Amphiregulin in a Leptomeningeal Carcinomatosis Model of ALK-Rearranged Lung Cancer. J Thorac Oncol. (2020) 15:752–65. doi: 10.1016/j.jtho.2020.01.001
118. Fujita S, Masago K, Katakami N, Yatabe Y. Transformation to SCLC after Treatment with the ALK Inhibitor Alectinib. J Thorac Oncol. (2016) 11:e67–72. doi: 10.1016/j.jtho.2015.12.105
119. Yoda S, Lin JJ, Lawrence MS, Burke BJ, Friboulet L, Langenbucher A, et al. Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer. Cancer Discovery. (2018) 8:714–29. doi: 10.1158/2159-8290.Cd-17-1256
120. Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discovery. (2014) 4:662–73. doi: 10.1158/2159-8290.Cd-13-0846
121. Cho BC, Kim DW, Bearz A, Laurie SA, McKeage M, Borra G, et al. ASCEND-8: A Randomized Phase 1 Study of Ceritinib, 450 mg or 600 mg, Taken with a Low-Fat Meal versus 750 mg in Fasted State in Patients with Anaplastic Lymphoma Kinase (ALK)-Rearranged Metastatic Non-Small Cell Lung Cancer (NSCLC). J Thorac Oncol. (2017) 12:1357–67. doi: 10.1016/j.jtho.2017.07.005
122. Redaelli S, Ceccon M, Zappa M, Sharma GG, Mastini C, Mauri M, et al. Lorlatinib Treatment Elicits Multiple On- and Off-Target Mechanisms of Resistance in ALK-Driven Cancer. Cancer Res. (2018) 78:6866–80. doi: 10.1158/0008-5472.Can-18-1867
123. Recondo G, Mezquita L, Facchinetti F, Planchard D, Gazzah A, Bigot L, et al. Diverse Resistance Mechanisms to the Third-Generation ALK Inhibitor Lorlatinib in ALK-Rearranged Lung Cancer. Clin Cancer Res. (2020) 26:242–55. doi: 10.1158/1078-0432.Ccr-19-1104
124. Mizuta H, Okada K, Araki M, Adachi J, Takemoto A, Kutkowska J, et al. Gilteritinib overcomes lorlatinib resistance in ALK-rearranged cancer. Nat Commun. (2021) 12:1261. doi: 10.1038/s41467-021-21396-w
125. Wang L, Lui VWY. Emerging Roles of ALK in Immunity and Insights for Immunotherapy. Cancers. (2020) 12:426. doi: 10.3390/cancers12020426
126. Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. (2012) 30:863–70. doi: 10.1200/jco.2011.35.6345
127. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. (2007) 131:1190–203. doi: 10.1016/j.cell.2007.11.025
128. Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. (2012) 18:4570–9. doi: 10.1158/1078-0432.CCR-12-0550
129. Matsuura S, Shinmura K, Kamo T, Igarashi H, Maruyama K, Tajima M, et al. CD74-ROS1 fusion transcripts in resected non-small cell lung carcinoma. Oncol Rep. (2013) 30:1675–80. doi: 10.3892/or.2013.2630
130. Cui M, Han Y, Li P, Zhang J, Ou Q, Tong X, et al. Molecular and clinicopathological characteristics of ROS1-rearranged non-small-cell lung cancers identified by next-generation sequencing. Mol Oncol. (2020) 14:2787–95. doi: 10.1002/1878-0261.12789
131. Lin JJ, Choudhury NJ, Yoda S, Zhu VW, Johnson TW, Sakhtemani R, et al. Spectrum of Mechanisms of Resistance to Crizotinib and Lorlatinib in ROS1 Fusion-Positive Lung Cancer. Clin Cancer Res. (2021) 27:2899–909. doi: 10.1158/1078-0432.CCR-21-0032
132. Gainor JF, Tseng D, Yoda S, Dagogo-Jack I, Friboulet L, Lin JJ, et al. Patterns of Metastatic Spread and Mechanisms of Resistance to Crizotinib in ROS1-Positive Non-Small-Cell Lung Cancer. JCO Precis Oncol. (2017) 2017:PO.17.00063. doi: 10.1200/PO.17.00063
133. Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci U S A. (2015) 112:3493–8. doi: 10.1073/pnas.1420785112
134. Facchinetti F, Loriot Y, Kuo MS, Mahjoubi L, Lacroix L, Planchard D, et al. Crizotinib-Resistant ROS1 Mutations Reveal a Predictive Kinase Inhibitor Sensitivity Model for ROS1- and ALK-Rearranged Lung Cancers. Clin Cancer Res. (2016) 22:5983–91. doi: 10.1158/1078-0432.CCR-16-0917
135. Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. (2013) 368:2395–401. doi: 10.1056/NEJMoa1215530
136. Gou W, Zhou X, Liu Z, Wang L, Shen J, Xu X, et al. CD74-ROS1 G2032R mutation transcriptionally up-regulates Twist1 in non-small cell lung cancer cells leading to increased migration, invasion, and resistance to crizotinib. Cancer Lett. (2018) 422:19–28. doi: 10.1016/j.canlet.2018.02.032
137. Zhu YC, Lin XP, Li XF, Wu LX, Chen HF, Wang WX, et al. Concurrent ROS1 gene rearrangement and KRAS mutation in lung adenocarcinoma: A case report and literature review. Thorac Cancer. (2018) 9:159–63. doi: 10.1111/1759-7714.12518
138. Dziadziuszko R, Le AT, Wrona A, Jassem J, Camidge DR, Varella-Garcia M, et al. An Activating KIT Mutation Induces Crizotinib Resistance in ROS1-Positive Lung Cancer. J Thorac Oncol. (2016) 11:1273–81. doi: 10.1016/j.jtho.2016.04.001
139. Harada G, Yang SR, Cocco E, Drilon A. Rare molecular subtypes of lung cancer. Nat Rev Clin Oncol. (2023) 20:229–49. doi: 10.1038/s41571-023-00733-6
140. Watanabe J, Furuya N, Fujiwara Y. Appearance of a BRAF Mutation Conferring Resistance to Crizotinib in Non-Small Cell Lung Cancer Harboring Oncogenic ROS1 Fusion. J Thorac Oncol. (2018) 13:e66–9. doi: 10.1016/j.jtho.2017.11.125
141. Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations. Cancer Discovery. (2018) 8:1227–36. doi: 10.1158/2159-8290.Cd-18-0484
142. Choudhury NJ, Schneider JL, Patil T, Zhu VW, Goldman DA, Yang SR, et al. Response to Immune Checkpoint Inhibition as Monotherapy or in Combination With Chemotherapy in Metastatic ROS1-Rearranged Lung Cancers. JTO Clin Res Rep. (2021) 2:100187. doi: 10.1016/j.jtocrr.2021.100187
143. Lara MS, Gubens MA, Bacaltos B, Daran L, Lim SL, Li T, et al. Phase 1 Study of Ceritinib Combined With Trametinib in Patients With Advanced ALK- or ROS1-Positive NSCLC. JTO Clin Res Rep. (2022) 3:100436. doi: 10.1016/j.jtocrr.2022.100436
144. Awad MM, Leonardi GC, Kravets S, Dahlberg SE, Drilon AE, Noonan S, et al. Impact of MET inhibitors on survival among patients (pts) with MET exon 14 mutant (METdel14) non-small cell lung cancer (NSCLC). J Clin Oncol. (2017) 35:8511–1. doi: 10.1200/JCO.2017.35.15_suppl.8511
145. Rodon J, Postel-Vinay S, Hollebecque A, Nuciforo P, Azaro A, Cattan V, et al. First-in-human phase I study of oral S49076, a unique MET/AXL/FGFR inhibitor, in advanced solid tumours. Eur J Cancer. (2017) 81:142–50. doi: 10.1016/j.ejca.2017.05.007
146. Sato H, Schoenfeld AJ, Siau E, Lu YC, Tai H, Suzawa K, et al. MAPK Pathway Alterations Correlate with Poor Survival and Drive Resistance to Therapy in Patients with Lung Cancers Driven by ROS1 Fusions. Clin Cancer Res. (2020) 26:2932–45. doi: 10.1158/1078-0432.CCR-19-3321
147. Leicht DT, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M, et al. Raf kinases: Function, regulation and role in human cancer. Biochim Biophys Acta (BBA) - Mol Cell Res. (2007) 1773:1196–212. doi: 10.1016/j.bbamcr.2007.05.001
148. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. (2012) 150:1107–20. doi: 10.1016/j.cell.2012.08.029
149. Marchetti A, Felicioni L, Malatesta S, Grazia Sciarrotta M, Guetti L, Chella A, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. (2011) 29:3574–9. doi: 10.1200/jco.2011.35.9638
150. Roviello G, D’Angelo A, Sirico M, Pittacolo M, Conter FU, Sobhani N. Advances in anti-BRAF therapies for lung cancer. Invest New Drugs. (2021) 39:879–90. doi: 10.1007/s10637-021-01068-8
151. Planchard D, Kim TM, Mazieres J, Quoix E, Riely G, Barlesi F, et al. Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. (2016) 17:642–50. doi: 10.1016/s1470-2045(16)00077-2
152. Liu X, Zhong D. [Research Progress of Targeted Therapy for BRAF Mutation in Advanced Non-small Cell Lung Cancer]. Zhongguo Fei Ai Za Zhi. (2018) 21:635–40. doi: 10.3779/j.issn.1009-3419.2018.08.10
153. Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. (2015) 51:2792–9. doi: 10.1016/j.ejca.2015.08.022
154. Huo KG, Notsuda H, Fang Z, Liu NF, Gebregiworgis T, Li Q, et al. Lung Cancer Driven by BRAF(G469V) Mutation Is Targetable by EGFR Kinase Inhibitors. J Thorac Oncol. (2022) 17:277–88. doi: 10.1016/j.jtho.2021.09.008
155. Yao Z, Torres Neilawattie M, Tao A, Gao Y, Luo L, Li Q, et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell. (2015) 28:370–83. doi: 10.1016/j.ccell.2015.08.001
156. Rudin CM, Hong K, Streit M. Molecular characterization of acquired resistance to the BRAF inhibitor dabrafenib in a patient with BRAF-mutant non-small-cell lung cancer. J Thorac Oncol. (2013) 8:e41–2. doi: 10.1097/JTO.0b013e31828bb1b3
157. Kim SM, Kim H, Jang KW, Kim MH, Sohn J, Yun MR, et al. EGFR-Mediated Reactivation of MAPK Signaling Induces Acquired Resistance to GSK2118436 in BRAF V600E-Mutant NSCLC Cell Lines. Mol Cancer Ther. (2016) 15:1627–36. doi: 10.1158/1535-7163.Mct-15-0375
158. Joshi M, Rice SJ, Liu X, Miller B, Belani CP. Trametinib with or without vemurafenib in BRAF mutated non-small cell lung cancer. PloS One. (2015) 10:e0118210. doi: 10.1371/journal.pone.0118210
159. Tissot C, Couraud S, Tanguy R, Bringuier PP, Girard N, Souquet PJ. Clinical characteristics and outcome of patients with lung cancer harboring BRAF mutations. Lung Cancer. (2016) 91:23–8. doi: 10.1016/j.lungcan.2015.11.006
160. Bearz A, De Carlo E, Doliana R, Schiappacassi M. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Third-Generation EGFR Tyrosine Kinase Inhibitor. J Thorac Oncol. (2017) 12:e181–2. doi: 10.1016/j.jtho.2017.07.017
161. Nguyen-Ngoc T, Bouchaab H, Adjei AA, Peters S. BRAF Alterations as Therapeutic Targets in Non–Small-Cell Lung Cancer. J Thorac Oncol. (2015) 10:1396–403. doi: 10.1097/JTO.0000000000000644
162. Chen SH, Zhang Y, Van Horn RD, Yin T, Buchanan S, Yadav V, et al. Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer Discovery. (2016) 6:300–15. doi: 10.1158/2159-8290.Cd-15-0896
163. La Monica S, Minari R, Cretella D, Bonelli M, Fumarola C, Cavazzoni A, et al. Acquired BRAF G469A Mutation as a Resistance Mechanism to First-Line Osimertinib Treatment in NSCLC Cell Lines Harboring an EGFR Exon 19 Deletion. Target Oncol. (2019) 14:619–26. doi: 10.1007/s11523-019-00669-x
164. Meng P, Koopman B, Kok K, Ter Elst A, Schuuring E, van Kempen LC, et al. Combined osimertinib, dabrafenib and trametinib treatment for advanced non-small-cell lung cancer patients with an osimertinib-induced BRAF V600E mutation. Lung Cancer. (2020) 146:358–61. doi: 10.1016/j.lungcan.2020.05.036
165. Zhou F, Zhao W, Chen X, Zhang J, Zhou C. Response to the combination of dabrafenib, trametinib and osimertinib in a patient with EGFR-mutant NSCLC harboring an acquired BRAF(V600E) mutation. Lung Cancer. (2020) 139:219–20. doi: 10.1016/j.lungcan.2019.10.014
166. Huang Y, Gan J, Guo K, Deng Y, Fang W. Acquired BRAF V600E Mutation Mediated Resistance to Osimertinib and Responded to Osimertinib, Dabrafenib, and Trametinib Combination Therapy. J Thorac Oncol. (2019) 14:e236–7. doi: 10.1016/j.jtho.2019.05.040
167. Mauclet C, Collard P, Ghaye B, Hoton D, Nana FA. Tumor response to EGFR/BRAF/MEK co-inhibition in a patient with EGFR mutated lung adenocarcinoma developing a BRAF(V600) mutation as an acquired resistance mechanism. Lung Cancer. (2021) 159:42–4. doi: 10.1016/j.lungcan.2021.06.025
168. Li S, Lin X, Sun S, Li S, Zhou C. Response to osimertinib plus trametinib in a heavily treated epidermal growth factor receptor (EGFR)-positive NSCLC harboring a rare, acquired rapidly accelerated fibrosarcoma B-type (BRAF) p.D594N mutation: a case report. Anticancer Drugs. (2022) 33:963–5. doi: 10.1097/cad.0000000000001367
169. Sun M, Wang X, Xu Y, Sun C, Guo Y, Qiu S, et al. Combined targeting of EGFR and BRAF triggers regression of osimertinib resistance by using osimertinib and vemurafenib concurrently in a patient with heterogeneity between different lesions. Thorac Cancer. (2022) 13:514–6. doi: 10.1111/1759-7714.14295
170. Subbiah V, Baik C, Kirkwood JM. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer. (2020) 6:797–810. doi: 10.1016/j.trecan.2020.05.009
171. Ettinger DS, Wood DE, Aggarwal C, Aisner DL, Akerley W, Bauman JR, et al. NCCN Guidelines Insights: Non–Small Cell Lung Cancer, Version 1.2020: Featured Updates to the NCCN Guidelines. J Natl Compr Cancer Network J Natl Compr Canc Netw. (2019) 17:1464–72. doi: 10.6004/jnccn.2019.0059
172. Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol. (2016) 12:192–202. doi: 10.1038/nrendo.2016.11
173. Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. (1994) 367:375–6. doi: 10.1038/367375a0
174. Kato S, Subbiah V, Marchlik E, Elkin SK, Carter JL, Kurzrock R. RET Aberrations in Diverse Cancers: Next-Generation Sequencing of 4,871 Patients. Clin Cancer Res. (2017) 23:1988–97. doi: 10.1158/1078-0432.Ccr-16-1679
175. Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer. (2014) 14:173–86. doi: 10.1038/nrc3680
176. Tsuta K, Kohno T, Yoshida A, Shimada Y, Asamura H, Furuta K, et al. RET-rearranged non-small-cell lung carcinoma: a clinicopathological and molecular analysis. Br J OF Cancer. (2014) 110:1571–8. doi: 10.1038/bjc.2014.36
177. Subbiah V, Yang D, Velcheti V, Drilon A, Meric-Bernstam F. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J Clin Oncol. (2020) 38:1209–21. doi: 10.1200/jco.19.02551
178. Drilon A, Rogers E, Zhai D, Deng W, Zhang X, Lee D, et al. 506P - TPX-0046 is a novel and potent RET/SRC inhibitor for RET-driven cancers. Ann Oncol. (2019) 30:v190–1. doi: 10.1093/annonc/mdz244.068
179. Subbiah V, Velcheti V, Tuch BB, Ebata K, Busaidy NL, Cabanillas ME, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol. (2018) 29:1869–76. doi: 10.1093/annonc/mdy137
180. Subbiah V, Gainor JF, Rahal R, Brubaker JD, Kim JL, Maynard M, et al. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discovery. (2018) 8:836–49. doi: 10.1158/2159-8290.Cd-18-0338
181. Rosen EY, Johnson ML, Clifford SE, Somwar R, Kherani JF, Son J, et al. Overcoming MET-Dependent Resistance to Selective RET Inhibition in Patients with RET Fusion-Positive Lung Cancer by Combining Selpercatinib with Crizotinib. Clin Cancer Res. (2021) 27:34–42. doi: 10.1158/1078-0432.Ccr-20-2278
182. Lin JJ, Liu SV, McCoach CE, Zhu VW, Tan AC, Yoda S, et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann Oncol. (2020) 31:1725–33. doi: 10.1016/j.annonc.2020.09.015
183. Shimizu K, Goldfarb M, Suard Y, Perucho M, Li Y, Kamata T, et al. Three human transforming genes are related to the viral ras oncogenes. Proc Natl Acad Sci. (1983) 80:2112–6. doi: 10.1073/pnas.80.8.2112
184. Stuart DD, Shao W, Mishina Y, Feng Y, Caponigro G, Cooke VG, et al. Abstract DDT01-04: Pharmacological profile and anti-tumor properties of LXH254, a highly selective RAF kinase inhibitor. Cancer Res. (2018) 78:DDT01–04-DDT01-04. doi: 10.1158/1538-7445.Am2018-ddt01-04
185. Park S, Kim TM, Cho SY, Kim S, Oh Y, Kim M, et al. Combined blockade of polo-like kinase and pan-RAF is effective against NRAS-mutant non-small cell lung cancer cells. Cancer Lett. (2020) 495:135–44. doi: 10.1016/j.canlet.2020.09.018
186. Yu C, Li B, Wang J, Zhang Z, Li S, Lei S, et al. miR-145-5p Modulates Gefitinib Resistance by Targeting NRAS and MEST in Non-Small Cell Lung Cancer. Ann Clin Lab Sci. (2021) 51:625–37.
187. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. (2004) 304:554. doi: 10.1126/science.1096502
188. Massacesi C, Di Tomaso E, Urban P, Germa C, Quadt C, Trandafir L, et al. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. (2016) 9:203–10. doi: 10.2147/ott.S89967
189. Scheffler M, Bos M, Gardizi M, König K, Michels S, Fassunke J, et al. PIK3CA mutations in non-small cell lung cancer (NSCLC): genetic heterogeneity, prognostic impact and incidence of prior malignancies. Oncotarget. (2015) 6:1315–26. doi: 10.18632/oncotarget.2834
190. Huang A, Fritsch C, Wilson C, Reddy A, Liu M, Lehar J, et al. Abstract 3749: Single agent activity of PIK3CA inhibitor BYL719 in a broad cancer cell line panel. Cancer Res. (2012) ;72:3749–9. doi: 10.1158/1538-7445.Am2012-3749
191. Daher S, Zer A, Tschernichovsky R, Yacobi R, Barshack I, Tsabari S, et al. Driver mutation characteristics of phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) in advanced non-small cell lung cancer. Lung Cancer. (2023) 178:229–36. doi: 10.1016/j.lungcan.2023.02.023
192. Suzawa K, Offin M, Schoenfeld AJ, Plodkowski AJ, Odintsov I, Lu D, et al. Acquired MET Exon 14 Alteration Drives Secondary Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor in EGFR-Mutated Lung Cancer. JCO Precis Oncol. (2019) 3):1–8. doi: 10.1200/po.19.00011
193. Huang T, Ren K, Ding G, Yang L, Wen Y, Peng B, et al. miR−10a increases the cisplatin resistance of lung adenocarcinoma circulating tumor cells via targeting PIK3CA in the PI3K/Akt pathway. Oncol Rep. (2020) 43:1906–14. doi: 10.3892/or.2020.7547
194. Chen F, Liu J, Song X, DuCote TJ, Byrd AL, Wang C, et al. EZH2 inhibition confers PIK3CA-driven lung tumors enhanced sensitivity to PI3K inhibition. Cancer Lett. (2022) 524:151–60. doi: 10.1016/j.canlet.2021.10.010
195. Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell. (2016) 30:229–42. doi: 10.1016/j.ccell.2016.06.004
196. Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, et al. mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. (2013) 5:196ra99. doi: 10.1126/scitranslmed.3005747
197. Chang DY, Ma WL, Lu YS. Role of Alpelisib in the Treatment of PIK3CA-Mutated Breast Cancer: Patient Selection and Clinical Perspectives. Ther Clin Risk Manage. (2021) 17:193–207. doi: 10.2147/tcrm.S251668
198. Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. (2014) 26:136–49. doi: 10.1016/j.ccr.2014.05.020
199. Pahuja KB, Nguyen TT, Jaiswal BS, Prabhash K, Thaker TM, Senger K, et al. Actionable Activating Oncogenic ERBB2/HER2 Transmembrane and Juxtamembrane Domain Mutations. Cancer Cell. (2018) 34:792–806.e5. doi: 10.1016/j.ccell.2018.09.010
200. Pillai RN, Behera M, Berry LD, Rossi MR, Kris MG, Johnson BE, et al. HER2 mutations in lung adenocarcinomas: A report from the Lung Cancer Mutation Consortium. CANCER. (2017) 123:4099–105. doi: 10.1002/cncr.30869
201. Kim EK, Kim KA, Lee CY, Shim HS. The frequency and clinical impact of HER2 alterations in lung adenocarcinoma. PloS One. (2017) 12:e0171280. doi: 10.1371/journal.pone.0171280
202. Tomizawa K, Suda K, Onozato R, Kosaka T, Endoh H, Sekido Y, et al. Prognostic and predictive implications of HER2/ERBB2/neu gene mutations in lung cancers. Lung Cancer. (2011) 74:139–44. doi: 10.1016/j.lungcan.2011.01.014
203. Yang S, Wang Y, Zhao C, Li X, Liu Q, Mao S, et al. Exon 20 YVMA insertion is associated with high incidence of brain metastasis and inferior outcome of chemotherapy in advanced non-small cell lung cancer patients with HER2 kinase domain mutations. Trans Lung Cancer Res. (2021) 10:753–65. doi: 10.21037/tlcr
204. Li BT, Ross DS, Aisner DL, Chaft JE, Hsu M, Kako SL, et al. HER2 Amplification and HER2 Mutation Are Distinct Molecular Targets in Lung Cancers. J Thorac Oncol. (2016) 11:414–9. doi: 10.1016/j.jtho.2015.10.025
205. Fang W, Zhao S, Liang Y, Yang Y, Yang L, Dong X, et al. Mutation Variants and Codiantsgoi. as Genomic Modifiers of Response to Afatinib in HER2inibsoi Lung Adenocarcinoma. Oncologist. (2019) 25:e545–54. doi: 10.1634/theoncologist.2019-0547
206. Dziadziuszko R, Smit EF, Dafni U, Wolf J, Wasąg B, Biernat W, et al. Afatinib in NSCLC With HER2 Mutations: Results of the Prospective, Open-Label Phase II NICHE Trial of European Thoracic Oncology Platform (ETOP). J Thorac Oncol. (2019) 14:1086–94. doi: 10.1016/j.jtho.2019.02.017
207. Song Z, Lv D, Chen S, Huang J, Wang L, Xu S, et al. Efficacy and Resistance of Afatinib in Chinese Non-Small Cell Lung Cancer Patients With HER2 Alterations: A Multicenter Retrospective Study. Article. Front Oncol. (2021) 11:657283. doi: 10.3389/fonc.2021.657283
208. Fan Y, Chen J, Zhou C, Wang H, Shu Y, Zhang J, et al. Afatinib in patients with advanced non-small cell lung cancer harboring HER2 mutations, previously treated with chemotherapy: A phase II trial. Lung Cancer. (2020) 147:209–13. doi: 10.1016/j.lungcan.2020.07.017
209. Kris MG, Camidge DR, Giaccone G, Hida T, Li BT, O'Connell J, et al. Targeting HER2 aberrations as actionable drivers in lung cancers: phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann Oncol. (2015) 26:1421–7. doi: 10.1093/annonc/mdv186
210. Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature. (2018) 554:189–94. doi: 10.1038/nature25475
211. Cornelissen R, Garassino MC, Le X, Clarke J, Tchekmedyian N, Goldman J, et al. MA11.04 Updated Efficacy, Safety and Dosing Management of Poziotinib in Previously Treated EGFR and HER2 Exon 20 NSCLC Patients. J Thorac Oncol. (2021) 16:S173–4. doi: 10.1016/j.jtho.2021.01.249
212. Peters S, Curioni-Fontecedro A, Nechushtan H, Shih J-Y, Liao W-Y, Gautschi O, et al. Activity of Afatinib in Heavily Pretreated Patients With ERBB2 Mutation–Positive Advanced NSCLC: Findings From a Global Named Patient Use Program. J Thorac Oncol. (2018) 13:1897–905. doi: 10.1016/j.jtho.2018.07.093
213. Li BT, Michelini F, Misale S, Cocco E, Baldino L, Cai Y, et al. HER2-Mediated Internalization of Cytotoxic Agents in ERBB2 Amplified or Mutant Lung Cancers. Cancer Discovery. (2020) 10:674–87. doi: 10.1158/2159-8290.Cd-20-0215
214. Iwama E, Zenke Y, Sugawara S, Daga H, Morise M, Yanagitani N, et al. Trastuzumab emtansine for patients with non–small cell lung cancer positive for human epidermal growth factor receptor 2 exon-20 insertion mutations. Eur J Cancer. (2022) 162:99–106. doi: 10.1016/j.ejca.2021.11.021
215. Li BT, Smit EF, Goto Y, Nakagawa K, Udagawa H, Mazières J, et al. Trastuzumab Deruxtecan in HER2-Mutant Non–Small-Cell Lung Cancer. New Engl J Med. (2021) 386:241–51. doi: 10.1056/NEJMoa2112431
216. Li BT, Smit EF, Goto Y, Nakagawa K, Udagawa H, Mazières J, et al. Trastuzumab Deruxtecan in HER2-Mutant Non-Small-Cell Lung Cancer. N Engl J Med. (2022) 386:241–51. doi: 10.1056/NEJMoa2112431
217. Mazières J, Barlesi F, Filleron T, Besse B, Monnet I, Beau-Faller M, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: results from the European EUHER2 cohort. Ann Oncol. (2016) 27:281–6. doi: 10.1093/annonc/mdv573
218. La Monica S, Cretella D, Bonelli M, Fumarola C, Cavazzoni A, Digiacomo G, et al. Trastuzumab emtansine delays and overcomes resistance to the third-generation EGFR-TKI osimertinib in NSCLC EGFR mutated cell lines. J Exp Clin Cancer Res. (2017) 36:174. doi: 10.1186/s13046-017-0653-7
219. Chuang JC, Stehr H, Liang Y, Das M, Huang J, Diehn M, et al. ERBB2-Mutated Metastatic Non-Small Cell Lung Cancer: Response and Resistance to Targeted Therapies. J Thorac Oncol. (2017) 12:833–42. doi: 10.1016/j.jtho.2017.01.023
220. Zhang P, Nie X, Wang B, Li L. Combined therapy with osimertinib and afatinib in a lung adenocarcinoma patient with EGFR T790M mutation and multiple HER2 alterations after resistance to icotinib: A case report. Thorac Cancer. (2018) 9:1774–7. doi: 10.1111/1759-7714.12889
221. Ding G, Wang J, Ding P, Wen Y, Yang L. Case report: HER2 amplification as a resistance mechanism to crizotinib in NSCLC with MET exon 14 skipping. Cancer Biol Ther. (2019) 20:837–42. doi: 10.1080/15384047.2019.1566049
222. Wang M, Zhang D, Wang G, Xia J, Chen S, Li S, et al. HER2 amplification as a potential mechanism of acquired resistance to afatinib in an advanced non-small-cell lung cancer patient. Lung Cancer. (2021) 151:106–7. doi: 10.1016/j.lungcan.2020.11.004
223. Tsui DCC, Aisner D, Nijmeh H, Bao L, Menter A, Camidge DR. Tumor Shrinkage With Combination of Alectinib and Trastuzumab in a Patient With ALK-Rearranged Non-small Cell Lung Cancer Harboring HER2-Amplification as an Acquired Resistance Mechanism to ALK Inhibitor Therapy. Clin Lung Cancer. (2022) 23:e99–e103. doi: 10.1016/j.cllc.2021.06.012
224. Takeda T, Yamamoto H, Suzawa K, Tomida S, Miyauchi S, Araki K, et al. YES1 activation induces acquired resistance to neratinib in HER2-amplified breast and lung cancers. Cancer Sci. (2020) 111:849–56. doi: 10.1111/cas.14289
225. Sung M, Tan X, Lu B, Golas J, Hosselet C, Wang F, et al. Caveolae-Mediated Endocytosis as a Novel Mechanism of Resistance to Trastuzumab Emtansine (T-DM1). Mol Cancer Ther. (2018) 17:243–53. doi: 10.1158/1535-7163.Mct-17-0403
226. Bon G, Pizzuti L, Laquintana V, et al. Loss of HER2 and decreased T-DM1 efficacy in HER2 positive advanced breast cancer treated with dual HER2 blockade: the SePHER Study. J Exp Clin Cancer Res. (2020) 39:279. doi: 10.1186/s13046-020-01797-3
227. Raghav KP, Gonzalez-Angulo AM, Blumenschein GR Jr. Role of HGF/MET axis in resistance of lung cancer to contemporary management. Transl Lung Cancer Res. (2012) 1:179–93. doi: 10.3978/j.issn.2218-6751.2012.09.04
228. Skead G, Govender D. Gene of the month: MET. J Clin Pathol. (2015) 68:405–9. doi: 10.1136/jclinpath-2015-203050
229. Heist RS, Shim HS, Gingipally S, Mino-Kenudson M, Le L, Gainor JF, et al. MET Exon 14 Skipping in Non-Small Cell Lung Cancer. ONCOLOGIST. (2016) 21:481–6. doi: 10.1634/theoncologist.2015-0510
230. Lee M, Jain P, Wang F, Ma PC, Borczuk A, Halmos B. MET alterations and their impact on the future of non-small cell lung cancer (NSCLC) targeted therapies. Expert Opin Ther Targets. (2021) 25:249–68. doi: 10.1080/14728222.2021.1925648
231. Pasquini G, Giaccone G. C-MET inhibitors for advanced non-small cell lung cancer. Expert Opin Investig Drugs. (2018) 27:363–75. doi: 10.1080/13543784.2018.1462336
232. Zhang J, Jiang X, Jiang Y, Guo M, Zhang S, Li J, et al. Recent advances in the development of dual VEGFR and c-Met small molecule inhibitors as anticancer drugs. Eur J Med Chem. (2016) 108:495–504. doi: 10.1016/j.ejmech.2015.12.016
233. Jiang W, Yang N, Zhang Y. Novel MET Exon 14 Skipping Treatment-Naïve Lung Adenocarcinoma Presented Primary Resistance to Crizotinib. J Thorac Oncol. (2018) 13:e124–6. doi: 10.1016/j.jtho.2018.02.030
234. Jin W, Shan B, Liu H, Zhou S, Li W, Pan J, et al. Acquired Mechanism of Crizotinib Resistance in NSCLC with MET Exon 14 Skipping. J Thorac Oncol. (2019) 14:e137–9. doi: 10.1016/j.jtho.2019.04.021
235. Juchum M, Günther M, Laufer SA. Fighting cancer drug resistance: Opportunities and challenges for mutation-specific EGFR inhibitors. Drug Resist Update. (2015) 20:12–28. doi: 10.1016/j.drup.2015.05.002
236. Shi P, Oh Y-T, Zhang G, Yao W, Yue P, Li Y, et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. (2016) 380:494–504. doi: 10.1016/j.canlet.2016.07.021
237. Peng L, Zhu L, Sun Y, Stebbing J, Selvaggi G, Zhang Y, et al. Targeting ALK Rearrangements in NSCLC: Current State of the Art. Front Oncol. (2022) 12:863461. doi: 10.3389/fonc.2022.863461
238. Lurienne L, Cervesi J, Duhalde L, de Gunzburg J, Andremont A, Zalcman G, et al. NSCLC Immunotherapy Efficacy and Antibiotic Use: A Systematic Review and Meta-Analysis. J Thorac Oncol. (2020) 15:1147–59. doi: 10.1016/j.jtho.2020.03.002
239. Duma N, Santana-Davila R, Molina JR. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clinic Proc. (2019) 94:1623–40. doi: 10.1016/j.mayocp.2019.01.013
240. Genova C, Dellepiane C, Carrega P, Sommariva S, Ferlazzo G, Pronzato P, et al. Therapeutic Implications of Tumor Microenvironment in Lung Cancer: Focus on Immune Checkpoint Blockade. Front Immunol. (2021) 12:799455. doi: 10.3389/fimmu.2021.799455
241. Ulas EB, Dickhoff C, Schneiders FL, Senan S, Bahce I. Neoadjuvant immune checkpoint inhibitors in resectable non-small-cell lung cancer: a systematic review. ESMO Open. (2021) 6:100244. doi: 10.1016/j.esmoop.2021.100244
242. Long K, Suresh K. Pulmonary toxicity of systemic lung cancer therapy. Respirology. (2020) 25 Suppl 2:72–9. doi: 10.1111/resp.13915
243. Giannone G, Ghisoni E, Genta S, Scotto G, Tuninetti V, Turinetto M, et al. Immuno-Metabolism and Microenvironment in Cancer: Key Players for Immunotherapy. Int J Mol Sci. (2020) 21(12):4414. doi: 10.3390/ijms21124414
244. Zhou F, Qiao M, Zhou C. The cutting-edge progress of immune-checkpoint blockade in lung cancer. Cell Mol Immunol. (2021) 18:279–93. doi: 10.1038/s41423-020-00577-5
245. Cascone T, Fradette J, Pradhan M, Gibbons DL. Tumor Immunology and Immunotherapy of Non-Small-Cell Lung Cancer. Cold Spring Harb Perspect Med. (2022) 12(5):a037895. doi: 10.1101/cshperspect.a037895
246. Reckamp KL, Redman MW, Dragnev KH, Minichiello K, Villaruz LC, Faller B, et al. Phase II Randomized Study of Ramucirumab and Pembrolizumab Versus Standard of Care in Advanced Non-Small-Cell Lung Cancer Previously Treated With Immunotherapy-Lung-MAP S1800A. J Clin Oncol. (2022) 40:2295–306. doi: 10.1200/JCO.22.00912
247. Ahern E, Solomon BJ, Hui R, Pavlakis N, O’Byrne K, Hughes BGM. Neoadjuvant immunotherapy for non-small cell lung cancer: right drugs, right patient, right time? J Immunother Cancer. (2021) 9(6):e002248. doi: 10.1136/jitc-2020-002248
248. Petricevic B, Kabiljo J, Zirnbauer R, Walczak H, Laengle J, Bergmann M. Neoadjuvant immunotherapy in gastrointestinal cancers - The new standard of care? Semin Cancer Biol. (2022) 86:834–50. doi: 10.1016/j.semcancer.2022.05.015
249. Tokaz MC, Baik CS, Houghton AM, Tseng D. New Immuno-oncology Targets and Resistance Mechanisms. Curr Treat Options Oncol. (2022) 23:1201–18. doi: 10.1007/s11864-022-01005-8
250. Derosa L, Routy B, Thomas AM, Iebba V, Zalcman G, Friard S, et al. Intestinal Akkermansia muciniphila predicts clinical response to PD-1 blockade in patients with advanced non-small-cell lung cancer. Nat Med. (2022) 28:315–24. doi: 10.1038/s41591-021-01655-5
251. Pradhan M, Chocry M, Gibbons DL, Sepesi B, Cascone T. Emerging biomarkers for neoadjuvant immune checkpoint inhibitors in operable non-small cell lung cancer. Transl Lung Cancer Res. (2021) 10:590–606. doi: 10.21037/tlcr-20-573
252. Xia L, Liu Y, Wang Y. PD-1/PD-L1 Blockade Therapy in Advanced Non-Small-Cell Lung Cancer: Current Status and Future Directions. Oncologist. (2019) 24:S31–41. doi: 10.1634/theoncologist.2019-IO-S1-s05
253. Wessolly M, Stephan-Falkenau S, Streubel A, Wiesweg M, Borchert S, Mairinger E, et al. Digital gene expression analysis of NSCLC-patients reveals strong immune pressure, resulting in an immune escape under immunotherapy. BMC Cancer. (2022) 22:46. doi: 10.1186/s12885-021-09111-w
254. Nassar AH, Adib E, Abou Alaiwi S, El Zarif T, Groha S, Akl EW, et al. Ancestry-driven recalibration of tumor mutational burden and disparate clinical outcomes in response to immune checkpoint inhibitors. Cancer Cell. (2022) 40(10):1161–72.e5. doi: 10.1016/j.ccell.2022.08.022
255. Gemelli M, Noonan DM, Carlini V, Pelosi G, Barberis M, Ricotta R, et al. Overcoming Resistance to Checkpoint Inhibitors: Natural Killer Cells in Non-Small Cell Lung Cancer. Front Oncol. (2022) 12:886440. doi: 10.3389/fonc.2022.886440
256. Zhou C, Wang Y, Zhao J, Chen G, Liu Z, Gu K, et al. Efficacy and Biomarker Analysis of Camrelizumab in Combination with Apatinib in Patients with Advanced Nonsquamous NSCLC Previously Treated with Chemotherapy. Clin Cancer Res. (2021) 27:1296–304. doi: 10.1158/1078-0432.CCR-20-3136
257. Long H, Jia Q, Wang L, Fang W, Wang Z, Jiang T, et al. Tumor-induced erythroid precursor-differentiated myeloid cells mediate immunosuppression and curtail anti-PD-1/PD-L1 treatment efficacy. Cancer Cell. (2022) 40(6):674–93.e7. doi: 10.1016/j.ccell.2022.04.018
258. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. (2018) 118(1):9–16. doi: 10.1038/bjc.2017.434
259. Zhang F, Wang J, Xu Y, Cai S, Li T, Wang G, et al. Co-occurring genomic alterations and immunotherapy efficacy in NSCLC. NPJ Precis Oncol. (2022) 6:4. doi: 10.1038/s41698-021-00243-7
261. Le I, Dhandayuthapani S, Chacon J, Eiring AM, Gadad SS. Harnessing the Immune System with Cancer Vaccines: From Prevention to Therapeutics. Vaccines (Basel). (2022) 10(5):816. doi: 10.3390/vaccines10050816
262. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
263. Mougel A, Terme M, Tanchot C. Therapeutic Cancer Vaccine and Combinations With Antiangiogenic Therapies and Immune Checkpoint Blockade. Front Immunol. (2019) 10:467. doi: 10.3389/fimmu.2019.00467
264. Donninger H, Li C, Eaton JW, Yaddanapudi K. Cancer Vaccines: Promising Therapeutics or an Unattainable Dream. Vaccines (Basel). (2021) 9(6):668. doi: 10.3390/vaccines9060668
265. Nemunaitis J, Dillman RO, Schwarzenberger PO, Senzer N, Cunningham C, Cutler J, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol. (2006) 24:4721–30. doi: 10.1200/jco.2005.05.5335
266. Giaccone G, Bazhenova LA, Nemunaitis J, Tan M, Juhász E, Ramlau R, et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur J Cancer. (2015) 51:2321–9. doi: 10.1016/j.ejca.2015.07.035
267. Raina D, Kosugi M, Ahmad R, Panchamoorthy G, Rajabi H, Alam M, et al. Dependence on the MUC1-C oncoprotein in non-small cell lung cancer cells. Mol Cancer Ther. (2011) 10:806–16. doi: 10.1158/1535-7163.Mct-10-1050
268. Butts C, Murray N, Maksymiuk A, Goss G, Marshall E, Soulières D, et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol. (2005) 23:6674–81. doi: 10.1200/jco.2005.13.011
269. Butts C, Maksymiuk A, Goss G, Soulières D, Marshall E, Cormier Y, et al. Updated survival analysis in patients with stage IIIB or IV non-small-cell lung cancer receiving BLP25 liposome vaccine (L-BLP25): phase IIB randomized, multicenter, open-label trial. J Cancer Res Clin Oncol. (2011) 137:1337–42. doi: 10.1007/s00432-011-1003-3
270. Butts C, Murray RN, Smith CJ, Ellis PM, Jasas K, Maksymiuk A, et al. A multicenter open-label study to assess the safety of a new formulation of BLP25 liposome vaccine in patients with unresectable stage III non-small-cell lung cancer. Clin Lung Cancer. (2010) 11:391–5. doi: 10.3816/CLC.2010.n.101
271. Zajac P, Schultz-Thater E, Tornillo L, Sadowski C, Trella E, Mengus C, et al. MAGE-A Antigens and Cancer Immunotherapy. Front Med (Lausanne). (2017) 4:18. doi: 10.3389/fmed.2017.00018
272. Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. (2016) 17:822–35. doi: 10.1016/s1470-2045(16)00099-1
273. Saavedra D, Crombet T. CIMAvax-EGF: A New Therapeutic Vaccine for Advanced Non-Small Cell Lung Cancer Patients. Front Immunol. (2017) 8:269. doi: 10.3389/fimmu.2017.00269
274. Neninger Vinageras E, de la Torre A, Osorio Rodríguez M, Catalá Ferrer M, Bravo I, Mendoza del Pino M, et al. Phase II randomized controlled trial of an epidermal growth factor vaccine in advanced non-small-cell lung cancer. J Clin Oncol. (2008) 26:1452–8. doi: 10.1200/jco.2007.11.5980
275. Hernandez AM, Vazquez AM. Racotumomab-alum vaccine for the treatment of non-small-cell lung cancer. Expert Rev Vaccines. (2015) 14:9–20. doi: 10.1586/14760584.2015.984691
276. Limacher JM, Quoix E. TG4010: A therapeutic vaccine against MUC1 expressing tumors. Oncoimmunology. (2012) 1:791–2. doi: 10.4161/onci.19863
277. Rochlitz C, Figlin R, Squiban P, Salzberg M, Pless M, Herrmann R, et al. Phase I immunotherapy with a modified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1-positive advanced cancer. J Gene Med. (2003) 5:690–9. doi: 10.1002/jgm.397
278. Ramlau R, Quoix E, Rolski J, Pless M, Lena H, Lévy E, et al. A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV Non-small cell lung cancer. J Thorac Oncol. (2008) 3:735–44. doi: 10.1097/JTO.0b013e31817c6b4f
279. Horton BL, Morgan DM, Momin N, Zagorulya M, Torres-Mejia E, Bhandarkar V, et al. Lack of CD8+ T cell effector differentiation during priming mediates checkpoint blockade resistance in non-small cell lung cancer. Sci Immunol. (2021) 6:eabi8800. doi: 10.1126/sciimmunol.abi8800
280. Passaro A, Brahmer J, Antonia S, Mok T, Peters S. Managing Resistance to Immune Checkpoint Inhibitors in Lung Cancer: Treatment and Novel Strategies. J Clin Oncol. (2022) 40:598–610. doi: 10.1200/JCO.21.01845
281. Awad MM, Govindan R, Balogh KN, Spigel DR, Garon EB, Bushway ME, et al. Personalized neoantigen vaccine NEO-PV-01 with chemotherapy and anti-PD-1 as first-line treatment for non-squamous non-small cell lung cancer. Cancer Cell. (2022) 40(9):1010–26.e11. doi: 10.1016/j.ccell.2022.08.003
282. Hack SP, Zhu AX, Wang Y. Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front Immunol. (2020) 11:598877. doi: 10.3389/fimmu.2020.598877
283. Yang M, Li J, Gu P, Fan X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact Mater. (2021) 6:1973–87. doi: 10.1016/j.bioactmat.2020.12.010
284. Socinski MA, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, et al. IMpower150 Final Overall Survival Analyses for Atezolizumab Plus Bevacizumab and Chemotherapy in First-Line Metastatic Nonsquamous NSCLC. J Thorac Oncol. (2021) 16:1909–24. doi: 10.1016/j.jtho.2021.07.009
285. Taylor MH, Lee C-H, Makker V, Rasco D, Dutcus CE, Wu J, et al. Phase IB/II Trial of Lenvatinib Plus Pembrolizumab in Patients With Advanced Renal Cell Carcinoma, Endometrial Cancer, and Other Selected Advanced Solid Tumors. J Clin Oncol. (2020) 38:1154–63. doi: 10.1200/JCO.19.01598
286. Tolaney SM, Garrett-Mayer E, White J, Blinder VS, Foster JC, Amiri-Kordestani L, et al. Updated Standardized Definitions for Efficacy End Points (STEEP) in Adjuvant Breast Cancer Clinical Trials: STEEP Version 2.0. J Clin Oncol. (2021) 39:2720–31. doi: 10.1200/JCO.20.03613
287. He K, Berz D, Gadgeel SM, Iams WT, Bruno DS, Blakely CM, et al. MRTX-500 Phase 2 Trial: Sitravatinib With Nivolumab in Patients With Nonsquamous NSCLC Progressing On or After Checkpoint Inhibitor Therapy or Chemotherapy. J Thorac Oncol. (2023) 27(12):1637–52. doi: 10.1016/j.jtho.2023.02.016
288. Lythgoe MP, Prasad V. Repositioning canakinumab for non-small cell lung cancer-important lessons for drug repurposing in oncology. Br J Cancer. (2022) 127:785–7. doi: 10.1038/s41416-022-01893-5
289. Diwanji R, O’Brien NA, Choi JE, Nguyen B, Laszewski T, Grauel AL, et al. Targeting the IL1β Pathway for Cancer Immunotherapy Remodels the Tumor Microenvironment and Enhances Antitumor Immune Responses. Cancer Immunol Res. (2023) 11:777–91. doi: 10.1158/2326-6066.CIR-22-0290
290. Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. (2020) 30:966–79. doi: 10.1038/s41422-020-00395-4
291. Spigel D, Jotte R, Nemunaitis J, Shum M, Schneider J, Goldschmidt J, et al. Randomized Phase 2 Studies of Checkpoint Inhibitors Alone or in Combination With Pegilodecakin in Patients With Metastatic NSCLC (CYPRESS 1 and CYPRESS 2). J Thorac Oncol. (2021) 16:327–33. doi: 10.1016/j.jtho.2020.10.001
292. Paz-Ares L, Kim TM, Vicente D, Felip E, Lee DH, Lee KH, et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Second-Line Treatment of Patients With NSCLC: Results From an Expansion Cohort of a Phase 1 Trial. J Thorac Oncol. (2020) 15:1210–22. doi: 10.1016/j.jtho.2020.03.003
293. Hakozaki T, Richard C, Elkrief A, Hosomi Y, Benlaïfaoui M, Mimpen I, et al. The Gut Microbiome Associates with Immune Checkpoint Inhibition Outcomes in Patients with Advanced Non-Small Cell Lung Cancer. Cancer Immunol Res. (2020) 8:1243–50. doi: 10.1158/2326-6066.CIR-20-0196
294. Zappasodi R, Serganova I, Cohen IJ, Maeda M, Merghoub T. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature (2021) 591(7851):652–658. doi: 10.1038/s41586-021-03326-4
295. Penter L, Zhang Y, Savell A, Huang T, Wu CJ. Molecular and cellular features of CTLA-4 blockade for relapsed myeloid malignancies after transplantation. Blood (2021) 137(23):3212–7. doi: 10.1182/blood.2021010867
296. Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nature Med. (2018) 24(2):1845–51. doi: 10.1038/s41591-018-0232-2
297. Houston Thompson R, Allison JP, Kwon ED. Anti-cytotoxic T lymphocyte antigen-4 (CTLA-4) immunotherapy for the treatment of prostate cancer. Nature Immunol. (2006) 24:442–7. doi: 10.1016/j.urolonc.2005.08.011
298. Egen JG, Kuhns MS, Allison JP, Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol. (2002) 3:611–8611–8. doi: 10.1038/ni0702-611
299. Ren Z, Guo J, Liao J, Luan Y, Liu Z, Sun Z, et al. CTLA-4 limits anti-CD20-mediated tumor regression. J Autoimmun. (2016) 45(100):49–57. doi: 10.1016/j.jaut.2013.06.006
300. Walker LSK. Treg and CTLA-4: Two intertwining pathways to immune tolerance. N Engl J Med. (2013) 45:49–57. doi: 10.1016/j.jaut.2013.06.006
301. Hodi FS, O’Day SJ, Mcdermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. Trends Immunol. (2010) 363:711–23. doi: 10.1016/j.it.2018.10.009
302. Liu Y, Zheng P. How Does an Anti-CTLA-4 Antibody Promote Cancer Immunity? Cancer Immunol Immunother. (2018) 39:953–6. doi: 10.1016/j.it.2018.10.009
303. Bengsch F, Knoblock DM, Liu A, Mcallister F, Beatty GL. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. JCI Immunother. (2017) 28(5):501–2. doi: 10.1007/s00262-017-2053-4
304. Pol J, Kroemer G. Anti-CTLA-4 immunotherapy: uncoupling toxicity and efficacy. Nature. (2018) 552(7685):S61. doi: 10.1038/s41422-018-0031-9
305. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. (2017) 541(7637):321–30. doi: 10.1038/nature21349
306. Somasundaram A, Burns TF. The next generation of immunotherapy: keeping lung cancer in check. J Hematol Oncol. (2017) 10(2):85–96. doi: 10.1186/s13045-017-0456-5
307. Sundar R, Cho BC, Brahmer JR, Soo RA. Nivolumab in NSCLC: latest evidence and clinical potential. J Ther Adv Med Oncol. (2015) 167(2):397–404.e9. doi: 10.1177/1758834014567470
308. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cancer Immunol Res. (2016) 2(7):632–42. doi: 10.1016/j.cell.2016.08.069
309. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. (2014) 2(7):632–42. doi: 10.1158/2326-6066.CIR-14-0053
310. Tobin RP, Jordan KR, Robinson WA, Dana D, Borges VF, Rene G, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int Immunopharmacol. (2018) 63:282–91. doi: 10.1016/j.intimp.2018.08.007
311. Fei Z, Evans KS, Christine X, Nicholas DV, Balamayooran T, Alisha H, et al. Stromal Fibroblasts Mediate Anti-PD-1 Resistance via MMP-9 and Dictate TGF-β Inhibitor Sequencing in Melanoma. Cancer Immunol Res. (2018) 6:canimm.0086.2018. doi: 10.1158/2326-6066.CIR-18-0086
312. Clavijo PE, Friedman J, Robbins Y, Moore EC, Smith E, Zauderer M, et al. Semaphorin4D Inhibition Improves Response to Immune-Checkpoint Blockade via Attenuation of MDSC Recruitment and Function. Cancer Immunol Res. (2019) 7:282–91. doi: 10.1158/2326-6066.Cir-18-0156
313. Gubens MA, Sequist LV, Stevenson JP, Powell SF, Villaruz LC, Gadgeel SM, et al. Pembrolizumab in combination with ipilimumab as second-line or later therapy for advanced non-small-cell lung cancer: KEYNOTE-021 cohorts D and H. J Lung Cancer. (2019) 130:59–66. doi: 10.1016/j.lungcan.2018.12.015
314. Garon EB, Cho BC, Reinmuth N, Lee KH, Rizvi NA. Patient-Reported Outcomes with Durvalumab With or Without Tremelimumab Versus Standard Chemotherapy as First-Line Treatment of Metastatic Non-Small Cell Lung Cancer (MYSTIC). Clin Lung Cancer. (2021) 22(4):301–312.e8. doi: 10.1016/j.cllc.2021.02.010
315. Gaudreau PO, Lee JJ, Heymach JV, Gibbons DL. Phase I/II Trial of Immunotherapy With Durvalumab and Tremelimumab With Continuous or Intermittent MEK Inhibitor Selumetinib in NSCLC: Early Trial Report. Clin Lung Cancer. (2020) 21:384–8. doi: 10.1016/j.cllc.2020.02.019
316. Saleh R, Elkord E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin Cancer Biol. (2020) 65:13–27. doi: 10.1016/j.semcancer.2019.07.017
317. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. (2021) 21:298–312. doi: 10.1038/s41568-021-00339-z
318. Yang L, Li A, Lei Q, Zhang Y. Tumor-intrinsic signaling pathways: key roles in the regulation of the immunosuppressive tumor microenvironment. J Hematol Oncol. (2019) 12:125. doi: 10.1186/s13045-019-0804-8
319. Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front Oncol. (2020) 10:188. doi: 10.3389/fonc.2020.00188
320. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
321. Enderlin Vaz da Silva Z, Lehr HA, Velin D. In vitro and in vivo repair activities of undifferentiated and classically and alternatively activated macrophages. Pathobiology. (2014) 81:86–93. doi: 10.1159/000357306
322. Mazzarella L, Duso BA, Trapani D, Belli C, D'Amico P, Ferraro E, et al. The evolving landscape of ‘next-generation’ immune checkpoint inhibitors: A review. Eur J Cancer. (2019) 117:14–31. doi: 10.1016/j.ejca.2019.04.035
323. Tesi RJ. MDSC; the Most Important Cell You Have Never Heard Of. Trends Pharmacol Sci. (2019) 40:4–7. doi: 10.1016/j.tips.2018.10.008
324. Igney FH, Krammer PH. Immune escape of tumors: apoptosis resistance and tumor counterattack. J Leukoc Biol. (2002) 71:907–20. doi: 10.1189/jlb.71.6.907
325. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. (2007) 25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609
326. Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. (2005) 8:369–80. doi: 10.1016/j.ccr.2005.10.012
327. Yasumoto K, Hanagiri T, Takenoyama M. Lung cancer-associated tumor antigens and the present status of immunotherapy against non-small-cell lung cancer. Gen Thorac Cardiovasc Surg. (2009) 57:449–57. doi: 10.1007/s11748-008-0433-6
328. Lahiri A, Maji A, Potdar PD, Singh N, Parikh P, Bisht B, et al. Lung cancer immunotherapy: progress, pitfalls, and promises. Mol Cancer. (2023) 22:40. doi: 10.1186/s12943-023-01740-y
329. Remy-Ziller C, Thioudellet C, Hortelano J, Gantzer M, Nourtier V, Claudepierre MC, et al. Sequential administration of MVA-based vaccines and PD-1/PD-L1-blocking antibodies confers measurable benefits on tumor growth and survival: Preclinical studies with MVA-βGal and MVA-MUC1 (TG4010) in a murine tumor model. Hum Vaccin Immunother. (2018) 14:140–5. doi: 10.1080/21645515.2017.1373921
330. Oliveres H, Caglevic C, Passiglia F, Taverna S, Smits E, Rolfo C. Vaccine and immune cell therapy in non-small cell lung cancer. J Thorac Dis. (2018) 10:S1602–s1614. doi: 10.21037/jtd.2018.05.134
331. Braiteh F, LoRusso P, Balmanoukian A, Klempner S, Camidge DR, Hellmann M, et al. Abstract CT169: A phase Ia study to evaluate RO7198457, an individualized Neoantigen Specific immunoTherapy (iNeST), in patients with locally advanced or metastatic solid tumors. Cancer Res. (2020) 80:CT169–9. doi: 10.1158/1538-7445.AM2020-CT169
332. Oosting LT, Franke K, Martin MV, Kloosterman WP, Jamieson JA, Glenn LA, et al. Development of a Personalized Tumor Neoantigen Based Vaccine Formulation (FRAME-001) for Use in a Phase II Trial for the Treatment of Advanced Non-Small Cell Lung Cancer. Pharmaceutics. (2022) 14(7):1515. doi: 10.3390/pharmaceutics14071515
333. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
334. Kim CG, Sang YB, Lee JH, Chon HJ. Combining Cancer Vaccines with Immunotherapy: Establishing a New Immunological Approach. Int J Mol Sci. (2021) 22(15):8035. doi: 10.3390/ijms22158035
335. Ruan J, Duan Y, Li F, Wang Z. Enhanced synergistic anti-Lewis lung carcinoma effect of a DNA vaccine harboring a MUC1-VEGFR2 fusion gene used with GM-CSF as an adjuvant. Clin Exp Pharmacol Physiol. (2017) 44:71–8. doi: 10.1111/1440-1681.12654
336. Creelan BC, Antonia S, Noyes D, Hunter TB, Simon GR, Bepler G, et al. Phase II trial of a GM-CSF-producing and CD40L-expressing bystander cell line combined with an allogeneic tumor cell-based vaccine for refractory lung adenocarcinoma. J Immunother. (2013) 36:442–50. doi: 10.1097/CJI.0b013e3182a80237
337. Lee JM, Lee MH, Garon E, Goldman JW, Salehi-Rad R, Baratelli FE, et al. Phase I Trial of Intratumoral Injection of CCL21 Gene-Modified Dendritic Cells in Lung Cancer Elicits Tumor-Specific Immune Responses and CD8(+) T-cell Infiltration. Clin Cancer Res. (2017) 23:4556–68. doi: 10.1158/1078-0432.CCR-16-2821
338. Papachristofilou A, Hipp MM, Klinkhardt U, Früh M, Sebastian M, Weiss C, et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J Immunother Cancer. (2019) 7:38. doi: 10.1186/s40425-019-0520-5
339. Sebastian M, Schröder A, Scheel B, Hong HS, Muth A, von Boehmer L, et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol Immunother. (2019) 68:799–812. doi: 10.1007/s00262-019-02315-x
340. Rodriguez PC, Popa X, Martínez O, Mendoza S, Santiesteban E, Crespo T, et al. A Phase III Clinical Trial of the Epidermal Growth Factor Vaccine CIMAvax-EGF as Switch Maintenance Therapy in Advanced Non-Small Cell Lung Cancer Patients. Clin Cancer Res. (2016) 22:3782–90. doi: 10.1158/1078-0432.Ccr-15-0855
341. Shi T, Song X, Wang Y, Liu F, Wei J. Combining Oncolytic Viruses With Cancer Immunotherapy: Establishing a New Generation of Cancer Treatment. Front Immunol. (2020) 11:683. doi: 10.3389/fimmu.2020.00683
342. Pateras IS, Kotsakis A, Avgeris M, Baliou E, Kouroupakis P, Patsea E, et al. Clinical Activity of an hTERT-Specific Cancer Vaccine (Vx-001) in “Immune Desert” NSCLC. Cancers (Basel). (2021) 13(7):1658. doi: 10.3390/cancers13071658
343. Gridelli C, Ciuleanu T, Domine M, Szczesna A, Bover I, Cobo M, et al. Clinical activity of a htert (vx-001) cancer vaccine as post-chemotherapy maintenance immunotherapy in patients with stage IV non-small cell lung cancer: final results of a randomised phase 2 clinical trial. Br J Cancer. (2020) 122:1461–6. doi: 10.1038/s41416-020-0785-y
344. Blanco B, Domínguez-Alonso C, Alvarez-Vallina L. Bispecific Immunomodulatory Antibodies for Cancer Immunotherapy. Clin Cancer Res. (2021) 27:5457–64. doi: 10.1158/1078-0432.Ccr-20-3770
345. Ruotsalainen TM, Halme M, Tamminen K, Szopinski J, Niiranen A, Pyrhönen S, et al. Concomitant chemotherapy and IFN-alpha for small cell lung cancer: a randomized multicenter phase III study. J Interferon Cytokine Res. (1999) 19:253–9. doi: 10.1089/107999099314180
346. Marincola FM, White DE, Wise AP, Rosenberg SA. Combination therapy with interferon alfa-2a and interleukin-2 for the treatment of metastatic cancer. J Clin Oncol. (1995) 13:1110–22. doi: 10.1200/jco.1995.13.5.1110
347. Pan Y, Hao Y, Han H, Chen T, Ding H, Labbe KE, et al. Nemvaleukin alfa, a novel engineered IL-2 fusion protein, drives antitumor immunity and inhibits tumor growth in small cell lung cancer. J Immunother Cancer. (2022) 10(9):e004913. doi: 10.1136/jitc-2022-004913
348. Dhupkar P, Gordon N. Interleukin-2: Old and New Approaches to Enhance Immune-Therapeutic Efficacy. Adv Exp Med Biol. (2017) 995:33–51. doi: 10.1007/978-3-319-53156-4_2
349. Lopes JE, Fisher JL, Flick HL, Wang C, Sun L, Ernstoff MS, et al. ALKS 4230: a novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. J Immunother Cancer. (2020) 8(1):e000673. doi: 10.1136/jitc-2020-000673
350. Zhao L, Wang WJ, Zhang JN, Zhang XY. 5-Fluorouracil and interleukin-2 immunochemotherapy enhances immunogenicity of non-small cell lung cancer A549 cells through upregulation of NKG2D ligands. Asian Pac J Cancer Prev. (2014) 15:4039–44. doi: 10.7314/apjcp.2014.15.9.4039
351. Orditura M, Romano C, De Vita F, Galizia G, Lieto E, Infusino S, et al. Behaviour of interleukin-2 serum levels in advanced non-small-cell lung cancer patients: relationship with response to therapy and survival. Cancer Immunol Immunother. (2000) 49:530–6. doi: 10.1007/s002620000150
352. Tian C, Lu S, Fan Q, Zhang W, Jiao S, Zhao X, et al. Prognostic significance of tumor-infiltrating CD8+ or CD3+ T lymphocytes and interleukin-2 expression in radically resected non-small cell lung cancer. Chin Med J (Engl). (2015) 128:105–10. doi: 10.4103/0366-6999.147828
353. Zhang X, Zeng Y, Qu Q, Zhu J, Liu Z, Ning W, et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. (2017) 22:1026–33. doi: 10.1007/s10147-017-1161-7
354. Gao Y, Yang J, Cai Y, Fu S, Zhang N, Fu X, et al. IFN-γ-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int J Cancer. (2018) 143:931–43. doi: 10.1002/ijc.31357
355. Kremenovic M, Chan AA, Feng B, Bäriswyl L, Robatel S, Gruber T, et al. BCG hydrogel promotes CTSS-mediated antigen processing and presentation, thereby suppressing metastasis and prolonging survival in melanoma. J Immunother Cancer. (2022) 10(6):e004133. doi: 10.1136/jitc-2021-004133
356. Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Pérez-Gracia JL, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer. (2019) 120:6–15. doi: 10.1038/s41416-018-0328-y
357. Firdaus FZ, Skwarczynski M, Toth I. Developments in Vaccine Adjuvants. Methods Mol Biol. (2022) 2412:145–78. doi: 10.1007/978-1-0716-1892-9_8
Keywords: non-small cell lung cancer, targeted therapy, immunotherapy, mechanisms of drug resistance, promising strategies
Citation: Xiang Y, Liu X, Wang Y, Zheng D, Meng Q, Jiang L, Yang S, Zhang S, Zhang X, Liu Y and Wang B (2024) Mechanisms of resistance to targeted therapy and immunotherapy in non-small cell lung cancer: promising strategies to overcoming challenges. Front. Immunol. 15:1366260. doi: 10.3389/fimmu.2024.1366260
Received: 05 January 2024; Accepted: 18 March 2024;
Published: 09 April 2024.
Edited by:
Haiyang Wu, Tianjin Medical University, ChinaReviewed by:
Federica Riccardo, University of Turin, ItalyYongbin He, University of North Carolina at Chapel Hill, United States
An He, Charité University Medicine Berlin, Germany
Copyright © 2024 Xiang, Liu, Wang, Zheng, Meng, Jiang, Yang, Zhang, Zhang, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Liu, MTI0OTQ3NjA5QHFxLmNvbQ==; Bo Wang, Ym93YW5nNTgxQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship