
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 22 July 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1363373
This article is part of the Research Topic Neuroimmune Communication in Inflammatory and Allergic Diseases View all 8 articles
 Magdalena Figat1*
Magdalena Figat1* Aleksandra Wiśniewska2
Aleksandra Wiśniewska2 Jacek Plichta1
Jacek Plichta1 Joanna Miłkowska-Dymanowska3
Joanna Miłkowska-Dymanowska3 Sebastian Majewski3
Sebastian Majewski3 Michał S. Karbownik4Piotr Kuna1
Michał S. Karbownik4Piotr Kuna1 Michał G. Panek1
Michał G. Panek1Introduction: Chronic obstructive lung diseases, such as asthma and COPD, appear to have a more extensive impact on overall functioning than previously believed. The latest data from clinical trials suggests a potential link between cognitive deterioration and chronic obstructive inflammatory lung disease. This raises the question of whether these diseases affect cognitive functions and whether any relevant biomarker may be identified.
Methods: This prospective observational study included 78 patients divided equally into asthma, COPD, and control groups (n=26, 27 and 25 respectively). The participants underwent identical examinations at the beginning of the study and after at least 12 months. The test battery comprised 16 questionnaires (11 self-rated, 5 observer-rated, assessing cognition and mental state), spirometry, and blood samples taken for PKA and CREB mRNA evaluation.
Results: A 2.3-fold increase in CREB mRNA was observed between examinations (p=0.014) for all participants; no distinctions were observed between the asthma, COPD, and control groups. Pooled, adjusted data revealed a borderline interaction between diagnosis and CREB expression in predicting MMSE (p=0.055) in COPD, CREB expression is also associated with MMSE (β=0.273, p=0.034) like with the other conducted tests (β=0.327, p=0.024) from COPD patients. No correlations were generally found for PKA, although one significant negative correlation was found between the first and second time points in the COPD group (β=-0.4157, p=0.049),.
Discussion: Chronic obstructive lung diseases, such as asthma and COPD, may have some linkage to impairment of cognitive functions. However, the noted rise in CREB mRNA expression might suggest a potential avenue for assessing possible changes in cognition, especially in COPD; such findings may reveal additional transcription factors linked to cognitive decline.
Both asthma and chronic obstructive pulmonary disease (COPD) are chronic inflammatory lung diseases characterized most notably by obturation, inflammation, chronicity and heterogeneity. These are fundamental features in both conditions.
In asthma, upon exposure to allergens or during hyperventilation, immunoglobulin E (IgE) triggers the degranulation of mast cells (1) and enhances the secretion of Interleukin-6 (IL-6) (2). This IL-6 regulates and activates CD4+ T-helper 2 (Th2) lymphocytes, prompting them to produce Th2 cytokines (2–4): IL-4, IL-5 and IL-13. Production of Th2 cytokines might be regulated by epithelial alarmins, for instance IL-25 and IL-33, which could have crucial role in non-atopic asthma (1). One of these cytokines, IL-5, stimulates the secretion of eosinophils (1), which are directed to the airways by eotaxin and the RANTES chemokine (Regulated upon Activation, Normal T cell Expressed and Secreted), also known as CCL5 (5). The inflammation in asthma has a chronic nature; this may be due to the activity of the NF-κB transcription factor, which amplifies and prolongs the inflammatory response (6). This results in increased local secretion of eosinophil-specific chemokines and reduced production of anti-inflammatory interleukin-10 (IL-10) (1, 6).
In COPD, exposure to cigarette or biomass smoke activates NF-κB and p38-mitogen activated protein kinase (MAPK) (7), increasing the presence of neutrophils and macrophages in the airways (1, 7). These cells release numerous mediators, including elevated levels of IL-8 (8), IL-1β, IL-6, tumor necrosis factor-α (TNF-α), and elastic enzymes (1, 8). Neutrophils promote neutrophilic inflammation and serine protease activity (1), both of which are implicated in common complications of COPD such as emphysema (1, 9) and mucus hypersecretion (5).
However, another important mediator of inflammation in chronic inflammatory lung diseases has been less widely investigated. In untreated or steroid-dependent asthmatic patients, inflammation severity has been found to correlate with increased expression of cAMP response element binding protein (CREB) (10). Similarly, CREB expression may also be elevated in COPD patients who respond poorly to inhaled corticosteroid (ICS) therapy (11). Furthermore, CREB activation appears to enhance airway smooth muscle (ASM) proliferation (12, 13).
Both NF-κB and CREB are integral to the cAMP/PKA/CREB signaling pathway (6) (Figure 1). Upon stimulation of G protein-coupled receptors, such as glucagon-like peptide-1 receptor (GLP-1R) (14) or β2-adrenoreceptor (6), CREB level is decreased as cAMP levels increase (15). Notably, after stimulating GLP-1R with the exogenous analogue exendin-4 (16), NF-κB levels decreased (17), suggesting that elevated protein kinase A (PKA) expression may result in NF-κB depletion (17). Collectively, glucagon-like peptide-1 (GLP-1) appears to mitigate chronic lung inflammation by inactivating PKA-dependent NF-κB (18), reducing IL-8 mRNA expression in ASM cells (19). PKA stimulation may also influence the ability of asthma medications to inhibit abnormal ASM growth (20). Currently, no studies specify typical PKA expression levels in asthma or COPD.
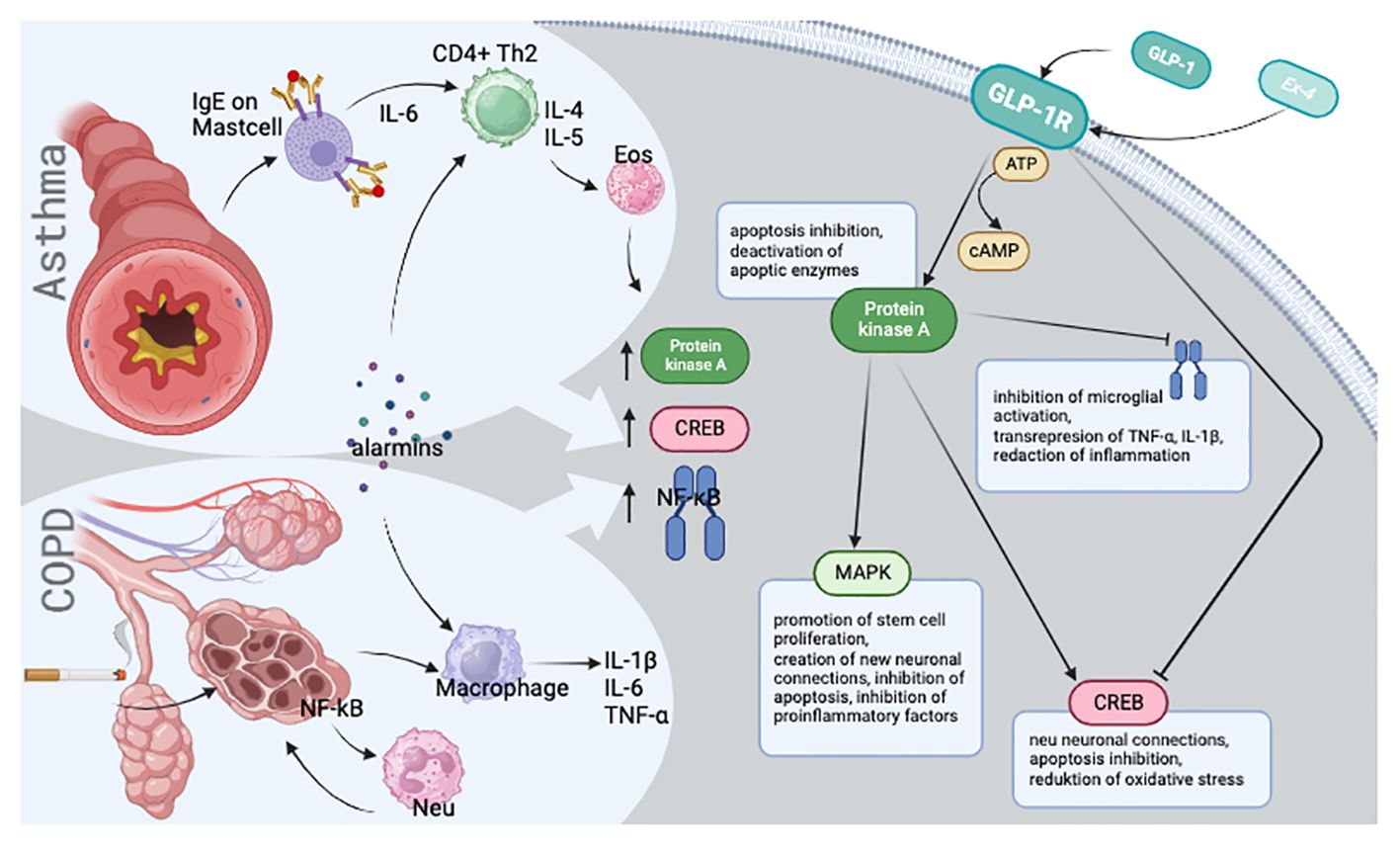
Figure 1 Visual presentation of the simplified disease pathomechanism and molecular background.
GLP-1 moderates eosinophil activation (21) and mitigates bronchial hyperresponsiveness post-sensitization in asthma (21). Notably, GLP-1R expression is markedly diminished in the ASM of COPD patients, yet its overexpression significantly inhibits the release of inflammatory mediators (22, 23). While GLP-1 is known to reduce inflammation in both cases, it is not known whether GLP-1 levels rise or fall during chronic inflammatory diseases; this may be due to the poor availability of tissue suitable for study, as GLP-1 is be expressed in peripheral blood, but in the pancreas tissue, which requires biopsy.
According to the outside-in theory (24, 25), inflammation may not be confined solely to the lungs but could manifest throughout the body. This theory proposes that IL-6 may play a role in brain tissue reactivity among dementia patients (26) by promoting neuroinflammation. IL-1β is believed to amplify p38-MAPK activity, potentially resulting in Tau-hyperphosphorylation (26). Additionally, the TNF-α released from microglia (27), in conjunction with IL-6, has been linked to late-onset dementia and a greater risk of dementia (28, 29).
Several prospective studies have found a potential link between chronic inflammatory lung diseases and neurodegeneration (30–32). This relationship has also been identified in diffusion-weighted magnetic resonance imaging (30). However, although the molecular background remains unknown, the pulmonary conditions may double the risk of mild cognitive impairment or dementia as compared to the general population (33). Both threats predominantly emerge during immaturity (34) or during midlife (33). Prior research primarily sought epidemiological correlations without pinpointing the molecular basis, while recent approaches have focused on assessing cognitive changes in response to asthma medication adjustments (34, 35) or lung disease progression following anti-dementia treatments (36), with little interest in molecular mechanisms. Further research could enhance our understanding of the pathophysiology of inflammation in both scenarios, and open the door to more effective treatment strategies for each phenotype (chronic inflammatory lung diseases and neurodegenerative diseases). However, to better understand the molecular background, the first step is to evaluate the linkage between chronic obstructive lung diseases and cognitive deterioration.
The aim of the study.
The objective of this study was to:
1. Characterize the participants in each group with regard to epidemiological self-reported data, quality of life and disease; these are assessed by survey.
2. Determine cognitive functions using psychiatric assessment tests, and identify any relationships between cognition and the studied chronic inflammatory lung diseases.
3. Identify any relationships between cognitive evaluation and disease-specific questionnaires or spirometric parameters.
4. Examine the correlation between the PKA mRNA and CREB mRNA expression and the cognition assessment.
The study was approved by the Bioethics Committee, resolution number: RNN/287/19/KE (13th June, 2019).
Three groups of participants were established. The first comprised outpatients and inpatients diagnosed with severe asthma according to GINA 2023 Guidelines, who were recruited from the Department of Internal Medicine, Asthma, and Allergy, N. Barlicki University Clinical Hospital No. 1 in Lodz, Poland. The second group consisted of patients with COPD, classified grade GOLD 2 or 3 as B or E categories in GOLD 2024 Standards (37); they were recruited from the Specialist Outpatient Clinic of Pulmonary Diseases and Allergology or received treatment from the Department of Pulmonology and Allergology at the same hospital. The third group functioned as a control, composed of healthy volunteer subjects chosen from the general population. These individuals neither had, nor were in the process of being diagnosed with, any chronic lung diseases or neurodegenerative disorders. Clear and stringent inclusion and exclusion criteria were set for all participants.
The size of the groups was limited by financial constraints. The project was intended to serve as a pilot study to generate initial findings. No power calculation was conducted prior to the study.
The following inclusion criteria were applied: over 18 years old, possess a diagnosis of either asthma (for Group 1) or COPD (for Group 2) confirmed based on GINA2023 (38) or GOLD 2023 (39) Guidelines established by a specialist, provide informed consent, and have both a blood sample available for molecular research and spirometry results on hand. In addition, the following exclusion criteria were applied: age under 18 years, no informed consent, could not be classified into one of the established groups, presence of severe obstructive airway diseases other than asthma or COPD, presence of a past medical history of neurodegenerative disease such as Alzheimer’s disease, Parkinson’s disease or dementia.
The volunteers for the control group had to meet all following eligibility criteria: age over 18 years old, signed informed consent and agreement to provide a blood sample for molecular analysis and undergo spirometry. The following exclusion criteria were also applied: presence of chronic obstructive or inflammatory lung diseases, diagnosis of neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, dementia, multiple sclerosis, amyotrophic lateral sclerosis, mild cognitive impairment or any other degenerative cerebral disorder.
A non-fasting blood sample was taken, and spirometry was performed as per ERS (40) technical standards. Additionally, the patient underwent a series of psychometric tests and fulfilled numerous questionnaires. In total, each participant completed sixteen questionnaires. The first part comprised eleven self-reported questions, scales and scores: 1) demographic data; several asthma-specific questionnaires recommended by the Global Strategy for Asthma Management and Prevention: 2) the Asthma Control Test TM (ACT TM) (39, 40) and 3) the Asthma Control Questionnaire (ACQ) (41, 42) and 4) the standardized Asthma Quality of Life Questionnaire (AQLQ(S)) (43).
The COPD patients completed several specific tools in line with Global Initiative for Chronic Lung Disease (GOLD) guidelines: 5) the COPD Assessment Test (CAT) (44, 45), 6) the modified Medical Research Council dyspnea scale (mMRC) (46), and 7) the St. George’s Respiratory Questionnaire specifically tailored for COPD patients (SGRQ-C) (47, 48).
Another three tests were used to describe the quality of life: 8) the 36-item Short Form Health Survey (SF-36) (49, 50), 9) The Index of Independence in Activities of Daily Living (ADL) (51, 52), and 10) The Lawton Instrumental Activities of Daily Living (IADL) (53).
The SF-36 results can be categorized into eight domains, and there are various methodologies for interpreting them. One renowned method is based on Taft’s Hypothesis (41), which calculates the result according to a Physical Component Summary (PCS) and a Mental Component Summary (MCS); in this case, scores above the expected range indicate not an improvement in health, but a decline in another health aspect (41). As this interpretation can be confusing, the present study adopted a different interpretation described in the Methodology of quality of life assessment (42) used in pharmacological care: this method organizes the eight domains into two components, using the median of each as a summary for physical and mental summaries.
The last self-reported test in the first part, 11) the GDS, counts to the indirect evaluation of cognitive functions.
The second part consist of observer-rated scales performed by the medical staff: 12) the Mini-Mental State Examination (MMSE) (43), which evaluates the cognitive aspects of mental functions (44), including mental tracking, expressive and receptive language, visual construction, immediate and delayed free verbal recall, calculation, and temporal and spatial orientation (45); 13) the Abbreviated Mental Test Score (AMTS) which assesses potential cognitive impairment (46), covering areas such as temporal and spatial orientation, visual memory, and semantic knowledge (45); 14) the Hachinski Ischaemic Score (HIS), which differentiates between senile or vascular origins of cognitive dysfunction (47–49); 15) the Clock Drawing Test (CDT), which is a cognitive screening tool used for the early detection, prediction, or monitoring of cognitive impairment (50, 51).
To evaluate deterioration in mental state, which can be both an indicator of cognitive decline and a hallmark of neurodegeneration (52–54), the following scales were implemented: 11) the 15-item Geriatric Depression Scale (GDS) (mentioned above), a self-rated tool that effectively screens for depression (55–57) and 16) the Hamilton Depression Rating Scale (HAM-D), which an observer-rated measure of depression (58, 59).
The testing procedure, comprising spirometry, blood sampling, and the sixteen tests, was conducted twice: first upon admission to the project and then again after a period of more than 12 months from the initial examination.
Venous blood samples from each patient were collected into tubes containing ethylenediaminetetraacetic acid (EDTA) and stored at -80°C.
Full blood was chosen as the most easily-available source of nucleated cells, i.e. white blood cells. Each full blood sample was pipetted into a separate 15 ml Falcon tube, at 2 ml of sample per tube, and 10 ml of RBCL cell lysis medium (A&A Biotechnology RBCL medium, REF 213-250, LOT 280222, Poland) were added to each tube. The tube contents were mixed and incubated on ice for 15 min. Tubes were centrifuged for 10 min at 3000 rpm and the supernatants discarded.
The remainder of the RNA extraction protocol was carried out with use of the A&A Biotechnology Total RNA Mini (REF 031-100, A&A Biotechnology, Poland) kit. Briefly, 800 µl of phenosol (a solution containing chaotropic salts and phenol) was added to each tube and cells lysed by repetitive pipetting. The tube contents were then transferred to sterile 1.5 ml Eppendorf tubes and incubated for 5 min at 50°C. Following this, 200 µl of chloroform was added, the samples were mixed gently, incubated for 3 min at RT, and then centrifuged for 10 min at 12 000 RPM. The supernatant was transferred to a new sterile 1.5 ml tube, and 250 µl of isopropanol was added to each tube.
The samples were then transferred to minicolumns placed in 2 ml tubes included in the RNA extraction kit, and centrifuged for 1 min at 12 000 RPM. The minicolumns were transferred to new 2 ml tubes and 700 µl of A1 wash solution were added to each minicolumn.
The samples were centrifuged for 1 min at 12 000 RPM. The filtrate was discarded from the tubes, the minicolumns were placed in the same tubes, 700 µl of A1 wash solution were added. The samples were centrifuged for 1 min at 12 000 RPM. The filtrate was discarded from the tubes, and the minicolumns were placed in the same tubes. Following this, 200 µl of A1 wash solution was added to each minicolumn. The samples were centrifuged for 2 min at 12 000 RPM.
The minicolumns were transferred to new sterile 1.5 ml elution tubes and 100 µl of sterile water was added directly onto the minicolumn silicon matrices. The samples were incubated for 3 min at room temperature (RT), centrifuged for 1 min at 12 000 RPM and the extracted RNA samples were stored at - 80°C until use.
RNA concentrations were measured with the use of a Picodrop Spectrophotometer (60), which can detect RNA concentrations of 2 – 3000 μg/mL in 0.5-2.0 μL of solution without dilution (60).
Reverse transcription was performed using the ImProm-II Reverse Transcription System (REF A3800, LOT 0000437833, Promega, USA).
The RNA samples were thawed on ice, diluted with nuclease-free water to final concentrations of 100 ng/µl, and 10µl of each sample was combined with 1.2 µl of Random Hex Primers in thin-walled PCR tubes. The tubes were placed into a preheated 70°C heat block for 5 minutes and immediately chilled on ice for another 5 minutes. The tubes were then centrifuged for 10 seconds and kept on ice until the later stages.
The 7.8 µl aliquots of the reverse transcription reaction mix (ImProm-II™ 5X Reaction Buffer 4.0 µl; dNTP Mix 1 µl; MgCl2 2.4 µl; ImProm-II™ Reverse Transcriptase 0.4 µl) were added to each PCR tube containing RNA samples and Random Hex primers (final volume 19 µl).
Annealing: The tubes were placed in a heat block at 25°C for 5 minutes.
Extension: The tubes were moved to a heat block at 42°C for one hour.
Reverse transcriptase inactivation: The tubes were moved to a heat block at 70°C for 15 minutes.
The resulting cDNA samples were stored at -20°C until subsequent stages of the assay.
The qPCR assay used the following reagents (all from Applied Biosystem, USA): Applied Biosystems TaqMan Universal PCR Master Mix REF 4304437 LOT 2208187; CREB-1 probe Hs00231713_m1 CREB1 LOT 1883523; PRKACB probe Hs01086757_m1 PRKACB LOT 1963364; Applied Biosystems Euk 18S rRNA (20X) REF 4333760F LOT 2003209.
The cDNA samples were thawed and kept on ice.
The reaction mixes (Master Mix 7.5 µl; Nuclease-free water 6.25 µl; Experimental or 18S probe 0.75 µl.) were aliquoted into PCR tubes, 14.5 µl per tube.
Each experimental probe and the 18S rRNA (61) were assayed in duplicate reactions.
To each PCR tube was added 0.5 µl of patient cDNA. The PCR tubes were centrifuged for 10 seconds and placed in a Bio-Rad CFX96 Touch Real-Time PCR Detection System.
The following qPCR protocol was used: 50°C for 2 min, 95°C for 10 min, followed by 44 cycles comprising 95°C for 15 s, 60°C for 1 min and plate read (45 PCR cycles total).
After completing all the steps, the threshold cycle (CT) values for each sample were calculated using Mx-Pro software. For instances where the difference between the two technical CT replicates (for PKA, CREB, and the housekeeping reference gene, 18S) was ≤ 1 (61), an average was computed. The CT for each gene was then determined using the formula: ΔCT = CT, GENE– CT, 18S. If the difference between the values exceeded 1, the RT-PCR was performed again, and the same calculation was also repeated. The final ΔCT was derived as the mean of the initial examination and the repeated RT-PCR value. When 45 cycles were insufficient to detect the examined gene, the value was computed as the difference between the number of completed cycles and the CT, 18S. This method was employed in only 15 measurements. The acquired data were then entered into the database and subjected to statistical analysis.
Categorical variables were described as numbers and frequencies. They were compared between groups with the use of Pearson’s chi-square test or logistic regression. Ordinal variables were described as medians with 1st and 3rd quartiles and compared between groups with the Mann-Whitney U-test. Continuous variables were described as means and standard deviations, and were compared between groups with the Student’s t-test or one-way analysis of variance.
Cognitive and depression parameters were compared between patient groups as raw values and as values adjusted for potential confounding factors, viz. time between the 1st and 2nd examination, age, education level, residence, mental exercises (reading books, crosswords, Sudoku), underwent aesthesia, smoking pack years, BMI ≥25 kg/m2, therapy with systemic steroids. These covariates were included as linearly linked to the modeled parameters.
The analysis between two time-points was performed using the paired t-test. Linear regression was used to test for correlation between two continuous variables. P-values below 0.05 were considered statistically significant. The analyses were performed with STATISTICA 13.3 (StatSoft, Tulsa, OK, USA).
The study involved a total of 78 participants, divided into three distinct groups.
The asthma group comprised 11 women (42%) and 15 men (58%), mean age 55 years, while the COPD group consisted of 13 women (48%) and 14 men (52%), mean age 66 years. The age difference is consistent with the understanding that COPD typically manifests later in life compared to asthma (62, 63). Given potential confounding factors (64), such as age, all results were age-adjusted. The patients in both groups were administered medication according to the prevailing GINA and GOLD guidelines. This medication is summarized in Supplementary Materials, Table 1.
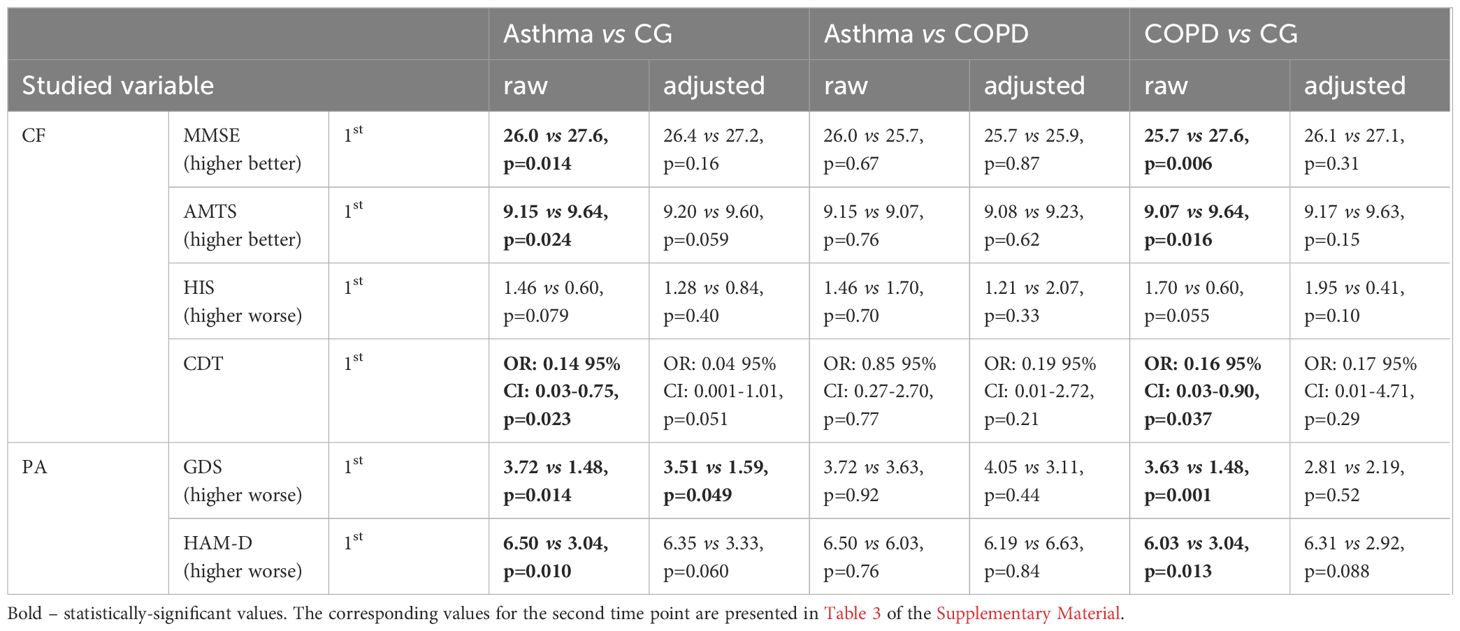
Table 1 Mean results in both groups at the first time point.
The control group was composed of 14 women (56%) and 11 men (44%) with a mean age of approximately 53 years. Most of the individuals in this group were overweight or obese (56%). No other of reported diagnoses revealed statistically significant.
The participants were further evaluated through additional questionnaires and examinations. The most interesting results are presented in Supplementary materials, Table 1. The spirometry results confirmed the previously established diagnoses among the participants and the grouping arrangement. For the control group, the spirometry primarily confirmed the absence of any lung disease.
Of the original 78 participants, 66 were examined at the second time point. Of the 12 remainders, three, one from each group, passed away, seven withdrew (8.9% overall dropout rate) and two were lost to follow-up.
In the raw analyses, the asthmatic and COPD patients presented poorer cognitive function than the control group, as revealed by MMSE, AMTS and CDT, and a higher prevalence of depressive symptoms according to the GDS and HAM-D. However, after adjusting for potential confounders, only borderline differences were found between asthmatic patients and controls with regard to depressive symptoms (GDS p=0.049), suggesting that asthma itself might predispose to depression (Table 1; Figure 2).
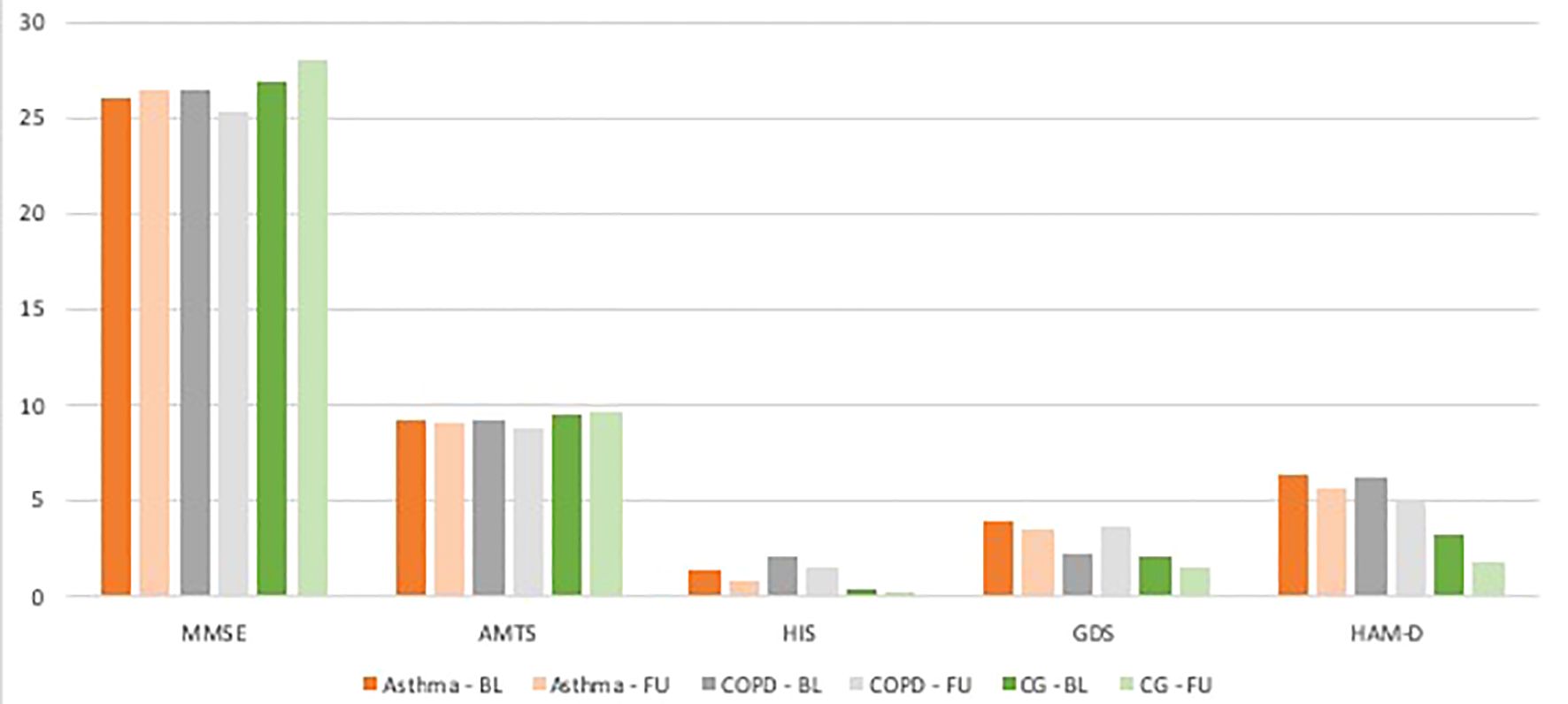
Figure 2 Mean adjusted results for each test within every group at the baseline and follow up. BL - baseline, FU - follow up.
No notable differences were found between the asthma and COPD groups in terms of cognitive function tests or psychiatric assessments: no significant differences in raw and adjusted MMSE, AMTS, HIS, CTD, GDS, and HAM-D scored were observed between the two groups.
Before comparing the two time points, a test-retest analysis was conducted. The correlation of values between the two time points was calculated for MMSE (r=0.66), AMTS (r=0.56), HIS (r=0.57), CDT (r=0.73), GDS (r=0.77), and HAM-D (r=0.62). A significant result (p<0.05) was noted for the HAM-D results (p=0.036). This value suggests an improvement in psychiatric state over the test period.
The results of the questionnaires were assessed both within the entire study population and within each group at the first measurement point.
Regarding the asthma-specific tests, the ACT TM exhibited a moderate significant correlation with MMSE (R=0.598), AMTS (R=0.349), HIS (R=-0.405), GDS (R=-0.470), and HAM-D (R=- 0.380) in the study population. Within the asthma group, the only significant correlations of ACT TM were observed with direct assessment of cognitive functions (MMSE R=0.705, AMTS R=0.460, HIS R=-0.643). The ACQ correlated negatively with MMSE (R=-0.630) and AMTS (R=-0.362) in the general population, but positively with HIS (R=0.417), GDS (R=0.425), and HAM-D (R=0.369); however, in the asthma group, the ACQ only displayed a significant correlation with neurocognitive assessment (R=-0.612, R=-0.498, R=0.622 for MMSE, AMTS and HIS, respectively). Lastly, the AQLQ(S) was found to correlate with all questionnaires in the overall study population; within the asthma group, it correlated with HIS (R=-0.467), GDS (R=-0.511) and HAM-D (R=-0.489).
The COPD-specific tests, namely CAT, mMRC and SGRQ-C, were found to correlate with MMSE, AMTS, GDS, and HAM-D in the broader study population. In the COPD subgroup, the only significant relationships were found between CAT and MMSE (R=-0.542), HIS (R=0.433) or HAM-D (R=0.453) (Table 2).
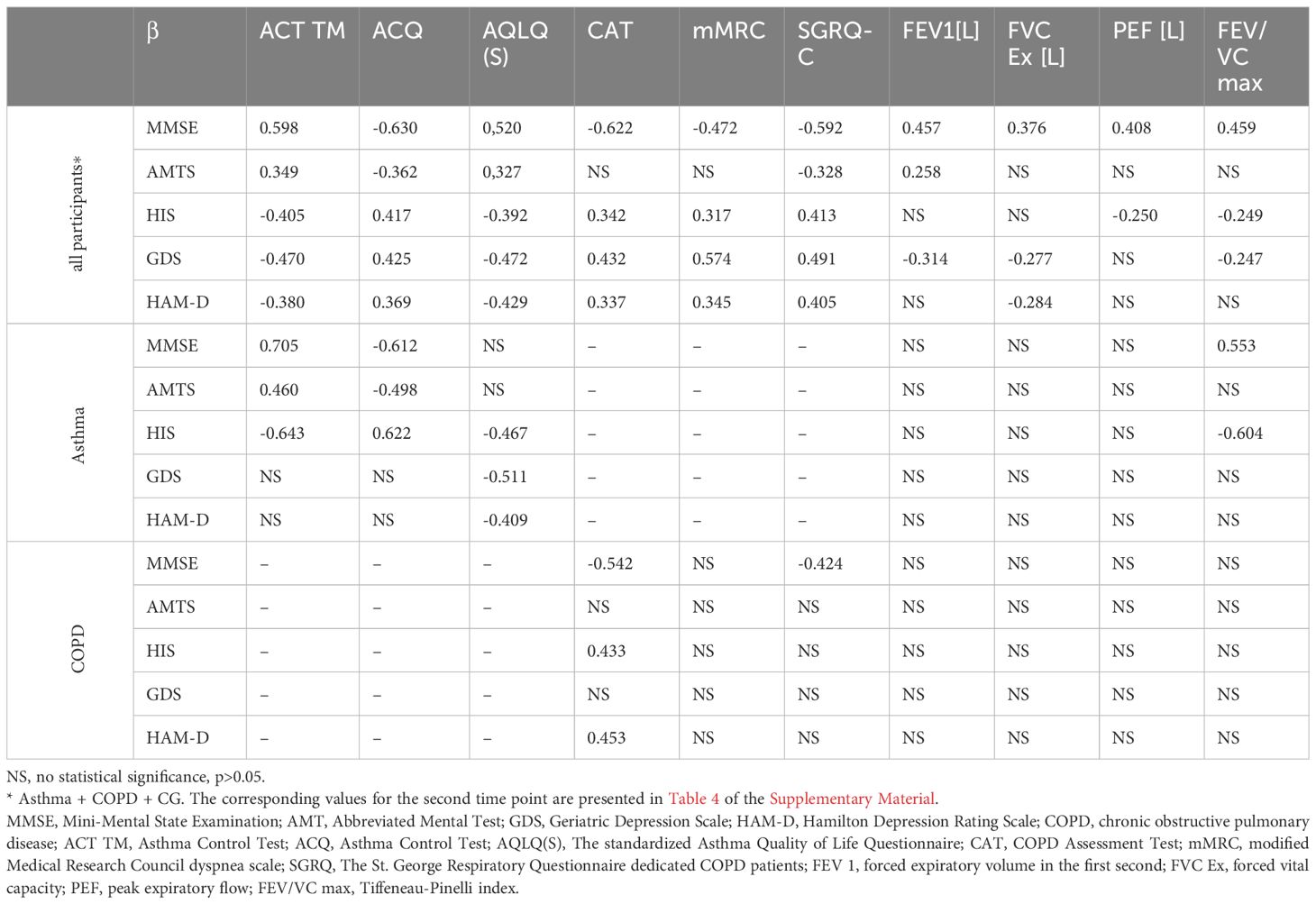
Table 2 The correlations at the first time point.
Furthermore, a baseline only 17 participants were able to accurately draw the clock in the asthma group, and only 13 patients in the COPD group. In contrast, 25 individuals in the control group executed the task correctly. Both comparisons with controls were significant (asthma vs. CG p=0.0055, COPD vs. CG p=0.0006).
Functional lung examination parameters, such as forced expiratory volume in the first second (FEV1), forced vital capacity (FVC Ex), peak expiratory flow (PEF), and the Tiffeneau-Pinelli index (FEV/VC max), correlated with all neuropsychiatric tests (MMSE, AMTS, GDS, and HAM-D) in the broader study population. However, the only notable relationship within the asthma group was between FEV/VC max and MMSE (R=0.553) or HIS (R=-0.604), and no correlation was found in the COPD group.
No correlation was found between the first and second time points for CREB mRNA expression at Supplementary Materials, Table 2; however, a 2.3-fold increase in CREB mRNA expression was observed between the two time points in study population (p=0.014). This change did not differ significantly between groups. When data from both time points were combined, a significant association was found between CREB and MMSE (β=0.166, p=0.048) (Table 3). In a model adjusted for covariates, a borderline interaction was noted between diagnosis and CREB in predicting MMSE (p=0.055). CREB mRNA level was significantly associated with MMSE in the COPD group (β=0.273, p=0.034), but this association was not significant in other groups. Among the other tests, CREB mRNA expression was significantly associated with AMTS, but only in the COPD group (p=0.018, β=0.333), and this association remained significant in the covariate-adjusted model (p=0.024, β=0.327).
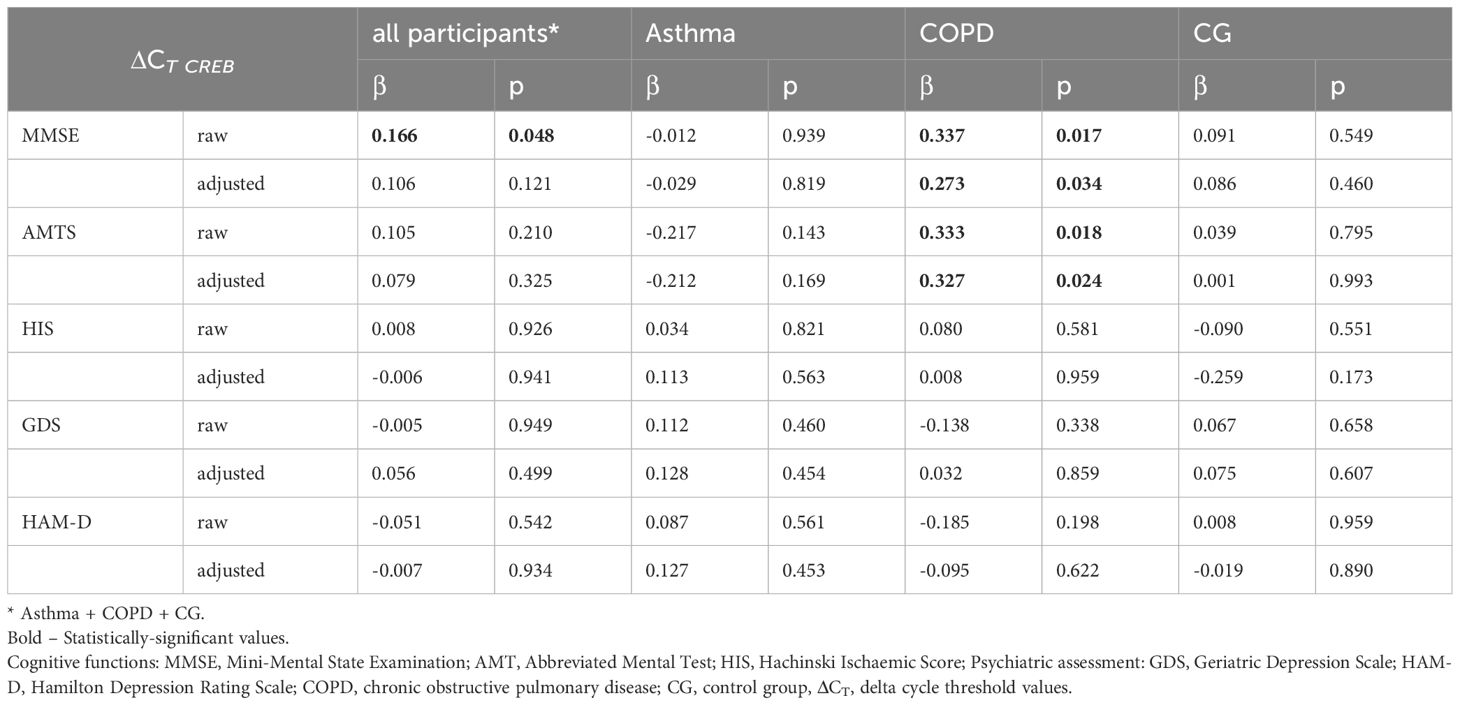
Table 3 The relationships between pooled results from cognitive function tests and psychiatric assessment scores from both time-points and mRNA expression of the CREB gene.
A logistic regression was conducted to identify whether the CDT results (Table 4), as an indicator of cognitive dysfunction, depended on the mRNA expression for both genes. All results obtained were statistically insignificant (p>0.05). Contrary to the previously described associations between CREB and MMSE or AMTS, no relationship was found between CREB and CDT in any group. In addition, no significant associations were found between CDT and PKA mRNA expression.
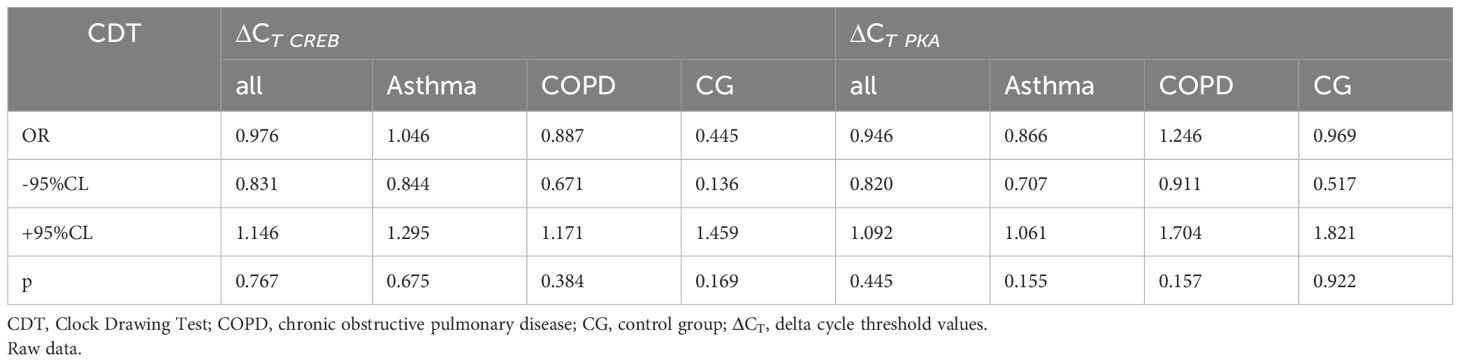
Table 4 The results of logistic regression of the associations between pooled CDT test results and ΔCT CREB and ΔCT PKAexpression between research cohorts.
PKA mRNA expression was compared between the two time points. A significant correlation was observed only in the COPD group (p=0.049, β=-0.4157). Intriguingly, no significant change in PKA mRNA expression was noted between these points, mirroring the findings for CREB expression. In addition, no differences in PKA mRNA expression were identified between groups. Additionally, PKA mRNA level was not linked to any cognitive function test or psychiatric assessment (Table 5). Even when pooling the results, no statistical significance emerged. Similarly, the covariate-adjusted model yielded no significant findings.
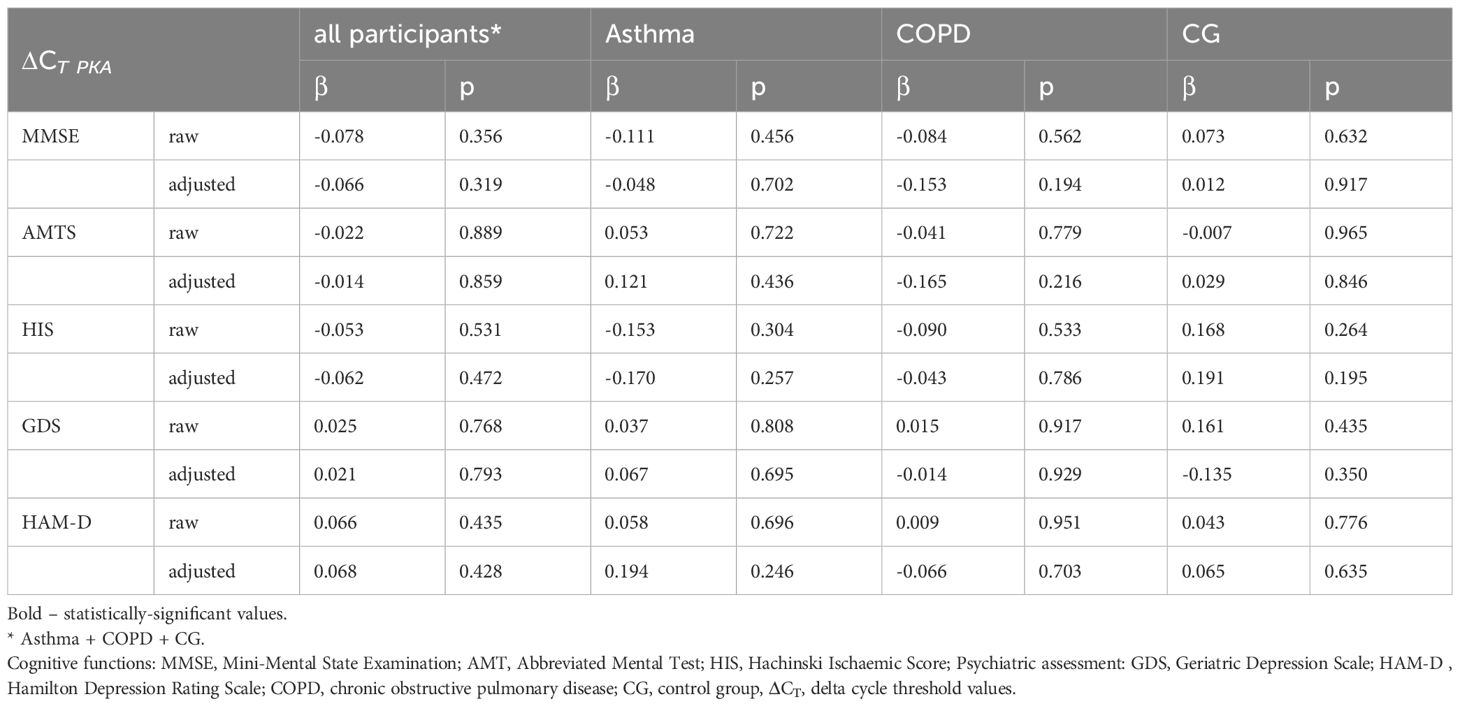
Table 5 Summary of correlations between combined cognitive test results and psychiatric evaluations in groups organized by ΔCT PKA expression level in the studied cohorts.
Remarkably, a relationship between PKA and CREB mRNA expression was evident at both the initial time point (p=0.000, β=0.610) and the later one (p=0.001, β=0.411). At baseline, a significant association between PKA and CREB was observed in the COPD group (p=0.000, β=0.929); however, at the second time point, a significant association was noted for both the asthma group (p=0.016, β=0.506) and the COPD group (p=0.028, β=0.458).
At first glance, chronic obstructive inflammatory pulmonary diseases appear to be a heterogeneous group with diversified endotypes. For example, asthma and COPD are characterized by different risk factors, and while inflammation may be the primary underlying mechanism in both diseases, the two differ with regard to the cells involved.
Several studies have examined whether inflammation might be a key factor influencing neurodegeneration in the hippocampus, and their findings suggest that better respiratory function in mid-life may be linked to a reduced risk of developing neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, mild cognitive impairment, frontotemporal dementia or lewy body dementia in later life (31). Indeed, other atopic conditions such as eczema and rhinitis have also been associated with a modest increase in the risk of neurodegenerative dementia which was not attributed to vascular disease or genetic factors (65); however, the frequency of asthma was not found to be higher among a cohort of dementia patients compared to controls (66).
A national cohort study also indicated a relationship between poor pulmonary function and cognitive impairment (31), which was also supported by pathomorphological examinations. Also, chronic asthma has been shown to lead to damage in the hippocampal ultrastructure of mice (67). While a temporary state of hypoxia directly followed by reoxia has been found to increase activation of MAPK (68, 69), there is currently no data about the effect of long-term hypoxia accompanied by inflammation on brain tissue, nor on the molecular link between asthma or COPD, being typical chronic obstructive lung diseases, and cognitive dysfunction. As such, the present article is one of the first to look for the association between proteins and these conditions in the neurodegenerative processes; it is also the first to prospectively monitor patients with chronic inflammatory diseases and consider the results in the light of data from sensitive tests for the diagnosis of dementia.
The demographic data collected at the study onset revealed initial distinctions among the groups. Notably, those with asthma tended to have lower educational attainment compared to controls, as well as lower quality of life in several domains based on the SF-36. Also, the asthma patients were more frequently employed and less prone to smoking than the COPD patients.
Further disparities emerged when cognitive functions were assessed by questionnaires. For example, a notable difference was found between the asthma and control groups concerning the deterioration of cerebrovascular parameters, as determined by AMTS and CDT; this suggests that asthma significantly hampers higher cognitive functions. Even after adjusting for the previously mentioned covariates, the results remained markedly lower for the asthma group compared to the controls. The presence of inferior cognitive function in asthma patients compared to controls raises the possibility that this defect may be associated with chronic obstructive lung disease. Indeed, no significant distinction in cognitive ability was noted between the asthma and COPD groups, suggesting that the nature of inflammation in chronic obstructive pulmonary disease does not play a pivotal role in neurodegenerative progression. This insight is clinically significant.
Additionally, no clear differences in cognitive functions, assessed using the MMSE tool, were found between the asthma and CG groups. Furthermore, while asthma and COPD progress along different inflammatory pathways, neither seems to influence cognitive dysfunction or neurodegeneration; in addition, neither condition appears to be associated with cognitive disturbances or increased risk of neurodegeneration. To elucidate these connections, more comprehensive research is warranted.
Among the asthma group, diminished cognitive function seems to manifest primarily as depression. The psychiatric status of the studied patients varied over the observation period, with no clear improvement in cognition identified based on the HAM-D outcomes. Interestingly, no association of mental status with any specific disease could be discerned from these results. Perplexingly, no notable shift in cognitive function was observed between the first and second time points for either asthma or COPD patients. One possible explanation for this static observation could be the relatively short duration of the study, suggesting that a more extended observation period might provide clearer insights.
Two molecular factors associated with the development of Parkinson’s disease (70), were selected for study as potential indicators of neurodegeneration in chronic obstructive inflammatory pulmonary disease: PKA and CREB. These were identified by a detailed analysis of the GLP-1 agonist mechanism (16). PKA mRNA levels did not exhibit any correlation with cognitive dysfunction. In contrast, CREB mRNA emerged as a potential marker of cognitive decline, specifically in COPD patients. Limitations of this study include the consideration of only two signaling factors in the GLP-1R signaling pathway. Analyzing all possible biomarkers exceeds the scope of this work and will be included in future prospective studies. Another limitation to note is the pilot nature of the study. Despite estimating the minimum sample size using an ANOVA model with a medium effect size (Cohen’s f = 0.25), the feasible number of subjects was recruited. This could lead to underpowered findings and increases the risk of type II errors. Consequently, detecting more subtle effects and differences might be hinder. Additionally, due to the small size of the groups, age-matched controls could not be provided. To minimize the impact of age discrepancies, all analyses were age-adjusted.
There is a need for more extensive and prolonged research to identify more potential markers of neurodegeneration among those associated with inflammation, a more extensive and prolonged study might be necessary.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Bioethics committee of Medical University of Lodz. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
MF: Conceptualization, Investigation, Methodology, Project administration, Resources, Visualization, Writing – original draft, Writing – review & editing. AW: Investigation, Resources, Writing – review & editing. JP: Investigation, Resources, Writing – review & editing. JD: Project administration, Resources, Writing – review & editing. SM: Project administration, Resources, Writing – review & editing. MK: Data curation, Formal analysis, Validation, Writing – review & editing. PK: Funding acquisition, Project administration, Writing – review & editing. MP: Conceptualization, Methodology, Project administration, Supervision, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research project was funded through the second edition of the “Student-Scientist” scholarship from the Medical University in Lodz, Poland.
We offer our acknowledgements to all of those who supported us in any respect during the completion of this project. This study was financially supported by grants from the Department of Internal Diseases, Asthma and Allergy, II Chair of Internal Diseases, Medical University of Lodz and from the Medical University of Lodz, Poland; these were awarded as part of the financial framework for the development of young scientists and PhD students.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1363373/full#supplementary-material
1. Barnes PJ. Cellular and molecular mechanisms of asthma and COPD. Clin Sci. (2017) 131:1541–58. doi: 10.1042/CS20160487
2. Renauld JC. New insights into the role of cytokines in asthma. J Clin Pathol. (2001) 54:577. doi: 10.1136/jcp.54.8.577
3. Kleniewska P, Pawliczak R. The participation of oxidative stress in the pathogenesis of bronchial asthma. Biomed Pharmacother. (2017) 94:100–8. doi: 10.1016/j.biopha.2017.07.066
4. Lambrecht BN, Hammad H, Fahy JV. The cytokines of asthma. Immunity. (2019) 50:975–91. doi: 10.1016/j.immuni.2019.03.018
5. Barnes PJ. Mechanisms in COPD: differences from asthma. Chest. (2000) 117:10S–4S. doi: 10.1378/chest.117.2_suppl.10S
6. Barnes PJ, Adcock IM. Transcription factors and asthma. Eur Respir J. (1998) 12:221–34. doi: 10.1183/09031936.98.12010221
8. Brightling C, Greening N. Airway inflammation in COPD: progress to precision medicine. Eur Respir J. (2019) 54. doi: 10.1183/13993003.00651-2019. SERIES CONTROVERSIES IN COPD.
9. Sethi S, Mahler DA, Marcus P, Owen CA, Yawn B, Rennard S. Inflammation in COPD: implications for management. Am J Med. (2012) 125:1162–70. doi: 10.1016/j.amjmed.2012.06.024
10. Chiappara G, Chanez P, Bruno A, Pace E, Pompeo F, Bousquet J, et al. Variable p-CREB expression depicts different asthma phenotypes. Allergy. (2007) 62:787–94. doi: 10.1111/j.1398-9995.2007.01417.x
11. Hołownia A, Drost EM, Braszko JJ, Mroz RM, Chyczewska E, Noparlik J, et al. Cytoplasm-nuclear trafficking of CREB and CREB phosphorylation at Ser133 during therapy of chronic obstructive pulmonary disease. J Physiol Pharmacol: an Off J Polish Physiol Soc. (2007) 58 Suppl 5(Pt 2):437–44. Participation of D1-4 dopamine receptors in the pro-cognitive effects of angiotensin IV View project Mechanism of cell death (apoptosis) in mesothelioma cells View project CYTOPLASM-NUCLEAR TRAFFICKING OF CREB AND CREB PHOSPHORYLATION AT SER133 DURING THERAPY OF CHRONIC.
12. Musa NL, Ramakrishnan M, Li J, Kartha S, Liu P, Pestell RG, et al. Forskolin inhibits cyclin D1 expression in cultured airway smooth-muscle cells. Am J Respir Cell Mol Biol. (1999) 20:352–8. doi: 10.1165/ajrcmb.20.2.3160
13. Wu Y, Lu Y, Zou F, Fan X, Li X, Zhang H, et al. PTEN participates in airway remodeling of asthma by regulating CD38/Ca2+/CREB signaling. Aging (Albany NY). (2020) 12:16326. doi: 10.18632/aging.v12i16
14. Luciani P, Deledda C, Benvenuti S, Cellai I, Squecco R, Monici M, et al. Differentiating effects of the glucagon-like peptide-1 analogue exendin-4 in a human neuronal cell model. Cell Mol Life Sci. (2010) 67:3711–23. doi: 10.1007/s00018-010-0398-3
15. Klinger S, Poussin C, Debril MB, Dolci W, Halban PA, Thorens B. Increasing GLP-1-induced β-cell proliferation by silencing the negative regulators of signaling cAMP response element modulator-α and DUSP14. Diabetes. (2008) 57:584–93. doi: 10.2337/db07-1414
16. Figat M, Kardas G, Kuna P, Panek MG. Beneficial influence of exendin-4 on specific organs and mechanisms favourable for the elderly with concomitant obstructive lung diseases. Brain Sci. from MDPI (2022) 12:1090. doi: 10.3390/brainsci12081090
17. Chaudhuri A, Ghanim H, Vora M, Sia CL, Korzeniewski K, Dhindsa S, et al. Exenatide exerts a potent antiinflammatory effect. J Clin Endocrinol Metab. (2012) 97:198–207. doi: 10.1210/jc.2011-1508
18. Zhu T, Wu XL, Zhang W, Xiao M. Glucagon like peptide-1 (GLP-1) modulates OVA-induced airway inflammation and mucus secretion involving a protein kinase A (PKA)-dependent nuclear factor-κB (NF-κB) signaling pathway in mice. Int J Mol Sci. (2015) 16:20195–211. doi: 10.3390/ijms160920195
19. Oldenburger A, Roscioni SS, Jansen E, Menzen MH, Halayko AJ, Timens W, et al. Anti-inflammatory role of the cAMP effectors epac and PKA: implications in chronic obstructive pulmonary disease. PloS One. (2012) 7:e31574. doi: 10.1371/journal.pone.0031574
20. Yan H, Deshpande DA, Misior AM, Miles MC, Saxena H, Riemer EC, et al. Anti-mitogenic effects of β-agonists and PGE2 on airway smooth muscle are PKA dependent. FASEB J. (2011) 25:389–97. doi: 10.1096/FJ.10-164798
21. Mitchell PD, Salter BM, Oliveria JP, El-Gammal A, Tworek D, Smith SG, et al. Glucagon-like peptide-1 receptor expression on human eosinophils and its regulation of eosinophil activation. Clin Exp Allergy. (2017) 47:331–8. doi: 10.1111/cea.12860
22. Sun YH, He L, Yan MY, Zha RQ, Li B, Wang F, et al. Overexpression of GLP-1 receptors suppresses proliferation and cytokine release by airway smooth muscle cells of patients with chronic obstructive pulmonary disease via activation of ABCA1. Mol Med Rep. (2017) 16:929–36. doi: 10.3892/MMR.2017.6618/HTML
23. Skurikhin EG, Pershina OV, Pakhomova AV, Pan ES, Krupin VA, Ermakova NN, et al. Endothelial progenitor cells as pathogenetic and diagnostic factors, and potential targets for GLP-1 in combination with metabolic syndrome and chronic obstructive pulmonary disease. Int J Mol Sci. (2019) 20:1105. doi: 10.3390/ijms20051105
24. Peters MC, Fahy JV. Metabolic consequences of obesity as an “outside in” mechanism of disease severity in asthma. Eur Respir J. (2016) 48:291. doi: 10.1183/13993003.01132-2016
25. Peters M, McGrath K, Hawkins G, Hastie A, Levy B, Israel E, et al. Plasma IL6 levels, metabolic dysfunction, and asthma severity: a cross-sectional analysis of two cohorts. Lancet Respir Med. (2016) 4:574. doi: 10.1016/S2213-2600(16)30048-0
26. Meraz-Ríos MA, Toral-Rios D, Franco-Bocanegra D, Villeda-Hernández J, Campos-Peña V. Inflammatory process in alzheimer’s disease. Front Integr Neurosci. (2013) 0:59/BIBTEX. doi: 10.3389/FNINT.2013.00059/BIBTEX
27. Schwab C, McGeer PL. Inflammatory aspects of alzheimer disease and other neurodegenerative disorders. J Alzheimer’s Dis. (2008) 13:359–69. doi: 10.3233/JAD-2008-13402
28. Sue W, Griffin T, Barger SW, Chairman V, Reynolds DW. Neuroinflammatory cytokines-the common thread in alzheimer’s pathogenesis. US Neurol. (2010) 6(2):19–27.
29. Vural P, Değirmenciolu S, Parildar-Karpuzoğlu H, Doǧru-Abbasoǧlu S, Hanagasi H, Karadaǧ B, et al. The combinations of TNFalpha-308 and IL-6 -174 or IL-10 -1082 genes polymorphisms suggest an association with susceptibility to sporadic late-onset Alzheimer’s disease. Acta Neurol Scand. (2009) 120:396–401. doi: 10.1111/j.1600-0404.2009.01230.x
30. Rosenkranz MA, Dean DC, Bendlin BB, Jarjour NN, Esnault S, Zetterberg H, et al. Neuroimaging and biomarker evidence of neurodegeneration in asthma. J Allergy Clin Immunol. (2021) 149(2):589–98.e6. doi: 10.1016/J.JACI.2021.09.010
31. Peng YH, Wu BR, Su CH, Liao WC, Muo CH, Hsia TC, et al. Adult asthma increases dementia risk: a nationwide cohort study. J Epidemiol Community Health (1978). (2015) 69:123–8. doi: 10.1136/jech-2014-204445
32. Chen MH, Li CT, Tsai CF, Lin WC, Chang WH, Chen TJ, et al. Risk of dementia among patients with asthma: a nationwide longitudinal study. J Am Med Dir Assoc. (2014) 15:763–7. doi: 10.1016/j.jamda.2014.06.003
33. Rusanen M, Ngandu T, Laatikainen T, Tuomilehto J, Soininen H, Kivipelto M. Chronic obstructive pulmonary disease and asthma and the risk of mild cognitive impairment and dementia: a population based CAIDE study. Curr Alzheimer Res. (2013) 10:549–55. doi: 10.2174/1567205011310050011
34. Bozek A, Jarzab J. Improved activity and mental function related to proper antiasthmatic treatment in elderly patients with Alzheimer’s disease. Allergy Asthma Proc. (2011) 32:341–5. doi: 10.2500/aap.2011.32.3459
35. Bożek A, Bednarski P, Jarzab J. Allergic rhinitis, bronchial asthma and other allergies in patients with Alzheimer’s disease: unnoticed issue. Adv Dermatol Allergol. (2016) 5:353–8. doi: 10.5114/ada.2016.62842
36. Lampela P, Tolppanen AM, Koponen M, Tanskanen A, Tiihonen J, Hartikainen S, et al. Asthma and chronic obstructive pulmonary disease as a comorbidity and association with the choice of antidementia medication among persons with alzheimer’s disease. J Alzheimers Dis. (2020) 73:1243–51. doi: 10.3233/JAD-190850
37. Venkatesan P. GOLD COPD report: 2024 update. Lancet Respir Med. (2024) 12:15–6. doi: 10.1016/S2213-2600(23)00461-7
38. Venkatesan P. 2023 GINA report for asthma. Lancet Respir Med. (2023) 11:589. doi: 10.1016/S2213-2600(23)00230-8
39. Agustí A, Celli BR, Criner GJ, Halpin D, Anzueto A, Barnes P, et al. Global initiative for chronic obstructive lung disease 2023 report: GOLD executive summary. Am J Respir Crit Care Med. (2023) 207:819–37. doi: 10.1164/RCCM.202301-0106PP/SUPPL_FILE/DISCLOSURES.PDF
40. Stanojevic S, Kaminsky DA, Miller M, Thompson B, Aliverti A, Barjaktarrevic I, et al. Early View Task force report ERS/ATS technical standard on interpretive strategies for routine lung function tests ERS/ATS Technical Standard on Interpretive Strategies for Routine Lung Function Tests. Eur Respir J. (2022) 60. doi: 10.1183/13993003.01499-2021
41. Ware JE, Kosinski M. Interpreting SF-36 summary health measures: A response. Qual Life Res. (2001) 10:405–13. doi: 10.1023/A:1012588218728/METRICS
42. Metodyka oceny jakości życia . Turska, Wioletta: FBC. Available online at: https://fbc.pionier.net.pl/details/nnlkd26 (Accessed April 13, 2023).
43. Pezzotti P, Scalmana S, Mastromattei A, Di Lallo D. The accuracy of the MMSE in detecting cognitive impairment when administered by general practitioners: A prospective observational study. BMC Fam Pract. (2008) 9:1–11. doi: 10.1186/1471-2296-9-29/TABLES/2
44. Folstein MF, Folstein SE, McHugh PR. Mini-mental state”: A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. (1975) 12:189–98. doi: 10.1016/0022-3956(75)90026-6
45. Cullen B, O’Neill B, Evans JJ, Coen RF, Lawlor BA. A review of screening tests for cognitive impairment. J Neurol Neurosurg Psychiatry. (2007) 78:790. doi: 10.1136/jnnp.2006.095414
46. Piotrowicz K, Romanik W, Skalska A, Gryglewska B, Szczerbińska K, Derejczk J, et al. The comparison of the 1972 Hodkinson’s Abbreviated Mental Test Score (AMTS) and its variants in screening for cognitive impairment. Aging Clin Exp Res. (2019) 31:561–6. doi: 10.1007/S40520-018-1009-7/TABLES/2
47. Pantoni L, Inzitari D. Hachinski’s ischemic score and the diagnosis of vascular dementia: A review. Ital J Neurological Sci. (1993) 14:7. doi: 10.1007/BF02339212
48. Rosen WG, Terry RD, Fuld PA, Katzman R, Peck A. Pathological verification of ischemic score in differentiation of dementias. Ann Neurol. (1980) 7:486–8. doi: 10.1002/ana.410070516
49. Moroney JT, Bagiella E, Desmond DW, Hachinski VC, Mölsä PK, Gustafson L, et al. Meta-analysis of the Hachinski Ischemic Score in pathologically verified dementias. Neurology. (1997) 49:1096–105. doi: 10.1212/WNL.49.4.1096
50. Shulman KI, Shedletsky R, Silver IL. The challenge of time: Clock-drawing and cognitive function in the elderly. Int J Geriatr Psychiatry. (1986) 1:135–40. doi: 10.1002/gps.930010209
51. Paganini-Hill A, Clark LJ, Henderson VW, Birge SJ. Clock drawing: analysis in a retirement community. J Am Geriatr Soc. (2001) 49:941–7. doi: 10.1046/j.1532-5415.2001.49185.x
52. Korczyn AD, Halperin I. Depression and dementia. J Neurol Sci. (2009) 283:139–42. doi: 10.1016/j.jns.2009.02.346
53. Leonard BE. Inflammation, depression and dementia: Are they connected? Neurochem Res. (2007) 32:1749–56. doi: 10.1007/S11064-007-9385-Y/TABLES/1
54. Enache D, Winblad B, Aarsland D. Depression in dementia: Epidemiology, mechanisms, and treatment. Curr Opin Psychiatry. (2011) 24:461–72. doi: 10.1097/YCO.0b013e32834bb9d4
55. Almeida OP, Almeida SA, Almeida OP. Short versions of the geriatric depression scale: a study of their validity for the diagnosis of a major depressive episode according to ICD-10 and DSM-IV. Int J OF GERIATRIC Psychiatry Int J Geriat Psychiatry. (1999) 14:858–65. doi: 10.1002/(ISSN)1099-1166
56. D’Ath P, Katona P, Mullan E, Evans S, Katona C. Screening, detection and management of depression in elderly primary care attenders. I: The acceptability and performance of the 15 item Geriatric Depression Scale (GDS15) and the development of short versions. Fam Pract. (1994) 11:260–6. doi: 10.1093/fampra/11.3.260
57. Brown LM, Schinka JA. Development of initial validation of a 15-item informant version of the Geriatric Depression Scale. Int J Geriatr Psychiatry. (2005) 20:911–8. doi: 10.1002/(ISSN)1099-1166
58. Nixon N, Guo B, Garland A, Kaylor-Hughes C, Nixon E, Morriss R. The bi-factor structure of the 17-item Hamilton Depression Rating Scale in persistent major depression; dimensional measurement of outcome. PloS One. (2020) 15(10):e0241370. doi: 10.1371/journal.pone.0241370
59. Cole JC, Motivala SJ, Dang J, Lucko A, Lang N, Levin MJ, et al. Structural validation of the hamilton depression rating scale. J Psychopathol Behav Assess. (2004) 26:4. doi: 10.1023/B:JOBA.0000045340.38371.04
60. Green MR, Sambrook J. Quantifying and storing RNA. Cold Spring Harb Protoc. (2020) 2020:pdb.top101709. doi: 10.1101/pdb.top101709
61. Xiao J, Li X, Liu J, Fan X, Lei H, Li C. Identification of reference genes in blood before and after entering the plateau for SYBR green RT-qPCR studies. PeerJ. (2017) 5:e3726. doi: 10.7717/peerj.3726
62. Buist AS, McBurnie MA, Vollmer WM, Gillespie S, Burney P, Mannino DM, et al. International variation in the prevalence of COPD (the BOLD Study): a population-based prevalence study. Lancet. (2007) 370:741–50. doi: 10.1016/S0140-6736(07)61377-4
63. Abramson MJ, Perret JL, Dharmage SC, McDonald VM, McDonald CF. Distinguishing adult-onset asthma from COPD: a review and a new approach. Int J Chron Obstruct Pulmon Dis. (2014) 9:945. doi: 10.2147/COPD.S46761
64. Covariate selection, in: Developing a Protocol for Observational Comparative Effectiveness Research: A User’s Guide. NCBI Bookshelf. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK126194/ (Accessed March 29, 2022).
65. Eriksson UK, Gatz M, Dickman PW, Fratiglioni L, Pedersen NL. Asthma, eczema, rhinitis and the risk for dementia. Dement Geriatr Cognit Disord. (2008) 25:148–56. doi: 10.1159/000112729
66. Kim SY, Min C, Oh DJ, Choi HG. Risk of neurodegenerative dementia in asthma patients: a nested case-control study using a national sample cohort. BMJ Open. (2019) 9(10):e030227. doi: 10.1136/bmjopen-2019-030227
67. Guo RB, Sun PL, Zhao AP, Gu J, Ding X, Qi J, et al. Chronic asthma results in cognitive dysfunction in immature mice. Exp Neurol. (2013) 247:209–17. doi: 10.1016/j.expneurol.2013.04.008
68. Padmasekar M, Lingwal N, Samikannu B, Chen C, Sauer H, Linn T. Exendin-4 protects hypoxic islets from oxidative stress and improves islet transplantation outcome. Endocrinology. (2013) 154:1424–33. doi: 10.1210/en.2012-1983
69. Lu K, Chang G, Ye L, Zhang P, Li Y, Zhang D. Protective effects of extendin-4 on hypoxia/reoxygenation-induced injury in H9c2 cells. Mol Med Rep. (2015) 12:3007–16. doi: 10.3892/mmr.2015.3682
Keywords: neurodegeneration, asthma, COPD, cognition, PKA, CREB, inflammation, brainlungs crosstalk
Citation: Figat M, Wiśniewska A, Plichta J, Miłkowska-Dymanowska J, Majewski S, Karbownik MS, Kuna P and Panek MG (2024) Potential association between obstructive lung diseases and cognitive decline. Front. Immunol. 15:1363373. doi: 10.3389/fimmu.2024.1363373
Received: 30 December 2023; Accepted: 05 July 2024;
Published: 22 July 2024.
Edited by:
Hongxiang Chen, Huazhong University of Science and Technology, ChinaReviewed by:
Gonzalo Emiliano Aranda-Abreu, Universidad Veracruzana, MexicoCopyright © 2024 Figat, Wiśniewska, Plichta, Miłkowska-Dymanowska, Majewski, Karbownik, Kuna and Panek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magdalena Figat, bWFnZGFsZW5hLmZpZ2F0QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.