 Subburaj Ilangumaran
Subburaj Ilangumaran Yirui Gui
Yirui Gui Akhil Shukla
Akhil Shukla Sheela Ramanathan
Sheela Ramanathan- Department of Immunology and Cell Biology, Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, QC, Canada
Suppressor of cytokine signaling 1 (SOCS1) is a potent regulator immune cell responses and a proven tumor suppressor. Inhibition of SOCS1 in T cells can boost antitumor immunity, whereas its loss in tumor cells increases tumor aggressivity. Investigations into the tumor suppression mechanisms so far focused on tumor cell-intrinsic functions of SOCS1. However, it is possible that SOCS1 expression in tumor cells also regulate antitumor immune responses in a cell-extrinsic manner via direct and indirect mechanisms. Here, we discuss the evidence supporting the latter, and its implications for antitumor immunity.
1 SOCS1-dependent checkpoints in innate and adaptive immune responses
SOCS1 is a negative feedback regulator of cytokine-induced Janus kinase (JAK)-Signal transducer and activation of transcription (STAT) signaling pathway and is the founding member of the SOCS protein family that contains eight members, namely, SOCS1 to SOCS7 and CISH (1–4). All SOCS family proteins have a central Src homology 2 (SH2) domain and a conserved SOCS box at the carboxy terminus. The SH2 domain interacts with phosphorylated JAKs and other phospho-tyrosine containing proteins, whereas the SOCS box promotes ubiquitination of many SOCS1-binding proteins for subsequent degradation by proteasomes (5, 6). Even though SOCS1 was discovered as an inhibitor of IL-6 signaling, generation of SOCS1-deficient mice revealed its indispensable role in attenuating IFNγ signaling and consequent inflammatory responses (1–3, 7, 8). Reversal of IFNγ-driven perinatal lethality in SOCS1-deficient mice through ablation of the Rag2 gene indicated that T lymphocytes are the major producer of IFNγ in these mice (7). SOCS1-dependent regulation of T cell activation is manifested predominantly in the CD8+ T cell compartment than in the CD4+ compartment (9–12). CD8+ T cells isolated from SOCS1-deficient mice display a memory-like phenotype and show increased sensitivity to the IL-2 family cytokines, especially IL-15 (11–15). T regulatory cells, which rely on IL-2 for survival and homeostasis, naturally downmodulate SOCS1 expression through constitutive expression of miR-155 that targets the Socs1 transcript (16).
Aberrant activation of CD8+ T cells and their exuberant production of IFNγ in SOCS1-deficient mice could arise from the compound effect of (i) excess production of inflammatory cytokines from macrophages and dendritic cells that activate T cells, and (ii) increased cytokine-driven proliferation and consequent priming for antigen stimulation. SOCS1 is essential to control Toll-like receptor 4 (TLR4) signaling induced by bacterial lipopolysaccharides (LPS), and thus macrophages and dendritic cells from SOCS1 deficient mice show increased production of inflammatory cytokines (17–20). Some of the inflammatory cytokines such as IL-16 and IL-21 can synergize with cytokines that promote T cell homeostasis such as IL-15 and IL-7 to induce antigen non-specific activation of naïve CD8+ T cells that acquire increased sensitivity to autoantigens (14, 21). We have shown that CD8+ T cells from SOCS1 deficient mice can elicit autoinflammatory disease upon recognition of cognate autoantigens (21–23). SOCS1 deficiency in myeloid and lymphoid cells has been shown to increase susceptibility to experimental inflammatory and autoimmune diseases (24, 25). Moreover, haploinsufficiency for the SOCS1 gene is associated with inflammatory syndrome and autoimmunity in human (26, 27).
The SOCS1-dependent checkpoint in T cells has been exploited to attenuate inflammatory diseases in experimental settings. A SOCS1-mimetic peptide Tkip, which resembles the peptide sequence surrounding the JAK-binding sequence of SOCS1, and similar peptides has been shown to inhibit inflammatory diseases such as experimental psoriasis and lung inflammation (28–35). In these settings, the SOCS1-mimetic peptide likely operates on both myeloid and T cells to attenuate their inflammatory responses. SOCS1-mimetic peptides can also impact parenchymal cells, as SOCS1 released from alveolar macrophages was reported to attenuate activation of airway epithelial cells (36).
SOCS1 can also modulate antitumor immunity in different ways depending on its expression level in antitumor lymphocytes and in tumor cells. It restrains the effector functions of tumor reactive CD8+ T cells as transduction of with Socs1-targeting microRNA miR-155 into these cells enhances their cytokine responsiveness resulting in improved efficiency to control tumors (37, 38). On the other hand, many lines of evidence suggest that SOCS1 expression in cancer cells can positively impact the development of antitumor immune responses, for which we present evidence in the following sections.
2 Cell-intrinsic tumor suppression mechanisms of SOCS1
Following the seminal finding that the SOCS1 gene is repressed in hepatocellular carcinoma by promoter CpG methylation, similar epigenetic and miRNA-mediated SOCS1 loss has been reported in many cancers including, neuroblastoma, myeloid leukemias and colorectal, pancreatic, breast, prostate and ovarian cancers (24, 39–49). Genetic studies demonstrating the susceptibility of SOCS1-deficient mice to develop radiation-induced leukemias and to experimental induction of hepatocellular carcinoma and colorectal cancer confirm that SOCS1 is a bona fide tumor suppressor (50–54). The tumor suppressor function of SOCS1 can be attributed to various mechanisms, which may operate in various combinations in diverse cancers in a context-dependent manner. These include attenuation of cytokine- and growth factor- induced oncogenic signaling via JAK and receptor tyrosine kinases (RTK) (55–58), potentiation of p53-mediated tumor suppressor functions (59, 60), inhibition of the paradoxical oncogenic functions of tumor suppressors such as cyclin-dependent kinase inhibitor 1A (CDKN1A; commonly known as p21Cip1/WAF1) and NFE2 Like BZIP Transcription Factor 2 (NEF2L2; commonly known as Nuclear factor erythroid 2-related factor 2 or NRF2) (52, 53, 61, 62). Moreover, SOCS1 expressed in mesenchymal cells can inhibit tumor promoting inflammatory cytokine signaling that establishes a tumor-promoting microenvironment (51, 54, 63). The cell-intrinsic tumor suppressor functions of SOCS1 were mostly gleaned from studies using overexpressed SOCS1, as endogenous SOCS1 is induced following exposure to cytokines, growth factors and myriad of other stimuli, and its expression regulated at the transcriptional and post-translational level. Among the potential tumor suppressor mechanisms of SOCS1, only the regulation of the oncogenic function of p21 is genetically proven (53), and all other mechanisms remain to be tested.
3 Evidence for modulation of antitumor immunity by SOCS1 expressed in tumor cells
The idea that SOCS1 expressed in tumor cells may influence the induction of antitumor immune response came from one of our unpublished observations. While studying the tumor suppressor functions of SOCS1, we compared the murine hepatocellular carcinoma cell line Hepa 1-6 expressing wildtype SOCS1 (Hepa-SOCS1), an SH2 domain mutant of SOCS1 (R105K) that does not inhibit IFNγ signaling (Hepa-SOCS1R) or a control vector (Hepa-Vector) (64) for their ability to form tumors. Consistent with the ability of SOCS1 to inhibit growth factor-induced RTK signaling, Hepa-SOCS1 cells showed appreciably reduced growth compared to Hepa-Vector or Hepa-SOCS1R cells in vitro, and this growth reduction was significant at higher cell densities (Figure 1A). Upon subcutaneous implantation in immuno-deficient NOD.scid.gamma (NSG) mice or immuno-competent C57BL/6 mice, Hepa-Vector cells formed large tumors, while Hepa-SOCS1 cells grew poorly in both hosts, consistent with the inhibition of mitogenic signals by SOCS1. On the other hand, Hepa-SOCS1R cells showed retarded growth in C57BL/6 mice but formed large tumors in NSG mice (Figures 1B, C). The possibility that the SOCS1R105K mutant might play a dominant negative role is unlikely because Hepa-SOCS1R cells did not grow more robustly than Hepa-Vector cells. Moreover, Hepa-SOCS1 and Hepa-SOCS1R tumors growing in C57BL/6 mice displayed more prominent inflammatory immune cell infiltrations adjacent to necrotic areas than Hepa-Vector tumors (Figure 1D). These data indicated that blocking the ability of SOCS1 to inhibit mitogenic cytokine and growth factor signaling using the R105K mutation has unmasked a hitherto unappreciated potential of SOCS1 in promoting antitumor immunity. We hypothesize that SOCS1 expressed in tumor cells could impact antitumor immune responses through multiple mechanisms that are discussed below.
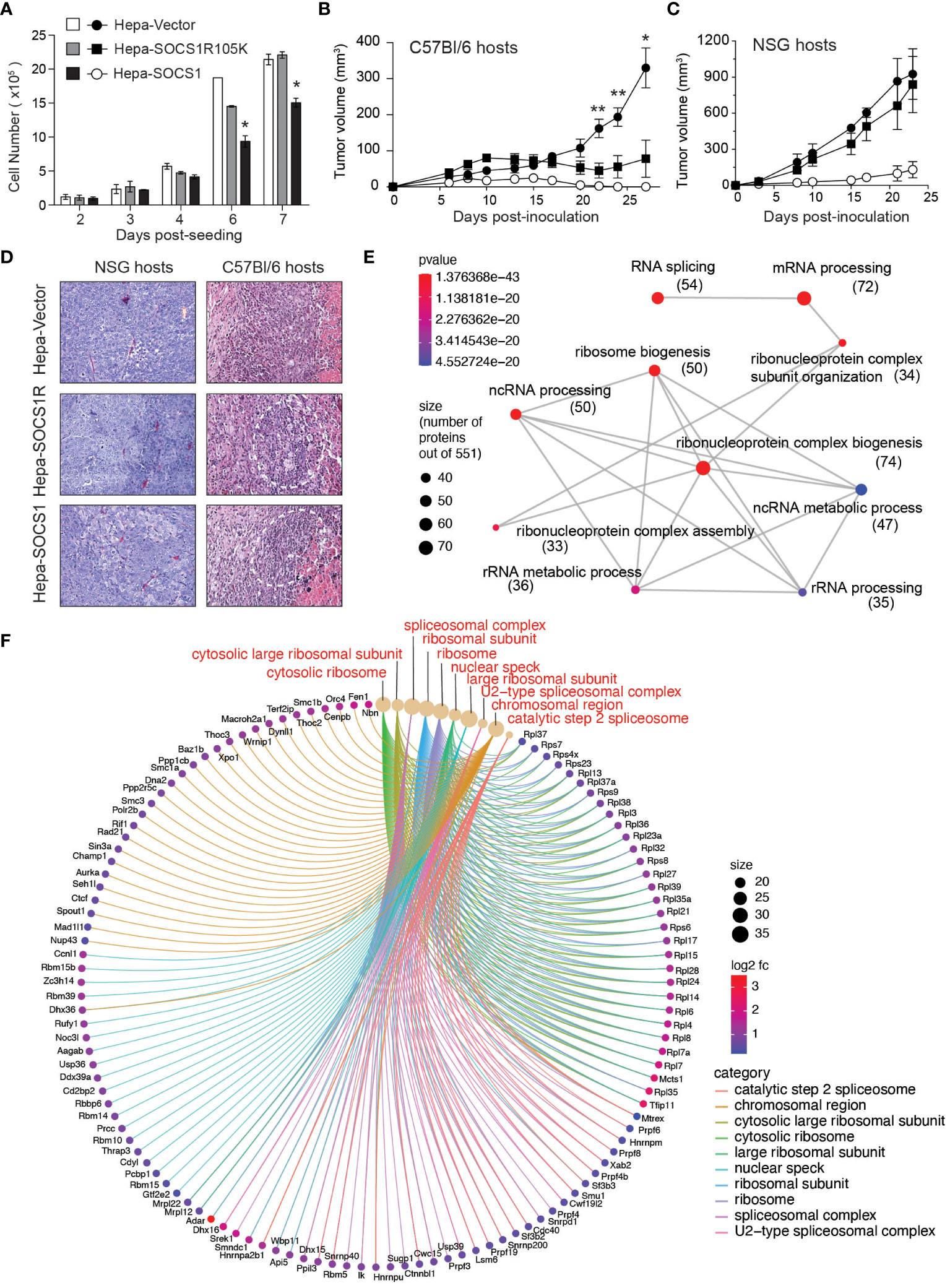
Figure 1 SOCS1R105K mutant unravels the potential role of SOCS1 in promoting antitumor immunity. (A) 1 ×105 Hepa-Vector, Hepa-SOCS1 or Hepa-SOCS1R105K cells were plated on 100 mm Petri dishes in triplicates and cultured by replenishing medium every 2 days. Cells were trypsinized on the indicated days and counted. Representative data from one of the two experiments are shown (mean ± standard error of mean; *p <0.05, Student’s t-test). (B, C) 5 ×106 Hepa-Vector, Hepa-SOCS1 or Hepa-SOCS1R105K cells were implanted subcutaneously in C57BL/6 (B) or NSG (C) mice (n=7-8) and tumor growth monitored as detailed in (56) Reference. Mean ± standard error of mean; *p <0.05, **p <0.01, Student’s t-test. (D) Representative hemoxylin and eosin-stained sections of tumors resected from NSG and C57BL/6 mice. In C57BL/6 hosts, all tumors had a central necrotic area and a reactive rim composed of infiltrating immune cells (encircles with dotted lines) that were more pronounced in Hepa-SOCS1 and Hepa-SOCS1R105K tumors than in Hepa-vector tumors. Data shown in A–C were generated from experiments approved by the Université de Sherbrooke Ethics Committee for Animal Care and Use. (E, F) Proteomes of Hepa-Vector and Hepa-SOCS1 cells were studied by tandem mass spectrometry, as detailed in (65) Reference. Proteins that were significantly upregulated in Hepa-SOCS1 cells (≥ 0.5 fold change and ≥1.3 -log10 p-Value; 551 proteins) were subjected to gene ontology analysis. Enrichment plot for biological pathways (emapplot, E) and cellular compartments (cnetplot, F) were made using the SRplot (https://www.bioinformatics.com.cn/en) data analysis tool. Number of proteins within the biological pathways are indicated in parenthesis. The mass spectrometry data used to generate Figures 1E, F are deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/archive/) with the dataset identifier PXD047908.
4 Potential anti-tumor immune functions of SOCS1 expressed in tumor cells
4.1 Promoting tumor antigen processing and presentation
Even though silencing SOCS1 in dendritic cells has been shown to enhance antigen presentation and antitumor immunity (66), SOCS1 may play an opposite role in cancer cells. Aside from binding and inhibiting JAKs and RTKs, SOCS1 functions as a substrate adaptor for protein ubiquitination that facilitates degradation by proteasomes (5, 6, 67). SOCS1 is known to interact with several signaling proteins via the central SH2 domain although other regions of SOCS1 may also be involved. The carboxy terminal of SOCS1 harbors the SOCS box domain, which is conserved among SOCS proteins and is composed of the BC box and a Cullin box. Whereas the Cullin box interacts with Cullin 5, the BC box interacts with elongin B and elongin C and brings in the RING-finger-domain-only protein RBX2 to assemble the Cullin5-RING-ubiquitin ligase CRL5SOCS1 (67–69). Others and we have shown that the ubiquitin-ligase function of SOCS1 promotes ubiquitination and proteasomal degradation of several oncogenic signaling proteins (5, 20, 52, 56, 70–76). Indeed, the list of signaling proteins regulated by SOCS1 is so diverse that the SOCS1-dependent proteasomal degradation may have a fundamental anti-tumor function that is coupled to inhibition of the oncogenic signaling pathways (Figure 2A).
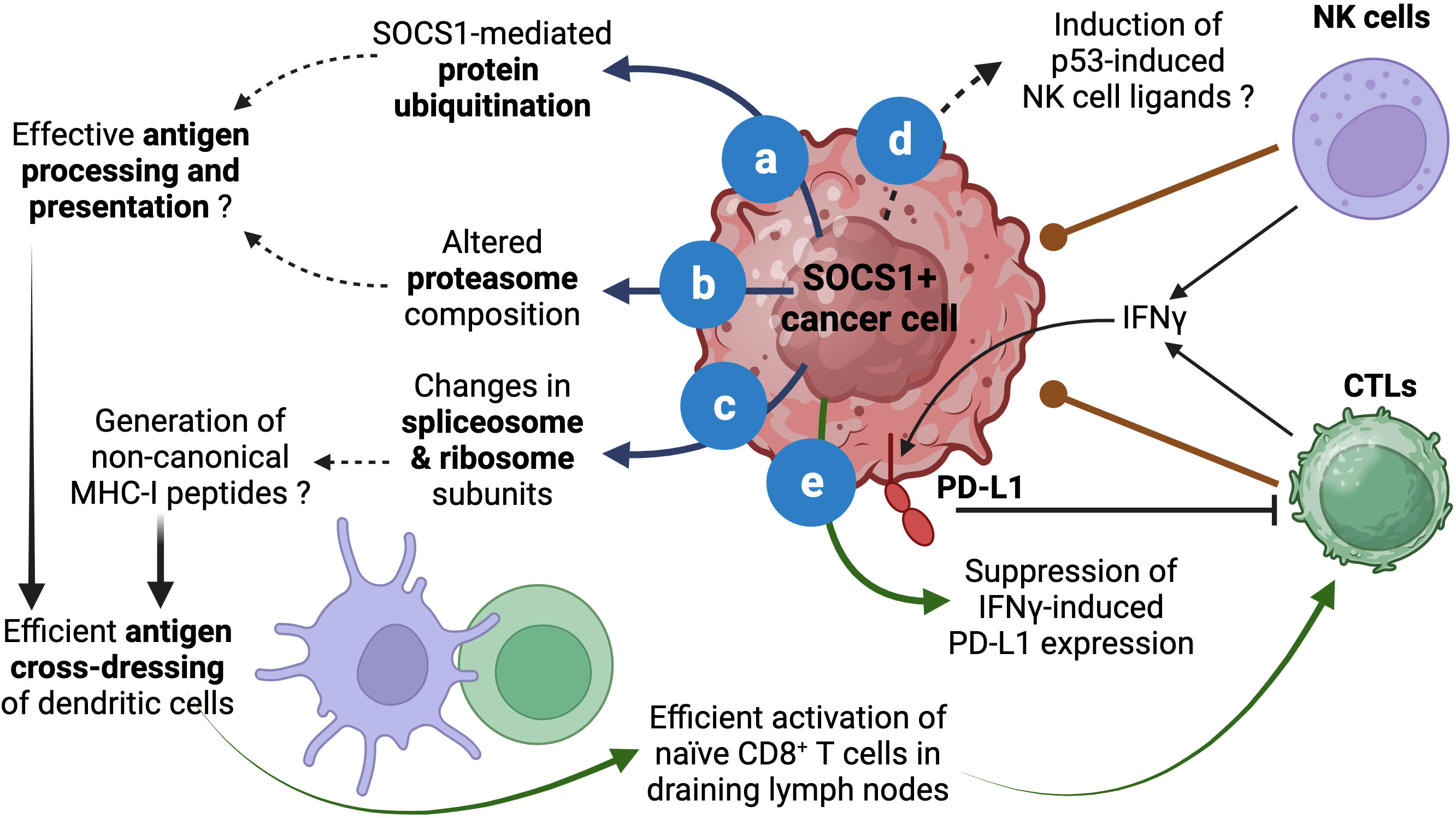
Figure 2 Proposed roles of SOCS1 in enhancing antitumor immunity. The role of SOCS1 in promoting protein ubiquitination and proteasomal degradation has been well documented (A). Stable SOCS1 expression in cancer cells alters proteosome composition (B). Both (A, B) can modulate the generation of antigenic peptides for loading MHC-I. SOCS1 expression also modulates spliceosome and ribosome subunits (C), which could impact protein translation and could potentially contribute to the generation of non-canonical MHC-I binding peptides. These events could increase cross-dressing of dendritic cells by tumor cell derived MHC-I:peptide complexes and enhance activation of naïve antitumor CD8+ T cells and their differentiation towards CTLs. The ability of SOCS1 in activating p53 and senescence induction could potentially modulate p53-driven expression of NK cell ligands (D), which could impact NK-cell mediated tumor cell killing. As a critical regulator of IFNγ, SOCS1 can inhibit adaptive immunosuppression mediated by IFNγ-induced PD-L1 expression in tumor cells (E). Reported data are indicated by solid arrows. Proposed functions that need to be experimentally tested are indicated by dotted arrows. Figure created with Biorender.com.
In addition to binding proteins and targeting them for ubiquitination, SOCS1 has the potential to modulate both the cellular ubiquitination system and the proteasome composition. In a proteomic study using Hepa-vector and Hepa-SOCS1 cells, we observed that SOCS1 increased the expression of certain E2 Ub conjugation enzymes, which transfer the Ub moiety to the E3 ligase for eventual transfer to the substrate protein (77, 78). SOCS1 also caused a similar modification in the proteasome subunit composition (77, 79). These findings raise the possibility that SOCS1, induced by diverse growth stimuli, may also perform additional tasks besides attenuating signal transduction. These changes in ubiquitination and proteasome machinery, which may influence cellular protein homeostasis, can alter the quality and quantity of peptides generated by proteasomes (Figure 2B).
In normal and cancer cells, proteasomes play a crucial role in normal protein turn over as well as in degrading misfolded proteins (78, 80). This process generates peptides that are presented by MHC class-I molecules to facilitate cancer immune surveillance by CD8+ T lymphocytes (81–83). In the cancer immunity cycle, the initial activation naïve antitumor CD8+ T cells that recognize potential cancer antigens occurs when dendritic cells that take up tumor cell fragments process and present tumor antigens as MHC-I:peptide complex to naïve CD8+ T cells in tumor draining lymph nodes by a process called antigen cross-presentation (84–89). However, emerging evidence indicate that antigen presentation by ‘cross-dressing’ is the main activation mechanism of tumor-reactive CD8+ T cells. This process involves the uptake of MHC:peptide complexes from live tumor cells by professional antigen presenting cells via trogocytosis (90–94). We propose that by promoting ubiquitin-dependent proteasomal degradation, SOCS1 expression in tumor cells can potentially increase antigen processing and presentation and thereby increase the efficiency of the cross-dressing pathway and promote antitumor immunity (Figure 2).
4.2 Generation of non-canonical MHC-I peptides
The tumor immune surveillance concept postulates that the immune system is constantly on the lookout for cancer cells by recognizing abnormally expressed cellular proteins, which fall into two main groups (95–98): (i) tumor-specific neoantigens, which arise from mutations and give rise to antigenic peptides encompassing the mutated protein sequence that were never encountered by the immune system (99); (ii) tumor-associated antigens, which arise from aberrant expression of embryonic proteins due to the dedifferentiation process associated with tumor progression, and thus can elicit immune responses (100). With the advent of genome sequencing, identification of tumor neoantigens arising from mutations in a tumor specimen has become a manageable task, and this has led to the development of tumor vaccines based on neo-antigenic peptides and CAR-T cells reactive to them for cellular therapy (99, 101). These approaches have demonstrated feasibility and success, but arguments highlighting the limitations of this approach are also raised (102–106). Firstly, in order to be presented to CD8+ T cells, the mutant peptide must reside within an amino acid sequence that would permit binding to MHC-I. Secondly, generation of MHC-I binding peptides at steady state is coupled to protein biosynthesis and this process is estimated to be limited in efficiency, i.e out of two million peptides generated per second only 150 peptides get loaded onto MHC-I, likely dictated by the ability to bind the peptide binding grove of MHC-I (107). Third, it has been recognized for a long time that MHC-I associated peptides can arise from non-canonical protein sequences, including altered reading frames (defective ribosomal products, DRiPs) and non-coding regions of the messenger RNA and RNAs arising from intronic sequences (108–111). Such peptides would not be present in the canonical, annotated protein coding sequence databases used to identify mutations, which represents only 2% of the genome. In fact, only a small fraction of the MHC-I immunopeptidome is derived from protein-coding sequences (112). Mass spectrometry analysis of peptides eluted from MHC-I molecules of human cancers have revealed that non-canonical MHC-I peptides are far more abundant than mutated peptides and they originate from chromosomal regions that are distinct from mutational hotspots and not shared by embryonic cells (111, 113–115). Immunization with a candidate non-canonical peptide has been shown to elicit protective antitumor immunity (116). Even though current approaches to identify the non-canonical MHC-I peptides rely on mass spectrometry-based detection of peptides eluted from MHC-I molecules, approaches involving generation of databases containing transcriptomes of the entire genome and predicted proteomes of six reading frames (three on each DNA strand) are being employed in a proteogenomic approach to predict potential non-canonical MHC-I peptides (117, 118).
We have observed that Hepa-SOCS1 cells displayed marked alterations in the constituents of spliceosome, a multiprotein complex involved in processing RNA transcripts to mediate splicing, remove intronic sequences and generate mRNA for translation (77). In a recent study using shotgun proteomics employing data-independent acquisition, we observed that SOCS1-expressing Hepa cells showed upregulation of several hundred proteins (65). Pathway analysis of significantly upregulated proteins revealed that these proteins are enriched for the biological processes related to RNA splicing, mRNA processing, non-coding RNA (ncRNA) processing, rRNA processing and ribonucleoprotein complex biogenesis (Figure 1E), and showed significant enrichment within the cellular compartments of spliceosome, U2-spliceosome, catalytic step 2 spliceosome, nuclear speck and chromosomal region (Figure 1F), suggesting a profound impact of SOCS1 on pre-mRNA processing that could generate alternate splice forms. In addition, SOCS1-expressing Hepa cells displayed significant upregulation of protein subunits of ribosome and ribonucleoprotein complex (Figures 1E, F), suggesting potential impact of SOCS1 on protein translation as well. Moreover, non-canonical MHC-I peptides can arise from fusion of unrelated peptides during proteasomal processing (119, 120). Whether the marked alterations in the composition of proteasome subunits in SOCS1 expressing cells impact the generation of proteasome-spliced peptides that bind MHC-I needs to be tested. These postulated alterations in RNA processing, their translation to polypeptides and proteasomal degradation promoted by SOCS1 could promote the generation of non-canonical antigenic peptides (Figure 2C), which in turn would impact the MHC-I peptidome, potentially contributing to tumor immune surveillance. As SOCS1 expression at steady state is very low and is highly induced by mitogenic cytokines, growth factors and oncogenic growth signaling (6, 8), SOCS1 is very well poised to play a key role tumor immune surveillance. This could be an important factor determining the widespread repression of the SOCS1 gene in diverse tumors.
4.3 Immune ligand expression by p53
Another potential influence of SOCS1 in modulating antitumor immune responses could occur via activating p53. In a cellular senescence model, oncogene-induced SOCS1 promotes p53 activation to induce senescence associated genes (59, 121). The p53 tumor suppressor plays multiple roles in mediating its functions. Depending on the extent of DNA damage, p53 facilitates DNA damage repair, or induce senescence or apoptosis to control tumor growth (122). The p53-induced senescence is also associated with the induction of natural killer (NK) cell activation ligands (RAE1, MULT1/ULBP1, H60A), chemokines and cytokines (CSF1, CCL2, CXCL1, IL-15), which can impact antitumor immune responses (123–126). In addition to CD8+ T cells, which play a key role in tumor immune surveillance, NK cells also play important roles in tumor control and tumor immune surveillance (127–130). As SOCS1 also harbors a nuclear localization signal and is required for activating p53-dependent senescence (59, 121, 131), it is not unlikely that SOCS1 may promote the induction of p53-dependent immune function-related genes and contribute to immune cell-mediated tumor control by inducing NK cell ligands and chemokines (Figure 2D).
4.4 Inhibition of immune checkpoint ligand expression
Activated, cytotoxic CD8+ T cells generated in draining lymph nodes emigrate, enter circulation, infiltrate tumors, engage tumor cells expressing cognate tumor antigenic peptide and release their cytotoxic granules to kill tumors (84, 132). At the same time these cytotoxic T lymphocytes also release effector cytokines, including IFNγ. IFNγ is a potent stimulator of the checkpoint ligand PD-L1, which engages PD1 on activated T cells and delivers an inhibitory signal to dampen their effector functions (133–135). Indeed, this adaptive immunosuppression mediated by IFNγ-induced PD-L1 expression is an important obstacle to efficient immune cell mediated tumor killing as it can inhibit cytotoxic CD8+ T and NK cells (136–139), although PD-L1 expression in tumors is considered a predictive biomarker for the effectiveness of immune checkpoint inhibitor therapy (140, 141). Given that SOCS1 is a non-redundant inhibitor of IFNγ signaling (7, 8), SOCS1 expression in cancer cells would dampen PD-L1-mediated checkpoint inhibition. Indeed, Naka and colleagues have shown that intra-tumoral delivery of SOCS1 by adenoviral vectors resulted in reduced PD-L1 expression that was correlated increased CD8+ T cell activation and improved tumor growth control (142). Thus, relieving the blockade on IFNγ signaling that upregulates the checkpoint ligand (Figure 2E) could be another reason why cancer cells choose to repress SOCS1 expression by diverse mechanisms.
5 Restoring SOCS1 expression in cancers
The cell-intrinsic tumor suppressor functions of SOCS1 and its potential contribution to antitumor immune responses raised the possibility of restoring SOCS1 expression to control tumor growth. The SOCS1 mimetic peptide Tkip has been shown to inhibit prostate cancer growth in vitro (55), indicating the potential utility of SOCS1-mediated inhibition in tumor growth control. However, it will be a challenging task to introduce SOCS1 mimetic peptides into all cancer cells. Moreover, such peptide mimetics that inhibit JAK kinases are unlikely to mediate the complex functions of SOCS1 in mediating protein degradation, activating p53, or induing the expression of several genes involved in RNA splicing and translation discussed above. SOCS1 gene therapy using adenoviral vectors in experimental models has been reported to reduce tumor growth by inhibiting oncogenic signaling and improving CD8+ T cell responses (142). As SOCS1 expression is highly regulated (6), stable constitutive expression of SOCS1 may lead to unintended consequences. As the SOCS1 gene repression mainly occurs via CpG methylation in diverse cancers, demethylating agents such as 5-azacytidine and decitabine could be potentially useful to restore regulated SOCS1 expression (41, 143). Even though demethylating agents do have off-target effects, 5-aza is an already approved anticancer drug (144, 145). SOCS1 inhibition by miR155 also occurs commonly in several cancers (46, 146, 147). Even though miR-155 sponges and miR-155 targeting oligos can be used to restore SOCS1 expression in vitro settings (148, 149), miR-155 targets many signaling molecules (150) and the effectiveness of such approaches in vivo could be challenging. Mouse cancer models with Socs1 gene repression remain scanty. Development of such models, also expressing surrogate tumor antigens, would help test the effectiveness of restoring endogenous SOCS1 expression and testing its impact on antitumor immune responses.
6 Discussion
The role of SOCS1 as a tumor suppressor has been well established and some of the underlying mechanisms include attenuation of mitogenic cytokine and growth factor signaling via the JAK-STAT and RTK pathways, inhibition of oncogenic signaling proteins by promoting their ubiquitination and proteasomal degradation, prevention of the oncogenic potential of tumor suppressors such as p21 and inhibition of NRF2-mediated tumor cell adaptation to elevated oxidative stress associated with neoplastic growth. Here, we have presented evidence that support potential cell-extrinsic role of SOCS1 in facilitating tumor immune surveillance and immune cell mediated tumor growth control via promoting tumor antigen processing and presentation, generation of non-canonical MHC-I peptides, expression of immune cell ligands and inhibition of immune checkpoint ligand expression (Figure 2). Validating these hypotheses will provide a compelling argument for the use of epigenetic modifying drugs to reverse SOCS1 gene repression to restore cell-intrinsic tumor suppressor functions as well as cell-extrinsic impact on antitumor immunity.
Data availability statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author/s.
Ethics statement
The animal study was approved by Université de Sherbrooke Ethics Committee for Animal Care and Use. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SI: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing. YG: Methodology, Writing – review & editing, Data curation, Formal Analysis, Investigation. AS: Data curation, Formal Analysis, Methodology, Writing – review & editing, Investigation. SR: Conceptualization, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by a Project grant from the Canadian Institutes of Health Research (CIHR) to SI (PJT-153174).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature (1997) 387:917–21. doi: 10.1038/43206
2. Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature (1997) 387:921–4. doi: 10.1038/43213
3. Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature (1997) 387:924–9. doi: 10.1038/43219
4. Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol (2002) 2:410–6. doi: 10.1038/nri818
5. De Sepulveda P, Ilangumaran S, Rottapel R. Suppressor of cytokine signaling-1 inhibits VAV function through protein degradation. J Biol Chem (2000) 275:14005–8. doi: 10.1074/jbc.C000106200
6. Ilangumaran S, Ramanathan S, Rottapel R. Regulation of the immune system by SOCS family adaptor proteins. Semin Immunol (2004) 16:351–65. doi: 10.1016/j.smim.2004.08.015
7. Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, et al. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell (1999) 98:609–16. doi: 10.1016/S0092-8674(00)80048-3
8. Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell (1999) 98:597–608. doi: 10.1016/S0092-8674(00)80047-1
9. Chong MM, Cornish AL, Darwiche R, Stanley EG, Purton JF, Godfrey DI, et al. Suppressor of cytokine signaling-1 is a critical regulator of interleukin-7-dependent CD8+ T cell differentiation. Immunity (2003) 18:475–87. doi: 10.1016/S1074-7613(03)00078-5
10. Ilangumaran S, Ramanathan S, Ning T, La Rose J, Reinhart B, Poussier P, et al. Suppressor of cytokine signaling 1 attenuates IL-15 receptor signaling in CD8+ thymocytes. Blood (2003) 102:4115–22. doi: 10.1182/blood-2003-01-0175
11. Ilangumaran S, Ramanathan S, La Rose J, Poussier P, Rottapel R. Suppressor of cytokine signaling 1 regulates IL-15 receptor signaling in CD8+CD44high memory T lymphocytes. J Immunol (2003) 171:2435–45. doi: 10.4049/jimmunol.171.5.2435
12. Cornish AL, Davey GM, Metcalf D, Purton JF, Corbin JE, Greenhalgh CJ, et al. Suppressor of cytokine signaling-1 has IFN-gamma-independent actions in T cell homeostasis. J Immunol (2003) 170:878–86. doi: 10.4049/jimmunol.170.2.878
13. Gagnon J, Ramanathan S, Leblanc C, Ilangumaran S. Regulation of IL-21 signaling by suppressor of cytokine signaling-1 (SOCS1) in CD8(+) T lymphocytes. Cell signalling (2007) 19:806–16. doi: 10.1016/j.cellsig.2006.10.003
14. Gagnon J, Ramanathan S, Leblanc C, Cloutier A, McDonald PP, Ilangumaran S. IL-6, in synergy with IL-7 or IL-15, stimulates TCR-independent proliferation and functional differentiation of CD8+ T lymphocytes. J Immunol (2008) 180:7958–68. doi: 10.4049/jimmunol.180.12.7958
15. Gagnon J, Chen XL, Forand-Boulerice M, Leblanc C, Raman C, Ramanathan S, et al. Increased antigen responsiveness of naive CD8 T cells exposed to IL-7 and IL-21 is associated with decreased CD5 expression. Immunol Cell Biol (2010) 88:451–60. doi: 10.1038/icb.2009.109
16. Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity (2009) 30:80–91. doi: 10.1016/j.immuni.2008.11.010
17. Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity (2002) 17:583–91. doi: 10.1016/S1074-7613(02)00446-6
18. Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity (2002) 17:677–87. doi: 10.1016/S1074-7613(02)00449-1
19. Hanada T, Yoshida H, Kato S, Tanaka K, Masutani K, Tsukada J, et al. Suppressor of cytokine signaling-1 is essential for suppressing dendritic cell activation and systemic autoimmunity. Immunity (2003) 19:437–50. doi: 10.1016/S1074-7613(03)00240-1
20. Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol (2006) 7:148–55. doi: 10.1038/ni1299
21. Ramanathan S, Dubois S, Chen XL, Leblanc C, Ohashi PS, Ilangumaran S. Exposure to IL-15 and IL-21 enables autoreactive CD8 T cells to respond to weak antigens and cause disease in a mouse model of autoimmune diabetes. J Immunol (2011) 186:5131–41. doi: 10.4049/jimmunol.1001221
22. Ramanathan S, Dubois S, Gagnon J, Leblanc C, Mariathasan S, Ferbeyre G, et al. Regulation of cytokine-driven functional differentiation of CD8 T cells by suppressor of cytokine signaling 1 controls autoimmunity and preserves their proliferative capacity toward foreign antigens. J Immunol (2010) 185:357–66. doi: 10.4049/jimmunol.1000066
23. Rodriguez GM, D'Urbano D, Bobbala D, Chen XL, Yeganeh M, Ramanathan S, et al. SOCS1 prevents potentially skin-reactive cytotoxic T lymphocytes from gaining the ability to cause inflammatory lesions. J Invest Dermatol (2013) 133:2013–22. doi: 10.1038/jid.2013.86
24. Trengove MC, Ward AC. SOCS proteins in development and disease. Am J Clin Exp Immunol (2013) 2:1–29.
25. Liang Y, Xu WD, Peng H, Pan HF, Ye DQ. SOCS signaling in autoimmune diseases: molecular mechanisms and therapeutic implications. Eur J Immunol (2014) 44:1265–75. doi: 10.1002/eji.201344369
26. Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol (2020) 146:1194–1200.e1. doi: 10.1016/j.jaci.2020.07.033
27. Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun (2020) 11:5341. doi: 10.1038/s41467-020-18925-4
28. Flowers LO, Johnson HM, Mujtaba MG, Ellis MR, Haider SM, Subramaniam PS. Characterization of a peptide inhibitor of Janus kinase 2 that mimics suppressor of cytokine signaling 1 function. J Immunol (2004) 172:7510–8. doi: 10.4049/jimmunol.172.12.7510
29. Mujtaba MG, Flowers LO, Patel CB, Patel RA, Haider MI, Johnson HM. Treatment of mice with the suppressor of cytokine signaling-1 mimetic peptide, tyrosine kinase inhibitor peptide, prevents development of the acute form of experimental allergic encephalomyelitis and induces stable remission in the chronic relapsing/remitting form. J Immunol (2005) 175:5077–86. doi: 10.4049/jimmunol.175.8.5077
30. Waiboci LW, Ahmed CM, Mujtaba MG, Flowers LO, Martin JP, Haider MI, et al. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: implications for the development of a SOCS-1 antagonist. J Immunol (2007) 178:5058–68. doi: 10.4049/jimmunol.178.8.5058
31. Jager LD, Dabelic R, Waiboci LW, Lau K, Haider MS, Ahmed CM, et al. The kinase inhibitory region of SOCS-1 is sufficient to inhibit T-helper 17 and other immune functions in experimental allergic encephalomyelitis. J Neuroimmunol (2011) 232:108–18. doi: 10.1016/j.jneuroim.2010.10.018
32. Ahmed CM, Massengill MT, Brown EE, Ildefonso CJ, Johnson HM, Lewin AS. A cell penetrating peptide from SOCS-1 prevents ocular damage in experimental autoimmune uveitis. Exp Eye Res (2018) 177:12–22. doi: 10.1016/j.exer.2018.07.020
33. Doti N, Scognamiglio PL, Madonna S, Scarponi C, Ruvo M, Perretta G, et al. New mimetic peptides of the kinase-inhibitory region (KIR) of SOCS1 through focused peptide libraries. Biochem J (2012) 443:231–40. doi: 10.1042/BJ20111647
34. Madonna S, Scarponi C, Doti N, Carbone T, Cavani A, Scognamiglio L, et al. Therapeutical potential of a peptide mimicking the SOCS1 kinase inhibitory region in skin immune responses. Eur J Immunol (2013) 43:1883–95.doi: 10.1002/eji.201343370
35. Sharma J, Larkin J. Therapeutic implication of SOCS1 modulation in the treatment of autoimmunity and cancer. Front Pharmacol (2019) 10:324. doi: 10.3389/fphar.2019.00324
36. Bourdonnay E, Zaslona Z, Penke LR, Speth JM, Schneider DJ, Przybranowski S, et al. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J Exp Med (2015) 212:729–42. doi: 10.1084/jem.20141675
37. Dudda JC, Salaun B, Ji Y, Palmer DC, Monnot GC, Merck E, et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity (2013) 38:742–53. doi: 10.1016/j.immuni.2012.12.006
38. Ji Y, Wrzesinski C, Yu Z, Hu J, Gautam S, Hawk NV, et al. miR-155 augments CD8+ T-cell antitumor activity in lymphoreplete hosts by enhancing responsiveness to homeostatic gammac cytokines. Proc Natl Acad Sci U.S.A (2015) 112:476–81. doi: 10.1073/pnas.1422916112
39. Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE, et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth- suppression activity. Nat Genet (2001) 28:29–35. doi: 10.1038/ng0501-29
40. Zardo G, Tiirikainen MI, Hong C, Misra A, Feuerstein BG, Volik S, et al. Integrated genomic and epigenomic analyses pinpoint biallelic gene inactivation in tumors. Nat Genet (2002) 32:453–8. doi: 10.1038/ng1007
41. Galm O, Yoshikawa H, Esteller M, Osieka R, Herman JG. SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood (2003) 101:2784–8. doi: 10.1182/blood-2002-06-1735
42. Sutherland KD, Lindeman GJ, Choong DY, Wittlin S, Brentzell L, Phillips W, et al. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene (2004) 23:7726–33. doi: 10.1038/sj.onc.1207787
43. Goel A, Xicola RM, Nguyen TP, Doyle BJ, Sohn VR, Bandipalliam P, et al. Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology (2010) 138:1854–62. doi: 10.1053/j.gastro.2010.01.035
44. Shiovitz S, Bertagnolli MM, Renfro LA, Nam E, Foster NR, Dzieciatkowski S, et al. CpG island methylator phenotype is associated with response to adjuvant irinotecan-based therapy for stage III colon cancer. Gastroenterology (2014) 147:637–45. doi: 10.1053/j.gastro.2014.05.009
45. Weniger MA, Melzner I, Menz CK, Wegener S, Bucur AJ, Dorsch K, et al. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene (2006) 25:2679–84. doi: 10.1038/sj.onc.1209151
46. Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu H, et al. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res (2010) 70:3119–27. doi: 10.1158/0008-5472.CAN-09-4250
47. Kobayashi N, Uemura H, Nagahama K, Okudela K, Furuya M, Ino Y, et al. Identification of miR-30d as a novel prognostic maker of prostate cancer. Oncotarget (2012) 3:1455–71. doi: 10.18632/oncotarget.696
48. Inagaki-Ohara K, Kondo T, Ito M, Yoshimura A. SOCS, inflammation, and cancer. Jak-Stat (2013) 2:e24053. doi: 10.4161/jkst.24053
49. Sasi W, Sharma AK, Mokbel K. The role of suppressors of cytokine signalling in human neoplasms. Mol Biol Int (2014) 2014:630797. doi: 10.1155/2014/630797
50. Metcalf D, Mifsud S, Di Rago L, Nicola NA, Hilton DJ, Alexander WS. Polycystic kidneys and chronic inflammatory lesions are the delayed consequences of loss of the suppressor of cytokine signaling-1 (SOCS-1). Proc Natl Acad Sci U.S.A (2002) 99:943–8. doi: 10.1073/pnas.022628499
51. Yoshida T, Ogata H, Kamio M, Joo A, Shiraishi H, Tokunaga Y, et al. SOCS1 is a suppressor of liver fibrosis and hepatitis-induced carcinogenesis. J Exp Med (2004) 199:1701–7. doi: 10.1084/jem.20031675
52. Yeganeh M, Gui Y, Kandhi R, Bobbala D, Tobelaim WS, Saucier C, et al. Suppressor of cytokine signaling 1-dependent regulation of the expression and oncogenic functions of p21(CIP1/WAF1) in the liver. Oncogene (2016) 35:4200–11. doi: 10.1038/onc.2015.485
53. Khan MGM, Boufaied N, Yeganeh M, Kandhi R, Petkiewicz S, Sharma A, et al. SOCS1 deficiency promotes hepatocellular carcinoma via SOCS3-dependent CDKN1A induction and NRF2 activation. Cancers (Basel) (2023) 15:905. doi: 10.3390/cancers15030905
54. Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med (2006) 203:1391–7. doi: 10.1084/jem.20060436
55. Flowers LO, Subramaniam PS, Johnson HM. A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene (2005) 24:2114–20. doi: 10.1038/sj.onc.1208437
56. Gui Y, Yeganeh M, Donates YC, Tobelaim WS, Chababi W, Mayhue M, et al. Regulation of MET receptor tyrosine kinase signaling by suppressor of cytokine signaling 1 in hepatocellular carcinoma. Oncogene (2015) 34:5718–28. doi: 10.1038/onc.2015.20
57. Gui Y, Khan MGM, Bobbala D, Dubois C, Ramanathan S, Saucier C, et al. Attenuation of MET-mediated migration and invasion in hepatocellular carcinoma cells by SOCS1. World J Gastroenterol (2017) 23:6639–49. doi: 10.3748/wjg.v23.i36.6639
58. Villalobos-Hernandez A, Bobbala D, Kandhi R, Khan MG, Mayhue M, Dubois CM, et al. SOCS1 inhibits migration and invasion of prostate cancer cells, attenuates tumor growth and modulates the tumor stroma. Prostate Cancer Prostatic Dis (2017) 20:36–47. doi: 10.1038/pcan.2016.50
59. Calabrese V, Mallette FA, Deschenes-Simard X, Ramanathan S, Gagnon J, Moores A, et al. SOCS1 links cytokine signaling to p53 and senescence. Mol Cell (2009) 36:754–67. doi: 10.1016/j.molcel.2009.09.044
60. Saint-Germain E, Mignacca L, Huot G, Acevedo M, Moineau-Vallee K, Calabrese V, et al. Phosphorylation of SOCS1 inhibits the SOCS1-p53 tumor suppressor axis. Cancer Res (2019) 79:3306–19. doi: 10.1158/0008-5472.CAN-18-1503
61. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer (2009) 9:400–14. doi: 10.1038/nrc2657
62. Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med (2016) 22:578–93. doi: 10.1016/j.molmed.2016.05.002
63. Kandhi R, Menendez A, Ramanathan S, Ilangumaran S. Regulation of high-fat diet-induced liver fibrosis by SOCS1 expression in hepatic stellate cells. J Clin Exp Hepatol (2024) 14:101280. doi: 10.1016/j.jceh.2023.09.001
64. Gui Y, Yeganeh M, Ramanathan S, Leblanc C, Pomerleau V, Ferbeyre G, et al. SOCS1 controls liver regeneration by regulating HGF signaling in hepatocytes. J Hepatol (2011) 55:1300–8. doi: 10.1016/j.jhep.2011.03.027
65. Shukla A, Khan MGM, Cayarga AA, Namvarpour M, Chowdhury MMH, Levesque D, et al. The tumor suppressor SOCS1 diminishes tolerance to oxidative stress in hepatocellular carcinoma. Cancers (Basel) (2024) 16:292. doi: 10.3390/cancers16020292
66. Shen L, Evel-Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol (2004) 22:1546–53. doi: 10.1038/nbt1035
67. Okumura F, Joo-Okumura A, Nakatsukasa K, Kamura T. The role of cullin 5-containing ubiquitin ligases. Cell Div (2016) 11:1. doi: 10.1186/s13008-016-0016-3
68. Babon JJ, Sabo JK, Zhang JG, Nicola NA, Norton RS. The SOCS box encodes a hierarchy of affinities for Cullin5: implications for ubiquitin ligase formation and cytokine signalling suppression. J Mol Biol (2009) 387:162–74. doi: 10.1016/j.jmb.2009.01.024
69. Okumura F, Matsuzaki M, Nakatsukasa K, Kamura T. The role of elongin BC-containing ubiquitin ligases. Front Oncol (2012) 2:10. doi: 10.3389/fonc.2012.00010
70. Frantsve J, Schwaller J, Sternberg DW, Kutok J, Gilliland DG. Socs-1 inhibits TEL-JAK2-mediated transformation of hematopoietic cells through inhibition of JAK2 kinase activity and induction of proteasome- mediated degradation. Mol Cell Biol (2001) 21:3547–57. doi: 10.1128/MCB.21.10.3547-3557.2001
71. Kamizono S, Hanada T, Yasukawa H, Minoguchi S, Kato R, Minoguchi M, et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J Biol Chem (2001) 276:12530–8. doi: 10.1074/jbc.M010074200
72. Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem (2002) 277:42394–8. doi: 10.1074/jbc.C200444200
73. Kamio M, Yoshida T, Ogata H, Douchi T, Nagata Y, Inoue M, et al. SOCS1 inhibits HPV-E7-mediated transformation by inducing degradation of E7 protein. Oncogene (2004) 23:3107–15. doi: 10.1038/sj.onc.1207453
74. Strebovsky J, Walker P, Lang R, Dalpke AH. Suppressor of cytokine signaling 1 (SOCS1) limits NFkappaB signaling by decreasing p65 stability within the cell nucleus. FASEB J (2011) 25:863–74. doi: 10.1096/fj.10-170597
75. Bunda S, Kommaraju K, Heir P, Ohh M. SOCS-1 mediates ubiquitylation and degradation of GM-CSF receptor. PloS One (2013) 8:e76370. doi: 10.1371/journal.pone.0076370
76. Parrillas V, Martinez-Munoz L, Holgado BL, Kumar A, Cascio G, Lucas P, et al. Suppressor of cytokine signaling 1 blocks mitosis in human melanoma cells. Cell Mol Life Sci CMLS (2013) 70:545–58. doi: 10.1007/s00018-012-1145-8
77. Santharam MA, Shukla A, Ihsan AU, Cloutier M, Levesque D, Ramanathan S, et al. SILAC proteomics implicates SOCS1 in modulating cellular macromolecular complexes and the ubiquitin conjugating enzyme UBE2D involved in MET receptor tyrosine kinase downregulation. Biochimie (2021) 182:185–96. doi: 10.1016/j.biochi.2021.01.012
78. Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J (1998) 17:7151–60. doi: 10.1093/emboj/17.24.7151
79. Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci (2009) 85:12–36. doi: 10.2183/pjab.85.12
80. Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta (2014) 1843:182–96. doi: 10.1016/j.bbamcr.2013.06.031
81. Kloetzel PM. Antigen processing by the proteasome. Nat Rev Mol Cell Biol (2001) 2:179–87. doi: 10.1038/35056572
82. Yewdell JW. Plumbing the sources of endogenous MHC class I peptide ligands. Curr Opin Immunol (2007) 19:79–86. doi: 10.1016/j.coi.2006.11.010
83. Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol (2016) 37:724–37. doi: 10.1016/j.it.2016.08.010
84. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
85. Hoffmann TK, Meidenbauer N, Dworacki G, Kanaya H, Whiteside TL. Generation of tumor-specific T-lymphocytes by cross-priming with human dendritic cells ingesting apoptotic tumor cells. Cancer Res (2000) 60:3542–9.
86. den Haan JM, Bevan MJ. Antigen presentation to CD8+ T cells: cross-priming in infectious diseases. Curr Opin Immunol (2001) 13:437–41. doi: 10.1016/S0952-7915(00)00238-7
87. Freigang S, Egger D, Bienz K, Hengartner H, Zinkernagel RM. Endogenous neosynthesis vs. cross-presentation of viral antigens for cytotoxic T cell priming. Proc Natl Acad Sci U.S.A (2003) 100:13477–82. doi: 10.1073/pnas.1835685100
88. Shen L, Rock KL. Priming of T cells by exogenous antigen cross-presented on MHC class I molecules. Curr Opin Immunol (2006) 18:85–91. doi: 10.1016/j.coi.2005.11.003
89. Yatim N, Cullen S, Albert ML. Dying cells actively regulate adaptive immune responses. Nat Rev Immunol (2017) 17:262–75. doi: 10.1038/nri.2017.9
90. Dolan BP, Gibbs KD Jr., Ostrand-Rosenberg S. Tumor-specific CD4+ T cells are activated by "cross-dressed" dendritic cells presenting peptide-MHC class II complexes acquired from cell-based cancer vaccines. J Immunol (2006) 176:1447–55. doi: 10.4049/jimmunol.176.3.1447
91. Wakim LM, Bevan MJ. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature (2011) 471:629–32. doi: 10.1038/nature09863
92. Nakayama M. Antigen presentation by MHC-dressed cells. Front Immunol (2014) 5:672. doi: 10.3389/fimmu.2014.00672
93. Das Mohapatra A, Tirrell I, Benechet AP, Pattnayak S, Khanna KM, Srivastava PK. Cross-dressing of CD8alpha(+) dendritic cells with antigens from live mouse tumor cells is a major mechanism of cross-priming. Cancer Immunol Res (2020) 8:1287–99. doi: 10.1158/2326-6066.CIR-20-0248
94. MacNabb BW, Tumuluru S, Chen X, Godfrey J, Kasal DN, Yu J, et al. Dendritic cells can prime anti-tumor CD8(+) T cell responses through major histocompatibility complex cross-dressing. Immunity (2022) 55:982–997 e8. doi: 10.1016/j.immuni.2022.04.016
95. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410:1107–11. doi: 10.1038/35074122
96. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity (2004) 21:137–48. doi: 10.1016/j.immuni.2004.07.017
97. Boon T, Cerottini JC, Van den Eynde B, van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol (1994) 12:337–65. doi: 10.1146/annurev.iy.12.040194.002005
98. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14:135–46. doi: 10.1038/nrc3670
99. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi: 10.1126/science.aaa4971
100. Weon JL, Potts PR. The MAGE protein family and cancer. Curr Opin Cell Biol (2015) 37:1–8. doi: 10.1016/j.ceb.2015.08.002
101. Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer (2017) 17:209–22. doi: 10.1038/nrc.2016.154
102. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19:747–52. doi: 10.1038/nm.3161
103. Khodadoust MS, Olsson N, Wagar LE, Haabeth OA, Chen B, Swaminathan K, et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature (2017) 543:723–7. doi: 10.1038/nature21433
104. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature (2017) 547:222–6. doi: 10.1038/nature23003
105. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature (2017) 547:217–21. doi: 10.1038/nature22991
106. Apavaloaei A, Hardy MP, Thibault P, Perreault C. The origin and immune recognition of tumor-specific antigens. Cancers (Basel) (2020) 12:2607. doi: 10.3390/cancers12092607
107. Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat Rev Immunol (2003) 3:952–61. doi: 10.1038/nri1250
108. Malarkannan S, Horng T, Shih PP, Schwab S, Shastri N. Presentation of out-of-frame peptide/MHC class I complexes by a novel translation initiation mechanism. Immunity (1999) 10:681–90. doi: 10.1016/S1074-7613(00)80067-9
109. Yewdell JW. DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. Trends Immunol (2011) 32:548–58. doi: 10.1016/j.it.2011.08.001
110. Dersh D, Holly J, Yewdell JW. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol (2021) 21:116–28. doi: 10.1038/s41577-020-0390-6
111. Chen J, Brunner AD, Cogan JZ, Nunez JK, Fields AP, Adamson B, et al. Pervasive functional translation of noncanonical human open reading frames. Science (2020) 367:1140–6. doi: 10.1126/science.aay0262
112. Pearson H, Daouda T, Granados DP, Durette C, Bonneil E, Courcelles M, et al. MHC class I-associated peptides derive from selective regions of the human genome. J Clin Invest (2016) 126:4690–701. doi: 10.1172/JCI88590
113. Zhao Q, Laverdure JP, Lanoix J, Durette C, Cote C, Bonneil E, et al. Proteogenomics uncovers a vast repertoire of shared tumor-specific antigens in ovarian cancer. Cancer Immunol Res (2020) 8:544–55. doi: 10.1158/2326-6066.CIR-19-0541
114. Erhard F, Dolken L, Schilling B, Schlosser A. Identification of the cryptic HLA-I immunopeptidome. Cancer Immunol Res (2020) 8:1018–26. doi: 10.1158/2326-6066.CIR-19-0886
115. Chong C, Muller M, Pak H, Harnett D, Huber F, Grun D, et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat Commun (2020) 11:1293. doi: 10.1038/s41467-020-14968-9
116. Laumont CM, Vincent K, Hesnard L, Audemard E, Bonneil E, Laverdure JP, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med (2018) 10:eaau5516. doi: 10.1126/scitranslmed.aau5516
117. Laumont CM, Daouda T, Laverdure JP, Bonneil E, Caron-Lizotte O, Hardy MP, et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat Commun (2016) 7:10238. doi: 10.1038/ncomms10238
118. Apavaloaei A, Hesnard L, Hardy MP, Benabdallah B, Ehx G, Theriault C, et al. Induced pluripotent stem cells display a distinct set of MHC I-associated peptides shared by human cancers. Cell Rep (2022) 40:111241. doi: 10.1016/j.celrep.2022.111241
119. Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science (2016) 354:354–8. doi: 10.1126/science.aaf4384
120. Ebstein F, Textoris-Taube K, Keller C, Golnik R, Vigneron N, Van den Eynde BJ, et al. Proteasomes generate spliced epitopes by two different mechanisms and as efficiently as non-spliced epitopes. Sci Rep (2016) 6:24032. doi: 10.1038/srep24032
121. Saint-Germain E, Mignacca L, Vernier M, Bobbala D, Ilangumaran S, Ferbeyre G. SOCS1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging (Albany NY) (2017) 9:2137–62. doi: 10.18632/aging.101306
122. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell (2009) 137:413–31. doi: 10.1016/j.cell.2009.04.037
123. Hoglund P. DNA damage and tumor surveillance: one trigger for two pathways. Sci STKE (2006) 2006:pe2. doi: 10.1126/stke.3172006pe2
124. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature (2007) 445:656–60. doi: 10.1038/nature05529
125. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol (2008) 8:59–73. doi: 10.1038/nri2216
126. Li H, Lakshmikanth T, Garofalo C, Enge M, Spinnler C, Anichini A, et al. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle (2011) 10:3346–58. doi: 10.4161/cc.10.19.17630
127. Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol (2003) 21:807–39. doi: 10.1146/annurev.immunol.21.120601.141135
128. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous Malignancy. Immunity (2008) 28:571–80. doi: 10.1016/j.immuni.2008.02.016
129. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer (2016) 16:7–19. doi: 10.1038/nrc.2015.5
130. Meza Guzman LG, Keating N, Nicholson SE. Natural killer cells: tumor surveillance and signaling. Cancers (Basel) (2020) 12:952. doi: 10.3390/cancers12040952
131. Baetz A, Koelsche C, Strebovsky J, Heeg K, Dalpke AH. Identification of a nuclear localization signal in suppressor of cytokine signaling 1. FASEB J (2008) 22:4296–305. doi: 10.1096/fj.08-116079
132. Mellman I, Chen DS, Powles T, Turley SJ. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity (2023) 56:2188–205. doi: 10.1016/j.immuni.2023.09.011
133. Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U.S.A (2003) 100:5336–41. doi: 10.1073/pnas.0931259100
134. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res (2004) 64:1140–5. doi: 10.1158/0008-5472.CAN-03-3259
135. Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett (2006) 580:755–62. doi: 10.1016/j.febslet.2005.12.093
136. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med (2013) 5:200ra116. doi: 10.1126/scitranslmed.3006504
137. McGray AJ, Hallett R, Bernard D, Swift SL, Zhu Z, Teoderascu F, et al. Immunotherapy-induced CD8+ T cells instigate immune suppression in the tumor. Mol Ther (2014) 22:206–18. doi: 10.1038/mt.2013.255
138. Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med (2017) 214:895–904. doi: 10.1084/jem.20160801
139. Ribas A. Adaptive immune resistance: how cancer protects from immune attack. Cancer Discovery (2015) 5:915–9. doi: 10.1158/2159-8290.CD-15-0563
140. Ribas A, Hu-Lieskovan S. What does PD-L1 positive or negative mean? J Exp Med (2016) 213:2835–40. doi: 10.1084/jem.20161462
141. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer (2016) 16:275–87. doi: 10.1038/nrc.2016.36
142. Nakagawa S, Serada S, Kakubari R, Hiramatsu K, Sugase T, Matsuzaki S, et al. Intratumoral delivery of an adenoviral vector carrying the SOCS-1 gene enhances T-cell-mediated antitumor immunity by suppressing PD-L1. Mol Cancer Ther (2018) 17:1941–50. doi: 10.1158/1535-7163.MCT-17-0822
143. Kim MH, Kim MS, Kim W, Kang MA, Cacalano NA, Kang SB, et al. Suppressor of cytokine signaling (SOCS) genes are silenced by DNA hypermethylation and histone deacetylation and regulate response to radiotherapy in cervical cancer cells. PloS One (2015) 10:e0123133. doi: 10.1371/journal.pone.0123133
144. Nervi C, De Marinis E, Codacci-Pisanelli G. Epigenetic treatment of solid tumours: a review of clinical trials. Clin Epigenet (2015) 7:127. doi: 10.1186/s13148-015-0157-2
145. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer (2008) 123:8–13. doi: 10.1002/ijc.23607
146. Yan XL, Jia YL, Chen L, Zeng Q, Zhou JN, Fu CJ, et al. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology (2013) 57:2274–86. doi: 10.1002/hep.26257
147. Zhao XD, Zhang W, Liang HJ, Ji WY. Overexpression of miR -155 promotes proliferation and invasion of human laryngeal squamous cell carcinoma via targeting SOCS1 and STAT3. PloS One (2013) 8:e56395. doi: 10.1371/journal.pone.0056395
148. Mignacca L, Saint-Germain E, Benoit A, Bourdeau V, Moro A, Ferbeyre G. Sponges against miR-19 and miR-155 reactivate the p53-Socs1 axis in hematopoietic cancers. Cytokine (2016) 82:80–6. doi: 10.1016/j.cyto.2016.01.015
149. Seto AG, Beatty X, Lynch JM, Hermreck M, Tetzlaff M, Duvic M, et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br J Haematol (2018) 183:428–44. doi: 10.1111/bjh.15547
Keywords: SOCS1, tumor suppressor, growth control, antigen presentation, tumor immunogenicity, checkpoint inhibition
Citation: Ilangumaran S, Gui Y, Shukla A and Ramanathan S (2024) SOCS1 expression in cancer cells: potential roles in promoting antitumor immunity. Front. Immunol. 15:1362224. doi: 10.3389/fimmu.2024.1362224
Received: 27 December 2023; Accepted: 31 January 2024;
Published: 13 February 2024.
Edited by:
Howard M. Johnson, University of Florida, United StatesReviewed by:
Zuzanna Urban-Wójciuk, University of Gdansk, PolandChulbul M. Ahmed, University of Florida, United States
Copyright © 2024 Ilangumaran, Gui, Shukla and Ramanathan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Subburaj Ilangumaran, U3ViYnVyYWouSWxhbmd1bWFyYW5AVXNoZXJicm9va2UuY2E=