
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 11 March 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1360342
This article is part of the Research Topic Oxidative Metabolism in Inflammation View all 13 articles
 Natalia Rodriguez RodriguezTrinisia FortuneEsha HegdeMatthew Paltiel WeinsteinAislinn M. Keane
Natalia Rodriguez RodriguezTrinisia FortuneEsha HegdeMatthew Paltiel WeinsteinAislinn M. Keane Jesse F. Mangold
Jesse F. Mangold Talia H. Swartz*
Talia H. Swartz*Human Immunodeficiency Virus Type 1 (HIV-1) presents significant challenges to the immune system, predominantly characterized by CD4+ T cell depletion, leading to Acquired Immunodeficiency Syndrome (AIDS). Antiretroviral therapy (ART) effectively suppresses the viral load in people with HIV (PWH), leading to a state of chronic infection that is associated with inflammation. This review explores the complex relationship between oxidative phosphorylation, a crucial metabolic pathway for cellular energy production, and HIV-1, emphasizing the dual impact of HIV-1 infection and the metabolic and mitochondrial effects of ART. The review highlights how HIV-1 infection disrupts oxidative phosphorylation, promoting glycolysis and fatty acid synthesis to facilitate viral replication. ART can exacerbate metabolic dysregulation despite controlling viral replication, impacting mitochondrial DNA synthesis and enhancing reactive oxygen species production. These effects collectively contribute to significant changes in oxidative phosphorylation, influencing immune cell metabolism and function. Adenosine triphosphate (ATP) generated through oxidative phosphorylation can influence the metabolic landscape of infected cells through ATP-detected purinergic signaling and contributes to immunometabolic dysfunction. Future research should focus on identifying specific targets within this pathway and exploring the role of purinergic signaling in HIV-1 pathogenesis to enhance HIV-1 treatment modalities, addressing both viral infection and its metabolic consequences.
Human Immunodeficiency Virus Type 1 (HIV-1) presents a chronic, intractable challenge, primarily characterized by extensive CD4+ T cell depletion (1–5). This pathology manifests in both lymphoid tissues and peripheral blood, culminating in profound immunodeficiency and progression to Acquired Immunodeficiency Syndrome (AIDS). Despite the efficacy of Antiretroviral Therapy (ART) in viral load suppression, HIV-1 infection is associated with chronic inflammation and, together with the direct effects of ART, is associated with metabolic dysregulation, enhanced inflammatory response, gene expression modulation, and biochemical pathway alterations (6–9).
We explore the confluence of oxidative phosphorylation and cellular metabolic transformations in the context of HIV-1 infection and ART in PWH. Key focus areas include the nuances of mitochondrial dysfunction, specifically alterations in oxidative phosphorylation and association with immune cell dysregulation. This underscores a potential nexus between mitochondrial functionality and immunological response. Additionally, we examine the long-term impacts of ART and the potential for more nuanced HIV-1 treatment modalities. This review aims to enrich the understanding of the intricate interplay between oxidative phosphorylation and HIV-1 pathogenesis, steering future research and therapeutic interventions in this critical domain.
Oxidative phosphorylation, a crucial biochemical process, involves the reduction of oxygen to generate ATP (10). Oxidative phosphorylation is the final stage of aerobic respiration, following glycolysis and the citric acid cycle. The efficiency of oxidative phosphorylation relies on successive oxidative/reductive reactions, notably the transfer of electrons by NADH and FADH2 to oxygen, the ultimate electron acceptor (11). The electron transport chain (ETC) facilitates this electron movement within mitochondria. In order of oxidative/reductive reactions, the complexes are I, II, coenzyme Q, III, cytochrome C, and IV. This process pumps protons across the mitochondrial intermembrane space, generating a proton electrochemical gradient that powers ATP synthase (complex V) (12). ATP synthase facilitates ATP biosynthesis with its rotating F0 and F1 components. The F1 component binds nucleotides at its catalytic sites, occupied by Mg-ADP and phosphate. Rotation, driven by F0 subunit reionization, alters F1 directionality, initiating ATP synthesis from ADP and phosphite (13). ATP generated through oxidative phosphorylation, the final and most efficient stage of aerobic respiration in the electron transport chain, serves as the primary energy source for cells, far surpassing the yields from glycolysis and the citric acid cycle.
The regulation of oxidative phosphorylation is complex and multifaceted. The mitochondrial membrane potential is at the center of the regulation, formed by the proton gradient and linked to both ATP and free radical production (14, 15). The most basic regulation of oxidative phosphorylation is allosteric control by negative feedback of substrates or intermediaries. A high NADH/NAD+ ratio slows the Krebs cycle, ETC, and oxidative phosphorylation, as does a high ATP/ADP ratio, in which ATP binds directly and inhibits cytochrome c (Cyt-c) and cytochrome oxidase (complex IV) (15, 16).
Oxidative phosphorylation complexes and Cyt-c are targeted for phosphorylation by regulatory kinases, including protein kinase C, cAMP-dependent tyrosine kinases, and EGFR (17–19). The formation of super-complexes (SCs) or respirasomes, consisting of various combinations of the ETC complexes, increases respiratory efficiency and decreases ROS production. SC abundance, along with expression of specific isoforms of Cyt-c or cytochrome oxidase, allow for regulation of ROS formation and energetic needs at tissue-level specificity (15). The fusion and fission of the mitochondrial network contribute to oxidative phosphorylation efficiency regulation, with highly connected networks promoting efficiency and curbing excess ROS production (20, 21).
Metabolic reprogramming is key in both cancer and HIV-1 infections. In cancer, this is seen as the Warburg effect, characterized by increased glucose uptake and lactate production, even when oxygen is available (22). Similarly, HIV-1 uses metabolic reprogramming to gather free nucleotides, amino acids, and lipids for viral replication and assembly (23). Studies reveal that CD4+ T cells with higher oxidative phosphorylation and glycolysis are more prone to HIV-1 infection, with infected cells showing elevated metabolic activity (24). HIV-1 also boosts glycolysis in CD4+ T cells by upregulating GLUT1 expression. The HIV-1 glycoprotein gp120 is implicated in this process, possibly by activating surface signaling molecules like CXCR4 and CCR5 and increasing the expression of glycolytic enzymes (25). Additionally, hexokinase activity is heightened in HIV-1 infected CD4+ T cells, a change dependent on viral replication (26, 27). However, the precise viral mechanisms promoting glycolysis upregulation by HIV-1 are not fully understood.
There is growing interest in understanding the impact of HIV on Mitochondrial-associated ER membranes (MAMs) due to their crucial role in metabolic reprogramming. MAMs serve as structures linking the endoplasmic reticulum (ER) and mitochondria, facilitating oxidative protein production and mitochondrial biogenesis. The viral HIV-1 Tat protein disrupts MAMs in neuronal cell lines upon infection by phosphorylating the mitochondrial protein PTPIP51 and disrupting its localization to MAMs, thus reducing calcium signaling and increasing ROS accumulation (28). Furthermore, HIV-1 Vpr has been shown to disrupt MAMs by decreasing the expression of critical MAM-associated proteins Mfn2 and Drp1 (29). The targeting of MAMs by HIV-1 induces oxidative stress that contributes to HIV-associated metabolic reprogramming, but further research is needed to establish other key mechanisms involved.
HIV-1 impacts host cell metabolism, initially enhancing glycolysis and fatty acid synthesis for viral replication (30–33). This increased metabolic activity, particularly in glycolysis and glucose transport, makes cells like T-cells and monocytes more susceptible to HIV-1 infection (24, 34, 35). Upregulation of the GLUT-1 transporter and mitochondrial oxidative damage, linked to ROS production, highlights the role of metabolic reprogramming in HIV-1 infection and potential research avenues (31, 35, 36). People with HIV (PWH) face systemic metabolic challenges due to both HIV-1 and antiretroviral therapy (ART), increasing metabolic disease risks (37, 38). Studies have shown that HIV-1 influences metabolic aging, with oxidative phosphorylation and pyruvate metabolism downregulation noted in PWH (39). Furthermore, HIV-1 affects the expression of electron transport chain (ETC) components and causes mitochondrial damage, as seen in the upregulation of Complex-IV subunit, contributing to oxidative stress (40, 41).
ART classes have long been associated with mitochondria dysfunction (42). Both Nucleoside/Nucleotide Reverse Transcriptase Inhibitors (NRTIs) and Non-nucleotide Reverse Transcriptase Inhibitors (NNRTIs) are associated with dyslipidemia in PWH and alter adipocyte differentiation. NRTIs induce lipodystrophy by promoting mitochondrial dysfunction and adipocyte death and interfering with mitochondrial DNA (mtDNA) synthesis (42). This inhibits Pol-gamma, increases ROS production, and reduces ETC activity and oxidative phosphorylation (37, 42–46). Additionally, protease inhibitors (PIs) target GLUT4, impairing glucose uptake into adipocytes and promoting insulin resistance (37, 47, 48).
HIV-1 infection exacerbates oxidative stress and mitochondrial damage, affecting cell metabolism and promoting diseases in PWH, especially those on ART. Gp120 upregulates CYP2E1, proline oxidase (POX), NOX2, and NOX4 to enhance ROS production (49–57). Nef interacts with the NADPH oxidases without affecting the NOX expression (57). The HIV-1 proteins Vpr and Tat both contribute to mitochondrial dysfunction: Vpr reduces membrane potential and triggers apoptosis by binding to ANT, part of the mitochondrial permeability transition pore, while Tat elevates free calcium levels in the cytoplasm by interacting with NADPH oxidases, leading to increased mitochondrial calcium uptake and reactive oxygen species (ROS) production (54, 55, 58–64). These changes increase the risk of metabolic diseases in PWH, with ART compounds like NRTIs and PIs exacerbating (65, 66).
Purinergic receptors, particularly the P1 and P2 subtypes, are integral to inflammatory responses and the progression of HIV-1. The P2X receptors, activated by ATP, are crucial in initiating immune responses and impacting the HIV-1 viral life cycle (67–71). The enzyme CD39 regulates ATP availability, influencing both the progression of AIDS and the function of T cells by modulating ATP and adenosine levels, affecting P2X receptor signaling (72–74).
The P2X receptors are implicated in cell entry and infection by the virus, and their inhibition can significantly reduce HIV-1 infection in various cell types (70, 75–85). Chronic inflammation in HIV is driven by factors like immunosenescence and persistent viral replication, even with antiretroviral therapy (ART). It is exacerbated by factors such as organ fibrosis and co-infections (4, 9, 86–88).
Furthermore, extracellular ATP and its interaction with P2X receptors regulate HIV infection and inflammation. This signaling leads to the production of inflammatory cytokines and the activation of the NLRP3 inflammasome, contributing to CD4+ T lymphocyte depletion and apoptosis (77, 89–92). HIV-1 interacts with Pannexin-1, a membrane channel, suggesting a link between ATP production and inflammatory signaling in HIV-1 infection (93–96). Circulating levels of ATP have been proposed as a biomarker of cognitive decline in PWH, predicting central nervous system compromise and suggesting the use of Pannexin-1 or purinergic receptor inhibitors for clinical intervention (97).
The progression of HIV-1-associated neurocognitive disorders (HAND) in PWH has been linked to purinergic signaling and oxidative stress. The central nervous system serves as a reservoir of HIV-1, leading to neuroinflammation and neurodegeneration that progress to HIV-1 encephalitis and HAND that persist despite the use of effective ART (98–100). Within the CNS, HIV-1 induces the release of extracellular ATP in infected cells to support viral replication, and ATP interacts with purinergic receptors to produce a proinflammatory response (83, 101, 102). The purinergic receptor subtype P2X is particularly interesting, notably P2X7, which is widely expressed in various brain cells, including astrocytes, oligodendrocytes, microglia, and neurons (102). In astrocytes, P2X7 activation induced by gp120 leads to Cx43 hemichannels and pannexin-1 opening, resulting in increased ATP release and nitric oxide production. This process is speculated to propagate gp120-mediated signaling to neighboring cells (95). In the peripheral nervous system (PNS), P2X4 is involved in gp120-induced lysosomal exocytosis and ATP release in Schwann Cells, increasing cytosolic calcium and generating ROS in dorsal root ganglia neurons (101). Although ART mitigates the neuropathology of HIV-1 infection in PWH, it does not eliminate it, with studies showing possible neurotoxicity of ART. ART-related neurotoxicity in the brain is thought to be linked to the loss of mtDNA due to NRTIs inhibiting mtDNA polymerase gamma (103, 104). Additionally, ART contributes to astrocyte autophagy, de-acidification of endolysosomes, and the promotion of amyloidogenesis, all associated with the pathogenesis of HAND (105).
Extracellular ATP and P2X receptors are integral in HIV-1 infection, influencing the virus’s life cycle and contributing to immunopathogenesis, neurodegeneration, and chronic inflammation. Their interaction with HIV-1 is key in disease progression, affecting viral replication, cytokine release, and cell death (75, 77, 78, 81, 83–85). This insight reveals the potential of targeting purinergic signaling in developing novel therapies for HIV-1, reducing inflammation and related comorbidities, and represents a significant area for future research.
The metabolic dynamics of immune cells, particularly CD4+ T cells, CD8+ T cells, and macrophages, are critical in understanding HIV-1 pathophysiology. CD4+ T cells infected with HIV-1 shift towards increased glycolysis, facilitating viral replication. A study identified a link between oxidative phosphorylation in these cells and higher viral loads, with NLRX1 and FASTKD5 as key factors. Metformin, a complex I inhibitor, was found to suppress HIV-1 replication in CD4+ T cells (106). In PWH on ART, there’s an upregulation of oxidative phosphorylation compared to elite controllers, and complex IV inhibition was linked to increased HIV-1 reactivation in a latency model (107).
CD8+ T cells in chronic HIV-1 infection experience metabolic exhaustion, diminishing their functionality and virus control ability. A combination therapy comprised of a mitochondrial superoxide scavenger, a small-molecule inhibitor of mitochondrial fission, and IL-15 can increase the frequency of IFNy and TNFa poly-functional CD8+ T cells and decrease the frequency of exhaustion markers (108).
In HIV-infected macrophages, there is notable support for prolonged HIV-1 replication and survival, shielding virions from ART and neutralizing antibodies. These macrophages show metabolic plasticity, shifting between oxidative phosphorylation and glycolysis based on their polarization state. HIV-1 infection alters macrophage metabolism, promoting a pro-inflammatory state and increasing glycolytic activity, which is linked to chronic inflammation seen in HIV-1 infection (109). A study demonstrated that superoxide dismutase (SOD) mimetic drugs could inhibit myeloid cell-driven bystander cell death in vitro (110).
Figure 1 highlights the impact of HIV-1 infection on mitochondrial oxidative phosphorylation. It contrasts the normal mitochondrial function with the altered state during HIV-1 infection, depicting how HIV-1 infection leads to an upregulation in the transcription of oxidative phosphorylation genes, resulting in greater electron transport chain efficiency. This heightened activity leads to increased production of ROS, a hallmark of cellular stress. Consequently, there’s an enhancement in mitochondrial membrane potential and a notable increase in ATP production compared to baseline conditions. These alterations signify a hyperactivated mitochondrial state triggered by HIV-1 infection, underlining the complex interplay between the virus and the host’s cellular metabolism.
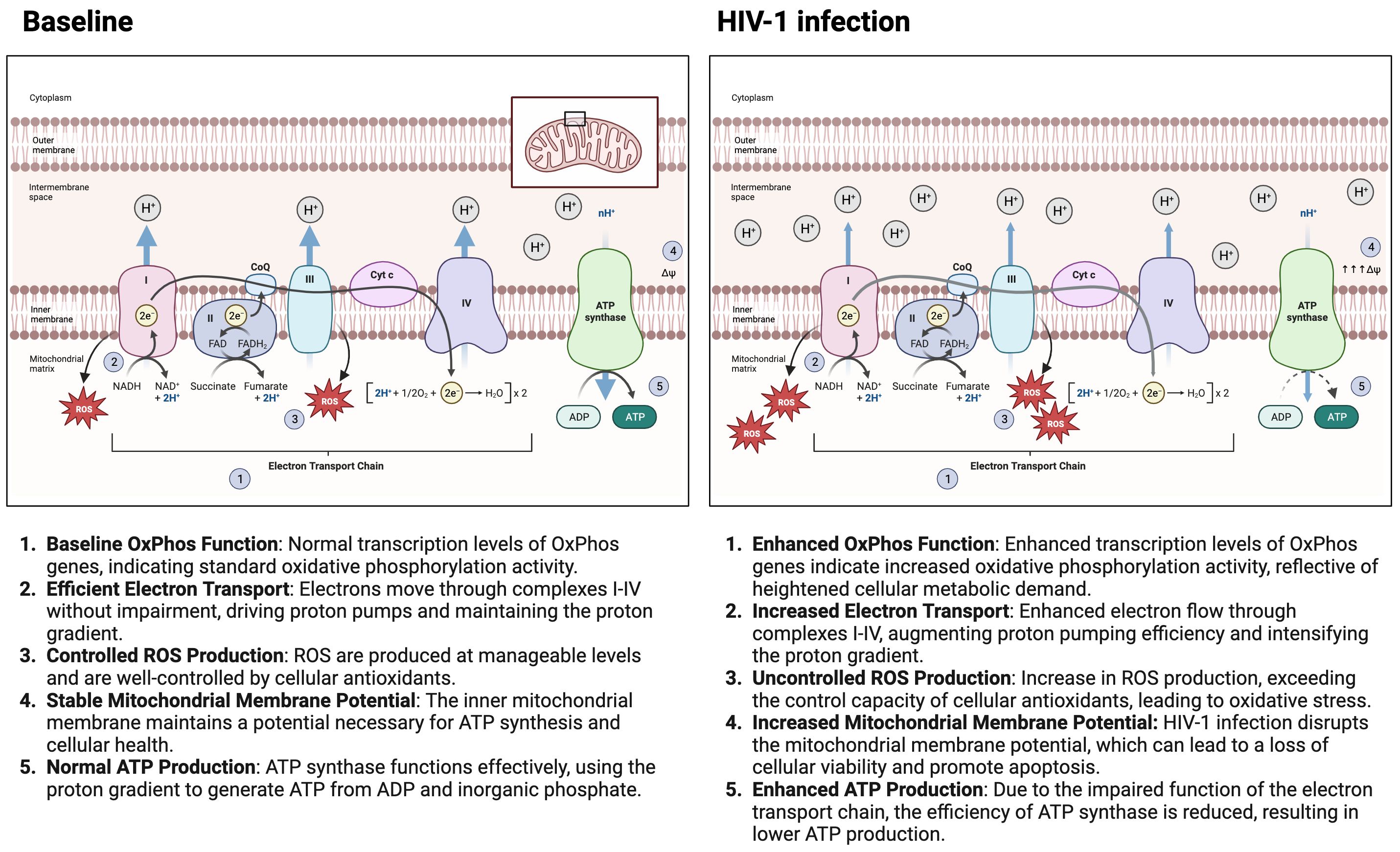
Figure 1 HIV-1 Infection impact on mitochondrial oxidative phosphorylation. The figure contrasts mitochondrial function under normal conditions with the altered state during HIV-1 infection. It shows that HIV-1 infection leads to increased transcription of oxidative phosphorylation genes, higher electron transport efficiency, and elevated reactive oxygen species (ROS) production, resulting in an enhanced mitochondrial membrane potential and increased ATP production compared to the baseline state. These changes indicate a hyperactivated mitochondrial state in response to HIV-1 infection. Made with biorender.com.
Therapeutic interventions targeting mitochondrial dysfunction and oxidative stress in HIV immunopathogenesis are emerging as a crucial field of research. Metformin, a diabetes medication, has shown varying effects on HIV-1 replication in studies by Rezai et al. and Guo et al. (106, 111). There is an additive effect of risk of metformin-associated lactic acidosis when used with NRTIs like tenofovir, which calls for caution in its clinical application (112, 113). Research on the antioxidant properties of plant flavonoids, such as naringin, is gaining attention for their potential to mitigate oxidative damage induced by NRTIs (114, 115). Statins, known for their anti-inflammatory and lipid-lowering effects, are being explored for their benefits in HIV management, with studies like SATURN-HIV and REPRIEVE indicating their impact on cardiovascular risk and mitochondrial function in PWH on ART (116–118).
Given that HIV-1 infection induces metabolic reprogramming like that observed in cancer cells, there is growing interest in exploring anti-cancer drug treatments as potential interventions to target these metabolic effects of HIV-1 (119). In neurons, the viral protein gp120 reduces ATP output via oxidative phosphorylation while increasing glycolysis production and PKM2 expression. Tepp-46, a selective PKM2 tetrameric stabilizer and a potential small molecule therapeutics for lung cancer, has demonstrated the ability to reverse the metabolic reprogramming induced by gp120 in neuronal cells (120). Further, studies have linked dyslipidemia in PWH on ART with mitochondrial oxidative stress, suggesting an association between lipid profiles and the function of mitochondrial electron transport chain complexes (121). Atovaquone, a mitochondrial complex III inhibitor, though not tested in chronic inflammation, is used for treating parasitic and fungal infections (81).
Advancements in understanding HIV-1 pathogenesis highlight the role of purinergic signaling, particularly the involvement of extracellular ATP and P2X receptors. These receptors play a significant role in the inflammatory response and immune modulation in HIV-1 infection (112, 113). This growing body of research underlines the importance of understanding the metabolic and mitochondrial aspects of HIV-1 infection and ART. The goal is to improve treatment outcomes by mitigating adverse effects and exploring new therapeutic avenues that address both HIV-1 management and broader metabolic and cardiovascular health.
The interaction between HIV-1 infection and metabolic activity significantly shapes the chronic inflammation commonly associated with HIV. The activation of oxidative phosphorylation is a key driver in chronic inflammation. This shift is evident in studies documenting the increased activity of mitochondrial respiratory chain complexes in HIV-1 infected cells (122–132). Such changes suggest that HIV-1 exploits the host’s mitochondrial machinery to its advantage, potentially exacerbating the inflammatory response. While ART effectively controls viral replication, it does not fully rectify this metabolic dysregulation, adding another layer to the complexity. Long-term ART use has been associated with continued mitochondrial dysfunction (133, 134), indicating that the impact of HIV on oxidative phosphorylation is a persistent challenge.
HIV-1 infection significantly disrupts host cell metabolism, increasing glycolysis and fatty acid synthesis to enhance viral replication, leading to oxidative stress and mitochondrial damage. This is further complicated by antiretroviral therapy (ART), which contributes to metabolic conditions like dyslipidemia and insulin resistance, intensifying mitochondrial dysfunction (135–137).
Studies show that both HIV-1 infection and prolonged ART use increase the risk of oxidative phosphorylation dysregulation and mitochondrial disruption in people with HIV (PWH) (122–132). Reduced activity of respiratory chain complexes and altered mitochondrial functioning in ART-naïve PWH have been linked to neurocognitive impairment (125). ART’s impact on oxidative phosphorylation in PWH also suggests a reduction in mitochondrial function associated with chronic/controlled HIV-1 (133). Interestingly, pre-exposure prophylaxis (PreP) also shows reduced mitochondrial function in healthy individuals (134). Gender-specific differences in response to long-term ART have been observed, indicating possible variations in immunological recovery (138).
Recent studies highlight the role of purinergic signaling, particularly the interaction between extracellular ATP and P2X receptors, in HIV-1 pathogenesis (112, 113). These receptors, activated by ATP released from stressed cells, influence the HIV-1 life cycle and contribute to immunometabolic dysfunction (96, 106, 109). The dual role of these receptors in potentially inhibiting or facilitating HIV replication is mediated through CD39, which modulates extracellular ATP levels (72, 139–142).
Understanding the interplay between HIV-1 infection, ART, oxidative phosphorylation, and purinergic signaling is crucial for developing comprehensive HIV-1 treatment strategies. This involves addressing the viral challenges and the broader metabolic and mitochondrial dysfunctions. Future research should focus on identifying specific targets influenced by HIV-1 and ART and exploring the impact of purinergic signaling on these pathways. Such targeted exploration may lead to innovative therapeutic approaches addressing the infection and its metabolic and mitochondrial consequences.
NR: Conceptualization, Data curation, Investigation, Methodology, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. TF: Writing – original draft. EH: Writing – original draft. MW: Writing – original draft. AK: Writing – original draft. JM: Writing – original draft. TS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work is supported by T32GM146636 (JM), NIDA R01 DA052255 (TS), NIDA R01 DA054526 (TS), NIMH R01 MH134319 (TS), and NIAID R21 AI162223 (TS).
We are especially grateful to our colleagues at the Icahn School of Medicine at Mount Sinai Chen/Swartz laboratory for their continuous support and constructive criticism during the writing and editing process. Their expertise and perspectives have been instrumental in refining the content and focus of this review. Our heartfelt thanks to all the patients and volunteers who have participated in HIV research. Their involvement has been fundamental in advancing our understanding of the disease and developing effective treatment strategies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
HIV-1, Human Immunodeficiency Virus Type 1; ART, Antiretroviral Therapy; PBMC, Peripheral Blood Mononuclear Cells; ROS, Reactive Oxygen Species; ETC, Electron Transport Chain; NRTIs, Nucleoside/Nucleotide Reverse Transcriptase Inhibitors; NNRTIs, Non-nucleoside Reverse Transcriptase Inhibitors; PIs, Protease Inhibitors; ATP, Adenosine Triphosphate; NADH, Nicotinamide Adenine Dinucleotide (Reduced Form); FADH2, Flavin Adenine Dinucleotide (Reduced Form); Cyt-c, Cytochrome c; TCA, Tricarboxylic Acid Cycle; GLUT1, Glucose Transporter 1; SMO, Spermine Oxidase; EGFR, Epidermal Growth Factor Receptor; mTFA, Mitochondrial Transcription Factor A.
1. Ellis RJ, Iudicello JE, Heaton RK, Isnard S, Lin J, Routy JP, et al. Markers of gut barrier function and microbial translocation associate with lower gut microbial diversity in people with HIV. Viruses. (2021) 13(1):1891. doi: 10.3390/v13101891
2. Muñoz-Arias I, Doitsh G, Yang Z, Sowinski S, Ruelas D, Greene WC. Blood-derived CD4 T cells naturally resist pyroptosis during abortive HIV-1 infection. Cell Host Microbe. (2015) 18:463–70. doi: 10.1016/j.chom.2015.09.010
3. Okoye AA, Picker LJ. CD4(+) T-cell depletion in HIV infection: mechanisms of immunological failure. Immunol Rev. (2013) 254:54–64. doi: 10.1111/imr.12066
4. Lederman MM, Calabrese L, Funderburg NT, Clagett B, Medvik K, Bonilla H, et al. Immunologic failure despite suppressive antiretroviral therapy is related to activation and turnover of memory CD4 cells. J Infect Dis. (2011) 204:1217–26. doi: 10.1093/infdis/jir507
5. Bandera A, Ferrario G, Saresella M, Marventano I, Soria A, Zanini F, et al. CD4+ T cell depletion, immune activation and increased production of regulatory T cells in the thymus of HIV-infected individuals. PloS One. (2010) 5:e10788. doi: 10.1371/journal.pone.0010788
6. Tenorio AR, Zheng Y, Bosch RJ, Krishnan S, Rodriguez B, Hunt PW, et al. Soluble markers of inflammation and coagulation but not T-cell activation predict non-AIDS-defining morbid events during suppressive antiretroviral treatment. J Infect Dis. (2014) 210:1248–59. doi: 10.1093/infdis/jiu254
7. Deeks SG, Tracy R, Douek DC. Systemic effects of inflammation on health during chronic HIV infection. Immunity. (2013) 39:633–45. doi: 10.1016/j.immuni.2013.10.001
8. Klatt NR, Chomont N, Douek DC, Deeks SG. Immune activation and HIV persistence: implications for curative approaches to HIV infection. Immunol Rev. (2013) 254:326–42. doi: 10.1111/imr.12065
9. Deeks SG. HIV infection, inflammation, immunosenescence, and aging. Annu Rev Med. (2011) 62:141–55. doi: 10.1146/annurev-med-042909-093756
10. Wilson DF. Oxidative phosphorylation: regulation and role in cellular and tissue metabolism. J Physiol. (2017) 595:7023–38. doi: 10.1113/JP273839
11. Miranda-Quintana RA, Martínez González M, Ayers PW. Electronegativity and redox reactions. Phys Chem Chem Phys. (2016) 18:22235–43. doi: 10.1039/C6CP03213C
12. Matlin KS. The heuristic of form: mitochondrial morphology and the explanation of oxidative phosphorylation. J Hist Biol. (2016) 49:37–94. doi: 10.1007/s10739-015-9418-3
13. Nirody JA, Budin I, Rangamani P. ATP synthase: Evolution, energetics, and membrane interactions. J Gen Physiol. (2020) 152(11):e201912475. doi: 10.1085/jgp.201912475
14. Hroudová J, Fišar Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural Regener Res. (2013) 8:363–75. doi: 10.3969/j.issn.1673-5374.2013.04.009
15. Hüttemann M, Lee I, Pecinova A, Pecina P, Przyklenk K, Doan JW. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J Bioenerg Biomembr. (2008) 40:445–56. doi: 10.1007/s10863-008-9169-3
16. Ferguson-Miller S, Brautigan DL, Margoliash E. Correlation of the kinetics of electron transfer activity of various eukaryotic cytochromes c with binding to mitochondrial cytochrome c oxidase. J Biol Chem. (1976) 251:1104–15. doi: 10.1016/S0021-9258(17)33807-3
17. Guo D, Nguyen T, Ogbi M, Tawfik H, Ma G, Yu Q, et al. Protein kinase C-epsilon coimmunoprecipitates with cytochrome oxidase subunit IV and is associated with improved cytochrome-c oxidase activity and cardioprotection. Am J Physiol Heart Circ Physiol. (2007) 293:H2219–30. doi: 10.1152/ajpheart.01306.2006
18. Boerner JL, Demory ML, Silva C, Parsons SJ. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol Cell Biol. (2004) 24:7059–71. doi: 10.1128/MCB.24.16.7059-7071.2004
19. Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, et al. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem. (2005) 280:6094–100. doi: 10.1074/jbc.M411335200
20. Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell. (2015) 57:521–36. doi: 10.1016/j.molcel.2015.01.003
21. Lopez-Fabuel I, Le Douce J, Logan A, James AM, Bonvento G, Murphy MP, et al. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc Natl Acad Sci U.S.A. (2016) 113:13063–8. doi: 10.1073/pnas.1613701113
22. Vaupel P, Schmidberger H, Mayer A. The Warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol. (2019) 95:912–9. doi: 10.1080/09553002.2019.1589653
23. Sanchez EL, Lagunoff M. Viral activation of cellular metabolism. Virology. (2015) 479-480:609–18. doi: 10.1016/j.virol.2015.02.038
24. Valle-Casuso JC, Angin M, Volant S, Passaes C, Monceaux V, Mikhailova A, et al. Cellular metabolism is a major determinant of HIV-1 reservoir seeding in CD4. Cell Metab. (2019) 29:611–626.e5. doi: 10.1016/j.cmet.2018.11.015
25. Valentín-Guillama G, López S, Kucheryavykh YV, Chorna NE, Pérez J, Ortiz-Rivera J, et al. HIV-1 envelope protein gp120 promotes proliferation and the activation of glycolysis in glioma cell. Cancers (Basel). (2018) 10(9):301. doi: 10.3390/cancers10090301
26. Kavanagh Williamson M, Coombes N, Juszczak F, Athanasopoulos M, Khan MB, Eykyn TR, et al. Upregulation of glucose uptake and hexokinase activity of primary human CD4+ T cells in response to infection with HIV-1. Viruses. (2018) 10(3):114. doi: 10.3390/v10030114
27. Hegedus A, Kavanagh Williamson M, Huthoff H. HIV-1 pathogenicity and virion production are dependent on the metabolic phenotype of activated CD4+ T cells. Retrovirology. (2014) 11:98. doi: 10.1186/s12977-014-0098-4
28. Arjona SP, Allen CNS, Santerre M, Gross S, Soboloff J, Booze R, et al. Disruption of Mitochondrial-associated ER membranes by HIV-1 tat protein contributes to premature brain aging. CNS Neurosci Ther. (2023) 29:365–77. doi: 10.1111/cns.14011
29. Huang CY, Chiang SF, Lin TY, Chiou SH, Chow KC. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PloS One. (2012) 7:e33657. doi: 10.1371/journal.pone.0033657
30. Crater JM, Nixon DF, Furler O’Brien RL. HIV-1 replication and latency are balanced by mTOR-driven cell metabolism. Front Cell Infect Microbiol. (2022) 12:1068436. doi: 10.3389/fcimb.2022.1068436
31. Hollenbaugh JA, Munger J, Kim B. Metabolite profiles of human immunodeficiency virus infected CD4+ T cells and macrophages using LC-MS/MS analysis. Virology. (2011) 415:153–9. doi: 10.1016/j.virol.2011.04.007
32. Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. (2012) 13:223–8. doi: 10.1038/ni.2236
33. Ono A, Waheed AA, Freed EO. Depletion of cellular cholesterol inhibits membrane binding and higher-order multimerization of human immunodeficiency virus type 1 Gag. Virology. (2007) 360:27–35. doi: 10.1016/j.virol.2006.10.011
34. Rahman AN, Liu J, Mujib S, Kidane S, Ali A, Szep S, et al. Elevated glycolysis imparts functional ability to CD8. Life Sci Alliance. (2021) 4(11):e202101081. doi: 10.26508/lsa.202101081
35. Palmer CS, Cherry CL, Sada-Ovalle I, Singh A, Crowe SM. Glucose metabolism in T cells and monocytes: new perspectives in HIV pathogenesis. EBioMedicine. (2016) 6:31–41. doi: 10.1016/j.ebiom.2016.02.012
36. Loisel-Meyer S, Swainson L, Craveiro M, Oburoglu L, Mongellaz C, Costa C, et al. Glut1-mediated glucose transport regulates HIV infection. Proc Natl Acad Sci U.S.A. (2012) 109:2549–54. doi: 10.1073/pnas.1121427109
37. Ahmed D, Roy D, Cassol E. Examining relationships between metabolism and persistent inflammation in HIV patients on antiretroviral therapy. Mediators Inflammation. (2018) 2018:6238978. doi: 10.1155/2018/6238978
38. Wester CW, Okezie OA, Thomas AM, Bussmann H, Moyo S, Muzenda T, et al. Higher-than-expected rates of lactic acidosis among highly active antiretroviral therapy-treated women in Botswana: preliminary results from a large randomized clinical trial. J Acquir Immune Defic Syndr. (2007) 46:318–22. doi: 10.1097/QAI.0b013e3181568e3f
39. Mikaeloff F, Gelpi M, Escos A, Knudsen AD, Høgh J, Benfield T, et al. Transcriptomics age acceleration in prolonged treated HIV infection. Aging Cell. (2023) 22:e13951. doi: 10.1111/acel.13951
40. Tripathy MK, Mitra D. Differential modulation of mitochondrial OXPHOS system during HIV-1 induced T-cell apoptosis: up regulation of Complex-IV subunit COX-II and its possible implications. Apoptosis. (2010) 15:28–40. doi: 10.1007/s10495-009-0408-9
41. Kallianpur KJ, Walker M, Gerschenson M, Shikuma CM, Gangcuangco LMA, Kohorn L, et al. Systemic mitochondrial oxidative phosphorylation protein levels correlate with neuroimaging measures in chronically HIV-infected individuals. AIDS Res Hum Retroviruses. (2020) 36:83–91. doi: 10.1089/aid.2019.0240
42. Maagaard A, Kvale D. Long term adverse effects related to nucleoside reverse transcriptase inhibitors: clinical impact of mitochondrial toxicity. Scand J Infect Dis. (2009) 41:808–17. doi: 10.3109/00365540903186181
43. Feeney ER, van Vonderen MG, Wit F, Danner SA, van Agtmael MA, Villarroya F, et al. Zidovudine/lamivudine but not nevirapine in combination with lopinavir/ritonavir decreases subcutaneous adipose tissue mitochondrial DNA. AIDS. (2012) 26:2165–74. doi: 10.1097/QAD.0b013e328358b279
44. McComsey GA, Daar ES, O’Riordan M, Collier AC, Kosmiski L, Santana JL, et al. Changes in fat mitochondrial DNA and function in subjects randomized to abacavir-lamivudine or tenofovir DF-emtricitabine with atazanavir-ritonavir or efavirenz: AIDS Clinical Trials Group study A5224s, substudy of A5202. J Infect Dis. (2013) 207:604–11. doi: 10.1093/infdis/jis720
45. Maagaard A, Kvale D. Mitochondrial toxicity in HIV-infected patients both off and on antiretroviral treatment: a continuum or distinct underlying mechanisms? J Antimicrob Chemother. (2009) 64:901–9. doi: 10.1093/jac/dkp316
46. Lewis W, Dalakas MC. Mitochondrial toxicity of antiviral drugs. Nat Med. (1995) 1:417–22. doi: 10.1038/nm0595-417
47. Murata H, Hruz PW, Mueckler M. Indinavir inhibits the glucose transporter isoform Glut4 at physiologic concentrations. AIDS. (2002) 16:859–63. doi: 10.1097/00002030-200204120-00005
48. Murata H, Hruz PW, Mueckler M. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J Biol Chem. (2000) 275:20251–4. doi: 10.1074/jbc.C000228200
49. Hileman CO, Kalayjian RC, Azzam S, Schlatzer D, Wu K, Tassiopoulos K, et al. Plasma citrate and succinate are associated with neurocognitive impairment in older people with HIV. Clin Infect Dis. (2021) 73:e765–72. doi: 10.1093/cid/ciab107
50. Banki K, Hutter E, Gonchoroff NJ, Perl A. Molecular ordering in HIV-induced apoptosis. Oxidative stress, activation of caspases, and cell survival are regulated by transaldolase. J Biol Chem. (1998) 273:11944–53. doi: 10.1074/jbc.273.19.11944
51. Deshmane SL, Mukerjee R, Fan S, Del Valle L, Michiels C, Sweet T, et al. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J Biol Chem. (2009) 284:11364–73. doi: 10.1074/jbc.M809266200
52. Shah A, Kumar S, Simon SD, Singh DP, Kumar A. HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome P450 2E1. Cell Death Dis. (2013) 4:e850. doi: 10.1038/cddis.2013.374
53. Foga IO, Nath A, Hasinoff BB, Geiger JD. Antioxidants and dipyridamole inhibit HIV-1 gp120-induced free radical-based oxidative damage to human monocytoid cells. J Acquir Immune Defic Syndr Hum Retrovirol. (1997) 16:223–9. doi: 10.1097/00042560-199712010-00001
54. Mataramvura H, Bunders MJ, Duri K. Human immunodeficiency virus and antiretroviral therapy-mediated immune cell metabolic dysregulation in children born to HIV-infected women: potential clinical implications. Front Immunol. (2023) 14:1182217. doi: 10.3389/fimmu.2023.1182217
55. Lecoeur H, Borgne-Sanchez A, Chaloin O, El-Khoury R, Brabant M, Langonné A, et al. HIV-1 Tat protein directly induces mitochondrial membrane permeabilization and inactivates cytochrome c oxidase. Cell Death Dis. (2012) 3:e282. doi: 10.1038/cddis.2012.21
56. Fields JA, Serger E, Campos S, Divakaruni AS, Kim C, Smith K, et al. HIV alters neuronal mitochondrial fission/fusion in the brain during HIV-associated neurocognitive disorders. Neurobiol Dis. (2016) 86:154–69. doi: 10.1016/j.nbd.2015.11.015
57. Vilhardt F, Plastre O, Sawada M, Suzuki K, Wiznerowicz M, Kiyokawa E, et al. The HIV-1 Nef protein and phagocyte NADPH oxidase activation. J Biol Chem. (2002) 277:42136–43. doi: 10.1074/jbc.M200862200
58. Halestrap AP, Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem. (2003) 10:1507–25. doi: 10.2174/0929867033457278
59. Gu Y, Wu RF, Xu YC, Flores SC, Terada LS. HIV Tat activates c-Jun amino-terminal kinase through an oxidant-dependent mechanism. Virology. (2001) 286:62–71. doi: 10.1006/viro.2001.0998
60. Capone C, Cervelli M, Angelucci E, Colasanti M, Macone A, Mariottini P, et al. A role for spermine oxidase as a mediator of reactive oxygen species production in HIV-Tat-induced neuronal toxicity. Free Radic Biol Med. (2013) 63:99–107. doi: 10.1016/j.freeradbiomed.2013.05.007
61. Rozzi SJ, Borelli G, Ryan K, Steiner JP, Reglodi D, Mocchetti I, et al. PACAP27 is protective against tat-induced neurotoxicity. J Mol Neurosci. (2014) 54:485–93. doi: 10.1007/s12031-014-0273-z
62. Haughey NJ, Mattson MP. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J Acquir Immune Defic Syndr. (2002) 31 Suppl 2:S55–61. doi: 10.1097/00126334-200210012-00005
63. Ferri KF, Jacotot E, Blanco J, Esté JA, Kroemer G. Mitochondrial control of cell death induced by HIV-1-encoded proteins. Ann N Y Acad Sci. (2000) 926:149–64. doi: 10.1111/j.1749-6632.2000.tb05609.x
64. Avdoshina V, Fields JA, Castellano P, Dedoni S, Palchik G, Trejo M, et al. The HIV protein gp120 alters mitochondrial dynamics in neurons. Neurotox Res. (2016) 29:583–93. doi: 10.1007/s12640-016-9608-6
65. Elbim C, Pillet S, Prevost MH, Preira A, Girard PM, Rogine N, et al. The role of phagocytes in HIV-related oxidative stress. J Clin Virol. (2001) 20:99–109. doi: 10.1016/S1386-6532(00)00133-5
66. Elbim C, Pillet S, Prevost MH, Preira A, Girard PM, Rogine N, et al. Redox and activation status of monocytes from human immunodeficiency virus-infected patients: relationship with viral load. J Virol. (1999) 73:4561–6. doi: 10.1128/JVI.73.6.4561-4566.1999
67. Burnstock G. P2X ion channel receptors and inflammation. Purinergic Signal. (2016) 12:59–67. doi: 10.1007/s11302-015-9493-0
68. Kopp R, Krautloher A, Ramírez-Fernández A, Nicke A. P2X7 interactions and signaling - making head or tail of it. Front Mol Neurosci. (2019) 12:183. doi: 10.3389/fnmol.2019.00183
69. Di Virgilio F, Schmalzing G, Markwardt F. The elusive P2X7 macropore. Trends Cell Biol. (2018) 28:392–404. doi: 10.1016/j.tcb.2018.01.005
70. Pacheco PA, Faria RX, Ferreira LG, Paixão IC. Putative roles of purinergic signaling in human immunodeficiency virus-1 infection. Biol Direct. (2014) 9:21. doi: 10.1186/1745-6150-9-21
71. Dubey RC, Mishra N, Gaur R. G protein-coupled and ATP-sensitive inwardly rectifying potassium ion channels are essential for HIV entry. Sci Rep. (2019) 9:4113. doi: 10.1038/s41598-019-40968-x
72. Nikolova M, Carriere M, Jenabian MA, Limou S, Younas M, Kök A, et al. CD39/adenosine pathway is involved in AIDS progression. PloS Pathog. (2011) 7:e1002110. doi: 10.1371/journal.ppat.1002110
73. Goepfert C, Imai M, Brouard S, Csizmadia E, Kaczmarek E, Robson SC. CD39 modulates endothelial cell activation and apoptosis. Mol Med. (2000) 6:591–603. doi: 10.1007/BF03401797
74. Savio LEB, de Andrade Mello P, Figliuolo VR, de Avelar Almeida TF, Santana PT, Oliveira SDS, et al. CD39 limits P2X7 receptor inflammatory signaling and attenuates sepsis-induced liver injury. J Hepatol. (2017) 67:716–26. doi: 10.1016/j.jhep.2017.05.021
75. Soare AY, Malik HS, Durham ND, Freeman TL, Alvarez R, Patel F, et al. P2X1 selective antagonists block HIV-1 infection through inhibition of envelope conformation-dependent fusion. J Virol. (2020) 94(6):e01622–19. doi: 10.1128/JVI.01622-19
76. Séror C, Melki MT, Subra F, Raza SQ, Bras M, Saïdi H, et al. Extracellular ATP acts on P2Y2 purinergic receptors to facilitate HIV-1 infection. J Exp Med. (2011) 208:1823–34. doi: 10.1084/jem.20101805
77. Soare AY, Durham ND, Gopal R, Tweel B, Hoffman KW, Brown JA, et al. P2X antagonists inhibit HIV-1 productive infection and inflammatory cytokines interleukin-10 (IL-10) and IL-1β in a human tonsil explant model. J Virol. (2019) 93:e01186–18. doi: 10.1128/JVI.01186-18
78. Swartz TH, Dubyak GR, Chen BK. Purinergic receptors: key mediators of HIV-1 infection and inflammation. Front Immunol. (2015) 6:585. doi: 10.3389/fimmu.2015.00585
79. Giroud C, Marin M, Hammonds J, Spearman P, Melikyan GB. P2X1 receptor antagonists inhibit HIV-1 fusion by blocking virus-coreceptor interactions. J Virol. (2015) 89:9368–82. doi: 10.1128/JVI.01178-15
80. Marin M, Du Y, Giroud C, Kim JH, Qui M, Fu H, et al. High-throughput HIV-cell fusion assay for discovery of virus entry inhibitors. Assay Drug Dev Technol. (2015) 13:155–66. doi: 10.1089/adt.2015.639
81. Swartz TH, Esposito AM, Durham ND, Hartmann BM, Chen BK. P2X-selective purinergic antagonists are strong inhibitors of HIV-1 fusion during both cell-to-cell and cell-free infection. J Virol. (2014) 88:11504–15. doi: 10.1128/JVI.01158-14
82. Hazleton JE, Berman JW, Eugenin EA. Purinergic receptors are required for HIV-1 infection of primary human macrophages. J Immunol. (2012) 188:4488–95. doi: 10.4049/jimmunol.1102482
83. Soare AY, Freeman TL, Min AK, Malik HS, Osota EO, Swartz TH. P2RX7 at the host-pathogen interface of infectious diseases. Microbiol Mol Biol Rev. (2021) 85(1):e00055–20. doi: 10.1128/MMBR.00055-20
84. Esposito AM, Soare AY, Patel F, Satija N, Chen BK, Swartz TH. A high-throughput cre-lox activated viral membrane fusion assay to identify inhibitors of HIV-1 viral membrane fusion. J Vis Exp. (2018). doi: 10.3791/58074-v
85. Esposito AM, Cheung P, Swartz TH, Li H, Tsibane T, Durham ND, et al. A high throughput Cre-lox activated viral membrane fusion assay identifies pharmacological inhibitors of HIV entry. Virology. (2016) 490:6–16. doi: 10.1016/j.virol.2015.10.013
86. Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis. (2003) 187:1534–43. doi: 10.1086/374786
87. Massanella M, Negredo E, Perez-Alvarez N, Puig J, Ruiz-Hernandez R, Bofill M, et al. CD4 T-cell hyperactivation and susceptibility to cell death determine poor CD4 T-cell recovery during suppressive HAART. AIDS. (2010) 24:959–68. doi: 10.1097/QAD.0b013e328337b957
88. Massanella M, Fromentin R, Chomont N. Residual inflammation and viral reservoirs: alliance against an HIV cure. Curr Opin HIV AIDS. (2016) 11:234–41. doi: 10.1097/COH.0000000000000230
89. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U.S.A. (2009) 106:20388–93. doi: 10.1073/pnas.0908698106
90. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. (2006) 440:228–32. doi: 10.1038/nature04515
91. Hung SC, Choi CH, Said-Sadier N, Johnson L, Atanasova KR, Sellami H, et al. P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PloS One. (2013) 8:e70210. doi: 10.1371/journal.pone.0070210
92. Sun L, Ma W, Gao W, Xing Y, Chen L, Xia Z, et al. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. (2019) 10:542. doi: 10.1038/s41419-019-1761-4
93. Malik S, Eugenin EA. Role of Connexin and Pannexin containing channels in HIV infection and NeuroAIDS. Neurosci Lett. (2019) 695:86–90. doi: 10.1016/j.neulet.2017.09.005
94. Orellana JA, Velasquez S, Williams DW, Sáez JC, Berman JW, Eugenin EA. Pannexin1 hemichannels are critical for HIV infection of human primary CD4+ T lymphocytes. J Leukoc Biol. (2013) 94:399–407. doi: 10.1189/jlb.0512249
95. Gajardo-Gómez R, Santibañez CA, Labra VC, Gómez GI, Eugenin EA, Orellana JA. HIV gp120 protein increases the function of connexin 43 hemichannels and pannexin-1 channels in astrocytes: repercussions on astroglial function. Int J Mol Sci. (2020) 21(7):2503. doi: 10.3390/ijms21072503
96. Freeman TL, Zhao C, Schrode N, Fortune T, Shroff S, Tweel B, et al. HIV-1 activates oxidative phosphorylation in infected CD4 T cells in a human tonsil explant model. Front Immunol. (2023) 14:1172938. doi: 10.3389/fimmu.2023.1172938
97. Velasquez S, Prevedel L, Valdebenito S, Gorska AM, Golovko M, Khan N, et al. Circulating levels of ATP is a biomarker of HIV cognitive impairment. EBioMedicine. (2020) 51:102503. doi: 10.1016/j.ebiom.2019.10.029
98. Heaton RK, Clifford DB, Franklin DR, Woods SP, Ake C, Vaida F, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. (2010) 75:2087–96. doi: 10.1212/WNL.0b013e318200d727
99. Zayyad Z, Spudich S. Neuropathogenesis of HIV: from initial neuroinvasion to HIV-associated neurocognitive disorder (HAND). Curr HIV/AIDS Rep. (2015) 12:16–24. doi: 10.1007/s11904-014-0255-3
100. Veenstra M, León-Rivera R, Li M, Gama L, Clements JE, Berman JW. Mechanisms of CNS viral seeding by HIV. mBio. (2017) 8(5):e01280–17. doi: 10.1128/mBio.01280-17
101. Datta G, Miller NM, Afghah Z, Geiger JD, Chen X. HIV-1 gp120 promotes lysosomal exocytosis in human schwann cells. Front Cell Neurosci. (2019) 13:329. doi: 10.3389/fncel.2019.00329
102. Yu Y, Ugawa S, Ueda T, Ishida Y, Inoue K, Kyaw Nyunt A, et al. Cellular localization of P2X7 receptor mRNA in the rat brain. Brain Res. (2008) 1194:45–55. doi: 10.1016/j.brainres.2007.11.064
103. Kakuda TN. Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity. Clin Ther. (2000) 22:685–708. doi: 10.1016/S0149-2918(00)90004-3
104. Robertson K, Liner J, Meeker RB. Antiretroviral neurotoxicity. J Neurovirol. (2012) 18:388–99. doi: 10.1007/s13365-012-0120-3
105. Cheney L, Guzik H, Macaluso FP, Macian F, Cuervo AM, Berman JW. HIV Nef and antiretroviral therapy have an inhibitory effect on autophagy in human astrocytes that may contribute to HIV-associated neurocognitive disorders. Cells. (2020) 9(6):1426. doi: 10.3390/cells9061426
106. Guo H, Wang Q, Ghneim K, Wang L, Rampanelli E, Holley-Guthrie E, et al. Multi-omics analyses reveal that HIV-1 alters CD4. Nat Immunol. (2021) 22:423–33. doi: 10.1038/s41590-021-00898-1
107. Ambikan AT, Svensson-Akusjärvi S, Krishnan S, Sperk M, Nowak P, Vesterbacka J, et al. Genome-scale metabolic models for natural and long-term drug-induced viral control in HIV infection. Life Sci Alliance. (2022) 5(9):e202201405. doi: 10.26508/lsa.202201405
108. Alrubayyi A, Moreno-Cubero E, Hameiri-Bowen D, Matthews R, Rowland-Jones S, Schurich A, et al. Functional restoration of exhausted CD8 T cells in chronic HIV-1 infection by targeting mitochondrial dysfunction. Front Immunol. (2022) 13:908697. doi: 10.3389/fimmu.2022.908697
109. Castellano P, Prevedel L, Valdebenito S, Eugenin EA. HIV infection and latency induce a unique metabolic signature in human macrophages. Sci Rep. (2019) 9:3941. doi: 10.1038/s41598-019-39898-5
110. Aquaro S, Caliò R, Balzarini J, Bellocchi MC, Garaci E, Perno CF. Macrophages and HIV infection: therapeutical approaches toward this strategic virus reservoir. Antiviral Res. (2002) 55:209–25. doi: 10.1016/S0166-3542(02)00052-9
111. Rezaei S, Timani KA, He JJ. Metformin treatment leads to increased HIV transcription and gene expression through increased CREB phosphorylation and recruitment to the HIV LTR promoter. Aging Dis. (2023). doi: 10.14336/AD.2023.0705
112. Ortiz-Brizuela E, Pérez-Patrigeon S, Recillas-Gispert C, Gómez-Pérez FJ. Lactic acidosis complicating metformin and non-nucleoside reverse transcriptase inhibitor combination therapy: A smoldering threat in the post-HAART era. Rev Invest Clin. (2015) 67:273–4.
113. Chaparala S, Da Silva RC, Papadopoulos JP. Severe lactic acidosis due to acute intoxication by emtricitabine/tenofovir alafenamide. Cureus. (2021) 13:e19008. doi: 10.7759/cureus.19008
114. Adebiyi OO, Adebiyi OA, Owira PM. Naringin reverses hepatocyte apoptosis and oxidative stress associated with HIV-1 nucleotide reverse transcriptase inhibitors-induced metabolic complications. Nutrients. (2015) 7:10352–68. doi: 10.3390/nu7125540
115. Oluwafeyisetan A, Olubunmi A, Peter O. Naringin ameliorates HIV-1 nucleoside reverse transcriptase inhibitors- induced mitochondrial toxicity. Curr HIV Res. (2016) 14:506–16. doi: 10.2174/1570162X14666160520114639
116. Funderburg NT, Jiang Y, Debanne SM, Labbato D, Juchnowski S, Ferrari B, et al. Rosuvastatin reduces vascular inflammation and T-cell and monocyte activation in HIV-infected subjects on antiretroviral therapy. J Acquir Immune Defic Syndr. (2015) 68:396–404. doi: 10.1097/QAI.0000000000000478
117. Grinspoon SK, Fitch KV, Zanni MV, Fichtenbaum CJ, Umbleja T, Aberg JA, et al. Pitavastatin to prevent cardiovascular disease in HIV infection. N Engl J Med. (2023) 389:687–99. doi: 10.1056/NEJMoa2304146
118. Morrison JT, Longenecker CT, Mittelsteadt A, Jiang Y, Debanne SM, McComsey GA. Effect of rosuvastatin on plasma coenzyme Q10 in HIV-infected individuals on antiretroviral therapy. HIV Clin Trials. (2016) 17:140–6. doi: 10.1080/15284336.2016.1184863
119. Kang S, Tang H. HIV-1 infection and glucose metabolism reprogramming of T cells: another approach toward functional cure and reservoir eradication. Front Immunol. (2020) 11:572677. doi: 10.3389/fimmu.2020.572677
120. Allen CNS, Arjona SP, Santerre M, De Lucia C, Koch WJ, Sawaya BE. Metabolic reprogramming in HIV-associated neurocognitive disorders. Front Cell Neurosci. (2022) 16:812887. doi: 10.3389/fncel.2022.812887
121. Parikh NI, Gerschenson M, Bennett K, Gangcuangco LM, Lopez MS, Mehta NN, et al. Lipoprotein concentration, particle number, size and cholesterol efflux capacity are associated with mitochondrial oxidative stress and function in an HIV positive cohort. Atherosclerosis. (2015) 239:50–4. doi: 10.1016/j.atherosclerosis.2014.12.005
122. Perrin S, Cremer J, Roll P, Faucher O, Ménard A, Reynes J, et al. HIV-1 infection and first line ART induced differential responses in mitochondria from blood lymphocytes and monocytes: the ANRS EP45 “Aging”. Study PloS One. (2012) 7:e41129. doi: 10.1371/journal.pone.0041129
123. Torres RA, Lewis W. Aging and HIV/AIDS: pathogenetic role of therapeutic side effects. Lab Invest. (2014) 94:120–8. doi: 10.1038/labinvest.2013.142
124. Arts EJ, Hazuda DJ. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med. (2012) 2:a007161. doi: 10.1101/cshperspect.a007161
125. Miró O, López S, Martínez E, Pedrol E, Milinkovic A, Deig E, et al. Mitochondrial effects of HIV infection on the peripheral blood mononuclear cells of HIV-infected patients who were never treated with antiretrovirals. Clin Infect Dis. (2004) 39:710–6. doi: 10.1086/423176
126. Brinkman K, ter Hofstede HJ, Burger DM, Smeitink JA, Koopmans PP. Adverse effects of reverse transcriptase inhibitors: mitochondrial toxicity as common pathway. AIDS. (1998) 12:1735–44. doi: 10.1097/00002030-199814000-00004
127. Apostolova N, Blas-García A, Esplugues JV. Mitochondrial toxicity in HAART: an overview of in vitro evidence. Curr Pharm Des. (2011) 17:2130–44. doi: 10.2174/138161211796904731
128. Fiala M, Murphy T, MacDougall J, Yang W, Luque A, Iruela-Arispe L, et al. HAART drugs induce mitochondrial damage and intercellular gaps and gp120 causes apoptosis. Cardiovasc Toxicol. (2004) 4:327–37. doi: 10.1385/CT:4:4
129. Kaur H, Minchella P, Alvarez-Carbonell D, Purandare N, Nagampalli VK, Blankenberg D, et al. Contemporary antiretroviral therapy dysregulates iron transport and augments mitochondrial dysfunction in HIV-infected human microglia and neural-lineage cells. Int J Mol Sci. (2023) 24(15):12242. doi: 10.20944/preprints202307.1492.v1
130. Wallace J, Gonzalez H, Rajan R, Narasipura SD, Virdi AK, Olali AZ, et al. Anti-HIV drugs cause mitochondrial dysfunction in monocyte-derived macrophages. Antimicrob Agents Chemother. (2022) 66:e0194121. doi: 10.1128/aac.01941-21
131. López S, Negredo E, Garrabou G, Puig J, Ruiz L, Sanjurjo E, et al. Longitudinal study on mitochondrial effects of didanosine-tenofovir combination. AIDS Res Hum Retroviruses. (2006) 22:33–9. doi: 10.1089/aid.2006.22.33
132. Massanella M, Singhania A, Beliakova-Bethell N, Pier R, Lada SM, White CH, et al. Differential gene expression in HIV-infected individuals following ART. Antiviral Res. (2013) 100:420–8. doi: 10.1016/j.antiviral.2013.07.017
133. Gangcuangco LMA, Mitchell BI, Siriwardhana C, Kohorn LB, Chew GM, Bowler S, et al. Mitochondrial oxidative phosphorylation in peripheral blood mononuclear cells is decreased in chronic HIV and correlates with immune dysregulation. PloS One. (2020) 15:e0231761. doi: 10.1371/journal.pone.0231761
134. Bowman ER, Cameron C, Richardson B, Kulkarni M, Gabriel J, Kettelhut A, et al. Exposure of leukocytes to HIV preexposure prophylaxis decreases mitochondrial function and alters gene expression profiles. Antimicrob Agents Chemother. (2020) 65(1):e01755–20. doi: 10.1128/AAC.01755-20
135. Butterfield TR, Landay AL, Anzinger JJ. Dysfunctional immunometabolism in HIV infection: contributing factors and implications for age-related comorbid diseases. Curr HIV/AIDS Rep. (2020) 17:125–37. doi: 10.1007/s11904-020-00484-4
136. Schank M, Zhao J, Moorman JP, Yao ZQ. The impact of HIV- and ART-induced mitochondrial dysfunction in cellular senescence and aging. Cells. (2021) 10(1):174. doi: 10.3390/cells10010174
137. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol. (2023) 23:159–73. doi: 10.1038/s41577-022-00760-x
138. Zhang L, Wang Z, Chen Y, Zhang C, Xie S, Cui Y. Label-free proteomic analysis of PBMCs reveals gender differences in response to long-term antiretroviral therapy of HIV. J Proteomics. (2015) 126:46–53. doi: 10.1016/j.jprot.2015.05.033
139. Ravimohan S, Tamuhla N, Nfanyana K, Ni H, Steenhoff AP, Gross R, et al. Elevated pre-antiretroviral therapy CD39+CD8+ T cell frequency is associated with early mortality in advanced human immunodeficiency virus/tuberculosis co-infection. Clin Infect Dis. (2017) 64:1453–6. doi: 10.1093/cid/cix155
140. Shahbaz S, Okoye I, Blevins G, Elahi S. Elevated ATP via enhanced miRNA-30b, 30c, and 30e downregulates the expression of CD73 in CD8+ T cells of HIV-infected individuals. PloS Pathog. (2022) 18:e1010378. doi: 10.1371/journal.ppat.1010378
141. Song JW, Huang HH, Zhang C, Yang HG, Zhang JY, Xu RN, et al. Expression of CD39 is correlated with HIV DNA levels in naïve tregs in chronically infected ART naïve patients. Front Immunol. (2019) 10:2465. doi: 10.3389/fimmu.2019.02465
Keywords: HIV-1, oxidative phosphorylation, antiretroviral therapy (ART), mitochondrial dysfunction, immune metabolism
Citation: Rodriguez NR, Fortune T, Hegde E, Weinstein MP, Keane AM, Mangold JF and Swartz TH (2024) Oxidative phosphorylation in HIV-1 infection: impacts on cellular metabolism and immune function. Front. Immunol. 15:1360342. doi: 10.3389/fimmu.2024.1360342
Received: 22 December 2023; Accepted: 26 February 2024;
Published: 11 March 2024.
Edited by:
Pedro Gonzalez-Menendez, University of Oviedo, SpainReviewed by:
Nabab Khan, Yale University, United StatesCopyright © 2024 Rodriguez, Fortune, Hegde, Weinstein, Keane, Mangold and Swartz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Talia H. Swartz, dGFsaWEuc3dhcnR6QG1zc20uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.