
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 16 February 2024
Sec. Microbial Immunology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1357967
 Ahmed Adel Baz1,2,3,4,5
Ahmed Adel Baz1,2,3,4,5 Huafang Hao1,2,3,4Shimei Lan1,2,3,4Zhangcheng Li1,2,3,4Shuang Liu1,2,3,4
Huafang Hao1,2,3,4Shimei Lan1,2,3,4Zhangcheng Li1,2,3,4Shuang Liu1,2,3,4 Shengli Chen1,2,3,4*
Shengli Chen1,2,3,4* Yuefeng Chu1,2,3,4*
Yuefeng Chu1,2,3,4*Neutrophils are innate immune cells that have a vital role in host defense systems. Neutrophil extracellular traps (NETs) are one of neutrophils’ defense mechanisms against pathogens. NETs comprise an ejected lattice of chromatin associated with histones, granular proteins, and cytosolic proteins. They are thought to be an efficient strategy to capture and/or kill bacteria and received intensive research interest in the recent years. However, soon after NETs were identified, it was observed that certain bacteria were able to evade NET entrapment through many different mechanisms. Here, we outline the recent progress of NETs in bacterial infections and the strategies employed by bacteria to evade or withstand NETs. Identifying the molecules and mechanisms that modulate NET release will improve our understanding of the functions of NETs in infections and provide new avenues for the prevention and treatment of bacterial diseases.
Neutrophils are the most prevalent type of leukocytes circulating in the blood and are crucial for maintaining health. They differentiate from hematopoietic stem cells and are discharged from the bone marrow when terminally mature. They are the first cells to leave circulation and migrate to the infection site during an immunological challenge. There, they eliminate bacteria, communicate damage status to other immune cells, and are involved in the healing process (1). Much is known about the antimicrobial response of neutrophils. They are highly skilled in phagocytosis, which is their first vital mechanism for the eradication of disease-causing pathogens and elimination of dead cells and tissue debris (2, 3). The second mechanism for eliminating pathogens is degranulation, which involves regulation of the immune system response via the release of various granules (4–6).
Approximately 20 years ago, a new mechanism by which neutrophils fight infections was termed neutrophil extracellular traps (NETs) (7). This involves the loosening of chromatin in the neutrophil nucleus and forming complexes with granular and cytoplasmic proteins, which are then released into the extracellular environment. There, NETs encapsulate and kill microorganisms, and block their dissemination (7, 8).
NETs are a special form of programmed cell death, which is distinct from other cell death forms (apoptosis, necroptosis, and pyroptosis). In general, the complex process of NET formation begins with the recognition of microorganism, triggering the activation of the NETs pathway. This, in turn, leads to the breakdown of the nuclear membrane and the release of decondensed nuclear DNA into the cytoplasm. As this DNA mingles with cellular components including the histones, granules and cytoplasmic proteins, the process culminates in the rupture of the plasma membrane and the ultimate release of the NET structure into the extracellular space. Simultaneously, these traps are capable of degrading virulent bacterial factors (9–11).
NET release is triggered by various stimuli. A range of bacterial species induce NET release. For Gram-positive bacteria, such as Staphylococcus aureus (7, 12, 13), Streptococcus pyogenes (14, 15), Streptococcus pneumoniae (16), Streptococcus agalactiae (17), Streptococcus sanguinis (18), Streptococcus suis (19). For Gram-negative bacteria, such as Escherichia coli (20–22), Salmonella typhimurium (7), Shigella flexneri (7), Haemophilus influenzae (23), Pseudomonas aeruginosa (24, 25), Yersinia pseudotuberculosis (26), Photorhabdus luminescens (27). In addition, other bacterial species, including Mycobacterium tuberculosis (28, 29), Mycobacterium canettii (28), Mycobacterium avium (30), and Mycoplasma bovis (31). Furthermore, bacterial components like lipopolysaccharide (LPS) (7), flagella (25), bacterial toxin nigericin (17), and calcium ionophore A2318 (17) are also involved in NETs stimulation. Additionally, diverse inflammatory mediators including interferon (IFN)-α (32), IFN-γ (32), interleukin (IL)-8 (7), IL-1β (33, 34), IL-2 (35), IL-6 (36), IL-10 (36), tumor necrosis factor (TNF)-α (33, 37), granulocyte-macrophage colony-stimulating factor (GM-CSF) (38), transforming growth factor (TGF)-β (34), and complement factor 5a (C5a) (32, 38) have been shown to stimulate NET release.
Moreover, Neutrophil receptors play a crucial role in activation of neutrophil and formation of NETs. The specific engagement of cell receptors by extracellular signaling molecules activates diverse intracellular signaling cascades and regulate NETs functions. In response to different bacterial triggers, NETs can release by the activation of different receptors including toll-like receptors (TLRs) (39–44), nucleotide-binding oligomerization domain-like receptors (NLRs) (45), C-type lectin receptors (CLRs) (46), complement receptors (CRs) (47), Fc receptors (47–50) and other neutrophil receptors like protease activated receptor-2 (PAR-2) (51).
The composition of NETs varies depending on the stimuli, and is primarily made up of DNA and histone proteins (H1, H2A, H2B, H3, and H4), which are originated from the nucleus followed by granular proteins that are normally stored in distinctive neutrophil granules in the cytoplasm of neutrophil such as neutrophil elastase (NE), myeloperoxidase (MPO), defensins, and cathepsin G from primary granules, lactoferrin, cathelicidins, and lysozyme C from secondary granules, and gelatinase from tertiary granules, as well as other proteins such as calprotectin and proteinase 3 are located in the cytoplasm of neutrophils (7, 52–54) (Supplementary Table S1).
NETs were first reported in 2004 and were primarily described as a cell death process in activated neutrophil (7). When DNA leaks extracellularly, not all NET developments result in cell death, and not every neutrophil death necessarily results in NET formation. “NETosis” is often employed throughout research for referring to NET formation, which can be categorized to as “lytic” and “non-lytic” forms.
The “lytic” mechanism of NET formation is the interaction between stimuli and cell receptors, which triggers Raf-MEK-ERK signaling and activates the NADPH oxidase complex (55). This complex produces peroxide ions in neutrophils, increasing levels of cytosolic reactive oxygen species (ROS). MPO detects this rise and is often paired with other proteases to form the azurosome. MPO trigger the activation and translocation of NE from azurophilic granules to the nucleus, where elastase proteolytically processes histones to disrupt chromatin packaging (56, 57). MPO then binds to chromatin and synergizes with NE to decondense chromatin independently of its enzymatic activity (57). The nucleus expands along with its chromatin, and cell lysis occurs because of gasdermin D, which can be activated by serine proteases in neutrophil inducing pore formation in the cell membrane (58). NETs are extruded into the extracellular space after membrane rupture and cell death within 3–8 h of neutrophil activation (9, 59).
The “non-lytic” or vital mechanism, involves increasing the concentration of cytosolic calcium through calcium ionophores, which activate the protein arginine deiminase 4 (PAD4) that leads to citrullates arginine histone residues, and reduces their positive charge. Histones then begin to lose their electrostatic attraction to DNA, and the chromatin expands along with the nucleus (37, 60). This oxidase-independent mechanism occurs within minutes of gram-positive skin infections; the cells release NETs during crawling without lysis, which prevents systemic bacterial dissemination (13, 61). However, vital NET formation might be more closely associated with infection than previously thought because soon after releasing NETs, neutrophils are still viable and can carry out additional host response processes, such as phagocytosis, chemotaxis, and microbial killing (62). In addition to non-lytic NETs with nuclear DNA release, NETs composed of mitochondrial DNA, NE, and are first described in GM-CSF-primed neutrophils stimulated in vitro with LPS or C5a (38, 63) since this process would not require neutrophil lysis, which would enable phagocytosis to continue (10).
However, divergent views have persisted in this field. NETs can form in the absence of PAD4 and citrullinated histone 3 (64, 65). It can also occur during leukotoxic hypercitrullination (LTH), defective mitophagy, and organ injury (66). Other types of NETs have also been described, including cloudy NETs, spiky NETs, aggregated NETs, and bicarbonate-induced aggregated NETs (67). Therefore, we cautiously take opinions about these studies into consideration
NETs have various effects on the morphology of bacteria, including physically trapping and immobilizing them, leading to changes in their morphology as they become entangled in the web-like structure of the NETs (7). Additionally, they can release DNA, enzymes, and antimicrobial proteins that can damage the cell walls of bacteria and/or membranes, leading to changes in their morphology as they become structurally compromised (68–70). Furthermore, NETs are involved in the disruption of bacterial biofilms, which can also lead to changes in the morphology of the bacteria (71).
Along with the impact of NETs on the shape of bacteria, they can kill and limit the growth of bacteria and prevent their spread in the environment. They possess antimicrobial properties with components such as DNA, histones, granules, and cytoplasmic proteins that have bactericidal effects (7, 68, 72–75). Interestingly, NETs play an important role in the defense against bacteria, even in the absence of microbicidal activity. These infections are ensnared but not eliminated by NETs (28, 76–79), indicating that NET-mediated microbial trapping alone plays a substantial role in immune defense.
As mentioned above, the formation of NETs and their effect on bacterial infection are mostly reported in vitro conditions. However, there is limited research describing the stimulation of NETs in response to bacterial infection in vivo. It is difficult to verify NETs in vivo because it calls for specialized technical knowledge. Besides, it is hard to assess the kind of stimulus, its dosage, and its exposure duration in vivo. Researchers may use genetically modified animal models to study the role of specific proteins or signaling pathways in the regulation of NET formation during bacterial infection (80).
Furthermore, the composition of NETs determines their efficiency. Mice exposed to low concentrations of cathepsin G are more susceptible to infection with the Gram-positive bacterium S. aureus (81), whereas those exposed to low concentrations of NE are more susceptible to Gram-negative bacteria such as E. coli, K. pneumoniae, and other Enterobacteriaceae (70, 81). Additionally, the antimicrobial response of NETs is influenced by the environment in which they are developed; NETs under static conditions show limited bacterial killing, whereas those under dynamic conditions show enhanced bacterial trapping and reduced killing (78).
Although NETs protect the host against microbes, there are diseases and conditions that can interfere with the release of NETs. Some examples of diseases that can affect NET release or neutrophil function are autoimmune diseases, chronic inflammatory conditions and sepsis, which can alter the function of neutrophils and their ability to release NETs. In these conditions, the dysregulation of neutrophil function and NET release can contribute to the pathology of the disease and affect the body’s ability to fight infections and maintain immune homeostasis (82, 83).
As mentioned before, the release of NETs is essential for defense against pathogens; the evasion of NETs appears to be a widespread strategy to allow pathogen proliferation and dissemination and is currently a topic of intense research interest. Here, we review the current knowledge of evasion strategies used by bacteria to dysregulate NET formation and functions.
Specific molecules and pathways that inhibit NET release. Thus, NET inhibition mechanisms in bacterial infections are summarized in this section of the review (Table 1).
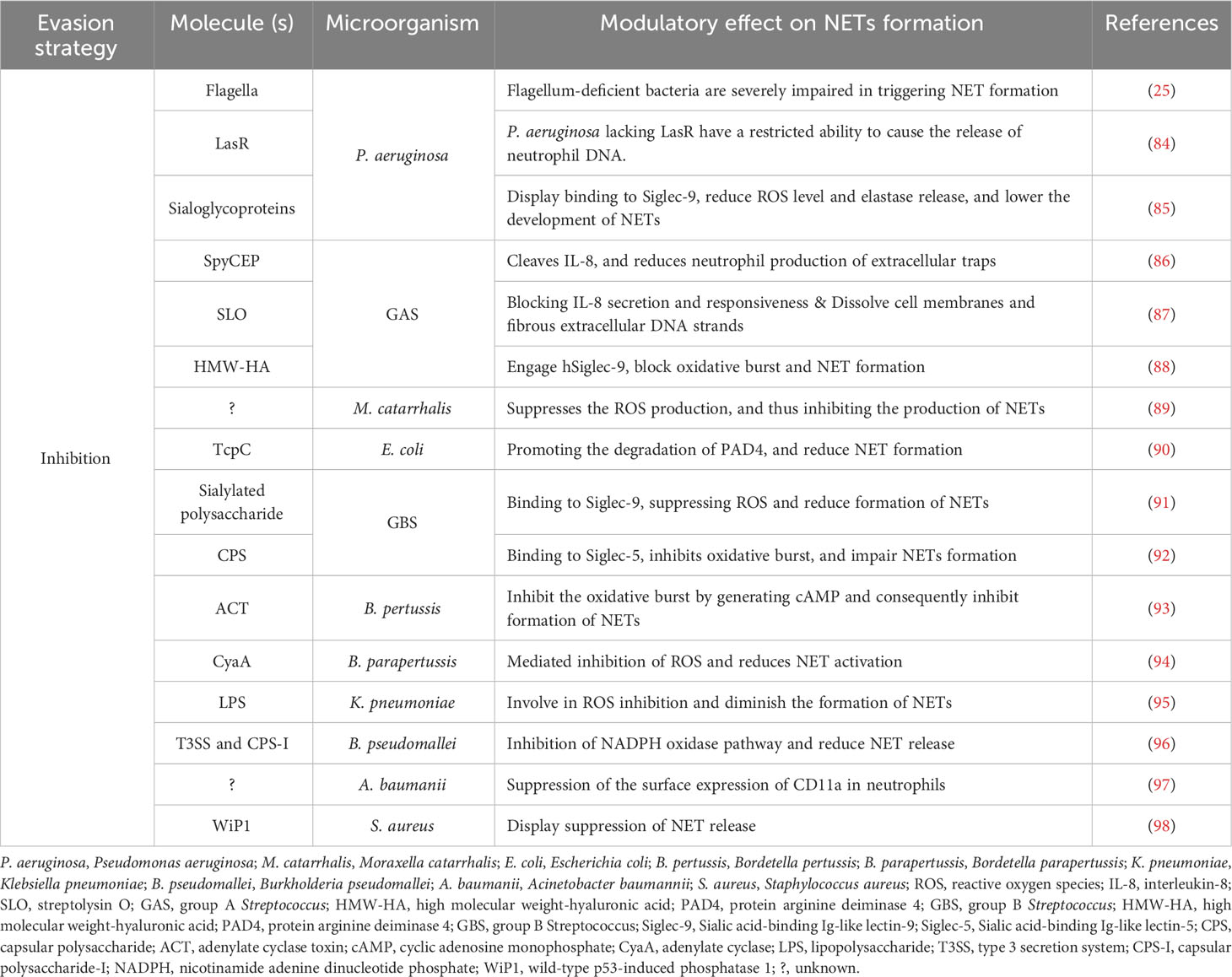
Table 1 Factors associated with the inhibition of NET release in bacterial infections.
Suppression of phenotypes that trigger NET release is a way to prevent their formation. Flagellum is the main bacterial component required to trigger maximal NET release, and flagellum-deficient bacteria remain seriously impaired in triggering NET formation (25). In addition, LasR-deficient P. aeruginosa strains harbor a limited capability to trigger neutrophil DNA release due to reduce the expression of elastase LasB and proteases LasA. The neutrophil NADPH oxidase pathway is required to decrease NET release caused by LasR-deficient strains, but it is not dependent on downstream quorum sensing pathways, LPS synthesis, or bacterial motility (84).
IL-8 is a prominent neutrophil chemoattractant and activator that induce NET formation (7). However, group A Streptococcus (GAS) protease SpyCEP (also called ScpC) cleaves IL-8 and reduces neutrophil production of extracellular traps (86). In addition, streptolysin O toxin (SLO) prevents the release of IL-8 and elastase from neutrophils, blocks NET formation, and inhibits NETs by dissolving cell membranes and fibrous extracellular DNA strands (87).
Neutrophil surface receptors have been linked to suppression of NET formation. GAS expresses a high-molecular-weight hyaluronic acid capsule (HMW-HA). hSiglec-9 specifically binds to HMW-HA through a region of its terminal Ig-like V-set domain distinct from the Sia-binding site. HMW-HA recognition by hSiglec-9 blocks the oxidative burst and limits NET formation, thereby promoting bacterial survival (88). Moreover, the sialylated capsular polysaccharide of Group B Streptococcus (GBS) interacts with neutrophil Siglec-9, dampening neutrophil responses in a Sia-and Siglec-9-dependent manner, causing a reduction in the neutrophil oxidative burst, diminished formation of NETs, and increased bacterial survival (91). In addition, β protein from GBS inhibits human leukocyte phagocytosis, oxidative burst, and extracellular trap formation by binding to hSiglec-5 in a Sia-independent manner (92). However, engagement of Siglec−14 by β−protein antagonizes the repressive effects of Siglec−5 by activating mitogen-activated protein kinase (MAPK) signaling (99). Moraxella catarrhalis limits ROS production by possibly binding to immunosuppressive Siglecs receptors (Siglec-5 and Siglec-9) and consequently suppressing NET generation (89). Furthermore, soluble Siglec-9 exhibited strong binding with α2-3-linked sialoglycoproteins adsorbed by P. aeruginosa. The interaction between P. aeruginosa (+Sias) and siglec-9 on neutrophils decreases the amount of ROS and the release of elastase, which in turn decreases the formation of NETs (85). Moreover, Acinetobacter baumannii inhibits the formation of NETs by suppressing the surface expression of CD11a in neutrophils, thereby escaping host immune responses and contributing to the development of A. baumannii infections (97).
NET formation is related to the production of ROS, both cytosolic and mitochondrial (55, 100). The adenylate cyclase toxin (ACT) of Bordetella pertussis inhibits formation of NETs by generating cyclic adenosine monophosphate (cAMP), consequently inhibiting oxidative burst (93). Bordetella parapertussis also expresses CyaA in a Bvg-regulated manner. This toxin is released into the extracellular space, which mediates the inhibition of ROS and reduces NET activation in human-derived neutrophils (94).
K. pneumoniae ST258 is a poor inducer of ROS generation, and consequently inhibits NET formation, suggesting that the polysaccharide part of LPS is responsible for this inhibition and results in increased bacterial survival (95). Burkholderia pseudomallei modifies the magnitude of NET formation via the action of its type 3 protein secretion system (T3SS), encoded by the bsa locus, and capsular polysaccharide I (CPS-I) encoded by the wcb operon, which attenuates NET release by inhibiting the NADPH oxidase pathway. B. pseudomallei mutants defective in the virulence-associated T3SS or CPS-I induced elevated levels of NETs. NET induction by these mutants is associated with increased bacterial killing (96).
Wild-type p53-induced phosphatase 1 (Wip1) suppresses NET release in S. aureus in mice model. Inhibition of Wip1 significantly suppresses the activity of S. aureus and accelerates abscess healing in S. aureus-induced abscess model mice by enhancing NET formation (98). Moreover, uropathogenic E. coli secretes a multifunctional virulence factor called TcpC, which primarily inhibits NETs by serving as an E3 ligase, promoting the degradation of PAD4. TcpC not only inhibits the citrullination of chromatin histones, but also affects the transcription of related genes in the nucleus and represents an additional NET evasion function of this bacterial derived virulence factor (90).
Studies investigating the production of NET-degrading molecules by bacteria have focused largely but not exclusively on the nucleases (Table 2; Figure 1).
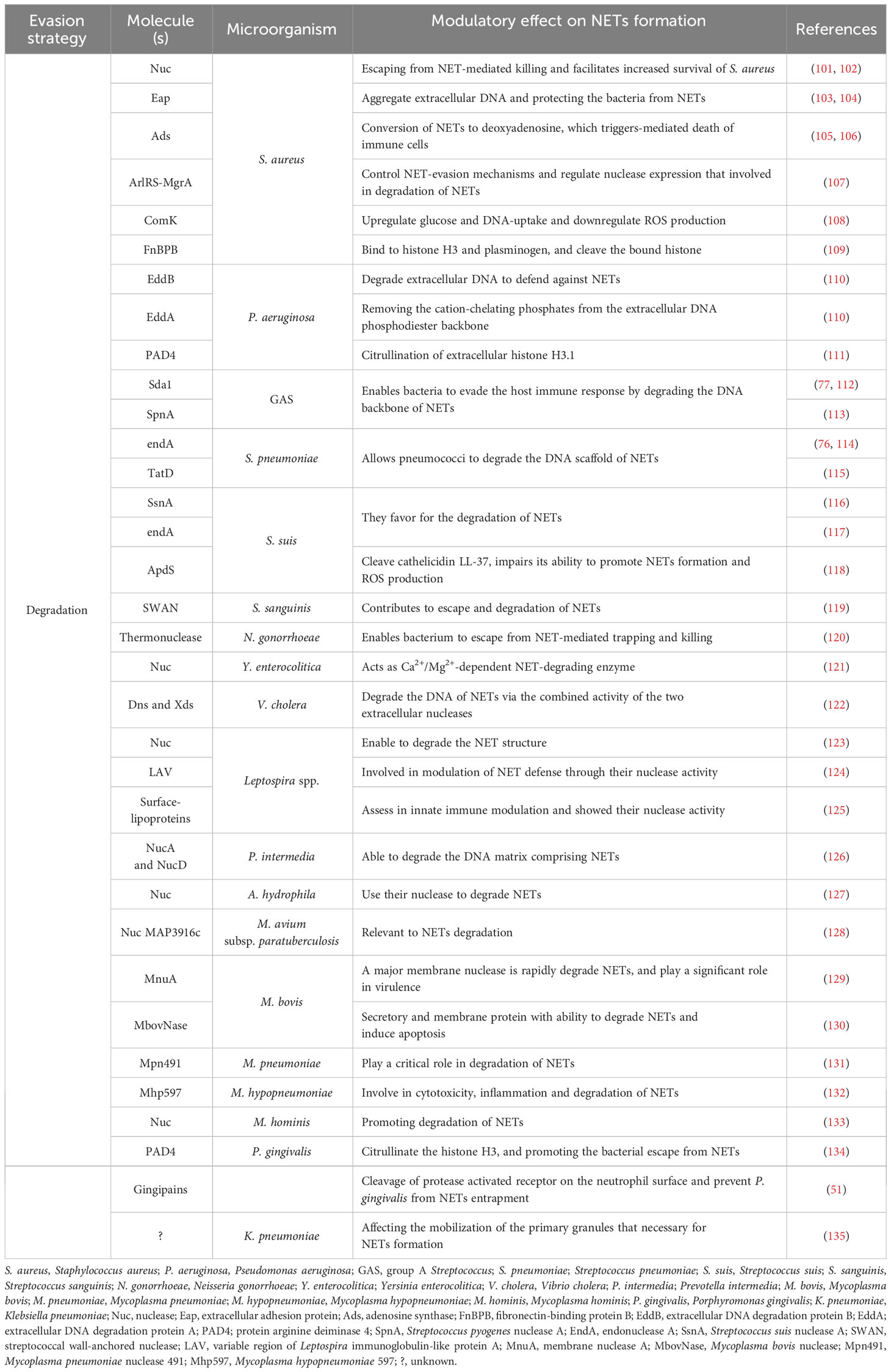
Table 2 Lists the molecules for degradation of NET release in bacterial infections.
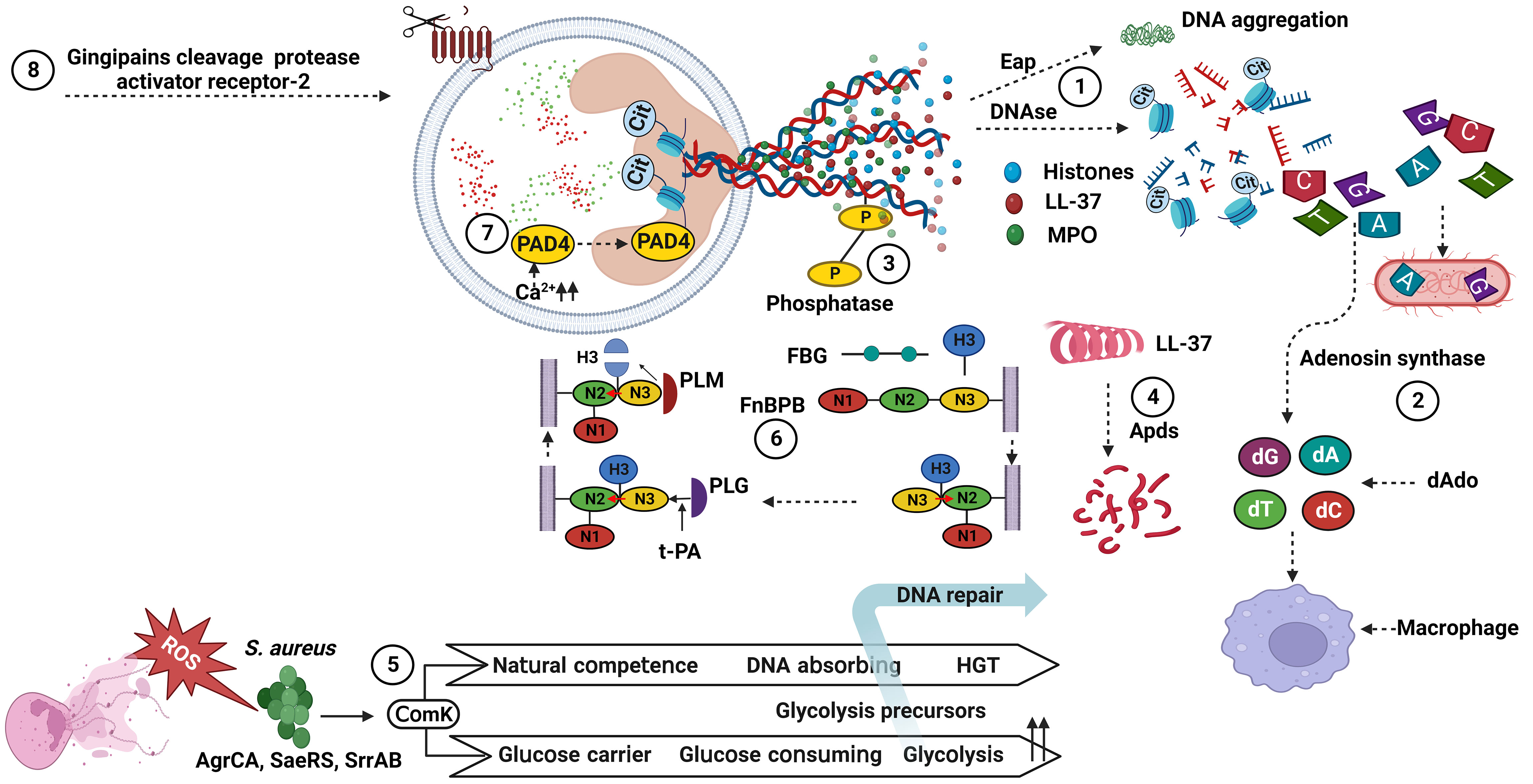
Figure 1 Bacterial factors associated with the degradation of NET components. (1) DNAses cleave NET-associated DNA, and Eap bind to the termini of linearized DNA and expresses an intrinsic DNA aggregation activity. (2) Adenosine synthase act together with the nuclease for generating deoxyadenosine fragments that are apoptotic for macrophages. (3) Phosphatase remove phosphate group from DNA, showing a cation chelating effect. (4) Apds cleave N-terminal amino acid from cathelicidin LL-37 and lose its helical structure. (5) ComK increase glucose and DNA uptake and downregulate ROS production. (6) FnBPB, in preference to FBG, H3 binds to the tunnel between the N2 and N3 of FnBPB, and then causes a conformational shift that allow the complex to stabilize by insertion of the terminal residues of the N3 extension (red arrow). Next, PLG binds to a N3 site, activated by t-PA to PLM, and cleaves FnBPB-bound H3. (7) PAD4 change histone arginines into histone citrullines, and losing its interactions with bacterial membranes. (8) Gingipains, cysteine protease, cleave protease-activated receptor-2 on the neutrophil surface. dAdo, deoxyadenosine; ROS, reactive oxygen species; HGT, horizontal gene transfer; PAD4, protein arginine deiminases 4; MPO, myeloperoxidase; FnBPB, fibronectin binding protein B; FBG, fibrinogen; H3, histones 3; PLG, plasminogen; t-PA, tissue plasminogen activator; PLM, plasmin; Eap, extracellular adhesion protein; Cit, citrullin. S. aureus, Staphylococcus aureus. Created with BioRender.com.
DNA forms a NET framework that holds multiple enzymes in close proximity, sometimes allowing for synergistic interactions among them (68). DNA is hydrolyzed by nucleases that belong to a group of hydrolases, which are further classified as endonucleases and exonucleases. These enzymes are involve in replication or repair of genetic material to maintain chromosome (136). However, extracellular nucleases have been reported in some bacterial species for the degradation of NET scaffold DNA, enabling bacteria to evade the NET antimicrobial mechanism, promoting pathogenicity, and dissemination to other sites in the host. For example, nuclease (Nuc) (101, 102) and extracellular adherence protein (Eap) (103, 104) are produced by S. aureus and interfere with the antimicrobial activity of NETs. These proteins help S. aureus to escape from NET-mediated killing, impede its removal, and increase infection-related mortality. Moreover, S. aureus escapes this defense by converting NETs to deoxyadenosine (dAdo) via the action of Nuc and adenosine synthase A (AdsA), which triggers caspase-3-mediated death of macrophages (105, 106). Moreover, S. aureus synchronizes gene expression during skin infection through the ArlRS-MgrA regulatory system, which regulates nuclease expression. This cascade is required for both the appropriate structuring of the abscess and evasion of the host innate immune system, both of which are necessary for S. aureus virulence. In contrast, mutants lacking MgrA and ArlRS have reduced capacity to avoid NET function (107). In addition, S. aureus expresses the competence regulator (ComK) when exposed to ROS. ComK upregulates the expression of genes encoding the transport machinery for glucose and DNA uptake, providing extra nutrients to increase the fermentation possibility of bacteria that are unable to respire and a source of nucleotides to repair DNA damage from ROS. Bacteria may use competence-related genes to better withstand NETs, because NETs are a source of both nucleotides and ROS (108).
P. aeruginosa encodes an operon of two secreted enzymes, a DNase and a predicted alkaline phosphatase. DNase (eddB) contributes to the degradation of NET to defend P. aeruginosa against NET-mediated killing, whereas eddA has both alkaline phosphatase and phosphodiesterase (PDase) activities that do not cause DNA degradation similar to that of DNase, but its protective function is likely a result of removing cation-chelating phosphates from the extracellular DNA phosphodiester backbone (110).
GAS is a leading cause of severe invasive disease in humans, and has evolved numerous virulence factors that aid in blocking NET function through the expression and secretion of the extracellular nuclease Sda1, which is advantageous for promoting bacterial dissemination throughout the host organism and evasion of the host innate immune response (77, 112). Similarly, S. pyogenes nuclease A (SpnA), a cell wall-anchored DNase, shows a unique protein architecture and promotes survival in human blood and in neutrophil killing assays, enable for the destruction of NETs and is believed to be an important immune evasion mechanism (113). A pneumococcal nuclease (EndA) acts as a virulence determinant, counteracting host-mediated trapping by NETs, thereby promoting bacterial spread from local sites to the lungs and then to the blood stream (76). Furthermore, the competence-independent activity of EndA is important for the virulence of Streptococcus pneumoniae, which mediates the rapid degradation of extracellular DNA and NETs (114). In addition, the secretion of nucleotide sequence-independent endodeoxyribonuclease TatD from S. pneumoniae is a novel potential extracellular DNase that plays a key role in evading NET-mediated bactericidal activity (115).
Streptococcus suis induces NET formation in porcine neutrophils and is entrapped, but not killed by NETs. The amount of NETs reduces over time due to the expression of nuclease A (SsnA), indicating that SsnA is a specific NET evasion factor in S. suis (116). In this sense, NET degradation is mediated not only by the known secreted SsnA but also by a putative endonuclease A of S. suis that is homologous to pneumococcal EndA (117). Actinobacillus pleuropneumoniae does not produce its own NET-degrading nucleases, but is hijacking other nucleases from host or from other co-infecting bacteria such as S. suis as a source for nicotinamide adenine dinucleotide (NAD) needed for efficient growth in the presence of NETs (137).
Moreover, Streptococcus sanguinis utilizes cell surface nuclease with a cell-wall anchor domain, termed streptococcal wall-anchored nuclease (SWAN), and contributes to bacterial resistance against the bactericidal activity of NETs (119). Neisseria gonorrhoeae encodes a putatively secreted thermonuclease that is implicated in biofilm remodeling and degrades the NET matrix to help N. gonorrhoeae from killing by neutrophils (120).
Yersinia enterocolitica O:3, O:8 and O:9 are able to induce NETs in human blood-derived neutrophils, but the amount of NETs is reduced at a later time, suggesting that the degradation of NETs has occurred and postulates that Y. enterocolitica produces Ca2+/Mg2+-dependent NET-degrading nuclease (121). Vibrio cholera induces the formation of NETs upon contact with neutrophils, while V. cholerae in return to expresses two extracellular nucleases, Dns and Xds, in the presence of NETs, and rapidly degrades the DNA component of the NETs by the combined activity of the two nucleases (122). Leptospira spp. is able to induce NETs using human ex vivo and murine in vivo models, resulting in the release of NETs. However, Leptospira spp. exerts nuclease activity and degrades DNA, resulting in a significantly reduced amount of NETs (123). In addition, the domain of the variable region of Leptospira immunoglobulin-like protein A (LAV) is involved in immune modulation; LAV has a nuclease activity and is demonstrated in the evasion of Leptospira from NETs (124). Additionally, surface-exposed lipoproteins in Leptospira are important for modulating host immune responses, and most of Len family surface proteins exhibit nuclease activity and are linked to NETs degradation (125).
Oral periodontopathogenic Prevotella intermedia produces two nucleases, NucA and NucD, which require Mg2+ and Ca2+ for their nuclease activity and contribute to NETs degradation (126). The fish pathogen Aeromonas hydrophila combats NETs using nuclease activity, while treatment of cells with β-glucan significantly protects NETs against bacterial degradation (127). Mycobacterium avium subsp. paratuberculosis encodes an extracellular nonspecific DNase that can destroy the NETs and promotes bacterial survival in vitro and in vivo (128).
Mycoplasma are the smallest bacteria that can infect and cause serious disorders in humans and various animal species (138, 139). Nucleases are a crucial component of Mycoplasma pathogenesis, facilitating bacterial growth and persistence in the host by digesting host nucleic acids and producing free nucleotide precursors (140–142). Membrane-associated or secreted nucleases have been found in many Mycoplasma species (129, 132, 143–146), and are homologous to staphylococcal nuclease (101). Mycoplasma lipoproteins are a major determinants of NET release during the innate immune response (39). Interestingly, most of the described Mycoplasma nucleases are able to escape from NET entrapment and killing by digesting NET’s DNA backbone, reducing structure stability, and enhancing NET elimination (129–131, 133).
Although DNA has been thoroughly examined, it is not the only NET component that is susceptible to degradation. PAD4 capable of deaminating arginine to citrulline and has been linked to NET formation in various context (147). PAD4 is involved in NET-mediated bacterial trapping and killing. Histones with cationic residues interact with negatively charged bacterial membranes and disrupt them (148). Thus, the loss of the positive charge of histones can lower their antibacterial function due to PAD4 activity. For example, Porphyromonas gingivalis produces its own PAD, Porphyromonas peptidylarginine deiminase (PPAD), which citrullinates histone H3, thereby facilitating bacterial escape from NETs, whereas PAD-mutant P. gingivalis is more prone to NET-mediated killing than its wild-type counterpart (134). Moreover, histone 3.1 displays bactericidal activity against P. aeruginosa. This bactericidal effect is reduced following citrullination by PAD4 or proteolysis by NE (111).
As previously mentioned, host defense peptides are key components of the innate immune system and act as the principal first line of defense against invading pathogens (149). The cysteine protease ApdS from S. suis cleaves cathelicidin LL-37 and impairs its ability to promote NETs formation and ROS production (118). K. pneumoniae affects the mobilization of primary granules and their components, which harbor proteins with more potent bactericidal properties and those related to NETs (135). Gingipain is a cysteine protease responsible for the virulence of P. gingivalis and is dependent on proteolytic activation of protease-activated receptor-2 (PAR-2). Intriguingly, P. gingivalis and purified Arg-specific gingipains (Rgp) induce NETs that not only lack bactericidal activity, but also stimulate the growth of bacterial species otherwise susceptible to killing in NETs. Taken together, gingipains play a dual role in NETs; they are potent direct inducers of NETs formation; however, their activity prevents P. gingivalis entrapment and subsequent killing (51). S. aureus produces a cell-wall-anchored protein known as fibronectin-binding protein B (FnBPB) that is the main histone receptor and bind to all types of histones. FnBPB provides a dual immune-evasion function that captures histones, and prevents them from reaching the bacterial membrane, and simultaneously binds plasminogen, thereby promoting its conversion to plasmin to destroy the bound histone (109).
In addition to inhibition of NET release and the degradation of NETs components, certain bacteria are resistant to the antimicrobial activity of NETs (Table 3).
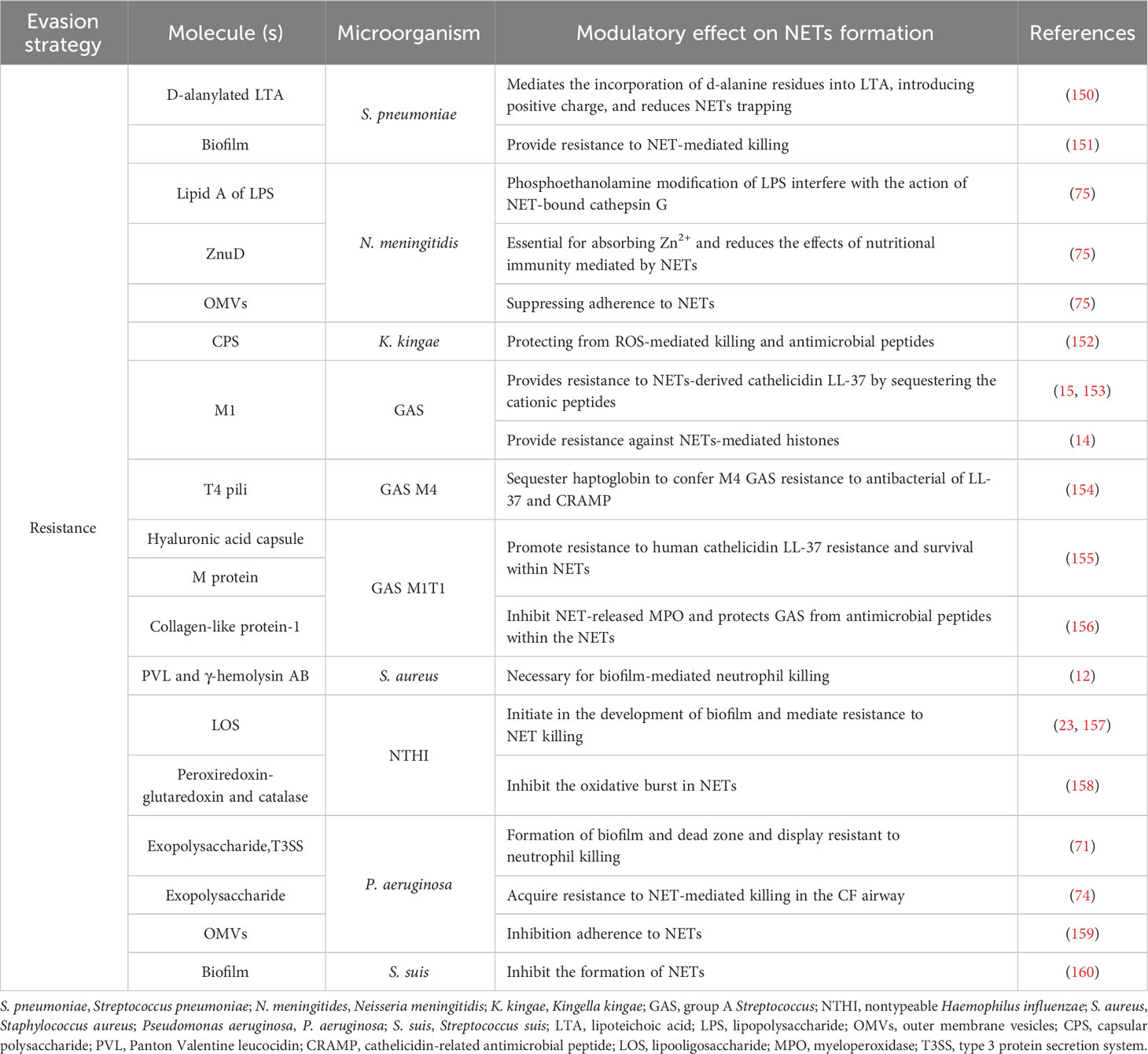
Table 3 Molecules involved in the resistance of NET components in bacterial infections.
Capsule is a surface structure of the organism and plays a critical role in virulence, principally by interfering with host clearance mechanisms. The thickness of the pneumococcal capsule plays a crucial role in determining the extent of NET formation and may contribute to pneumonia severity (161, 162). Interestingly, the electrostatic charge of the capsule helps evade NET-mediated killing by repelling interactions with antimicrobial proteins in the DNA framework. In this context, S. pneumoniae contains a dlt operon that mediates the incorporation of d-alanine residues into lipoteichoic acid (LTA), thereby introducing a positive charge that reduces trapping by NETs in vitro (150). Kingella kingae polysaccharide capsule and exopolysaccharide function distinctly to promote neutrophil evasion. The K. kingae polysaccharide capsule prevents ROS production and neutrophil association, whereas the K. kingae exopolysaccharide reduces neutrophil phagocytosis and sensitivity to antimicrobial peptides (152). In addition, the hyaluronic acid capsule and M protein of the GAS serotype M1T1 promote resistance to human cathelicidin LL-37, which may be an important contributor to the NET-resistance phenotype (155).
Microbial biofilm communities are microbes embedded in a matrix of self-produced polysaccharides and other molecules, such as lipids, proteins, and nucleic acids that can bind to various surfaces, which complicates the immunological and therapeutic response (163). Biofilm formation by P. aeruginosa is a major cause of bacterial keratitis and is facilitated by bacterial Psl exopolysaccharide and T3SS. NETs are stimulated by high expression of T3SS and form a barrier, known as dead-zone, confining bacteria to the external corneal environment, preventing them from spreading to the brain. Once formed, ocular biofilms advance eye pathology because they are resistant to neutrophil killing and antibiotics (71). Furthermore, P. aeruginosa acquires resistance to NET-mediated killing in cystic fibrosis airways. This resistance correlates with the development of excess exopolysaccharide production (which characterizes the mucoid phenotype) (74). In addition, studies conducted in vitro have demonstrated that P. aeruginosa can profit from the released DNA of NETs and incorporate it into the extracellular matrix, potentially strengthening the matrix’s resistance to antibiotics and host antimicrobial peptides (164–167).
Clinical isolates of H. influenzae share a common tendency to produce biofilms, and the factors that facilitate biofilm formation include the expression of certain lipooligosaccharide (LOS) glycoforms (168–170), type IV pili (171), and double-stranded DNA release (172). Nontypeable Haemophilus influenzae (NTHi) promotes resistance to killing within NETs structures through LOS moieties that promote biofilm formation (23, 157). Methicillin-resistant S. aureus (MRSA) biofilms rapidly skew neutrophils toward NETs formation through the combined activity of the Panton-Valentine leukocidin (PVL) and γ-hemolysin AB, which are important for biofilm-mediated neutrophil killing. Through this response, S. aureus can persist because the antimicrobial activity of the released NETs is ineffective in eradicating bacterial biofilm (12). S. aureus nuclease-mediated NET degradation promotes the persistence of biofilm bacteria entrapped in NETs (173). S. suis serotype 2 induces NETs release and can be captured by the NETs, whereas biofilm formation inhibit NETs release through the biofilm extracellular matrix and enables survival by allowing the pathogen to persist and resist the host immune system (160). In addition, pneumococci produce biofilms in vivo and observe web-or net-like structures surrounded by dense matrix material, which is intertwined with the formation of NETs (151).
OMVs are nanostructures released by pathogenic and non-pathogenic bacteria in vivo and in vitro and can act as decoys for NET capture (174). Ocular keratitis is often associated with P. aeruginosa infection. Neutrophils release NETs in response to both cytotoxic and invasive clinical isolates of P. aeruginosa. Cytotoxic strains are less prone to NET capture than invasive strains because they release OMVs that inhibit NET adherence (159). Furthermore, Neisseria meningitidis releases OMVs as potent NET inducers. NETs are unable to kill NET- bound meningococci, but they slow down their proliferation rate. The bacteriostatic effect of NETs is counteracted by spontaneously released OMVs from N. meningitidis, which reduces their adherence to NETs (75).
NET-releasing proteins attach to negatively charged phospholipids in pathogen membranes with electrostatic affinity, which promotes death (175). Nonetheless, certain bacteria modify their cell surfaces, which reduces their affinity for attachment to NET-releasing peptides. For example, the surface-associated protein M1 protein contributes to GAS virulence by interfering with NET-mediated killing, which sequester and neutralize cathelicidin LL-37 through the N-terminal hypervariable (HV) region and A repeat region that encode the type-specific immunologic epitopes of the M1 serotype (15, 153). In addition, the N-terminal portion of the M1 protein binds and inactivates histones before they reach their cell wall target of action and mediate resistance against released extracellular histones in NETs, allowing the pathogen to tolerate high concentrations of histones and promote survival in NETs (14). Streptococcal collagen-like protein 1 (Scl-1) in GAS serotype M1T1 confers resistance to NET-mediated killing, which may be in large part due to the antimicrobial peptides present within the NETs; Scl-1 has an additional role in suppressing the release of MPO, which ultimately limits the production of NETs (156). Haptoglobin is an abundant acute-phase protein produced upon infection, which binds to human neutrophils and monocytes and inhibits their functions (176, 177). The T4 antigen, the pilus backbone protein of GAS M4, sequester the host haptoglobin. Coating M4 GAS with haptoglobin causes a reduction in susceptibility to cathelicidin LL-37 and murine cathelicidin-related antimicrobial peptide (CRAMP) and promotes resistance to NET-mediated killing (154).
Modification of lipid A of meningococcal LPS with phosphoethanolamine shields N. meningitidis from the action of NET-bound cathepsin G (75). In a process known as nutritional immunity, host organisms restrict the availability of trace nutrients in the blood or secretions to prevent the growth of invading microorganisms (178). The outer membrane receptor ZnuD of N. meningitidis is crucial for Zn2+ uptake at very low concentrations (179). N. meningitidis utilizes high affinity absorption systems for critical ions on mucosal surfaces, producing ZnuD, which absorbs Zn2+; contribute to survival within NETs (75).
Finally, the initiation of the oxidative burst is important for the induction of NETs, and oxidants contribute to microbicidal activity within the NETs (9, 180). Noticeable H. influenzae expresses the bifunctional peroxiredoxin-glutaredoxin (encoded by pdgX) and catalase (encoded by hktE), which confer resistance of NTHI to oxidative killing and thus promote the survival of NTHI within NET structures and persistence in vivo in the lung and middle ear. In addition, exogenous catalase partially rescued NTHI from NET-mediated killing in vitro. The expression of both peroxiredoxin-glutaredoxin and catalase is a mechanism by which NTHI combats the effects of NETs (158).
Neutrophils are type of white blood cell that play a crucial role in the immune system’s defense against infections. There are different subsets of neutrophils that have different functions and responses to various stimuli. NET release is a form of neutrophil immune response that, depending on the context in which it is examined, can have both pathogenic and physiological effects. They can entangle and immobilize bacteria, preventing their spread, and facilitating their destruction by other immune cells. Additionally, dysregulation of NET release has several implications for the immune system and overall health. On the other hand, impaired NET formation may lead to reduced ability to fight off infections. Therefore, maintaining a balanced and controlled NET release is essential for the proper functioning of the defense system and overall health. In recent years, there has been a significant increase in research focused on the role NETs in response to pathogens and the mechanisms involved in the modulation of their release. Bacteria have developed various mechanisms to evade or resist NET release, allowing them to escape the immune system’s defenses and several key findings and advancements have emerged from these studies. In this review, we have discussed the strategies used by various bacteria to counteract NET-mediated antimicrobial effects as such inhibiting NET release, deactivating their components, degrading their net-like framework, or blocking their contact, resulting in infection dissemination and immune system inactivation. Bacteria possess a vast array of mechanism for modulating NET activity, and ongoing investigations are expected to uncover new molecules and pathways involved in controlling NET release. Current understanding of different inflammatory mediators in modulate NET release is quite limited and need be further elucidated. Overall, the recent focus on NETs and their modulation has provided a deeper understanding of the complex interplay between neutrophils, pathogens, and the immune system. This knowledge has the potential to identify pharmacological targets and drive the development of therapeutic and diagnostic approaches to counteract bacterial evasion strategies and combat the role of NETs in inflammatory and autoimmune diseases. It is crucial to understand that various factors affect whether NETs are advantageous or harmful, with the dose and the timing of NET release and clearance being critical factors. A better understanding of the roles of NETs and their effects on hosts will make it possible to inhibit the adverse attributes without affecting the beneficial ones, which will ultimately enable strategies related to NETs to be used in disease treatment.
AA: Investigation, Visualization, Writing – original draft. HH: Investigation, Visualization, Writing – review & editing. SLan: Investigation, Visualization, Writing – review & editing. ZL: Investigation, Visualization, Writing – review & editing. SLiu: Investigation, Visualization, Writing – review & editing. SC: Conceptualization, Funding acquisition, Investigation, Supervision, Visualization, Writing – review & editing. YC: Funding acquisition, Project administration, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by grants from the National Key Research and Development Plan of China (2022YFD1800903), The National Natural Science Foundation of China (32373019 and 31402223), the Chinese Academy of Agricultural Science and Technology Key Project (CAAS-ASTIP-JBGS-20210701). The funders had no role in study design, data collection, analysis, publishing decisions, or manuscript preparation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1357967/full#supplementary-material
1. Gierlikowska B, Stachura A, Gierlikowski W, Demkow U. Phagocytosis, degranulation and extracellular traps release by neutrophils-the current knowledge, pharmacological modulation and future prospects. Front Pharmacol (2021) 12:666732. doi: 10.3389/fphar.2021.666732
2. Schumann J. It is all about fluidity: Fatty acids and macrophage phagocytosis. Eur J Pharmacol (2016) 785(15):18–23. doi: 10.1016/j.ejphar.2015.04.057
3. Jaumouillé V, Waterman CM. Physical constraints and forces involved in phagocytosis. Front Immunol (2020) 11:1097. doi: 10.3389/fimmu.2020.01097
4. Mortaz E, Alipoor SD, Adcock IM, Mumby S, Koenderman L. Update on neutrophil function in severe inflammation. Front Immunol (2018) 9:2171. doi: 10.3389/fimmu.2018.02171
5. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi: 10.1038/nri3399
6. Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes infection. (2003) 5(14):1317–27. doi: 10.1016/j.micinf.2003.09.008
7. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
8. Manda A, Pruchniak MP, Araźna M, Demkow UA. Neutrophil extracellular traps in physiology and pathology. Central-European J Immunol (2014) 39(1):116–21. doi: 10.5114/ceji.2014.42136
9. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176(2):231–41. doi: 10.1083/jcb.200606027
10. Yipp BG, Kubes P. NETosis: How vital is it? Blood (2013) 122(16):2784–94. doi: 10.1182/blood-2013-04-457671
11. Zawrotniak M, Rapala-Kozik M. Neutrophil extracellular traps (NETs) - formation and implications. Acta Biochim Polonica. (2013) 60(3):277–84. doi: 10.18388/abp.2013_1983
12. Bhattacharya M, Berends ETM, Chan R, Schwab E, Roy S, Sen CK, et al. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc Natl Acad Sci U S A. (2018) 115(28):7416–21. doi: 10.1073/pnas.1721949115
13. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (2010) 185(12):7413–25. doi: 10.4049/jimmunol.1000675
14. Döhrmann S, LaRock CN, Anderson EL, Cole JN, Ryali B, Stewart C, et al. Group A streptococcal M1 protein provides resistance against the antimicrobial activity of histones. Sci Rep (2017) 7:43039. doi: 10.1038/srep43039
15. Lauth X, von Köckritz-Blickwede M, McNamara CW, Myskowski S, Zinkernagel AS, Beall B, et al. M1 protein allows Group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. J innate immunity. (2009) 1(3):202–14. doi: 10.1159/000203645
16. Mori Y, Yamaguchi M, Terao Y, Hamada S, Ooshima T, Kawabata S. α-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J Biol Chem (2012) 287(13):10472–81. doi: 10.1074/jbc.M111.280321
17. Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. (2017) 6:e24437. doi: 10.7554/eLife.24437
18. Sumioka R, Nakata M, Okahashi N, Li Y, Wada S, Yamaguchi M, et al. Streptococcus sanguinis induces neutrophil cell death by production of hydrogen peroxide. PloS One (2017) 12(2):e0172223. doi: 10.1371/journal.pone.0172223
19. Dai J, Lai L, Tang H, Wang W, Wang S, Lu C, et al. Streptococcus suis synthesizes deoxyadenosine and adenosine by 5'-nucleotidase to dampen host immune responses. Virulence. (2018) 9(1):1509–20. doi: 10.1080/21505594.2018.1520544
20. Pieterse E, Rother N, Yanginlar C, Hilbrands LB, van der Vlag J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front Immunol (2016) 7:484. doi: 10.3389/fimmu.2016.00484
21. Grinberg N, Elazar S, Rosenshine I, Shpigel NY. Beta-hydroxybutyrate abrogates formation of bovine neutrophil extracellular traps and bactericidal activity against mammary pathogenic Escherichia coli. Infection Immun (2008) 76(6):2802–7. doi: 10.1128/IAI.00051-08
22. Krivošíková K, Šupčíková N, Gaál Kovalčíková A, Janko J, Pastorek M, Celec P, et al. Neutrophil extracellular traps in urinary tract infection. Front pediatrics. (2023) 11:1154139. doi: 10.3389/fped.2023.1154139
23. Hong W, Juneau RA, Pang B, Swords WE. Survival of bacterial biofilms within neutrophil extracellular traps promotes nontypeable Haemophilus influenzae persistence in the chinchilla model for otitis media. J innate immunity. (2009) 1(3):215–24. doi: 10.1159/000205937
24. Rada B, Jendrysik MA, Pang L, Hayes CP, Yoo DG, Park JJ, et al. Pyocyanin-enhanced neutrophil extracellular trap formation requires the NADPH oxidase. PloS One (2013) 8(1):e54205. doi: 10.1371/journal.pone.0054205
25. Floyd M, Winn M, Cullen C. Swimming motility mediates the formation of neutrophil extracellular traps induced by flagellated pseudomonas aeruginosa. PloS Pathog (2016) 12(11):e1005987. doi: 10.1371/journal.ppat.1005987
26. Gillenius E, Urban CF. The adhesive protein invasin of Yersinia pseudotuberculosis induces neutrophil extracellular traps via β1 integrins. Microbes infection. (2015) 17(5):327–36. doi: 10.1016/j.micinf.2014.12.014
27. Altincicek B, Stötzel S, Wygrecka M, Preissner KT, Vilcinskas A. Host-derived extracellular nucleic acids enhance innate immune responses, induce coagulation, and prolong survival upon infection in insects. J Immunol (2008) 181(4):2705–12. doi: 10.4049/jimmunol.181.4.2705
28. Ramos-Kichik V, Mondragón-Flores R, Mondragón-Castelán M, Gonzalez-Pozos S, Muñiz-Hernandez S, Rojas-Espinosa O, et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (2009) 89(1):29–37. doi: 10.1016/j.tube.2008.09.009
29. Braian C, Hogea V, Stendahl O. Mycobacterium tuberculosis- induced neutrophil extracellular traps activate human macrophages. J innate immunity. (2013) 5(6):591–602. doi: 10.1159/000348676
30. Ladero-Auñon I, Molina E, Holder A, Kolakowski J, Harris H, Urkitza A, et al. Bovine Neutrophils Release Extracellular Traps and Cooperate With Macrophages in Mycobacterium avium subsp. paratuberculosis clearance In Vitro. Front Immunol (2021) 12:645304. doi: 10.3389/fimmu.2021.645304
31. Gondaira S, Nishi K, Fujiki J, Iwano H, Watanabe R, Eguchi A, et al. Innate immune response in bovine neutrophils stimulated with Mycoplasma bovis. Vet Res (2021) 52(1):58. doi: 10.1186/s13567-021-00920-2
32. Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, et al. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem (2004) 279(42):44123–32. doi: 10.1074/jbc.M405883200
33. Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PloS One (2012) 7(10):e48111. doi: 10.1371/journal.pone.0048111
34. Long J, Sun Y, Liu S, Yang S, Chen C, Zhang Z, et al. Targeting pyroptosis as a preventive and therapeutic approach for stroke. Cell Death discovery. (2023) 9(1):155. doi: 10.1038/s41420-023-01440-y
35. Lv M, Wang Y, Yu J, Kong Y, Zhou H, Zhang A, et al. Grass carp Il-2 promotes neutrophil extracellular traps formation via inducing ROS production and autophagy in vitro. Fish shellfish Immunol (2024) 144:109261. doi: 10.1016/j.fsi.2023.109261
36. Mikhalchik E, Basyreva LY, Gusev SA, Panasenko OM, Klinov DV, Barinov NA, et al. Activation of neutrophils by mucin-vaterite microparticles. Int J Mol Sci (2022) 23(18):10579. doi: 10.3390/ijms231810579
37. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol (2009) 184(2):205–13. doi: 10.1083/jcb.200806072
38. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death differentiation. (2009) 16(11):1438–44. doi: 10.1038/cdd.2009.96
39. Cacciotto C, Cubeddu T, Addis MF, Anfossi AG, Tedde V, Tore G, et al. Mycoplasma lipoproteins are major determinants of neutrophil extracellular trap formation. Cell Microbiol (2016) 18(12):1751–62. doi: 10.1111/cmi.12613
40. Ma F, Chang X, Wang G, Zhou H, Ma Z, Lin H, et al. Streptococcus Suis Serotype 2 Stimulates Neutrophil Extracellular Traps Formation via Activation of p38 MAPK and ERK1/2. Front Immunol (2018) 9:2854. doi: 10.3389/fimmu.2018.02854
41. Tamarozzi F, Turner JD, Pionnier N, Midgley A, Guimaraes AF, Johnston KL, et al. Wolbachia endosymbionts induce neutrophil extracellular trap formation in human onchocerciasis. Sci Rep (2016) 6:35559. doi: 10.1038/srep35559
42. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13(4):463–9. doi: 10.1038/nm1565
43. Wan T, Zhao Y, Fan F, Hu R, Jin X. Dexamethasone Inhibits S. aureus-Induced Neutrophil Extracellular Pathogen-Killing Mechanism, Possibly through Toll-Like Receptor Regulation. Front Immunol (2017) 8:60. doi: 10.3389/fimmu.2017.00060
44. Hook JS, Patel PA, O'Malley A, Xie L, Kavanaugh JS, Horswill AR, et al. Lipoproteins from staphylococcus aureus drive neutrophil extracellular trap formation in a TLR2/1- and PAD-dependent manner. J Immunol (2021) 207(3):966–73. doi: 10.4049/jimmunol.2100283
45. Alyami HM, Finoti LS, Teixeira HS, Aljefri A, Kinane DF, Benakanakere MR. Role of NOD1/NOD2 receptors in Fusobacterium nucleatum mediated NETosis. Microbial pathogenesis. (2019) 131:53–64. doi: 10.1016/j.micpath.2019.03.036
46. Chen ST, Li FJ, Hsu TY, Liang SM, Yeh YC, Liao WY, et al. CLEC5A is a critical receptor in innate immunity against Listeria infection. Nat Commun (2017) 8(1):299. doi: 10.1038/s41467-017-00356-3
47. Palmer LJ, Damgaard C, Holmstrup P, Nielsen CH. Influence of complement on neutrophil extracellular trap release induced by bacteria. J periodontal Res (2016) 51(1):70–6. doi: 10.1111/jre.12284
48. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Differential use of human neutrophil fc receptors for inducing neutrophil extracellular trap formation. J Immunol Res (2016) 2016:2908034. doi: 10.1155/2016/2908034
49. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Transforming growth factor-β-activated kinase 1 is required for human fcγRIIIb-induced neutrophil extracellular trap formation. Front Immunol (2016) 7:277. doi: 10.3389/fimmu.2016.00277
50. Diago-Navarro E, Calatayud-Baselga I, Sun D, Khairallah C, Mann I, Ulacia-Hernando A, et al. Antibody-based immunotherapy to treat and prevent infection with hypervirulent klebsiella pneumoniae. Clin Vaccine Immunol (2017) 24(1):e00456-16. doi: 10.1128/CVI.00456-16
51. Bryzek D, Ciaston I, Dobosz E, Gasiorek A. Triggering NETosis via protease-activated receptor (PAR)-2 signaling as a mechanism of hijacking neutrophils function for pathogen benefits. PloS pathogens. (2019) 15(5):e1007773. doi: 10.1371/journal.ppat.1007773
52. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PloS Pathog (2009) 5(10):e1000639. doi: 10.1371/journal.ppat.1000639
53. Brinkmann V. Neutrophil extracellular traps in the second decade. J innate Immun (2018) 10(5-6):414–21. doi: 10.1159/000489829
54. Dwyer M, Shan Q, D'Ortona S, Maurer R, Mitchell R, Olesen H, et al. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J innate immunity. (2014) 6(6):765–79. doi: 10.1159/000363242
55. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol (2011) 7(2):75–7. doi: 10.1038/nchembio.496
56. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep (2014) 8(3):883–96. doi: 10.1016/j.celrep.2014.06.044
57. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol (2010) 191(3):677–91. doi: 10.1083/jcb.201006052
58. Sollberger G, Choidas A. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol (2018) 3(26):eaar6689. doi: 10.1126/sciimmunol.aar6689
59. Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. (2019) 133(20):2178–85. doi: 10.1182/blood-2018-11-844530
60. Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol (2012) 3:307. doi: 10.3389/fimmu.2012.00307
61. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med (2012) 18(9):1386–93. doi: 10.1038/nm.2847
62. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med (2017) 23(3):279–87. doi: 10.1038/nm.4294
63. Dunham-Snary KJ, Surewaard BG, Mewburn JD, Bentley RE, Martin AY, Jones O, et al. Mitochondria in human neutrophils mediate killing of Staphylococcus aureus. Redox Biol (2022) 49:102225. doi: 10.1016/j.redox.2021.102225
64. Konig MF, Andrade F. A critical reappraisal of neutrophil extracellular traps and NETosis mimics based on differential requirements for protein citrullination. Front Immunol (2016) 7:461. doi: 10.3389/fimmu.2016.00461
65. Zhu S, Yu Y, Ren Y, Xu L, Wang H, Ling X, et al. The emerging roles of neutrophil extracellular traps in wound healing. Cell Death Dis (2021) 12(11):984. doi: 10.1038/s41419-021-04294-3
66. Tanikawa C, Espinosa M, Suzuki A, Masuda K, Yamamoto K, Tsuchiya E, et al. Regulation of histone modification and chromatin structure by the p53-PADI4 pathway. Nat Commun (2012) 3:676. doi: 10.1038/ncomms1676
67. Daniel C, Leppkes M, Muñoz LE, Schley G, Schett G, Herrmann M. Extracellular DNA traps in inflammation, injury and healing. Nat Rev Nephrology. (2019) 15(9):559–75. doi: 10.1038/s41581-019-0163-2
68. Halverson TW, Wilton M, Poon KK, Petri B, Lewenza S. DNA is an antimicrobial component of neutrophil extracellular traps. PloS pathogens. (2015) 11(1):e1004593. doi: 10.1371/journal.ppat.1004593
69. Richards RC, O'Neil DB, Thibault P, Ewart KV. Histone H1: an antimicrobial protein of Atlantic salmon (Salmo salar). Biochem Biophys Res Commun (2001) 284(3):549–55. doi: 10.1006/bbrc.2001.5020
70. Weinrauch Y, Drujan D, Shapiro SD, Weiss J, Zychlinsky A. Neutrophil elastase targets virulence factors of enterobacteria. Nature. (2002) 417(6884):91–4. doi: 10.1038/417091a
71. Thanabalasuriar A, Scott BNV, Peiseler M, Willson ME, Zeng Z, Warrener P, et al. Neutrophil extracellular traps confine pseudomonas aeruginosa ocular biofilms and restrict brain invasion. Cell Host Microbe (2019) 25(4):526–36.e4. doi: 10.1016/j.chom.2019.02.007
72. Marin-Esteban V, Turbica I, Dufour G, Semiramoth N, Gleizes A, Gorges R, et al. Afa/Dr diffusely adhering Escherichia coli strain C1845 induces neutrophil extracellular traps that kill bacteria and damage human enterocyte-like cells. Infection immunity. (2012) 80(5):1891–9. doi: 10.1128/IAI.00050-12
73. Li H, Liu L, Wang J, Zhao W. The emerging role of neutrophil extracellular traps in endometritis. Front Immunol (2023) 14:1153851. doi: 10.3389/fimmu.2023.1153851
74. Young RL, Malcolm KC, Kret JE, Caceres SM, Poch KR, Nichols DP, et al. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PloS One (2011) 6(9):e23637. doi: 10.1371/journal.pone.0023637
75. Lappann M, Danhof S, Guenther F, Olivares-Florez S, Mordhorst IL, Vogel U. In vitro resistance mechanisms of Neisseria meningitidis against neutrophil extracellular traps. Mol Microbiol (2013) 89(3):433–49. doi: 10.1111/mmi.12288
76. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol (2006) 16(4):401–7. doi: 10.1016/j.cub.2006.01.056
77. Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol (2006) 16(4):396–400. doi: 10.1016/j.cub.2005.12.039
78. Azzouz L, Cherry A, Riedl M, Khan M, Pluthero FG, Kahr WHA, et al. Relative antibacterial functions of complement and NETs: NETs trap and complement effectively kills bacteria. Mol Immunol (2018) 97(1):71–81. doi: 10.1016/j.molimm.2018.02.019
79. Douda DN, Jackson R, Grasemann H, Palaniyar N. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J Immunol (2011) 187(4):1856–65. doi: 10.4049/jimmunol.1004201
80. Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun (2015) 6:6673. doi: 10.1038/ncomms7673
81. Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. (2002) 416(6878):291–7. doi: 10.1038/416291a
82. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: Potential anti-NETs therapeutics. Clin Rev Allergy Immunol (2021) 61(2):194–211. doi: 10.1007/s12016-020-08804-7
83. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol (2018) 18(2):134–47. doi: 10.1038/nri.2017.105
84. Skopelja-Gardner S, Theprungsirikul J, Lewis KA, Hammond JH, Carlson KM, Hazlett HF, et al. Regulation of pseudomonas aeruginosa-mediated neutrophil extracellular traps. Front Immunol (2019) 10:1670. doi: 10.3389/fimmu.2019.01670
85. Khatua B, Bhattacharya K, Mandal C. Sialoglycoproteins adsorbed by Pseudomonas aeruginosa facilitate their survival by impeding neutrophil extracellular trap through siglec-9. J leukocyte Biol (2012) 91(4):641–55. doi: 10.1189/jlb.0511260
86. Zinkernagel AS, Timmer AM, Pence MA, Locke JB, Buchanan JT, Turner CE, et al. The IL-8 protease SpyCEP/ScpC of group A Streptococcus promotes resistance to neutrophil killing. Cell Host Microbe (2008) 4(2):170–8. doi: 10.1016/j.chom.2008.07.002
87. Uchiyama S, Döhrmann S, Timmer AM, Dixit N, Ghochani M, Bhandari T, et al. Streptolysin O rapidly impairs neutrophil oxidative burst and antibacterial responses to group A streptococcus. Front Immunol (2015) 6:581. doi: 10.3389/fimmu.2015.00581
88. Secundino I, Lizcano A, Roupé KM, Wang X, Cole JN, Olson J, et al. Host and pathogen hyaluronan signal through human siglec-9 to suppress neutrophil activation. J Mol Med (2016) 94(2):219–33. doi: 10.1007/s00109-015-1341-8
89. Nicchi S, Giusti F, Carello S, Utrio Lanfaloni S, Tavarini S, Frigimelica E, et al. Moraxella catarrhalis evades neutrophil oxidative stress responses providing a safer niche for nontypeable Haemophilus influenzae. iScience. (2022) 25(3):103931. doi: 10.1016/j.isci.2022.103931
90. Ou Q, Fang JQ, Zhang ZS, Chi Z, Fang J, Xu DY, et al. TcpC inhibits neutrophil extracellular trap formation by enhancing ubiquitination mediated degradation of peptidylarginine deiminase 4. Nat Commun (2021) 12(1):3481. doi: 10.1038/s41467-021-23881-8
91. Carlin AF, Uchiyama S, Chang YC, Lewis AL, Nizet V, Varki A. Molecular mimicry of host sialylated glycans allows a bacterial pathogen to engage neutrophil Siglec-9 and dampen the innate immune response. Blood. (2009) 113(14):3333–6. doi: 10.1182/blood-2008-11-187302
92. Carlin AF, Chang YC, Areschoug T, Lindahl G, Hurtado-Ziola N, King CC, et al. Group B Streptococcus suppression of phagocyte functions by protein-mediated engagement of human Siglec-5. J Exp Med (2009) 206(8):1691–9. doi: 10.1084/jem.20090691
93. Eby JC, Gray MC, Hewlett EL. Cyclic AMP-mediated suppression of neutrophil extracellular trap formation and apoptosis by the Bordetella pertussis adenylate cyclase toxin. Infection immunity. (2014) 82(12):5256–69. doi: 10.1128/IAI.02487-14
94. Gorgojo J, Scharrig E, Gómez RM, Harvill ET, Rodríguez ME. Bordetella parapertussis circumvents neutrophil extracellular bactericidal mechanisms. PloS One (2017) 12(1):e0169936. doi: 10.1371/journal.pone.0169936
95. Castillo LA, Birnberg-Weiss F, Rodriguez-Rodrigues N, Martire-Greco D, Bigi F, Landoni VI, et al. Klebsiella pneumoniae ST258 negatively regulates the oxidative burst in human neutrophils. Front Immunol (2019) 10:929. doi: 10.3389/fimmu.2019.00929
96. Riyapa D, Buddhisa S, Korbsrisate S, Cuccui J, Wren BW, Stevens MP, et al. Neutrophil extracellular traps exhibit antibacterial activity against burkholderia pseudomallei and are influenced by bacterial and host factors. Infection immunity. (2012) 80(11):3921–9. doi: 10.1128/IAI.00806-12
97. Kamoshida G, Kikuchi-Ueda T, Nishida S, Tansho-Nagakawa S, Ubagai T, Ono Y. Pathogenic bacterium acinetobacter baumannii inhibits the formation of neutrophil extracellular traps by suppressing neutrophil adhesion. Front Immunol (2018) 9:178. doi: 10.3389/fimmu.2018.00178
98. Chen Y, Zhao C, Guo H, Zou W, Zhang Z, Wei D, et al. Wip1 inhibits neutrophil extracellular traps to promote abscess formation in mice by directly dephosphorylating Coronin-1a. Cell Mol Immunol (2023) 20(8):941–54. doi: 10.1038/s41423-023-01057-2
99. Ali SR, Fong JJ, Carlin AF, Busch TD, Linden R, Angata T, et al. Siglec-5 and Siglec-14 are polymorphic paired receptors that modulate neutrophil and amnion signaling responses to group B Streptococcus. J Exp Med (2014) 211(6):1231–42. doi: 10.1084/jem.20131853
100. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22(2):146–53. doi: 10.1038/nm.4027
101. Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Köckritz-Blickwede M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J innate immunity. (2010) 2(6):576–86. doi: 10.1159/000319909
102. Herzog S, Dach F, de Buhr N, Niemann S, Schlagowski J, Chaves-Moreno D, et al. High nuclease activity of long persisting staphylococcus aureus isolates within the airways of cystic fibrosis patients protects against NET-mediated killing. Front Immunol (2019) 10:2552. doi: 10.3389/fimmu.2019.02552
103. Eisenbeis J, Saffarzadeh M, Peisker H, Jung P, Thewes N, Preissner KT, et al. The staphylococcus aureus extracellular adherence protein eap is a DNA binding protein capable of blocking neutrophil extracellular trap formation. Front Cell infection Microbiol (2018) 8:235. doi: 10.3389/fcimb.2018.00235
104. Chavakis T, Wiechmann K, Preissner KT, Herrmann M. Staphylococcus aureus interactions with the endothelium: the role of bacterial "secretable expanded repertoire adhesive molecules" (SERAM) in disturbing host defense systems. Thromb haemostasis. (2005) 94(2):278–85. doi: 10.1160/TH05-05-0306
105. Thammavongsa V, Missiakas DM, Schneewind O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science. (2013) 342(6160):863–6. doi: 10.1126/science.1242255
106. Thammavongsa V, Kern JW, Missiakas DM, Schneewind O. Staphylococcus aureus synthesizes adenosine to escape host immune responses. J Exp Med (2009) 206(11):2417–27. doi: 10.1084/jem.20090097
107. Kwiecinski JM, Kratofil RM, Parlet CP, Surewaard BGJ, Kubes P, Horswill AR. Staphylococcus aureus uses the ArlRS and MgrA cascade to regulate immune evasion during skin infection. Cell Rep (2021) 36(4):109462. doi: 10.1016/j.celrep.2021.109462
108. Cordero M, García-Fernández J. The induction of natural competence adapts staphylococcal metabolism to infection. Nat Commun (2022) 13(1):1525. doi: 10.1038/s41467-022-29206-7
109. Pietrocola G, Nobile G, Alfeo MJ, Foster TJ, Geoghegan JA, De Filippis V, et al. Fibronectin-binding protein B (FnBPB) from Staphylococcus aureus protects against the antimicrobial activity of histones. J Biol Chem (2019) 294(10):3588–602. doi: 10.1074/jbc.RA118.005707
110. Wilton M, Halverson TWR, Charron-Mazenod L, Parkins MD, Lewenza S. Secreted Phosphatase and Deoxyribonuclease Are Required by Pseudomonas aeruginosa To Defend against Neutrophil Extracellular Traps. Infection Immun (2018) 86(9):e00403-18. doi: 10.1128/IAI.00403-18
111. Tanner L, Bhongir RKV, Karlsson CAQ, Le S, Ljungberg JK, Andersson P, et al. Citrullination of extracellular histone H3.1 reduces antibacterial activity and exacerbates its proteolytic degradation. J cystic fibrosis Off J Eur Cystic Fibrosis Soc (2021) 20(2):346–55. doi: 10.1016/j.jcf.2020.07.010
112. Moon AF, Krahn JM, Lu X, Cuneo MJ, Pedersen LC. Structural characterization of the virulence factor Sda1 nuclease from Streptococcus pyogenes. Nucleic Acids Res (2016) 44(8):3946–57. doi: 10.1093/nar/gkw143
113. Chang A, Khemlani A, Kang H, Proft T. Functional analysis of Streptococcus pyogenes nuclease A (SpnA), a novel group A streptococcal virulence factor. Mol Microbiol (2011) 79(6):1629–42. doi: 10.1111/j.1365-2958.2011.07550.x
114. Zhu L, Kuang Z, Wilson BA, Lau GW. Competence-independent activity of pneumococcal EndA mediates degradation of extracellular dna and nets and is important for virulence. PloS One (2013) 8(7):e70363. doi: 10.1371/annotation/75f51a45-a2de-4893-94cf-0045056a4d7c
115. Jhelum H, Sori H, Sehgal D. A novel extracellular vesicle-associated endodeoxyribonuclease helps Streptococcus pneumoniae evade neutrophil extracellular traps and is required for full virulence. Sci Rep (2018) 8(1):7985. doi: 10.1038/s41598-018-25865-z
116. de Buhr N, Neumann A, Jerjomiceva N, von Köckritz-Blickwede M, Baums CG. Streptococcus suis DNase SsnA contributes to degradation of neutrophil extracellular traps (NETs) and evasion of NET-mediated antimicrobial activity. Microbiology. (2014) 160(2):385–95. doi: 10.1099/mic.0.072199-0
117. de Buhr N, Stehr M, Neumann A, Naim HY, Valentin-Weigand P, von Köckritz-Blickwede M, et al. Identification of a novel DNase of Streptococcus suis (EndAsuis) important for neutrophil extracellular trap degradation during exponential growth. Microbiol . (2015) 161(4):838–50. doi: 10.1099/mic.0.000040
118. Xie F, Zan Y, Zhang Y, Zheng N, Yan Q, Zhang W, et al. The cysteine protease ApdS from Streptococcus suis promotes evasion of innate immune defenses by cleaving the antimicrobial peptide cathelicidin LL-37. J Biol Chem (2019) 294(47):17962–77. doi: 10.1074/jbc.RA119.009441
119. Morita C, Sumioka R, Nakata M, Okahashi N, Wada S, Yamashiro T, et al. Cell wall-anchored nuclease of Streptococcus sanguinis contributes to escape from neutrophil extracellular trap-mediated bacteriocidal activity. PloS One (2014) 9(8):e103125. doi: 10.1371/journal.pone.0103125
120. Juneau RA, Stevens JS, Apicella MA, Criss AK. A thermonuclease of Neisseria gonorrhoeae enhances bacterial escape from killing by neutrophil extracellular traps. J Infect diseases. (2015) 212(2):316–24. doi: 10.1093/infdis/jiv031
121. Möllerherm H, Neumann A, Schilcher K, Blodkamp S, Zeitouni NE, Dersch P, et al. Yersinia enterocolitica-mediated degradation of neutrophil extracellular traps (NETs). FEMS Microbiol Lett (2015) 362(23):fnv192. doi: 10.1093/femsle/fnv192
122. Seper A, Hosseinzadeh A, Gorkiewicz G, Lichtenegger S, Roier S, Leitner DR, et al. Vibrio cholerae evades neutrophil extracellular traps by the activity of two extracellular nucleases. PloS pathogens. (2013) 9(9):e1003614. doi: 10.1371/journal.ppat.1003614
123. Scharrig E, Carestia A, Ferrer MF, Cédola M, Pretre G, Drut R, et al. Neutrophil extracellular traps are involved in the innate immune response to infection with leptospira. PloS Negl Trop Dis (2015) 9(7):e0003927. doi: 10.1371/journal.pntd.0003927
124. Kumar A, Varma VP, Sridhar K, Abdullah M, Vyas P, Ashiq Thalappil M, et al. Deciphering the role of leptospira surface protein ligA in modulating the host innate immune response. Front Immunol (2021) 12:807775. doi: 10.3389/fimmu.2021.807775
125. Kumar A, Varma VP, Faisal SM. Screening of surface-exposed lipoproteins of leptospira involved in modulation of host innate immune response. Front Microbiol (2022) 13:761670. doi: 10.3389/fmicb.2022.761670
126. Doke M, Fukamachi H, Morisaki H, Arimoto T, Kataoka H, Kuwata H. Nucleases from Prevotella intermedia can degrade neutrophil extracellular traps. Mol Oral Microbiol (2017) 32(4):288–300. doi: 10.1111/omi.12171
127. Brogden G, von Köckritz-Blickwede M, Adamek M, Reuner F, Jung-Schroers V, Naim HY, et al. β-Glucan protects neutrophil extracellular traps against degradation by Aeromonas hydrophila in carp (Cyprinus carpio). Fish shellfish Immunol (2012) 33(4):1060–4. doi: 10.1016/j.fsi.2012.08.009
128. Zang X, Dang G, Cai Z, Shao M, Tang Y, Cao J, et al. Extracellular DNase MAP3916c attacks the neutrophil extracellular traps and is needed for Mycobacterium avium subsp. paratuberculosis virulence. Veterinary Microbiol (2022) 273:109529. doi: 10.1016/j.vetmic.2022.109529
129. Mitiku F, Hartley CA, Sansom FM, Coombe JE, Mansell PD, Beggs DS, et al. The major membrane nuclease MnuA degrades neutrophil extracellular traps induced by Mycoplasma bovis. Veterinary Microbiol (2018) 218:13–9. doi: 10.1016/j.vetmic.2018.03.002
130. Zhang H, Zhao G, Guo Y, Menghwar H, Chen Y, Chen H, et al. Mycoplasma bovis MBOV_RS02825 encodes a secretory nuclease associated with cytotoxicity. Int J Mol Sci (2016) 17(5):628. doi: 10.3390/ijms17050628
131. Yamamoto T, Kida Y, Sakamoto Y, Kuwano K. Mpn491, a secreted nuclease of Mycoplasma pneumoniae, plays a critical role in evading killing by neutrophil extracellular traps. Cell Microbiol (2017) 19(3):e12666. doi: 10.1111/cmi.12666
132. Li P, Zhang Y, Li X, Zhou W, Li X, Jiang F, et al. Mycoplasma hyopneumoniae Mhp597 is a cytotoxicity, inflammation and immunosuppression associated nuclease. Veterinary Microbiol (2019) 235:53–62. doi: 10.1016/j.vetmic.2019.05.011
133. Cacciotto C, Dessì D, Cubeddu T, Cocco AR, Pisano A, Tore G, et al. MHO_0730 as a surface-exposed calcium-dependent nuclease of mycoplasma hominis promoting neutrophil extracellular trap formation and escape. J Infect diseases. (2019) 220(12):1999–2008. doi: 10.1093/infdis/jiz406
134. Stobernack T, du Teil Espina M, Mulder LM, Palma Medina LM, Piebenga DR, Gabarrini G, et al. A secreted bacterial peptidylarginine deiminase can neutralize human innate immune defenses. mBio. (2018) 9(5):10–128. doi: 10.1128/mBio.01704-18
135. Birnberg-Weiss F, Castillo LA, Pittaluga JR, Martire-Greco D, Gómez SA, Landoni VI, et al. Modulation of neutrophil extracellular traps release by Klebsiella pneumoniae. J leukocyte Biol (2021) 109(1):245–56. doi: 10.1002/JLB.4MA0620-099R
136. Yang W. Nucleases: diversity of structure, function and mechanism. Q Rev biophysics. (2011) 44(1):1–93. doi: 10.1017/S0033583510000181
137. de Buhr N, Bonilla MC, Pfeiffer J, Akhdar S, Schwennen C, Kahl BC, et al. Degraded neutrophil extracellular traps promote the growth of Actinobacillus pleuropneumoniae. Cell Death Dis (2019) 10(9):657. doi: 10.1038/s41419-019-1895-4
138. Cacciotto C, Alberti A. Eating the enemy: mycoplasma strategies to evade neutrophil extracellular traps (NETs) promoting bacterial nucleotides uptake and inflammatory damage. Int J Mol Sci (2022) 23(23):15030. doi: 10.3390/ijms232315030
139. Benedetti F, Curreli S, Zella D. Mycoplasmas-host interaction: Mechanisms of inflammation and association with cellular transformation. Microorganisms. (2020) 8(9):1351. doi: 10.3390/microorganisms8091351
140. Masukagami Y, De Souza DP, Dayalan S, Bowen C, O'Callaghan S, Kouremenos K, et al. Comparative Metabolomics of Mycoplasma bovis and Mycoplasma gallisepticum Reveals Fundamental Differences in Active Metabolic Pathways and Suggests Novel Gene Annotations. mSystems. (2017) 2(5):e00055-17. doi: 10.1128/mSystems.00055-17
141. Yiwen C, Yueyue W, Lianmei Q, Cuiming Z, Xiaoxing Y. Infection strategies of mycoplasmas: Unraveling the panoply of virulence factors. Virulence. (2021) 12(1):788–817. doi: 10.1080/21505594.2021.1889813
142. Yueyue W, Feichen X, Yixuan X, Lu L, Yiwen C, Xiaoxing Y. Pathogenicity and virulence of Mycoplasma genitalium: Unraveling Ariadne's Thread. Virulence. (2022) 13(1):1161–83. doi: 10.1080/21505594.2022.2095741
143. Yacoub E, Ben Abdelmoumen Mardassi B. Mm19, a mycoplasma meleagridis major surface nuclease that is related to the RE_AlwI superfamily of endonucleases. PloS One (2016) 11(3):e0152171. doi: 10.1371/journal.pone.0152171
144. Li L, Krishnan M, Baseman JB, Kannan TR. Molecular cloning, expression, and characterization of a Ca2+-dependent, membrane-associated nuclease of Mycoplasma genitalium. J bacteriology (2010) 192(19):4876–84. doi: 10.1128/JB.00401-10
145. Cacciotto C, Addis MF, Coradduzza E, Carcangiu L, Nuvoli AM, Tore G, et al. Mycoplasma agalactiae MAG_5040 is a Mg2+-dependent, sugar-nonspecific SNase recognised by the host humoral response during natural infection. PloS One (2013) 8(2):e57775. doi: 10.1371/journal.pone.0057775
146. Xu J, Teng D, Jiang F, Zhang Y, El-Ashram SA, Wang H, et al. Mycoplasma gallisepticum MGA_0676 is a membrane-associated cytotoxic nuclease with a staphylococcal nuclease region essential for nuclear translocation and apoptosis induction in chicken cells. Appl Microbiol Biotechnol (2015) 99(4):1859–71. doi: 10.1007/s00253-014-6185-6
147. Tilvawala R, Thompson PR. Peptidyl arginine deiminases: detection and functional analysis of protein citrullination. Curr Opin Struct Biol (2019) 59:205–15. doi: 10.1016/j.sbi.2019.01.024
148. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med (2010) 207(9):1853–62. doi: 10.1084/jem.20100239
149. Pachón-Ibáñez ME, Smani Y, Pachón J, Sánchez-Céspedes J. Perspectives for clinical use of engineered human host defense antimicrobial peptides. FEMS Microbiol Rev (2017) 41(3):323–42. doi: 10.1093/femsre/fux012
150. Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, et al. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol (2007) 9(5):1162–71. doi: 10.1111/j.1462-5822.2006.00857.x
151. Reid SD, Hong W, Dew KE, Winn DR, Pang B, Watt J, et al. Streptococcus pneumoniae forms surface-attached communities in the middle ear of experimentally infected chinchillas. J Infect diseases. (2009) 199(6):786–94. doi: 10.1086/597042
152. Muñoz VL, Porsch EA, St Geme JW 3rd. Kingella kingae surface polysaccharides promote resistance to neutrophil phagocytosis and killing. mBio. (2019) 10(3):e00631–19. doi: 10.1128/mBio.00631-19
153. LaRock CN, Döhrmann S, Todd J, Corriden R, Olson J, Johannssen T, et al. Group A streptococcal M1 protein sequesters cathelicidin to evade innate immune killing. Cell Host Microbe (2015) 18(4):471–7. doi: 10.1016/j.chom.2015.09.004
154. Chen YH, Li SH, Yang YC, Hsu SH, Nizet V. T4 pili promote colonization and immune evasion phenotypes of nonencapsulated M4 streptococcus pyogenes. mBio (2020) 11(4):e01580–20. doi: 10.1128/mBio.01580-20
155. Cole JN, Pence MA, von Köckritz-Blickwede M, Hollands A, Gallo RL, Walker MJ, et al. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. mBio (2010) 1(4):e00191–10. doi: 10.1128/mBio.00191-10
156. Döhrmann S, Anik S, Olson J, Anderson EL, Etesami N, No H, et al. Role for streptococcal collagen-like protein 1 in M1T1 group A Streptococcus resistance to neutrophil extracellular traps. Infection immunity. (2014) 82(10):4011–20. doi: 10.1128/IAI.01921-14
157. Juneau RA, Pang B, Weimer KE, Armbruster CE, Swords WE. Nontypeable Haemophilus influenzae initiates formation of neutrophil extracellular traps. Infection immunity. (2011) 79(1):431–8. doi: 10.1128/IAI.00660-10
158. Juneau RA, Pang B, Armbruster CE, Murrah KA, Perez AC, Swords WE. Peroxiredoxin-glutaredoxin and catalase promote resistance of nontypeable Haemophilus influenzae 86-028NP to oxidants and survival within neutrophil extracellular traps. Infection immunity. (2015) 83(1):239–46. doi: 10.1128/IAI.02390-14
159. Shan Q, Dwyer M, Rahman S, Gadjeva M. Distinct susceptibilities of corneal Pseudomonas aeruginosa clinical isolates to neutrophil extracellular trap-mediated immunity. Infection immunity. (2014) 82(10):4135–43. doi: 10.1128/IAI.02169-14
160. Ma F, Yi L, Yu N, Wang G, Ma Z, Lin H, et al. Streptococcus suis serotype 2 biofilms inhibit the formation of neutrophil extracellular traps. Front Cell infection Microbiol (2017) 7:86. doi: 10.3389/fcimb.2017.00086
161. Paton JC, Trappetti C. Streptococcus pneumoniae capsular polysaccharide. Microbiol spectrum. (2019) 7(2):10–128. doi: 10.1128/microbiolspec.GPP3-0019-2018
162. Moorthy AN, Rai P, Jiao H, Wang S, Tan KB, Qin L, et al. Capsules of virulent pneumococcal serotypes enhance formation of neutrophil extracellular traps during in vivo pathogenesis of pneumonia. Oncotarget. (2016) 7(15):19327–40. doi: 10.18632/oncotarget.8451
163. Rather MA, Gupta K, Mandal M. Microbial biofilm: formation, architecture, antibiotic resistance, and control strategies. Braz J Microbiol (2021) 52(4):1701–18. doi: 10.1007/s42770-021-00624-x
164. Chiang WC, Nilsson M, Jensen P, Høiby N, Nielsen TE, Givskov M, et al. Extracellular DNA shields against aminoglycosides in Pseudomonas aeruginosa biofilms. Antimicrobial Agents chemotherapy. (2013) 57(5):2352–61. doi: 10.1128/AAC.00001-13
165. Tseng BS, Zhang W, Harrison JJ, Quach TP, Song JL, Penterman J, et al. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ Microbiol (2013) 15(10):2865–78. doi: 10.1111/1462-2920.12155
166. Mulcahy H, Charron-Mazenod L, Lewenza S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PloS pathogens. (2008) 4(11):e1000213. doi: 10.1371/journal.ppat.1000213
167. Walker TS, Tomlin KL, Worthen GS, Poch KR, Lieber JG, Saavedra MT, et al. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infection immunity. (2005) 73(6):3693–701. doi: 10.1128/IAI.73.6.3693-3701.2005
168. Swords WE, Moore ML, Godzicki L, Bukofzer G, Mitten MJ, VonCannon J. Sialylation of lipooligosaccharides promotes biofilm formation by nontypeable Haemophilus influenzae. Infection Immun (2004) 72(1):106–13. doi: 10.1128/IAI.72.1.106-113.2004
169. Hong W, Pang B, West-Barnette S, Swords WE. Phosphorylcholine expression by nontypeable Haemophilus influenzae correlates with maturation of biofilm communities in vitro and in vivo. J bacteriology (2007) 189(22):8300–7. doi: 10.1128/JB.00532-07
170. Greiner LL, Watanabe H, Phillips NJ, Shao J, Morgan A, Zaleski A, et al. Nontypeable Haemophilus influenzae strain 2019 produces a biofilm containing N-acetylneuraminic acid that may mimic sialylated O-linked glycans. Infection immunity. (2004) 72(7):4249–60. doi: 10.1128/IAI.72.7.4249-4260.2004
171. Jurcisek JA, Bookwalter JE, Baker BD, Fernandez S, Novotny LA, Munson RS Jr., et al. The PilA protein of non-typeable Haemophilus influenzae plays a role in biofilm formation, adherence to epithelial cells and colonization of the mammalian upper respiratory tract. Mol Microbiol (2007) 65(5):1288–99. doi: 10.1111/j.1365-2958.2007.05864.x
172. Jurcisek JA, Bakaletz LO. Biofilms formed by nontypeable Haemophilus influenzae in vivo contain both double-stranded DNA and type IV pilin protein. J bacteriology. (2007) 189(10):3868–75. doi: 10.1128/JB.01935-06
173. Bhattacharya M, Berends ETM, Zheng X, Hill PJ, Chan R, Torres VJ, et al. Leukocidins and the nuclease nuc prevent neutrophil-mediated killing of staphylococcus aureus biofilms. Infection immunity. (2020) 88(10):e00372–20. doi: 10.1128/IAI.00372-20
174. Lieberman LA. Outer membrane vesicles: A bacterial-derived vaccination system. Front Microbiol (2022) 13:1029146. doi: 10.3389/fmicb.2022.1029146
175. Cole JN, Nizet V. Bacterial evasion of host antimicrobial peptide defenses. Microbiol Spectr. (2016) 4(1):10.1128/microbiolspec.VMBF-0006-2015. doi: 10.1128/microbiolspec.VMBF-0006-2015
176. Yang H, Wang H, Levine YA, Gunasekaran MK, Wang Y, Addorisio M, et al. Identification of CD163 as an antiinflammatory receptor for HMGB1-haptoglobin complexes. JCI Insight (2018) 3(24):e126617. doi: 10.1172/jci.insight.126617
177. Arredouani MS, Kasran A, Vanoirbeek JA, Berger FG, Baumann H, Ceuppens JL. Haptoglobin dampens endotoxin-induced inflammatory effects both in vitro and in vivo. Immunology (2005) 114(2):263–71. doi: 10.1111/j.1365-2567.2004.02071.x
178. Murdoch CC, Skaar EP. Nutritional immunity: the battle for nutrient metals at the host-pathogen interface. Nat Rev Microbiol (2022) 20(11):657–70. doi: 10.1038/s41579-022-00745-6
179. Stork M, Bos MP, Jongerius I, de Kok N, Schilders I, Weynants VE, et al. An outer membrane receptor of Neisseria meningitidis involved in zinc acquisition with vaccine potential. PloS pathogens. (2010) 6(7):e1000969. doi: 10.1371/journal.ppat.1000969
Keywords: neutrophil, neutrophil extracellular traps, bacterial infection, mycoplasma, NETs evasion, DNase
Citation: Baz AA, Hao H, Lan S, Li Z, Liu S, Chen S and Chu Y (2024) Neutrophil extracellular traps in bacterial infections and evasion strategies. Front. Immunol. 15:1357967. doi: 10.3389/fimmu.2024.1357967
Received: 19 December 2023; Accepted: 26 January 2024;
Published: 16 February 2024.
Edited by:
Yong Huang, Northwest A&F University, ChinaReviewed by:
Soraya Mezouar, Aix-Marseille University, FranceCopyright © 2024 Baz, Hao, Lan, Li, Liu, Chen and Chu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengli Chen, Y2hlbnNoZW5nbGlAY2Fhcy5jbg==; Yuefeng Chu, Y2h1eXVlZmVuZ0BjYWFzLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.