
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 March 2024
Sec. T Cell Biology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1356869
This article is part of the Research Topic Immunoregulatory Cells: Balancing Immune Responses During Pathogen Infections View all 5 articles
 Xinyong Liu1†
Xinyong Liu1† Longwang Chen2†
Longwang Chen2† Wei Peng1
Wei Peng1 Hongsheng Deng1Hongying Ni1Hongjie Tong1Hangbo Hu1Shengchao Wang1
Hongsheng Deng1Hongying Ni1Hongjie Tong1Hangbo Hu1Shengchao Wang1 Jin Qian1Andong Liang3*
Jin Qian1Andong Liang3* Kun Chen1*
Kun Chen1*Sepsis is a multi-organ dysfunction characterized by an unregulated host response to infection. It is associated with high morbidity, rapid disease progression, and high mortality. Current therapies mainly focus on symptomatic treatment, such as blood volume supplementation and antibiotic use, but their effectiveness is limited. Th17/Treg balance, based on its inflammatory property, plays a crucial role in determining the direction of the inflammatory response and the regression of organ damage in sepsis patients. This review provides a summary of the changes in T-helper (Th) 17 cell and regulatory T (Treg) cell differentiation and function during sepsis, the heterogeneity of Th17/Treg balance in the inflammatory response, and the relationship between Th17/Treg balance and organ damage. Th17/Treg balance exerts significant control over the bloom and wanes in host inflammatory response throughout sepsis.
Sepsis is defined as an initial hyperinflammatory response to systemic infection caused by an invasive pathogen (1, 2). This response involves hemodynamic changes and leads to subsequent immunosuppression and organ dysfunction, eventually triggering multiple organ failure, secondary infection, and mortality (1, 2). According to data from Germany, approximately 48.9 million people suffer from sepsis each year, with sepsis-related deaths accounting for about 20% of all deaths (3, 4). A global observational study in 2017 recorded 11 million sepsis-related deaths, accounting for 19.7% of the total mortality rate for that specific year (5). In mainland China, sepsis patients make up a significant proportion of intensive care unit (ICU) admissions, with a 90-day mortality rate of 35.5%, imposing a substantial economic burden (6). From 1990 to 2017, the incidence of sepsis decreased by 37.0% and the mortality by 52.8% (5). Despite extensive investigations into the pathophysiology of sepsis in recent years, which have made substantial contributions to drug development and organ support therapy, sepsis continues to prevail as the primary driver of mortality in critically ill patients (7, 8).
Sepsis presents an extremely heterogeneous feature, due to its complex types of pathogen infections and differential organismal response ability (9). The inflammatory and immunosuppressive states of sepsis occur concurrently (10). In the early stages of sepsis, besides the fact that Toll-like receptor (TLR) located on the cell membrane surfaces of antigen-presenting cells (APC) (e.g., macrophage, dendritic cell (DC), and neutrophil) recognize either pathogen-associated molecular marker (PAMP) or damage-associated molecular patterns (DAMP) for initiation of inflammation, a massive decrease in T cells via apoptosis has been observed (11, 12). During the initial exposure phase to severe infection, TLR recognizes and binds PAMP, activating the innate immune system and complement systems (13). Then, large amounts of complement component 3a (C3a) and complement component 5a (C5a) are produced and secreted, prompting the release of pro-inflammatory cytokines, thereby triggering a cytokine storm (13). A prolonged hyperinflammatory response triggers cellular damage and promotes the release of DAMP, further bolstering the production and removal of inflammatory mediators (13). In addition, the hyperinflammatory response provokes coagulation and endothelial cell activation, resulting in disseminated intravascular coagulation and endothelial leakage (14, 15). Subsequently, the adaptive immune system takes effect, and the T cell receptor (TCR) activates T helper cells (Th, e.g., Th1, Th17) and cytotoxic T cells, releasing inflammatory factors and amplifying the inflammatory response to facilitate the clearance of the infection (16). Tregs have the opposite function to Th17, suppressing excessive T-cell responses and promoting self-tolerance under inflammatory conditions (17). The balance between Th subsets and Tregs is important for clearing the infection. When a compensatory anti-inflammatory response fails to balance an explosive pro-inflammatory response, sepsis is exacerbated and secondary infections are triggered, leading to organ dysfunction and death. Furthermore, in the distant phase of sepsis, the continued expression of adaptive anti-inflammatory markers alters the function of innate immune cells and induces lymphocyte exhaustion, which leads to prolonged immunosuppression (14, 18). This phase can last for several years (19).
Th17 and regulatory T (Treg) cells are two subsets of CD4+ T cells that participate in the immune response through the defense, immune surveillance and immune regulation, and act to promote or suppress the inflammatory response (20). Th17 are differentiated mainly from T cells stimulated by transforming growth factor-β (TGF-β) (21), IL-6 (22), IL-21 (23), IL-23 (24) and IL-1β (25), which further secrete IL-17 and various inflammatory factors, such as IL-6, IL-22, IL-23, granulocyte-colony stimulating factor (GCSF) and tumor necrosis factor-alpha (TNF-α), to induce recruitment of neutrophils (26–28). Forkhead box protein 3 (Foxp3), a transcription factor, is involved in the Tregs differentiation. Tregs mainly secret IL-10 and TGF-β to maintain self-tolerance and suppress inflammation (29–31). In sepsis, Foxp3 inhibits T-cell function, while IL-17 promotes its function, and the two thereby oppose and interact with each other (32–35). As such, during the inflammatory response, the alterations in the function and quantity of Th17 and Tregs can effectively modulate the immune response (Figure 1). In sepsis, the altered immune function of T lymphocytes is an essential driver of increased mortality and poor prognosis (36). A study has confirmed that Th17, a central component in inflammatory diseases, is significantly elevated in septic patients, which correlates with the intensive inflammatory response and severity of the disease (37). Meanwhile, some experimental and clinical trials have shown that sepsis enhances Treg function, which acts on the innate and adaptive immune system, suppressing immune function and leading to immune paralysis and, ultimately, septic death (38–40). Compared to survivors of sepsis, deceased patients have a higher Th17/Treg ratio in the circulation, shifting the balance toward Th17 (41). Restoration of the Th17/Treg balance affects the prognosis of the disease and contributes to the treatment of sepsis.
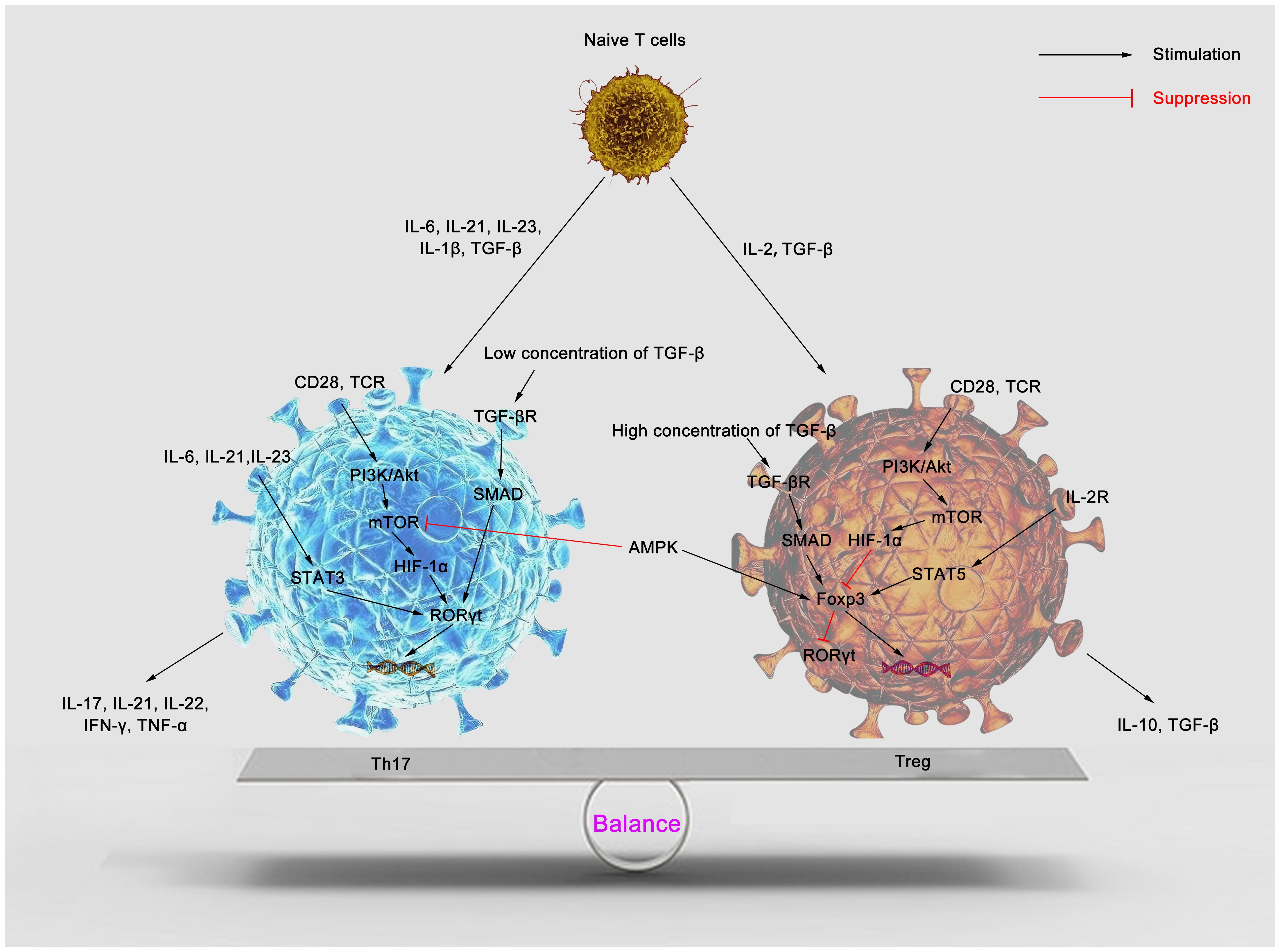
Figure 1 Cytokines regulate Th17/Treg balance. Naïve T cells can differentiate into Th17 and Tregs in response to specific cytokines. IL-1β, IL-6, IL-21, IL-23 and TGF-β induce Th17 differentiation. IL-6, IL-21 and IL-23 promote STAT3 phosphorylation, induce RORγt gene expression, and then stimulate CD4+ T cells to differentiate into Th17. mTOR is activated through the PI3K/Akt pathway to promote Th17 differentiation. A low TGF-β concentration activates SMAD and synergizes with IL-6 to promote Th17 production. Numerous mature Th17 secrete pro-inflammatory factors such as IL-17, IL-21, IL-22, IFN-γ, TNF-α, etc., promoting inflammation. Treg differentiation is mediated by IL-2 and TGF-β. IL-2/STAT5 pathway and high concentrations of TGF-β promote the expression of the Foxp3 gene and contribute to Treg differentiation. PI3K/Akt/mTOR pathway inhibits the expression of Foxp3 to suppress Treg differentiation. AMPK inhibits mTOR activity and promotes Foxp3 expression, which facilitates Treg differentiation. Many Tregs secrete IL-10 and TGF-β and induce host anti-inflammatory responses. Th17, T helper 17; Treg, regulatory T; IL, interleukin; TGF-β, transforming growth factor-β; TCR, T cell receptor; PI3K, phosphatidylinositol 3-kinase; Akt, protein kinase B; STAT3, signal transducer and activator of transcription 3; STAT5, signal transducer and activator of transcription 5; mTOR, mammalian target of rapamycin; RORγt, retinoid acid-related orphan receptor γt; HIF-1α, hypoxia inducible factor 1α; IFN-γ, interferon-γ; TNF-α, tumor necrosis factor-α; Foxp3, forkhead box protein P3; AMPK, AMP-activated protein kinase.
The immune response is designed to clear infectious agents, but the excessive pro-inflammatory response will lead to multiple organ damage (42–44). Meanwhile, the compensatory anti-inflammatory response, aimed at suppressing the excessive inflammation, can lead to immunosuppression (45). These two key events in the inflammatory response are interactive and concomitant (46). Th17 are pro-inflammatory, while Tregs are anti-inflammatory (47). In recent years, Th17/Treg balance has been increasingly recognized as critical in preventing excessive immune activation (48–51), autoimmune responses (52–54), and the pathogenesis of metabolic syndrome (55, 56). This review discusses the changes in Th17/Treg balance and its role in the pathophysiology of sepsis.
Treatment of sepsis is currently focused on continuous fluid resuscitation, organ support, and anti-infective therapy with antibiotics (57). To date, novel effective strategies that directly target the physiological mechanisms are not available (58). The essence of sepsis is an uncontrolled immune response eroded by the host in the fight against pathogens or stresses, leading to multi-organ dysfunction (59). Clinical and animal studies on sepsis still rely on the characterization of systemic inflammatory response syndromes (60–63). Numerous studies have confirmed that excessive inflammatory response and cytokine storm during sepsis are the leading causes of poor prognosis (64–66). Our previous study confirmed that the inflammatory response in septic mice was enhanced by inhibiting SUMOylation of peritoneal macrophages and increasing NF-κB p65 activity, inducing inflammatory storm, which markedly inhibited intra-abdominal bacterial clearance and greatly exacerbated organ damage and mortality (67). Another study, which enrolled 3,250 subjects, found that serum concentrations of IL-1β, TNF-α, and interferon (IFN)-γ were elevated in patients with sepsis (68). The mean TNF-α concentration in sepsis was increased approximately 10-fold compared to the mean concentration in healthy participants (68). Elevated TNF-α concentration was associated with increased 28-day sepsis mortality (68). Weber and Ebihara et al. also found that the levels of inflammatory factors IL-3, IL-6, and growth differentiation factor (GDF)-15 were significantly elevated in sepsis and correlated with prognosis (64, 69). Overexpression of inflammatory factors leads to an aggressive inflammatory response in the host. Failure to control the intensity of the inflammatory response promptly triggered a persistent and excessive inflammatory response, which severely compromised the prognosis of patients with sepsis. However, drugs targeting inflammatory mediators, like IL-1β, TNF-α, or TLR, to suppress inflammation are ineffective in improving survival in sepsis patients (70–72).
Th17 and Tregs antagonize each other during the inflammatory reaction and the immune response (73). Th17 produces many pro-inflammatory factors, which recruit neutrophils and promote inflammation. In contrast, Tregs produces anti-inflammatory factors that inhibit the activity of immune cells, thereby suppressing the inflammatory response to avoid overactivation of inflammation (74). Together, Th17 and Tregs direct the immune process of sepsis.
Th17 can be induced to differentiate by cytokines IL-1β, IL-6, IL-21, IL-23, and TGF-β (Table 1) (89). Signal transducer and activator of transcription 3 (STAT3) and SMAD are activated by cytokines, which in turn induce IL-17 and the transcription factor retinoic acid-related orphan receptor-γt (RORγt) to differentiate Th17 (90, 91). Th17 mediates inflammatory response and promotes host clearance of infected lesions (92). The main mechanisms include the secretion of pro-inflammatory cytokines (e.g., IL-17, IL-21, IL-22, IFN-γ), aggregation of neutrophils, activation of innate immune cells, and enhancement of B cell function (93–95). Various studies have shown that Th17 and the secreted cytokine IL-17 are inextricably linked to the pathogenesis of sepsis (35, 96–99). Mohammad et al. confirmed that sepsis led to upregulation of IL-2-inducible T-cell kinase (ITK) protein (35). ITK regulated the expression of transcription factors such as nuclear factor of activated T cells cytoplasmic 1 (NFATc1) and STAT3 in naïve CD4+ T cells (Th0), promoting their transformation into Th17 (96, 97). Inhibition of ITK expression using ITK inhibitors significantly reduced the number of IL-17A+CD4+ T cells, and decreased the levels of inflammatory factors IL-6 and monocyte chemotactic factor 1 (MCP-1) (35). That shows the involvement of Th17 in the inflammatory response in sepsis. In addition, overexpression of calcium release-activated calcium modulator 1 (Orai1) inhibited the growth of the Th17 population in sepsis and reduced mortality and organ damage in septic mice (98). Meanwhile, Li et al. have shown that the percentages of Th17 and the levels of IL-17, IL-22 were independent risk factors for septic lung injury (99). Th17 and the cytokines it releases are involved in the pathogenesis and prognosis of sepsis due to their pro-inflammatory properties.
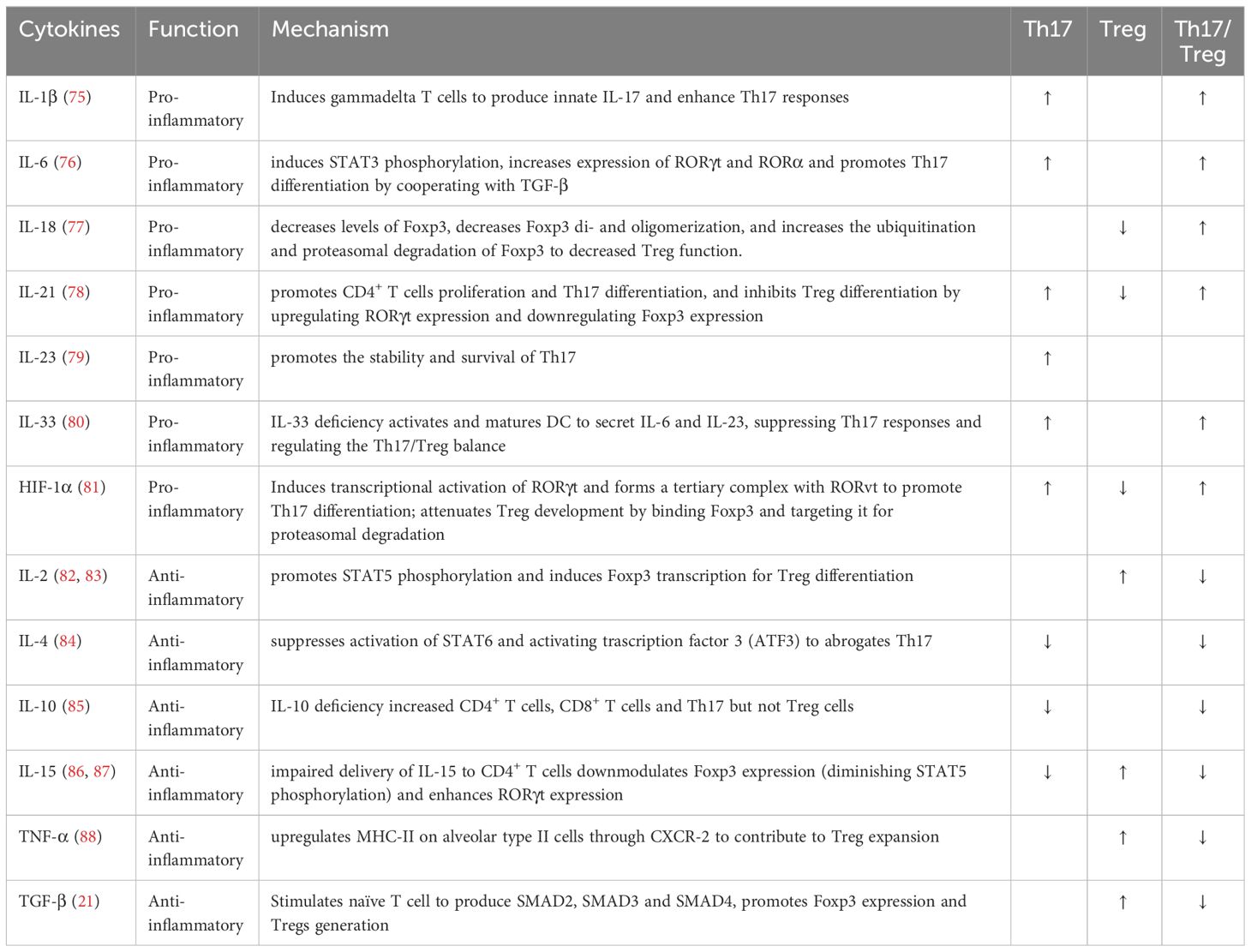
Table 1 The role of cytokines in Th17/Treg balance.
Tregs contribute to the maintenance of immune homeostasis and immune tolerance (100). Tregs differentiate from naïve T cells in the thymus or peripheral lymphoid tissue to protect organisms from autoimmune diseases and promote tissue repair (Table 1) (101). The main mechanisms of Tregs include: (1) contact-dependent mechanism: expression of co-stimulatory receptors, including cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), programmed cell death 1 (PD-1), T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), T cell immunoglobulin and ITIM domain (TIGIT), lymphocyte-activation gene 3 (LAG-3), ICOS, GITR, to prevent activation and maturation of DC or to prevent activated T cell response (102–106); (2) contact-independent mechanism: secretion of anti-inflammatory factors (IL-10, IL-35, TGF-β) to inhibit T cell proliferation (21, 107, 108); (3) CD25/IL-2 depletion mechanism: CD25, as the α-chain of the IL-2 receptor, is highly expressed on Tregs, which cause IL-2 deprivation and thus effector T cells death (109); (4) modification of the Foxp3 gene to enhance Treg stability (110–112); and (5) metabolic shifts in Treg from glycolysis to oxidative phosphorylation to enhance anti-inflammatory properties (113, 114). Many studies have shown that Tregs significantly inhibit inflammation and attenuate septic lung injury (115, 116). In the acute phase of sepsis, high-mobility group box 1 protein (HMGB1)-induced activation of chromosome ten (PTEN) reduced TGF-β release from macrophages, leading to decreased Treg differentiation (117). Neutralization of HMGB1 or knockdown of PTEN in macrophages promoted Tregs differentiation and inhibits Th17 differentiation, which alleviated the inflammatory response (117). In addition, Chinese medicinal preparations, such as Berberine and Tanshinone IIA, can inhibit septic inflammation by promoting Tregs differentiation (118, 119). The above studies confirmed that a high proportion of Treg contributed to the abatement of inflammation in septic mice.
However, Treg exacerbated the long-term death of sepsis. Patients and experimental mice that survived sepsis had more significant amounts of Tregs and higher concentrations of IL-33 (74, 120, 121). IL-33, present in the nucleus as a nuclear factor, is rapidly released during cell stress or damage (122). IL-33, in combination with ST2, induces the production of inflammatory factors by DC, macrophages, Th2, Tregs, mast cells, and innate lymphoid cell type 2 (ILC2) (123, 124), promotes the proliferation of Tregs and enhances the inhibitory capacity of Tregs (125, 126). The IL-33/ST2 signaling pathway mediates Treg amplification and exerts immunosuppressive effects. High levels of IL-33 have been found in survivors of sepsis, which in part contributes to a state of persistent immune paralysis. This leads to a depressed immune response and a much higher probability of infection, when the host is re-invaded by the similar or different pathogens. In addition, in a colitis model, IL-33/ST2 signaling was confirmed that increased the frequency of Tregs and ILC2 (127). Siede et al. demonstrated that IL-33/ST2 signaling promotes the expression of TGF-β, IL-10, and Th2-associated cytokines (IL-5/IL-13), and negatively affects effector T cells through the contact-independent mechanism (126). Neutralization of IL-33 significantly inhibited the expansion of Tregs and reduced mortality in mice affected by secondary infection (74). Moreover, most Tregs circulating in the blood originate from the spleen (128). In the immunosuppressive stage, removal of the spleen or use of drugs (e.g., granulocyte-macrophage colony-stimulating factor (GM-CSF), Poria cocos polysaccharides) to reduce the proliferation of Tregs can improve the survival rate and attenuate organ damage in septic mice (129–131). These results reflect the opposite and conflicting effects of Tregs in different stages of sepsis.
Briefly, there are differences in the function and expression levels of Th17 and Tregs in patients with sepsis. The balance of Th17 and Tregs in the body is highly relevant to the prognosis of patients with sepsis. A prospective observational study found heavily reduced numbers of circulating lymphocytes and inverted Th17/Treg ratios in patients with sepsis (34). Gupta et al. also found a high Treg proportion and a low Th17 proportion in post-traumatic sepsis patients, with a significant imbalance of Th17/Treg in cell-mediated immune response and disturbance (41). Zhou et al. found that the level of Th17/Treg ratio was higher in patients with sepsis-induced acute kidney injury (AKI) than in patients without it. Moreover, they confirmed that Th17/Treg imbalance was associated with the occurrence and severity of AKI in sepsis patients (132). Another study has demonstrated a strong positive correlation between the Th17/Treg ratio and the SOFA score: the higher the Th17/Treg ratio, the higher the SOFA score and the worse the prognosis of the patient (41). In a prospective clinical trial, septic patients who initiated enteral nutrition within 48 h had a significantly shorter duration of mechanical ventilation and the ICU hospitalization duration, which was shown to be achieved through modulating Th17/Treg imbalance and inhibiting the IL-23/IL-17 axis (133). Therefore, Th17/Treg imbalance is an essential factor in the pathogenesis of sepsis. Regulating Th17/Treg balance can effectively treat sepsis.
Meanwhile, many animal studies have validated the effect of Th17/Treg imbalance in cecal ligation and puncture (CLP) models of sepsis. It was confirmed that Th17/Treg imbalance caused organ damage and negatively affected survival in septic mice (134–141). Decreased macrophage recruitment and Th17/Treg imbalance were observed in the low-lethality non-severe sepsis model (134). The novel cytokine Metrnl administration reduced the peritoneal bacterial load while improving survival by promoting peritoneal macrophage recruitment and restoring Th17/Treg balance (135). Calcitriol treatment resulted in a more rational Th17/Treg balance in septic obese mice and activation of the renin-angiotensin system (RAS) anti-inflammatory pathway to attenuate acute lung injury (ALI) (136, 137). Nadeem et al. demonstrated that ALI in septic mice was attenuated via modulation of the Th17/Treg immune response and reduction of oxidative stress (138). Restoration of Th17/Treg balance also alleviated lung capillary leak in septic mice (139). Administration of arginine maintained Th17/Treg homeostasis and attenuated hepatic inflammation in sepsis (140). Mesenchymal stem cell (MSC) controlled sepsis-induced inflammatory responses by regulating Th17/Treg balance, which reduced tissue damage, protected organ function, and ultimately improved the survival rate in aged septic rats (141). Luo et al. also demonstrated that MSC modulated the Th17/Treg balance by decreasing IL-17 and RORγt levels and inducing IL-10 and Fxop3 expression via the Gal/Tim-3 pathway (142). As a result, modulation of Th17/Treg balance by various means in the CLP model significantly improved the prognosis of septic mice. This provides strong evidence for further clinical studies. However, the reports of clinical trials of related measures are fewer at present.
Under physiological conditions, CD4+ T cells can differentiate into multiple subtypes, such as Th1, Th2, Th9, Tfh, Th17, Treg, and other T-cell subtypes, which play corresponding immune functions (142). Differing from most somatic cells, T cells remain interconvertible between their subtypes due to instability and plasticity (143–145). Among all Th cell subtypes, Th17 and Treg were considered to be the most plastic subtypes (146, 147). Under appropriate cytokine stimulation or pathogenic conditions, Th17, which exerts a pro-inflammatory effect, can be converted to Treg, suppressing the immune response (148, 149). Jian and his colleagues obtained some Evidence from in vitro experiments. They stimulated tumor-infiltrating T lymphocytes (TILs) with OKT3 and mononuclear cells to acquire Th17 clones. After amplifying this subpopulation three times, Foxp3+ cells grew remarkably, while IL-17+ cells decreased (150), indicating partial transformation of Th17 to Treg. They are currently thought to result from epigenetic alterations and gene expression profiles reprogramming (148, 150).
In addition, due to prolonged exposure to a pro-inflammatory environment, Treg undergoes Foxp3 expression quiescence and is reprogrammed to Th17-like Treg, exerting pro-inflammatory effects and losing its inhibitory function (151). IL-1β and IL-6 stimulation increase the expression of STAT3 and RORγt in the Treg. This promotes the conversion of Treg to Th17-like Treg, releasing pro-inflammatory IL-17A (152). IL-23 exacerbates this phenomenon (152). TGF-β serves as a key factor in inducing Treg differentiation. Yang et al. used TGF-β to prompt Foxp3 expression. Then intervention with IL-6 was performed, and the results showed that the expression of Foxp3 was significantly downregulated, while the expression of IL-17 was increased (153). Similarly, inhibition of indoleamine 2,3-dioxygenase (IDO), a plasmacytoid DC-produced tryptophan-catabolizing enzyme that maintains Treg/Th17 balance, increased IL-6 expression and promoted the transformation of Treg into Th17 (154). Another study has shown that using single-cell RNA sequencing to focus on T cell subtype alteration in chronically exposed hypercholesterolemic environments (155). The results confirmed the continuous increase of Treg in atherosclerotic mice, which were partially transformed into Th17-like cells with highly pro-inflammatory properties (155). These findings are indicative of the instability and plasticity between Th17 and Treg, which are capable of transforming into each other.
Reports on Treg in sepsis are mixed. Some studies have demonstrated that increased circulating Treg proportion contributed to lymphocyte dysfunction and significantly reduced survival (156), while suppression of Treg percentage improved survival in sepsis (157). In contrast, other studies have found that adoptive transfer of Treg or pharmacologic modulation of Treg response protected septic mice from death (158, 159). Apart from the excessive inflammatory response and immune paralysis associated with sepsis, the underlying cause of this conflicting result may lie in the dysregulation of plasticity between Treg and Th17, leading to different immune effects of Treg.
Sepsis undergoes a sustained initial phase of hyperinflammation followed by a protracted period of immunosuppression (72, 160). Patients surviving sepsis often die from secondary infections due to immunosuppression and organ damage (161). Uncontrolled lymphocyte apoptosis is thought to be the major cause of immunosuppression (10, 160). As stated by Cao et al., sepsis is a life-and-death race between the pathogen and the host immune response, and the competition between the two determines the prognosis of septic patients (160, 162). In this process, Th17 and Tregs’ effects change during sepsis.
In the acute phase of sepsis, the proportion of Th17 is markedly elevated in the peripheral blood of the host, accompanied by an expansion in Tregs (130, 132). Invading bacteria activate T cell function, induce Th17 differentiation, and trigger an adaptive immune response to clear the bacteria (163, 164). However, persistent infection leads to excessive inflammatory activation and promotes Treg differentiation to avoid organ damage from hyper-inflammation (15, 165). Studies have shown that patients suffering from sepsis have an increased percentage of Tregs in the peripheral blood, which occurs mainly in the early stages of sepsis. Persistently high Treg counts are a vital factor in the poor prognosis of patients (38, 166). Furthermore, it has been verified in a sepsis mouse model that Treg percentage showed a significant augmentation following CLP, a process that lasted up to 7 days (167). Control of Tregs and Th17 ratio and maintenance of Th17/Treg balance can effectively affect the prognosis of sepsis. Lu et al. discovered that mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) overexpression promoted the polarization of CD4+ T cells towards Th17 and increased the Th17/Treg ratio, which in turn promoted systemic inflammation, elevated levels of oxidative stress, and exacerbated organ damage (168). Meanwhile, receiving MSC intravenous treatment, septic mice showed a lower Th17/Treg ratio in peripheral blood and spleen, protecting organs from inflammatory damage and increasing the 72 h survival rate (140). However, another study has reported that increasing the intestinal Th17 population reduced intestinal inflammation and bacterial translocation, protected mice from endotoxemia-induced intestinal injury, and enhanced survival (169). This may be a hyperacute phase response triggered when intestinal bacteria invade the host, thus helping the host eliminate the bacteria before they enter the bloodstream in large numbers, avoiding a storm of inflammatory factors. Meanwhile, it cannot be ruled out that due to Th17 plasticity, pathogen stimulation and alteration of the cytokine environment promoted the reprogramming of Th17 to Treg, which suppressed the inflammatory response in the gut (148, 149). However, more experiments are needed to clarify. The above study confirmed that the ratio of Th17/Treg dramatized the prognosis of sepsis. In the early phase of sepsis, excessive inflammatory response causes organ damage. Increasing the proportion of Tregs, decreasing the proportion of Th17, and correcting the Th17/Treg balance are essential factors for restoring immune homeostasis, reducing inflammatory organ damage, and decreasing mortality.
When pathogens are effectively cleared, immune balance can be restored; conversely, immune regulation is imbalanced, anti-inflammatory responses are enhanced, and the host is susceptible to a state of immunosuppression or even organ failure (58). In the later stages of sepsis, the proportion of Treg increased, accompanied by a decrease in the proportion of Th17, which led to a lower Th17/Treg ratio (34). This was related to the initial inflammatory factor storm causing inflammatory factor depletion and severe T cell apoptosis (34). Lymphopenia coupled with increased apoptosis and decreased proliferation of lymphocytes, and more circulating Treg are associated with persistent organ dysfunction, secondary infections and long-term mortality (170, 171). Therefore, in the late stage of sepsis, restoring T cell function and promoting inflammatory response can help enhance host immunity and effectively avoid immunosuppression, reducing the late mortality of sepsis patients. In a two-stroke model of sepsis, methicillin-resistant Staphylococcus aureus (MRSA) inoculation was used to trigger secondary pneumonia (172). The presence of long-term immune dysfunction in septic mice was confirmed by higher bacterial counts in bronchoalveolar lavages, spleen and kidney homogenates, increased T-cell apoptosis, enhanced Treg ratio, accompanied by severe lung injury, and a markedly higher 20-day mortality rate (172). Treatment with citrulline restored T-cell mitochondrial activity, reduced the number of Tregs, and reversed the mortality rate in mice (172). In addition, during the immunosuppressive phase of sepsis, IL-33, GM-CSF or tumor necrosis factor receptor antibody (DTA-1) administration can inhibit Treg amplification and restore Th17/Treg balance, favoring the survival of sepsis (120, 130, 173).
Currently, many studies are exploring the mechanisms of Th17/Treg imbalance at different stages of sepsis. In the early stage, pathogens invade the organism and activate innate immune cells to phagocytose and clear them, triggering the innate immune response and complement system to produce various pro-inflammatory factors, e.g., IL-1β, IL-6, IL-12, and TNF-α (174, 175). The study has demonstrated that complement C5a induced DC transfer from the peritoneal cavity to peripheral blood and lymph nodes, inducing the expansion Th17 (176). Co-incubation of human DC with gram-positive bacteria cell wall peptidoglycan polymers stimulates the production of IL-23 and IL-1β by DC, thereby inducing the CD4+ T cells to differentiate to Th17 (177). Inflammatory factors stimulate T-lymphocyte proliferation and differentiation, increasing the proportion of Th17. The persistently increased Th17 continues to secrete pro-inflammatory cytokines, further inducing cytokine storm and Th17/Treg imbalance. Furthermore, most patients who survive sepsis develop a prolonged immunocompromised state, which is mainly caused by immunosuppression in the late stage of sepsis (178, 179). The excessive inflammatory response prompts overexpression of immune checkpoint molecules (e.g., PD-1, TIM-3, CTLA-4) (58). These negative co-stimulatory factors cause Th17 decrease and T-cell depletion by inhibiting T-lymphocyte activation and proliferation or by inducing apoptosis (180–183). The relative increase in Treg during sepsis was shown to be due to the high resistance of Treg to apoptosis and preferential loss of other subtypes (Th1, Th2, Th17, Tfh), which ultimately leads to a Th17/Treg balance toward Treg (156, 184–187). In addition, inflammation-related markers are also associated with Treg amplification. C3aR/C5aR-mediated inflammatory activity impairs Treg function (188). Excessive inflammatory response in the sepsis leads to complement C3a depletion, which is thought to be associated with Treg amplification (189). Administration of exogenous complement C3a inhibited Treg expansion in sepsis animal model (190). In addition, cytokine IL-33 also upregulates Treg and contributes to sepsis-induced immunosuppression by promoting M2 macrophage polarization and IL-10 secretion (74). The above pathways increase the Treg proportion in late spies, inducing Th17/Treg balance biased toward Treg.
However, the great instability and plasticity of Th17/Treg left multiple possibilities for immune alterations in the septic process. Under certain conditions, Th17 and Treg underwent functional changes through internal reprogramming to express other T-cell related cytokines. Th17 can be transformed into Th1, Th2, Treg, and Tfh. In contrast, Treg can also be transformed into Th1-like, Th2-like, and Th17-like, Tfh-like Treg, which was confirmed in many studies in the tumor microenvironment, autoimmune disease, allergic asthma, and multiple sclerosis (MS) (90, 146, 191–197). Both Th17 and Treg required TGF-β to regulate their differentiation (90, 198). Treg inhibited cell differentiation into Th17 by inhibiting RORyt activity through Foxp3 expression (22). This restriction can be lifted by IL-6 and other cytokines, and undergone reprogramming of Th17 and Treg (22). In the early stages of sepsis, the Th17/Treg balance favored Th17 in the presence of pathogen stimulation and a strong inflammatory microenvironment. However, there was still no evidence to confirm whether Treg lost its suppressive function or reprogrammed to Th17, further promoting a hyperinflammatory response. Similarly, in advanced sepsis, both Th17 and Tregs showed decreased numbers due to lymphocyte apoptosis. Th17/Treg favored Treg, and showed an overall immunosuppressive state. Nevertheless, whether this state was due to the plasticity and instability of Th17 leading to the transformation of Th17 to Treg remained unknown in the field of sepsis research. Overall, it can be concluded that the Th17/Treg balance has diametrically opposing effects on host prognosis at different stages of sepsis, and the underlying phenotypic and functional alterations were unclear.
Most studies have focused on the function and characterization of Th17/Treg in the circulation or spleen (34, 41, 132, 133, 137). However, Th17 and Treg are prevalent not only within the immune organs but also in the lungs, kidneys, liver, heart, brain, and muscle (199–204). The pathophysiological features of sepsis vary from organ to organ due to the functional and structural differences of each organ. Th17/Treg balance has tissue characterization on each organ.
The lung is the most susceptible and is the first organ to be engaged in sepsis (205). Sepsis often causes pulmonary inflammation that fails to subside, inducing ALI/acute respiratory distress syndrome (ARDS) with a high morbidity and mortality rate (206, 207). The pathology is characterized primarily by a massive inflammatory cell infiltrate, loss of barrier integrity and increased permeability of alveolar capillaries, leading to tissue damage in inflammation (207, 208). Many studies have shown that restoration of Th17/Treg balance has a positive effect on sepsis-induced lung injury (137, 138, 209, 210). Xia et al. demonstrated that Maresin 1, an emerging specific pro-inflammatory mediator, increased the number of Tregs and decreased the Th17 count, which regulated the Th17/Treg balance (210). Altered T-cell balance significantly inhibited excessive inflammatory response, promoted inflammatory regression, reduced septic lung damage and improved lung function (210). On the other hand, Nadeem et al. also acted to regulate the Th17/Treg balance by inhibiting the expression of ITK, an essential regulator of Th17 differentiation, reducing Th17 and expanding Tregs (137). This behavior reversed the level of airway inflammation and oxidative stress in septic lungs (137). In addition, Traditional Chinese medicine (TCM) also played a positive role in modulating Th17/Treg balance. Our previous study confirmed that berberine attenuated pulmonary edema and hypoxemia in septic mice by regulating Th17/Treg homeostatic (209). Paclitaxel also alleviated lung injury and prolonged survival in septic mice by altering the Th17 and Treg populations.
Besides the lungs, the kidneys are often vulnerable to sepsis. Many studies have focused on the interaction of Th17 or Treg with sepsis-induced acute kidney injury (SAKI) (211–214). A high level of IL-17 and tissue infiltration of Th17 often accompanies SAKI (215). Inhibition of Th17 differentiation and function can effectively alleviate renal damage (216). Additionally, several animal studies confirmed that Tregs protected the kidney from inflammation (214, 217, 218). However, other studies suggested that Treg exacerbated renal damage, which could be attenuated by depletion of Treg and inhibition of IL-10 (219, 220). This called for consideration of the impact of Treg plasticity on SAKI during different phases of sepsis. Alterations in the phenotype and function of Treg can partially contribute to the inflammatory response towards the two opposite outcomes. Treg exerting anti-inflammatory effects effectively alleviates renal inflammation in SAKI (213). Nevertheless, Treg with pro-inflammatory properties, such as Th17-like Treg, can exacerbate immunopathologic changes in the kidney, but further studies were necessary to explain this. Furthermore, contradictory results encouraged us to concentrate on the impact of Th17/Treg balance on SAKI. Zhou et al. investigated Th17/Treg balance and found that patients with SAKI had an elevated Th17 ratio and Th17/Treg imbalance (132). The Th17/Treg balance axis tended to favor the Th17 lineage (132). The Th17/Treg ratio was an independent risk factor for SAKI (132). Therefore, perhaps it is not Th17 or Tregs, but rather the Th17/Treg balance that influenced the pathological process of SAKI.
Variation in the immune function affects regression in septic patients. The differentiation and function of Th17 and Treg, and bias in Th17/Treg balance, determine different grades of inflammatory response and organ damage. Many studies are still limited to pathophysiological alterations of Th17 or Treg in sepsis, along with conflicting results. This forced us to focus on Th17/Treg balance rather than solely on Th17 or Treg. However, the current established studies have limited insights into Th17/Treg balance in sepsis. This review summarizes the pathophysiology of Th17 and Treg in sepsis, and describes the effects of Th17/Treg balance in different inflammatory stages and organs. Since Th17/Treg balance controls the shifts in pro-inflammatory and anti-inflammatory responses, amelioration of sepsis by clarifying the optimal point of Th17/Treg balance may be an effective therapeutic intervention.
XL: Funding acquisition, Investigation, Resources, Visualization, Writing – original draft, Writing – review & editing. LC: Funding acquisition, Investigation, Resources, Visualization, Writing – original draft, Writing – review & editing. WP: Data curation, Visualization, Writing – original draft. HD: Data curation, Visualization, Writing – original draft. HN: Data curation, Visualization, Writing – original draft. HT: Data curation, Visualization, Writing – original draft. HH: Data curation, Visualization, Writing – original draft. SW: Data curation, Visualization, Writing – original draft. JQ: Data curation, Visualization, Writing – original draft. AL: Conceptualization, Project administration, Visualization, Writing – review & editing. KC: Conceptualization, Funding acquisition, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Fundamental Research Project of Jinhua Hospital Affiliated to Zhejiang University (No. JY2022-5-02).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. (2013) 369:840–51. doi: 10.1056/NEJMra1208623
2. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
3. Fleischmann C, Thomas-Rueddel DO, Hartmann M, Hartog CS, Welte T, Heublein S, et al. Hospital incidence and mortality rates of sepsis. Dtsch Arztebl Int. (2016) 113:159–66. doi: 10.3238/arztebl.2016.0159
4. Fleischmann-Struzek C, Schwarzkopf D, Reinhart K. [Sepsis incidence in Germany and worldwide: Current knowledge and limitations of research using health claims data]. Med Klin Intensivmed Notfmed. (2022) 117:264–8. doi: 10.1007/s00063-021-00777-5
5. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality 1990-2017: analysis for the global burden of disease study. Lancet. (2020) 395:200–11. doi: 10.1016/s0140-6736(19)32989-7
6. Xie J, Wang H, Kang Y, Zhou L, Liu Z, Qin B, et al. The epidemiology of sepsis in chinese ICUs: A national cross-sectional survey. Crit Care Med. (2020) 48:e209–18. doi: 10.1097/ccm.0000000000004155
7. Karakike E, Kyriazopoulou E, Tsangaris I, Routsi C, Vincent JL, Giamarellos-Bourboulis EJ. The early change of SOFA score as a prognostic marker of 28-day sepsis mortality: analysis through a derivation and a validation cohort. Crit Care. (2019) 23:387. doi: 10.1186/s13054-019-2665-5
8. Busch LM, Kadri SS. Antimicrobial treatment duration in sepsis and serious infections. J Infect Dis. (2020) 222:S142–s155. doi: 10.1093/infdis/jiaa247
9. Wang W, Liu CF. Sepsis heterogeneity. World J Pediatr. (2023) 19:919–27. doi: 10.1007/s12519-023-00689-8
10. Torres LK, Pickkers P, van der Poll T. Sepsis-induced immunosuppression. Annu Rev Physiol. (2022) 84:157–81. doi: 10.1146/annurev-physiol-061121-040214
11. Nedeva C, Menassa J, Puthalakath H. Sepsis: inflammation is a necessary evil. Front Cell Dev Biol. (2019) 7:108. doi: 10.3389/fcell.2019.00108
12. Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE Jr., Hui JJ, Chang KC, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. (2001) 166:6952–63. doi: 10.4049/jimmunol.166.11.6952
13. Tang D, Wang H, Billiar TR, Kroemer G, Kang R. Emerging mechanisms of immunocoagulation in sepsis and septic shock. Trends Immunol. (2021) 42:508–22. doi: 10.1016/j.it.2021.04.001
14. Hamers L, Kox M, Pickkers P. Sepsis-induced immunoparalysis: mechanisms, markers, and treatment options. Minerva Anestesiol. (2015) 81:426–39.
15. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. (2017) 17:407–20. doi: 10.1038/nri.2017.36
16. Nedeva C. Inflammation and cell death of the innate and adaptive immune system during sepsis. Biomolecules. (2021) 11(7):1011. doi: 10.3390/biom11071011
17. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
18. Davies K, McLaren JE. Destabilisation of T cell-dependent humoral immunity in sepsis. Clin Sci (Lond). (2024) 138:65–85. doi: 10.1042/cs20230517
19. Ong DSY, Bonten MJM, Spitoni C, Verduyn Lunel FM, Frencken JF, Horn J, et al. Epidemiology of multiple herpes viremia in previously immunocompetent patients with septic shock. Clin Infect Dis. (2017) 64:1204–10. doi: 10.1093/cid/cix120
20. Brady J, Horie S, Laffey JG. Role of the adaptive immune response in sepsis. Intensive Care Med Exp. (2020) 8:20. doi: 10.1186/s40635-020-00309-z
21. Wang J, Zhao X, Wan YY. Intricacies of TGF-β signaling in Treg and Th17 cell biology. Cell Mol Immunol. (2023) 20(9):1002–22. doi: 10.1038/s41423-023-01036-7
22. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
23. Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. (2008) 454:350–2. doi: 10.1038/nature07021
24. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
25. Besnard AG, Togbe D, Couillin I, Tan Z, Zheng SG, Erard F, et al. Inflammasome-IL-1-Th17 response in allergic lung inflammation. J Mol Cell Biol. (2012) 4:3–10. doi: 10.1093/jmcb/mjr042
26. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol. (2017) 18:612–21. doi: 10.1038/ni.3742
27. Vecellio M, Hake VX, Davidson C, Carena MC, Wordsworth BP, Selmi C. The IL-17/IL-23 axis and its genetic contribution to psoriatic arthritis. Front Immunol. (2020) 11:596086. doi: 10.3389/fimmu.2020.596086
28. Xu S, Cao X. Interleukin-17 and its expanding biological functions. Cell Mol Immunol. (2010) 7:164–74. doi: 10.1038/cmi.2010.21
29. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev. (2014) 13:668–77. doi: 10.1016/j.autrev.2013.12.004
30. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
31. Pawlak M, Ho AW, Kuchroo VK. Cytokines and transcription factors in the differentiation of CD4(+) T helper cell subsets and induction of tissue inflammation and autoimmunity. Curr Opin Immunol. (2020) 67:57–67. doi: 10.1016/j.coi.2020.09.001
32. Liu P, Xiao Z, Yan H, Lu X, Zhang X, Luo L, et al. Baicalin suppresses Th1 and Th17 responses and promotes Treg response to ameliorate sepsis-associated pancreatic injury via the RhoA-ROCK pathway. Int Immunopharmacol. (2020) 86:106685. doi: 10.1016/j.intimp.2020.106685
33. Brunialti MK, Santos MC, Rigato O, MaChado FR, Silva E, Salomao R. Increased percentages of T helper cells producing IL-17 and monocytes expressing markers of alternative activation in patients with sepsis. PloS One. (2012) 7:e37393. doi: 10.1371/journal.pone.0037393
34. Li J, Li M, Su L, Wang H, Xiao K, Deng J, et al. Alterations of T helper lymphocyte subpopulations in sepsis, severe sepsis, and septic shock: a prospective observational study. Inflammation. (2015) 38:995–1002. doi: 10.1007/s10753-014-0063-3
35. Algahtani MM, Alshehri S, Alqarni SS, Ahmad SF, Al-Harbi NO, Alqarni SA, et al. Inhibition of ITK signaling causes amelioration in sepsis-associated neuroinflammation and depression-like state in mice. Int J Mol Sci. (2023) 24(9):8101. doi: 10.3390/ijms24098101
36. He W, Xiao K, Fang M, Xie L. Immune cell number, phenotype, and function in the elderly with sepsis. Aging Dis. (2021) 12:277–96. doi: 10.14336/ad.2020.0627
37. Liu Y, Wang X, Yu L. Th17, rather than Th1 cell proportion, is closely correlated with elevated disease severity, higher inflammation level, and worse prognosis in sepsis patients. J Clin Lab Anal. (2021) 35:e23753. doi: 10.1002/jcla.23753
38. Monneret G, Debard AL, Venet F, Bohe J, Hequet O, Bienvenu J, et al. Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit Care Med. (2003) 31:2068–71. doi: 10.1097/01.CCM.0000069345.78884.0F
39. Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, et al. Human CD4+CD25+ regulatory T lymphocytes inhibit lipopolysaccharide-induced monocyte survival through a Fas/Fas ligand-dependent mechanism. J Immunol. (2006) 177:6540–7. doi: 10.4049/jimmunol.177.9.6540
40. Huang LF, Yao YM, Dong N, Yu Y, He LX, Sheng ZY. Association between regulatory T cell activity and sepsis and outcome of severely burned patients: a prospective, observational study. Crit Care. (2010) 14:R3. doi: 10.1186/cc8232
41. Gupta DL, Bhoi S, Mohan T, Galwnkar S, Rao DN. Coexistence of Th1/Th2 and Th17/Treg imbalances in patients with post traumatic sepsis. Cytokine. (2016) 88:214–21. doi: 10.1016/j.cyto.2016.09.010
42. Shahbazi R, Yasavoli-Sharahi H, Alsadi N, Ismail N, Matar C. Probiotics in treatment of viral respiratory infections and neuroinflammatory disorders. Molecules. (2020) 25(21):4891. doi: 10.3390/molecules25214891
43. Kanashiro A, Hiroki CH, da Fonseca DM, Birbrair A, Ferreira RG, Bassi GS, et al. The role of neutrophils in neuro-immune modulation. Pharmacol Res. (2020) 151:104580. doi: 10.1016/j.phrs.2019.104580
44. Suresh RV, Bradley EW, Higgs M, Russo VC, Alqahtani M, Huang W, et al. Nlrp3 increases the host’s susceptibility to tularemia. Front Microbiol. (2021) 12:725572. doi: 10.3389/fmicb.2021.725572
45. Wei X, Zhang L, Zhang R, Wu R, Petitte JN, Hou Y, et al. Targeting the TLR2 receptor with a novel thymopentin-derived peptide modulates immune responses. Front Immunol. (2021) 12:620494. doi: 10.3389/fimmu.2021.620494
46. Cavaillon JM. During sepsis and COVID-19, the pro-inflammatory and anti-inflammatory responses are concomitant. Clin Rev Allergy Immunol. (2023) 65(2):183–7. doi: 10.1007/s12016-023-08965-1
47. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
48. Lu Y, Liu X, Lin S, Chen P, Chen L, Zhao G. Stanniocalcin-1 attenuates collagen-induced arthritis through inhibiting STAT3 phosphorylation and Th17 response. Scandinavian J Immunol. (2023) 97:e13226. doi: 10.1111/sji.13226
49. Zhang H, Yuan Z, Zhu Y, Yuan Z, Wang J, Nong C, et al. Th17/Treg imbalance mediates hepatic intolerance to exogenous lipopolysaccharide and exacerbates liver injury in triptolide induced excessive immune response. J Ethnopharmacol. (2022) 295:115422. doi: 10.1016/j.jep.2022.115422
50. Ma B, Chen D, Liu Y, Zhao Z, Wang J, Zhou G, et al. Yanghe decoction suppresses the experimental autoimmune thyroiditis in rats by improving NLRP3 inflammasome and immune dysregulation. Front Pharmacol. (2021) 12:645354. doi: 10.3389/fphar.2021.645354
51. Shi C, Zhang H, Wang X, Jin B, Jia Q, Li Y, et al. Cinnamtannin D1 attenuates autoimmune arthritis by regulating the balance of Th17 and treg cells through inhibition of aryl hydrocarbon receptor expression. Pharmacol Res. (2020) 151:104513. doi: 10.1016/j.phrs.2019.104513
52. Aso K, Kono M, Kanda M, Kudo Y, Sakiyama K, Hisada R, et al. Itaconate ameliorates autoimmunity by modulating T cell imbalance via metabolic and epigenetic reprogramming. Nat Commun. (2023) 14:984. doi: 10.1038/s41467-023-36594-x
53. Lee GR. The balance of Th17 versus treg cells in autoimmunity. Int J Mol Sci. (2018) 19(3):730. doi: 10.3390/ijms19030730
54. DiToro D, Harbour SN, Bando JK, Benavides G, Witte S, Laufer VA, et al. Insulin-like growth factors are key regulators of T helper 17 regulatory T cell balance in autoimmunity. Immunity. (2020) 52:650–667.e610. doi: 10.1016/j.immuni.2020.03.013
55. Tao L, Liu H, Gong Y. Role and mechanism of the Th17/Treg cell balance in the development and progression of insulin resistance. Mol Cell Biochem. (2019) 459:183–8. doi: 10.1007/s11010-019-03561-4
56. Li L, Xia Y, Ji X, Wang H, Zhang Z, Lu P, et al. MIG/CXCL9 exacerbates the progression of metabolic-associated fatty liver disease by disrupting Treg/Th17 balance. Exp Cell Res. (2021) 407:112801. doi: 10.1016/j.yexcr.2021.112801
57. Shorr AF, Zilberberg MD. Sepsis and septic shock: evolving evidence, evolving paradigms. Semin Respir Crit Care Med. (2022) 43:39–45. doi: 10.1055/s-0041-1740975
58. Liu D, Huang SY, Sun JH, Zhang HC, Cai QL, Gao C, et al. Sepsis-induced immunosuppression: mechanisms, diagnosis and current treatment options. Mil Med Res. (2022) 9:56. doi: 10.1186/s40779-022-00422-y
59. Seymour CW, Liu VX, Iwashyna TJ, Brunkhorst FM, Rea TD, Scherag A, et al. Assessment of clinical criteria for sepsis: for the third international consensus definitions for sepsis and septic shock (Sepsis-3). Jama. (2016) 315:762–74. doi: 10.1001/jama.2016.0288
60. Schertz AR, Lenoir KM, Bertoni AG, Levine BJ, Mongraw-Chaffin M, Thomas KW. Sepsis Prediction Model for Determining Sepsis vs SIRS, qSOFA, and SOFA. JAMA Netw Open. (2023) 6:e2329729. doi: 10.1001/jamanetworkopen.2023.29729
61. Wang Z, Wang Z. The role of macrophages polarization in sepsis-induced acute lung injury. Front Immunol. (2023) 14:1209438. doi: 10.3389/fimmu.2023.1209438
62. Qiu X, Lei YP, Zhou RX. SIRS, SOFA, qSOFA, and NEWS in the diagnosis of sepsis and prediction of adverse outcomes: a systematic review and meta-analysis. Expert Rev Anti Infect Ther. (2023) 21:891–900. doi: 10.1080/14787210.2023.2237192
63. Miao R, Huang J. MCC950 improves lipopolysaccharide−induced systemic inflammation in mice by relieving pyroptosis in blood neutrophils. Exp Ther Med. (2023) 26:417. doi: 10.3892/etm.2023.12117
64. Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A, et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science. (2015) 347:1260–5. doi: 10.1126/science.aaa4268
65. Fu X, Liu Z, Wang Y. Advances in the study of immunosuppressive mechanisms in sepsis. J Inflammation Res. (2023) 16:3967–81. doi: 10.2147/jir.S426007
66. Chen J, Wei H. Immune intervention in sepsis. Front Pharmacol. (2021) 12:718089. doi: 10.3389/fphar.2021.718089
67. Liu X, Chen L, Zhang C, Dong W, Liu H, Xiao Z, et al. Ginkgolic acid promotes inflammation and macrophage apoptosis via SUMOylation and NF-κB pathways in sepsis. Front Med (Lausanne). (2022) 9:1108882. doi: 10.3389/fmed.2022.1108882
68. Gharamti AA, Samara O, Monzon A, Montalbano G, Scherger S, DeSanto K, et al. Proinflammatory cytokines levels in sepsis and healthy volunteers, and tumor necrosis factor-alpha associated sepsis mortality: A systematic review and meta-analysis. Cytokine. (2022) 158:156006. doi: 10.1016/j.cyto.2022.156006
69. Ebihara T, Matsumoto H, Matsubara T, Togami Y, Nakao S, Matsuura H, et al. Cytokine elevation in severe COVID-19 from longitudinal proteomics analysis: comparison with sepsis. Front Immunol. (2021) 12:798338. doi: 10.3389/fimmu.2021.798338
70. Cohen J, Opal S, Calandra T. Sepsis studies need new direction. Lancet Infect Dis. (2012) 12:503–5. doi: 10.1016/s1473-3099(12)70136-6
71. Angus DC. The search for effective therapy for sepsis: back to the drawing board? Jama. (2011) 306:2614–5. doi: 10.1001/jama.2011.1853
72. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. (2013) 13:862–74. doi: 10.1038/nri3552
73. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. (2010) 140:845–58. doi: 10.1016/j.cell.2010.02.021
74. Nascimento DC, Melo PH, Piñeros AR, Ferreira RG, Colón DF, Donate PB, et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun. (2017) 8:14919. doi: 10.1038/ncomms14919
75. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. (2009) 31:331–41. doi: 10.1016/j.immuni.2009.08.001
76. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
77. Akimova T, Zhang T, Christensen LM, Wang Z, Han R, Negorev D, et al. Obesity-related IL-18 impairs T-regulatory cell function and promotes lung ischemia-reperfusion injury. Am J Respir Crit Care Med. (2021) 204:1060–74. doi: 10.1164/rccm.202012-4306OC
78. Shi Y, Chen Z, Zhao Z, Yu Y, Fan H, Xu X, et al. IL-21 induces an imbalance of Th17/Treg cells in moderate-to-severe plaque psoriasis patients. Front Immunol. (2019) 10:1865. doi: 10.3389/fimmu.2019.01865
79. Wu B, Wan Y. Molecular control of pathogenic Th17 cells in autoimmune diseases. Int Immunopharmacol. (2020) 80:106187. doi: 10.1016/j.intimp.2020.106187
80. Cheng L, Jiao Y, Jiang W, Zhang X, Zhang L, Jia G. IL-33 deficiency attenuates lung inflammation by inducing Th17 response and impacting the Th17/Treg balance in LPS-induced ARDS mice via dendritic cells. J Immunol Res. (2022) 2022:9543083. doi: 10.1155/2022/9543083
81. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
82. Zhang X, Miao M, Zhang R, Liu X, Zhao X, Shao M, et al. Efficacy and safety of low-dose interleukin-2 in combination with methotrexate in patients with active rheumatoid arthritis: a randomized, double-blind, placebo-controlled phase 2 trial. Signal Transduct Target Ther. (2022) 7:67. doi: 10.1038/s41392-022-00887-2
83. Xie Y, Jin C, Sang H, Liu W, Wang J. Ivermectin protects against experimental autoimmune encephalomyelitis in mice by modulating the Th17/Treg balance involved in the IL-2/STAT5 pathway. Inflammation. (2023) 46:1626–38. doi: 10.1007/s10753-023-01829-y
84. Guenova E, Skabytska Y, Hoetzenecker W, Weindl G, Sauer K, Tham M, et al. IL-4 abrogates T(H)17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc Natl Acad Sci U.S.A. (2015) 112:2163–8. doi: 10.1073/pnas.1416922112
85. Zhang B, Wang J, Liu M, Zhao Q, Yu G, Zhang B, et al. IL-10 regulates Th17 response to inhibit hepatobiliary injury caused by Clonorchis sinensis infection in C57BL/6J mice. Front Cell Infect Microbiol. (2022) 12:994838. doi: 10.3389/fcimb.2022.994838
86. Tosiek MJ, Fiette L, El Daker S, Eberl G, Freitas AA. IL-15-dependent balance between Foxp3 and RORγt expression impacts inflammatory bowel disease. Nat Commun. (2016) 7:10888. doi: 10.1038/ncomms10888
87. Pandiyan P, Yang XP, Saravanamuthu SS, Zheng L, Ishihara S, O’Shea JJ, et al. The role of IL-15 in activating STAT5 and fine-tuning IL-17A production in CD4 T lymphocytes. J Immunol. (2012) 189:4237–46. doi: 10.4049/jimmunol.1201476
88. Guo N, Wen Y, Wang C, Kang L, Wang X, Liu X, et al. Lung adenocarcinoma-related TNF-α-dependent inflammation upregulates MHC-II on alveolar type II cells through CXCR-2 to contribute to Treg expansion. FASEB J. (2020) 34:12197–213. doi: 10.1096/fj.202000166RR
89. Mills KH. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol. (2008) 38:2636–49. doi: 10.1002/eji.200838535
90. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. (2008) 453:236–40. doi: 10.1038/nature06878
91. Yoon JH, Sudo K, Kuroda M, Kato M, Lee IK, Han JS, et al. Phosphorylation status determines the opposing functions of Smad2/Smad3 as STAT3 cofactors in TH17 differentiation. Nat Commun. (2015) 6:7600. doi: 10.1038/ncomms8600
92. Bouch RJ, Zhang J, Miller BC, Robbins CJ, Mosher TH, Li W, et al. Distinct inflammatory Th17 subsets emerge in autoimmunity and infection. J Exp Med. (2023) 220(10):e20221911. doi: 10.1084/jem.20221911
93. Chen P, Tang X. Gut microbiota as regulators of Th17/Treg balance in patients with myasthenia gravis. Front Immunol. (2021) 12:803101. doi: 10.3389/fimmu.2021.803101
94. Fossiez F, Banchereau J, Murray R, Van Kooten C, Garrone P, Lebecque S. Interleukin-17. Int Rev Immunol. (1998) 16:541–51. doi: 10.3109/08830189809043008
95. Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. (1998) 160:3513–21. doi: 10.4049/jimmunol.160.7.3513
96. Adam N, Kandelman S, Mantz J, Chrétien F, Sharshar T. Sepsis-induced brain dysfunction. Expert Rev Anti Infect Ther. (2013) 11:211–21. doi: 10.1586/eri.12.159
97. Lechner KS, Neurath MF, Weigmann B. Role of the IL-2 inducible tyrosine kinase ITK and its inhibitors in disease pathogenesis. J Mol Med (Berl). (2020) 98:1385–95. doi: 10.1007/s00109-020-01958-z
98. Chen L, Ke H, Zhang Y, Jin P, Liu X, Hong G, et al. Orai1 overexpression improves sepsis-induced T-lymphocyte immunosuppression and acute organ dysfunction in mice. Heliyon. (2022) 8:e12082. doi: 10.1016/j.heliyon.2022.e12082
99. Li G, Zhang L, Han N, Zhang K, Li H. Increased Th17 and Th22 cell percentages predict acute lung injury in patients with sepsis. Lung. (2020) 198:687–93. doi: 10.1007/s00408-020-00362-1
100. Scott DW. Driving CARs to BARs: the winding road to specific regulatory T cells for tolerance. Front Immunol. (2021) 12:742719. doi: 10.3389/fimmu.2021.742719
101. Darrigues J, van Meerwijk JPM, Romagnoli P. Age-dependent changes in regulatory T lymphocyte development and function: A mini-review. Gerontology. (2018) 64:28–35. doi: 10.1159/000478044
102. Vaddi PK, Osborne DG, Nicklawsky A, Williams NK, Menon DR, Smith D, et al. CTLA4 mRNA is downregulated by miR-155 in regulatory T cells, and reduced blood CTLA4 levels are associated with poor prognosis in metastatic melanoma patients. Front Immunol. (2023) 14:1173035. doi: 10.3389/fimmu.2023.1173035
103. McGee MC, Zhang T, Magazine N, Islam R, Carossino M, Huang W. PD-1 and ICOS counter-regulate tissue resident regulatory T cell development and IL-10 production during flu. Front Immunol. (2022) 13:984476. doi: 10.3389/fimmu.2022.984476
104. Yang R, Sun L, Li CF, Wang YH, Yao J, Li H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun. (2021) 12:832. doi: 10.1038/s41467-021-21099-2
105. Wegrzyn AS, Kedzierska AE, Obojski A. Identification and classification of distinct surface markers of T regulatory cells. Front Immunol. (2022) 13:1055805. doi: 10.3389/fimmu.2022.1055805
106. Scott EN, Gocher AM, Workman CJ, Vignali DAA. Regulatory T cells: barriers of immune infiltration into the tumor microenvironment. Front Immunol. (2021) 12:702726. doi: 10.3389/fimmu.2021.702726
107. Jiao J, Zhao X, Wang Y, Liang N, Li J, Yang X, et al. Normal mesenchymal stem cells can improve the abnormal function of T cells in psoriasis via upregulating transforming growth factor-β receptor. J Dermatol. (2022) 49:988–97. doi: 10.1111/1346-8138.16490
108. Li Q, Li X, Quan H, Wang Y, Qu G, Shen Z, et al. IL-10(-/-) Enhances DCs Immunity Against Chlamydia psittaci Infection via OX40L/NLRP3 and IDO/Treg Pathways. Front Immunol. (2021) 12:645653. doi: 10.3389/fimmu.2021.645653
109. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. (2007) 8:1353–62. doi: 10.1038/ni1536
110. Ciudad M, Ouandji S, Lamarthée B, Cladière C, Ghesquière T, Nivet M, et al. Regulatory T-cell dysfunctions are associated with increase in tumor necrosis factor α in autoimmune hemolytic anemia and participate in Th17 polarization. Haematologica. (2023) 109(2):444–57. doi: 10.3324/haematol.2023.282859
111. Yang J, Wei P, Barbi J, Huang Q, Yang E, Bai Y, et al. The deubiquitinase USP44 promotes Treg function during inflammation by preventing FOXP3 degradation. EMBO Rep. (2020) 21:e50308. doi: 10.15252/embr.202050308
112. Santinon F, Batignes M, Mebrek ML, Biton J, Clavel G, Hervé R, et al. Involvement of tumor necrosis factor receptor type II in FoxP3 stability and as a marker of treg cells specifically expanded by anti-tumor necrosis factor treatments in rheumatoid arthritis. Arthritis Rheumatol. (2020) 72:576–87. doi: 10.1002/art.41134
113. Liu Q, Zhu F, Liu X, Lu Y, Yao K, Tian N, et al. Non-oxidative pentose phosphate pathway controls regulatory T cell function by integrating metabolism and epigenetics. Nat Metab. (2022) 4:559–74. doi: 10.1038/s42255-022-00575-z
114. Chen K, Tang L, Nong X. Artesunate targets cellular metabolism to regulate the Th17/Treg cell balance. Inflammation Res. (2023) 72:1037–50. doi: 10.1007/s00011-023-01729-9
115. D’Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. (2009) 119:2898–913. doi: 10.1172/jci36498
116. Kalathil SG, Lugade AA, Pradhan V, Miller A, Parameswaran GI, Sethi S, et al. T-regulatory cells and programmed death 1+ T cells contribute to effector T-cell dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2014) 190:40–50. doi: 10.1164/rccm.201312-2293OC
117. Zhou M, Fang H, Du M, Li C, Tang R, Liu H, et al. The modulation of regulatory T cells via HMGB1/PTEN/β-catenin axis in LPS induced acute lung injury. Front Immunol. (2019) 10:1612. doi: 10.3389/fimmu.2019.01612
118. Gao M, Ou H, Jiang Y, Wang K, Peng Y, Zhang H, et al. Tanshinone IIA attenuates sepsis-induced immunosuppression and improves survival rate in a mice peritonitis model. BioMed Pharmacother. (2019) 112:108609. doi: 10.1016/j.biopha.2019.108609
119. Qiu D, Zhang W, Song Z, Xue M, Zhang Y, Yang Y, et al. Berberine suppresses cecal ligation and puncture induced intestinal injury by enhancing Treg cell function. Int Immunopharmacol. (2022) 106:108564. doi: 10.1016/j.intimp.2022.108564
120. Nascimento DC, Alves-Filho JC, Sônego F, Fukada SY, Pereira MS, Benjamim C, et al. Role of regulatory T cells in long-term immune dysfunction associated with severe sepsis. Crit Care Med. (2010) 38:1718–25. doi: 10.1097/CCM.0b013e3181e78ad0
121. Cavassani KA, Carson WFT, Moreira AP, Wen H, Schaller MA, Ishii M, et al. The post sepsis-induced expansion and enhanced function of regulatory T cells create an environment to potentiate tumor growth. Blood. (2010) 115:4403–11. doi: 10.1182/blood-2009-09-241083
122. Hirsiger S, Simmen HP, Werner CM, Wanner GA, Rittirsch D. Danger signals activating the immune response after trauma. Mediators Inflamm. (2012) 2012:315941. doi: 10.1155/2012/315941
123. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity. (2015) 42:1005–19. doi: 10.1016/j.immuni.2015.06.006
124. Rostan O, Arshad MI, Piquet-Pellorce C, Robert-Gangneux F, Gangneux JP, Samson M. Crucial and diverse role of the interleukin-33/ST2 axis in infectious diseases. Infect Immun. (2015) 83:1738–48. doi: 10.1128/iai.02908-14
125. Lei S, Jin J, Zhao X, Zhou L, Qi G, Yang J. The role of IL-33/ST2 signaling in the tumor microenvironment and Treg immunotherapy. Exp Biol Med (Maywood). (2022) 247:1810–8. doi: 10.1177/15353702221102094
126. Siede J, Fröhlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 receptor-expressing regulatory T cells are highly activated, Th2 biased and suppress CD4 T cell proliferation through IL-10 and TGFβ Release. PloS One. (2016) 11:e0161507. doi: 10.1371/journal.pone.0161507
127. Ngo Thi Phuong N, Palmieri V, Adamczyk A, Klopfleisch R, Langhorst J, Hansen W, et al. IL-33 drives expansion of type 2 innate lymphoid cells and regulatory T cells and protects mice from severe, acute colitis. Front Immunol. (2021) 12:669787. doi: 10.3389/fimmu.2021.669787
128. Han Y, Ouyang X, Chen Y, Lai S, Sun H, Wu N, et al. scRNA-seq profiling of neonatal and adult thymus-derived CD4+ T cells by a T cell origin-time tracing model. J Mol Cell Biol. (2023) 14(12):mjac072. doi: 10.1093/jmcb/mjac072
129. Wu Y, Li D, Wang H, Wan X. Protective effect of poria cocos polysaccharides on fecal peritonitis-induced sepsis in mice through inhibition of oxidative stress, inflammation, apoptosis, and reduction of treg cells. Front Microbiol. (2022) 13:887949. doi: 10.3389/fmicb.2022.887949
130. Sehgal R, Maiwall R, Rajan V, Islam M, Baweja S, Kaur N, et al. Granulocyte-macrophage colony-stimulating factor modulates myeloid-derived suppressor cells and treg activity in decompensated cirrhotic patients with sepsis. Front Immunol. (2022) 13:828949. doi: 10.3389/fimmu.2022.828949
131. Chen H, Huang N, Tian H, Li J, Li B, Sun J, et al. Splenectomy provides protective effects against CLP-induced sepsis by reducing TRegs and PD-1/PD-L1 expression. Int J Biochem Cell Biol. (2021) 136:105970. doi: 10.1016/j.biocel.2021.105970
132. Zhou X, Yao J, Lin J, Liu J, Dong L, Duan M. Th17/Regulatory T-cell imbalance and acute kidney injury in patients with sepsis. J Clin Med. (2022) 11(14):4027. doi: 10.3390/jcm11144027
133. Sun JK, Zhang WH, Chen WX, Wang X, Mu XW. Effects of early enteral nutrition on Th17/Treg cells and IL-23/IL-17 in septic patients. World J Gastroenterol. (2019) 25:2799–808. doi: 10.3748/wjg.v25.i22.2799
134. Chen X, Chen X, Yang Y, Luo N, Yang J, Zhong L, et al. Protective role of the novel cytokine Metrnl/interleukin-41 in host immunity defense during sepsis by promoting macrophage recruitment and modulating Treg/Th17 immune cell balance. Clin Immunol. (2023) 254:109690. doi: 10.1016/j.clim.2023.109690
135. Yeh CL, Wu JM, Chen KY, Wu MH, Yang PJ, Lee PC, et al. Potential therapeutic implications of calcitriol administration and weight reduction on CD4 T cell dysregulation and renin angiotensin system-associated acute lung injury in septic obese mice. BioMed Pharmacother. (2023) 165:115127. doi: 10.1016/j.biopha.2023.115127
136. Yeh CL, Wu JM, Chen KY, Wu MH, Yang PJ, Lee PC, et al. Effects of different routes and forms of vitamin D administration on CD4(+) T cell homeostasis and renin-angiotensin system-associated lung injury in obese mice complicated with polymicrobial sepsis. BioMed Pharmacother. (2022) 156:113961. doi: 10.1016/j.biopha.2022.113961
137. Nadeem A, Al-Harbi NO, Ahmad SF, Al-Harbi MM, Alhamed AS, Alfardan AS, et al. Blockade of interleukin-2-inducible T-cell kinase signaling attenuates acute lung injury in mice through adjustment of pulmonary Th17/Treg immune responses and reduction of oxidative stress. Int Immunopharmacol. (2020) 83:106369. doi: 10.1016/j.intimp.2020.106369
138. Shen M, Lin B, Qian F, Zhao L, Xi Y, Qian Y. Taxifolin ameliorates sepsis-induced lung capillary leak through inhibiting the JAK/STAT3 pathway. Allergol Immunopathol (Madr). (2022) 50:7–15. doi: 10.15586/aei.v50i2.550
139. Yeh CL, Tanuseputero SA, Wu JM, Tseng YR, Yang PJ, Lee PC, et al. Intravenous arginine administration benefits CD4(+) T-cell homeostasis and attenuates liver inflammation in mice with polymicrobial sepsis. Nutrients. (2020) 12(4):1047. doi: 10.3390/nu12041047
140. Wang L, Deng Z, Sun Y, Zhao Y, Li Y, Yang M, et al. The study on the regulation of th cells by mesenchymal stem cells through the JAK-STAT signaling pathway to protect naturally aged sepsis model rats. Front Immunol. (2022) 13:820685. doi: 10.3389/fimmu.2022.820685
141. Luo C, Luo F, Che L, Zhang H, Zhao L, Zhang W, et al. Mesenchymal stem cells protect against sepsis-associated acute kidney injury by inducing Gal-9/Tim-3 to remodel immune homeostasis. Ren Fail. (2023) 45:2187229. doi: 10.1080/0886022x.2023.2187229
142. Zhu J. T helper cell differentiation, heterogeneity, and plasticity. Cold Spring Harb Perspect Biol. (2018) 10(10):a030338. doi: 10.1101/cshperspect.a030338
143. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. (2015) 523:221–5. doi: 10.1038/nature14452
144. Liu ZZ, Sun GQ, Hu XH, Kwak-Kim J, Liao AH. The transdifferentiation of regulatory T and Th17 cells in autoimmune/inflammatory diseases and its potential implications in pregnancy complications. Am J Reprod Immunol. (2017) 78(2):e12657. doi: 10.1111/aji.12657
145. Di Y, Zhang M, Chen Y, Sun R, Shen M, Tian F, et al. Catalpol inhibits tregs-to-th17 cell transdifferentiation by up-regulating let-7g-5p to reduce STAT3 protein levels. Yonsei Med J. (2022) 63:56–65. doi: 10.3349/ymj.2022.63.1.56
146. Cerboni S, Gehrmann U, Preite S, Mitra S. Cytokine-regulated Th17 plasticity in human health and diseases. Immunology. (2021) 163:3–18. doi: 10.1111/imm.13280
147. Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. (2019) 16:634–43. doi: 10.1038/s41423-019-0220-6
148. Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. Malignant B cells skew the balance of regulatory T cells and TH17 cells in B-cell non-Hodgkin’s lymphoma. Cancer Res. (2009) 69:5522–30. doi: 10.1158/0008-5472.Can-09-0266
149. Meng L, Bai Z, He S, Mochizuki K, Liu Y, Purushe J, et al. The notch ligand DLL4 defines a capability of human dendritic cells in regulating Th1 and Th17 differentiation. J Immunol. (2016) 196:1070–80. doi: 10.4049/jimmunol.1501310
150. Ye J, Su X, Hsueh EC, Zhang Y, Koenig JM, Hoft DF, et al. Human tumor-infiltrating Th17 cells have the capacity to differentiate into IFN-γ+ and FOXP3+ T cells with potent suppressive function. Eur J Immunol. (2011) 41:936–51. doi: 10.1002/eji.201040682
151. Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol. (2009) 21:281–5. doi: 10.1016/j.coi.2009.05.007
152. Gouirand V, Habrylo I, Rosenblum MD. Regulatory T cells and inflammatory mediators in autoimmune disease. J Invest Dermatol. (2022) 142:774–80. doi: 10.1016/j.jid.2021.05.010
153. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. (2008) 29:44–56. doi: 10.1016/j.immuni.2008.05.007
154. Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. (2009) 113:6102–11. doi: 10.1182/blood-2008-12-195354
155. Wolf D, Gerhardt T, Winkels H, Michel NA, Pramod AB, Ghosheh Y, et al. Pathogenic autoimmunity in atherosclerosis evolves from initially protective apolipoprotein B(100)-reactive CD4(+) T-regulatory cells. Circulation. (2020) 142:1279–93. doi: 10.1161/circulationaha.119.042863
156. Venet F, Chung CS, Kherouf H, Geeraert A, Malcus C, Poitevin F, et al. Increased circulating regulatory T cells (CD4(+)CD25 (+)CD127 (-)) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med. (2009) 35:678–86. doi: 10.1007/s00134-008-1337-8
157. Hiraki S, Ono S, Tsujimoto H, Kinoshita M, Takahata R, Miyazaki H, et al. Neutralization of interleukin-10 or transforming growth factor-β decreases the percentages of CD4+ CD25+ Foxp3+ regulatory T cells in septic mice, thereby leading to an improved survival. Surgery. (2012) 151:313–22. doi: 10.1016/j.surg.2011.07.019
158. Heuer JG, Zhang T, Zhao J, Ding C, Cramer M, Justen KL, et al. Adoptive transfer of in vitro-stimulated CD4+CD25+ regulatory T cells increases bacterial clearance and improves survival in polymicrobial sepsis. J Immunol. (2005) 174:7141–6. doi: 10.4049/jimmunol.174.11.7141
159. Li X, Zhu Z, Xia Z, Xu B. FGF15 protects septic mice by inhibiting inflammation and modulating treg responses. J Inflammation Res. (2022) 15:6187–97. doi: 10.2147/jir.S387613
160. Cao C, Yu M, Chai Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. (2019) 10:782. doi: 10.1038/s41419-019-2015-1
161. Marques A, Torre C, Pinto R, Sepodes B, Rocha J. Treatment advances in sepsis and septic shock: modulating pro- and anti-inflammatory mechanisms. J Clin Med. (2023) 12(8):2892. doi: 10.3390/jcm12082892
162. Hotchkiss RS, Opal S. Immunotherapy for sepsis–a new approach against an ancient foe. N Engl J Med. (2010) 363:87–9. doi: 10.1056/NEJMcibr1004371
163. Wang B, Dileepan T, Briscoe S, Hyland KA, Kang J, Khoruts A, et al. Induction of TGF-beta1 and TGF-beta1-dependent predominant Th17 differentiation by group A streptococcal infection. Proc Natl Acad Sci U.S.A. (2010) 107:5937–42. doi: 10.1073/pnas.0904831107
164. Morita Y, Masters EA, Schwarz EM, Muthukrishnan G. Interleukin-27 and its diverse effects on bacterial infections. Front Immunol. (2021) 12:678515. doi: 10.3389/fimmu.2021.678515
165. Zhao J, Liu Y, Hu JN, Peng M, Dong N, Zhu XM, et al. Autocrine regulation of interleukin-3 in the activity of regulatory T cells and its effectiveness in the pathophysiology of sepsis. J Infect Dis. (2021) 223:893–904. doi: 10.1093/infdis/jiaa441
166. Leng FY, Liu JL, Liu ZJ, Yin JY, Qu HP. Increased proportion of CD4(+)CD25(+)Foxp3(+) regulatory T cells during early-stage sepsis in ICU patients. J Microbiol Immunol Infect. (2013) 46:338–44. doi: 10.1016/j.jmii.2012.06.012
167. Hu ZQ, Yao YM, Chen W, Bian JL, Zhao LJ, Chen LW, et al. Partial depletion of regulatory T cells enhances host inflammatory response against acute pseudomonas aeruginosa infection after sepsis. Inflammation. (2018) 41:1780–90. doi: 10.1007/s10753-018-0821-8
168. Wang Y, Liu Z, Zhang M, Yu B, Ai F. Mucosa-associated lymphoid tissue lymphoma translocation protein 1 exaggerates multiple organ injury, inflammation, and immune cell imbalance by activating the NF-κB pathway in sepsis. Front Microbiol. (2023) 14:1117285. doi: 10.3389/fmicb.2023.1117285
169. Gu L, Jiang J, Liu Z, Liu Q, Liao J, Zeng Q, et al. Intestinal recruitment of CCR6-expressing Th17 cells by suppressing miR-681 alleviates endotoxemia-induced intestinal injury and reduces mortality. Inflammation Res. (2023) 72:715–29. doi: 10.1007/s00011-023-01697-0
170. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. (2011) 306:2594–605. doi: 10.1001/jama.2011.1829
171. Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol. (2018) 14:121–37. doi: 10.1038/nrneph.2017.165
172. Reizine F, Grégoire M, LesouHaitier M, Coirier V, Gauthier J, Delaloy C, et al. Beneficial effects of citrulline enteral administration on sepsis-induced T cell mitochondrial dysfunction. Proc Natl Acad Sci U.S.A. (2022) 119(8):e2115139119. doi: 10.1073/pnas.2115139119
173. de Lima MHF, Hiroki CH, de Fátima Borges V, Cebinelli GCM, Santos J, Rosa MH, et al. Sepsis-induced immunosuppression is marked by an expansion of a highly suppressive repertoire of FOXP3+ T-regulatory cells expressing TIGIT. J Infect Dis. (2022) 225:531–41. doi: 10.1093/infdis/jiab405
174. Lazzaro A, De Girolamo G, Filippi V, Innocenti GP, Santinelli L, Ceccarelli G, et al. The interplay between host defense, infection, and clinical status in septic patients: A narrative review. Int J Mol Sci. (2022) 23(2):803. doi: 10.3390/ijms23020803
175. López-Lera A, Corvillo F, Nozal P, Regueiro JR, Sánchez-Corral P, López-Trascasa M. Complement as a diagnostic tool in immunopathology. Semin Cell Dev Biol. (2019) 85:86–97. doi: 10.1016/j.semcdb.2017.12.017
176. Ma N, Xing C, Xiao H, Wang Y, Wang K, Hou C, et al. C5a regulates IL-12+ DC migration to induce pathogenic Th1 and Th17 cells in sepsis. PloS One. (2013) 8:e69779. doi: 10.1371/journal.pone.0069779
177. Turner S, Raisley B, Roach K, Bajaña S, Munroe ME, James JA, et al. Gram-positive bacteria cell wall peptidoglycan polymers activate human dendritic cells to produce IL-23 and IL-1β and promote T(H)17 cell differentiation. Microorganisms. (2023) 11(1):173. doi: 10.3390/microorganisms11010173
178. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. (2013) 13:260–8. doi: 10.1016/s1473-3099(13)70001-x
179. Patil NK, Bohannon JK, Sherwood ER. Immunotherapy: A promising approach to reverse sepsis-induced immunosuppression. Pharmacol Res. (2016) 111:688–702. doi: 10.1016/j.phrs.2016.07.019
180. Yao YM, Huang LF. [The potential role of regulatory T cells in postburn sepsis]. Zhonghua Shao Shang Za Zhi. (2011) 27:81–3. doi: 10.3760/cma.j.issn.1009-2587.2011.02.001
181. Liu Q, An L, Qi Z, Zhao Y, Li C. Increased expression of programmed cell death-1 in regulatory T cells of patients with severe sepsis and septic shock: an observational clinical study. Scand J Immunol. (2017) 86:408–17. doi: 10.1111/sji.12612
182. Gao DN, Yang ZX, Qi QH. Roles of PD-1, Tim-3 and CTLA-4 in immunoregulation in regulatory T cells among patients with sepsis. Int J Clin Exp Med. (2015) 8:18998–9005.
183. Jayaraman P, Jacques MK, Zhu C, Steblenko KM, Stowell BL, Madi A, et al. TIM3 Mediates T Cell Exhaustion during Mycobacterium tuberculosis Infection. PloS Pathog. (2016) 12:e1005490. doi: 10.1371/journal.ppat.1005490
184. Tatura R, Zeschnigk M, Adamzik M, Probst-Kepper M, Buer J, Kehrmann J. Quantification of regulatory T cells in septic patients by real-time PCR-based methylation assay and flow cytometry. PloS One. (2012) 7:e49962. doi: 10.1371/journal.pone.0049962
185. Hein F, Massin F, Cravoisy-Popovic A, Barraud D, Levy B, Bollaert PE, et al. The relationship between CD4+CD25+CD127- regulatory T cells and inflammatory response and outcome during shock states. Crit Care. (2010) 14:R19. doi: 10.1186/cc8876
186. Venet F, Pachot A, Debard AL, Bohé J, Bienvenu J, Lepape A, et al. Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25- lymphocytes. Crit Care Med. (2004) 32:2329–31. doi: 10.1097/01.CCM.0000145999.42971.4B
187. Tatura R, Zeschnigk M, Hansen W, Steinmann J, Vidigal PG, Hutzler M, et al. Relevance of Foxp3+ regulatory T cells for early and late phases of murine sepsis. Immunology. (2015) 146:144–56. doi: 10.1111/imm.12490
188. Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med. (2013) 210:257–68. doi: 10.1084/jem.20121525
189. Yuan Y, Yan D, Han G, Gu G, Ren J. Complement C3 depletion links to the expansion of regulatory T cells and compromises T-cell immunity in human abdominal sepsis: a prospective pilot study. J Crit Care. (2013) 28:1032–8. doi: 10.1016/j.jcrc.2013.09.007
190. Yuan Y, Ren J, Cao S, Zhang W, Li J. Exogenous C3 protein enhances the adaptive immune response to polymicrobial sepsis through down-regulation of regulatory T cells. Int Immunopharmacol. (2012) 12:271–7. doi: 10.1016/j.intimp.2011.11.022
191. Guéry L, Hugues S. Th17 cell plasticity and functions in cancer immunity. BioMed Res Int. (2015) 2015:314620. doi: 10.1155/2015/314620
192. Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood. (2013) 121:2402–14. doi: 10.1182/blood-2012-09-378653
193. Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, et al. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci U.S.A. (2010) 107:14751–6. doi: 10.1073/pnas.1003852107
194. Nyirenda MH, Morandi E, Vinkemeier U, Constantin-Teodosiu D, Drinkwater S, Mee M, et al. TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: a novel mechanism of reduced regulatory T cell function in multiple sclerosis. J Immunol. (2015) 194:5761–74. doi: 10.4049/jimmunol.1400472
195. Saigusa R, Asano Y, Taniguchi T, Hirabayashi M, Nakamura K, Miura S, et al. Fli1-haploinsufficient dermal fibroblasts promote skin-localized transdifferentiation of Th2-like regulatory T cells. Arthritis Res Ther. (2018) 20:23. doi: 10.1186/s13075-018-1521-3
196. Ayyoub M, Deknuydt F, Raimbaud I, Dousset C, Leveque L, Bioley G, et al. Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the T(H)17 lineage-specific transcription factor RORgamma t. Proc Natl Acad Sci U.S.A. (2009) 106:8635–40. doi: 10.1073/pnas.0900621106
197. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. (2009) 10:1000–7. doi: 10.1038/ni.1774
198. Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. (2006) 24:677–88. doi: 10.1016/j.immuni.2006.06.002
199. Lourenço JD, Ito JT, Martins MA, Tibério I, Lopes F. Th17/Treg imbalance in chronic obstructive pulmonary disease: clinical and experimental evidence. Front Immunol. (2021) 12:804919. doi: 10.3389/fimmu.2021.804919
200. Luo Y, Guo J, Zhang P, Cheuk YC, Jiang Y, Wang J, et al. Mesenchymal stem cell protects injured renal tubular epithelial cells by regulating mTOR-mediated Th17/Treg axis. Front Immunol. (2021) 12:684197. doi: 10.3389/fimmu.2021.684197
201. Hann A, Oo YH, Perera M. Regulatory T-cell therapy in liver transplantation and chronic liver disease. Front Immunol. (2021) 12:719954. doi: 10.3389/fimmu.2021.719954
202. Delgobo M, Weiß E, Ashour D, Richter L, Popiolkowski L, Arampatzi P, et al. Myocardial milieu favors local differentiation of regulatory T cells. Circ Res. (2023) 132:565–82. doi: 10.1161/circresaha.122.322183
203. Saito M, Fujinami Y, Ono Y, Ohyama S, Fujioka K, Yamashita K, et al. Infiltrated regulatory T cells and Th2 cells in the brain contribute to attenuation of sepsis-associated encephalopathy and alleviation of mental impairments in mice with polymicrobial sepsis. Brain Behav Immun. (2021) 92:25–38. doi: 10.1016/j.bbi.2020.11.010
204. Huang T, Huang J, Liao Z, Lan H, Jian X, Gu R, et al. Regenerating myofiber directs Tregs and Th17 responses in inflamed muscle through the intrinsic TGF-β signaling-mediated IL-6 production. Am J Physiol Endocrinol Metab. (2022) 323:E92–e106. doi: 10.1152/ajpendo.00247.2021
205. Fan EKY, Fan J. Regulation of alveolar macrophage death in acute lung inflammation. Respir Res. (2018) 19:50. doi: 10.1186/s12931-018-0756-5
206. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med. (2017) 377:562–72. doi: 10.1056/NEJMra1608077
207. Bos LDJ, Ware LB. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet. (2022) 400:1145–56. doi: 10.1016/s0140-6736(22)01485-4
208. Luyt CE, Bouadma L, Morris AC, Dhanani JA, Kollef M, Lipman J, et al. Pulmonary infections complicating ARDS. Intensive Care Med. (2020) 46:2168–83. doi: 10.1007/s00134-020-06292-z
209. Chen L, Liu X, Wang X, Lu Z, Ye Y. Berberine alleviates acute lung injury in septic mice by modulating Treg/Th17 homeostasis and downregulating NF-κB signaling. Drug Des Devel Ther. (2023) 17:1139–51. doi: 10.2147/dddt.S401293
210. Xia H, Wang F, Wang M, Wang J, Sun S, Chen M, et al. Maresin1 ameliorates acute lung injury induced by sepsis through regulating Th17/Treg balance. Life Sci. (2020) 254:117773. doi: 10.1016/j.lfs.2020.117773
211. Bartsch P, Kilian C, Hellmig M, Paust HJ, Borchers A, Sivayoganathan A, et al. Th17 cell plasticity towards a T-bet-dependent Th1 phenotype is required for bacterial control in Staphylococcus aureus infection. PloS Pathog. (2022) 18:e1010430. doi: 10.1371/journal.ppat.1010430
212. Nadeem A, Ahmad SF, Al-Harbi NO, Ibrahim KE, Sarawi W, Attia SM, et al. Role of ITK signaling in acute kidney injury in mice: Amelioration of acute kidney injury associated clinical parameters and attenuation of inflammatory transcription factor signaling in CD4+ T cells by ITK inhibition. Int Immunopharmacol. (2021) 99:108028. doi: 10.1016/j.intimp.2021.108028
213. Chen L, Lu Y, Zhao L, Hu L, Qiu Q, Zhang Z, et al. Curcumin attenuates sepsis-induced acute organ dysfunction by preventing inflammation and enhancing the suppressive function of Tregs. Int Immunopharmacol. (2018) 61:1–7. doi: 10.1016/j.intimp.2018.04.041
214. Shih JM, Shih YM, Pai MH, Hou YC, Yeh CL, Yeh SL. Fish oil-based fat emulsion reduces acute kidney injury and inflammatory response in antibiotic-treated polymicrobial septic mice. Nutrients. (2016) 8:165. doi: 10.3390/nu8030165
215. Maravitsa P, Adamopoulou M, Pistiki A, Netea MG, Louis K, Giamarellos-Bourboulis EJ. Systemic over-release of interleukin-17 in acute kidney injury after septic shock: Clinical and experimental evidence. Immunol Lett. (2016) 178:68–76. doi: 10.1016/j.imlet.2016.08.002
216. Yang XY, Song J, Hou SK, Fan HJ, Lv Q, Liu ZQ, et al. Ulinastatin ameliorates acute kidney injury induced by crush syndrome inflammation by modulating Th17/Treg cells. Int Immunopharmacol. (2020) 81:106265. doi: 10.1016/j.intimp.2020.106265
217. Herrnstadt GR, Niehus CB, Ramcke T, Hagenstein J, Ehnold LI, Nosko A, et al. The CCR6/CCL20 axis expands RORγt(+) Tregs to protect from glomerulonephritis. Kidney Int. (2023) 104:74–89. doi: 10.1016/j.kint.2023.02.027
218. Chen J, Xu C, Yang K, Gao R, Cao Y, Liang L, et al. Inhibition of ALKBH5 attenuates I/R-induced renal injury in male mice by promoting Ccl28 m6A modification and increasing Treg recruitment. Nat Commun. (2023) 14:1161. doi: 10.1038/s41467-023-36747-y
219. Lee SY, Lee YS, Choi HM, Ko YS, Lee HY, Jo SK, et al. Distinct pathophysiologic mechanisms of septic acute kidney injury: role of immune suppression and renal tubular cell apoptosis in murine model of septic acute kidney injury. Crit Care Med. (2012) 40:2997–3006. doi: 10.1097/CCM.0b013e31825b912d
Keywords: sepsis, Th17, Treg, balance, pathophysiology
Citation: Liu X, Chen L, Peng W, Deng H, Ni H, Tong H, Hu H, Wang S, Qian J, Liang A and Chen K (2024) Th17/Treg balance: the bloom and wane in the pathophysiology of sepsis. Front. Immunol. 15:1356869. doi: 10.3389/fimmu.2024.1356869
Received: 16 December 2023; Accepted: 20 February 2024;
Published: 15 March 2024.
Edited by:
Carlos Jimenez-Cortegana, University of Seville, SpainReviewed by:
Sandip Ashok Sonar, University of Arizona, United StatesCopyright © 2024 Liu, Chen, Peng, Deng, Ni, Tong, Hu, Wang, Qian, Liang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andong Liang, bGFubmRvbmdAMTYzLmNvbQ==; Kun Chen, MTM5NTc5NzA3MDdAc2luYS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.