 Ariane Hammitzsch1*†
Ariane Hammitzsch1*† Andreas Ossadnik1†
Andreas Ossadnik1† Quirin Bachmann1Helga Merwald-Fraenk2Georg Lorenz3Matthias Witt4Franziska Wiesent2Heinrich Mühlhofer5Davide Simone6Paul Bowness7
Quirin Bachmann1Helga Merwald-Fraenk2Georg Lorenz3Matthias Witt4Franziska Wiesent2Heinrich Mühlhofer5Davide Simone6Paul Bowness7 Uwe Heemann1Martin Arbogast8
Uwe Heemann1Martin Arbogast8 Philipp Moog1‡Christoph Schmaderer1‡
Philipp Moog1‡Christoph Schmaderer1‡- 1Department of Nephrology, Klinikum Rechts der Isar, School of Medicine, Technical University of Munich, Munich, Germany
- 2Amedes Holding AG, Ambulatory Healthcare Center (MVZ) Endokrinologikum München, Munich, Germany
- 3Department of Nephrology and Rheumatology, Klinik Augustinum München, Munich, Germany
- 4Rheumazentrum Bad Abling Erding, Erding, Germany
- 5Clinic and Policlinic of Orthopaedics and Sports’ Orthopaedics, Klinikum Rechts der Isar, School of Medicine, Technical University of Munich, Munich, Germany
- 6Kennedy Institute of Rheumatology, University of Oxford, Oxford, United Kingdom
- 7Botnar Research Centre, Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences, Medical Sciences Division, University of Oxford, Oxford, United Kingdom
- 8Department of Rheumatic Orthopedics and Hand Surgery, Klinik Oberammergau, Waldburg-Zeil Kliniken GmbH und Co KG, Oberammergau, Germany
Objectives: IL26 levels are elevated in the blood and synovial fluid of patients with inflammatory arthritis. IL26 can be produced by Th17 cells and locally within joints by tissue-resident cells. IL26 induces osteoblast mineralization in vitro. As osteoproliferation and Th17 cells are important factors in the pathogenesis of axial spondyloarthritis (axSpA), we aimed to clarify the cellular sources of IL26 in spondyloarthritis.
Methods: Serum, peripheral blood mononuclear cells (n = 15–35) and synovial tissue (n = 3–9) of adult patients with axSpA, psoriatic arthritis (PsA) and rheumatoid arthritis (RA) and healthy controls (HCs, n = 5) were evaluated by ELISA, flow cytometry including PrimeFlow assay, immunohistochemistry and immunofluorescence and quantitative PCR.
Results: Synovial tissue of axSpA patients shows significantly more IL26-positive cells than that of HCs (p < 0.01), but numbers are also elevated in PsA and RA patients. Immunofluorescence shows co-localization of IL26 with CD68, but not with CD3, SMA, CD163, cadherin-11, or CD90. IL26 is elevated in the serum of RA and PsA (but not axSpA) patients compared with HCs (p < 0.001 and p < 0.01). However, peripheral blood CD4+ T cells from axSpA and PsA patients show higher positivity for IL26 in the PrimeFlow assay compared with HCs. CD4+ memory T cells from axSpA patients produce more IL26 under Th17-favoring conditions (IL-1β and IL-23) than cells from PsA and RA patients or HCs.
Conclusion: IL26 production is increased in the synovial tissue of SpA and can be localized to CD68+ macrophage-like synoviocytes, whereas circulating IL26+ Th17 cells are only modestly enriched. Considering the osteoproliferative properties of IL26, this offers new therapeutic options independent of Th17 pathways.
Introduction
Axial spondyloarthritis (axSpA) and psoriatic arthritis (PsA) belong to a group of immune-mediated inflammatory diseases with primarily axial but also peripheral arthritis, with a prevalence of approximately 0.54% in Europe (1, 2). These diseases additionally present with extra-articular manifestations like uveitis, inflammatory bowel disease and psoriasis. As the onset of disease predominates in the third decade, socioeconomic and personal effects are high and long-lasting, demanding efficient therapy. The common denominators of axSpA and PsA are inflammation of the entheses and osteoproliferation along the tendons and capsules. Presumably, inflammation precedes new bone formation and early halting of inflammation by therapies like non-steroidal anti-inflammatory drugs (NSAIDs), anti-TNF, anti-IL-17A/F and Janus kinase inhibition (JAKi) can avoid osteoproliferation and ultimately ankylosis. However, the precise mechanisms and players of this osteoproliferation have not been satisfactorily elucidated yet.
The efficacy of anti-IL-17A/F treatment in clinical studies together with genetic and functional evidence underlines the role of the IL-23/IL-17A pathway in the pathogenesis of SpA (3–5). Main players of the IL-23/IL-17A pathway in SpA are “pathogenic” Th17 cells, producing IL-17A, IL-17F, IL-22, GM-CSF and IFNγ (6). Pathogenic Th17 cells arise from Th17 cells, which are responsible for the maintenance of tissue homeostasis at mucosal barriers and host defense against fungi and extracellular bacteria, in the presence of IL-23 (7–9). IL26, a member of the IL-10 family, is also produced by pathogenic Th17 cells (10, 11). It uniquely combines proinflammatory with antimicrobial, DNA-binding and DNA-shuttling properties (12, 13). Elevated systemic levels and locally increased expression of IL26 have been reported in chronic inflammatory diseases like Crohn’s disease, rheumatoid arthritis, SpA, psoriasis and ANCA-associated vasculitis (11, 13–16). IL26 signaling is mediated by multiple pathways, including a classical cytokine receptor (IL-10R2 and IL-20R1) leading to activation of JAK1 and TYK2, and direct cell membrane penetration. IL26 expression has also been reported in CD8+ T cells, mucosal-associated invariant T (MAIT) cells, NKp44+ NK cells, γδT cells, innate lymphoid cells type 3 (ILC3), CD68+ macrophage-like and synoviolin+ fibroblast-like synoviocytes and synovial myofibroblasts (15–21). Effects have been observed on human intestinal and bronchial epithelial cells, endothelial cells, fibroblasts, monocytes, NK and B cells, plasmacytoid dendritic cells and neutrophils (12, 15, 16, 22–28).
IL26 also increases mineralization in human osteoblasts, and inhibits receptor activator of nuclear factor κB ligand-induced osteoclastogenesis and bone-resorbing activity of mature osteoclasts, making it an interesting therapeutic target for preventing osteoproliferation (16, 24). Additionally, the capacity of IL26 to bind DNA released during inflammation-induced tissue damage renders IL26 a key driver of chronic inflammation. This provides another interesting feature of anti-IL26 therapy for axSpA and PsA because, with current therapeutic options (NSAIDs, anti-TNF, anti-IL-17A/F, and JAKi), approximately 40% of patients will not enter sustained remission. Therefore, the need for new therapies remains high in these patients.
In this study we comparatively investigate the expression and cellular sources of IL26 in the peripheral blood and peripheral synovial tissue of anti-TNF therapy-naive patients with axSpA, PsA, and RA. We demonstrate significantly increased local expression of IL26 in axSpA, potentially implicating a significant role of IL26 in osteoproliferation and a new therapeutic target.
Patients and methods
Patient samples
Venous blood (max. 50 ml) and synovial tissue were obtained from patients with axSpA (n = 29/9, modified New York criteria), PsA [n = 20/3, Classification Criteria for Psoriatic Arthritis (CASPAR)], and RA (n = 15/7, American College of Rheumatology/European League against Rheumatism 2010 criteria) and from healthy controls (HCs, n = 35/5), after obtaining informed written consent, with ethical permission (100/18 S, Ethical Committee of the Faculty of Medicine, Technical University Munich and 06/Q1606/139 Oxford University Hospitals NHS Foundation Trust). The study has been registered within the German Clinical Trials Register (DRKS00014672).
Healthy donors were sex- and age-matched to the axSpA group. All research was performed in accordance with the relevant guidelines and regulations including the Helsinki Declaration.
Serum was isolated from a 4.7-ml serum tube (Sarstedt, Nümbrecht, Germany), aliquoted and stored at −80°C until analysis.
Cell purification and cell culture
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density-gradient centrifugation (4 × 8 ml, CPT tube, BD, Franklin Lakes, USA). CD4+ T cells were then negatively, and CD14+ cells positively selected via magnetic beads (BioLegend, San Diego, USA, >90.96% purity on average), and cultured under stimulatory conditions as described in the online Supplementary Material and Methods. Cells were cryopreserved in FCS with 10% DMSO (Sigma-Aldrich, Burlington, USA) in liquid nitrogen until further analysis.
Synovial tissue acquisition and preparation
Synovial tissue was obtained during orthopedic surgery or arthroscopy and processed as described in the online Supplementary Materials and Methods.
ELISA
Serum levels of IL26 were analyzed with a modified IL26 ELISA kit (Elabscience, Wuhan, China). For detailed modifications, see the online Supplementary Materials and Methods.
Immunohistochemistry
Tissue sections were prepared and stained with the DAKO Envision+ System HRP Kit (Dako, Santa Clara, USA), and images were acquired and quantitatively analyzed as described in the online Supplementary Material and Methods.
Immunofluorescence
Immunofluorescence was performed according to an adapted protocol from Dakin S et al. with the incorporation of the Tyramide SuperBoost Kit (Invitrogen, Waltham, USA) (29). For detailed information, see the online Supplementary Material and Methods and Supplementary Table S1.
Flow cytometry and PrimeFlow assay
PBMCs were stimulated and stained for surface phenotyping of IL-1R1 and chemokine receptors and detection of IL26-positive cells in combination with surface markers, and intracellular cytokines as described in the online Supplementary Materials and Methods and Supplementary Tables S2, S3.
qPCR
qPCR was performed on CD4+ T cells (1 × 106) after 6 days of in vitro culture as further described in the online Supplementary Materials and Methods and Supplementary Table S4.
Statistical analysis
GraphPad Prism software version 5 was used for statistical analysis. Statistical tests were applied as stated in the figure legends. Significance was defined as p ≤ 0.05.
Results
Serum levels of IL26 are significantly elevated in PsA and RA, but not in axSpA
Two studies have reported elevated levels of IL26 in the serum and synovial fluid of RA and SpA patients (15, 16). However, SpA patients were not further classified, the numbers were low (n = 9–15) and treatment was either unclear or included anti-TNFα. Therefore, we compared the serum levels of IL26 from anti-TNF-naive patients with axSpA, PsA and RA with age- and sex-matched HCs. Detailed patient characteristics are shown in Table 1. Serum levels of IL26 were not significantly elevated in axSpA patients compared with HCs (Figure 1, mean 187.2 pg/ml, SEM 60.78 pg/ml and mean 131.8 pg/ml, SEM 25.19 pg/ml). However, significantly increased serum levels of IL26 were evident in patients with PsA and RA (mean 649.3 pg/ml, SEM 153.2 pg/ml and mean 970.5 pg/ml, SEM 238.2 pg/ml; p < 0.01 and p < 0.001). Corresponding synovial fluid for comparative analysis was not available. Correlation analysis of IL26 serum levels with markers of disease activity and inflammation revealed a weak, yet significant positive correlation only with CRP levels in axSpA patients (Supplementary Figure S1, r = 0.467, 95% CI [r] = 0.088 to 0.728, r2 = 0.218, p < 0.05).
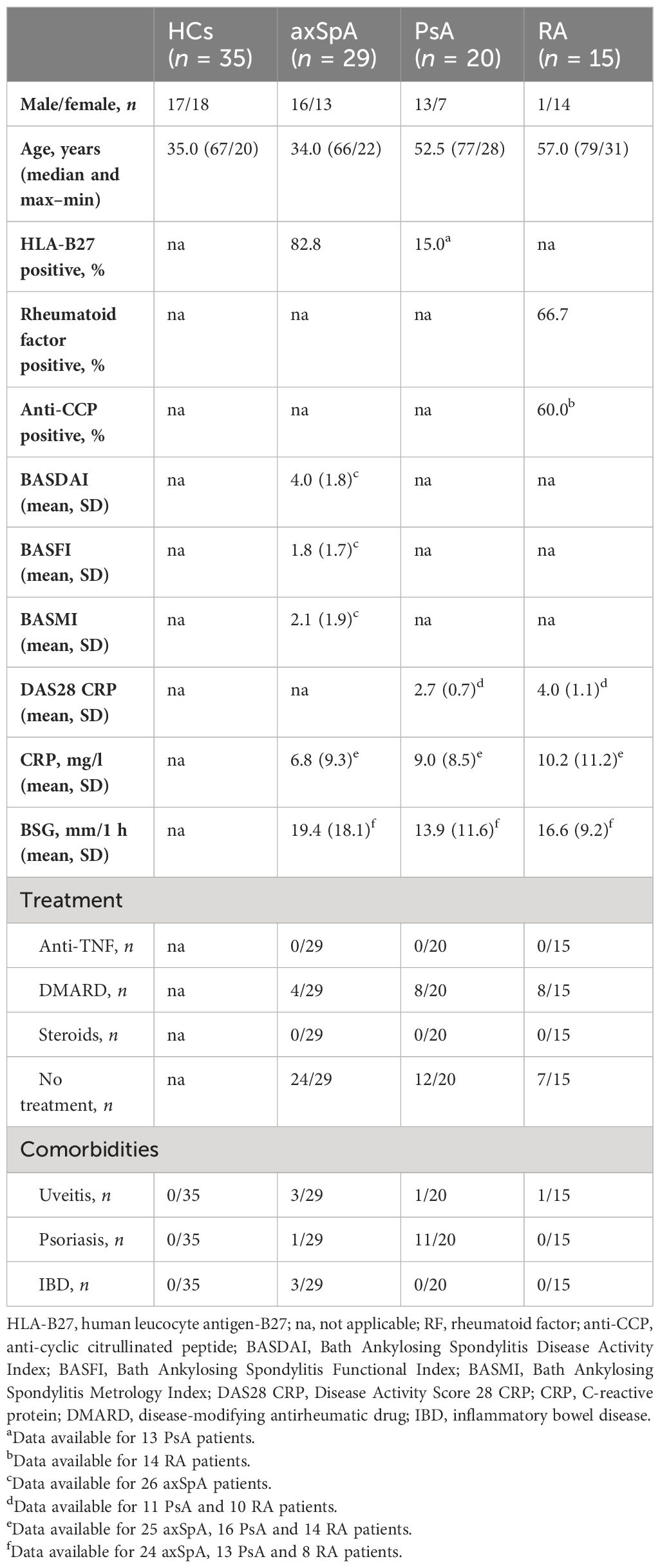
Table 1 Study population characteristics—blood samples.
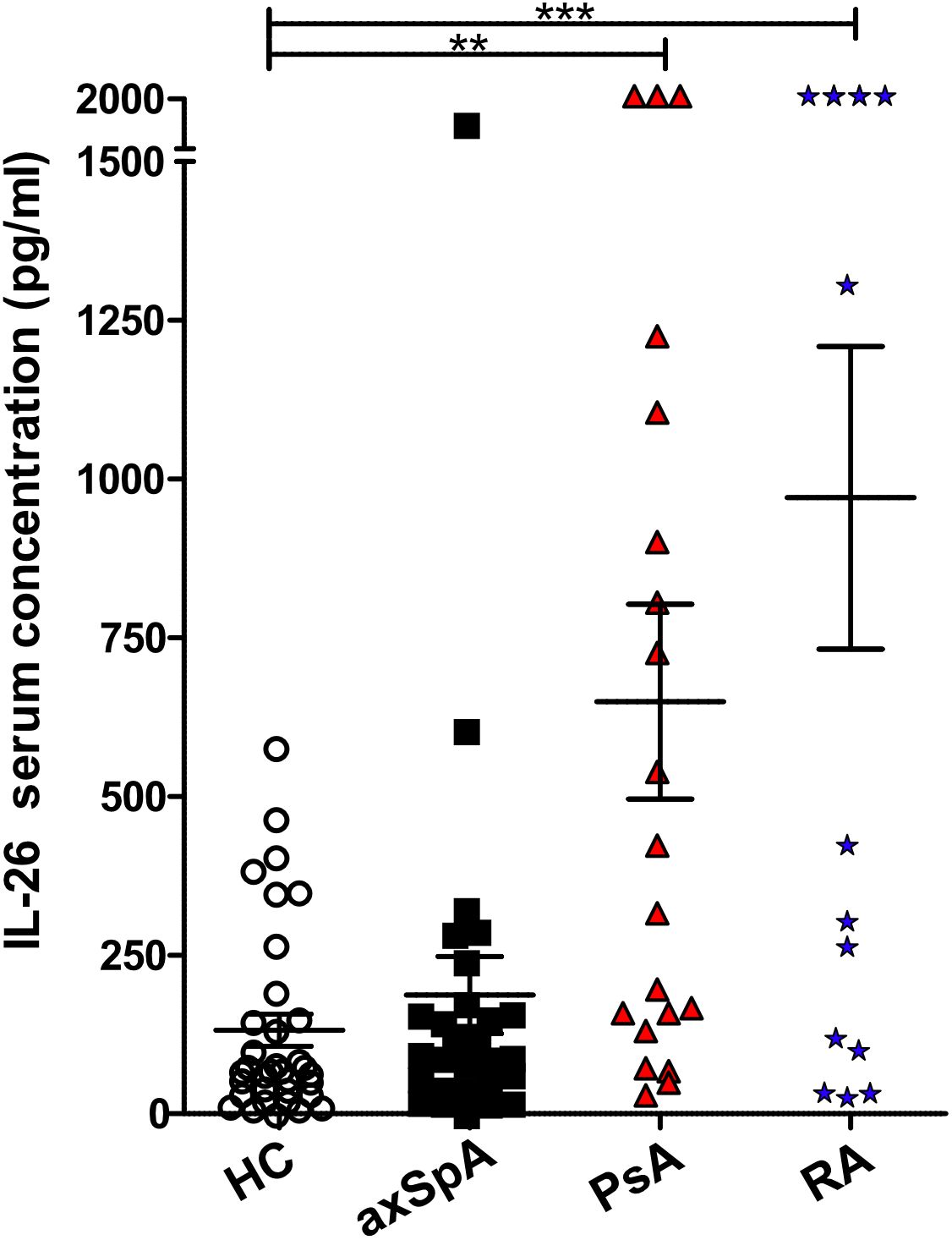
Figure 1 Increased IL-26 levels in the serum of psoriatic arthritis (PsA) and rheumatoid arthritis (RA) patients. IL-26 serum levels were quantified by ELISA in 35 HCs (white), 29 axial spondyloarthritis (axSpA) (black), 20 PsA (red) and 15 RA (blue) patients. Preincubation with heat-aggregated bovine IgG and signal amplification with biotinyl-tyramide were applied in order to minimize false results through interfering antibodies (30). Statistical analysis: mean ± SEM, **p < 0.01, ***p < 0.001 (one-way ANOVA followed by Dunnett post-test for multiple comparisons).
No significant enrichment of IL26-producing Th17 cells in the peripheral blood of axSpA and PsA patients
As Th17 cells reportedly are the main producers of IL26 in the peripheral blood of HCs (12), we analyzed the cellular sources of IL26 in the peripheral blood by flow cytometry. Because of a lack of a reliable anti-IL26 antibody for flow cytometry, we analyzed Th17 cells from the peripheral blood of axSpA and PsA patients and HCs for IL26 expression with the PrimeFlow assay (in-situ hybridization utilizing branched DNA technology with target-specific probes compatible with conventional flow cytometry). Total PBMCs were stimulated for 4 h in vitro with PMA and ionomycin prior to analysis. Figures 2A, B illustrate higher, yet not significantly higher percentages of IL26+ cells in total CD4+ T cells in axSpA (mean 4.74%, SEM 1.70%) and PsA (mean 4.42%, SEM 0.64%) patients compared with HCs (mean 3.70%, SEM 0.85%; ns). The percentages of IL26+ cells of CD4+CD26+CD161+ cells, a surface pattern known to characterize CD4+ T cells that harbor the majority of IL-17A-producing cells, are also slightly increased in axSpA (mean 8.36%, SEM 2.63%) and PsA (mean 7.62%, SEM 1.03%) patients compared with HCs (mean 6.63%, SEM 1.80%; ns) (31). For the gating strategy, see Supplementary Figure S2A.
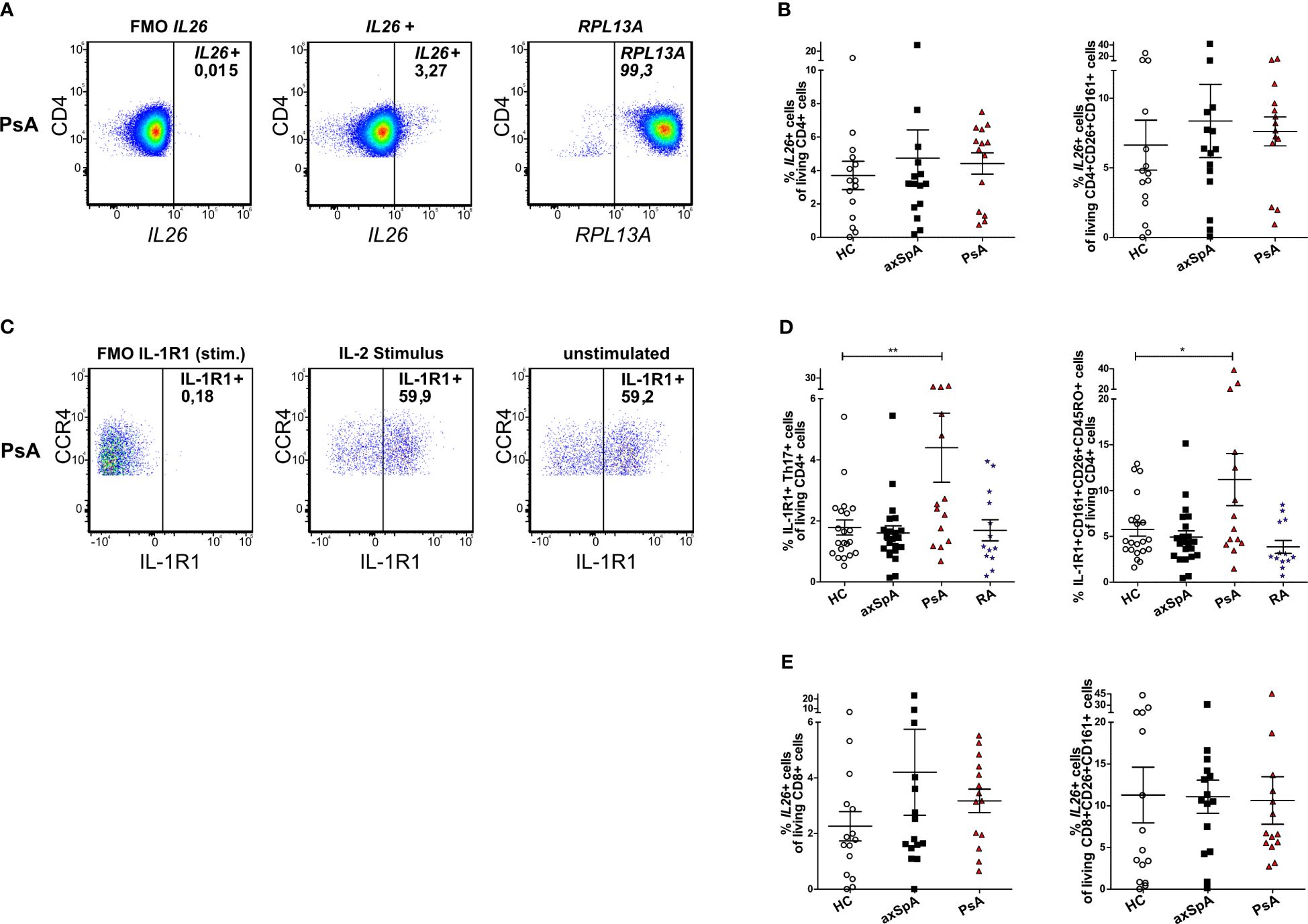
Figure 2 IL26-expressing cells are only slightly increased in circulating CD4+ and CD8+ T cells from patients with axSpA and PsA. (A) Representative flow cytometry plots of total IL26+CD4+ T cells with the PrimeFlow assay in a PsA patient. RPLA13A represents the positive control for the assay. (B) Percentages of total IL26+CD4+ T cells (left) and percentages of IL26+ cells of CD26+CD161+CD4+ T cells (right) for HCs (n = 15; white) and axSpA (n = 15; black) and PsA (n = 14; red) patients gated from PBMCs stimulated in vitro with PMA and ionomycin in the PrimeFlow assay. (C) Representative flow cytometry plots of total IL-1R1+ Th17 cells in a PsA patient comparing IL-2 stimulation to unstimulated conditions. (D) Percentages of IL-1R1+ Th17 cells (Th17 cells gated as CD4+CD45RO+CD161+CCR6+CCR4+CXCR3−) of CD4+ cells from HCs (n = 21; white) and axSpA (n = 22; black), PsA (n = 14; red), and RA (n = 13; blue) patients after in vitro stimulation with IL-2 (1,000 IU/ml) overnight (left) and percentages of IL-1R1+CD26+CD161+ cells of CD4+ cells (right). (E) Percentages of total IL26+CD8+ T cells (left) and percentages of IL26+ cells of CD26+CD161+CD8+ T cells (right) for HCs (n = 15; white) and axSpA (n = 15; black) and PsA (n = 14; red) patients gated from PBMCs stimulated in vitro with PMA and ionomycin. Statistical analysis: mean ± SEM, *p < 0.05, **p < 0.01 (one-way ANOVA followed by Dunnett post-test for multiple comparisons).
As these experiments showed only a trend for higher percentages of IL26+ Th17 cells in axSpA and PsA, an alternative strategy for the identification of IL26-producing Th17 cells was applied using surface staining for IL-1R1 as a surrogate marker in an extended set of patient samples including approximately half of the PsA samples and one-third of the axSpA and HC samples from the PrimeFlow assay. IL26 secretion is restricted to IL-1R1+ Th17 cells and can be elicited by IL-1β or TCR activation (32). Th17 cells from axSpA, PsA and RA patients and HCs were characterized as CD4+CD45RO+CD161+CCR6+CCR4+CXCR3− cells and analyzed for IL-1R1 (Figures 2C, D; for the gating strategy, see Supplementary Figure S2B). Percentages of these IL-1R1+ Th17 cells were only significantly increased in PsA patients (mean 4.40%, SEM 1.13%) compared with HCs (mean 1.78%, SEM 0.25%; p < 0.01, Figure 2D), but not in axSpA (mean 1.60%, SEM 0.23%) and RA (mean 1.69%, SEM 0.35%) patients. This finding was confirmed when analyzing the percentages of IL-1R1+CD161+CD26+CD45RO+ cells of living CD4+ cells in the same set of experiments (PsA: mean 11.20%, SEM 2.84% vs. HCs: mean 5.76%, SEM 0.74%; p < 0.05, Figure 2D) with no differences for axSpA (mean 4.93%, SEM 0.68%) and RA (mean 3.86%, SEM 0.71%).
As CD8+ T cells are another source of IL-17A, mainly by Tc17 (approximately 90%) and MAIT cells (approximately 4%), and are enriched in the synovial fluid from PsA and other SpA patients, where Tc17 cells show increased expression of IL26, we further investigated this cell type (21). Percentages of IL26+ cells in total CD8+ T cells tended to be higher in patients with axSpA (mean 4.21%, SEM 1.55%) and PsA (mean 3.17%, SEM 0.42%) compared with HCs (mean 2.26%, SEM 0.53%; ns, Figure 2E left). This increase could not be observed for percentages of IL26+ cells in CD8+CD26+CD161+ cells, a surface antigen pattern harboring most of IL-17A-producing CD8+ T cells (HCs: mean 11.29%, SEM 3.33% vs. axSPA: mean 11.09, SEM 1.99% and PsA: mean 10.63, SEM 2.85; ns; Figure 2E right) (21).
Taken together, these results do not show a clear enrichment of IL26+ Th17 and Tc17 cells in the circulation of patients with axSpA and PsA compared with HCs in our cohort of patients with moderate disease activity.
CD4+ T cells from axSpA and PsA are primed for IL26 production by IL-1β and IL-23 in vitro
Inflammation- and Th17 immunity-promoting conditions are enriched in joint tissue and are likely to increase IL26 expression. Therefore, we investigated the capacity of CD4+ T cells from axSpA and PsA patients to express IL26 at the mRNA level. Ex-vivo expression of IL26 was detectable in higher percentages of axSpA (63.1%) and PsA (77.8%) CD4+ T cells compared with HCs (45%; ns; Supplementary Figure S3A). In order to evaluate if CD4+ T cells could be prompted to express IL26 under Th17-favoring conditions, we incubated them for 6 days with different cytokine combinations of IL-2, IL-1β, IL-23, IL-6, and TNFα. After 6 days, levels of IL26 expression were highest with a combination of classical Th17-favoring cytokines IL-1β and IL-23 relative to basal stimulation with IL-2 only (Figure 3A; HCs, axSpA, PsA: p < 0.001; RA: p < 0.01). Addition of TNFα had no additive effect compared with the combination of IL-2, IL-1β and IL-23, but remained significant compared with IL-2 (HCs, axSpA, PsA: p < 0.001; RA: p < 0.01). The inductive effect of IL-1β and IL-23 could also be observed for Th17-type cytokines like IL17A, IL22, and IFNG, but not for IL4 and IL1R1 (Figure 3A, Supplementary Figure S3C; p < 0.05 to p < 0.001). RORC expression was only significantly induced in PsA by the addition of IL-1β and IL-23 (Supplementary Figure S3C; p < 0.05). Comparing the inductive effect of the combination of IL-2, IL-1β, and IL-23 between the disease groups and HCs, we found a trend toward a higher relative expression in axSpA for IL26, IL17A, and RORC (Figure 3B). IL22, IFNG, and IL1R1 expression levels were not different between the groups, but the relative expression of IL4 was significantly increased in RA (Supplementary Figure S3B; p < 0.05).
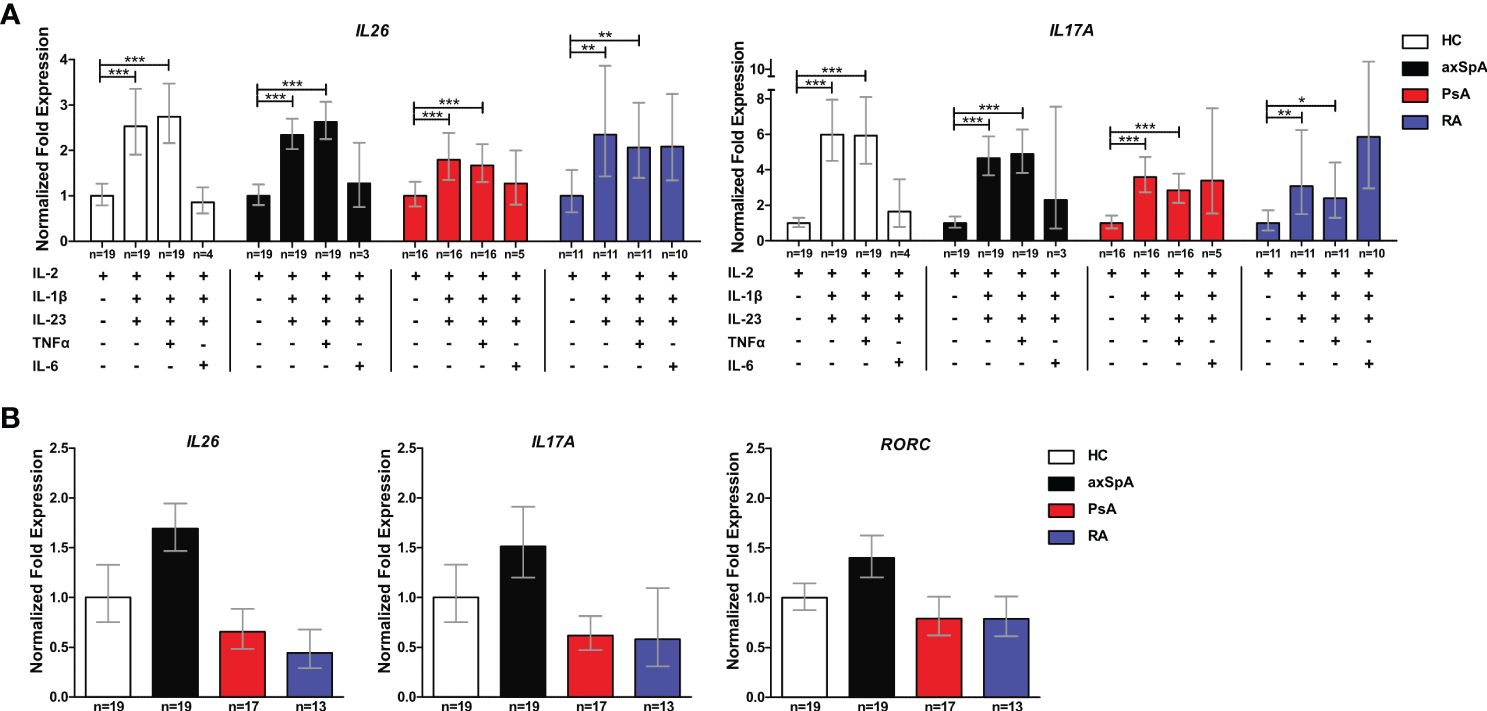
Figure 3 CD4+ T cells from axSpA patients are prone to IL26 expression under typical Th17-favoring conditions in vitro. (A) Total CD4+ T cells from HCs (n = 20; white) and axSpA (n = 19; black), PsA (n = 18; red), and RA n = 13; blue) patients were incubated with different Th17-favoring cytokine combinations in vitro for 6 days, and the expression of IL26 and IL17A was analyzed by qPCR in triplicates. Expression was normalized to two control genes (ACT and RPL13A) and is shown as fold expression in relation to basal stimulation with IL-2 only. (B) CD4+ T cells from the same cohort as in (A) were stimulated with IL-2, IL-1β, and IL-23 for 6 days in vitro, and fold expression (triplicates each) of the respective genes is shown in relation to HCs after normalizations for ACT and RLP13A. Statistical analysis: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001 [(A) repeated-measures ANOVA and (B) one-way ANOVA followed by Dunnett post-test for multiple comparisons].
These data demonstrate that CD4+ T cells from patients with axSpA are primed to express IL26 upon encountering IL-1β and IL-23, classical cytokines for the induction of pathogenic Th17 cells.
IL26 expression is highest in the peripheral joint tissue of axSpA and co-localizes with CD68+ synoviocytes
Given the importance of local actions of IL26 (high expression in RA joint tissue and synovial fluid of inflammatory arthritis), we deemed it of particular importance to examine the local production of IL26 within the peripheral synovial tissue (15, 16). We performed comparative immunohistochemistry for IL26 in peripheral synovial tissue from patients with axSpA, PsA and RA, and HCs. For detailed patient characteristics, see Table 2. Immunohistochemistry revealed significantly elevated expression of IL26 in the lining and sublining layers of axSpA tissue compared with HCs by automated quantification (open source tool “IHC profiler,” Figures 4A, B; mean 22.88%, SEM 3.58% vs. mean 5.25%, SEM 0.58%; p < 0.01) (33). However, IL26 expression was also increased in tissue from PsA and RA patients (mean 18.14%, SEM 5.84% and mean 11.71%, SEM 3.33%; ns). In order to further characterize the IL26-expressing cells in SpA tissue, immunofluorescence was performed. In axSpA, co-localization of IL26 could only be shown with CD68, a marker for macrophage-like synoviocytes (Figure 5A, panel 4: CD68+IL26), but not for either the pan-T-cell marker CD3, the alternative macrophage-like synoviocyte marker CD163, the myofibroblast marker SMA, or IL-1R1, a marker of IL26-producing CD4+ T cells. The same staining pattern was apparent in PsA tissue (Supplementary Figure S4). IL26 did also not co-localize with either cadherin-11, a marker of lining fibroblast-like synoviocytes, or CD90, a marker of sublining fibroblast-like synoviocytes (Figure 5B).
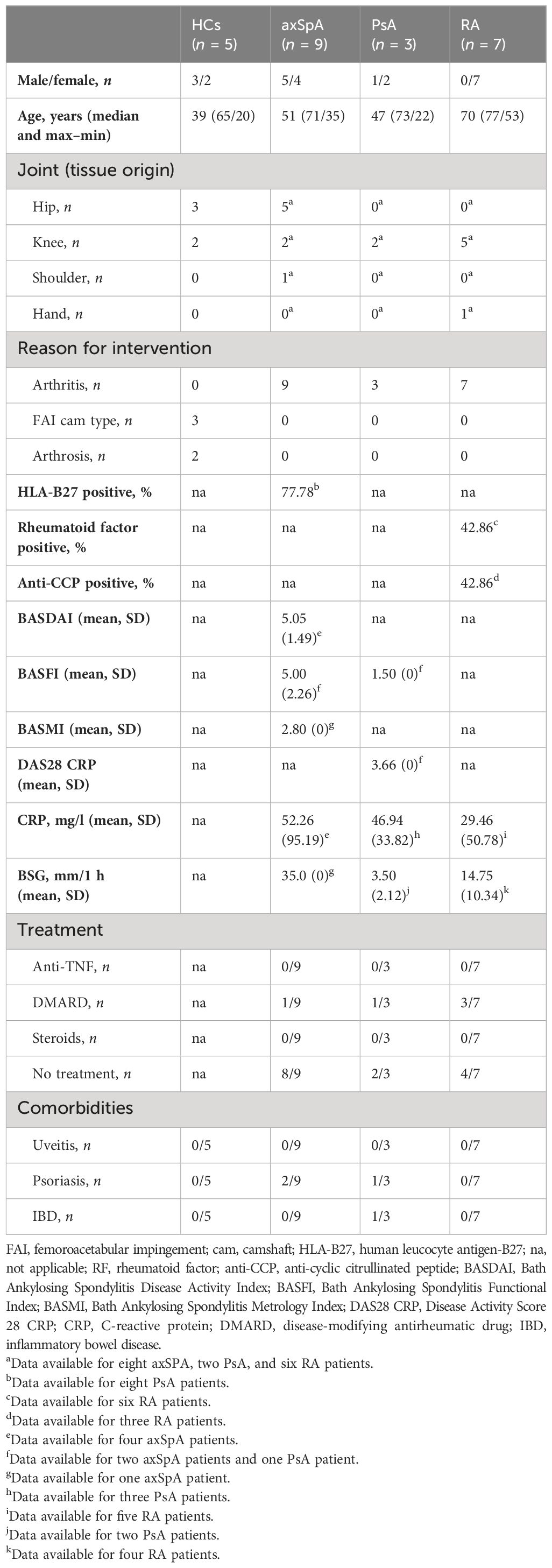
Table 2 Study population characteristics—synovial tissue samples.
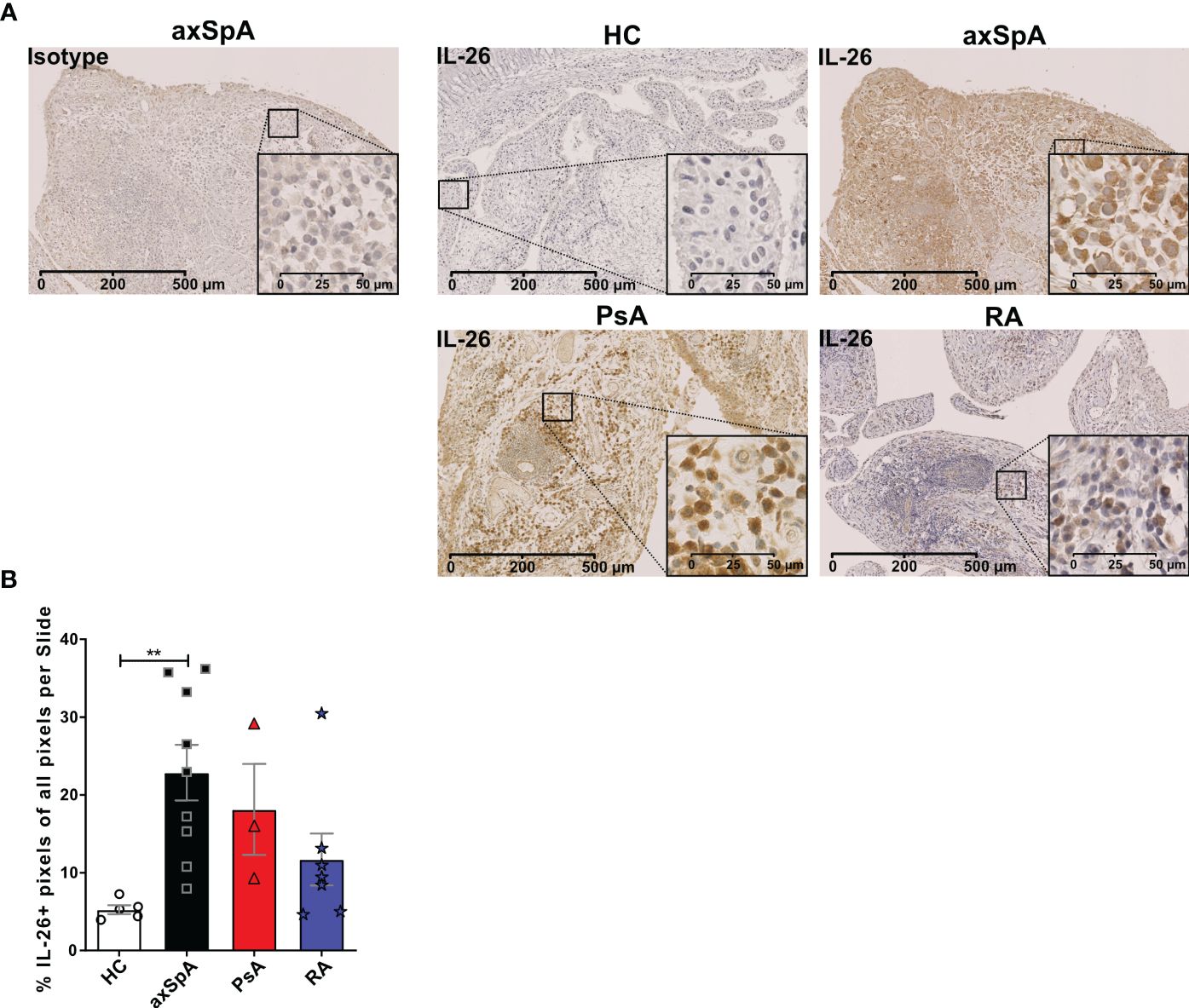
Figure 4 IL-26 expression in peripheral joints is increased in axSpA. (A) Representative slides of IL-26 expression by immunohistochemistry in peripheral synovial tissue from HCs (n = 5; white) and axSpA (n = 9; black), PsA (n = 3; red), and RA (n = 7; blue) patients. Isotype staining in 3,3′diaminobenzidine (brown) is shown on the far left. 20× (scale bar 500 μm) and inlay 80× (scale bar 50 μm). (B) Semiquantitative analysis of IL-26+ pixels in peripheral joint tissue sections from the cohort described in (A). Statistical analysis (B): mean ± SEM, **p < 0.01 (Kruskal–Wallis test).
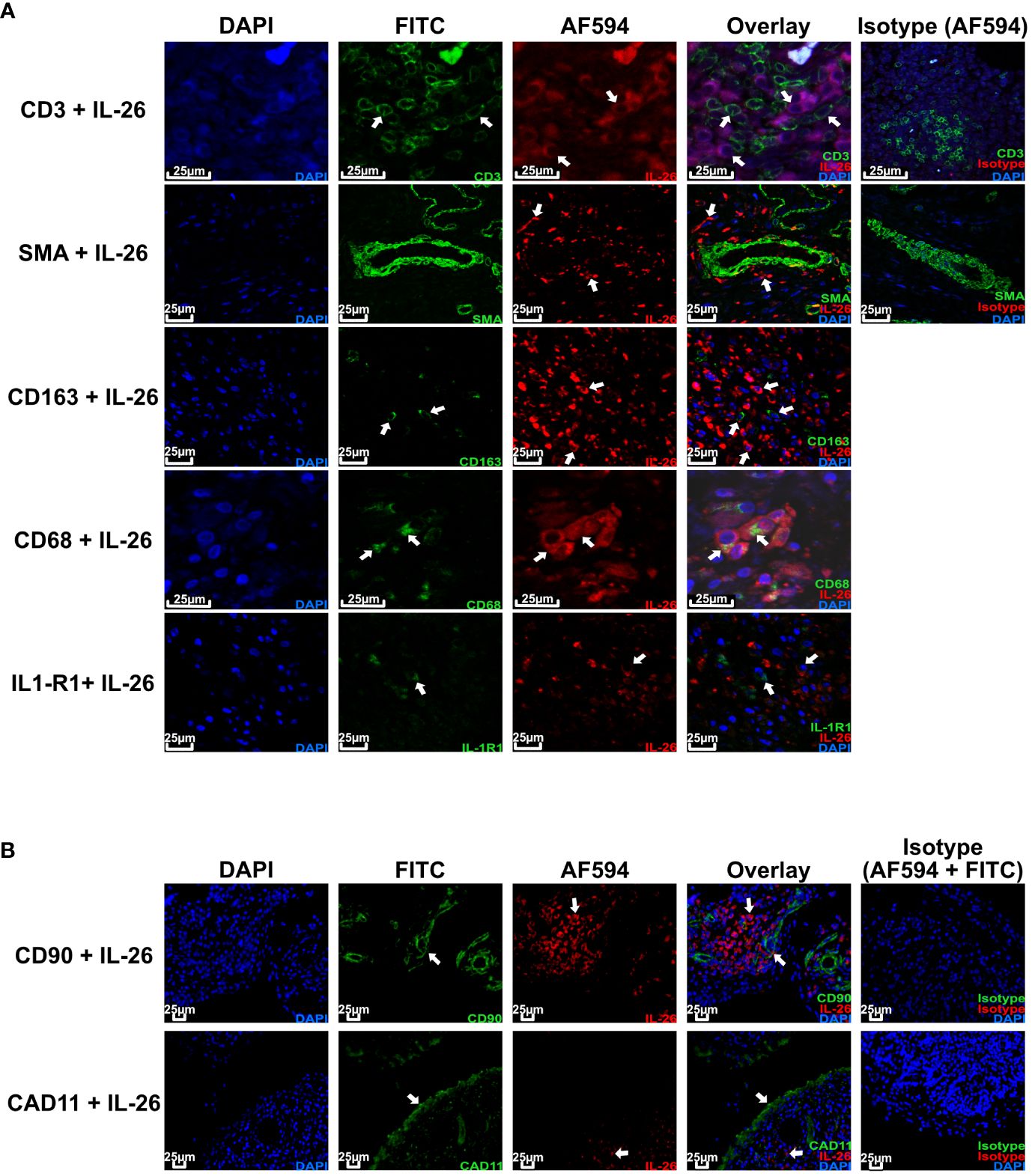
Figure 5 IL-26 co-localizes with CD68+ macrophage-like synoviocytes in axSpA. Representative confocal (A) and immunofluorescence images (B) of peripheral joint tissue from axSpA stained for IL-26 (red) and CD3/SMA/CD163/CD68/IL-1R1/Cadherin-11/CD90 (green). Blue shows DAPI nuclear stain. Scale bar 25 µm. Arrows indicate cells of interest for co-localization of the respective cell surface markers and IL-26.
This finding confirms the local action of IL26 in axSpA and PsA in the context of CD68+ macrophage-like synoviocytes, and not CD3+ T cells or fibroblast-like synoviocytes.
Discussion
This is so far the largest comparative study of IL26 in inflammatory arthritis with a focus on axSpA and PsA, clearly showing the increased expression of IL26 in peripheral joints.
Our data uniquely add to previous findings of increased IL26 expression in peripheral joints of axSpA by demonstrating IL26 expression in axSpA as well as in PsA peripheral synovial tissue by immunohistochemistry and immunofluorescence (16). In our hands, IL26 co-localizes with CD68+ macrophage-like synoviocytes in the sublining of the synovium. This is of special interest as efficacy of 12 weeks of secukinumab (ACR20 response rate of 65%) in peripheral SpA was accompanied by a significant reduction of synovial sublining CD68+ macrophages and CD15+ neutrophils (34). Compared with RA, state-of-the-art analysis of synovial tissue-resident macrophages (STMs) is not available from SpA yet. In human RA joint tissue, two populations of STMs are present: MerTK−CD206− STMs that induce proinflammatory responses in synovial fibroblasts and MerTK+CD206+ STMs that induce resolution of inflammation and repair mechanisms (35). These MerTK+CD206+ STMs are predominant in RA in remission, and a proportion of less than 47.5% is an independent predictor for disease flares after tapering or discontinuation of treatment. It remains to be elucidated if IL26-positive CD68+ synovial macrophages stem from circulating monocytes or from synovial tissue-resident macrophages, which both have been demonstrated in the K/BxN serum-transfer arthritis model (36). An ex vivo study on synovial fluid from PsA showed an expansion of intermediate monocytes and macrophages (37). These cells demonstrated expression of MRC1, the gene encoding CD206, which was shown to be an important marker for STMs in RA. At the moment, we cannot exclude that the IL26+ synovial myeloid cells are not producing IL26 themselves, but rather have shuttled extracellular DNA from cell death due to excessive inflammation in complex with IL26 into their cytosol (13). Possible local factors inducing IL26 production in resident CD68+ macrophage-like synoviocytes are not yet clear and need to be investigated. Studies with human alveolar macrophages have revealed bacterial products such as endotoxins and also environmental toxins (e.g., water-soluble tobacco smoke components) as inducers of IL26 (22, 38). Potential inducers of IL26 in CD68+ macrophage-like synoviocytes could be damage-associated molecular pattern (DAMP) molecules, such as HMGB1—either released after cell death or actively secreted—activating TLR4 and subsequently NF-κB (39). The latter was shown to be responsible for IL-1β-induced IL26 production in human memory CD4+ T cells (32). Due to the lack of synovial fluid samples from patients with PsA and axSpA, we were not able to further investigate the sources of IL26 in this compartment, which might differ from the actual synovial tissue. CD3+ T cells and fibroblast-like synoviocytes identified by cadherin-11 and CD90 are not involved in the increased expression of IL26 in the synovium of axSpA and PsA in our study, confirming the findings from SpA synovial fluid by Heftdal et al. (16). This contrasts with the findings from RA joint tissue, where IL26 co-localizes with CD3, RORγt, and synoviolin (15). However, IL26 supports Th17 immunity by upregulating IL-17A secretion from Th17 cells and inducing plasticity of memory non-Th17 cells toward Th17 cells through upregulation of IL-1β secretion from macrophages (15). In context of the DNA-binding capacity of IL26, it remains to be elucidated if IL26 in the peripheral joints of SpA patients also represents extracellular traps and, therefore, a further mechanism by which IL26 perpetuates chronic inflammation. IL26 has been shown to enhance NET-induced secretion of IL-6 from human monocytes and of CXCL8 from human neutrophils (13). As only a small fraction of IL26+ synovial cells are CD68+ in axSpA and PsA, further detailed investigations of local producers of IL26 in axSpA and PsA are warranted. Candidates include mainly CD3-negative cell types like ILC3, fibroblast-like synoviocytes and neutrophils. One caveat of our study is the fact that most of the joint samples analyzed were acquired during joint replacement surgery and, therefore, most likely represent a chronic state of inflammation with low activity in axSpA and PsA. In addition, these peripheral samples might not represent the mechanisms of inflammation and bone remodeling in the axial joints of SpA patients. This, however, is a common problem of SpA research, and the previously examined facet joints from SpA patients were also from established, chronic diseases. Nevertheless, IL26 is also present in the bone marrow of these joints (16).
Despite significantly elevated expression of IL26 in peripheral joint tissue in axSpA and PsA, we did not observe a relevant increase of IL26 in the serum of axSpA patients. This is congruent with the results of a previous study on plasma levels of IL26 in SpA patients (16). However, IL26 serum levels were significantly increased in PsA and RA patients compared with HCs. This could be explained by the fact that PsA and RA represent inflammatory arthritis with peripheral arthritis, whereas axSpA is predominately an axial disease. In our cohort of axSpA patients, only 27.6% had peripheral disease compared with 95% of PsA patients. The significantly elevated percentages of IL-1R1+ Th17 cells in PsA patients identified by chemokine receptor profile might also explain the significantly higher levels of serum IL26 in this group compared with HCs. Considering the significant enhancement of IL26 in the peripheral joint tissue of axSpA patients, this points to a distinct role of IL26 at the site of inflammation in axSpA compared with PsA and RA. Additional local factors involved in this distinct role of IL26 in axSpA joints need to be identified in the future. Differences between the serum levels of cytokines and local expression are not uncommon and have been described for example for IL-20 in patients with rheumatoid arthritis (40). This discrepancy has also been observed in patients with IBD, where increased protein and mRNA expression levels of IL26 in the gut were evident compared with HCs, but IL26 serum levels and IL26 expression in PBMCs were lower (41). This would fit with our observation of local IL26 attributable to tissue-resident CD68+ macrophage-like synoviocytes in SpA and not extravasated CD3+ T cells. A recent study in HCs demonstrated a large proportion of IL26+IL-17A− cells within circulating Th17 cells (identified through chemokine receptor staining) via PrimeFlow (42). This population seems to be a Th17 intermediate as it differentiates into IL-17A+ Th17 cells upon TGF-β1 exposure. Such intermediates and the described differentiation were also shown in psoriatic skin lesions. Therefore, the lack of IL26+CD3+ cells in our peripheral tissue samples could be attributed to a loss of IL26 expression in chronic or long-standing disease. Additionally, the only slightly elevated percentages of circulating IL26+ Th17 cells in axSpA and PsA patients approximately corresponded with the results from the serum measurements of IL26, further strengthening the local role of IL26 in SpA. However, the reduced sample number in the PrimeFlow assay of IL26 (approx. 15 vs. 30 samples) might be responsible for not showing significantly elevated percentages of IL26+ Th17 cells. Moreover, the slightly increased percentages of IL26+CD8+ T cells in axSpA and PsA could be an alternative source of peripheral IL26. This could reflect the fact that, like IL-17A, IL26 can be produced by different cell types as part of a pathologic type 3 immunity. Nevertheless, we found memory CD4+ T cells from axSpA patients to be more prone to IL26 expression upon encountering typical Th17-favoring cytokine combinations like IL-2, IL-1β, and IL-23 in vitro. In addition, circulating CD4+ T cells from axSpA and PsA more often expressed IL26 ex vivo compared with HCs, being in congruence with previously reported data showing IL26 expression exclusively in peripheral CD4+ T cells (12).
IL26 could play a major part in the chronicity of inflammation in axSpA and PsA as IL26 can increase the secretion of IL-1β, IL-6, TNFα, and CCL20 (Th17 cell recruitment) from CD14+ myeloid cells as shown from RA synovial fluid (15). In this context, therapies targeting IL26 could be of great relevance in patients failing current therapies. Due to the employment of JAK1 and TYK2 downstream of the IL26 receptor, current JAK inhibitors like upadacitinib should reduce IL26 responses to some extent. However, specifically targeting IL26 could still have superior effects to current JAK1 or future TYK2 inhibition as several other signaling options have been described for IL26. For example, IL26/DNA complexes can enter plasmacytoid dendritic cells and activate TLR9 or activate monocytes by inflammasome-mediated activation of the STING (stimulator of interferon genes) pathway (12, 13). It also remains possible that IL26 alone due to its chemical properties could penetrate the cell membrane and illicit proinflammatory responses (13, 43). This aspect also remains interesting as it is not clear through which pathway IL26 induces bone mineralization in human osteoblasts (16). So far, the specific receptor chain for IL26, IL-20R1, has been detected in human amniotic fluid-derived stem cells, which can be differentiated into osteocytes, and in the mouse osteoblast precursor cell line MC3T3-E1 (44, 45), but not in primary human osteoblasts or osteoblast precursors. So, the exact role and mechanisms of IL26 in SpA-associated osteoproliferation remain unclear and warrant further investigation. Inhibiting RORγt, one of the key transcription factors of Th17 cells, significantly downregulates the expression of IL26 in axSpA Th17 cells (46), but unfortunately, none of the candidate compounds has been successfully trialed so far (47). As IL-17A increases IL26 secretion from RA synovial fibroblasts, IL-17A blockers like sekukinumab could also exert some effects on synovial pathophysiology in axSpA and PsA (15). Monoclonal antibodies targeting IL26 significantly suppressed imiquimod-induced skin inflammation in human IL26 transgenic mice without apparent effects on antimicrobial activity (48). In this study, the concentrations of anti-IL26 mAb used were much lower than the concentrations reported necessary for the antimicrobial effects of IL26. This allows for further evaluation of anti-IL26 mAb as a therapeutic target in inflammatory diseases like psoriasis, PsA and axSpA. As IL26 secretion from human memory CD4+ T cells induced by IL-1β/IL-1R1 was nearly abrogated by pharmacological inhibition of NF-κB activation, this provides a further target for blocking IL26-induced pathology assuming a role of the aforementioned IL26+IL-17− Th17 intermediates in the active or early stages of the disease (32).
In summary, we have shown that different cells are responsible for local and systemic production of IL26 in axSpA and PsA providing evidence for the first time that CD68+ macrophage-like synoviocytes could be a local source of IL26 in SpA peripheral joints. Regarding its role in chronicity of inflammation and osteoproliferation, IL26 provides an interesting therapeutic approach for axSpA and PsA by simultaneously targeting the two key modules of its pathophysiology.
Limitations of the study
Nevertheless, the results of this study must be interpreted bearing a few limitations in mind. The most obvious limitation is the fact that we were unable to obtain corresponding peripheral blood and synovial fluid for the analysis of IL26 levels and producers. This might have helped to prove that IL26 is indeed enhanced at the site of joint inflammation in axSpA. However, it has already been shown that the levels of IL26 are significantly increased in SpA synovial fluid compared with matching plasma (16). Our study unfortunately can also not provide any information on IL26 expression and producers in axial joints. This is an issue that is hard to overcome as biopsies of axial joints or iliosacral joints are not routinely performed and usually involve CT localization exposing patients to radiation. Another limitation is the small sample size used for phenotyping of IL26-expressing T cells and IL-1R1+ T cells by flow cytometry. Therefore, a significant enrichment of IL26+CD4+ or IL26+CD8+ T cells as well as IL-1R1+ T cells in axSpA and PsA might not have become evident, despite the clear increase in local IL26 levels in synovial tissue. Additional studies involving larger numbers of patients are necessary to clarify the role of circulating IL26-producing cells. Further limitations are the low disease activity of the PBMC cohort and the long-standing disease phenotype of the synovial tissue samples. We were unable to provide single-cell RNA sequencing of unfixed synovial tissue from patients with active peripheral disease or from affected axial joints. Such data will however be of great interest for further delineating the producers of IL26 in SpA in the future.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by 100/18 S, Ethical Committee of the Faculty of Medicine, Technical University Munich and 06/Q1606/139 Oxford University Hospitals NHS Foundation Trust. The study has been registered within the German Clinical Trials Register (DRKS00014672). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
AH: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AO: Formal analysis, Investigation, Visualization, Writing – original draft. QB: Writing – review & editing, Resources. HM-F: Writing – review & editing, Resources. GL: Writing – review & editing, Resources. MW: Writing – review & editing, Resources. FW: Writing – review & editing, Resources. HM: Writing – review & editing, Resources. DS: Writing – review & editing, Resources. PB: Supervision, Writing – review & editing, Resources. UH: Resources, Writing – review & editing. MA: Writing – review & editing, Resources. PM: Resources, Supervision, Writing – review & editing. CS: Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. AH received funding from Hans and Klementia Langmatz-Stiftung (charitable trust). AO received funding from DGIM (German Society of Internal Medicine) within the Peter-Scriba-Promotionsstipendium. PB was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC).
Conflict of interest
Travel and congress support: Boehringer Ingelheim AH, Lilly AH, UCB AH, AbbVie QB, medac QB, Novartis QB, Pfizer QB, FW, galápagos FW, Biogen FW.
Speakers´ fees: Novartis AH, DS, FW, MSD DS, UCB HM-F, FW, Pfizer FW, GSK FW, AbbVie FW, AstraZeneca FW, Sanofi-Aventis FW.
Research support: Regeneron PB, Novartis PB, GSK PB.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1355824/full#supplementary-material
References
1. Stolwijk C, van Onna M, Boonen A, van Tubergen A. Global prevalence of spondyloarthritis: A systematic review and meta-regression analysis. Arthritis Care Res (Hoboken). September. (2016) 68:1320–31. doi: 10.1002/acr.22831
2. Dougados M, Baeten D. Spondyloarthritis. Lancet. (2011) 377:2127–37. doi: 10.1016/S0140-6736(11)60071-8
3. Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med. (2015) 373:2534–48. doi: 10.1056/NEJMoa1505066
4. Australo-Anglo-American Spondyloarthritis Consortium (TASC), eveille JD, Sims AM, Danoy P, Evans DM, Leo P. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat Genet. (2010) 42:123–7. doi: 10.1038/ng.513
5. Shen H, Goodall JC, Hill Gaston JS. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. (2009) 60:1647–56. doi: 10.1002/art.24568
6. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. (2012) 13:991–9. doi: 10.1038/ni.2416
7. Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO. Late developmental plasticity in the T helper 17 lineage. Immunity. (2009) 30:92–107. doi: 10.1016/j.immuni.2008.11.005
8. Blaschitz C, Raffatellu M. Th17 cytokines and the gut mucosal barrier. J Clin Immunol. (2010) 30:196–203. doi: 10.1007/s10875-010-9368-7
9. Hernández-Santos N, Gaffen SL. Th17 cells in immunity to Candida albicans. Cell Host Microbe. (2012) 11:425–35. doi: 10.1016/j.chom.2012.04.008
10. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. (2007) 8:950–7. doi: 10.1038/ni1497
11. Pène J, Chevalier S, Preisser L, Vénéreau E, Guilleux MH, Ghannam S. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol. (2008) 180:7423–30. doi: 10.4049/jimmunol.180.11.7423
12. Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D. T(H)17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol. (2015) 16:970–9. doi: 10.1038/ni.3211
13. Poli C, Augusto JF, Dauvé J, Adam C, Preisser L, Larochette V. IL26 confers proinflammatory properties to extracellular DNA. J Immunol. (2017) 198:3650–61. doi: 10.4049/jimmunol.1600594
14. Dambacher J, Beigel F, Zitzmann K, De Toni EN, Göke B, Diepolder HM. The role of the novel Th17 cytokine IL26 in intestinal inflammation. Gut. (2009) 58:1207–17. doi: 10.1136/gut.2007.130112
15. Corvaisier M, Delneste Y, Jeanvoine H, Preisser L, Blanchard S, Garo E. IL26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PloS Biol. (2012) 10:e1001395. doi: 10.1371/journal.pbio.1001395
16. Heftdal LD, Andersen T, Jæhger D, Woetmann A, Østgård R, Kenngott EE. Synovial cell production of IL26 induces bone mineralization in spondyloarthritis. J Mol Med (Berl). (2017) 95:779–87. doi: 10.1007/s00109-017-1528-2
17. Cols M, Rahman A, Maglione PJ, Garcia-Carmona Y, Simchoni N, Ko HBM. Expansion of inflammatory innate lymphoid cells in patients with common variable immune deficiency. J Allergy Clin Immunol. (2016) 137:1206–1215.e6. doi: 10.1016/j.jaci.2015.09.013
18. Meermeier EW, Zheng CL, Tran JG, Soma S, Worley AH, Weiss DI. Human lung-resident mucosal-associated invariant T cells are abundant, express antimicrobial proteins, and are cytokine responsive. Commun Biol. (2022) 5:942. doi: 10.1038/s42003-022-03823-w
19. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JKM. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. (2009) 457:722–5. doi: 10.1038/nature07537
20. Jaeger N, Gamini R, Cella M, Schettini JL, Bugatti M, Zhao S. Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat Commun. (2021) 12:1921. doi: 10.1038/s41467-021-22164-6
21. Steel KJA, Srenathan U, Ridley M, Durham LE, Wu SY, Ryan SE. Polyfunctional, proinflammatory, tissue-Resident memory phenotype and function of synovial interleukin-17A+CD8+ T cells in psoriatic arthritis. Arthritis Rheumatol. (2020) 72:435–47. doi: 10.1002/art.41156
22. Che KF, Tengvall S, Levänen B, Silverpil E, Smith ME, Awad M. Interleukin-26 in antibacterial host defense of human lungs. Effects on neutrophil mobilization. Am J Respir Crit Care Med. (2014) 190:1022–31. doi: 10.1164/rccm.201404-0689OC
23. Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J Med Chem. (2017) 60:1247–61. doi: 10.1021/acs.jmedchem.6b01751
24. Peng YJ, Wang CY, Lin YH, Lin GJ, Huang SH, Shyu JF. Interleukin 26 suppresses receptor activator of nuclear factor κB ligand induced osteoclastogenesis via down-regulation of nuclear factor of activated T-cells, cytoplasmic 1 and nuclear factor κB activity. Rheumatology. (2016) 55:2074–83. doi: 10.1093/rheumatology/kew302
25. Miot C, Beaumont E, Duluc D, Le Guillou-Guillemette H, Preisser L, Garo E. IL26 is overexpressed in chronically HCV-infected patients and enhances TRAIL-mediated cytotoxicity and interferon production by human NK cells. Gut. (2015) 64:1466–75. doi: 10.1136/gutjnl-2013-306604
26. Hummelshoj L, Ryder LP, Poulsen LK. The role of the interleukin-10 subfamily members in immunoglobulin production by human B cells. Scand J Immunol. (2006) 64:40–7. doi: 10.1111/j.1365-3083.2006.01773.x
27. Itoh T, Hatano R, Komiya E, Otsuka H, Narita Y, Aune TM. Biological effects of IL26 on T cell-mediated skin inflammation, including psoriasis. J Invest Dermatol. (2019) 139:878–89. doi: 10.1016/j.jid.2018.09.037
28. Ohnuma K, Hatano R, Aune TM, Otsuka H, Iwata S, Dang NH. Regulation of pulmonary graft-versus-host disease by IL26+CD26+CD4 T lymphocytes. J Immunol. (2015) 194:3697–712. doi: 10.4049/jimmunol.1402785
29. Dakin SG, Martinez FO, Yapp C, Wells G, Oppermann U, Dean BJF. Inflammation activation and resolution in human tendon disease. Sci Transl Med. (2015) 7:311ra173. doi: 10.1126/scitranslmed.aac4269
30. Kragstrup TW, Vorup-Jensen T, Deleuran B, Hvid M. A simple set of validation steps identifies and removes false results in a sandwich enzyme-linked immunosorbent assay caused by anti-animal IgG antibodies in plasma from arthritis patients. Springerplus. (2013) 2:263. doi: 10.1186/2193-1801-2-263
31. Bengsch B, Seigel B, Flecken T, Wolanski J, Blum HE, Thimme R. Human th17 cells express high levels of enzymatically active dipeptidylpeptidase IV (CD26). J Immunol. (2012) 188:5438–47. doi: 10.4049/jimmunol.1103801
32. Weiss DI, Ma F, Merleev AA, Maverakis E, Gilliet M, Balin SJ. IL-1β Induces the rapid secretion of the antimicrobial protein IL26 from th17 cells. J Immunol. (2019) 203:911–21. doi: 10.4049/jimmunol.1900318
33. Varghese F, Bukhari AB, Malhotra R, De A. IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PloS One. (2014) 9:e96801. doi: 10.1371/journal.pone.0096801
34. Mens LJJ, Sande MGH, Menegatti S, Chen S, Blijdorp ICJ, Jong HM. Brief report: interleukin-17 blockade with secukinumab in peripheral spondyloarthritis impacts synovial immunopathology without compromising systemic immune responses. Arthritis Rheumatol. (2018) 70:1994–2002. doi: 10.1002/art.40581
35. Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med. (2020) 26:1295–306. doi: 10.1038/s41591-020-0939-8
36. Culemann S, Grüneboom A, Nicolás-Ávila JÁ, Weidner D, Lämmle KF, Rothe T. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature. (2019) 572:670–5. doi: 10.1038/s41586-019-1471-1
37. Yager N, Cole S, Lledo Lara A, Maroof A, Penkava F, Knight JC. Ex vivo mass cytometry analysis reveals a profound myeloid proinflammatory signature in psoriatic arthritis synovial fluid. Ann Rheum Dis. (2021) 80:1559–67. doi: 10.1136/annrheumdis-2021-220280
38. Che KF, Tufvesson E, Tengvall S, Lappi-Blanco E, Kaarteenaho R, Levänen B. The neutrophil-mobilizing cytokine interleukin-26 in the airways of long-term tobacco smokers. Clin Sci (Lond). (2018) 132:959–83. doi: 10.1042/CS20180057
39. Erlandsson Harris H, Andersson U. Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol. (2004) 34:1503–12. doi: 10.1002/eji.200424916
40. Hsu YH, Li HH, Hsieh MY, Liu MF, Huang KY, Chin LS. Function of interleukin-20 as a proinflammatory molecule in rheumatoid and experimental arthritis. Arthritis Rheum. (2006) 54:2722–33. doi: 10.1002/art.22039
41. Song D, Lai L, Lu J, Tong J, Ran Z. Interleukin-26 expression in inflammatory bowel disease and its immunoregulatory effects on macrophages. Front Med 6. April. (2022) 9:797135. doi: 10.3389/fmed.2022.797135
42. Fries A, Saidoune F, Kuonen F, Dupanloup I, Fournier N, Guerra De Souza AC. Differentiation of IL26+ TH17 intermediates into IL-17A producers via epithelial crosstalk in psoriasis. Nat Commun. (2023) 14:3878. doi: 10.1038/s41467-023-39484-4
43. Sapay N, Guermeur Y, Deléage G. Prediction of amphipathic in-plane membrane anchors in monotopic proteins using a SVM classifier. BMC Bioinf. (2006) 7:255. doi: 10.1186/1471-2105-7-255
44. Hsu YH, Chiu YS, Chen WY, Huang KY, Jou IM, Wu PT. Anti-IL-20 monoclonal antibody promotes bone fracture healing through regulating IL-20-mediated osteoblastogenesis. Sci Rep. (2016) 6:24339. doi: 10.1038/srep24339
45. Hsu YH, Chen WY, Chan CH, Wu CH, Sun ZJ, Chang MS. Anti–IL-20 monoclonal antibody inhibits the differentiation of osteoclasts and protects against osteoporotic bone loss. J Exp Med. (2011) 208:1849–61. doi: 10.1084/jem.20102234
46. de Wit J, Al-Mossawi MH, Hühn MH, Arancibia-Cárcamo CV, Doig K, Kendrick B. RORγt inhibitors suppress T(H)17 responses in inflammatory arthritis and inflammatory bowel disease. J Allergy Clin Immunol. (2016) 137:960–3. doi: 10.1016/j.jaci.2015.09.048
47. Zeng J, Li M, Zhao Q, Chen M, Zhao L, Wei S. Small molecule inhibitors of RORγt for Th17 regulation in inflammatory and autoimmune diseases. J Pharm Anal. (2023) 13:545–62. doi: 10.1016/j.jpha.2023.05.009
Keywords: IL26, Th17 cells, synvovial tissue, axial spondyloarthritis, psoriatic arthritis
Citation: Hammitzsch A, Ossadnik A, Bachmann Q, Merwald-Fraenk H, Lorenz G, Witt M, Wiesent F, Mühlhofer H, Simone D, Bowness P, Heemann U, Arbogast M, Moog P and Schmaderer C (2024) Increased interleukin-26 in the peripheral joints of patients with axial spondyloarthritis and psoriatic arthritis, co-localizing with CD68-positive synoviocytes. Front. Immunol. 15:1355824. doi: 10.3389/fimmu.2024.1355824
Received: 14 December 2023; Accepted: 01 April 2024;
Published: 10 May 2024.
Edited by:
Michele Maria Luchetti Gentiloni, Marche Polytechnic University, ItalyReviewed by:
Sadiq Umar, University of Illinois Chicago, United StatesJose Antonio Lopez-Escamez, University of Sydney, Australia
Copyright © 2024 Hammitzsch, Ossadnik, Bachmann, Merwald-Fraenk, Lorenz, Witt, Wiesent, Mühlhofer, Simone, Bowness, Heemann, Arbogast, Moog and Schmaderer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ariane Hammitzsch, QXJpYW5lLkhhbW1pdHpzY2hAbXJpLnR1bS5kZQ==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship