 Satoshi Yamaga
Satoshi Yamaga Monowar Aziz
Monowar Aziz Atsushi Murao
Atsushi Murao Max Brenner
Max Brenner Ping Wang
Ping Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 25 January 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1353990
This article is part of the Research TopicCommunity Series in Translational Insights into Mechanisms and Therapy of Organ Dysfunction in Sepsis and Trauma, volume IIIView all 20 articles
The heightened risk of ionizing radiation exposure, stemming from radiation accidents and potential acts of terrorism, has spurred growing interests in devising effective countermeasures against radiation injury. High-dose ionizing radiation exposure triggers acute radiation syndrome (ARS), manifesting as hematopoietic, gastrointestinal, and neurovascular ARS. Hematopoietic ARS typically presents with neutropenia and thrombocytopenia, while gastrointestinal ARS results in intestinal mucosal injury, often culminating in lethal sepsis and gastrointestinal bleeding. This deleterious impact can be attributed to radiation-induced DNA damage and oxidative stress, leading to various forms of cell death, such as apoptosis, necrosis and ferroptosis. Damage-associated molecular patterns (DAMPs) are intrinsic molecules released by cells undergoing injury or in the process of dying, either through passive or active pathways. These molecules then interact with pattern recognition receptors, triggering inflammatory responses. Such a cascade of events ultimately results in further tissue and organ damage, contributing to the elevated mortality rate. Notably, infection and sepsis often develop in ARS cases, further increasing the release of DAMPs. Given that lethal sepsis stands as a major contributor to the mortality in ARS, DAMPs hold the potential to function as mediators, exacerbating radiation-induced organ injury and consequently worsening overall survival. This review describes the intricate mechanisms underlying radiation-induced release of DAMPs. Furthermore, it discusses the detrimental effects of DAMPs on the immune system and explores potential DAMP-targeting therapeutic strategies to alleviate radiation-induced injury.
The increasing threat of unexpected exposure to ionizing radiation implicates the urgent unmet need for developing medical countermeasures. The global communities must be united to confront a broad range of nuclear threats. These encompass the detonation of sophisticated and improvised nuclear weapons, potential terrorist deployment of radiologic dispersal devices (i.e., dirty bombs), attacks on nuclear power plants, and the occurrence of accidents at these facilities caused by technical failure or natural disasters as happened in Chernobyl, Ukraine in 1986 and Fukushima, Japan in 2011 (1–4). Exposure to high doses of radiation can cause serious health problems known as acute radiation syndrome (ARS), consisting of three major sub-syndromes, hematopoietic (H), gastrointestinal (GI), and neurovascular (NV) ARS (5, 6). Medical countermeasures for ARS are classified into radioprotectors administered before radiation exposure, radiation mitigators administered shortly after radiation exposure and before symptoms appear, and radiation therapeutics administered after symptoms of radiation exposure appear (7). At the moment, the U.S. Food and Drug Administration (FDA) has approved two radioprotectors to be used in the context of radiotherapy: the free radical scavenger Amifostine and the recombinant human keratinocyte growth factor Palifermin (8). The FDA has also approved six radiomitigating medical countermeasures: the granulocyte colony-stimulating factors (G-CSF) Neupogen (filgrastim), Neulasta (pegfilgrastim), Udenyka (pegfilgrastim-cbqv), and Stimufend (pegfilgrastim-fpgk), the granulocyte-macrophage colony-stimulating factor (GM-CSF) Leukine (sargramostim), and thrombopoietin receptor agonist Nplate (romiplostim) (9). These radiomitigators are not approved as radioprotectors and only mitigate hematopoietic dysfunction to some extent. There are no FDA-approved medical countermeasures available for GI-ARS or NV-ARS. The major mechanisms of medical countermeasures currently under development include free radical scavenging, DNA damage reduction, DNA repair promotion, apoptosis inhibition, and GI recovery, in addition to the promotion of hematopoiesis (10, 11).
Exposure to ionizing radiation induces the release of damage-associated molecular patterns (DAMPs) by DNA damage and several types of cell death (12). A growing body of evidence suggests that DAMPs play pro-inflammatory roles and are associated with disease severity and organ damage in several diseases (13, 14). DAMPs hold the potential to function as mediators that exacerbate radiation-induced organ injuries, ultimately worsening survival outcomes. This review aims to comprehensively outline the mechanisms underlying radiation injury, with a particular focus on the involvement of DAMPs. Furthermore, we delve into potential therapeutic interventions aimed at mitigating radiation-induced DAMPs and their associated consequences.
The hematopoietic system emerges as the most vulnerable organ to ARS, with discernible symptoms of H-ARS manifesting following exposure to total body irradiation (TBI) exceeding 2 gray (Gy) in humans (15). Depending on the dose of ionizing radiation, death of hematopoietic stem and progenitor cells and apoptosis of lymphocytes occur (16). Notably, lymphopenia becomes apparent within 6-24 hours after radiation exposure (17). Subsequent to this initial event of the loss of lymphocytes, neutropenia, thrombocytopenia, and anemia ensue, depending on the radiation dose, based on the half-life of these cells and cell fragments in the circulation (18). GI-ARS generally occurs in individuals following exposure to TBI exceeding 5 Gy (19). Radiation exposure damages progenitor cells in the crypts and differentiated cells in the villi cannot turn over due to the loss of the progenitor and stem cells (20, 21). Injury to the intestinal mucosal barrier results in the infiltration of antigens and bacteria into the intestinal wall from the lumen. This breach gives rise to various complications, including the onset of secretory diarrhea resulting in severe protein loss and electrolyte dysfunction, bacterial translocation from the intestinal lumen into the circulation, and intestinal hemorrhage (22). These complications collectively contribute to the lethal sepsis and bleeding along with neutropenia and thrombocytopenia due to H-ARS (23). NV-ARS occurs after TBI with over 10 Gy in humans (19). Damage to the central nervous system involves disruptions in capillary circulation, blood-brain barrier integrity, interstitial edema, and the onset of acute inflammation. NV-ARS results in death within a few days of radiation exposure (19). Human data following radiation exposure indicates that the LD50/60, estimating the radiation dose causing lethality in 50% of a population within 60 days, ranges from 3.3-4.5 Gy without medical management to 6-7 Gy with medical intervention, including antimicrobial agents and blood transfusions (17, 23). In addition to ionizing radiation, nuclear explosions also release energy in the form of heat and shock waves (3). Thus, in situations involving nuclear explosions, 60-70% of victims may suffer from a radiation-combined injury (RCI) that includes burns and trauma (24). As a result, RCI can be lethal at low radiation doses due to infections (and possibly increased release of DAMPs, as described later in this review) in patients with burns and trauma (22, 25).
Oxidative stress causes DNA damage by reactive oxygen species (ROS) generated after the ionization of water in the cells. Ionizing radiation can also induce direct DNA damage including single- and double-strand breaks (26). The DNA damage response induces a reversible cell cycle arrest to give cells time to repair damage (12). When DNA damage cannot be resolved by the repair process, the DNA damage response converts into a signal pathway of apoptotic cell death or senescence, most often through p53-dependent mechanisms (27). Mitotic catastrophe can occur when cells undergo DNA damage during mitosis or enter mitosis with DNA damage. Mitotic catastrophe causes the formation of cells with multi-micronuclei or two nuclei (28). These cells are unable to complete the cell cycle, ultimately leading to their entry into senescence or their demise through apoptosis or necrosis (27). Exposure to ionizing radiation can affect not only nuclear DNA, but also other cellular organelles including mitochondria, Golgi apparatus, and lysosomes (29). Mitochondria serve as the primary generators of ROS within the cells. Radiation-induced mitochondrial DNA damage induces ROS generation, leading to a decrease in mitochondrial membrane potential and inducing mitochondria outer membrane permeabilization (MOMP) (29). The subsequent release of cytochrome c and activation of caspase-9 lead to apoptosis (30).
Recent studies have unveiled non-apoptotic signaling pathways of programmed cell death such as necroptosis, pyroptosis, and ferroptosis as additional forms of ionizing radiation-induced cell death. For instance, oxidative stress induced by ionizing radiation activates the inflammasome pathway, causing pyroptosis and contributing to organ damage (31, 32). Necroptosis in the intestine has been shown to contribute to mortality after lethal radiation exposure (33). Furthermore, the accumulation of ROS resulting from radiation exposure has been linked to the induction of ferroptosis, correlating with H-ARS, GI-ARS, and radiation-induced lung injury (34–36).
The immune system initiates an immune response against invasive pathogens based on the distinction of ‘self’ from ‘non-self’ (37). Innate immune cells express pattern recognition receptors, such as Toll-like receptors (TLRs), recognizing pathogen-associated molecular patterns (PAMPs) (37). Pathogen-independent immune responses are also initiated by recognition of DAMPs released from stressed or damaged cells (38). DAMPs activate innate immune cells such as neutrophils, macrophages, and dendritic cells (39). Consequently, various cytokines and chemokines are released, activating adaptive immune responses (40). DAMPs also activate non-immune cells such as epithelial cells, endothelial cells, and fibroblasts, leading to the release of inflammatory mediators (37). Typical endogenous DAMPs include extracellular cold-inducible RNA-binding protein (eCIRP), high mobility group box 1 (HMGB1), histones, heat shock proteins (HSPs), ATP, and nuclear and mitochondrial DNA (mtDNA), extracellular RNA (exRNA), and uric acid (38). The major DAMP-recognizing pattern recognition receptors include TLRs, NOD-like receptors (NLRs), RIG-I-like receptors, and C-type lectin receptors (41). Following the recognition of DAMPs, TLRs interact with myeloid differentiation factor 88 and TIR-domain-containing adaptor-inducing beta interferon to phosphorylate downstream mitogen-activated protein kinases, leading to activation of the transcription factors, activator protein-1 and nuclear factor kappa B (NF-κB) (37). DAMPs also transduce signals through the receptor for advanced glycation end products (RAGE), triggering receptor expressed on myeloid cells-1 (TREM-1), G-protein-coupled receptors, transient receptor potential, and P2X7 receptor channels (37).
The release of DAMPs has been observed in radiation injury and a number of other diseases including sepsis, trauma, ischemia-reperfusion injury, brain injuries, and radiation exposure (39, 42–45). In sepsis, host recognition of DAMPs is a critical contributor to an excessive immune response. Circulating DAMPs correlate with disease severity, and their inhibition has been shown to improve outcomes in experimental models of sepsis (38, 39). In severe trauma, the escalation of the innate immune response due to exposure to DAMPs leads to coagulopathy and excessive inflammation, resulting in multiple organ dysfunction, such as intestinal barrier dysfunction and acute lung injury (46). In intestinal ischemia-reperfusion injury, circulating mtDNA correlates with inflammatory responses and contributes to intestinal injury (43). Excessive inflammatory signaling induced by DAMPs can exhaust hematopoiesis and induce dysfunction of hematopoietic stem cells (47). Furthermore, the DAMPs-mediated neuroinflammation after traumatic brain injury, subarachnoid hemorrhage, and cerebral ischemia exacerbates secondary damage of the central nerve system by promoting cerebral edema or triggering vascular spasm and microthrombi (48). Given that these clinical complications are frequently observed in ARS, it is evident that the release of DAMPs can exacerbate the outcomes in ARS.
Radiation-induced cell damage can signal the immune system to potential danger through the release of DAMPs, akin to the molecular pattern recognition seen in microorganisms. Inflammation resulting from an excess of DAMP release has the potential to worsen radiation-induced tissue and organ damage, emphasizing the significance of DAMP modulation as potential mitigation for radiation injury (49). Here, we provide a summary of the major DAMPs released during radiation injury, and explore interventions aimed at enhancing outcomes (Table 1).
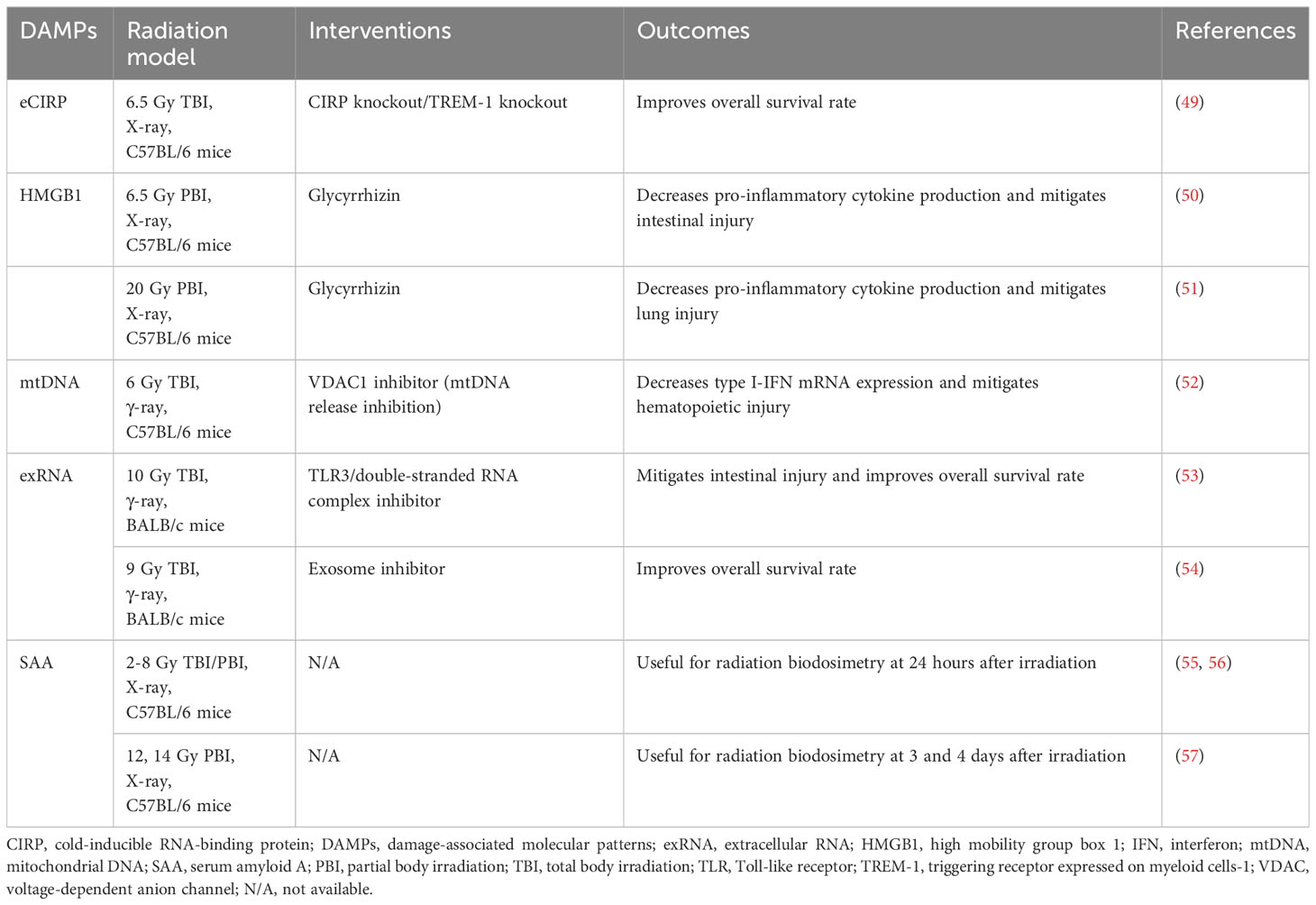
Table 1 Targeting DAMPs in radiation injury.
Intracellular CIRP is an RNA chaperone that functions as a cellular stress response protein (58). Once outside of the cell, eCIRP plays an important role as an endogenous inflammatory mediator acting as a new DAMP in radiation injury, sepsis, hemorrhagic shock, and ischemia-reperfusion injury (58, 59). eCIRP induces the production of proinflammatory cytokines and chemokines in macrophages via the TLR4/MD2 complex and NF-κB signaling axis. Accordingly, it contributes to tissue damage by upregulating ICAM-1 expression and promoting excessive NET formation in neutrophils (58, 60). eCIRP can also bind to TREM-1, worsening organ damage and prognosis by promoting inflammation (61). eCIRP has been shown to suppress the bacterial phagocytosis of macrophages by promoting the formation of STAT3-βPIX complex and suppressing the activation of Rac1 (62). We have recently demonstrated that high-dose ionizing radiation induces eCIRP release in the peritoneal cavity of irradiated mice and in the cell culture supernatant of irradiated mouse peritoneal cells (49). Moreover, the radiation-induced eCIRP release upregulated the expression of TREM-1 on macrophages, and mice knocked out for CIRP or TREM-1 had improved 30-day survival rates after 6.5 Gy TBI (49). Therefore, targeting the eCIRP-TREM-1 axis holds promise as a therapeutic strategy for post-radiation injury, potentially attenuating lethal infections and sepsis (49).
HMGB1 is a nuclear protein that acts as a DNA chaperone (63). As a DAMP, HMGB1 binds to pattern recognition receptors, including RAGE, TLR2, TLR4, TLR9, and TREM-1, to induce inflammatory responses (64, 65). In a murine model of sepsis, HMGB1, released from macrophages, has been shown to function as a late lethal mediator (66). In acute lung injury and hepatic ischemia-reperfusion injury models, administration of anti-HMGB1 antibody reduces lung neutrophil influx and pulmonary edema and mitigates liver damage (67, 68). Exposure to ionizing radiation increases in both cellular mRNA expression and serum protein levels of HMGB1 in a dose-dependent manner in experimental mice model (69). Glycyrrhizin, one of the inhibitors of HMGB1, reduced the release of serum inflammatory cytokines in abdominal irradiated mice (6.5 Gy) through the downregulation of the HMGB1/TLR4 signaling pathway and significantly attenuated intestinal injury (50). In thoracic irradiated mice, glycyrrhizin also reduced inflammatory cytokines in the bronchoalveolar lavage fluid and mitigated radiation-induced lung injury by inhibiting downstream transcription factors related to the HMGB1/TLR4 axis (51).
mtDNA is susceptible to ionizing radiation compared to nuclear DNA due to the lack of protective histones and limited repair mechanism (29). Mitochondrial dysfunction resulting from failure of DNA repair leads to the release of mitochondrial components including cytochrome c, ATP, mtDNA, and mtRNA (29). The mtDNA in the cytosol is recognized by the cyclic GMP-AMP synthase (cGAS) to activate the stimulator of interferon genes (STING) signaling pathway, resulting in the production of type I interferons (IFNs) (65, 70). Excessive production of type I IFNs can cause exhaustion in hematopoietic stem cells (71, 72). In the 6 Gy TBI model, mtDNA released into the cytosol has been reported to activate the cGAS-STING pathway and induced type I IFN production, and inhibition of the mtDNA release ameliorated hematopoietic tissue injury (52). The mtDNA released into the cytoplasm can also bind to and activate the NLRP3 inflammasome, which leads to the release of inflammatory cytokines such as IL-1β and IL-18 followed by pyroptosis (31). Previous studies have shown that pyroptosis in intestinal cells occurred through NLRP3 activation, inducing intestinal injury in the abdominal irradiation model (73, 74). These studies suggest that mtDNA exacerbates ARS pathophysiology and could be a potential therapeutic target for GI-ARS by mitigating pyroptosis and inflammation.
exRNA from host cells is carried along microvesicles and exosomes which protect exRNA from enzyme degradation (75). exRNA acts as a pivotal signaling molecule to mediate communication between cells, but can also serve as DAMPs (75, 76). TLR3 has been known to recognize not only double strand RNA released from virus-infected cells but also endogenous exRNA released from necrotic cells, promoting inflammation and organ dysfunction in autoimmune diseases and sepsis (77–79). It has been shown that large exRNA released from the host small intestine after ionizing radiation exposure induced extensive crypt cell death via TLR3 and inhibition of exRNA binding to TLR3 improved survival in 10 Gy TBI by preventing GI-ARS (53). In the mouse model after 9 Gy TBI, the release of microRNAs (miRNAs) via exosomes worsened intestinal injury by promoting apoptosis and DNA damage of intestinal cells (54). These findings implicate a potential detrimental role for exRNA in ARS.
In a radiological event, mass screenings are necessary to triage exposed and non-exposed individuals and to determine the severity of the radiation dose to the exposed population (55). Established biomarkers for estimating radiation dose in exposed individuals are quantification of chromosomal aberrations and γ-H2AX, a marker of DNA double-strand breaks, but these tests are only available in laboratories with trained personnel. Serum amyloid A (SAA) is one of the proteins associated with acute phase reactions resulting from inflammation, infection, trauma, or other events (80). SAA has also been reported as a radiation-responsive biomarker in TBI and partial-body irradiation (PBI) models (55–57, 81). In the mice, after 2-8 Gy TBI, SAA is significantly increased at 24 hours after irradiation compared to controls (55, 56). In the mouse PBI model of GI-ARS, SAA levels significantly increase 3 and 4 days after irradiation with 12 Gy and 14 Gy, respectively (57). Although the serum concentration of SAA increases within a few hours after irradiation and returns to the baseline within 7 days after irradiation, combinations of SAA with other biomarkers provide greater accuracy for the assessment of radiation injury especially, at the acute phase (56, 81).
The pathways of DAMP release can be categorized into two primary pathways: i) passive release and ii) active release (38). Passive release is primarily linked to cell death processes, including apoptosis, necrosis/necroptosis, pyroptosis, and ferroptosis. Concurrently, DAMPs are actively released through secretion mechanisms such as exocytosis in the forms of lysosomes and exosomes (38). Below, we have outlined the universal approaches to DAMP release. Given that radiation injury is directly associated with these cell death events or initiates pathways relevant to the active release of DAMPs, it is reasonable to infer that these processes are linked to the mechanisms governing DAMPs release in ARS (Figure 1).
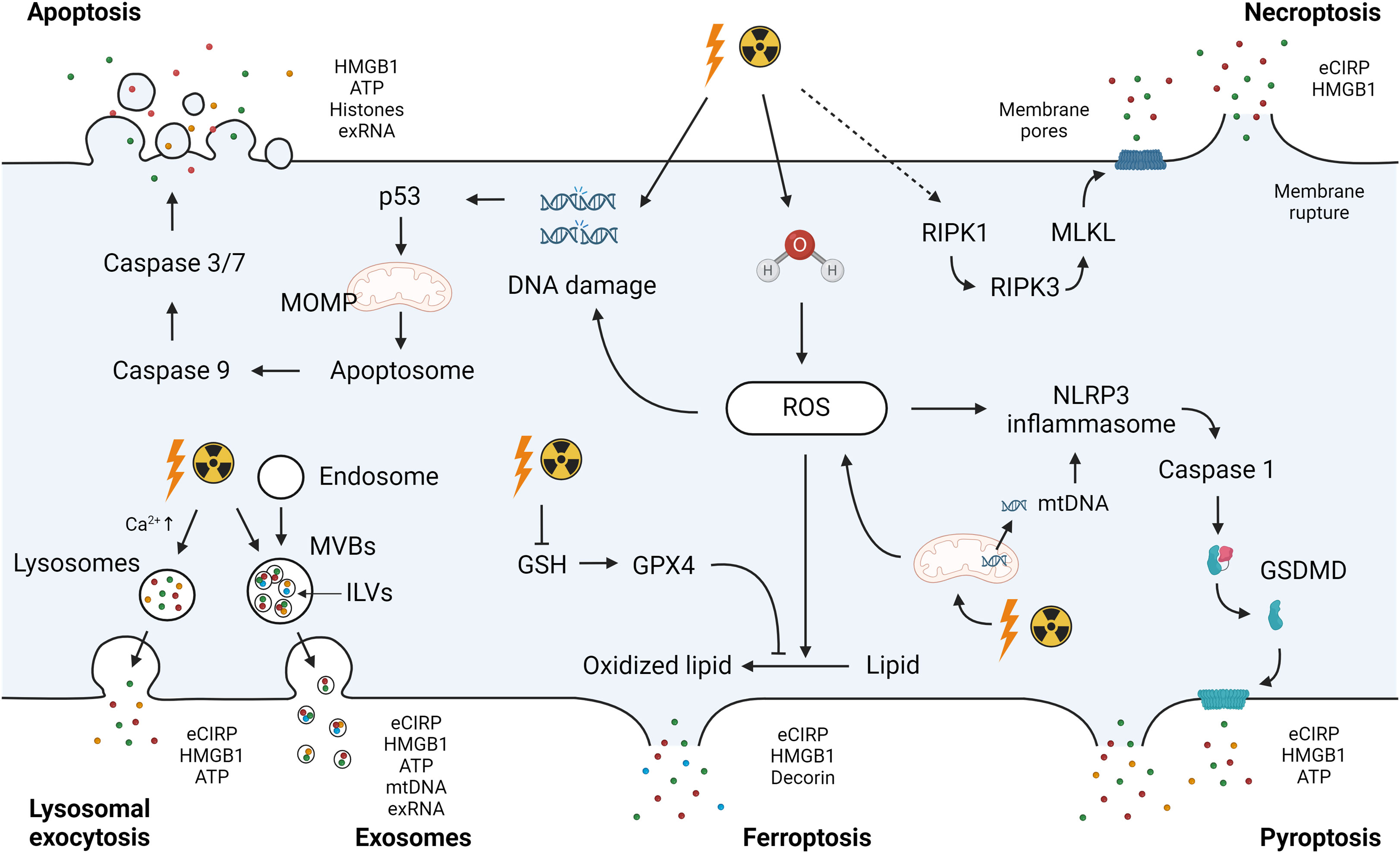
Figure 1 Radiation-induced DAMPs release. Exposure to ionizing radiation causes direct DNA damage or ROS-induced DNA damage to activate the intrinsic pathway of apoptosis. Mitochondrial damage by ionizing radiation amplifies ROS production and contributes to DNA damage. ROS accumulation after radiation exposure due to GPX4 impairment and GSH depletion induces the oxidation of polyunsaturated fatty acids in membrane phospholipids, leading to ferroptosis. Radiation-induced ROS leads to activation of the NLRP3 inflammasome. The NLRP3 inflammasome complex activates caspase-1 to cleave GSDMD and form a membrane pore with the GSDMD-N domain, inducing pyroptosis. Ionizing radiation causes the activation of RIPK1, which recruits RIPK3 to phosphorylate MLKL. Phosphorylated MLKL forms MLKL oligomers, leading to necroptosis. DAMPs including eCIRP, HMGB1, ATP, histones, exRNA, and mtDNA can be released as a passive release mechanism after these cell deaths. Mitochondrial dysfunction results in the release of mitochondrial components including mtDNA into the cytosol. Secretion mechanisms such as exocytosis in the forms of lysosomes and exosomes cause active release of DAMPs. DAMPs, damage-associated molecular patterns; ROS, reactive oxygen species; GPX4, glutathione peroxidase 4; GSH, glutathione; NLRP3, NLR family pyrin domain containing 3; GSDMD, Gasdermin D; RIPK, receptor-interacting serine/threonine-protein kinase; MLKL, mixed lineage kinase domain like pseudokinase; eCIRP, extracellular cold-inducible RNA-binding protein; HMGB1, high mobility group box 1; exRNA, extracellular RNA; mtDNA, mitochondrial DNA; MOMP, mitochondria outer membrane permeabilization; MVBs, multivesicular bodies; ILVs, intraluminal vesicles.
Apoptosis is a form of programmed cell death, initiated by two distinct p53-dependent signaling pathways: the extrinsic and intrinsic apoptosis (82). In the extrinsic receptor-mediated pathway, extracellular death receptors are activated by ligand binding and death-inducible signaling complex (DISC) is formed at the intracellular tail of the receptor to activate pro-caspase-8, or pro-caspase-10, with adapter proteins such as Fas-associated via death domain. The extrinsic apoptosis is also induced by dependence receptors, which mediate lethal signals in the absence of their ligand, and pro-caspase-9 is activated (83, 84). Activated caspase-8 (or caspase-10) and caspase-9 eventually activate executioner caspase-3 and caspase-7 to induce apoptosis (83). On the other hand, severe DNA damage induced by radiation mainly activates the intrinsic pathway (82). A transcription factor p53 induces pro-apoptosis Bcl family proteins including BAX, NOXA and PUMA (85, 86). These proteins mediate MOMP, resulting in the release of mitochondrial proteins into the cytosol (82). Cytochrome c binds to apoptotic peptidase-activating factor 1 and pro-caspase-9 to form apoptosomes that ultimately activate caspase-3 and caspase-7 via the activation of caspase-9 (12, 82). During apoptosis, DAMPs such as HMGB1, ATP, histones, and exRNA are released into the extracellular space (87–90).
Necrosis is an accidental cell death caused by external stimuli such as physical and chemical stress and heat (91). In necrotic cell death, the increased cell expansion disrupts the plasma membrane. As a result, cytoplasmic contents and pro-inflammatory molecules are released into the extracellular space (92). DAMPs, including HMGB1, HSP, and ATP, are shown to be released from necrotic cells (91, 93, 94). Necroptosis is a form of programmed cell death dependent on the activation of receptor-interacting serine/threonine-protein kinase 1 (RIPK1). RIPK1 activation can be caused by the stimulation of tumor necrosis factor (TNF) receptor and other death receptors such as Fas. Activated RIPK1 recruits RIPK3 and this complex phosphorylates mixed lineage kinase domain-like pseudokinase (MLKL) (95). Phosphorylated MLKL forms MLKL oligomers and translocates to the plasma membrane, resulting in inducing membrane permeabilization and cell disruption (95). The inhibition of necroptosis has been shown to decrease HMGB1 release in an LPS-induced intestinal injury model, demonstrating the contribution of necroptosis to the release of HMGB1 (96). eCIRP has also been demonstrated to be released by necroptosis in a sepsis model (97).
Pyroptosis is a lytic cell death process that allows the release of potential immunostimulatory molecules. This programmed cell death can be induced by the canonical and non-canonical pathways (98). In the canonical pathway, pathogen-derived molecules are recognized by inflammasomes. For example, cell stressors including bacteria, viruses, and DAMPs induce potassium efflux, leading to NIMA-related kinase 7 binding to NLR family pyrin domain containing 3 (NLRP3). Subsequently, NLRP3 oligomerization is induced to activate pro-caspase 1 through the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD) (99). Caspase-1 processes and activates pro-IL-1β and pro-IL-18 and cleaves Gasdermin D (GSDMD) to form a membrane pore with the GSDMD-N domain. In the non-canonical pathway, endotoxin from Gram-negative bacteria directly activates pro-caspase-4 or pro-caspase-5 in humans (or pro-caspase-11 in mice), which also cleave GSDMD. Small DAMPs such as eCIRP (17-19 kDa) are then released through GSDMD’s pores along with IL-1β and IL-18 (100, 101). The release of larger DAMPs, such as HMGB1, can ensue when irreversible GSDMD pores cause terminal cell rupture (102, 103).
Ferroptosis is a form of programmed cell death characterized by the iron-dependent accumulation of lipid peroxidation (104). At the steady state, intracellular glutathione peroxidase 4 (GPX4) utilizes glutathione (GSH) to reduce lipid peroxidation generated by ROS. However, ROS accumulation due to the impairment of GPX4 and depletion of GSH causes the oxidation of polyunsaturated fatty acids in membrane phospholipids through an intracellular iron-dependent mechanism, leading to ferroptosis (83, 104). DAMPs released by ferroptosis include eCIRP, HMGB1, and decorin, a small leucine-rich proteoglycan of the extracellular matrix (105–107). Decorin, similar to HMGB1, binds to TLR4 and RAGE to induce pro-inflammatory signaling (107, 108).
Exosomes are nano-sized membrane vesicles released upon fusion of multivesicular bodies (MVBs) with the plasma membrane and contain a variety of proteins, lipids, and nucleic acids (109). The endosomal membranes in early endosomes invaginate to form intraluminal vesicles (ILVs) in the lumen of the vesicular organelles (110). ILVs mature into MVBs by the endosome sorting complex required for transporting (ESCRT)-dependent or ESCRT-independent mechanism. MVBs are then transported to the plasma membrane dependent on their interaction with actin and the microtubule cytoskeleton to be released into the extracellular environment as exosomes (110). Exosomal DAMPs include HMGB1, HSPs, ATP, histones, mtDNA, and eCIRP (111–116). Increased DAMPs in exosomes induce inflammation and exacerbate organ dysfunction in a plethora of disorders including sepsis (117).
Lysosomal exocytosis is another mechanism of active release of DAMPs. After stimulation of a cell receptor, an increase in intracellular Ca2+ is detected by synaptotagmin, which is an intracellular calcium sensor on lysosomes. The lysosomes are mobilized to the microtubule organizing center to be associated with the kinesin and transported to the site of secretion (118). The lysosomes dock and fuse with the plasma membrane through soluble N-ethylmaleimide-sensitive factor attachment proteins receptor (SNARE) complex Rab proteins, leading to secretion of the soluble components (118, 119). HMGB1, ATP, and eCIRP have been shown to be released by lysosomal exocytosis (58, 120, 121).
Sepsis from enteric microorganisms in GI-ARS is the major cause of morbidity and mortality in radiation injury (19). The breakdown of the mucosal barrier due to disruption of the intestinal epithelium leads to severe secretory diarrhea and translocation of bacteria into the systemic circulation, resulting in severe sepsis (20). Furthermore, damage to the intestinal mucosa and sepsis of enteric origin occur even after TBI in the H-ARS dose range (122). DAMPs generated by the disruption of the intestinal mucosal barrier in sepsis are transported through the mesenteric lymphatics into the systemic circulation, developing multiple organ failure such as acute lung injury and further deteriorating intestinal barrier injury (123). In RCI with burn and trauma, the pathophysiological role of DAMPs remains unexplored. In severe burns, DAMPs are released from the injured tissue, inducing systemic inflammatory response (124). Immunological exhaustion due to the excessive inflammatory response leads to inhibition of the innate and acquired immune systems, resulting in increased susceptibility to bacterial infections not only from the skin but also from the lung or gut microbiota to cause severe sepsis (124, 125). Furthermore, DAMPs release in trauma is associated with the development of organ injuries such as acute lung injury and acute renal failure (126, 127). DAMPs are also involved in the activation of coagulation pathways, leading to coagulopathy during trauma (124). Additionally, DAMPs have been suggested to induce post-traumatic immunosuppression (42). In the context of RCI, DAMPs may play a critically detrimental role in multiple organ failure and compromised immune systems, thereby exacerbating the prognosis of the patients.
Medical countermeasures for ARS are currently limited and only available for H-ARS (128). We described DAMPs as exacerbating factors and potential therapeutic targets for ARS. Pharmacological strategies targeting DAMPs to inhibit immune responses include the uses of monoclonal antibodies, peptides, decoy receptors, and small molecules (129). CIRP-derived small peptides that inhibit eCIRP binding to the TLR4-MD2 complex and binding to TREM-1 have been developed and shown to attenuate sepsis, acute kidney injury, and hepatic ischemia-reperfusion injury (61, 130–132). A molecule that directly neutralizes eCIRP has also been identified to improve acute lung injury and survival in sepsis (133). Additionally, we have recently described an engineered oligopeptide that effectively promotes eCIRP clearance from the circulation leading to markedly improved outcomes in sepsis (134). Anti-HMGB1 antibodies have been to be effective in various experimental models such as abdominal sepsis and hemorrhagic shock (63, 135, 136). Small molecules that bind to and block HMGB1, such as glycyrrhizin, and decoy receptors that block HMGB1-RAGE signaling, such as soluble RAGE, also improved several inflammatory disease models (137, 138). exRNAs include non-coding RNA (ncRNA) such as miRNAs, long non-coding RNAs, and circular RNAs as well as messenger RNAs. Recently, miRNAs have been identified as potential biomarkers for various diseases, including radiation injury (139). In experimental radiation models, serum miRNA profiling has been shown to be able to identify exposure to radiation and predict the extent of ARS and the probability of survival, potentially facilitating timely intervention after radiation exposure (140, 141). The high stability and reproducibility of serum miRNAs make them attractive candidates as biomarkers of radiation injury (141). The function of specific ncRNAs as DAMPs in radiation injury remains unclear and may be the subject for future research. In the context of RCI resulting from nuclear explosions, more than half of the patients are accompanied by burns and trauma, resulting in more complicated and severe radiation injury (24). FDA-approved countermeasures for H-ARS alone have failed to improve survival in RCI (24). DAMPs released in burn and trauma injury have potential links with the mechanical injury, ischemia/reperfusion injury, metabolic acidosis, and hypoxia, culminating in multiple organ failure (124). These findings underscore the potential of DAMPs-targeting strategies in addressing the challenges posed by RCI.
In conclusion, exposure to ionizing radiation causes direct DNA damage and oxidative stress, leading to cell death. The subsequent release of DAMPs has the potential to worsen ARS across various body compartments. However, there is a scarcity of studies specifically addressing DAMPs in the context of radiation injury. A more profound comprehension of the role of DAMPs in radiation injury could pave the way for the development of innovative therapeutic strategies to mitigate the impact of nuclear and radiological threats and accidents.
SY: Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. MA: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. AM: Investigation, Methodology, Writing – original draft, Writing – review & editing. MB: Formal Analysis, Investigation, Supervision, Visualization, Writing – review & editing, Funding acquisition. PW: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was partially supported by the National Institutes of Health (NIH) grants U01AI170018, U01AI133655, and R35GM118337.
Figure 1 was created using BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ARS, acute radiation syndrome; ASC, apoptosis-associated speck-like protein containing a CARD; cGAS, cyclic GMP-AMP synthase; DAMPs, damage-associated molecular patterns; exRNA, extracellular RNA; GI-ARS, gastrointestinal-ARS; GPX4, glutathione peroxidase 4; GSDMD, Gasdermin D; GSH, glutathione; HSPs, heat shock proteins; H-ARS, hematopoietic-ARS; HMGB1, high mobility group box 1; IFNs, interferons; MOMP, mitochondria outer membrane permeabilization; mtDNA, mitochondrial DNA; MLKL, mixed lineage kinase domain like pseudokinase; MVBs, multivesicular bodies; NLRP3, NLR family pyrin domain containing 3; NLRs, NOD-like receptors; NF-κB, nuclear factor kappa B; NV-ARS, neurovascular-ARS; PAMPs, pathogen-associated molecular patterns; RCI, radiation-combined injury; ROS, reactive oxygen species; RAGE, receptor for advanced glycation end products; RIPK, receptor-interacting serine/threonine-protein kinase; SAA, serum amyloid A; STING, stimulator of interferon genes; TLRs, Toll-like receptors; TBI, total body irradiation; TREM-1, triggering receptor expressed on myeloid cells-1; TNF, tumor necrosis factor.
1. Christodouleas JP, Forrest RD, Ainsley CG, Tochner Z, Hahn SM, Glatstein E. Short-term and long-term health risks of nuclear-power-plant accidents. N Engl J Med (2011) 364(24):2334–41. doi: 10.1056/NEJMra1103676
2. Hasegawa A, Tanigawa K, Ohtsuru A, Yabe H, Maeda M, Shigemura J, et al. Health effects of radiation and other health problems in the aftermath of nuclear accidents, with an emphasis on Fukushima. Lancet (2015) 386(9992):479–88. doi: 10.1016/S0140-6736(15)61106-0
3. Obrador E, Salvador-Palmer R, Villaescusa JI, Gallego E, Pellicer B, Estrela JM, et al. Nuclear and radiological emergencies: biological effects, countermeasures and biodosimetry. Antioxid (Basel) (2022) 11(6):1098. doi: 10.3390/antiox11061098
4. Hirohashi N, Shime N, Fujii T. Beyond the unthinkable: are we prepared for rare disasters? Anaesth Crit Care Pain Med (2023) 42(4):101266. doi: 10.1016/j.accpm.2023.101266
5. Dainiak N. Medical management of acute radiation syndrome and associated infections in a high-casualty incident. J Radiat Res (2018) 59(suppl_2):ii54–64. doi: 10.1093/jrr/rry004
6. Dorr H, Meineke V. Acute radiation syndrome caused by accidental radiation exposure - therapeutic principles. BMC Med (2011) 9:126. doi: 10.1186/1741-7015-9-126
7. Singh VK, Newman VL, Romaine PL, Wise SY, Seed TM. Radiation countermeasure agents: an update (2011-2014). Expert Opin Ther Pat (2014) 24(11):1229–55. doi: 10.1517/13543776.2014.964684
8. Aliper AM, Bozdaganyan ME, Sarkisova VA, Veviorsky AP, Ozerov IV, Orekhov PS, et al. Radioprotectors.Org: an open database of known and predicted radioprotectors. Aging (Albany NY) (2020) 12(15):15741–55. doi: 10.18632/aging.103815
9. U.S. Food and Drug Administration. Radiological and Nuclear Emergency Preparedness Information from FDA (2023). Available at: https://www.fda.gov/emergency-preparedness-and-response/mcm-issues/radiological-and-nuclear-emergency-preparedness-information-fda (Accessed November 22, 2023).
10. Singh VK, Seed TM, Olabisi AO. Drug discovery strategies for acute radiation syndrome. Expert Opin Drug Discovery (2019) 14(7):701–15. doi: 10.1080/17460441.2019.1604674
11. Liu L, Liang Z, Ma S, Li L, Liu X. Radioprotective countermeasures for radiation injury (Review). Mol Med Rep (2023) 27(3):66. doi: 10.3892/mmr.2023.12953
12. Rodriguez-Ruiz ME, Vitale I, Harrington KJ, Melero I, Galluzzi L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat Immunol (2020) 21(2):120–34. doi: 10.1038/s41590-019-0561-4
13. Zhou M, Aziz M, Wang P. Damage-associated molecular patterns as double-edged swords in sepsis. Antioxid Redox Signal (2021) 35(15):1308–23. doi: 10.1089/ars.2021.0008
14. Hu Q, Ren H, Ren J, Liu Q, Wu J, Wu X, et al. Released mitochondrial DNA following intestinal ischemia reperfusion induces the inflammatory response and gut barrier dysfunction. Sci Rep (2018) 8(1):7350. doi: 10.1038/s41598-018-25387-8
15. Shao L, Luo Y, Zhou D. Hematopoietic stem cell injury induced by ionizing radiation. Antioxid Redox Signal (2014) 20(9):1447–62. doi: 10.1089/ars.2013.5635
16. Heylmann D, Rodel F, Kindler T, Kaina B. Radiation sensitivity of human and murine peripheral blood lymphocytes, stem and progenitor cells. Biochim Biophys Acta (2014) 1846(1):121–9. doi: 10.1016/j.bbcan.2014.04.009
17. Dainiak N, Waselenko JK, Armitage JO, MacVittie TJ, Farese AM. The hematologist and radiation casualties. Hematol Am Soc Hematol Educ Program (2003), 473–96. doi: 10.1182/asheducation-2003.1.473
18. Dainiak N. Hematologic consequences of exposure to ionizing radiation. Exp Hematol (2002) 30(6):513–28. doi: 10.1016/s0301-472x(02)00802-0
19. Dainiak N, Gent RN, Carr Z, Schneider R, Bader J, Buglova E, et al. Literature review and global consensus on management of acute radiation syndrome affecting nonhematopoietic organ systems. Disaster Med Public Health Prep (2011) 5(3):183–201. doi: 10.1001/dmp.2011.73
20. Moussa L, Usunier B, Demarquay C, Benderitter M, Tamarat R, Semont A, et al. Bowel radiation injury: complexity of the pathophysiology and promises of cell and tissue engineering. Cell Transplant (2016) 25(10):1723–46. doi: 10.3727/096368916X691664
21. Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, et al. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature (2019) 569(7754):121–5. doi: 10.1038/s41586-019-1154-y
22. Kiang JG, Olabisi AO. Radiation: A poly-traumatic hit leading to multi-organ injury. Cell Biosci (2019) 9:25. doi: 10.1186/s13578-019-0286-y
23. Waselenko JK, MacVittie TJ, Blakely WF, Pesik N, Wiley AL, Dickerson WE, et al. Medical management of the acute radiation syndrome: recommendations of the strategic national stockpile radiation working group. Ann Intern Med (2004) 140(12):1037–51. doi: 10.7326/0003-4819-140-12-200406150-00015
24. Kiang JG, Blakely WF. Combined radiation injury and its impacts on radiation countermeasures and biodosimetry. Int J Radiat Biol (2023) 99(7):1055–65. doi: 10.1080/09553002.2023.2188933
25. Jacob A, Shah KG, Wu R, Wang P. Ghrelin as a novel therapy for radiation combined injury. Mol Med (2010) 16(3-4):137–43. doi: 10.2119/molmed.2009.00154
26. Nikitaki Z, Hellweg CE, Georgakilas AG, Ravanat JL. Stress-induced DNA damage biomarkers: applications and limitations. Front Chem (2015) 3:35. doi: 10.3389/fchem.2015.00035
27. Adjemian S, Oltean T, Martens S, Wiernicki B, Goossens V, Vanden Berghe T, et al. Ionizing radiation results in a mixture of cellular outcomes including mitotic catastrophe, senescence, methuosis, and iron-dependent cell death. Cell Death Dis (2020) 11(11):1003. doi: 10.1038/s41419-020-03209-y
28. Huang X, Tran T, Zhang L, Hatcher R, Zhang P. DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proc Natl Acad Sci U.S.A. (2005) 102(4):1065–70. doi: 10.1073/pnas.0409130102
29. Shimura T. Mitochondrial signaling pathways associated with DNA damage responses. Int J Mol Sci (2023) 24(7):6128. doi: 10.3390/ijms24076128
30. Averbeck D, Rodriguez-Lafrasse C. Role of mitochondria in radiation responses: epigenetic, metabolic, and signaling impacts. Int J Mol Sci (2021) 22(20):11047. doi: 10.3390/ijms222011047
31. Cheng H, Chen L, Huang M, Hou J, Chen Z, Yang X. Hunting down Nlrp3 inflammasome: an executioner of radiation-induced injury. Front Immunol (2022) 13:967989. doi: 10.3389/fimmu.2022.967989
32. Liu YG, Chen JK, Zhang ZT, Ma XJ, Chen YC, Du XM, et al. Nlrp3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis (2017) 8(2):e2579. doi: 10.1038/cddis.2016.460
33. Huang Z, Epperly M, Watkins SC, Greenberger JS, Kagan VE, Bayir H. Necrostatin-1 rescues mice from lethal irradiation. Biochim Biophys Acta (2016) 1862(4):850–6. doi: 10.1016/j.bbadis.2016.01.014
34. Zhang X, Tian M, Li X, Zheng C, Wang A, Feng J, et al. Hematopoietic protection and mechanisms of ferrostatin-1 on hematopoietic acute radiation syndrome of mice. Int J Radiat Biol (2021) 97(4):464–73. doi: 10.1080/09553002.2021.1876956
35. Li X, Zhuang X, Qiao T. Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem Biophys Res Commun (2019) 519(2):240–5. doi: 10.1016/j.bbrc.2019.08.165
36. Wang L, Wang A, Fu Q, Shi Z, Chen X, Wang Y, et al. Ferroptosis plays an important role in promoting ionizing radiation-induced intestinal injuries. Biochem Biophys Res Commun (2022) 595:7–13. doi: 10.1016/j.bbrc.2022.01.068
37. Gong T, Liu L, Jiang W, Zhou R. Damp-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20(2):95–112. doi: 10.1038/s41577-019-0215-7
38. Murao A, Aziz M, Wang H, Brenner M, Wang P. Release mechanisms of major damps. Apoptosis (2021) 26(3-4):152–62. doi: 10.1007/s10495-021-01663-3
39. Denning NL, Aziz M, Gurien SD, Wang P. Damps and nets in sepsis. Front Immunol (2019) 10:2536. doi: 10.3389/fimmu.2019.02536
40. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10(12):826–37. doi: 10.1038/nri2873
41. Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol (2016) 16(1):35–50. doi: 10.1038/nri.2015.8
42. Vourc’h M, Roquilly A, Asehnoune K. Trauma-induced damage-associated molecular patterns-mediated remote organ injury and immunosuppression in the acutely ill patient. Front Immunol (2018) 9:1330. doi: 10.3389/fimmu.2018.01330
43. Zhang X, Wu J, Liu Q, Li X, Li S, Chen J, et al. Mtdna-sting pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis (2020) 11(12):1050. doi: 10.1038/s41419-020-03239-6
44. Paudel YN, Angelopoulou E, Piperi C, Othman I, Shaikh MF. Hmgb1-mediated neuroinflammatory responses in brain injuries: potential mechanisms and therapeutic opportunities. Int J Mol Sci (2020) 21(13):4609. doi: 10.3390/ijms21134609
45. Ratikan JA, Micewicz ED, Xie MW, Schaue D. Radiation takes its toll. Cancer Lett (2015) 368(2):238–45. doi: 10.1016/j.canlet.2015.03.031
46. Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol (2018) 19(4):327–41. doi: 10.1038/s41590-018-0064-8
47. Jost PJ, Hockendorf U. Necroinflammation emerges as a key regulator of hematopoiesis in health and disease. Cell Death Differ (2019) 26(1):53–67. doi: 10.1038/s41418-018-0194-4
48. Balanca B, Desmurs L, Grelier J, Perret-Liaudet A, Lukaszewicz AC. Damps and rage pathophysiology at the acute phase of brain injury: an overview. Int J Mol Sci (2021) 22(5):2439. doi: 10.3390/ijms22052439
49. Yamaga S, Murao A, Ma G, Brenner M, Aziz M, Wang P. Radiation upregulates macrophage trem-1 expression to exacerbate injury in mice. Front Immunol (2023) 14:1151250. doi: 10.3389/fimmu.2023.1151250
50. Zhang XM, Hu X, Ou JY, Chen SS, Nie LH, Gao L, et al. Glycyrrhizin ameliorates radiation enteritis in mice accompanied by the regulation of the Hmgb1/Tlr4 pathway. Evid Based Complement Alternat Med (2020) 2020:8653783. doi: 10.1155/2020/8653783
51. Zheng L, Zhu Q, Xu C, Li M, Li H, Yi PQ, et al. Glycyrrhizin mitigates radiation-induced acute lung injury by inhibiting the Hmgb1/Tlr4 signalling pathway. J Cell Mol Med (2020) 24(1):214–26. doi: 10.1111/jcmm.14703
52. Guan H, Zhang W, Xie D, Nie Y, Chen S, Sun X, et al. Cytosolic release of mitochondrial DNA and associated Cgas signaling mediates radiation-induced hematopoietic injury of mice. Int J Mol Sci (2023) 24(4):4020. doi: 10.3390/ijms24044020
53. Takemura N, Kawasaki T, Kunisawa J, Sato S, Lamichhane A, Kobiyama K, et al. Blockade of Tlr3 protects mice from lethal radiation-induced gastrointestinal syndrome. Nat Commun (2014) 5:3492. doi: 10.1038/ncomms4492
54. Li H, Jiang M, Zhao SY, Zhang SQ, Lu L, He X, et al. Exosomes are involved in total body irradiation-induced intestinal injury in mice. Acta Pharmacol Sin (2021) 42(7):1111–23. doi: 10.1038/s41401-021-00615-6
55. Sproull M, Kramp T, Tandle A, Shankavaram U, Camphausen K. Serum amyloid a as a biomarker for radiation exposure. Radiat Res (2015) 184(1):14–23. doi: 10.1667/RR13927.1
56. Sproull M, Kramp T, Tandle A, Shankavaram U, Camphausen K. Multivariate analysis of radiation responsive proteins to predict radiation exposure in total-body irradiation and partial-body irradiation models. Radiat Res (2017) 187(2):251–8. doi: 10.1667/RR14558.1
57. Kumar VP, Wuddie K, Tsioplaya A, Weaver A, Holmes-Hampton GP, Ghosh SP. Development of a multi-organ radiation injury model with precise dosimetry with focus on Gi-Ars. Radiat Res (2023) 201(1):19–34. doi: 10.1667/RADE-23-00068.1
58. Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W, et al. Cold-inducible Rna-binding protein (Cirp) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med (2013) 19(11):1489–95. doi: 10.1038/nm.3368
59. Aziz M, Brenner M, Wang P. Extracellular Cirp (Ecirp) and inflammation. J Leukoc Biol (2019) 106(1):133–46. doi: 10.1002/JLB.3MIR1118-443R
60. Ode Y, Aziz M, Wang P. Cirp increases Icam-1(+) phenotype of neutrophils exhibiting elevated inos and nets in sepsis. J Leukoc Biol (2018) 103(4):693–707. doi: 10.1002/JLB.3A0817-327RR
61. Denning NL, Aziz M, Murao A, Gurien SD, Ochani M, Prince JM, et al. Extracellular Cirp as an endogenous trem-1 ligand to fuel inflammation in sepsis. JCI Insight (2020) 5(5):e134172. doi: 10.1172/jci.insight.134172
62. Zhou M, Aziz M, Yen HT, Ma G, Murao A, Wang P. Extracellular Cirp dysregulates macrophage bacterial phagocytosis in sepsis. Cell Mol Immunol (2023) 20(1):80–93. doi: 10.1038/s41423-022-00961-3
63. Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. Hmgb1 as biomarker and drug target. Pharmacol Res (2016) 111:534–44. doi: 10.1016/j.phrs.2016.06.031
64. Yang H, Wang H, Andersson U. Targeting inflammation driven by Hmgb1. Front Immunol (2020) 11:484. doi: 10.3389/fimmu.2020.00484
65. Nofi CP, Wang P, Aziz M. Chromatin-associated molecular patterns (Camps) in sepsis. Cell Death Dis (2022) 13(8):700. doi: 10.1038/s41419-022-05155-3
66. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. Hmg-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285(5425):248–51. doi: 10.1126/science.285.5425.248
67. Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. Hmg-1 as a mediator of acute lung inflammation. J Immunol (2000) 165(6):2950–4. doi: 10.4049/jimmunol.165.6.2950
68. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor Hmgb1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med (2005) 201(7):1135–43. doi: 10.1084/jem.20042614
69. Kim HC, Oh H, You JS, Chung YE. Glycyrrhizin ameliorating sterile inflammation induced by low-dose radiation exposure. Sci Rep (2021) 11(1):18356. doi: 10.1038/s41598-021-97800-8
70. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol (2017) 18(5):488–98. doi: 10.1038/ni.3704
71. Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med (2009) 15(6):696–700. doi: 10.1038/nm.1973
72. Xia P, Wang S, Ye B, Du Y, Li C, Xiong Z, et al. A circular Rna protects dormant hematopoietic stem cells from DNA sensor Cgas-mediated exhaustion. Immunity (2018) 48(4):688–701 e7. doi: 10.1016/j.immuni.2018.03.016
73. Wu D, Han R, Deng S, Liu T, Zhang T, Xie H, et al. Protective effects of flagellin a N/C against radiation-induced Nlr pyrin domain containing 3 inflammasome-dependent pyroptosis in intestinal cells. Int J Radiat Oncol Biol Phys (2018) 101(1):107–17. doi: 10.1016/j.ijrobp.2018.01.035
74. Hu L, Chen H, Zhang X, Feng Z, Zhang H, Meng Q. Rosiglitazone ameliorates radiation-induced intestinal inflammation in rats by inhibiting Nlrp3 inflammasome and Tnf-alpha production. J Radiat Res (2020) 61(6):842–50. doi: 10.1093/jrr/rraa062
75. Wu D, Tao T, Eshraghian EA, Lin P, Li Z, Zhu X. Extracellular Rna as a kind of communication molecule and emerging cancer biomarker. Front Oncol (2022) 12:960072. doi: 10.3389/fonc.2022.960072
76. Preissner KT, Fischer S, Deindl E. Extracellular Rna as a versatile damp and alarm signal that influences leukocyte recruitment in inflammation and infection. Front Cell Dev Biol (2020) 8:619221. doi: 10.3389/fcell.2020.619221
77. Chattopadhyay S, Sen GC. Dsrna-activation of Tlr3 and Rlr signaling: gene induction-dependent and independent effects. J Interferon Cytokine Res (2014) 34(6):427–36. doi: 10.1089/jir.2014.0034
78. Brentano F, Schorr O, Gay RE, Gay S, Kyburz D. Rna released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via toll-like receptor 3. Arthritis Rheum (2005) 52(9):2656–65. doi: 10.1002/art.21273
79. Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, et al. Tlr3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med (2008) 205(11):2609–21. doi: 10.1084/jem.20081370
81. Ossetrova NI, Sandgren DJ, Blakely WF. Protein biomarkers for enhancement of radiation dose and injury assessment in nonhuman primate total-body irradiation model. Radiat Prot Dosimetry (2014) 159(1-4):61–76. doi: 10.1093/rpd/ncu165
82. Speidel D. Transcription-independent P53 apoptosis: an alternative route to death. Trends Cell Biol (2010) 20(1):14–24. doi: 10.1016/j.tcb.2009.10.002
83. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
84. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ (2012) 19(1):107–20. doi: 10.1038/cdd.2011.96
85. Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a Bh3-only member of the Bcl-2 family and candidate mediator of P53-induced apoptosis. Science (2000) 288(5468):1053–8. doi: 10.1126/science.288.5468.1053
86. Nakano K, Vousden KH. Puma, a novel proapoptotic gene, is induced by P53. Mol Cell (2001) 7(3):683–94. doi: 10.1016/s1097-2765(01)00214-3
87. Jiang W, Bell CW, Pisetsky DS. The relationship between apoptosis and high-mobility group protein 1 release from murine macrophages stimulated with lipopolysaccharide or polyinosinic-polycytidylic acid. J Immunol (2007) 178(10):6495–503. doi: 10.4049/jimmunol.178.10.6495
88. Wu D, Ingram A, Lahti JH, Mazza B, Grenet J, Kapoor A, et al. Apoptotic release of histones from nucleosomes. J Biol Chem (2002) 277(14):12001–8. doi: 10.1074/jbc.M109219200
89. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature (2009) 461(7261):282–6. doi: 10.1038/nature08296
90. Reich CF 3rd, Pisetsky DS. The content of DNA and Rna in microparticles released by Jurkat and Hl-60 cells undergoing in vitro apoptosis. Exp Cell Res (2009) 315(5):760–8. doi: 10.1016/j.yexcr.2008.12.014
91. Nicotera P, Leist M, Ferrando-May E. Intracellular Atp, a switch in the decision between apoptosis and necrosis. Toxicol Lett (1998) 102-103:139–42. doi: 10.1016/s0378-4274(98)00298-7
92. Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol (2004) 16(6):663–9. doi: 10.1016/j.ceb.2004.09.011
93. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein Hmgb1 by necrotic cells triggers inflammation. Nature (2002) 418(6894):191–5. doi: 10.1038/nature00858
94. Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the Nf-Kappa B pathway. Int Immunol (2000) 12(11):1539–46. doi: 10.1093/intimm/12.11.1539
95. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517(7534):311–20. doi: 10.1038/nature14191
96. Liu Y, Xu Q, Wang Y, Liang T, Li X, Wang D, et al. Necroptosis is active and contributes to intestinal injury in a piglet model with lipopolysaccharide challenge. Cell Death Dis (2021) 12(1):62. doi: 10.1038/s41419-020-03365-1
97. Reilly B, Tan C, Murao A, Nofi C, Jha A, Aziz M, et al. Necroptosis-mediated Ecirp release in sepsis. J Inflammation Res (2022) 15:4047–59. doi: 10.2147/JIR.S370615
98. Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol (2017) 17(5):333–40. doi: 10.1038/nri.2016.153
99. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ (2019) 26(1):99–114. doi: 10.1038/s41418-018-0212-6
100. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity (2018) 48(1):35–44 e6. doi: 10.1016/j.immuni.2017.11.013
101. Tan C, Reilly B, Jha A, Murao A, Lee Y, Brenner M, et al. Active release of Ecirp via gasdermin D channels to induce inflammation in sepsis. J Immunol (2022) 208(9):2184–95. doi: 10.4049/jimmunol.2101004
102. Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin Hmgb1 in endotoxemia. J Immunol (2010) 185(7):4385–92. doi: 10.4049/jimmunol.1000803
103. Vasudevan SO, Behl B, Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunol (2023) 69:101781. doi: 10.1016/j.smim.2023.101781
104. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
105. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of Hmgb1 in ferroptosis. Biochem Biophys Res Commun (2019) 510(2):278–83. doi: 10.1016/j.bbrc.2019.01.090
106. Shimizu J, Murao A, Nofi C, Wang P, Aziz M. Extracellular Cirp promotes Gpx4-mediated ferroptosis in sepsis. Front Immunol (2022) 13:903859. doi: 10.3389/fimmu.2022.903859
107. Liu J, Zhu S, Zeng L, Li J, Klionsky DJ, Kroemer G, et al. Dcn released from ferroptotic cells ignites ager-dependent immune responses. Autophagy (2022) 18(9):2036–49. doi: 10.1080/15548627.2021.2008692
108. Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhao JG, et al. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through Pdcd4 and Microrna-21. Sci Signal (2011) 4(199):ra75. doi: 10.1126/scisignal.2001868
109. Henne WM, Stenmark H, Emr SD. Molecular mechanisms of the membrane sculpting Escrt pathway. Cold Spring Harb Perspect Biol (2013) 5(9):a016766. doi: 10.1101/cshperspect.a016766
110. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci (2018) 75(2):193–208. doi: 10.1007/s00018-017-2595-9
111. Collett GP, Redman CW, Sargent IL, Vatish M. Endoplasmic reticulum stress stimulates the release of extracellular vesicles carrying danger-associated molecular pattern (Damp) molecules. Oncotarget (2018) 9(6):6707–17. doi: 10.18632/oncotarget.24158
112. Gupta S, Knowlton AA. Hsp60 trafficking in adult cardiac myocytes: role of the exosomal pathway. Am J Physiol Heart Circ Physiol (2007) 292(6):H3052–6. doi: 10.1152/ajpheart.01355.2006
113. Sakaki H, Tsukimoto M, Harada H, Moriyama Y, Kojima S. Autocrine regulation of macrophage activation via exocytosis of Atp and activation of P2y11 receptor. PloS One (2013) 8(4):e59778. doi: 10.1371/journal.pone.0059778
114. Nair RR, Mazza D, Brambilla F, Gorzanelli A, Agresti A, Bianchi ME. Lps-challenged macrophages release microvesicles coated with histones. Front Immunol (2018) 9:1463. doi: 10.3389/fimmu.2018.01463
115. Guescini M, Genedani S, Stocchi V, Agnati LF. Astrocytes and glioblastoma cells release exosomes carrying Mtdna. J Neural Transm (Vienna) (2010) 117(1):1–4. doi: 10.1007/s00702-009-0288-8
116. Murao A, Tan C, Jha A, Wang P, Aziz M. Exosome-mediated Ecirp release from macrophages to induce inflammation in sepsis. Front Pharmacol (2021) 12:791648. doi: 10.3389/fphar.2021.791648
117. Murao A, Brenner M, Aziz M, Wang P. Exosomes in sepsis. Front Immunol (2020) 11:2140. doi: 10.3389/fimmu.2020.02140
118. Blott EJ, Griffiths GM. Secretory lysosomes. Nat Rev Mol Cell Biol (2002) 3(2):122–31. doi: 10.1038/nrm732
119. Buratta S, Tancini B, Sagini K, Delo F, Chiaradia E, Urbanelli L, et al. Lysosomal exocytosis, exosome release and secretory autophagy: the autophagic- and endo-lysosomal systems go extracellular. Int J Mol Sci (2020) 21(7):2576. doi: 10.3390/ijms21072576
120. Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, et al. The nuclear protein Hmgb1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep (2002) 3(10):995–1001. doi: 10.1093/embo-reports/kvf198
121. Zhang Z, Chen G, Zhou W, Song A, Xu T, Luo Q, et al. Regulated Atp release from astrocytes through lysosome exocytosis. Nat Cell Biol (2007) 9(8):945–53. doi: 10.1038/ncb1620
122. Wang J, Shao L, Hendrickson HP, Liu L, Chang J, Luo Y, et al. Total body irradiation in the “Hematopoietic” Dose range induces substantial intestinal injury in non-human primates. Radiat Res (2015) 184(5):545–53. doi: 10.1667/RR14191.1
123. Assimakopoulos SF, Triantos C, Thomopoulos K, Fligou F, Maroulis I, Marangos M, et al. Gut-origin sepsis in the critically ill patient: pathophysiology and treatment. Infection (2018) 46(6):751–60. doi: 10.1007/s15010-018-1178-5
124. Pantalone D, Bergamini C, Martellucci J, Alemanno G, Bruscino A, Maltinti G, et al. The role of damps in burns and hemorrhagic shock immune response: pathophysiology and clinical issues. Review Int J Mol Sci (2021) 22(13):7020. doi: 10.3390/ijms22137020
125. Zhang P, Zou B, Liou YC, Huang C. The pathogenesis and diagnosis of sepsis post burn injury. Burns Trauma (2021) 9:tkaa047. doi: 10.1093/burnst/tkaa047
126. Cohen MJ, Brohi K, Calfee CS, Rahn P, Chesebro BB, Christiaans SC, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care (2009) 13(6):R174. doi: 10.1186/cc8152
127. Zhang BF, Wang PF, Cong YX, Lei JL, Wang H, Huang H, et al. Anti-high mobility group box-1 (Hmgb1) antibody attenuates kidney damage following experimental crush injury and the possible role of the tumor necrosis factor-alpha and C-jun N-terminal kinase pathway. J Orthop Surg Res (2017) 12(1):110. doi: 10.1186/s13018-017-0614-z
128. Singh VK, Seed TM. Radiation countermeasures for hematopoietic acute radiation syndrome: growth factors, cytokines and beyond. Int J Radiat Biol (2021) 97(11):1526–47. doi: 10.1080/09553002.2021.1969054
129. Land WG. Use of damps and samps as therapeutic targets or therapeutics: A note of caution. Mol Diagn Ther (2020) 24(3):251–62. doi: 10.1007/s40291-020-00460-z
130. Zhang F, Brenner M, Yang WL, Wang P. A cold-inducible Rna-binding protein (Cirp)-derived peptide attenuates inflammation and organ injury in septic mice. Sci Rep (2018) 8(1):3052. doi: 10.1038/s41598-017-13139-z
131. McGinn J, Zhang F, Aziz M, Yang WL, Nicastro J, Coppa GF, et al. The protective effect of a short peptide derived from cold-inducible Rna-binding protein in renal ischemia-reperfusion injury. Shock (2018) 49(3):269–76. doi: 10.1097/SHK.0000000000000988
132. Borjas T, Jacob A, Yen H, Patel V, Coppa GF, Aziz M, et al. Inhibition of the interaction of Trem-1 and Ecirp attenuates inflammation and improves survival in hepatic ischemia/reperfusion. Shock (2022) 57(2):246–55. doi: 10.1097/SHK.0000000000001894
133. Murao A, Jha A, Ma G, Chaung W, Aziz M, Wang P. A synthetic Poly(a) tail targeting extracellular cirp inhibits sepsis. J Immunol (2023) 211(7):1144–53. doi: 10.4049/jimmunol.2300228
134. Nofi CP, Tan C, Ma G, Kobritz M, Prince J, Wang H, et al. A novel opsonic Ecirp inhibitor for lethal sepsis. J Leukoc Biol (2023). doi: 10.1093/jleuko/qiad119
135. Suda K, Kitagawa Y, Ozawa S, Saikawa Y, Ueda M, Ebina M, et al. Anti-high-mobility group box chromosomal protein 1 antibodies improve survival of rats with sepsis. World J Surg (2006) 30(9):1755–62. doi: 10.1007/s00268-005-0369-2
136. Yang R, Harada T, Mollen KP, Prince JM, Levy RM, Englert JA, et al. Anti-Hmgb1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med (2006) 12(4-6):105–14. doi: 10.2119/2006-00010.Yang
137. Zhao F, Fang Y, Deng S, Li X, Zhou Y, Gong Y, et al. Glycyrrhizin protects rats from sepsis by blocking Hmgb1 signaling. BioMed Res Int (2017) 2017:9719647. doi: 10.1155/2017/9719647
138. Raman KG, Sappington PL, Yang R, Levy RM, Prince JM, Liu S, et al. The role of rage in the pathogenesis of intestinal barrier dysfunction after hemorrhagic shock. Am J Physiol Gastrointest Liver Physiol (2006) 291(4):G556–65. doi: 10.1152/ajpgi.00055.2006
139. May JM, Bylicky M, Chopra S, Coleman CN, Aryankalayil MJ. Long and short non-coding Rna and radiation response: A review. Transl Res (2021) 233:162–79. doi: 10.1016/j.trsl.2021.02.005
140. Acharya SS, Fendler W, Watson J, Hamilton A, Pan Y, Gaudiano E, et al. Serum micrornas are early indicators of survival after radiation-induced hematopoietic injury. Sci Transl Med (2015) 7(287):287ra69. doi: 10.1126/scitranslmed.aaa6593
Keywords: DAMPs, radiation, necrosis, apoptosis, eCIRP, HMGB1
Citation: Yamaga S, Aziz M, Murao A, Brenner M and Wang P (2024) DAMPs and radiation injury. Front. Immunol. 15:1353990. doi: 10.3389/fimmu.2024.1353990
Received: 11 December 2023; Accepted: 15 January 2024;
Published: 25 January 2024.
Edited by:
Pietro Ghezzi, University of Urbino Carlo Bo, ItalyReviewed by:
Vidya P. Kumar, Armed Forces Radiobiology Research Institute, United StatesCopyright © 2024 Yamaga, Aziz, Murao, Brenner and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Wang, cHdhbmdAbm9ydGh3ZWxsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.