 Milica Lazarević
Milica Lazarević- Department of Immunology, Institute for Biological Research “Siniša Stanković” - National Institute of Republic of Serbia, University of Belgrade, Belgrade, Serbia
Complete Freund’s adjuvant (CFA) is used as a standard adjuvant for the induction of experimental autoimmune encephalomyelitis (EAE), the most commonly used animal model in multiple sclerosis studies. Still, CFA induces glial activation and neuroinflammation on its own and provokes pain. In addition, as CFA contains Mycobacteria, an immune response against bacterial antigens is induced in parallel to the response against central nervous system antigens. Thus, CFA can be considered as a confounding factor in multiple sclerosis–related studies performed on EAE. Here, we discuss the effects of CFA in EAE in detail and present EAE variants induced in experimental animals without the use of CFA. We put forward CFA-free EAE variants as valuable tools for studying multiple sclerosis pathogenesis and therapeutic approaches.
1 Introduction
Experimental autoimmune encephalomyelitis (EAE) is an animal model of the central nervous system (CNS) autoimmune diseases. It is mostly used to study the pathogenesis of multiple sclerosis and to explore therapeutic approaches for the disease (1). EAE was invented by Rivers and colleagues in 1933 as a consequence of studying the neurological side effects of vaccination against rabies (2). For more details on the timeline of the origin and development of EAE, readers are referred to a comprehensive review by Alan G. Baxter (3). Complete Freund’s adjuvant (CFA) was introduced as an essential component for EAE induction as early as 1947 (4, 5), just 5 years after its invention (6). CFA is used in most variants of EAE, including the most prominent ones, such as myelin oligodendrocyte glycoprotein (MOG)35–55–induced chronic EAE in C57BL/6 mice, proteolipid protein (PLP)139–151–induced relapsing-remitting EAE in Swiss Jim Lambert (SJL) mice, and myelin basic protein (MBP)–induced monophasic acute EAE in Lewis rats. Moreover, T-cell lines produced for adoptive transfer in passive EAE are originating from animals immunized with CNS antigens emulsified in CFA (7).
CFA is a suspension of desiccated heat-inactivated mycobacterium (most commonly used is M. tuberculosis, followed by M. butyricum) in paraffin oil and mannide monooleate. It has been regularly used as an adjuvant of choice for induction of autoimmune disorders in experimental animals (8). CFA extends injected autoantigen lifetime, galvanizes the effective delivery of the antigens to the immune system, and stimulates the innate compartment of the immune system (8), which, in turn, results in the effective induction of autoimmune disorders in experimental animals, including EAE. Still, in studies where EAE is used as a multiple sclerosis model, CFA introduces numerous confounding effects, including immune reactivity against antigens unrelated to the CNS, activation of glia independent of the autoimmune process, and induction of pain (Figure 1). These and other confounding effects of CFA are discussed in detail in the following sections.
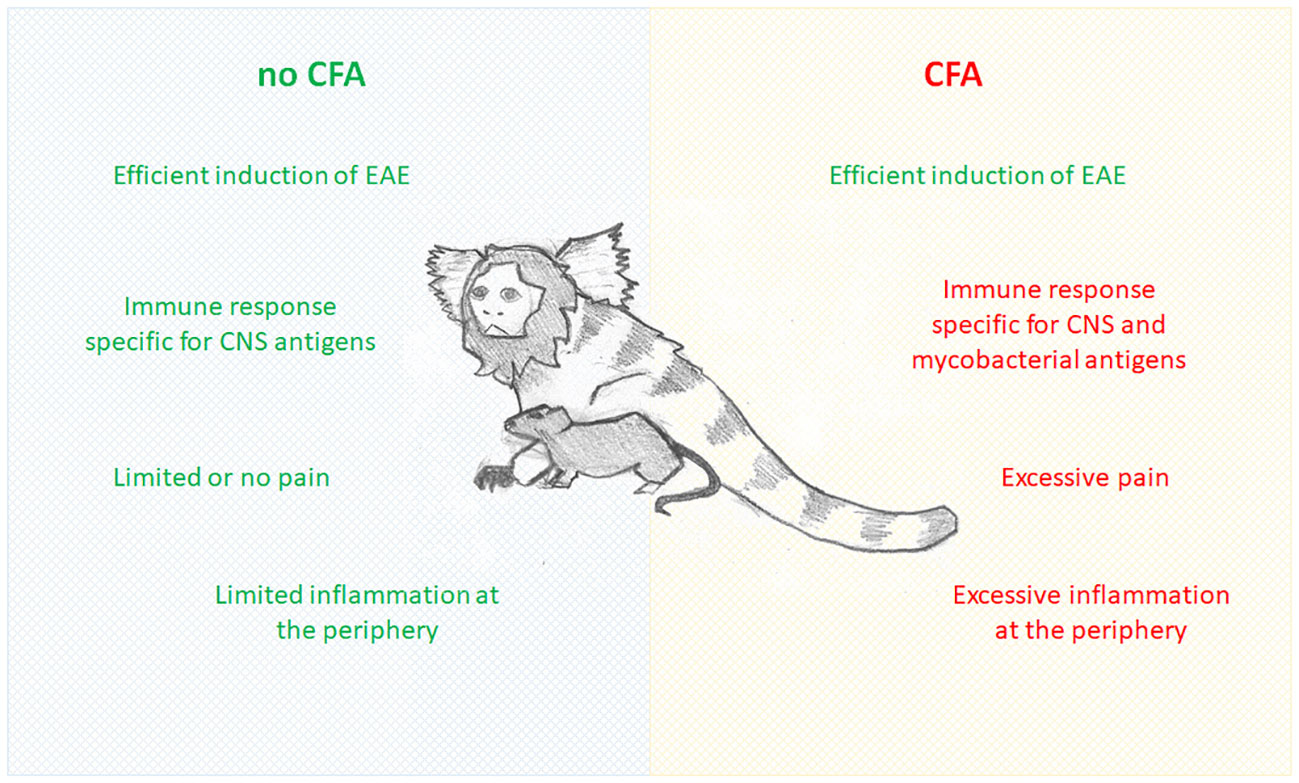
Figure 1 Comparison of EAE induced without and with CFA.
2 Confounding effects of CFA in EAE
2.1 Mycobacteria
One of the confounding effects of CFA arising from the presence of mycobacteria is the stimulation of lymphocytes specific for non-CNS antigens that contribute to the inflammatory response in EAE (8). As an example, it has been reported that lymphocytes obtained from the lymph nodes draining the site of immunization or from the CNS of EAE rats immunized with MBP + CFA exhibited reactivity against mycobacterial purified protein derivative (PPD) (9). Moreover, the addition of a PPD-specific T-cell line to MBP-specific T cells in passive EAE induction resulted in increased blood-brain barrier disruption, suggesting that PPD-specific T cells contribute to the CNS autoimmunity in EAE (10). On the contrary, reactivity to mycobacterial antigens homologous to mammalian proteins, such as heat-shock proteins (HSPs), may interfere with EAE induction. Indeed, prophylactic application of mycobacterial HSP was shown to inhibit the development of EAE (11, 12).
Mycobacteria are intracellular bacteria, and the dominant immune response against them is performed by activated T helper 1 (Th1) cells. Therefore, there is a concern that CFA skews immune response toward Th1 and away from Th17 arm. Both Th populations are involved in multiple sclerosis pathogenesis, and each population has been associated with different clinical expressions of the disease and/or different mechanisms of effector actions within the CNS (13, 14). Still, CFA does not contain live mycobacteria, and it is more likely that it shapes immune response through mycobacterial pathogen-associated molecular patterns, such as muramyl dipeptide (MDP), trehalose dimycolate (TDM), and lipoarabinomannan (LAM), which are recognized by nucleotide oligomerization domain (NOD), C-type lectin-type receptors, or Toll-like receptor 2 (TLR2), respectively (8, 15–17). Whereas TLR2 stimulation enhances production of cytokines interleukin (IL)-12, interferon (IFN)--γ, and IL-18, promoting Th1 differentiation, TDM induces production of TNF, IL-6, and CXCL2, promoting development of both Th1 and Th17 cells (8, 17). Similar to TDM-induced stimulation, acute NOD2 activation by MDP results in induction of TNF, IL-12, IL-6, IL-8, and IL-10 (18). Th1-dominated cytokine pattern is also induced through TLR9 stimulation by unmethylated 5′-C-phosphate-G-3′ (CpG) oligodeoxynucleotides and TLR4 stimulation by mycobacterial HSP (19–21). Thus, it seems that CFA is able to produce both Th1 and Th17 responses. As an example, both Th1 and Th17 T cells are present in the CNS in EAE induced in Dark Agouti (DA) rats with spinal cord homogenate (SCH) + CFA (22). Still, predominance of Th1 response cannot be excluded as a confounding factor of CFA.
The other important question is if mycobacteria are involved in the aetiology and pathogenesis of multiple sclerosis. There were studies on the role of mycobacterial HSP70 in multiple sclerosis (23, 24), not only on the human zoonotic infection with M. avium subsp. paratuberculosis as a cause of multiple sclerosis (25, 26) but also on the protective role of vaccination with attenuated strain of M. bovis (BCG vaccine) on disease activity in patients with multiple sclerosis (27, 28). In short, it seems that M. avium subsp. paratuberculosis might be a causal agent in genetically predisposed individuals that appear to be restricted to certain human populations, such as those of Sardinia and Japan. As for BCG, epidemiological data support its protective role in multiple sclerosis, but the mechanisms behind its effects are still elusive. For more details on the possible role of mycobacteria in multiple sclerosis etiopathogenesis, readers are referred to a review by Cossu and colleagues (29). Thus, it seems that there is insufficient evidence on the role of mycobacteria in multiple sclerosis pathogenesis to support the use of CFA in EAE.
In addition, one should be careful when studying gut/lung microbiota role in the pathogenesis of the CNS autoimmunity using EAE induced with CFA, as there are studies showing that CFA is affecting microbiota in experimental animals (30, 31). Overall, as different findings demonstrated diverse relationship between mycobacteria and multiple sclerosis, the presence of mycobacterial components in CFA during EAE induction could potentially serve as a disease-modifying factor. This could pose challenges in conclusively identifying the precise pathological mechanisms driving the CNS autoimmunity and in evaluating potential mechanisms for multiple sclerosis therapy.
2.2 Pain, glial activation, and neuroinflammation
CFA is known to induce pain in experimental animals (8). Continual release of antigens from the oily deposit induces a delayed hypersensitivity reaction characterized by intense inflammation and hyperalgesia at the injection site (8). Noted responses to CFA include local acute and chronic inflammation, granulomatous reactions, skin ulceration, local abscess, and sloughing. Systemic reactions include diffuse systemic granulomas resulting from the migration of the oil emulsion, adjuvant-related arthritis, and chronic wasting disease (32). Inflammatory pain is influenced by various modulators, including neurotransmitters, receptors, ion channels, and signaling pathways (33). In a study exploring effects of CFA on various metabolites, a decrease of arginine levels and an increase in histidine, phenylalanine, and tyrosine levels were found in response to CFA injection (34). These changes in amino acid levels have been associated with alterations in neurotransmitter levels and, subsequently, with potentiation of chronic inflammatory pain (34). Additional mechanisms involved in CFA-induced pain include the production and release of prostaglandin E2, NO, leukotriene B2, TNF, IL-2, and IL-17 (35). These mediators contribute to synovitis, polyarticular inflammation, bone resorption, periosteal bone proliferation, and consequently to joint degeneration (36).
CFA is typically used to induce peripheral inflammation that can subsequently affect the CNS. Peripheral inflammation induced by CFA leads to the release of inflammatory mediators, including cytokines that can affect the blood-brain barrier and influence communication between the immune system and the CNS. Indeed, it was previously demonstrated that the permeability of the blood-brain barrier increased after CFA administration (37). This can affect both glial cells and neurons and ultimately induce pain. Accordingly, CFA-induced pain in experimental animals is paralleled with the activation of glia and the production of inflammatory mediators in the spinal cord (38). Microglia and astrocytes are recognized as active participants in the initiation and maintenance of pain facilitation triggered by inflammation and damage to peripheral tissues, peripheral nerves, spinal nerves, and spinal cord (39–42). Upon activation, glial cells release a variety of mediators, including proinflammatory cytokines that can enhance pain transmission by activating and sensitizing neurons (41–44). In turn, activated neurons can have reciprocal effects on glial cells, thus maintaining persistent inflammation and prolonged pain sensitization (45). Specifically, increased expression of IL-1β and IL-1RI was found in glial cells and sensory neurons in an articular arthritis model induced with CFA (46). Furthermore, intraplantar administration of CFA led to elevated expression of microglial markers (Mac-1, CD11b/c, TLR4, and CD14) in the spinal cord and brain during all stages of inflammation (47). In contrast to microglia, increased expression of astrocytic markers, glial fibrillary acidic protein (GFAP) and S100 calcium-binding protein B (S100B), was detected only at the later stages, indicating delayed astrocytic activation. Having in mind all before mentioned, immunization with CFA represents a standard model for studying pain (48).
Considering the wellbeing of experimental animals, the ability of CFA to cause inflammatory pain in experimental animals is problematic per se and should be avoided to prevent unnecessary suffering and additional distress in animals. Furthermore, the enduring inflammatory pain induced by CFA interferes with various classical tests assessing exploratory behavior, stress coping, and naturalistic behavior. A systematic review and meta-analysis, encompassing numerous experiments with hundreds of mice and rats, distinctly revealed that CFA markedly reduces exploratory behavior and heightens immobility in the tail suspension test (49). The most pronounced negative impact was observed in naturalistic behaviors like burrowing and wheel running. Finally, pain induced by CFA interferes with the studies of pain caused by CNS autoimmunity and, consequently, with translation to multiple sclerosis. Neuropathic pain is a common symptom in patients with multiple sclerosis, affecting between 28 and 87% of individuals (50, 51). It stems from damage of the central or peripheral somatosensory systems, including the hyperexcitability of neurons within pain pathways (52). As CFA induces pain on its own, EAE induced with CFA is not a reliable animal model for analyzing the mechanism underlying chronic neuropathic pain frequently registered in patients with multiple sclerosis.
3 EAE models without CFA
EAE can be induced in monkeys and rats with IFA (incoplete Freund’s adjuvant) as adjuvant or even without adjuvant at all. Although EAE cannot be induced in mice without adjuvant, transgenic mice develop EAE without induction (vide infra). The list of actively induced EAE variants in experimental animals without adjuvant or with IFA is provided in Table 1. Importantly, the idea of using CFA-free animal models in the study of CNS autoimmunity is not novel, having been proposed by pioneers in the field, Levine and Wenk, some 60 years ago (65). These authors were able to show that EAE can be induced by intracutaneous injection of neural tissue homogenates without adjuvant in various strains of rats, with Lewis rats being the most susceptible. In addition, they tested SCH of different origins, including various rodents, dogs, bovines, guinea pigs, and even humans. Their results showed that guinea pig and rat SCH were by far the most efficient.
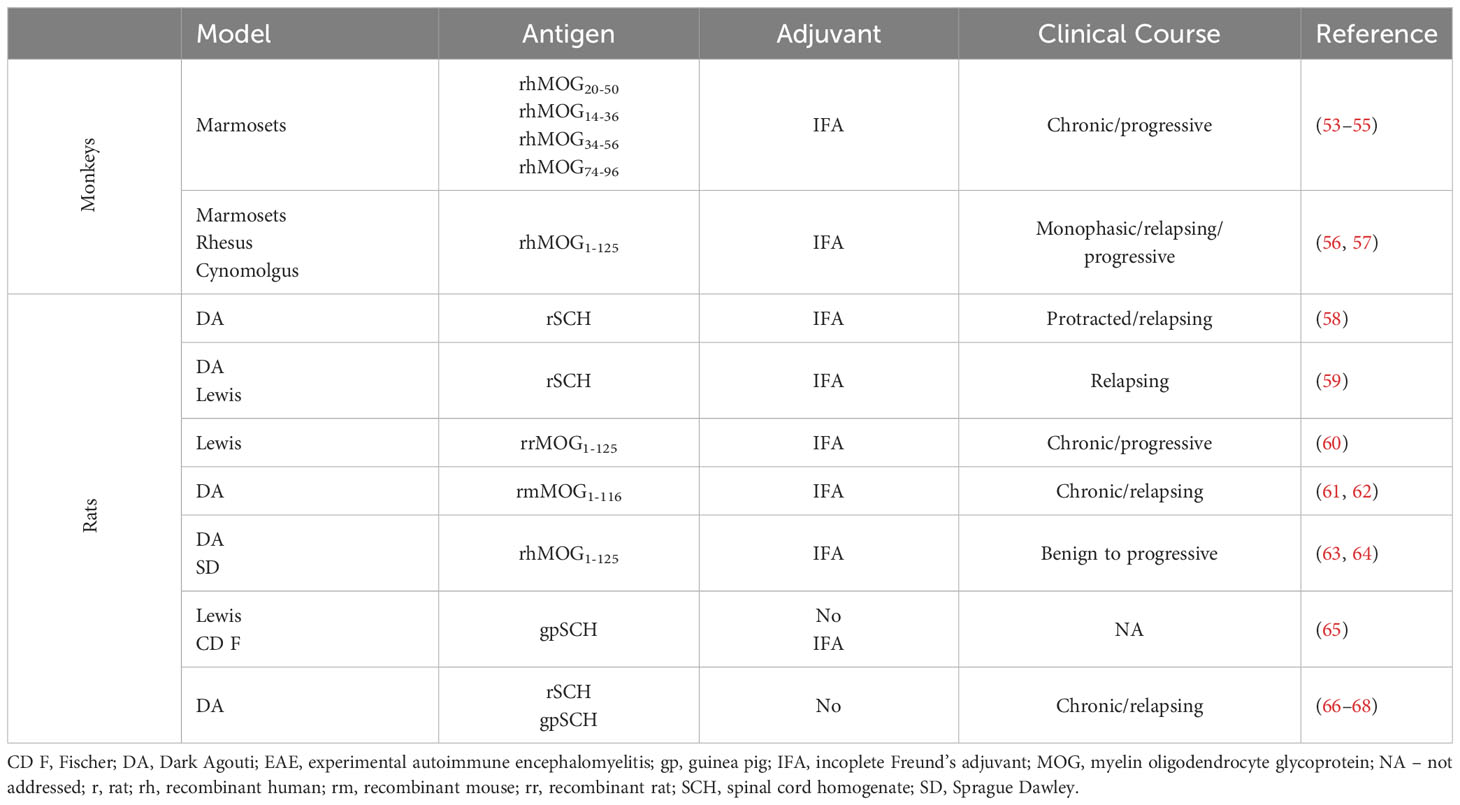
Table 1 Active EAE models with IFA or no adjuvant.
DA rats were not investigated in their study, but we were able to convincingly demonstrate that these rats are also highly susceptible to CFA-free EAE (66–68). Classically, EAE in DA rats is induced with SCH (either guinea pig or rat) emulsified in CFA (69–71). Moreover, if immunization of DA rats is performed with SCH mixed with carbonyl iron, then strong and highly lethal EAE is induced (72). Still, EAE is easily induced in DA rats by rat SCH immunization even without adjuvant. The incidence higher than 90% and the clinical course that is prolonged in comparison to SCH + CFA immunization was determined in DA rats immunized with SCH (67). Infiltrates were observed in both the white and the gray matter of the spinal cord and also in the brain (68). This model has proven useful for evaluating the therapeutic efficacy of novel agents (73, 74). We are currently investigating cellular and molecular mechanisms behind the pathogenesis of CNS autoimmunity in this model in detail. We are particularly interested in the effects of immune cells on various brain structures, such as the cortex, the cerebellum, and the hippocampus. Moreover, antigen specificity of T and B cells activated by immunization with SCH in DA rats is underway.
Furthermore, DA rats immunized with rat SCH and IFA develop severe protracted and relapsing EAE characterized by extensive demyelinating inflammatory lesions and autoreactivity to the rat-specific neuro antigens MOG, MBP69–87, and MBP87–101 (58). Axonal loss in DA rats immunized with MOG in IFA correlates with clinical severity and number of relapses (61). A similar mechanism of inflammatory demyelination involving different components of endoplasmic reticulum stress has been proposed for demyelination in both the MOG + IFA rat EAE model and in multiple sclerosis (62). Recently, a novel low-dose MOG + IFA–induced EAE rat model was introduced in DA and SD rats to investigate pain with minimal motor impairment/disability and to find a potential treatment for multiple sclerosis–related pain (63, 64). The high susceptibility of DA rats to EAE was also confirmed by the induction of severe disease after injection of an encephalitogen in Titermax, an adjuvant consisting of the block copolymer CRL-8300, squalene, and a sorbitan monooleate (75).
EAE in marmosets immunized with rhMOG peptides in IFA is characterized by demyelinating cortical gray matter and prevalent white matter lesions (53–55). Interestingly, the cross-reactivity of MOG-specific T cells with the effector memory cells that control latent CMV infection was determined in rhesus monkeys (76). Although, this cross-reactivity was not investigated in marmosets, the possibility exists that CMV infection, which is common in these monkeys, contributes to the rhMOG peptide + IFA–induced EAE model. The variable clinical presentation of EAE in monkeys is related to inflammation and demyelination in the CNS (56), whereas the occurrence of an early anti-rhMOG IgM response is associated with a more severe disease course (76). However, the purity of the antigen is critical for the development of a mild form of EAE using IFA, as low traces of LPS in rhMOG can increase the severity of clinical and histologic features of the disease in cynomolgus monkeys (57).
Finally, resistance of mice to EAE induction with MOG35–55 or PLP139–151 in IFA can be abolished by addition of the bacterial component peptidoglycan from S. aureus (77) or by co-injection of pertussis toxin (78, 79), respectively. EAE can also be induced in mice with MOG35–55 and quillaja bark saponin as an adjuvant followed by pertussis toxin injections (80, 81). Microbial products seem to be essential for efficient antigen-presenting cell activity and, consequently, for the induction of CNS autoimmunity in mice (77–79). Thus, it can be concluded that active EAE induction in mice is not possible without adjuvant or with IFA as the only adjuvant. However, there are transgenic mouse models that develop EAE without any immunization, such as double-transgenic TCRMOG35–55 × IgHMOG C57BL/6 or non-obese diabetic (NOD) mice (82–84) and TCRMOG92–106 transgenic SJL mice (85, 86). These models are very useful for studying the pathogenesis of CNS autoimmunity yet with limitations introduced by intrinsic restriction in the specificity of their antigen recognition receptors.
Importantly, different models presented in Table 1 cover the full spectrum of multiple sclerosis expression, from benign/mild, through the most common relapsing-remitting, and to the chronic and progressive forms. For a comprehensive understanding of the complexity of clinical manifestations of multiple sclerosis and their significance for the pathogenesis and treatment of the disease, please refer to a review by Confavreux and Vukusic (87). This versatility of the CFA-free animal models makes them useful for studying and addressing specific questions related to different clinical forms of multiple sclerosis.
4 Discussion
Numerous variants of EAE are available for the investigation of multiple sclerosis. Given the diversity in the pathogenesis of multiple sclerosis as well as the elusive etiology of the disease, each of the EAE variants is a valuable tool for the studies. We strongly support the use of CFA-free EAE variants as complementary models in multiple sclerosis studies. This approach can help to eliminate the potential effects of CFA, which were described in detail above. Interestingly, there were important early reports showing that EAE can be induced in rats by immunization with an emulsion made of CFA and lung homogenate (88). In addition, a demyelinating disease was induced in Syrian hamsters and guinea pigs by liver homogenate emulsified in CFA (89). It seems unlikely that CNS and lung or liver share some common antigens that could serve as autoantigens in CNS autoimmunity. It is more likely that CFA leads to neuroinflammation independent of the presence of CNS antigens in the reported studies. This is one more reason to consider CFA as a confounding factor in multiple sclerosis studies based on EAE. Thus, we propose that CFA-free animal models should be used more frequently in the exploration of multiple sclerosis pathogenesis and therapeutic opportunities. Contemporary studies of multiple sclerosis should provide a means to prevent demyelination and neurodegeneration and to promote remyelination and neuroregeneration. We strongly suggest CFA-free EAE variants for such studies as they are superior to CFA-based EAE variants, which are discredited by the weaknesses introduced into the model by the use of CFA, as discussed above.
Author contributions
ML: Writing – original draft, Writing – review & editing. SS: Writing – original draft, Writing – review & editing. NN: Writing – original draft, Writing – review & editing. MD: Writing – original draft, Writing – review & editing. ĐM: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors declare financial support from the Ministry of Science, Technological Development, and Innovations, Republic of Serbia (Contract No. 451-03-47/2023-01/200007 and 451-03-66/2024-03/200007).
Acknowledgments
The authors are thankful to Katarina Miljković for artistic contribution to Figure 1 of the article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Krishnamoorthy G, Wekerle H. EAE: an immunologist's magic eye. Eur J Immunol (2009) 39:2031–5. doi: 10.1002/eji.200939568
2. Rivers TM, Sprunt DH, Berry GP. Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J Exp Med (1933) 58:39–53. doi: 10.1084/jem.58.1.39
3. Baxter AG. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol (2007) 7:904–12. doi: 10.1038/nri2190
4. Morgan IM. Allergic encephalomyelitis in monkeys in response to injection of normal monkey nervous tissue. J Exp Med (1947) 85:131–40. doi: 10.1084/jem.85.1.131
5. Freund J, Stern ER, Pisani TM. Isoallergic encephalomyelitis and radiculitis in Guinea pigs after one injection of brain and Mycobacteria in water-in-oil emulsion. J Immunol (1947) 57:179–94.
6. Freund J, McDermott K. Sensitisation to horse serum by means of adjuvants. Proc Soc Exp Biol (1942) 49:548–53. doi: 10.3181/00379727-49-13625
7. Ben-Nun A, Wekerle H, Cohen IR. The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol (1981) 11:195–9. doi: 10.1002/eji.1830110307
8. Billiau A, Matthys P. Modes of action of Freund's adjuvants in experimental models of autoimmune diseases. J Leukoc Biol (2001) 70:849–60. doi: 10.1189/jlb.70.6.849
9. Lyman WD, Abrams GA, Raine CS. Experimental autoimmune encephalomyelitis: isolation and characterization of inflammatory cells from the central nervous system. J Neuroimmunol (1989) 25:195–201. doi: 10.1016/0165-5728(89)90137-9
10. Namer IJ, Steibel J, Poulet P, Armspach JP, Mohr M, Mauss Y, et al. Blood-brain barrier breakdown in MBP-specific T cell induced experimental allergic encephalomyelitis. A quantitative in vivo MRI study. Brain (1993) 116:147–59. doi: 10.1093/brain/116.1.147
11. van Eden W, van der Zee R, Paul AG, Prakken BJ, Wendling U, Anderton SM, et al. Do heat shock proteins control the balance of T-cell regulation in inflammatory diseases? Immunol Today (1998) 19:303–7. doi: 10.1016/s0167-5699(98)01283-3
12. Birnbaum G, Kotilinek L, Schlievert P, Clark HB, Trotter J, Horvath E, et al. Heat shock proteins and experimental autoimmune encephalomyelitis (EAE): I. Immunization with a peptide of the myelin protein 2',3' cyclic nucleotide 3' phosphodiesterase that is cross-reactive with a heat shock protein alters the course of EAE. J Neurosci Res (1996) 44:381–96. doi: 10.1002/(SICI)1097-4547(19960515)44:4<381::AID-JNR10>3.0.CO;2-5
13. Arellano G, Acuña E, Reyes LI, Ottum PA, De Sarno P, Villarroel L, et al. Th1 and Th17 cells and associated cytokines discriminate among clinically isolated syndrome and multiple sclerosis phenotypes. Front Immunol (2017) 8:753. doi: 10.3389/fimmu.2017.00753
14. Krishnarajah S, Becher B. TH cells and cytokines in encephalitogenic disorders. Front Immunol (2022) 13:822919. doi: 10.3389/fimmu.2022.822919
15. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278:8869–72. doi: 10.1074/jbc.C200651200
16. Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med (2009) 206:2879–88. doi: 10.1084/jem.20091750
17. Miyake Y, Toyonaga K, Mori D, Kakuta S, Hoshino Y, Oyamada A, et al. C-type lectin MCL is an FcRγ-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity (2013) 38:1050–62. doi: 10.1016/j.immuni.2013.03.010
18. Zheng S, Abraham C. NF-κB1 inhibits NOD2-induced cytokine secretion through ATF3-dependent mechanisms. Mol Cell Biol (2013) 33:4857–71. doi: 10.1128/MCB.00797-13
19. Chu RS, Targoni OS, Krieg AM, Lehmann PV, Harding CV. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J Exp Med (1997) 186:1623–31. doi: 10.1084/jem.186.10.1623
20. Kol A, Lichtman AH, Finberg RW, Libby P, Kurt-Jones EA. Cutting edge: heat shock protein (HSP) 60 activates the innate immune response: CD14 is an essential receptor for HSP60 activation of mononuclear cells. J Immunol (2000) 164:13–7. doi: 10.4049/jimmunol.164.1.13
21. Ohashi K, Burkart V, Flohé S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol (2000) 164:558–61. doi: 10.4049/jimmunol.164.2.558
22. Momcilović M, Miljković Z, Popadić D, Miljković D, Mostarica-Stojković M. Kinetics of IFN-gamma and IL-17 expression and production in active experimental autoimmune encephalomyelitis in Dark Agouti rats. Neurosci Lett (2008) 447:148–52. doi: 10.1016/j.neulet.2008.09.082
23. Salvetti M, Ristori G, Buttinelli C, Fiori P, Falcone M, Britton W, et al. The immune response to mycobacterial 70-kDa heat shock proteins frequently involves autoreactive T cells and is quantitatively disregulated in multiple sclerosis. J Neuroimmunol (1996) 65:143–53. doi: 10.1016/0165-5728(96)00013-6
24. Cossu D, Masala S, Frau J, Cocco E, Marrosu MG, Sechi LA. Anti Mycobacterium avium subsp. paratuberculosis heat shock protein 70 antibodies in the sera of Sardinian patients with multiple sclerosis. J Neurol Sci (2013) 335:131–3. doi: 10.1016/j.jns.2013.09.011
25. Cossu D, Masala S, Frau J, Mameli G, Marrosu MG, Cocco E, et al. Antigenic epitopes of MAP2694 homologous to T-cell receptor gamma-chain are highly recognized in multiple sclerosis Sardinian patients. Mol Immunol (2014) 57:138–40. doi: 10.1016/j.molimm.2013.09.001
26. Otsubo S, Cossu D, Eda S, Otsubo Y, Sechi LA, Suzuki T, et al. Seroprevalence of IgG1 and IgG4 class antibodies against Mycobacterium avium subsp. paratuberculosis in Japanese population. Foodborne Pathog Dis (2015) 12:851–6. doi: 10.1089/fpd.2015.1956
27. Ristori G, Buzzi MG, Sabatini U, Giugni E, Bastianello S, Viselli F, et al. Use of Bacille Calmette-Guèrin (BCG) in multiple sclerosis. Neurology (1999) 53:1588–9. doi: 10.1212/wnl.53.7.1588
28. Ristori G, Romano S, Cannoni S, Visconti A, Tinelli E, Mendozzi L, et al. Effects of Bacille Calmette-Guerin after the first demyelinating event in the CNS. Neurology (2014) 82:41–8. doi: 10.1212/01.wnl.0000438216.93319.ab
29. Cossu D, Yokoyama K, Hattori N. Conflicting role of mycobacterium species in multiple sclerosis. Front Neurol (2017) 8:216. doi: 10.3389/fneur.2017.00216
30. Johanson DM 2nd, Goertz JE, Marin IA, Costello J, Overall CC, Gaultier A. Experimental autoimmune encephalomyelitis is associated with changes of the microbiota composition in the gastrointestinal tract. Sci Rep (2020) 10:15183. doi: 10.1038/s41598-020-72197-y
31. Khadka S, Omura S, Sato F, Tsunoda I. Adjuvant injections altered the ileal and fecal microbiota differently with changes in immunoglobulin isotypes and antimycobacterial antibody responses. Int J Mol Sci (2023) 24:2818. doi: 10.3390/ijms24032818
32. Almarestani L, Fitzcharles MA, Bennett GJ, Ribeiro-da-Silva A. Imaging studies in Freund's complete adjuvant model of regional polyarthritis, a model suitable for the study of pain mechanisms, in the rat. Arthritis Rheumatol (2011) 63:1573–81. doi: 10.1002/art.30303
33. Jiao Q, Ren Y, Ariston Gabrie AN, Wang Q, Wang Y, Du L, et al. Advances of immune checkpoints in colorectal cancer treatment. BioMed Pharmacother (2020) 123:109745. doi: 10.1016/j.biopha.2019.109745
34. Zhang W, Lyu J, Xu J, Zhang P, Zhang S, Chen Y, et al. The related mechanism of complete Freund's adjuvant-induced chronic inflammation pain based on metabolomics analysis. BioMed Chromatogr (2021) 35:e5020. doi: 10.1002/bmc.5020
35. Nisar A, Akhter N, Singh G, Masood A, Malik A, Banday B, et al. Modulation of T-helper cytokines and inflammatory mediators by Atropa accuminata. Royle in adjuvant induced arthritic tissues. J Ethnopharmacol (2015) 162:215–24. doi: 10.1016/j.jep.2014.08.008
36. Bendele A. Animal models of rheumatoid arthritis. J Musculoskelet Neuronal Interact (2001) 1:377–85.
37. Reiber H, Suckling AJ, Rumsby MG. The effect of Freund's adjuvants on blood-cerebrospinal fluid barrier permeability. J Neurol Sci (1984) 63:55–61. doi: 10.1016/0022-510x(84)90108-4
38. Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci (2004) 20:467–73. doi: 10.1111/j.1460-9568.2004.03514.x
39. Sweitzer SM, Colburn RW, Rutkowski M, DeLeo JA. Acute peripheral inflammation induces moderate glial activation and spinal IL-1beta expression that correlates with pain behavior in the rat. Brain Res (1999) 829:209–21. doi: 10.1016/s0006-8993(99)01326-8
40. Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci (2001) 24:450–5. doi: 10.1016/s0166-2236(00)01854-3
41. Raghavendra V, Tanga F, Rutkowski MD, DeLeo JA. Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain (2003) 104:655–64. doi: 10.1016/S0304-3959(03)00138-6
42. Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther (2003) 306:624–30. doi: 10.1124/jpet.103.052407
43. Sommer C. Painful neuropathies. Curr Opin Neurol (2003) 16:623–8. doi: 10.1097/01.wco.0000093106.34793.06
44. Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov (2003) 2:973–85. doi: 10.1038/nrd1251
45. Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology (2018) 129:343–66. doi: 10.1097/ALN.0000000000002130
46. Li M, Shi J, Tang JR, Chen D, Ai B, Chen J, et al. Effects of complete Freund's adjuvant on immunohistochemical distribution of IL-1beta and IL-1R I in neurons and glia cells of dorsal root ganglion. Acta Pharmacol Sin (2005) 26:192–8. doi: 10.1111/j.1745-7254.2005.00522.x
47. Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA (2003) 100:8514–9. doi: 10.1073/pnas.1432609100
48. Coderre TJ, Laferrière A. The emergence of animal models of chronic pain and logistical and methodological issues concerning their use. J Neural Transm (Vienna) (2020) 127:393–406. doi: 10.1007/s00702-019-02103-y
49. Burek DJ, Massaly N, Yoon HJ, Doering M, Morón JA. Behavioral outcomes of complete Freund adjuvant-induced inflammatory pain in the rodent hind paw: a systematic review and meta-analysis. Pain (2022) 163:809–19. doi: 10.1097/j.pain.0000000000002467
50. Ehde DM, Jensen MP, Engel JM, Turner JA, Hoffman AJ, Cardenas DD. Chronic pain secondary to disability: a review. Clin J Pain (2003) 19:3–17. doi: 10.1097/00002508-200301000-00002
51. Solaro C, Trabucco E, Messmer Uccelli M. Pain and multiple sclerosis: pathophysiology and treatment. Curr Neurol Neurosci Rep (2013) 13:320. doi: 10.1007/s11910-012-0320-5
52. Mirabelli E, Elkabes S. Neuropathic pain in multiple sclerosis and its animal models: focus on mechanisms, knowledge gaps and future directions. Front Neurol (2021) 12:793745. doi: 10.3389/fneur.2021.793745
53. Jagessar SA, Kap YS, Heijmans N, van Driel N, van Straalen L, Bajramovic JJ, et al. Induction of progressive demyelinating autoimmune encephalomyelitis in common marmoset monkeys using MOG34-56 peptide in incomplete freund adjuvant. J Neuropathol Exp Neurol (2010) 69:372–85. doi: 10.1097/NEN.0b013e3181d5d053
54. Jagessar SA, Heijmans N, Blezer EL, Bauer J, Blokhuis JH, Wubben JA, et al. Unravelling the T-cell-mediated autoimmune attack on CNS myelin in a new primate EAE model induced with MOG34-56 peptide in incomplete adjuvant. Eur J Immunol (2012) 42:217–27. doi: 10.1002/eji.201141863
55. Jagessar SA, Heijmans N, Blezer EL, Bauer J, Weissert R, 't Hart BA. Immune profile of an atypical EAE model in marmoset monkeys immunized with recombinant human myelin oligodendrocyte glycoprotein in incomplete Freund's adjuvant. J Neuroinflammation (2015) 12:169. doi: 10.1186/s12974-015-0378-5
56. Haanstra KG, Jagessar SA, Bauchet AL, Doussau M, Fovet CM, Heijmans N, et al. Induction of experimental autoimmune encephalomyelitis with recombinant human myelin oligodendrocyte glycoprotein in incomplete Freund's adjuvant in three non-human primate species. J Neuroimmune Pharmacol (2013) 8:1251–64. doi: 10.1007/s11481-013-9487-z
57. Stimmer L, Confais J, Jong A, Veth J, Fovet CM, Horellou P, et al. Recombinant myelin oligodendrocyte glycoprotein quality modifies evolution of experimental autoimmune encephalitis in macaques. Lab Invest (2021) 101:1513–22. doi: 10.1038/s41374-021-00646-x
58. Lorentzen JC, Issazadeh S, Storch M, Mustafa MI, Lassman H, Linington C, et al. Protracted, relapsing and demyelinating experimental autoimmune encephalomyelitis in DA rats immunized with syngeneic spinal cord and incomplete Freund's adjuvant. J Neuroimmunol (1995) 63:193–205. doi: 10.1016/0165-5728(95)00153-0
59. Kjellén P, Issazadeh S, Olsson T, Holmdahl R. Genetic influence on disease course and cytokine response in relapsing experimental allergic encephalomyelitis. Int Immunol (1998) 10:333–40. doi: 10.1093/intimm/10.3.333
60. Muhallab S, Dahlman I, Wallström E. Disparate MHC class II haplotypes in myelin oligodendrocyte glycoprotein- and myelin basic protein-induced experimental autoimmune encephalomyelitis. J Neuroimmunol (2005) 161:155–61. doi: 10.1016/j.jneuroim.2005.01.002
61. Papadopoulos D, Pham-Dinh D, Reynolds R. Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp Neurol (2006) 197:373–85. doi: 10.1016/j.expneurol.2005.10.033
62. Ní Fhlathartaigh M, McMahon J, Reynolds R, Connolly D, Higgins E, Counihan T, et al. Calreticulin and other components of endoplasmic reticulum stress in rat and human inflammatory demyelination. Acta Neuropathol Commun (2013) 1:37. doi: 10.1186/2051-5960-1-37
63. Kwilasz AJ, Green Fulgham SM, Duran-Malle JC, Schrama AEW, Mitten EH, Todd LS, et al. Toll-like receptor 2 and 4 antagonism for the treatment of experimental autoimmune encephalomyelitis (EAE)-related pain. Brain Behav Immun (2021) 93:80–95. doi: 10.1016/j.bbi.2020.12.016
64. Kwilasz AJ, Clements MA, Larson TA, Harris KM, Litwiler ST, Woodall BJ, et al. Involvement of TLR2-TLR4, NLRP3, and IL-17 in pain induced by a novel Sprague-Dawley rat model of experimental autoimmune encephalomyelitis. Front Pain Res (Lausanne) (2022) 3:932530. doi: 10.3389/fpain.2022.932530
65. Levine S, Wenk EJ. Induction of experimental allergic encephalomyelitis in rats without the aid of adjuvant. Ann N Y Acad Sci (1965) 122:209–26. doi: 10.1111/j.1749-6632.1965.tb20204.x
66. Stosic-Grujicic S, Ramic Z, Bumbasirevic V, Harhaji L, Mostarica-Stojkovic M. Induction of experimental autoimmune encephalomyelitis in Dark Agouti rats without adjuvant. Clin Exp Immunol (2004) 136:49–55. doi: 10.1111/j.1365-2249.2004.02418.x
67. Lazarević M, Djedovic N, Stanisavljević S, Dimitrijević M, Stegnjaić G, Krishnamoorthy G, et al. Complete Freund's adjuvant-free experimental autoimmune encephalomyelitis in Dark Agouti rats is a valuable tool for multiple sclerosis studies. J Neuroimmunol (2021) 354:577547. doi: 10.1016/j.jneuroim.2021.577547
68. Lazarević M, Stegnjaić G, Jevtić B, Despotović S, Ignjatović Đ, Stanisavljević S, et al. Increased regulatory activity of intestinal innate lymphoid cells type 3 (ILC3) prevents experimental autoimmune encephalomyelitis severity. J Neuroinflamm (2024) 21(1):26. doi: 10.1186/s12974-024-03017-7
69. Mostarica-Stojković M, Petrović M, Lukić ML. Resistance to the induction of EAE in AO rats: its prevention by the pre-treatment with cyclophosphamide or low dose of irradiation. Clin Exp Immunol (1982) 50:311–7.
70. Badovinac V, Mostarica-Stojković M, Dinarello CA, Stosić-Grujicić S. Interleukin-1 receptor antagonist suppresses experimental autoimmune encephalomyelitis (EAE) in rats by influencing the activation and proliferation of encephalitogenic cells. J Neuroimmunol (1998) 85:87–95. doi: 10.1016/s0165-5728(98)00020-4
71. Miljkovic D, Stosic-Grujicic S, Markovic M, Momcilovic M, Ramic Z, Maksimovic-Ivanic D, et al. Strain difference in susceptibility to experimental autoimmune encephalomyelitis between Albino Oxford and Dark Agouti rats correlates with disparity in production of IL-17, but not nitric oxide. J Neurosci Res (2006) 84:379–88. doi: 10.1002/jnr.20883
72. Miljković D, Momčilović M, Stanojević Z, Rašić D, Mostarica-Stojković M. It is still not for the old iron: adjuvant effects of carbonyl iron in experimental autoimmune encephalomyelitis induction. J Neurochem (2011) 118:205–14. doi: 10.1111/j.1471-4159.2011.07303.x
73. Stegnjaić G, Tsiailanis AD, Lazarević M, Gkalpinos VK, Djedovic N, Antoniou T, et al. Phenethyl ester of gallic acid ameliorates experimental autoimmune encephalomyelitis. Molecules (2022) 27:8770. doi: 10.3390/molecules27248770
74. Stegnjaić G, Lazarević M, Diamantis DA, Djedović N, Jevtić B, Stanisavljević S, et al. Phenethyl ester of rosmarinic acid ameliorates experimental autoimmune encephalomyelitis. Immunol Lett (2022) 251-252:9–19. doi: 10.1016/j.imlet.2022.09.006
75. Lukic ML, Mensah-Brown E, Galadari S, Shahin A. Lack of apoptosis of infiltrating cells as the mechanism of high susceptibility to EAE in DA rats. Dev Immunol (2001) 8:193–200. doi: 10.1155/2001/32636
76. Brok HP, Boven L, van Meurs M, Kerlero de Rosbo N, Celebi-Paul L, Kap YS, et al. The human CMV-UL86 peptide 981-1003 shares a crossreactive T-cell epitope with the encephalitogenic MOG peptide 34-56, but lacks the capacity to induce EAE in rhesus monkeys. J Neuroimmunol (2007) 182:135–52. doi: 10.1016/j.jneuroim.2006.10.010
77. Visser L, Jan de Heer H, Boven LA, van Riel D, van Meurs M, Melief MJ, et al. Proinflammatory bacterial peptidoglycan as a cofactor for the development of central nervous system autoimmune disease. J Immunol (2005) 174:808–16. doi: 10.4049/jimmunol.174.2.808
78. Hofstetter HH, Shive CL, Forsthuber TG. Pertussis toxin modulates the immune response to neuroantigens injected in incomplete Freund's adjuvant: induction of Th1 cells and experimental autoimmune encephalomyelitis in the presence of high frequencies of Th2 cells. J Immunol (2002) 169:117–25. doi: 10.4049/jimmunol.169.1.117
79. Hofstetter HH, Forsthuber TG. Kinetics of IL-17- and interferon-gamma-producing PLPp-specific CD4 T cells in EAE induced by coinjection of PLPp/IFA with pertussis toxin in SJL mice. Neurosci Lett (2010) 476:150–5. doi: 10.1016/j.neulet.2010.04.018
80. Démosthènes A, Sion B, Giraudet F, Moisset X, Daulhac L, Eschalier A, et al. In-depth characterization of somatic and orofacial sensitive dysfunctions and interfering-symptoms in a relapsing-remitting experimental autoimmune encephalomyelitis mouse model. Front Neurol (2022) 12:789432. doi: 10.3389/fneur.2021.789432
81. Peiris M, Monteith GR, Roberts-Thomson SJ. Cabot PJ. A model of experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice for the characterisation of intervention therapies. J Neurosci Methods (2007) 163:245–54. doi: 10.1016/j.jneumeth.2007.03.013
82. Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest (2006) 116:2385–92. doi: 10.1172/JCI28330
83. Bettelli E, Baeten D, Jäger A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest (2006) 116:2393–402. doi: 10.1172/JCI28334
84. Anderson AC, Chandwaskar R, Lee DH, Sullivan JM, Solomon A, Rodriguez-Manzanet R, et al. A transgenic model of central nervous system autoimmunity mediated by CD4+ and CD8+ T and B cells. J Immunol (2012) 188:2084–92. doi: 10.4049/jimmunol.1102186
85. Pöllinger B, Krishnamoorthy G, Berer K, Lassmann H, Bösl MR, Dunn R, et al. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med (2009) 206:1303–16. doi: 10.1084/jem.20090299
86. Salvador F, Deramoudt L, Leprêtre F, Figeac M, Guerrier T, Boucher J, et al. A spontaneous model of experimental autoimmune encephalomyelitis provides evidence of MOG-specific B cell recruitment and clonal expansion. Front Immunol (2022) 13:755900. doi: 10.3389/fimmu.2022.755900
87. Confavreux C, Vukusic S. The clinical course of multiple sclerosis. Handb Clin Neurol (2014) 122:343–69. doi: 10.1016/B978-0-444-52001-2.00014-5
88. Allegretti N, Vitale B. Neural tissue and pulmonary lesions in normal and irradiated rats injected with homogenized homologous lung tissue mixed with Freund's adjuvant. Nature (1961) 189:673. doi: 10.1038/189673a0
Keywords: multiple sclerosis, experimental autoimmune encephalomyelitis, complete Freund’s adjuvant, antigen, pain
Citation: Lazarević M, Stanisavljević S, Nikolovski N, Dimitrijević M and Miljković Đ (2024) Complete Freund’s adjuvant as a confounding factor in multiple sclerosis research. Front. Immunol. 15:1353865. doi: 10.3389/fimmu.2024.1353865
Received: 11 December 2023; Accepted: 29 January 2024;
Published: 15 February 2024.
Edited by:
Marisa Mariel Fernandez, Universidad de Buenos Aires, ArgentinaReviewed by:
Anne Cooke, University of Cambridge, United KingdomSocorro Miranda-Hernandez, James Cook University, Australia
Copyright © 2024 Lazarević, Stanisavljević, Nikolovski, Dimitrijević and Miljković. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Đorđe Miljković, georgije_zw@yahoo.com