 Christina A. Capobianco
Christina A. Capobianco Kurt D. Hankenson
Kurt D. Hankenson Alexander J. Knights
Alexander J. Knights
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 22 February 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1352819
This article is part of the Research Topic Interdependencies and Interfaces in Bone Regeneration - The Immune System at its Core View all 10 articles
Bone fracture repair is a complex, multi-step process that involves communication between immune and stromal cells to coordinate the repair and regeneration of damaged tissue. In the US, 10% of all bone fractures do not heal properly without intervention, resulting in non-union. Complications from non-union fractures are physically and financially debilitating. We now appreciate the important role that immune cells play in tissue repair, and the necessity of the inflammatory response in initiating healing after skeletal trauma. The temporal dynamics of immune and stromal cell populations have been well characterized across the stages of fracture healing. Recent studies have begun to untangle the intricate mechanisms driving the immune response during normal or atypical, delayed healing. Various in vivo models of fracture healing, including genetic knockouts, as well as in vitro models of the fracture callus, have been implemented to enable experimental manipulation of the heterogeneous cellular environment. The goals of this review are to (1): summarize our current understanding of immune cell involvement in fracture healing (2); describe state-of-the art approaches to study inflammatory cells in fracture healing, including computational and in vitro models; and (3) identify gaps in our knowledge concerning immune-stromal crosstalk during bone healing.
Unlike most tissues in the body, bone has the unique ability to regenerate - this process is dependent on carefully orchestrated crosstalk between immune and stromal cells. Although the term ‘osteoimmunology’ was coined over twenty years ago to describe the role of immune cells in normal and pathological bone remodeling, there is much that remains unknown about mechanisms guiding immune-stromal cell interactions during the process of bone repair (1). With 600,000 yearly cases of malunion or non-union fractures in the US, there is a critical need to understand both restorative and detrimental properties of immune-stromal crosstalk during the fracture healing response (2).
Fracture repair involves recruitment of immune cells in a temporal and spatial manner that influences the proliferation and differentiation of stromal cells. During the initial stages of long bone callus formation, a fracture hematoma forms, followed by inflammation and stromal progenitor cell recruitment as illustrated in Figure 1. Bone formation occurs next via direct, osteoblast-mediated mechanisms (intramembranous ossification) and via indirect, chondrocyte-mediated mechanisms (endochondral ossification) (3). The majority of pre-clinical fracture studies occur in rodents due to feasibility, reproducibility, and similarities in dynamics of fracture healing to that of humans (4).
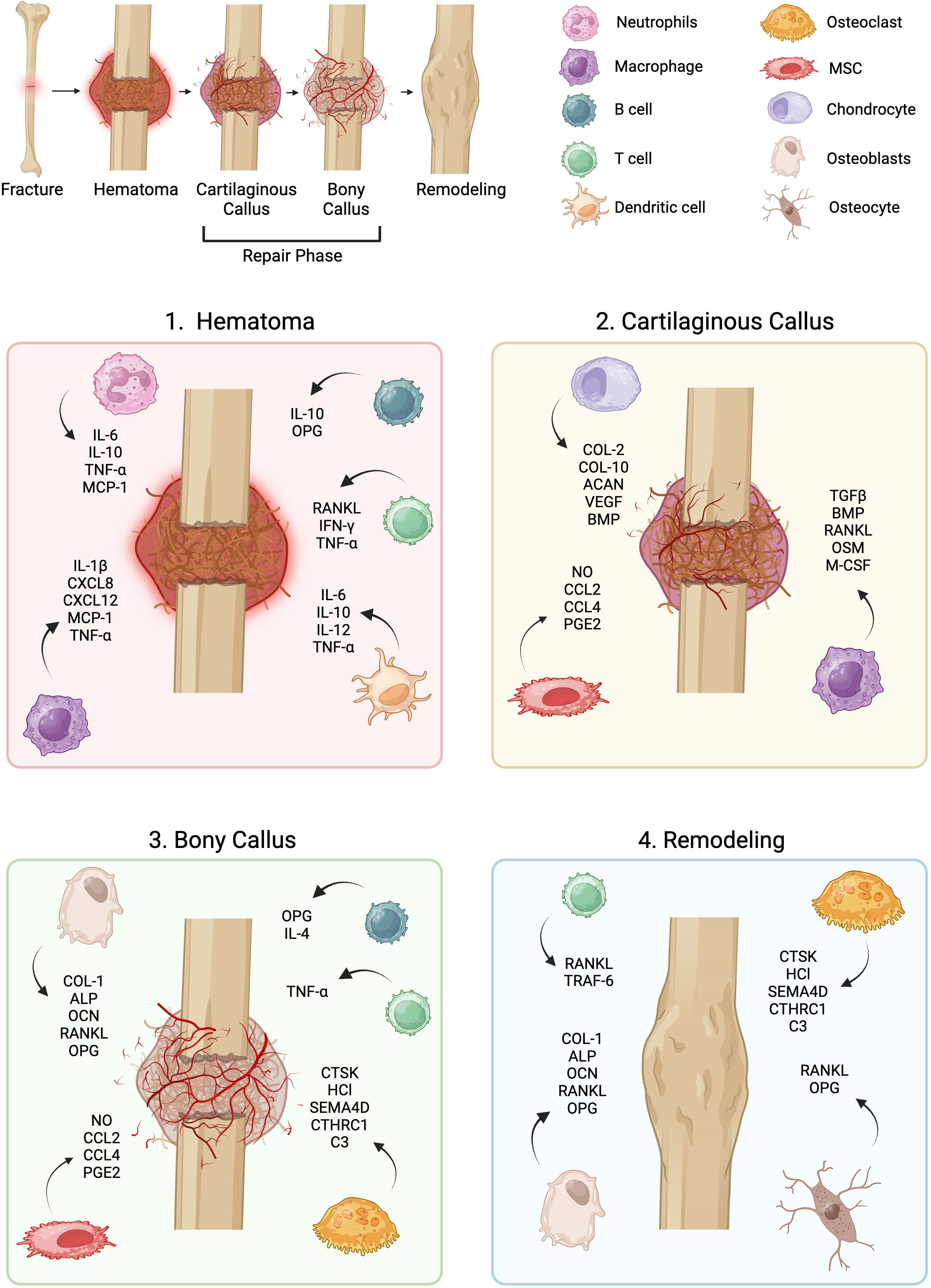
Figure 1 Overview of fracture repair. Fracture repair occurs across distinct phases, each of which involves dynamic stromal-immune cell interactions: the hematoma phase, repair phase, and remodeling phase. MSC, mesenchymal stromal cell.
This phase occurs over the first 1-5 days post fracture in humans (5, 6).
In the first 24 hours of fracture healing, a hematoma forms and is infiltrated by granulocytic cells (predominantly neutrophils) that act as ‘first responders’ (7, 8). These cells recruit monocytes via secretion of cytokines like interleukins (IL-) 1, 6, and 10; tumor necrosis factor alpha (TNF-α); and monocyte chemoattractant protein 1 (MCP-1) (9–14). Neutrophils have also been implicated in contributing to the initial fibrin-rich clot. Within 48 hours of fracture, neutrophils make up the vast majority of cells present at the injury site and synthesize a fibronectin-containing extracellular matrix (ECM) (15). Fibronectin binds fibrin and provides binding sites for other ECM proteins, cells, and growth factors (16). Neutrophil depletion by anti-Ly6G antibody treatment impairs fracture healing, highlighting the essential role of neutrophils in the early inflammatory response (17). While neutrophil infiltration is key to the formation of the hematoma, sustained neutrophil activation leads to diminished osteogenic activity, reduced callus mineralization, and impaired/delayed healing (18, 19).
Upon recruitment, systemically-derived monocytes differentiate into macrophages and dendritic cells. Dendritic cells are present during the early phases of fracture healing, and express inflammatory cytokines (IL-6, IL-12, TNF-a, IL-10) (20–24). Furthermore, CD8+ dendritic cells are known to stimulate CD8+ T cells (20). Early on, macrophages remove cellular debris and secrete inflammatory cytokines including IL-1, TNF-α, IL-6, chemokine (C-X-C motif) ligand (CXCL) 8, CXCL12, and MCP-1 (25–27). Macrophage polarization occurs along a spectrum but is often simplified into 3 subclasses: a naïve, pro-inflammatory, or pro-regenerative phenotype. While macrophages are present throughout the healing process, macrophage depletion studies have identified that their presence is most critical in the immediate aftermath of injury during the pro-inflammatory phase (28–31). Polarized macrophages have been shown to exhibit plasticity in their ability to revert back to a naïve resting state in vitro (32) and through predictive modeling (33). Inflammatory macrophages demonstrate reduced inducible nitric oxide synthase (iNOS) signaling as time progresses after pro-inflammatory stimulation, eventually returning to a naïve state. While this observation may hold for inflammatory macrophages in tissue repair, it has yet to be described in the context of fracture healing. Macrophage-derived cytokines IL-1β and TNF-α also stimulate fibroblast proliferation within the fracture callus (34). Some studies posit that cytokines, such as TNF-α, secreted by pro-inflammatory macrophages, induce bone morphogenetic protein (BMP) 2, the transcription factor RUNX2, and expression of alkaline phosphatase in mesenchymal stromal cells (MSC) (35, 36). However, other studies suggest that later pro-regenerative macrophages secrete BMP2 and oncostatin M (OSM) to promote ECM mineralization, underscoring the importance of temporal dynamics in fracture healing (37, 38). It has also been demonstrated that during this initial phase pro-inflammatory macrophages secrete vascular endothelial growth factor (VEGF) to stimulate neovascularization. As the pro-inflammatory to pro-regenerative shift occurs, pro-regenerative macrophages secrete platelet-derived growth factor (PDGF) (39). Importantly, although the acute inflammatory phase following hematoma formation is critical for fracture healing, chronic inflammation and persistence of pro-inflammatory macrophages impairs fracture healing (40, 41).
Little is known about the function of natural killer (NK) cells during fracture repair; however, it is hypothesized that they likely assist in debridement of the fracture callus and recruit macrophages to the injury site (9). Early work suggested that NK cell activity was suppressed in fracture patients; whereas recent studies indicate an important role for NK cells in MSC recruitment to the fracture site through neutrophil activating peptide 2 secretion, and in regulation of osteoclastogenesis (42–44). Different classes of NK cells regulate progenitor cell survival during digit tip regeneration that may be comparable to events during fracture healing (45). NK cells also show interdependency with MSC, where MSC secretion of IL-10, transforming growth factor beta (TGF-β), and prostaglandin E2 (PGE2), has been linked to suppression of NK cells (46–48).
Lymphocytes arrive as the initial inflammatory phase wanes. T cells express the pro-osteoclastogenic cytokine, receptor activator of nuclear factor κB ligand (RANKL), whereas B cells express osteoprotegerin (OPG), which blocks RANKL activity, inhibiting osteoclastogenesis (49). Spatio-temporal studies of T and B cells in fracture healing have established increased T cells in the bone marrow immediately after injury, with a significant increase in CD4+ T cells compared to CD8+ T cells. Following this initial spike in T and B cells, they retreat from the injury site, reappearing later during bone formation and remodeling (49). Notably, Reinke et al. determined that CD8+ T cells release interferon ɣ (IFN-ɣ) and TNF-α, and that their persistence throughout the fracture repair process greatly impairs osteoblast differentiation and healing (50). To prevent this, IgM+ CD27+ regulatory B cells release IL-10, suppressing IFN-ɣ, TNF-α, and IL-2 signals from CD8+ T cells to promote resolution of the inflammatory response (51).
The repair phase occurs between 5 and 21 days in humans and consists of the formation of a cartilaginous soft callus that then converts to a hard bony callus (5). During the repair phase, bone will heal by endochondral ossification, where it goes through a cartilaginous intermediate, or direct intramembranous ossification where MSC differentiate into osteoblasts and deposit a mineralized ECM (31, 52). Both processes are necessary for fracture repair, however the amount that each contributes to healing depends on fracture stabilization and mechanical forces (53). During the soft and hard callus phases, MSC, chondrocytes, osteoblasts, macrophages, osteoclasts, T cells, and B cells are the dominant cell populations (5, 9).
Bone-resident macrophages regulate bone formation and play a key role in MSC differentiation. Activated macrophages release the cytokines TGFβ, BMP, and OSM to induce MSC differentiation (54). Chang et al. coined the term ‘osteomacs’ to define a discrete F4/80pos Mac-2neg/lo TRACPneg macrophage population found on the periosteum and endosteum lining the bone (55, 56). Osteomacs promote intramembranous ossification and have been shown to exert control over osteoblast maintenance. Within calvarial cultures, the removal of osteomacs results in decreased mineralization, reduced osteocalcin (OCN) induction, and a limited TNF-α response to LPS, demonstrating an integral role in bone homeostasis and osteoblast function (55, 57). Studies have further demonstrated the importance of the osteomac population in a murine tibia fracture model, where depletion resulted in decreased bone formation (56). During the latter part of the repair phase, inflammatory macrophages, described as F4/80pos Mac-2pos TRACPneg differentiate into osteoclasts through macrophage colony-stimulating factor (M-CSF) and RANKL signaling (56). Osteoclasts can induce osteoblast differentiation through secretion of soluble factors like including collagen triple helix repeat-containing protein 1 (CTHRC1) and complement component C (C3) (58, 59). In contrast to osteomac depletion, depletion of osteoclasts, which resorb cartilaginous ECM through catabolic activity, did not impair bone formation (56). Notably an MSC-derived population of septoclasts have also been recently implicated in cartilage resorption during fracture healing as well as developmental ossification, potentially augmenting this activity. However, septoclast importance in bone remodeling post-fracture is still under investigation (60).
During fracture repair, T and B cells infiltrate the fracture site and assist in osteoblast maturation and retention. In this ‘second-wave’ lymphocytes are absent from the cartilaginous regions of the fracture callus, however they are present near the regions of woven bone (49). Konnecke et al. reported that B cells maintain bone homeostasis through the production of OPG to reduce osteoclastogenesis, and physically interact with osteoblasts to influence their differentiation and function (49). Numerous studies have likewise described T cells as critical for fracture repair (61–65). T cells secrete TNF-α to induce osteogenesis and are necessary for normal deposition of collagen I by osteoblasts during fracture healing (61). T cell depletion further exhibited similar premature mineral deposition as seen in Rag1-deficient mice (which lack mature lymphocytes), pointing toward a T cell-osteoblast interaction pathway (61).
MSC derive from various sources including the periosteum and bone marrow (66). In the healing callus they begin to differentiate into chondrocytes and osteoblasts. MSC modulate the immune environment by secreting regulatory molecules including nitric oxide (NO) (67), chemokine ligand (CCL) 2 and 4, and PGE2, to recruit macrophages which trigger MSC chondrogenic and osteogenic differentiation (54, 68, 69). Current literature suggests that skeletal MSC derive from multiple sources including the periosteum, endosteum, bone marrow, and vasculature (66). Periosteal-derived MSC at the callus edges have increased osteoblastogenic potential and undergo intramembranous ossification, secreting collagen 1 (COL-1), OCN and alkaline phosphatase (ALP) (70). On the other hand, bone marrow-derived MSC at the fracture site are more predisposed toward endochondral ossification, depositing collagens 2 (COL-2) and 10 (COL-10) as well as sulfated glycosaminoglycans such as aggrecan (ACAN) (10, 71, 72). Under injury conditions, periosteal-derived MSC have also been shown to contribute to endochondral ossification (71). During this process, the cartilaginous callus begins to stimulate vascular infiltration as hypertrophic chondrocytes secrete angiogenic factors VEGF (73), PDGF (74), and placental growth factor (PGF) (75). Vascular infiltration has been demonstrated to be crucial for the replacement of the cartilaginous callus by bone (76). Although immune-derived cues may direct MSC differentiation pathways, recognized contributors to this spatial phenomenon of MSC becoming either osteoblasts or chondrocytes are mechanical cues and hypoxia (40, 77, 78).
This phase typically takes around 18 weeks but can last for up to 1 year under typical fracture healing conditions in humans (5, 79). During fracture remodeling, the initial fracture callus is replaced with mature mineralized tissue and normal bone structure is restored. This coordinated response to injury is the last stage of fracture repair and is the longest, and the least well-studied (80). During the remodeling phase, inflammatory cells (other than osteoclasts) are dramatically reduced, and remodeling is driven by continuous local and systemic cell signaling (81). Bone remodeling occurs as a function of the stresses that bone receives due to forces acting upon it, including muscle actions (82, 83). The ability of bone to remodel post-fracture declines with age in humans. Indeed, children are more likely than adults to experience overgrowth of mineralized tissue, resulting in ectopic bone formation (84). Studies in mice have corroborated the age-related decline in fracture healing potential in humans, showing significant delays in bone remodeling and decreased bone recovery in elderly mice post-fracture (85, 86).
Although osteoclast activity is present early on in fracture repair, it is most prominent in the remodeling phase (87). Osteoclasts work in a balance with osteoblasts and osteocytes to first degrade immature woven bone which is then replaced with more mature bone. Osteoclasts create a reversal zone where the bone surface is eroded, leaving a canopy where osteoprogenitors are found. The basic multicellular unit -an assembly of osteoblasts, osteoclasts, and capillaries- is a prominent hallmark of bone remodeling (81, 88). Osteoclast differentiation is positively regulated by RANKL signaling and negatively regulated by OPG (89). Osteoclasts dissolve bone through secretion of cathepsin K (CTSK) and hydrochloric acid, and degrade ECM via secreted matrix metalloproteinases (90, 91).
MSC differentiate into osteoblasts, which deposit mineral in equilibrium with osteoclast activity (21). Osteoprogenitors and osteoblasts constitute the canopy around blood vessels, serving as the main source of cells contributing to bone formation. A bone remodeling compartment forms near capillaries and sinusoids, providing access to osteoprogenitors including bone lining cells and pericytes (88). Pericytes encircle capillaries, however evidence suggests that these pericytes can migrate to the bone surface and differentiate into mature osteoblasts (92, 93). Osteoblasts secrete RANKL and OPG to modulate osteoclastogenesis (94).
T cells regulate osteoblast-osteoclast equilibrium by secretion of RANKL (95). Although T cell expression of RANKL may drive osteoclastogenesis during bone remodeling, T cells also drive degradation of TNF receptor associated factor 6 (TRAF6), acting as a negative feedback mechanism for osteoclast activity (96).
Osteocytes make up 90% of healthy adult bone and function in response to changes in their microenvironment, such as mechanical deformation, to initiate remodeling responses via RANKL and OPG production (97).
The mechanisms by which immune and stromal cells orchestrate fracture repair are not fully understood. To interrogate these complex biological interactions, various models of fracture healing have been developed. Herein follows an overview of models of in vivo fracture healing, in vitro fracture models, and computational models, to replicate both typical and impaired fracture healing.
Animal models most faithfully recapitulate the physiological environment and allow for manipulation of cell responses through genetic knockouts and pharmacological or environmental intervention. Selective ablation of immune cell types in mice has contributed heavily to our understanding of the immune system in fracture healing. Fracture models of comorbidities illustrating immune disruption in fracture healing has been thoroughly reviewed (98–100). Numerous studies have utilized transgenic cre drivers such as LysM-Cre, Mrp8-Cre, and Lck-Cre, as well as Macrophage-Fas Induced Apoptosis (MAFIA) mice to generate immune cell-type specific targeting (101–111). Closed long bone fractures in rodent models are often employed to study fracture healing (112). Factors such as age, ischemia, osteoporosis, and immune deficiency are then incorporated to examine causes of impaired healing (57, 98, 113–117). Fracture in aged populations exhibit increased pro-inflammatory macrophage recruitment as well as increased apoptotic markers in human (118) and mouse (119) systems. Lopez et al. demonstrated that anti-inflammatory modulation of the aged fracture rescues callus formation and healing in aged mice (119). The ischemic fracture model exhibits distinctly smaller callus formation and increased fibrosis (114). Ovariectomy produces postmenopausal osteoporosis in mice, leading to chronic inflammation and increased catabolic activity within bone. Fracture following ovariectomy demonstrates delayed callus mineralization, and remodeling (120, 121). Macrophage populations also exhibit increased IFN-ɣ, nitric oxide, and IL-6 expression (57, 122). Interestingly, MSC isolated from osteoporotic patients do not have impaired potential to regenerate bone, emphasizing the critical role of the immune environment in vivo (123). Multiple studies have revealed that fracture healing is greatly impaired in immunodeficient mice, underscoring the necessity of the immune response in fracture repair (101, 124). While the importance of the innate immune system is indisputable, studies have contested the importance of the adaptive response; Toben et al. demonstrated that eradication of the adaptive immune response using RAG1-/- mice accelerated fracture healing and improved bone quality (125). However others have stressed the immunoregulatory importance of adaptive immune cells (particularly T cells) in guiding the repair response and enabling osteoblast activity (63, 126). This emphasizes the complexity of the immune response in fracture repair and the necessity for diverse models to better dissect these pathways.
While the gold standard of preclinical studies is animal models, these models may have limited transferability due to differences in timeline, physiologic structure, pharmacologic response, and variation in specific gene pathways across species, supporting the need for in vitro models using human cells and tissues to complement animal work (4, 127, 128). In vitro models have been developed over the past decade to create a more physiologically-relevant system for studying human fracture. Along with reducing the number of animals necessary to carry out fracture research, the use of human cells carries additional translational transferability. Pfeiffenberger et al. extensively developed a human-based fracture gap model to interrogate immune-stromal crosstalk in vitro (128, 129). While other models, in particular co-culture models (130), focus on later stages of regeneration, this approach uses coagulation of human peripheral blood and MSC to model hematoma development and its progression through fracture repair (129). The hematoma is combined with scaffold-free bone-like constructs made from mesenchymal condensation and allows for manipulation of molecular and environmental cues such as oxygen availability. Hoff et al. developed a human hematoma model using tissue from total hip arthroplasties to monitor and characterize the immune response under bioenergetically-controlled conditions. Cells were exposed to hypoxia with limited nutrients, generating an inflammatory response representative of that seen in fracture after the first 24 hours (131). Increased vascular endothelial growth factor and IL-8 secretion under hypoxia in this model resulted in a decreased granulocytes and increased lymphocytes, as seen in vivo (131). Sridharan et al. investigated the interaction of MSC and macrophages in different collagen scaffolds functionalized with hydroxyapatite particles of varying shapes and sizes (132). This emphasized the ability of microenvironmental stimuli to modulate the immune system and presents a unique opportunity to study these interactions in a cell-specific manner. The hydroxyapatite scaffold polarized macrophages toward a pro- or anti-inflammatory phenotype depending upon changes in scaffold particle size and shape, and the authors also demonstrated that macrophage presence increased osteogenesis. Importantly, these studies demonstrate comparable results from an in vitro human hematoma model with that shown in vivo. In vitro models present a powerful tool to understand discrete mechanisms of fracture healing selective to specific cell populations.
Only recently has computational modeling of fracture healing incorporated intrinsic and extrinsic effects of the immune system, to ascertain their influence on mechanical and biological properties of the callus (133, 134). Computational models are a powerful complementary tool for guiding hypotheses when integrated with in vivo and in vitro experiments. State-of-the-art in silico models encompass continuous, discrete, or hybrid models to interrogate the complex spatiotemporal aspects of fracture healing. No model can holistically capture these processes; however, a corpus of literature is available that aims to help researchers build their own in silico models to study the spatiotemporal effects of the immune response on fracture repair (133). Continuous models function at the tissue and cellular level; these models use partial differential equations to create a continuous overview of a given scenario to study inflammation, bone mechanics, and bone repair. Discrete models study specific individual behaviors at the subcellular level, using agent-based approaches or cellular automata models to understand mechanistic processes in response to their environments (133).
Hybrid models aim to bridge the gap from subcellular mechanisms to the tissue. According to Lafuente-Gracia et al., to address the physiologic processes of the inflammatory response, a compartment model is required, where each compartment is assigned its own equation and set of agents (molecules or cells) and transitions (biological processes like phagocytosis or differentiation) between compartments (133).
Kojouharov et al. developed a mathematical model of the early inflammatory response in fracture healing using nonlinear ordinary differential equations (135). It was then elaborated on further in subsequent papers to consider unactivated (M0), classically activated (M1) and alternatively activated (M2) macrophages as separate variables (136) as well as migration due to molecular factors (137). This study was one of the first to incorporate both the primary hematoma formation and the inflammatory response, by identifying the primary entities involved in the early fracture - bone debris, pro- and anti-inflammatory cytokines, macrophages, MSC and osteoblasts. Informing the computational model with the known progression from fracture hematoma to cartilaginous fracture callus to repair, the authors developed a model for differentiation and cytokine production that includes known events such as initial MSC density, debridement rate, proliferation rate, and synthesis of cartilage and bone (135, 136). The model also maintains assumptions such as the inability of M1 and M2 to dedifferentiate back to M0 (136). This model provides an instrument for studying normal and impaired fracture repair, and for extrapolating mechanistic pathways that may otherwise be overlooked and could be adapted clinically to infer the effects of pharmacologics on fracture repair. This group has most recently extended their model to study the direct effects of phagocytes and inflammatory cytokines on macrophage and MSC cell migration during the initial inflammatory and repair phases (137).
Ghiasi et al. also developed a computational model of human fracture with a specific emphasis on the initial inflammatory stage of fracture healing, however they approached it from a mechanobiology perspective (138). This model employs a finite element-based approach that simulates the processes of fracture healing, and the entities present, such as MSC and debris. Both the Kojouharov and Ghiasi models incorporate initial fracture size and cellular density; however, Kojouharov et al. placed emphasis on the cytokines released, while Ghiasi et al. emphasized the Young’s modulus of the granulation tissue along with stresses and mechanical responses that shape hematoma formation and influence callus formation (135, 136, 138).
Most recently, Borgiani et al. developed the COMMBINI model, an agent-based computational model to understand macrophage dynamics that occur during the early inflammatory phase (up to 5 days post-injury) (139). This model utilizes deep-learning algorithms on immunofluorescent stained slides to generate spatial information about different macrophage populations. It uniquely addresses phenotype-specific cell activities (eg. cell proliferation, migration, phagocytosis, apoptosis) and incorporates polarization and cytokine signaling. While the COMMBINI includes neutrophils, the focus of the model is on macrophages, subdivided into categories M0, M1, and M2. To understand the inflammatory phase of fracture repair in guiding healing, the model focuses on expression of key pro-inflammatory and pro-regenerative cytokines like TNFα, IL10, TGFβ, and IFNγ (139). While valuable and an important primary step, the field recognizes that macrophages exist on a spectrum of functionality, more nuanced than discrete M0, M1, or M2 states.
Fracture repair is a complex process orchestrated by immune and stromal cells to regenerate bone tissue. Inducing ischemia results in aberrant repair and regeneration of tissues, underscoring the importance of systemic immune cells to guide healing (140, 141). Studies involving the effect of limb ischemia on fracture healing date back to the 1960s and while the importance of the immune response during fracture repair is well acknowledged, immune alterations in fracture healing under ischemic conditions remain unclear (142). This is true for other impaired healing conditions as well, for instance in aged models or diabetic models where there is increased systemic inflammation. Numerous methods for inducing and modulating fracture repair have been developed to study tissue healing and remodeling in vivo – including the integral roles of immune cells. In vitro systems allow for the study of mechanisms in discrete phases and specific cell interactions, with the advantage of utilizing human cells. Computational models enhance our study of fracture healing by expanding upon our understanding of networks underlying the fracture microenvironment and simulating the healing response. Importantly, they serve as a tool to study pharmacologic intervention in fracture repair, in conjunction with in vivo and in vitro models. Used together, these models provide a powerful and holistic approach for interrogating immune dynamics and mechanisms in normal and impaired fracture healing, and will continue to evolve and incorporate more complex variables.
CC: Conceptualization, Writing – original draft, Writing – review & editing. KH: Conceptualization, Writing – review & editing. AK: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. CC was funded by a T32 from the National Institutes of Health (T32TR004371). AK was funded by a K99/R00 from the National Institutes of Health (K99AR081894).
We apologize to the authors of important and relevant publications that we unfortunately could not incorporate into this manuscript, given the limitations of the mini-review format.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Buza JA, Einhorn T. Bone healing in 2016. Clin cases Miner Bone Metab (2016) 13:101–5. doi: 10.11138/ccmbm/2016.13.2.101
3. Hankenson KD, Dishowitz M, Gray C, Schenker M. Angiogenesis in bone regeneration. Injury (2011) 42:556–61. doi: 10.1016/j.injury.2011.03.035
4. Gao H, Huang J, Wei Q, He C. Advances in animal models for studying bone fracture healing. Bioengineering (2023) 10:201. doi: 10.3390/bioengineering10020201
5. Muire PJ, Mangum LH, Wenke JC. Time course of immune response and immunomodulation during normal and delayed healing of musculoskeletal wounds. Front Immunol (2020) 11:1056. doi: 10.3389/fimmu.2020.01056
6. Marsell R, Einhorn TA. The biology of fracture healing. Injury (2011) 42:551–5. doi: 10.1016/j.injury.2011.03.031
7. Claes L, Recknagel S, Ignatius A. Fracture healing under healthy and inflammatory conditions. Nat Rev Rheumatol (2012) 8:133–43. doi: 10.1038/nrrheum.2012.1
8. Chung R, Cool JC, Scherer MA, Foster BK, Xian CJ. Roles of neutrophil-mediated inflammatory response in the bony repair of injured growth plate cartilage in young rats. J Leukoc Biol (2006) 80:1272–80. doi: 10.1189/jlb.0606365
9. Baht GS, Vi L, Alman BA. The role of the immune cells in fracture healing. Curr Osteoporos Rep (2018) 16:138–45. doi: 10.1007/s11914-018-0423-2
10. Bahney CS, Zondervan RL, Allison P, Theologis A, Ashley JW, Ahn J, et al. Cellular biology of fracture healing. J Orthop Res (2019) 37:35–50. doi: 10.1002/jor.24170
11. Hurst SM, Wilkinson TS, Mcloughlin RM, Jones S, Horiuchi S, Yamamoto N, et al. IL-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity (2001) 14:705–14. doi: 10.1016/S1074-7613(01)00151-0
12. Kasama T, Strieter RM, Standiford TJ, Burdick MD, Kunkel SL. Expression and regulation of human neutrophil-derived macrophage inflammatory protein 1 alpha. J Exp Med (1993) 178:63–72. doi: 10.1084/jem.178.1.63
13. Kasten KR, Muenzer JT, Caldwell CC. Neutrophils are significant producers of IL-10 during sepsis. Biochem Biophys Res Comm (2010) 393:28–31. doi: 10.1016/j.bbrc.2010.01.066
14. Feiken E, Rømer J, Eriksen J, Lund LR. Neutrophils express tumor necrosis factor-alpha during mouse skin wound healing. J Invest Dermatol (1995) 105:120–3. doi: 10.1111/1523-1747.ep12313429
15. Bastian OW, Koenderman L, Alblas J, Leenen LP, Blokhuis TJ. Neutrophils contribute to fracture healing by synthesizing fibronectin+ extracellular matrix rapidly after injury. Clin Immunol (2016) 164:78–84. doi: 10.1016/j.clim.2016.02.001
16. Singh P, Carraher C, Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Ann Rev Cell Dev Biol (2010) 26:397–419. doi: 10.1146/annurev-cellbio-100109-104020
17. Kovtun A, Bergdolt S, Wiegner R, Radermacher P, Huber-Lang M, Ignatius A. The crucial role of neutrophil granulocytes in bone fracture healing. Eur Cell Mater (2016) 32:152–62. doi: 10.22203/eCM.v032a10
18. Bastian OW, Croes M, Alblas J, Koenderman L, Leenen LPH, Blokhuis TJ. Neutrophils inhibit synthesis of mineralized extracellular matrix by human bone marrow-derived stromal cells In Vitro. Front Immunol (2018) 9:945. doi: 10.3389/fimmu.2018.00945
19. Bastian OW, Mrozek MH, Raaben M, Leenen LPH, Koenderman L, Blokhuis TJ. Serum from the human fracture hematoma contains a potent inducer of neutrophil chemotaxis. Inflammation (2018) 41:1084–92. doi: 10.1007/s10753-018-0760-4
20. Bucher CH, Berkmann JC, Burkhardt L-M, Paschke C, Schlundt C, Lang A, et al. Local immune cell contributions to fracture healing in aged individuals – A novel role for interleukin 22. Exp Mol Med (2022) 54:1262–76. doi: 10.1038/s12276-022-00834-9
21. Ehnert S, Relja B, Schmidt-Bleek K, Fischer V, Ignatius A, Linnemann C, et al. Effects of immune cells on mesenchymal stem cells during fracture healing. World J Stem Cells (2021) 13:1667–95. doi: 10.4252/wjsc.v13.i11.1667
22. Piqueras B, Connolly J, Freitas H, Palucka AK, Banchereau J. Upon viral exposure, myeloid and plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood (2006) 107:2613–8. doi: 10.1182/blood-2005-07-2965
23. Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat Immunol (2017) 18:74–85. doi: 10.1038/ni.3632
24. Dong X, Swaminathan S, Bachman L-A, Croatt A-J, Nath K-A, Griffin M-D. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia–reperfusion injury. Kidney Int (2007) 71:619–28. doi: 10.1038/sj.ki.5002132
25. Currie HN, Loos MS, Vrana JA, Dragan K, Boyd JW. Spatial cytokine distribution following traumatic injury. Cytokine (2014) 66:112–8. doi: 10.1016/j.cyto.2014.01.001
26. Schmidt-Bleek K, Schell H, Kolar P, Pfaff M, Perka C, Buttgereit F, et al. Cellular composition of the initial fracture hematoma compared to a muscle hematoma: A study in sheep. J Orthop Res (2009) 27:1147–51. doi: 10.1002/jor.20901
27. Horst K, Eschbach D, Pfeifer R, Hübenthal S, Sassen M, Steinfeldt T, et al. Local inflammation in fracture hematoma: results from a combined trauma model in pigs. Mediators Inflammation (2015) 2015:1–8. doi: 10.1155/2015/126060
28. Pajarinen J, Lin T, Gibon E, Kohno Y, Maruyama M, Nathan K, et al. Mesenchymal stem cell-macrophage crosstalk and bone healing. Biomaterials (2019) 196:80–9. doi: 10.1016/j.biomaterials.2017.12.025
29. Burnett SH, Kershen EJ, Zhang J, Zeng L, Straley SC, Kaplan AM, et al. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J Leukoc Biol (2004) 75:612–23. doi: 10.1189/jlb.0903442
30. Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods (1994) 174:83–93. doi: 10.1016/0022-1759(94)90012-4
31. Raggatt LJ, Wullschleger ME, Alexander KA, Wu ACK, Millard SM, Kaur S, et al. Fracture healing via periosteal callus formation requires macrophages for both initiation and progression of early endochondral ossification. Am J Pathol (2014) 184:3192–204. doi: 10.1016/j.ajpath.2014.08.017
32. Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol (2015) 53:676–88. doi: 10.1165/rcmb.2015-0012OC
33. Weinstock LD, Forsmo JE, Wilkinson A, Ueda J, Wood LB. Experimental control of macrophage pro-inflammatory dynamics using predictive models. Front Bioeng Biotechnol (2020) 8. doi: 10.3389/fbioe.2020.00666
34. Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol (1975) 78:71–100.
35. Wang M, Crisostomo PR, Herring C, Meldrum KK, Meldrum DR. Human progenitor cells from bone marrow or adipose tissue produce VEGF, HGF, and IGF-I in response to TNF by a p38 MAPK-dependent mechanism. Am J Physiol Regul Integr Comp Physiol (2006) 291:R880–4. doi: 10.1152/ajpregu.00280.2006
36. Egea V, Von Baumgarten L, Schichor C, Berninger B, Popp T, Neth P, et al. TNF-α respecifies human mesenchymal stem cells to a neural fate and promotes migration toward experimental glioma. Cell Death Differ (2011) 18:853–63. doi: 10.1038/cdd.2010.154
37. Lacey DC, Simmons PJ, Graves SE, Hamilton JA. Proinflammatory cytokines inhibit osteogenic differentiation from stem cells: implications for bone repair during inflammation. Osteoarthr Cartil (2009) 17:735–42. doi: 10.1016/j.joca.2008.11.011
38. Nicolaidou V, Wong MM, Redpath AN, Ersek A, Baban DF, Williams LM, et al. Monocytes induce STAT3 activation in human mesenchymal stem cells to promote osteoblast formation. PloS One (2012) 7:e39871. doi: 10.1371/journal.pone.0039871
39. Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW, et al. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials (2014) 35:4477–88. doi: 10.1016/j.biomaterials.2014.02.012
40. Maruyama M, Rhee C, Utsunomiya T, Zhang N, Ueno M, Yao Z, et al. Modulation of the inflammatory response and bone healing. Front Endocrinol (2020) 11:386. doi: 10.3389/fendo.2020.00386
41. Lin TH, Gibon E, Loi F, Pajarinen J, Córdova LA, Nabeshima A, et al. Decreased osteogenesis in mesenchymal stem cells derived from the aged mouse is associated with enhanced NF-κB activity. J Orthop Res (2017) 35:281–8. doi: 10.1002/jor.23270
42. Hauser CJ. Suppression of natural killer cell activity in patients with fracture/soft tissue injury. Arch Surg (1997) 132:1326. doi: 10.1001/archsurg.1997.01430360072013
43. Almeida RC, Caires HR, Vasconcelos DP, Barbosa MA. NAP-2 secreted by human NK cells can stimulate mesenchymal stem/stromal cell recruitment. Stem Cell Rep (2016) 6:466–73. doi: 10.1016/j.stemcr.2016.02.012
44. Söderström K, Stein E, Colmenero P, Purath U, Müller-Ladner U, Teixeira De Matos C, et al. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. PNAS (2010) 107:13028–33. doi: 10.1073/pnas.1000546107
45. Dastagir N, Beal Z, Godwin J. Tissue origin of cytotoxic natural killer cells dictates their differential roles in mouse digit tip regeneration and progenitor cell survival. Stem Cell Rep (2022) 17:633–48. doi: 10.1016/j.stemcr.2022.01.006
46. Liu W, Gao Y, Li H, Wang H, Ye M, Jiang G, et al. Intravenous transplantation of mesenchymal stromal cells has therapeutic effects in a sepsis mouse model through inhibition of septic natural killer cells. Int J Biochem Cell Biol (2016) 79:93–103. doi: 10.1016/j.biocel.2016.08.013
47. Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, Papamichail M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells (2006) 24:74–85. doi: 10.1634/stemcells.2004-0359
48. Petri RM, Hackel A, Hahnel K, Dumitru CA, Bruderek K, Flohe SB, et al. Activated tissue-resident mesenchymal stromal cells regulate natural killer cell immune and tissue-regenerative function. Stem Cell Rep (2017) 9:985–98. doi: 10.1016/j.stemcr.2017.06.020
49. Könnecke I, Serra A, El Khassawna T, Schlundt C, Schell H, Hauser A, et al. T and B cells participate in bone repair by infiltrating the fracture callus in a two-wave fashion. Bone (2014) 64:155–65. doi: 10.1016/j.bone.2014.03.052
50. Reinke S, Geissler S, Taylor WR, Schmidt-Bleek K, Juelke K, Schwachmeyer V, et al. Terminally differentiated CD8+ T cells negatively affect bone regeneration in humans. Sci Transl Med (2013) 5:177ra36. doi: 10.1126/scitranslmed.3004754
51. Sun G, Wang Y, Ti Y, Wang J, Zhao J, Qian H. Regulatory B cell is critical in bone union process through suppressing proinflammatory cytokines and stimulating Foxp3 in Treg cells. Clin Exp Pharmacol Physiol (2017) 44:455–62. doi: 10.1111/1440-1681.12719
52. Yahara Y, Nguyen T, Ishikawa K, Kamei K, Alman BA. The origins and roles of osteoclasts in bone development, homeostasis and repair. Development (2022) 149:8. doi: 10.1242/dev.199908
53. Le AX, Miclau T, Hu D, Helms JA. Molecular aspects of healing in stabilized and non-stabilized fractures. J Orthop Res (2001) 19:78–84. doi: 10.1016/S0736-0266(00)00006-1
54. Medhat D, Rodríguez CI, Infante A. Immunomodulatory effects of MSCs in bone healing. Int J Mol Sci (2019) 20:5467. doi: 10.3390/ijms20215467
55. Chang MK, Raggatt LJ, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol (2008) 181:1232–44. doi: 10.4049/jimmunol.181.2.1232
56. Alexander KA, Chang MK, Maylin ER, Kohler T, Müller R, Wu AC, et al. Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J Bone Miner Res (2011) 26:1517–32. doi: 10.1002/jbmr.354
57. Chen L, Cheng S, Sun K, Wang J, Liu X, Zhao Y, et al. Changes in macrophage and inflammatory cytokine expressions during fracture healing in an ovariectomized mice model. BMC Musculoskelet Disord (2021) 22:494. doi: 10.1186/s12891-021-04360-z
58. Takeshita S, Fumoto T, Matsuoka K, Park K-A, Aburatani H, Kato S, et al. Osteoclast-secreted CTHRC1 in the coupling of bone resorption to formation. J Clin Invest (2013) 123:3914–24. doi: 10.1172/JCI69493
59. Matsuoka K, Park KA, Ito M, Ikeda K, Takeshita S. Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J Bone Miner Res (2014) 29:1522–30. doi: 10.1002/jbmr.2187
60. Sivaraj KK, Majev P-G, Jeong H-W, Dharmalingam B, Zeuschner D, Schröder S, et al. Mesenchymal stromal cell-derived septoclasts resorb cartilage during developmental ossification and fracture healing. Nat Commun (2022) 13:571. doi:10.1038/s41467-022-28142-w
61. El Khassawna T, Serra A, Bucher CH, Petersen A, Schlundt C, Könnecke I, et al. T lymphocytes influence the mineralization process of bone. Front Immunol (2017) 8:562. doi: 10.3389/fimmu.2017.00562
62. Levy S, Feduska JM, Sawant A, Gilbert SR, Hensel JA, Ponnazhagan S. Immature myeloid cells are critical for enhancing bone fracture healing through angiogenic cascade. Bone (2016) 93:113–24. doi: 10.1016/j.bone.2016.09.018
63. Nam D, Mau E, Wang Y, Wright D, Silkstone D, Whetstone H, et al. T-Lymphocytes Enable Osteoblast Maturation via IL-17F during the Early Phase of Fracture Repair. PloS One (2012) 7:e40044. doi: 10.1371/journal.pone.0040044
64. Zaiss MM, Axmann R, Zwerina J, Polzer K, Gückel E, Skapenko A, et al. Treg cells suppress osteoclast formation: A new link between the immune system and bone. Arthritis Rheumatol (2007) 56:4104–12. doi: 10.1002/art.23138
65. Liu Y-J. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell (2001) 106:259–62. doi: 10.1016/S0092-8674(01)00456-1
66. Colnot C. Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J Bone Miner Res (2009) 24:274–82. doi: 10.1359/jbmr.081003
67. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell (2008) 2:141–50. doi: 10.1016/j.stem.2007.11.014
68. Rafei M, Hsieh J, Fortier S, Li M, Yuan S, Birman E, et al. Mesenchymal stromal cell–derived CCL2 suppresses plasma cell immunoglobulin production via STAT3 inactivation and PAX5 induction. Blood (2008) 112:4991–8. doi: 10.1182/blood-2008-07-166892
69. Suzdaltseva Y, Goryunov K, Silina E, Manturova N, Stupin V, Kiselev SL. Equilibrium among inflammatory factors determines human MSC-mediated immunosuppressive effect. Cells (2022) 11:1210. doi: 10.3390/cells11071210
70. Wang T, Zhang X, Bikle DD. Osteogenic differentiation of periosteal cells during fracture healing. J Cell Physiol (2017) 232:913–21. doi: 10.1002/jcp.25641
71. Debnath S, Yallowitz AR, Mccormick J, Lalani S, Zhang T, Xu R, et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature (2018) 562:133–9. doi: 10.1038/s41586-018-0554-8
72. Knuth C, Andres Sastre E, Fahy N, Witte-Bouma J, Ridwan Y, Strabbing E, et al. Collagen type X is essential for successful mesenchymal stem cell-mediated cartilage formation and subsequent endochondral ossification. Eur Cell Mater (2019) 38:106–22. doi: 10.22203/eCM.v038a09
73. Gerber H-P, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med (1999) 5:623–8. doi: 10.1038/9467
74. Andrew JG, Hoyland JA, Freemont AJ, Marsh DR. Platelet-derived growth factor expression in normally healing human fractures. Bone (1995) 16:455–60. doi: 10.1016/8756-3282(95)90191-4
75. Maes C. Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J Clin Invest (2006) 116:1230–42. doi: 10.1172/JCI26772
76. Bahney CS, Hu DP, Miclau T, Marcucio RS. The multifaceted role of the vasculature in endochondral fracture repair. Front Endocrinol (2015) 6. doi: 10.3389/fendo.2015.00004
77. Palomares KTS, Gleason RE, Mason ZD, Cullinane DM, Einhorn TA, Gerstenfeld LC, et al. Mechanical stimulation alters tissue differentiation and molecular expression during bone healing. J Orthop Res (2009) 27:1123–32. doi: 10.1002/jor.20863
78. Hirao M, Tamai N, Tsumaki N, Yoshikawa H, Myoui A. Oxygen tension regulates chondrocyte differentiation and function during endochondral ossification. J Biol Chem (2006) 281:31079–92. doi: 10.1074/jbc.M602296200
79. Veitch SW, Findlay SC, Hamer AJ, Blumsohn A, Eastell R, Ingle BM. Changes in bone mass and bone turnover following tibial shaft fracture. Osteoporos Int (2006) 17:364–72. doi: 10.1007/s00198-005-2025-y
80. Naik P. Remodelling in children’s fractures and limits of acceptability. Indian J Orthop (2021) 55:549–59. doi: 10.1007/s43465-020-00320-2
81. Bolamperti S, Villa I, Rubinacci A. Bone remodeling: an operational process ensuring survival and bone mechanical competence. Bone Res (2022) 10:48. doi: 10.1038/s41413-022-00219-8
82. Klein P, Schell H, Streitparth F, Heller M, Kassi J-P, Kandziora F, et al. The initial phase of fracture healing is specifically sensitive to mechanical conditions. J Orthop Res (2003) 21:662–9. doi: 10.1016/S0736-0266(02)00259-0
83. Mencio GA, Frick SL, Green NE. Green’s skeletal trauma in children. Sixth edition. ed. Philadelphia, PA. Elsevier/Saunders (2020) 1–15. doi: 10.1016/B978-0-323-18773-2.00001-9
84. Shapiro F. Fractures of the femoral shaft in children: the overgrowth phenomenon. Acta Orthop Scand (1981) 52:649–55. doi: 10.3109/17453678108992162
85. Lu C, Miclau T, Hu D, Hansen E, Tsui K, Puttlitz C, et al. Cellular basis for age-related changes in fracture repair. J Orthop Res (2005) 23:1300–7. doi: 10.1016/j.orthres.2005.04.003.1100230610
86. Emami AJ, Toupadakis CA, Telek SM, Fyhrie DP, Yellowley CE, Christiansen BA. Age dependence of systemic bone loss and recovery following femur fracture in mice. J Bone Miner Res (2019) 34:157–70. doi: 10.1002/jbmr.3579
87. Schell H, Lienau J, Epari DR, Seebeck P, Exner C, Muchow S, et al. Osteoclastic activity begins early and increases over the course of bone healing. Bone (2006) 38:547–54. doi: 10.1016/j.bone.2005.09.018
88. Parfitt AM. The bone remodeling compartment: A circulatory function for bone lining cells. J Bone Miner Res (2001) 16:1583–5. doi: 10.1359/jbmr.2001.16.9.1583
89. Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell (2002) 3:889–901. doi: 10.1016/S1534-5807(02)00369-6
90. Drake FH, Dodds RA, James IE, Connor JR, Debouck C, Richardson S, et al. Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. J Biol Chem (1996) 271:12511–6. doi: 10.1074/jbc.271.21.12511
91. Blair HC, Teitelbaum SL, Ghiselli R, Gluck S. Osteoclastic bone resorption by a polarized vacuolar proton pump. Science (1989) 245:855–7. doi: 10.1126/science.2528207
92. Díaz-Flores L, Gutiérrez R, Madrid JF, Varela H, Valladares F, Acosta E, et al. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol (2009) 24:909–69. doi: 10.14670/HH-24.909
93. Supakul S, Yao K, Ochi H, Shimada T, Hashimoto K, Sunamura S, et al. Pericytes as a source of osteogenic cells in bone fracture healing. Int J Mol Sci (2019) 20:1079. doi: 10.3390/ijms20051079
94. Udagawa N, Koide M, Nakamura M, Nakamichi Y, Yamashita T, Uehara S, et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J Bone Miner Metab (2021) 39:19–26. doi: 10.1007/s00774-020-01162-6
95. Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol (2006) 169:987–98. doi: 10.2353/ajpath.2006.060180
96. Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature (2000) 408:600–5. doi: 10.1038/35046102
97. Morrell AE, Brown GN, Robinson ST, Sattler RL, Baik AD, Zhen G, et al. Mechanically induced Ca2+ oscillations in osteocytes release extracellular vesicles and enhance bone formation. Bone Res (2018) 6:6. doi: 10.1038/s41413-018-0007-x
98. Haffner-Luntzer M, Hankenson KD, Ignatius A, Pfeifer R, Khader BA, Hildebrand F, et al. Review of animal models of comorbidities in fracture-healing research. J Orthop Res (2019) 37:2491–8. doi: 10.1002/jor.24454
99. Haffner-Luntzer M, Kovtun A, Rapp AE, Ignatius A. Mouse models in bone fracture healing research. Curr Mol Biol Rep (2016) 2:101–11. doi: 10.1007/s40610-016-0037-3
100. Vantucci CE, Roy K, Guldberg RE. Immunomodulatory strategies for immune dysregulation following severe musculoskeletal trauma. J Immunol Regener Med (2018) 2:21–35. doi: 10.1016/j.regen.2018.07.001
101. Rapp AE, Bindl R, Recknagel S, Erbacher A, Müller I, Schrezenmeier H, et al. Fracture healing is delayed in immunodeficient NOD/scid−IL2Rγ cnull mice. PloS One (2016) 11:e0147465. doi: 10.1371/journal.pone.0147465
102. Shi J, Hua L, Harmer D, Li P, Ren G. Cre driver mice targeting macrophages. Methods Mol Biol (2018) 1784:263–75. doi: 10.1007/978-1-4939-7837-3_24
103. Zhen G, Dan Y, Wang R, Dou C, Guo Q, Zarr M, et al. An antibody against Siglec-15 promotes bone formation and fracture healing by increasing TRAP+ mononuclear cells and PDGF-BB secretion. Bone Res (2021) 9:47. doi: 10.1038/s41413-021-00161-1
104. Deng L, Zhou J-F, Sellers RS, Li J-F, Nguyen AV, Wang Y, et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am J Pathol (2010) 176:952–67. doi: 10.2353/ajpath.2010.090622
105. Dai X-M, Zong X-H, Akhter MP, Stanley ER. Osteoclast deficiency results in disorganized matrix, reduced mineralization, and abnormal osteoblast behavior in developing bone. J Bone Miner Res (2004) 19:1441–51. doi: 10.1359/JBMR.040514
106. Keshvari S, Caruso M, Teakle N, Batoon L, Sehgal A, Patkar OL, et al. CSF1R-dependent macrophages control postnatal somatic growth and organ maturation. PloS Genet (2021) 17:e1009605. doi: 10.1371/journal.pgen.1009605
107. Hume DA, Batoon L, Sehgal A, Keshvari S, Irvine KM. CSF1R as a Therapeutic Target in Bone Diseases: Obvious but Not so Simple. Curr Osteoporos Rep (2022) 20:516–31. doi: 10.1007/s11914-022-00757-4
108. Yoshida H, Hayashi S-I, Kunisada T, Ogawa M, Nishikawa S, Okamura H, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature (1990) 345:442–4. doi: 10.1038/345442a0
109. Vi L, Baht GS, Whetstone H, Ng A, Wei Q, Poon R, et al. Macrophages promote osteoblastic differentiation in vivo: implications in fracture repair and bone homeostasis. J Bone Miner Res (2015) 30:1090–102. doi: 10.1002/jbmr.2422
110. Sinder BP, Pettit AR, Mccauley LK. Macrophages: their emerging roles in bone. J Bone Miner Res (2015) 30:2140–9. doi: 10.1002/jbmr.2735
111. Dallas SL, Xie Y, Shiflett LA, Ueki Y. Mouse cre models for the study of bone diseases. Curr Osteoporos Rep (2018) 16:466–77. doi: 10.1007/s11914-018-0455-7
112. Garcia P, Histing T, Holstein JH, Klein M, Laschke MW, Matthys R, et al. Rodent animal models of delayed bone healing and non-union formation: a comprehensive review. Eur Cell Mater (2013) 26:1–12. doi: 10.22203/eCM.v026a01
113. Hebb JH, Ashley JW, Mcdaniel L, Lopas LA, Tobias J, Hankenson KD, et al. Bone healing in an aged murine fracture model is characterized by sustained callus inflammation and decreased cell proliferation. J Orthop Res (2017) 36:149–58. doi: 10.1002/jor.23652
114. Lu C, Miclau T, Hu D, Marcucio RS. Ischemia leads to delayed union during fracture healing: A mouse model. J Orthop Res (2007) 25:51–61. doi: 10.1002/jor.20264
115. Tang G, Charo DN, Wang R, Charo IF, Messina L. CCR2-/- knockout mice revascularize normally in response to severe hindlimb ischemia. J Vasc Surg (2004) 40:786–95. doi: 10.1016/j.jvs.2004.07.012
116. Miedel E, Dishowitz MI, Myers MH, Dopkin D, Yu YY, Miclau TS, et al. Disruption of thrombospondin-2 accelerates ischemic fracture healing. J Orthop Res (2013) 31:935–43. doi: 10.1002/jor.22302
117. Retzepi M, Donos N. The effect of diabetes mellitus on osseous healing. Clin Oral Implants Res (2010) 21:673–81. doi: 10.1111/j.1600-0501.2010.01923.x
118. Vester H, Huber-Lang MS, Kida Q, Scola A, Van Griensven M, Gebhard F, et al. The immune response after fracture trauma is different in old compared to young patients. Immun Ageing (2014) 11:20. doi: 10.1186/s12979-014-0020-x
119. Lopez EM, Leclerc K, Ramsukh M, Parente PE, Patel K, Aranda CJ, et al. Modulating the systemic and local adaptive immune response after fracture improves bone regeneration during aging. Bone (2022) 157:116324. doi: 10.1016/j.bone.2021.116324
120. Batoon L, Millard SM, Raggatt LJ, Wu AC, Kaur S, Sun LWH, et al. Osteal macrophages support osteoclast-mediated resorption and contribute to bone pathology in a postmenopausal osteoporosis mouse model. J Bone Miner Res (2021) 36:2214–28. doi: 10.1002/jbmr.4413
121. Cline-Smith A, Axelbaum A, Shashkova E, Chakraborty M, Sanford J, Panesar P, et al. Ovariectomy activates chronic low-grade inflammation mediated by memory T cells, which promotes osteoporosis in mice. J Bone Miner Res (2020) 35:1174–87. doi: 10.1002/jbmr.3966
122. Dou C, Ding N, Zhao C, Hou T, Kang F, Cao Z, et al. Estrogen deficiency-mediated M2 macrophage osteoclastogenesis contributes to M1/M2 ratio alteration in ovariectomized osteoporotic mice. J Bone Miner Res (2018) 33:899–908. doi: 10.1002/jbmr.3364
123. Laguna E, Pérez-Núñez MI, Del Real Á, Menéndez G, Sáinz-Aja JA, López-Delgado L, et al. Effects of systemic or local administration of mesenchymal stem cells from patients with osteoporosis or osteoarthritis on femoral fracture healing in a mouse model. Biomolecules (2022) 12:722. doi: 10.3390/biom12050722
124. Zwingenberger S, Niederlohmann E, Vater C, Rammelt S, Matthys R, Bernhardt R, et al. Establishment of a femoral critical-size bone defect model in immunodeficient mice. J Surg Res (2013) 181:e7–e14. doi: 10.1016/j.jss.2012.06.039
125. Toben D, Schroeder I, El Khassawna T, Mehta M, Hoffmann J-E, Frisch J-T, et al. Fracture healing is accelerated in the absence of the adaptive immune system. J Bone Miner Res (2011) 26:113–24. doi: 10.1002/jbmr.185
126. Aurora R, Silva MJ. T cells heal bone fractures with help from the gut microbiome. J Clin Invest (2023) 133:8. doi: 10.1172/JCI167311
127. Van Norman GA. Limitations of animal studies for predicting toxicity in clinical trials: is it time to rethink our current approach? JACC Basic Transl Sci (2019) 4:845–54. doi: 10.1016/j.jacbts.2019.10.008
128. Pfeiffenberger M, Damerau A, Ponomarev I, Bucher CH, Chen Y, Barnewitz D, et al. Functional scaffold-free bone equivalents induce osteogenic and angiogenic processes in a human in vitro fracture hematoma model. J Bone Miner Res (2021) 36:1189–201. doi: 10.1002/jbmr.4267
129. Pfeiffenberger M, Bartsch J, Hoff P, Ponomarev I, Barnewitz D, Thöne-Reineke C, et al. Hypoxia and mesenchymal stromal cells as key drivers of initial fracture healing in an equine in vitro fracture hematoma model. PloS One (2019) 14:e0214276. doi: 10.1371/journal.pone.0214276
130. Borciani G, Montalbano G, Baldini N, Cerqueni G, Vitale-Brovarone C, Ciapetti G. Co-culture systems of osteoblasts and osteoclasts: Simulating in vitro bone remodeling in regenerative approaches. Acta Biomater (2020) 108:22–45. doi: 10.1016/j.actbio.2020.03.043
131. Hoff P, Maschmeyer P, Gaber T, Schütze T, Raue T, Schmidt-Bleek K, et al. Human immune cells’ behavior and survival under bioenergetically restricted conditions in an in vitro fracture hematoma model. Cell Mol Immunol (2013) 10:151–8. doi: 10.1038/cmi.2012.56
132. Sridharan R, Genoud KJ, Kelly DJ, O’Brien FJ. Hydroxyapatite particle shape and size influence MSC osteogenesis by directing the macrophage phenotype in collagen-hydroxyapatite scaffolds. ACS Appl Bio Mater (2020) 3:7562–74. doi: 10.1021/acsabm.0c00801
133. Lafuente-Gracia L, Borgiani E, Nasello G, Geris L. Towards in silico models of the inflammatory response in bone fracture healing. Front Bioeng Biotechnol (2021) 9:703725. doi: 10.3389/fbioe.2021.703725
134. Ural A. Advanced modeling methods—Applications to bone fracture mechanics. Curr Osteoporos Rep (2020) 18:568–76. doi: 10.1007/s11914-020-00615-1
135. Kojouharov HV, Trejo I, Chen-Charpentier BM. Modeling the effects of inflammation in bone fracture healing, in: AIP Conf, (2017) 1895:1. doi: 10/1063/1.5007359.
136. Trejo I, Kojouharov H, Chen-Charpentier B. Modeling the macrophage-mediated inflammation involved in the bone fracture healing process. Math Comput Appl (2019) 24:12. doi: 10.3390/mca24010012
137. Trejo I. Kojouharov HV. A mathematical model to study the fundamental functions of phagocytes and inflammatory cytokines during the bone fracture healing process. Lett Biomath (2020) 7:171–89. doi: 10.30707/LiB7.1.1647875326.052507
138. Ghiasi MS, Chen JE, Rodriguez EK, Vaziri A, Nazarian A. Computational modeling of human bone fracture healing affected by different conditions of initial healing stage. BMC Musculoskelet Disord (2019) 20:562. doi: 10.1186/s12891-019-2854-z
139. Borgiani E, Nasello G, Ory L, Herpelinck T, Groeneveldt L, Bucher CH, et al. COMMBINI: an experimentally-informed COmputational Model of Macrophage dynamics in the Bone INjury Immunoresponse. Front Immunol (2023) 14. doi: 10.3389/fimmu.2023.1231329
140. Tammaro A, Kers J, Scantlebery AML, Florquin S. Metabolic flexibility and innate immunity in renal ischemia reperfusion injury: the fine balance between adaptive repair and tissue degeneration. Front Immunol (2020) 11:1346. doi: 10.3389/fimmu.2020.01346
141. Abbasi-Habashi S, Jickling GC, Winship IR. Immune modulation as a key mechanism for the protective effects of remote ischemic conditioning after stroke. Front Neurol (2021) 12:746486. doi: 10.3389/fneur.2021.746486
Keywords: fracture healing, osteoimmunology, inflammation, bone, crosstalk
Citation: Capobianco CA, Hankenson KD and Knights AJ (2024) Temporal dynamics of immune-stromal cell interactions in fracture healing. Front. Immunol. 15:1352819. doi: 10.3389/fimmu.2024.1352819
Received: 08 December 2023; Accepted: 06 February 2024;
Published: 22 February 2024.
Edited by:
Yasser M. El-Sherbiny, Nottingham Trent University, United KingdomReviewed by:
Elisabeth Seebach, Heidelberg University Hospital, GermanyCopyright © 2024 Capobianco, Hankenson and Knights. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander J. Knights, YWtuaWdodHNAdW1pY2guZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.