
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 22 February 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1348131
This article is part of the Research TopicCommunity Series in Autoimmune Diabetes: Molecular Mechanisms and Neoantigens, Volume IIView all 7 articles
 Janet M. Wenzlau1
Janet M. Wenzlau1 Orion J. Peterson2,3
Orion J. Peterson2,3 Anthony N. Vomund2,3
Anthony N. Vomund2,3 James E. DiLisio1
James E. DiLisio1 Anita Hohenstein1
Anita Hohenstein1 Kathryn Haskins1*
Kathryn Haskins1* Xiaoxiao Wan2,3*
Xiaoxiao Wan2,3*There is accumulating evidence that pathogenic T cells in T1D recognize epitopes formed by post-translational modifications of β-cell antigens, including hybrid insulin peptides (HIPs). The ligands for several CD4 T-cell clones derived from the NOD mouse are HIPs composed of a fragment of proinsulin joined to peptides from endogenous β-cell granule proteins. The diabetogenic T-cell clone BDC-6.9 reacts to a fragment of C-peptide fused to a cleavage product of pro-islet amyloid polypeptide (6.9HIP). In this study, we used a monoclonal antibody (MAb) to the 6.9HIP to determine when and where HIP antigens are present in NOD islets during disease progression and with which immune cells they associate. Immunogold labeling of the 6.9HIP MAb and organelle-specific markers for electron microscopy were employed to map the subcellular compartment(s) in which the HIP is localized within β-cells. While the insulin B9-23 peptide was present in nearly all islets, the 6.9HIP MAb stained infiltrated islets only in NOD mice at advanced stages of T1D development. Islets co-stained with the 6.9HIP MAb and antibodies to mark insulin, macrophages, and dendritic cells indicate that 6.9HIP co-localizes within insulin-positive β-cells as well as intra-islet antigen-presenting cells (APCs). In electron micrographs, the 6.9HIP co-localized with granule structures containing insulin alone or both insulin and LAMP1 within β-cells. Exposing NOD islets to the endoplasmic reticulum (ER) stress inducer tunicamycin significantly increased levels of 6.9HIP in subcellular fractions containing crinosomes and dense-core granules (DCGs). This work demonstrates that the 6.9HIP can be visualized in the infiltrated islets and suggests that intra-islet APCs may acquire and present HIP antigens within islets.
A broad variety of self-antigens have been implicated in autoimmune diabetes, but only a subset of these is likely to be involved in disease initiation, with subsequent epitope spreading to other antigens. In the NOD mouse, substantial evidence supports proinsulin as the predominant instigating antigen (1–3). Efforts to define which proinsulin-derived epitopes are most pertinent to disease etiology have included a wide variety of approaches, including genetics, biochemistry, confocal and electron microscopy (EM), and targeted mass spectrometry. These studies have revealed different forms of insulin-derived epitopes, including native epitopes of insulin B-chain and C-peptide, as well as a class of modified antigens, hybrid insulin peptides (HIPs) resulting from peptide fusion, all of which contribute to the activation of pathogenic CD4 T cells.
Insulin secretory granules are considered a repository of antigenic targets involved in autoimmune diabetes (4). However, the subcellular localization of immunogenic insulin-derived epitopes in β-cell granules remains largely unexplored. The primary secretory granule in β-cells is the insulin dense-core granule (DCG), containing a crystal core of insulin. The DCG has diverse biological functions, such as proinsulin processing and insulin secretion. Maintaining insulin homeostasis in β-cells is achieved by fusing excessive DCGs with lysosomes for catabolic reduction. This crinophagic pathway generates a minor set of β-cell granules, termed crinophagic bodies or crinosomes, which possess lysosomal activities to catabolize secretory proteins in DCGs (5–7).
The subcellular distribution of insulin-derived antigens that elicit the most robust T-cell responses within β-cells has been probed using differential centrifugation to fractionate DCGs and crinosomes with subsequent mass spectrometry to identify the peptides therein (8). The DCG fraction obtained at a high speed (25,000 × g) primarily contained intact insulin, long B-chain peptides, and some C-peptides, whereas the granule fraction obtained at a lower speed (5,000 × g), representing crinosomes, was enriched for shorter insulin peptides, including shorter B-chain peptides associated with the predominant epitope B9-23 and diverse C-peptide sequences. Further investigation of crinosome-derived peptides, initially by manual investigation of unassigned spectra, revealed a few HIP sequences (8), suggesting the presence of HIPs in crinosomes. These results were later confirmed in a subsequent study (9) by searching the crinosome peptidome against an in silico HIP database (10). This approach also identified HIPs in the secretome (peptide contents exocytosed from β-cells upon glucose stimulation) (9), raising the possibility that HIPs are also present in DCGs.
HIPs are the product of a unique post-translational modification whereby insulin peptide fragments are fused to sequences from endogenous proteolytically processed β-cell secretory granule proteins. One such HIP, the 6.9HIP, is composed of a fragment of C-peptide fused to IAPP2 (DLQTLAL/NAARD) and is the antigenic ligand of diabetogenic CD4+ T-cell clone BDC-6.9 from the NOD mouse (11). T cells reactive to the human counterpart of this HIP and others have been identified in PBMCs from T1D patients (12) and distinct HIP-reactive T cells have been cultured from the islets of deceased T1D organ donors (13–16). Importantly, a nearly identical sequence to the 6.9HIP (LQTLAL/NAARD) was identified in the MHC-II (I-Ag7) peptidomes of both pancreatic islets and draining lymph nodes in NOD mice (9), indicating that 6.9HIP is a bona fide neoepitope that is presented in vivo by MHC-II.
Mapping the intracellular site of HIP formation is challenging, yet essential for putative intervention strategies. Crinosomes and DCGs are both favorable environments for HIP formation due to high concentrations of the peptide components of HIPs and enzymes that function at low pH. Proposed mechanisms for HIP formation via enzymes cathepsin D and cathepsin L have been reported from in vitro studies (17, 18). A monoclonal antibody (MAb) specific for the insulin B-chain sequences B9-23, but not native insulin, has been employed to localize the chief epitope B9-23 to crinosomes (8). The B9-23 MAb co-localizes with the lysosomal marker LAMP1 in vesicles distinct from DCGs, providing a punctate staining pattern with immunofluorescent microscopy. Diabetogenic effector T cells reactive to the B12-20 epitope are responsive to the contents of these crinosomes, suggesting that other critical antigens could be present in the same organelles (8, 19). In the current study, we developed a MAb to the C-peptide/IAPP2 (6.9) HIP, previously identified by mass spectrometry, to determine whether such rare post-translationally modified peptides can be detected through microscopic analysis of antibody staining. The primary goals were to establish in which cellular compartment the 6.9HIPs are localized and with what type of antigen-presenting cells (APCs) they may be associated.
NOD/ShiLtJ (NOD) mice were originally obtained from The Jackson Laboratory. All mice were bred, maintained, and used in experiments in our pathogen-free animal facility in accordance with the guidelines of the Division of Comparative Medicine at Washington University School of Medicine (Association for Assessment and Accreditation of Laboratory Animal Care accreditation no. A3381-01). NOD.IAPP−/− mice were previously bred in the Haskins mouse colony by backcrossing C57BL6.IAPP−/− mice (20) onto the NOD background (21).
BALB/c mice were immunized three times, 2 weeks apart with 100 µg/each of the NOD 6.9HIP (SLDQLALNAARDPN) conjugated to KLH and combined 1:1 with CFA, IFA, and IFA, respectively. Sera were monitored for MAb titer with enzyme-linked immunosorbent assays (ELISAs) and radio binding assays [as in (22)] using a recombinant probe expressing the NOD 6.9HIP. A MAb targeting the NOD 6.9HIP sequence was developed as previously described (23). Specificity of the 6.9HIP MAb was determined using Western blots and in-house ELISAs, using anti-mouse HRP 1:10,000 (Pierce) and quantitation with o-phenylenediamine dihydrochloride substrate at 490 nm as per vendor recommendations (Sigma).
Mouse HIPs were cloned in-frame with fusion partner NUS in vector pET43a (Novagen) for expression in bacteria. Induced proteins were purified on Ni-Agarose (Qiagen) according to the manufacturer’s instructions. 6.9HIP and control peptides (>95% purity, Genscript or Peptide 2.0) were resuspended at a concentration of 10 mg/mL according to solubility recommendations.
Ninety-six-well ELISA plates were coated with 6.9HIP peptide (LQTLAL/NAARD, 2 μM) in carbonate buffer overnight at 4°C. Plates were washed and subsequently blocked with DMEM/5% fetal bovine serum (FBS). The 6.9HIP MAb was serially diluted and directly added to the coated plate. Horseradish peroxide (HRP)-conjugated goat anti-mouse IgG (1:10,000) antibody was then added for 2 h at 4°C, the responses were measured using the OptEIA TMB Substrate (BD), and the data [optical density (OD) at 450 nm] were collected using an iMark Microplate Reader (Bio-Rad Laboratories).
ELISA plates were coated with 6.9HIP peptide (2 μM) in carbonate buffer overnight at 4°C. Plates were washed and subsequently blocked with DMEM/5% FBS. Soluble competitive inhibitors (synthetic peptides) were pre-incubated with the 6.9HIP MAb (50 ng/mL) for 30 min and the mixture was incubated with the plate-bound antigens for 2 h at 37°C. In the absence of soluble competitive inhibitors, the concentration of the MAb resulted in approximately 50% binding to the plate-bound antigen. HRP-conjugated goat anti-mouse IgG (1:10,000) antibody was then added for 2 h at 4°C, the responses were quantified using the OptEIA TMB Substrate (BD), and the data (OD at 450 nm) were collected using an iMark Microplate Reader (Bio-Rad Laboratories).
Mouse peritoneal cavities were opened to clamp the common bile duct leading to the duodenum. Type XI collagenase (0.4 mg/mL in serum-free DMEM; Sigma-Aldrich) digestion buffer was injected through the bile duct to inflate the pancreas, which was removed and incubated in digestion buffer at 37°C (12 min) and then shaken vigorously for 90 s. Pancreata were washed with serum-free (SF) DMEM (3×) through a stainless-steel strainer. The flow-through, containing islets, was then filtered through a 70-µm cell strainer. Retained islets were then flushed into a Petri dish for hand-picking. The NOD.IAPP−/− islets were prepared similarly by infusion of pancreata with CIzyme RI (VitaCyte) through the bile duct followed by gravity filtration washes (as above). Islets were enriched with gradient centrifugation using Lympholyte 1.1 (Cedarlane Laboratories) where they concentrate at the interface and were hand-dissected. Pure hand-picked islets were dispersed using Cell Dissociation Solution Non-Enzymatic (Sigma-Aldrich) for 10 min at 37°C.
Intact islets were fixed in 4% formaldehyde, permeabilized, and blocked in 0.2% saponin/3% bovine serum albumin (BSA). Primary 6.9HIP or AIP antibody (2 µg/mL) was used to stain islets overnight in 0.2% saponin/3% BSA. Anti-mouse-AF594 secondary antibody (4 µg/mL) was permitted to bind islets for 1 h in saponin/BSA. Islets were washed with saponin/BSA and mounted on slides with prolong diamond antifade mounting media (Invitrogen). Islets were blocked with saponin/BSA and then stained with antibodies to cell surface markers (CD11c-488 or CD4-BV480, SIRPα-AF488, and F4/80-AF594) at 2 µg/mL for 1 h followed by washing with saponin/BSA for imaging. For staining with conjugated 6.9HIP MAb, islets were fixed in 4% formaldehyde, permeabilized and blocked with saponin/BSA, and stained with 6.9HIP-AF647 at 0.033 µg/mL overnight in saponin/BSA. Islets were washed and mounted as above for imaging using an inverted Leica SP8 confocal scanning microscope with Leica’s LAS X software. The microscope was equipped with a 63× 1.40 numerical aperture (NA) oil-immersion objective and a white light laser. Optical sections were taken every 0.75 µm. Images shown were taken with Imaris 9.0 software.
For immunolocalization at the ultrastructural level, islets were fixed in 4% paraformaldehyde/0.05% glutaraldehyde (Polysciences) in 100 mM PIPES/0.5 mM MgCl2, pH 7.2, for 1 h at 4°C. Samples were then embedded in 10% gelatin and infiltrated overnight with 2.3 M sucrose/20% polyvinyl pyrrolidone in PIPES/MgCl2 at 4°C. Samples were trimmed, frozen in liquid nitrogen, and sectioned with a Leica Ultracut UCT7 cryo-ultramicrotome (Leica Microsystems). Ultrathin sections of 50 nm were blocked with 5% FBS/5% normal goat serum for 30 min and subsequently incubated with indicated primary antibodies for 1 h at room temperature. Following washes in block buffer, sections were then incubated with goat anti-mouse IgG conjugated to 18-nm colloidal gold, anti-rat IgG conjugated to 12-nm colloidal gold, and goat anti-rabbit IgG antibody conjugated to 6-nm colloidal gold (Jackson ImmunoResearch Laboratories) for 1 h. Sections were stained with 0.3% uranyl acetate/2% methyl cellulose and viewed on a JEOL 1200 EX transmission electron microscope (JEOL) equipped with an AMT 8-megapixel digital camera and AMT Image Capture Engine V602 software (Advanced Microscopy Techniques). All labeling experiments were conducted in parallel with controls omitting the primary antibody.
T-cell hybridomas were generated by fusion of activated lymph node cells elicited by 6.9HIP peptide immunization with BW5147 fusion partner following standard protocols. The T-cell hybridoma clones were screened for reactivity with the 6.9HIP peptide using the C3.g7 APC line in antigen assays. IL-2 production was assessed via IL-2 ELISA.
Whole islets were isolated from NOD mice as described and dispersed for 3 min at 37°C using non-enzymatic cell dissociation buffer (Millipore Sigma). Cells were collected by centrifugation, washed with PBS, and counted. Cells were resuspended in DMEM/10% FBS low-glucose media (1 g/L glucose, Gibco, D10F-LG) and split into 1.0 mL aliquots in microfuge tubes for treatment overnight at 37°C in D10F-LG or D10F-LG containing 100 µM tauroursodeoxycholic acid (TUDCA; Millipore Sigma). Tunicamycin (Millipore Sigma) was added at 1 µg/mL to appropriate samples and was incubated for 2 h at 37°C, after which cells were collected, washed, and counted for antigen assays.
Islet cells were lysed in 1.0 mL of phosphate-buffered saline (PBS) by passage through a cell homogenizer (Isobiotec) 5×. The cell lysate was spun 2× for 10 min at 500 × g at 4°C to remove cell debris. The supernatants were then sequentially spun 2× for 10 min at 5,000 × g at 4°C, and the cell pellets were resuspended in media and pooled as the crinosome fraction. The supernatant was spun for 30 min at 25,000 × g at 4°C, and the pellet was resuspended in media as the DCG fraction. For presentation, the isolated crinosomes (5,000 × g pellet) or DCGs (25,000 × g pellet) were offered to the C3.g7 APC line (B cell lymphoma expressing I-Ag7; 5 × 104/well) in a 96-well culture plate. After a 2 h incubation, CD4+ T-cell hybridomas (5 × 104/well) were added.
APCs were cultured overnight with T-cell hybridomas (6.9-11, 6.9-51, and 6.9-61) recognizing the 6.9HIP. The T-cell responses were tested for IL-2 production measured by IL-2 ELISA (Bectin Dickinson) coupled with streptavidin-poly HRP (Thermo Fisher). The responses were quantified using the OptEIA TMB Substrate (Bectin Dickinson), and the data (OD at 450 nm) were collected using an iMark Microplate Reader (Bio-Rad Laboratories).
Within the islets, HIPs are present at very low concentrations (13). To evaluate the specificity of the putative 6.9HIP (SLDQLAL/NAARDPN) MAb, we generated a set of NUS/HIP recombinant fusion proteins (expressed in the vector pET43) including the NOD 6.9HIP, the BALB/c 6.9HIP, the 2.5HIP, and a human C-peptide/IAPP2 HIP and queried the ability of 6.9HIP MAb to bind to each using Western analysis (Figures 1A, B). Results showed that the 6.9MAb bound specifically to the NOD 6.9HIP fusion protein and not to the 2.5HIP fusion protein, which shares the identical seven-amino-acid C-peptide region (Figures 1A, B, blue). In a similar experiment, fusion proteins 6.9HIP BALB/c and 6.9HIP NOD, which differ by one amino acid within the IAPP2 segment (R>G), and a human peptide (Hu CpepG3/IAPP2) with significant differences in both C-peptide and IAPP2 regions were all detected by blotting with an anti-HIS antibody, indicating approximately equal protein loading (Figure 1B, left) but only the 6.9HIP NOD peptide was detected by the 6.9HIP MAb (Figure 1B, right).
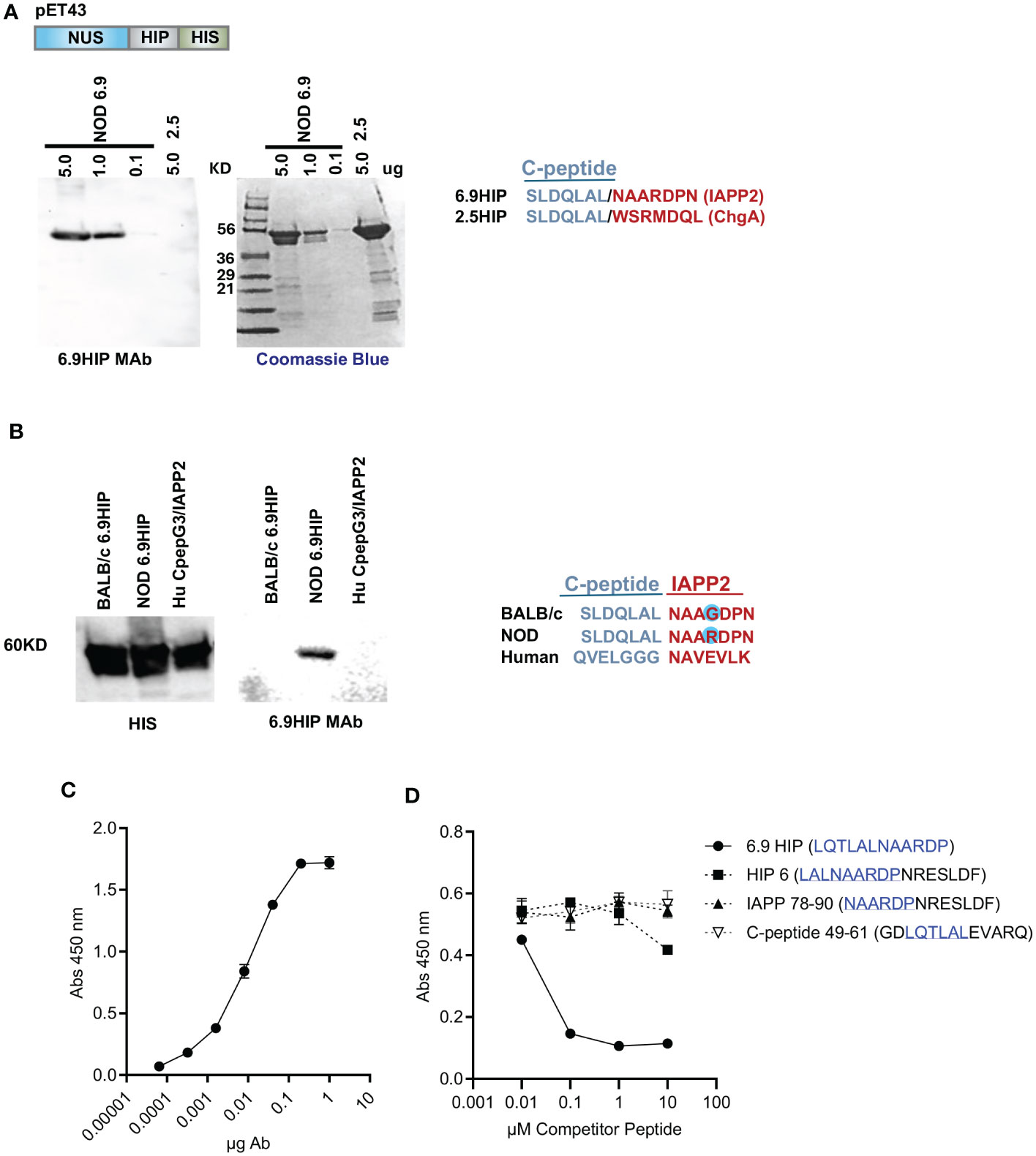
Figure 1 Characterization of 6.9HIP MAb. (A) Western blot (with 6.9MAb) and Coomassie blue-stained gel with titration of recombinant NUS-6.9HIP (5.0–0.1 mg) and NUS-2.5HIP (5 mg) fusion proteins. HIP sequences are aligned on the right. (B) Western blots with recombinant BALB/c 6.9HIP, NOD 6.9HIP, and HuC-pepG3/IAPP2 HIP proteins probed with HIS and 6.9HIP primary antibodies and goat anti-mouse-HRP secondary antibodies. HIP sequences are aligned below. (C) ELISA showing the binding of the 6.9HIPMAb to plate-bound 6.9HIP peptide. (D) Competitive ELISA showing the inhibition of the 6.9MAb:6.9HIP binding by indicated competitor peptides. The peptide sequences related to the 6.9HIP are underlined.
ELISA confirmed that reactivity of 6.9HIP MAb against plate-bound 6.9HIP peptide (LQTLAL/NAARDP) was strictly dose-dependent (Figure 1C). To test the level of specificity, a competitive ELISA was performed, in which the 6.9HIP MAb was preincubated with different concentrations of one of four competitor peptides, including the synthetic 6.9HIP and three other peptides containing a portion of the 6.9HIP (see Figure 1D for details). The 6.9HIP MAb:peptide mixtures were then incubated with the plate-bound 6.9HIP peptide, and the binding was assayed by ELISA. As shown in Figure 1D, only the 6.9HIP MAb:6.9HIP peptide combination blocked the binding of the 6.9HIP MAb to plate-bound 6.9HIP, indicating the high specificity of the MAb for 6.9HIP.
MAb AIP, specific for the insulin B9-23 peptide, has previously been used to identify the location of the B9-23 peptide in pancreatic islets by confocal imaging (8). We performed intracellular staining with unconjugated AIP MAb in non-sectioned, intact islets obtained from 6-week-old male NOD or NOD.Rag1-/- mice. After extensive washing, the signal was visualized by a fluorochrome-conjugated anti-mouse IgG F(ab′)2 secondary antibody. In young (6 weeks) NOD (Figure 2A) and NOD.Rag1-/- (Figure 2B) mice, we observed strong signals from the AIP antibody, which manifested a speckled pattern of staining, indicating the presence of the B9-23 peptide under conditions of low islet inflammation. It should be noted that the secondary anti-mouse IgG F(ab′)2 antibody also recognized endogenous IgG deposited onto islet-resident macrophages known to express Fcγ receptors at a high level (24, 25). This macrophage staining (Figure 2A, blue arrow) was distinguishable from the peptide staining (speckled pattern; Figure 2A red arrow) and was only observed in islets from WT NOD (Figure 2A) but not NOD.Rag1-/- mice that lack endogenous IgG (Figure 2B).
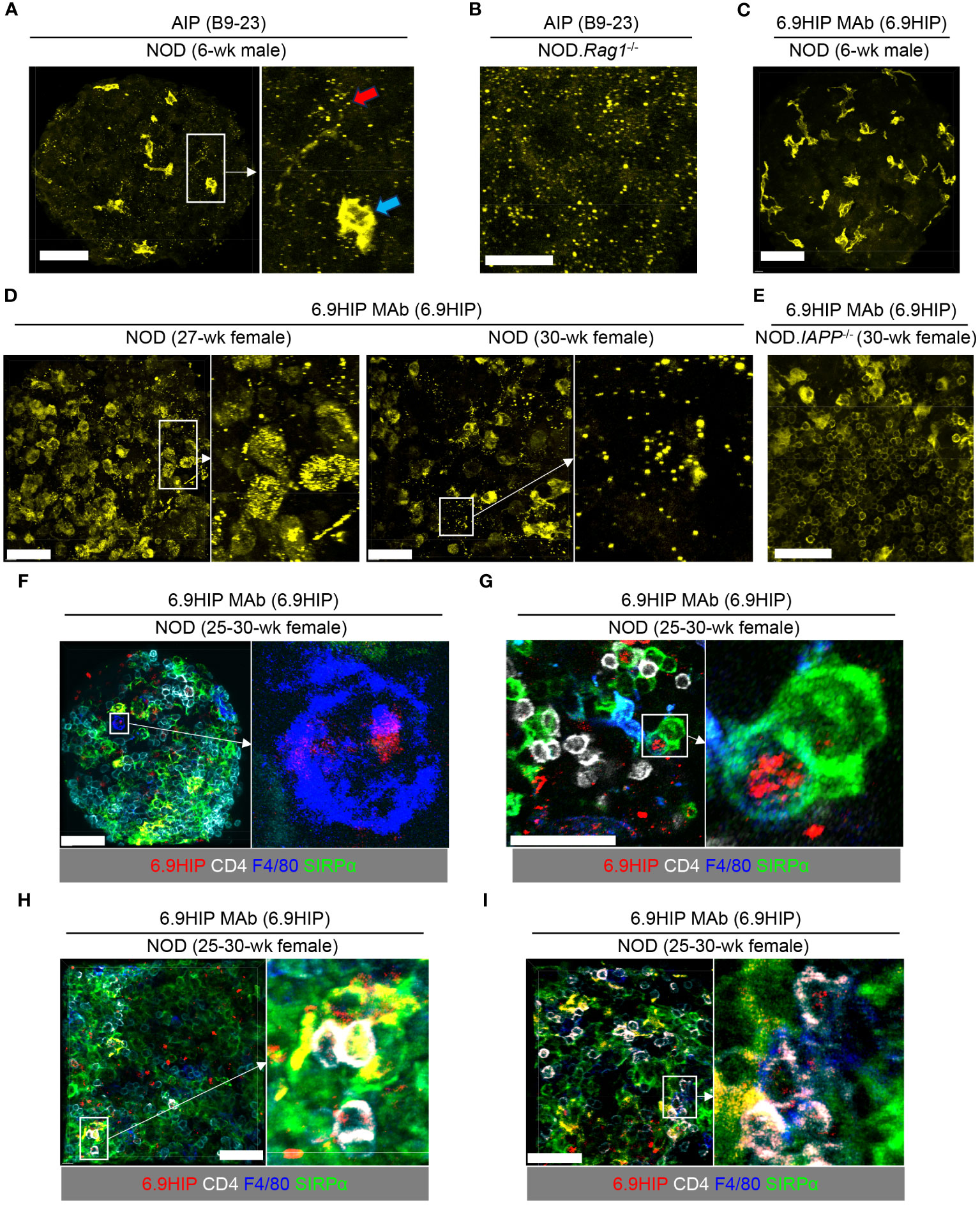
Figure 2 Visualization of 6.9HIP in inflamed islets by confocal microscopy. (A–E) Intact islets isolated from NOD mice of indicated sex and age were stained with unconjugated AIP (A, B) or 6.9HIP MAbs (C–E) followed by a secondary fluorochrome-conjugated anti-mouse IgG F(ab)2 antibody. (A) A representative islet (left) from 6-week-old male NOD mice stained with the AIP antibody. A selected region (rectangle) is enlarged in inset (right), showing punctate peptide (B9-23) staining (red arrow), and staining of intra-islet phagocytes by secondary antibody (blue arrow). (B) A representative islet from NOD.Rag1-/- mice stained with the AIP antibody. (C) A representative islet from 6-week-old male NOD mice stained with the 6.9HIP MAb. (D) Two representative islets from 27- and 30-week-old female NOD mice stained with the 6.9HIP MAb. Enlarged images on the right show the punctate staining of the 6.9HIP distributed within the islets. (E) A representative islet from 30-week-old female NOD.IAPP-/- mice stained with the 6.9HIP MAb. (F–I) Intact islets isolated from 25- to 30-week-old female NOD mice were stained with fluorochrome-conjugated antibodies to 6.9HIP (red), CD4 (white), F4/80 (blue), and Sirpα (green). (F, G) Representative islets showing the co-localization of 6.9HIP with F4/80+ macrophages (F) or Sirpα+ cDC2 (G). (H, I) Representative islets showing the presence of 6.9HIP when CD4+ T cells interact with Sirpα+ cDC2 (H) or F4/80+ macrophages (I). White scale bars in all images are 50 µm.
We then employed the 6.9MAb to visualize the 6.9HIP in islets. We initially used the same staining protocol as described above. To ask whether 6.9HIP could be detected when islet infiltration is minimal, we examined islets isolated from 6-week-old male WT NOD mice. In contrast to results seen in Figures 2A, B for detection of B9-23, we did not observe detectable peptide staining in these islets with the 6.9HIP MAb (Figure 2C), suggesting the minimal presence of the 6.9HIP in young mice. Considering that 6.9HIP-specific CD4+ T cells have been shown to accumulate in pancreatic islets in NOD mice at advanced stages of diabetes (26), we hypothesized that levels of 6.9HIP might increase in islets from aged NOD mice. To test this, we stained islets from 25- to 30-week-old non-diabetic female NOD mice with the 6.9HIP MAb. In contrast to results seen in young mice, we found a prominent presence of 6.9HIP in infiltrated islets at both 25- and 30-week time points (Figure 2D). Like B9-23, the 6.9HIP also exhibited a punctate staining profile within the islets (Figure 2D). These results suggest that autoinflammatory responses in aged NOD mice can promote the formation of the 6.9HIP in pancreatic islets.
To further confirm the specificity of the 6.9HIP staining, we examined islets isolated from age/sex-matched NOD mice lacking IAPP (NOD.IAPP-/-), in which 6.9HIP should be absent. Although islets from NOD.IAPP-/- mice also displayed background staining of intra-islet phagocytes, no detectable punctate staining of 6.9HIP was observed (Figure 2E), confirming the specificity of the 6.9HIP observed in WT NOD mice. To ask if 6.9HIP could be taken up by intra-islet APCs for antigen presentation to T cells, we used fluorochrome-conjugated 6.9HIP MAb to stain infiltrated islets from 25- to 30-week-old non-diabetic female NOD mice, in conjunction with antibody markers that identify CD4+ T cells (anti-CD4), type 2 conventional dendritic cells (cDC2 and anti-SIRPa), and islet macrophages (anti-F4/80). We observed co-localization of 6.9HIP with islet macrophages (F4/80+, Figure 2F) and cDC2 dendritic cells (SIRPα+, Figure 2G) in different islets. Such localization was also noted when islet-infiltrating CD4+ T cells were in close contact with cDC2 dendritic cells (Figure 2H) and islet macrophages (Figure 2I). We examined a total of 150 islet macrophages and 321 cDC2. Among these, 26 macrophages (17.3%) and 47 cDC2 (14.6%) showed co-localization with 6.9HIP, supporting that 6.9HIP is a low-abundance antigen. Additionally, we did not observe detectable 6.9HIP signals in islets of 27-week-old female NOD.Rag1-/- mice (Supplementary Figure 1A), whereas the presence of B9-23 was evident (Supplementary Figure 1B). These results further confirm the association of 6.9HIP with islet inflammation. Overall, our data support the hypothesis that the 6.9HIP can be presented by different intra-islet APCs to CD4+ T cells in inflamed islets.
Visualization of 6.9HIP in pancreatic islets by confocal microscopy prompted us to further analyze its localization in β-cells at the ultrastructural level. To achieve this, we employed immunogold EM with the 6.9HIP MAb. This method utilizes secondary antibodies conjugated with colloidal gold of different sizes, enabling the visualization of subcellular locations of specific markers (identified by primary antibodies) by EM. Specifically, 6.9HIP was visualized by staining with the 6.9HIP MAb, followed by the addition of a secondary anti-mouse IgG antibody conjugated to 18-nm gold. To test the hypothesis that 6.9HIP might be localized in β-cell granules, we also included a rabbit anti-insulin antibody paired with secondary anti-rabbit IgG (6-nm) and a rat anti-LAMP1 antibody paired with secondary anti-rat IgG (12-nm). These combinations allowed us to identify granule structures associated with the DCGs, lysosomes, and crinosomes.
Given that the success of immunogold EM depends highly on the antibody clone used, we sought to determine the specificity of the 6.9HIP MAb using infiltrated islets from non-diabetic female NOD (30-week-old) mice. We used a rigorous approach in which the 6.9HIP MAb was used as the only primary antibody but was followed by the addition of all three secondary antibodies: anti-mouse IgG (18-nm), anti-rat IgG (12-nm), and anti-rabbit IgG (6-nm). The majority of the observed spots were of the 18-nm size, indicating the binding of the 6.9HIP MAb specifically with the anti-mouse IgG (Supplementary Figure 2A). Spots with a 12- or 6-nm size were only occasionally observed (data not shown). Using the same approach, we tested the primary anti-insulin (Supplementary Figure 2B) or anti-LAMP1 (Supplementary Figure 2C) antibody in islets isolated from the same mice. Both primary antibodies showed specific interactions with their respective secondary antibodies with a rare background. Overall, these results validated the high specificity of the 6.9HIP MAb and confirmed its suitability for use in immunogold EM.
We then performed triple labeling using all three primary antibodies to assess the localization of the 6.9HIP in different β-cell granules. Within the same β-cell, we identified granule compartments containing insulin alone (DCGs), LAMP1 alone (lysosomes), or both insulin and LAMP1 (crinosomes) (Figures 3A–D). In β-cells from non-diabetic female NOD mice (30 weeks old), we observed granules co-labeled with insulin and 6.9HIP, indicating the localization of 6.9HIP within DCGs (Figures 3A, B). We also found granules that were simultaneously labeled with insulin, LAMP1, and 6.9HIP, signifying the presence of 6.9HIP within crinosomes (Figure 3C). None of the LAMP1 single-positive granules contained 6.9HIP, suggesting minimal presence of 6.9HIP in regular lysosomes. Notably, nearly all the identified spots with different sizes were localized within vesicular structures, with minimal staining in other organelles such as nucleus, mitochondria, or endoplasmic reticulum (ER) (Figures 3A–D). These results revealed that 6.9HIPs are specifically localized in both DCGs and crinosomes in β-cells.
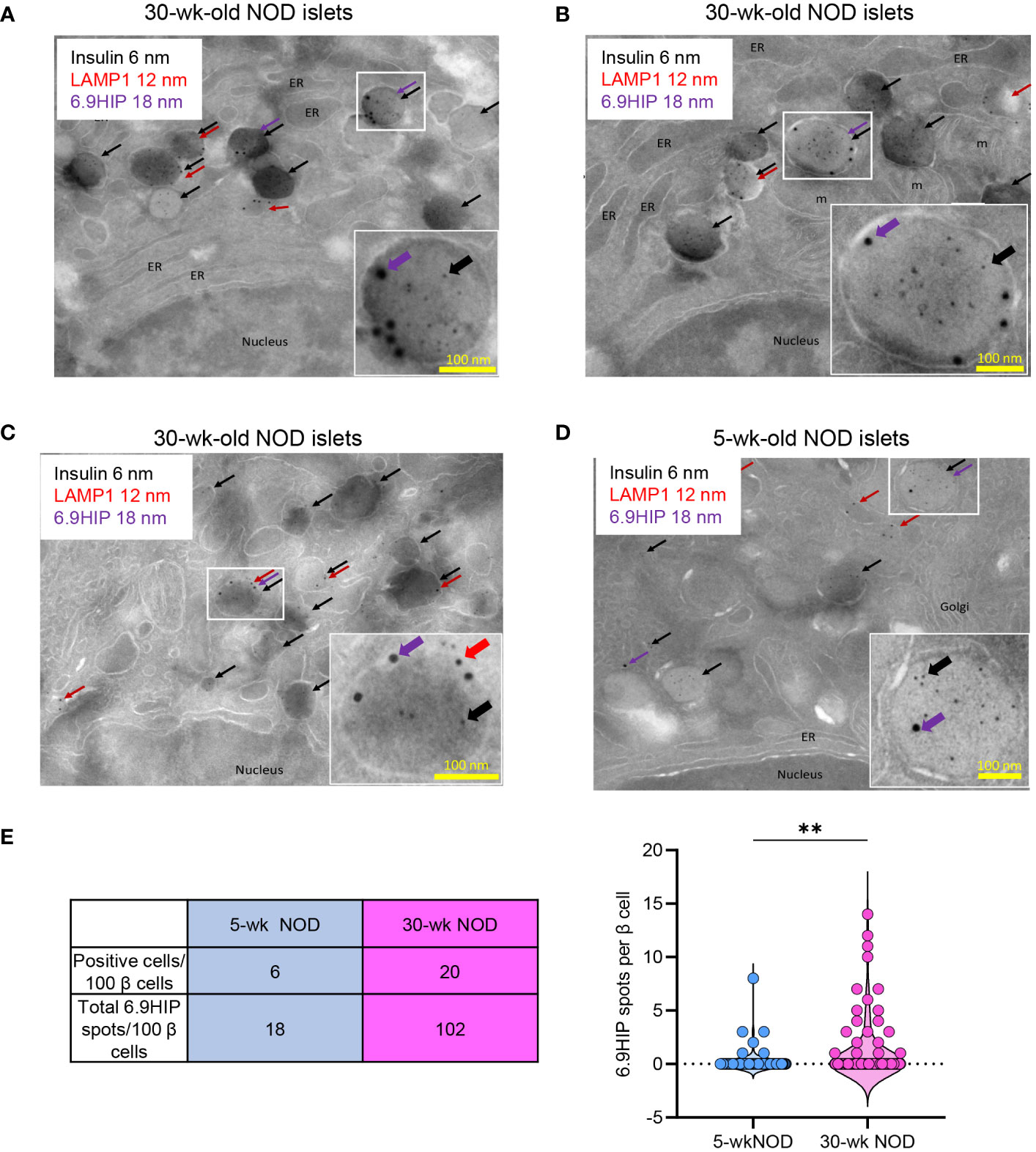
Figure 3 Immunogold electron microscopy reveals that 6.9HIP is localized in two sets of β-cell granules. Islets from 30-week-old (A–C) or 5-week-old (D) NOD mice were triple labeled with primary rabbit anti-insulin, rat anti-LAMP1, and mouse anti-6.9HIP antibodies, followed by staining with secondary antibodies conjugated to colloidal gold, including anti-rabbit 6-nm (insulin, black), anti-rat 12-nm (LAMP1, red), and anti-mouse 18-nm (6.9HIP, purple). Each image in (A–D) shows representative β-cells. The selected regions are illustrated in the enlarged inserts depicting granules containing 6.9HIP in the presence of insulin alone or insulin and LAMP1. ER = endoplasmic reticulum, m = mitochondria. (E) The table summarizes the numbers of β-cells containing at least one 6.9HIP spot and the total numbers of 6.9HIP spots quantified from 100 β-cells from either 30- or 5-week-old NOD mice. The violin plot shows the number of 6.9HIP spots from 100 individual β-cells from each condition. Data summarize results from two independent experiments. Each experiment used 200–300 islets isolated from two 5-week-old or five 30-week-old NOD mice. **p < 0.01; Mann–Whitney test.
In line with our confocal imaging results, the presence of 6.9HIP was less frequent in β-cells from 5-week-old female NOD mice (see Figure 3D showing 6.9HIP in DCGs). We examined 100 β-cells for the presence of 6.9HIP in 5- or 30-week-old female NOD mice. Of the 100 β-cells examined, only 6 contained at least one 6.9HIP spot in 5-week-old mice, as compared to 20 β-cells in 30-week-old mice (Figure 3E). Furthermore, the number of the 6.9HIP spots across all 100 β-cells was significantly higher in the older mice (102 total in 30-week-old vs. 18 in 5-week-old mice) (Figure 3E). These results provide additional evidence to support the important role of autoimmune reactions in generating 6.9HIP in β-cell granules.
The diabetic autoimmune process is strongly associated with ER stress in β-cells (27–31). The observation of enhanced 6.9HIP presence in inflamed islets led us to examine the impact of ER stress signals on 6.9HIP formation in crinosomes and DCGs. We employed an antigen presentation system designed to quantitatively measure the level of 6.9HIPs in crinosome and DCG subcellular fractions purified from primary islets.
Specifically, we prepared dispersed islet cells from 4- to 6-week-old male NOD mice, followed by equilibration in media containing the physiological concentration of glucose (5.5 mM) for 16 h. The islet cells were then left untreated or briefly exposed to the ER stress inducer, tunicamycin (1 µg/mL), for 2 h, a treatment protocol that does not induce β-cell death (data not shown). To confirm the specificity of ER stress, we pretreated one aliquot of islet cells with TUDCA, a well-established ER stress inhibitor during the equilibration period before tunicamycin exposure. TUDCA has been demonstrated to attenuate ER stress and inhibit diabetes development in NOD mice (29). Islet cells under the three conditions were collected, washed, and then subjected to differential centrifugation to isolate subcellular fractions containing crinosomes or DCGs, using our previously established protocol (8). The two fractions were then offered to the C3.g7 APC line (B-cell lymphoma expressing I-Ag7) for antigen presentation to 6.9HIP-reactive, I-Ag7-restricted CD4 T-cell hybridomas. After 24 h, ELISA was used to determine culture IL-2 concentrations for a relative measure of 6.9HIP presence under each condition (see Figure 4A for details).
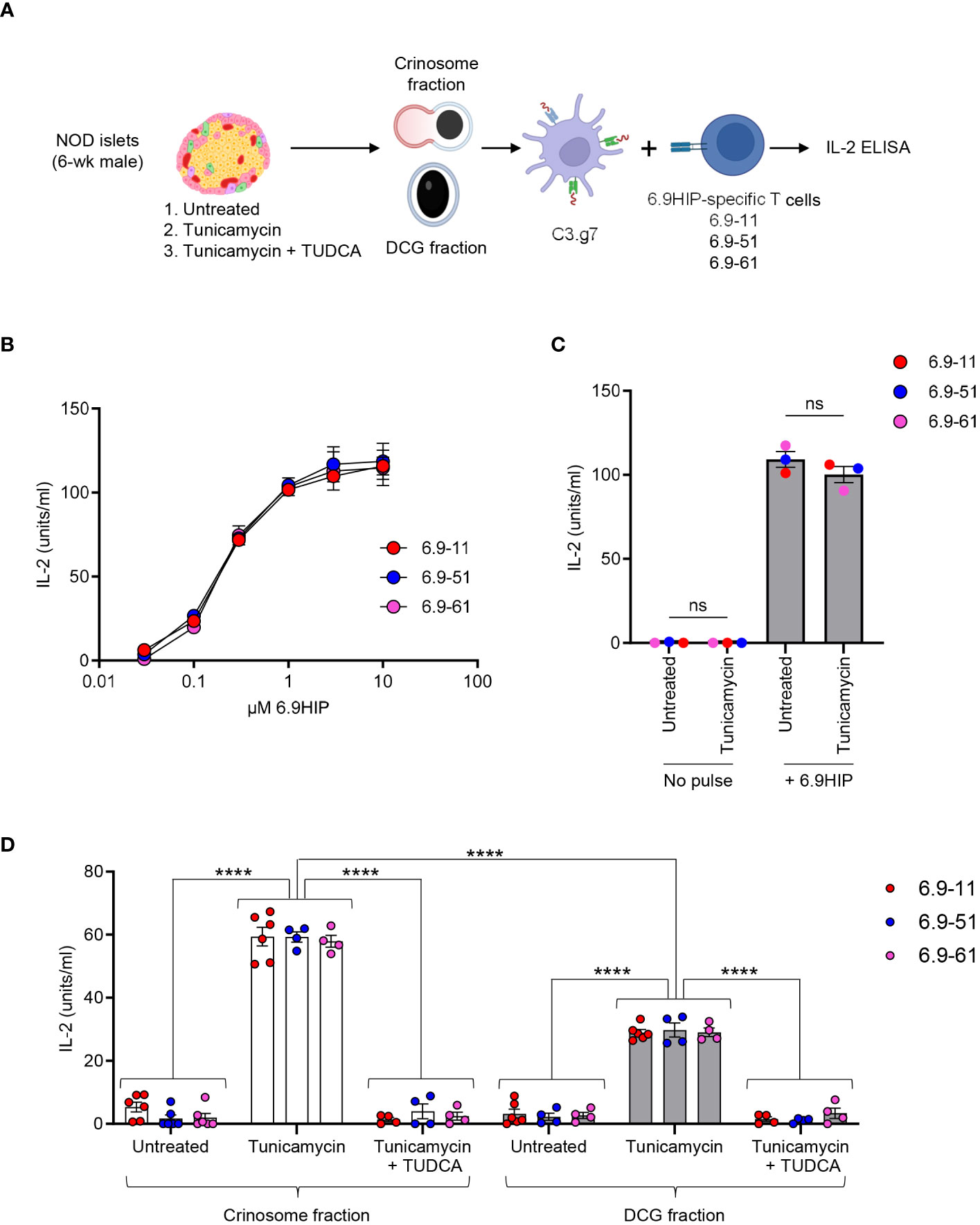
Figure 4 ER stress promotes the production of 6.9HIP in β-cell granules. (A) Schematic for assessing the level of 6.9HIP in crinosome and DCG fractions isolated from islets of 6-week-old male NOD mice in the presence or absence of tunicamycin and TUDCA. (B) Responses of three 6.9HIP-reactive CD4 T-cell hybridomas to C3.g7 APCs pulsed with indicated concentrations of the 6.9HIP peptide. (C) Responses of three 6.9HIP-reactive T-cell hybridomas that were untreated or exposed to tunicamycin for 2 h, in the absence or presence of exogenous pulse with the 6.9HIP peptide (10 µM). (D) Responses of the three 6.9HIP-reactive T cells to either the crinosome or the DCG subcellular fractions isolated from islets that were untreated, treated with tunicamycin alone, or treated with both tunicamycin and TUDCA. Data summarize results from three independent experiments; each point represents a biological replicate of a granule fraction. ns, not significant; ****p < 0.001; two-way ANOVA with Sidak’s multiple comparisons test.
For this study, we generated three 6.9HIP-specific CD4 T-cell hybridomas (6.9-11, 6.9-51, and 6.9-61), each showing dose-responsive reactivity to C3.g7 pulsed with serially diluted concentrations of the synthetic 6.9HIP peptide in vitro (Figure 4B). In the absence of peptide pulse, treatment of each T-cell hybridoma with tunicamycin did not induce any response (Figure 4C). In contrast, exogenous pulsing with 6.9HIP elicited strong and similar responses from all three hybridomas, regardless of tunicamycin exposure (Figure 4C). Thus, the T-cell hybridomas were specific for the 6.9HIP peptide and did not react non-specifically to tunicamycin.
All three 6.9HIP-specific T-cell hybridomas responded at background levels to either the crinosome or the DCG fraction from untreated islets from young male NOD mice (Figure 4D), confirming that 6.9HIP production is minimal when islet inflammation is low. In contrast, all 3 hybridomas exhibited significantly stronger responses to either crinosomes or DCGs from tunicamycin-treated islet cells (Figure 4D). Of note, although crinosomes are anticipated to be much lower in number than DCGs, the level of the T-cell response triggered by the tunicamycin treatment was significantly higher in the crinosome compared to the DCG fraction (Figure 4D). Furthermore, TUDCA completely blocked the tunicamycin-induced increase in the presentation of the 6.9HIP in both crinosomes and DCGs (Figure 4D), validating the specificity of the observed effects by tunicamycin. Collectively, these data demonstrated that ER stress promoted the concentration of 6.9HIP in both crinosomes and DCGs from islets of NOD mice, with a more pronounced effect observed in crinosomes.
There is increasing evidence that HIPs may be important antigenic peptides in type 1 diabetes. There have been multiple reports of their role in the NOD mouse, demonstrating that HIPs are present in β-cells and are presented to T cells, and that HIP-reactive T cells are key players in disease (11–14, 16, 26, 32). Other studies have shown that HIPs, when presented in tolerogenic fashion, can prevent the adoptive transfer of diabetogenic T cells and protect against destruction of islet isografts in diabetic NOD mice (33, 34). HIPs have also been detected in human islets and HIP-specific T cells with inflammatory phenotypes have been isolated from the PBMCs of T1D patients (12, 35). T cells reactive to HIPs may also serve as important biomarkers of disease as they can be detected in PBMCs taken from subjects at risk for developing T1D (R. Baker et al., unpublished). The accumulating information on HIPs as autoantigens in diabetes thus points to the importance of understanding how these peptides contribute to disease.
In this report, we have described the localization of a HIP within specific islet β-cell subcellular compartments, particularly the DCGs and crinosomes. Although staining with the antibody to insulin B9-23 showed that this peptide could be detected in nearly all of the islets, evidence for the presence of the 6.9HIP was obvious only in the infiltrated islets and in later stages of disease. Micrographs of islets stained with the 6.9HIP MAb, together with antibodies to detect insulin, macrophages, and dendritic cells, indicate that 6.9HIP co-localizes in intra-islet APCs as well as within insulin-positive β-cells. By EM, the 6.9HIP was seen to co-localize with granule structures containing insulin alone or both insulin and LAMP1 within β-cells. Through antigen presentation assays, we could demonstrate that exposure of NOD islets to the ER stress inducer tunicamycin significantly increased the levels of 6.9HIP in subcellular fractions containing DCGs and crinosomes.
These findings are significant for a number of reasons. Firstly, where HIPs are located in the islets has been a looming question since the discovery of these peptides was first reported (13). Although previous biochemical and mass spectrometric analyses have all pointed to the β-cell granules as the origin of HIPs, there has also been evidence of HIPs in APCs and, as shown here, in crinosomes. It is likely that HIPs are produced in the β-cell granules as the pH and densely packed protein environment, as well as the presence of key proteases necessary for formation of HIPs, are all conducive to their formation. Although it is not known exactly how HIPs get taken up into other cellular bodies, the multiple stresses in β-cells incurred during disease-induced inflammation could provide various pathways for these peptides to get processed from granules into other organelles, leading to apoptosis and presentation as antigen by intra-islet APCs. Our results showing that inducing stress in NOD islets leads to increased 6.9HIP levels is in line with the last point.
Here, we used the 6.9HIP MAb to analyze the localization of the 6.9HIP by confocal microscopy and immunogold EM. A key challenge in such studies is the generation of peptide-specific MAbs with satisfactory specificity and applicability. While our study is limited to analysis of a single HIP sequence, such information could potentially be extended to other HIPs, such as the 2.5HIP recognized by the highly pathogenic BDC-2.5 CD4 T-cell clone. Because tetramer-binding CD4 T cells reactive to either 2.5HIP or 6.9HIP were shown to accumulate in the islets as diabetes advances (26), the production of the 2.5HIP may also be driven by islet inflammation, as observed by the increased levels of 6.9HIP in stressed islets in the current study. It is, however, important to acknowledge that different HIPs may exhibit variations in their subcellular locations, modes of presentation, and dynamics of T-cell recognition. Despite these potential differences, disease-relevant HIPs like the 2.5HIP and 6.9HIP are likely to share key characteristics in their involvement in the autoimmune processes of T1D. Understanding these commonalities and differences may be crucial for elucidating the mechanisms of disease progression in T1D and developing targeted therapies.
The discovery that 6.9HIP is predominantly localized within DCGs and crinosomes in β-cells provides new insights into our understanding of T1D pathogenesis. This specific localization suggests that the mechanisms governing the release and processing of HIPs might be intricately linked to those controlling insulin secretion and processing. This connection also sheds light on how HIPs, despite their low abundance, become accessible to the immune system. Our previous studies have demonstrated that islet-resident macrophages are capable of taking up intact secretory granules from β-cells (36), highlighting their prominent role in presenting insulin-derived epitopes in pancreatic islets (36–38). This finding offers intriguing possibilities about the pathways through which HIPs are acquired and presented by intra-islet APCs. One potential mechanism is through direct uptake of granules by islet macrophages possibly during phagocytosis of dying β-cells. Such a process could promote shuttling of granules directly into endocytic compartments for processing and presentation by intra-islet APCs. Alternatively, HIPs might be exocytosed along with insulin peptides as β-cells degranulate when responding to glucose stimulation (8, 19, 39). This exocytosis process could provide intra-islet APCs with access to extracellular HIPs for direct peptide loading onto MHC-II molecules either at the cell surface or within the recycling endosomes, as previously seen with degraded insulin peptides (8, 40–43). This pathway suggests a more dynamic interplay between β-cells and APCs in the islet, where the secretion of β-cell granules directly influences the antigenic landscape encountered by the immune system. In summary, our data suggest that the localization of HIPs in β-cell granules may provide a guided mode of presentation to facilitate efficient recognition of HIPs by the immune system as low-abundance antigens.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was approved by Division of Comparative Medicine at Washington University School of Medicine Association for Assessment and Accreditation of Laboratory Animal Care accreditation no. A3381-01. The study was conducted in accordance with the local legislation and institutional requirements.
JW: Conceptualization, Investigation, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. OP: Investigation, Methodology, Validation, Writing – review & editing. AV: Investigation, Methodology, Validation, Writing – review & editing. JD: Investigation, Methodology, Writing – review & editing. AH: Investigation, Methodology, Validation, Writing – review & editing. KH: Funding acquisition, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. XW: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work is supported by the Diabetes Research Center at Washington University (P30 DK020579 to XW), the Juvenile Diabetes Research Foundation (5-CDA-2022-1175-A-N to XW), the National Institute of Health (R01AI62591 and R01DK134437 to XW; R21AI132907 to JW and KH and R01DK81166 to KH), and the University of Colorado Diabetes Research Center award P30DK116073.
The authors thank Wandy Beatty for assisting immunogold EM analysis. We also thank Neetu Srivastava and Cheryl Lichti for critical evaluation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1348131/full#supplementary-material
Supplementary Figure 1 | Validation of the 6.9HIP MAb in NOD.Rag1-/- mice. Intact islets from NOD.Rag1-/- mice (27-week-old) were stained for 6.9HIP (A) or B9-23 (B) along with the macrophage marker F4/80. The islets were then visualized by confocal microscopy. Data are representative of two independent experiments using 4 mice. White scale bars are 50 µM.
Supplementary Figure 2 | Validation of primary antibodies for immunogold electron microscopy. NOD mouse (30-week-old) islets were single-labeled with the primary mouse anti-6.9HIP (A), rat anti-LAMP1 (B), or rabbit anti-insulin (C) antibody, followed by addition of all three secondary antibodies conjugated to colloidal gold, including anti-rabbit (6-nm), anti-rat (12-nm), and anti-mouse (18-nm). Each enlarged image shows a granule that contains 6.9HIP (18-nm; A), LAMP1 (12-nm; B), or insulin (6-nm; C).
1. Zhang L, Nakayama M, Eisenbarth GS. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol. (2008) 20:111–8. doi: 10.1016/j.coi.2007.11.005.
2. Unanue ER. Antigen presentation in the autoimmune diabetes of the NOD mouse. Annu Rev Immunol. (2014) 32:579–608. doi: 10.1146/annurev-immunol-032712-095941.
3. Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. (2005) 435:220–3. doi: 10.1038/nature03523.
4. Roep BO. There is something about insulin granules. Diabetes. (2020) 69:2575–7. doi: 10.2337/dbi20-0039.
5. Smith RE, Farquhar MG. Lysosome function in the regulation of the secretory process in cells of the anterior pituitary gland. J Cell Biol. (1966) 31:319–47. doi: 10.1083/jcb.31.2.319.
6. Halban PA, Wollheim CB. Intracellular degradation of insulin stores by rat pancreatic islets in vitro. An alternative pathway for homeostasis of pancreatic insulin content. J Biol Chem. (1980) 255:6003–6. doi: 10.1016/S0021-9258(18)43686-1.
7. Orci L, Ravazzola M, Amherdt M, Yanaihara C, Yanaihara N, Halban P, et al. Insulin, not C-peptide (proinsulin), is present in crinophagic bodies of the pancreatic β-cell. J Cell Biol. (1984) 98:222–8. doi: 10.1083/jcb.98.1.222.
8. Wan X, Zinselmeyer BH, Zakharov PN, Vomund AN, Taniguchi R, Santambrogio L, et al. Pancreatic islets communicate with lymphoid tissues via exocytosis of insulin peptides. Nature. (2018) 560:107–11. doi: 10.1038/s41586-018-0341-6.
9. Wan X, Vomund AN, Peterson OJ, Chervonsky AV, Lichti CF, Unanue ER. The MHC-II peptidome of pancreatic islets identifies key features of autoimmune peptides. Nat Immunol. (2020) 21:455–63. doi: 10.1038/s41590-020-0623-7.
10. Wiles TA, Powell R, Michel R, Beard KS, Hohenstein A, Bradley B, et al. Identification of hybrid insulin peptides (HIPs) in mouse and human islets by mass spectrometry. J Proteome Res. (2019) 18:814–25. doi: 10.1021/acs.jproteome.8b00875.
11. Wiles TA, Delong T, Baker RL, Bradley B, Barbour G, Powell RL, et al. An insulin-IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non-obese diabetic mouse. J Autoimmun. (2017) 78:11–8. doi: 10.1016/j.jaut.2016.10.007.
12. Baker RL, Rihanek M, Hohenstein AC, Nakayama M, Michels A, Gottlieb PA, et al. Hybrid insulin peptides are autoantigens in type 1 diabetes. Diabetes. (2019) 68:1830–40. doi: 10.2337/db19-0128.
13. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science. (2016) 351:711–4. doi: 10.1126/science.aad2791.
14. Arribas-Layton D, Guyer P, Delong T, Dang M, Chow IT, Speake C, et al. Hybrid insulin peptides are recognized by human T cells in the context of DRB1*04:01. Diabetes. (2020) 69:1492–502. doi: 10.2337/db19-0620.
15. Mitchell AM, Alkanani AA, McDaniel KA, Pyle L, Waugh K, Steck AK, et al. T-cell responses to hybrid insulin peptides prior to type 1 diabetes development. Proc Natl Acad Sci U.S.A. (2021) 118(6):e2019129118. doi: 10.1073/pnas.2019129118.
16. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med. (2016) 22:1482–7. doi: 10.1038/nm.4203.
17. Crawford SA, Wiles TA, Wenzlau JM, Powell RL, Barbour G, Dang M, et al. Cathepsin D drives the formation of hybrid insulin peptides relevant to the pathogenesis of type 1 diabetes. Diabetes. (2022) 71:2793–803. doi: 10.2337/db22-0303.
18. Reed B, Crawford F, Hill RC, Jin N, White J, Krovi SH, et al. Lysosomal cathepsin creates chimeric epitopes for diabetogenic CD4 T cells via transpeptidation. J Exp Med. (2021) 218(2):e20192135. doi: 10.1084/jem.20192135.
19. Vomund AN, Zinselmeyer BH, Hughes J, Calderon B, Valderrama C, Ferris ST, et al. Beta cells transfer vesicles containing insulin to phagocytes for presentation to T cells. Proc Natl Acad Sci U S A. (2015) 112:E5496–502. doi: 10.1073/pnas.1515954112.
20. Gebre-Medhin S, Mulder H, Pekny M, Westermark G, Tornell J, Westermark P, et al. Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochem Biophys Res Commun. (1998) 250:271–7. doi: 10.1006/bbrc.1998.9308.
21. Delong T, Baker RL, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Islet amyloid polypeptide is a target antigen for diabetogenic CD4+ T cells. Diabetes. (2011) 60:2325–30. doi: 10.2337/db11-0288.
22. Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A. (2007) 104:17040–5. doi: 10.1073/pnas.0705894104.
23. Holzlohner P, Hanack K. Generation of murine monoclonal antibodies by hybridoma technology. J Vis Exp. (2017) 119(2):e54832. doi: 10.3791/54832.
24. Ferris ST, Zakharov PN, Wan X, Calderon B, Artyomov MN, Unanue ER, et al. The islet-resident macrophage is in an inflammatory state and senses microbial products in blood. J Exp Med. (2017) 214:2369–85. doi: 10.1084/jem.20170074.
25. Zakharov PN, Hu H, Wan X, Unanue ER. Single-cell RNA sequencing of murine islets shows high cellular complexity at all stages of autoimmune diabetes. J Exp Med. (2020) 217(6):e20192362. doi: 10.1084/jem.20192362.
26. Baker RL, Jamison BL, Wiles TA, Lindsay RS, Barbour G, Bradley B, et al. CD4 T cells reactive to hybrid insulin peptides are indicators of disease activity in the NOD mouse. Diabetes. (2018) 67:1836–46. doi: 10.2337/db18-0200.
27. Sahin GS, Lee H, Engin F. An accomplice more than a mere victim: The impact of beta-cell ER stress on type 1 diabetes pathogenesis. Mol Metab. (2021) 54:101365. doi: 10.1016/j.molmet.2021.101365.
28. Marre ML, James EA, Piganelli JD. beta cell ER stress and the implications for immunogenicity in type 1 diabetes. Front Cell Dev Biol. (2015) 3:67. doi: 10.3389
29. Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL, et al. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci Transl Med. (2013) 5:211ra156. doi: 10.1126/scitranslmed.3006534.
30. Lee H, Lee YS, Harenda Q, Pietrzak S, Oktay HZ, Schreiber S, et al. Beta cell dedifferentiation induced by IRE1alpha deletion prevents type 1 diabetes. Cell Metab. (2020) 31:822–36 e5. doi: 10.1016/j.cmet.2020.03.002.
31. Lee H, Sahin GS, Chen CW, Sonthalia S, Canas SM, Oktay HZ, et al. Stress-induced beta cell early senescence confers protection against type 1 diabetes. Cell Metab. (2023) 35(12):2200–15. doi: 10.1016/j.cmet.2023.10.014.
32. Tran MT, Faridi P, Lim JJ, Ting YT, Onwukwe G, Bhattacharjee P, et al. T cell receptor recognition of hybrid insulin peptides bound to HLA-DQ8. Nat Commun. (2021) 12:5110. doi: 10.1038/s41467-021-25404-x.
33. Jamison BL, DiLisio JE, Beard KS, Neef T, Bradley B, Goodman J, et al. Tolerogenic delivery of a hybrid insulin peptide markedly prolongs islet graft survival in the NOD mouse. Diabetes. (2022) 71:483–96. doi: 10.2337/db20-1170.
34. Jamison BL, Neef T, Goodspeed A, Bradley B, Baker RL, Miller SD, et al. Nanoparticles containing an insulin-ChgA hybrid peptide protect from transfer of autoimmune diabetes by shifting the balance between effector T cells and regulatory T cells. J Immunol. (2019) 203:48–57. doi: 10.4049/jimmunol.1900127.
35. Wiles TA, Hohenstein A, Landry LG, Dang M, Powell R, Guyer P, et al. Characterization of human CD4 T cells specific for a C-peptide/C-peptide hybrid insulin peptide. Front Immunol. (2021) 12:668680. doi: 10.3389/fimmu.2021.668680.
36. Zinselmeyer BH, Vomund AN, Saunders BT, Johnson MW, Carrero JA, Unanue ER. The resident macrophages in murine pancreatic islets are constantly probing their local environment, capturing beta cell granules and blood particles. Diabetologia. (2018) 61:1374–83. doi: 10.1007/s00125-018-4592-4.
37. Carrero JA, McCarthy DP, Ferris ST, Wan X, Hu H, Zinselmeyer BH, et al. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc Natl Acad Sci U S A. (2017) 114:E10418–E27. doi: 10.1073/pnas.1713543114.
38. Srivastava N, Hu H, Vomund AN, Peterson OJ, Baker RL, Haskins K, et al. Chromogranin A deficiency confers protection from autoimmune diabetes via multiple mechanisms. Diabetes. (2021) 70:2860–70. doi: 10.2337/db21-0513.
39. Vomund AN, Lichti CF, Peterson OJ, Arbelaez AM, Wan X, Unanue ER. Blood leukocytes recapitulate diabetogenic peptide-MHC-II complexes displayed in the pancreatic islets. J Exp Med. (2021) 218(6):e20202530. doi: 10.1084/jem.20202530.
40. Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med. (2011) 208:2375–83. doi: 10.1084/jem.20111502.
41. Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. (2010) 11:350–4. doi: 10.1038/ni.1850.
42. Wan X, Thomas JW, Unanue ER. Class-switched anti-insulin antibodies originate from unconventional antigen presentation in multiple lymphoid sites. J Exp Med. (2016) 213:967–78. doi: 10.1084/jem.20151869.
Keywords: autoimmune diabetes, type 1 diabetes, hybrid insulin peptide, antigen, post-translational modification, NOD, islets, β-cells
Citation: Wenzlau JM, Peterson OJ, Vomund AN, DiLisio JE, Hohenstein A, Haskins K and Wan X (2024) Mapping of a hybrid insulin peptide in the inflamed islet β-cells from NOD mice. Front. Immunol. 15:1348131. doi: 10.3389/fimmu.2024.1348131
Received: 01 December 2023; Accepted: 05 February 2024;
Published: 22 February 2024.
Edited by:
Arnaud Zaldumbide, Leiden University Medical Center (LUMC), NetherlandsReviewed by:
Stuart Ian Mannering, University of Melbourne, AustraliaCopyright © 2024 Wenzlau, Peterson, Vomund, DiLisio, Hohenstein, Haskins and Wan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathryn Haskins, a2F0aWUuaGFza2luc0BjdWFuc2NodXR6LmVkdQ==; Xiaoxiao Wan, d2FueEB3dXN0bC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.