 Raju S. Rajmani
Raju S. Rajmani Avadhesha Surolia
Avadhesha Surolia- 1Molecular Biophysics Unit, Indian Institute of Science, Bengaluru, Karnataka, India
- 2Dr. Reddy's Institute of Life Sciences, Hyderabad, Telangana, India
It is essential to understand the interactions and relationships between Mycobacterium tuberculosis (Mtb) and macrophages during the infection in order to design host-directed, immunomodulation-dependent therapeutics to control Mtb. We had reported previously that ornithine acetyltransferase (MtArgJ), a crucial enzyme of the arginine biosynthesis pathway of Mtb, is allosterically inhibited by pranlukast (PRK), which significantly reduces bacterial growth. The present investigation is centered on the immunomodulation in the host by PRK particularly the activation of the host’s immune response to counteract bacterial survival and pathogenicity. Here, we show that PRK decreased the bacterial burden in the lungs by upregulating the population of pro-inflammatory interstitial macrophages (IMs) and reducing the population of Mtb susceptible alveolar macrophages (AMs), dendritic cells (DCs), and monocytes (MO). Additionally, we deduce that PRK causes the host macrophages to change their metabolic pathway from fatty acid metabolism to glycolytic metabolism around the log phage of bacterial multiplication. Further, we report that PRK reduced tissue injury by downregulating the Ly6C-positive population of monocytes. Interestingly, PRK treatment improved tissue repair and inflammation resolution by increasing the populations of arginase 1 (Arg-1) and Ym1+Ym2 (chitinase 3-like 3) positive macrophages. In summary, our study found that PRK is useful not only for reducing the tubercular burden but also for promoting the healing of the diseased tissue.
Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis is one of the most persistent and lethal pathogens in human history and is still one of the world’s leading causes of death. It infects around 10.0-12.0 million, and kills about 1.6 million people respectively, annually (1, 2). The emergence of drug-resistant strains, co-infection with HIV, and socioeconomic circumstances in developing nations exacerbate further the lethality associated with Mtb (3–5). The sobering realization that Mtb has become resistant to every frontline medication used to treat tuberculosis highlights the urgent need to develop new treatment regimens and medications (6). Additionally, a six-month-long course of treatment for tuberculosis that includes both first and second lines of treatment with different antibiotics increases the risk of the emergence of drug-tolerant bacteria (7, 8). Therefore, the development of novel medications with the ability to shorten the treatment has become important for the fight against tuberculosis. Nonetheless, several FDA-approved repurposed medications are in various phases of clinical trials to treat tuberculosis infection for reducing the duration that patients must undergo therapy for its treatment (9).
A novel strategy for developing anti-tuberculosis (anti-TB) drugs is to use host-directed methods to target the pathogen’s intracellular survival. Additionally, this approach may be successful in combating drug-resistant strains and reducing the likelihood that new drug-resistant strains will emerge (10, 11). Mtb is a facultative intracellular parasite that primarily targets the lung, where it infects the lung resident macrophages as well as other immune cells for pathogenicity, propagation, and dissemination (12). Moreover, macrophages are the most prevalent host cells at infection sites and have been linked to the management and advancement of disease (13). Additionally, macrophages also play important roles in wound healing, homeostasis, tissue integrity, and inflammation regulation (14, 15). The lungs contain two main types of macrophages: tissue-resident alveolar macrophages (AMs) and interstitial macrophages (IMs), which are derived from monocytes. AMs are an M2-type population with anti-inflammatory properties that foster an environment favorable to Mtb replication and propagation (16–20). On the other hand, IMs are associated with an immune environment that is more hostile to microbes (21). Hence, comprehending how Mtb and macrophages interact is crucial for deciphering the mechanism behind this interaction as well as for the creation of targeted therapies that specifically target the Mtb-host interface (11, 22). A number of host factors have been found to support the intracellular survival of Mycobacterium tuberculosis within the macrophage (23, 24). To create targeted therapies for the Mtb-host interface and to understand the mechanisms behind the Mtb-macrophage interaction, it is crucial to comprehend the interaction and the co-evolutionary relationship between Mtb and macrophages (22).
Pranlukast (PRK) is both clinically and commercially available FDA-approved medication for the treatment of asthma (25). Pranlukast, an allosteric inhibitor of MtArgJ (Mtb ornithine acetyltransferase) has previously been shown to inhibit the survival and virulence of Mtb (26), and by specifically inhibiting the function of cysteinyl leukotriene receptor 1 (CysLT1R), pranlukast possesses anti-inflammatory properties (25).
The goal of the current study was to understand host immunomodulation by PRK better, specifically how Pranlukast (PRK) stimulates the host’s immune system to combat bacterial survival, pathogenicity, and possible host-pathogen interaction. After confirming that PRK has no negative effects on mice’s health and is not harmful to vital organs like the liver, kidney, or lungs, we conducted the infection and treatment experiment to investigate the activity of PRK in acute as well as chronic Mtb infection models of mice. Here, we demonstrate that PRK reduced the bacterial burden in the lungs by decreasing the number of Mtb-susceptible alveolar macrophages (AMs), dendritic cells (DCs), and monocytes (MO) and increasing the population of pro-inflammatory interstitial macrophages (IMs). Additionally, we surmise that PRK causes the host macrophages to change their metabolic dependence from fatty acid metabolism to glycolytic metabolism during the log phage of bacterial multiplication. Further, we observed that PRK treatment increased the populations of arginase 1 (Arg-1) and Ym1+Ym2 (chitinase 3-like 3) positive macrophage, which in turn improved tissue repair and inflammation resolution. Significantly, we show that Pranlukast (PRK) reduced tissue injury by downregulating Ly6C-positive populations of monocytes. Additionally, within the present study, we found that PRK treatment in Mtb-infected mice consistently reduces bacterial burden in the acute model of infection compared to the chronic model, indicating PRK’s bactericidal action. Ultimately, we noted that PRK had a positive impact on tissue pathology repair as well as in tuberculosis load reduction.
Materials and methods
Bacterial culture
Mycobacterium tuberculosis (H37Rv strain; a virulent laboratory strain of M. tuberculosis, American Type Culture Collection) was cultured in Middlebrook 7H9 broth media (Difco) supplemented with 10% albumin-dextrose-catalase (ADC) (Becton, Dickinson), 0.4% glycerol, and 0.05% Tween 80 and the cultures were grown to mid-log phase (OD600nm 0.4–0.7).
Ethics and animal husbandry
The work plans for the animal experiments were examined and approved by the Indian Institute of Science, Bangalore’s Institute Animals Ethical Committee (IAEC), and the Institute Biosafety Committee (IBSC). The experiment was carried out in compliance with the guidelines set forth by the Committee for the Purpose of Control and Supervision of Experiments on Animals, or CPCSEA. A required number of 4-to 6-week-old female BALB/C mice were procured from the Central Animal Facility at IISc, Bangalore. Before being infected, the animals were allowed to acclimatize for two weeks at the ABSL-3 laboratory.
In vivo toxicity experiments
The toxicity of PRK was assessed in 4-6 weeks-old female BALB/C mice. The mice were divided into two groups (n = 5), one for PRK treatment and the other for PBS treatment as a control. PRK was orally administered @ 40mg/kg/b/wt, weekly at 5 doses for up to 4 weeks, and during this period mice were monitored for health and clinical signs, and weight was taken at weekly intervals for 4 weeks. On the 4th week of post-treatment 500ul of blood was collected from all the animals through the retro-orbital puncture. Before this, the animals were given an inhalation anesthetic treatment consisting of isoflurane (2–3%). Serum was collected from blood and subsequently, blood serum tests were performed to measure the levels of blood urea nitrogen(BUN), serum glutamic pyruvic transaminase (SGPT), and alkaline phosphatase (ALP) level. All animals of both groups were humanely sacrificed by the cervical dislocation method to collect the lungs and liver for histopathological evaluation.
Study design for the chronic and acute infection models
Chronic model- For the chronic model of infection, BALB/c mice were infected via aerosol through a Madison chamber aerosol generation instrument calibrated to deliver 100 CFU. After aerosol infection mice were housed for establishment of infection for 4 weeks, post which they were randomly grouped (n=6) into “H37Rv Control” and “PRK treated”, and treatment was initiated (PRK; 5 doses weekly@ 40mg/kg.b.wt) through oral route. 8 weeks post-infection, the mice were humanely sacrificed by cervical dislocation method to assess the bacterial load in the lungs.
Acute model
To assess the bactericidal activity of PRK, we established acute model of infection, in an acute model of Infection, 4-6 weeks female BALB/C mice were infected with 500 bacilli (≈ 500 CFU) of H37Rv, and after one week of post-infection, (≈ 104 CFU), mice were randomly grouped as mentioned in chronic model and treatment started as in chronic models for four weeks (PRK; 5 doses weekly@ 40mg/kg.b.wt through oral route). After that, the mice were humanely sacrificed by the cervical dislocation method to access the bacterial load in the lungs.
Infection of animals
On the day of infection, the Mid-log phase (OD600nm 0.4–0.7) grown bacterial cultures (H37Rv) were passed through different gauges of needles from lower to higher (23G, 26G, 30G) for making a single cell suspension and turbidity of the culture was measured. Animals were infected through aerosol means using the Madison Aerosol Chamber for Infection. Infected animals (n=5) were sacrificed by the cervical dislocation method, post-24 hours of infection to assess the infection dose. The remaining animals were, humanely sacrificed by cervical dislocation method at the appropriate time point of the study plan. Lungs and spleens were harvested and weighed for assessing CFU. One part of the right upper lobe of the lungs was fixed in 10% neutral buffered formalin for histopathology, the remaining tissues were homogenized in 2ml of PBS and homogenate of lungs in appropriate dilution were plated on 7H11 agar media supplemented with OADC (10% oleic acid, albumin, dextrose, and catalase)and PANTA (polymyxin B, amphotericin B, nalidixic acid, trimethoprim, and azlocillin)for enumeration of CFU. All experiments were conducted in the BSL-3 laboratory of IISC. Bangalore.
Histological examination and scoring
After sacrificing animals, the right upper lobe of the lung tissues was infused with 10% neutral buffered formalin and preserved until processed for histopathological assessment. At the time of tissue processing, the tissues were embedded in paraffin and sections of 4µm thickness from formalin-fixed and paraffin-embedded tissues were cut onto glass slides and stained with hematoxylin and eosin (H&E) for histopathological evaluation and imaging. The number of granulomas and granuloma areas with specific pathology was assessed.
Granuloma score
Granuloma scoring was performed by observing and manually counting individual granulomas. we developed a scientific method for scoring the formation of granuloma with specific pathological markers (27). Granulomas with necrosis were given a score of 5, granulomas with no necrosis were given a score of 2.5, and granulomas with fibrosis were given a score of 1. These scores were summed up to provide the total granuloma scores (Table 1).

Table 1 Methodology for granuloma scoring.
Histopathology scores
The histopathological evaluation was done by the histopathology scoring system for the lung tissues, we developed a scientific method using Mitchison’s virulence scoring system with some modification, considering the number and areas of granulomas, necrosis, alveolar consolidation along with the type of infiltration of immune cells. The histopathology scores were graded as 0-3, (Severe pathology -3; Moderate pathology -2; Minor/minimum pathology -1; No pathology -0) (8, 28).
Processing of lungs for immune cell isolation
The lungs were removed aseptically, minced, and digested in a 2% FBS/DMEM solution with 0.2 mg/ml liberase and 0.1 mg/ml of DNase (Roche). The mixture was then incubated for 30 minutes at 37°C while being shaken. Subsequently, the pre-digested lung samples underwent processing in a Miltenyi Biotec mild MACS dissociator, following the usual lung dissociation technique. After the lung material was processed, it was put through a 70 µm cell strainer. The filtered samples were then centrifuged at 1600 rpm for 5 minutes at 4°C, and the supernatant was discarded. Next, the pellet containing the red blood cells (RBCs) was lysed with freshly made RBC lysis buffer and allowed to sit at room temperature (RT) for 5 to 10 minutes. To counteract the lysis buffer’s effects, 10 ml of PBS was added to each sample. The lung cell suspensions were then treated for five minutes on ice with Fc block (BD Bioscience), after which the cells were counted and stained for cell surface markers with a cocktail of antibodies and incubated in the dark at room temperature for 30 minutes. For the intracellular antigens, BD Biosciences’ fix and perm buffer were used to fix and permeabilize the cells. The samples were then treated with antibodies in accordance with the BD bioscience protocol. Samples were acquired on a BD FACSAria™ Fusion flow cytometer (BD Biosciences, San Jose, CA). The antibodies used in the present investigation are listed in Supplementary Table 1.
Analysis
The Flow cytometry data were analyzed using BD FACSDiva™ version 8.0.1 and FlowJo 10.8.0 software (BD Biosciences). The gating sequence included the following order: live (SSC-A+ FSC-A+), FSC-H vs FSC-A to exclude doublets (singlet gate), SSC-A+CD45+ (leucocytes), and so on.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism software (version 8.0), and values were presented as mean ± SD. Data information: Statistical significance between experimental groups was determined by a two-tailed, unpaired Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and P > 0.5; n.s. not significant).
Results
This study reports the induction of significant immunomodulation inside the host by Pranlukast (PRK) treatment, resulting in significant sterilization of bacterial burden in both acute and chronic infection animal models of tuberculosis. Further, addresses the potential beneficial effect of PRK on the healing of the lung parenchyma.
Pranlukast does not exhibit toxicity in mice
To evaluate the toxicity effect of PRK in BALB/c mice, PRK was given orally at a dose of 40 mg/kg/b/wt, five times a week for a maximum of four weeks (Figure 1A). Throughout this time, the mice’s health and clinical signs were tracked, and their weight was recorded every week for four weeks. Throughout the whole experimental period, all the cohorts’ animals remained healthy and alive. There were no appreciable variations in body weights between the PBS and PRK groups (Figure 1B). During the entire experiment no clinical symptoms such as weight loss or piloerection were seen. Blood urea nitrogen (BUN), serum alkaline phosphatase (ALP), and serum glutamic pyruvic transaminase (SGPT) levels were measured by blood chemistry analysis. Compared to the PBS-treated groups, the enzymatic levels of SGPT and ALP, which serve as indices of liver function, showed insignificant changes in the PRK-treated group (Figures 1C, D). Their absolute values (for all animals combined) were mainly contained within the published reference limits (29) suggesting that PRK has no hepatotoxic effects. Insignificant alterations in BUN levels, which are indicators of kidney function, were also observed in all animals in the PRK-treated cohorts compared to the PBS-treated cohort (Figure 1E). These findings suggest that PRK did not have any nephrotoxic consequences. Additionally, we found no histological abnormalities in the liver or lungs of the PRK-treated groups (Figure 1F), suggesting that PRK is a safe drug for treatment.
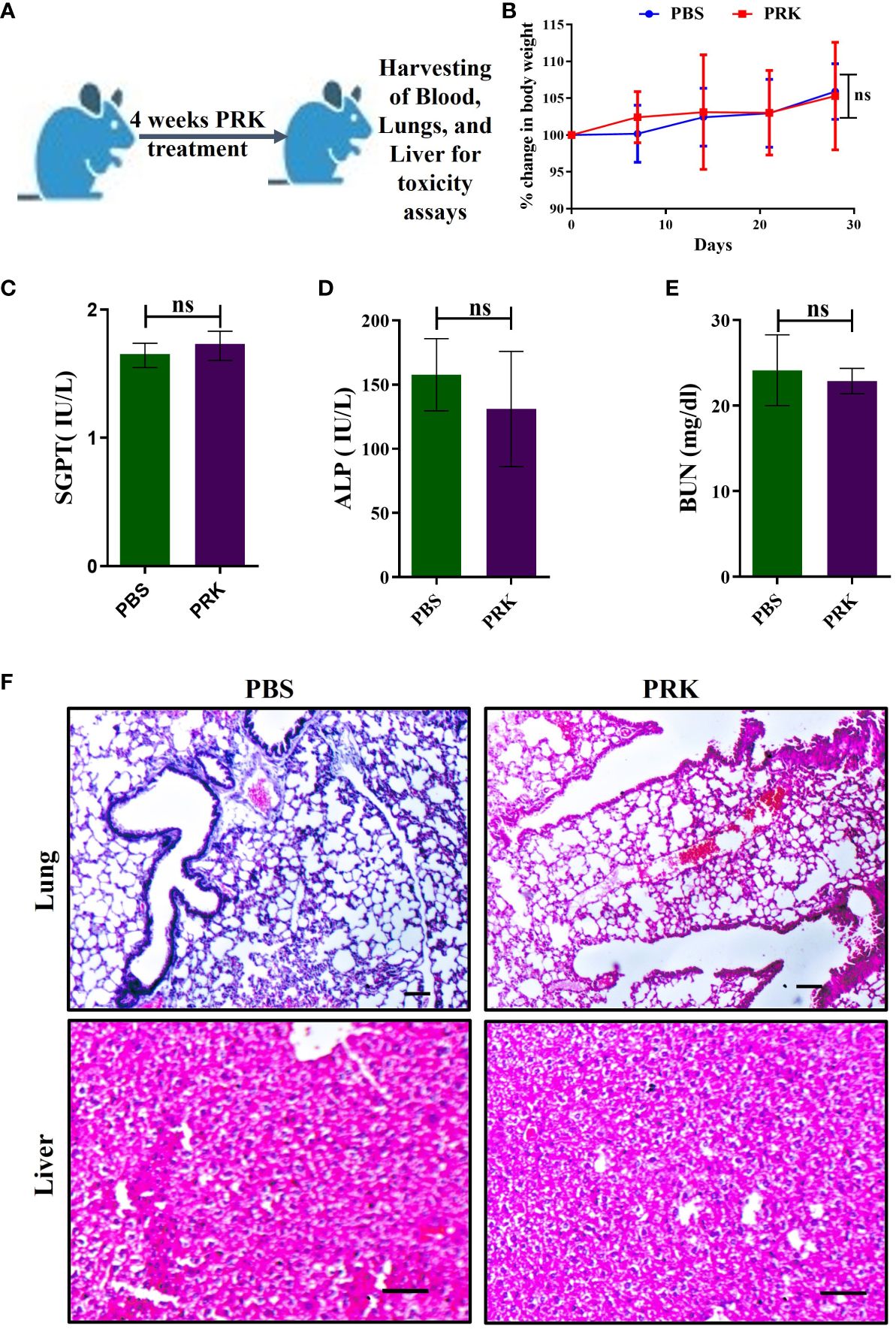
Figure 1 PRK shows no toxicity in mice. (A) Schematic of PRK toxicity experiment in mice. (B) Percentage of weight gain and loss in PRK-treated and PBS control mice (n=4) of toxicity experiment. Comparative values of SGPT (C), ALP (D), and BUN (E) of the PRK treated and PBS control groups, showing non-significant(ns) changes (P > 0.05 calculated by Student’s unpaired t-test). These parameters indicate that the PRK shows no significant hepatotoxic and nephrotoxic effects. (F) Representative histopathology (H&E-stained) of the lung and liver of PRK-treated and PBS control groups showed no histological abnormalities upon PRK treatment (Scale bars 50um). ns, non-significant.
PRK worked better in the acute model of infection and treatment than in the chronic model
After establishing that PRK has no adverse effect on the health of mice. We performed the infection and treatment experiment to check the efficacy of PRK in chronic as well as acute Mtb infection models of mice. For the chronic model of infection, 4-6 weeks female BALB/c mice were infected via aerosol through a Madison chamber aerosol generation instrument calibrated to deliver 100 CFU, then mice were housed for 4 weeks of incubation for the establishment of infection, and after 4 weeks of post-infection, the mice were randomly grouped (n=6) as H37Rv Control group and PRK Treatment group, (PRK; 5 doses weekly@ 40mg/kg.b.wt through oral route), and on the 8th week of post-infection (p.i.), the mice were humanely sacrificed for accessing bacterial load in the lungs. For the acute model of infection, 4 -6 weeks female BALB/C mice were infected with 500 bacilli (≈ 500 CFU) of H37Rv through the aerosolization in the Madison aerosol chamber, and after one week of post-infection, mice were randomly grouped as H37Rv infected control and PRK treated group (n=6), and treatment started as in chronic models for four weeks (Figure 2A). After 4 weeks of treatment in both infection models, the mice were humanely sacrificed to assess bacterial load in the lungs and spleens. In the acute model, the differences in bacterial load between H37Rv control and PRK treated were 1 log, whereas the differences in bacterial load in the chronic model between H37Rv control and PRK treated were 0.8 log in the lungs of mice (Figures 2C, E). Similarly, the bacterial load between H37Rv control and PRK-treated groups of spleens in the acute infection model was 1.1 logs, whereas the differences in bacterial load in the chronic model between H37Rv control and PRK-treated groups were 0.4 logs. (Figures 2D, F). The gross pathology (Figure 2B) of H37Rv infected control and PRK treated groups in acute and chronic models of infection and treatment, respectively in the lungs were found consistent with the CFU count. We observed severe pathology with multiple small and large granulomas, alveolar consolidation, and infiltration of immune cells in the H37Rv infected control of the acute infection model of lungs (Figure 2G). Similarly, we observed severe to moderate pathology in the H37Rv-infected chronic model of mice lungs (Figure 2H). In PRK treatment groups, in both acute and chronic models of infection, we observed minimum pathology with large areas with normal alveolar space (Figures 2G, H). Granuloma scores and histopathology scores were observed to be consistent with bacterial infection and PRK treatment in the acute H37Rv control and acute PRK treated groups (Figure 2I, J) as well as the chronic H37Rv control and chronic PRK treated groups (Figures 2K, L), respectively. Thus, PRK works better in the acute model of bacterial infection than in the chronic model of infection in reducing the bacterial burden. Taken together, these results indicate the bactericidal properties of PRK. The results of acute and chronic infection model are now summarized in Table 2.
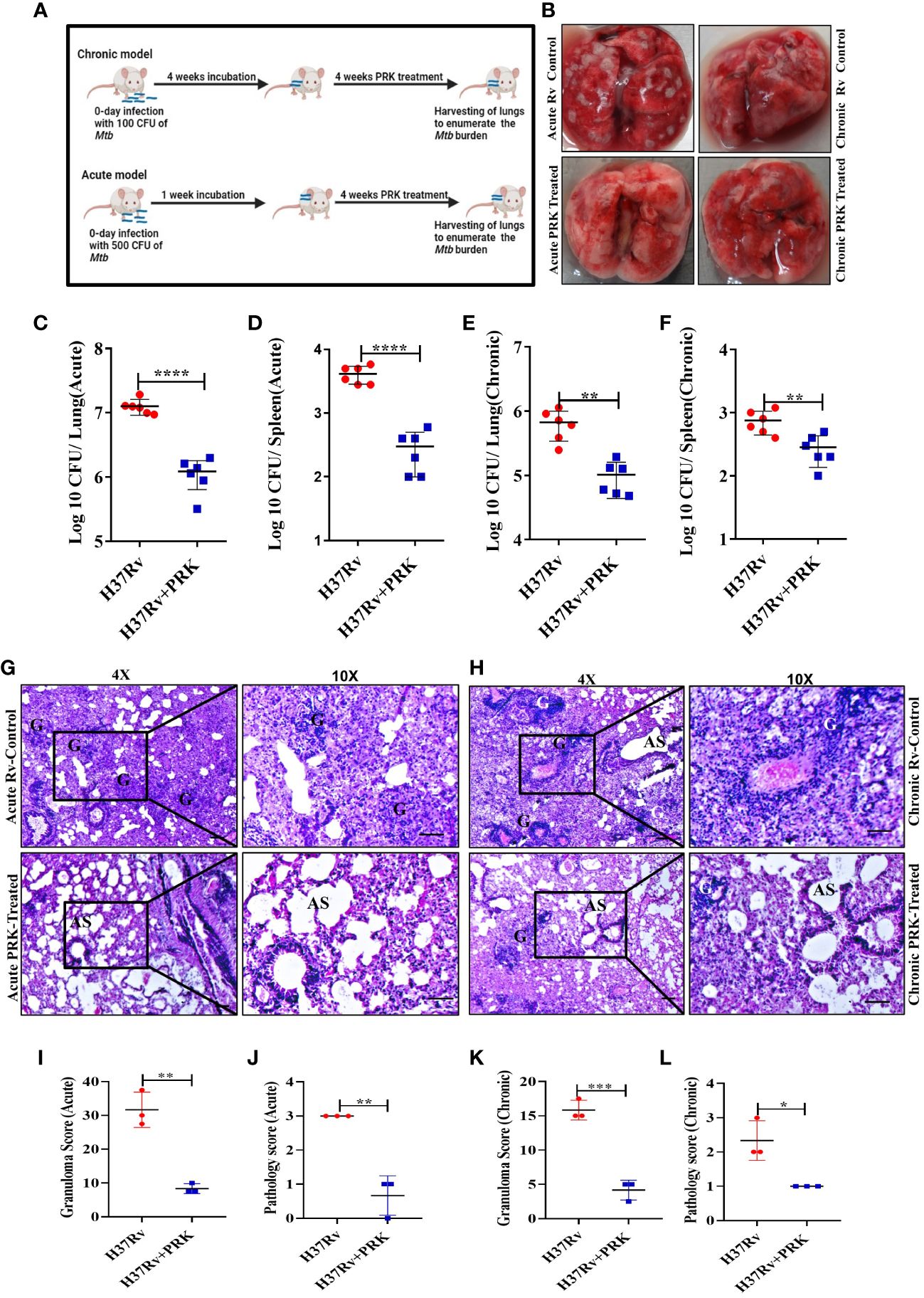
Figure 2 PRK performed better in the acute model of infection and treatment than in the chronic. (A) Schematic of Mtb infection and drug treatment in BALB/C mice. (B) Representative gross morphology of lungs of Mtb infected and PRK treated cohort of acute and chronic mice models of infection, the white spot-on lungs correspond to tubercular granulomas. Bacterial count in the lungs (C) and spleens (D) after treatment with PRK in the acute infection and treatment model of mice. Bacterial count in the lungs (E) and spleens (F) after treatment with PRK in chronic infection and treatment model of mice. H&E-stained representative images of histopathology of the lungs of the H37Rv infected control and PRK treated in the acute (G) and chronic (H) model of infection and treatment respectively, G-granuloma and AS represent Alveolar space (Scale bars 50um). Histopathological analysis, granuloma score (I), and histopathology score (J) of H37Rv infected and PRK treated groups of acute infection model (n=3). Histopathological analysis, granuloma score (K), and histopathology score (L) of H37Rv infected and PRK treated groups of chronic infection model(n=3). Data information: Statistical significance between experimental groups was determined by a two-tailed, unpaired Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and n.s. not significant). Mean and standard error (SD) were determined from six biological replicates (n = 6) for CFU and n=3 for histopathological analysis; Bars indicate means ± SD.
Table 2 Colony forming unit (CFU) in chronic and acute model.
PRK treatment reduced alveolar macrophages, monocytes, and dendritic cells population
There are two primary types of macrophages found in the lung, the tissue-resident alveolar macrophages (AMs; CD11c+, Siglec F+), and the interstitial macrophages (IMs; CD11b+, Ly6C+, and MHC-II+), which are derived from monocytes (16, 17, 19, 30). Nevertheless, it has been previously noted that the immune cell population most vulnerable to Mtb infection is constituted by permissive monocytes, dendritic cells, and alveolar macrophages (18, 19, 31, 32). Additionally, Huang et al. (19) reported that AMs’ anti-inflammatory properties foster an atmosphere that is favorable for Mtb replication and propagation. On the other hand, IMs promote inflammation and regulate the spread and replication of Mtb. In the present study, we found that in the acute model of mice infection and treatment, PRK-treated groups had lower populations of double positive, SSC-A+CD11c+ macrophage phenotypes (Figures 3A, B), which are representative of resident alveolar macrophages (AMs), double positive F480+Ly6c+ monocytes (Figure 3C), and dendritic cells (DCs) (Figure 3D). Because the alveolar macrophage populations rely on the metabolism of fatty acids for energy, they could change into inflammatory foamy macrophages, whose surroundings are favorable for Mtb replication and spread (19). The decline in the bacterial load in PRK treatment groups is apparently related to a reduction in the alveolar macrophage population. Similar patterns were also seen in the chronic model of mouse infection, where the populations of macrophages that correspond to alveolar macrophages (AMs); SSC-A+CD11c+ (Figures 4A, B), CD64+CD11c+ (Figure 4C), CD11chigh (Figure 4E), and SiglecFhigh (Figure 4D) were reduced in a somewhat non-significant manner in the cohort that received PRK treatment. It was earlier reported that macrophages generated from monocytes with CD11chigh expression are extremely receptive to Mtb infection (33) (Figure 3A). Taken together, our findings are also consistent with a prior study.
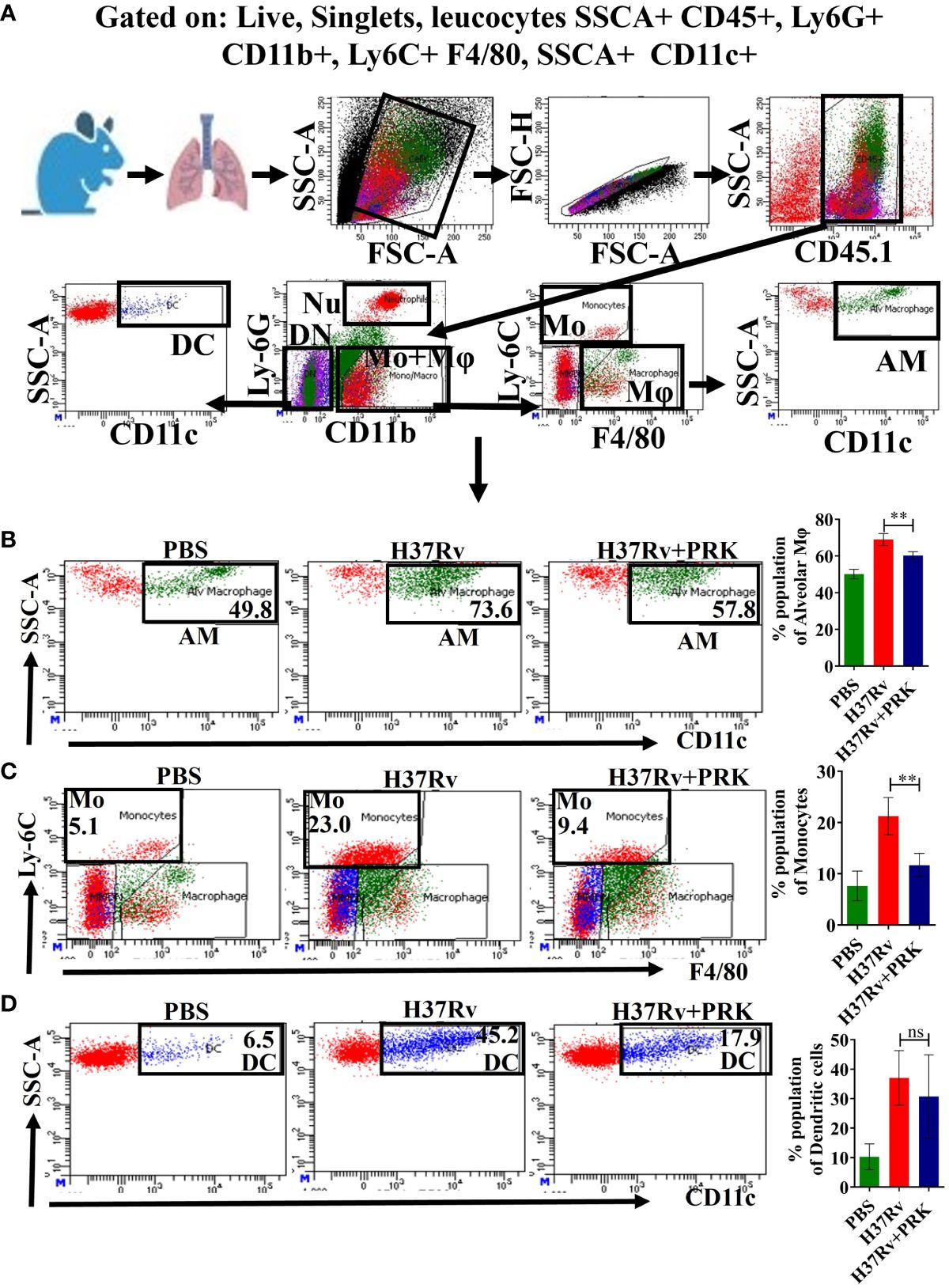
Figure 3 PRK treatment significantly reduced Mtb permissive alveolar macrophages (AMs), and monocytes (Mo) populations in an acute model of infection and treatment. (A) General gating strategy used for Flow cytometry analysis of the lung immune cells obtained from PBS Control, H37Rv infected, and H37Rv+PRK treated cohorts of mice (n=4). Identification of lung alveolar macrophages (AMs; SSC-A+CD11C+) (B), monocytes (Mo; Ly-6C+ F4/80+) (C), and dendritic cells (DCs; SSC-A+CD11C+) (D) of the acute infection and treatment model of mice (harvesting lungs 5 weeks post-infection), showing representative FACS dot plot with the percentage populations (% of parent cells acquired) of AMs, Mo, and DCs, respectively. Statistical significance between experimental groups was determined by a two-tailed, unpaired Student’s t-test (**P < 0.01, and n.s. not significant). Mean and standard error (SD) determined from four biological replicates (n = 4) for FACS analysis; Bars indicate means ± SD.
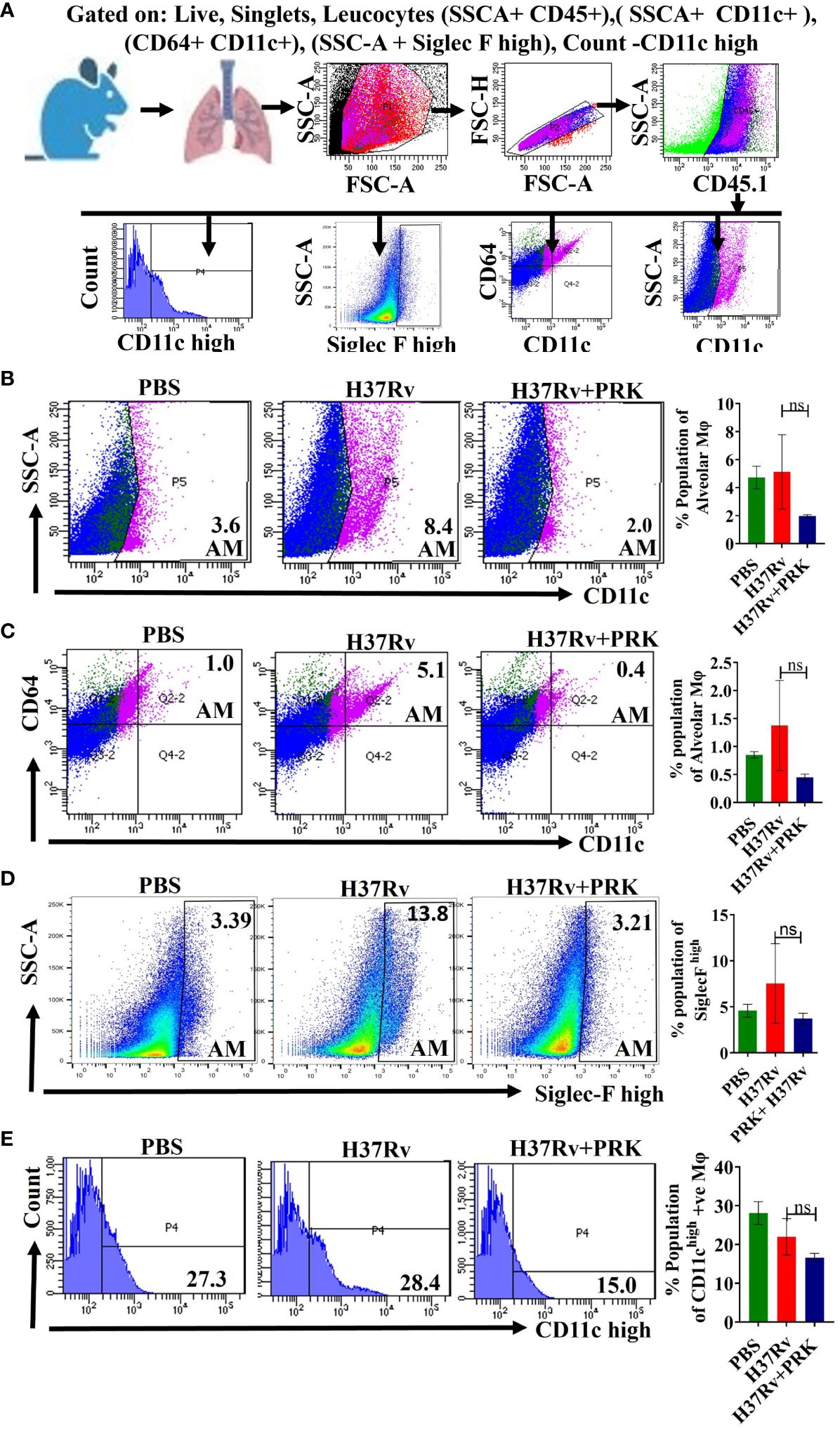
Figure 4 PRK treatment reduced in a somewhat insignificant manner of the alveolar macrophage (AMs) populations in the chronic infection and treatment model. (A) General gating strategy used for flow cytometry analysis of lung Immune cells obtained from PBS Control, H37Rv infected, and H37Rv+PRK treated mice (n=4).Identification of lung alveolar macrophages (AMs; SSC-A+CD11C+ (B); CD64+ CD11C+ (C), Siglec Fhigh (D), and CD11chigh (E) macrophage population in the chronic infection and treatment model of mice (harvesting lungs 8 weeks post-infection), showing representative FACS dot plot with the percentage populations (% of parent cells acquired) of macrophages. Data information: Statistical significance between experimental groups was determined by a two-tailed, unpaired Student’s t-test (P > 0.05; n.s. not significant). Mean and standard error (SD) determined from four biological replicates (n = 4) for FACS analysis; Bars indicate means ± SD.
PRK up-regulates CD11b+ IMs populations in acute infection and treatment models of mice
The expression of phenotypic markers CD11c and CD11b on macrophages after Mtb infection is important in characterizing interstitial macrophages (IMs) (32). Here, we report that the CD11b+ (Figures 5A, B) and CD11b+Ly6G+ (Figure 5C) macrophage population corresponding to the interstitial macrophage (IMs) and neutrophils, significantly increased in the PRK-treated cohort as compared to the H37Rv infected control in the acute model of infection and treatment. Since interstitial macrophages (IMs) follow glycolytically active pathways, their enrichment is not supportive of the growth of Mtb (19). Interestingly, we were not able to observe a similar pattern of interstitial macrophage (CD11b+) upregulation in chronic models of infection and treatment, but the CD11b+ population significantly reduced in chronic infection and treatment models (Figure 5D). Nevertheless, despite the decline in IMs (CD11b+) population, we saw a noteworthy decrease in the bacterial load both in the lungs and the spleens in the chronic model of infection and treatment (Figures 2E, F). Furthermore, Huang et al. (19) reported that depletion of AMs reduced bacterial burden (Figure 5A), whereas depletion of IMs increased bacterial burden. On the other hand, we note that post-infection lifetime appears to be a determining factor for both IMs populations and bacterial load, rather than an arithmetic proportion under all settings. IMs population and bacterial burden in the acute model of infection and treatment were shown to be in arithmetic proportion in our experiment; however, in the chronic model of infection and treatment, IMs were not elevated, despite a reduction in bacterial burden. These findings lead us to the conclusion that PRK alters the metabolic transition of host macrophages from fatty acid metabolism to glycolytic metabolism at the log phage of the bacterial infection in the host.
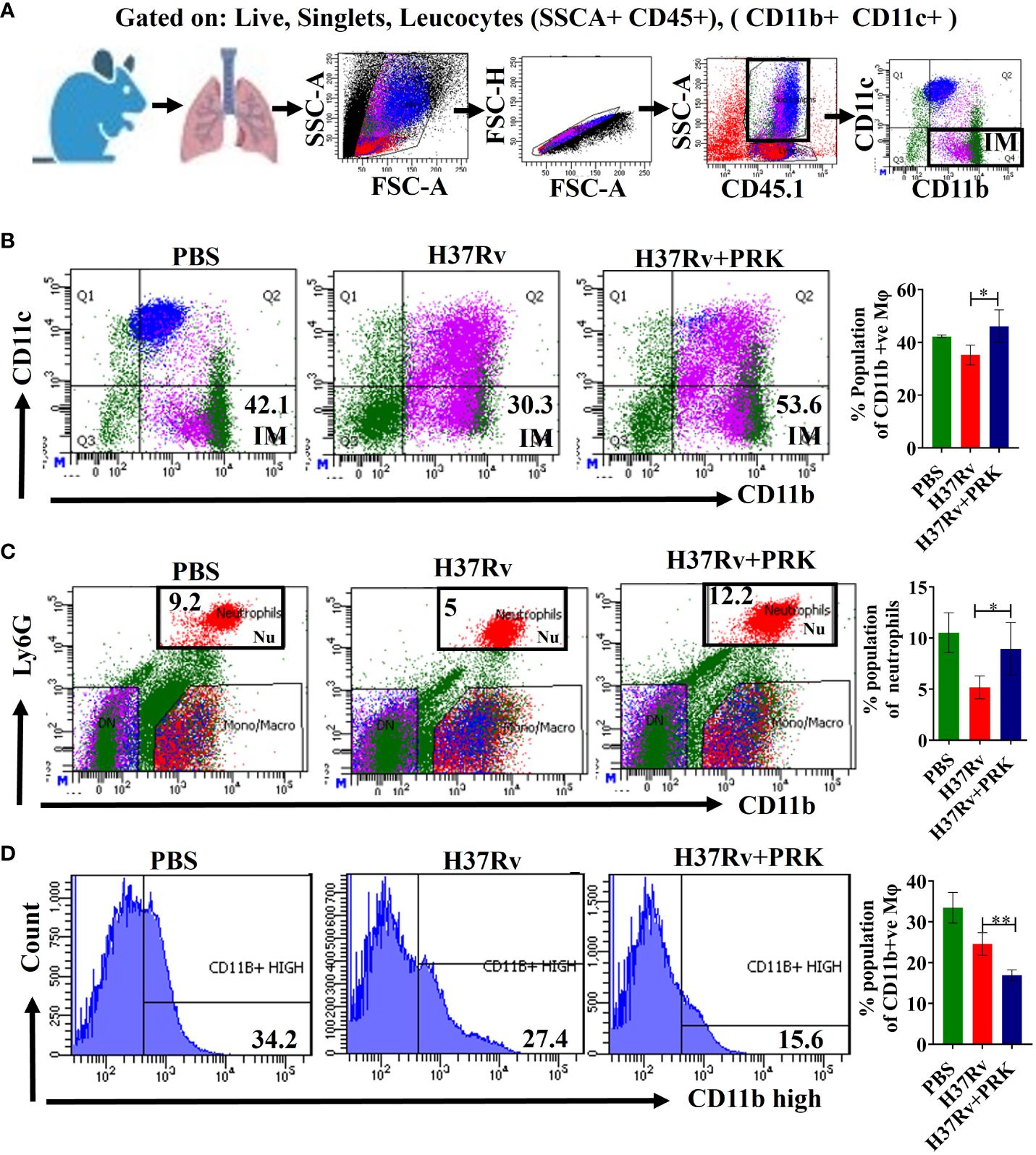
Figure 5 PRK up-regulates CD11b+ IMs populations in acute infection and treatment models of mice. (A) General gating strategy used for Flow cytometry analysis of lung Immune cells obtained from PBS Control, H37Rv infected, and H37Rv +PRK treated mice (n=4). Identification of lung interstitial macrophages (IMs; CD11b+) (B) and neutrophils (Ly6G+CD11b+) (C) of the acute infection and treatment model of mice (harvesting lungs 5 weeks post-infection), showing representative FACS dot plot with the percentage populations (% of parent cells acquired) of IMs. (D) Identification of lung interstitial macrophages (IMs; CD11bhigh) of the chronic infection and treatment model of mice (harvesting lungs 8 weeks post-infection), showing representative FACS plot with the percentage populations (% of parent cells acquired) of CD11bhigh macrophages. Data information: Statistical significance between experimental groups was determined by a two-tailed, unpaired Student’s t-test (*P < 0.05, **P < 0.01). Mean and standard error (SD) determined from four biological replicates (n = 4) for FACS analysis; Bars indicate means ± SD.
Pralukast reduced tissue injury by downregulating the Ly6C-positive population
The expression of the TREM-2 marker on Ly6c+ macrophages determine their activity in a proportional manner (34, 35). Furthermore, Mtb-infected mice showed monocytosis at 2- and 4 weeks post-infection, as evidenced by a rise in Ly6Chigh, CD11b+, and CD115+ cells in the blood (19). Normally, in the acute phase of tissue damage, Ly6Chigh inflammatory monocyte precursors move to wounded areas and cause inflammation. Nonetheless, monocyte-derived macrophages aid in the resolution of inflammation and tissue healing if the sources of tissue injury are removed (36, 37) Consistent with the above, we observed an upregulation of Ly6C+ and Ly6Chigh inflammatory monocytes after 5 and 8 weeks of post-infection in H37Rv control groups of acute and chronic infection model, respectively; into contrast, we saw a notable decrease in Ly6C+ and Ly6Chigh inflammatory monocytes in the groups that received PRK treatment in both acute and chronic model of infection and treatment (Figures 6A–C).
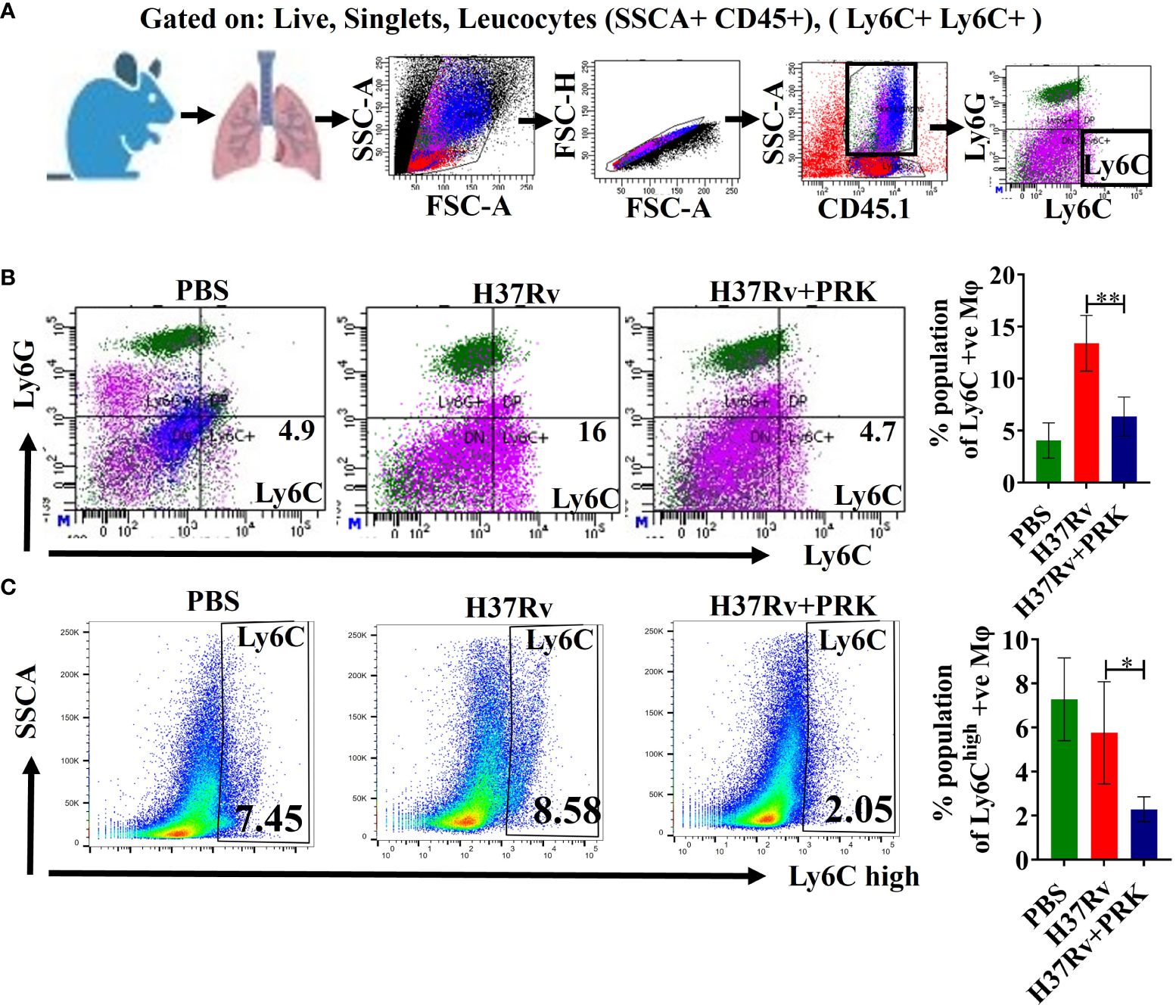
Figure 6 PRK reduced tissue injury by downregulating the Ly6C-positive population. (A) General gating strategy used for flow cytometry analysis of lung Immune cells obtained from PBS Control, H37Rv infected, and H37Rv +PRK treated mice (n=4). (B) Identification of lung Ly6C+ macrophages of the acute infection and treatment model of mice (harvesting lungs 5 weeks post-infection), showing representative FACS dot plot with the percentage populations (% of parent cells acquired) of Ly6C+ macrophages. (C) Identification of lung Ly6Chigh macrophages of the chronic infection and treatment model of mice (harvesting lungs 8 weeks post-infection), showing representative FACS plot with the percentage populations (% of parent cells acquired) of Ly6Chigh macrophages. Data information: Statistical significance between experimental groups was determined by a two-tailed, unpaired Student’s t-test (*P < 0.05, **P < 0.01). Mean and standard error (SD) determined from four biological replicates (n = 4) for FACS analysis; Bars indicate means ± SD.
PRK stimulates Ym1+Ym2 and Arginase1-positive populations of lung macrophage to heal lung pathology
Microbial infection results in tissue damage and acute inflammation by the activation and assembly of multiple immune cell types at the injury site. Additionally, following the elimination of the tissue injury’s source, the activated and assembled immune cells coordinate the reduction of inflammation, tissue repair, and regeneration (30, 35, 38–40). The primary movers in tissue regeneration and repair following microbial infection are tissue-resident macrophages and inflammatory monocytes. They achieve this by phenotypically altering their recognition and activity during the resolution phases of inflammation and injury (14, 35). In mice, the expression of the proteins Ym1 (chitinase 3-like 3) and arginase-1 (Arg-1) consistently indicates the presence of tissue repair-associated macrophage phenotypes that are variable in activation (15, 37, 38, 41–43). Here, we report that, in the chronic model of infection and therapy, PRK-treated mice showed an increase in the Ym1+Ym2 (Figures 7A, B) and arginase-1 (Arg-1) (Figure 7C) positive population of macrophage in comparison to the H37Rv infected group. Since Arginase -1 (Arg-1) and Ym1+Ym2 positive macrophages are associated with the healing of lung tissues, our observation of their upregulation highlights a salutary role of PRK not only in reducing the tubercular burden but also in the subsequent healing of the tissue pathology. Given that the repair of lung tissues is linked to Arginase-1 (Arg-1) and Ym1+Ym2 positive macrophages, our observation of their upregulation underscores a protective function of PRK in both lowering the tubercular burden and promoting the healing of tissue disease that follows. The results of immune response in acute and chronic infection model are now summarized in Table 3. (Supplementary material; Supplementary Figure 1).
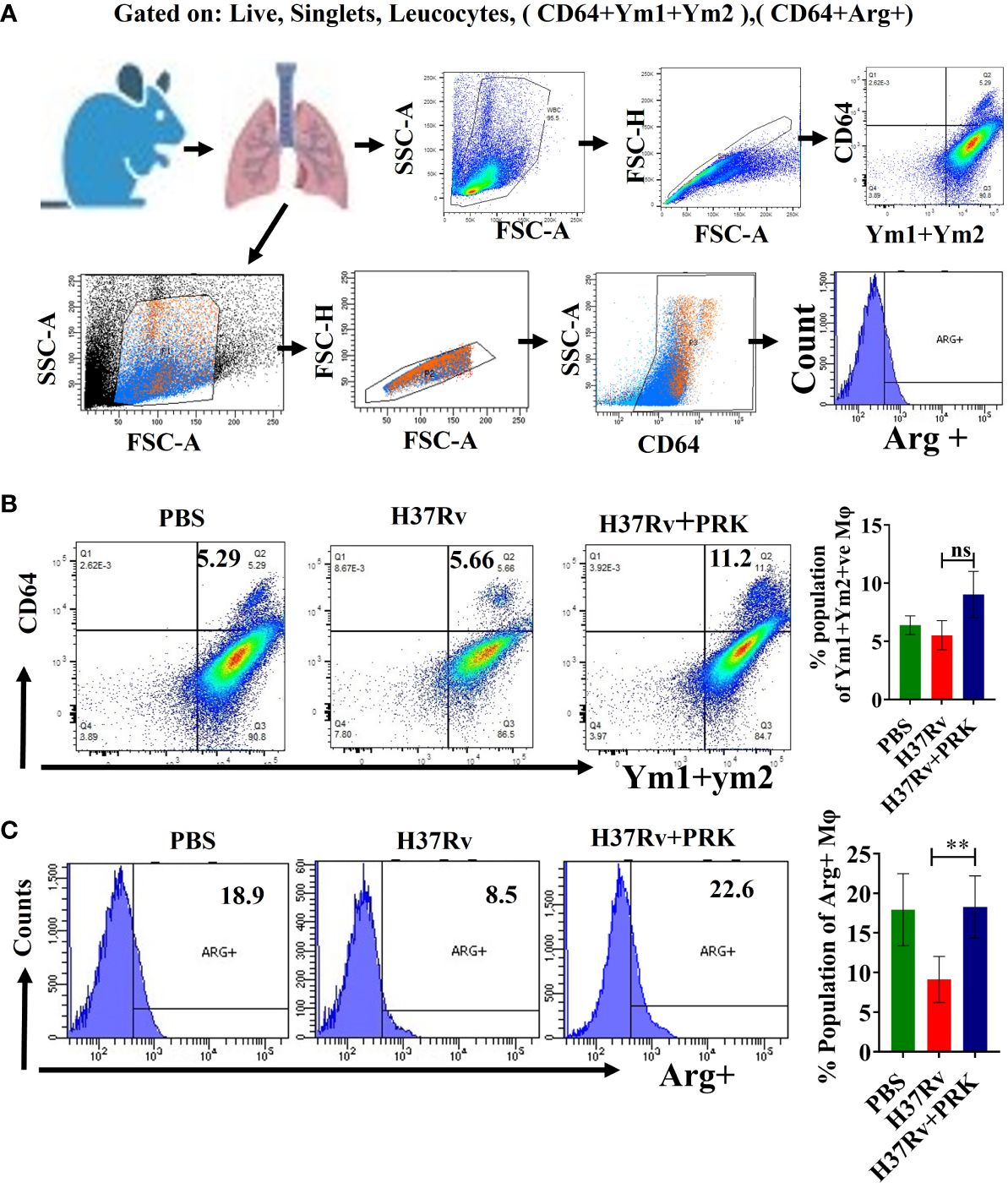
Figure 7 PRK stimulates Ym1+Ym2 and Arginase1-positive populations of lung macrophage to heal lung pathology (A) General gating strategy used for flow cytometry analysis of lung Immune cells obtained from PBS Control, H37Rv infected, and H37Rv +PRK treated mice. (B) Identification of lung Ym1+Ym2 +(chitinase-3 like 3) macrophages of the chronic infection and treatment model of mice (harvesting lungs 8 weeks post-infection), showing representative FACS dot plot with the percentage populations (% of parent cells acquired) of Ym1+Ym2 + macrophages (n=3) (C) Identification of the lung arginase-1 (Arg-1), Arg+ macrophages of the chronic infection and treatment model of mice (harvesting lungs 8 weeks post-infection), showing representative FACS plot with the percentage populations (% of parent cells acquired) of Arg+ macrophages (n=4). Data information: Statistical significance between experimental groups was determined by a two-tailed, unpaired Student’s t-test (**P < 0.01, and n.s. not significant). Mean and standard error (SD) determined from three and four biological replicates (n = 3/4) for FACS analysis; Bars indicate means ± SD.

Table 3 Immune response in PRK treated group as compared to H37Rv control group.
Discussion
We previously reported on the direct killing impact of pranlukast (PRK) on Mtb by the allosteric inhibition of Mtb ornithine acetyltransferase, a crucial enzyme in the arginine biosynthesis pathway resulting in the inhibition of arginine production in the bacteria (26). In the current study, we examined the host immune effectors that lower the bacterial burden and promote lung healing in PRK-treated mice. We carried out the infection and treatment experiment to examine the activity of PRK in acute as well as chronic Mtb infection models of mice after establishing that PRK shows no detrimental influence on mice’s health and no toxicity on the essential organs such as the liver, kidney, and lungs. We found that PRK functions more effectively during the acute stage of bacterial infection than it does during the chronic stage. The CFU count has a strong correlation with the macroscopic pathology and histopathology of the infected lungs.
Furthermore, in the acute model of mice infection, we found that PRK-treated groups significantly downregulated CD11c+ macrophage phenotypes corresponding to resident alveolar macrophage (AMs) and dendritic cells (DCs), as well as monocyte populations. A decline in the alveolar macrophage population may be the cause of the bacterial load reduction in PRK treatment groups since these macrophages are dependent on fatty acid metabolism and can develop into pro-inflammatory foamy macrophages, whose milieu is favorable for Mtb growth and dissemination. A comparable result was also noted in the chronic model of mouse infection, where we found that the PRK-treated group had down-regulated populations of CD11c+, CD64+CD11c+, CD11chigh, and Siglec Fhigh, which correlate to alveolar macrophages.
Furthermore, we found that in the acute model of infection and treatment, the PRK-treated group exhibited a significant increase in the CD11b+ and CD11b+Ly6G+ macrophage population, which corresponds to interstitial macrophages (IMs) and neutrophils, as compared to the H37Rv infected control. Interstitial macrophage (IMs) enrichment is detrimental to Mtb proliferation because IMs adhere to glycolytically active pathways (19, 44). However, CD11b+ populations were dramatically reduced in the chronic infection and treatment model, which is interesting because we were not able to find a comparable pattern of Interstitial macrophage (CD11b+) upregulation in these models. However, in the chronic model of infection and therapy, we observed a significant decrease in the bacterial burden despite the decline in IMs (CD11b+) population. Moreover, Huang et al. (19) found that whereas IM depletion increased bacterial burden, AM depletion decreased it. However, rather than being an arithmetic proportion in all cases, we see that post-infection lifetime seems to be a determining factor for both IMs populations and bacterial load. In our experiment, we found that in the acute model of infection and treatment, IMs populations and bacterial burden were in arithmetic proportion; in contrast, despite a decrease in bacterial burden, IMs were not enhanced in the chronic model of infection and treatment.
After analyzing these data, we may draw the conclusion that PRK causes the host macrophages to switch from fatty acid metabolism to glycolytic metabolism in the vicinity of the log phage bacterial infection. Since blood monocytes are the source of IMs and Mtb infection upregulates blood monocytes (Ly6chigh), monocytosis results, which is dependent on the quantity of infecting bacteria. In the current study, the day 1 bacterial infection in the chronic mode of infection and treatment was lower (100 CFU) than the acute infection model (500 CFU). However, after 4 weeks of infection, the former has a lower bacterial load than the acute model of infection, and the bacterial load further decreases with PRK treatment, leading to a reduction in monocytosis. As a result, there were fewer IMs in the chronic infection and therapy mode. This is in line with earlier studies showing that lung macrophage population alteration can have a significant impact on bacterial load, both favorably and unfavorably (45, 46). Furthermore, these earlier investigations suggest that lung macrophages’ permissiveness to bacterial growth may play a role in the course of the disease in addition to immunological control. The inflammatory response is first and foremost essential for the removal of germs. However, the initial inflammatory response is probably switched to tissue repair and regeneration processes if the sources of tissue injury are removed (14, 35). Nevertheless, according to (Murray & Wynn (47), macrophages are essential for both the development and remission of inflammation. Tissue-resident macrophages and invading monocytes jointly cause inflammation during the acute inflammatory phase by secreting inflammatory cytokines and chemokines. On the other hand, during the phase of inflammatory recovery, these cells also support tissue regeneration and repair (14, 19). Further, Huang et al. (19) reported that Mtb-infected mice developed monocytosis, as evidenced by increased numbers of Ly6Chigh CD11b+ CD115+ cells in the blood at 2- and 4 weeks post-infection. In the acute phase of tissue damage, Ly6Chigh inflammatory monocyte precursors normally move to wounded areas and cause inflammation. However, monocyte-derived macrophages aid in the resolution of inflammation and tissue healing if the underlying causes of tissue damage are removed (36, 37). Here we document a comparable pattern of elevation of Ly6Chigh inflammatory monocyte following 5 and 8 weeks of Mtb post-infection in H37Rv control groups, while a notable decrease in Ly6C+ inflammatory monocyte was noted in the groups treated with PRK. Consistent with the above findings, we first saw an increase in the populations of proinflammatory macrophages (IMs) in the PRK-treated groups in the acute model of infection. However, in the chronic model of mice infection, the Ym1+Ym2 and Arg-1-positive population of macrophage increased in PRK-treated mice as compared to the H37Rv-infected control group. We believe that the emergence of Ym1+Ym2 and Arg-1 positive subpopulation of lung macrophage contributes to the resolution of inflammation and tissue repair in the PRK treatment group as compared to the untreated infected controls. This is reminiscent of the role of the expression of Arg-1 positive populations of macrophages in the wound-healing process as reported by (37, 48). Our findings are consistent with previous research demonstrating that manipulation of the lung macrophage population can have a significant impact on the bacterial burden (45, 46). PRK not only lowers the tubercular load but also augments the tissue repair process consequently, it augurs well for induction in anti-tubercular therapeutics.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was approved by Institute Animal Ethics Committee- CAF/ETHICS/899/2022. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
RR: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AS: Conceptualization, Funding acquisition, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The study is supported by the grants BT/PR24443/MED/29/1220/2017 & BT/PR39006/BRB/10/1883/2020 from the Dept. of Biotechnology, Govt. of India to AS.
Acknowledgments
We thank Dr. Amit Singh for the Mtb strains, we thank Vasista Adiga from the CIDR FACS facility at IISc for flow cytometry experiments. We acknowledge the DBT-IISc-supported BSL3 facility for carrying out experiments on Mtb strains.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1347045/full#supplementary-material
References
2. WHO. WHO consolidated guidelines on tuberculosis. module 4: treatment-drug-resistant tuberculosis treatment 2022 Update. Geneva, Switzerland: World Health Organization (2022).
3. O’Brien RJ, Nunn PP. The need for new drugs against tuberculosis. Obstacles, opportunities, and next steps. Am J Respir Crit Care Med. (2001) 163:1055–8. doi: 10.1164/ajrccm.163.5.2007122
4. Sharma S, Rajmani RS, Kumar A, Bhaskar A, Singh A, Manivel V, et al. Differential proteomics approach to identify putative protective antigens of Mycobacterium tuberculosis presented during early stages of macrophage infection and their evaluation as DNA vaccines. Indian J Exp Biol. (2015) 53:429–39.
5. Wilson JW, Tsukayama DT. Extensively drug-resistant tuberculosis: principles of resistance, diagnosis, and management. Mayo Clin Proc. (2016) 91:482–95. doi: 10.1016/j.mayocp.2016.01.014
6. Boshoff HIM, Warner DF, Gold B. Editorial: drug-resistant mycobacterium tuberculosis. Front Cell Infect Microbiol. (2023) 13:1215294. doi: 10.3389/fcimb.2023.1215294
7. Günther G. Multidrug-resistant and extensively drug-resistant tuberculosis: a review of current concepts and future challenges. Clin Med (Lond). (2014) 14:279–85. doi: 10.7861/clinmedicine.14-3-279
8. Mishra R, Kohli S, Malhotra N, Bandyopadhyay P, Mehta M, Munshi M, et al. Targeting redox heterogeneity to counteract drug tolerance in replicating Mycobacterium tuberculosis. Sci Transl Med. (2019) 11:518. doi: 10.1126/scitranslmed.aaw6635
9. Zumla A, Nahid P, Cole ST. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discovery. (2013) 12:388–404. doi: 10.1038/nrd4001
10. Kellam P. Attacking pathogens through their hosts. Genome Biol. (2006) 7:201. doi: 10.1186/gb-2006-7-1-201
11. Chandra P, Rajmani RS, Verma G, Bhavesh NS, Kumar D. Targeting drug-sensitive and -resistant strains of mycobacterium tuberculosis by inhibition of src family kinases lowers disease burden and pathology. mSphere. (2016) 1:2. doi: 10.1128/mSphere.00043-15
12. Daffé M, Etienne G. The capsule of Mycobacterium tuberculosis and its implications for pathogenicity. Tuber Lung Dis. (1999) 79:153–69. doi: 10.1054/tuld.1998.0200
13. Flynn JL, Chan J, Lin PL. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol. (2011) 4:271–8. doi: 10.1038/mi.2011.14
14. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
15. Gordon S, Plüddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. (2014) 262:36–55. doi: 10.1111/imr.12223
16. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three unique interstitial macrophages in the murine lung at steady state. Am J Respir Cell Mol Biol. (2017) 57:66–76. doi: 10.1165/rcmb.2016-0361OC
17. Sabatel C, Radermecker C, Fievez L, Paulissen G, Chakarov S, Fernandes C, et al. Exposure to bacterial cpG DNA protects from airway allergic inflammation by expanding regulatory lung interstitial macrophages. Immunity. (2017) 46:457–73. doi: 10.1016/j.immuni.2017.02.016
18. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. (2018) 24:439–446.e4. doi: 10.1016/j.chom.2018.08.001
19. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med. (2018) 215:1135–52. doi: 10.1084/jem.20172020
20. Pisu D, Huang L, Grenier JK, Russell DG. Dual RNA-seq of mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell Rep. (2020) 30:335–350.e4. doi: 10.1016/j.celrep.2019.12.033
21. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
22. Liu CH. M. tuberculosis and macrophages: co-existence and co-evolution. J Pulm Respir Med. (2016) 04. doi: 10.4172/2161-105X.1000e133
23. Jayaswal S, Kamal MA, Dua R, Gupta S, Majumdar T, Das G, et al. Identification of host-dependent survival factors for intracellular Mycobacterium tuberculosis through an siRNA screen. PloS Pathog. (2010) 6:e1000839. doi: 10.1371/journal.ppat.1000839
24. Kumar D, Nath L, Kamal M, Varshney A, Jain A, Singh S, et al. Genome-wide analysis of the host intracellular network that regulates survival of mycobacterium tuberculosis. Cell. (2010) 140:731–43. doi: 10.1016/j.cell.2010.02.012
25. Barnes NC, Pujet JC. Pranlukast, a novel leukotriene receptor antagonist: results of the first European, placebo controlled, multicentre clinical study in asthma. Thorax. (1997) 52:523–7. doi: 10.1136/thx.52.6.523
26. Mishra A, Mamidi AS, Rajmani RS, Ray A, Roy R, Surolia A. An allosteric inhibitor of Mycobacterium tuberculosis ArgJ: Implications to a novel combinatorial therapy. EMBO Mol Med. (2018) 10:e8038. doi: 10.15252/emmm.201708038
27. Khan MZ, Bhaskar A, Upadhyay S, Kumari P, Rajmani RS, Jain P, et al. Protein kinase G confers survival advantage to Mycobacterium tuberculosis during latency-like conditions. J Biol Chem. (2017) 292:16093–108. doi: 10.1074/jbc.M117.797563
28. Mitchison DA, Wallace JG, Bhatia AL, Selkon JB, Subbaiah TV, Lancaster MC. A comparison of the virulence in Guinea-pigs of South Indian and British tubercle bacilli. Tubercle. (1960) 41:1–22. doi: 10.1016/s0041-3879(60)80019-0
29. Ostergaard GH, Hansen N, Ottesen JL. Physiological, hematological and clinical chemistry parameters, including conversion factors. In: CRC Handbook of laboratory animal science (Boca Raton, Florida, US: Taylor and Francis group) (2011). p. 667–709.
30. Ikeda N, Asano K, Kikuchi K, Uchida Y, Ikegami H, Takagi R, et al. Emergence of immunoregulatory Ym1 + Ly6C hi monocytes during recovery phase of tissue injury. Sci Immunol. (2018) 3:28. doi: 10.1126/sciimmunol.aat0207
31. Wolf AJ, Linas B, Trevejo-Nunãez GJ, Kincaid E, Tamura T, Takatsu K, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol. (2007) 179:2509–19. doi: 10.4049/jimmunol.179.4.2509
32. Srivastava S, Ernst JD, Desvignes L. Beyond macrophages: the diversity of mononuclear cells in tuberculosis. Immunol Rev. (2014) 262:179–92. doi: 10.1111/imr.12217
33. Lee J, Boyce S, Powers J, Baer C, Sassetti CM, Behar SM. CD11cHi monocyte-derived macrophages are a major cellular compartment infected by Mycobacterium tuberculosis. PLoS Pathog. (2020) 16(6):e1008621. doi: 10.1371/journal.ppat.1008621
34. Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. (2015) 212:287–95. doi: 10.1084/jem.20142322
35. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
36. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo J-L, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204:3037–47. doi: 10.1084/jem.20070885
37. Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE. The phenotype of murine wound macrophages. J Leukoc Biol. (2010) 87:59–67. doi: 10.1189/jlb.0409236
38. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
39. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. (2008) 214:199–210. doi: 10.1002/path.2277
40. Goren I, Pfeilschifter J, Frank S. Uptake of neutrophil-derived Ym1 protein distinguishes wound macrophages in the absence of interleukin-4 signaling in murine wound healing. Am J Pathol. (2014) 184:3249–61. doi: 10.1016/j.ajpath.2014.08.011
41. Raes G, De Baetselier P, Noël W, Beschin A, Brombacher F, Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol. (2002) 71:597–602. doi: 10.1189/jlb.71.4.597
42. Raes G, Van den Bergh R, De Baetselier P, Ghassabeh GH, Scotton C, Locati M, et al. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J Immunol. (2005) 174:6561; author reply 6561–2. doi: 10.4049/jimmunol.174.11.6561
43. Welch JS, Escoubet-Lozach L, Sykes DB, Liddiard K, Greaves DR, Glass CK. TH2 cytokines and allergic challenge induce Ym1 expression in macrophages by a STAT6-dependent mechanism. J Biol Chem. (2002) 277:42821–9. doi: 10.1074/jbc.M205873200
44. Podinovskaia M, Lee W, Caldwell S, Russell DG. Infection of macrophages with Mycobacterium tuberculosis induces global modifications to phagosomal function. Cell Microbiol. (2013) 15:843–59. doi: 10.1111/cmi.12092
45. Leemans JC, Thepen T, Weijer S, Florquin S, van Rooijen N, van de Winkel JG, et al. Macrophages play a dual role during pulmonary tuberculosis in mice. J Infect Dis. (2005) 191:65–74. doi: 10.1086/426395
46. Antonelli LRV, Gigliotti Rothfuchs A, Gonçalves R, Roffê E, Cheever AW, Bafica A, et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest. (2010) 120:1674–82. doi: 10.1172/JCI40817
47. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
Keywords: Mycobacterium tuberculosis (MTB), Pranlukast (PRK), immunomodulation, alveolar macrophages (AMs), interstitial macrophages (IMs)
Citation: Rajmani RS and Surolia A (2024) Antimycobacterial and healing effects of Pranlukast against MTB infection and pathogenesis in a preclinical mouse model of tuberculosis. Front. Immunol. 15:1347045. doi: 10.3389/fimmu.2024.1347045
Received: 30 November 2023; Accepted: 19 April 2024;
Published: 02 May 2024.
Edited by:
Laurence Rahme, Harvard Medical School, United StatesReviewed by:
Debora Decote-Ricardo, Federal Rural University of Rio de Janeiro, BrazilAnagha Malur, East Carolina University, United States
Brenna Carey, Cincinnati Children’s Research Foundation, United States
Copyright © 2024 Rajmani and Surolia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raju S. Rajmani, cmFqdS5yYWptYW5pQHlhaG9vLmlu; Avadhesha Surolia, c3Vyb2xpYUBpaXNjLmFjLmlu