
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 24 July 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1346446
This article is part of the Research Topic Community Series in Regulators of Immune System Function in Autoimmunity and Aging - Molecular and Cellular Research: Volume II View all 3 articles
 Ling Wang1†Zhengwei Zhang1†Haichao Zhang1Minmin Zhou1
Ling Wang1†Zhengwei Zhang1†Haichao Zhang1Minmin Zhou1 Cheng Huang2Wenjiang Xia1*
Cheng Huang2Wenjiang Xia1* Jun Li2*
Jun Li2* Hongmei You3*
Hongmei You3*The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) signaling pathway is one of the fundamental mechanisms of the body’s defense, which responds to the abnormal presence of double-stranded DNA in the cytoplasm to establish an effective natural immune response. In addition to detecting microbial infections, the cGAS pathway may be triggered by any cytoplasmic DNA, which is absent from the normal cytoplasm, and only conditions such as senescence and mitochondrial stress can lead to its leakage and cause sterile inflammation. A growing body of research has shown that the cGAS-STING pathway is strongly associated with sterile inflammation. In this study, we reviewed the regulatory mechanisms and biological functions of the cGAS-STING pathway through its involvement in aseptic inflammation in liver disease, kidney disease, and cellular senescence.
A thorough summary of the many functions of the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway in malignancies and autoimmune disorders has recently been published. Nonetheless, a wealth of data suggests that the cGAS-STING pathway is strongly expressed in several non-immune-related tissues, including the liver (1), kidney (2), and lungs (3), in addition to most immune-related tissues. Endogenous chemicals activate cGAS-STING, which causes inflammation in various disorders. Oxidative stress and apoptosis cause mitochondrial DNA leaking in nonalcoholic fatty liver disease (NAFLD) (4), nonalcoholic steatohepatitis (NASH) (4), alcoholic liver disease (ALD) (5), and renal diseases (6); this “foreign” DNA is detected by cGAS-STING signaling and subsequently triggers the immune system. Both acute kidney injury and chronic kidney disease may be influenced by incomplete and functional mitochondria as well as mitochondrial DNA (mtDNA) copy number abnormalities (mtDNA-cn) (7). In response to cellular stress, mtDNA is released into the cytoplasm and detected by various DNA-sensing pathways, including cGAS-STING signaling (8). Compensatory increases in hepatic mitochondrial respiration, mitochondrial leakage, and hepatic STING protein levels have been observed in patients with NAFLD (9). Patients with alcohol use disorders and persistently alcohol-fed animals with an activated cGAS-STING pathway had considerably higher serum levels of mtDNA-enriched MPs (9, 10).
Cellular senescence, a permanent state of cell cycle termination, is one of the main causes of senescence-associated sterile inflammation and disease. It is primarily characterized by morphometabolic changes, permanent withdrawal from the cell cycle, resistance to cell death, and emergence of inflammatory senescence-associated secretory phenotypes (SASP) (11). The cGAS-STING pathway has been shown to be a significant signaling mechanism that stimulates SASP production in cells, and there is a substantial correlation between it and cellular senescence (12, 13). Because senescent cells express the nuclear marker lamin B1 (14), which is broken down by autophagy, the cytoplasmic DNA of these cells is derived from cytoplasmic chromatin CCFS. When lamin B1 autophagy brings DNA into the cytoplasm, cGAS recognizes the aberrant DNA and the cGAS-STING pathway is initiated, allowing cells to secrete SASP (15). Furthermore, β-amyloid buildup in the brains of patients with Alzheimer’s disease has been linked to double-stranded DNA (dsDNA) breaks and oxidative damage to mtDNA (16, 17). These events facilitate the release of mtDNA into the cytoplasm and activate the cGAS-STING pathway.
In this review, we emphasize the reaction of the cGAS-STING signaling mechanism to self-abnormal DNA and describe the current developments in kidney disease, liver disease, and cellular senescence.
Recognition of foreign DNA is a crucial component of immunity in many species. The cGAS-STING pathway, which has emerged as a critical mechanism for connected DNA sensing and triggers innate immune defense programs, plays a major role in this process in mammalian cells (18). cGAS contains two primary DNA-binding structural domains and a nucleotidyl transferase structural domain. cGAS is self-repressed in the absence of DNA 2’ 3’ cyclic GMP-AMP (cGAMP) is produced when dsDNA binds to conjugated cGAS, activating its enzymatic activity (19). This isomer of cGAMP is known as “2’ 3’ ′-cGAMP” and functions as a second messenger by binding to the endoplasmic reticulum (ER) membrane adaptor STING (20, 21). STING proteins are activated by cGAMP synthase and move from the ER to the Golgi apparatus, where they undergo higher order zwitterionization to form tetramers (22). STING is directly activated by bacterial CDNs (23, 24). Interferon regulatory factor 3 (IRF3) and TANK-binding kinase 1 (TBK1) are believed to be recruited by palmitoylation of Golgi STING (25). STING tetramerization causes the TBK1 dimer to be recruited and activated, and TBK1 transphosphorylates STING’s c-terminal structural domain, activating IRF3 (26). Following its translocation to the nucleus, IRF3 stimulates the production of type I interferon (IFN-I) and immune-stimulated genes (ISG) (24). Additionally, STING may trigger inflammatory cytokines, including interleukin (IL)-6 and tumor necrosis factor (TNF), and activate nuclear factor kappa B (NF-κB) signaling (9) (Figure 1).
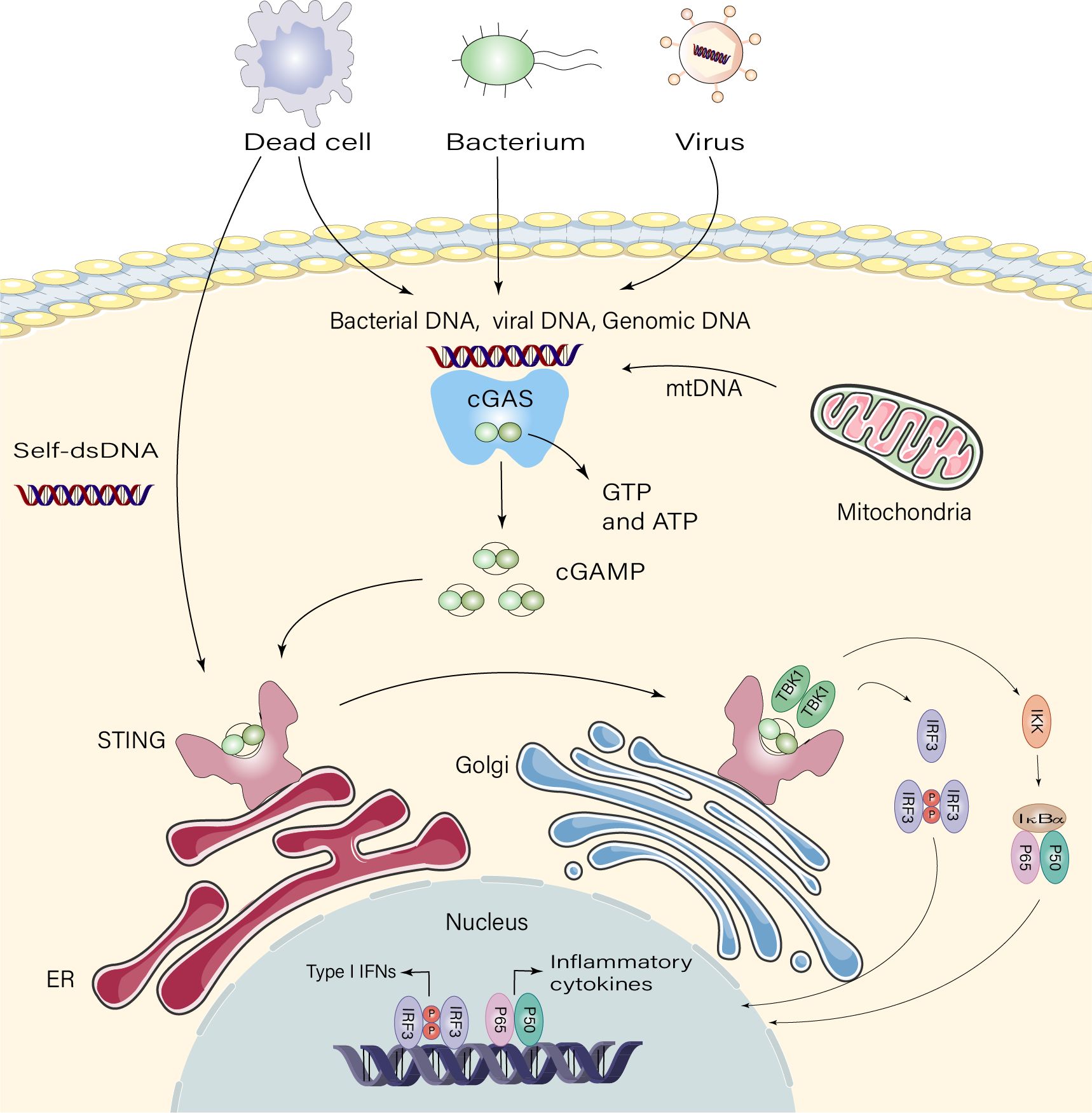
Figure 1 Signaling pathway of cGAS-STING. Dead cells, viruses, and damaged mitochondria release DNA that binds and activates the cyclic GMP-AMP synthase(cGAS), which catalyzes the synthesis of 2’3′-cGAMP from ATP and GTP. 2′3′-cGAMP binds to the endoplasmic reticulum adaptor STING, and cGAMP binds to the interferon gene-stimulating factor (STING) dimer on the endoplasmic reticulum (ER) membrane. STING then activates inhibitor of kappa B kinase (IKK) and TANK-binding kinase 1 (TBK1). TBK1 phosphorylates STING, which recruits TBK1, promoting autophosphorylation of TBK1, phosphorylation of STING, and recruitment of interferon regulatory factor 3 (IRF3). Phosphorylation of IRF3 by TBK1 dimerizes and translocates IRF3 to the nucleus, thereby inducing type I interferon, and gene expression of several other inflammatory mediators, pro-apoptotic genes and chemokines.
The spectrum of NAFLD includes a range of conditions, from simple accumulation of fat in the liver to inflammation, fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) (27). NAFLD is divided into two subtypes: non-progressive non-alcoholic fatty liver (NAFL) and progressive non-alcoholic steatohepatitis (NASH) (28). These data suggest that NAFLD and NASH are promoted by the innate immune response. Therefore, it is unclear how the innate immune response contributes to NALFD at a fundamental level (29, 30). Obesity and NAFLD are strongly linked, and metabolic disorders are mostly influenced by persistent sterile inflammation of the adipose tissue (31). Chronic inflammation in adipose tissue is the primary cause of insulin resistance caused by obesity. Mouse adipose tissues showed significantly higher expression levels of cGAS and STING when the mouse were obese (31). In the mitochondrial matrix, the oxidoreductase-like protein (DsbA-L) functions as a chaperone-like protein. The targeted knockdown (KD) of this protein may compromise mitochondrial function and promote mtDNA release (32). A persistent sterile inflammatory response is triggered by the activation of the cGAS-STING signaling pathway by mtDNA escape generated by a high-fat diet (HFD). DsbA-L overexpression specific to the adipose tissue or STING KD shielded animals from obesity induced by a HFD. This suggests that by blocking the activation of the cGAS-cGAMP-STING pathway caused by mtDNA release, DsbA-L, a crucial regulator of mitochondrial integrity, shielded mice from inflammation and metabolic failure caused by obesity. This further implies that obesity-induced chronic inflammation may be ameliorated by focusing on the cGAS-cGAMP-STING axis in adipose tissue (32). The level of STING protein in the liver tissue of NAFLD patients also shows an increasing trend. (29). During the development of NAFLD/NASH, there is a notable increase in mtDNA levels in the blood and hepatocytes of humans and mice (33). One of the endogenous damage-associated molecular patterns, mtDNA, activates cGAS-STING signaling, which results in hepatocyte abnormalities (34). The STING-irf3 pathway connects the activated STING protein with Bcl-2-associated X protein (Bax) in the mitochondria, initiating the mitochondrial apoptotic pathway and resulting in apoptosis in an IFN-independent manner (35). IRF3 phosphorylation is inhibited by STING deficiency, and IRF3 phosphorylation is further inhibited by the absence of Bax interaction and activation of caspases 3 and PARP, which reduces hepatocyte death (36). In addition to apoptosis, inflammation is essential in the conversion of NAFLD to NASH, inflammation as well as apoptosis are essential (37). Activation of the hepatic cGAS-STING-TBK1 pathway may cause inflammation and interfere with cellular metabolism. These effects may include increased glycolysis and systemic lipid metabolism (38). Lipotoxic activation of TBK1 and consequent phosphorylation of p62 in hepatocytes are mediated by cGAS and STING. Accumulation of protein inclusions in hepatocytes is one of the main pathophysiological indicators of NASH. Large protein inclusions in hepatocytes and lipotoxicity-induced ubiquitinated protein aggregation depend on TBK1-mediated p62 phosphorylation (39, 40). Notably, Luo et al. (29) confirmed that plasma ALT and hepatic triglyceride levels were significantly lower in HFD wild-type (WT) mice than in HFD-fed STINGgt mice. In addition, hepatic inflammation, collagen deposition, and the expression levels of α-SMA and Col1α1 were significantly lower after inhibition of cGAS-STING compared to HFD-WT mice. The findings of Iracheta-Vellve et al (40). were also consistent. According to this research, a novel therapeutic target for stopping the onset and progression of NAFLD may be the cGAS-STING signaling pathway.
Hepatic inflammation, intestinal leakiness, and aberrant oxidative stress are associated with ALD, a global health concern (41). Numerous studies have indicated that sterile inflammation leads to alcoholic liver injury, and that lipogenesis and inflammation are both triggered by the cGAS-STING signaling pathway (42, 43). Hepatic RNA-seq analysis of patients showed a positive correlation between cGAS-IRF3 pathway expression and ALD severity (9). Connexin 32 (Cx32) plays a crucial role in ALD as a regulator of the 2’ 3’ cGAMP intercellular transfer produced by cGAS. Inhibition of Cx32 expression prevents the cytoplasmic sensor cGAS from inducing IRF3 activation in alcohol-damaged hepatocytes and the surrounding parenchyma via the gap junction intercellular communication pathway (44, 45). IRF3 has also been implicated in ALD. Petrasek et al. (46) reported that phosphorylated IRF3 and STING mediate ethanol-induced ER stress in hepatocytes during the early phases of ALD. Activation of IRF3, which is associated with the pro-apoptotic molecule Bax, results in hepatocyte death. Their results indicated that the first phases of ALD are driven by hepatocyte-specific IRF3 and not by macrophage-dependent inflammation, even though macrophages are required for the development of alcoholic steatohepatitis. IRF3 is independent of IFN-I and inflammatory cytokines, and it plays a unique role as a signaling molecule in the crosstalk between ER stress and hepatocyte death in ALD. To further validate the role of cGAS-STING in ALD, researchers conducted experimental observations. They found that mice with a STING gene knockout (KO) did not show obvious hepatocyte apoptosis after alcohol consumption compared to WT mice. In addition, these KO mice exhibited a reduced degree of fat degeneration and did not exhibit IRF3 upregulation. These results suggest that inhibition of the GAS-STING pathway has a significant protective effect against alcoholic liver injury and is of great significance for a deeper understanding of the mechanism underlying the development of this disease (46). Excessive build-up of the extracellular matrix (ECM) caused by long-term damage is known as liver fibrosis. Oxidative stress and inflammation accelerate liver damage (47). Without adequate therapy, liver fibrosis progresses to cirrhosis. Several causes of liver fibrosis have been documented for decades and several possible treatments have been suggested. However, there is currently no effective therapeutic strategy (48). TGF-β causes healthy HSCs to release mtDNA from voltage-dependent anion channels (VDACS) (49) and form an mtDNA cap on the outer membrane of the mitochondria, which activates the cGAS-STING-IRF3 pathway (50). Parkin is an essential regulator of mitochondria. Patients with liver fibrosis have elevated levels of circulating mtDNA, hepatic parkin, and VDAC1 (51). Parkin KD worsened hepatic mtDNA release and STING signaling activation and reduced autophagy and apoptosis in mice with liver fibrosis. Furthermore, mtDNA release into the cytoplasm was inhibited and VDAC1 oligomerization was stopped by parkin site-specific ubiquitination of VDAC1 at lysine 53. Liver fibrosis was inhibited by parkin-mediated ubiquitination of VDAC1 at certain locations, which stopped VDAC1 oligomerization and mtDNA release (52). Hepatocytes that accumulate microbial DNA also show high levels of phosphorylated STING and cGAS. Activation of the cGAS-STING signaling pathway leads to the overexpression of NLRP3 and enhances hepatic cellular pyroptosis, which is involved in liver fibrosis. In mouse models, acute and chronic fibrosis induced by carbon tetrachloride can be alleviated by the lack of STING, which reduces liver cell death, inflammation, and fibrosis (18). Furthermore, the effect of microbial DNA on the inflammatory response of insulin-sensitized cells was mitigated by cGAS KD (53). These results highlight the significance of Cgas-STING signaling in hepatocyte responses caused by microbial DNA. However whether the cGAS-STING pathway contributes to hepatic fibrosis by inducing cellular pyroptosis, reactive oxygen species (ROS) accumulation, or an inflammatory response needs to be further explored.
High morbidity and mortality rates are associated with acute kidney injury (AKI). Elevations in serum creatinine levels and urine output were used to classify the severity of AKI (54). Acute kidney injury is a complex clinical condition that changes depending on the type of damage. However, the disruption of energy metabolism is shared by several acute renal injury disorders (54). Given that the energy metabolism pathways are important causes of AKI, they provide novel therapeutic options.
Similar to the elevated levels of STING observed in the kidneys of patients with AKI, the nephrotoxic drug cisplatin enhanced the production of cGAS and STING in injured tubules of mice. According to these results, the BCL-2-like protein 4 (BAX) hole in the outer mitochondrial membrane is the mechanism through which cisplatin promotes mtDNA leakage into the cell membrane (35). In a cisplatin-induced mouse model, tubular inflammation in AKI was activated via the cGAS-STING pathway, and in AKI mice, tubular inflammation was reduced by STING inhibition. By preventing mtDNA replication, the expression of the cGAS-STING pathway and cisplatin-induced inflammation were significantly reduced. According to previous studies, cisplatin exposure causes mitochondrial malfunction in tubular cells, which in turn leads to inflammatory reactions via cGAS-STING signaling induced by cytoplasmic mtDNA (6). The biological role of Myr, a flavonoid abundantly distributed in a variety of plants, was identified by Qi et al. (55). It was discovered that the HS15-Myr nanomicelles reduced cisplatin-induced AKI by blocking the cGAS-STING signaling pathway. By controlling mitochondrial processes, such as mitophagy and mitochondrial fission, PGAM5 plays an important role in cellular activity (56, 57). In the kidneys of mice with AKI and human renal biopsy samples, PGAM5 levels were elevated. PGAM5 dephosphorylates Bax, which causes mtDNA leakage, which is linked to renal injury and the inflammatory response in AKI. To reduce the inflammation in AKI, PGAM5 KD prevented the release of mtDNA and the interaction of the cellular DNA receptor cGAS with mtDNA in cultured renal tubular epithelial cells (2). cGAS-STING KO by small interfering RNA resulted in a cisplatin-induced decrease in the phosphorylation of the downstream target proteins TBK1 and P65 in HK-2 cells, which ameliorated renal tubular inflammation and the extent of neutrophil infiltration in the tubular interstitium in mice with AKI (6). This suggests that the activation of the cGAS-STING pathway plays an important role in acute kidney injury. However, in an AKI mouse model, STING KO did not completely eliminate damage or inflammatory responses, indicating that upstream mitochondrial dysfunction may lead to the activation of other downstream pathways that participate in cisplatin-induced AKI.
Chronic kidney disease (CKD) is caused by inflammation, cellular and ECM deposition, and nephron loss (58). However, persistent activation of this pathway worsens chronic kidney disease and may result in renal cancer (59).
A mouse model of CKD showed that the renal cortex and glomeruli activate the cGAS-STING signaling pathway. Mitrofanova et al. employed terminally differentiated murine and human podocytes as in vitro models to examine the presence and function of various elements involved in the cGAS-STING signaling pathway (60). For in vivo investigations, they employed db/db mice as a representative model of type 2 diabetes (T2D) accompanied by diabetic kidney disease (DKD), and Col4a3-/- mice as an experimental model for kidney disease linked to Alport syndrome (AS). The results indicated that the cGAS-STING signaling pathway was expressed in both human and murine podocytes. Proteinuria and podocyte loss were observed in WT mice following STING activation. In addition, activation of the cGAS-STING signaling pathway was observed in both the kidney cortices and glomeruli of db/db mice and mice with AS. Furthermore, this activation was correlated with the presence of albuminuria. Notably, genetic or pharmacological suppression of STING effectively improved glomerular injury and kidney failure induced by diabetes or streptozotocin as well as AS-associated conditions (60). Further research is necessary to determine the exact mechanisms underlying this phenomenon. Innate immunological protection against infection is facilitated by the cGAS-STING signaling pathway, and controlling the degree of STING suppression is crucial for the management of CKD.
DKD is a common complication that typically develops in patients several years after diabetes onset (60). DKD causes serious damage to the kidneys, including decreased glomerular filtration rate, proteinuria, and ultimately, kidney failure. Although there are currently some treatments for DKD, such as interventions to control blood pressure, blood sugar, and lipid levels, additional treatment targets and methods are needed to effectively manage this severe complication. In recent years, increasing evidence has shown that chronic low-grade systemic inflammation and oxidative stress play key roles in the development of DKD (61). Previous studies have shown that an increase in PCSK9 is associated with renal inflammation in mice fed a HFD, and with HGPA-induced inflammation in HK-2 cells. The cGAS/STING pathway is involved in the development of DKD. Further experiments showed that PCSK9 triggered mtDNA damage in DKD and activated the cGAS-STING pathway, leading to a series of adverse immune reactions and renal tissue damage. The use of the STING inhibitor C-176 in renal tubular epithelial cells HK-2 significantly reduced the expression levels of p-TBK1/NF-κB, thereby alleviating kidney inflammation caused by chronic kidney injury and immune-mediated kidney injury (62). STING KO mice have also been studied. WT and STING KO mice were used to establish a streptozotocin-induced diabetes model. Compared to the WT mice in the STZ group, the increase in the levels of urinary albumin and urinary Kim-1 was significantly reduced in the STING KO mice with neuropathic pain (63). These results suggest that the cGAS-STING/p-TBK1/NF-κB axis plays a key role in the development of diabetic nephropathy. In conclusion, these findings provide new insights for the treatment and prevention of immune-related chronic kidney damage, and provide important references for further exploration of the mechanism of action of the cGAS-STING/p-TBK1/NF-κB axis in other immune-related diseases.
Various types of kidney disorders are associated with tissue damage, which has been primarily attributed to the effect of IFN-I, a cytokine widely employed in the field of clinical oncology for many years, (64) on multiple renal cell types, thereby influencing their biology and function. Augmented IFN-I signaling is detected during viral infections and autoimmune conditions, all of which may contribute to kidney impairment and share histopathological and pathogenic characteristics (65). It is worth noting that STING pathway activation serves as a pivotal mechanism for sensing innate immunity, resulting in the production of IFN-I within the tumor microenvironment. As CD8+ T cells are associated with tumors exhibiting an IFN-I signature, the potential for utilizing intratumoral STING agonists as a viable cancer treatment shows promise (66). The host STING pathway is at the interface of cancer and immunity. Activation of the STING pathway in the tumor microenvironment by antigen-presenting cells results in the generation of IFN-I and facilitates the spontaneous development of CD8+ T-cell responses against tumors, which exhibited therapeutic effects in multiple murine cancer models (67, 68). Xing et al. (69) reported that TRIM29, a protein abundantly present in human AECs and induced in myeloid dendritic cells upon exposure to dsDNA, is utilized by DNA viruses such as the Epstein-Barr virus, which inhibits the innate immune response of the host (70). Previous studies have indicated that TRIM29 acts as a crucial suppressor of the innate immune response to DNA viruses by specifically interacting with STING, thereby facilitating the prolonged presence of DNA viruses (70). Similarly, Fang et al. reported that the suppression or removal of TRIM18 in macrophages from humans or mice resulted in an increased production of IFN-I when exposed to dsRNA and dsDNA, as well as during infection with RNA and DNA viruses. From a mechanistic standpoint, this study revealed that TRIM18 plays a role in recruiting PPM1A to dephosphorylate TBK1. Deactivation of TBK1 prevents its interaction with upstream adaptors of mitochondrial antiviral signaling and STING, ultimately leading to the attenuation of antiviral signaling during viral infections (71). Interestingly, a study conducted in animals has provided evidence suggesting the involvement of TRIM18 in kidney damage and fibrosis in mice with diabetes (72). Inhibition of TRIM18 has been shown to effectively prevents inflammation, epithelial-mesenchymal transition, and fibrosis in HK-2 cells induced by high glucose levels (72). Previous research has indicated that TRIM29 facilitates podocyte pyroptosis via the NF‐kB/NLRP3 inflammasome signaling pathway and aggravates diabetic nephropathy (70). However, increasing evidence has revealed that pyroptosis is a double-edged sword in human cancers. Cancer cell pyroptosis contributes to the suppression of tumor progression (73), whereas pyroptosis in normal tissues contributes to the side effects of chemotherapy. Thus, further validation of the upstream and downstream relationship between TRIM29/TRIM18 and STING in various renal diseases is necessary to determine whether TRIM29/TRIM18 can effectively regulate STING, thereby controlling acute and chronic kidney injury, as well as renal cancer. However, it is imperative to further investigate the extent and proportion of pyroptotic cell death mediated by TRIM29 in both normal kidney and renal cancer cells. Undoubtedly, this remains a substantial undertaking requiring gradual and comprehensive exploration.
Irreversible cell-cycle arrest, known as cellular senescence, restricts the ability of injured cells to proliferate (74). Genetic instability is a defining feature of the aging process (75). The SASP, which is characterized by the secretion of pro-inflammatory cytokines, chemokines, the ECM, and growth regulators by senescent cells, has recently been demonstrated to be induced by DNA damage (76). cGAS-STING promotes inflammatory senescence by increasing SASP secretion (12). This indicates a surprising relationship between the two seemingly unconnected channels.
Senescent cells are subtly influenced by the SASP to undergo self-destruction and cause neighboring cells to undergo senescence. As a result, age-related illnesses such as Alzheimer’s disease (AD) can be affected by cellular senescence caused by the SASP (77). AD is a neurodegenerative disorder. The condition is defined by the buildup of amyloid β peptide (Aβ) plaques in the brain, the tangle of phosphorylated Tau proteins and neurogenic fibers, and gradual cognitive deterioration in patients (78). Damage to mtDNA, the only organelle with its own genome, is linked to several neurodegenerative illnesses. Unlike genomic DNA, the mitochondrial genome is far more vulnerable to oxidative damage and lacks a strong defense mechanism against damage (12). Consequently, clearance of damaged mitochondria by autophagy is crucial (79). However, in the brains of AD model mice, the interaction between cGAS and dsDNA increased more than 4-fold compared to WT animals because of defective mitochondrial autophagy in senescent cells (80). Furthermore, the brains of patients with AD and senescent animals exhibit markedly increased levels of STING, TBK1, and IRF3 phosphorylation, suggesting that the cGAS-STING pathway is activated (77). In the brain, microglia and astrocytes operate as glial cells, promoting the survival and functionality of neuronal cells, exhibiting neurotrophic effects, and upholding the overall homeostasis of the brain (81). However, in illnesses, the inherent homeostatic qualities of glial cells are jeopardized, releasing inflammatory mediators that create a harmful inflammatory brain environment (21). Microglia, astrocytes, and cortical neurons all exhibit activation of the cGAS-STING pathway; however, only microglia show signs of inflammation. Activated microglia in AD model mice generated IL-1β and TNF-α, which changed astrocytes into neurotoxic A1-type astrocytes (81). ROS, reactive nitrogen species (RNS), and pro-inflammatory cytokines are produced in concert by activated astrocytes and microglia. Inducible nitric oxide synthase (iNOS) causes RNS. Nitrosative stress and the activation of additional neurodegenerative pathways are caused by elevated levels of nitric oxide and its highly reactive derivative peroxynitrite (82). cGAS KO in the brains of AD mice with reduced Aβ levels and a better proportion of microglia near Aβ plaques suggests that deletion of cGAS promotes Aβ clearance by microglia and ameliorates AD-associated cognitive decline. Deficiency of cGAS in Aβ42 peptide-treated primary microglia leads to the inhibition of the cGAS-STING pathway, decreased phosphorylation levels of TBK1 protein, and reduced expression of IFNβ, CXCL10, and CCL5. This also results in the inhibition of neurotoxic A1-type astrocytes, thus alleviating the brain damage caused by Aβ. In addition, in vitro experiments have shown that cGAS inhibitor RU.521 and STING inhibitor H-151 can protect neurons from Aβ damage; in vivo experiments have demonstrated that they significantly reduce the levels of Aβ in the brain and decrease the number of activated microglia and astrocytes, thereby improving cognitive decline associated with AD. These results provide important clues for finding new approaches to treat AD, and offer valuable information to gain a deeper understanding of the pathogenesis of AD. Hopefully, through further research and exploration, more effective and safer treatment methods can be developed to address this common but incurable neurodegenerative disease.
In addition to AD, Parkinson’s disease (PD) is significantly influenced by cGAS-STING in glial cells (83). Damaged dsDNA produced by dying dopaminergic neurons stimulates cGAS/STING (84). PD is characterized by an inflammatory response resulting in dopaminergic neuronal damage and death. et al. Barazzuol et al. (85). discovered that a mutation in the protein parkin, phosphatase, and tensin homolog (PTEN)-induced kinase (PINK) catalyzes a burst of Mfn2 phosphorylation. This in turn causes the Mfn2 complex to break down from the outer mitochondrial membrane in a p97-dependent manner, separating the mitochondria from the ER and promoting mitochondrial autophagy (86). The VDAC pore of the mitochondrial outer membrane allows the release of mtDNA and mediates activation of the cGAS-STING pathway. Alternatively, mtDNA can protrude through the inner membrane of the mitochondria and be released by the BAX/BAK complex (87). When cGAS-STING is activated in astrocytes, p16INK4a and p-p65 expression are upregulated, and STING proteins directly bind to YY1, facilitating YY1 nuclear translocation and boosting LCN2 transcription (88). Increased SASP secretion accelerates neurodegenerative lesions, and cGAS-STING-LCN2 stimulates senescence-associated β-galactosidase activity and astrocyte senescence, making them larger and flatter (89) (Figure 2). The loss of cGAS in PD mice leads to the blockade of LCN, and a significant downregulation of P161NK4a, p-STING, and p-p65 expression, while significantly improving the behavior of MPTP-treated mice and reversing dopamine (DA) levels in PD model mice. These findings indicate that cGAS plays an important role in the development of PD.
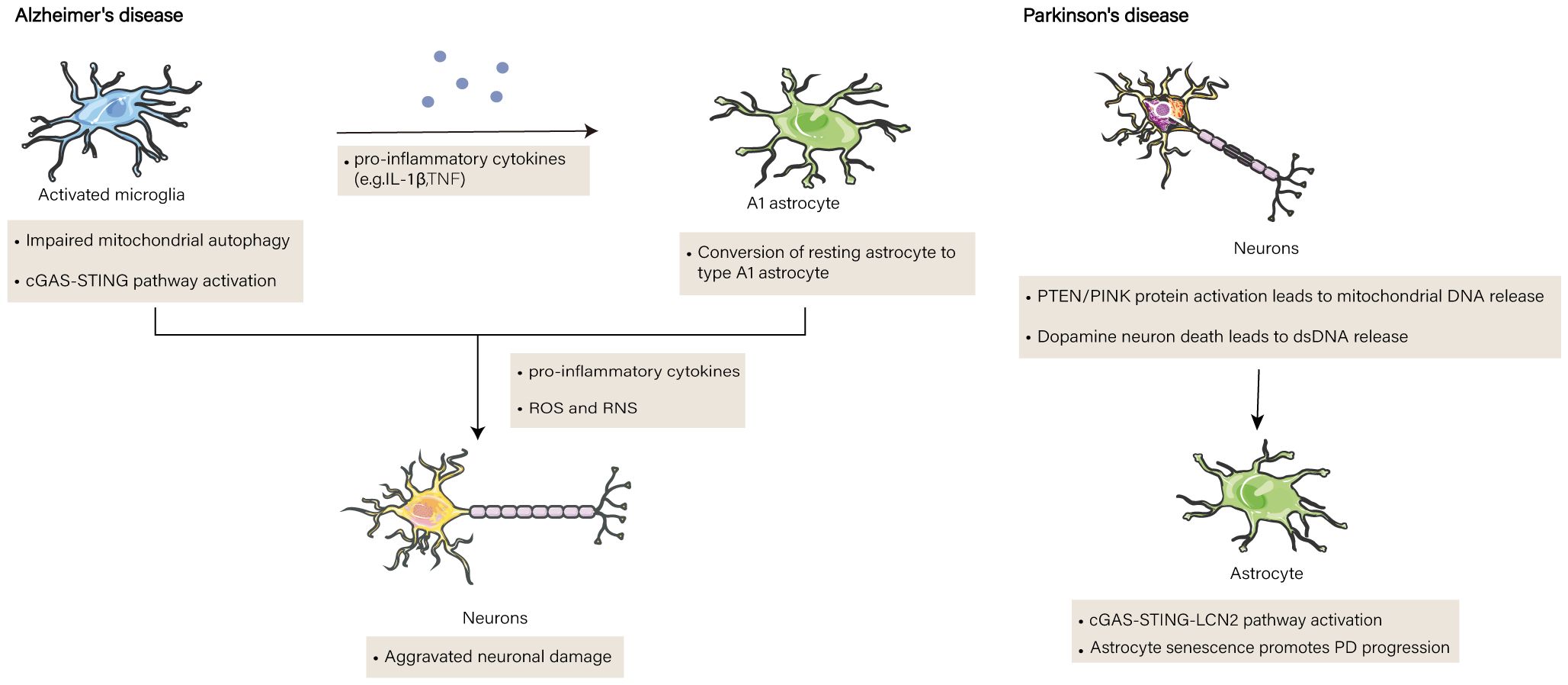
Figure 2 The function of cGAS-STING in brain diseases. Impaired mitochondrial autophagy in senescent cells in the brains of Alzheimer’s patients results in the release of mitochondrial DNA (mtDNA) into microglia, where the cGAS-STING pathway on microglia is activated and leads to the release of pro-inflammatory factors. Pro-inflammatory factors induce the transformation of astrocytes into type A1 astrocytes. The two cells synergize to produce pro-inflammatory cytokines, reactive oxygen species (ROS) and reactive nitrogen species(RNS), ultimately leading to neurodegeneration. In addition to the release of double-stranded DNA (dsDNA) from dead dopamine neurons in Parkinson’s disease mice, an increase in phosphatase and tensin homolog/PTEN induced putative kinase 1 (PTEN/PINK1) proteins induces the release of mtDNA from the voltage-dependent anion channel (VDAC) and bcl2-associated X protein/bcl2 antagonist/killer (BAX/BAK) channels, both of which activate the cGAS-STING signaling pathway, and the STING proteins increase the transcription oflipocalin 2 (LCN2), thereby secretion of senescence-associated secretory phenotype(SASP), and contribute to the aggravation of senescence in astrocytes. and promote astrocyte senescence and aggravate the disease process.
Atherosclerosis (AS) is characterized by accumulation of vascular smooth muscle cells (VSMCs), vascular plaque formation, and cholesterol overload (macrophage accumulation). Additionally, senescence of VSMCs is linked to the formation of micronuclei, activation of cytoplasmic sensing by cGAS-STING, and induction of multiple pro-inflammatory cytokines (90). During AS, VSMCs exhibit signs of senescence and DNA damage. IFN-I is induced by IRF3, nuclear DNA in the cytoplasm triggers the cGAS-STING cytoplasmic DNA sensing pathway, and NF-κB triggers a pro-inflammatory response (91). The activation of the TBK1 and cGAS-STING pathways is linked to this pro-inflammatory phenotype, as cGAS deletion eliminates the induction of IL-1α and IL-8 and raises NF-κB phosphorylation. Furthermore, IQ motif-containing GTPase-activating protein 1 (IQGAP1) functions as a scaffolding protein that controls mitochondrial activity and encourages the generation of ROS, which results in oxidative stress and mitochondrial malfunction, and is linked to the development of atherogenesis (92). Induction of mtDNA release into the cytoplasm and activation of the DNA sensor cGAS-STING to trigger endothelial cell pyroptosis to generate AS are the results of ROS generation promoted by IQGAP1. Moreover, TDP43, a nuclear (DNA)/RNA-binding protein that has been demonstrated to be localized to the mitochondria, increases the production of SASP by releasing mtDNA and initiating the cGAS-STING signaling pathway (93). The use of the cGAS inhibitor RU.521 and STING inhibitor C-176 in human umbilical vein endothelial cells reversed IQGAP1-induced elevated levels of NLRP3, caspase-1, and inflammatory cytokines (92). Moreover, studies have found that a HFD leads to increased expression of cGAMP and STING in ApoE-/- mice. Treatment of mice with the specific STING antagonist C-167 reduced plaque damage and decreased lipid accumulation. Additionally, stimulation-induced downstream molecules IRF3 and various typical inflammatory markers such as IL-6, IL-1β, and TNF-α were found to be elevated in AS plaques (94). Genetic associations in humans suggest that cellular senescence plays a protective role against AS (95). This suggests that senescence may play a dual role: the growth of atheromatous plaques is limited by macrophage and monocyte proliferation blockade; however, the disease process can also be induced by cell-secreted SASP factors (96).
Cellular senescence is closely associated with aging and age-related diseases. While youthful stromal cells regenerate senescent stem cells and the senescent milieu “ages” young stem cells, the accumulation of senescent cells is the primary cause of senescence-associated tissue deterioration. Cells use a process called mechanical signal transduction to detect the structural, mechanical, and physical characteristics of their surroundings. Guarnieri et al. (97) discovered that by blocking the cGAS-STING signaling pathway, stromal cells with intact YAP/TAZ mechanotransduction may postpone senescence. SASP and IFN-I-stimulated genes were secreted in greater amounts by WI-38 fibroblasts when YAP/TAZ was knocked down; however, the levels of these genes were reversed upon cGAS/STING KD. Both young and old YAP/TAZ-KO animals had noticeable and severely malformed nuclei, and the absence of YAP/TAZ caused an increase in cGAS at cell nuclear membrane discontinuities. YAP/TAZ mediates suppression of SASP gene expression and cGAS-STING activation by shielding the shell membrane via ACTR2 and lamin B1. Furthermore, the serine/threonine kinase,PINK1, regulates mitochondrial dysfunction (98). Renal tubular injury and an increase in senescence and SASP are the results of PINK1 deficiency (99). In the renal tubular epithelial cells of 24-month-old Pink1-/- mice, these abnormalities were particularly noticeable. According to a gene expression study by RNA sequencing, PINK1 deficiency was linked to an elevated inflammatory response, and the activation of cGAS-STING was noteworthy in 24-month-old Pink1-/- mice. Pink1 deficiency has been linked to mitochondrial malfunction, which can activate the cGAS-STING pathway and induce aging-related inflammatory responses. Pink1 is also associated with renal aging (86).
The non-traditional cGAS-STING signaling pathway plays a crucial role in cellular senescence, in contrast to the classical cGAS-STING pathway. Significant phosphorylation of the elF2a S51 site and inhibition of cap-dependent mRNA translation processes can result from the activation of cGAS-STING signaling during cellular senescence. This function is independent of the classical STING complex, which consists of the key kinase TBK1 and signaling protein IRF3, in various species (100). Consequently, elF2α is phosphorylated by PERK in response to DNA sensing, which greatly reduces mRNA translation across the cell but specifically increases the inflammatory translational program. Parallel to but much earlier than the traditional STING-TBK1-IRF3 signaling in the Golgi apparatus, STING-PERK-elF2α signaling is immediately triggered in the ER. Taken together, these findings show that the cGAS-STING pathway plays a critical role in the regulation of cellular senescence, and that blocking this pathway might be beneficial for treating disorders associated with cellular senescence (Figure 3).

Figure 3 Signaling pathways upstream of cGAS-STING in aging. Deletion of yes-associated protein/transcriptional coactivator with pdz-binding motif (YAP/TAZ) protein in the nucleus of mouse dermal fibroblasts inhibits laminB1 and actin related protein 2 (ACTR2) proteins at the nuclear membrane, leading to nuclear deformation and leakage of nuclear DNA; deletion of PINK protein in the mitochondria of senescent mouse renal tubular cells leads to mitochondrial autophagy damage and leakage of mitochondrial DNA. DNA binds to cGAS to produce cGAMP, and STING binds to cGAMP, part of which interacts with protein kinase R-like endoplasmic reticulum kinase (PERK), leading to significant phosphorylation of el2α and inhibition of the cap-dependent mRNA translation process, which specifically promotes part of inflammation and cellular senescence. The other part is involved in cellular senescence by activating TBK1, which secretes SASP through the classical pathway.
In this review, we provide an overview of the current developments in our understanding of the function of the cGAS-STING pathway in kidney, liver, and cell senescence disorders. In addition, the sterile inflammatory SASP, which is a characteristic of senescence and contributes to aging, is related to cGAS-STING. This contrasts with the inflammation that arises in response to pathogens such as bacteria and viruses. Finally, while pathway inhibitors of cGAS-STING are an effective way to treat chronic inflammation, the diversity of endogenous cytoplasmic DNA functions must be considered. For example, inhibition of CCF activation downstream of cGAS-STING signaling in chronic inflammation may be beneficial during aging, but it may also impair micronuclei-initiated cell-intrinsic immune detection and tumor suppressor signaling. Overall, these studies provided new ideas and targets for the treatment of aging and metabolic diseases. Therefore, the development of cGAS and STING proteins has received considerable attention in recent years. Several targeted drugs are currently undergoing clinical trials and are expected to be used for disease treatment in the future.
GTPase-activating protein SH3 domain-binding protein 1 (G3BP1) is a key protein in the sensing and activation of cGAS with DNA, and its deletion causes inefficient binding of cGAS to DNA and inhibits cGAS-dependent IFN production (101). Liu et al. (102) found that the natural small molecule epigallocatechin gallate (EGCG), identified in green tea polyphenols, could interfere with the binding of G3BP1 to cGAS, thus preventing cGAS from recognizing abnormal DNA and resulting in the inability of downstream pathways to be activated. The effectiveness of EGCG inhibitors has been demonstrated in animal models of autoimmune diseases and in patients with Aicardi-Goutières syndrome (AGS). The team also found that in the cells of patients with AGS as well as in mouse models, aspirin acetylated lysine residues (Lys384, Lys394, and Lys414) at positions 384, 394, and 414, respectively, of cGAS, which kept the cGAS protein inactive, prevented cGAS from activating downstream signaling pathways, and inhibited autoimmune responses (103). Recently, Chu et al. (104) reported that perilla aldehyde inhibits the auto-intrinsic immunity induced by cytoplasmic DNA by inhibiting cGAS activity. Mice treated with perilla aldehyde – a natural monoterpene extracted from Perilla frutescens – in the AGS disease model showed reduced expression levels of several ISG, such as Cxcl10, Isg15, Isg56, and Ifit3, and significantly attenuated autoimmune inflammation. Existing chemicals can serve as valuable bases for the development of clinical drug candidates. cGAS-STING-mediated sterile inflammation in liver and kidney diseases and aging warrants further exploration in clinical settings.
JL: Conceptualization, Supervision, Writing – review & editing. LW: Writing – original draft. ZZ: Writing – review & editing. HZ: Supervision, Writing – review & editing. MZ: Supervision, Writing – review & editing. CH: Writing – review & editing. WX: Conceptualization, Supervision, Writing – review & editing. HY: Conceptualization, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by the National Natural Science Foundation of China (U19A2001 and 81970534).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bai J, Liu F. cGAS–STING signaling and function in metabolism and kidney diseases. J Mol Cell Biol. (2021) 13:728–38. doi: 10.1093/jmcb/mjab066
2. Li J, Sun X, Yang N, Ni J, Xie H, Guo H, et al. Phosphoglycerate mutase 5 initiates inflammation in acute kidney injury by triggering mitochondrial DNA release by dephosphorylating the pro-apoptotic protein Bax. Kidney Int. (2023) 103:115–33. doi: 10.1016/j.kint.2022.08.022
3. Benmerzoug S, Rose S, Bounab B, Gosset D, Duneau L, Chenuet P, et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat Commun. (2018) 9. doi: 10.1038/s41467-018-07425-1
4. Donne R, Saroul-Ainama M, Cordier P, Hammoutene A, Kabore C, Stadler M, et al. Replication stress triggered by nucleotide pool imbalance drives DNA damage and cGAS-STING pathway activation in NAFLD. Dev Cell. (2022) 57:1728–1741.e6. doi: 10.1016/j.devcel.2022.06.003
5. Ma X, Chen A, Melo L, Clemente-Sanchez A, Chao X, Ahmadi AR, et al. Loss of hepatic DRP1 exacerbates alcoholic hepatitis by inducing megamitochondria and mitochondrial maladaptation. Hepatology. (2023) 77:159–75. doi: 10.1002/hep.32604
6. Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. (2019) 29(5):1261–1273. doi: 10.1016/j.celrep.2019.09.050
7. Jin L, Yu B, Armando I, Han F. Mitochondrial DNA-mediated inflammation in acute kidney injury and chronic kidney disease. Oxid Med Cell Longev. (2021) 2021. doi: 10.1155/2021/9985603
8. Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. (2019) 30:784–99.e5. doi: 10.1016/j.cmet.2019.08.003
9. Luther J, Khan S, Gala MK, Kedrin D, Sridharan G, Goodman RP, et al. Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation. (2020) 117(28):16704. doi: 10.1073/pnas.1911870117/-/DCSupplemental
10. Wang L, You HM, Meng HW, Pan XY, Chen X, Bi YH, et al. STING-mediated inflammation contributes to Gao binge ethanol feeding model. J Cell Physiol. (2022) 237:1471–85. doi: 10.1002/jcp.30606
11. Regulski MJ. Cellular Senescence: What, Why, and How (2017). Available online at: www.woundsresearch.com.
12. Loo TM, Miyata K, Tanaka Y, Takahashi A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. (2020) 111:304–11. doi: 10.1111/cas.14266
13. Guo Q, Chen X, Chen J, Zheng G, Xie C, Wu H, et al. STING promotes senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the NF-κB signaling pathway. Cell Death Dis. (2021) 12. doi: 10.1038/s41419-020-03341-9
14. Qi X, Zheng S, Ma M, Lian N, Wang H, Chen L, et al. Curcumol suppresses CCF-mediated hepatocyte senescence through blocking LC3B–lamin B1 interaction in alcoholic fatty liver disease. Front Pharmacol. (2022) 13:912825. doi: 10.3389/fphar.2022.912825
15. Frediani E, Scavone F, Laurenzana A, Chillà A, Tortora K, Cimmino I, et al. Olive phenols preserve lamin B1 expression reducing cGAS/STING/NFκB-mediated SASP in ionizing radiation-induced senescence. J Cell Mol Med. (2022) 26:2337–50. doi: 10.1111/jcmm.17255
16. Santos RX, Correia SC, Zhu X, Smith MA, Moreira PI, Castellani RJ, et al. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxid Redox Signal. (2013) 18:2444–57. doi: 10.1089/ars.2012.5039
17. Wang X, Wang W, Li L, Perry G, Lee Hg, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. (2014) 1842:1240–7. doi: 10.1016/j.bbadis.2013.10.015
18. Ablasser A, Chen ZJ. CGAS in action: Expanding roles in immunity and inflammation. Sci (1979). (2019) 363. doi: 10.1126/science.aat8657
19. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. (2016) 17:1142–9. doi: 10.1038/ni.3558
20. Cheng Z, Dai T, He X, Zhang Z, Xie F, Wang S, et al. The interactions between cGAS-STING pathway and pathogens. Signal Transduct Target Ther. (2020) 5. doi: 10.1038/s41392-020-0198-7
21. Paul BD, Snyder SH, Bohr VA. Signaling by cGAS–STING in neurodegeneration, neuroinflammation, and aging. Trends Neurosci. (2021) 44:83–96. doi: 10.1016/j.tins.2020.10.008
22. Shang G, Zhang C, Chen ZJ, Bai Xc, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature. (2019) 567:389–93. doi: 10.1038/s41586-019-0998-5
23. Won JK, Bakhoum SF. The cytosolic DNA-sensing cGAS–sting pathway in cancer. Cancer Discovery. (2020) 10:26–39. doi: 10.1158/2159-8290.CD-19-0761
24. Jiang M, Jiang M, Chen P, Chen P, Wang L, Li W, et al. CGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol. (2020) 13. doi: 10.1186/s13045-020-00916-z
25. Li W, Lu L, Lu J, Wang X, Yang C, Jin J, et al. cGAS-STING-mediated DNA sensing maintains CD8 + T cell stemness and promotes antitumor T cell therapy (2020). Available online at: http://stm.sciencemag.org/.
26. Zhang C, Shang G, Gui X, Zhang X, Bai Xc, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature. (2019) 567:394–8. doi: 10.1038/s41586-019-1000-2
27. Isaacs S. Nonalcoholic fatty liver disease. Endocrinol Metab Clin North Am. (2023) 52:149–64. doi: 10.1016/j.ecl.2022.06.007
28. Bedogni G, Palmese F, Foschi FG. Nonalcoholic fatty liver disease: An update. Curr Opin Lipidol. (2023) 34:114–8. doi: 10.1097/MOL.0000000000000874
29. Luo X, Li H, Ma L, Zhou J, Guo X, Woo SL, et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology. (2018) 155:1971–84.e4. doi: 10.1053/j.gastro.2018.09.010
30. Deng ZB, Liu Y, Liu C, Xiang X, Wang J, Cheng Z, et al. Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology. (2009) 50:1412–20. doi: 10.1002/hep.23148
31. Bai J, Cervantes C, He S, He J, Plasko GR, Wen J, et al. Mitochondrial stress-activated cGAS-STING pathway inhibits thermogenic program and contributes to overnutrition-induced obesity in mice. Commun Biol. (2020) 3. doi: 10.1038/s42003-020-0986-1
32. Bai J, Cervantes C, Liu J, He S, Zhou H, Zhang B, et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc Natl Acad Sci U.S.A. (2017) 114:12196–201. doi: 10.1073/pnas.1708744114
33. Lv J, Xing C, Chen Y, Bian H, Lv N, Wang Z, et al. The STING in non-alcoholic fatty liver diseases: potential therapeutic targets in inflammation-carcinogenesis pathway. Pharmaceuticals. (2022) 15. doi: 10.3390/ph15101241
34. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest. (2019) 129:546–55. doi: 10.1172/JCI121842
35. Wolf P, Schoeniger A, Edlich F. Pro-apoptotic complexes of BAX and BAK on the outer mitochondrial membrane. Biochim Biophys Acta Mol Cell Res. (2022) 1869. doi: 10.1016/j.bbamcr.2022.119317
36. Qiao JT, Cui C, Qing L, Wang LS, He TY, Yan F, et al. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism. (2018) 81:13–24. doi: 10.1016/j.metabol.2017.09.010
37. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
38. Shaker ME. The contribution of sterile inflammation to the fatty liver disease and the potential therapies. Biomedicine Pharmacotherapy. (2022) 148. doi: 10.1016/j.biopha.2022.112789
39. Hasan M, Gonugunta VK, Dobbs N, Ali A, Palchik G, Calvaruso MA, et al. Chronic innate immune activation of TBK1 suppresses mTORC1 activity and dysregulates cellular metabolism. Proc Natl Acad Sci U.S.A. (2017) 114:746–51. doi: 10.1073/pnas.1611113114
40. Cho CS, Park HW, Ho A, Semple IA, Kim B, Jang I, et al. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1–mediated p62/sequestosome 1 phosphorylation. Hepatology. (2018) 68:1331–46. doi: 10.1002/hep.29742
41. Iracheta-Vellve A, Petrasek J, Gyongyosi B, Satishchandran A, Lowe P, Kodys K, et al. Endoplasmic reticulum stress-induced hepatocellular death pathways mediate liver injury and fibrosis via stimulator of interferon genes. J Biol Chem. (2016) 291:26794–805. doi: 10.1074/jbc.M116.736991
42. Cai Y, Xu MJ, Koritzinsky EH, Zhou Z, Wang W, Cao H, et al. Mitochondrial DNA–enriched microparticles promote acute-on-chronic alcoholic neutrophilia and hepatotoxicity. JCI Insight. (2017) 2. doi: 10.1172/jci.insight.92634
43. Xu D, Tian Y, Xia Q, Ke B. The cGAS-STING pathway: novel perspectives in liver diseases. Front Immunol. (2021) 12:682736. doi: 10.3389/fimmu.2021.682736
44. Patel SJ, King KR, Casali M, Yarmush ML. DNA-triggered innate immune responses are propagated by gap junction communication. Proc Natl Acad Sci U S A. (2009) 106(31):12867–72. doi: 10.1073/pnas.0809292106
45. Segretain D, Falk MM. Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochim Biophys Acta Biomembr. (2004) 1662:3–21. doi: 10.1016/j.bbamem.2004.01.007
46. Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci U.S.A. (2013) 110:16544–9. doi: 10.1073/pnas.1308331110
47. Lin CY, Adhikary P, Cheng K. Cellular protein markers, therapeutics, and drug delivery strategies in the treatment of diabetes-associated liver fibrosis. Adv Drug Delivery Rev. (2021) 174:127–39. doi: 10.1016/j.addr.2021.04.008
48. Lin Y, Dong MQ, Liu ZM, Xu M, Huang ZH, Liu HJ, et al. A strategy of vascular-targeted therapy for liver fibrosis. Hepatology. (2022) 76:660–75. doi: 10.1002/hep.32299
49. Guimarães EL, Best J, Dollé L, Najimi M, Sokal E, Van Grunsven LA. Mitochondrial uncouplers inhibit hepatic stellate cell activation. BMC Gastroenterol. (2012) 12:68. doi: 10.1186/1471-230X-12-68
50. Arumugam S, Li B, Lakshmi S, Boodapati SLT, Nathanson MH, Sun B, et al. Mitochondrial DNA and the STING pathway are required for hepatic stellate cell activation. Hepatology (2023) 78(5):1448–1461. doi: 10.1097/HEP.0000000000000388
51. Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. MITOCHONDRIAL BIOLOGY VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease . Available online at: http://science.sciencemag.org/.
52. Wu NN, Wang L, Wang L, Xu X, Lopaschuk GD, Zhang Y, et al. Site-specific ubiquitination of VDAC1 restricts its oligomerization and mitochondrial DNA release in liver fibrosis. Exp Mol Med. (2023) 55:269–80. doi: 10.1038/s12276-022-00923-9
53. Luo Z, Ji Y, Gao H, Gomes Dos Reis FC, Bandyopadhyay G, Jin Z, et al. CRIg+ Macrophages prevent gut microbial DNA–containing extracellular vesicle–induced tissue inflammation and insulin resistance. Gastroenterology. (2021) 160:863–74. doi: 10.1053/j.gastro.2020.10.042
54. Mercado MG, Smith DK, Guard EL. Acute Kidney Injury: Diagnosis and Management.Am Fam Physician. (2019) 100(11):687–694.
55. Qi X, Wang J, Fei F, Gao X, Wu X, Shi D, et al. Myricetin-loaded nanomicelles protect against cisplatin-induced acute kidney injury by inhibiting the DNA damage-cGAS-STING signaling pathway. Mol Pharm. (2023) 20:136–46. doi: 10.1021/acs.molpharmaceut.2c00520
56. Sekine S, Yao A, Hattori K, Sugawara S, Naguro I, Koike M, et al. The ablation of mitochondrial protein phosphatase pgam5 confers resistance against metabolic stress. EBioMedicine. (2016) 5:82–92. doi: 10.1016/j.ebiom.2016.01.031
57. Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. (2012) 148:228–43. doi: 10.1016/j.cell.2011.11.030
58. Rayego-Mateos S, Valdivielso JM. New therapeutic targets in chronic kidney disease progression and renal fibrosis. Expert Opin Ther Targets. (2020) 24:655–70. doi: 10.1080/14728222.2020.1762173
59. Nastase MV, Zeng-Brouwers J, Wygrecka M, Schaefer L. Targeting renal fibrosis: Mechanisms and drug delivery systems. Adv Drug Delivery Rev. (2018) 129:295–307. doi: 10.1016/j.addr.2017.12.019
60. Mitrofanova A, Fontanella A, Tolerico M, Mallela S, David JM, Zuo Y, et al. Activation of stimulator of IFN genes (STING) causes proteinuria and contributes to glomerular diseases. J Am Soc Nephrol. (2022) 33:2153–73. doi: 10.1681/ASN.2021101286
61. Zang N, Cui C, Guo X, Song J, Hu H, Yang M, et al. cGAS-STING activation contributes to podocyte injury in diabetic kidney disease. iScience. (2022) 25. doi: 10.1016/j.isci.2022.105145
62. Feng Z, Liao X, Peng J, Quan J, Zhang H, Huang Z, et al. PCSK9 causes inflammation and cGAS/STING pathway activation in diabetic nephropathy. FASEB J. (2023) 37. doi: 10.1096/fj.202300342RRR
63. Myakala K, Wang XX, Shults NV, Krawczyk E, Jones BA, Yang X, et al. NAD metabolism modulates inflammation and mitochondria function in diabetic kidney disease. J Biol Chem. (2023) 299. doi: 10.1016/j.jbc.2023.104975
64. Rajsbaum R, García-Sastre A, Versteeg GA. TRIMmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J Mol Biol. (2014) 426:1265–84. doi: 10.1016/j.jmb.2013.12.005
65. Lodi L, Mastrolia MV, Bello F, Rossi GM, Angelotti ML, Crow YJ, et al. Type I interferon–related kidney disorders. Kidney Int. (2022) 101:1142–59. doi: 10.1016/j.kint.2022.02.031
66. Corrales L, McWhirter SM, Dubensky TW, Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest. (2016) 126:2404–11. doi: 10.1172/JCI86892
67. Baird JR, Friedman D, Cottam B, Dubensky TW, Kanne DB, Bambina S, et al. Radiotherapy combined with novel STING-targeting oligonucleotides results in regression of established tumors. Cancer Res. (2016) 76:50–61. doi: 10.1158/0008-5472.CAN-14-3619
68. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. (2014) 41:830–42. doi: 10.1016/j.immuni.2014.10.017
69. Fu J, Kanne DB, Leong M, Glickman LH, Mcwhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade . Available online at: www.ScienceTranslationalMedicine.org.
70. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. (2017) 8. doi: 10.1038/s41467-017-00101-w
71. Chaudhuri D, Sasaki K, Karkar A, Sharif S, Lewis K, Mammen MJ, et al. Corticosteroids in COVID-19 and non-COVID-19 ARDS: a systematic review and meta-analysis. Intensive Care Med. (2021) 47:521–37. doi: 10.1007/s00134-021-06394-2
72. Chen Q, Gao C, Wang M, Fei X, Zhao N. TRIM18-regulated STAT3 signaling pathway via PTP1B promotes renal epithelial–mesenchymal transition, inflammation, and fibrosis in diabetic kidney disease. Front Physiol. (2021) 12:709506. doi: 10.3389/fphys.2021.709506
73. Hou J, Hsu JM, Hung MC. Molecular mechanisms and functions of pyroptosis in inflammation and antitumor immunity. Mol Cell. (2021) 81:4579–90. doi: 10.1016/j.molcel.2021.09.003
74. De Galarreta MR, Lujambio A. DNA sensing in senescence. Nat Cell Biol. (2017) 19:1008–9. doi: 10.1038/ncb3603
75. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. (2014) 28:99–114. doi: 10.1101/gad.235184.113
76. Zhao Y, Simon M, Seluanov A, Gorbunova V. DNA damage and repair in age-related inflammation. Nat Rev Immunol. (2023) 23:75–89. doi: 10.1038/s41577-022-00751-y
77. Hou Y, Wei Y, Lautrup S, Yang B, Wang Y, Cordonnier S, et al. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer's disease via cGAS-STING. Proc Natl Acad Sci U S A. (2021) 118(37):e2011226118. doi: 10.1073/pnas.2011226118/-/DCSupplemental
78. Bloom GS. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. (2014) 71:505–8. doi: 10.1001/jamaneurol.2013.5847
79. Chen G, Kroemer G, Kepp O. Mitophagy: an emerging role in aging and age-associated diseases. Front Cell Dev Biol. (2020) 8:200. doi: 10.3389/fcell.2020.00200
80. Xie X, Ma G, Li X, Zhao J, Zhao Z, Zeng J. Activation of innate immune cGAS-STING pathway contributes to Alzheimer’s pathogenesis in 5×FAD mice. Nat Aging. (2023) 3:202–12. doi: 10.1038/s43587-022-00337-2
81. Kwon O, Song J, Yang Y, Kim S, Kim JY, Seok M, et al. SGK1 inhibition in glia ameliorates pathologies and symptoms in Parkinson disease animal models. EMBO Mol Med. (2021) 13. doi: 10.15252/emmm.202013076
82. Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dementia. (2016) 12:719–32. doi: 10.1016/j.jalz.2016.02.010
83. Microglial cGAS-STING links innate immunity and Alzheimer's disease. Nat Aging. (2023) 3(2):155–6. doi: 10.1038/s43587-022-00350-5
84. Ma C, Liu Y, Li S, Ma C, Huang J, Wen S, et al. Microglial cGAS drives neuroinflammation in the MPTP mouse models of Parkinson’s disease. CNS Neurosci Ther. (2023) 29:2018–35. doi: 10.1111/cns.14157
85. Barazzuol L, Giamogante F, Brini M, Calì T. PINK1/Parkin mediated mitophagy, Ca2+ Signalling, and ER-mitochondria contacts in Parkinson’s disease. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21051772
86. Koyano F, Yamano K, Kosako H, Kimura Y, Kimura M, Fujiki Y, et al. Parkin-mediated ubiquitylation redistributes MITOL/March5 from mitochondria to peroxisomes. EMBO Rep. (2019) 20. doi: 10.15252/embr.201947728
87. Csordás G, Weaver D, Hajnóczky G. Endoplasmic reticulum–mitochondrial contactology: structure and signaling functions. Trends Cell Biol. (2018) 28:523–40. doi: 10.1016/j.tcb.2018.02.009
88. Jiang SY, Tian T, Yao H, Xia XM, Wang C, Cao L, et al. The cGAS-STING-YY1 axis accelerates progression of neurodegeneration in a mouse model of Parkinson's disease via LCN2-dependent astrocyte senescence. Cell Death Differ. (2023) 30(10):2280–2292. doi: 10.1038/s41418-023-01216-y
89. Gaikwad S, Puangmalai N, Bittar A, Montalbano M, Garcia S, McAllen S, et al. Tau oligomer induced HMGB1 release contributes to cellular senescence and neuropathology linked to Alzheimer’s disease and frontotemporal dementia. Cell Rep. (2021) 36. doi: 10.1016/j.celrep.2021.109419
90. Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. (2006) 47. doi: 10.1016/j.jacc.2005.09.068
91. Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han X, et al. Research progress on the relationship between atherosclerosis and inflammation. Biomolecules. (2018) 8. doi: 10.3390/biom8030080
92. An C, Sun F, Liu C, Huang S, Xu T, Zhang C, et al. IQGAP1 promotes mitochondrial damage and activation of the mtDNA sensor cGAS-STING pathway to induce endothelial cell pyroptosis leading to atherosclerosis. Int Immunopharmacol. (2023) 123. doi: 10.1016/j.intimp.2023.110795
93. Huangfu N, Wang Y, Xu Z, Zheng W, Tao C, Li Z, et al. TDP43 exacerbates atherosclerosis progression by promoting inflammation and lipid uptake of macrophages. Front Cell Dev Biol. (2021) 9:687169. doi: 10.3389/fcell.2021.687169
94. Wan X, Tian J, Hao P, Zhou K, Zhang J, Zhou Y, et al. cGAS-STING pathway performance in the vulnerable atherosclerotic plaque. Aging Dis. (2022) 13:1606–14. doi: 10.14336/AD.2022.0417
95. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. (2011) 12:204–12. doi: 10.1038/ni.2001
96. Pedro-Botet J, Climent E, Benaiges D. Atherosclerosis and inflammation. New therapeutic approaches Arteriosclerosis e inflamación. Nuevos enfoques terapéuticos. (2020) 155(6):256–62. p. doi: 10.1016/j.medcli.2020.04.024
97. Sladitschek-Martens HL, Guarnieri A, Brumana G, Zanconato F, Battilana G, Xiccato RL, et al. YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS–STING. Nature. (2022) 607:790–8. doi: 10.1038/s41586-022-04924-6
98. Harbauer AB, Hees JT, Wanderoy S, Segura I, Gibbs W, Cheng Y, et al. Neuronal mitochondria transport Pink1 mRNA via synaptojanin 2 to support local mitophagy. Neuron. (2022) 110:1516–31.e9. doi: 10.1016/j.neuron.2022.01.035
99. Quinn PMJ, Moreira PI, Ambrósio AF, Alves CH. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol Commun. (2020) 8:189. doi: 10.1186/s40478-020-01062-w
100. McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol. (2018) 217:65–77. doi: 10.1083/jcb.201708092
101. Jin G, Zhang Z, Wan J, Wu X, Liu X, Zhang W. G3BP2: structure and function. Pharmacol Res. (2022) 186. doi: 10.1016/j.phrs.2022.106548
102. Liu ZS, Cai H, Xue W, Wang M, Xia T, Li WJ, et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol. (2019) 20:18–28. doi: 10.1038/s41590-018-0262-4
103. Dai J, Huang YJ, He X, Zhao M, Wang X, Liu ZS, et al. Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell. (2019) 176:1447–60.e14. doi: 10.1016/j.cell.2019.01.016
Keywords: cGAS-STING, sterile inflammation, liver diseases, kidney diseases, cellular senescence
Citation: Wang L, Zhang Z, Zhang H, Zhou M, Huang C, Xia W, Li J and You H (2024) The effects of cGAS-STING inhibition in liver disease, kidney disease, and cellular senescence. Front. Immunol. 15:1346446. doi: 10.3389/fimmu.2024.1346446
Received: 29 November 2023; Accepted: 27 June 2024;
Published: 24 July 2024.
Edited by:
Poornima Paramasivan, Independent Researcher, Nottingham, United KingdomReviewed by:
Junji Xing, Houston Methodist Research Institute, United StatesCopyright © 2024 Wang, Zhang, Zhang, Zhou, Huang, Xia, Li and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongmei You, eXlvdWhvbmdtZWlAMTYzLmNvbQ==; Jun Li, bGpAYWhtdS5lZHUuY24=; Wenjiang Xia, MjAyODY5NTY3N0BxcS5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.