
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 May 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1341079
This article is part of the Research Topic Identification of Potential Therapeutic Targets for the Tumor Microenvironment of Gastrointestinal Tumor View all 7 articles
 Chiara Musiu1†
Chiara Musiu1† Francesca Lupo2†
Francesca Lupo2† Antonio Agostini3,4†
Antonio Agostini3,4† Gabriella Lionetto5
Gabriella Lionetto5 Michele Bevere6
Michele Bevere6 Salvatore Paiella5Carmine Carbone4
Salvatore Paiella5Carmine Carbone4 Vincenzo Corbo2
Vincenzo Corbo2 Stefano Ugel1*‡
Stefano Ugel1*‡ Francesco De Sanctis1*‡
Francesco De Sanctis1*‡Despite the efforts, pancreatic ductal adenocarcinoma (PDAC) is still highly lethal. Therapeutic challenges reside in late diagnosis and establishment of peculiar tumor microenvironment (TME) supporting tumor outgrowth. This stromal landscape is highly heterogeneous between patients and even in the same patient. The organization of functional sub-TME with different cellular compositions provides evolutive advantages and sustains therapeutic resistance. Tumor progressively establishes a TME that can suit its own needs, including proliferation, stemness and invasion. Cancer-associated fibroblasts and immune cells, the main non-neoplastic cellular TME components, follow soluble factors-mediated neoplastic instructions and synergize to promote chemoresistance and immune surveillance destruction. Unveiling heterotypic stromal-neoplastic interactions is thus pivotal to breaking this synergism and promoting the reprogramming of the TME toward an anti-tumor milieu, improving thus the efficacy of conventional and immune-based therapies. We underscore recent advances in the characterization of immune and fibroblast stromal components supporting or dampening pancreatic cancer progression, as well as novel multi-omic technologies improving the current knowledge of PDAC biology. Finally, we put into context how the clinic will translate the acquired knowledge to design new-generation clinical trials with the final aim of improving the outcome of PDAC patients.
Pancreatic ductal adenocarcinoma (PDAC), with an 11% 5-year overall survival, represents a great clinical challenge (1). Increased insurgency related to improved life expectancy at birth and metabolic co-morbidities, as well as limited surgical chances, frequent relapses and high tumor resilience represent peculiar features that underscore an unmet clinical need and project this disease toward the second leading cancer-dependent death cause, in the USA, by 2040 (2). High lethality is mainly attributed to late diagnosis. Most of the patients face no symptoms until an advanced or metastatic evolutionary disease stage, whereas the pancreas anatomical position does not allow for routine screening test; in addition, germline mutations and known risk factors (e.g. pancreatitis) contribute to a dismal amount of PDAC. Late diagnosis restricts surgery to only 10-20% of patients; nonetheless almost 80% of them will relapse within 2 years underscoring that micro metastases are already present at resection (3).
Tumor microenvironment (TME) represents a critical tumor component, composed of normal cells of different ontogeny and function which can support or dampen tumor proliferation, stemness and invasion as well as sustain primary and acquired resistance to therapy (4). This complex landscape includes immune cells (from both innate and adaptive arms), fibroblasts, endothelial cells, dense extracellular matrix (ECM)and additional factors (e.g. cytokines, growth factors, extracellular vesicles) which establish multiple heterotypic interactions defining and leveraging neoplastic phenotypes. Tumor cells play a pivotal role in orchestrating TME by secreting a multitude of soluble factors altering physiological processes to a pathological state, including hematopoiesis (5), inflammation, angiogenesis (6) and hijacking the recruitment and polarization of specific stromal subsets (7) with the final aim to support cancer outgrowth. Notably, TME composition and function are highly heterogeneous between patients with similar histopathological characteristics and even within the same patient (8). Indeed, histological and molecular analysis unveil the coexistence of multiple “subTMEs” which are linked to tumor differentiation, immune infiltrate, response to treatment and clinical outcome (9). These subTMEs can be categorized as “reactive”, when infiltrated by heterogenous cancer-associated fibroblast and immune cell subsets and associated with more aggressive basal epithelial phenotype; or matrix-rich “deserted”, in which the infiltration of activated CAF and immune cells is less prominent, and this phenotype is usually associated with resistance to chemotherapy. Notably, the coexistence of multiple and heterogenous subTMEs is not casual and provides malignant tumor advantages (9).
Fibroblasts represent a peculiar component of PDAC TME. They can be activated to CAFs by a plethora of stimuli, including extracellular matrix composition and stiffness, DNA damage, cytokines such as TGFβ, IL6, IL1β and tyrosine kinase receptors such as PDGF and FGF receptors (10). CAFs can support tumor progression both directly, by matrix remodeling or tumor metabolic reprogramming and indirectly by acting on other stromal components by restraining immune responses and promoting angiogenesis (10). CAF targeting represents a clinical challenge because of their heterogeneity in both composition and pro-tumor/anti-tumor functions. CAFs are indeed categorized according to phenotypic markers and function: the ones exhibiting a matrix-producing contractile matrix producing function (myCAFs) shares a myofibroblast phenotype; fibroblasts with immunomodulatory ability, named inflammatory CAFs (iCAFs), express inflammatory cytokines, such as IL6. my-CAFs are induced by TGFβ and are phenotypically characterized by elevated α-SMA expression. Fibroblast activation protein α (FAP)+-CAFs and antigen-presenting CAFs (apCAFs) are both endowed with immunomodulatory properties. Notably, most of the CAF subsets are shared in both pancreatic cancer and other solid tumors associated with metastasis and worse prognosis (11), strengthening the relationship of peculiar TME components in supporting invasive abilities and spreading.
From an immunological point of view pancreatic cancer is moderately antigenic (12) and poorly immunogenic (13), a hallmark of cancer progression (14). Indeed, neoplastic cells abrogate cancer immune surveillance by both dampening the generation and maturation of functional dendritic cells (15) and promoting differentiation of both monocytes and granulocytes toward the generation of immune-suppressive myeloid-derived suppressor cells (MDSCs). Pancreatic tumor immune microenvironment (TIME) is typically cold, characterized by low immune infiltration, especially of tumor-infiltrating lymphocytes (TILs) and natural killers (NK). Indeed, tumor cells promote stepwise preferential recruitment of MDSCs and tumor-associated macrophages (TAMs) with an anti-inflammatory phenotype in the tumor core to achieve immune privileged and unrestricted proliferation (13). KRAS oncogenic activation, very common in PDAC, is pivotal in establishing a highly suppressive TIME by triggering the release of cytokine and chemokines which in turn promote the recruitment and polarization of myeloid and lymphoid cells with immune regulatory properties (16–18). These myeloid immune regulatory cells sculpt a T lymphocyte-hostile TME by depleting essential amino acids (Arginine, Tryptophan, Cysteine) through the coordinated action of arginase (18–20), iNOS (18), IDO1 (21, 22) by generating toxic metabolites (reactive oxygen-ROS and nitrogen species-RNS), secreting immune suppressive cytokines (IL10, TGFβ) and by ligand-receptor interactions (e.g. PDL1-PD1, FASL-FAS) (23). Conversely, increased infiltration of T lymphocytes, as well as the presence of B lymphocytes functionally organized in specific tridimensional structures, named tertiary lymphoid structures (TLSs) are associated with improved overall survival in PDAC (24, 25).
Although poor surgical chances represent a further hurdle to comprehensively solve PDAC complexity, preclinical and clinical studies demonstrated TME’s ability to support cancer progression and the prominence of combining tumor cell targeting with TME manipulation (acting on both immunologic and fibroblast compartments) to promote its conversion from a tumor-prone to an anti-tumor milieu. However, our limited understanding of the multiple heterotypic interactions among the three main compartments, coupled with the challenged of deciphering the complex heterogeneity of “subTME” composition represents the main hurdle hindering the development of novel therapeutic approaches. In this review, we envision how last-generation multi-omic technologies provided critical insights about the contribution of both CAFs and immune cells in modulating PDAC biology and current tools that can be employed to model this heterogeneity and to predict the efficacy of new therapeutic strategies.
PDAC represents the most common types of pancreatic cancer (90% of cases) followed by neuroendocrine tumors, acinar carcinoma and pancreatoblastoma (26). PDAC derives from 2 different evolutive trajectories driven by different genetic pathways: intraductal papillary mucinous neoplasms (IPMN) are macroscopic lesions derived from the main duct or its branches, whereas pancreatic intraepithelial neoplasia (PanIN) are the most frequent early microscopic tumor lesions. Low-grade PanINs develop following proto-oncogene KRAS mutation and activation in ductal epithelial cells or in acinar cells triggering a de-differentiation process called acinar to ductal metaplasia (27). KRAS as well as G-protein alpha subunit Gas (GNAS) mutation and activation, together with loss of tumor-suppressor gene RING-type E3 ubiquitin ligase (RNF43) can drive IPMN (28, 29). Early lesions begin an evolutive path characterized by the stepwise acquisition of new features, including loss of oncosuppressor genes (e.g. CDKN2A, TP53, SMAD4), high grade atypia, PDAC and invasion abilities (30). Notably, preclinical models have been established resembling stepwise evolution of human PanIN and IPMN to PDAC at both genetic and histopathological levels (31, 32) allowing to study of early lesions, which are rarely identified in the clinic. KRAS mutation and constitutive activation are observed in the majority of PDAC. Its relevance in triggering and shaping neoplastic evolution has been confirmed in preclinical models of mutant KRAS reversible expression in which switching off the gene induces regression of both primary (33) and metastatic lesions (34) as well as promotes ductal to acinar re-differentiation (35). Accordingly, novel therapeutic strategies based on targeting mutant KRAS, including inhibitors of KRASG12C (36) KRASG12D (37), KRASG12D degraders (NCT05382559), as well as a transgenic TCR recognizing KRAS neoantigen (38) have been recently developed and currently tested in both mouse and human studies with encouraging results. Nonetheless, it has been demonstrated that neoplastic cells may acquire a KRAS independent proliferative instructions through the activation of other proto-oncogenes (39).
The biological and functional heterogeneity of PDAC can be orchestrated by tumor-cell intrinsic factors, revealing a cancer cell-driven immunosuppression. The production of the chemokine CXCL1 of cancer cells arrests T cell infiltration, thus generating a non-T- cell-inflamed TME that affects immunotherapy sensitivity (40). In human PDAC, the presence of ephrin-A receptor 2 (EPHA2) led to the upregulation of prostaglandin endoperoxide synthase 2 (PTGS2) in cancer cells, with a subsequent T cell exclusion from PDAC TME (41). The inhibition of EPHA2/PTGS2 signaling rescues T cell infiltration and may increase tumor responsiveness to immunotherapy (41). PDAC cells can shape the TIME also at epigenetic levels (42). Lysine demethylase 3A (KDM3A) regulates anti-tumor immunity through epidermal growth factor receptor (EGFR) expression in cancer cells. By acting on Krueppel-like factor 5 (KLF5) and SMAD family member 4 (SMAD4), KDM3A leads to EGFR upregulation followed by the deficiency of intratumoral T cells (42). Acquired resistance to immunotherapy response has been associated with epithelial-to-mesenchymal transition (EMT). The silencing of interferon regulatory factor 6 (Irf6) in cancer cells by EMT-transcription factors (EMT-TFs) ZEB1 and SNAIL minimize the pro-apoptotic effects of tumor necrosis factor (TNF) α (43). Hence, PDAC undertakes several mechanisms to remain refractory to immunotherapy.
Whole transcriptomic studies have been employed to leverage the characterization of both neoplastic cell and tumor contexture in PDAC. Although the limitation of achieving a bulk signature that averages the contribution of all cells in the analysis, this approach allowed to improve the characterization of tumors with apparently similar histological features. By integrating different clinical specimens, molecular technologies, and bioinformatics pipelines, a less differentiated molecular subtype of PDAC (named basal-like or quasi-mesenchymal or squamous) was identified (44, 45). The squamous subtype was associated with worse clinical outcomes (44–46), whereas the more differentiated classical subtype can be eventually subcategorized in aberrantly differentiated endocrine exocrine (ADEX), pancreatic progenitor, and immunogenic (associated with increased leucocyte infiltration) type of tumors (45). Then, laser microdissection was employed to distinguish the molecular profiles of neoplastic and stromal compartments with the final aim of establishing a computational model that may improve the definition of classical to basal subtypes and their contribution to clinical outcome in whole transcriptomic datasets (47). There is increasing evidence of TAMs role in PDAC recurrence occur often in defined cellular signaling pathways and participate in sculpting molecular subtype and increasing tumor heterogeneity (48, 49). Furthermore, integrating molecular signatures with morphological PDAC features already improved taxonomy of PDAC generating a solid soil for designing more effective therapeutic strategies (50). Nevertheless, molecular fingerprinting identification of PDAC subtypes, has not been yet employed in the clinic, for therapeutic purposes. The main reasons relate to TME plasticity that can evolve according to the stage of progression and therapeutic strategy. Moreover, several subTMEs can reside in the same tumor, and whole transcriptomics cannot take into account either TME heterogeneity or TME-neoplastic interactions. To this aim, recently, single-cell RNAseq (scRNA-seq) and first-generation spatial transcriptomic technologies were introduced. These tools allowed to increase further the resolution of both stroma and tumor cells characterization, by unveiling the spatial organization of TME as well as the cellular function and beginning the definition of heterotypic interactions between neoplastic and stromal cells (51, 52), which will be discussed in the next sections.
The advent of omics, such as scRNA-seq or high-dimensional spatial analysis, has provided an unprecedented depth of knowledge on the TME complexity and heterogeneity in PDAC. A recent phenotypic and spatial immune atlas of human PDAC identifying leukocyte composition within histopathologically defined regions of surgical resections from PDAC patients allowed a new classification of PDAC based on the precise quantification of leukocyte profiles into hypoinflammed, myeloid enriched and lymphoid enriched (8). Indeed, parallel use of CyTOF, single-cell RNA sequencing, and multiplex IHC techniques has demonstrated a complex network of interaction between neoplastic and normal cell types, highlighting an inverse correlation between myeloid populations and effector CD8+ T lymphocytes (CTLs) (53). Multiple immune cell subsets have been shown to impact tumor biology (54). Innate immune cells represent the largest leukocyte subset detected in PDAC tumors. Myeloid cells include mostly TAMs, MDSCs and neutrophils. Importantly, the phenotype of tumor-infiltrating myeloid cells is a determinant of treatment outcome (55). TAMs arise from both infiltrating monocytes and tissue-resident macrophages (56). A limited number of studies suggest that depending on their ontogeny, TAMs have both overlapping and distinct functions in shaping the TME. Whereas monocyte-derived TAMs are more potent at sampling tumor antigens, embryonically-derived TAMs exert unique fibrosis-modulating functions which led to the production and remodeling of the extracellular matrix (ECM) (56). Indeed, TAMs show high plasticity and may be engaged in either tumor-promoting or tumor-suppressive fashion. Whereas in vitro TAMs have been classified into two opposite polarization states, M1-like macrophages with anti-tumor activity and M2-like macrophages with protumor properties, their phenotype in vivo reflects the complexity of polarization signals present within the TME (57). The presence of prostaglandin E2 (PGE2) and tumor necrosis factor (TNF) within the TME led to the conversion of tumor-infiltrating monocytes in interleukin-1β (IL-1β)-expressing TAMs (58). This population is transcriptionally enriched in inflammatory response, leukocyte recruitment and angiogenesis genes. The spatial proximity of IL-1β+ TAMs and cancer cells drive the acquisition of inflammatory and pathogenic properties of a subset of PDAC cells expressing an IL-1β response signature during the early stages of tumor development. The persistent inflammatory signaling in epithelial cells accelerated tumorigenesis and is associated with poor outcomes for PDAC patients (58). Hence, myeloid cells have a key role in the establishment of an immunosuppressive TME and disease progression. In a genetically engineered mouse model of PDAC, the inhibition of CSF1R+ TAMs resulted in a marked increase of CD8+ effector cells together with the reduction of collagen and hypoxia, offering a way to specifically target macrophages in PDAC (59). Toll-like receptor 9 (TLR-9) based immunotherapy was able to locally activate the immune system, converting the immune hostile into an immune permissive TME sensitizing PDAC to immunotherapy (60). In preclinical models, the CXCR1/2 inhibition was able to arrest the recruitment of macrophages and their polarization toward a tumor-promoting phenotype and increase the efficacy of anti-PD-1 (61).
Accordingly, several studies highlight the potential therapeutic benefit of redirecting myeloid cells towards antitumor and antistromal properties (55, 62). There is increasing evidence of TAMs role in PDAC recurrence. The different recurrence patterns are driven by spatially restricted tumor- and stroma-associated immune drivers, resulting in different immune cell populations and integrin networks. The spatial analysis of PDAC patients with liver recurrences emphasized an innate immune response characterized by the presence of immunosuppressive CD68+ TAMs localized close to tumor cells and a reduced number of CD8+ T cells (63). By contrast, PDAC with lung and local recurrences displayed a mix of adaptive and innate immune response. When compared to PDAC with liver recurrences, lung relapses shown an upregulation of integrin ITGAM (CD11b), which is known to inhibit immune suppression and promote antitumor immune response. Therefore, CD11b targeting could be beneficial in patients with recurrent PDACs (63).
The second major immunosuppressive cell type in PDAC is represented by MDSCs, a heterogeneous cell population composed of mature and immature cells of myeloid origin characterized by immune regulatory properties (64). During tumorigenesis, MDSCs disrupt tumor immunosurveillance by suppressing CD8+ T cell-mediated antitumor immunity (65–67). MDSCs can be divided into two main subgroups according to the expression of selective surface markers, the monocytic lineage (M-MDSCs) and the polymorphonuclear lineage (PMN-MDSCs); however, in humans exists an “early immature” MDSCs (eMDSC) subset (64, 68, 69). Cell plasticity and longer half-life are typical features of the monocytic-MDSC subset (70), since this population is able to differentiate into TAMs (71, 72). Tumor-released soluble factors induce an imbalanced myelopoiesis that ultimately supports MDSC generation. Several proinflammatory signals (e.g. TNFα) activates alternative molecular pathways that differentiate normal monocytes/neutrophils in MDSCs. For instance, the antiapoptotic molecule cellular FLICE (FADD-like IL-1-converting enzyme)-inhibitory protein(c-FLIP) is a main driver for M-MDSCs expansion (73, 74). Indeed c-FLIP can activate the transcription of several immunosuppression and inflammation-associated genes (i.e., Il10, Il6, Cd274, and Cd273), by alternative activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-KB) and STAT3 activation (75, 76). Interestingly, the frequency of c-FLIP-expressing, PDL-1+ monocytes isolated from PDAC patients in combination with high levels of serum IL-6, has been identified as a negative independent prognostic factor for both overall survival and disease-free survival (DFS) (75). Tumor cells orchestrate MDSCs recruitment via multiple tumor-secreted factors. For instance, the intratumor levels of Regnase-1, an RNA-binding protein with endoribonuclease activity, have been negatively associated with tumor-infiltrating myeloid cells and clinical outcome in PDAC patients (77). Interestingly, Regnase-1 deletion regulates a variety of cytokines and chemokines (i.e., CXCL1, CXCL2, CSF2, and TGFβ) involved in the recruitment and education of MDSCs thus promoting PDAC progression (77). Moreover, MDSCs recruitment in the pancreatic tumor site occurs via tumor-secreted factors (e.g. CXCR2 ligands, GM-CSF and CXCL5) thus blocking the recruitment and priming of T cells (78, 79). By employing mass cytometry, crosstalk between cancer cells and PMN-MDSCs was uncovered in human PDAC (80). In myeloid-enriched and T cell-excluded contexts, PMN-MDSCs amplify inflammation and promote immune tolerance by TNF production. The PMN-MDSCs-derived TNF rewire CXCL1 overproduction by cancer cells that in turns led to dysfunction and spatial exclusion of T cells from tumor core (80). Besides soluble mediators, myeloid recruitment can occur following alterations in the expression of cell-surface molecules, such as integrins. Both MDSCs and TAMs share the expression of CD11b/CD18 integrin heterodimer (Mac-1), a key player that regulates the adhesion and migration of myeloid cells in inflamed tissues. Leukadherin-1, a small molecule activator of CD11b, was found to reduce infiltration of CD11b+ myeloid cells and repolarization of TAMs with a concomitant increase of CD8+ T cells and activated DCs in genetically engineered KPC mouse model (81). Interestingly, the activation of the antitumor response by CD11b agonist sensitizes normally resistant tumors to immunotherapies (82).
Dendritic cells (DCs) are critical for antigen cross-presentation and tumor-specific T-cell immunity. Emerging evidence demonstrates that PDAC itself can promote an immunosuppressive TME. In an engineered model neoantigen of PDAC, the disruption of immune surveillance by type I conventional dendritic cells (cDC1) led to the arrest of CD8+ T cells and TH1 activity, accelerating the neoplastic progression (15). cDC1 dysfunction begins in the early stages of PDAC, where elevated serum IL-6 affects cDC1 quantitatively and functionally, resulting in a DC semi-maturation state and a defective T cell priming (83). The use of CD40 agonist and Fms-related tyrosine kinase 3 ligand (Flt3L) enabled tumor growth control by increasing the number and activity of cDCs (83). However, more studies on the DC subset in PDAC are needed to open new therapeutic options potentially. Among the main components of the adaptive immune system, regulatory T (Treg) cells are the most abundant CD4+ T cell population. In humans, Tregs infiltrate starting from preneoplastic lesions to established cancer and their high prevalence has been associated with poor prognosis in PDAC (84). Despite being known as an immune suppressive population, the role of Tregs is debatable in PDAC. Various mechanisms have been proposed to define Treg as a tumor-supportive subset that led to CD8+ T cell suppression. For instance, tumor-infiltrating Tregs can promote immune tolerance by restraining the immunogenic functions of tumor-associated dendritic cells (DCs) necessary for CD8+ activation (85). In contrast, Treg depletion in a genetically engineered mouse model of PDAC accelerates tumor progression due to compensatory myeloid infiltration (86). Tregs are a key source of TGFβ ligands and their depletion results in the differentiation of inflammatory fibroblast subsets (e.g. myCAF) that increase the secretion of chemoattractants for suppressive myeloid cells (86). Other T cell populations within the TME play pro-tumorigenic roles, including Th17, Th22, CD4 and γδ T cells (87–89). During PanIN formation, CD4+ T cells are recruited within the tumor to arrest CD8+ effector functions thus contributing to tumorigenesis (90). Despite PDAC being poorly infiltrated by CD8+ T cells, scRNA-seq technology enabled a better understanding of this subset. A minority of CD8+ T cells show an exhausted transcriptional profile which escalates in advanced stages of the disease (53). Interestingly, tumors presenting both a high number of neoantigens and a strong CTL infiltrate are associated with long-term survivors, highlighting the presence of functional T cells controlling disease progression (91). Recently, the expression of immune checkpoint TIGIT was associated with a combination of immune populations (exhausted CD8+ T cell, Treg and NK). The CD155/TIGIT axis is essential to support immune evasion; when combined with PD-1 blockade plus CD40 activation, the CD155/TIGIT targeting stimulates a robust anti-tumor response in preclinical models of PDAC (92). Albeit less studied than T cells, B cells infiltrate and accumulate during PanIN and PDAC lesions both in mice and humans. B cells can act as either anti-tumorigenic or pro-tumorigenic populations depending on their localization and functional organization (93). Tumor-infiltrating B cells reside in TLSs, functional immune-responsive niches that have recently gained strong attention in PDAC (25, 94). When present within tumors, TLS usually associated with a most beneficial outcome (95). However, B cell fate depends on TLS maturity (96). In mature TLS, anti-tumorigenic B cells contribute to cancer immunosurveillance by producing anti-tumor antibodies and presenting tumor antigens to T cells (97). By contrast, immature TLS can originate regulatory B cells (Bregs), a pro-tumorigenic subset dispersed inside the TME and characterized by the secretion of anti-inflammatory cytokines (e.g., IL-10 and IL-35) which promote tumor progression (98, 99). In PDAC, Breg cells can restrict the activity of effector T cells while boosting Tregs, MDSCs and TAMs. For instance, Breg - FcRγ+ TAMs crosstalk drives a tumor-promoting macrophage phenotype supporting tumor growth (100). The spatial organization of immune cells within the TME influences survival and response to therapy in several tumor types, including PDAC (101, 102). By comparing transcriptional data of PDAC tissue samples from resected long-term and short-term survivors, a higher infiltration of B cells was observed in long-term survivors. Spatial data of long-term survivors revealed the proximity of CD20+ B cells and T cells with an activated effector phenotype, highlighting the importance of studying the differences in immune infiltration in a specific location (101).
In summary, many differences in the immune landscape and other stromal components co-exist within individual tumors and the understanding of the spatial and dynamic relationships among diverse cell types is necessary to design accurate treatment interventions (Figure 1).
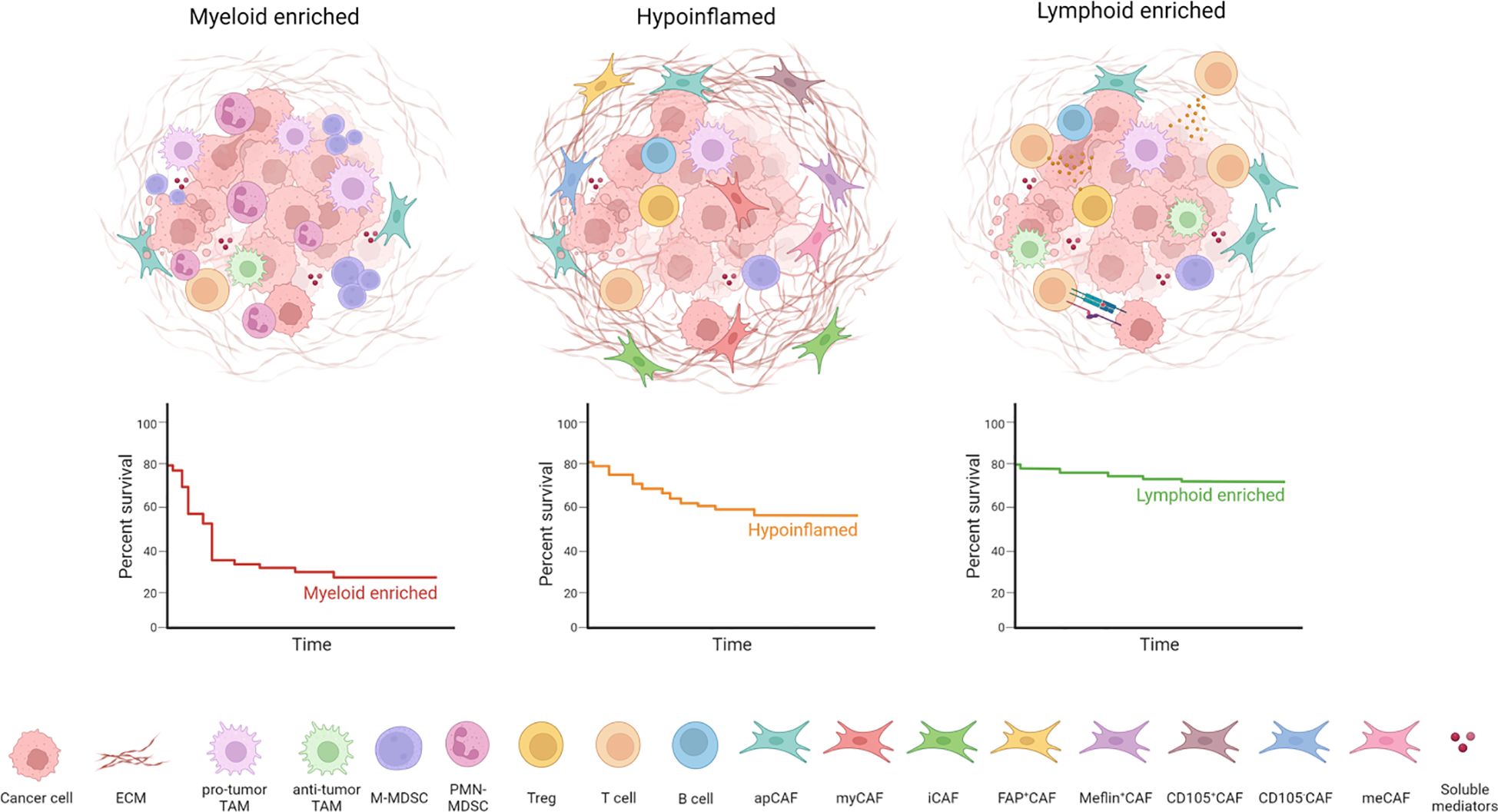
Figure 1 PDAC TME heterogeneity and organization. PDAC can be divided into different subtypes according to TME composition: tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) characterize the myeloid-enriched subtype; several cancer-associated fibroblasts (CAFs) and extracellular matrix (ECM) components determine the desmoplastic stroma typical of hypoinflammed subtype; infiltration of T cells is associated with the lymphoid-enriched subtype. The lymphoid enriched subtype holds a significant survival advantage compared to the myeloid-enriched and hypoinflamed subtype.
One of the hallmarks of PDAC is an intense desmoplastic reaction with abundant ECM deposition which is mainly contributed by CAFs (103–105). CAFs are often the most abundant cell type in PDAC and represent a very heterogeneous population of diverse phenotypes and functions that concur to define a highly immunosuppressive TME (106, 107). The heterogeneity of CAFs emerges as the consequence of biochemical and physical signals that define sub-microenvironments with distinctive immune features in PDAC (9).
The origin of CAFs is still not fully understood, but several cellular sources have been proposed. Pancreatic stellate cells (PSCs) are specialized cells that play a crucial role in maintaining pancreas homeostasis. In their quiescent state, PSCs have a star-shaped morphology containing vitamin A lipid droplets. In response to various signals (i.e., injury or inflammation), PSCs can undergo activation, thereby losing vitamin A droplets and adopting a myofibroblast-like phenotype (108). PSCs have been considered for long the only source of CAFs, but recent studies have identified other potential sources of CAFs, including Fabp4+ fibroblasts, Gli1+ fibroblasts and Wt1+ fibroblasts (109–111).
Several attempts have been made to target PDAC stroma, which has been originally considered a physical barrier to drug delivery. Inhibition of the Hedgehog (HH) signaling pathway with the HH pathway inhibitor IPI-926, along with gemcitabine treatment, improved overall survival in mouse models of PDAC by increasing tumor vasculature (112). However, clinical trials using this same pharmacological scheme (gemcitabine + HH pathway inhibitor, Vismodegib) in patients with metastatic pancreatic cancer did not improve overall survival (113, 114). Subsequent preclinical studies established that genetic or pharmacological inhibition of the HH pathway promoted the emergence of poorly differentiated neoplastic phenotypes (115–117). Alternatively, the enzymatic ablation of hyaluronic acid (HA), a major component of PDAC desmoplastic stroma, restored interstitial fluid pressure in autochthonous PDAC mouse models, allowing a more efficient drug delivery associated with increased survival (118). Moreover, the combination of PEGPH20 (a clinically formulated PEGylated human recombinant PH20 hyaluronidase) and gemcitabine resulted in the inhibition of PDAC tumor growth and higher survival compared to gemcitabine treatment alone (119). Despite encouraging preclinical results, this approach was ultimately unsuccessful in clinical practice (120). The exposure of PSCs and PDAC cell lines to all-trans retinoic acid (ATRA) or vitamin D analog (calcipotriol) led to a quiescent cellular identity, in vivo reduced tumor growth and increased cell death (121, 122). However, calcipotriol also hampers T-cell effector functions promoting the upregulation of PD-L1, potentially compromising T-cell mediated anti-tumor response (123). To date, the effect of calcipotriol has been investigated on autoimmune diseases and a few cancer types, including breast cancer and squamous cell carcinoma. Its effect on PDAC requires still clinical investigation. The failure in translating preclinical findings based on agnostic CAFs targeting into clinical application underlined the necessity of mapping out the different phenotypes and functions of this heterogeneous stromal population. Indeed, subsequent studies have identified several CAF subtypes, with both pro-tumorigenic and anti-tumorigenic features, highlighting how stromal heterogeneity in PDAC can hinder therapeutic approaches.
Based on the antagonistic activity of TGFβ and IL1, two main phenotypes of CAF exist. myCAFs are located proximal to cancer cells where the local gradient of TGF-β reduces expression of IL1R, while iCAFs are located far from cancer cells where they sense and respond to IL1R/JAK/STAT signaling (124). If iCAFs are invariably considered pro-tumorigenic (124), myCAFs have been described with both tumor-promoting (125–127) and restraining functions (115, 117, 124). Noteworthy, these CAF subpopulations are interconvertible and able to change their phenotype according to different extracellular stimuli (128). For these reasons, the possibility of converting pro-tumorigenic CAFs into anti-tumorigenic CAFs might represent a promising therapeutic strategy. Recently, a third CAF population has been identified and named apCAFs, showing several features of the antigen presentation machinery, such as the ability to present antigens to CD4+ T cells, probably modulating the immune response in PDAC (129). However, since apCAFs do not express costimulatory molecules (e.g., CD80, CD86 or CD40), they cannot properly function as antigen-presenting cells. Moreover, a recent study has reported a mesothelial origin for this CAF subtype, with a potential role in immunosuppression (111). Accordingly, apCAFs induce CD8 T cell death by FASL-FAS and PDL2-PD1 interaction in breast cancer (130) and promote CD4 naïve T lymphocyte differentiation towards T regulatory cells (111). Other subpopulations of CAFs have been proposed, based on the expression of specific markers. Single-cell mass cytometric analysis has revealed that the cell surface marker CD105 can effectively distinguish two CAF populations that are functionally distinct and non-interconvertible: tumor-permissive CD105+ CAFs and tumor-suppressive CD105- CAFs (131). The cell surface protein Meflin has been described as a marker for CAFs with tumor suppressor activity. The presence of Meflin+ CAFs is associated with a favorable prognosis in both human and KPC mice, whereas genetic ablation of Meflin+ CAFs leads to the development of poorly differentiated tumors in vivo (132, 133). FAP+CAFs can inhibit CD8+ T cells infiltration by expressing high levels of CXCL12 (134). The depletion of this CAFs subpopulation sensitizes these tumors to immune checkpoint therapy (106). Through single-cell RNA sequencing, an additional CAFs population expressing leucine-rich repeat containing 15 (LRRC15) and induced by TGF-β has been identified. This myCAFs subpopulation is associated with poor response to anti-PD-L1 therapy in PDAC patients (135). In vivo depletion of this population restrains tumor growth, reducing total fibroblast content and enhancing intratumoral CTL infiltration (136). In addition, CAFs exhibiting a highly activated metabolic state (meCAFs) have been recently identified and associated with poor prognosis and conversely to a better response to PD-1 blockade treatment in PDAC patients (137). Although CAF heterogeneity has been deeply investigated, it is still difficult to compare CAF phenotypes across species (e.g., mouse and human). Human studies investigating the presence and function of apCAFs are not available yet. Extending the definition of human CAF subsets and their functional roles is mandatory to guide the development of new therapeutic strategies.
Overall, deeper decoding of functions and features of CAFs with new technologies at single-cell resolution may foster new opportunities for stroma-targeting drugs.
Spatial biology is an interdisciplinary field that combines multi-omics and imaging technologies to study the biological processes and cellular interactions inside the structures of the tissue architecture. ScRNA-seq integrated with other omics data like proteomics and Assay for Transposase-Accessible Chromatin with Sequencing (ATAC-Seq), has contributed to the discovery of rare cellular populations in both development and disease. Spatial technologies add topological information to scRNA-seq thus identifying a niche with peculiar cellular composition unraveling new pathological models and novel therapeutic targets. The resolution and complexity of these technologies have become determinants for the study of the intricated TME that sets PDAC apart from numerous other solid tumors. Xue et al. revealed novel tumor-promoting functions of Schwann cells in PDAC by employing both single-cell RNA-seq and spatial transcriptomics (138). They found that Schwann cells affect both tumor and stromal compartments toward tumor outgrowth. PDAC cells located in this area displayed typical Basal-like markers and other invasiveness markers such as metalloproteases and EMT markers. Indeed, SC-CM supports CAF polarization toward iCAF by IL1 and supports tumor proliferation. Yousuf et al. provided a holistic view of immune dysfunction in PDAC (139). They found increased amounts of inhibitory and exhausted T cells in the tumor, with consistent downregulation of cytotoxicity-related genes in CTLs placed in the tumor core compared to the ones placed at the tumor border. Infiltrating NKT cells showed an extraordinarily exhausted phenotype too, identified by the high expression of immune checkpoint proteins TIM3, LAG3, and CTLA4. Performing cell-to-cell interaction and ligand-receptor analyses on spatial transcriptomics data they identified TIGIT expression on T and NK cells correlating with the expression of its ligands PVR and PVRL2 on myeloid and tumor cells. Spatial technologies also provided new insight into the tissue remodeling and cellular population dynamics that happen during the therapy in PDAC patients. Hwang et al. integrated single nuclei RNA-seq and digital spatial profiling to characterize the cellular subtypes and spatial communities in naïve and neoadjuvant-treated PDAC patients (52). They validated the existence of previously identified clusters (44–46) exhibiting a consistent molecular pattern characterized by both classical and squamous features. Additionally, they discovered a novel cluster prevalent among patients who underwent chemotherapy, linked to poorer clinical outcomes, and labeled as “neural-like progenitor” (NRP). This study sheds light on the inter- and intratumoral diversity of pancreatic cancer, identifying treatment-associated remodeling and clinically relevant prognostication. The refined molecular and cellular taxonomy of treated PDAC samples identified in this study shed more light on the complex dynamics that happen in response to treatments highlighting novel pathways and cellular populations that may be targeted to improve standard treatment efficacy. Another study attempted to decipher insights driving chemoresistance in PDAC, integrating bulk-sequencing technologies, proteomics/phosphoproteomics, single-cell sequencing with spatial transcriptomics and high-resolution cellular imaging (51). The authors identified an iCAF increase in chemotherapy-treated patients compared to untreated ones. Moreover, they showed that specific pathways implicated in therapy resistance were upregulated in iCAFs according to the treatment. While Heat shock and AP-1 genes were more highly expressed in FOLFIRINOX samples; metallothioneins were more highly expressed in Gemcitabine + nab-paclitaxel samples. These genes are implicated in chemoresistance, suggesting a strategy to deplete iCAF via IL1β-R or JAK-STAT pathway inhibition to improve therapeutic efficacy. Besides spatial transcriptomics, other omics technologies can supply spatial definition and cues of heterotypic interactions. Proteomics has the advantage of more effectively associating data with cell phenotype. Le large et al. employed a combination of microdissection and Nanoscale liquid chromatography coupled with tandem mass spectrometry (nano LC-MS/MS) to perform spatial proteomics of both tumor and stroma regions in PDAC patients (140). They found several proteins associated with prognosis. Tumor cells from patients with poor outcomes were enriched in proteins involved in DNA replication and included in signatures of proliferation and contractility. Stroma from patients with worse survival was rich instead of proteins involved in focal adhesion processes. Moreover, they identified EPHA2 as a possible novel therapeutic target in PDAC since is highly expressed in PDAC and inhibitors are already available. Brouwer et al. employed single-cell mass cytometry and multiplex spatial IF to compare the local and systemic immunophenotype integrating samples from primary tumors, and peripheral and portal vein blood (141). They confirmed preferential infiltration of B lymphocytes and T regs in primary tumors that were confirmed as well in portal vein blood but not in circulation, and paucity of CTLs. Leveraging both mass cytometry and multiplex spatial IF they identified only in primary tumors a substantial fraction of innate lymphoid 1 like cells producing high levels of IFNγ and characterized as CD127–CD103+CD39+CD45RO+ cells.
These studies have paved the way for more detailed multi-omics profiling of the complex interactions occurring in pancreatic TME. Moreover, they identified novel pancreatic and stromal subsets enriched in patients non responding to chemotherapy, supplying a framework to better stratify patients and to identify new therapeutic targets. Since we are still in the early phase of the spatial technology era, it is easy to envision that these technologies may lead to innovative discoveries in the years to come, hopefully resulting in significant clinical improvements for pancreatic cancer patients. Nevertheless, these technologies still have significant limitations. Indeed, further development is needed both technically and, especially, computationally to improve the integration of a larger number of samples and thus achieve the identification of reliable clinical markers.
Advancements in TME deconvolution at single-cell level are crucial to identifying unprecedented biochemical and functional neoplastic-stromal relationships. These technologies allow to identification of potential target candidates that can be enrolled on an in vitro and in vivo preclinical screening pipeline to be then eventually tested in clinical trials. Thus, the development of tools that can recapitulate in vitro (although in a simplistic way) the tridimensional organization of tumor and stromal support in providing therapeutic resistance is mandatory. Patient-derived organoids (PDO) can suit these needs. Organoids are in vitro self-organized 3D tissues deriving from stem cells (pluripotent, fetal, or adult). Noteworthy, the organoids can faithfully recapitulate the key functional, structural, and biological complexity of an organ in vivo, hence, also called “mini-organs” (142–144). To enable the ex vivo survival and expansion of the epithelial compartment, including stem-like cells, the organoid technology relies on two crucial components: exogenous supplementation of stromal-niche factors and extracellular matrix gels (i.e., hydrogel, Matrigel) (145). Different culture systems have been described and they can be broadly divided into Wnt -dependent (146–148) and -independent (149, 150) cultures systems (Table 1).
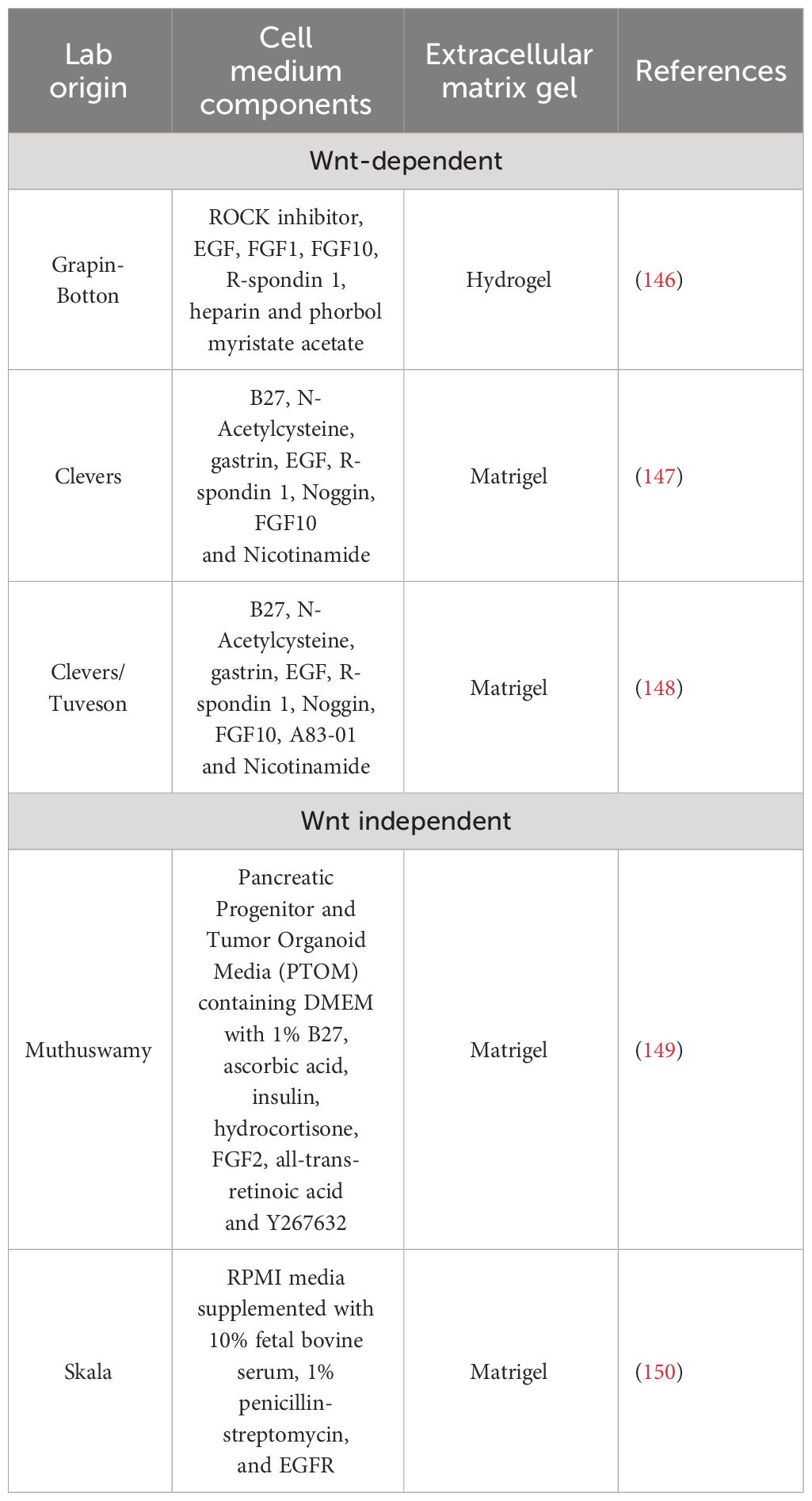
Table 1 Different protocols to generate and propagate pancreatic organoids.
The culture conditions do not support the long-term propagation of native components of the tumor microenvironment. The air-liquid interface system favors the retention of native components but only to a certain extent (151). Hence, the majority of organoid culture systems lack stromal and immunological components that are key determinants of tumor biology. PDAC organotypic cultures lack TME components (e.g., fibroblasts, immune cells, endothelial cells) and several efforts have been made to better shape recapitulate the complexity of the disease by establishing co-cultures of organoids with other cell types (152, 153). Moreover, the selection of the proper culture condition is necessary to control the expression of the more appropriate transcriptional profile. This calls for important efforts in the field to further improve the culture systems by leveraging our increased understanding of physical and chemical interactions occurring with the tumor microenvironment. In pancreatic cancer as well as in other tumor types, organoid-based coculture systems have been used to recreate ex vivo relevant bi-directional interactions. These systems can be used to perform compartmentalized studies where individual subsets of interactions are interrogated to advance our understanding of cancer biology. It is however unlikely that organoid-based system will be able to recapitulate the cellular and physiological complexity of the native tissue. Nonetheless, it is now well established that PDOs retain the main genomic features of the parental tissue (148, 154, 155) and are exquisitely suited to understand how the microenvironment affects neoplastic cell phenotypes (156, 157). Even if the PDOs are originally established from tissue specimens which are representative of given in vivo subTME, exposure of PDOs to relevant TME cues might be able, at least in principle, to approximate in vivo cell states. That is essential to proper modulate pharmacological responses which are known to be affected by microenvironmental components. As proof of that, transcriptional cell states which are enriched in tissues from post-treatment tumors can be found ex vivo when PDOs are subjected to the same treatments. Hence, improving the quality of patients’ lives by acting on micro- and macroenvironmental factors holds promise in PDAC treatment (52, 158). These findings emphasize the potential of PDOs as an effective platform for drug screening in PDAC. Another interesting aspect of the organoid technology applied to pancreatic cancer is the possibility of establishing models from different disease stages (e.g., PanINs, overt carcinomas, metastatic diseases) as well as normal ductal cells, which will give the possibility of testing pharmacological sensitivities/toxicity in different settings (148, 159).
However, even if PDOs are developed from epithelial tumor cells isolated from surgical resection or tumor biopsies, which are not representative of the whole tumor, PDOs are highly dynamic cultures and are influenced by changes in TME components cell medium compositions and drug treatment (157). To obtain a PDO-derived preclinical tool for pharmacological testing and drug treatments, overcoming the limitation given by the poor representation of TME components, several approaches have been explored, including co-culture systems and on a chip-platforms (160–163).
Considering the precision oncology revolution we are experiencing, the current standard of care for PDAC has been only marginally involved. Only small subsets of patients harbor unique, actionable genetic alterations – such as homologous recombination deficiency (BRCA1/2, PALB2), mismatch repair deficiency, rare fusions (NRG1, NTRK) – granting them access to potentially beneficial targeted therapies (164). For the others, the mainstay of treatment relies on multiagent combination chemotherapeutic regimens (FOLFIRINOX and Gemcitabine/nab-paclitaxel), according to disease stage and patient fitness (165).
However, major preclinical research efforts to decode the complex interplay between cancer cells and the TME have identified new molecular vulnerabilities, opening the door to novel targeted therapies whose efficacy is currently under investigation in several clinical trials (Figure 2; Table 2).
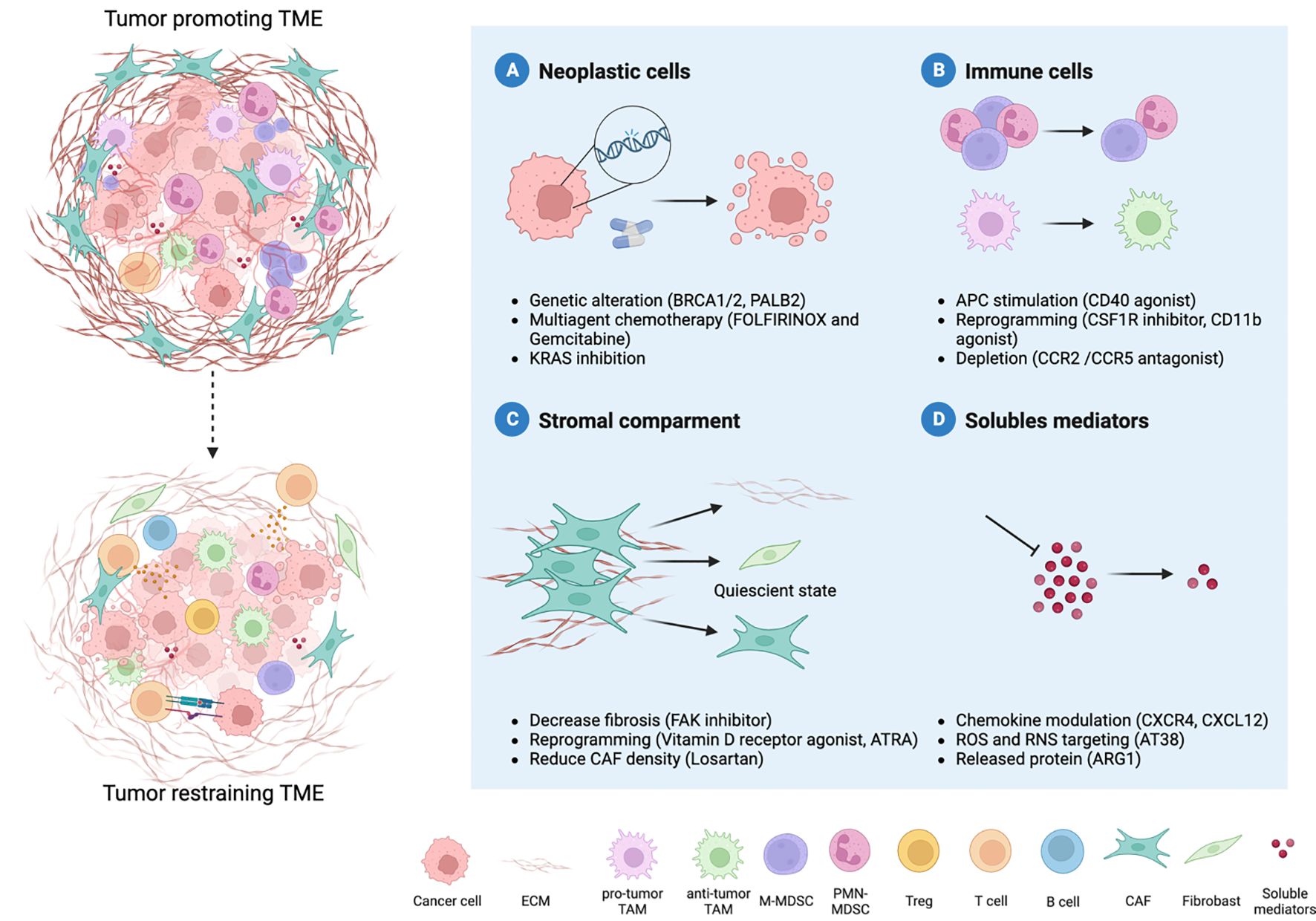
Figure 2 Therapy-induced reprogramming of PDAC TME. The combination of different therapeutic strategies can revert the hostile microenvironment towards an anti-tumor one. Four major therapeutic interventions are emerging to improve PDAC treatment: neoplastic cells, immune cells, stromal compartments and soluble mediators. Several approaches are focused on neoplastic cells and their genetic alterations (A). Moreover, the microenvironment that surrounds PDAC cells holds key insight into novel treatment options. The reprogramming of myeloid cells can relieve immune suppression (B) and coupled with increased antigen presentation allow the recruitment and priming of T cells. Another strategy is represented by the modulation of the desmoplastic stroma, such as cancer-associated fibroblasts (CAFs) (C). Given the double role of CAFs in the TME, reverting the tumor-promoting into quiescent cells represent the best strategy. Indeed, multiple cell types co-exist within the TME, and their communication occurs through soluble mediators (metabolites, cytokines, chemokines) (D). Targeting soluble mediators can hamper immune cells recruitment and pro-tumor differentiation, as well as deposition of extracellular matrix, thus limiting tumor progression.
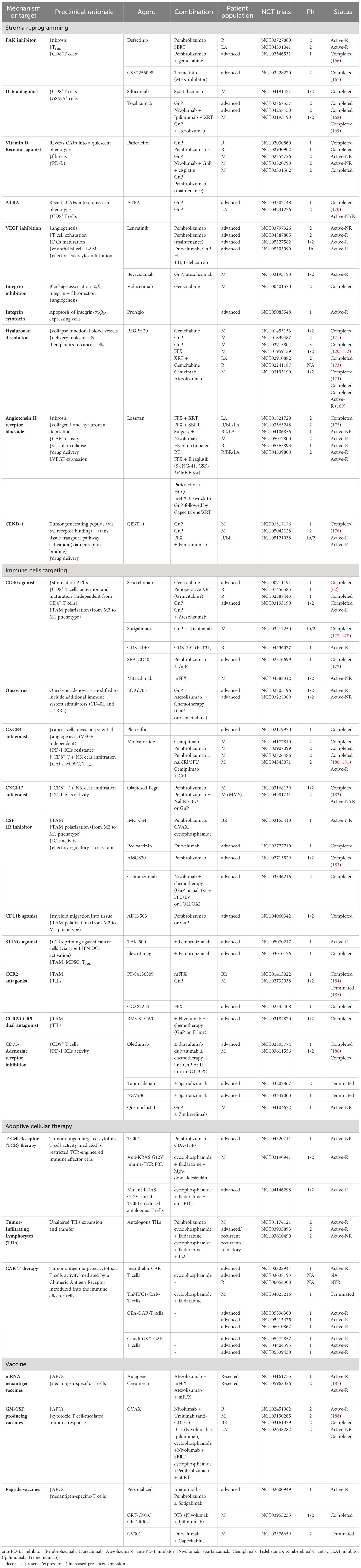
Table 2 Selected trials in PDAC by mechanisms or target.
Lessons learned from the first testing of stroma-depleting drugs have shaped the understanding of CAF biology and their double-edge role in promoting or restraining tumor progression (115). Therefore, subsequent trials have aimed at reverting tumor-supportive cells into quiescence or reprogramming into an anti-tumor phenotype. The disappointing results of single-agent immune checkpoint inhibitors (ICIs) in unselected patient populations paved the way for developing multimodal combinatorial strategies to overcome therapeutic resistance in PDAC and maximize clinical benefit (189). However, improved clinical activity compared to standard-of-care treatments has yet to be demonstrated. Current boundaries to these combinatorial approaches include limited availability of preclinical tumor models that faithfully represent the complex interaction between cancer-, immune- and stromal cells in the TME, the absence of early detection diagnostic and predictive and prognostic biomarkers to detect early disease and identify patients with the greatest chance of response and the likelihood that even minimal variation in dosing schedule, treatment duration and/or drug sequencing might alter the anti-tumor efficacy of the combination (190). Considering the improved definition of PDAC subtypes and increased availability of drugs targeting neoplastic and stromal (immune and fibroblast) cells, the “traditional” Phase 1 to 3 trial progression might not be appropriate (191). Thus, new protocol designs have emerged (192), including “basket” trials (in which patients with solid tumors of different histology but with shared features - e.g. specific biomarkers - are put together in the same study), and “umbrella” platforms (in which patients with same pathology are divided in different arms according to the presence of peculiar biomarkers). For instance, MORPHEUS-PDAC (NCT03193190), is an umbrella trial designed to evaluate ten experimental arms with various Atezolizumab combinations with two comparators arms (GnP or mFOLFOX) in the metastatic setting; whereas Precision Promise (NCT04229004), PIONEER-Panc (NCT04481204), REVOLUTION (NCT04787991) and the GVAX immunotherapy (NCT02451982) trials will test multiple investigational combinations in parallel against standard of care arms.
Continue efforts in integrated pre-clinical and clinical research and collaborations between stakeholders are essential to identify new effective treatments and to implement a biomarker-based selection of cytotoxic chemotherapy, targeted therapy, and immunotherapy to improve the survival of PDAC patients.
PDAC therapy is undergoing a paradigm shift by applying new small-drugs and immunological approaches. Characterize the genetic profile of cancer cells (intrinsic properties) but also the immune contexture of tumor microenvironment (extrinsic properties) is mandatory to optimize the effectiveness of these approaches, since cancer and stromal cells are continuously in communication and are capable of bidirectional influence, reshaping each other’s cell properties (58, 193, 194). It is currently unclear how PDAC immune landscape evolves as tumors acquire additional genomic alterations as well as the drivers shaping TME immune complexity in primary tumors versus distant metastases (8) but both information are essential for patient stratification. Therefore, one of the major goals in the near future is the definition of new biomarker able to take into account both intrinsic and extrinsic PDAC features. To maximize and speed up the bench to patients ‘bed translation, it is essential to integrate better preclinical and clinical research. Essential achievements in improving the knowledge of PDAC evolution has been obtained by generating PDAC-disease models (31, 195), PDAC in vitro screening cell systems (148, 196), new chemical drugs and inhibitors (197), TME datasets (52, 139, 198) and genomic/molecular tumor profiles (45, 91, 199), as well as advance in biotechnology and clinical research allowed to employ cutting-edge approaches, including personalized RNA neoantigen vaccines (200) and selective inhibitors in PDAC treatment (201, 202). However, results from immunotherapy clinical trials in PDAC have thus far been modest, with clinical benefit in a small subset of MMR-D PDAC. In a comprehensive report from 2022, a revision was conducted on all interventional studies for PDAC completed between 2010 and 2020, as documented in the ClinicalTrials.gov registry— the US National Library of Medicine’s database for clinical trials. The findings revealed that out of 551 trials included in the study, 165 (30%) lacked available results. Among the remaining 297 trials with accessible results published in full-text articles, the median duration between the primary completion date and the date of publication was 47.6 months, with a 95% confidence interval of 39.6 to 61.9 months (203). Collectively, these results pinpoint that clinical trial results, including clinical trials with negative outcomes, should be published in a timely manner to support inform and improve future trial design changing both parameters for patient recruitment and combination therapies.
Emerging evidence point out that the state of well-being of the patient influences treatment response in cancer patients, including PDAC (204). Lifestyle interventions, such as exercise and nutrition, can improve the quality of life of PDAC patients by meeting their physiological needs (204). For instance, aerobic exercise can amplify the immune-cell mediated cytotoxicity on pancreatic cancer cells to reduce PDAC growth and enhance sensitivity to both immunotherapy and standard-of-care chemotherapy (205). Also, dietary interventions can influence immune responses since the growth and viability of cancer cells depend on nutrient availability. A strong anti-tumoral effect was described in KPC mice undergoing ketogenetic diet (206). When combined with gemcitabine, a ketogenetic diet contributes to increment the effectiveness of chemotherapy (206). Hence, improving the quality of patients’ lives by acting on micro- and macroenvironmental factors holds promise in PDAC treatment.
Pursuing high-risk research with high-reward potential may advance the development of promising personalized combination therapies. To this aim, integrating acknowledged (e.g. chemo and radiotherapy-based) and novel neoplastic targeting approaches [including passive and active immunotherapeutic strategies (200, 207–210)] with TME targeting solutions (211) and additional research fields, which could not be addressed in this review, such as cancer metabolism (212), vessel remodeling (213, 214), CAR T cell therapies (215) have good promises as potential strategies for PDAC therapy. We aspire to transform PDAC from a silent, formidable threat into a manageable and treatable disease in the near future.
CM: Writing – original draft, Writing – review & editing. FL: Writing – original draft, Writing – review & editing. AA: Writing – original draft, Writing – review & editing. GL: Writing – original draft, Writing – review & editing. MB: Writing – original draft, Writing – review & editing. SP: Writing – original draft, Writing – review & editing. CC: Writing – original draft, Writing – review & editing. VC: Writing – original draft, Writing – review & editing. SU: Writing – original draft, Writing – review & editing. FS: Writing – original draft, Writing – review & editing, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was jointly supported by PRIN programs of the Italian Ministry of Education, University, and Research (MIUR, PI: De Sanctis F., CUP: B39J22001200001); AIRC (PI: Carbone C Grant No. 23681; PI: Corbo V. Grant No. 18178, PI: Ugel S. Grant No. 21509 and 28730); EU (MSCA project PRECODE, PI: Corbo V. Grant No 861196); PNRR programs of the Italian MUR (Project “National Center for Gene Therapy and Drugs based on RNA Technology”, application code CN00000041, Mission 4, Component 2 Investment 1.4, funded from the European Union - NextGenerationEU, MUR Directorial Decree No. 1035 of 17 June 2022, CUP B33C22000630001; PI: Ugel S). CM was supported by an AIRC fellowship for Italy (Call 2022), and was supported by Fondazione Umberto Veronesi (Call 2023). MB was supported by AIRC fellowships for Italy (28054; 29829).
The authors acknowledge the colleagues of the Immunology section, Pancreatic Surgery Unit, Pathologic Anatomy unit of the University of Verona, and Oncology unit of Policlinico Gemelli for providing feedback on this manuscript. We would like to thank Biorender for providing a platform for creative artwork.
Paiella S. receives consultancy fees from AlphaTau.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. (2023) 73:17–48. doi: 10.3322/caac.21763
2. Rahib L, Wehner MR, Matrisian LM, Nead KT. Estimated projection of US cancer incidence and death to 2040. JAMA Netw Open. (2021) 4:e214708. doi: 10.1001/jamanetworkopen.2021.4708
3. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. (2012) 148:349–61. doi: 10.1016/j.cell.2011.11.025
4. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
5. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. (2015) 125:3365–76. doi: 10.1172/JCI80006
6. De Sanctis F, Ugel S, Facciponte J, Facciabene A. The dark side of tumor-associated endothelial cells. Semin Immunol. (2018) 35:35–47. doi: 10.1016/j.smim.2018.02.002
7. Hofer F, Di Sario G, Musiu C, Sartoris S, De Sanctis F, Ugel S. A complex metabolic network confers immunosuppressive functions to myeloid-derived suppressor cells (MDSCs) within the tumour microenvironment. Cells. (2021) 10. doi: 10.3390/cells10102700
8. Liudahl SM, Betts CB, Sivagnanam S, Morales-Oyarvide V, da Silva A, Yuan C, et al. Leukocyte heterogeneity in pancreatic ductal adenocarcinoma: phenotypic and spatial features associated with clinical outcome. Cancer Discovery. (2021) 11:2014–31. doi: 10.1158/2159-8290.CD-20-0841
9. Grunwald BT, Devisme A, Andrieux G, Vyas F, Aliar K, McCloskey CW, et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell. (2021) 184:5577–5592e18. doi: 10.1016/j.cell.2021.09.022
10. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
11. Zhao Z, Zhu Y. FAP, CD10, and GPR77-labeled CAFs cause neoadjuvant chemotherapy resistance by inducing EMT and CSC in gastric cancer. BMC Cancer. (2023) 23:507. doi: 10.1186/s12885-023-11011-0
12. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
13. Balachandran VP, Beatty GL, Dougan SK. Broadening the impact of immunotherapy to pancreatic cancer: challenges and opportunities. Gastroenterology. (2019) 156:2056–72. doi: 10.1053/j.gastro.2018.12.038
14. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
15. Hegde S, Krisnawan VE, Herzog BH, Zuo C, Breden MA, Knolhoff BL, et al. Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer. Cancer Cell. (2020) 37:289–307e9. doi: 10.1016/j.ccell.2020.02.008
16. Ischenko I, D'Amico S, Rao M, Li J, Hayman MJ, Powers S, et al. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat Commun. (2021) 12:1482. doi: 10.1038/s41467-021-21736-w
17. Hamarsheh S, Gross O, Brummer T, Zeiser R. Immune modulatory effects of oncogenic KRAS in cancer. Nat Commun. (2020) 11:5439. doi: 10.1038/s41467-020-19288-6
18. De Sanctis F, Lamolinara A, Boschi F, Musiu C, Caligola S, Trovato R, et al. Interrupting the nitrosative stress fuels tumor-specific cytotoxic T lymphocytes in pancreatic cancer. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2021-003549
19. Cane S, Barouni RM, Fabbi M, Cuozzo J, Fracasso G, Adamo A, et al. Neutralization of NET-associated human ARG1 enhances cancer immunotherapy. Sci Transl Med. (2023) 15:eabq6221. doi: 10.1126/scitranslmed.abq6221
20. Trovato R, Fiore A, Sartori S, Cane S, Giugno R, Cascione L, et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J Immunother Cancer. (2019) 7:255. doi: 10.1186/s40425-019-0734-6
21. Blair AB, Kleponis J, Thomas DL 2nd, Muth ST, Murphy AG, Kim V, et al. IDO1 inhibition potentiates vaccine-induced immunity against pancreatic adenocarcinoma. J Clin Invest. (2019) 129:1742–55. doi: 10.1172/JCI124077
22. Serafini M, Torre E, Aprile S, Grosso ED, Gesu A, Griglio A, et al. Discovery of highly potent benzimidazole derivatives as indoleamine 2,3-dioxygenase-1 (IDO1) inhibitors: from structure-based virtual screening to in vivo pharmacodynamic activity. J Med Chem. (2020) 63:3047–65. doi: 10.1021/acs.jmedchem.9b01809
23. De Sanctis F, Solito S, Ugel S, Molon B, Bronte V, Marigo I. MDSCs in cancer: Conceiving new prognostic and therapeutic targets. Biochim Biophys Acta. (2016) 1865:35–48. doi: 10.1016/j.bbcan.2015.08.001
24. Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. (2017) 8:15095. doi: 10.1038/ncomms15095
25. Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer. (2015) 112:1782–90. doi: 10.1038/bjc.2015.145
26. Park W, Chawla A, O'Reilly EM. Pancreatic cancer: A review. JAMA. (2021) 326:851–62. doi: 10.1001/jama.2021.13027
27. Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. (2003) 3:565–76. doi: 10.1016/S1535-6108(03)00140-5
28. Patra KC, Kato Y, Mizukami Y, Widholz S, Boukhali M, Revenco I, et al. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol. (2018) 20:811–22. doi: 10.1038/s41556-018-0122-3
29. Hosein AN, Dangol G, Okumura T, Roszik J, Rajapakshe K, Siemann M, et al. Loss of rnf43 accelerates kras-mediated neoplasia and remodels the tumor immune microenvironment in pancreatic adenocarcinoma. Gastroenterology. (2022) 162:1303–1318 e18. doi: 10.1053/j.gastro.2021.12.273
30. Halbrook CJ, Lyssiotis CA, Pasca di Magliano M, Maitra A. Pancreatic cancer: Advances and challenges. Cell. (2023) 186:1729–54. doi: 10.1016/j.cell.2023.02.014
31. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. (2005) 7:469–83. doi: 10.1016/j.ccr.2005.04.023
32. Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. (2007) 11:229–43. doi: 10.1016/j.ccr.2007.01.017
33. Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. (2012) 122:639–53. doi: 10.1172/JCI59227
34. Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, et al. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PloS One. (2012) 7:e49707. doi: 10.1371/journal.pone.0049707
35. Collins MA, Yan W, Sebolt-Leopold JS, Pasca di Magliano M. MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology. (2014) 146:822–834 e7. doi: 10.1053/j.gastro.2013.11.052
36. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. (2020) 383:1207–17. doi: 10.1056/NEJMoa1917239
37. Kemp SB, Cheng N, Markosyan N, Sor R, Kim IK, Hallin J, et al. Efficacy of a small-molecule inhibitor of krasG12D in immunocompetent models of pancreatic cancer. Cancer Discovery. (2023) 13:298–311. doi: 10.1158/2159-8290.CD-22-1066
38. Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih YP, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N Engl J Med. (2022) 386:2112–9. doi: 10.1056/NEJMoa2119662
39. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. (2014) 158:185–97. doi: 10.1016/j.cell.2014.06.003
40. Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity. (2018) 49:178–193 e7. doi: 10.1016/j.immuni.2018.06.006
41. Markosyan N, Li J, Sun YH, Richman LP, Lin JH, Yan F, et al. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J Clin Invest. (2019) 129:3594–609. doi: 10.1172/JCI127755
42. Li J, Yuan S, Norgard RJ, Yan F, Sun YH, Kim IK, et al. Epigenetic and transcriptional control of the epidermal growth factor receptor regulates the tumor immune microenvironment in pancreatic cancer. Cancer Discovery. (2021) 11:736–53. doi: 10.1158/2159-8290.CD-20-0519
43. Kim IK, Diamond M, Yuan S, Kemp S, Li Q, Lin J, et al. Plasticity-induced repression of Irf6 underlies acquired resistance to cancer immunotherapy. Res Sq. (2023) 15(1):1532. doi: 10.1038/s41467-024-46048-7
44. Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. (2011) 17:500–3. doi: 10.1038/nm.2344
45. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. (2016) 531:47–52. doi: 10.1038/nature16965
46. Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. (2015) 47:1168–78. doi: 10.1038/ng.3398
47. Maurer C, Holmstrom SR, He J, Laise P, Su T, Ahmed A, et al. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut. (2019) 68:1034–43. doi: 10.1136/gutjnl-2018-317706
48. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. (2008) 321:1801–6. doi: 10.1126/science.1164368
49. Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O'Kane GM, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet. (2020) 52:231–40. doi: 10.1038/s41588-019-0566-9
50. N Kalimuthu S, Wilson GW, Grant RC, Seto M, O'Kane G, Vajpeyi R, et al. Morphological classification of pancreatic ductal adenocarcinoma that predicts molecular subtypes and correlates with clinical outcome. Gut. (2020) 69:317–28. doi: 10.1136/gutjnl-2019-318217
51. Cui Zhou D, Jayasinghe RG, Chen S, Herndon JM, Iglesia MD, Navale P, et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat Genet. (2022) 54:1390–405. doi: 10.1038/s41588-022-01157-1
52. Hwang WL, Jagadeesh KA, Guo JA, Hoffman HI, Yadollahpour P, Reeves JW, et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat Genet. (2022) 54:1178–91. doi: 10.1038/s41588-022-01134-8
53. Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat Cancer. (2020) 1:1097–112. doi: 10.1038/s43018-020-00121-4
54. Bronte V, Cingarlini S, Marigo I, De Santo C, Gallina G, Dolcetti L, et al. Leukocyte infiltration in cancer creates an unfavorable environment for antitumor immune responses: a novel target for therapeutic intervention. Immunol Invest. (2006) 35:327–57. doi: 10.1080/08820130600754994
55. Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNgamma and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discovery. (2016) 6:400–13. doi: 10.1158/2159-8290.CD-15-1032
56. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. (2017) 47:323–338 e6. doi: 10.1016/j.immuni.2017.07.014
57. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. (2011) 11:762–74. doi: 10.1038/nri3070
58. Caronni N, La Terza F, Vittoria FM, Barbiera G, Mezzanzanica L, Cuzzola V, et al. IL-1beta(+) macrophages fuel pathogenic inflammation in pancreatic cancer. Nature. (2023) 623:415–22. doi: 10.1038/s41586-023-06685-2
59. Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, et al. CSF1R(+) macrophages sustain pancreatic tumor growth through T cell suppression and maintenance of key gene programs that define the squamous subtype. Cell Rep. (2018) 23:1448–60. doi: 10.1016/j.celrep.2018.03.131
60. Carbone C, Piro G, Agostini A, Delfino P, De Sanctis F, Nasca V, et al. Intratumoral injection of TLR9 agonist promotes an immunopermissive microenvironment transition and causes cooperative antitumor activity in combination with anti-PD1 in pancreatic cancer. J Immunother Cancer. (2021) 9. doi: 10.1136/jitc-2021-002876
61. Piro G, Carbone C, Agostini A, Esposito A, De Pizzol M, Novelli R, et al. CXCR1/2 dual-inhibitor ladarixin reduces tumour burden and promotes immunotherapy response in pancreatic cancer. Br J Cancer. (2023) 128:331–41. doi: 10.1038/s41416-022-02028-6
62. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. (2011) 331:1612–6. doi: 10.1126/science.1198443
63. Karamitopoulou E, Wenning AS, Acharjee A, Zlobec I, Aeschbacher P, Perren A, et al. Spatially restricted tumour-associated and host-associated immune drivers correlate with the recurrence sites of pancreatic cancer. Gut. (2023) 72:1523–33. doi: 10.1136/gutjnl-2022-329371
64. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
65. Thyagarajan A, Alshehri MSA, Miller KLR, Sherwin CM, Travers JB, Sahu RP. Myeloid-derived suppressor cells and pancreatic cancer: implications in novel therapeutic approaches. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11111627
66. Sarhan D, Eisinger S, He F, Bergsland M, Pelicano C, Driescher C, et al. Targeting myeloid suppressive cells revives cytotoxic anti-tumor responses in pancreatic cancer. iScience. (2022) 25:105317. doi: 10.1016/j.isci.2022.105317
67. Oweida AJ, Mueller AC, Piper M, Milner D, Van Court B, Bhatia S, et al. Response to radiotherapy in pancreatic ductal adenocarcinoma is enhanced by inhibition of myeloid-derived suppressor cells using STAT3 anti-sense oligonucleotide. Cancer Immunol Immunother. (2021) 70:989–1000. doi: 10.1007/s00262-020-02701-w
68. Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. (2010) 40:22–35. doi: 10.1002/eji.200939903
69. Mandruzzato S, Brandau S, Britten CM, Bronte V, Damuzzo V, Gouttefangeas C, et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: results from an interim study. Cancer Immunol Immunother. (2016) 65:161–9. doi: 10.1007/s00262-015-1782-5
70. Ugel S, Peranzoni E, Desantis G, Chioda M, Walter S, Weinschenk T, et al. Immune tolerance to tumor antigens occurs in a specialized environment of the spleen. Cell Rep. (2012) 2:628–39. doi: 10.1016/j.celrep.2012.08.006
71. Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. (2010) 70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672
72. Kwak T, Wang F, Deng H, Condamine T, Kumar V, Perego M, et al. Distinct populations of immune-suppressive macrophages differentiate from monocytic myeloid-derived suppressor cells in cancer. Cell Rep. (2020) 33:108571. doi: 10.1016/j.celrep.2020.108571
73. Haverkamp JM, Smith AM, Weinlich R, Dillon CP, Qualls JE, Neale G, et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity. (2014) 41:947–59. doi: 10.1016/j.immuni.2014.10.020
74. Adamo A, Frusteri C, Pilotto S, Caligola S, Belluomini L, Poffe O, et al. Immune checkpoint blockade therapy mitigates systemic inflammation and affects cellular FLIP-expressing monocytic myeloid-derived suppressor cells in non-progressor non-small cell lung cancer patients. Oncoimmunology. (2023) 12:2253644. doi: 10.1080/2162402X.2023.2253644
75. Fiore A, Ugel S, De Sanctis F, Sandri S, Fracasso G, Trovato R, et al. Induction of immunosuppressive functions and NF-kappaB by FLIP in monocytes. Nat Commun. (2018) 9:5193. doi: 10.1038/s41467-018-07654-4
76. Musiu C, Caligola S, Fiore A, Lamolinara A, Frusteri C, Del Pizzo FD, et al. Fatal cytokine release syndrome by an aberrant FLIP/STAT3 axis. Cell Death Differ. (2022) 29:420–38. doi: 10.1038/s41418-021-00866-0
77. Okabe J, Kodama T, Sato Y, Shigeno S, Matsumae T, Daiku K, et al. Regnase-1 downregulation promotes pancreatic cancer through myeloid-derived suppressor cell-mediated evasion of anticancer immunity. J Exp Clin Cancer Res. (2023) 42:262. doi: 10.1186/s13046-023-02831-w
78. Vonderheide RH, Bear AS. Tumor-derived myeloid cell chemoattractants and T cell exclusion in pancreatic cancer. Front Immunol. (2020) 11:605619. doi: 10.3389/fimmu.2020.605619
79. Chen Y, Kim J, Yang S, Wang H, Wu CJ, Sugimoto H, et al. Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell. (2021) 39:548–565 e6. doi: 10.1016/j.ccell.2021.02.007
80. Bianchi A, De Castro Silva I, Deshpande NU, Singh S, Mehra S, Garrido VT, et al. Cell-autonomous cxcl1 sustains tolerogenic circuitries and stromal inflammation via neutrophil-derived TNF in pancreatic cancer. Cancer Discovery. (2023) 13:1428–53. doi: 10.1158/2159-8290.CD-22-1046
81. DeNardo DG, Galkin A, Dupont J, Zhou L, Bendell J. GB1275, a first-in-class CD11b modulator: rationale for immunotherapeutic combinations in solid tumors. J Immunother Cancer. (2021) 9. doi: 10.1136/jitc-2021-003005
82. Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aau9240
83. Lin JH, Huffman AP, Wattenberg MM, Walter DM, Carpenter EL, Feldser DM, et al. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J Exp Med. (2020) 217. doi: 10.1084/jem.20190673
84. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. (2006) 12:5423–34. doi: 10.1158/1078-0432.CCR-06-0369
85. Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. (2017) 20:558–71. doi: 10.1016/j.celrep.2017.06.062
86. Zhang Y, Lazarus J, Steele NG, Yan W, Lee HJ, Nwosu ZC, et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discovery. (2020) 10:422–39. doi: 10.1158/2159-8290.CD-19-0958
87. McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell. (2014) 25:621–37. doi: 10.1016/j.ccr.2014.03.014
88. Perusina Lanfranca M, Zhang Y, Girgis A, Kasselman S, Lazarus J, Kryczek I, et al. Interleukin 22 signaling regulates acinar cell plasticity to promote pancreatic tumor development in mice. Gastroenterology. (2020) 158:1417–1432 e11. doi: 10.1053/j.gastro.2019.12.010
89. Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, et al. gammadelta T cells support pancreatic oncogenesis by restraining alphabeta T cell activation. Cell. (2016) 166:1485–1499 e15. doi: 10.1016/j.cell.2016.07.046
90. Zhang Y, Yan W, Mathew E, Bednar F, Wan S, Collins MA, et al. CD4+ T lymphocyte ablation prevents pancreatic carcinogenesis in mice. Cancer Immunol Res. (2014) 2:423–35. doi: 10.1158/2326-6066.CIR-14-0016-T
91. Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nat. (2017) 551:512–6. doi: 10.1038/nature24462
92. Freed-Pastor WA, Lambert LJ, Ely ZA, Pattada NB, Bhutkar A, Eng G, et al. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell. (2021) 39:1342–1360 e14. doi: 10.1016/j.ccell.2021.07.007
93. Gupta SL, Khan N, Basu S, Soni V. B-cell-based immunotherapy: A promising new alternative. Vaccines (Basel). (2022) 10. doi: 10.3390/vaccines10060879
94. J Gunderson A, Rajamanickam V, Bui C, Bernard B, Pucilowska J, Ballesteros-Merino C, et al. Germinal center reactions in tertiary lymphoid structures associate with neoantigen burden, humoral immunity and long-term survivorship in pancreatic cancer. Oncoimmunology. (2021) 10:1900635. doi: 10.1080/2162402X.2021.1900635
95. Schumacher TN, Thommen DS. Tertiary lymphoid structures in cancer. Science. (2022) 375:eabf9419. doi: 10.1126/science.abf9419
96. Fridman WH, Siberil S, Pupier G, Soussan S, Sautes-Fridman C. Activation of B cells in Tertiary Lymphoid Structures in cancer: Anti-tumor or anti-self? Semin Immunol. (2023) 65:101703. doi: 10.1016/j.smim.2022.101703
97. Senturk ZN, Akdag I, Deniz B, Sayi-Yazgan A. Pancreatic cancer: Emerging field of regulatory B-cell-targeted immunotherapies. Front Immunol. (2023) 14:1152551. doi: 10.3389/fimmu.2023.1152551
98. Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Discovery. (2016) 6:247–55. doi: 10.1158/2159-8290.CD-15-0843
99. Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TF, et al. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer Res. (2013) 73:2468–79. doi: 10.1158/0008-5472.CAN-12-3450
100. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discovery. (2016) 6:270–85. doi: 10.1158/2159-8290.CD-15-0827
101. Aziz HM, Saida L, de Koning W, Stubbs AP, Li Y, Sideras K, et al. Spatial genomics reveals a high number and specific location of B cells in the pancreatic ductal adenocarcinoma microenvironment of long-term survivors. Front Immunol. (2022) 13:995715. doi: 10.3389/fimmu.2022.995715
102. Li R, He Y, Zhang H, Wang J, Liu X, Liu H, et al. Identification and validation of immune molecular subtypes in pancreatic ductal adenocarcinoma: implications for prognosis and immunotherapy. Front Immunol. (2021) 12:690056. doi: 10.3389/fimmu.2021.690056
103. Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. (2007) 6:1186–97. doi: 10.1158/1535-7163.MCT-06-0686
104. Caligiuri G, Tuveson DA. Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell. (2023) 41:434–49. doi: 10.1016/j.ccell.2023.02.015
105. Sherman MH, Beatty GL. Tumor microenvironment in pancreatic cancer pathogenesis and therapeutic resistance. Annu Rev Pathol. (2023) 18:123–48. doi: 10.1146/annurev-pathmechdis-031621-024600
106. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U.S.A. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
107. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. (2008) 68:918–26. doi: 10.1158/0008-5472.CAN-07-5714
108. Wu Y, Zhang C, Jiang K, Werner J, Bazhin AV, D'Haese JG. The role of stellate cells in pancreatic ductal adenocarcinoma: targeting perspectives. Front Oncol. (2020) 10:621937. doi: 10.3389/fonc.2020.621937
109. Helms EJ, Berry MW, Chaw RC, DuFort CC, Sun D, Onate MK, et al. Mesenchymal lineage heterogeneity underlies nonredundant functions of pancreatic cancer-associated fibroblasts. Cancer Discovery. (2022) 12:484–501. doi: 10.1158/2159-8290.CD-21-0601
110. Garcia PE, Adoumie M, Kim EC, Zhang Y, Scales MK, El-Tawil YS, et al. Differential contribution of pancreatic fibroblast subsets to the pancreatic cancer stroma. Cell Mol Gastroenterol Hepatol. (2020) 10:581–99. doi: 10.1016/j.jcmgh.2020.05.004
111. Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. (2022) 40:656–673 e7. doi: 10.1016/j.ccell.2022.04.011
112. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. (2009) 324:1457–61. doi: 10.1126/science.1171362
113. Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N, et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res. (2014) 20:5937–45. doi: 10.1158/1078-0432.CCR-14-1269
114. Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized phase ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol. (2015) 33:4284–92. doi: 10.1200/JCO.2015.62.8719
115. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
116. Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U.S.A. (2014) 111:E3091–100. doi: 10.1073/pnas.1411679111
117. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
118. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. (2012) 21:418–29. doi: 10.1016/j.ccr.2012.01.007
119. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. (2013) 62:112–20. doi: 10.1136/gutjnl-2012-302529
120. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, et al. HALO 202: randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol. (2018) 36:359–66. doi: 10.1200/JCO.2017.74.9564
121. Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology. (2011) 141:1486–97. doi: 10.1053/j.gastro.2011.06.047
122. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. (2014) 159:80–93. doi: 10.1016/j.cell.2014.08.007
123. Gorchs L, Ahmed S, Mayer C, Knauf A, Fernandez Moro C, Svensson M, et al. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci Rep. (2020) 10:17444. doi: 10.1038/s41598-020-74368-3
124. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. (2017) 214:579–96. doi: 10.1084/jem.20162024
125. Hupfer A, Brichkina A, Koeniger A, Keber C, Denkert C, Pfefferle P, et al. Matrix stiffness drives stromal autophagy and promotes formation of a protumorigenic niche. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2105367118
126. Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun. (2017) 8:16031. doi: 10.1038/ncomms16031
127. Vennin C, Chin VT, Warren SC, Lucas MC, Herrmann D, Magenau A, et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci Transl Med. (2017) 9. doi: 10.1126/scitranslmed.aai8504
128. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discovery. (2019) 9:282–301. doi: 10.1158/2159-8290.CD-18-0710
129. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discovery. (2019) 9:1102–23. doi: 10.1158/2159-8290.CD-19-0094
130. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat Commun. (2018) 9:948. doi: 10.1038/s41467-018-03347-0
131. Hutton C, Heider F, Blanco-Gomez A, Banyard A, Kononov A, Zhang X, et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell. (2021) 39:1227–1244 e20. doi: 10.1016/j.ccell.2021.06.017
132. Maeda K, Enomoto A, Hara A, Asai N, Kobayashi T, Horinouchi A, et al. Identification of meflin as a potential marker for mesenchymal stromal cells. Sci Rep. (2016) 6:22288. doi: 10.1038/srep22288
133. Mizutani Y, Kobayashi H, Iida T, Asai N, Masamune A, Hara A, et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. (2019) 79:5367–81. doi: 10.1158/0008-5472.CAN-19-0454
134. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. (2010) 330:827–30. doi: 10.1126/science.1195300
135. Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15(+) myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discovery. (2020) 10:232–53. doi: 10.1158/2159-8290.CD-19-0644
136. Krishnamurty AT, Shyer JA, Thai M, Gandham V, Buechler MB, Yang YA, et al. LRRC15(+) myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature. (2022) 611:148–54. doi: 10.1038/s41586-022-05272-1
137. Wang Y, Liang Y, Xu H, Zhang X, Mao T, Cui J, et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discovery. (2021) 7:36. doi: 10.1038/s41421-021-00271-4
138. Xue M, Zhu Y, Jiang Y, Han L, Shi M, Su R, et al. Schwann cells regulate tumor cells and cancer-associated fibroblasts in the pancreatic ductal adenocarcinoma microenvironment. Nat Commun. (2023) 14:4600. doi: 10.1038/s41467-023-40314-w
139. Yousuf S, Qiu M, Voith von Voithenberg L, Hulkkonen J, Macinkovic I, Schulz AR, et al. Spatially resolved multi-omics single-cell analyses inform mechanisms of immune dysfunction in pancreatic cancer. Gastroenterology. (2023) 165:891–908 e14. doi: 10.1053/j.gastro.2023.05.036
140. Le Large TY, Mantini G, Meijer LL, Pham TV, Funel N, van Grieken NC, et al. Microdissected pancreatic cancer proteomes reveal tumor heterogeneity and therapeutic targets. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.138290
141. Brouwer TP, de Vries NL, Abdelaal T, Krog RT, Li Z, Ruano D, et al. Local and systemic immune profiles of human pancreatic ductal adenocarcinoma revealed by single-cell mass cytometry. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-004638
142. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. (2009) 459:262–5. doi: 10.1038/nature07935
143. Huch M, Koo BK. Modeling mouse and human development using organoid cultures. Development. (2015) 142:3113–25. doi: 10.1242/dev.118570
144. Simian M, Bissell MJ. Organoids: A historical perspective of thinking in three dimensions. J Cell Biol. (2017) 216:31–40. doi: 10.1083/jcb.201610056
145. Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. (2014) 345:1247125. doi: 10.1126/science.1247125
146. Greggio C, De Franceschi F, Figueiredo-Larsen M, Gobaa S, Ranga A, Semb H, et al. Artificial three-dimensional niches deconstruct pancreas development in vitro. Development. (2013) 140:4452–62. doi: 10.1242/dev.096628
147. Huch M, Bonfanti P, Boj SF, Sato T, Loomans CJ, van de Wetering M, et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. (2013) 32:2708–21. doi: 10.1038/emboj.2013.204
148. Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. (2015) 160:324–38. doi: 10.1016/j.cell.2014.12.021
149. Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med. (2015) 21:1364–71. doi: 10.1038/nm.3973
150. Walsh AJ, Castellanos JA, Nagathihalli NS, Merchant NB, Skala MC. Optical imaging of drug-induced metabolism changes in murine and human pancreatic cancer organoids reveals heterogeneous drug response. Pancreas. (2016) 45:863–9. doi: 10.1097/MPA.0000000000000543
151. Li X, Ootani A, Kuo C. An air-liquid interface culture system for 3D organoid culture of diverse primary gastrointestinal tissues. Methods Mol Biol. (2016) 1422:33–40. doi: 10.1007/978-1-4939-3603-8_4
152. Grebenyuk S, Ranga A. Engineering organoid vascularization. Front Bioeng Biotechnol. (2019) 7:39. doi: 10.3389/fbioe.2019.00039
153. Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q, et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer. (2018) 18:335. doi: 10.1186/s12885-018-4238-4
154. Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discovery. (2018) 8:1112–29. doi: 10.1158/2159-8290.CD-18-0349
155. Seppälä TT, Zimmerman JW, Sereni E, Plenker D, Suri R, Rozich N, et al. Patient-derived organoid pharmacotyping is a clinically tractable strategy for precision medicine in pancreatic cancer. Ann Surg. (2020) 272:427–35. doi: 10.1097/SLA.0000000000004200
156. Raghavan S, Winter PS, Navia AW, Williams HL, DenAdel A, Lowder KE, et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell. (2021) 184:6119–6137 e26. doi: 10.1016/j.cell.2021.11.017
157. Malinova A, Schreyer D, Fiorini E, Pasini D, Bevere M, D’Agosto S, et al. ecDNA amplification of MYC drives intratumor copy-number heterogeneity and adaptation to stress in PDAC. bioRxiv. (2023). 2023.09.27.559717. doi: 10.1101/2023.09.27.559717
158. Zhou X, An J, Kurilov R, Brors B, Hu K, Peccerella T, et al. Persister cell phenotypes contribute to poor patient outcomes after neoadjuvant chemotherapy in PDAC. Nat Cancer. (2023) 4:1362–81. doi: 10.1038/s43018-023-00628-6
159. Filippini D, Agosto S, Delfino P, Simbolo M, Piro G, Rusev B, et al. Immunoevolution of mouse pancreatic organoid isografts from preinvasive to metastatic disease. Sci Rep. (2019) 9:12286. doi: 10.1038/s41598-019-48663-7
160. Geyer M, Gaul LM, D Agosto SL, Corbo V, Queiroz K. The tumor stroma influences immune cell distribution and recruitment in a PDAC-on-a-chip model. Front Immunol. (2023) 14:1155085. doi: 10.3389/fimmu.2023.1155085
161. Haque MR, Wessel CR, Leary DD, Wang C, Bhushan A, Bishehsari F. Patient-derived pancreatic cancer-on-a-chip recapitulates the tumor microenvironment. Microsyst Nanoeng. (2022) 8:36. doi: 10.1038/s41378-022-00370-6
162. Haque MR, Rempert TH, Al-Hilal TA, Wang C, Bhushan A, Bishehsari F. Organ-chip models: opportunities for precision medicine in pancreatic cancer. Cancers (Basel). (2021) 13. doi: 10.3390/cancers13174487
163. Lai Benjamin FL, Lu Rick X, Hu Y, Davenport HL, Dou W, Wang EY, et al. Recapitulating pancreatic tumor microenvironment through synergistic use of patient organoids and organ-on-a-chip vasculature. Adv Funct Mater. (2020) 30. doi: 10.1002/adfm.202000545
164. Casolino R, Corbo V, Beer P, Hwang CI, Paiella S, Silvestri V, et al. Germline aberrations in pancreatic cancer: implications for clinical care. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14133239
165. Tempero MA, Malafa MP, Al-Hawary M, Behrman SW, Benson AB, Cardin DB, et al. Pancreatic adenocarcinoma, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2021) 19:439–57. doi: 10.6004/jnccn.2021.0017
166. Wang-Gillam A, Lim KH, McWilliams R, Suresh R, Lockhart AC, Brown A, et al. Defactinib, pembrolizumab, and gemcitabine in patients with advanced treatment refractory pancreatic cancer: A phase I dose escalation and expansion study. Clin Cancer Res. (2022) 28:5254–62. doi: 10.1158/1078-0432.CCR-22-0308
167. Aung KL, McWhirter E, Welch S, Wang L, Lovell S, Stayner LA, et al. A phase II trial of GSK2256098 and trametinib in patients with advanced pancreatic ductal adenocarcinoma. J Gastrointest Oncol. (2022) 13:3216–26. doi: 10.21037/jgo
168. Chen IM, Donia M, Chamberlain CA, Jensen AWP, Draghi A, Theile S, et al. Phase 2 study of ipilimumab, nivolumab, and tocilizumab combined with stereotactic body radiotherapy in patients with refractory pancreatic cancer (TRIPLE-R). Eur J Cancer. (2023) 180:125–33. doi: 10.1016/j.ejca.2022.11.035
169. Ko AH, Kim KP, Siveke JT, Lopez CD, Lacy J, O'Reilly EM, et al. Atezolizumab plus PEGPH20 versus chemotherapy in advanced pancreatic ductal adenocarcinoma and gastric cancer: MORPHEUS phase ib/II umbrella randomized study platform. Oncologist. (2023) 28:553–e472. doi: 10.1093/oncolo/oyad022
170. Kocher HM, Basu B, Froeling FEM, Sarker D, Slater S, Carlin D, et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat Commun. (2020) 11:4841. doi: 10.1038/s41467-020-18636-w
171. Arias-Lorza AM, Costello JR, Hingorani SR, Von Hoff DD, Korn RL, Raghunand N. Tumor response to stroma-modifying therapy: magnetic resonance imaging findings in early-phase clinical trials of pegvorhyaluronidase alpha (PEGPH20). Res Sq. (2023). doi: 10.21203/rs.3.rs-3314770/v1
172. Wang S, Bager CL, Karsdal MA, Chondros D, Taverna D, Willumsen N. Blood-based extracellular matrix biomarkers as predictors of survival in patients with metastatic pancreatic ductal adenocarcinoma receiving pegvorhyaluronidase alfa. J Transl Med. (2021) 19:39. doi: 10.1186/s12967-021-02701-z
173. Van Cutsem E, Tempero MA, Sigal D, Oh DY, Fazio N, Macarulla T, et al. Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol. (2020) 38:3185–94. doi: 10.1200/JCO.20.00590
174. Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, et al. Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J Clin Oncol. (2019) 37:1062–9. doi: 10.1200/JCO.18.01295
175. Murphy JE, Wo JY, Ryan DP, Clark JW, Jiang W, Yeap BY, et al. Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: A phase 2 clinical trial. JAMA Oncol. (2019) 5:1020–7. doi: 10.1001/jamaoncol.2019.0892
176. Dean A, Gill S, McGregor M, Broadbridge V, Jarvelainen HA, Price T. Dual alphaV-integrin and neuropilin-1 targeting peptide CEND-1 plus nab-paclitaxel and gemcitabine for the treatment of metastatic pancreatic ductal adenocarcinoma: a first-in-human, open-label, multicentre, phase 1 study. Lancet Gastroenterol Hepatol. (2022) 7:943–51. doi: 10.1016/S2468-1253(22)00167-4
177. O'Hara MH, O'Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH, et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol. (2021) 22:118–31. doi: 10.1016/S1470-2045(20)30532-5
178. Padron LJ, Maurer DM, O'Hara MH, O'Reilly EM, Wolff RA, Wainberg ZA, et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat Med. (2022) 28:1167–77. doi: 10.1038/s41591-022-01829-9
179. Coveler AL, Smith DC, Phillips T, Curti BD, Goel S, Mehta AN, et al. Phase 1 dose-escalation study of SEA-CD40: a non-fucosylated CD40 agonist, in advanced solid tumors and lymphomas. J Immunother Cancer. (2023) 11. doi: 10.1136/jitc-2022-005584
180. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. (2020) 26:878–85. doi: 10.1038/s41591-020-0880-x
181. Bockorny B, Macarulla T, Semenisty V, Borazanci E, Feliu J, Ponz-Sarvise M, et al. Motixafortide and pembrolizumab combined to nanoliposomal irinotecan, fluorouracil, and folinic acid in metastatic pancreatic cancer: the COMBAT/KEYNOTE-202 trial. Clin Cancer Res. (2021) 27:5020–7. doi: 10.1158/1078-0432.CCR-21-0929
182. Suarez-Carmona M, Williams A, Schreiber J, Hohmann N, Pruefer U, Krauss J, et al. Combined inhibition of CXCL12 and PD-1 in MSS colorectal and pancreatic cancer: modulation of the microenvironment and clinical effects. J Immunother Cancer. (2021) 9. doi: 10.1136/jitc-2021-002505
183. Razak AR, Cleary JM, Moreno V, Boyer M, Calvo Aller E, Edenfield W, et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J Immunother Cancer. (2020) 8. doi: 10.1136/jitc-2020-001006
184. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. (2016) 17:651–62. doi: 10.1016/S1470-2045(16)00078-4
185. Noel M, O'Reilly EM, Wolpin BM, Ryan DP, Bullock AJ, Britten CD, et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. (2020) 38:800–11. doi: 10.1007/s10637-019-00830-3
186. Bendell J, LoRusso P, Overman M, Noonan AM, Kim DW, Strickler JH, et al. First-in-human study of oleclumab, a potent, selective anti-CD73 monoclonal antibody, alone or in combination with durvalumab in patients with advanced solid tumors. Cancer Immunol Immunother. (2023) 72:2443–58. doi: 10.1007/s00262-023-03430-6
187. Wienbeck M, Blasberg M. Effects of an enkephalin analog on motility of the small and large intestine in the cat. Z Gastroenterol. (1986) 24:179–87.
188. Parsons PJ, LeRoy AF. Determination of cis-diamminedichloroplatinum(II) in human plasma using ion-pair chromatography with electrochemical detection. J Chromatogr. (1986) 378:395–408. doi: 10.1016/S0378-4347(00)80735-5
189. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
190. Skorupan N, Palestino Dominguez M, Ricci SL, Alewine C. Clinical strategies targeting the tumor microenvironment of pancreatic ductal adenocarcinoma. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14174209
191. Hu ZI, O'Reilly EM. Therapeutic developments in pancreatic cancer. Nat Rev Gastroenterol Hepatol. (2023) 14(17):4209. doi: 10.3390/cancers14174209
192. Bogin V. Master protocols: New directions in drug discovery. Contemp Clin Trials Commun. (2020) 18:100568. doi: 10.1016/j.conctc.2020.100568
193. Picard FSR, Lutz V, Brichkina A, Neuhaus F, Ruckenbrod T, Hupfer A, et al. IL-17A-producing CD8(+) T cells promote PDAC via induction of inflammatory cancer-associated fibroblasts. Gut. (2023) 72:1510–22. doi: 10.1136/gutjnl-2022-327855
194. Lacroix M, Riscal R, Arena G, Linares LK, Le Cam L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol Metab. (2020) 33:2–22. doi: 10.1016/j.molmet.2019.10.002
195. Burdziak C, Alonso-Curbelo D, Walle T, Reyes J, Barriga FM, Haviv D, et al. Epigenetic plasticity cooperates with cell-cell interactions to direct pancreatic tumorigenesis. Sci 380. (2023) 380(6645):eadd5327. doi: 10.1126/science.add5327
196. Driehuis E, Gracanin A, Vries RGJ, Clevers H, Boj SF. Establishment of pancreatic organoids from normal tissue and tumors. STAR Protoc. (2020) 1:100192. doi: 10.1016/j.xpro.2020.100192
197. Zhang Z, Rohweder PJ, Ongpipattanakul C, Basu K, Bohn MF, Dugan EJ, et al. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell. (2022) 40:1060–1069 e7. doi: 10.1016/j.ccell.2022.07.005
198. Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and Malignant progression in pancreatic ductal adenocarcinoma. Cell Res. (2019) 29:725–38. doi: 10.1038/s41422-019-0195-y
199. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. (2015) 518:495–501. doi: 10.1038/nature14169
200. Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. (2023) 618:144–50. doi: 10.1038/s41586-023-06063-y
201. Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol. (2020) 21:271–82. doi: 10.1016/S1470-2045(19)30691-6
202. Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discovery. (2017) 7:963–72. doi: 10.1158/2159-8290.CD-17-0507
203. Pellat A, Boutron I, Ravaud P. Availability of results of trials studying pancreatic adenocarcinoma over the past 10 years. Oncologist. (2022) 27:e849–55. doi: 10.1093/oncolo/oyac156
204. Frenkel M, David A, Sapire K, Hausner D. Complementary and integrative medicine in pancreatic cancer. Curr Oncol Rep. (2023) 25:231–42. doi: 10.1007/s11912-023-01370-z
205. Kurz E, Hirsch CA, Dalton T, Shadaloey SA, Khodadadi-Jamayran A, Miller G, et al. Exercise-induced engagement of the IL-15/IL-15Ralpha axis promotes anti-tumor immunity in pancreatic cancer. Cancer Cell. (2022) 40:720–737 e5. doi: 10.1016/j.ccell.2022.05.006
206. Cortez NE, Rodriguez Lanzi C, Hong BV, Xu J, Wang F, Chen S, et al. A ketogenic diet in combination with gemcitabine increases survival in pancreatic cancer KPC mice. Cancer Res Commun. (2022) 2:951–65. doi: 10.1158/2767-9764.CRC-22-0256
207. Sandri S, Bobisse S, Moxley K, Lamolinara A, De Sanctis F, Boschi F, et al. Feasibility of telomerase-specific adoptive T-cell therapy for B-cell chronic lymphocytic leukemia and solid Malignancies. Cancer Res. (2016) 76:2540–51. doi: 10.1158/0008-5472.CAN-15-2318
208. Sandri S, De Sanctis F, Lamolinara A, Boschi F, Poffe O, Trovato R, et al. Effective control of acute myeloid leukaemia and acute lymphoblastic leukaemia progression by telomerase specific adoptive T-cell therapy. Oncotarget. (2017) 8:86987–7001. doi: 10.18632/oncotarget.v8i50
209. Mazzocco M, Martini M, Rosato A, Stefani E, Matucci A, Dalla Santa S, et al. Autologous cellular vaccine overcomes cancer immunoediting in a mouse model of myeloma. Immunology. (2015) 146:33–49. doi: 10.1111/imm.12477
210. Pierini S, Fang C, Rafail S, Facciponte JG, Huang J, De Sanctis F, et al. A tumor mitochondria vaccine protects against experimental renal cell carcinoma. J Immunol. (2015) 195:4020–7. doi: 10.4049/jimmunol.1500281
211. De Sanctis F, Adamo A, Cane S, Ugel S. Targeting tumour-reprogrammed myeloid cells: the new battleground in cancer immunotherapy. Semin Immunopathol. (2023) 45:163–86. doi: 10.1007/s00281-022-00965-1
212. Nwosu ZC, Ward MH, Sajjakulnukit P, Poudel P, Ragulan C, Kasperek S, et al. Uridine-derived ribose fuels glucose-restricted pancreatic cancer. Nature. (2023) 618:151–8. doi: 10.1038/s41586-023-06073-w
213. Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. (2020) 17:487–505. doi: 10.1038/s41575-020-0300-1
214. Ugel S, Facciponte JG, De Sanctis F, Facciabene A. Targeting tumor vasculature: expanding the potential of DNA cancer vaccines. Cancer Immunol Immunother. (2015) 64:1339–48. doi: 10.1007/s00262-015-1747-8
Keywords: PDAC - pancreatic ductal adenocarcinoma, immunotherapy, TME (tumor microenvironment), cancer associated fibroblast (CAF), MDSC (myeloid-derived suppressor cells), TILs (tumor infiltrating lymphocytes), immunosuppression
Citation: Musiu C, Lupo F, Agostini A, Lionetto G, Bevere M, Paiella S, Carbone C, Corbo V, Ugel S and De Sanctis F (2024) Cellular collusion: cracking the code of immunosuppression and chemo resistance in PDAC. Front. Immunol. 15:1341079. doi: 10.3389/fimmu.2024.1341079
Received: 19 November 2023; Accepted: 02 May 2024;
Published: 16 May 2024.
Edited by:
Louis M. Weiner, Georgetown University, United StatesReviewed by:
Willem de Koning, Erasmus Medical Center, NetherlandsCopyright © 2024 Musiu, Lupo, Agostini, Lionetto, Bevere, Paiella, Carbone, Corbo, Ugel and De Sanctis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco De Sanctis, ZnJhbmNlc2NvLmRlc2FuY3Rpc0B1bml2ci5pdA==; Stefano Ugel, U3RlZmFuby51Z2VsQHVuaXZyLml0
†These authors share first authorship
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.