 Magdalena Makuch
Magdalena Makuch Mariia Stepanechko
Mariia Stepanechko Małgorzata Bzowska
Małgorzata Bzowska- Department of Immunology, Faculty of Biochemistry, Biophysics, and Biotechnology, Jagiellonian University, Kraków, Poland
Macrophages are highly plastic cells ubiquitous in various tissues, where they perform diverse functions. They participate in the response to pathogen invasion and inflammation resolution following the immune response, as well as the maintenance of homeostasis and proper tissue functions. Macrophages are generally considered long-lived cells with relatively strong resistance to numerous cytotoxic factors. On the other hand, their death seems to be one of the principal mechanisms by which macrophages perform their physiological functions or can contribute to the development of certain diseases. In this review, we scrutinize three distinct pro-inflammatory programmed cell death pathways – pyroptosis, necroptosis, and ferroptosis – occurring in macrophages under specific circumstances, and explain how these cells appear to undergo dynamic yet not always final changes before ultimately dying. We achieve that by examining the interconnectivity of these cell death types, which in macrophages seem to create a coordinated and flexible system responding to the microenvironment. Finally, we discuss the complexity and consequences of pyroptotic, necroptotic, and ferroptotic pathway induction in macrophages under two pathological conditions – atherosclerosis and cancer. We summarize damage-associated molecular patterns (DAMPs) along with other microenvironmental factors, macrophage polarization states, associated mechanisms as well as general outcomes, as such a comprehensive look at these correlations may point out the proper methodologies and potential therapeutic approaches.
1 Introduction
Macrophages, ever since Elie Metchnikoff’s pioneering discovery of their phagocytic capabilities in the late 19th century, have occupied a central role in immunology. Present in nearly every tissue, these versatile professional phagocytes perform multifaceted functions in antimicrobial defense, shaping the immune response through antigen presentation and cytokine secretion. However, it is worth noting that macrophages’ roles extend well beyond direct participation in antimicrobial defense, and over the past three decades research has unveiled a multitude of fascinating functions of these cells. We are now gaining a deeper understanding of the diverse and highly specialized roles that macrophages play as pivotal contributors to the clearance of senescent or dead cells, tissue repair, regeneration, and the maintenance of tissue integrity and homeostasis (1).
One of the distinctive characteristics of macrophages is their remarkable plasticity and adaptability, allowing them to fulfill the unique demands of the surrounding tissue microenvironment. The tissue-specific diversity of macrophages derives from phenomena such as their embryonic origin or recruitment from circulating monocyte pools, and prior encounters with conditions like inflammation or injury. All these experiences leave a lasting imprint on cell identity (2–4). Consequently, this led to the development of macrophage classification, which is based on their possible mechanisms of activation and subsequent roles in either promoting or resolving inflammation. Macrophage polarization states were divided into M1 and M2 phenotypes, commonly referred to as classically and alternatively activated. The M1 type is induced by LPS and IFNγ, and associated with enhanced phagocytic activity, antigen presentation, increased reactive oxygen species (ROS) production, and the release of pro-inflammatory cytokines. Conversely, following the downstream signals of cytokines such as IL-4 and IL-13 macrophages polarize into the M2 phenotype, which results in an anti-inflammatory response that promotes angiogenesis, tissue remodeling, and repair (5). Such a view, however, is regarded now as too dichotomic and rather applicable to polarized murine macrophages, which are easier to distinguish (6). In contrast, the macrophage polarization scheme derived from human studies seems to be much more complex, dependent on various contexts (7), and reversible (8). A good illustration is the possibility of differentiating M2a, M2b, M2c, and M2d subsets. They occur upon induction with various stimuli, yet all are characterized by the ability to secrete high levels of anti-inflammatory IL-10 (7, 9). Indeed, M2a type is induced by IL-4/IL-13, whereas M2b by immune complexes and LPS/IL-1β. On the other hand, IL-10/TGFβ and glucocorticoids polarize macrophages toward M2c, while TLR+adenosine A2 receptor agonists induce M2d phenotype (7, 9). The challenges linked to the classification of macrophage phenotypes have been extensively discussed (5, 10). We highlight that M1- and M2-like macrophages are not mutually exclusive and frequently coexist. Moreover, the diverse nature of stimuli, the interaction of various pathways, and the lack of specific markers suggest that macrophages may form mixed phenotypes, rather than stable subsets (5). Later in the text, we describe the role of macrophage cell death types in their polarization as well as the progression of two human diseases – atherosclerosis and cancer. We decided to use the simplified phenotype classification according to that employed by the authors. Nevertheless, we emphasize that this represents a form of simplification.
Macrophage states are the results of various phenomena that cells encounter throughout their life. All of them profoundly influence macrophage physiology and can finally culminate in cell death. Noteworthy, there are various, other than apoptosis, forms of programmed cell death (PCD), which are controlled by multiple discrete yet interconnected signaling pathways (11, 12). In the first part of this review, we describe the mechanisms of three different pro-inflammatory PCD modalities – pyroptosis, necroptosis, and ferroptosis – with a particular emphasis on their occurrence in macrophages under specific circumstances. All these cell deaths have profound implications not only for the affected cell but, more importantly, for the microenvironment, tissue homeostasis, prolongation of inflammation, and progression of diseases. In the second part of our review, we meticulously examine two such maladies – atherosclerosis and cancer. Our attention is directed toward them, as there is a considerable amount of data showing a significant impact of both macrophage death and inflammation in their pathogenesis and treatment. Moreover, evidence from both diseases implied a connection between the occurrence of specific inflammatory PCD and macrophage polarization state. For the first time in one place we gather information on specific triggering factors, microenvironmental conditions, pathways, and their crosstalk, which result in the dance of macrophages with death. Understanding the mechanisms underlying macrophage pyroptosis, necroptosis, and ferroptosis is of paramount significance since they may constitute attractive targets for potential therapies.
2 Pyroptosis, necroptosis, and ferroptosis: mechanisms in macrophages
Pyroptosis, necroptosis, and ferroptosis represent distinctive forms of programmed cell death differing from silent apoptosis, as they are recognized for their pro-inflammatory and immunogenic roles. Morphologically, both pyroptosis and necroptosis involve cell swelling, although the first one generally results in milder cellular content leakage compared to the second one. In both cases, however, cell membrane damage is induced by oligomerization of pore-forming effector protein – GSDMD in pyroptosis and MLKL in necroptosis. It signifies a similar effector mechanism leading to the release of intracellular material, such as pro-inflammatory cytokines IL-1β and IL-18 and/or damage-associated molecular patterns (DAMPs) (13). By contrast, little information is available regarding the integrity of cell membrane in ferroptosis. It is thought, however, that at the late stage of this cell death plasma membrane rupture may occur after prior cell swelling (14). Nevertheless, both the progression and execution of ferroptosis are underpinned by disturbances in bioenergetic metabolism as well as the disruption of redox homeostasis, which closely resembles the mechanisms observed in necroptosis (15). Another common feature of these two PCDs is that macrophages seem to exhibit varying susceptibilities to undergo them, depending on the cell polarization state. Further elaboration on this aspect is provided at the end of this section.
2.1 Pyroptosis
The name ‘pyroptosis’ derives from Greek, where pyro refers to fire or fever, and ptosis means failing. It was first described in 1986 by Friedlander, who observed that primary mouse macrophages treated with anthrax lethal toxin (LT) underwent cell death accompanied by the release of cellular content (16). Later, pyroptosis was described by Zychlinksy et al., who indicated that Shigella flexneri infection results in suicide in macrophages, however, this cell death was initially referred to as apoptosis (17). These findings were further examined by Chen et al., who in 1996 reported that S. flexneri-infected macrophages were characterized by activation of interleukin-1β-converting enzyme (ICE, caspase-1) (18). ICE was discovered for the first time in 1989 (19) and subsequently associated with the ability to process interleukin-1β precursor into its mature form (20, 21). Such a significant role of ICE in the described form of programmed cell death led Cookson et al. to propose the term pyroptosis (22).
The characteristics of pyroptosis include the activation of inflammasomes and their sensors (also known as pattern recognition receptors, PRRs) as well as the loss of plasma membrane integrity (23). A canonical inflammasome is a multimolecular complex assembled in the cytoplasm after PRRs activation, which can recognize pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (24). PRRs include Nod-like receptor (NLR) family, the DNA receptor Absent in Melanoma 2 (AIM2), and the Pyrin receptor (23). NLRP3 and AIM2 inflammasomes require two signals for their activation (Figure 1). The first signal (also known as priming signal) is usually obtained through PAMPs detection by Toll-like receptors (TLR) or induction of cytokine receptors (e.g., IL-1R), and results in NF-κB activation and upregulated transcription of NLRP3, adapter apoptosis-associated speck-like protein (ASC), pro-IL-1β, and pro-IL-18 (25). Interestingly, it has recently been shown that exosomes released from TNF-α-stimulated neutrophils can also act as a priming signal for macrophages (26). The second signal (also known as activation signal) can be triggered by a variety of stimuli including ions efflux/influx, oxidized mitochondrial DNA, lysosomal rupture, reactive oxygen species (ROS), ATP (27), and myoglobin (28). After activation, inflammasome sensors oligomerize and subsequently bind ASC, which contains a caspase activation recruitment domain (CARD) necessary to recruit pro-caspase-1 and assemble inflammasome (23, 24). Such canonical inflammasomes constitute a platform for the autoproteolysis of caspase-1, which activated cleaves pro-IL-1β and pro-IL-18 to their mature forms (Figure 1).
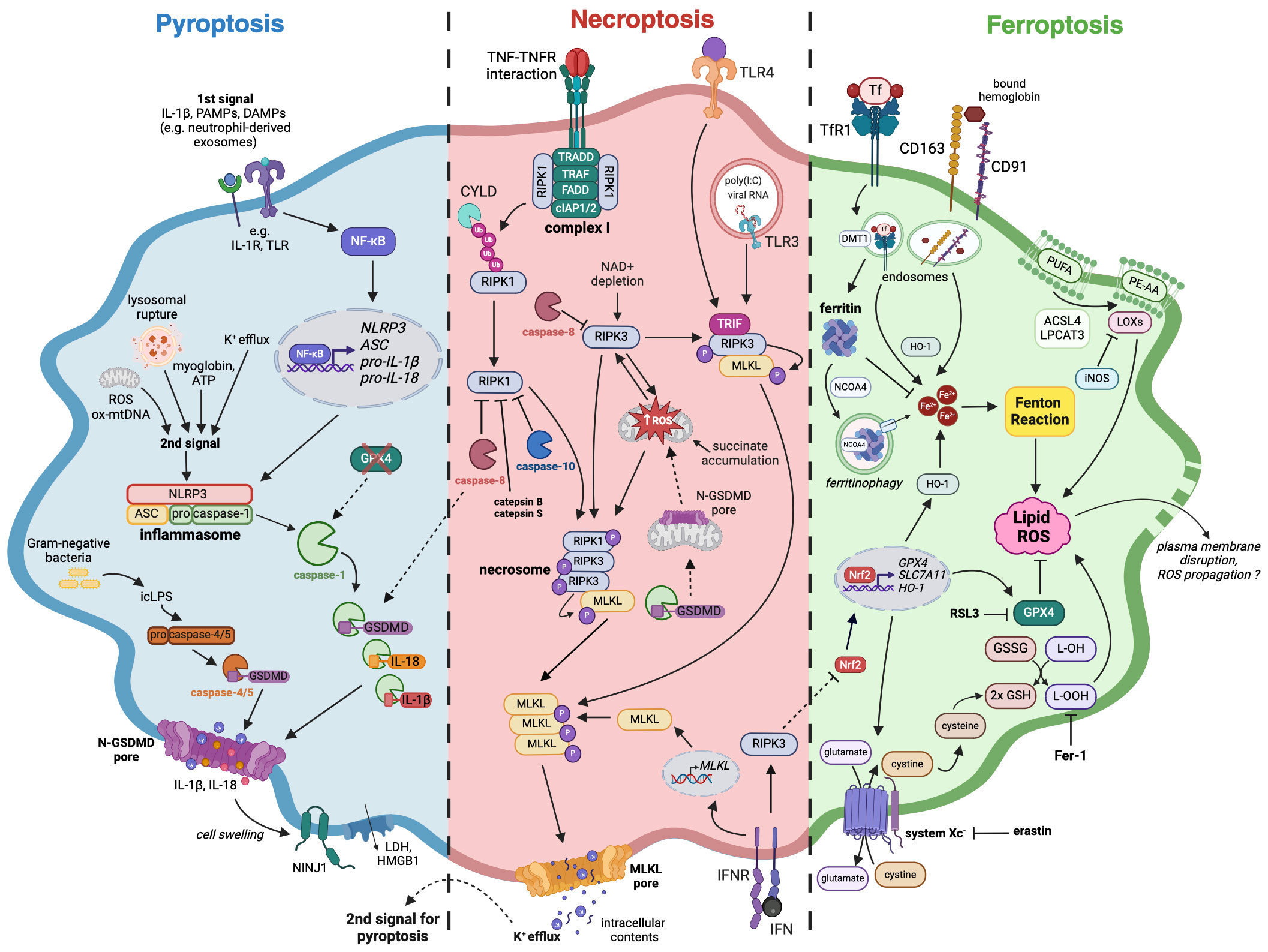
Figure 1 The mechanisms of pyroptosis, necroptosis, and ferroptosis in macrophages. A large number of studies confirmed that macrophages are capable of following different immunogenic programmed cell death pathways upon various stimuli. Pyroptosis (blue) induction requires two signals, which eventually lead to the assembly of inflammasome complex and activation of caspase-1, followed by IL-18, IL-1β as well as GSDMD cleavage and maturation. Necroptosis (red) can arise after TNF or TLR stimulation as a switch from apoptosis when there is a simultaneous caspases inhibition. Necrosome is created as a result of RIPK1 and RIPK3 autoactivation or direct interaction of TRIF adaptor protein with RIPK3, followed by phosphorylation of the effector protein, which is MLKL. Both pyroptosis and necroptosis, despite requiring different stimuli for their activation, are characterized by the presence of membrane pores, formed by N-GSDMD and pMLKL respectively. Additionally, cell swelling-associated events that happen in pyroptosis can activate NINJ1, leading to plasma membrane rupture. The subsequent release of cellular contents like HMGB1 further stimulates immune response. Ferroptosis (green) occurs as an outcome of iron homeostasis disruption and an increase in the labile iron pool that can undergo Fenton reaction, leading to lipid peroxidation and subsequent cell death. An iron-storing protein ferritin binds free iron, thereby impeding it from entering the reaction. Amino acid antitransporter system Xc- and GPX4 can alleviate oxidative stress and prevent ferroptosis while their blockade by erastin and RSL3 respectively diminishes cellular antioxidative defense. The loss of cell membrane integrity during ferroptosis has not been well documented, however, it is thought that plasma membrane disruption may occur. Dashed lines indicate crosstalk between pyroptotic, necroptotic, and ferroptotic signaling pathways. Pyroptosis: ASC – apoptosis-associated speck-like protein containing a CARD, DAMPs – danger-associated molecular patterns, GSDMD – gasdermin D, HMGB1 – High mobility group protein B1, icLPS – intracellular lipopolysaccharides, IL-1β – interleukin-1β, IL-18 – interleukin-18, IL-1R – interleukin-1 receptor, LDH – lactate dehydrogenase, NF-κB – Nuclear factor kappa-light-chain-enhancer of activated B cells, NINJ1 – Ninjurin 1, N-GSDMD – GSDMD N-terminal domain, NLRP3 – NLR Family Pyrin Domain Containing 3, ox-mtDNA – oxidized mitochondrial DNA, PAMPs – pathogen-associated molecular patterns, pro-IL-1β – pro-interleukin-1β, pro-IL-18 – pro-interleukin-18, ROS – reactive oxygen species, TLR – Toll-like receptor; Necroptosis: cIAP1/2 – cellular inhibitor of apoptosis protein 1/2, CYLD – cylindromatosis protein, FADD – Fas-associated death domain, IFN – interferon, IFNR – interferon receptor, MLKL – mixed lineage kinase domain-like protein, polyIC – Polyinosinic-polycytidylic acid, RIPK1 – receptor-interacting protein kinase 1, RIPK3 – receptor-interacting protein kinase 3, TLR3 – Toll-like receptor 3, TLR4 – Toll-like receptor 4, TRADD – TNFR-associated death domain, TRAF – TNFR-associated factor, TRIF – TIR-domain-containing adapter-inducing interferon β; Ferroptosis: ACSL4 – acyl-CoA synthase long chain family member 4, DMT-1 – divalent metal transporter 1, Fer-1 – ferrostatin-1, GSH – glutathione, GSSG – glutathione disulfide, GPX4 – glutathione peroxidase 4, HO-1 – heme oxygenase 1, iNOS – inducible nitric oxide synthase, L-OH – lipid alcohol, L-OOH – lipid peroxide, LOXs – lipoxygenases, LPCAT3 – lysophosphatidylcholine acyltransferase 3, Nrf2 – nuclear factor erythroid 2-related factor 2, NCOA4 – nuclear receptor coactivator 4, PE-AA – Phosphatidylethanolamine (PE)-linked arachidonic acid, PUFA – polyunsaturated fatty acids, RSL3 – RAS-selective lethal 3, SLC7A11 – solute carrier family 7 member 11, TfR1 – transferrin receptor 1.
Furthermore, caspase-1 cleaves gasdermin D to form N-terminal GSDMD (N-GSDMD) able to oligomerize and perforate cell membrane by creating a transmembrane pore. It subsequently leads to cell swelling, lysis, and pyroptosis accompanied by the release of cellular contents that strongly activate inflammatory responses (24). Gasdermin D pore diameter is about ~21 nm (29), which makes it quite selective about large molecules, but non-selective about smaller cellular contents. However, recent findings indicated the importance of charge displayed by the cargo transported through GSDMD pore. In their study, Xia et al. demonstrated that gasdermin D pore seems to favor the passage of mature IL-1β and IL-18 rather than their proforms. Such phenomenon originates from the fact that IL-1β precursor contains an acidic domain, which is removed by active caspase-1, enabling mature interleukin to cross the negatively charged conduit of the GSDMD pore (30). Moreover, Kayagaki et al. demonstrated that cell swelling, which occurs in macrophage pyroptosis and necrosis, can lead to cell-surface Ninjurin 1 (NINJ1) protein activation (31). Consequently, there is an induction of plasma membrane rupture, followed by the release of lactate dehydrogenase (LDH) and DAMPs such as high mobility group protein B1 (HMGB1), which further intensify inflammation (31).
Pyroptosis can also derive from the non-canonical pathway, which can be activated by intracellular LPS (icLPS) recognized by caspase-11 (in mice) or caspase-4/5 (in humans) by their CARD domain (24). The interaction between icLPS and caspase-4/5/11 results in autoproteolytic activation of caspases, which also cleave GSDMD (Figure 1). Subsequently, N-GSDMD oligomerizes and forms membrane pores, leading to K+ efflux (23). Interestingly, such ion fluctuation is the signal for NLRP3 inflammasome assembling and caspase-1 activation, which eventually lead to pyroptosis and the release of IL-1β and IL-18. Although caspases-4/5/11 are not capable of cleaving pro-IL-1β and pro-IL-18, they mediate their maturation processing through NLRP3/caspase-1 pathway in some cell types (24).
A growing body of evidence suggests that certain cells with activated inflammasomes and undergoing cell death sometimes do not experience cell lysis. For example, DiPeso et al. showed that pyroptotic cell death in bone marrow-derived macrophages (BMDMs) is not always accompanied by cell rupture, therefore lysis prevention does not couple with inhibition of cell death after a pyroptotic stimulus (32). These data suggest that pyroptosis does not necessarily result in cell lysis, though cell swelling and leaking are still important consequences of GSDMD pores formation (32). Moreover, inflammasome activation and pyroptosis induction in macrophages seem to be reversible. The latest findings demonstrated that endosomal sorting complexes required for transport III (ESCRT III) machinery in macrophages was able to dampen GSDMD pore-mediated cell death and IL-1β release, even though inflammasomes were activated in both canonical and non-canonical ways (33). The authors suggest a potential role of ESCRTs in removing GSDMD pores from the macrophage membrane, thereby indicating that pyroptosis in these cells can be restricted (33).
2.2 Necroptosis
The elucidation of necroptosis arose from the initial observation that TNF, a cytokine traditionally associated with the ability to induce apoptosis (34), could also trigger necrotic cell death when various caspases were concurrently inhibited (35). Subsequently, Holler et al. proved that such necrotic cell death could also be initiated by Fas as well as TNF-related apoptosis-inducing ligand (TRAIL), and that active receptor-interacting protein kinase 1 (RIPK1) seemed to be vital for this process (36, 37) This RIPK1-dependent necrotic cell death was termed necroptosis (36), and, despite morphological similarities, was distinguished from necrosis, which is an accidental cell death resulting from irreversible cellular injury. Over time researchers identified other significant molecules downstream RIPK1, including receptor-interacting protein kinase 3 (RIPK3) (38–40) and mixed lineage kinase domain-like protein (MLKL), which serves as the ultimate executor of necroptosis (41).
To describe the exact mechanism of necroptosis, it is inevitable to mention first the TNF-mediated activation and apoptosis pathways. The interaction between TNF and tumor necrosis factor receptor (TNFR) leads to the formation of complex I (Figure 1), which is situated at the cell membrane and consists of RIPK1, Fas-associated death domain (FADD), TNFR-associated death domain (TRADD), TNFR-associated factors (TRAFs) as well as cellular inhibitor of apoptosis protein 1 (cIAP1) and 2 (cIAP2). Within complex I, RIPK1 undergoes extensive ubiquitination by TRAFs, cIAPs, and linear ubiquitin chain assembly complex (LUBAC), inhibiting its kinase activity and promoting nuclear factor kappa B (NFκB)-mediated cell survival (42). However, a reduction in RIPK1 ubiquitination, either by deubiquitinating enzyme cylindromatosis protein (CYLD) or inhibition of cIAPs with SMAC mimetic (SM), leads to RIPK1 dissociation from complex I. Subsequently, RIPK1 binds to RIPK3, FADD, TRADD, and caspase-8, thereby forming complex II. Active caspase-8 cleaves downstream caspase-3/7 and RIPK1, inducing cell apoptosis while simultaneously inhibiting necroptosis (43). In the absence of caspase-8 activity, RIPK1 and RIPK3 initiate necroptosis – an alternative, caspase-independent pathway of PCD (Figure 1). RIPK1 and RIPK3 interact by their RIP homotypic interaction motifs (RHIMs), undergo autophosphorylation, and form a complex called necrosome. It recruits and phosphorylates MLKL, which oligomerizes and forms pores in the cell membrane, leading to the release of intracellular contents and eventual cell death (41, 44).
Induction of necroptosis by TNF and pan-caspase inhibitor is observed in primary human monocyte-derived macrophages (hMDMs), the U937 monocytic cell line, primary murine peritoneal macrophages (PMs), BMDMs, and the J774A.1 cell line. Interestingly, human THP-1 and murine RAW264.7 cell lines exhibit resistance to described canonical necroptosis stimulation (45, 46) and require additional degradation of cIAPs.
Caspase-8 serves as a pivotal regulator of cell fate, with options spanning across survival, apoptosis, or necroptosis (23) — a role that has been firmly established, also in macrophages (47, 48). The current thinking is that caspase-8 homodimers promote cell death, whereas cleavage events mediated by heterodimers of caspase-8 and cFLIP dictate life (49). Moreover, activation of the death receptor Fas on murine macrophages induces caspase-8-dependent processing of caspase-3 and apoptosis, whereas stimulation of TNFR1, TLR3, or TLR4 does not. However, in the absence of caspase-8, either TNF or TLR3 and TLR4 agonists are lethal to murine macrophages, which undergo cell death mediated by RIPK3 and MLKL (50). Active caspase-8 appears to suppress necroptosis by cleaving RIPK1 and limiting its interaction with RIPK3 (47, 51). Recently, it was also demonstrated that caspase-10 cleaved RIPK1 more efficiently than caspase-8, thus U937 cells lacking caspase-10 were more sensitive to death-inducing stimuli (52). Cathepsins B and S, common proteolytic enzymes in myeloid cells, are also able to cleave RIPK1, thereby efficiently mitigating TNF- or LPS-induced necroptosis in BMDMs (53).
When caspase activity is suppressed, necroptosis can also be triggered upon TLRs stimulation, commonly in human and murine macrophages (Figure 1). For TLR2, TLR5, and TLR9 this process depends on TNF release and the autocrine or paracrine activation of TNFR1. In contrast, TLR3 and TLR4 stand out as they rely on the TRIF adaptor protein (TIR-domain-containing adapter-inducing interferon β) in their signaling cascade. Upon exposure to poly-I:C (a ligand for TLR3) or LPS (a ligand for TLR4), the TRIF protein directly interacts with RIPK3 through its RHIM domain, leading to the necrosome formation and the activation of MLKL (54–57). Contrarily to endothelial cells and fibroblasts, RIPK1 in macrophages is implicated in the mechanism of necroptosis induced by TLR3 and TLR4 (54, 55). Interestingly, RIPK1’s ubiquitination pattern, which modulates its interactions with proteins like TRADD, FADD, and TNFR1, exerts dual effects by promoting TNF- but simultaneously restraining TLR-induced necroptosis (58). Furthermore, c-Jun N-terminal kinases (JNKs) serve as additional regulators that facilitate necroptosis in response to TNF and TLR ligands. Strikingly, regardless of their kinase activity, JNKs’ scaffolding function limits TRIF oligomerization and hinders TLR-induced necroptosis (59).
The oligomerization and attachment of phosphorylated MLKL to the cell membrane during necroptosis is not equivalent of cell death. Indeed, similarly to pyroptosis, necroptosis-induced membrane disruptions can be repaired by the ESCRT III machinery, which was confirmed in immortalized macrophages and BMDMs (60, 61). This repair process is more effective in bone marrow-derived dendritic cells than in BMDMs, correlating with their different sensitivities to LPS- and caspase inhibitor-induced necroptosis (61). Surprisingly, once pMLKL translocates to the membrane, phosphatidylserine is externalized, a feature traditionally linked to apoptosis. This precedes the release of vesicles called necroptotic bodies (62). Noteworthy, MLKL protein, via its interaction with ESCRT III machinery, also facilitates endosomal vesicle trafficking independently of necroptotic stimuli (61) as well as plays a role in lipid handling (63) and phagolysosome fusion in macrophages (64).
Reactive oxygen species are firmly implicated in necroptosis across diverse cell types, including macrophages (45, 65–72). Accumulating evidence indicates that ROS act by oxidizing cysteine residues within RIPK1 and MLKL, consequently promoting protein aggregation and necrosome assembly (54, 68). Additionally, activated RIPK3 amplifies the generation of ROS (Figure 1), thereby establishing a positive feedback loop that favors necroptosis (67). TNF elicits RIPK3-dependent mitochondrial ROS generation, partially through the activation of the pyruvate dehydrogenase complex (PDC), which regulates oxidative respiration (66). Moreover, excessive TNF triggers robust glutaminolysis, succinate accumulation, and subsequent generation of mtROS via reverse electron transport (RET) at mitochondrial complex I (73). RET, along with NAD+ depletion, plays confirmed roles in TNF-, SMAC mimetic-, and caspase inhibitor-induced necroptosis in BMDMs (69, 74). The triggering receptor expressed on myeloid cells 1 (TREM-1) activation through mTOR-mediated signaling promotes Drp1-dependent mitochondrial fission, mitophagy, and necroptosis of alveolar macrophages during acute lung injury (75). Importantly, elevated TREM-1 expression on myeloid cells has been reported in certain tumors and correlated with worse prognosis (76).
2.3 Ferroptosis
Ferroptosis was first described in 2012 by Dixon et al. as iron-dependent cell death that could be triggered by small molecule erastin and inhibited by ferrostatin-1 (Fer-1) (77). Generally, this cell death type is associated with the accumulation of free iron (Fe2+), lipid peroxidation (LPO), ROS production, and changes in mitochondrial morphology. Furthermore, ferroptosis can be regulated by amino acid antitransporter system Xc-, glutathione peroxidase 4 (GPX4), or nuclear factor erythroid 2–related factor 2 (Nrf2) (78). It is believed that ferroptosis, contrary to earlier described pyroptosis and necroptosis, does not result in plasma membrane rupture, leaving it intact (79). Nevertheless, recent findings suggest that cell membrane rupture indeed takes place and is preceded by cell swelling (14, 80) as well as an increase in cytosolic level of Ca2+ (80). Interestingly, Riegman et al. showed that ferroptosis could propagate in vitro through the spreading of cell swelling in a lipid peroxide- and iron-dependent manner (14), thus characterizing additional unique features of this cell death pathway. Extensive reviews on either the mechanism of ferroptosis or its differences from other types of regulated cell death may be found elsewhere (78, 81, 82).
As for macrophages, their ferroptosis was first described in murine models of hemochromatosis (83) and transfusion (84). The first study revealed that ferric citrate overload led to ferroptosis induction in murine BMDMs in vitro. It was accompanied by the upregulation of Slc7a11 (a member of Xc- system) and increased levels of ROS together with nuclear Nrf2 (Figure 1). Moreover, Slc7a11 knock-out was associated with higher susceptibility to iron-induced cell death, which was also shown to be independent of autophagy (83). The latter research described ferroptosis occurring in red pulp macrophages performing elevated erythrophagocytosis. This phenomenon was accompanied by increased ROS and LPO levels in vivo – the effects which were aborted by the Fer-1 use in vitro (84).
Since ferroptosis is not the only cell death modality that macrophages can undergo, and elevated ROS and LPO levels can arise in different scenarios, it is important to distinguish ferroptosis-related events from others. Wiernicki et al. revealed that in murine BMDMs membrane lipid peroxidation level was incomparably higher during ferroptosis than in other types of programmed cell death, with the prevailing fraction of phosphatidylethanolamine peroxide species (85). Furthermore, lipid ROS increased prior to cell permeabilization, unlike in non-canonical pyroptosis, and could be prevented by administrating Fer-1 (85).
Ferroptosis is considered one of the inflammatory cell death types (86), however, the knowledge on whether its occurrence in immune cells, macrophages in particular, affects the production of inflammatory mediators and downstream signaling pathways is still scarce. A study conducted by Xia et al. shed light on this topic as it demonstrated that murine MHV-A549-infected ferroptotic macrophages secreted IL-6, whereas the level of this cytokine significantly decreased after Fer-1 treatment (87). This data suggests that ferroptosis may enhance the inflammatory response of macrophages, however, further research is needed to validate the results.
2.4 The differential susceptibility of M1 and M2 macrophages to necroptosis and ferroptosis
The basal expression levels of cIAP family proteins, along with other components of cell death pathways, are closely associated with the variable susceptibility of macrophages to cell death during their differentiation process (88), and polarization state (89–91). The degradation of cIAPs prompted by the administration of SMAC mimetics results in either RIPK1-dependent apoptosis or necroptosis, when caspase-8 is concurrently inhibited (92, 93). M1-like murine and human macrophages treated with SM show increased susceptibility to necroptosis compared to their M2-like counterparts (89, 90). This vulnerability is attributed to classical inducers of the M1 subset, namely IFNγ and LPS treatment, which in BMDMs upregulate MLKL, Z-DNA/RNA binding protein 1 (ZBP1), and RIPK3 – the key molecules involved in necroptosis (89). In human macrophages, both type I and type II IFNs upregulate MLKL while downregulating miR324-5p, a direct regulator of MLKL expression, rendering them more susceptible to various necroptosis inducers (94). During influenza virus (IAV) infection, diminished miR324-5p expression in hMDMs contributes to controlling viral spread (94). MLKL expression is maintained at high levels by IFN signaling to ensure swift necroptosis in both BMDMs and hMDMs (Figure 1), leading to its classification as a constitutive IFN-regulated effector of necroptosis (CIREN) (94, 95). These pro-necroptotic effects of IFNs have implications in autoimmunity (95) and murine models of sterile sepsis induced by TNF (48). Furthermore, IFNβ suppresses IL-4-induced M2 polarization of BMDMs by controlling α-ketoglutarate/succinate ratio (96). However, there is also evidence that type I IFNs promote M2 polarization (97), and the resolution of bacterial inflammation by enhancing efferocytosis and reprogramming of macrophages to a pro-resolving phenotype (98).
Intensive macrophage cell death is also observed as a result of simultaneous caspase and TGF-activated kinase 1 (TAK1) inhibition, which prevents the phosphorylation of RIPK1 (46, 91, 99). Notably, this treatment selectively triggers necroptosis in M2 macrophages, while M1 macrophages, which rely on alternative survival signals (AURKA and GSK3β), are largely resistant (91).
In the case of ferroptosis, interesting results were provided by two different studies characterizing the capacity of both primary human and murine macrophages to handle iron. They revealed that M2-polarized macrophages displayed a higher rate of iron internalization and release when compared to M1 cells (100, 101). Additionally, Agoro et al. demonstrated that iron overload in vitro promoted M2 phenotype and decreased the LPS-induced pro-inflammatory response of M1 macrophages (102). These findings suggested that the activation state of macrophages determined their susceptibility to ferroptosis, which was later proved in mice (103, 104). Indeed, in a study performed by Kapralov et al. both inflammatory macrophages and microglia showed higher resistance to ferroptosis inducer RSL3 compared to non-polarized (M0) or M2 cells under both in vitro and in vivo conditions. Mechanistically, M1 cells had a higher content of inducible nitric oxide synthase and increased production of NO•, which could inhibit 15-lipoxygenase – an enzyme capable of producing lipid peroxides (Figure 1). Additionally, NO• could also replace GPX4 in the cellular anti-ferroptotic machinery (103). Interestingly, 15-lipoxygenase plays a reverse function in the absence of ferroptosis inducer and is associated with the generation of pro-resolving lipid mediator RvD1 in satiated macrophages (105). In another study, Piattini et al. demonstrated that GPX4 loss in M2 cells led to their ferroptosis, while GPX4-deficient inflammatory macrophages maintained their functions and cell survival. Although NO• supplementation alleviated ferroptosis in the M2 subset, iNOS inhibition in M1 cells could not induce lipid peroxidation. Thus, these results suggest that some other antioxidant mechanisms were responsible for controlling ROS levels in GPX4-deficient inflammatory macrophages (104). Noteworthy, the increased activity of heme oxygenase 1 (HO-1) might serve as another factor affecting M2 macrophage susceptibility to ferroptosis due to its potential to release Fe2+ from heme (Figure 1), as was implied by Li et al. (106). Contrarily, Fuhrmann et al. demonstrated that a hypoxic environment prevented hMDMs from RSL3-induced ferroptosis via decreasing nuclear receptor coactivator 4 (NCOA4)-dependent ferritinophagy and increasing mitochondrial ferritin level (107). Although the polarization state of macrophages was not assessed, hypoxia is known to promote the M2 phenotype (108). Therefore, it indicates that different M2-polarizing stimuli may have a distinct impact on the ferroptosis pathway, or that there is a varying connection between phenotype and susceptibility to ferroptosis in murine and human cells.
3 Cross-interactions among pyroptosis, necroptosis, and ferroptosis in macrophages
One of the earliest observations on pyroptosis-necroptosis crosstalk was that the release of the mature form of IL-1β can derive from ripoptosome and RIPK3-mediated NLRP3 activation (109, 110). Essential for this process seemed to be MLKL, which once activated was able to create pores in the cell membrane. This led to potassium efflux, thereby triggering the activation of inflammasome and IL-1β processing (Figure 1). Moreover, the subsequent IL-1β release seemed to be GSDMD-independent (111). Now, the evidence supporting these observations in macrophages is still growing (112–115). For example, Polykratis et al. have recently shown that RIPK3-MLKL-mediated necroptosis in macrophages was also accompanied by NLRP3 activation and IL-1β secretion (115). Interestingly, all these findings indicate the contribution of necroptosis signaling in pyroptosis. A reverse scenario has been recently described (116), in which disrupted mitochondrial homeostasis and mtROS enabled N-GSDMD to switch to a mitochondrial initiator of necroptosis (Figure 1). Moreover, Feng et al. challenged the classical model stating that cell death accompanying inflammasome activation should always be pyroptosis. They demonstrated that the bacteriolysis of Staphylococcus aureus USA300 strain in infected macrophages induces AIM2-mediated necroptosis (117).
At the crossroads of apoptosis, necroptosis, and pyroptosis, the pivotal regulator, caspase-8, orchestrates both necrotic cell death and the induction of pro-inflammatory gene expression, including NLRP3 (118–121). Upon activation of death receptors, TLR3, TLR4 as well as intracellular nucleic acid sensors RIG-I and ZBP1 (122, 123), caspase-8 is recruited to intracellular signaling complexes. Its functional role depends on the interplay with other partners, and even subtle alterations in the composition of caspase-8 complexes can give rise to contradicting physiological outcomes, such as the switching between macrophage death modalities or between cell death and cell activation (119). Notably, in the absence of an X‐linked inhibitor of apoptosis (XIAP), extrinsic caspase-8 promotes pyroptotic GSDMD processing, resulting in the demise of macrophages lacking both inflammasome and apoptosis signaling components (caspase-1, -3, -7, -11 and BID) (124). In addition to RIPK1, caspase-8 can cleave RIPK3 (125). Primed caspase-8-deficient macrophages exhibit elevated IL-1β production due to unrestricted activity of RIPK3 (126). Furthermore, caspase-8 can assemble an ASC-caspase-1 complex, culminating in caspase-1-dependent cell death and the release of IL-1β and IL-18 (119, 120).
Emerging evidence indicates that macrophages exhibit a flexible response to stimuli, leading to the induction of various cell death pathways (127). It has already been demonstrated that Salmonella Typhimurium infection in macrophages can induce pyroptosis (128) and necroptosis (129). Moreover, such evidence has recently been shown for human respiratory syncytial virus (RSV) (72). Bedient et al. revealed that macrophages infected with RSV activated NLRP3-ASC inflammasome, caspase-1, as well as RIPK3-MLKL. Intriguingly, ASC/NLRP3 deficiency in macrophages led to a substantial loss (72-75%) of lytic cell death, while inhibition of either caspase-1 or RIPK3 individually resulted in 46% and 54% loss, respectively (72). Considering the association of RIPK3-MLKL with NLRP3 activation, it appears that ASC-NLRP3 inflammasome contributes to both pyroptosis and necroptosis in RSV-infected macrophages. Furthermore, both types of lytic cell death may promote the amplification of lung inflammation during RSV infection (72).
Some supporting evidence showing that, indeed, the simultaneous induction of pyroptotic and necroptotic pathways in macrophages can drive inflammation has recently arisen in case of sepsis. Chen et al. demonstrated that mice with double depletion of RIPK3/GSDMD or MLKL/GSDMD exhibited higher protection against septic shock and multi-organ injury (130). A bone marrow transplantation from Ripk3-/-Gsdmd-/- to WT mice enhanced the protection against sepsis in such animals, suggesting that both myeloid and non-myeloid cells contributed to the disease progression. In vitro experiments from the same study revealed that macrophages lacking RIPK3 and GSDMD were resistant to both pyroptosis and necroptosis as well as cytokine production and release (130). All these findings suggest that pyroptotic macrophages together with necroptotic ones may contribute to inflammation and sepsis progression. However, it remains unclear whether these cell death pathways mutually influenced each other or if they were simply simultaneously activated without further crosstalk. Moreover, the possible occurrence of pyroptosis and necroptosis in other immune cells and their subsequent impact on sepsis progression cannot be excluded.
The concurrent occurrence of macrophage pyroptosis and necroptosis, along with its impact on exacerbating tissue damage in sepsis, can also be implied from the later studies by Russo et al. They focused on galactin-1, which increased levels can be found in sera from human patients with sepsis (131). The authors showed that galactin-1 is released by both pyroptotic and necroptotic macrophages, which is associated with LPS-induced lethality (131). Thus, targeting both lytic cell death types in macrophages may constitute a novel promising therapy to alleviate septic conditions. Interestingly, there are findings which show that Bcl-2, as well as necrosulfonamide (NSA), can attenuate both pyroptosis and necroptosis in macrophages (132, 133).
The relationship between ferroptosis and pyroptosis or necroptosis is not yet fully established. A study conducted by Kang et al. on the sepsis model demonstrated that GPX4 knock-out in macrophages, followed by increased lipid peroxidation, resulted in GSDMD cleavage mediated by both caspase-1 and caspase-11 (134). Since GPX4 is an antioxidant defense enzyme, its ablation could make macrophages more susceptible to ferroptosis and subsequent ROS generation could also activate inflammasome leading to pyroptosis. Thus, GPX4 knock-out can exacerbate the development of sepsis by possibly activating both cell death pathways. To further test this hypothesis, the expression levels of other ferroptosis markers (e.g. SLC7A11) as well as mitochondria morphology in GPX4-deficient macrophages should be assessed. Nuclear translocation of Nrf2, which is another regulator of ferroptosis, is downregulated in Salmonella Typhimurium-infected macrophages via IFN-1/RIPK3/PGAM5 axis (Figure 1). This leads to ROS-mediated mitochondrial dysfunction which can be a sign of cells undergoing ferroptosis (129). Interestingly, these events occur simultaneously with the induction of the classic necroptotic pathway, suggesting that both cell death modalities can be activated in infected macrophages. Mycobacterium tuberculosis, an intracellular pathogen known to cause macrophages to undergo necroptosis (135), can also prompt them to exhibit hallmarks of ferroptosis, that can be abolished by Fer-1 (136). While it is still not clear whether ferroptosis and necroptosis take place concurrently in the infected macrophages, ferroptosis might potentiate other cell death modalities to support the release of bacteria.
In this section, we explored the crosstalk between PCDs in macrophages. Interestingly, there is a clear involvement of pyro-, necro-, and ferroptosis in intracellular bacterial and viral infections as well as sepsis. In these settings, the initiation of multiple death programs seems to intensify the release of pathogens which can support disease progression. On the other hand, the inhibition of macrophage death can hamper these processes. Therefore, research into the molecules able to simultaneously inhibit lytic PCDs seems to be a worthwhile pursuit. At the same time, more extensive studies are required to discern the role of dying macrophages in diverse infectious conditions, beyond those mentioned above. In the following sections of this review, we will focus on discussing macrophage death within the context of more investigated illnesses – namely atherosclerosis and cancer.
4 Cell Death in atherosclerotic plaques: pyroptosis, necroptosis, and ferroptosis in macrophages
Atherosclerosis, the most prevalent form of cardiovascular disease, is primarily characterized by persistent low-grade inflammation as well as the gradual buildup of lipids in the intimal space of medium and large arteries (137). Among diverse cell types, macrophages affect the pathological progression of atherosclerosis, exerting both favorable and detrimental functions. They participate in the uptake of proatherogenic, oxidized low-density lipoproteins (oxLDLs), thus transforming into foam cells, which facilitate the development of atherosclerotic plaques (138, 139). In the early stages of the disease, this internalization may represent a beneficial adaptive mechanism of macrophages to manage lipid deposits. However, as the plaque advances, the regulation of cellular turnover becomes disrupted (140), and foam cells undergo both apoptotic and nonapoptotic forms of cell death. When inadequately cleared (141), these cells contribute to the formation of an acellular necrotic core (NC), fostering instability of advanced lesions. The destabilization of plaques is undesirable, as it poses the risk of atheroma rupture, bleeding, and thrombosis. For a long time, the necrotic debris within lesions was attributed solely to the passive lysis of uncleared apoptotic cells. However, research has revealed the involvement of various other forms of PCD in the formation of necrotic core and plaque destabilization (139, 142). Once developed NC advances along with the disease progression and its size correlates with the instability of the plaque (143). Thus, the occurrence of PCDs may influence disease outcomes. Here, we explore how three cellular death modalities in macrophages – pyroptosis, necroptosis, and ferroptosis – affect atherosclerosis progression. We discuss the occurrence of lytic PCDs in human plaques as well as possible triggers and modulators of these pathways in the atherosclerotic milieu. Finally, we analyze macrophage cell death in the context of its subpopulations observed in lesions.
4.1 Evidence for pyroptosis, necroptosis, and ferroptosis activation in macrophages within human atherosclerotic plaques
4.1.1 Pyroptosis: a flame in atherosclerosis
Epidemiological studies reveal the presence of pyroptotic markers in human carotid atherosclerotic plaques (144–146) with NLRP3 emerging as a pivotal component implicated in atherosclerosis progression. Zheng et al. demonstrated high aortic expression of NLRP3 in patients with coronary atherosclerosis, which correlated with both disease severity and atherosclerotic risk factors such as smoking, hypertension, diabetes mellitus, and dyslipidemia (146). Subsequently, Shi et al. confirmed strong expression of NLRP3, ASC, caspase-1, IL-1β, and IL-18 in unstable atherosclerotic plaques, as opposed to stable ones (147). Notably, the NLRP3 inflammasomes were primarily localized in macrophages and foam cells (148) and associated with cholesterol crystals inside and outside these cells (147). The formation of cholesterol crystals is considered a significant factor contributing to atherosclerotic plaque expansion and rupture (149). They are abundantly present within foam cells and have already been shown to trigger pyroptosis in macrophages (150).
Similarly, activated caspase-1, together with IL-1β and IL-18 were identified in human advanced atherosclerotic plaques as opposed to non-atherosclerotic vessels and early-stage disease lesions. It was also demonstrated that cleaved caspase-1 co-localized with macrophages around the NC, while cleavage of apoptotic caspases was not observed (148, 151). Furthermore, Puylavert et al. showed that NT-GSDMD was localized in macrophage- and VSMC-rich regions of the plaques (152). However, this study did not include a comparison with plaque-free arteries, nor did it establish a correlation with the stage of the disease (152). Immunofluorescence and single-cell RNA sequencing analyses revealed that atherosclerotic macrophages localized in the NC of plaque exhibited higher expression of GSDME (145). This elevated expression was accompanied by increased levels of IL-1β and caspase-3 as compared to the non-necrotic areas within the same plaque (145).
4.1.2 Necroptosis: unveiling molecular triggers
The necroptotic pathway is also known to be activated in human atherosclerotic plaques, especially in macrophages, and is associated with the formation of NCs. For instance, by a comprehensive gene expression analysis Karunakaran et al. identified elevated levels of RIPK3 and MLKL mRNAs in human atheroma compared to disease-free control arteries (71). A notable increase was observed in symptomatic plaques in comparison to asymptomatic individuals (71). Moreover, MLKL phosphorylation was confirmed in clinical atherosclerotic samples, specifically in close proximity to the NC in advanced plaques, but not in early lesions (71). The upstream molecule, RIPK1, was highly expressed in human macrophages, whereas exhibited a low presence in vascular smooth muscle cells-rich regions in human carotid atheromas (153). Another study reported increased levels of the RIPK1-RIPK3 complex in atherosclerotic plaques with NCs (144). However, contradictory gene expression data indicated higher levels of RIPK1 in early atherosclerotic lesions than in regions with more severe plaques (154).
4.1.3 Ferroptosis: connecting dots
The direct evidence of ferroptosis induction within human atherosclerotic plaques remains limited. The involvement of ferroptosis in atherosclerosis can be inferred indirectly, as indicated by the accumulation of ROS, lipid peroxidation in macrophages, atheroma hemorrhage, and iron deposition, all of which are notable features of advanced atherosclerotic plaque (139). However, a recent analysis of single-cell RNA sequencing data from human atheromas revealed that macrophages associated with symptomatic plaques upregulated FTH1 and FTL genes encoding heavy and light chains of ferritin, respectively (155). These results align with the previous data on increased colocalization of iron, ferritin accumulation, and macrophage hemoglobin scavenger receptor (CD163) or hepcidin in advanced human plaques (156, 157). Collectively, these results indicate that there is a subset of macrophages in human atheromas that retain bound iron which should decrease their likelihood of undergoing ferroptosis. In this research topic, Sarad et al. reported a subtype-specific expression of core ferroptosis genes (e.g. Cp, Hells, Slc40a1) in inflammatory versus tissue-resident macrophages (158). This observation suggests a link between ferroptosis and inflammatory microenvironment appearing at a very early stage of atherogenesis. Their findings indicate that Nrf2 deficiency in aortic macrophages leads to subtype-specific transcriptomic changes associated with cell death pathways (158).
4.2 Triggers and modulators of macrophage pyroptosis, necroptosis, and ferroptosis within atherosclerotic milieu
A literature analysis revealed that the inflammatory forms of LDLs, particularly oxLDLs, serve as triggers for all three types of death in macrophages. They also modify cell susceptibility to other stimuli. These observations imply a nuanced interplay among pyroptotic, necroptotic, and ferroptotic pathways in the context of atherosclerosis progression. However, the majority of findings in this field originate from in vitro studies employing diverse macrophage types, including cell lines. Additionally, these studies often involve various forms of oxLDLs with imprecisely controlled compositions. Furthermore, such investigations frequently focus on the pathway associated with only one of the three cell death modalities. In the next section, we take a closer look at the role of oxLDLs along with other molecules, conditions, and processes implicated in the induction and regulation of macrophage death within the context of atherosclerosis. The existing knowledge is also organized and summarized in Table 1.
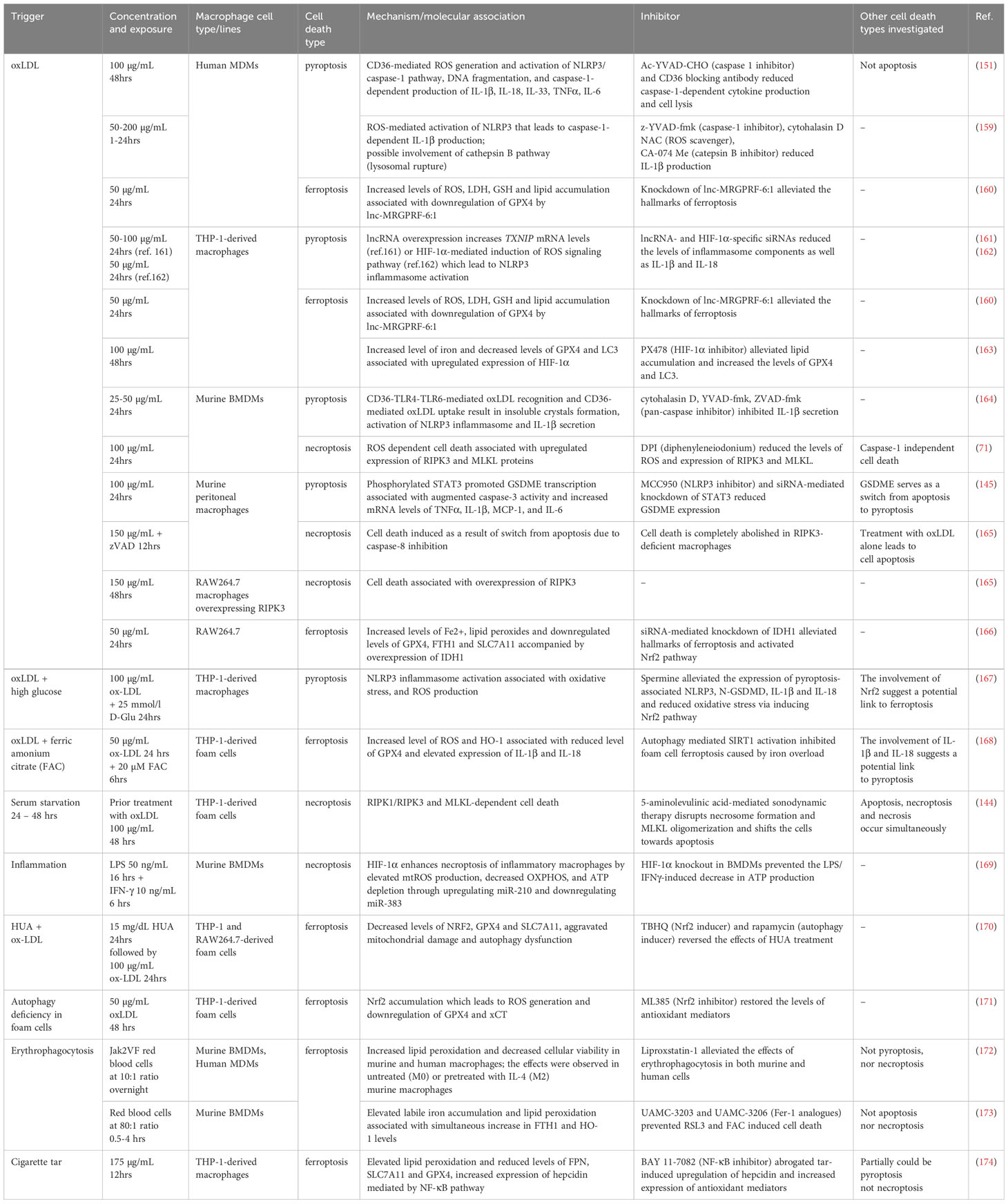
Table 1 Lytic PCDs in macrophages in the context of atherosclerosis: inducers and mechanisms.
4.2.1 OxLDL-induced pyroptosis in macrophages
It has been demonstrated that in hMDMs, oxLDL induced the expression of NLRP3, caspase-1, and ASC, leading to caspase-1 activation, macrophage lysis, and the release of IL-1β and IL-18 (151, 159). It was later proven that oxLDL-induced pyroptosis in hMDMs relied on CD36 and was associated with robust ROS generation and NLRP3/caspase-1 pathway activation. Noteworthy, although apoptotic caspases (caspase-3, -6, -8, and -9) were also activated in oxLDL-treated cells, only the use of caspase-1 inhibitor led to a significant suppression of cell lysis (151). In contrast, Nogieć et al. found neither the release of IL-1β nor an increased accumulation of its precursor form in non-stimulated human monocyte-derived foam cells (hMDFCs) treated with oxLDL (175). This discrepancy is likely due to a time- and/or dose-dependent issue, as the treatment involved a relatively low concentration of oxLDLs for an extended incubation period, which should mimic in vivo conditions more accurately. Moreover, these cells tended to follow a necrotic pathway distinct from pyroptosis and did so more promptly than short-term-induced foam cells (175). Noteworthy, oxLDL is a known inducer of miR-155 expression (176, 177), which correlates with atherosclerosis (178). Given that hMDFCs, obtained after prolonged exposure to oxidized LDL, respond to inflammasome activation preferentially with pyroptosis combined with other types of necrotic death (175), it is plausible to think that miR-155 effector mechanism lies in switching between cell death pathways, regulating the immunogenic cell death burden.
Oxidized LDL may contribute to pyroptosis in macrophages also through the upregulation of the expression of GSDME (145). Interestingly, GSDME augments caspase-3 activity, which shifts the mode of macrophage cell death from apoptosis to pyroptosis, thereby promoting the progression of atherosclerosis. Moreover, GSDME deficiency significantly decreases serum levels of inflammatory factors, such as IL-1β, TNFα, and monocyte chemoattractant protein-1 (MCP-1), resulting in the suppression of both inflammation and disease development (145).
Recently, it was described that long non-coding RNAs (lncRNAs) can be involved in the regulation of oxLDL-induced pyroptosis (161, 162). Indeed, both linc00657 (161) and AC078850.1 (162) potentiated pyroptosis in THP-1-derived macrophages. Interestingly, the latter molecule acted (161) in conjunction with hypoxia-inducible factor 1-alpha (HIF-1α) to promote NLRP3-mediated pyroptosis and foam cell formation in atherosclerosis cases (162).
As mentioned earlier, CD36 has been documented to play a role in oxLDL-mediated macrophage pyroptosis (151). CD36 is a scavenger receptor that cooperates with the TLRs heterodimer TLR4-TLR6 in the recognition of oxLDLs as well as mediates its uptake from the surrounding environment. It appears to function as a dual signal in NLRP3 inflammasome-mediated macrophage pyroptosis activation since macrophages from transgenic mice with CD36 knock-out exhibited reduced levels of Il1a, Il1b and Nlrp3 mRNA, as well as lower IL-1β serum level. Furthermore, such animals were protected from atherosclerosis (164).
4.2.2 Hyperglycemia-induced pyroptosis in macrophages
Qiu et al. generated interesting results related to pyroptotic pathway inhibition for atherosclerosis treatment (167). They focused on spermine, a natural cellular metabolite that exerted protective properties against macrophage pyroptosis induced by hyperglycemia and oxLDL via activation of Nrf2 signaling pathway and the inhibition of NLRP3 inflammasome. Furthermore, spermine significantly reduced the generation of ROS, and release of IL-1β and LDH (167). Nie et al. found that under diabetic conditions, the expression of GSDMD and caspase-1 in macrophages was increased, and the recognition of cytoplasmic double-stranded DNA by AIM2 inflammasome might play an important role in initiating pyroptosis in macrophages under hyperglycemia (179). While these studies may not have focused directly on atherosclerosis, they are still worth mentioning, since diabetes promotes pro-inflammatory gene expression in macrophages and contributes to atherosclerotic lesions development (137, 180).
4.2.3 OxLDL-induced necroptosis in macrophages: beyond caspase inhibition
Caspase inhibition in peritoneal murine macrophages was shown to switch oxLDL-induced apoptosis to necroptosis, with complete abolition of necrotic cell death in RIPK3-deficient murine peritoneal macrophages (165). Furthermore, oxLDLs alone could induce cell death in the RAW264.7 macrophage cell line overexpressing RIPK3. Additionally, in vivo studies revealed increased RIPK3 levels in atherosclerotic macrophages during lesion development and the degree of necroptosis was found to correlate with the level of RIPK3 deletion in tested animals (165). Importantly, Karunakaran et al. reported that in BMDMs, atherogenic forms of LDL increased both RIPK3 and MLKL expression through direct activation of their promoters, confirming that oxLDLs could induce necroptotic cell death even without synthetic caspase inhibitors (71). Overall, these findings suggest that oxLDLs can independently lead macrophages to undergo necroptosis when the expression of RIPK3 in atherosclerotic plaques reaches a certain threshold. Interestingly, the induction of necroptosis by oxLDLs was independent of inflammasome activation, as cells deficient in caspase-1 or treated with caspase-1 inhibitors still underwent necroptotic cell death in response to oxLDL. Moreover, they did so to the same extent as wild-type or untreated cells (71).
4.2.4 Impact of hypoxia, nutrient deficiency, and HDL intervention on macrophage necroptosis
Hypoxic and nutrient-deficient conditions in atherosclerotic plaque can contribute to its growth, instability, and eventual rupture. For instance, serum starvation could induce necrotic types of cell death in THP-1-derived foam cells. Although apoptosis, necroptosis, and necrosis were observed under insufficient nutrient supply, results following pre-treatment with necrostatin-1 suggested that nearly half of the deceased cells were attributed to necroptosis (144). Furthermore, researchers demonstrated a significant increase in RIPK1 and RIPK3 protein levels, RIPK1-RIPK3 complexes, and MLKL oligomerization (144). The interplay of hypoxia, macrophages, and atherosclerosis was assessed by Karshovska et al. (169). They showed that the additional deletion of HIF-1α in myeloid cells from ApoE-deficient mice reduced atherosclerosis and NC formation by limiting macrophage necroptosis. In vitro experiments on inflammatory HIF-1α-deficient BMDMs from the same study confirmed increased oxidative phosphorylation and ATP levels, alongside reduced ROS production and necroptosis (169).
Recently, it has been elucidated that anti-atherogenic high-density lipoproteins (HDLs) may attenuate NC development in atherosclerotic plaques. Kluck et al. demonstrated that the HDL treatment of both THP-1 and murine peritoneal macrophages suppressed necroptosis induced by TNF or oxLDL in the presence of the pan-caspases inhibitor. This protective effect was contingent on the HDL receptor SR-B1 and activation of Akt kinase (181).
4.2.5 OxLDL-induced ferroptosis in macrophages
A growing body of evidence suggests that oxLDLs can also trigger ferroptosis in macrophages. Li et al. demonstrated that oxLDL treatment in RAW264.7 macrophages increased Fe2+ levels, induced lipid peroxidation, and downregulated the protein levels of GPX4, FTH1, and SLC7A11 (166). Foam cells were also characterized by overexpression of isocitrate dehydrogenase (IDH1), whereas the inhibition of IDH1 activated Nrf2 pathway, thereby reducing macrophage ferroptosis and foam cell development (166). Interestingly, a high level of uric acid (HUA) appeared to contribute to the oxLDL-induced foam cell formation from THP-1 and RAW264.7 cells in a similar manner as IDH1 (170). Namely, HUA inhibited Nrf2 signaling pathway and promoted ferroptosis. It also impaired mitochondrial function and autophagy in macrophage-derived foam cells. These effects could be restored by ferroptosis inhibitor Fer-1 (170).
4.2.6 Regulating ferroptosis in macrophages in the context of atherosclerosis
Significantly, IL-37 can mitigate ferroptosis-associated oxidative stress and Nrf2 inhibition. IL-37 enhances cellular viability and promotes the nuclear translocation of Nrf2 in THP-1 macrophages treated with oxLDLs (182). Long non-coding RNA lnc-MRGPRF-6:1 is another type of molecule that can contribute to foam cell ferroptosis. It appears that lnc-MRGPRF-6:1 may promote ferroptosis in THP-1 and hMDMs by suppressing GPX4 (160). Nevertheless, it remains to be elucidated whether this molecule only inhibits GPX4 mRNA or if it also downregulates the expression of Nrf2 located upstream.
Nrf2 is typically regarded as a master regulator of cellular antioxidant defense (183). However, recent findings by Peng et al. have shown that in the late stages of atherosclerosis, autophagy deficiency can lead to Nrf2 accumulation in THP-1 foam cells, resulting in the generation of ROS and the induction of ferroptosis (171). These data suggest that, in specific circumstances, Nrf2 may negatively influence overall cell survival and promote disease progression.
HIF-1α is a well-known transcription factor recognized as an essential mediator of oxygen homeostasis (184). HIF-1α is upregulated in human atherosclerotic macrophages and its inhibition alleviates lipid accumulation in oxLDL-induced THP-1-derived macrophages in vitro by suppressing ferroptosis and activating autophagy (163). It would be of interest to assess whether the Nrf2/HIF-1α ratio in macrophages changes as a result of ferroptosis occurrence and plaque formation, and if a higher proportion could serve as a prognostic factor for patients.
Remarkably, ferroptosis is not limited to foam cells. It also occurs in M0 and M2 macrophages following the erythrophagocytosis of Jak2VF red blood cells, as demonstrated by Liu et al. (172). The Jak2VF mutation is found in human patients with atherosclerosis, whereas in murine models is linked to accelerated disease due to increased ferroptosis of macrophages (172). Cigarette tar is another factor that promotes the development of atherosclerotic lesions and is associated with the upregulation of hepcidin in plaque macrophages. In vitro studies on THP-1 have shown that tar induced ferroptosis via NF-κB-activated hepcidin/FPN/SLC7A11 pathway. It has been further observed that disrupting this signaling either pharmacologically or genetically can alleviate the progression of atherosclerosis in vivo (174).
4.3 Pyroptosis, necroptosis, and ferroptosis in the context of macrophage subsets in atherosclerosis
The incontrovertible involvement of macrophages in atherosclerosis development is acknowledged. However, the temporal changes that heterogenic populations of macrophages undergo functionally within the dynamic and complex milieu of atherosclerotic plaques are less recognized (185). The spatiotemporal distribution, phenotypic diversity, and death of macrophages within lesions assume a crucial role in shaping the microenvironment and influencing the fate of atheromas. As the macrophage heterogeneity in atherosclerotic plaque has been extensively reviewed elsewhere (186), we aim to discuss them in the context of necrotic PCDs occurring within the atherosclerotic microenvironment.
4.3.1 M1 and M2 polarization and cell fate in atherosclerosis
M1 macrophages typically dominate in the rupture-prone shoulder regions of the plaque and adjacent to the NC (187). The buildup of lipids not only leads to foam cell formation but also fosters M1-like polarization of the cells and establishes a mutually reinforcing relationship with them, creating a self-sustaining loop. Much like M1 macrophages, foam cells are commonly situated in close proximity to the NC. In contrast, M2 macrophages, recognized for their anti-inflammatory and repair functions, primarily reside in more stable regions of plaques, near newly formed blood vessels, and in areas of ongoing repair (187). Hence, managing the balance between pro-inflammatory M1 and anti-inflammatory M2 macrophages through repolarization or targeted depletion may offer significant therapeutic advantages.
Although direct evidence is currently lacking, it is plausible that M1 macrophages may demonstrate heightened sensitivity to pyroptosis. Specifically, the activation of the inflammasomes and the subsequent release of pro-inflammatory cytokines, which are distinctive features of pyroptosis, might align with the functional pro-inflammatory characteristics of M1 macrophages.
The data obtained by Hao et al. and Ali et al. implied that under conditions of inflammation and stress, M1 macrophages may be predisposed toward necroptosis (89, 90). Given the persistent inflammatory milieu characteristic of atherosclerosis, it is conceivable that cells undergoing necroptosis manifest a pro-inflammatory phenotype. This hypothesis was substantiated to some extent in a study conducted by Karshovska et al., wherein the heightened susceptibility of inflammatory macrophages to necroptosis was demonstrated to be dependent on HIF-1α-mediated metabolic reprogramming toward an inflammatory state (169).
Although the differential susceptibility of macrophages to ferroptosis is not yet elucidated in the context of atherosclerosis, the general viewpoint is that murine M1 macrophages, in comparison with M0 and M2, exhibit increased resistance to pharmacologically-induced ferroptosis (103). This resistance seems to hinge on elevated NO production and the secure sequestration of iron through ferritin within M1 macrophages (100). On the other hand, alternatively activated macrophages appear to be more prone to ferroptosis in the absence of GPX4, indicating a diminished antioxidant capacity in these cells (104). Moreover, iron overload, while seemingly ineffectual on macrophages, leads to ferroptosis upon their exposure to oxLDLs (168). The demise of M2 cells from the atherosclerotic plaque could potentially worsen disease progression by downregulating the processes associated with these macrophages such as secretion of anti-inflammatory cytokines, efferocytosis of cellular debris, or tissue repair (188).
The lack of data on macrophage polarization in the context of cellular death in atherosclerotic milieu renders it challenging to anticipate which subpopulations of macrophages are sensitive to either of the lytic PCDs and how it could be leveraged for therapeutic purposes. This intricacy is further compounded by the ability of oxidized phospholipids to induce phenotypic switching in both murine M1 and M2 macrophages, giving rise to the so-called Mox phenotype (189). The existence of macrophages associated with high levels of iron adds another level of complexity to this issue.
4.3.2 Mox and hemoglobin-associated phenotypes: antioxidant defense, potential ferroptosis resistance, and iron homeostasis
Mox macrophages, described by Kadl et al., were approximated to constitute one-third of total macrophages present in mouse atherosclerotic plaques (189). Mox phenotype is distinguished by Nrf2-mediated upregulation of the expression of antioxidant enzymes, including HO-1, thioredoxin reductase 1, and sulfiredoxin-1 (189). Although their resilience against ferroptosis remains unexplored, it is plausible that the Mox subset may confer increased resistance to this particular form of PCD. Interestingly, in vitro experiments involving the exposure of macrophages to oxidized phospholipids resulted in elevated levels of HO-1, ferritin, and hepcidin (190), suggesting that a microenvironment rich in oxLDL could promote iron retention in macrophages, preventing them from undergoing ferroptosis.
Oxidative stress within macrophages can be induced by various compounds present in atherosclerotic plaques such as hemoglobin found at sites of intraplaque hemorrhage. Boyle et al. identified a hemorrhage-associated subpopulation of macrophages, termed Mhem, characterized by high hemoglobin content (191). Correspondingly, a macrophage subset named M(Hb) was identified for its ability to engulf hemoglobin-haptoglobin complexes (192). Both Mhem and M(Hb) macrophages are predominantly located around human hematomas and are notably absent in stable and hemorrhage-free plaques. They exhibit elevated expression levels of CD163 and Nrf2-dependent HO-1 (191, 192). The characteristic iron retention by these subsets can be attributed to TLR-mediated signaling that induces hepcidin expression, an effect similar to one achieved by LPS stimulation (193, 194). Due to the upregulation of cholesterol efflux proteins and resistance to foam cell transformation, Mhem and M(Hb) macrophages are considered atheroprotective. Additionally, as a result of increased expression of ferroportin, they have low intracellular levels of iron and ROS (192). These features can be a sign of adaptation to the atherosclerotic microenvironment and may protect them against cell death. However, they can contribute to elevated iron levels in the plaque, the subsequent accumulation of which in other macrophage populations activates pro-inflammatory M1 phenotype and promotes the demise of cells possibly via ferroptosis (100). Interestingly, Mhem and M(Hb) murine counterparts have not been described yet. However, recent studies on murine models have shown that erythrophagocytosis by macrophages induces non-canonical ferroptosis that manifests by increased HO-1 expression and contributes to plaque destabilization (173).
4.4 Section summary
Here, we have discussed the involvement of three modes of macrophage death in atherosclerosis development and progression. Noteworthy, recent single-cell studies in human atherosclerotic plaques confirmed the presence of two inflammatory subsets characterized by the expression of inflammasome components, IL-1β, and TNF (195). Additionally, a macrophage cluster with unique upregulation of genes responsible for iron storage and metabolism was identified (155), further linking the atherosclerotic environment to specific types of cell death. However, there is currently a lack of spatiotemporal analysis regarding the respective contributions of intraplaque pyroptosis, necroptosis, and ferroptosis to atherogenesis. Neverthless, targeting lytic PCDs within the macrophages may hold promise for alleviating the disease. In this regard, the efficacy has recently been demonstrated for baicalin – a natural flavonoid isolated from medicinal herbs (68, 196), and dimethyl fumarate – an anti-inflammatory agent approved for the treatment of relapsing-remitting multiple sclerosis (69, 197). However, further studies are necessary to validate whether the results obtained in vitro or in murine models can be translated to the human body. Moreover, extensive research is needed to determine which signaling pathway should be inhibited at various stages of atherosclerotic plaque and NC development. An alternative approach might involve shifting macrophages undergoing regulated necrotic cell death toward regulated non-necrotic death modalities, e.g. apoptosis and autophagy. Another avenue involves enhancing the clearance of necrotic cells through the modulation of macrophage metabolism, potentially utilizing agents such as resolvins (198). Nonetheless, there is a long road ahead before any of these concepts can be developed into therapies. Even then, targeting macrophage death to resolve inflammatory microenvironment should come along with dietary changes and a reduction in the intake of LDLs, which while oxidized aggravate atherosclerosis and serve as potent inducers of macrophage necrotic PCDs.
5 Tumor microenvironment: pyroptosis, necroptosis, and ferroptosis in macrophages
5.1 The impact of macrophages and pro-inflammatory cell death on tumor microenvironment
Macrophages constitute a highly prevalent type of leukocytes within the tumor microenvironment (TME) that are able to survive in regions with low oxygen levels, known as hypoxic areas (199). Such tumor-associated macrophages (TAMs) are also regarded to follow the paradigm of M1- and M2-like phenotypes (200, 201). In general, due to their pro-inflammatory characteristics, M1-like macrophages are typically identified as tumor-suppressing and associated with extended overall patient survival (200). For instance, they can accomplish this by recruiting CD8+ T and NK cells as well as secreting cytokines, which further enhance anti-cancer immune response (201). On the contrary, anti-inflammatory M2-like TAMs contribute to tumor development by promoting angiogenesis as well as the production of factors that stimulate cancer cell proliferation and survival (202). These macrophages are more frequently found in TME than M1-like type and correlate with poor prognosis (200).
The elimination of tumor-supporting TAMs constitutes a potential approach for cancer therapies, as there is much evidence indicating their role in promoting tumor growth (203). In this context, the induction of a pro-inflammatory type of macrophage death seems to be particularly appealing. It not only eliminates cells with often pro-tumorigenic functions but also leads to the release of molecules that support an anti-tumor immune response. Indeed, DAMPs and cytokines produced during immunogenic PCDs were documented to trigger antigen-presenting cell maturation and/or activation (204), antigen-specific CD8+ T cell response (205) as well as chemotaxis and cytotoxicity of NK cells (206). Thus far, numerous studies have concentrated on the induction of pyroptosis, necroptosis, and ferroptosis specifically in tumor cells for potential cancer therapy, as discussed elsewhere (207–210). In contrast, the amount of research focusing on these PCDs in macrophages within TME is significantly lower. Moreover, their impact on tumorigenesis seems to vary under different conditions, thus being intricate. Here, we gather factors triggering macrophage pyroptosis, necroptosis, and ferroptosis in TME. We also compare the mechanisms of these processes, their links to TAMs polarization as well as the overall effects on tumor progression and organize these data in Table 2. Our approach aims to provide a comprehensive overview and summarize current knowledge in this field.
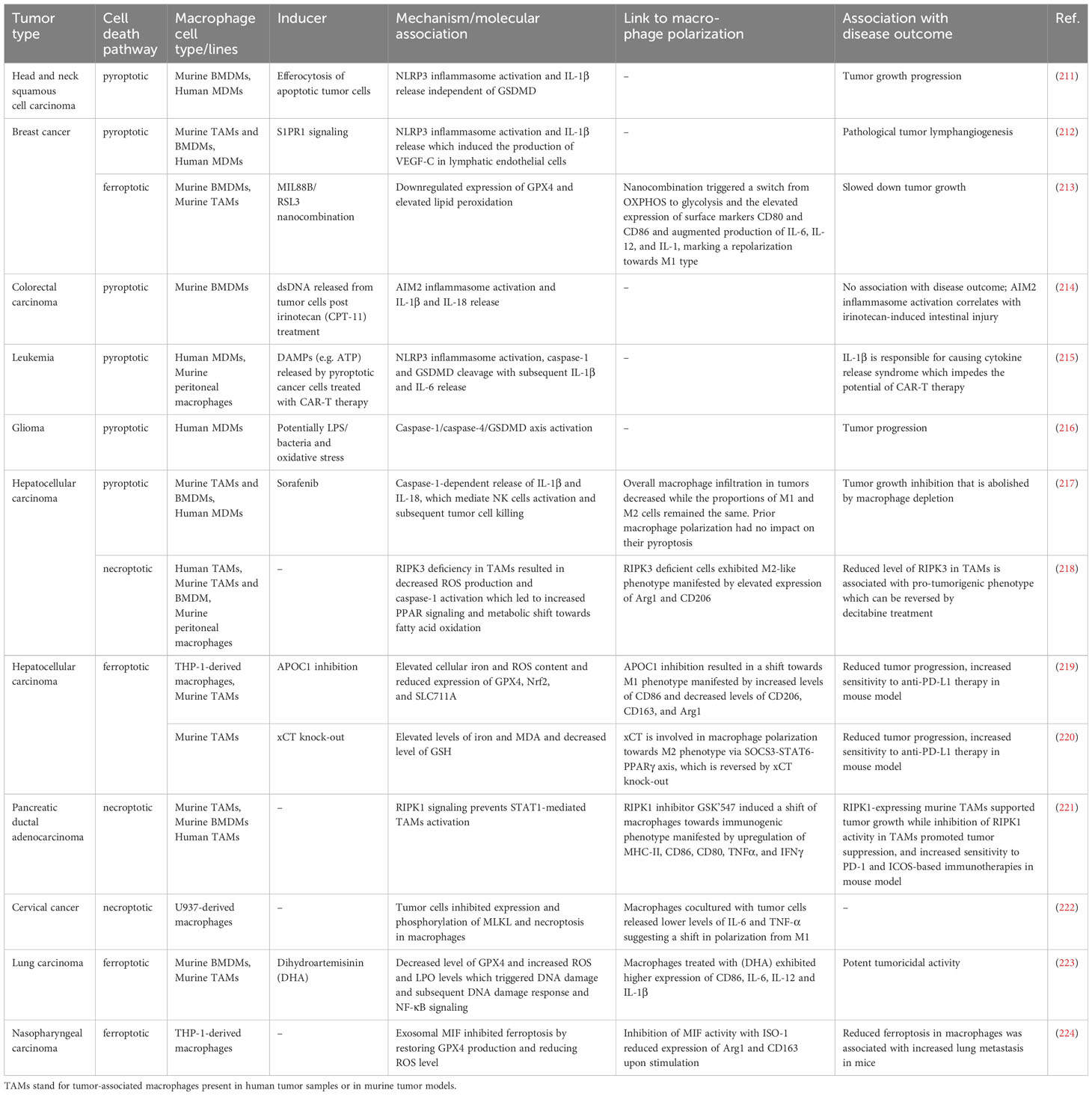
Table 2 Lytic PCDs in macrophages in the context of cancer: mechanisms and outcomes.
5.2 Macrophage pyroptosis in TME
5.2.1 Factors inducing pyroptosis in TAMs
Looking for factors that trigger TAMs pyroptosis and have an impact on the TME and immune response, seems to be reasonable. Some studies demonstrate that either cancer cells or tumor-derived molecules can act as DAMPs causing inflammasome activation and pyroptosis in macrophages (211, 212, 214, 215, 225). Interestingly, it was also reported that pyroptosis and/or inflammasome activation in TAMs can arise as a side effect of various anticancer therapies (214, 215, 225, 226) or may be triggered by factors other than DAMPs (216, 227). In this section, we take a closer look at all this evidence.
5.2.1.1 Tumor cells and their DAMPs
Cancer cells can act as factors activating pyroptotic molecules in macrophages. Lang et al. demonstrated that efferocytosis of apoptotic cancer cells by TAMs both in vitro and in vivo led to NLRP3 inflammasome activation, caspase-1 processing, and IL-1β secretion in those macrophages (211). This phenomenon resulted in immunosuppression and the promotion of tumor growth. Interestingly, in GSDMD KO mice a decreased tumor progression was not observed, which indicated the lack of a tumor-supportive role of GSDMD in models used in this study (211). Noteworthy, this research also included bulk RNA sequencing of CD11b+ myeloid cells sorted from human head & neck squamous cell carcinoma (HNSCC) tumors. Such analysis demonstrated an increase in NLRP3 and IL1B gene expression in tumor-infiltrating myeloid cells, which was consistent with transcriptomic results from in vitro experiments. Moreover, tumor-derived CD11b+ cells were characterized by upregulated expression of efferocytosis-associated genes. These findings support the authors’ hypothesis that efferocytosis-induced NLRP3 inflammasome activation in tumor-associated macrophages may promote the pro-tumorigenic effects within TME.
The contrary impact of pyroptotic macrophages on tumor growth was proposed by Wang et al. (228). They suggested that inhibition of sphingosine kinase 1 (SPHK1) activity in TME cells along with induction of pyroptosis in TAMs might contribute to the suppression of tumor development. The concept was based on the findings that sphingosine, the substrate for SPHK1, can derive from cancer cells and act as a DAMP. Its accumulation can lead to pyroptosis in tumor-associated macrophages and further induction of anti-tumor immune response (228). While no publication delves deeper into and supports their hypothesis, there is a study focusing on related molecules. Weichand et al. showed that the SPHK1 product, which is sphingosine-1-phosphate, and its receptor play a prominent role in tumor progression (212). The authors demonstrated that S1P receptor 1 (S1PR1) signaling in TAMs promoted lymphangiogenesis via NLRP3-dependent IL-1β secretion. It was suggested that IL-1β, released from macrophages after NLRP3 activation, evoked the production of VEGF-C in lymphatic endothelial cells (212). Interestingly, S1PR1 knock-out in TAMs strongly reduced IL-1β level and lymphangiogenesis, though it did not affect tumor development in methylcholanthrene (MCA)-induced fibrosarcoma murine model (212). The above results show that there is a strong need for further research into sphingosine and its phosphorylated form in the context of TME, as well as other cancer-derived molecules potentially influencing macrophage pyroptosis.
5.2.1.2 DAMPs released as a side effect of anticancer therapy
Inflammasome activation in TAMs was reported to be a possible outcome of anticancer treatment as many antitumor therapies can lead to the release of tumor-derived DAMPs (214, 215, 225, 226). For example, HCT-116 cells treated with irinotecan (CPT-11) secreted nuclear double-stranded DNA in exosomes that were subsequently engulfed by macrophages leading to the activation of AIM2 inflammasome and IL-1β release (214). Although the inhibition of AIM2 alleviated CPT-11-induced intestinal toxicity, it did not influence the drug’s anticancer efficacy (214). This data suggests that inflammasome activation in macrophages did not contribute to the antitumor immune response, but it triggered unwanted effects of CPT-11 treatment instead. It remains unclear whether IL-1β production was associated with macrophage pyroptosis.
Chemotherapy is not the only example of antitumor therapy, in which macrophage inflammasome activation seems to be a side effect. Recent findings indicate that chimeric antigen receptor (CAR) T cell therapy may result in extensive pyroptosis in B leukemic and other target cells (215). This treatment is accompanied by the release of large amounts of DAMPs e.g. ATP that can be picked up by macrophages and can further activate NLRP3 inflammasome leading to caspase-1 and GSDMD cleavage, as well as IL-1β maturation and secretion. Such processes, however, result in cytokine release syndrome (CRS) and CAR-T-related toxicities rather than effective antitumor immune response and cancer regression (215). This topic has been further explored by Deng et al., who proposed HMGB1 as one of the crucial DAMPs responsible for the strong side effects of this therapy (225). Their concept is derived from the fact that patients treated with CAR-T cells are characterized by high levels of HMGB1 (225).
Radiotherapy is another example of anticancer treatment that has an established contribution to the induction of macrophage pyroptosis. Liu et al. demonstrated radiation-induced NLRP3 inflammasome activation, IL-1β production, and pyroptosis in BDMDs in a dose-dependent manner. Moreover, they showed that macrophages were tolerant to relatively low doses of radiation. The mentioned effects were downregulated by knockout of NLRP3 both in vitro and in vivo (226). These observations provoke an interesting question, whether radiation-induced macrophage pyroptosis is favorable, and may stimulate an anticancer immune response, or mediate severe tissue damage instead. Nonetheless, it has not been further discussed in the literature.
5.2.1.3 Factors other than DAMPs
Since macrophage pyroptosis can be induced by a range of factors, DAMPs may not be the exclusive initiators of this process in TME. A recent study on the correlation between the occurrence of TAMs pyroptosis and cancer progression utilizing single-cell RNA-sequencing analysis demonstrated that human glioma-infiltrating macrophages, MDMs in particular, had significant co-expression of caspase-1 and GSDMD genes (216). Moreover, MDMs from high-grade gliomas showed increased transcripts of pyroptotic genes and their elevated infiltration was a characteristic of the high-risk group. Authors proposed LPS/bacteria and oxidative stress as potential initiators of MDMs pyroptosis since the signaling pathways responding to such factors were activated during macrophages’ progression toward the pyroptosis fate (216). It is particularly intriguing, considering evidence of the presence of bacteria and LPS in human gliomas (229). Since these components have already been shown to trigger macrophage pyroptosis, we hypothesize that bacterial LPS found in gliomas may induce pyroptosis in infiltrating MDMs. Its subsequent influence on the tumor microenvironment can promote cancer progression and lead to shorter overall patient survival. On the other hand, the observed oxidative stress signaling may be increased due to hypoxia, a common feature of the tumor microenvironment (230). Hypoxia has been recently proposed as a second signal to induce pyroptosis in LPS-primed macrophages leading to NLRP3 inflammasome activation, pro-caspase-1 and GSDMD processing, as well as IL-1β and LDH release (227). Thus, both studies together shed new light on the role of bacteria along with oxidative stress in inducing macrophage pyroptosis and promoting tumor progression.
5.2.2 Advantageous induction of macrophage pyroptosis in anticancer therapy
Targeting TAMs pyroptosis may diminish the number of often pro-tumorigenic macrophages and promote antitumor immunity simultaneously. Several studies have suggested that the induction of macrophage pyroptosis can stimulate an anticancer immune response, thereby being favorable (217, 228, 231). Okondo et al. demonstrated that Val-boroPro, a nonselective inhibitor of post-proline cleaving serine proteases and a potential anticancer agent, triggered pyroptosis in monocytes and macrophages (231). Such a phenomenon may be vital for the high efficacy of Val-boroPro against cancers, as it was shown that the depletion of phagocytic cells significantly reduced Val-boroPro’s antitumor activity (232).
Another study by Hage et al. showed that macrophage pyroptosis is essential for the antitumoral effects of sorafenib, a multitarget kinase inhibitor approved for the treatment of hepatocellular carcinoma (HCC) (217). The efficacy of sorafenib depended on the release of interleukins 1β and 18 from pyroptotic macrophages, as well as subsequent induction of natural killer cell-mediated cytotoxicity against HCC (217).
5.3 Macrophage necroptosis in TME
Necroptosis in cancer cells was reported to be a tumor-suppressive mechanism, particularly serving as an alternative cell death pathway in apoptosis-resistant cells (233). However, there is still a lack of consistent and comprehensive data addressing the role of necroptosis mediators in the function of tumor-infiltrating macrophages. It appears that the outcomes of targeting necroptosis in TAMs vary depending on the specific protein that is inhibited. Moreover, the potential benefit from this approach is more complex when considering the link between necroptotic molecules and M1- and M2-like macrophage polarization.
5.3.1 Outcomes of targeting different necroptotic molecules in TAMs
5.3.1.1 RIPK1 inhibition in TAMs leads to M1-like polarization and tumor suppression
An elevated expression of RIPK1 was observed in both malignant epithelial cells and tumor-associated macrophages within human pancreatic ductal adenocarcinoma (PDA) (221). It was established that the pivotal factor for PDA progression was the presence of RIPK1 in the extra-tumoral compartment. Subsequently, it was elucidated that the selective inhibition of RIPK1 using GSK’547 induced a repolarization of TAMs toward an immunogenic phenotype, that led to the activation of adaptive immune responses and tumor suppression. Notably, such an outcome was substantiated through experiments in mice and organotypic models of human PDA. The inhibition of RIPK1 enhanced the effectiveness of PD-1 and ICOS-based immunotherapies in PDA-bearing mice (221). In the context of this review, it is worth noting that the tolerogenic phenotype exhibited by tumor-associated macrophages in PDA was reliant on RIPK1 but independent of RIPK3 and necroptosis (221).
5.3.1.2 RIPK3 deficiency in TAMs leads to M2-like phenotype and tumorigenesis
In contrast to the role of RIPK1 as a central regulator of immune tolerance in PDA, the decreased expression of RIPK3 in macrophages associated with HCC correlated with tumorigenesis as observed in both murine models and human samples. Such a reduction in RIPK3 levels was also associated with the accumulation and polarization of TAMs toward an M2-like phenotype (218). Furthermore, in this study macrophages were subjected to treatment with decitabine, a hypomethylating drug evaluated in a clinical trial for liver metastasis and colorectal cancer therapy. As a result, the expression of RIPK3 in such macrophages was augmented which effectively reversed the immunosuppressive activities of TAMs within the HCC tumor microenvironment (218).
5.3.1.3 Decreased MLKL leads to the reduction in both necroptosis and pro-inflammatory mediators
A high level of MLKL in peripheral blood mononuclear cells from cervical cancer patients was associated with improved overall survival (222). Notably, when the U937 macrophages were cocultured with cervical cancer cells, the decreased expression and phosphorylation of MLKL were observed along with reduced necroptosis. This was accompanied by the downregulation of pro-inflammatory mediators, suggesting a shift from M1 polarization and the potential establishment of a more immunosuppressive environment. Importantly, the overexpression of RIPK3 in macrophages reversed the impact of cancer cells on macrophage polarization and necroptosis (222).
5.3.2 M1- vs M2-like macrophages susceptibility to necroptosis in TME
M1 and M2 macrophages display differential sensitivity to necroptosis-inducing stimuli, such as SMAC mimetics or TAK1 kinase inhibitors. These agents hold significant potential in the field of cancer therapy since cancer cells acquire resistance to apoptosis. Nevertheless, it is necessary to consider that necroptosis of tumor cells within the TME can result in TAMs-mediated immunosuppression (234, 235). Moreover, elevated susceptibility of M1 macrophages to the pro-necroptotic effects of SMAC mimetics may pose additional challenges for therapies based on them (89, 90). On the contrary, the alteration in the M1/M2 macrophage ratio, which stems from the increased susceptibility of M2 populations to necroptosis induction by TAK1 kinase inhibitors, provides a rationale for triggering necroptosis in anticancer therapy (91).
5.4 Macrophage ferroptosis in TME
So far, a large body of literature showed that ferroptosis activation in TME is linked to the depletion of M2 TAMs (236), their repolarization toward M1 type in vitro (213, 219, 223) as well as decreased tumor progression and metastasis (213, 219, 220, 223, 224). While no direct evidence of macrophage reprogramming in vivo has been obtained yet, it still seems that inducing ferroptosis in tumor microenvironment and/or tumor-associated macrophages may be advantageous.
5.4.1 Ferroptosis induction reduces the number of M2-like macrophages and leads to M1-like phenotype repolarization
The evidence that ferroptosis-related genes affect the polarization state of macrophages has been previously provided by Hu et al. Their bioinformatical analysis of gene expression in HNSCC identified – suppressor of cytokine signaling 1 (SOCS1) and ferroptosis inhibitor FTH1 as prognostic factors. In this study, among tumor-infiltrating immune cells the former gene was associated with M1 macrophages, and the expression of the latter was mostly correlated with M2 type (237). Moreover, FTH1 mRNA was elevated in lymph node metastasis. The authors further implicated that induction of ferroptosis may serve as an anticancer strategy capable of improving the efficacy of available immunotherapies in HNSCC. The experiments performed by Zhao et al. on a murine HNSCC xenograft model proved that ferroptosis inducer RSL3 successfully initiated this form of PCD in the grafts and also significantly reduced the number of M2 TAMs in the microenvironment (236). Although the number of M1 TAMs was not assessed, it is possible that RSL3 treatment led to macrophage repolarization. The supporting evidence provided by Gu et al. (213) showed that MIL88B nanoparticles containing RSL3 elicited ferroptotic stress in M2-polarized macrophages manifested by inhibition of GPX4 expression and high lipid peroxidation levels. Such a nanocombination also triggered metabolic reprogramming by switching from oxidative phosphorylation to glycolysis and induced M1 repolarization from M2 cells in vitro. Moreover, in vivo experiments showed an increase in the percentage of tumoricidal and metastasis-suppressing M1 cells in the breast tumor model (213).
The induction of macrophage ferroptosis constitutes a promising approach for resolving immunosuppressive microenvironment in HCC. Hao et al. discovered that APOC1 protein was overexpressed in HCC tissues and TAMs, while inhibition of APOC1 gene shifted macrophage phenotype toward M1 (219). It was achieved by the induction of the ferroptotic pathway evidenced by increased iron and ROS content in the cells as well as decreased expression of GPX4, Nrf2, and SLC7A11. APOC1 knock-out in TAMs was shown to reduce proliferation, migration, and invasion of tumor cells. Moreover, it boosted sensitivity to anti-PD-1 therapy (219). Similarly, xCT-specific knock-down in murine model of HCC also inhibited M2-type polarization in TAMs via activating ferroptosis pathway (220). x-CT mediated macrophage ferroptosis additionally enhanced PD-L1 expression in these cells, thus improving the efficacy of antitumor treatment. Erastin-containing nanoparticles constructed by the researchers were effective in inducing macrophage ferroptosis and combined with anti-PD-L1 performed better than either of the monotherapies (220).
Lastly, dihydroartemisinin (DHA) is an antimalarial drug that was additionally proved to shift the polarization status of M2 macrophages toward M1 type both in vitro and in vivo in murine lung carcinoma model (223). As previously, repolarization was linked to macrophage ferroptosis manifested by increased levels of ROS and LPO. This, in turn, led to DNA damage and the associated cellular response which in turn activated NF-κB signaling pathway (223).
5.4.2 Cancer-related molecules inhibiting TAMs ferroptosis
Noteworthy, cancer cells may target ferroptosis in macrophages to abolish its anticancer effects as was demonstrated previously (224). In this study exosomal macrophage migration inhibitory factor derived from CNE-2 tumor cells inhibited macrophage ferroptosis in lung metastasis, thus promoting disease spreading (224). In addition, an analysis of single-cell sequencing and transcriptome data obtained from melanoma and HNSCC tumors (238) indicated that CD86hi (M2-like) TAMs displayed an increased level of ferroptosis (238). Noteworthy, these cells were considered major contributors to immune therapy resistance (173). This implies that in some cases cancer cells may harness macrophage ferroptosis to promote disease development; hence, further studies are necessary to untangle this relationship.
5.5 Section summary
The data discussed above indicates that targeting macrophage pyroptosis for the preferred induction of antitumor immunity rather than driving the unwanted vicious circle of inflammation and toxicity can pose a challenge. Although the occurrence of TAMs pyroptosis within the tumor microenvironment has been extensively documented, it is still hard to reach a clear conclusion. The impact of macrophage pyroptosis on tumor development seems to depend not only on its inducer but also on the cancer type. The whole scenario can become even more intricate, considering the fact that certain tumors, e.g. gastric and colorectal cancer, are inflammation-driven (239). A more evident conclusion can be drawn for necroptosis. Based on the limited dataset, it seems that within TME the principal regulators of necroptosis primarily influence macrophage phenotype in a manner independent of cell death. Nevertheless, these findings suggest a potential therapeutic strategy that involves manipulating necroptosis components to target the polarization and metabolism of TAMs. So far, studies on macrophage ferroptosis in TME suggest that its induction may resolve the immunosuppressive microenvironment, promote M1-like polarization, and reduce tumor growth. Since it may improve the effectiveness of cancer immunotherapies, it should be an attractive candidate for further research.
6 Concluding remarks
Given the unique role of macrophages, the demise of these cells acquired exceptional significance (240–244). Despite the distinct molecular pathways through which pyroptosis, necroptosis, and ferroptosis are executed, the data summarized in this review unequivocally highlight remarkable flexibility and crosstalk among these mechanisms. It becomes evident that diverse modes of cell death can be initiated in response to the same stimulus, as exemplified by the uptake of oxLDLs by macrophages. It is still an open question whether the observed phenomenon represents the cumulative effect of various cell death modalities taking place in the mixture of cells or if it involves a mixed-type response within an individual cell, akin to the concept of PANoptosis (245). Differential susceptibility to death pathways induced by oxLDLs can also be caused by macrophage polarization states, which govern cell functions and phenotype. However, various subsets of plaque-associated macrophages described only in humans or in mice limit the ability to compare the processes occurring in the atherosclerotic milieu of the two species. Nevertheless, the occurrence of either lytic PCD aggravates the disease, as opposed to cancer, where the impact of macrophage death on tumor growth is contextually variable and depends on both the type of tumor and macrophage PCD modality. Thus, the research into the mechanisms governing macrophage death in atherosclerosis, e.g. various non-coding RNAs, could open new horizons for treating cancers in which targeting macrophages is of interest. Similarly, looking into the negative regulators of macrophage death in tumors could create new avenues for alleviating atherosclerosis.
The main limitation of the studies discussed in the review lies in the utilization of a dichotomic in vitro model of macrophage polarization, which dramatically simplifies the heterogeneity of TAMs or macrophages in atherosclerotic plaques observed in patients (76, 155, 195, 246). A further validation of macrophage activation subsets and their origin in humans is required to assess the susceptibility of tumor- and atherosclerosis-associated macrophages to various PCD modes. Nonetheless, it is conceivable that with time these discoveries, coupled with advanced techniques providing detailed insights into the spatial organization of tumors and atherosclerotic plaques at the single-cell level, will facilitate the development of innovative therapeutic approaches.
Author contributions
MM: Visualization, Writing – original draft, Writing - review & editing. MS: Visualization, Writing – original draft, Writing - review & editing. MB: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The open-access publication has been supported by scientific subsidy from Ministry of Education and Science (Warsaw, Poland).
Acknowledgments
Dr. Krzysztof Guzik (Department of Immunology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University) is kindly acknowledged for comments and suggestions regarding the manuscript. The figure was created with BioRender.com through a purchased license.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gordon S, Plüddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol. (2017) 15:53. doi: 10.1186/s12915-017-0392-4
2. Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
3. Netea MG, Schlitzer A, Placek K, Joosten LAB, Schultze JL. Innate and adaptive immune memory: an evolutionary continuum in the host’s response to pathogens. Cell Host Microbe. (2019) 25:13–26. doi: 10.1016/j.chom.2018.12.006
4. Sun J-X, Xu X-H, Jin L. Effects of metabolism on macrophage polarization under different disease backgrounds. Front Immunol. (2022) 13:880286. doi: 10.3389/fimmu.2022.880286
5. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
6. Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. (2014) 5:532. doi: 10.3389/fimmu.2014.00532
7. Vogel DYS, Glim JE, Stavenuiter AWD, Breur M, Heijnen P, Amor S, et al. Human macrophage polarization in vitro: Maturation and activation methods compared. Immunobiology. (2014) 219:695–703. doi: 10.1016/j.imbio.2014.05.002
8. Xu W, Zhao X, Daha MR, van Kooten C. Reversible differentiation of pro- and anti-inflammatory macrophages. Mol Immunol. (2013) 53:179–86. doi: 10.1016/j.molimm.2012.07.005
9. Li H, Cao Z, Wang L, Liu C, Lin H, Tang Y, et al. Macrophage subsets and death are responsible for atherosclerotic plaque formation. Front Immunol. (2022) 13:843712. doi: 10.3389/fimmu.2022.843712
10. Murray PJ. Macrophage polarization. Annu Rev Physiol. (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
11. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
12. Kist M, Vucic D. Cell death pathways: intricate connections and disease implications. EMBO J. (2021) 40:e106700. doi: 10.15252/embj.2020106700
13. Flores-Romero H, Ros U, Garcia-Saez AJ. Pore formation in regulated cell death. EMBO J. (2020) 39:e105753. doi: 10.15252/embj.2020105753
14. Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol. (2020) 22:1042–8. doi: 10.1038/s41556-020-0565-1
15. Florean C, Song S, Dicato M, Diederich M. Redox biology of regulated cell death in cancer: A focus on necroptosis and ferroptosis. Free Radic Biol Med. (2019) 134:177–89. doi: 10.1016/j.freeradbiomed.2019.01.008
16. Friedlander AM. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem. (1986) 261:7123–6. doi: 10.1016/S0021-9258(17)38364-3
17. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. (1992) 358:167–9. doi: 10.1038/358167a0
18. Chen Y, Smith MR, Thirumalai K, Zychlinsky A. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO J. (1996) 15:3853–60.
19. Black RA, Kronheim SR, Merriam JE, March CJ, Hopp TP. A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J Biol Chem. (1989) 264:5323–6.
20. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. (1992) 356:768–74. doi: 10.1038/356768a0
21. Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science. (1992) 256:97–100. doi: 10.1126/science.1373520
22. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. (2001) 9:113–4. doi: 10.1016/s0966-842x(00)01936-3
23. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. (2021) 18:1106–21. doi: 10.1038/s41423-020-00630-3
24. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. (2021) 6:128. doi: 10.1038/s41392-021-00507-5
25. Rastogi S, Briken V. Interaction of mycobacteria with host cell inflammasomes. Front Immunol. (2022) 13:791136. doi: 10.3389/fimmu.2022.791136
26. Jiao Y, Zhang T, Zhang C, Ji H, Tong X, Xia R, et al. Exosomal miR-30d-5p of neutrophils induces M1 macrophage polarization and primes macrophage pyroptosis in sepsis-related acute lung injury. Crit Care Lond Engl. (2021) 25:356. doi: 10.1186/s13054-021-03775-3
27. Zha Q-B, Wei H-X, Li C-G, Liang Y-D, Xu L-H, Bai W-J, et al. ATP-induced inflammasome activation and pyroptosis is regulated by AMP-activated protein kinase in macrophages. Front Immunol. (2016) 7:597. doi: 10.3389/fimmu.2016.00597
28. Li N, Chen J, Geng C, Wang X, Wang Y, Sun N, et al. Myoglobin promotes macrophage polarization to M1 type and pyroptosis via the RIG-I/Caspase1/GSDMD signaling pathway in CS-AKI. Cell Death Discovery. (2022) 8:90. doi: 10.1038/s41420-022-00894-w
29. Santa Cruz Garcia AB, Schnur KP, Malik AB, Mo GCH. Gasdermin D pores are dynamically regulated by local phosphoinositide circuitry. Nat Commun. (2022) 13:52. doi: 10.1038/s41467-021-27692-9
30. Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature. (2021) 593:607–11. doi: 10.1038/s41586-021-03478-3
31. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. (2021) 591:131–6. doi: 10.1038/s41586-021-03218-7
32. DiPeso L, Ji DX, Vance RE, Price JV. Cell death and cell lysis are separable events during pyroptosis. Cell Death Discovery. (2017) 3:17070. doi: 10.1038/cddiscovery.2017.70
33. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. (2018) 362:956–60. doi: 10.1126/science.aar7607
34. Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol Baltim Md 1950. (1988) 141:2629–34.
35. Ray CA, Pickup DJ. The mode of death of pig kidney cells infected with cowpox virus is governed by the expression of the crmA gene. Virology. (1996) 217:384–91. doi: 10.1006/viro.1996.0128
36. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. (2005) 1:112–9. doi: 10.1038/nchembio711
37. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. (2000) 1:489–95. doi: 10.1038/82732
38. Zhang D-W, Shao J, Lin J, Zhang N, Lu B-J, Lin S-C, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. (2009) 325:332–6. doi: 10.1126/science.1172308
39. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. (2009) 137:1100–11. doi: 10.1016/j.cell.2009.05.021
40. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. (2009) 137:1112–23. doi: 10.1016/j.cell.2009.05.037
41. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
42. Welz P-S, Wullaert A, Vlantis K, Kondylis V, Fernández-Majada V, Ermolaeva M, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. (2011) 477:330–4. doi: 10.1038/nature10273
43. Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. (2015) 25:347–53. doi: 10.1016/j.tcb.2015.01.001
44. Chen X, Li W, Ren J, Huang D, He W-T, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. (2014) 24:105–21. doi: 10.1038/cr.2013.171
45. Butler RE, Krishnan N, Garcia-Jimenez W, Francis R, Martyn A, Mendum T, et al. Susceptibility of Mycobacterium tuberculosis-infected host cells to phospho-MLKL driven necroptosis is dependent on cell type and presence of TNFα. Virulence. (2017) 8:1820–32. doi: 10.1080/21505594.2017.1377881
46. Place DE, Samir P, Malireddi RS, Kanneganti T-D. Integrated stress response restricts macrophage necroptosis. Life Sci Alliance. (2022) 5:e202101260. doi: 10.26508/lsa.202101260
47. Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose-Girma M, Dohse M, et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature. (2019) 574:428–31. doi: 10.1038/s41586-019-1548-x
48. Legarda D, Justus SJ, Ang RL, Rikhi N, Li W, Moran TM, et al. CYLD proteolysis protects macrophages from TNF-mediated auto-necroptosis induced by LPS and licensed by type I IFN. Cell Rep. (2016) 15:2449–61. doi: 10.1016/j.celrep.2016.05.032
49. Muendlein HI, Jetton D, Connolly WM, Eidell KP, Magri Z, Smirnova I, et al. cFLIPL protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science. (2020) 367:1379–84. doi: 10.1126/science.aay3878
50. Newton K. Multitasking kinase RIPK1 regulates cell death and inflammation. Cold Spring Harb Perspect Biol. (2020) 12:a036368. doi: 10.1101/cshperspect.a036368
51. Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature. (2020) 577:103–8. doi: 10.1038/s41586-019-1828-5
52. Cho M, Dho SH, Shin S, Lee Y, Kim Y, Lee J, et al. Caspase-10 affects the pathogenesis of primary biliary cholangitis by regulating inflammatory cell death. J Autoimmun. (2022) 133:102940. doi: 10.1016/j.jaut.2022.102940
53. McComb S, Shutinoski B, Thurston S, Cessford E, Kumar K, Sad S. Cathepsins limit macrophage necroptosis through cleavage of rip1 kinase. J Immunol. (2014) 192:5671–8. doi: 10.4049/jimmunol.1303380
54. He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc Natl Acad Sci. (2011) 108:20054–9. doi: 10.1073/pnas.1116302108
55. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL *. J Biol Chem. (2013) 288:31268–79. doi: 10.1074/jbc.M113.462341
56. McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB, et al. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci U.S.A. (2014) 111:E3206–3213. doi: 10.1073/pnas.1407068111
57. Zhang H, Wu X, Li X, Li M, Li F, Wang L, et al. Crucial roles of the RIP homotypic interaction motifs of RIPK3 in RIPK1-dependent cell death and lymphoproliferative disease. Cell Rep. (2020) 31:107650. doi: 10.1016/j.celrep.2020.107650
58. Li X, Zhang M, Huang X, Liang W, Li G, Lu X, et al. Ubiquitination of RIPK1 regulates its activation mediated by TNFR1 and TLRs signaling in distinct manners. Nat Commun. (2020) 11:6364. doi: 10.1038/s41467-020-19935-y
59. Cao M, Chen F, Xie N, Cao M-Y, Chen P, Lou Q, et al. c-Jun N-terminal kinases differentially regulate TNF- and TLRs-mediated necroptosis through their kinase-dependent and -independent activities. Cell Death Dis. (2018) 9:1–16. doi: 10.1038/s41419-018-1189-2
60. Gong Y-N, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. (2017) 169:286–300.e16. doi: 10.1016/j.cell.2017.03.020
61. Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity. (2017) 47:51–65.e7. doi: 10.1016/j.immuni.2017.06.001
62. Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, et al. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PloS Biol. (2017) 15:e2002711. doi: 10.1371/journal.pbio.2002711
63. Rasheed A, Robichaud S, Nguyen M-A, Geoffrion M, Wyatt H, Cottee ML, et al. Loss of MLKL (Mixed lineage kinase domain-like protein) decreases necrotic core but increases macrophage lipid accumulation in atherosclerosis. Arterioscler Thromb Vasc Biol. (2020) 40:1155–67. doi: 10.1161/ATVBAHA.119.313640
64. Wager CML, Bonifacio JR, Simper J, Naoun AA, Arnett E, Schlesinger LS. Activation of transcription factor CREB in human macrophages by Mycobacterium tuberculosis promotes bacterial survival, reduces NF-kB nuclear transit and limits phagolysosome fusion by reduced necroptotic signaling. PloS Pathog. (2023) 19:e1011297. doi: 10.1371/journal.ppat.1011297
65. Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. (2013) 4:e716–6. doi: 10.1038/cddis.2013.238
66. Yang Z, Wang Y, Zhang Y, He X, Zhong C-Q, Ni H, et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat Cell Biol. (2018) 20:186–97. doi: 10.1038/s41556-017-0022-y
67. Zhang Y, Su SS, Zhao S, Yang Z, Zhong C-Q, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. (2017) 8:14329. doi: 10.1038/ncomms14329
68. Huang Y-T, Liang Q-Q, Zhang H-R, Chen S-Y, Xu L-H, Zeng B, et al. Baicalin inhibits necroptosis by decreasing oligomerization of phosphorylated MLKL and mitigates caerulein-induced acute pancreatitis in mice. Int Immunopharmacol. (2022) 108:108885. doi: 10.1016/j.intimp.2022.108885
69. Shi F-L, Ni S-T, Luo S-Q, Hu B, Xu R, Liu S-Y, et al. Dimethyl fumarate ameliorates autoimmune hepatitis in mice by blocking NLRP3 inflammasome activation. Int Immunopharmacol. (2022) 108:108867. doi: 10.1016/j.intimp.2022.108867
70. González-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, et al. Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PloS Pathog. (2015) 11:e1005337. doi: 10.1371/journal.ppat.1005337
71. Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. (2016) 2:e1600224. doi: 10.1126/sciadv.1600224
72. Bedient L, Pokharel SM, Chiok KR, Mohanty I, Beach SS, Miura TA, et al. Lytic cell death mechanisms in human respiratory syncytial virus-infected macrophages: roles of pyroptosis and necroptosis. Viruses. (2020) 12:932. doi: 10.3390/v12090932
73. Roca FJ, Whitworth LJ, Prag HA, Murphy MP, Ramakrishnan L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science. (2022) 376:eabh2841. doi: 10.1126/science.abh2841
74. Pajuelo D, Gonzalez-Juarbe N, Tak U, Sun J, Orihuela CJ, Niederweis M. NAD+ Depletion triggers macrophage necroptosis, a cell death pathway exploited by mycobacterium tuberculosis. Cell Rep. (2018) 24:429–40. doi: 10.1016/j.celrep.2018.06.042
75. Zhong W-J, Zhang J, Duan J-X, Zhang C-Y, Ma S-C, Li Y-S, et al. TREM-1 triggers necroptosis of macrophages through mTOR-dependent mitochondrial fission during acute lung injury. J Transl Med. (2023) 21:179. doi: 10.1186/s12967-023-04027-4
76. Pittet MJ, Michielin O, Migliorini D. Clinical relevance of tumour-associated macrophages. Nat Rev Clin Oncol. (2022) 19:402–21. doi: 10.1038/s41571-022-00620-6
77. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of non-apoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
78. Kinowaki Y, Taguchi T, Onishi I, Kirimura S, Kitagawa M, Yamamoto K. Overview of ferroptosis and synthetic lethality strategies. Int J Mol Sci. (2021) 22:9271. doi: 10.3390/ijms22179271
79. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. (2016) 23:369–79. doi: 10.1038/cdd.2015.158
80. Pedrera L, Espiritu RA, Ros U, Weber J, Schmitt A, Stroh J, et al. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. (2021) 28:1644–57. doi: 10.1038/s41418-020-00691-x
81. Li J, Cao F, Yin H, Huang Z, Lin Z, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. (2020) 11:1–13. doi: 10.1038/s41419-020-2298-2
82. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. (2022) 185:2401–21. doi: 10.1016/j.cell.2022.06.003
83. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. (2017) 66:449–65. doi: 10.1002/hep.29117
84. Youssef LA, Rebbaa A, Pampou S, Weisberg SP, Stockwell BR, Hod EA, et al. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood. (2018) 131:2581–93. doi: 10.1182/blood-2017-12-822619
85. Wiernicki B, Dubois H, Tyurina YY, Hassannia B, Bayir H, Kagan VE, et al. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. (2020) 11:922. doi: 10.1038/s41419-020-03118-0
86. Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. The emerging role of ferroptosis in inflammation. BioMed Pharmacother Biomedecine Pharmacother. (2020) 127:110108. doi: 10.1016/j.biopha.2020.110108
87. Xia H, Zhang Z, You F. Inhibiting ACSL1-related ferroptosis restrains murine coronavirus infection. Viruses. (2021) 13:2383. doi: 10.3390/v13122383
88. Rijal D, Ariana A, Wight A, Kim K, Alturki NA, Aamir Z, et al. Differentiated macrophages acquire a pro-inflammatory and cell death–resistant phenotype due to increasing XIAP and p38-mediated inhibition of RipK1. J Biol Chem. (2018) 293:11913–27. doi: 10.1074/jbc.RA118.003614
89. Hao Q, Kundu S, Kleam J, Zhao ZJ, Idell S, Tang H. Enhanced RIPK3 kinase activity-dependent lytic cell death in M1 but not M2 macrophages. Mol Immunol. (2021) 129:86–93. doi: 10.1016/j.molimm.2020.11.001
90. Ali H, Caballero R, Dong SXM, Gajnayaka N, Vranjkovic A, Ahmed D, et al. Selective killing of human M1 macrophages by Smac mimetics alone and M2 macrophages by Smac mimetics and caspase inhibition. J Leukoc Biol. (2021) 110:693–710. doi: 10.1002/JLB.4A0220-114RR
91. Varga Z, Molnár T, Mázló A, Kovács R, Jenei V, Kerekes K, et al. Differences in the sensitivity of classically and alternatively activated macrophages to TAK1 inhibitor-induced necroptosis. Cancer Immunol Immunother. (2020) 69:2193–207. doi: 10.1007/s00262-020-02623-7
92. Wong WW-L, Vince JE, Lalaoui N, Lawlor KE, Chau D, Bankovacki A, et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood. (2014) 123:2562–72. doi: 10.1182/blood-2013-06-510743
93. McComb S, Cheung HH, Korneluk RG, Wang S, Krishnan L, Sad S. cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. (2012) 19:1791–801. doi: 10.1038/cdd.2012.59
94. Dou X, Yu X, Du S, Han Y, Li L, Zhang H, et al. Interferon-mediated repression of miR-324-5p potentiates necroptosis to facilitate antiviral defense. EMBO Rep. (2022) 23:e54438. doi: 10.15252/embr.202154438
95. Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, et al. Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ. (2019) 26:332–47. doi: 10.1038/s41418-018-0122-7
96. Ming-Chin Lee K, Achuthan AA, De Souza DP, Lupancu TJ, Binger KJ, Lee MKS, et al. Type I interferon antagonism of the JMJD3-IRF4 pathway modulates macrophage activation and polarization. Cell Rep. (2022) 39:110719. doi: 10.1016/j.celrep.2022.110719
97. Yang L, Guo P, Wang P, Wang W, Liu J. IL-6/ERK signaling pathway participates in type I IFN-programmed, unconventional M2-like macrophage polarization. Sci Rep. (2023) 13:1827. doi: 10.1038/s41598-022-23721-9
98. Kumaran Satyanarayanan S, El Kebir D, Soboh S, Butenko S, Sekheri M, Saadi J, et al. IFN-β is a macrophage-derived effector cytokine facilitating the resolution of bacterial inflammation. Nat Commun. (2019) 10:3471. doi: 10.1038/s41467-019-10903-9
99. Maeda K, Nakayama J, Taki S, Sanjo H. TAK1 limits death receptor fas-induced proinflammatory cell death in macrophages. J Immunol Baltim Md 1950. (2022) 209:1173–9. doi: 10.4049/jimmunol.2200322
100. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol. (2010) 40:824–35. doi: 10.1002/eji.200939889
101. Corna G, Campana L, Pignatti E, Castiglioni A, Tagliafico E, Bosurgi L, et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica. (2010) 95:1814–22. doi: 10.3324/haematol.2010.023879
102. Agoro R, Taleb M, Quesniaux VFJ, Mura C. Cell iron status influences macrophage polarization. PloS One. (2018) 13:e0196921. doi: 10.1371/journal.pone.0196921
103. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. (2020) 16:278–90. doi: 10.1038/s41589-019-0462-8
104. Piattini F, Matsushita M, Muri J, Bretscher P, Feng X, Freigang S, et al. Differential sensitivity of inflammatory macrophages and alternatively activated macrophages to ferroptosis. Eur J Immunol. (2021) 51:2417–29. doi: 10.1002/eji.202049114
105. Rostoker R, Yaseen H, Schif-Zuck S, Lichtenstein RG, Rabinovich GA, Ariel A. Galectin-1 induces 12/15-lipoxygenase expression in murine macrophages and favors their conversion toward a pro-resolving phenotype. Prostaglandins Other Lipid Mediat. (2013) 107:85–94. doi: 10.1016/j.prostaglandins.2013.08.001
106. Li Y, Yang Y, Guo T, Weng C, Yang Y, Wang Z, et al. Heme oxygenase-1 determines the cell fate of ferroptotic death of alveolar macrophages in COPD. Front Immunol. (2023) 14:1162087. doi: 10.3389/fimmu.2023.1162087
107. Fuhrmann DC, Mondorf A, Beifuß J, Jung M, Brüne B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. (2020) 36:101670. doi: 10.1016/j.redox.2020.101670
108. Raggi F, Pelassa S, Pierobon D, Penco F, Gattorno M, Novelli F, et al. Regulation of human macrophage M1–M2 polarization balance by hypoxia and the triggering receptor expressed on myeloid cells-1. Front Immunol. (2017) 8:1097. doi: 10.3389/fimmu.2017.01097
109. Vince JE, Wong WW-L, Gentle I, Lawlor KE, Allam R, O’Reilly L, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. (2012) 36:215–27. doi: 10.1016/j.immuni.2012.01.012
110. Kang T-B, Yang S-H, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. (2013) 38:27–40. doi: 10.1016/j.immuni.2012.09.015
111. Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SWG, et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1β Independently of gasdermin-D. J Immunol Baltim Md 1950. (2017) 198:2156–64. doi: 10.4049/jimmunol.1601757
112. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. (2015) 6:6282. doi: 10.1038/ncomms7282
113. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
114. Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U.S.A. (2017) 114:E961–9. doi: 10.1073/pnas.1613305114
115. Polykratis A, Martens A, Eren RO, Shirasaki Y, Yamagishi M, Yamaguchi Y, et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat Cell Biol. (2019) 21:731–42. doi: 10.1038/s41556-019-0324-3
116. Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ, et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell. (2022) 185:3214–3231.e23. doi: 10.1016/j.cell.2022.06.038
117. Feng S, Yang Y, Liu Z, Chen W, Du C, Hu G, et al. Intracellular bacteriolysis contributes to pathogenicity of Staphylococcus aureus by exacerbating AIM2-mediated inflammation and necroptosis. Virulence. (2022) 13:1684–96. doi: 10.1080/21505594.2022.2127209
118. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. (2018) 362:1064–9. doi: 10.1126/science.aau2818
119. Newton K, Wickliffe KE, Maltzman A, Dugger DL, Reja R, Zhang Y, et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature. (2019) 575:679–82. doi: 10.1038/s41586-019-1752-8
120. Fritsch M, Günther SD, Schwarzer R, Albert M-C, Schorn F, Werthenbach JP, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. (2019) 575:683–7. doi: 10.1038/s41586-019-1770-6
121. Gitlin AD, Heger K, Schubert AF, Reja R, Yan D, Pham VC, et al. Integration of innate immune signalling by caspase-8 cleavage of N4BP1. Nature. (2020) 587:275–80. doi: 10.1038/s41586-020-2796-5
122. Yuan J, Ofengeim D. A guide to cell death pathways. Nat Rev Mol Cell Biol. (2023), 1–17. doi: 10.1038/s41580-023-00689-6
123. Ai Y, Meng Y, Yan B, Zhou Q, Wang X. The biochemical pathways of apoptotic, necroptotic, pyroptotic, and ferroptotic cell death. Mol Cell. (2024) 84:170–9. doi: 10.1016/j.molcel.2023.11.040
124. Hughes SA, Lin M, Weir A, Huang B, Xiong L, Chua NK, et al. Caspase-8-driven apoptotic and pyroptotic crosstalk causes cell death and IL-1β release in X-linked inhibitor of apoptosis (XIAP) deficiency. EMBO J. (2023) 42:e110468. doi: 10.15252/embj.2021110468
125. Newton K, Wickliffe KE, Maltzman A, Dugger DL, Webster JD, Guo H, et al. Caspase cleavage of RIPK3 after Asp333 is dispensable for mouse embryogenesis. Cell Death Differ. (2024) 31:254–62. doi: 10.1038/s41418-023-01255-5
126. Kim S, Lu HC, Steelman AJ, Li J. Myeloid caspase-8 restricts RIPK3-dependent proinflammatory IL-1β production and CD4 T cell activation in autoimmune demyelination. Proc Natl Acad Sci. (2022) 119:e2117636119. doi: 10.1073/pnas.2117636119
127. Doerflinger M, Deng Y, Whitney P, Salvamoser R, Engel S, Kueh AJ, et al. Flexible usage and interconnectivity of diverse cell death pathways protect against intracellular infection. Immunity. (2020) 53:533–547.e7. doi: 10.1016/j.immuni.2020.07.004
128. Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. (2007) 9:2562–70. doi: 10.1111/j.1462-5822.2007.01036.x
129. Hos NJ, Ganesan R, Gutiérrez S, Hos D, Klimek J, Abdullah Z, et al. Type I interferon enhances necroptosis of Salmonella Typhimurium-infected macrophages by impairing antioxidative stress responses. J Cell Biol. (2017) 216:4107–21. doi: 10.1083/jcb.201701107
130. Chen H, Li Y, Wu J, Li G, Tao X, Lai K, et al. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. (2020) 27:2568–85. doi: 10.1038/s41418-020-0524-1
131. Russo AJ, Vasudevan SO, Méndez-Huergo SP, Kumari P, Menoret A, Duduskar S, et al. Intracellular immune sensing promotes inflammation via gasdermin D-driven release of a lectin alarmin. Nat Immunol. (2021) 22:154–65. doi: 10.1038/s41590-020-00844-7
132. Shi C-S, Kehrl JH. Bcl-2 regulates pyroptosis and necroptosis by targeting BH3-like domains in GSDMD and MLKL. Cell Death Discovery. (2019) 5:151. doi: 10.1038/s41420-019-0230-2
133. Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. (2018) 3:eaat2738. doi: 10.1126/sciimmunol.aat2738
134. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. (2018) 24:97–108.e4. doi: 10.1016/j.chom.2018.05.009
135. Ramon-Luing LA, Olvera Y, Flores-Gonzalez J, Palacios Y, Carranza C, Aguilar-Duran Y, et al. Diverse cell death mechanisms are simultaneously activated in macrophages infected by virulent mycobacterium tuberculosis. Pathogens. (2022) 11:492. doi: 10.3390/pathogens11050492
136. Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, et al. A major role for ferroptosis in Mycobacterium tuberculosis–induced cell death and tissue necrosis. J Exp Med. (2019) 216:556–70. doi: 10.1084/jem.20181776
137. Björkegren JLM, Lusis AJ. Atherosclerosis: recent developments. Cell. (2022) 185:1630–45. doi: 10.1016/j.cell.2022.04.004
138. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J Cell Physiol. (2018) 233:2116–32. doi: 10.1002/jcp.25930
139. Martinet W, Coornaert I, Puylaert P, De Meyer GRY. Macrophage death as a pharmacological target in atherosclerosis. Front Pharmacol. (2019) 10:306. doi: 10.3389/fphar.2019.00306
140. Schrijvers DM, De Meyer GRY, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. (2005) 25:1256–61. doi: 10.1161/01.ATV.0000166517.18801.a7
141. Thorp EB. Mechanisms of failed apoptotic cell clearance by phagocyte subsets in cardiovascular disease. Apoptosis Int J Program Cell Death. (2010) 15:1124–36. doi: 10.1007/s10495-010-0516-6
142. Liu C, Jiang Z, Pan Z, Yang L. The function, regulation and mechanism of programmed cell death of macrophages in atherosclerosis. Front Cell Dev Biol. (2022) 9:809516. doi: 10.3389/fcell.2021.809516
143. Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. (2006) 47:C13–8. doi: 10.1016/j.jacc.2005.10.065
144. Tian F, Yao J, Yan M, Sun X, Wang W, Gao W, et al. 5-aminolevulinic acid-mediated sonodynamic therapy inhibits RIPK1/RIPK3-dependent necroptosis in THP-1-derived foam cells. Sci Rep. (2016) 6:21992. doi: 10.1038/srep21992
145. Wei Y, Lan B, Zheng T, Yang L, Zhang X, Cheng L, et al. GSDME-mediated pyroptosis promotes the progression and associated inflammation of atherosclerosis. Nat Commun. (2023) 14:929. doi: 10.1038/s41467-023-36614-w
146. Zheng F, Xing S, Gong Z, Xing Q. NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ. (2013) 22:746–50. doi: 10.1016/j.hlc.2013.01.012
147. Shi X, Xie W-L, Kong W-W, Chen D, Qu P. Expression of the NLRP3 inflammasome in carotid atherosclerosis. J Stroke Cerebrovasc Dis. (2015) 24:2455–66. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.024
148. Rajamäki K, Mäyränpää MI, Risco A, Tuimala J, Nurmi K, Cuenda A, et al. p38δ MAPK. Arterioscler Thromb Vasc Biol. (2016) 36:1937–46. doi: 10.1161/ATVBAHA.115.307312
149. Vedre A, Pathak DR, Crimp M, Lum C, Koochesfahani M, Abela GS. Physical factors that trigger cholesterol crystallization leading to plaque rupture. Atherosclerosis. (2009) 203:89–96. doi: 10.1016/j.atherosclerosis.2008.06.027
150. Rajamäki K, Lappalainen J, Öörni K, Välimäki E, Matikainen S, Kovanen PT, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PloS One. (2010) 5:e11765. doi: 10.1371/journal.pone.0011765
151. Lin J, Shou X, Mao X, Dong J, Mohabeer N, Kushwaha KK, et al. Oxidized low density lipoprotein induced caspase-1 mediated pyroptotic cell death in macrophages: implication in lesion instability? PloS One. (2013) 8:e62148. doi: 10.1371/journal.pone.0062148
152. Puylaert P, Van Praet M, Vaes F, Neutel CHG, Roth L, Guns P-J, et al. Gasdermin D deficiency limits the transition of atherosclerotic plaques to an inflammatory phenotype in apoE knock-out mice. Biomedicines. (2022) 10:1171. doi: 10.3390/biomedicines10051171
153. Coornaert I, Puylaert P, Marcasolli G, Grootaert MOJ, Vandenabeele P, Meyer GRYD, et al. Impact of myeloid RIPK1 gene deletion on atherogenesis in ApoE-deficient mice. Atherosclerosis. (2021) 322:51–60. doi: 10.1016/j.atherosclerosis.2021.02.021
154. Karunakaran D, Nguyen M-A, Geoffrion M, Vreeken D, Lister Z, Cheng HS, et al. RIPK1 expression associates with inflammation in early atherosclerosis in humans and can be therapeutically silenced to reduce NF-κB activation and atherogenesis in mice. Circulation. (2021) 143:163–77. doi: 10.1161/CIRCULATIONAHA.118.038379
155. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
156. Xiao L, Luo G, Guo X, Jiang C, Zeng H, Zhou F, et al. Macrophage iron retention aggravates atherosclerosis: Evidence for the role of autocrine formation of hepcidin in plaque macrophages. Biochim Biophys Acta BBA - Mol Cell Biol Lipids. (2020) 1865:158531. doi: 10.1016/j.bbalip.2019.158531
157. Li W, Xu L-H, Yuan X-M. Macrophage hemoglobin scavenger receptor and ferritin accumulation in human atherosclerotic lesions. Ann N Y Acad Sci. (2004) 1030:196–201. doi: 10.1196/annals.1329.025
158. Sarad K, Stefańska M, Kraszewska I, Szade K, Sluimer JC, Błyszczuk P, et al. Single-cell transcriptomics reveals subtype-specific molecular profiles in Nrf2-deficient macrophages from murine atherosclerotic aortas. Front Immunol. (2023) 14:1249379. doi: 10.3389/fimmu.2023.1249379
159. Jiang Y, Wang M, Huang K, Zhang Z, Shao N, Zhang Y, et al. Oxidized low-density lipoprotein induces secretion of interleukin-1β by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem Biophys Res Commun. (2012) 425:121–6. doi: 10.1016/j.bbrc.2012.07.011
160. You Z, Ye X, Jiang M, Gu N, Liang C. lnc-MRGPRF-6:1 Promotes ox-LDL-Induced Macrophage Ferroptosis via Suppressing GPX4. Mediators Inflammation. (2023) 2023:5513245. doi: 10.1155/2023/5513245
161. Liang Y, Xu X-D, Xu X, Cai Y-B, Zhu Z-X, Zhu L, et al. Linc00657 promoted pyroptosis in THP-1-derived macrophages and exacerbated atherosclerosis via the miR-106b-5p/TXNIP/NLRP3 axis. Int J Biol Macromol. (2023) 253:126953. doi: 10.1016/j.ijbiomac.2023.126953
162. Tian Y, Luo Q, Huang K, Sun T, Luo S. Long noncoding RNA AC078850.1 induces NLRP3 inflammasome-mediated pyroptosis in atherosclerosis by upregulating ITGB2 transcription via transcription factor HIF-1α. Biomedicines. (2023) 11:1734. doi: 10.3390/biomedicines11061734
163. Hu G, Yuan Z, Wang J. Autophagy inhibition and ferroptosis activation during atherosclerosis: Hypoxia-inducible factor 1α inhibitor PX-478 alleviates atherosclerosis by inducing autophagy and suppressing ferroptosis in macrophages. BioMed Pharmacother Biomedecine Pharmacother. (2023) 161:114333. doi: 10.1016/j.biopha.2023.114333
164. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. (2013) 14:812–20. doi: 10.1038/ni.2639
165. Lin J, Li H, Yang M, Ren J, Huang Z, Han F, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. (2013) 3:200–10. doi: 10.1016/j.celrep.2012.12.012
166. Li B, Wang C, Lu P, Ji Y, Wang X, Liu C, et al. IDH1 promotes foam cell formation by aggravating macrophage ferroptosis. Biology. (2022) 11:1392. doi: 10.3390/biology11101392
167. Qiu Y, Li L, Guo X, Liu J, Xu L, Li Y. Exogenous spermine inhibits high glucose/oxidized LDL-induced oxidative stress and macrophage pyroptosis by activating the Nrf2 pathway. Exp Ther Med. (2022) 23:310. doi: 10.3892/etm.2022.11239
168. Su G, Yang W, Wang S, Geng C, Guan X. SIRT1-autophagy axis inhibits excess iron-induced ferroptosis of foam cells and subsequently increases IL-1Β and IL-18. Biochem Biophys Res Commun. (2021) 561:33–9. doi: 10.1016/j.bbrc.2021.05.011
169. Karshovska E, Schober A. HIF-1α promotes necroptosis in macrophages by micrornas. Atherosclerosis. (2017) 263:e11. doi: 10.1016/j.atherosclerosis.2017.06.063
170. Yu W, Liu W, Xie D, Wang Q, Xu C, Zhao H, et al. High level of uric acid promotes atherosclerosis by targeting NRF2-mediated autophagy dysfunction and ferroptosis. Oxid Med Cell Longev. (2022) 2022:e9304383. doi: 10.1155/2022/9304383
171. Peng Q, Liu H, Luo Z, Zhao H, Wang X, Guan X. Effect of autophagy on ferroptosis in foam cells via Nrf2. Mol Cell Biochem. (2022) 477:1597–606. doi: 10.1007/s11010-021-04347-3
172. Liu W, Östberg N, Yalcinkaya M, Dou H, Endo-Umeda K, Tang Y, et al. Erythroid lineage Jak2V617F expression promotes atherosclerosis through erythrophagocytosis and macrophage ferroptosis. J Clin Invest. (2022) 132:e155724. doi: 10.1172/JCI155724
173. Puylaert P, Roth L, Van Praet M, Pintelon I, Dumitrascu C, van Nuijs A, et al. Effect of erythrophagocytosis-induced ferroptosis during angiogenesis in atherosclerotic plaques. Angiogenesis. (2023) 26:505–22. doi: 10.1007/s10456-023-09877-6
174. Bao X, Luo X, Bai X, Lv Y, Weng X, Zhang S, et al. Cigarette tar mediates macrophage ferroptosis in atherosclerosis through the hepcidin/FPN/SLC7A11 signaling pathway. Free Radic Biol Med. (2023) 201:76–88. doi: 10.1016/j.freeradbiomed.2023.03.006
175. Nogieć A, Bzowska M, Demczuk A, Varol C, Guzik K. Phenotype and response to PAMPs of human monocyte-derived foam cells obtained by long-term culture in the presence of oxLDLs. Front Immunol. (2020) 11:1592. doi: 10.3389/fimmu.2020.01592
176. Li X, Kong D, Chen H, Liu S, Hu H, Wu T, et al. miR-155 acts as an anti-inflammatory factor in atherosclerosis-associated foam cell formation by repressing calcium-regulated heat stable protein 1. Sci Rep. (2016) 6:21789. doi: 10.1038/srep21789
177. Chen T, Huang Z, Wang L, Wang Y, Wu F, Meng S, et al. MicroRNA-125a-5p partly regulates the inflammatory response, lipid uptake, and ORP9 expression in oxLDL-stimulated monocyte/macrophages. Cardiovasc Res. (2009) 83:131–9. doi: 10.1093/cvr/cvp121
178. Fitzsimons S, Oggero S, Bruen R, McCarthy C, Strowitzki MJ, Mahon NG, et al. microRNA-155 is decreased during atherosclerosis regression and is increased in urinary extracellular vesicles during atherosclerosis progression. Front Immunol. (2020) 11:576516. doi: 10.3389/fimmu.2020.576516
179. Nie L, Zhao P, Yue Z, Zhang P, Ji N, Chen Q, et al. Diabetes induces macrophage dysfunction through cytoplasmic dsDNA/AIM2 associated pyroptosis. J Leukoc Biol. (2021) 110:497–510. doi: 10.1002/JLB.3MA0321-745R
180. Edgar L, Akbar N, Braithwaite AT, Krausgruber T, Gallart-Ayala H, Bailey J, et al. Hyperglycemia induces trained immunity in macrophages and their precursors and promotes atherosclerosis. Circulation. (2021) 144:961–82. doi: 10.1161/CIRCULATIONAHA.120.046464
181. Kluck GEG, Qian AS, Sakarya EH, Quach H, Deng YD, Trigatti BL. Apolipoprotein A1 protects against necrotic core development in atherosclerotic plaques: PDZK1-dependent high-density lipoprotein suppression of necroptosis in macrophages. Arterioscler Thromb Vasc Biol. (2023) 43:45–63. doi: 10.1161/ATVBAHA.122.318062
182. Xu J, Han X, Xia N, Zhao Q, Cheng Z. IL−37 suppresses macrophage ferroptosis to attenuate diabetic atherosclerosis via the NRF2 pathway. Exp Ther Med. (2023) 25:289. doi: 10.3892/etm.2023.11988
183. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. (2013) 53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320
184. Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. (2000) 88:1474–80. doi: 10.1152/jappl.2000.88.4.1474
185. Peled M, Fisher EA. Dynamic aspects of macrophage polarization during atherosclerosis progression and regression. Front Immunol. (2014) 5:579. doi: 10.3389/fimmu.2014.00579
186. Wu J, He S, Song Z, Chen S, Lin X, Sun H, et al. Macrophage polarization states in atherosclerosis. Front Immunol. (2023) 14:1185587. doi: 10.3389/fimmu.2023.1185587
187. Stöger JL, Gijbels MJJ, van der Velden S, Manca M, van der Loos CM, Biessen EAL, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. (2012) 225:461–8. doi: 10.1016/j.atherosclerosis.2012.09.013
188. Bi Y, Chen J, Hu F, Liu J, Li M, Zhao L. M2 macrophages as a potential target for antiatherosclerosis treatment. Neural Plast. (2019) 2019:6724903. doi: 10.1155/2019/6724903
189. Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. (2010) 107:737–46. doi: 10.1161/CIRCRESAHA.109.215715
190. Marques L, Negre-Salvayre A, Costa L, Canonne-Hergaux F. Iron gene expression profile in atherogenic Mox macrophages. Biochim Biophys Acta BBA - Mol Basis Dis. (2016) 1862:1137–46. doi: 10.1016/j.bbadis.2016.03.004
191. Boyle JJ, Johns M, Kampfer T, Nguyen AT, Game L, Schaer DJ, et al. Activating transcription factor 1 directs mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res. (2012) 110:20–33. doi: 10.1161/CIRCRESAHA.111.247577
192. Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. (2012) 59:166–77. doi: 10.1016/j.jacc.2011.10.852
193. Theurl I, Theurl M, Seifert M, Mair S, Nairz M, Rumpold H, et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood. (2008) 111:2392–9. doi: 10.1182/blood-2007-05-090019
194. Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood. (2006) 107:3727–32. doi: 10.1182/blood-2005-06-2259
195. Depuydt MAC, Prange KHM, Slenders L, Örd T, Elbersen D, Boltjes A, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. (2020) 127:1437–55. doi: 10.1161/CIRCRESAHA.120.316770
196. Li C-G, Yan L, Mai F-Y, Shi Z-J, Xu L-H, Jing Y-Y, et al. Baicalin inhibits NOD-like receptor family, pyrin containing domain 3 inflammasome activation in murine macrophages by augmenting protein kinase A signaling. Front Immunol. (2017) 8:1409. doi: 10.3389/fimmu.2017.01409
197. Shi F, Yuan L, Wong T, Li Q, Li Y, Xu R, et al. Dimethyl fumarate inhibits necroptosis and alleviates systemic inflammatory response syndrome by blocking the RIPK1-RIPK3-MLKL axis. Pharmacol Res. (2023) 189:106697. doi: 10.1016/j.phrs.2023.106697
198. Hosseini Z, Marinello M, Decker C, Sansbury BE, Sadhu S, Gerlach BD, et al. Resolvin D1 enhances necroptotic cell clearance through promoting macrophage fatty acid oxidation and oxidative phosphorylation. Arterioscler Thromb Vasc Biol. (2021) 41:1062–75. doi: 10.1161/ATVBAHA.120.315758
199. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. (2004) 104:2224–34. doi: 10.1182/blood-2004-03-1109
200. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. (2021) 22:6995. doi: 10.3390/ijms22136995
201. Ji ZZ, Chan MK-K, Chan AS-W, Leung K-T, Jiang X, To K-F, et al. Tumour-associated macrophages: versatile players in the tumour microenvironment. Front Cell Dev Biol. (2023) 11:1261749. doi: 10.3389/fcell.2023.1261749
202. Yin M, Li X, Tan S, Zhou HJ, Ji W, Bellone S, et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J Clin Invest. (2016) 126:4157–73. doi: 10.1172/JCI87252
203. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discovery. (2018) 17:887–904. doi: 10.1038/nrd.2018.169
204. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
205. Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med. (2013) 210:491–502. doi: 10.1084/jem.20122006
206. Maltez VI, Tubbs AL, Cook KD, Aachoui Y, Falcone EL, Holland SM, et al. Inflammasomes coordinate pyroptosis and natural killer cell cytotoxicity to clear infection by a ubiquitous environmental bacterium. Immunity. (2015) 43:987–97. doi: 10.1016/j.immuni.2015.10.010
207. Jia Y, Wang X, Deng Y, Li S, Xu X, Qin Y, et al. Pyroptosis provides new strategies for the treatment of cancer. J Cancer. (2023) 14:140–51. doi: 10.7150/jca.77965
208. Wu S, Li T, Liu W, Huang Y. Ferroptosis and cancer: complex relationship and potential application of exosomes. Front Cell Dev Biol. (2021) 9:733751. doi: 10.3389/fcell.2021.733751
209. Chen Y, Fan Z, Hu S, Lu C, Xiang Y, Liao S. Ferroptosis: A new strategy for cancer therapy. Front Oncol. (2022) 12:830561. doi: 10.3389/fonc.2022.830561
210. Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther. (2022) 7:1–26. doi: 10.1038/s41392-022-01046-3
211. Lang C, Roy S, Wang Y, Graves D, Xu Y, Serezani CH, et al. Efferocytosis drives myeloid NLRP3 dependent inflammasome signaling secretion of IL-1β to promote tumor growth. Front Immunol. (2022) 13:993771. doi: 10.3389/fimmu.2022.993771
212. Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J Exp Med. (2017) 214:2695–713. doi: 10.1084/jem.20160392
213. Gu Z, Liu T, Liu C, Yang Y, Tang J, Song H, et al. Ferroptosis-strengthened metabolic and inflammatory regulation of tumor-associated macrophages provokes potent tumoricidal activities. Nano Lett. (2021) 21:6471–9. doi: 10.1021/acs.nanolett.1c01401
214. Lian Q, Xu J, Yan S, Huang M, Ding H, Sun X, et al. Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Res. (2017) 27:784–800. doi: 10.1038/cr.2017.54
215. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. (2020) 5:eaax7969. doi: 10.1126/sciimmunol.aax7969
216. Li L, Wu L, Yin X, Li C, Hua Z. Bulk and single-cell transcriptome analyses revealed that the pyroptosis of glioma-associated macrophages participates in tumor progression and immunosuppression. Oxid Med Cell Longev. (2022) 2022:1803544. doi: 10.1155/2022/1803544
217. Hage C, Hoves S, Strauss L, Bissinger S, Prinz Y, Pöschinger T, et al. Sorafenib induces pyroptosis in macrophages and triggers natural killer cell-mediated cytotoxicity against hepatocellular carcinoma. Hepatol Baltim Md. (2019) 70:1280–97. doi: 10.1002/hep.30666
218. Wu L, Zhang X, Zheng L, Zhao H, Yan G, Zhang Q, et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res. (2020) 8:710–21. doi: 10.1158/2326-6066.CIR-19-0261
219. Hao X, Zheng Z, Liu H, Zhang Y, Kang J, Kong X, et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. (2022) 56:102463. doi: 10.1016/j.redox.2022.102463
220. Tang B, Zhu J, Wang Y, Chen W, Fang S, Mao W, et al. Targeted xCT-mediated Ferroptosis and Protumoral Polarization of Macrophages Is Effective against HCC and Enhances the Efficacy of the Anti-PD-1/L1 Response. Adv Sci. (2023) 10:2203973. doi: 10.1002/advs.202203973
221. Wang W, Marinis JM, Beal AM, Savadkar S, Wu Y, Khan M, et al. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell. (2018) 34:757–774.e7. doi: 10.1016/j.ccell.2018.10.006
222. Li L, Yu S, Zang C. Low necroptosis process predicts poor treatment outcome of human papillomavirus positive cervical cancers by decreasing tumor-associated macrophages M1 polarization. Gynecol Obstet Invest. (2018) 83:259–67. doi: 10.1159/000487434
223. Li L-G, Peng X-C, Yu T-T, Xu H-Z, Han N, Yang X-X, et al. Dihydroartemisinin remodels macrophage into an M1 phenotype via ferroptosis-mediated DNA damage. Front Pharmacol. (2022) 13:949835. doi: 10.3389/fphar.2022.949835
224. Chen W, Zuo F, Zhang K, Xia T, Lei W, Zhang Z, et al. Exosomal MIF derived from nasopharyngeal carcinoma promotes metastasis by repressing ferroptosis of macrophages. Front Cell Dev Biol. (2021) 9:791187. doi: 10.3389/fcell.2021.791187
225. Deng T, Tang C, Zhang G, Wan X. DAMPs released by pyroptotic cells as major contributors and therapeutic targets for CAR-T-related toxicities. Cell Death Dis. (2021) 12:129. doi: 10.1038/s41419-021-03428-x
226. Liu Y-G, Chen J-K, Zhang Z-T, Ma X-J, Chen Y-C, Du X-M, et al. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis. (2017) 8:e2579. doi: 10.1038/cddis.2016.460
227. Watanabe S, Usui-Kawanishi F, Karasawa T, Kimura H, Kamata R, Komada T, et al. Glucose regulates hypoxia-induced NLRP3 inflammasome activation in macrophages. J Cell Physiol. (2020) 235:7554–66. doi: 10.1002/jcp.29659
228. Wang X, Yang Y, Cai W-Q, Lu Y. The relationship of sphingosine kinase 1 with pyroptosis provides a new strategy for tumor therapy. Front Immunol. (2020) 11:574990. doi: 10.3389/fimmu.2020.574990
229. Zhao J, He D, Lai HM, Xu Y, Luo Y, Li T, et al. Comprehensive histological imaging of native microbiota in human glioma. J Biophotonics. (2022) 15:e202100351. doi: 10.1002/jbio.202100351
230. McGarry T, Biniecka M, Veale DJ, Fearon U. Hypoxia, oxidative stress and inflammation. Free Radic Biol Med. (2018) 125:15–24. doi: 10.1016/j.freeradbiomed.2018.03.042
231. Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat Chem Biol. (2017) 13:46–53. doi: 10.1038/nchembio.2229
232. Walsh MP, Duncan B, Larabee S, Krauss A, Davis JPE, Cui Y, et al. Val-boroPro accelerates T cell priming via modulation of dendritic cell trafficking resulting in complete regression of established murine tumors. PloS One. (2013) 8:e58860. doi: 10.1371/journal.pone.0058860
233. Yan J, Wan P, Choksi S, Liu Z-G. Necroptosis and tumor progression. Trends Cancer. (2022) 8:21–7. doi: 10.1016/j.trecan.2021.09.003
234. Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. (2016) 532:245–9. doi: 10.1038/nature17403
235. Sprooten J, De Wijngaert P, Vanmeerbeek I, Martin S, Vangheluwe P, Schlenner S, et al. Necroptosis in immuno-oncology and cancer immunotherapy. Cells. (2020) 9:1823. doi: 10.3390/cells9081823
236. Zhao Y-Y, Lian J-X, Lan Z, Zou K-L, Wang W-M, Yu G-T. Ferroptosis promotes anti-tumor immune response by inducing immunogenic exposure in HNSCC. Oral Dis. (2021) 29:933-41. doi: 10.1111/odi.14077
237. Hu Z-W, Wen Y-H, Ma R-Q, Chen L, Zeng X-L, Wen W-P, et al. Ferroptosis driver SOCS1 and suppressor FTH1 independently correlate with M1 and M2 macrophage infiltration in head and neck squamous cell carcinoma. Front Cell Dev Biol. (2021) 9:727762. doi: 10.3389/fcell.2021.727762
238. Wang L, He T, Liu J, Tai J, Wang B, Chen Z, et al. Pan-cancer analysis reveals tumor-associated macrophage communication in the tumor microenvironment. Exp Hematol Oncol. (2021) 10:31. doi: 10.1186/s40164-021-00226-1
239. Kolb R, Liu G-H, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in cancer: a double-edged sword. Protein Cell. (2014) 5:12–20. doi: 10.1007/s13238-013-0001-4
240. Mázló A, Jenei V, Burai S, Molnár T, Bácsi A, Koncz G. Types of necroinflammation, the effect of cell death modalities on sterile inflammation. Cell Death Dis. (2022) 13:1–12. doi: 10.1038/s41419-022-04883-w
241. Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity. (2015) 42:145. doi: 10.1016/j.immuni.2014.12.020
242. Cooper KN, Potempa J, Bagaitkar J. Dying for a cause: The pathogenic manipulation of cell death and efferocytic pathways. Mol Oral Microbiol. (2023). doi: 10.1111/omi.12436
243. Sai K, Parsons C, House JS, Kathariou S, Ninomiya-Tsuji J. Necroptosis mediators RIPK3 and MLKL suppress intracellular Listeria replication independently of host cell killing. J Cell Biol. (2019) 218:1994. doi: 10.1083/jcb.201810014
244. Gonzalez-Juarbe N, Bradley KM, Riegler AN, Reyes LF, Brissac T, Park S-S, et al. Bacterial pore-forming toxins promote the activation of caspases in parallel to necroptosis to enhance alarmin release and inflammation during pneumonia. Sci Rep. (2018) 8:5846. doi: 10.1038/s41598-018-24210-8
245. Wang Y, Kanneganti T-D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J. (2021) 19:4641–57. doi: 10.1016/j.csbj.2021.07.038
Keywords: macrophages, cell death, pyroptosis, necroptosis, ferroptosis, tumor-associated macrophages, atherosclerosis, cancer
Citation: Makuch M, Stepanechko M and Bzowska M (2024) The dance of macrophage death: the interplay between the inevitable and the microenvironment. Front. Immunol. 15:1330461. doi: 10.3389/fimmu.2024.1330461
Received: 30 October 2023; Accepted: 26 February 2024;
Published: 20 March 2024.
Edited by:
Philippe Saas, Etablissement Français du Sang AuRA, FranceReviewed by:
Xiao Wang, University of Illinois Chicago, United StatesAmiram Ariel, University of Haifa, Israel
Copyright © 2024 Makuch, Stepanechko and Bzowska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Małgorzata Bzowska, bWFsZ29yemF0YS5iem93c2thQHVqLmVkdS5wbA==
†These authors have contributed equally to this work and share first authorship