 Ivan Talucci1,2†
Ivan Talucci1,2† Friederike A. Arlt3,4†
Friederike A. Arlt3,4† Kai O. Kreissner1
Kai O. Kreissner1 Mahoor Nasouti1,3Anna-Lena Wiessler5
Mahoor Nasouti1,3Anna-Lena Wiessler5 Ramona Miske6Swantje Mindorf6Inga Dettmann6Mehrnaz Moniri1Markus Bayer1Peter Broegger Christensen7
Ramona Miske6Swantje Mindorf6Inga Dettmann6Mehrnaz Moniri1Markus Bayer1Peter Broegger Christensen7 Ilya Ayzenberg8Andrea Kraft9
Ilya Ayzenberg8Andrea Kraft9 Matthias Endres4,10,11,12,13
Matthias Endres4,10,11,12,13 Lars Komorowski6
Lars Komorowski6 Carmen Villmann5
Carmen Villmann5 Kathrin Doppler2
Kathrin Doppler2 Harald Prüss3,4*
Harald Prüss3,4* Hans M. Maric1*
Hans M. Maric1*- 1Rudolf Virchow Center for Integrative and Translational Bioimaging; University of Würzburg, Würzburg, Germany
- 2Department of Neurology, University Hospital Würzburg, Würzburg, Germany
- 3Department of Neurology and Experimental Neurology, Charité-Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität Berlin, Berlin, Germany
- 4German Center for Neurodegenerative Diseases (DZNE), Berlin, Germany
- 5Institute for Clinical Neurobiology, University of Wuerzburg, Würzburg, Germany
- 6Institute for Experimental Immunology, affiliated to EUROIMMUN Medizinische Labordiagnostika AG, Lübeck, Germany
- 7Department of Neurology, Aalborg University Hospital, Aalborg, Denmark
- 8Department of Neurology, St. Josef-Hospital, Ruhr-University Bochum, Bochum, Germany
- 9Department of Neurology, Hospital Martha-Maria, Halle, Germany
- 10Klinik und Hochschulambulanz für Neurologie, Charité-Universitätsmedizin, Berlin, Germany
- 11Center for Stroke Research, Berlin, Germany
- 12German Centre for Cardiovascular Research (DZHK), Berlin, Germany
- 13German Center for Mental Health (DZPG), Berlin, Germany
Introduction: Subgroups of autoantibodies directed against voltage-gated potassium channel (Kv) complex components have been associated with immunotherapy-responsive clinical syndromes. The high prevalence and the role of autoantibodies directly binding Kv remain, however, controversial. Our objective was to determine Kv autoantibody binding requirements and to clarify their contribution to the observed immune response.
Methods: Binding epitopes were studied in sera (n = 36) and cerebrospinal fluid (CSF) (n = 12) from a patient cohort positive for Kv1.2 but negative for 32 common neurological autoantigens and controls (sera n = 18 and CSF n = 5) by phospho and deep mutational scans. Autoantibody specificity and contribution to the observed immune response were resolved on recombinant cells, cerebellum slices, and nerve fibers.
Results: 83% of the patients (30/36) within the studied cohort shared one out of the two major binding epitopes with Kv1.2-3 reactivity. Eleven percent (4/36) of the serum samples showed no binding. Fingerprinting resolved close to identical sequence requirements for both shared epitopes. Kv autoantibody response is directed against juxtaparanodal regions in peripheral nerves and the axon initial segment in central nervous system neurons and exclusively mediated by the shared epitopes.
Discussion: Systematic mapping revealed two shared autoimmune responses, with one dominant Kv1.2-3 autoantibody epitope being unexpectedly prevalent. The conservation of the molecular binding requirements among these patients indicates a uniform autoantibody repertoire with monospecific reactivity. The enhanced sensitivity of the epitope-based (10/12) compared with that of the cell-based detection (7/12) highlights its use for detection. The determined immunodominant epitope is also the primary immune response visible in tissue, suggesting a diagnostic significance and a specific value for routine screening.
Highlights
● Kv autoantibodies are prevalent but of controversial diagnostic significance.
● Kv-exclusive autoimmunity in 30 patients is characterized by a uniform and monospecific autoantibody repertoire.
● A single immunodominant Kv1.2/1.3 autoantibody epitope has been identified.
● The dominant epitope manifests as the only immune response in all tested tissues and cells.
Introduction
Neurological diseases associated with autoantibodies are increasingly recognized as new medical entities (1) but frequently remain to be fully resolved at the molecular level. The identification of exact underlying disease-defining autoantibody epitopes is critical for understanding and addressing the root cause of these clinical entities. Advanced peptide microarray technologies demonstrated a remarkable success in resolving comprehensive linear epitope landscapes from raw patient samples (2, 3). Here, we used a peptide microarray–based readout (4) for analyzing the largest so far studied cohort with Kv1.2-exclusive immune response in molecular detail.
Autoantibodies directed against Kv1 channel complexes have been identified in several neurological diseases, including autoimmune encephalitis, limbic encephalitis, and Morvan’s syndrome (5, 6). Within this group, anti-leucine-rich glioma inactivated 1 (LGI1) and anti-Contactin-associated protein-like 2 (Caspr2) autoantibodies are among the most prevalent, and these have been associated with clinical syndromes that are immunotherapy-responsive (7, 8). Previous studies also identified highly prevalent intracellular binding of Kv1 antibodies, many of which are targeting intracellular epitopes. Only a fraction of the intracellular positive Kv1 patients (27%) showed a sustained immunotherapy benefit (7). The subgroup of Kv1 channels play a critical role in regulating neurotransmission in both the central and peripheral nervous system by controlling the flux of potassium ions from the neuron during the action potential. Kv1.3 was related not only to astrocyte activation in experimental autoimmune encephalitis but also to CD4+ T-cell differentiation during inflammatory immune–mediated disease (9). Pharmacological and knockout blockade of this channel has been shown to suppress these functions (10, 11). Kv1.2 knockout mice show seizures in early developmental stages (12). Furthermore, Kv1.2 dysfunction has been associated not only with epilepsy (13–15) and developmental disorders (16, 17) but also with multiple sclerosis (18) and neuroinflammatory disorders due to its crucial role in T-lymphocytes (19). The role of Kv1.2 in autoimmune disorders remains to be fully explored (20). Kv1.2 autoantibodies were shown to exacerbate an epileptic phenotype in rodent models (21), and cases of Kv1 autoantibody–associated limbic encephalitis were reported to be immunotherapy-sensitive (22). In vitro evidence suggests that autoantibodies can potentially reach their intracellular epitopes through Fc receptor–mediated internalization (23, 24), consequently leading to a smoldering autoimmunity. The high prevalence and the functional role of anti–Kv1.2 autoantibodies in vivo and their association with specific clinical phenotypes are currently undefined, thereby potentially limiting diagnostic and therapeutic options. This is in part due to the lack of molecular knowledge on the involved epitopes and their contribution to the observed immune response.
Here, we report the screening, mapping, and validation of two Kv1.2 epitopes in 32 patients, thereby providing detailed molecular information on Kv1.2 autoimmunity and a basis for the development of diagnostic approaches.
Methods
Collection of human sera and CSFs
The study comprised sera from 18 controls and 36 patients, along with cerebrospinal fluid (CSF) from five controls and 12 patients. The patients exhibited diverse neuropsychiatric disease phenotypes, including neurodegenerative disorders and autoimmune encephalitis. Two patients (ID 20 and 21), included in this work, have been previously published (20); their CSF samples were not available. Data of all patients were collected at the Charité, Berlin, and all participants gave an informed written consent. All patient sera underwent cell-based screening at Euroimmun (EUROIMMUN Medizinische Labordiagnostika AG). Immunoglobulin G (IgG) detection was conducted using a biochip array with acetone-fixed recombinant human embryonic kidney 293 (HEK293) cells separately expressing the following autoantigens: Aquaporin-4 (AQP4), Rho GTPase activating protein 26 (ARHGAP26), sodium-potassium ATPase catalytic subunit alpha-3 (ATP1A3), Contactin-1 (CNTN1), collapsin response mediator protein 5 (CRMP5 or CV2), Carbonic Anhydrase-Related Protein VIII (CARP VIII), contactin-associated protein-like 2 (CASPR2), Tr/Delta/Notch-like epidermal growth factor-related receptor (DNER/Tr), Dipeptidyl-Peptidase-like Protein-6 (DPPX), Flotilin 1/2, γ-aminobutyric acid type B (GABAB) receptor, glutamic acid decarboxylase 65 (GAD65), γ-aminobutyric acid type A (GABAA)receptor, glycine receptor, glutamate receptors [types α-Amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor, Glutamate Receptor ionotropic delta-2 (GLURD2), Metabotropic glutamate receptor 1 (mGluR1), Metabotropic glutamate receptor 5 (MGluR5), N-methyl-D-aspartate (NMDA)], Homer 3, Hu, Inositol 1,4,5-Trisphosphate Receptor Type 1 (ITPR1), IgLON family member 5 (IgLON5), Kv1.2, leucine-rich glioma inactivated 1 (LGI1), myelin oligodendrocyte glycoprotein (MOG), Neurochondrin (NCDN), Neurofascin 155 (NF155), Neurofascin 186 (NF186), Paraneoplastic antigen Ma2 (PNMA2), recoverin, Ri paraneoplastic antigen, Seizure Related 6 Homolog Like 2 (Sez6L2), Yo paraneoplastic antigen, Zic Family Member 4 (ZIC4). The initial laboratory pre-screening encompassed a cohort of 96 healthy controls, from which four individuals exhibited positive Kv1.2 autoantibodies (4.2% positive healthy donors). Subsequently, these samples were subjected to detailed peptide microarray–based evaluation.
Microarray synthesis and quality control
The complete Kv sequences (UniProtKB: Q09470, P16389, P22001, and P22459) were displayed in microarray format as 15-mer overlapping peptide libraries. Peptide arrays were synthesized using µSPOT (25), a SPOT-based (26) synthesis approach. In brief, custom-prepared discs containing 9-fluorenylmethyloxycarbonyl(Fmoc)-β-alanine linkers (average loading: 130 nmol/disc, 4 mm in diameter) were loaded in a MultiPep rSi robot (CEM GmbH, Kamp-Lintford, Germany) together natural amino acid (AA) building blocks and phospho-building blocks from IRIS (IRIS Biotech GmbH, Marktredwitz, Germany) and the freshly prepared reagents. Synthesis was carried out by deprotecting the Fmoc-group using 20% piperidine in dimethylformamide (DMF). Peptide chains were elongated using a coupling solution consisting of aAs (0.5 M) with Oxyma (1 M) and diisopropylmethanediimine (1 M) in DMF (1:1:1). Coupling steps were carried out three times (30 min), followed by capping (4% acetic anhydride in DMF). Discs were transferred into 96–deep-well plates for the workup. Side chains were deprotected using 90% trifluoracetic acid (TFA), 2% dichloromethane (DCM), 5% H2O, and 3% triisopropylsilane (TIPS) (150 μL per well) for 1 h at room temperature (RT). Afterwards, the deprotection solution was removed, and the discs were solubilized overnight (ON) at RT while shaking, using a solvation mixture containing 88.5% TFA, 4% trifluoromethanesulfonic acid, 5% H2O, and 2.5% TIPS (250 μL per well). The resulting peptide-cellulose conjugates were precipitated with ice-cold ether (700 μL per well) and spin down at 2,000×g for 10 min at 4°C, followed by two additional washes of the formed pellet with ice-cold ether. The resulting pellets were dissolved in DMSO (250 μL per well).
Liquid chromatography–mass spectrometry (LC-MS) (27) was carried out using peptide quality controls that were cleaved from the solid support. To ensure cleavage, a Rink amide linker (Iris) suitable for Solid Phase Peptide synthesis (SPPS) on cellulose support was introduced during the first coupling cycle. In an acidic environment, the quality controls were cleaved off the solid support. To isolate the quality controls, 150 µL of the supernatant was transferred to 1.5-mL reaction tubes, followed by the addition of 700 µL of diethyl ether. The samples were then vortexed, and the peptides were allowed to precipitate by incubation at −20°C overnight. After centrifugation at 13,300×g and 4°C for 10 min, the supernatant was discarded, and 500 µL of diethyl ether was added. The mixture was vortexed and centrifuged for 10 min, and the supernatant was decanted. This process was repeated twice, and the peptides were left to dry for 60 min. Finally, the Rink amides were dissolved in 50 µL of 50% acetonitrile and 0.1% formic acid (v/v) and vortexed briefly before centrifugation at 13,300×g and RT. For analysis, the quality controls were diluted 1:3 and analyzed via LC-MS (Agilent technologies).
Microarray printing and binding assay
Peptide-cellulose conjugate (PCC) solutions were mixed 2:1 with saline-sodium citrate buffer [150 mM NaCl and 15 mM trisodium citrate (pH 7.0)] and transferred to a 384-well plate. For transfer of the PCC solutions to white-coated CelluSpot blank slides (76 mm × 26 mm, Intavis AG Peptide Services GmbH and Co. KG), a SlideSpotter (CEM GmbH) was used. After completion of the printing procedure, slides were left to dry overnight.
The microarray slides were blocked for 60 min in 5% (w/v) skimmed milk powder (Carl Roth) 0.05% Tween 20 phosphate-buffered saline [PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 (pH7.4)]. After blocking, the slides were incubated for 30 min with either positive and negative serum or cerebrospinal fluid (dilution ranging from 1:300 to 1:3,000) or Kv1.2 monoclonal (NeuroMab clone: K14/16 at 0.5 µg/mL) in the blocking buffer and then washed three times with PBS 0.05% and Tween 20 for 1 min. IgG antibodies were detected using goat anti-human IgG-Horseradish peroxidase (HRP) (Thermo Fisher, Cat. No. 31410; 1:2,500) or goat anti-mouse IgG-HRP (Thermo Fisher, Cat. No. 31430; 1:5000). The chemiluminescence readout was detected with an Azure imaging system c400 (lowest sensitivity, exposure time of 60 s for serum and of 30 s for monoclonal) using SuperSignal West Femto maximum sensitive substrate (Thermo Scientific, GmbH, Schwerte, Germany). Microarray binding intensities were quantified using the MicroArray Rastering Tool – MARTin (28). Neutralization assays were performed by pre-incubating the sera in neutralizing solution, containing cleavable peptides or buffer for 30 min. The binding assay followed in the same manner.
Recombinant expression of Kv1 in human embryonic kidney 293 cells
Kv1.1, Kv1.2, and Kv1.6 were expressed in HEK293 cells, following a previously described protocol (Miske et al., 2023). To summarize, genomic DNA was extracted from HEK293 cells and utilized as a template for Kv1.2 coding sequence amplification via polymerase chain reaction (PCR). Respective DNA oligonucleotides were employed to introduce the required enzyme restriction sites (Table 1). Indicated enzymes were used to digest the resulting PCR fragments and subsequently ligated with NcoI/XhoI-linearized pTriEx-1 (Merck). Prior to transfection, HEK293 cells were plated on sterile poly-L-lysine–treated coverslips. Transient expression of Kv1-encoded proteins was accomplished through Polyethylenimine (PEI)-mediated transfection (PEI 25K™) following the manufacturer’s instructions (Polysciences, Europe). After 48 h of transfection, cells were fixed, permeabilized with acetone, and rinsed with PBS before conducting the immunofluorescence described below.
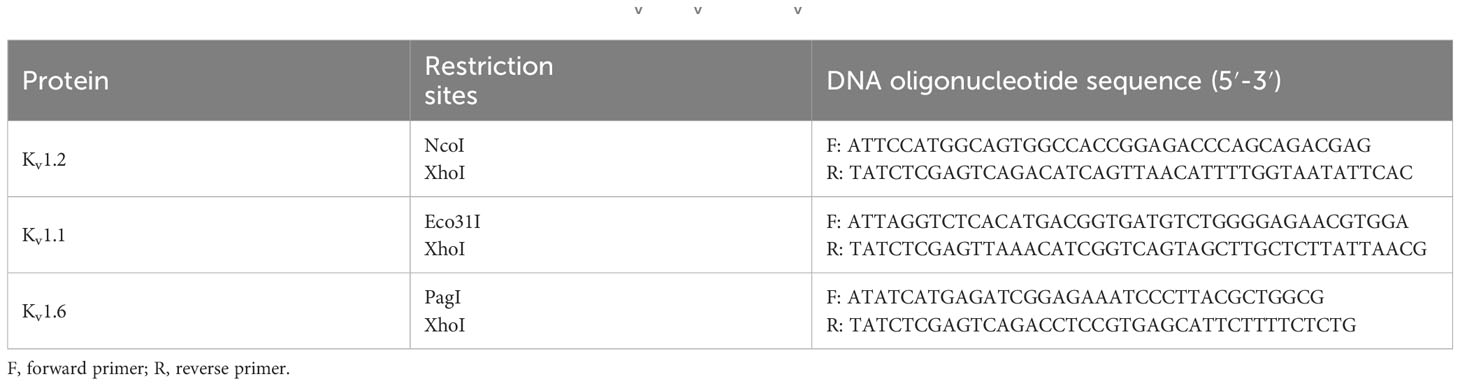
Table 1 DNA oligonucleotide primers for PCR amplification of Kv1.2, Kv1.1, and Kv1.6.
Mouse brain and sciatic nerve processing
All animal procedures were approved by the Landesamt für Gesundheit und Soziales (LaGeSo) Berlin, Germany (approval numbers T-CH 0009/22), and conducted in compliance with the German and international guidelines for care and humane use of animals. Unfixed brain processing and sciatic nerve teased fiber preparations were performed as previously described (29). In brief, male C57BL/6 mice were used at an age of 10–12 weeks. Unfixed brains were dissected and frozen in 2-methylbutan. Cryostat-cut 20-µm sections were mounted on glass slides and used for tissue-based immunofluorescence. Murine sciatic nerves were dissected from hind limbs, fixed in 4% paraformaldehyde, and washed in PBS. The epineurium was removed prior to teasing. Teased fibers were air-dried and stored at −20°C until further usage.
Cell- and tissue-based immunofluorescence binding assays
Slides and cells were post-fixed with acetone and washed with PBS. Serum, CSF, and/or commercial Kv1.1 (Abcam ab65790), Kv1.2 (K14/16 Neuromab, supplier Antibodies Incorporated 75-008), and Kv1.6 antibody (Antibodies Incorporated 75-012) incubation was done overnight at +4°C in a dilution of 1:300 (serum), 1:2 (CSF), and 1:200 (commercial) in PBS-T (containing 0.2% Tween 20 and 0.1% Triton X). Secondary antibodies against human IgG (Alexa488-labeled Dianova, 109-545-003; and Cy3-labeled Dianova, 109-165-003), mouse IgG (Alexa549-labeled goat anti-mouse, Jackson Research, 115-585-03; and Alexa-488-labeled, Dianova, 115-546-003) and rabbit IgG (Alexa488-labeled, Dianova, 111-546-003) were added in a dilution of 1:1,000 in PBS-T for 1 h at RT. Cell nuclei were stained using 4',6-diamidino-2-phenylindole (DAPI). Coverslips and glass slides were mounted in Fluoroshield and visualized using a widefield fluorescence microscope (LeicaSPE).
Cell and tissue neutralization of anti-Kv sera
Neutralization assays were adapted from established protocols (Miske et al., 2023). In brief, peptides were solubilized in PBS at a final concentration of 1 mg/mL. Neutralization assays were performed on transfected HEK293 cells and on mouse tissues. Sera were diluted in PBS-T to the abovementioned concentrations. The peptide antigen was added in a dilution of 1:10. Sera and peptides were thereby pre-incubated for 1 h at RT and subsequently added to slides and cells incubating for 1 h at RT. After washing with PBS-T, cells and slides were stained with secondary Alexa488-labeled antibodies for 30 min at RT. After washing with PBS-T, cell nuclei were stained with DAPI. Co-staining with the commercial Kv1.2 antibody (K14/16 NeuroMab) and secondary Alexa594-labeled antibody was accomplished through sequential staining rounds, thereby avoiding potential cross-reactivity of secondary antibodies to primary mouse or human antibodies.
Data availability
Data are available upon reasonable request to qualified investigators for the purposes of replicating procedures and results.
Results and discussion
Patient samples
Screening, mapping, and validation of Kv1.2 epitopes was based on 36 samples from patients (74.3% men, median disease onset age of 65, median high sera titer of 1:1,000, with four out of the 96 positive healthy controls positive in this readout) positive for Kv1.2 but negative for 33 common neurological autoantigens in cell-based assays. Patients showed heterogeneous neuropsychiatric phenotypes ranging from dementia, to epilepsy, autoimmune encephalitis, ischemic strokes, and peripheral neuropathies. Patient ID 19 had anti-AP3B2 IgG (titer 1:32) and patient ID 30 anti-NMDAR IgM (1:1,000) autoantibodies in serum; in the rest of the cohort, no other co-existing antibodies were detected.
Single–amino acid resolution mapping of two immunodominant Kv1.2 epitopes
To identify anti-Kv1.2 epitopes, the qualified patient samples were screened in peptide microarray format. Here, the entire primary sequence of Kv1.2 was displayed in the form of 20-mer peptides with 17-residue overlap (Figure 1A). First, the array approach was validated using the commercial anti-mouse Kv1.2 (NeuroMab clone K14/16) antibody. The microarray defined the sequence NEDFRE as the core motif (Figure 1B), thereby recapitulating the expected (463EGVNNSNEDFREENLKTA480) epitope. Next, 22 seropositive (Figure 1C) and 13 seronegative (control) (Figure 1D) sera were probed on the Kv1.2 array. Here, autoantibody binding was detected using anti-human IgG coupled to HRP for chemiluminescence detection. The screening identified two prominent intracellular (Figure 1E) epitopes: Epitope 1 (E1) (469NEDFREENLKTANCTLA485) and Epitope 2 (E2) (481NCTLANTNYVNITK495). Notably, none of the tested sera shared both epitopes. An additional array library with a single-AA shift of 15-mer peptides (Figure 1F) defined 478KTANCTLA485 as the E1 core motif, which was shared among 30 patients (Serum IDs 1–6, 8–10, 14–21, 23–34, and 36) and 485ANTNYVNITK495 as the minimal required E2 core motif, which was shared by two patients (Serum IDs 11 and 24) (Table 2).
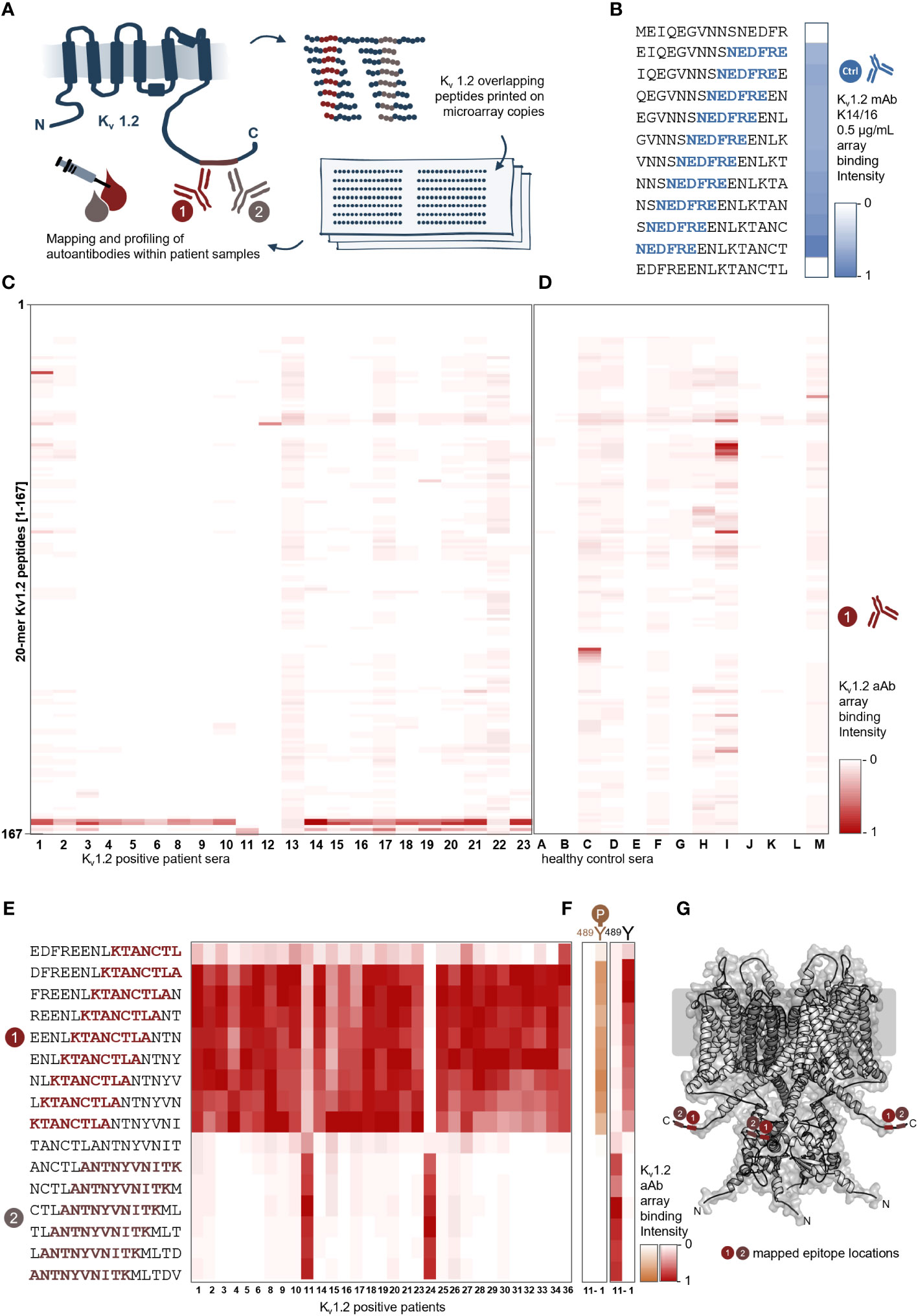
Figure 1 Peptide microarray screening of anti–Kv1.2-positive patient samples reveals two shared binding epitopes. (A) Scheme of the Kv1.2 microarray display. Kv1.2 complete primary sequence was displayed in microarray format as 20-mer peptides with 17-residue overlap, and autoantibody binding was detected using goat anti-human IgG coupled to HRP for chemiluminescence readout. (B) Array validation using a commercial antibody. The microarray reports the residues NEDFRE as the core motif for Anti-mouse Kv1.2 (NeuroMab clone K14/16) that was raised against immunogen (Fusion Kv1.2 residues: 428–499) and previously mapped for the following sequence EGVNNSNEDFREENLKTA. (C) Identification of two distinct epitopes from sera. Epitope mapping of 22 patient sera. The sera reactivity of 22 positive samples and (D) 13 negative samples over 161 peptides were analyzed and plotted as a heat map over the most prominent peptide binder [0-1]; each rectangle corresponds to a single patient sample reactivity. Two main epitopes were identified between the peptides 156–161. (E) Single–amino acid mapping resolves the two minimal motifs. A 15-mer library revealed a shortest motif for E1 (KTANCTLA) and E2 (ANTNYVNITK). (F) Phosphorylation dependency of the two autoantibody epitopes. Here, two different samples (left: sera 11; right: sera 1) were tested on unphosphorylated and phosphorylated Y-489 peptides. E2 from patient 11 is negatively affected by phosphorylation; in contrast, E1 from patient 1 is not affected. (G) Visualization of the identified core motifs. Cartoon model of the Kv1.2 tetramer highlighting the two intracellular epitopes.
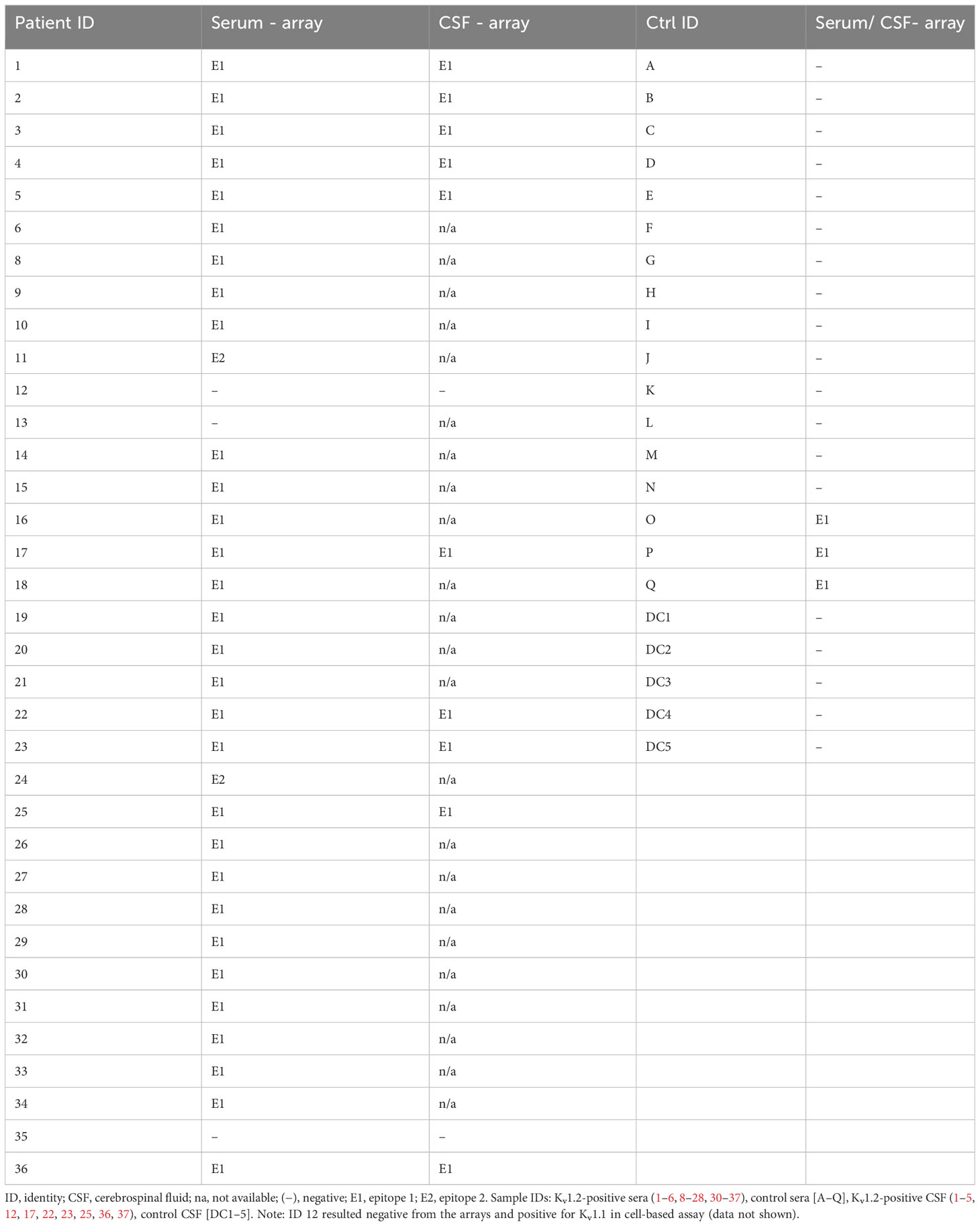
Table 2 Serum and cerebrospinal fluid peptide microarray samples used in this study.
Remarkably, the identified epitopes both overlap and include a validated phosphorylation site (389Tyr; PhosphoSitePlus: P16389). In order to delineate the differences in the mode of binding between these two antibodies and specifically their dependency on phosphorylation, we next probed the corresponding phospho-tyrosine Kv1.2 library. Whereas autoantibody binding of E1 was not affected, E2 binding was completely abolished upon phosphorylation (Figure 1G).
Autoantibody binding profiles hint toward a common molecular motif
To further resolve the binding requirements for autoantibodies, we conducted a deep mutational scan of the newly defined minimal binding epitopes. These libraries comprised all possible single-point variants of 18-mer peptides that harbored the minimal core motifs. The subsequent fingerprint analysis of the resulting 2 × 342 peptide variants (Figure 2A) confirmed both the previously defined minimal core motifs (Figure 1C) and the impact of the Tyr489 phosphorylation (Figure 1G).
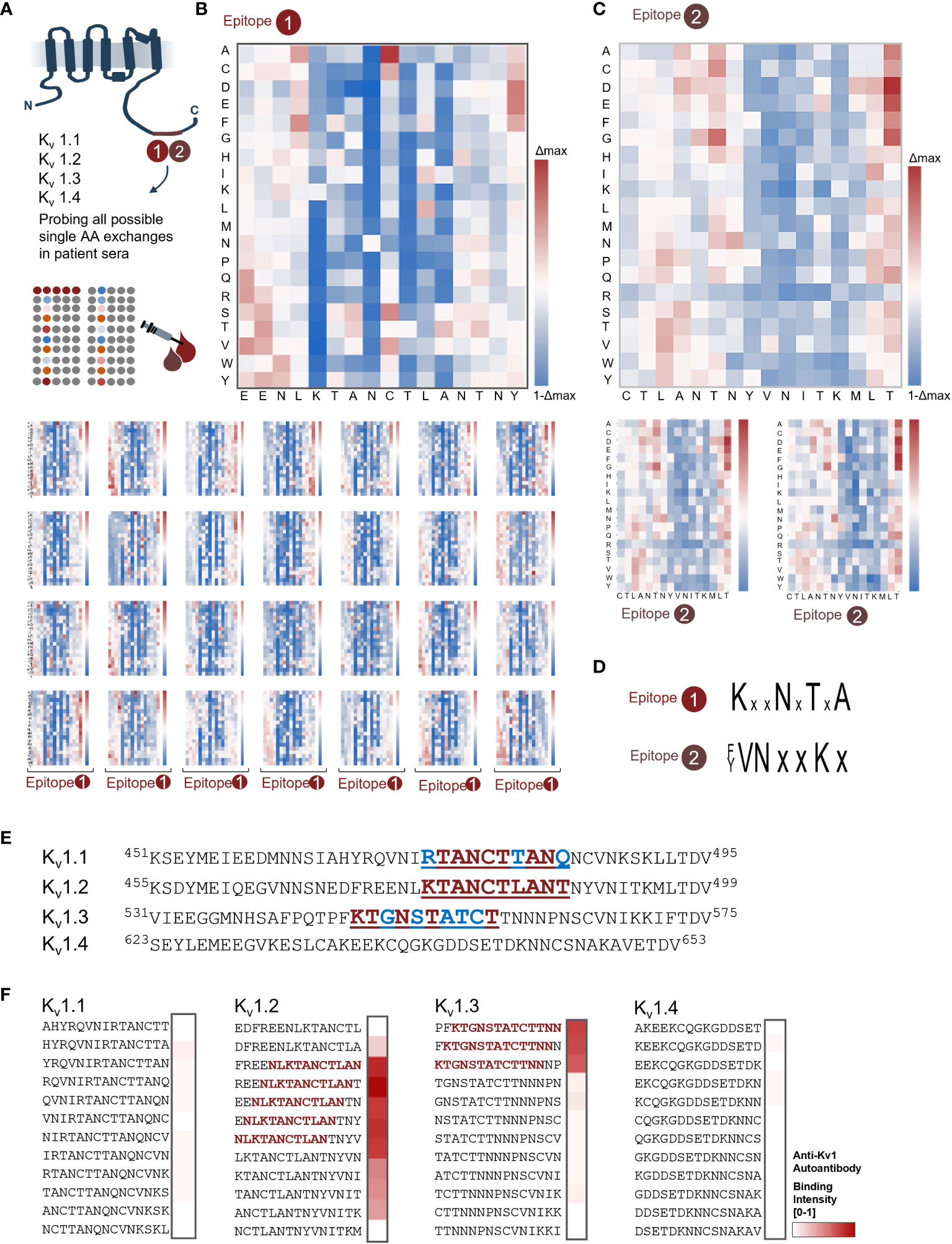
Figure 2 Deep mutational scans reveal common binding profiles for both shared epitopes. (A) Scheme of the Kv1.2 microarray profiling of E1 and E2 core motifs. From both epitopes, each possible positional substitution was generated (342 variants for each epitope) and printed in microarray format. (B) Fingerprint analysis for sera containing autoantibody in the E1 group. Heat map overview of the major epitope fingerprint from serum 1; the wt sequence was sequentially scanned from N to C terminal by exchanging each position into each proteinogenic amino acid. Subsequently, the residue binding contribution was depicted in blue-white-red shades, where white corresponds to no variation over the wt [1], blue shades depict a loss, and red shades a gain of binding intensity. The most conserved residues for E1 were K478, N481, and T483. (C) Fingerprint analysis revealed a shared profile in the E2 group. General overview of the binding profile for E2 (patient 11) shows matching conserved residues. Here, Y489, V490, N491, and K494 were the most conserved residues. (D) Recapitulated minimal motifs for E1 and E2. (E) Alignment of Kv1 channels shows high homology for the 1.1, 1.2, and 1.3 subunits. Residues “PQTP” on Kv1.3 are the known Cortactin interface, where the “TDV” sequence is a known PDZ-binding interface (F) C-terminal Kv1 peptides display reveals novel co-occurring autoantigen “sub-scenario” for E1. Combined Kv1.1, Kv1.2, Kv1.3, and Kv1.4 peptides were probed with patient 1 sera from the E1 group. In addition to the already mapped Kv1.2 epitope, the Kv1.3 peptides showed strong binding as depicted in the heat map. In line with the previous fingerprinting analysis, the R to K exchange in-between Kv1.1 and Kv1.2/1.3 abolishes binding.
Notably, for E1, all patient samples displayed a seemingly identical binding requirement for their Kv1.2 autoantibodies (Figures 2B, C). More precisely, within the mapped core motif 78KTANCTLA485 (Figure 1C) residues, 478Lys, 481Asn, and 483Thr were characterized by strict conservation with no tolerance toward any AA exchange (Figure 2B) for all tested patients (Figure 2C). For E2, both patients were fingerprinted (Figure 2D). Here, the fine-mapped core motif 485ANTNYVNITK495 (Figure 1C) was also recapitulated by both patients in the same way. In addition to the mapping, the fingerprint further highlights a strong conservation of the C-terminal part of the core motif 489Tyr to 495Lys and strict conservation of 490Val, 491Asn, and 494Lys. In line with the analysis of the phosphorylated peptides (Figure 1G), the phophomimetic exchange of Tyr489 with Glu resulted in complete loss of autoantibody recognition, thereby confirming our previous finding of orthogonal recognition of this Tyrosine depending on its phosphorylation status. In summary, the profiling highlights and substantiates a shared autoimmune response toward two distinct Kv1.2 epitopes. The unexpectedly high similarities in the relative binding responses within the patient cohort and the strict conservation of identical residues suggest a homogenous autoantibody repertoire within the tested patient group and further hint toward a shared molecular origin of the observed epitopes.
The resolved binding profiles prompted us to next explore Kv-isoform specificity of the immunodominant antibody. Comparing the alignment of Kv subfamily members 1.1 to 1.4 with the previous binding requirements highlights the conservation of critical residues within these four subfamily members (Figure 2E), specifically between Kv1.1, Kv1.2, and Kv1.3. We therefore displayed and probed overlapping C-terminal peptides of all four members of this subfamily in microarray format and found that autoantibody binding to E1 is maintained within Kv1.2 and Kv1.3 but not Kv1.1 and Kv1.4 (Figure 2F). The additional binding capacity for Kv1.3 is in line with the previously resolved binding requirements of E1 (Figure 2C). Vice versa, the lack of binding of Kv1.1 that shares high homology in this region highlights the need for experimental validation of putative binders.
Array detection complements cell-based assay
To resolve the predictive value of the specific epitope signals, we correlated seropositivity with the observed disease phenotype. To this end, the Euroimmun pre-screening determined 4% of the samples from healthy individuals as anti–Kv1.2-positive (four out of the 96 samples, data not shown). Our microarray readout recapitulated the reactivity and deep mutational scan analysis and attributed it toward E1, thus suggesting a similar broad occurrence (4%) of these mono-reactive autoantibodies within healthy individuals (Supplementary Figure 1). In this light, we focused on the CSF samples obtained from 12 patients exploring both the diagnostic value and the sensitivity of the array screening in comparison to cell-based screening and its dependence of key parameters of the peptide display (Figure 3). The CSF of first five patients was analyzed with longer peptides and larger offset (20-mers, offset of 3) (Figure 3A), and CSF from seven patients was screened using shorter peptides with shorter offset (15-mers, offset of 1) (Figure 3B) (Table 2). Complementation of the array-based screening with cell-based screens showed equal in several cases even superior sensitivity for both display variants (7/12 CBA and 10/12 array detected patients) (Figure 3C). Specifically, CSFs from patients 2, 25, and 36 were tested positive in microarray and negative in CBA under the tested conditions. In addition, patient 12, negative from the arrays, resulted positive for Kv1.2 and Kv1.1 in cell-based assay (data not shown), potentially bearing a conformational epitope. Importantly, the CBA-determined positivity showed an association with cognitive impairment (30). Here, the array showed improved sensitivity over the CBA-based assay. We therefore conclude that, for Kv autoimmunity patients, clinicians should consider combining array- and CBA-based testing for autoantibody confirmation, when an autoimmune pathogenesis is suspected.
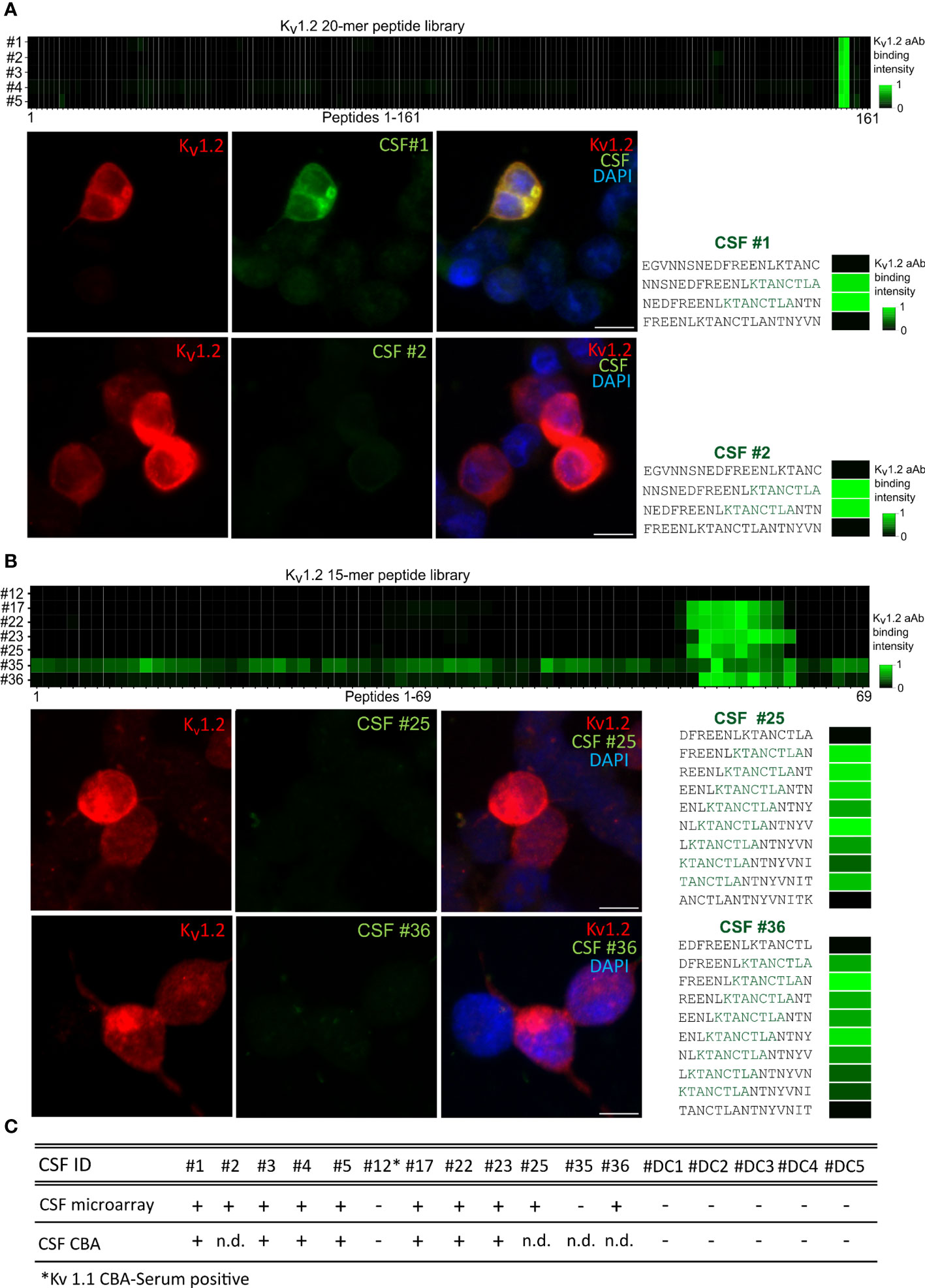
Figure 3 Higher sensitivity of the microarray over cell-based detection for CSF anti-Kv1.2. (A) Kv1.2 autoantibody detection within CSF by CBA and 20-mer peptides in array format. Top: Cerebrospinal fluid autoantibody binding of six positive samples on 161 Kv1.2 peptides. E1 has been detected between the overlapping peptides 156–157. Bottom: Comparison of HEK293 cell Kv 1.2 binding assay and 20-mer peptide array for CSF 1 and 2. (B) Kv1.2 autoantibody detection within CSF by CBA and 15-mer peptides in array format. Core binding motif resolved for seven additional CSF-positive patients, binding observed between the Kv1.2 peptides 55–62. Comparison of HEK293 cell Kv1.2 binding assay and 15-mer peptide array for patients 25 and 36. (C) Overview of the Kv1.2 autoantibody detection within CSF. CSFs from patients 2, 25, and 36 tested positive in microarray but negative in CBA under the conditions tested. Patient 12 was tested positive for Kv1.1 and Kv1.2 autoantibodies in serum CBA. n.d., non detected.
The identified Kv1.2 epitopes are the exclusive mediators of the observed immune response
A commonly assumed limitation of array-based antibody screenings is the risk of missing autoantigen contributions from certain conformational and discontinuous epitopes not represented by the linear peptide display. In addition, anti-Kv1.2 autoimmune response was previously observed to co-occur with unrelated neurological diseases (30, 31) as well as other Kv1 complex–directed (7, 8) autoantibodies. To clarify the isoform specificity, Kv1.1, Kv1.2, and Kv1.6 were expressed in HEK293 cells and probed against the serum IDs 1, 3, and 27 and then compared with the respective control antibodies (Supplementary Figure 2). In line with the deep mutational scans (Figure 2), Kv1.1 and Kv1.6 showed no binding, thus confirming Kv1.2 specificity of the serum antibodies. To resolve contributions from possible additional antibodies binding via discontinuous epitopes that could not be resolved using peptide microarrays, we next conducted neutralization experiments of the identified epitope/autoantigen on chips and on cells. Soluble peptides were synthesized and purified on a preparative scale. On-chip neutralization resulted in a strongly reduced signal, thereby supporting a specific and exclusive binding through the identified epitopes toward Kv1.2 in this format (Figures 4A, B). To search for additional, e.g., conformational, epitopes, we expressed Kv1.2 in HEK293 cells (Figures 4C, D) and tested the neutralization of the observed immune response for the two identified epitopes and the commercial control antibody (Figure 4C). Transfected HEK293 cells were stained with serum pre-treated with neutralizing peptides, non-neutralizing peptides, and buffer only (Figure 4C, D). Patient’s sera were selected on the basis of their different epitopes for neutralization. Consequently, no residual binding was detected for either neutralized sample bearing E1 (Figure 4C) or E2 (Figure 4D). The non-neutralizing peptide had no impact on autoantibody binding. Notably, because no residual binding was detected, we conclude that Kv1.2 autoimmunity is primarily mediated by autoantibodies that recognize the previously highlighted linear motif, without any contributions from additional linear or conformational epitopes.
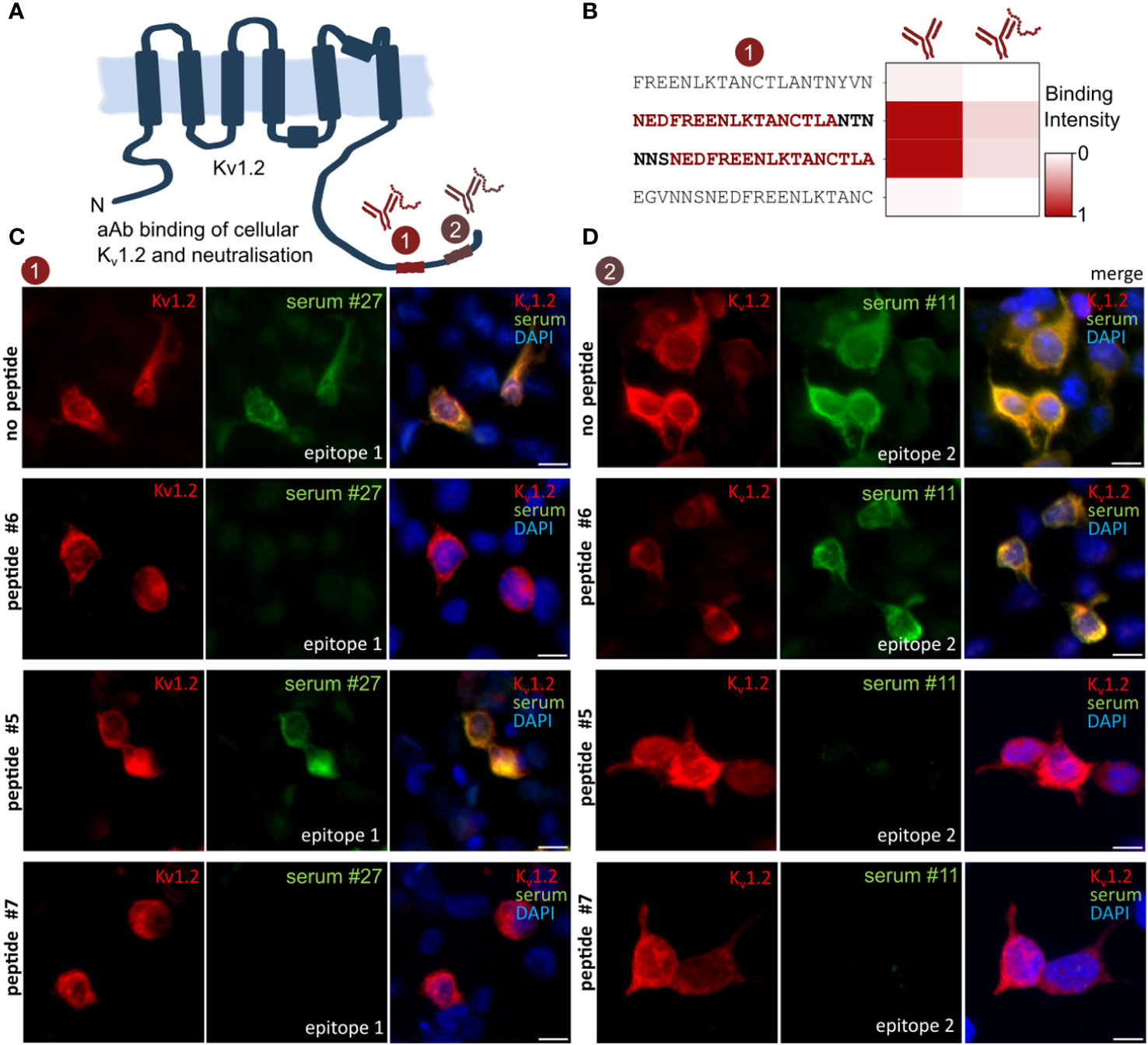
Figure 4 Anti–Kv1.2 Autoantibody Neutralization excludes the presence of additional epitopes. (A) Scheme of the identified Kv1.2 epitope landscape. (B) On-chip neutralization. Mapped peptides were synthesized and applied for pre-absorption experiments with patient sera. Upon incubation with peptide epitope, the microarray intensity has been neutralized. (C) HEK293 cell neutralization confirms E1 as solely mediator of the Kv binding. Transfected Kv1.2 cells were permeabilized and incubated with either commercial anti-Kv1.2 or patient sera, IgG binding was visualized with either anti-mouse IgG or anti-human. Peptides were designed based on the previous mappings (Figures 1, 2). Pre-incubation with the mapped peptide epitope results in a complete autoantibody neutralization, whereas the non-neutralizing peptide epitope did not affect the binding. Neutralizing peptide #6 and #7: EDFREENLKTANCTLANTNY and ENLKTANCTLANTNYVNITK. Non-neutralizing peptide #5: ANCTLANTNYVNITKMLTDV. (D) HEK293 cell neutralization of E2 patient. Neutralizing peptide #5 and #7: ANCTLANTNYVNITKMLTDV and ENLKTANCTLANTNYVNITK. Non-neutralizing peptide #6: EDFREENLKTANCTLANTNY. Scale bar, 10 µm.
Peptide-based autoantibody neutralization on nerves and brain sections
Prompted by the confirmation of the identified epitopes as sole driver of the observed immune response in transfected recombinant cells, we next explored autoantibody binding toward the native autoantigens in their cellular context. Here, we applied the neutralized sera on teased fibers (Figure 5A). Commercial antibody (K14/16) signals recapitulate the expected juxtaparanodal binding on teased fibers (Figure 5B). Sera applied together without peptide (Figure 5C) and with non-neutralizing peptide (Figure 5D) recapitulated the same labeling. In stark contrast, sera pre-treated with neutralizing peptide display a complete loss of binding signal (Figure 5E). Thus, complete anti-Kv1.2 epitope-specific neutralization was achieved using only the minimal Kv1.2 epitope. Remarkably, no residual autoantibody binding was detected in the nerve tissues, and the tested sera did not cross-react with additional autoantigens co-expressed peripherally. This corroborates the hypothesis of selective Kv1.2 autoimmunity without co-existing peripheral autoantibodies, thus contrasting previous observations (7, 8, 30, 31).
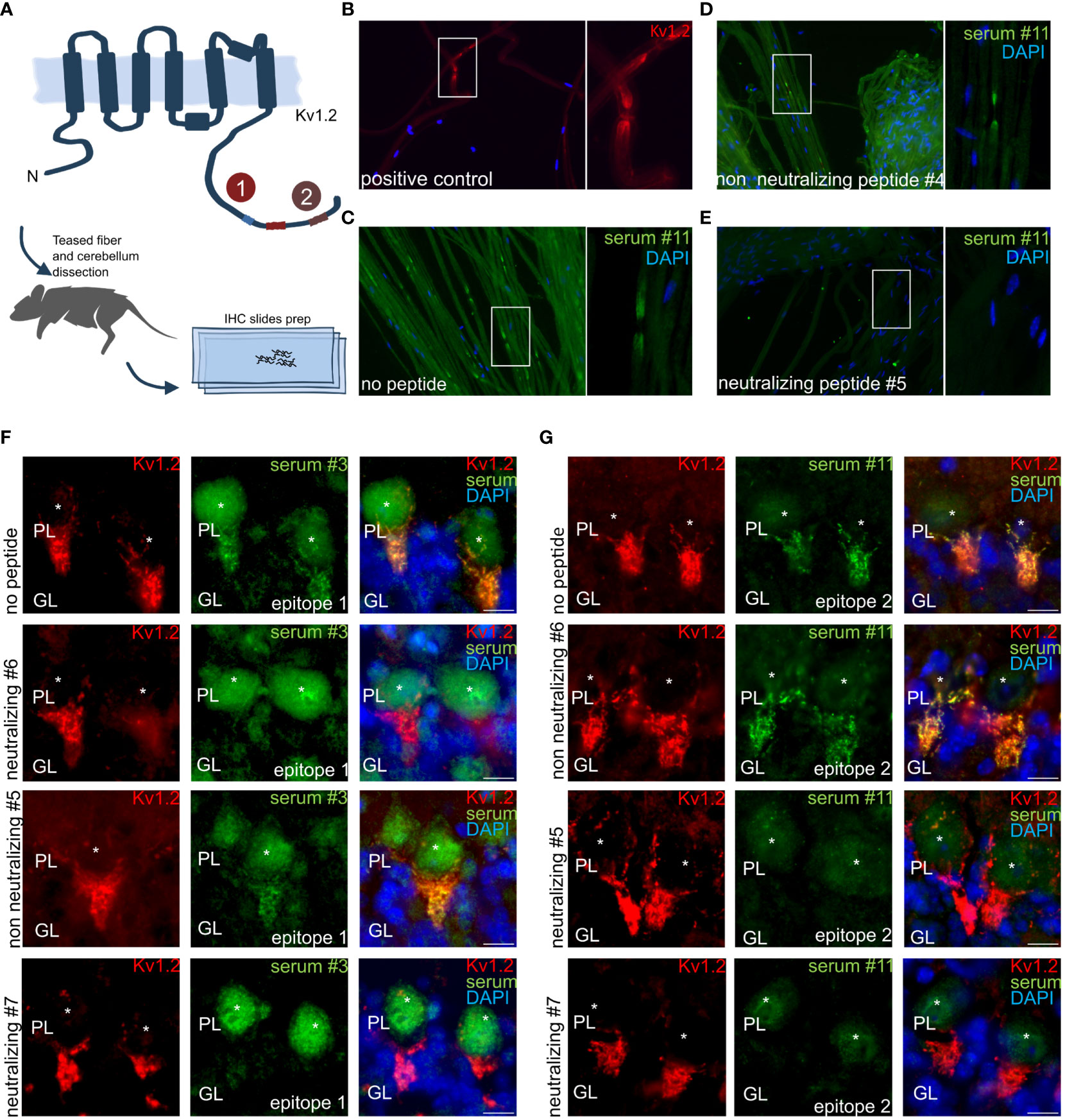
Figure 5 Anti–Kv1.2 Autoantibody Neutralization Exclude the Presence of Co-Existing Peripheral and Central Autoantigens. (A) Teased fibers and mouse cerebellum preparation scheme. Teased fibers commercial Kv1.2 staining confirm correct antigen expression. Incubation of permeabilized mouse teased fibers with (B) clone K14/16 or (C) patient sera. (D) Non-neutralizing peptides did not interfere in teased fibers. (E) Pre-incubation with the mapped peptide epitope results in a complete autoantibody neutralization for patient 11. Non neutralizing and neutralizing peptides for E2: VNNSNEDFREENLKTANCTL, ANCTLANTNYVNITKMLTDV. (F) E1 epitope neutralization of patient sera immune response in mouse hippocampus. Neutralizing peptide #6, #7: EDFREENLKTANCTLANTNY, ENLKTANCTLANTNYVNITK. Non-neutralizing peptide #5: ANCTLANTNYVNITKMLTDV. (G) Neutralizing peptide leads to complete binding loss in mouse hippocampus for E2. Neutralizing peptide #5, #7: ANCTLANTNYVNITKMLTDV, ENLKTANCTLANTNYVNITK. Non-neutralizing peptide #6: EDFREENLKTANCTLANTNY. Neutralisation assay on mouse cerebellum: PL: Purkinje cell layer, GL: granular layer. *indicates Purkinje cell soma. Scale bar, 10 µm.
The neutralization in peripheral nerves was complemented by neutralization in tissue slices from the central nervous system, specifically mouse cerebellum, where Kv1.2 expression is high at the axon initial segment (AIS) of Purkinje cells. MAb K14/16 served as a Kv1.2 antibody control for specific binding of autoantibodies. Here, the typical axonal initial segment stainings were observed on the Purkinje cell layer. E1 (Figure 5F) and E2 (Figure 5G) positive serum was applied in presence of several neutralizing and non-neutralizing peptide variants.
Taken together, the neutralization data in cell and tissues confirmed the high selectivity for the identified peptide epitopes, showing no detectable residual binding. Thus, leading to the exciting conclusion that a single, broadly shared epitope may contribute significantly to the often-reported Kv1 autoimmunity (7, 8, 30, 31), in some cases, even without coexisting autoantigens or other conformational epitopes. In addition, the disease association of the detection of the here defined immunodominant epitope in CSF suggests implications for diagnosis, possibly even the pathology of a subgroup of autoimmune neuropsychiatric phenotypes.
Conclusion
Among Kv1 complex–directed autoantibodies, anti-Kv1 are among the most prevalent (7, 8); compared with LGI1 and CASPAR2 autoantibodies subgroups, their association with clinical syndromes and their immunotherapy responsiveness, however, appears less clear. Despite the association of Kv1 subfamily autoantibodies to neurological autoimmune diseases (5, 6, 9) and their pathology (7, 8, 10, 11, 20–22) and their resulting diagnostic and therapeutic potential the involved Kv1 epitopes remained largely undefined. Here, we provide a first Kv1.2 autoantibody epitope landscape within a cohort of 36 Kv1.2-exclusive neuropsychiatric patients and 18 healthy controls. In contrast to structural (24) and recombinant protein-based approaches (32–34), the array approach (2–4) combined with cell-based and tissue-based studies enabled the high-throughput molecular characterization of the autoantibodies directly from patient samples. Our data depict an unexpectedly monospecific and uniform autoantibody repertoire with two shared responses including one immunodominant Kv1.2 and Kv1.3 autoantibody epitope common to most of the patients tested here. Binding to additional subfamily members Kv1.1, Kv1.4, and Kv1.6 has been excluded by array or cellular assays. Moreover, the notable similarity in binding responses and the preservation of the required residues between patients implies a shared molecular genesis, which may include viral or bacterial antigens. In line with a possibly elevated immunogenic potential, Kv positivity was reported in swine abattoir workers negative for both anti-LGI1 and anti-CASPR2 (35) as well as co-occurring with non-immunogenic neurological disease (30, 31). Sequences with high similarity to “EENLKTANCTLANTNYVNITK,” namely, “EESLKTGNAG” and “ANTIYVNITKMLT,” were previously reported (36, 37) as highly immunogenic. Autoantibody response is exclusively directed against Kv1.2 as substantiated by juxtaparanodal reactivity on teased nerves and AIS labeling in Purkinje cells. Importantly, the mapped epitopes enabled the complete neutralization of the observed reactivity, thereby establishing the outlined auto-antigen region as the primary mediator and even sole driver, of the observed autoimmune response. It remains to be seen whether these antibodies can bind in vivo (23, 24) to interfere with Kv channel function or protein-protein interactions such as the scaffolds PSD-95 and Cortactin, which are reported to bind near the N-terminal "PQTP" (38) or C-terminal "TDV" (39). On the other hand, a direct functional effect on the Kv1.2 channel has been shown to exacerbate a pro-epileptic state (21) and could potentially modulate the excitability of entire complex (40–42). The high prevalence of the here identified Kv1 epitope argues for future detailed studies on a possible intracellular action or indirect mechanisms such as T-cell cytotoxicity (43). Investigation of the relevant T-cell subpopulation involved and the HLA association, together with a possible tolerance mechanism, could shed light on the identified disease-specific antigen and why it is shared by several patients, similar to multiple sclerosis (44). CSF was previously reported to harbor enhanced diagnostic value in autoimmune neurological diseases (45, 46), but detection of low autoantibody titers remains challenging for conventional ELISA (enzyme-linked immunosorbent assay) and cell-based assay approaches that are limited in the density of the antigen display and further require the successful expression and immobilization of the antigens. Notably, the here reported prevalent and immunodominant Kv1 epitope achieves sensitivity and specificity for autoantibody detection in CSF. Significantly, the detected presence of autoantibodies in CSF associates with the clinical symptoms of cognitive impairment, thus highlighting its value for Kv1.2 autoantibody confirmation. This study provides a prevalent and immunodominant epitope together with the underlying autoantibody binding requirements in Kv1 autoimmunity in neuropsychiatric patients. Thus, setting the stage for future investigation of the molecular origin and a potential intracellular action of the here identified mono-specific Kv1 antibodies. These studies may focus on the analysis of clinical presentations, longitudinal samples, and immunotherapy responsiveness. Finally, the reported epitope also provides a means for isolating or even depleting the potentially disease-defining autoantibodies or their respective B cells.
In summary, our study defines an immunodominant epitope as single determinant in Kv1 autoimmunity and thus outstanding diagnostic potential.
Data availability statement
The original contributions presented in the study are included this articles/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ethics Committee of Charité University Medicine Berlin (EA1/258/18). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
IT: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft. FA: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. KK: Investigation, Methodology, Writing – review & editing. MN: Investigation, Writing – review & editing. A-LW:Data curation, Formal analysis, Investigation, Visualization, Writing – review & editing. RM: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Resources, Validation, Writing – review & editing. SM: Formal analysis, Investigation, Methodology, Writing – review & editing. ID: Formal analysis, Investigation, Methodology, Writing – review & editing. MM: Investigation, Writing – review & editing. MB: Investigation, Writing – review & editing. PC: Data curation, Formal analysis, Investigation, Methodology, Resources, Supervision, Validation, Writing – review & editing. IA: Formal analysis, Investigation, Resources, Writing – review & editing. AK: Formal analysis, Investigation, Methodology, Writing – review & editing. ME: Data curation, Formal analysis, Investigation, Methodology, Resources, Supervision, Writing – review & editing. LK: Formal analysis, Investigation, Methodology, Writing – review & editing. CV: Data curation, Funding acquisition, Supervision, Resources, Validation, Writing – review & editing. KD: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing. HP: Conceptualization, Data curation, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – original draft. HM: Conceptualization, Data curation, Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. HM and KD acknowledge funding by the Interdisziplinäres Zentrum für Klinische Forschung (IZKF) of Würzburg, project number A-F-N-419. HM further received funding from the Junior Group Leader program of the Rudolf Virchow Center, the excellent ideas programme of the University of Würzburg and the Emmy Noether Programme of the DFG (MA6957/1-1). KK acknowledges funding by the Graduate School of Life Sciences of the University of Würzburg. ME received funding from DFG under Germany´s Excellence Strategy – EXC-2049 – 390688087, Collaborative Research Center ReTune TRR 295-424778381, BMBF, DZNE, DZHK, EU, Corona Foundation, and Foundation Leducq. Plus: KFO 5023 (HP/ME). CV acknowledges funding by the Research unit SYNABS FOR3004 and KFO 5001 (CV/KD). HP acknowledges funding by the German Research Foundation (DFG; grants FOR3004, PR1274/3-1, PR1274/5-1, PR1274/9-1, Research Unit FOR3004 "Synabs", and Clinical Research Unit 5023/1 "BECAUSE-Y"), by the Helmholtz Association (HIL-A03 BaoBab), and by the German Federal Ministry of Education and Research (Connect-Generate 01GM1908D).
Acknowledgments
We thank Sonja Kachler for her contributions to the microarray production process and excellent technical assistance and Clemens Schulte for his contributions to the data visualization. We thank Heinz Terlau (University of Kiel, Germany) for kindly providing Kv1.1, Kv1.2, and Kv1.6 plasmids for eukaryotic expression.
Conflict of interest
MS, RM, ID and LK are employees of the Euroimmun AG, a company that develops, produces, and manufactures immunoassays for the detection of disease-associated antibodies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The remaining authors declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1329013/full#supplementary-material
Supplementary Figure 1 | Anti-Kv1.2 detection in three sera from healthy individuals highlights the presence of “naturally” occurring autoantibodies (A) Four additional samples (N, O, P, Q) coming from the EUROIMMUN screening were subjected to microarray analysis. Three sera resulted positive for E1; (B) subsequently, these samples were fully profiled. Here, the characteristic E1 fingerprint was recapitulated.
Supplementary Figure 2 | Binding of serum samples #1, #3, #27, healthy control (HC) serum and a commercial antibody against the according Kv channels to transfected HEK293 cells. Cells were transfected with either human Kv1.1 (A), Kv1.2 (B) or Kv1.6 (C) and stained with commercial antibodies (red). Binding of patient serum was visualized with anti-human-IgG-Cy3 antibody (green). Scale bar refers to 10 µm.
References
1. Prüss H. Autoantibodies in neurological disease. Nat Rev Immunol (2021) 21:798–813. doi: 10.1038/s41577-021-00543-w
2. Li Y, Ma M-l, Lei Q, Wang F, Hong W, Lai D-y, et al. Linear epitope landscape of the SARS-CoV-2 Spike protein constructed from 1,051 COVID-19 patients. Cell Rep (2021) 34:108915. doi: 10.1016/j.celrep.2021.108915
3. Longworth J, Dittmar G. An antigen microarray protocol for COVID-19 serological analysis. STAR Protoc (2021) 2:100815. doi: 10.1016/j.xpro.2021.100815
4. Talucci I, Maric HM. Peptide Microarrays for Studying Autoantibodies in Neurological Disease. In: Cretich M, Gori A, editors. Peptide Microarrays: Methods and Protocols. New York, NY: Springer US (2023). p. 17–25.
5. Barber PA, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K channel antibodies. Neurology (2000) 54:771. doi: 10.1212/WNL.54.3.771
6. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain (2010) 133:2734–48. doi: 10.1093/brain/awq213
7. Lang B, Makuch M, Moloney T, Dettmann I, Mindorf S, Probst C, et al. Intracellular and non-neuronal targets of voltage-gated potassium channel complex antibodies. J Neurology Neurosurg & Psychiatry (2017) 88:353. doi: 10.1136/jnnp-2016-314758
8. Agnes van S, Marco WJS, A.A.M.d.B. Marienke B, Mariska MPN, Esther SPH, Roelien HE, et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology (2016) 86:1692. doi: 10.1212/WNL.0000000000002637
9. Bozic I, Tesovic K, Laketa D, Adzic M, Jakovljevic M, Bjelobaba I, et al. Voltage gated potassium channel kv1.3 is upregulated on activated astrocytes in experimental autoimmune encephalomyelitis. Neurochemical Res (2018) 43:1020–34. doi: 10.1007/s11064-018-2509-8
10. Gocke AR, Lebson LA, Grishkan IV, Hu L, Nguyen HM, Whartenby KA, et al. Kv1.3 deletion biases T cells toward an immunoregulatory phenotype and renders mice resistant to autoimmune encephalomyelitis. J Immunol (2012) 188:5877–86. doi: 10.4049/jimmunol.1103095
11. Grishkan IV, Tosi DM, Bowman MD, Harary M, Calabresi PA, Gocke AR. Antigenic stimulation of kv1.3-deficient th cells gives rise to a population of foxp3-independent T cells with suppressive properties. J Immunol (Baltimore Md. 1950) (2015) 195:1399–407. doi: 10.4049/jimmunol.1403024
12. Robbins CA, Tempel BL. Kv1.1 and Kv1.2: Similar channels, different seizure models. Epilepsia (2012) 53:134–41. doi: 10.1111/j.1528-1167.2012.03484.x
13. Imbrici P, Conte E, Blunck R, Stregapede F, Liantonio A, Tosi M, et al. A novel KCNA2 variant in a patient with non-progressive congenital ataxia and epilepsy: functional characterization and sensitivity to 4-aminopyridine. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22189913
14. Pena SD, Coimbra RL. Ataxia and myoclonic epilepsy due to a heterozygous new mutation in KCNA2: proposal for a new channelopathy. Clin Genet (2015) 87:e1–3. doi: 10.1111/cge.12542
15. Syrbe S, Hedrich UBS, Riesch E, Djémié T, Müller S, Møller RS, et al. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet (2015) 47:393–9. doi: 10.1038/ng.3239
16. Döring JH, Schröter J, Jüngling J, Biskup S, Klotz KA, Bast T, et al. Refining genotypes and phenotypes in KCNA2-related neurological disorders. Int J Mol Sci (2021) 22(6). doi: 10.3390/ijms22062824
17. Masnada S, Hedrich UBS, Gardella E, Schubert J, Kaiwar C, Klee EW, et al. Clinical spectrum and genotype–phenotype associations of KCNA2-related encephalopathies. Brain (2017) 140:2337–54. doi: 10.1093/brain/awx184
18. Lioudyno V, Abdurasulova I, Negoreeva I, Stoliarov I, Kudriavtsev I, Serebryakova M, et al. A common genetic variant rs2821557 in KCNA3 is linked to the severity of multiple sclerosis. J Neurosci Res (2021) 99:200–8. doi: 10.1002/jnr.24596
19. Wang X, Li G, Guo J, Zhang Z, Zhang S, Zhu Y, et al. Kv1.3 channel as a key therapeutic target for neuroinflammatory diseases: state of the art and beyond. Front Neurosci (2020) 13:. doi: 10.3389/fnins.2019.01393
20. Timäus C, von Gottberg P, Hirschel S, Lange C, Wiltfang J, Hansen N. KCNA2 autoimmunity in progressive cognitive impairment: case series and literature review. Brain Sci (2021) 11. doi: 10.3390/brainsci11010089
21. Kirschstein T, Sadkiewicz E, Hund-Göschel G, Becker J, Guli X, Müller S, et al. Stereotactically injected kv1.2 and CASPR2 antisera cause differential effects on CA1 synaptic and cellular excitability, but both enhance the vulnerability to pro-epileptic conditions. Front Synap Neurosci. (2020) 12:. doi: 10.3389/fnsyn.2020.00013
22. Buckley C, Oger J, Clover L, Tüzün E, Carpenter K, Jackson M, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol (2001) 50:73–8. doi: 10.1002/ana.1097
23. Bünger I, Talucci I, Kreye J, Höltje M, Makridis KL, Foverskov Rasmussen H, et al. Synapsin autoantibodies during pregnancy are associated with fetal abnormalities. Brain Behavior Immun - Health (2023) 33:100678. doi: 10.1016/j.bbih.2023.100678
24. Noviello CM, Kreye J, Teng J, Prüss H, Hibbs RE. Structural mechanisms of GABAA receptor autoimmune encephalitis. Cell (2022) 185:2469–2477.e13. doi: 10.1016/j.cell.2022.06.025
25. Schulte C, Khayenko V, Maric HM. Peptide Microarray-Based Protein Interaction Studies Across Affinity Ranges: Enzyme Stalling, Cross-Linking, Depletion, and Neutralization. In: Cretich M, Gori A, editors. Peptide Microarrays: Methods and Protocols. New York, NY: Springer US (2023). p. 143–59.
26. Frank R. Spot-synthesis: an easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron (1992) 48:9217–32. doi: 10.1016/S0040-4020(01)85612-X
27. Schulte C, Khayenko V, Gupta AJ, Maric HM. Low-cost synthesis of peptide libraries and their use for binding studies via temperature-related intensity change. STAR Protoc (2021) 2:100605. doi: 10.1016/j.xpro.2021.100605
28. Kreissner KO, Faller B, Talucci I, Maric HM. MARTin-An open-source platform for microarray analysis Front Bioninform. (2023) 4:1329062. doi: 10.3389/fbinf.2024.1329062
29. Arlt FA, Miske R, Machule ML, Broegger Christensen P, Mindorf S, et al. KCNA2 IgG autoimmunity in neuropsychiatric diseases. Brain Behav Immun. (2014) 117:399–411. doi: 10.1016/j.bbi.2024.01.220
30. Matthew J, Sola O, Daniel du P, Angela V, Matthew B, Mark WH, et al. Gerstmann-straüssler-scheinker disease. Neurology (2014) 82:2107. doi: 10.1212/WNL.0000000000000500
31. Meghan R, Simon M, John C, Peter R, Angela V. Neuronal antibodies in patients with suspected or confirmed sporadic Creutzfeldt-Jakob disease. J Neurology Neurosurg & Psychiatry (2015) 86:692. doi: 10.1136/jnnp-2014-308695
32. Ng JK, Malotka J, Kawakami N, Derfuss T, Khademi M, Olsson T, et al. Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology (2012) 79:2241–8. doi: 10.1212/WNL.0b013e31827689ad
33. Ramberger M, Berretta A, Tan JMM, Sun B, Michael S, Yeo T, et al. Distinctive binding properties of human monoclonal LGI1 autoantibodies determine pathogenic mechanisms. Brain (2020) 143:1731–45. doi: 10.1093/brain/awaa104
34. Zou A, Ramanathan S, Dale RC, Brilot F. Single-cell approaches to investigate B cells and antibodies in autoimmune neurological disorders. Cell Mol Immunol (2021) 18:294–306. doi: 10.1038/s41423-020-0510-z
35. Meeusen JW, Klein CJ, Pirko I, Haselkorn KE, Kryzer TJ, Pittock SJ, et al. Potassium channel complex autoimmunity induced by inhaled brain tissue aerosol. Ann Neurol (2012) 71:417–26. doi: 10.1002/ana.22674
36. Jaenisch T, Heiss K, Fischer N, Geiger C, Bischoff FR, Moldenhauer G, et al. High-density peptide arrays help to identify linear immunogenic B-cell epitopes in individuals naturally exposed to malaria infection*[S]. Mol Cell Proteomics (2019) 18:642–56. doi: 10.1074/mcp.RA118.000992
37. Lagatie O, Verheyen A, Van Dorst B, Batsa Debrah L, Debrah A, Stuyver LJ. Linear epitopes in Onchocerca volvulus vaccine candidate proteins and excretory-secretory proteins. Parasite Immunol. (2018) 40. doi: 10.1111/pim.12587
38. Hajdu P, Martin GV, Chimote AA, Szilagyi O, Takimoto K, Conforti L. The C-terminus SH3-binding domain of Kv1.3 is required for the actin-mediated immobilization of the channel via cortactin. Mol Biol Cell (2015) 26:1640–51. doi: 10.1091/mbc.E14-07-1195
39. Kim E, Niethammer M, Rothschild A, Nung Jan Y, Sheng M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature (1995) 378:85–8. doi: 10.1038/378085a0
40. Extrémet J, El Far O, Ankri N, Irani SR, Debanne D, Russier M. An epitope-specific LGI1-autoantibody enhances neuronal excitability by modulating kv1.1 channel. Cells (2022) 11. doi: 10.3390/cells11172713
41. Lugarà E, Kaushik R, Leite M, Chabrol E, Dityatev A, Lignani G, et al. LGI1 downregulation increases neuronal circuit excitability. Epilepsia (2020) 61:2836–46. doi: 10.1111/epi.16736
42. Petit-Pedrol M, Sell J, Planagumà J, Mannara F, Radosevic M, Haselmann H, et al. LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain (2018) 141:3144–59. doi: 10.1093/brain/awy253
43. Tröscher AR, Mair KM, Verdú de Juan L, Köck U, Steinmaurer A, Baier H, et al. Temporal lobe epilepsy with GAD antibodies: neurons killed by T cells not by complement membrane attack complex. Brain (2023) 146:1436–52. doi: 10.1093/brain/awac404
44. Prinz JC. Immunogenic self-peptides - the great unknowns in autoimmunity: Identifying T-cell epitopes driving the autoimmune response in autoimmune diseases. Neurol Neuroimmunol Neuroinflamm. (2023) 13:. doi: 10.3389/fimmu.2022.1097871
45. Kunchok A, Zekeridou A, McKeon A. Autoimmune glial fibrillary acidic protein astrocytopathy. Curr Opin Neurol 32 (2019) 13. doi: 10.1097/WCO.0000000000000676
Keywords: autoantibodies, Kv channel, KCNA2, autoimmune encephalitis, epitope mapping, immunodominant antigen, dementia, critical
Citation: Talucci I, Arlt FA, Kreissner KO, Nasouti M, Wiessler A-L, Miske R, Mindorf S, Dettmann I, Moniri M, Bayer M, Broegger Christensen P, Ayzenberg I, Kraft A, Endres M, Komorowski L, Villmann C, Doppler K, Prüss H and Maric HM (2024) Molecular dissection of an immunodominant epitope in Kv1.2-exclusive autoimmunity. Front. Immunol. 15:1329013. doi: 10.3389/fimmu.2024.1329013
Received: 27 October 2023; Accepted: 22 January 2024;
Published: 11 April 2024.
Edited by:
Kelli M Money, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Andre Ortlieb Guerreiro Cacais, Karolinska Institutet (KI), SwedenDominique Debanne, INSERM U1072 Neurobiologie des canaux Ioniques et de la Synapse, France
Copyright © 2024 Talucci, Arlt, Kreissner, Nasouti, Wiessler, Miske, Mindorf, Dettmann, Moniri, Bayer, Broegger Christensen, Ayzenberg, Kraft, Endres, Komorowski, Villmann, Doppler, Prüss and Maric. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans M. Maric, aGFucy5tYXJpY0B1bmktd3VlcnpidXJnLmRl; Harald Prüss, SGFyYWxkLlBydWVzc0Bkem5lLmRl
†These authors have contributed equally to this work