 Bergithe E. Oftedal
Bergithe E. Oftedal Thea Sjøgren
Thea Sjøgren Anette S. B. Wolff
Anette S. B. Wolff- 1Department of Clinical Science, University of Bergen, Bergen, Norway
- 2Department of Medicine, Haukeland University Hospital, Bergen, Norway
Type I interferons (IFN-I) are key immune messenger molecules that play an important role in viral defense. They act as a bridge between microbe sensing, immune function magnitude, and adaptive immunity to fight infections, and they must therefore be tightly regulated. It has become increasingly evident that thymic irregularities and mutations in immune genes affecting thymic tolerance can lead to the production of IFN-I autoantibodies (autoAbs). Whether these biomarkers affect the immune system or tissue integrity of the host is still controversial, but new data show that IFN-I autoAbs may increase susceptibility to severe disease caused by certain viruses, including SARS-CoV-2, herpes zoster, and varicella pneumonia. In this article, we will elaborate on disorders that have been identified with IFN-I autoAbs, discuss models of how tolerance to IFN-Is is lost, and explain the consequences for the host.
Thymic development and function
The thymus is a primary lymphoid organ located in the thoracic cavity, behind the sternum and above the heart. It develops together with the parathyroid glands from the most anterior region of the foregut, the pharynx, from which they bud out and detach, before migrating to their final location. The thymus holds a highly specialized microenvironment with a unique capacity to support efficient development of self-tolerant T cells expressing a wide repertoire of T cell receptors (antigen receptors) (1–3).
The thymus has two equal lobes consisting of a cortex and a medulla. The cortical-medullary junction intersects these regions and contains the blood vessels responsible for both transporting hematopoietic progenitors from the bone marrow into the thymus, and mature T lymphocytes out of the thymus to peripheral lymphoid organs (1, 4). The cortex is the site of early-thymocyte development and is filled with double-positive thymocytes. Single-positive thymocytes are found in the medulla where late-thymocyte development occurs before they egress from the thymus (5–7). T cell differentiation and development has been extensively reviewed elsewhere (8–10). In addition to the mesenchymal, endothelial, and dendritic cells required for efficient T cell development, both compartments contain distinct populations of thymic epithelial cells (TECs) (1, 11, 12). TECs create a three-dimensional network in which thymocytes migrate through their development. The correct patterning and organization of the thymic stromal components are essential for optimal T cell development and thymus function. Thymic cross talk, the interactions between thymocytes and TECs, is required in the formation of the stromal microenvironment and likely influences both the cortex and the medulla (8, 9). Structural or developmental thymic defects can result in serious health consequences, including immunodeficiency and autoimmunity (1).
The importance of correct thymic development is illustrated by how loss of function (LOF) mutations in FOXN1 (forkhead family transcription factor), the main transcription factor required for correct TEC proliferation and differentiation, can cause immune related diseases. Nude mice deficient in Foxn1 have thymic abnormalities and they develop alopecia universalis and nail dystrophy (13–17). In humans, FOXN1 deficiency leads to a rare form of severe combined immunodeficiency (SCID) (18) with absent or low T cell numbers due to abnormal thymic stroma (18) (19). Abnormal thymic stromal phenotypes are associated with various inherited autoimmune diseases in humans or their disease-related mouse models, as seen in Table 1. Another important protein for thymic immune tolerance is the Autoimmune Regulator (AIRE), which regulates the expression of a panel of tissue-restricted antigens (TRAs) to be presented to developing T cells, in medullary TECs (mTECs) (95, 96). AIRE deficiency leads to autoimmune polyendocrine syndrome type I (APS-I), a rare disease characterized by endocrine autoimmunity and chronic mucocutaneous candidiasis (97). Other transcription factors with roles complementary to AIRE might exist to secure immune tolerance to a broader range of TRAs. Although FEZ Family Zinc Finger 2 (FEZF2) has been suggested as one candidate (98, 99), no human disease is directly linked to mutations in this gene. Other disorders that implicate disrupted thymic organization, and thereby incomplete central tolerance, include hypomorphic RAG 1 and 2 mutations (Omenn syndrome) (37), resulting in failure of variable (V), diversity (D), and joining (J) segment recombination in the generation of the highly diverse B and T cell receptors; DiGeorge syndrome (76) where patients have microdeletions of chromosome 22; Down syndrome with trisomy 21 (76), and the polygenic disorder systemic lupus erythematosus (SLE) (65, 66) (Table 1).
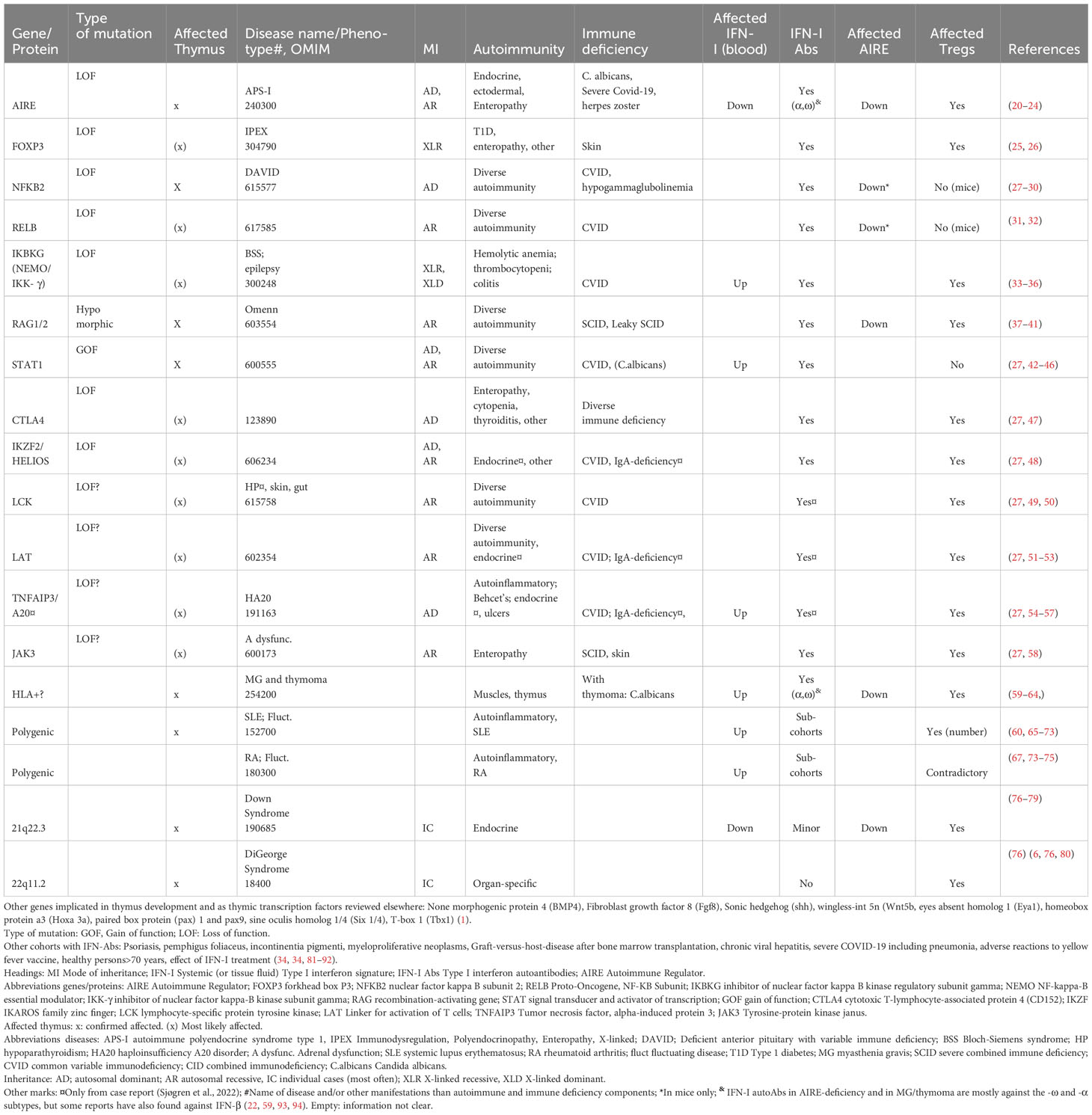
Table 1 Diseases/groups seen with interferon type 1 autoantibodies: genetic determinants and their consequences when mutated.
Interferons are targeted by immune cells in individuals with diseases relating to the thymus
Thymic tolerance is crucial to avoid autoimmunity against TRAs and immune mediators, including the type I interferons (IFN-Is), which are special immune signals in viral host defense (100, 101) (see Box 1). There is a growing consensus that mutations in genes compromising thymic tolerance mechanisms might lead to a dysregulation of IFN-Is (22, 25, 27, 33, 38, 103–111). In the following text, examples of thymic abnormalities, dysregulation of IFN-Is, IFN-I autoAbs, and their link to immunomodulated diseases will be discussed.
Box 1. Interferons
In humans, there are three classes of interferons (IFNs); I (13 αs, one -ω, one -β, one -ϵ, one - к), II (IFN-γ) and III (four IFN-λs). Each class has its own preferred receptor; IFN-Is bind to the IFN-α receptor (IFNAR1/IFNAR2) while IFN-γ recognizes the interferon-gamma receptor (IFNGR1/IFNGR2) and the IFNLR1/IL10RB is the receptor for IFN-λs. The different IFN/receptor-complexes are activated via phosphorylation cascades involving diverse Janus kinases (JAKs)/Signal Transducer and Activator of Transcription 1 (STAT)/Tyrosine kinase 2 (TYK)-reactions, ultimately leading to translocation of the STATs into the nucleus and expression of interferon stimulated genes (ISGs) (see (102) for review). These IGSs can then mediate direct antiviral effects through viral RNA degradation and viral translation inhibition and by boosting adaptive responses. IFNs are therefore powerful molecules to protect tissues from invaders, but their activity must be tightly regulated to avoid excess activity and tissue damage.
The different IFN subclasses have a variety of distinct functions, but their biological activity overlaps to different extents causing a redundancy of the system. Type I IFNs (IFN-I) are especially important in antiviral host defense but also possess antitumor, and anti-proliferative effects. However, details regarding the specific functions within each subtype (e.g. the different IFN-αs compared to IFN-ω within IFN-Is) are still not clear.
An apt example of the relationship between thymus malfunction and IFN-I autoAbs is AIRE LOF mutations (112, 113). Patients with APS-I are found to have high titers of neutralizing IFN-I autoAbs (against the subtypes -αs and -ω) already at infancy, before clinical manifestations occur (109, 114, 115), and these autoAbs are remarkably stable throughout life. Even a partial loss of AIRE, as seen in dominant negative mutations, can induce development of autoAbs in affected individuals (116, 117). Intriguingly, serological autoAbs against IFN-Is are also found in several other diseases with decreased or complete loss of thymic AIRE expression, which may or may not implicate proper thymic architecture organization. Examples include patients with LOF mutations in the non-canonical Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-KB) pathway (103, 105, 107, 118) (e.g. NFKB2, RELB Proto-Oncogene, NF-KB Subunit (RelB), MAP3K14/NF-κB-inducing kinase (NIK) and IKBKG (encoding NF-kappa-B essential modulator (NEMO) (34)) and hypomorphic RAG1/2 mutations (37–41) (Table 1). In thymoma patients, a rare condition characterized by thymus carcinoma, thymopoiesis continues despite the lack of AIRE. This represents an example of acquired AIRE-deficiency and is also associated with anti-IFN-αs and -ω (59–61). The clinical consequences overlap in patients with thymoma and APS-I, with the appearance of autoimmune endocrine manifestations and failure to clear Candida albicans infections. Both conditions present with APS-I like antibodies, highlighting the importance of AIRE expression to ensure proper thymic function.
Interestingly, sporadic cases with other mutations located in immune regulatory genes have also been found to acquire anti-IFN-I autoAbs. This includes STAT1 gain of function (GOF) mutations and rare (presumably LOF) variants in CTLA-4, IKZF2, FOXP3, LCK, LAT, TNFAIP3 and JAK3 (25, 27, 38, 105)(Table 1). Their encoded proteins are important in T cell activity and for the generation of central T cell tolerance in the thymus (Table 1, Figure 1). Although the physiological roles of these autoAbs is yet unknown, they serve as excellent markers of disease, suggesting that genomic analysis should be performed when detected in patients (27).
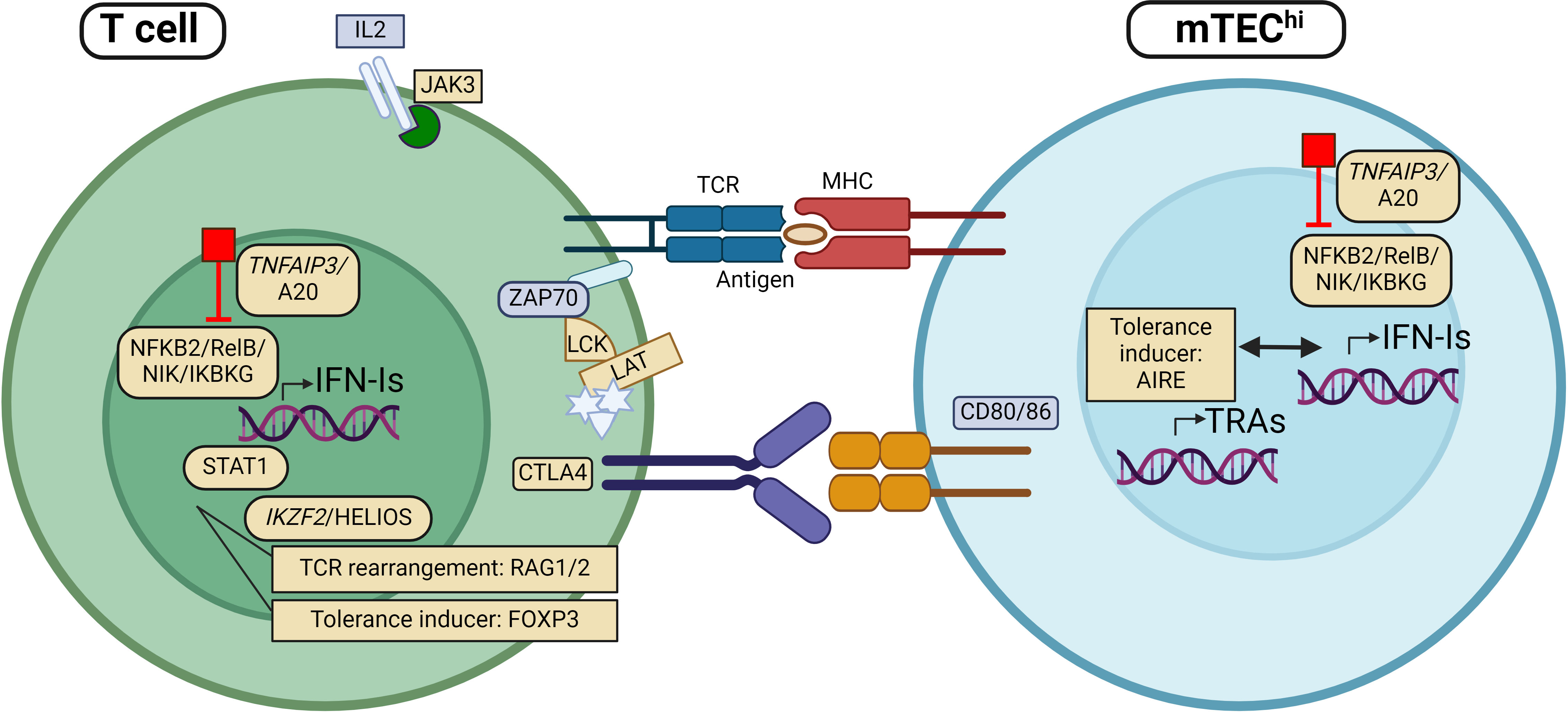
Figure 1 Patients with mutations in genes encoding proteins involved in regulation of type I interferon (IFN-I) expression, including TNFAIP3, NFKB2, RelB, NIK and IKBKG, have been found with autoantibodies against IFN-I (IFN-I autoAbs). The same applies to individuals with mutations in other genes implicated directly in thymic tolerance like AIRE, RAG and FOXP3. More recently, rare variants in genes encoding proteins with key functions in T cell regulation/activity, including JAK3, STAT1, HELIOS, CTLA4, LCK and LAT have been identified with IFN-I autoAbs. Genes (and proteins) that are found mutated in patients with IFN-I autoAbs are marked in yellow. APC, Antigen presenting cell; mTEC, medullary thymic epithelial cells; TCR, T cell receptor; MHC, major histocompatibility complex. Figure created with BioRender.com.
IFN-I autoAbs in other disease cohorts provide clues on their etiology and immune consequences
There is strong evidence that overproduction of IFN-Is is associated with pathological roles in several systemic (119, 120) and organ-specific autoimmune disorders (121–126). Even when administered as treatment of chronic active hepatitis, IFN-α may cause hyperthyroidism or autoimmune hepatitis (127–129). Several primary immune deficiencies, caused by mutations in genes directly involved in regulation of IFN-I responses, also present with dysregulated IFN-I profiles (130). However, these disorders rarely come with IFN-I autoAbs as long as thymic immune tolerance is intact (111) (Table 1). Very high levels of IFN-Is and/or chronic inflammation might still break peripheral immunological tolerance, and the first stories on IFN-I autoAbs were reported in various patient cohorts treated with IFN-β (131–133). A sudden endogenous increase of cytokine expression following e.g. septic shock or viral (e.g. severe COVID-19, acute viral hepatitis and chronic HIV) or bacterial infections have also been reported to induce anti-cytokine autoimmunity (reviewed elsewhere (134)). IFN-I autoAbs were subsequently identified in patients with chronic graft-versus-host disease following allogeneic bone marrow transplantation (81, 82), in pemphigus foliaceus (83) and further in cohorts of SLE patients (67, 135). The IFN-I autoAbs are less prevalent in other autoinflammatory disorders like Sjøgren’s disease (SS) and rheumatoid arthritis (RA), where reported frequencies depend on the cohorts, subtypes of disease, IFN-I autoAbs assays and “threshold level” (67, 135). The likelihood of developing IFN-I autoimmunity follows a spectrum from ~100% in AIRE deficiency, to various, but lower, frequencies in other genetic disorders that affect the development and function of the thymus, and as also reflected by dominant AIRE mutations Patients with systemic autoimmune disorders probably need additional environmental triggers, such as infections, drugs or vaccines, to develop IFN-I autoimmunity.
Recognized physiologic roles of autoAbs to IFN-Is
Recent studies have highlighted the role of IFN-I autoAbs in relation to the outcome of infectious diseases, such as COVID-19. Early in the COVID-19 pandemic, it was reported that patients with APS-I became critically ill upon being infected with the SARS-CoV2 virus (136–138). Pre-existing IFN-I autoAbs was the common denominator and it was established that the viral defense was affected in these patients. Although the severity of COVID-19 disease in APS-I has later been challenged (139, 140), perhaps as a consequence of evolving virus variants and/or vaccination, the fact that individuals with anti-IFN-I autoAbs are at risk of severe lung manifestations still stands. This is supported by the presence of high levels of neutralizing IFN-I autoAbs in about 10% of patients across different age groups suffering from life-threatening COVID-19 (34, 137). Later it was shown that neutralizing IFN-I autoAbs were present in serum from about 4% of uninfected individuals over 70 years of age, suggesting that thymic involution might play a role in initiating IFN-I autoimmunity (84). The host protection against other pathogens like herpes zoster and/or varicella pneumonia has also recently been found to be impaired in different patient cohorts with serological anti-IFN-I autoAbs like APS-I, Omenn’ syndrome and SLE (68, 141–143). Adverse reactions causing life-threatening disease following vaccination with a yellow fever live attenuated vaccine were furthermore recently noted with post-findings of IFN-I autoAbs in otherwise healthy individuals with a large age span (13-80 years) (85). These autoAbs were shown to inhibit the protective effect of IFN-Is against strains of the yellow fever vaccine in subsequent experiments (85). This indicates that IFN-I autoAbs can underlie development of critical disease following live virus vaccines in persons with immune deficiencies and/or known presence of IFN-I autoAbs. A systemic effect of presence of IFN-I autoAbs resulting in a compromised host immune efficacy is also indicated by the correlation of IFN-I autoAbs in APS-I patients (and others with high levels of neutralizing IFN-I autoAbs) with reduced peripheral expression of interferon stimulated genes (ISG) (20, 21, 92). However, it has also been suggested that these autoAbs can protect against type 1 diabetes (T1D) and thyroid disease, as reported in a few APS-I patients without IFN-α autoAbs (144, 145). The possible protective role of IFN-I autoAbs in distinct biological processes in inflammatory disorders is supported by the finding of an association of IFN-α autoAbs with decreased/normalized ISG profiles and fluctuating disease activity in the systemic autoimmune disorders SLE, SS and RA (67, 135, 146). These observations have led to the application of molecules of the IFN-I pathway as new therapeutic targets in subgroups of SLE (86–88), e.g. anifrolumab which blocks the activity of the IFN-I receptor. Notably, as IFN subtypes are redundant in their properties, untargeted IFN-I subtypes (which are not attacked by the hosts immune system in susceptible patients) may compensate and limit the biological consequences of IFN-I autoimmunity.
Thymus is involved in regulation of IFN-I tolerance
Thymic mechanisms are challenging to study in humans due to the inaccessibility and shrinking of the organ with age (involution). Therefore, most knowledge comes from mouse studies. Over the past years, it has become clear that Ifn-Is in mice are expressed at high levels in the thymus, and that Aire in mTECsis involved, either directly or indirectly, in regulating their expression (100, 147–149). Subsets of developed “post-/late-Aire+” mTECs produce relatively high amounts of Ifn-β at homeostatic conditions, and the Ifnα receptor is expressed by both thymocytes and antigen-presenting cells in the thymus, implicating that these cells have the potential to respond to local Ifn-I signaling. It is still unclear whether the Ifn-β expression is directly linked to the transcriptional activity of Aire, but studies have shown that knocking out Aire in mice leads to the loss of Ifn-β signals in the thymus (100, 150). This indicates that Ifn-Is can be direct responders of Aire’s transcription factor activity or that Aire can affect Ifn-I biology by its function in thymic architecture and thymopoiesis. Furthermore, as tonic Ifn-α expression has not been detected in mice thymi (100), there must be considerable differences in expression and mechanisms of tolerance achievement between the Ifn-I subtypes (151–153). To complicate the picture, Nf-kb recruitment is necessary for both Aire induction and as a sensor of viral exposure leading to activation of an Ifn-I immune response (100, 154, 155). Hence, Aire and Ifn-Is, at least Ifn-β, are connected by the Nf-kb pathway. IFN-I regulation is even more complex in humans than in mice, with additional IFN-α subtypes and IFN-ω (which is lacking in mice). In inborn or acquired AIRE deficiency, seen in APS-I patients and myasthenia gravis (MG) patients with thymoma (156, 157) respectively, expression of IFN-I will be out of control, leading to dysregulation of the feedback loop and probably high local levels of these cytokines in the thymus (156). The lack of AIRE will simultaneously interfere with proper immune tolerance against AIRE-regulated TRAs, likely also affecting IFN-I expression (100), leading to autoreactivity against them. The devastating combination of high local levels of IFN-Is with no generated immune tolerance might then become a self-driving loop continuing when the autoimmune naïve T cells exit the thymus. For MG with thymoma, further evidence for a link between AIRE and IFN-Is has been established as the expression of the α-subunit of the acetylcholine receptor (AChR), the main autoantigen in MG, is regulated by both AIRE and IFN-β in the thymus (155, 158). A plausible explanation for MG in some thymoma patients may then be a “bystander effect” where neoplastic cells fail to drive AChR and IFN-I generation in an AIRE free milieu, breaking tolerance against them, causing both anti-IFN and anti-AChR autoimmunity. Upregulation of IFN-Is is also responsible for the generation of anti-AChR autoAbs in early onset MG without thymoma. This is shown indirectly as upregulation of IFNs increases B cell infiltration, ectopic GC formation and ISG stimulation. However, patients with early onset MG without thymoma do not have anti-IFN-Is, despite having a high-IFN-I ISG profile (60), probably because of an intact tolerance mechanism. This starts a vicious circle where upregulation of IFN-Is again exaggerates ectopic B cell formation in the thymus of these patients. Regulatory T cells (Tregs) have also been studied in MG-patients in relation to IFNs. Of interest, IFN-I expression in Tregs may affect the development of these natural suppressor cells (159, 160) and IFN-I signaling within Tregs promotes their inherent suppressive role in anti-viral defense (161). It further protects the Treg population under inflammation (162). Several of the other conditions with IFN-autoAbs also have Treg disturbances (Table 1), but the role of the IFN-Treg symbiosis must be studied further before causative/functional links are fully established.
Indirect proof for the importance of the thymus in IFN tolerance is also the very concept of this article: the abundance of autoAbs against IFN-Is in AIRE deficiency, in defects of the NF-KB axis (27, 34) and in defects of genes encoding key factors of immune tolerance, like FOXP3, RAG and CTLA-4 (Table 1). There is, however, yet one big question left to answer: Why are the other type I IFNs (IFN-β, -ϵ, and –к) seldom targets of autoimmunity? One theory is that mechanisms that promote IFN-I production in mTECs are also likely to be induced in tolerance to IFNs and ISG, and type, timing, and expression levels of IFN-I is vital in thymopoiesis. When first targeted, IFN-Is can be implicated in thymic atrophy leading to the loss of developing thymocytes with increased survival of self-reactive cells (modulated by IFN-β) (163–165). IFN-Is also affect thymocyte selection and maturation, and development/maturation of mTECs are required for tolerance against these immune modulators (147, 151).
We hypothesize two, not mutually contradictory, alternative mechanisms for the generation of IFN-I autoAbs. (I) T cell driven hypothesis. In the thymus, IFNs are expressed by mTECs and AIRE is directly involved in their production and/or in the development of mTECs (100, 166). When thymic organogenesis or AIRE expression fails, tolerance against IFNs is disturbed, leading to autoimmunity towards IFN-I; (II) B cell driven hypothesis. High thymic and/or tissue levels of IFNs in affected individuals feed into the infectious environment of the organs in early development and could kickstart tissue cell destruction with autoimmune mechanisms. The initial cause of IFN-I secretion may be a virus or an unknown environmental trigger. When the IFNs reach high levels in a “still not achieved immune tolerance” situation, this will mediate an autoimmunization environment in both the thymus and other organs/periphery, resulting in peripheral break of tolerance. Then, instead of a direct break of tolerance, the immune system reacts by feedback loops when identifying the dangerous concentrations of cytokines in the wrong place at the wrong time, thereby stimulating the adaptive immune cells to start producing antibodies against IFNs to fight the uncontrolled situation. Either way, when tolerance against anti-IFNs is broken, this establishes an anti-IFN response that is maintained throughout life.
Conclusion
Defining the molecular mechanisms behind thymus-associated and/or IFN-I linked disorders has revolutionized our understanding on IFN-I regulation and messaging. New evidence of anti-IFN-I interference with viral disease outcome should warrant analysis of anti-IFN-Is in individuals with known autoimmune and/or immunodeficiency disorders before giving advice on vaccine modalities for disease prevention and before administrating treatment with IFN-α or -β for certain disorders. Furthermore, as it has become clear that anti-IFN-Is are often connected with inborn or acquired immunodeficiency and thymic disease, sequencing of positive IFN-I autoAbs cases should be considered to identify their underlying molecular cause.
Author contributions
BO: Conceptualization, Writing – original draft, Writing – review & editing. TS: Conceptualization, Writing – original draft, Writing – review & editing. AW: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We are grateful to Lars Breivik who critically read and commented on the review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
IFN-I, Type 1 interferons; IFN-I autoAbs, Type 1 interferon autoantibodies; TEC, thymic epithelial cells; ISG, Interferon stimulated genes; TRA, tissue restricted antigens.
References
1. Gordon J, Manley NR. Mechanisms of thymus organogenesis and morphogenesis. Development (2011) 138:3865–78. doi: 10.1242/dev.059998
2. Miller JF. Immunological function of the thymus. Lancet (1961) 2:748–9. doi: 10.1016/S0140-6736(61)90693-6
3. Boehm T. Thymus development and function. Curr Opin Immunol (2008) 20(2):178–84. doi: 10.1016/j.coi.2008.03.001
4. Muсoz JJZA. Thymus Ontogeny and Development. In: Passos G, editor. Thymus Transcriptome and Cell Biology, vol. 2019 . Springer Nature, Switzerland AG (2019). p. 19–34. doi: 10.1007/978-3-030-12040-5_2
5. Anderson G, Jenkinson WE, Jones T, Parnell SM, Kinsella FA, White AJ, et al. Establishment and functioning of intrathymic microenvironments. Immunol Rev (2006) 209:10–27. doi: 10.1111/j.0105-2896.2006.00347.x
6. Hollander G, Gill J, Zuklys S, Iwanami N, Liu C, Takahama Y. Cellular and molecular events during early thymus development. Immunol Rev (2006) 209:28–46. doi: 10.1111/j.0105-2896.2006.00357.x
7. Takahama Y. Journey through the thymus: stromal guides for T-cell development and selection. Nat Rev Immunol (2006) 6:127–35. doi: 10.1038/nri1781
8. Abramson J, Anderson G. Thymic epithelial cells. Annu Rev Immunol (2017) 35:85–118. doi: 10.1146/annurev-immunol-051116-052320
9. Fletcher AL, Calder A, Hince MN, Boyd RL, Chidgey AP. The contribution of thymic stromal abnormalities to autoimmune disease. Crit Rev Immunol (2011) 31:171–87. doi: 10.1615/CritRevImmunol.v31.i3.10
10. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14:377–91. doi: 10.1038/nri3667
11. Hollander GA, Wang B, Nichogiannopoulou A, Platenburg PP, van Ewijk W, Burakoff SJ, et al. Developmental control point in induction of thymic cortex regulated by a subpopulation of prothymocytes. Nature (1995) 373:350–3. doi: 10.1038/373350a0
12. Palmer DB, Viney JL, Ritter MA, Hayday AC, Owen MJ. Expression of the alpha beta T-cell receptor is necessary for the generation of the thymic medulla. Dev Immunol (1993) 3:175–9. doi: 10.1155/1993/56290
13. Schorpp M, Hofmann M, Dear TN, Boehm T. Characterization of mouse and human nude genes. Immunogenetics (1997) 46:509–15. doi: 10.1007/s002510050312
14. Nehls M, Pfeifer D, Schorpp M, Hedrich H, Boehm T. New member of the winged-helix protein family disrupted in mouse and rat nude mutations. Nature (1994) 372:103–7. doi: 10.1038/372103a0
15. Vaidya HJ, Briones Leon A, Blackburn CC. FOXN1 in thymus organogenesis and development. Eur J Immunol (2016) 46:1826–37. doi: 10.1002/eji.201545814
16. Flanagan SP. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet Res (1966) 8:295–309. doi: 10.1017/S0016672300010168
17. Pantelouris EM. Absence of thymus in a mouse mutant. Nature (1968) 217:370–1. doi: 10.1038/217370a0
18. Tasher D, Dalal I. The genetic basis of severe combined immunodeficiency and its variants. Appl Clin Genet (2012) 5:67–80. doi: 10.2147/TACG.S18693
19. Rota IA, Dhalla F. FOXN1 deficient nude severe combined immunodeficiency. Orphanet J Rare Dis (2017) 12:6. doi: 10.1186/s13023-016-0557-1
20. Kisand K, Link M, Wolff AS, Meager A, Tserel L, Org T, et al. Interferon autoantibodies associated with AIRE deficiency decrease the expression of IFN-stimulated genes. Blood (2008) 112:2657–66. doi: 10.1182/blood-2008-03-144634
21. Oftedal BE, Delaleu N, Dolan D, Meager A, Husebye ES, Wolff ASB. Systemic interferon type I and B cell responses are impaired in autoimmune polyendocrine syndrome type 1. FEBS Lett (2023) 597:1261–74. doi: 10.1002/1873-3468.14625
22. Meager A, Visvalingam K, Peterson P, Moll K, Murumagi A, Krohn K, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PloS Med (2006) 3:e289. doi: 10.1371/journal.pmed.0030289
23. Kekalainen E, Miettinen A, Arstila TP. Does the deficiency of Aire in mice really resemble human APECED? Nat Rev Immunol (2007) 7:1. doi: 10.1038/nri2136-c1
24. Kekalainen E, Tuovinen H, Joensuu J, Gylling M, Franssila R, Pontynen N, et al. A defect of regulatory T cells in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Immunol (2007) 178:1208–15. doi: 10.4049/jimmunol.178.2.1208
25. Rosenberg JM, Maccari ME, Barzaghi F, Allenspach EJ, Pignata C, Weber G, et al. Neutralizing anti-cytokine autoantibodies against interferon-alpha in immunodysregulation polyendocrinopathy enteropathy X-linked. Front Immunol (2018) 9:544. doi: 10.3389/fimmu.2018.00544
26. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci (2018) 1417:5–22. doi: 10.1111/nyas.13011
27. Sjogren T, Bratland E, Royrvik EC, Grytaas MA, Benneche A, Knappskog PM, et al. Screening patients with autoimmune endocrine disorders for cytokine autoantibodies reveals monogenic immune deficiencies. J Autoimmun (2022) 133:102917. doi: 10.1016/j.jaut.2022.102917
28. Le Voyer T, Parent AV, Liu X, Cederholm A, Gervais A, Rosain J, et al. I. Impaired thymic AIRE expression underlies autoantibodies against type I IFNs in humans with inborn errors of the alternative NF-kB pathway. Authorea (2023) 623(7988):803–13. doi: 10.1016/j.clim.2023.109369
29. Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front Immunol (2019) 10:297. doi: 10.3389/fimmu.2019.00297
30. Zhu M, Chin RK, Christiansen PA, Lo JC, Liu X, Ware C, et al. NF-kappaB2 is required for the establishment of central tolerance through an Aire-dependent pathway. J Clin Invest (2006) 116:2964–71. doi: 10.1172/JCI28326
31. Heino M, Peterson P, Sillanpaa N, Guerin S, Wu L, Anderson G, et al. RNA and protein expression of the murine autoimmune regulator gene (Aire) in normal, RelB-deficient and in NOD mouse. Eur J Immunol (2000) 30:1884–93. doi: 10.1002/1521-4141(200007)30:7<1884::AID-IMMU1884>3.0.CO;2-P
32. Zhou S, Wu W, Wang Z, Wang Z, Su Q, Li X, et al. RelB regulates the homeostatic proliferation but not the function of Tregs. BMC Immunol (2020) 21:37. doi: 10.1186/s12865-020-00366-9
33. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2022) 42:1473–507. doi: 10.1007/s10875-022-01289-3
34. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (2020) 370(6515):eabd4585. doi: 10.1126/science.abd4585
35. de Jesus AA, Hou Y, Brooks S, Malle L, Biancotto A, Huang Y, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest (2020) 130:1669–82. doi: 10.1172/JCI129301
36. Chen X, Willette-Brown J, Wu X, Hu Y, Howard OM, Hu Y, et al. IKKalpha is required for the homeostasis of regulatory T cells and for the expansion of both regulatory and effector CD4 T cells. FASEB J (2015) 29:443–54. doi: 10.1096/fj.14-259564
37. Poliani PL, Facchetti F, Ravanini M, Gennery AR, Villa A, Roifman CM, et al. Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: possible implications for the pathophysiology of Omenn syndrome. Blood (2009) 114:105–8. doi: 10.1182/blood-2009-03-211029
38. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest (2015) 125:4135–48. doi: 10.1172/JCI80477
39. Elnour IB, Ahmed S, Halim K, Nirmala V. Omenn’s Syndrome: A rare primary immunodeficiency disorder. Sultan Qaboos Univ Med J (2007) 7:133–8.
40. Gennery A. Recent advances in understanding RAG deficiencies. F1000Res (2019) 8. doi: 10.12688/f1000research.17056.1
41. Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol (2016) 16:234–46. doi: 10.1038/nri.2016.28
42. Scott O, Lindsay K, Erwood S, Mollica A, Roifman CM, Cohn RD, et al. STAT1 gain-of-function heterozygous cell models reveal diverse interferon-signature gene transcriptional responses. NPJ Genom Med (2021) 6:34. doi: 10.1038/s41525-021-00196-7
43. Kaleviste E, Saare M, Leahy TR, Bondet V, Duffy D, Mogensen TH, et al. Interferon signature in patients with STAT1 gain-of-function mutation is epigenetically determined. Eur J Immunol (2019) 49:790–800. doi: 10.1002/eji.201847955
44. Giovannozzi S, Demeulemeester J, Schrijvers R, Gijsbers R. Transcriptional profiling of STAT1 gain-of-function reveals common and mutation-specific fingerprints. Front Immunol (2021) 12:632997. doi: 10.3389/fimmu.2021.632997
45. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood (2016) 127:3154–64. doi: 10.1182/blood-2015-11-679902
46. Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol (2013) 131:1611–23. doi: 10.1016/j.jaci.2012.11.054
47. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med (2014) 20:1410–6. doi: 10.1038/nm.3746
48. Hetemaki I, Kaustio M, Kinnunen M, Heikkila N, Keskitalo S, Nowlan K, et al. Loss-of-function mutation in IKZF2 leads to immunodeficiency with dysregulated germinal center reactions and reduction of MAIT cells. Sci Immunol (2021) 6:eabe3454. doi: 10.1126/sciimmunol.abe3454
49. Hauck F, Randriamampita C, Martin E, Gerart S, Lambert N, Lim A, et al. Primary T-cell immunodeficiency with immunodysregulation caused by autosomal recessive LCK deficiency. J Allergy Clin Immunol (2012) 130:1144–52:e1111. doi: 10.1016/j.jaci.2012.07.029
50. Kagawa K, Sato S, Koyama K, Imakura T, Murakami K, Yamashita Y, et al. The lymphocyte-specific protein tyrosine kinase-specific inhibitor A-770041 attenuates lung fibrosis via the suppression of TGF-beta production in regulatory T-cells. PloS One (2022) 17:e0275987. doi: 10.1371/journal.pone.0275987
51. Bacchelli C, Moretti FA, Carmo M, Adams S, Stanescu HC, Pearce K, et al. Mutations in linker for activation of T cells (LAT) lead to a novel form of severe combined immunodeficiency. J Allergy Clin Immunol (2017) 139:634–642 e635. doi: 10.1016/j.jaci.2016.05.036
52. Keller B, Zaidman I, Yousefi OS, Hershkovitz D, Stein J, Unger S, et al. Early onset combined immunodeficiency and autoimmunity in patients with loss-of-function mutation in LAT. J Exp Med (2016) 213:1185–99. doi: 10.1084/jem.20151110
53. Chuck MI, Zhu M, Shen S, Zhang W. The role of the LAT-PLC-gamma1 interaction in T regulatory cell function. J Immunol (2010) 184:2476–86. doi: 10.4049/jimmunol.0902876
54. He T, Huang Y, Luo Y, Xia Y, Wang L, Zhang H, et al. Haploinsufficiency of A20 due to novel mutations in TNFAIP3. J Clin Immunol (2020) 40:741–51. doi: 10.1007/s10875-020-00792-9
55. Yan M, Li D, Aknai S, Zhu H, Abudureyim M. Mutation analysis of the TNFAIP3 in A20 haploinsufficiency: A case report. Med (Baltimore) (2021) 100:e25954. doi: 10.1097/MD.0000000000025954
56. Urbano PCM, He X, van Heeswijk B, Filho OPS, Tijssen H, Smeets RL, et al. TNFalpha-signaling modulates the kinase activity of human effector treg and regulates IL-17A expression. Front Immunol (2019) 10:3047. doi: 10.3389/fimmu.2019.03047
57. Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol (2015) 16:618–27. doi: 10.1038/ni.3172
58. Notarangelo LD, Mella P, Jones A, de Saint Basile G, Savoldi G, Cranston T, et al. Mutations in severe combined immune deficiency (SCID) due to JAK3 deficiency. Hum Mutat (2001) 18:255–63. doi: 10.1002/humu.1188
59. Hapnes L, Willcox N, Oftedal BE, Owe JF, Gilhus NE, Meager A, et al. Radioligand-binding assay reveals distinct autoantibody preferences for type I interferons in APS I and myasthenia gravis subgroups. J Clin Immunol (2012) 32:230–7. doi: 10.1007/s10875-011-9617-4
60. Meager A, Wadhwa M, Dilger P, Bird C, Thorpe R, Newsom-Davis J, et al. Anti-cytokine autoantibodies in autoimmunity: preponderance of neutralizing autoantibodies against interferon-alpha, interferon-omega and interleukin-12 in patients with thymoma and/or myasthenia gravis. Clin Exp Immunol (2003) 132:128–36. doi: 10.1046/j.1365-2249.2003.02113.x
61. Shiono H, Wong YL, Matthews I, Liu JL, Zhang W, Sims G, et al. Spontaneous production of anti-IFN-alpha and anti-IL-12 autoantibodies by thymoma cells from myasthenia gravis patients suggests autoimmunization in the tumor. Int Immunol (2003) 15:903–13. doi: 10.1093/intimm/dxg088
62. Burbelo PD, Browne SK, Sampaio EP, Giaccone G, Zaman R, Kristosturyan E, et al. Anti-cytokine autoantibodies are associated with opportunistic infection in patients with thymic neoplasia. Blood (2010) 116:4848–58. doi: 10.1182/blood-2010-05-286161
63. Campanati A, Benfaremo D, Luchetti MM, Ganzetti G, Gabrielli A, Offidani A. Certolizumab pegol for the treatment of psoriasis. Expert Opin Biol Ther (2017) 17:387–94. doi: 10.1080/14712598.2017.1283401
64. Wolff AS, Karner J, Owe JF, Oftedal BE, Gilhus NE, Erichsen MM, et al. Clinical and serologic parallels to APS-I in patients with thymomas and autoantigen transcripts in their tumors. J Immunol (2014) 193:3880–90. doi: 10.4049/jimmunol.1401068
65. Fujimaki S, Kihara I, Tanaka R. Systemic lupus erythematosus (SLE) and the thymus. Acta Med Biol (Niigata) (1968) 16:1–15.
66. Kammer GM, Perl A, Richardson BC, Tsokos GC. Abnormal T cell signal transduction in systemic lupus erythematosus. Arthritis Rheumatol (2002) 46:1139–54. doi: 10.1002/art.10192
67. Gupta S, Tatouli IP, Rosen LB, Hasni S, Alevizos I, Manna ZG, et al. Distinct functions of autoantibodies against interferon in systemic lupus erythematosus: A comprehensive analysis of anticytokine autoantibodies in common rheumatic diseases. Arthritis Rheumatol (2016) 68:1677–87. doi: 10.1002/art.39607
68. Mathian A, Breillat P, Dorgham K, Bastard P, Charre C, Lhote R, et al. Lower disease activity but higher risk of severe COVID-19 and herpes zoster in patients with systemic lupus erythematosus with pre-existing autoantibodies neutralising IFN-alpha. Ann Rheum Dis (2022) 81:1695–703. doi: 10.1136/ard-2022-222549
69. Panem S, Check IJ, Henriksen D, Vilcek J. Antibodies to alpha-interferon in a patient with systemic lupus erythematosus. J Immunol (1982) 129:1–3. doi: 10.4049/jimmunol.129.1.1
70. Burbelo PD, Browne S, Holland SM, Iadarola MJ, Alevizos I. Clinical features of Sjogren’s syndrome patients with autoantibodies against interferons. Clin Transl Med (2019) 8:1. doi: 10.1186/s40169-018-0218-1
71. Psarras A, Wittmann M, Vital EM. Emerging concepts of type I interferons in SLE pathogenesis and therapy. Nat Rev Rheumatol (2022) 18:575–90. doi: 10.1038/s41584-022-00826-z
72. Li W, Deng C, Yang H, Wang G. The regulatory T cell in active systemic lupus erythematosus patients: A systemic review and meta-analysis. Front Immunol (2019) 10:159. doi: 10.3389/fimmu.2019.00159
73. Muskardin TLW, Niewold TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol (2018) 14:214–28. doi: 10.1038/nrrheum.2018.31
74. Jiang Q, Yang G, Liu Q, Wang S, Cui D. Function and role of regulatory T cells in rheumatoid arthritis. Front Immunol (2021) 12:626193. doi: 10.3389/fimmu.2021.626193
75. Lubbers J, Brink M, van de Stadt LA, Vosslamber S, Wesseling JG, van Schaardenburg D, et al. The type I IFN signature as a biomarker of preclinical rheumatoid arthritis. Ann Rheum Dis (2013) 72:776–80. doi: 10.1136/annrheumdis-2012-202753
76. Marcovecchio GE, Bortolomai I, Ferrua F, Fontana E, Imberti L, Conforti E, et al. Thymic epithelium abnormalities in diGeorge and down syndrome patients contribute to dysregulation in T cell development. Front Immunol (2019) 10:447. doi: 10.3389/fimmu.2019.00447
77. Malle L, Patel RS, Martin-Fernandez M, Stewart OJ, Philippot Q, Buta S, et al. Autoimmunity in Down’s syndrome via cytokines, CD4 T cells and CD11c(+) B cells. Nature (2023) 615:305–14. doi: 10.1038/s41586-023-05736-y
78. Khor B, Buckner JH. Down syndrome: insights into autoimmune mechanisms. Nat Rev Rheumatol (2023) 19:401–2. doi: 10.1038/s41584-023-00970-0
79. Skogberg G, Lundberg V, Lindgren S, Gudmundsdottir J, Sandstrom K, Kampe O, et al. Altered expression of autoimmune regulator in infant down syndrome thymus, a possible contributor to an autoimmune phenotype. J Immunol (2014) 193:2187–95. doi: 10.4049/jimmunol.1400742
80. McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers (2015) 1:15071. doi: 10.1038/nrdp.2015.71
81. Prummer O, Bunjes D, Wiesneth M, Arnold R, Porzsolt F, Heimpel H. High-titre interferon-alpha antibodies in a patient with chronic graft-versus-host disease after allogeneic bone marrow transplantation. Bone Marrow Transplant (1994) 14:483–6.
82. Prummer O, Bunjes D, Wiesneth M, Arnold R, Porzsolt F, Heimpel H. Antibodies to interferon-alpha: a novel type of autoantibody occurring after allogeneic bone marrow transplantation. Bone Marrow Transplant (1996) 17:617–23.
83. Prummer O, Zillikens D, Porzsolt F. High-titer interferon-alpha antibodies in a patient with pemphigus foliaceus. Exp Dermatol (1996) 5:213–7. doi: 10.1111/j.1600-0625.1996.tb00119.x
84. Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, Manry J, et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci Immunol (2021) 6(62). doi: 10.1126/sciimmunol.abl4340
85. Bastard P, Michailidis E, Hoffmann HH, Chbihi M, Le Voyer T, Rosain J, et al. Auto-antibodies to type I IFNs can underlie adverse reactions to yellow fever live attenuated vaccine. J Exp Med (2021) 218(4). doi: 10.1084/jem.20202486
86. Fanouriakis A, Kostopoulou M, Andersen J, Aringer M, Arnaud L, Bae SC, et al. EULAR recommendations for the management of systemic lupus erythematosus: 2023 update. Ann Rheum Dis (2023) 83(1):15–29. doi: 10.1136/ard-2023-224762
87. Kalunian KC, Furie R, Morand EF, Bruce IN, Manzi S, Tanaka Y, et al. A randomized, placebo-controlled phase III extension trial of the long-term safety and tolerability of anifrolumab in active systemic lupus erythematosus. Arthritis Rheumatol (2023) 75:253–65. doi: 10.1002/art.42392
88. Vital EM, Merrill JT, Morand EF, Furie RA, Bruce IN, Tanaka Y, et al. Anifrolumab efficacy and safety by type I interferon gene signature and clinical subgroups in patients with SLE: post hoc analysis of pooled data from two phase III trials. Ann Rheum Dis (2022) 81:951–61. doi: 10.1136/annrheumdis-2021-221425
89. Bergman R, Ramon M, Wildbaum G, Avitan-Hersh E, Mayer E, Shemer A, et al. Psoriasis patients generate increased serum levels of autoantibodies to tumor necrosis factor-alpha and interferon-alpha. J Dermatol Sci (2009) 56:163–7. doi: 10.1016/j.jdermsci.2009.08.006
90. Borsani O, Bastard P, Rosain J, Gervais A, Sant'Antonio E, Vanni D, et al. Autoantibodies against type I IFNs in patients with Ph-negative myeloproliferative neoplasms. Blood (2022) 139:2716–20. doi: 10.1182/blood.2021014890
91. Troya J, Bastard P, Planas-Serra L, Ryan P, Ruiz M, de Carranza M, et al. Neutralizing autoantibodies to type I IFNs in >10% of patients with severe COVID-19 pneumonia hospitalized in Madrid, Spain. J Clin Immunol (2021) 41(5):914–22. doi: 10.1007/s10875-021-01036-0
92. van der Wijst MGP, Vazquez SE, Hartoularos GC, Bastard P, Grant T, Bueno R, et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci Transl Med (2021) 13:eabh2624. doi: 10.1126/scitranslmed.abh2624
93. Meloni A, Furcas M, Cetani F, Marcocci C, Falorni A, Perniola R, et al. Autoantibodies against type I interferons as an additional diagnostic criterion for autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab (2008) 93:4389–97. doi: 10.1210/jc.2008-0935
94. Wolff AS, Erichsen MM, Meager A, Magitta NF, Myhre AG, Bollerslev J, et al. Autoimmune polyendocrine syndrome type 1 in Norway: phenotypic variation, autoantibodies, and novel mutations in the autoimmune regulator gene. J Clin Endocrinol Metab (2007) 92:595–603. doi: 10.1210/jc.2006-1873
95. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science (2002) 298:1395–401. doi: 10.1126/science.1075958
96. Derbinski J, Pinto S, Rosch S, Hexel K, Kyewski B. Promiscuous gene expression patterns in single medullary thymic epithelial cells argue for a stochastic mechanism. Proc Natl Acad Sci U S A (2008) 105:657–62. doi: 10.1073/pnas.0707486105
97. Husebye ES, Anderson MS, Kampe O. Autoimmune polyendocrine syndromes. N Engl J Med (2018) 378:1132–41. doi: 10.1056/NEJMra1713301
98. Qi Y, Zhang R, Lu Y, Zou X, Yang W. Aire and Fezf2, two regulators in medullary thymic epithelial cells, control autoimmune diseases by regulating TSAs: Partner or complementer? Front Immunol (2022) 13:948259. doi: 10.3389/fimmu.2022.948259
99. Takaba H, Morishita Y, Tomofuji Y, Danks L, Nitta T, Komatsu N, et al. Fezf2 orchestrates a thymic program of self-antigen expression for immune tolerance. Cell (2015) 163:975–87. doi: 10.1016/j.cell.2015.10.013
100. Lienenklaus S, Cornitescu M, Zietara N, Lyszkiewicz M, Gekara N, Jablonska J, et al. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta. vivo J Immunol (2009) 183:3229–36. doi: 10.4049/jimmunol.0804277
101. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity (2006) 25:373–81. doi: 10.1016/j.immuni.2006.08.007
102. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi: 10.1038/nri3581
103. Haljasorg U, Bichele R, Saare M, Guha M, Maslovskaja J, Kond K, et al. A highly conserved NF-kappaB-responsive enhancer is critical for thymic expression of Aire in mice. Eur J Immunol (2015) 45:3246–56. doi: 10.1002/eji.201545928
104. Kong XF, Worley L, Rinchai D, Bondet V, Jithesh PV, Goulet M, et al. Three copies of four interferon receptor genes underlie a mild type I interferonopathy in down syndrome. J Clin Immunol (2020) 40:807–19. doi: 10.1007/s10875-020-00803-9
105. Le Voyer T, Parent AV, Liu X, Cederholm A, Gervais A, Rosain J, et al. Autoantibodies against type I IFNs in humans with alternative NF-kappaB pathway deficiency. Nature (2023) 623:803–13. doi: 10.1038/s41586-023-06717-x
106. Meager A, Peterson P, Willcox N. Hypothetical review: thymic aberrations and type-I interferons; attempts to deduce autoimmunizing mechanisms from unexpected clues in monogenic and paraneoplastic syndromes. Clin Exp Immunol (2008) 154:141–51. doi: 10.1111/j.1365-2249.2008.03739.x
107. Miller CN, Proekt I, von Moltke J, Wells KL, Rajpurkar AR, Wang H, et al. Thymic tuft cells promote an IL-4-enriched medulla and shape thymocyte development. Nature (2018) 559:627–31. doi: 10.1038/s41586-018-0345-2
108. Shiono H, Roxanis I, Zhang W, Sims GP, Meager A, Jacobson LW, et al. Scenarios for autoimmunization of T and B cells in myasthenia gravis. Ann N Y Acad Sci (2003) 998:237–56. doi: 10.1196/annals.1254.026
109. Wolff AS, Sarkadi AK, Marodi L, Karner J, Orlova E, Oftedal BE, et al. Anti-cytokine autoantibodies preceding onset of autoimmune polyendocrine syndrome type I features in early childhood. J Clin Immunol (2013) 33:1341–8. doi: 10.1007/s10875-013-9938-6
110. Zhang Q, Bastard P, Bolze A, Jouanguy E, Zhang SY, Effort CHG, et al. Life-threatening COVID-19: defective interferons unleash excessive inflammation. Med (N Y) (2020) 1:14–20. doi: 10.1016/j.medj.2020.12.001
111. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4570
112. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. The Finnish-German APECED Consortium. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Nat Genet (1997) 17:399–403. doi: 10.1038/ng1297-399
113. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional cloning of the APECED gene. Nat Genet (1997) 17:393–8. doi: 10.1038/ng1297-393
114. Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med (2010) 207:299–308. doi: 10.1084/jem.20091669
115. Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med (2010) 207(2):291–7. doi: 10.1084/jem.20091983
116. Oftedal BE, Assing K, Baris S, Safgren SL, Johansen IS, Jakobsen MA, et al. Dominant-negative heterozygous mutations in AIRE confer diverse autoimmune phenotypes. iScience (2023) 26:106818. doi: 10.1016/j.isci.2023.106818
117. Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, et al. Dominant mutations in the autoimmune regulator AIRE are associated with common organ-specific autoimmune diseases. Immunity (2015) 42:1185–96. doi: 10.1016/j.immuni.2015.04.021
118. Vrana B, Paschke H, Paschke A, Popp P, Schuurmann G. Performance of semipermeable membrane devices for sampling of organic contaminants in groundwater. J Environ Monit (2005) 7:500–8. doi: 10.1039/b411645c
119. Rich SA. Human lupus inclusions and interferon. Science (1981) 213:772–5. doi: 10.1126/science.6166984
120. Steinberg AD, Baron S, Talal N. The pathogenesis of autoimmunity in New Zealand mice, I. Induction of antinucleic acid antibodies by polyinosinic-polycytidylic acid. Proc Natl Acad Sci U S A (1969) 63:1102–7. doi: 10.1073/pnas.63.4.1102
121. Ferreira RC, Guo H, Coulson RM, Smyth DJ, Pekalski ML, Burren OS, et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes (2014) 63:2538–50. doi: 10.2337/db13-1777
122. Newby BN, Mathews CE. Type I interferon is a catastrophic feature of the diabetic islet microenvironment. Front Endocrinol (Lausanne) (2017) 8:232. doi: 10.3389/fendo.2017.00232
123. Tomer Y, Blackard JT, Akeno N. Interferon alpha treatment and thyroid dysfunction. Endocrinol Metab Clin North Am (2007) 36:1051–66. doi: 10.1016/j.ecl.2007.07.001
124. Ruiz Castro PA, Yepiskoposyan H, Gubian S, Calvino-Martin F, Kogel U, Renggli K, et al. Systems biology approach highlights mechanistic differences between Crohn’s disease and ulcerative colitis. Sci Rep (2021) 11:11519. doi: 10.1038/s41598-021-91124-3
125. Loberman-Nachum N, Sosnovski K, Di Segni A, Efroni G, Braun T, BenShoshan M, et al. Defining the celiac disease transcriptome using clinical pathology specimens reveals biologic pathways and supports diagnosis. Sci Rep (2019) 9:16163. doi: 10.1038/s41598-019-52733-1
126. Wolf J, Willscher E, Loeffler-Wirth H, Schmidt M, Flemming G, Zurek M, et al. Deciphering the transcriptomic heterogeneity of duodenal coeliac disease biopsies. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22052551
127. Roti E, Minelli R, Giuberti T, Marchelli S, Schianchi C, Gardini E, et al. Multiple changes in thyroid function in patients with chronic active HCV hepatitis treated with recombinant interferon-alpha. Am J Med (1996) 101:482–7. doi: 10.1016/S0002-9343(96)00259-8
128. Preziati D, La Rosa L, Covini G, Marcelli R, Rescalli S, Persani L, et al. Autoimmunity and thyroid function in patients with chronic active hepatitis treated with recombinant interferon alpha-2a. Eur J Endocrinol (1995) 132:587–93. doi: 10.1530/eje.0.1320587
129. Calabrese LH, Lenfant T, Calabrese C. Interferon therapy for COVID-19 and emerging infections: Prospects and concerns. Cleve Clin J Med (2020). doi: 10.3949/ccjm.87a.ccc066
130. Crow YJ, Stetson DB. The type I interferonopathies: 10 years on. Nat Rev Immunol (2022) 22:471–83. doi: 10.1038/s41577-021-00633-9
131. Vallbracht A, Treuner J, Flehmig B, Joester KE, Niethammer D. Interferon-neutralizing antibodies in a patient treated with human fibroblast interferon. Nature (1981) 289:496–7. doi: 10.1038/289496a0
132. Kappos L, Clanet M, Sandberg-Wollheim M, Radue EW, Hartung HP, Hohlfeld R, et al. Neutralizing antibodies and efficacy of interferon beta-1a: a 4-year controlled study. Neurology (2005) 65:40–7. doi: 10.1212/01.wnl.0000171747.59767.5c
133. Deisenhammer F. Interferon-beta: neutralizing antibodies, binding antibodies, pharmacokinetics and pharmacodynamics, and clinical outcomes. J Interferon Cytokine Res (2014) 34:938–45. doi: 10.1089/jir.2012.0135
134. Cheng A, Holland SM. Anti-cytokine autoantibodies: mechanistic insights and disease associations. Nat Rev Immunol (2023). doi: 10.1038/s41577-023-00933-2
135. Morimoto AM, Flesher DT, Yang J, Wolslegel K, Wang X, Brady A, et al. Association of endogenous anti-interferon-alpha autoantibodies with decreased interferon-pathway and disease activity in patients with systemic lupus erythematosus. Arthritis Rheumatol (2011) 63:2407–15. doi: 10.1002/art.30399
136. Beccuti G, Ghizzoni L, Cambria V, Codullo V, Sacchi P, Lovati E, et al. A COVID-19 pneumonia case report of autoimmune polyendocrine syndrome type 1 in Lombardy, Italy: letter to the editor. J Endocrinol Invest (2020) 43:1175–7. doi: 10.1007/s40618-020-01323-4
137. Bastard P, Orlova E, Sozaeva L, Levy R, James A, Schmitt MM, et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J Exp Med (2021) 218(7). doi: 10.1084/jem.20210554
138. Schidlowski L, Iwamura APD, Covid SUD, Condino-Neto A, Prando C. Diagnosis of APS-1 in two siblings following life-threatening COVID-19 pneumonia. J Clin Immunol (2022) 42:749–52. doi: 10.1007/s10875-022-01245-1
139. Meisel C, Akbil B, Meyer T, Lankes E, Corman VM, Staudacher O, et al. Mild COVID-19 despite autoantibodies against type I IFNs in autoimmune polyendocrine syndrome type 1. J Clin Invest (2021) 131(14). doi: 10.1172/JCI150867
140. Wolff ASB, Hansen L, Grytaas MA, Oftedal BE, Breivik L, Zhou F, et al. Vaccination prevents severe COVID-19 outcome in patients with neutralizing type 1 interferon autoantibodies. iScience (2023) 26:107084. doi: 10.1016/j.isci.2023.107084
141. Busnadiego I, Abela IA, Frey PM, Hofmaenner DA, Scheier TC, Schuepbach RA, et al. Critically ill COVID-19 patients with neutralizing autoantibodies against type I interferons have increased risk of herpesvirus disease. PloS Biol (2022) 20:e3001709. doi: 10.1371/journal.pbio.3001709
142. Hetemaki I, Laakso S, Valimaa H, Kleino I, Kekalainen E, Makitie O, et al. Patients with autoimmune polyendocrine syndrome type 1 have an increased susceptibility to severe herpesvirus infections. Clin Immunol (2021) 231:108851. doi: 10.1016/j.clim.2021.108851
143. Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol (2015) 35:119–24. doi: 10.1007/s10875-014-0121-5
144. Meyer S, Woodward M, Hertel C, Vlaicu P, Haque Y, Karner J, et al. AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell (2016) 166:582–95. doi: 10.1016/j.cell.2016.06.024
145. Bruserud O, Oftedal BE, Landegren N, Erichsen MM, Bratland E, Lima K, et al. A longitudinal follow-up of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab (2016) 101:2975–83. doi: 10.1210/jc.2016-1821
146. Bradford HF, Haljasmagi L, Menon M, McDonnell TCR, Sarekannu K, Vanker M, et al. Inactive disease in patients with lupus is linked to autoantibodies to type I interferons that normalize blood IFNalpha and B cell subsets. Cell Rep Med (2023) 4:100894. doi: 10.1016/j.xcrm.2022.100894
147. Martinez RJ, Hogquist KA. The role of interferon in the thymus. Curr Opin Immunol (2023) 84:102389. doi: 10.1016/j.coi.2023.102389
148. Laan M, Salumets A, Klein A, Reintamm K, Bichele R, Peterson H, et al. Post-aire medullary thymic epithelial cells and Hassall’s corpuscles as inducers of tonic pro-inflammatory microenvironment. Front Immunol (2021) 12:635569. doi: 10.3389/fimmu.2021.635569
149. Colantonio AD, Epeldegui M, Jesiak M, Jachimowski L, Blom B, Uittenbogaart CH. IFN-alpha is constitutively expressed in the human thymus, but not in peripheral lymphoid organs. PloS One (2011) 6:e24252. doi: 10.1371/journal.pone.0024252
150. Ferreirinha P, Ribeiro C, Morimoto J, Landry JJM, Matsumoto M, Meireles C, et al. A novel method to identify Post-Aire stages of medullary thymic epithelial cell differentiation. Eur J Immunol (2021) 51:311–8. doi: 10.1002/eji.202048764
151. Xing Y, Wang X, Jameson SC, Hogquist KA. Late stages of T cell maturation in the thymus involve NF-kappaB and tonic type I interferon signaling. Nat Immunol (2016) 17:565–73. doi: 10.1038/ni.3419
152. Martinez RJ, Breed ER, Worota Y, Ashby KM, Voboril M, Mathes T, et al. Type III interferon drives thymic B cell activation and regulatory T cell generation. Proc Natl Acad Sci U S A (2023) 120:e2220120120. doi: 10.1073/pnas.2220120120
153. Ardouin L, Luche H, Chelbi R, Carpentier S, Shawket A, Montanana Sanchis F, et al. Broad and largely concordant molecular changes characterize tolerogenic and immunogenic dendritic cell maturation in thymus and periphery. Immunity (2016) 45:305–18. doi: 10.1016/j.immuni.2016.07.019
154. Pfeffer LM. The role of nuclear factor kappaB in the interferon response. J Interferon Cytokine Res (2011) 31:553–9. doi: 10.1089/jir.2011.0028
155. Cufi P, Dragin N, Ruhlmann N, Weiss JM, Fadel E, Serraf A, et al. Central role of interferon-beta in thymic events leading to myasthenia gravis. J Autoimmun (2014) 52:44–52. doi: 10.1016/j.jaut.2013.12.016
156. Le Panse R, Cizeron-Clairac G, Cuvelier M, Truffault F, Bismuth J, Nancy P, et al. Regulatory and pathogenic mechanisms in human autoimmune myasthenia gravis. Ann N Y Acad Sci (2008) 1132:135–42. doi: 10.1196/annals.1405.019
157. Poea-Guyon S, Christadoss P, Le Panse R, Guyon T, De Baets M, Wakkach A, et al. Effects of cytokines on acetylcholine receptor expression: implications for myasthenia gravis. J Immunol (2005) 174:5941–9. doi: 10.4049/jimmunol.174.10.5941
158. Cufi P, Soussan P, Truffault F, Fetouchi R, Robinet M, Fadel E, et al. Thymoma-associated myasthenia gravis: On the search for a pathogen signature. J Autoimmun (2014) 52:29–35. doi: 10.1016/j.jaut.2013.12.018
159. ElTanbouly MA, Zhao Y, Nowak E, Li J, Schaafsma E, Le Mercier I, et al. VISTA is a checkpoint regulator for naive T cell quiescence and peripheral tolerance. Science (2020) 367(6475):eaay0524. doi: 10.1126/science.aay0524
160. Hemmers S, Schizas M, Azizi E, Dikiy S, Zhong Y, Feng Y, et al. IL-2 production by self-reactive CD4 thymocytes scales regulatory T cell generation in the thymus. J Exp Med (2019) 216:2466–78. doi: 10.1084/jem.20190993
161. Gangaplara A, Martens C, Dahlstrom E, Metidji A, Gokhale AS, Glass DD, et al. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PloS Pathog (2018) 14:e1006985. doi: 10.1371/journal.ppat.1006985
162. Pacella I, Spinelli FR, Severa M, Timperi E, Tucci G, Zagaglioni M, et al. ISG15 protects human Tregs from interferon alpha-induced contraction in a cell-intrinsic fashion. Clin Transl Immunol (2020) 9:e1221. doi: 10.1002/cti2.1221
163. Elsaesser HJ, Mohtashami M, Osokine I, Snell LM, Cunningham CR, Boukhaled GM, et al. Chronic virus infection drives CD8 T cell-mediated thymic destruction and impaired negative selection. Proc Natl Acad Sci U S A (2020) 117:5420–9. doi: 10.1073/pnas.1913776117
164. Demoulins T, Baron ML, Gauchat D, Kettaf N, Reed SJ, Charpentier T, et al. Induction of thymic atrophy and loss of thymic output by type-I interferons during chronic viral infection. Virology (2022) 567:77–86. doi: 10.1016/j.virol.2021.12.007
165. Savino W. The thymus is a common target organ in infectious diseases. PloS Pathog (2006) 2:e62. doi: 10.1371/journal.ppat.0020062
Keywords: type I interferons, autoimmunity, immune deficiencies, thymus, AIRE
Citation: Oftedal BE, Sjøgren T and Wolff ASB (2024) Interferon autoantibodies as signals of a sick thymus. Front. Immunol. 15:1327784. doi: 10.3389/fimmu.2024.1327784
Received: 25 October 2023; Accepted: 07 February 2024;
Published: 22 February 2024.
Edited by:
Hela Jaïdane, University of Monastir, TunisiaReviewed by:
Nichole Danzl, Bristol Myers Squibb, United StatesKristin Hogquist, University of Minnesota Twin Cities, United States
Copyright © 2024 Oftedal, Sjøgren and Wolff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anette S. B. Wolff, QW5ldHRlLmJvZUB1aWIubm8=