 Ann Mary Joseph
Ann Mary Joseph Ahmad Al Aiyan
Ahmad Al Aiyan Basel Al-Ramadi
Basel Al-Ramadi Shiv K. Singh
Shiv K. Singh Uday Kishore
Uday Kishore
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 February 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1323198
This article is part of the Research Topic Implications of Immune Landscape in Tumor Microenvironment View all 12 articles
One of the most deadly and aggressive cancers in the world, pancreatic ductal adenocarcinoma (PDAC), typically manifests at an advanced stage. PDAC is becoming more common, and by the year 2030, it is expected to overtake lung cancer as the second greatest cause of cancer-related death. The poor prognosis can be attributed to a number of factors, including difficulties in early identification, a poor probability of curative radical resection, limited response to chemotherapy and radiotherapy, and its immunotherapy resistance. Furthermore, an extensive desmoplastic stroma that surrounds PDAC forms a mechanical barrier that prevents vascularization and promotes poor immune cell penetration. Phenotypic heterogeneity, drug resistance, and immunosuppressive tumor microenvironment are the main causes of PDAC aggressiveness. There is a complex and dynamic interaction between tumor cells in PDAC with stromal cells within the tumour immune microenvironment. The immune suppressive microenvironment that promotes PDAC aggressiveness is contributed by a range of cellular and humoral factors, which itself are modulated by the cancer. In this review, we describe the role of innate and adaptive immune cells, complex tumor microenvironment in PDAC, humoral factors, innate immune-mediated therapeutic advances, and recent clinical trials in PDAC.
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest solid tumours in humans. It is the most frequent form of pancreatic cancer, 90% of all pancreas neoplasms, which is characterised by tubular adenocarcinoma of the ductal glands (1, 2). Pancreatic cancer and pancreatic ductal adenocarcinoma are sometimes used interchangeably. Only 11% of patients with PDAC survive for at least 5 years (3). Over 400,000 people die from PDAC every year, the seventh most common cancer-related cause of death worldwide (4). A usually poor prognosis is projected for the more than 450,000 patients who receive annual diagnosis (5). It is predicted that pancreatic cancer-related death will overtake lung cancer as the second most prevalent cause of cancer-related death in the United States by 2030 (6). Along with the aggressive tumour biology, the pancreas’ central placement within the abdominal cavity, the lack of a distinct organ capsule, and the abundance of nearby blood and lymphatic arteries all contribute to the tumor’s ability to spread locally and elsewhere such as liver, lung, bone and brain (7). The pancreas, a comparatively clean organ, with very few lymphocytes, is located in the retroperitoneum and has no direct contact with the outside world; instead, it communicates with the digestive tract solely through the pancreatic duct. Therefore, very few lymphocytes can be seen in healthy pancreatic tissue (8, 9). In contrast to other malignancies, the incidence of pancreatic cancer is still rising while survival rates are barely improving.
A few recent reviews in the field describe the immunosuppressive TiME in PDAC and the TME targeted therapeutic approaches (10), pro- and anti-tumour properties of immune cells (11), the effector immune cells with potential biomarkers and targets (12), and the need for reprogramming of the stroma for the development of new therapeutic strategies (13). In this review, we have examined the immune landscape in human PDAC more holistically and how that affects survival and treatment for PDAC patients. This review also includes some of the important areas such as humoral immune factors, its significance, and the coexistence of classical and basal-like phenotypes.
The main therapeutic modalities for PDAC are surgical resection, chemotherapy, and radiotherapy. In most cases, the only form of treatment that has a chance of being curative is radical surgical resection (14, 15). Less than 25–30% of all PDAC patients are considered candidates for partial pancreatectomy at the time of diagnosis (16). Nearly 80% of PDAC patients cannot have a curative resection due to the stromal microenvironment which plays a role in malignant transformation, local invasion, and distant metastasis (17, 18). The development of immune checkpoint blockade (ICB) therapy has revolutionized cancer treatment, but the PDAC immunotherapy regimen, whether used alone or in combination with chemotherapy, has not shown encouraging results in patients with metastatic PDAC (mPDAC) (19). Borderline resectable or locally advanced PDAC patients, have significantly better survival rates in those patients who received neoadjuvant therapy (20, 21). The neoadjuvant therapy regimen includes chemotherapy with 5-fluorouracil (FOLFIRINOX/FOLFOX), gemcitabine (gemcitabine/nab-paclitaxel) or chemoradiotherapy before surgery. The CA19-9 level is considered a specific biomarker for tumor resectability and overall survival (22).
Conventional cytotoxic therapies such as chemotherapy and radiation therapy have not increased the chances of survival for patients with pancreatic cancer. Since 2011, 5-fluorouracil/leucovorin with irinotecan and oxaliplatin (FOLFIRINOX) and nab-paclitaxel with gemcitabine have been the preferred treatments for mPDAC. Response rates for these treatments range between 23% and 31%, progression-free survival time ranges from 5.5 to 6.6 months, and overall survival times range from 8.5 to 11 months. The only targeted treatment for PDAC that the US Food and Drug Administration (FDA) has approved is erlotinib, an epidermal growth factor receptor (EGFR) inhibitor, in combination with gemcitabine hydrochloride in patients with metastatic, locally advanced, or unresectable PDAC. The absolute benefit of gemcitabine and erlotinib, however, is also negligible for up to 2 weeks (23).
Despite advancements in pancreatic cancer research, screening, and treatment strategies, PDAC has a poor prognosis and resistance to many treatments, including immunotherapy (24). A large matrix of stromal cells is strongly connected with the poor prognosis of PDAC (25). PDAC is characterised by a desmoplastic stroma, a fibrotic TME with respect to normal pancreatic tissue as illustrated in Figure 1. Additionally, different epigenetic modifications as well as mutations in protooncogenes and tumour suppressor genes are seen in the stromal cells surrounding the tumour as well as the tumour epithelium (26, 27). The compact dysplastic stroma of PDAC is a significant barrier to chemotherapeutic agents. Thus, stroma-targeting therapy has been recognised as a prospective approach to enhance the effectiveness of chemotherapy, and hence, patient survival rates (28).
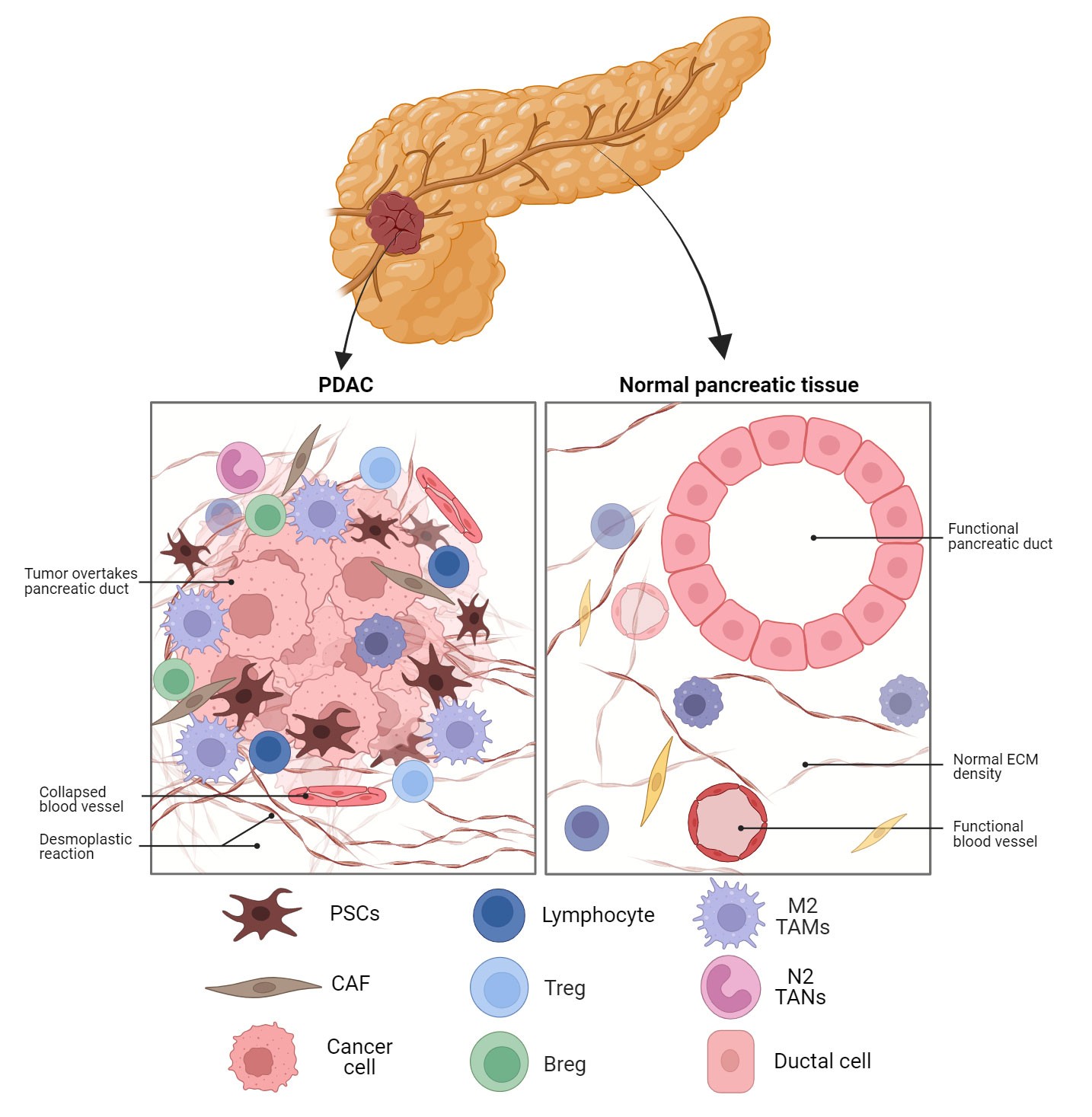
Figure 1 Schematic representation of the PDAC TME compared to normal pancreatic tissue. Fibroblast activated in the tissue to CAFs are the dominant cell type in PDAC along with M2 TAMs and MDSC. The dense desmoplastic reaction and collapsed blood vessels provide barriers to cytotoxic T cell infiltration. PDAC, pancreatic ductal adenocarcinoma; ECM, extracellular matrix; PSC, pancreatic stellate cell; CAF, cancer-associated fibroblast;Treg, regulatory T cell; Breg, regulatory B cell; TAM, tumour-associated macrophage; TAN, tumour-associated neutrophil.
A major component of the stroma in PDAC, hyaluronic acid (HA), interacts with cell surface receptors CD44 and receptor for HA-mediated motility (RHAMM) to promote tumour cell survival and to initiate signalling pathways associated with tumour cell proliferation, migration, and invasion (29–32). Hence, the targeting of HA is regarded as a promising therapeutic approach in the context of PDAC. PEGylated hyaluronidase (PEGPH20) refers to a PEGylated nanoscale complex that consists of recombinant human hyaluronidase (33, 34). Several studies have demonstrated that PEGPH20 has the ability to degrade HA, remodel tumour vasculature, and enhance the effectiveness of chemotherapeutic drugs (33, 35, 36). The study HALO-109-202, a phase II clinical trial, examined the effects of combining PEGPH20 with Abraxane (an albumin-bound paclitaxel nanocomplex) and gemcitabine in 279 patients diagnosed with mPDAC. The results demonstrated a significant increase in progression-free survival and overall survival among patients with elevated levels of HA (37). However, the phase III clinical study failed to considerably improve the PDAC patients’ overall survival.
Collagen represents another significant constituent within the extracellular matrix (ECM) of tumours. High levels of fibrillar collagens found in the stroma of PDAC play a critical role in promoting tumour cell survival and tumour progression. This process is mediated by the involvement of discoidin domain receptor 1 and 2 (DDR1 and DDR2). Huo et al. revealed that there was a significant correlation between elevated expression levels of DDR1 and an increased risk of unfavourable prognosis in PDAC patients (38). A small molecule inhibitor targeting DDR1 resulted in a decrease in fibrillar collagen deposition and an enhancement in the efficacy of chemotherapy in orthotopic mouse models of PDAC (39). KI-301690, a small molecule that disrupts DDR1 signaling, is a selective DDR1 inhibitor. A combination treatment with gemcitabine significantly inhibited the growth of pancreatic cancer cells (40).
Hedgehog (Hh) signalling pathway is typically characterised by an increased activity in PDAC via the activation of pancreatic stellate cells (41). This pathway has been shown to play a role in the regulation of stroma deposition (42). Multiple strategies have been developed with the aim of treating PDAC through the inhibition of the Hh signalling pathway, with the ultimate goal of eradicating the tumour stroma (43). Cyclopamine, a steroidal alkaloid of natural origin, has been found to effectively inhibit the Hh signalling pathway by binding to the Smoothened (SMO) protein (44). In the PDAC xenograft mouse model, it was found that the fibronectin content was decreased and tumour vascularization was increased. Co-administration of cyclopamine with paclitaxel-loaded nanoparticles resulted in a significant enhancement of tumour growth inhibition (45). The anti-tumor efficacy was mediated by increased tumor infiltration of CD8+ T cells without concomitant infiltration of immune suppressive cells, and by the coordinated action of Paclitaxel and IFN-γ (46). Another polymeric conjugate of docetaxel and cyclopamine has been examined for its anti-cancer effect in murine PDAC (47). This combination therapy resulted in greater inhibition of orthotopic pancreatic tumor growth.
The majority of PDAC patients have non-resectable tumours by the time they develop symptoms such as weight loss, abdominal pain and jaundice (48). Early detection of PDAC improves survival rates, but its low prevalence makes screening the general population impractical. Screening subgroups may include people with germline mutations, pancreatitis, mucinous pancreatic cysts, and elderly new-onset diabetics (49). For accurate diagnosis, high-resolution ultrasound, endoscopic ultrasound (EUS), computed tomography (CT), and magnetic resonance imaging (MRI) are needed.
Advanced PDAC has few treatment options, making early detection crucial for prognosis. Thus, developing diagnostic biomarkers for high-risk populations is important. CA19-9, the only FDA-approved biomarker for the diagnosis and monitoring of PDAC, is probably the most extensively validated biomarker that has diagnostic, prognostic and surveillance value (50, 51). CA125, CA72-4, CA50, CA199, and CA242 are other antigens used as biomarkers (52, 53). A single diagnostic potential for any of these biomarkers could not be established; however, when used along with CA 19-9, they may help distinguish between benign and malignant pancreatic lesions. Similarly, CA19-9, when combined with CEA, appears to have a better prognostic value, particularly in advanced PDAC (54).
Typically, PDAC is characterised by the presence of oncogenic mutations in genes such as KRAS and loss-of-function mutations in tumour suppressors such as TP53, CDNK2A, SMAD4, and BRCA2. These biomarkers and genomic mutations have the potential to function as targets or prognostic indicators, depending on the expression. PDAC originates from a series of precursor lesions, such as pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasm (IPMN), and mucinous cystic neoplasm (MCN) (55). In most cases, KRAS mutations emerge in PanIN-1 lesions and drive the initiation process, while CDKN2A mutations emerge in PanIN-2 and drive the disease forward. Mutations in TP53 and SMAD4, are frequently found in PanIN-3 and invasive tumours (56, 57). Approximately 95% of pancreatic tumours exhibit RAS mutations, with KRAS alterations being the most prevalent, accounting for 85% of cases. Additionally, KRAS stimulates the nuclear factor κB (NF-κB) pathway, which is linked to the development of a strong inflammatory response (58). Besides mutations in KRAS, inactivation of CDKN2A is observed in approximately 90% of PDAC cases, while SMAD4/DPC4 alterations are present in approximately 55% of cases (59). Also, a significant proportion of PDAC cases, ranging from approximately 50% to 70%, exhibit mutations in the TP53 gene (60). The SMAD4 gene is deactivated in approximately 60% of cases of PDAC (61). This gene plays a crucial role as an effector in the transforming growth factor β (TGF-β) signalling, which is also disrupted in 47% of PDAC cases (62, 63). Dysregulation of various critical processes-related signalling pathways, such as apoptosis and cell proliferation, occurs because of these mutations.
Genomic profiling at a large scale has shown that PDAC has two different histological types: “classical” and “basal-like”. As shown in Figure 2, the “Classical” or progenitor subtype was distinguished by the expression of epithelial markers and a good prognosis, while the “Basal-like,” squamous or quasi-mesenchymal subtype was characterised by the expression of mesenchymal markers and aggressive/metastatic properties. There is still disagreement over how to actually use the subtype classification for clinical decision-making in PDAC, despite the fact that these molecular subtypes of PDAC may offer new avenues for precision medicine approaches (65). Collisson et al. conducted transcriptome analyses on tissue samples of PDAC, as well as human and murine PDAC cell lines, and identified three distinct molecular subtypes of PDAC, namely the classical, quasi-mesenchymal, and exocrine-like subtypes (66). The classical subtype is distinguished by the activation of genes associated with epithelial and adhesion functions. In contrast, the quasi-mesenchymal subtype predominantly exhibits the expression of genes related to mesenchymal characteristics. Additionally, the exocrine-like subtype is characterised by the upregulation of genes associated with digestive enzymes. It is noteworthy that these subtypes exhibit relevance in terms of survival, as the classical subtype is associated with the most favorable prognosis, while the quasi-mesenchymal subtype is linked with the poorest prognosis (66). Moreover, it has been observed that PDAC cell lines belonging to the classical subtype exhibit resistance to gemcitabine therapy but show sensitivity to erlotinib.
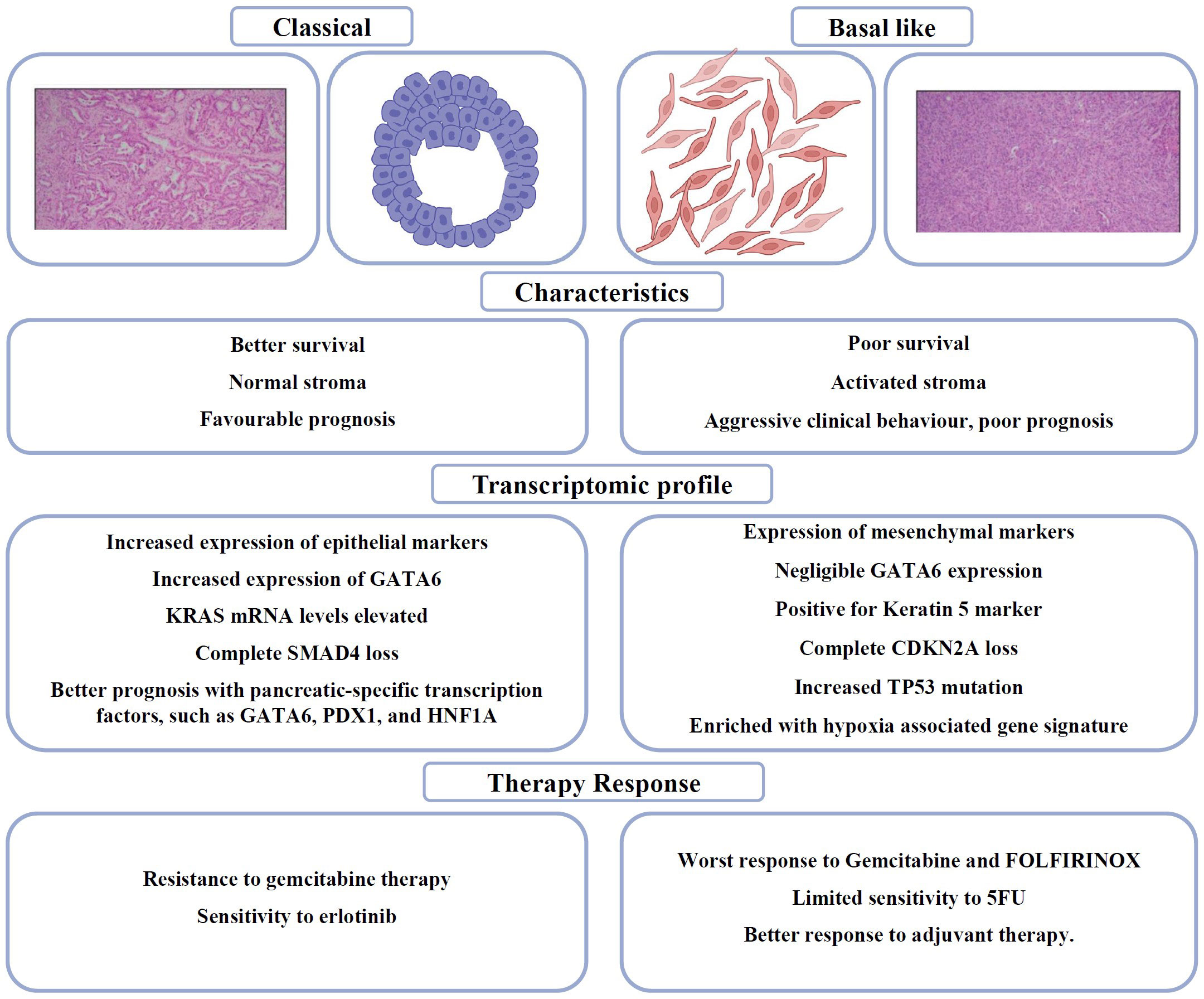
Figure 2 Schematic and H&E sections PDAC (Scale: 200 μm) to distinguish between Classical and Basal like subtypes in PDAC (64), summarising the clinicopathological differences, genetic signatures and therapy responses.
Moffitt et al. later achieved successful molecular subtyping of both the epithelial cells and stroma of PDAC, leading to the identification of two distinct subtypes: normal and activated PDAC stroma. Notably, the activated subtype was associated with a poorer prognosis. The two subtypes specific to tumour were denoted as Classical and Basal-Like (67). The classical subtype is distinguished by the presence of overlapping genetic signatures, such as GATA6. The Basal-like subtype is correlated with a more unfavorable prognosis compared to the Classical subtype. However, it exhibits a more favorable response to adjuvant therapy.
In 2018, Puleo et al. examined the influence of the tumor microenvironment (TME) in PDAC. They categorised PDAC into five distinct clinical subtypes: Pure-basal-like, Stroma-activated, Desmoplastic, Pure-classical, and Immune-classical. Yet another classification distinguishes PDAC into Basal-like A/B, Classical A/B and Hybrids. Basal-like tumors are more aggressive; Basal-like A is associated with metastatic disease, and Basal-like B with resectable disease. Classical A/B tumors are frequently found in the early stage while Hybrids reveal the presence of multiple expression signatures (68).
Recent studies observed the coexistence of basal-like and classical subtype in PDAC. The intratumoral coexistence, which is increased during disease progression, inversely affects the prognosis and treatment based on subtypes. A comprehensive study of the dichotomous role of AP1 transcription factors (JUNB/AP1 versus cJUN/AP1) in PDAC subtype heterogeneity sheds light on the plasticity and stability of classical and basal-like neoplastic cells (69). It also highlights the importance of anti-tumor necrosis factor α (TNF-α) with gemcitabine chemotherapy which may provide a valuable strategy for a better treatment response in PDAC. The co-expression of tumor subtypes has been observed in approximately 90% of tumors using a multiplex immunofluorescence pipeline, based on the protein expression of PDAC subtype markers (70). The extensive intratumoral heterogeneity needs further characterisation in terms of intrinsic and extrinsic factors that dictate subtype heterogeneity. This will open up new prognosis and treatment options for PDAC patients (71).
The cellular and humoural components make up the heterogeneous PDAC TME. In the cellular component, there are immune cells, endothelial cells, pancreatic stellate cells (PSCs), cancer-associated fibroblasts (CAFs) and myofibroblasts (Figure 3; Table 1). The humoural component is made up of collagen, fibronectin, and multiple soluble factors, including cytokines, chemokines, growth factors and complement components residing in the ECM (94–97). The interaction between these two components is essential for promoting tumour growth and the emergence of therapeutic intervention resistance. The development of an immunosuppressive TME, which allows the tumour to elude immune surveillance, is a feature frequently observed in PDAC.
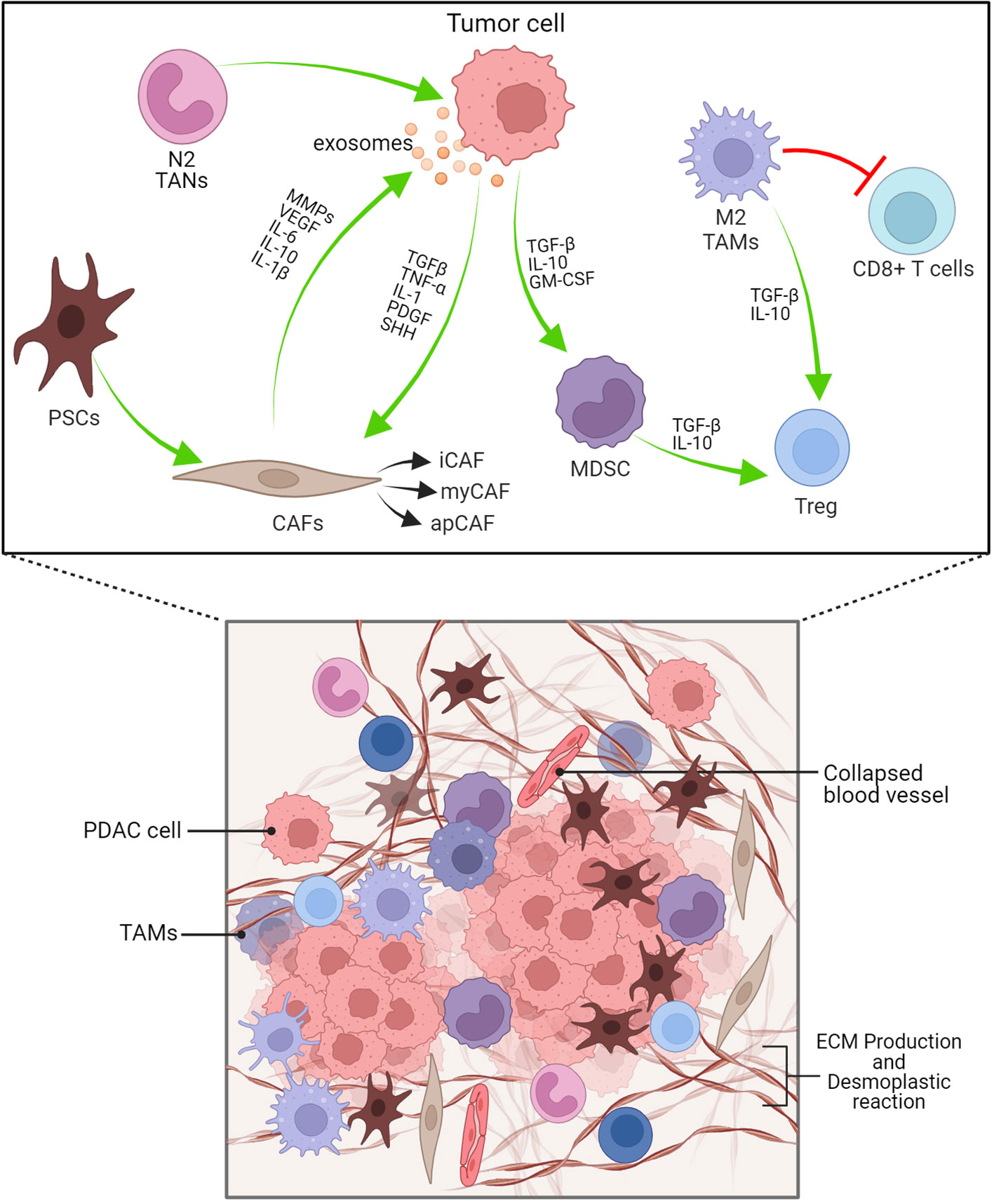
Figure 3 PDAC’s dense desmoplastic stroma and tumour microenvironment are depicted schematically here. The PDAC stroma is largely made up of CAFs, Macrophages, MDSCs and other immune cells. Exosomes produced from PDAC cells recruits and activates CAFs. Cytokines, TGF-β, IL-1, PDGF, SHH are cruicial for CAF activation. N2 TANs, myCAF, Tregs and Bregs have protumourigenic role. iCAF-inflammatory CAF, myCAF-myofibroblastic CAF, apCAF-antigen presenting CAF.
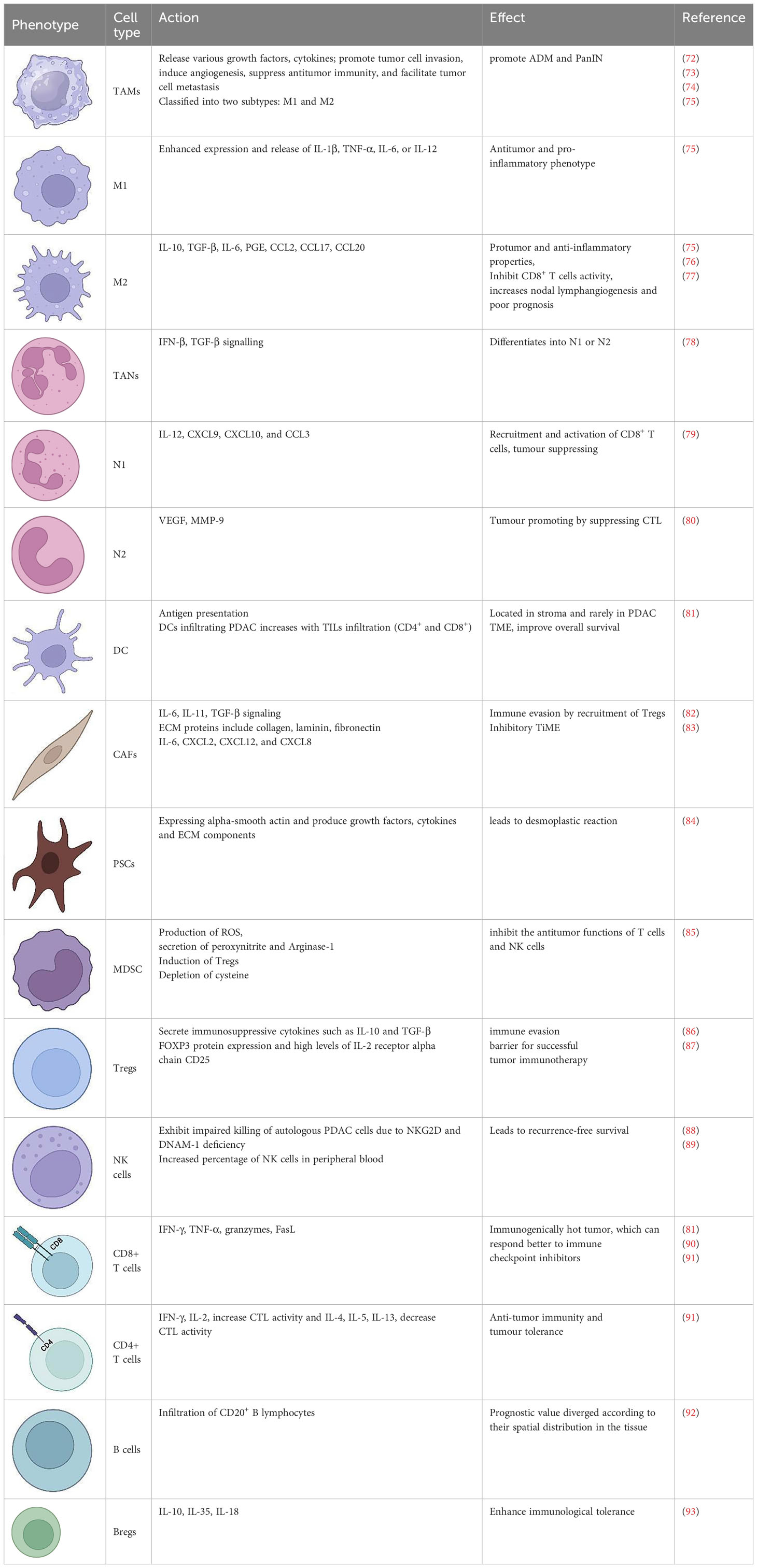
Table 1 The immune cells in the tumor microenvironment of pancreatic ductal adenocarcinoma.
The human pancreas is comprised of exocrine (acinar), epithelial (ductal), and endocrine (α, β, δ, ϵ) cells. The plasticity of the pancreas is believed to be responsible for maintaining its homeostasis and promoting regeneration. Both acinar and ductal cells in the healthy pancreas can give rise to PDAC, though acinar cells appear to be more prone to oncogenic transformation (98). Acinar cells undergo a plastic trans-differentiation process known as acinar to ductal metaplasia (ADM), which can progress to PanINs, and eventually, adenocarcinoma (99), in response to specific macro- and microenvironmental stimuli, such as tissue damage, inflammatory factors, or stress conditions (98, 100), and become more vulnerable to activating mutations in the proto-oncogene KRAS. PanINs are the most frequent precursor lesions that are linked to the development of invasive PDAC among premalignant lesions with distinct histopathological features such as microscopic mucinous pancreatic ductal lesions with flat to papillary, micropapillary, or cribriform formation with severe nuclear atypia, loss of polarity, macronucleoli, and abnormal mitotic figures (86).
The etiology of PDAC would not be complete without highlighting the importance of inflammatory signals for initiation and progression of tumorigenesis. Inflammation and increased immune cell infiltration are common risk factors for human pancreatic cancer. Tumor-promoting inflammation, (101) is an integral part of neoplastic progression in PDAC. Chronic inflammation of the pancreas, known as pancreatitis, is a significant risk factor for the development of PDAC (102); the importance of environmental factors that cause chronic inflammation (e.g., smoking, heavy alcohol consumption, diet, and obesity) in pancreatic cancer is well established (103, 104). Inflammation promotes tumour formation, growth, progression, and metastasis (105).
The TME inflammatory cells and cancer cells are known to secrete several cytokines, such as IL-6, IL-10, IL-13, VEGF, and TGF-β (106). The anti-inflammatory TGF-β and IL-10, as well as the pro-inflammatory IL-1, IL-6, IL-17, and TNF-α, play a significant role in PDAC. Depending on the cross-talk between cancer cells and inflammatory cells, the ratio of pro- to anti-inflammatory cytokines in the TME constantly changes. Ling et al. demonstrated in a genetically engineered mouse model that oncogenic KRAS leads to a constitutive activation of NF-κB through IL-1α and p62 (107). Consequently, cancer cell-intrinsic inflammatory signalling networks generate a protumorigenic TME via the expression of cytokines that promote angiogenesis and the recruitment of immune and stromal cells. Several interleukins, including IL-6, were among those dysregulated by the depletion of NF-κB signalling. IL-6 class cytokines (e.g., IL-6, LIF, OSM, IL-11) are among the few regarded as master regulators of inflammation associated with cancer (108). Blocking the inflammatory cytokine IL-6 may improve the efficacy of anti-PD-L1 therapy by modulating immunological features of PDAC in murine models (109). It may enhance T cell trafficking and alter the tumor’s T cell population, as the ability of a patient to respond to checkpoint inhibitors is significantly impacted by T cell infiltration into tumours (110).
The immunosuppressive TME and cell types, a hallmark of pancreatic cancer, are thought to promote tumour invasion and growth. Myeloid cells develop from hematopoietic stem cells in the bone marrow through myelopoiesis. They are characterised by the expression of CD45 and CD11b surface markers. Subsequently, they undergo differentiation into discrete subpopulations, namely macrophages, granulocytes, mast cells, and dendritic cells, all of which are integral constituents of the innate immune system. Myeloid cell abundance in tumours correlates with worse clinical outcomes (111, 112).
The macrophages present within the tumour are commonly known as tumor-associated macrophages (TAMs). Two main phenotypes of macrophages, known as M1 and M2, are known for their ability to display plasticity. M2 polarised macrophages display immunosuppressive traits and a restricted adaptive immune response. Induction of an M1-like phenotype is typically seen to enable adaptive immunosurveillance. TAMs inhibit T-lymphocyte responses (113) and secrete cytokines that promote the tumor phenotype and metastasis (73, 114). In addition to their ability to directly induce T-lymphocyte apoptosis (115), TAMs produce arginase-1(72), a metalloenzyme that metabolizes and depletes the environment of arginine, an essential compound for T-lymphocyte proliferation (116, 117). Another major TAM subpopulation includes SPP1+ and C1QC+ TAMs, which on further characterization, showed enrichment for epithelial-to-mesenchymal transition (EMT) and a high angiogenesis score in SPP1+ TAMs, while C1QC+ TAMs were enriched for antigen presentation and phagocytosis (118). Granulocytes can be further categorised into three subtypes: eosinophils, basophils, and neutrophils. In the context of the TME, it is common for neutrophils and monocytes to exist in an immature state, which is commonly referred to as immature myeloid cells or myeloid-derived suppressor cells (MDSCs).
Myeloid cells and other immune cells infiltrate the PDAC TME, resulting in a state of local inflammation (119) in which tumour cells interact with infiltrating immune cells. However, for a transformed cell to survive, it must attain an immunosuppressive phenotype, such as downregulation of MHC class I expression and upregulation of programmed cell death receptor ligand-1 (PD-L1) and CD47, which hinder the anti-tumor immune response by engaging and suppressing the activated T cells and relaying ‘don’t eat me’ signal to the phagocytic macrophages, respectively (120, 121). Constitutively active KRasG12D regulates autophagy-induced MHC class I downregulation, which is a major mechanism that PDAC cells employ to escape immune surveillance (122, 123). Table 1 provides the immune cell composition and its effect in the PDAC TME.
In addition to myeloid immune cells, fibroblasts in stromal components, known as Cancer-associated fibroblasts (CAFs), are an important TME component, including myofibroblastic CAFs (myCAFs), immunogenic CAFs (iCAFs), and antigen-presenting CAFs (apCAFs). PDAC has a unique fibrotic TME with desmoplastic stroma, abundant in ECM proteins produced by CAFs that represent a significant proportion of the cellular composition in the PDAC stroma, ranging from 15% to 85% of stromal cells (124) (Figure 3). CAFs, immune cells, cytokines and chemokines accumulate in the TME of primary and metastatic PDAC, exacerbating the development of an immunosuppressive phenotype (86, 125). CAFs create a physical and metabolic barrier via ECM proteins, thereby diminishing the effectiveness of therapeutic interventions against PDAC by increasing the interstitial tumor pressure that impairs vascular function (126–128). Additionally, CAFs facilitate tumour growth and invasion (129–132) and contribute to chemotherapy resistance by the presence of hyaluronan (18, 33, 127). The role of CAFs in immunosuppression (133), tumour metabolism (134), and secretion of inflammatory factors such as IL-1β, potential initiator of NF-κB signalling (135), have been studied. Therefore, the elimination of CAFs from the TME has the potential to serve as a possible therapeutic approach for the treatment of PDAC (131, 136).
Increasing desmoplasia, which frequently matches or exceeds the tumor’s epithelial component, is a hallmark of PDAC progression. The ECM provides physiological signals to neighboring cells in all tissues. The accumulation of ECM proteins is prevalent in solid tumours including PDAC and is referred to as a desmoplastic reaction (137). TME can modulate interstitial fluid pressure (33, 126) and reduce the density of blood vessels within tumours (33). Collagens, integrins, proteoglycans, glycoproteins, and proteases dominate the ECM of PDAC. These components interact with cancer cells through a variety of mechanisms (138, 139). Collagens Type I, III and IV are the most prevalent of these constituents. Collagens are active components in PDAC stroma with not for just structural support, but have a direct effect on the growth, survival, and spread of cancer cells (140); patients with higher level of fibrillar collagen have lower overall survival rate (141). αvβ6, an epithelial integrin, is upregulated in PDAC (142). Galectin-1 (GAL1), along with other glycoproteins such as periostin and fibulin, has been found to be upregulated in the PDAC TME and is poorly expressed in long-term (10 years) PDAC survivors (143).
CAFs, which originate primarily from PSCs and bone-marrow derived mesenchymal stem cells, are a major regulator of the ECM (144). PSCs are primarily located in the vicinity of pancreatic glands and possess the capability to produce ECM proteins, matrix metalloproteinases (MMPs), and MMP inhibitors, which play a crucial role in regulating ECM turnover (145). PSCs can be activated by pro-inflammatory cytokines, oxidative stress, hypoxia, hyperglycemia, and heightened interstitial pressure (146). Activated PSCs can secrete growth factors such as TGF-β1, PDGF and VEGF (147) to promote pancreatic cancer cell growth, decrease apoptosis, and increase invasion (148). PSCs are the primary source of collagen in tumour stroma, secreting ECM proteins such as α-smooth muscle actin and collagen. Reducing myofibroblasts and ECM in PDAC in vivo can inhibit tumour growth and improve chemotherapy sensitivity.
PDAC is immunologically heterogeneous; this heterogeneity exists between cells within PDAC. CD8+ cytotoxic T lymphocytes (CTL) and CD4+ T cells are the effector tumor infiltrating lymphocytes (TILs) observed in resected cancer tissue and are believed to participate in the host immune response against cancer which is considered a positive prognostic marker (149). Out of the total T lymphocytes (CD3), >80% are CD8+ T and CD4+ T Cells (150). The immune cells that target tumors are CTLs. CTLs use the Fas-FasL and perforin–granzyme pathways as major effector mechanisms of cytotoxicity; loss of Fas expression in PDAC tumours result in cancer immune evasion (151). PSCs produce elevated amounts of ECM, driving a fibrotic tissue that entraps infiltrated T cells, alongside immunosuppressive cytokines and expression of PDL-1. Pancreatic cancer cells avoid T cell killing by downregulating Fas, exhibiting low tumour mutational burden, expressing PDL-1 and secreting growth factors and cytokines that recruit immunosuppressive cells. CTLs are localised along the invasive margin of the tumour border or trapped in the surrounding fibrotic tissue but are not present within the tumour core. Moreover, infiltrated CD8+ T cells in PDAC tumours often display minimal signs of activation (152). MDSCs express PDL-1 and suppress T cells functions by several mechanisms, including depletion of arginase 1, the release of reactive oxygen species, and secretion of cytokines. Tregs directly suppress T cells, express cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and secrete cytokines such as TGF-β and IL-10. TAMs play a role in sequestering T cells at the periphery and secrete immunosuppressive cytokines (91). Tumour-derived cytokines and chemokines drive recruitment of myeloid cells to the TME. These cells, which include TAMs and MDSCs, block the recruitment and priming of T cells, resulting in T cell exclusion within the TME (153).
There is considerable infiltration of CD20+ B lymphocytes in the TME of human PDAC, unlike normal pancreatic tissue (92). There is a distinct spatial heterogeneity for B cells either in ectopic lymph nodes like tertiary lymphoid structures, or interspersed at the tumour–stroma interface. In addition, B cells produce anti-tumor antibodies and present tumor antigens to T cells to improve the cancer immunosurveillance. B cells in the TME respond to tumor-associated antigens by secreting IgG1 antibodies to activate the complement system, and phagocytosis by NK cells and macrophages (154). Alternatively, regulatory B cells (Bregs), dispersed inside the TME, contribute to the dampening of anti-tumor immune responses by secreting anti-inflammatory cytokines (IL-10 and IL-35), which promote tumor growth and metastasis (93). It appears that innate immune cells such as macrophages and neutrophils have a larger role to play in PDAC than the adaptive immune mechanisms.
In PDAC, regulatory T cells (Tregs) play a major role in tumour immune suppression. Through immunohistochemistry, they can be identified based on forkhead box protein 3 (FOXP3) expression and high levels of IL-2 receptor α chain CD25 in tumour tissues. There is sufficient evidence that Tregs are the primary barrier to an effective tumour immunotherapy (87). In fact, Tregs are significantly increased in the blood of PDAC patients as well as in the pancreatic tissue (155). They are recruited to tumour sites, where they inhibit antitumour cytotoxic response by binding to DCs and preventing DCs from activating CD8+T cells (156). From the premalignant to the invasive stages of PDAC, Tregs aid in suppressing the immune response against PDAC cells (157). In addition, it appears that a high Treg prevalence in PDAC is linked to a poor prognosis and weak PDAC differentiation (158). Single cell RNA seq studies revealed that activated TME is defined by the presence of Tregs, FGF, TAMs (SPP1+, GRN+), M2 like macrophages; in contrast, patients with normal stroma show M1-like macrophages, increased effector and exhausted T-cells (159).
Using the KC mouse model, a model where KRAS genetic changes are brought in for the development of pancreatic cancer, the immune cell infiltration at different stages of PDAC, including normal pancreas, PanINs, and invasive carcinoma, were examined (160, 161). Tregs and MDSCs predominated the immune infiltrate in the early PanIN stages. When the disease reached the PDAC stage, CD4+ and CD8+ cells were infrequently found and the existing CD8+ cells were not activated, suggesting an immunosuppressed TME (160). Strong inverse correlations between MDSCs and CD8+ T-lymphocytes at all disease stages imply that MDSCs are a key player in tumour immunosuppression (160).
Clinically, pancreatic cancer frequently contains T lymphocytes which surround the pancreatic lesion; CD8+ cells are elevated in the circulation of PDAC patients (162). PDAC has a high percentage of CD4+ Tregs, which support an immunosuppressive phenotype. They are typically found in the stromal regions of the tumour and rarely in conjunction with tumour epithelial cells (157). Treg accumulation is correlated with the progression of both the major preneoplastic lesions, PanINs and IPMN, in clinical samples of pre-malignant lesions (157). In murine models of PDAC, an association between Treg infiltration and the growth of pancreatic cancer is established. When syngeneic C57BL/6 mice are subcutaneously injected with mouse pancreatic tumour cells from Pan02, the spleen and tumour-draining lymph nodes of these mice exhibit a marked increase in Tregs (163). The CCR5 receptor, which is preferentially expressed by Tregs, is ligated by tumour cells in murine as well as human PDAC (164). Growth of PDAC is inhibited by CCR5 mediated blockade of Treg accumulation.
Tumor-infiltrating B lymphocytes in PDAC differentiate into Regulatory B cells (Bregs) that produce IL-10 or IL-35 with the help of other immune cells such as Tregs and MDSCs, cytokines IL-18, CAFs, tumor-associated antigens, damage-associated molecular patterns, hypoxia, pancreatic microbiota, and metabolites in the TME (165, 166). A high number of IL-10/IL-35-producing Bregs are observed in the PDAC stroma of KPC and KC murine models and PDAC patient samples (93). IL-18 promotes Breg differentiation and enhances immunological tolerance, leading to the development and metastasis of PDAC (46). In addition to IL-18, other chemokines such as CXCL13 and CCL21, are responsible for B-cell migration and accumulation within tumors (93).
Cancer stem cells (CSCs) in PDAC have the ability to self-renew, differentiate into numerous lineages, initiate tumourigenesis, and resist conventional cancer therapy. CSCs are characterized by specific cell surface markers, CD44+CD24+ESA+ (167). Pro-inflammatory cytokines are involved in the CSC self-renewal process (168). Following the development of pancreatitis, the number of CSCs in the circulation greatly increased. However, treatment with the anti-inflammatory drug, dexamethasone, lowers the level of CSCs in the circulation. Thus, inflammation plays an important role in the spread of pancreatic CSCs and perhaps even in PDAC metastasis.
Tumour-derived cytokines and chemokines in PDAC set up the immunosuppressive cellular network by attracting myeloid cells to the TME. TAMs and MDSCs contribute to T cell exclusion from the TME by inhibiting their recruitment and priming. By secreting cytokines, chemokines, and other factors such as GM-CSF, CSF-1, IL-3, CXCL12, and CCL2, TAMs and MDSCs can shape the TME in ways that promote or inhibit tumour growth and survival.
Acinar cell trans-differentiation into duct-like cells, known as acinar to ductal metaplasia (ADM), is the first histologically distinct event during PDAC pathogenesis (98, 99, 169). ADM is required for pancreatic regeneration by the acinar cells and is accompanied by a loss of polarity or contact between cells or with the ECM. However, pro-inflammatory cytokines prevent the acinar reversibility in the presence of oncogenic Kras and advance ADM to lesions PanIN (99). TNF-α and RANTES (Regulated on Activation Normal T Cell Expressed and Secreted) are two pro-inflammatory cytokines secreted by TAMs that cause ADM by triggering NF- κB signalling and the expression of MMPs (170, 171).
The functional relevance of the chemokines in PDAC and their association with the NF-κB pathway has been studied (172). TAMs also secrete IL-6 and promote STAT signaling resulting in tumour growth and progression (173, 174). The initial secretion of cytokines such as PDGF and TGF-β, recruits additional lymphoid and myeloid subsets into the TME, which then secrete more TGF-ß1, CTGF, high mobility group box protein 1 (HMGB1), IL-10, IL-1α, IL-1β, IL-8, TNF-α, and CCL18 depending upon their activation status (175), resulting in chronically inflamed tissues. Signaling through a family of G-protein coupled receptors is an additional important stimulus for the infiltration of these immune cells into the PDAC tissue (176).
Numerous chemokines are described in relation to PDAC pathogenesis and therapy resistance. PDAC cells produce chemokine CCL2 or monocyte chemotactic protein 1 (MCP1), a proinflammatory chemokine that binds to CCR2 and CCR4 under normal conditions (177). CCL2 is found to be highly expressed in Basal like subtype compared to Classical subtype and recruits TAMs to the TME (178). This basal expression is further increased when the cells are stimulated with IL-1, TNF-α or FAS ligand (179). Furthermore, the regulation of CCL2 expression in PDAC cells are attributed to the NF-κB pathway (177, 179, 180). CXCL8, or IL-8, is a chemokine produced by many cell types. IL-8 also binds to CXCR1 and CXCR2, with a higher affinity for CXCR1. In addition to angiogenic functions, IL-8 mediates phagocytosis and chemotaxis.
Immune cell fractionation in PDAC revealed a higher proportion of innate immune cells than adaptive immune cells (8). PDAC tissue contains an abundance of macrophages, MDSCs, DCs, and neutrophils. Single-cell RNA sequencing (RNA-seq) data revealed that macrophages are the predominant immune cells among the CD45+ population in PDAC (8). Neutrophils also contribute to significant portion of the immune cell infiltrate observed in PDAC (181). Neutrophils are transformed into tumour-associated neutrophils (TANs) after migrating into tumour tissues. TANs were identified as Ly6G+CD11b+ cells (182), and further classified as N1 (tumour suppressing) or N2 (tumour promoting) phenotype (100) and are associated with poor prognoses in PDAC (183). Neutrophils are recruited to the PDAC TME via multiple tumour-secreted chemokines including CXCL1, CXCL2, CXCL5, and CXCL8. They respond to these chemokines by the expression of the CXCR1 and CXCR2 CXC receptors. Tumour size in PDAC correlates with the level of CXCR2 expression (184). Myeloperoxidase+ (MPO+) neutrophils and CD11b+Ly6G+ MDSCs infiltration into tumours is reduced in CXCR2 knockout PKF [mice with conditional KrasG12D mutation and knockout of TGF-β receptor type II (Tgfbr2), (LSL-KrasG12D/+; Tgfbr2flox/flox, Ptf1a-Cre] mice compared to control animals (185). CXCL1, CXCL2, and CXCL5 secretion from tumour cells is elevated in the KPC (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx1-Cre) mouse model, in comparison to the normal pancreas (186). Another study demonstrated that CXCL5 has the greatest increase in human PDAC and correlated with both tumour-infiltrating CD15+ granulocytes and neutrophil elastase+ (NE+) granulocytes (187). Neutrophil depletion has been shown in multiple PDAC studies to reduce tumour growth and metastasis. Importantly, in wound healing and transwell assays in vitro, neutrophils derived from PDAC patients significantly promoted the migration and invasion of pancreatic cancer cells, whereas neutrophils derived from healthy individuals did not (188). In addition, Neutrophil to Lymphocyte Ratio correlates with a poor prognosis in patients with resectable and unresectable pancreatic cancer (187, 189–191). PDAC patient outcomes also correlate with the presence of neutrophils within the tumour. Neutrophil marker CD177 is inversely associated with overall survival in patients with PDAC (192). Patients with PDAC who have tumour-infiltrating neutrophils with high levels of CD66b+ have significantly lower survival rates (181). In human PDAC tissues, TAN-derived TGF-β induces EMT in human lung cancer tissues through the TGF-β/Smad pathway, contributing to carcinogenesis (193, 194). Another study indicates that inhibition of CXCR2 decreases TAN accumulation, and inhibits PDAC metastasis in mice (186, 195).
DCs are uncommon in the TME of pancreatic cancers and are located at the tumour’s periphery (196). Systemically, PDAC patients have decreased levels of blood DCs (197). Notably, higher levels of circulating DCs are associated with improved survival in PDAC patients (197, 198). In addition, surgical removal of the pancreatic tumour improved blood DC function, supporting a tumour-derived effect on immune function of DCs (199). During disease progression, the immune response of the host to pancreatic cancer is reported to shift from immune surveillance to immune tolerance. CXCL17 and intercellular adhesion molecule 2 (ICAM2) appear to mediate this polarisation (200). In addition, tumour-derived cytokines such as TGF-β, IL-10 and IL-6 have been shown to inhibit DC survival and proliferation (201). The proliferation of immature myeloid cells in the bloodstream and spleen may further compromise the immune response. The level of circulating MDSCs is increased in PDAC, which may promote tumour progression (202, 203). MDSCs inhibit DC activation in pancreatic cancer by producing nitric oxide (NO) (204).
Innate lymphoid cells (ILCs) are innate immune cells that bridge between innate and adaptive immune system. Group 2 ILCs (ILC2s) are activated by IL-33, which have differential roles in PDAC development and progression; ILC2 activation recruits T cells to boost anti-cancer immunity in PDAC tissues via recruitment of CD103+ DCs (205). However, yet another study demonstrated that IL-33-treated ILC2s produced IL-10 and played a protective role in islet allograft survival (206). These results indicate that ILC2s are a highly dynamic cell type and their phenotypes and functions are controlled by the TME. In the TME, immunosuppression is observed where hypoxia converts ILC2s to IL-10+ ILCregs, helping to form a tolerogenic state in pancreatic cancer (9). ILCs recruit CD8+ T and memory T cells in PDAC; ILCs are also able to help CD108+ B cells migrate to tumour locations (207).
PDAC is notoriously resistant to immunotherapy, such as cytokine therapy, adoptive T cell therapy, and checkpoint blockade strategies (208–210) Failure of these therapies has been attributed to a lack of CD8+ T cells and severe immunosuppression in the TME of PDAC (45, 211, 212). The presence of excessive fibrosis in the TME hinders the infiltration of adaptive immune cells (127).
At the early PanIN stages, Tregs and MDSCs dominate the immune infiltrate. As the disease progresses, CD4+ and CD8+ cells are inconsistently found; existing CD8+ cells display a lack of activation, suggesting an immune suppressed TME (160). At all stages of disease, there is a strong inverse correlation between MDSCs and CD8+ T-lymphocytes, suggesting that MDSCs are a mediator of tumour immunosuppression (160).
Conventional DCs (cDCs) have been identified as important mediators of antigen priming and T cell activity, with Batf3/Irf8-dependent CD103+ CD24+ cDC1s responsible for CD8+ CTL cross-priming. Moreover, Irf4-dependent CD11b+ CD172a+ cDC2s are implicated in the priming of CD4+ T helper cells (Th) (213). cDCs have also been implicated in T cell-dependent tumour killing and immunotherapy response (214–218). Nonetheless, it has been reported that the levels of circulating MDSCs are elevated in pancreatic cancer, which may promote tumour progression (202, 203); MDSCs produce NO, which inhibits DC activation (204). Depending on microenvironmental stimuli, DC can differentiate into distinct subpopulations, leading to proliferation of myeloid DCs that induce Th1 cell activation, or plasmacytoid DCs that facilitate immunosuppressive T cell development. Tumour-derived cytokines have been reported to induce a tolerogenic plasmacytoid DC phenotype (201). Furthermore, recent data suggest the existence of a specific subset CD11b+ DCs that foster an immunosuppressive TME, which favors metastatic progression through the expansion of Tregs and suppression of CD8+ T cells (219). These findings indicate that PDAC is characterized not only by a reduced number of DCs, but also by complex modulation of DC subpopulations, which affects tumour development.
The complement system is a crucial mechanism that connects innate immunity to adaptive immunity and aids the body in combating foreign pathogens and abnormal host cells (220). The complement system can be activated by three distinct pathways: classical, alternative, and lectin. The three pathways converge on the cleavage of complement component C3 into subunits C3a and C3b (C3 convertase) and C5 into fragments C5a and C5b (C5 convertase). As inflammatory mediators or anaphylatoxins, C3a and C5a, cause inflammation by causing histamine release and by activating immune cells such as neutrophils, eosinophils, and macrophages (221). Malignant tumours have increased complement protein expression (222). Activation of the complement system in the TME promotes tumourigenesis (222). PDAC tissue shows an upregulation of C3 and C5, producing more anaphylatoxins (223, 224), C3a and C5a, which upregulate inflammatory mediators and cytokines and cause direct stimulation of TNF-α and IL-1 (220). In addition, these anaphylatoxins increase the recruitment of macrophages in the TME (225).
The complement system has an important role to play in PDAC. The expression of complement regulatory proteins/receptors, CD46, CD55, and CD59, is well established in PDAC cell lines (226). Properdin, the only known up-regulator of the alternative pathway, is highly expressed in the early stages of PDAC; its decreased expression in samples from patients with late-stage PDAC has been reported (227). Neutrophils are known to secrete properdin, which is stored in their granules (228). Elevated properdin expression in PDAC patients with increased neutrophil infiltration is more likely to associate with classical subtype and higher overall and disease-free survival. Properdin induces apoptosis in basal-like pancreatic cancer cell lines, suggesting its anti-tumourigenic role in PDAC (229). Studies have also reported that properdin can recognize cancer cells and play a protective role during tumourigenesis (230). Alternatively, properdin level is strongly down-regulated in PDAC serum (231). The inhibition of complement activation promotes cancer cell immune evasion and seems to hamper the efficacy of cancer immunotherapy.
Existing therapies for patients with PDAC include surgical resection, chemoradiation therapy, and immunotherapy; however, only a small percentage of patients benefit from these treatments. Single-cell RNA-seq studies on the PDAC TME show innate immune cell dominance, which can be directly activated by many cytokines without antigen presentation, unlike adaptive immunity. Given the predominant infiltration, decreased antigenicity, and instant activation, innate immunity may be more important than adaptive immunity in the PDAC immune TME.
On the basis of a growing comprehension of the role of TME in PDAC, neutrophils have emerged as a possible therapeutic target. Targeting neutrophils in PDAC has shown encouraging results in a number of preclinical studies that utilised CXCR2 inhibitors or Ly6G antibodies (187). The preference for CXCR2 as a target may possibly arise from the fact that blocking CXCR2 affects not just the CXCL5/CXCR2 axis but also additional CXCR2 ligands, such as CXCL1-3 and CXCL6-8.
TAMs are one of the most important regulators in the PDAC TME. Depletion of TAMs could dramatically decrease tumourigenesis (232); inhibiting M2 macrophage polarisation is essential for preventing PDAC development, enhancing antitumour immunity, and even clinical treatment (233). New developments in macrophage adjustment have been put forth, such as blocking CSF-1/CSF-1R, CD40 agonists, and other agents, which are helpful in re-educating TAMs from their M2 state to M1. It is currently possible to effectively halt tumour growth and cure tumours owing to an expanding variety of macrophage-targeting strategies. When combined with standard therapy and immunotherapeutic drugs, the blocking of CSF-1/CSF-1R activation can be a potential strategy for treating PDAC by decreasing the TAM population (234). Reprogramming the M2 phenotype of TAMs can significantly change the immunological status of the TME and reactivate the immune system’s antitumour activity.
The phase II clinical testing of multiple antibodies against CSF1/1R in PDAC patients has been undertaken (235). The efficacy of cabiralizumab (Five Prime), a humanized IgG4 mAb against CSF1R, together with the anti-PD1 antibody, nivolumab, in patients with advanced/metastatic PDAC who progressed after first-line chemotherapy (NCT03336216) was evaluated (236). Similarly, another phase Ib/II trial evaluated a fully humanised IgG2 monoclonal anti–CSF1R antibody, AMG 820 (Amgen), in combination with pembrolizumab, on patients with metastatic PDAC (NCT02713529). Both trials failed to reach their effectiveness goals despite exhibiting target-specific alterations, such as the decrease in monocytes. The failure may have been related to the normal stroma association of the CSF1/CSF1R signaling and non-specific targeting based on the expression of CSF1R seen across all myeloid cells. In another phase Ib/II trial that included patients with metastatic PDAC, limited activity was observed with the anti–CSF1 antibody, lacnotuzumab (Novartis), given in combination with anti-PD1 spartalizumab (NCT02807844) (237).
HA is highly overexpressed by tumour cells and CAFs in PDAC; enzymatic depletion of HA using PEGylated hyaluronidase improves therapeutic effectiveness (37). Based on the expression levels of HA, clinical phase I/II study (NCT01839487) revealed robust response rates for patients. However, when combined together, nab-paclitaxel and gemcitabine failed to prolong progression free survival (238).
High levels of integrin molecule CD11b/CD18 on myeloid, cell surface, which is essential for their trafficking and cellular activities within inflammatory tissues, make them amenable to therapeutic targetting. ADH-503 is a small-molecule agonist that partially activates CD11b, causing TAMs to repolarize, fewer immunosuppressive myeloid cells to infiltrate the tumour, and improve DC responses. As a result, checkpoint inhibitors are now effective in PDAC models that were previously resistant to their effects and antitumour T cell immunity is improved. These results show that molecular inhibition of CD11b alters immunosuppressive myeloid cell responses and may overcome the limitations of existing clinical approaches to immunotherapy resistance (239).
DC vaccination has emerged as a novel strategy to prime host anti-tumour immunity (240). Specifically, the combination of a DC vaccine with gemcitabine led to eradication of orthotopic tumours and provided durable protection against PDAC in mouse models (241).
ECM plays a significant role in PDAC tumour growth, metastasis, and resistance to therapy. Accumulating preclinical studies with patient-derived specimens suggest that targeting the dense desmoplastic ECM proteins of PDAC may offer the potential for clinically useful treatments. In clinical practice, it has not yet been possible to successfully target the ECM to improve overall survival.
Antibodies against immune checkpoints, such as anti-PD-1/PD-L1 and anti-CTLA-4, brought transformation in the treatment of several malignancies, but failed to elicit effective anti-tumour response in PDAC patients (19). A phase II clinical trial (NCT02879318) assessed the safety and efficacy of combination chemotherapy (Gemcitabine and nab-paclitaxel) with immune checkpoint inhibitors (durvalumab; PD-L1 inhibitor) and tremelimumab (CTLA-4 inhibitor), which did not improve survival rate significantly (242). Modified FOLFIRINOX (Folinic acid, fluorouracil, irinotecan, and oxaliplatin) with Sintilimab (human IgG4 monoclonal antibody for PD-1) used in a clinical trial (NCT03977272) did not show any survival benefit (243). Another phase II trial (NCT032124250) evaluated the efficacy of nivolumab (anti-PD-1) and/or sotigalimab (CD40 agonistic antibody) with gemcitabine/nab-paclitaxel (chemotherapy) in patients with first-line metastatic PDAC (244). The overall survival rate was 57.7% in nivolumab/chemotherapy group compared to 48.1% observed in sotigalimab/chemotherapy and 41.3% in nivolumab/sotigalimab/chemotherapy treatment regimen. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting allogeneic pancreatic tumour cell (GVAX) immunotherapy and ipilimumab did not improve overall survival, but clear biologic effects on peripheral and intratumoural immune cells were observed, such as increase in T cell activation markers, peripheral T helper and cytotoxic effector memory cells, and decrease in naïve cytotoxic T cells and increase in M1 macrophage content (245). A Phase Ib/II study (NCT02331251) using gemcitabine, nab-paclitaxel, and pembrolizumab to evaluate the safety and efficacy in mPDAC improved the overall survival rate in naive chemotherapy patients (246).
A phase 1b clinical trial (NCT03307148) targeting PSCs with all-trans-retinoic-acid (ATRA) can reprogram pancreatic stroma to suppress PDAC growth (247). ATRA as a stromal-targeting agent with gemcitabine-nab-paclitaxel is safe and tolerable and will be evaluated in a phase II randomized controlled trial for locally advanced PDAC.
The clinicaltrials.gov registry provided with recent clinical trial data on PDAC having interventional therapy (Table 2). NCT02993731 is the largest cohort of patients with mPDAC administered nab-paclitaxel with gemcitabine. The addition of napabucasin to nab-paclitaxel with gemcitabine did not improve efficacy in patients with previously untreated mPDAC (248). CXCR2 antagonist that blocks neutrophil migration and reduces circulating neutrophil counts was studied in a clinical trial, (NCT02583477). In NCT02501902, the tolerability and antitumor activity of palbociclib plus nab-paclitaxel treatment in patients with PDAC did not meet the prespecified efficacy (249). The safety and efficacy of LMB‐100, an immunotoxin that targets mesothelin with and without nab‐paclitaxel was studied in NCT02810418 (250). This study resulted in increased numbers of active circulating CD4 and CD8 T cells, and identified specific changes in serum cytokines and peripheral CD4 T cell subsets associated with capillary leak syndrome, the major toxicity of immunotoxin therapies. NCT03611556 showed similar safety but a trend towards improved outcome (251). In NCT02289898, addition of demcizumab did not improve the efficacy in comparison with placebo (252). NCT01893801, provided encouraging results with high response rate and improved median survival (253). In NCT01658943, selumetinib plus MK-2206 did not improve overall survival in patients with mPDAC for whom gemcitabine-based chemotherapy had failed (254). The baseline immune status predicts PDAC disease course and overall survival in NCT01280058 (255). Tremelimumab monotherapy is ineffective for metastatic PDAC (NCT02527434) (256). New therapeutic options are being studied in the clinical trials: NCT02981342, NCT02558894, NCT02178709. The availability of these results and other ongoing research will help improve the future trials in PDAC patients.
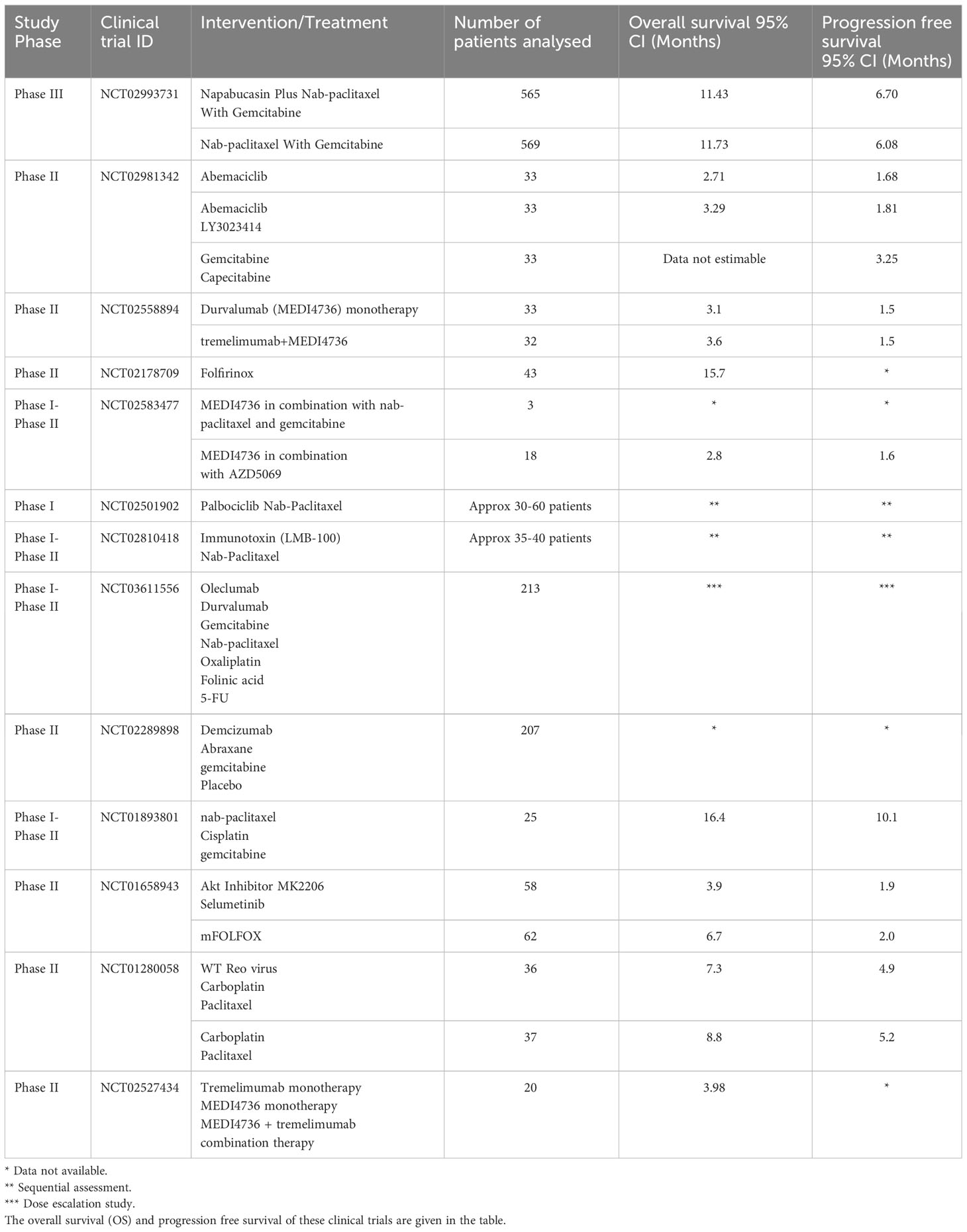
Table 2 The clinical trial data collected from clinicaltrials.gov with keywords as pancreatic ductal adenocarcinoma and interventional study.
The abundant desmoplastic stroma is inextricably linked to the immune landscape of human PDAC. This dense extracellular matrix contributes to the low immunogenicity of PDAC, thereby impeding the infiltration of effector T cells and fostering an immunosuppressive TME. There exists an urgent demand to enhance our understanding of the intricate interplay among tumour cells, immune cells and stromal components within the context of pancreatic cancer. Enhancing our understanding of these interactions will be essential for improving therapeutic approaches for human PDAC.
The communication between the tumour cells and the TME is mediated by many factors including extracellular vesicles (EVs). Exosomes which are EVs with a diameter of 30-150 nm are secreted by tumour cells as well as the stromal cells in the TiME during tumour progression (257). PDAC-derived EVs distinctly regulate angiogenesis by inducing cell proliferation, mobility and secretion of pro-angiogenic factors. Cancer-associated thrombosis is yet another complication in PDAC via the expression of tissue factors. PDAC derived exosomes regulate pancreatic functions including lipidosis and glucose intake inhibition. The immunosuppressive Treg expansion is also mediated by EVs in PDAC through upregulation of the expression of FOXO transcription factors and nuclear translocation in FOXP3+ Tregs (258). The chemoresistant cells produce exosomal cargos that aggravate chemoresistance in sensitive cells leading to anti-apoptotic effect. Advanced PDAC patient serum has exosomes that can enhance liver and lung metastasis (259). There exists a crosstalk between tumour cells and TME mediated by EVs. PDAC-derived small EVs induce the polarisation towards M2 macrophages and inhibit effector T-cell response that promote immunosuppression and anti-tumour immunity. The potential of exosomes to stimulate the immune system of PDAC patients can be used as nanocarriers of immunotherapeutic agents (260). Therefore, understanding about this crosstalk can help develop targeted immunotherapy (261).
The immunosuppression in PDAC is multi-factorial. PDAC is characterized by an abundance of MDSCs and M2 TAMs. In contrast, the presence of CD8+ T cells is significantly low. The varied role of TME in PDAC can be treated by a multi-modal strategy that targets tumour promoting properties and improve the survival rate (262). The chemotherapy with immunotherapy combination tried thus far did not improve survival in mPDAC. Hence, site specific delivery of immunotherapeutics is currently under development. The ongoing clinical trials that evaluate the combination immunotherapy may elucidate mechanisms to bring down the immune suppression by TiME. The clinical trials may be evaluated further for the infiltration of adaptive immune cells like effector T cells. Patient specific biomarker identification and targeted therapy may improve the clinical outcomes. More clinical studies targeting the TiME in PDAC can enhance the potency of chemotherapy/immunotherapy treatment regimen. In general, it is thought that conceptual breakthroughs in understanding the overall TME of PDAC could facilitate the development of novel therapeutic approaches that target numerous processes simultaneously, resulting in combined benefits.
AJ: Data curation, Formal analysis, Investigation, Visualization, Writing – original draft. AA: Software, Visualization, Writing – original draft, Writing – review & editing. BA-R: Funding acquisition, Writing – review & editing. SS: Conceptualization, Formal analysis, Investigation, Supervision, Writing – review & editing. UK: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. AJ, BA-R and UK are funded by UAEU (Grant Number- 12F043).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
PDAC, Pancreatic Ductal Adenocarcinoma; TME, Tumor microenvironment; ADM, acinar-to-ductal metaplasia; IL, Interleukin; CAF, Cancer Associated Fibroblast; TAMs, Tumor associated Macrophages; TANs, Tumor associated Neutrophils; ECM, Extracellular Matrix; Tregs, Regulatory T cells; Bregs, Regulatory B cells; MDSCs, Myeloid derived suppressor cells; TP53, Tumor Protein P53; CDKN2A, Cyclin-dependent kinase inhibitor 2A; SMAD4, Mothers against decapentaplegic homolog 4, or DPC4; BRCA2, BReast CAncer gene 2; EMT, Epithelial-to-Mesenchymal Transition; mPDAC, metastatic PDAC.
1. Cascinu S, Falconi M, Valentini V, Jelic S. ESMO Guidelines Working Group. Pancreatic cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol (2010) 21(Suppl 5):v55–8. doi: 10.1093/annonc/mdq165
2. Haeberle L, Esposito I. Pathology of pancreatic cancer. Transl Gastroenterol Hepatol (2019) 4:50. doi: 10.21037/tgh.2019.06.02
3. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistic. CA: A Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
4. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
5. Khalaf N, El-Serag HB, Abrams HR, Thrift AP. Burden of pancreatic cancer: from epidemiology to practice. Clin Gastroenterol Hepatol (2021) 19(5):876–84. doi: 10.1016/j.cgh.2020.02.054
6. Rahib L, Wehner MR, Matrisian LM, Nead KT. Estimated projection of US cancer incidence and death to 2040. JAMA Network Open (2021) 4(4):e214708. doi: 10.1001/jamanetworkopen.2021.4708
7. Zhang L, Jin R, Yang X, Ying D. A population-based study of synchronous distant metastases and prognosis in patients with PDAC at initial diagnosis. Front Oncol (2023) 13:1087700. doi: 10.3389/fonc.2023.1087700
8. Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat Cancer (2020) 1(11):1097–112. doi: 10.1038/s43018-020-00121-4
9. Ye L, Jin K, Liao Z, Xiao Z, Xu H, Lin X, et al. Hypoxia-reprogrammed regulatory group 2 innate lymphoid cells promote immunosuppression in pancreatic cancer. EBioMedicine (2022) 79:104016. doi: 10.1016/j.ebiom.2022.104016
10. Guo J, Wang S, Gao Q. An integrated overview of the immunosuppression features in the tumor microenvironment of pancreatic cancer. Front Immunol (2023) 14:1258538. doi: 10.3389/fimmu.2023.1258538
11. Rubin SJS, Sojwal RS, Gubatan J, Rogalla S. The tumor immune microenvironment in pancreatic ductal adenocarcinoma: neither hot nor cold. Cancers (Basel) (2022) 14(17):4236. doi: 10.3390/cancers14174236
12. Muller M, Haghnejad V, Schaefer M, Gauchotte G, Caron B, Peyrin-Biroulet L, et al. The immune landscape of human pancreatic ductal carcinoma: key players, clinical implications, and challenges. Cancers (Basel) (2022) 14(4):995. doi: 10.3390/cancers14040995
13. Wang K, He H. Pancreatic tumor microenvironment. Adv Exp Med Biol (2020) 1296:243–57. doi: 10.1007/978-3-030-59038-3_15
14. Nitecki SS, Sarr MG, Colby TV, van Heerden JA. Long-term survival after resection for ductal adenocarcinoma of the pancreas Is it really improving?. Ann Surg (1995) 221(1):59–66. doi: 10.1097/00000658-199501000-00007
15. Vera R, Díez L, Martín Pérez E, Plaza JC, Sanjuanbenito A, Carrato A. Surgery for pancreatic ductal adenocarcinoma. Clin Transl Oncol (2017) 19(11):1303–11. doi: 10.1007/s12094-017-1688-0
16. Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol (2010) 7(3):163–72. doi: 10.1038/nrclinonc.2009.236
17. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature (2004) 432(7015):332–7. doi: 10.1038/nature03096
18. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res (2008) 68(3):918–26. doi: 10.1158/0008-5472.CAN-07-5714
19. O’Reilly EM, Oh DY, Dhani N, Renouf DJ, Lee MA, Sun W, et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: A phase 2 randomized clinical trial. JAMA Oncol (2019) 5(10):1431–8. doi: 10.1001/jamaoncol.2019.1588
20. Pan L, Fang J, Tong C, Chen M, Zhang B, Juengpanich S, et al. Survival benefits of neoadjuvant chemo(radio)therapy versus surgery first in patients with resectable or borderline resectable pancreatic cancer: a systematic review and meta-analysis. World J Surg Onc (2019) 18(1):1. doi: 10.1186/s12957-019-1767-5
21. Chawla A, Molina G, Pak LM, Rosenthal M, Mancias JD, Clancy TE, et al. Neoadjuvant Therapy is Associated with Improved Survival in Borderline-Resectable Pancreatic Cancer. Ann Surg Oncol (2020) 27(4):1191–1200. doi: 10.1245/s10434-019-08087-z
22. Nasief H, Hall W, Zheng C, Tsai S, Wang L, Erickson B, et al. Improving treatment response prediction for chemoradiation therapy of pancreatic cancer using a combination of delta-radiomics and the clinical biomarker CA19-9. Front Oncol (2020) 9:1464. doi: 10.3389/fonc.2019.01464
23. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol (2007) 25(15):1960–6. doi: 10.1200/JCO.2006.07.9525
24. Nsingwane Z, Elebo N, Mhlambi N, Devar J, Omoshoro-jones J, Smith M, et al. Complement protein levels decrease with tumour severity in pancreatic ductal adenocarcinoma. HPB (2022) 24:S266. doi: 10.1016/j.hpb.2022.05.562
25. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (2011) 331(6024):1612–6. doi: 10.1126/science.1198443
26. Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis1. Cancer Res (2000) 60(9):2562–6.
27. Hu M, Yao J, Cai L, Bachman KE, van den Brûle F, Velculescu V, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet (2005) 37(8):899–905. doi: 10.1038/ng1596
28. Jiang B, Zhou L, Lu J, Wang Y, Liu C, You L, et al. Stroma-targeting therapy in pancreatic cancer: one coin with two sides? Front Oncol (2020) 10. doi: 10.3389/fonc.2020.576399
29. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer (2006) 6(8):583–92. doi: 10.1038/nrc1893
30. Sohr S, Engeland K. RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle (2008) 7(21):3448–60. doi: 10.4161/cc.7.21.7014
31. Toole BP, Slomiany MG. Hyaluronan: a constitutive regulator of chemoresistance and Malignancy in cancer cells. Semin Cancer Biol (2008) 18(4):244–50. doi: 10.1016/j.semcancer.2008.03.009
32. Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front Immunol (2015) 6:201. doi: 10.3389/fimmu.2015.00201
33. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut (2013) 62(1):112–20. doi: 10.1136/gutjnl-2012-302529
34. Singha NC, Nekoroski T, Zhao C, Symons R, Jiang P, Frost GI, et al. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol Cancer Ther (2015) 14(2):523–32. doi: 10.1158/1535-7163.MCT-14-0580
35. Thompson CB, Shepard HM, O’Connor PM, Kadhim S, Jiang P, Osgood RJ, et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther (2010) 9(11):3052–64. doi: 10.1158/1535-7163.MCT-10-0470
36. Whatcott CJ, Han H, Von Hoff DD. Orchestrating the tumor microenvironment to improve survival for patients with pancreatic cancer: normalization, not destruction. Cancer J (2015) 21(4):299–306. doi: 10.1097/PPO.0000000000000140
37. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, et al. HALO 202: randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol (2018) 36(4):359–66. doi: 10.1200/JCO.2017.74.9564
38. Huo Y, Yang M, Liu W, Yang J, Fu X, Liu D, et al. High expression of DDR1 is associated with the poor prognosis in Chinese patients with pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res (2015) 34(1):88. doi: 10.1186/s13046-015-0202-1
39. Aguilera KY, Huang H, Du W, Hagopian MM, Wang Z, Hinz S, et al. Inhibition of discoidin domain receptor 1 reduces collagen-mediated tumorigenicity in pancreatic ductal adenocarcinoma. Mol Cancer Ther (2017) 16(11):2473–85. doi: 10.1158/1535-7163.MCT-16-0834
40. Ko S, Jung KH, Yoon YC, Han BS, Park MS, Lee YJ, et al. A novel DDR1 inhibitor enhances the anticancer activity of gemcitabine in pancreatic cancer. Am J Cancer Res (2022) 12(9):4326–42.
41. Rucki AA, Zheng L. Pancreatic cancer stroma: understanding biology leads to new therapeutic strategies. World J Gastroenterol (2014) 20(9):2237–46. doi: 10.3748/wjg.v20.i9.2237
42. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature (2008) 455(7211):406–10. doi: 10.1038/nature07275
43. Onishi H, Katano M. Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer. World J Gastroenterol (2014) 20(9):2335–42. doi: 10.3748/wjg.v20.i9.2335
44. Heretsch P, Tzagkaroulaki L, Giannis A. Cyclopamine and hedgehog signaling: chemistry, biology, medical perspectives. Angew Chem Int Ed Engl (2010) 49(20):3418–27. doi: 10.1002/anie.200906967
45. Zhang B, Jiang T, Shen S, She X, Tuo Y, Hu Y, et al. Cyclopamine disrupts tumor extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials (2016) 103:12–21. doi: 10.1016/j.biomaterials.2016.06.048
46. Zhao Y, Shen M, Feng Y, He R, Xu X, Xie Y, et al. Regulatory B cells induced by pancreatic cancer cell-derived interleukin-18 promote immune tolerance via the PD-1/PD-L1 pathway. Oncotarget (2018) 9(19):14803–14. doi: 10.18632/oncotarget.22976
47. Almawash SA, Mondal G, Mahato RI. Coadministration of polymeric conjugates of docetaxel and cyclopamine synergistically inhibits orthotopic pancreatic cancer growth and metastasis. Pharm Res (2018) 35(1):17. doi: 10.1007/s11095-017-2303-3
48. Porta M, Fabregat X, Malats N, Guarner L, Carrato A, de Miguel A, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol (2005) 7(5):189–97. doi: 10.1007/BF02712816
49. Singhi AD, Koay EJ, Chari ST, Maitra A. Early detection of pancreatic cancer: opportunities and challenges. Gastroenterology (2019) 156(7):2024–40. doi: 10.1053/j.gastro.2019.01.259
50. Goonetilleke KS, Siriwardena AK. Systematic review of carbohydrate antigen (CA 19-9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur J Surg Oncol (2007) 33(3):266–70. doi: 10.1016/j.ejso.2006.10.004
51. Ballehaninna UK, Chamberlain RS. Serum CA 19-9 as a biomarker for pancreatic cancer-A comprehensive review. Indian J Surg Oncol (2011) 2(2):88–100. doi: 10.1007/s13193-011-0042-1
52. Wang Z, Tian YP. Clinical value of serum tumor markers CA19-9, CA125 and CA72-4 in the diagnosis of pancreatic carcinoma. Mol Clin Oncol (2014) 2(2):265–8. doi: 10.3892/mco.2013.226
53. Lei XF, Jia SZ, Ye J, Qiao YL, Zhao GM, Li XH, et al. Application values of detection of serum CA199, CA242 and CA50 in the diagnosis of pancreatic cancer. J Biol Regul Homeost Agents (2017) 31(2):383–8.
54. van Manen L, Groen JV, Putter H, Vahrmeijer AL, Swijnenburg RJ, Bonsing BA, et al. Elevated CEA and CA19-9 serum levels independently predict advanced pancreatic cancer at diagnosis. Biomarkers (2020) 25(2):186–93. doi: 10.1080/1354750X.2020.1725786
55. Maitra A, Fukushima N, Takaori K, Hruban RH. Precursors to invasive pancreatic cancer. Adv Anat Pathol (2005) 12(2):81–91. doi: 10.1097/01.pap.0000155055.14238.25
56. Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res (2000) 6(8):2969–72.
57. Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer (2016) 16(9):553–65. doi: 10.1038/nrc.2016.66
58. Sclabas GM, Fujioka S, Schmidt C, Evans DB, Chiao PJ. NF-kappaB in pancreatic cancer. Int J Gastrointest Cancer (2003) 33(1):15–26. doi: 10.1385/IJGC:33:1:15
59. Löhr M, Klöppel G, Maisonneuve P, Lowenfels AB, Lüttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia (2005) 7(1):17–23. doi: 10.1593/neo.04445
60. Scarpa A, Capelli P, Mukai K, Zamboni G, Oda T, Iacono C, et al. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol (1993) 142(5):1534–43.
61. Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science (1996) 271(5247):350–3. doi: 10.1126/science.271.5247.350
62. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature (2016) 531(7592):47–52. doi: 10.1038/nature16965
63. Dardare J, Witz A, Merlin JL, Gilson P, Harlé A. SMAD4 and the TGFβ Pathway in patients with pancreatic ductal adenocarcinoma. Int J Mol Sci (2020) 21(10):3534. doi: 10.3390/ijms21103534
64. Low RRJ, Lim WW, Nguyen PM, Lee B, Christie M, Burgess AW, et al. The diverse applications of pancreatic ductal adenocarcinoma organoids. Cancers (2021) 13(19):4979. doi: 10.3390/cancers13194979
65. Miyabayashi K, Baker LA, Deschênes A, Traub B, Caligiuri G, Plenker D, et al. Intraductal transplantation models of human pancreatic ductal adenocarcinoma reveal progressive transition of molecular subtypes. Cancer Discov (2020) 10(10):1566–89. doi: 10.1158/2159-8290.CD-20-0133
66. Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med (2011) 17(4):500–3. doi: 10.1038/nm.2344
67. Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet (2015) 47(10):1168–78. doi: 10.1038/ng.3398
68. Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet (2020) 52(2):231–40. doi: 10.1038/s41588-019-0566-9
69. Klein L, Tu M, Krebs N, Urbach L, Grimm D, Latif MU, et al. Spatial tumor immune heterogeneity facilitates subtype co-existence and therapy response via AP1 dichotomy in pancreatic cancer. bioRxiv (2023) 173. doi: 10.1101/2023.10.30.563552
70. Williams HL, Dias Costa A, Zhang J, Raghavan S, Winter PS, Kapner KS, et al. Spatially resolved single-cell assessment of pancreatic cancer expression subtypes reveals co-expressor phenotypes and extensive intratumoral heterogeneity. Cancer Res (2023) 83(3):441–55. doi: 10.1158/0008-5472.CAN-22-3050
71. Espinet E, Klein L, Puré E, Singh SK. Mechanisms of PDAC subtype heterogeneity and therapy response. Trends Cancer (2022) 8(12):1060–71. doi: 10.1016/j.trecan.2022.08.005
72. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi: 10.1158/0008-5472.CAN-04-0465
73. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PloS One (2009) 4(8):e6562. doi: 10.1371/journal.pone.0006562
74. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet (2016) 388(10039):73–85. doi: 10.1016/S0140-6736(16)00141-0
75. Helm O, Held-Feindt J, Grage-Griebenow E, Reiling N, Ungefroren H, Vogel I, et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int J Cancer (2014) 135(4):843–61. doi: 10.1002/ijc.28736
76. Eriksson E, Milenova I, Wenthe J, Moreno R, Alemany R, Loskog A. IL-6 signaling blockade during CD40-mediated immune activation favors antitumor factors by reducing TGF-β, collagen type I, and PD-L1/PD-1. J Immunol (2019) 202(3):787–98. doi: 10.4049/jimmunol.1800717
77. Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, et al. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res (2011) 167(2):e211–219. doi: 10.1016/j.jss.2009.05.026
78. Yin H, Gao S, Chen Q, Liu S, Shoucair S, Ji Y, et al. Tumor-associated N1 and N2 neutrophils predict prognosis in patients with resected pancreatic ductal adenocarcinoma: A preliminary study. MedComm (2020) (2022) 3(4):e183. doi: 10.1002/mco2.183
79. Wang X, Qiu L, Li Z, Wang XY, Yi H. Understanding the multifaceted role of neutrophils in cancer and autoimmune diseases. Front Immunol (2018) 9:2456. doi: 10.3389/fimmu.2018.02456
80. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer (2020) 20(9):485–503. doi: 10.1038/s41568-020-0281-y
81. Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas (2004) 28(1):e26–31. doi: 10.1097/00006676-200401000-00023
82. Gorchs L, Fernández Moro C, Bankhead P, Kern KP, Sadeak I, Meng Q, et al. Human pancreatic carcinoma-associated fibroblasts promote expression of co-inhibitory markers on CD4+ and CD8+ T-cells. Front Immunol (2019) 10:847. doi: 10.3389/fimmu.2019.00847
83. Zhang T, Ren Y, Yang P, Wang J, Zhou H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis (2022) 13(10):1–11. doi: 10.1038/s41419-022-05351-1
84. Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas (2004) 29(3):179–87.
85. Thyagarajan A, Alshehri MSA, Miller KLR, Sherwin CM, Travers JB, Sahu RP. Myeloid-derived suppressor cells and pancreatic cancer: implications in novel therapeutic approaches. Cancers (Basel) (2019) 11(11):1627. doi: 10.3390/cancers11111627
86. Ren B, Cui M, Yang G, Wang H, Feng M, You L, et al. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer (2018) 17:108. doi: 10.1186/s12943-018-0858-1
87. Curiel TJ. Tregs and rethinking cancer immunotherapy. J Clin Invest (2007) 117(5):1167–74. doi: 10.1172/JCI31202
88. Lim SA, Kim J, Jeon S, Shin MH, Kwon J, Kim TJ, et al. Defective localization with impaired tumor cytotoxicity contributes to the immune escape of NK cells in pancreatic cancer patients. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00496
89. Hoshikawa M, Aoki T, Matsushita H, Karasaki T, Hosoi A, Odaira K, et al. NK cell and IFN signatures are positive prognostic biomarkers for resectable pancreatic cancer. Biochem Biophys Res Commun (2018) 495(2):2058–65. doi: 10.1016/j.bbrc.2017.12.083
90. van der Leun AM, Thommen DS, Schumacher TN. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer (2020) 20(4):218–32. doi: 10.1038/s41568-019-0235-4
91. Goulart MR, Stasinos K, Fincham REA, Delvecchio FR, Kocher HM. T cells in pancreatic cancer stroma. World J Gastroenterol (2021) 27(46):7956–68. doi: 10.3748/wjg.v27.i46.7956
92. Castino GF, Cortese N, Capretti G, Serio S, Di Caro G, Mineri R, et al. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology (2016) 5(4):e1085147. doi: 10.1080/2162402X.2015.1085147
93. Senturk ZN, Akdag I, Deniz B, Sayi-Yazgan A. Pancreatic cancer: Emerging field of regulatory B-cell-targeted immunotherapies. Front Immunol (2023) 14:1152551. doi: 10.3389/fimmu.2023.1152551
94. Chu GC, Kimmelman AC, Hezel AF, DePinho RA. Stromal biology of pancreatic cancer. J Cell Biochem (2007) 101(4):887–907. doi: 10.1002/jcb.21209
95. Korc M. Pancreatic cancer–associated stroma production. Am J Surg (2007) 194(4, Supplement):S84–6. doi: 10.1016/j.amjsurg.2007.05.004
96. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res (2012) 18(16):4266–76. doi: 10.1158/1078-0432.CCR-11-3114
97. Kota J, Hancock J, Kwon J, Korc M. Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Letters (2017) 391:38–49. doi: 10.1016/j.canlet.2016.12.035
98. Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP, et al. Identification of sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 22(6):737–50. doi: 10.1016/j.ccr.2012.10.025
99. Storz P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol (2017) 14(5):296–304. doi: 10.1038/nrgastro.2017.12
100. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
101. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
102. Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, et al. Pancreatitis and the risk of pancreatic cancer. New Engl J Med (1993) 328(20):1433–7. doi: 10.1056/NEJM199305203282001
103. Xu M, Jung X, Hines OJ, Eibl G, Chen Y. Obesity and pancreatic cancer: overview of epidemiology and potential prevention by weight loss. Pancreas (2018) 47(2):158. doi: 10.1097/MPA.0000000000000974
104. Weissman S, Takakura K, Eibl G, Pandol SJ, Saruta M. The diverse involvement of cigarette smoking in pancreatic cancer development and prognosis. Pancreas (2020) 49(5):612. doi: 10.1097/MPA.0000000000001550
105. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
106. Yako YY, Kruger D, Smith M, Brand M. Cytokines as biomarkers of pancreatic ductal adenocarcinoma: A systematic review. PloS One (2016) 11(5):e0154016. doi: 10.1371/journal.pone.0154016
107. Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21(1):105–20. doi: 10.1016/j.ccr.2011.12.006
108. Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheumatic Dis (2011) 70(Suppl 1):i104–8. doi: 10.1136/ard.2010.140145
109. Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut (2018) 67(2):320–32. doi: 10.1136/gutjnl-2016-311585
110. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
111. Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin Cancer Res (2013) 19(13):3404–15. doi: 10.1158/1078-0432.CCR-13-0525
112. Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep (2017) 19(1):203–17. doi: 10.1016/j.celrep.2017.03.037
113. Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol (2001) 166(9):5398–406. doi: 10.4049/jimmunol.166.9.5398
114. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66(23):11238–46. doi: 10.1158/0008-5472.CAN-06-1278
115. Saio M, Radoja S, Marino M, Frey AB. Tumor-infiltrating macrophages induce apoptosis in activated CD8(+) T cells by a mechanism requiring cell contact and mediated by both the cell-associated form of TNF and nitric oxide. J Immunol (2001) 167(10):5583–93. doi: 10.4049/jimmunol.167.10.5583
116. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109(4):1568–73. doi: 10.1182/blood-2006-06-031856
117. Munder M, Choi BS, Rogers M, Kropf P. L-arginine deprivation impairs Leishmania major-specific T-cell responses. Eur J Immunol (2009) 39(8):2161–72. doi: 10.1002/eji.200839041
118. Werba G, Weissinger D, Kawaler EA, Zhao E, Kalfakakou D, Dhara S, et al. Single-cell RNA sequencing reveals the effects of chemotherapy on human pancreatic adenocarcinoma and its tumor microenvironment. Nat Commun (2023) 14(1):797. doi: 10.1038/s41467-023-36296-4
119. Komura T, Sakai Y, Harada K, Kawaguchi K, Takabatake H, Kitagawa H, et al. Inflammatory features of pancreatic cancer highlighted by monocytes/macrophages and CD4+ T cells with clinical impact. Cancer Sci (2015) 106(6):672–86. doi: 10.1111/cas.12663
120. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (2016) 352(6282):227–31. doi: 10.1126/science.aac9935
121. Coelho MA, de Carné Trécesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity (2017) 47(6):1083–1099.e6. doi: 10.1016/j.immuni.2017.11.016
122. El-Jawhari JJ, El-Sherbiny YM, Scott GB, Morgan RSM, Prestwich R, Bowles PA, et al. Blocking oncogenic RAS enhances tumour cell surface MHC class I expression but does not alter susceptibility to cytotoxic lymphocytes. Mol Immunol (2014) 58(2):160–8. doi: 10.1016/j.molimm.2013.11.020
123. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature (2020) 581(7806):100–5. doi: 10.1038/s41586-020-2229-5
124. Lafaro KJ, Melstrom LG. The paradoxical web of pancreatic cancer tumor microenvironment. Am J Pathology (2019) 189(1):44–57. doi: 10.1016/j.ajpath.2018.09.009
125. Diab M, El-Rayes BF. The heterogeneity of CAFs and immune cell populations in the tumor microenvironment of pancreatic adenocarcinoma. J Cancer Metastasis Treat (2022) 8:42. doi: 10.20517/2394-4722.2022.60
126. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (2009) 324(5933):1457–61. doi: 10.1126/science.1171362
127. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21(3):418–29. doi: 10.1016/j.ccr.2012.01.007
128. Steins A, van Mackelenbergh MG, van der Zalm AP, Klaassen R, Serrels B, Goris SG, et al. High-grade mesenchymal pancreatic ductal adenocarcinoma drives stromal deactivation through CSF-1. EMBO Rep (2020) 21(5):e48780. doi: 10.15252/embr.201948780
129. Ohuchida K, Mizumoto K, Murakami M, Qian LW, Sato N, Nagai E, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res (2004) 64(9):3215–22. doi: 10.1158/0008-5472.CAN-03-2464
130. Sada M, Ohuchida K, Horioka K, Okumura T, Moriyama T, Miyasaka Y, et al. Hypoxic stellate cells of pancreatic cancer stroma regulate extracellular matrix fiber organization and cancer cell motility. Cancer Lett (2016) 372(2):210–8. doi: 10.1016/j.canlet.2016.01.016
131. Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T, et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology (2017) 152(6):1492–1506.e24. doi: 10.1053/j.gastro.2017.01.010
132. Koikawa K, Ohuchida K, Ando Y, Kibe S, Nakayama H, Takesue S, et al. Basement membrane destruction by pancreatic stellate cells leads to local invasion in pancreatic ductal adenocarcinoma. Cancer Lett (2018) 425:65–77. doi: 10.1016/j.canlet.2018.03.031
133. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
134. Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell (2017) 31(1):5–19. doi: 10.1016/j.ccell.2016.12.006
135. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16(9):582–98. doi: 10.1038/nrc.2016.73
136. Kozono S, Ohuchida K, Eguchi D, Ikenaga N, Fujiwara K, Cui L, et al. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res (2013) 73(7):2345–56. doi: 10.1158/0008-5472.CAN-12-3180
137. Pandol S, Edderkaoui M, Gukovsky I, Lugea A, Gukovskaya A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol (2009) 7(11 Suppl):S44–47. doi: 10.1016/j.cgh.2009.07.039
138. Weniger M, Honselmann KC, Liss AS. The extracellular matrix and pancreatic cancer: A complex relationship. Cancers (2018) 10(9):316. doi: 10.3390/cancers10090316
139. Tian C, Clauser KR, Öhlund D, Rickelt S, Huang Y, Gupta M, et al. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc Natl Acad Sci USA (2019) 116(39):19609–18. doi: 10.1073/pnas.1908626116
140. Öhlund D, Franklin O, Lundberg E, Lundin C, Sund M. Type IV collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer (2013) 13:154. doi: 10.1186/1471-2407-13-154
141. Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med (2016) 22(5):497–505. doi: 10.1038/nm.4082
142. Sipos B, Hahn D, Carceller A, Piulats J, Hedderich J, Kalthoff H, et al. Immunohistochemical screening for beta6-integrin subunit expression in adenocarcinomas using a novel monoclonal antibody reveals strong up-regulation in pancreatic ductal adenocarcinomas in vivo and in vitro. Histopathology (2004) 45(3):226–36. doi: 10.1111/j.1365-2559.2004.01919.x
143. Pan S, Chen R, Tamura Y, Crispin DA, Lai LA, May DH, et al. Quantitative glycoproteomics analysis reveals changes in N-glycosylation level associated with pancreatic ductal adenocarcinoma. J Proteome Res (2014) 13(3):1293–306. doi: 10.1021/pr4010184
144. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20(3):174–86. doi: 10.1038/s41568-019-0238-1
145. Ferdek PE, Jakubowska MA. Biology of pancreatic stellate cells-more than just pancreatic cancer. Pflugers Arch (2017) 469(9):1039–50. doi: 10.1007/s00424-017-1968-0
146. Apte MV, Pirola RC, Wilson JS. Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol (2012) 3:344. doi: 10.3389/fphys.2012.00344
147. Korc M, Chandrasekar B, Shah GN. Differential binding and biological activities of epidermal growth factor and transforming growth factor alpha in a human pancreatic cancer cell line. Cancer Res (1991) 51(23 Pt 1):6243–9.
148. Kikuta K, Masamune A, Watanabe T, Ariga H, Itoh H, Hamada S, et al. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem Biophys Res Commun (2010) 403(3–4):380–4. doi: 10.1016/j.bbrc.2010.11.040
149. Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res (2019) 29(9):725–38. doi: 10.1038/s41422-019-0195-y
150. Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun (2017) 8(1):15095. doi: 10.1038/ncomms15095
151. Tesfaye AA, Kamgar M, Azmi A, Philip PA. The evolution into personalized therapies in pancreatic ductal adenocarcinoma: challenges and opportunities. Expert Rev Anticancer Ther (2018) 18(2):131–48. doi: 10.1080/14737140.2018.1417844
152. Stone ML, Beatty GL. Cellular determinants and therapeutic implications of inflammation in pancreatic cancer. Pharmacol Ther (2019) 201:202–13. doi: 10.1016/j.pharmthera.2019.05.012
153. Vonderheide RH, Bear AS. Tumor-derived myeloid cell chemoattractants and T cell exclusion in pancreatic cancer. Front Immunol (2020) 11:605619. doi: 10.3389/fimmu.2020.605619
154. Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Discovery (2016) 6(3):247–55. doi: 10.1158/2159-8290.CD-15-0843
155. Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol (2002) 169(5):2756–61. doi: 10.4049/jimmunol.169.5.2756
156. Krishnamoorthy M, Lenehan JG, Burton JP, Maleki Vareki S. Immunomodulation in pancreatic cancer. Cancers (2020) 12(11):3340. doi: 10.3390/cancers12113340
157. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res (2006) 12(18):5423–34. doi: 10.1158/1078-0432.CCR-06-0369
158. Tang Y, Xu X, Guo S, Zhang C, Tang Y, Tian Y, et al. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PloS One (2014) 9(3):e91551. doi: 10.1371/journal.pone.0091551
159. Oh K, Yoo YJ, Torre-Healy LA, Rao M, Fassler D, Wang P, et al. Coordinated single-cell tumor microenvironment dynamics reinforce pancreatic cancer subtype. Nat Commun (2023) 14(1):5226. doi: 10.1038/s41467-023-40895-6
160. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res (2007) 67(19):9518–27. doi: 10.1158/0008-5472.CAN-07-0175
161. Hackeng WM, Hruban RH, Offerhaus GJA, Brosens LAA. Surgical and molecular pathology of pancreatic neoplasms. Diagn Pathol (2016) 11(1):47. doi: 10.1186/s13000-016-0497-z
162. Fogar P, Sperti C, Basso D, Sanzari MC, Greco E, Davoli C, et al. Decreased total lymphocyte counts in pancreatic cancer: an index of adverse outcome. Pancreas (2006) 32(1):22. doi: 10.1097/01.mpa.0000188305.90290.50
163. Linehan DC, Goedegebuure PS. CD25+ CD4+ regulatory T-cells in cancer. Immunol Res (2005) 32(1–3):155–68. doi: 10.1385/IR:32:1-3:155
164. Tan MCB, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol (2009) 182(3):1746–55. doi: 10.4049/jimmunol.182.3.1746
165. Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TFE, et al. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer Res (2013) 73(8):2468–79. doi: 10.1158/0008-5472.CAN-12-3450
166. Arsenijevic T, Nicolle R, Bouchart C, D’Haene N, Demetter P, Puleo F, et al. Pancreatic cancer meets human microbiota: close encounters of the third kind. Cancers (Basel) (2021) 13(6):1231. doi: 10.3390/cancers13061231
167. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res (2007) 67(3):1030–7. doi: 10.1158/0008-5472.CAN-06-2030
168. Korkaya H, Liu S, Wicha MS. Regulation of cancer stem cells by cytokine networks: attacking cancers inflammatory roots. Clin Cancer Res (2011) 17(19):6125–9. doi: 10.1158/1078-0432.CCR-10-2743
169. Parte S, Nimmakayala RK, Batra SK, Ponnusamy MP. Acinar to ductal cell trans-differentiation: A prelude to dysplasia and pancreatic ductal adenocarcinoma. Biochim Biophys Acta (BBA) - Rev Cancer (2022) 1877(1):188669. doi: 10.1016/j.bbcan.2021.188669
170. Liou GY, Döppler H, Necela B, Krishna M, Crawford HC, Raimondo M, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κB and MMPs. J Cell Biol (2013) 202(3):563–77. doi: 10.1083/jcb.201301001
171. Poh AR, Ernst M. Tumor-associated macrophages in pancreatic ductal adenocarcinoma: therapeutic opportunities and clinical challenges. Cancers (2021) 13(12):2860. doi: 10.3390/cancers13122860
172. Geismann C, Schäfer H, Gundlach JP, Hauser C, Egberts JH, Schneider G, et al. NF-κB dependent chemokine signaling in pancreatic cancer. Cancers (2019) 11(10):1445. doi: 10.3390/cancers11101445
173. Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, et al. Stat3/socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell (2011) 19(4):456–69. doi: 10.1016/j.ccr.2011.03.009
174. Deschênes-Simard X, Mizukami Y, Bardeesy N. Macrophages in pancreatic cancer: Starting things off on the wrong track. J Cell Biol (2013) 202(3):403–5. doi: 10.1083/jcb.201307066
175. Chen Q, Wang J, Zhang Q, Zhang J, Lou Y, Yang J, et al. Tumour cell-derived debris and IgG synergistically promote metastasis of pancreatic cancer by inducing inflammation via tumour-associated macrophages. Br J Cancer (2019) 121(9):786–95. doi: 10.1038/s41416-019-0595-2
176. Sriram K, Salmerón C, Wiley SZ, Insel PA. GPCRs in pancreatic adenocarcinoma: Contributors to tumour biology and novel therapeutic targets. Br J Pharmacol (2020) 177(11):2434–55. doi: 10.1111/bph.15028
177. Takaya H, Andoh A, Shimada M, Hata K, Fujiyama Y, Bamba T. The expression of chemokine genes correlates with nuclear factor-kappaB activation in human pancreatic cancer cell lines. Pancreas (2000) 21(1):32–40. doi: 10.1097/00006676-200007000-00049
178. Tu M, Klein L, Spinet E, Georgomanolis T, Wegwitz F, Li X, et al. TNF-α-producing macrophages determine subtype identity and prognosis via AP1 enhancer reprogramming in pancreatic cancer. Nat Cancer (2021) 2(11):1185–203. doi: 10.1038/s43018-021-00258-w
179. Shimada M, Andoh A, Araki Y, Fujiyama Y, Bamba T. Ligation of the Fas antigen stimulates chemokine secretion in pancreatic cancer cell line PANC-1. J Gastroenterol Hepatol (2001) 16(9):1060–7. doi: 10.1046/j.1440-1746.2001.02583.x
180. Sun H, Zhao L, Pan K, Zhang Z, Zhou M, Cao G. “Integrated analysis of mRNA and miRNA expression profiles in pancreatic ductal adenocarcinoma”. Oncology Reports 37, no. 5 (2017), 2779–86. doi: 10.3892/or.2017.5526
181. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer (2013) 108(4):914–23. doi: 10.1038/bjc.2013.32
182. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep (2015) 10(4):562–73. doi: 10.1016/j.celrep.2014.12.039
183. Wang WQ, Liu L, Xu HX, Wu CT, Xiang JF, Xu J, et al. Infiltrating immune cells and gene mutations in pancreatic ductal adenocarcinoma. Br J Surg (2016) 103(9):1189–99. doi: 10.1002/bjs.10187
184. Qiao B, Luo W, Liu Y, Wang J, Liu C, Liu Z, et al. The prognostic value of CXC chemokine receptor 2 (CXCR2) in cancers: a meta-analysis. Oncotarget (2018) 9(19):15068–76. doi: 10.18632/oncotarget.23492
185. Sano M, Ijichi H, Takahashi R, Miyabayashi K, Fujiwara H, Yamada T, et al. Blocking CXCLs-CXCR2 axis in tumor-stromal interactions contributes to survival in a mouse model of pancreatic ductal adenocarcinoma through reduced cell invasion/migration and a shift of immune-inflammatory microenvironment. Oncogenesis (2019) 8(2):8. doi: 10.1038/s41389-018-0117-8
186. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell (2016) 29(6):832–45. doi: 10.1016/j.ccell.2016.04.014
187. Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut (2018) 67(6):1112–23. doi: 10.1136/gutjnl-2017-313738
188. Jin W, Yin H, Li H, Yu XJ, Xu HX, Liu L. Neutrophil extracellular DNA traps promote pancreatic cancer cells migration and invasion by activating EGFR/ERK pathway. J Cell Mol Med (2021) 25(12):5443–56. doi: 10.1111/jcmm.16555
189. Garcea G, Ladwa N, Neal CP, Metcalfe MS, Dennison AR, Berry DP. Preoperative neutrophil-to-lymphocyte ratio (NLR) is associated with reduced disease-free survival following curative resection of pancreatic adenocarcinoma. World J Surg (2011) 35(4):868–72. doi: 10.1007/s00268-011-0984-z
190. Stotz M, Gerger A, Eisner F, Szkandera J, Loibner H, Ress AL, et al. Increased neutrophil-lymphocyte ratio is a poor prognostic factor in patients with primary operable and inoperable pancreatic cancer. Br J Cancer (2013) 109(2):416–21. doi: 10.1038/bjc.2013.332
191. Suzuki R, Takagi T, Hikichi T, Konno N, Sugimoto M, Watanabe KO, et al. Derived neutrophil/lymphocyte ratio predicts gemcitabine therapy outcome in unresectable pancreatic cancer. Oncol Lett (2016) 11(5):3441–5. doi: 10.3892/ol.2016.4381
192. Wang Y, Fang T, Huang L, Wang H, Zhang L, Wang Z, et al. Neutrophils infiltrating pancreatic ductal adenocarcinoma indicate higher Malignancy and worse prognosis. Biochem Biophys Res Commun (2018) 501(1):313–9. doi: 10.1016/j.bbrc.2018.05.024
193. Aoyagi Y, Oda T, Kinoshita T, Nakahashi C, Hasebe T, Ohkohchi N, et al. Overexpression of TGF-beta by infiltrated granulocytes correlates with the expression of collagen mRNA in pancreatic cancer. Br J Cancer (2004) 91(7):1316–26. doi: 10.1038/sj.bjc.6602141
194. Hu P, Shen M, Zhang P, Zheng C, Pang Z, Zhu L, et al. Intratumoral neutrophil granulocytes contribute to epithelial-mesenchymal transition in lung adenocarcinoma cells. Tumour Biol (2015) 36(10):7789–96. doi: 10.1007/s13277-015-3484-1
195. Gong L, Cumpian AM, Caetano MS, Ochoa CE, de la Garza MM, Lapid DJ, et al. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol Cancer (2013) 12(1):154. doi: 10.1186/1476-4598-12-154
196. Dallal RM, Christakos P, Lee K, Egawa S, Son YI, Lotze MT. Paucity of dendritic cells in pancreatic cancer. Surgery (2002) 131(2):135–8. doi: 10.1067/msy.2002.119937
197. Tjomsland V, Sandström P, Spångeus A, Messmer D, Emilsson J, Falkmer U, et al. Pancreatic adenocarcinoma exerts systemic effects on the peripheral blood myeloid and plasmacytoid dendritic cells: an indicator of disease severity? BMC Cancer (2010) 10:87. doi: 10.1186/1471-2407-10-87
198. Yamamoto T, Yanagimoto H, Satoi S, Toyokawa H, Yamao J, Kim S, et al. Circulating myeloid dendritic cells as prognostic factors in patients with pancreatic cancer who have undergone surgical resection. J Surg Res (2012) 173(2):299–308. doi: 10.1016/j.jss.2010.09.027
199. Takahashi K, Toyokawa H, Takai S, Satoi S, Yanagimoto H, Terakawa N, et al. Surgical influence of pancreatectomy on the function and count of circulating dendritic cells in patients with pancreatic cancer. Cancer Immunol Immunother (2006) 55(7):775–84. doi: 10.1007/s00262-005-0079-5
200. Hiraoka N, Yamazaki-Itoh R, Ino Y, Mizuguchi Y, Yamada T, Hirohashi S, et al. CXCL17 and ICAM2 are associated with a potential anti-tumor immune response in early intraepithelial stages of human pancreatic carcinogenesis. Gastroenterology (2011) 140(1):310–21. doi: 10.1053/j.gastro.2010.10.009
201. Bellone G, Carbone A, Smirne C, Scirelli T, Buffolino A, Novarino A, et al. Cooperative induction of a tolerogenic dendritic cell phenotype by cytokines secreted by pancreatic carcinoma cells. J Immunol (2006) 177(5):3448–60. doi: 10.4049/jimmunol.177.5.3448
202. Basso D, Fogar P, Falconi M, Fadi E, Sperti C, Frasson C, et al. Pancreatic tumors and immature immunosuppressive myeloid cells in blood and spleen: role of inhibitory co-stimulatory molecules PDL1 and CTLA4. An in vivo and in vitro study. PloS One (2013) 8(1):e54824. doi: 10.1371/journal.pone.0054824
203. Karakhanova S, Link J, Heinrich M, Shevchenko I, Yang Y, Hassenpflug M, et al. Characterization of myeloid leukocytes and soluble mediators in pancreatic cancer: importance of myeloid-derived suppressor cells. Oncoimmunology (2015) 4(4):e998519. doi: 10.1080/2162402X.2014.998519
204. Markowitz J, Wang J, Vangundy Z, You J, Yildiz V, Yu L, et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4+ T cells in cancer and measurement of STAT1 nitration. Sci Rep (2017) 7(1):15424. doi: 10.1038/s41598-017-14970-0
205. Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature (2020) 579(7797):130–5. doi: 10.1038/s41586-020-2015-4
206. Huang Q, Ma X, Wang Y, Niu Z, Wang R, Yang F, et al. IL-10 producing type 2 innate lymphoid cells prolong islet allograft survival. EMBO Mol Med (2020) 12(11):e12305. doi: 10.15252/emmm.202012305
207. Sugimura R, Wang CY. The role of innate lymphoid cells in cancer development and immunotherapy. Front Cell Dev Biol (2022) 10:803563. doi: 10.3389/fcell.2022.803563
208. Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother (2010) 33(8):828–33. doi: 10.1097/CJI.0b013e3181eec14c
209. Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
210. Kunk PR, Bauer TW, Slingluff CL, Rahma OE. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J Immunother Cancer (2016) 4:14. doi: 10.1186/s40425-016-0119-z
211. Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut (2014) 63(11):1769–81. doi: 10.1136/gutjnl-2013-306271
212. Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterology (2015) 149(1):201–10. doi: 10.1053/j.gastro.2015.04.010
213. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol (2016) 37(12):855–65. doi: 10.1016/j.it.2016.09.006
214. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell (2016) 30(2):324–36. doi: 10.1016/j.ccell.2016.06.003
215. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity (2016) 44(4):924–38. doi: 10.1016/j.immuni.2016.03.012
216. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell (2017) 31(5):711–723.e4. doi: 10.1016/j.ccell.2017.04.003
217. de Mingo Pulido Á, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. TIM-3 regulates CD103+ Dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell (2018) 33(1):60–74.e6. doi: 10.1016/j.ccell.2017.11.019
218. Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4+ T cell immunity. Cell (2019) 177(3):556–71. doi: 10.1016/j.cell.2019.02.005
219. Kenkel JA, Tseng WW, Davidson MG, Tolentino LL, Choi O, Bhattacharya N, et al. An immunosuppressive dendritic cell subset accumulates at secondary sites and promotes metastasis in pancreatic cancer. Cancer Res (2017) 77(15):4158–70. doi: 10.1158/0008-5472.CAN-16-2212
220. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol (2015) 6:257. doi: 10.3389/fimmu.2015.00257
221. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
222. Zhang R, Liu Q, Li T, Liao Q, Zhao Y. Role of the complement system in the tumor microenvironment. Cancer Cell Int (2019) 19:300. doi: 10.1186/s12935-019-1027-3
223. Bettac L, Denk S, Seufferlein T, Huber-Lang M. Complement in pancreatic disease-perpetrator or savior? Front Immunol (2017) 8:15. doi: 10.3389/fimmu.2017.00015
224. Hussain N, Das D, Pramanik A, Pandey MK, Joshi V, Pramanik KC. Targeting the complement system in pancreatic cancer drug resistance: a novel therapeutic approach. Cancer Drug Resist (2022) 5(2):317–27. doi: 10.20517/cdr.2021.150
225. Ajona D, Ortiz-Espinosa S, Pio R. Complement anaphylatoxins C3a and C5a: Emerging roles in cancer progression and treatment. Semin Cell Dev Biol (2019) 85:153–63. doi: 10.1016/j.semcdb.2017.11.023
226. Ravindranath NMH, Shuler C. Cell-surface density of complement restriction factors (CD46, CD55, and CD59): oral squamous cell carcinoma versus other solid tumors. Oral Surg Oral Med Oral Pathol Oral Radiol Endod (2007) 103(2):231–9. doi: 10.1016/j.tripleo.2006.05.028
227. Mellby LD, Nyberg AP, Johansen JS, Wingren C, Nordestgaard BG, Bojesen SE, et al. Serum biomarker signature-based liquid biopsy for diagnosis of early-stage pancreatic cancer. J Clin Oncol (2018) 36(28):2887–94. doi: 10.1200/JCO.2017.77.6658
228. Varghese PM, Mukherjee S, Al-Mohanna FA, Saleh SM, Almajhdi FN, Beirag N, et al. Human properdin released by infiltrating neutrophils can modulate influenza A virus infection. Front Immunol (2021) 12:747654. doi: 10.3389/fimmu.2021.747654
229. Kishore U, Varghese PM, Mangogna A, Klein L, Tu M, Urbach L, et al. Neutrophil-derived complement factor P induces cytotoxicity in basal-like cells via caspase 3/7 activation in pancreatic cancer. bioRxiv (2023) 10:28. doi: 10.1101/2023.10.28.564512v1
230. Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer (2005) 5(7):526–42. doi: 10.1038/nrc1649
231. Wingren C, Sandström A, Segersvärd R, Carlsson A, Andersson R, Löhr M, et al. Identification of serum biomarker signatures associated with pancreatic cancer. Cancer Res (2012) 72(10):2481–90. doi: 10.1158/0008-5472.CAN-11-2883
232. Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut (2017) 66(1):124–36. doi: 10.1136/gutjnl-2016-312078
233. Yang S, Liu Q, Liao Q. Tumor-associated macrophages in pancreatic ductal adenocarcinoma: origin, polarization, function, and reprogramming. Front Cell Dev Biol (2021) 8:607209. doi: 10.3389/fcell.2020.607209
234. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer (2017) 5(1):53. doi: 10.1186/s40425-017-0257-y
235. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74(18):5057–69. doi: 10.1158/0008-5472.CAN-13-3723
236. Wang-Gillam A, O’Reilly EM, Bendell JC, Wainberg ZA, Borazanci EH, Bahary N, et al. A randomized phase II study of cabiralizumab (cabira) + nivolumab (nivo) ± chemotherapy (chemo) in advanced pancreatic ductal adenocarcinoma (PDAC). JCO (2019) 37(4_suppl):TPS465–5. doi: 10.1200/JCO.2019.37.4_suppl.TPS465
237. Ho WJ, Jaffee EM. Macrophage-targeting by CSF1/1R blockade in pancreatic cancers. Cancer Res (2021) 81(24):6071–3. doi: 10.1158/0008-5472.CAN-21-3603
238. Van Cutsem E, Tempero MA, Sigal D, Oh DY, Fazio N, Macarulla T, et al. Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol (2020) 38(27):3185–94. doi: 10.1200/JCO.20.00590
239. Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Trans Med (2019) 11(499):eaau9240. doi: 10.1126/scitranslmed.aau9240
240. Lamberti MJ, Nigro A, Mentucci FM, Rumie Vittar NB, Casolaro V, Dal Col J. Dendritic cells and immunogenic cancer cell death: A combination for improving antitumor immunity. Pharmaceutics (2020) 12(3):256. doi: 10.3390/pharmaceutics12030256
241. Konduri V, Li D, Halpert MM, Liang D, Liang Z, Chen Y, et al. Chemo-immunotherapy mediates durable cure of orthotopic KrasG12D/p53–/– pancreatic ductal adenocarcinoma. Oncoimmunology (2016) 5(9):e1213933. doi: 10.1080/2162402X.2016.1213933
242. Renouf DJ, Loree JM, Knox JJ, Topham JT, Kavan P, Jonker D, et al. The CCTG PA.7 phase II trial of gemcitabine and nab-paclitaxel with or without durvalumab and tremelimumab as initial therapy in metastatic pancreatic ductal adenocarcinoma. Nat Commun (2022) 13:5020. doi: 10.1038/s41467-022-32591-8
243. Fu Q, Chen Y, Huang D, Guo C, Zhang X, Xiao W, et al. Sintilimab plus modified FOLFIRINOX in metastatic or recurrent pancreatic cancer: the randomized phase ii cispd3 trial. Ann Surg Oncol (2023) 30(8):5071–80. doi: 10.1245/s10434-023-13383-w
244. Padrón LJ, Maurer DM, O’Hara MH, O’Reilly EM, Wolff RA, Wainberg ZA, et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat Med (2022) 28(6):1167–77. doi: 10.1038/s41591-022-01829-9
245. Wu AA, Bever KM, Ho WJ, Fertig EJ, Niu N, Zheng L, et al. A phase 2 study of allogeneic GM-CSF transfected pancreatic tumor vaccine (GVAX) with ipilimumab as maintenance treatment for metastatic pancreatic cancer. Clin Cancer Res (2020) 26(19):5129–39. doi: 10.1158/1078-0432.CCR-20-1025
246. Weiss GJ, Blaydorn L, Beck J, Bornemann-Kolatzki K, Urnovitz H, Schütz E, et al. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Invest New Drugs (2018) 36(1):96–102. doi: 10.1007/s10637-017-0525-1
247. Kocher HM, Basu B, Froeling FEM, Sarker D, Slater S, Carlin D, et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat Commun (2020) 11(1):4841. doi: 10.1038/s41467-020-18636-w
248. Bekaii-Saab T, Okusaka T, Goldstein D, Oh DY, Ueno M, Ioka T, et al. Napabucasin plus nab-paclitaxel with gemcitabine versus nab-paclitaxel with gemcitabine in previously untreated metastatic pancreatic adenocarcinoma: an adaptive multicentre, randomised, open-label, phase 3, superiority trial. eClinicalMedicine (2023) 58:101897. doi: 10.1016/j.eclinm.2023.101897
249. Hidalgo M, Garcia-Carbonero R, Lim KH, Messersmith WA, Garrido-Laguna I, Borazanci E, et al. A preclinical and phase ib study of palbociclib plus nab-paclitaxel in patients with metastatic adenocarcinoma of the pancreas. Cancer Res Commun (2022) 2(11):1326–33. doi: 10.1158/2767-9764.CRC-22-0072
250. Pegna GJ, Lee M, Peer CJ, Ahmad MI, Venzon DJ, Yu Y, et al. Systemic immune changes accompany combination treatment with immunotoxin LMB-100 and nab-paclitaxel. Cancer Med (2022) 12(4):4236–49. doi: 10.1002/cam4.5290
251. Coveler AL, Reilley M, Zalupski M, Macarulla T, Fountzilas C, Castanon Alvarez E, et al. Safety and clinical activity of oleclumab (O) ± durvalumab (D) + chemotherapy (CT) in patients (pts) with metastatic pancreatic ductal adenocarcinoma (mPDAC): A phase 1b/2 randomized study. JCO (2023) 41(16_suppl):4136–6. doi: 10.1200/JCO.2023.41.16_suppl.4136
252. Gracian AC, Dean A, Muñoz A, Hidalgo M, Pazo-Cid R, Martin M, et al. YOSEMITE: A 3 arm double-blind randomized phase 2 study of gemcitabine, paclitaxel protein-bound particles for injectable suspension, and placebo (GAP) versus gemcitabine, paclitaxel protein-bound particles for injectable suspension and either 1 or 2 truncated courses of demcizumab (GAD). Ann Oncol (2017) 28:v211. doi: 10.1093/annonc/mdx369.004
253. Jameson GS, Borazanci EH, Babiker HM, Poplin E, Niewiarowska AA, Gordon MS, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer. JCO (2017) 35(4_suppl):341–1. doi: 10.1200/JCO.2017.35.4_suppl.341
254. Chung V, McDonough S, Philip PA, Cardin D, Wang-Gillam A, Hui L, et al. Effect of selumetinib and MK-2206 vs oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG S1115 study randomized clinical trial. JAMA Oncol (2017) 3(4):516–22. doi: 10.1001/jamaoncol.2016.5383
255. Farren MR, Mace TA, Geyer S, Mikhail S, Wu C, Ciombor K, et al. Systemic immune activity predicts overall survival in treatment-naïve patients with metastatic pancreatic cancer. Clin Cancer Res (2016) 22(10):2565–74. doi: 10.1158/1078-0432.CCR-15-1732
256. Smith C, Zheng W, Dong J, Wang Y, Lai J, Liu X, et al. Tumor microenvironment in pancreatic ductal adenocarcinoma: Implications in immunotherapy. World J Gastroenterol (2022) 28(27):3297–313. doi: 10.3748/wjg.v28.i27.3297
257. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles (2018) 7(1):1535750. doi: 10.1080/20013078.2018.1535750
258. Shen T, Huang Z, Shi C, Pu X, Xu X, Wu Z, et al. Pancreatic cancer-derived exosomes induce apoptosis of T lymphocytes through the p38 MAPK-mediated endoplasmic reticulum stress. FASEB J (2020) 34(6):8442–58. doi: 10.1096/fj.201902186R
259. Chang CH, Pauklin S. Extracellular vesicles in pancreatic cancer progression and therapies. Cell Death Dis (2021) 12(11):1–12. doi: 10.1038/s41419-021-04258-7
260. Batista IA, Melo SA. Exosomes and the future of immunotherapy in pancreatic cancer. Int J Mol Sci (2019) 20(3):567. doi: 10.3390/ijms20030567
261. Fyfe J, Dye D, Razak NBA, Metharom P, Falasca M. Immune evasion on the nanoscale: Small extracellular vesicles in pancreatic ductal adenocarcinoma immunity. Semin Cancer Biol (2023) 96:36–47. doi: 10.1016/j.semcancer.2023.09.004
Keywords: PDAC, TME, immune surveillance, immune suppression, EMT, macrophages, TNF-α
Citation: Joseph AM, Al Aiyan A, Al-Ramadi B, Singh SK and Kishore U (2024) Innate and adaptive immune-directed tumour microenvironment in pancreatic ductal adenocarcinoma. Front. Immunol. 15:1323198. doi: 10.3389/fimmu.2024.1323198
Received: 17 October 2023; Accepted: 11 January 2024;
Published: 07 February 2024.
Edited by:
Juana Serrano Lopez, Health Research Institute Foundation Jimenez Diaz (IIS-FJD), SpainReviewed by:
Jean-René Pallandre, INSERM U1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique, FranceCopyright © 2024 Joseph, Al Aiyan, Al-Ramadi, Singh and Kishore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Uday Kishore, dWtpc2hvcmVAaG90bWFpbC5jb20=; dWRheS5raXNob3JlQHVhZXUuYWMuYWU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.