
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 18 September 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1323171
This article is part of the Research TopicInnate Immunity in VasculitisView all 5 articles
 Paula Uittenbogaard1†
Paula Uittenbogaard1† Stejara A. Netea2,3†
Stejara A. Netea2,3† Michael W. T. Tanck4
Michael W. T. Tanck4 Judy Geissler1Piotr Buda5Monika Kowalczyk-Domagała6Magdalena Okarska-Napierała7
Judy Geissler1Piotr Buda5Monika Kowalczyk-Domagała6Magdalena Okarska-Napierała7 Diana van Stijn2Carline E. Tacke2 US Kawasaki Disease Genetics Consortium
Diana van Stijn2Carline E. Tacke2 US Kawasaki Disease Genetics Consortium David P. Burgner8,9Chisato Shimizu10Jane C. Burns10
David P. Burgner8,9Chisato Shimizu10Jane C. Burns10 Irene M. Kuipers11
Irene M. Kuipers11 Taco W. Kuijpers1,2‡
Taco W. Kuijpers1,2‡ Sietse Q. Nagelkerke1,2*‡
Sietse Q. Nagelkerke1,2*‡Introduction: Kawasaki disease (KD) is a pediatric vasculitis that can result in coronary artery aneurysm (CAA) formation, which is a dangerous complication. Treatment with intravenous immunoglobulin (IVIg) significantly decreases the risk of CAA, possibly through competitive binding to Fc-gamma receptors (FcγRs), which reduces the binding of pathological immune complexes. However, ~20% of children have recrudescence of fever and have an increased risk of CAA. Therefore, we aimed to identify genetic markers at the FCGR2/3 locus associated with susceptibility to KD, IVIg resistance, or CAA.
Materials and methods: We investigated the association of single-nucleotide polymorphisms (SNPs) and copy number variations (CNVs) at the FCGR2/3 locus with KD susceptibility, IVIg resistance, and CAA risk using a family-based test (KD susceptibility) and case–control analyses (IVIg resistance and CAA risk) in different cohorts, adding up to a total of 1,167 KD cases. We performed a meta-analysis on IVIg resistance and CAA risk including all cohorts supplemented by previous studies identified through a systematic search.
Results: FCGR2A-p.166His was confirmed to be strongly associated with KD susceptibility (Z = 3.17, p = 0.0015). In case–control analyses, all of the investigated genetic variations at the FCGR2/3 locus were generally not associated with IVIg resistance or with CAA risk, apart from a possible association in a Polish cohort for the FCGR3B-NA2 haplotype (OR = 2.15, 95% CI = 1.15–4.01, p = 0.02). Meta-analyses of all available cohorts revealed no significant associations of the FCGR2/3 locus with IVIg resistance or CAA risk.
Discussion: FCGR2/3 polymorphisms are associated with susceptibility to KD but not with IVIg resistance and CAA formation. Currently known genetic variations at the FCGR2/3 locus are not useful in prediction models for IVIg resistance or CAA risk.
Kawasaki disease (KD) is a pediatric vasculitis involving the coronary arteries that mainly affects children under the age of 5 (1). The disease can result in coronary artery aneurysm (CAA) formation, which occurs in up to 25% of untreated cases, making it the most common cause of acquired heart disease in children in developed countries (2). The risk of CAA has significantly diminished since the introduction of treatment with human-pooled intravenous immunoglobulin (IVIg) (3). Although its exact mechanism of action remains uncertain, various mechanisms of action have been proposed (4–7). Similarly, there is the involvement of the Fc-gamma receptors (FcγRs), for instance, by competitive binding of IVIg to the FcγRs, thereby reducing the binding of pathological immune complexes and inhibiting cellular activation. In turn, IVIg-resistant patients have an increased risk of developing CAA (8, 9). IVIg resistance occurs in approximately 20% of treated patients (10), with up to 20% of affected children developing CAA despite timely treatment (9). This underlines the need to better understand the mechanisms behind poor treatment response and the importance of identifying prognostic markers that accurately predict children at risk for poor treatment response or of developing CAA. Although risk scores have been developed to predict IVIg resistance including various laboratory findings (11–14), these risk score systems have been developed for the Japanese population and have a limited predictive value in non-Japanese populations (15, 16). As has also been proposed in the KD guidelines of the American Heart Association (AHA) (17) and the European Initiative Single Hub and Access point for pediatric Rheumatology in Europe (SHARE) (18), better predictive models, possibly incorporating biomarkers or genetic variants, are needed for non-Asian populations.
The exact etiology of KD is unknown, but it is hypothesized to be a post-infectious hyperinflammatory reaction in children with a genetic susceptibility. The hypothesis of a post-infectious etiology was reinforced by the recent outbreak of multisystem inflammatory syndrome in children, which shares some clinical features with KD and is triggered by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (19, 20). Several findings point to a genetic susceptibility for KD. Firstly, the incidence of KD differs widely per country, with the incidence in European countries ranging between 10 and 15 per 100,000 children under the age of 5 per year (21). Over 10-fold higher incidence rates of KD have been reported in Asian countries, particularly in Japanese and Koreans, which persist when patients of these ethnicities migrate to other countries (22, 23). Secondly, as has been shown by twin studies and familial cases, siblings of patients with KD have a higher KD risk compared with the general population (24, 25).
Among other gene regions (encoding, e.g., caspase 3, human leukocyte antigen class II, B-cell lymphoid kinase, inositol 1,4,5-trisphosphate kinase C, and CD40 protein) (17), KD has been linked to the gene cluster of the FcγR family (26). This FCGR2/3 locus consists of genes encoding the five low- to medium-affinity FcγRs (FCGR2A, FCGR2B, FCGR2C, FCGR3A, and FCGR3B) that either activate or inhibit (FCGR2B) cellular immune functions after binding immunoglobulin G (IgG). Various single-nucleotide polymorphisms (SNPs), haplotypes, and copy number variations (CNVs) at the FCGR2/3 locus impact receptor function and expression levels (27–29).
Most studies on the association of KD with the FCGR2/3 locus have focused on the genetic association of the SNP FCGR2A-p.His166Arg (rs1801274, formerly known as FCGR2A-p.His131Arg) with KD susceptibility and have shown that FCGR2A-p.166His is a marker of increased susceptibility (30–33). This SNP in the FCGR2A gene can be typed with conventional methods, such as genome-wide association study (GWAS). However, the FCGR2/3 locus is notoriously difficult to genotype. Firstly, it comprises a large segmental duplication with over 98% homology shared between the duplicated regions (30). Secondly, CNV is common in this locus, resulting in a highly polymorphic region. There is a strong linkage disequilibrium (LD) between various variants (27). Most of the known variants on the FCGR2/3 locus cannot be included in GWAS analyses.
Comprehensive analysis of the FCGR2/3 locus can be achieved using the previously described multiplex ligation-dependent probe amplification (MLPA), which can reliably distinguish the currently known SNPs, haplotypes, and CNVs. These SNPs include FCGR2A-p.Gln62Trp, FCGR2A-p.His166Arg, FCGR2B-p.Ile232Thr, and FCGR3A-p.Val176Phe. Known haplotypes that can be distinguished include the FCGR2B and FCGR2C promoter haplotypes 2B.1/2B.2/2B.4, FCGR3B NA1/NA2/SH haplotypes (which encode the human neutrophil antigen), and FCGR2C Stop/ORF/non-classic ORF haplotypes. CNVs (both deletions and duplications) occur in the CNR1–CNR4 regions, generally involving either FCGR3A-FCGR2C or FCGR2C-FCGR3B, and can be distinguished. Using MLPA, Nagelkerke et al. found that, in addition to FCGR2A-p.His166Arg, the FCGR2C-ORF haplotype (rs759550223 and rs7677413) was significantly associated with susceptibility to KD in the European population (30). Although the association of FCGR2A-p.His166Arg appears to be independent of ethnicity, a clear ethnic variation was seen for the FCGR2C-ORF. This haplotype was associated with susceptibility to KD in Europeans but is virtually absent in Asian subjects, stressing the importance of accounting for ethnicity when investigating genetic associations in KD.
The association of FCGR2A-p.His166Arg with KD susceptibility raises the question of whether this SNP or other genetic variants at the FCGR2/3 locus may also impact the clinical outcomes of patients with KD, particularly the risk of IVIg resistance and CAA. The few studies that investigated such associations lack consensus and often only focus on one or several FcγRs in relatively small patient populations (31–36). This highlights the need to further fine-map genetic associations at the FCGR2/3 locus accounting for ethnic variations in extensive patient populations. In the present study, we, therefore, investigated the association of all currently known functionally relevant SNPs, haplotypes, and CNVs at the FCGR2/3 locus with susceptibility to KD, response to treatment, and risk of CAA development using a family-based test (KD susceptibility) and case–control analyses (CAA and IVIg response). Cases were recruited from four cohorts from different parts of Europe (the Netherlands and Poland), the United States, and Australia. Subsequently, we performed a meta-analysis for the association of FCGR2/3 genetic variations with IVIg resistance and CAA risk in all four cohorts, supplemented by studies identified through a systematic review.
We used the Quanto software to calculate the statistical power of our cohort size (37). We expected a total cohort of at least 800 subjects (children with KD) with an estimated prevalence of IVIg resistance of 0.22 and an estimated CAA prevalence of 0.30. We calculated the power to detect an odds ratio of 2 per minor allele for variants with minor allele frequencies (MAFs) ranging from 0.05 to 0.5, assuming an allelic genetic model and setting the significance threshold (p-value) at 0.05. For IVIg, the power to detect an OR of 2 was >80% for variants with an MAF of 0.05 and >90% for an MAF ≥0.1. For CAA, the power was >90% for the whole MAF range.
Complete KD cases, who had not received prior immunomodulatory or immunosuppressive treatment, were recruited from the Netherlands (n = 388), the United States (n = 447), Australia (n = 213), and Poland (n = 119) with diagnosis based on the standard diagnostic clinical criteria from the AHA (17). In total, 1,167 patients were included in the study. A total of 762 patients (701 complete trios and 61 incomplete trios) were included in the transmission disequilibrium test (TDT) analysis for susceptibility to KD.
This TDT analysis is an extension of our previously published TDT analysis (30) using the same trios supplemented by an additional 155 trios from the Netherlands.
As per the 2017 AHA guidelines, primary treatment consisted of a single IVIg infusion (2 g/kg) and aspirin (started at 30–50 mg kg−1 day−1 and reduced to 3–5 mg kg−1 day−1 once fever subsided for 48 h) (17).
For associations within the patient groups for susceptibility to CAA and response to IVIg treatment, we performed case–control analyses for all patients for which these clinical parameters, as well as self-reported ethnicity, were documented. For both these case–control analyses, there was considerable, but not complete, overlap with the patients in the TDT. Many of the KD cases were previously studied for general susceptibility to KD in our GWAS (33) and follow-up study (27). There was no overlap with the patients with KD described by Biezeveld et al. (28). In total, 872 patients of self-reported European descent and 60 patients of self-reported Asian descent were included in the case–control analyses. For an overview of the cohorts, see Supplementary Table 1.
Clinical information from the disease episode (i.e., sex, ethnicity, treatment response, and CAA development) was collected using electronic health records and processed in a combined database (REDCap and Castor). CAAs were defined as a coronary artery with a Z-score ≥2.5 (17). In the case–control analysis, cases were defined as patients with CAA, whereas controls were defined as patients without CAA. IVIg resistance was defined as persistent or recurrent fever >38°C at >36 h and <7 days after completing the initial IVIg infusion in patients treated with IVIg ≤10 days post-onset of fever (17). In this case–control analysis, cases were defined as patients having an adequate IVIg response, while controls were defined as patients with IVIg non-responsiveness. This case–control analysis only included patients treated with IVIg ≤10 days of the onset of the disease.
Genomic DNA from whole blood was isolated using the QIAamp Blood Mini kit (Qiagen, Hilden, Germany). Genomic DNA from saliva was isolated using the Oragene DNA self-collection kit (DNA Genotek, Ontario, Canada) or the Puregene DNA purification kit (Qiagen, Hilden, Germany).
SNPs and CNVs in the low-affinity FCGR genes were determined using an FCGR-specific MLPA assay as described previously (30, 38) (P110 and P111, MRC-Holland, Amsterdam, the Netherlands). The MLPA contained probes to detect all the currently known SNPs and the gene-specific probes to determine CNVs. DNA samples of three healthy individuals well typed for all CNVs and SNPs were included as a reference in all experiments. The results were analyzed using the program Genemarker version 3.0.0 (Soft Genetics LLC, State College, PA, USA), with consideration for the homology within the locus and taking into account known LD aiding in the construction of haplotypes, as described previously (29, 30).
For the family-based analysis using FBAT (see Statistical analysis), phased genotypes were required (i.e., separated into the paternal and maternal alleles for each individual). Because the MLPA results could not be phased over the full locus, we separated the genotypes for each SNP manually, assuming an “even distribution” of the copies over the columns. For example, if an individual had two allele copies (no CNV), then we assumed that each allele contained one copy unless our trio data suggested otherwise (rare occasions). Regarding CNVs of FCGR3B and the NA1 and NA2(SH) genotypes, previous evidence has shown that the NA1 and NA2(SH) genotypes are generally on the same allele (the duplication allele) (39). For the other genotypes, this was not found, and we assumed that the duplication allele contained the same genotypes (e.g., FCGR2C: ORF-ORF and Stop-Stop). In case of ambiguity, the genotypes were solved by using the trio information combined with the linkage information on the locus.
Firstly, we performed a (multi-marker) TDT (using an FBAT toolkit in R) in the parent-affected offspring trios to investigate the association between KD susceptibility and the various SNPs, CNVs, and haplotypes.
Thereafter, differences in the prevalence of variants were compared between the cohorts using Fisher’s exact test to assess which data could be pooled in further analyses. There were significant differences in the variants for one or more variants between all groups, except for the US and Australian cohorts. The latter cohorts showed no significant differences and were merged into a single cohort (data not shown).
Subsequently, case–control analyses of the association of each genetic variant (allelic model) with IVIg resistance and CAA development were conducted. Analyses were performed in R using one-sided Fisher’s exact tests and univariable logistic regression models. Variants that were significantly associated with IVIg resistance or CAA development in previous literature reports and/or in our own analyses were included in a meta-analysis of the findings found in all separate cohorts (meta package in R). To assess differences by ethnicity, we primarily separated the ethnic groups in the case–control analysis and then performed a meta-analysis with the complete dataset.
A p-value below 0.05 was considered statistically significant. Statistical analyses were performed in R version 4.1.
A systematic search in PubMed/Medline was conducted on March 2, 2023, to identify all studies that reported the relationship between SNPs in the FCGR2/3 locus and IVIg resistance and CAAs in patients with KD. The search strategy included the following search terms: (FCGR* OR FCγR* OR Fc-gamma OR CD64 OR CD32 OR CD16 OR rs1801274 OR rs759550223 OR rs76277413 OR rs149754834 OR rs201218628 OR rs143796418 OR rs396991 OR rs5030738 OR rs1050501) AND (“Mucocutaneous Lymph Node Syndrome” [Mesh] OR “Mucocutaneous Lymph Node Syndrome” OR MCLS OR “Kawasaki disease” OR KD OR “Kawasaki-disease” OR “Kawasaki Syndrome” OR “Acute inflammatory Vasculitis”). Studies solely reporting on DNA methylation status were excluded. We extracted data on the study characteristics (e.g., study design, number of patients enrolled in the study, number of patients fulfilling the review’s inclusion criteria, ethnicity of the patients, length of follow-up, and methods), outcome measures (e.g., definitions used and detection methods), and the main study results. A meta-analysis was performed with the pooled data of comparable studies with similar definitions (KD, IVIg resistance, and CAA). We collected all available allele-specific beta coefficients and standard errors (calculated using the odds ratios and 95% confidence intervals) from univariable logistic regressions performed to investigate the association between various allele frequencies within the FcγR locus and the risk of IVIg resistance and/or CAA development. When only the genotypic odds ratios were reported, then we calculated the allele-specific odds ratios by using the genotype frequencies. Lastly, we conducted a meta-analysis that included our own data and the data extracted from the studies identified in the systematic literature review. The meta-analysis was performed using the meta package (version 6.2-0) in R. Forest plots were made in R using the forest package.
Informed consent and written approval were obtained. All patients were included in accordance with the study protocol, the International Conference on Harmonization Good clinical Practice guidelines, and the provisions of the Declaration of Helsinki. Further information per cohort is listed below.
Subjects from the Netherlands were recruited at the Amsterdam UMC and participating hospitals in the broad Amsterdam region as part of the long-term observational Kawasaki disease study as approved by the Medical Ethical Board of the AMC, with the reference number 2012_155 (no. NL41023.018.12).
Subjects from the United States were recruited at Rady Children’s Hospital San Diego (n = 241), Boston Children’s Hospital (n = 81), Northwestern University (n = 37), University of Hawaii (n = 19), Children’s Hospital of Los Angeles (n = 6), and individuals across the US who directly contacted the University of California San Diego to participate (n = 63). The Human Research Protection Program of the University of California San Diego approved this research protocol (IRB 170790). All parents and subjects gave written consent or assent as appropriate.
Subjects from Australia (n = 213) were recruited in accordance with the protocol approved by the ethics committees of all participating tertiary pediatric hospitals.
The blood samples of 119 patients diagnosed with KD were collected between 2016 and 2020, isolated, and stored at the Department of Pediatrics, Nutrition and Metabolic Diseases, Children’s Memorial Health Institute in Warsaw, Poland. All children were born in Poland (Mazowieckie Voivodeship) and were of European ethnic descent. Patients were included as part of the project “Searching for molecular markers related to Kawasaki disease,” which has been approved by the Bioethics Committee at the Children’s Memorial Health Institute Review Board.
In total, 2,631 individuals were genotyped. A detailed description of the included patients is shown in Supplementary Table 1.
In our TDT analysis (FBAT), we included 762 families, with 701 complete trios and 61 incomplete trios. A significant association with susceptibility to KD of the FCGR2A-p.His166Arg SNP, which has been previously confirmed in meta-analyses, was found in our cohort in the single-marker FBAT analysis (Supplementary Table 2) and was also confirmed in the multi-marker FBAT (Table 1). Neither FCGR2C-ORF nor any of the other SNPs were significantly associated with susceptibility to KD (Table 1).
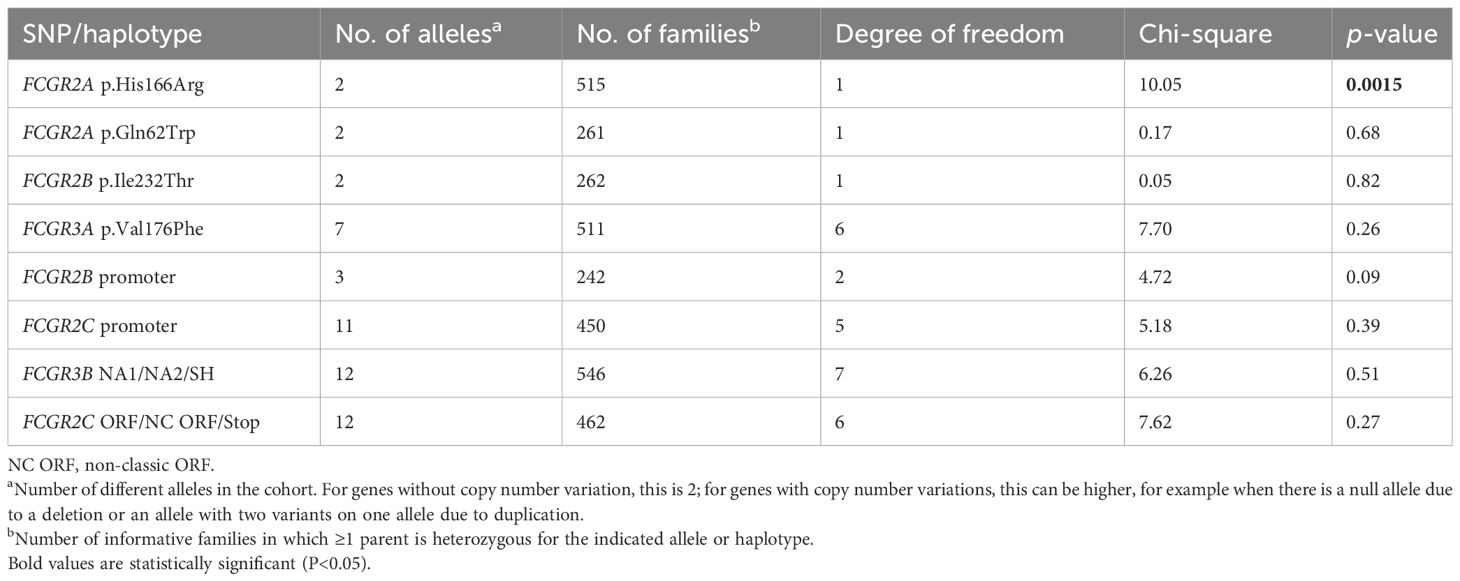
Table 1. Multi-marker transmission disequilibrium test for the different variants at the FCGR2/3 locus in a family-based association study.
We performed a case–control analysis including KD patients with adequate IVIg response as cases and patients with IVIg non-responsiveness as controls. None of the variants tested were associated with IVIg response in our case–control analysis for the combined US and Australian cohort (Supplementary Table 3), the Dutch cohort (Supplementary Table 4), or the Polish cohort (Supplementary Table 5). The subsequent meta-analysis of our different cohorts did not reveal associations either (Supplementary Figure 1).
Subsequently, we performed an additional meta-analysis based on studies identified through a systematic search (Supplementary Figure 1). Although four studies focused on the relationship between variants at the FCGR2/3 locus with IVIg resistance (31, 34, 35, 40), only a single study (34) could be included in our subsequent meta-analysis (Supplementary Table 6; Supplementary Figure 2). This study focused on SNPs in FCGR2B, including the SNP FCGR2B-p.Ile232Thr, which was not significantly associated with IVIg resistance in this study, nor in our meta-analysis. The study also reported that minor allele A at FCGR2B-c.-120T/A was more frequently detected in patients of European descent who responded to IVIg (OR = 3.23, 95% CI = 1.22–8.33, p = 0.01) (34). We also included the latter SNP in our meta-analysis as part of the FCGR2B promoter haplotype 2B.4 and could not reproduce the significant association with IVIg (Figure 1). While this SNP can also occur before FCGR2C, this is extremely rare [5/2,631 (0.19%) samples in the current study] and likely does not affect our analysis. The other studies could not be included in our meta-analysis because they 1) compared patients with IVIg resistance to healthy controls (31) and 2) did not report allelic frequencies (35, 40).
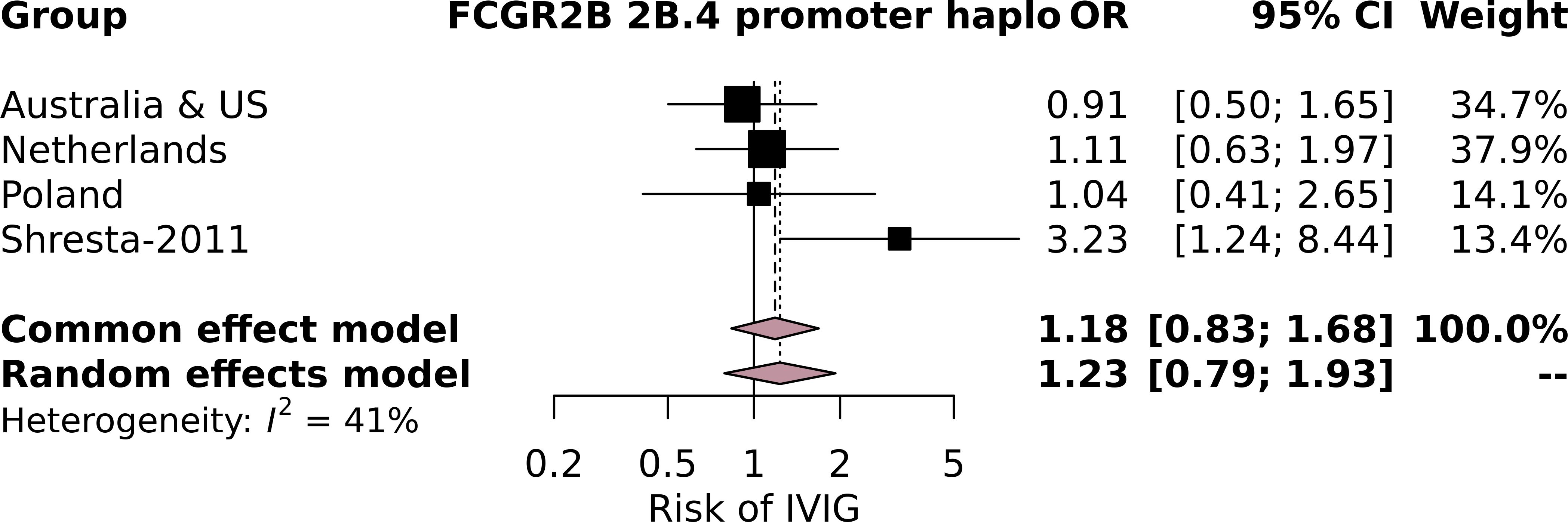
Figure 1. Forest plot of the meta-analysis investigating the association of the FCGR2B 2B.4 promoter haplotype, which contains the FCGR2B-c.-120T/A minor allele A, investigated in Shresta-2011, with intravenous immunoglobulin (IVIg) resistance in individuals of European descent in the current study cohorts and the cohort from Shresta-2011.
Although not included in the meta-analysis, one of these studies found over-transmission of FCGR3B-NA1 among IVIg non-responders (cases) compared to healthy controls (controls) (31). In another study, the copy numbers of FCGR2C in Hispanics and FCGR3B in the European population were found to be associated with IVIg resistance (35). No significant associations of FCGR2A-p.166His with IVIg resistance were found (31, 40).
We also performed a case–control analysis including KD patients with CAAs as cases and patients without CAAs as controls. On the basis of self-reported ethnicity, we investigated informative cases of European descent (the Netherlands: 65 cases, 291 controls; Poland: 43 cases, 76 controls; and Australia and USA: 105 cases, 262 controls) and Asian ethnicity (Australia and USA: 23 cases, 46 controls) separately. The various genotypes and allele frequencies of the CNVs and SNPs are shown in Supplementary Tables 7–9.
In the Polish cohort, the FCGR3B-NA2 haplotype was significantly associated with CAA risk (Fisher’s exact test: p = 0.05; single logistic regression: p = 0.02; OR = 2.15, 95% CI = 1.15–4.01). In the same cohort, having less than two copies of CNR1 was also associated with CAA risk using Fisher’s exact test (p = 0.05), but was not confirmed in the single logistic regression (OR = 0, 95% CI = 0.00–Inf, p = 0.99), presumably because of the low MAF of less than two copies (deletion) of this CNR.
None of the other SNPs or haplotypes were associated with CAA risk in any of the cohorts investigated (Supplementary Tables 7–9).
In our systematic literature review, we identified studies that focused on the relationship between variants at the FCGR2/3 locus and the risk of CAA (28, 31, 32, 36, 40, 41) (Supplementary Table 6; Supplementary Figure 2). Four of these focused on the association of FCGR2A-p.His166Arg with clinical outcomes. FCGR2A-p.166His was significantly associated with CAA risk in one (Taniuchi et al.) of the four studies (28, 32, 36, 41) in which it was investigated. In addition, over-transmission of FCGR3B-NA1 was described in one study among patients with CAAs (31) but was not confirmed in another study (32). None of the other variants investigated were associated with CAA risk.
Subsequently, we performed a meta-analysis including our own cohorts and the four studies that used similar definitions for cases (KD patients with CAAs) and controls (KD patients without CAAs). The variants investigated in the meta-analysis including the studies from the literature were FCGR2A-p.His166Arg, FCGR2B-p.Ile232Thr, FCGR3A-p.Phe176Val, and FCGR3B-NA1. None of the variants investigated were associated with CAA risk in our meta-analysis of the total population (Supplementary Figure 3). This included the FCGR3B-NA2 haplotype, which, although significantly associated in the current Polish cohort, was not associated in the meta-analysis. Finally, because a strong correlation of FCGR2A-p.166His with CAA risk was reported by Taniuchi et al. (32), we also performed a separate meta-analysis with the European (from all four current study cohorts) (28, 41) and Asian subpopulations (from the current Australian and US cohorts) (32, 36) to exclude an ethnicity-dependent association. No significant effect of FCGR2A-p.166His on CAA risk was found in either the European or the Asian subpopulation (Figures 2A, B, respectively).
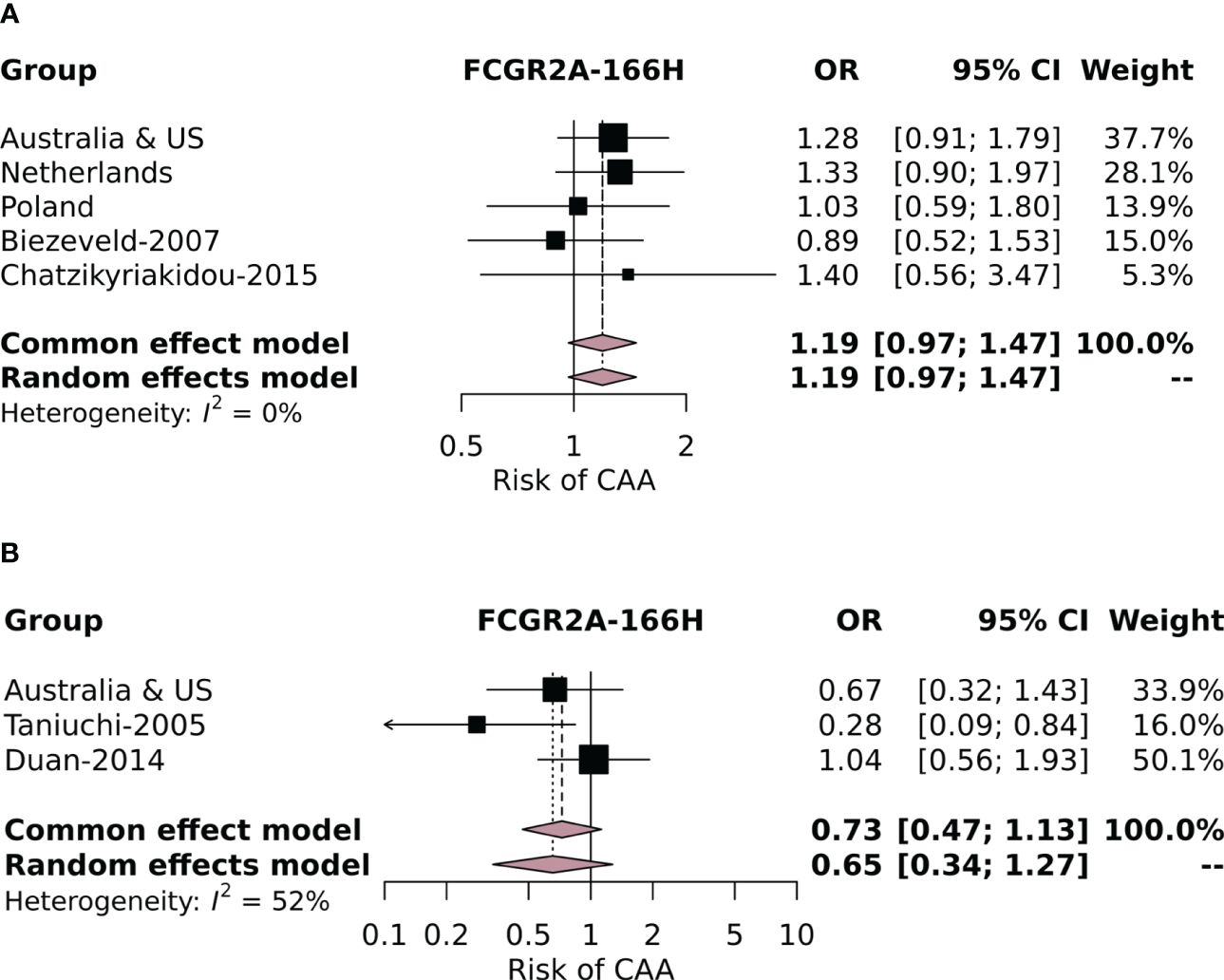
Figure 2. Forest plot of the meta-analysis investigating the association of the FCGR2A-p.His166Arg SNP with coronary artery aneurysm (CAA) risk in individuals of European descent in the current study cohorts and cohorts identified in the literature review (A) and in (B) Asian individuals from the Australian and US cohorts and the Asian cohorts identified in the literature review.
In the current study, we investigated the association of all currently known functionally relevant SNPs and CNVs at the FCGR2/3 locus with KD susceptibility, IVIg resistance, and CAA risk in an extensive cohort, taking into account ethnic differences and using a meta-analysis. We confirmed the established association of the FCGR2A-p.His166Arg SNP with susceptibility to KD, whereas none of the investigated genetic variations at the FCGR2/3 locus were associated with resistance to IVIg or risk of CAA.
Firstly, we reproduced the association of the FCGR2A-p.His166Arg SNP with susceptibility to KD in an extended single- and multi-marker TDT analysis, which corrected for ethnic differences (parents of the affected child were used as internal controls). This overall association reinforces the findings in our initial GWAS in KD (33) and the previous meta-analyses (42–44). The SNP results in an arginine (R) or histidine (H) at amino position 131 in the mature protein after cleaving of signal peptides, with the histidine variant being a risk factor for KD. It is important to note that FcγRIIa-His131 can bind human IgG2, whereas FcγRIIa-Arg131 cannot (45, 46). IgG2 is the second most prevalent IgG in the IVIg preparations; the distribution of the different IgG subclasses in IVIg varies slightly per preparation, and an IVIg infusion contains approximately 54%–70% IgG1, 29%–45% IgG2, 1%–4% IgG3, and 0%–0.5% IgG4 (47).
It has been previously suggested that the genetic association may only account for susceptibility to KD in Asian patients but not in European patients (43). A more recent meta-analysis (42) did find a significant association of the SNP with KD susceptibility in sub-analyses including solely patients of European descent, although methodological issues of the study resulted in the inclusion of two studies with the same study population (27, 33), which may have resulted in bias. Nevertheless, this study did not include the data of the TDT analysis that we have previously performed for FCGR2A-p.His166Arg in European patients only (48). In this cohort, the FCGR2A-p.166His was also significantly associated with susceptibility to KD (Z = 2.94, p = 0.003) (19). Therefore, we postulate that the association of the FCGR2A- p.His166Arg SNP with KD susceptibility is general and holds true in different ethnicities.
Interestingly, neither the FCGR2C-ORF haplotype nor any of the other FCGR2/3 variants were associated with KD susceptibility in our TDT analyses. This haplotype results in the expression of the activating FcγRIIc, which is usually not expressed because of a stop codon in exon 3 (38). In our previous study (30), the FCGR2C-ORF haplotype was not significantly associated with KD susceptibility in a TDT analysis, but it was associated with KD susceptibility in a case–control analysis in patients with European ancestry and remained significant in a meta-analysis combining the associations of the TDT and case–control analyses. In the current TDT cohort, which largely overlapped with our previous study (30), the SNP was again not significantly associated with KD susceptibility in the TDT analysis, indicating that FCGR2A SNP rs1801274 is the major gene variant for the clear association with the disease.
None of the known functional variations at the FCGR2/3 locus were associated with IVIg resistance or CAA risk in KD in our case–control analyses, apart from the Polish cohort in which the FCGR3B-NA2 haplotype was significantly associated with CAA risk. Notably, this cohort also had the highest proportion of patients with IVIg resistance (35% vs. 13%–26%), possibly leading to an overestimation of the effect of the FCGR3B-NA2 haplotype. In contrast to our own findings, several previous studies have reported associations between various genetic variations and IVIg resistance (31, 34, 35, 40) or CAA risk (28, 31, 32, 36, 40, 41). However, these studies relied on limited study populations [n = 177–358 vs. n = 653 in our cohort (IVIg); n = 41–424 per cohort vs. n = 911 (CAA)], which may have impacted the statistical power and heterogeneity of these studies. On the basis of our combined meta-analysis, we show that a prognostic value of genetic variations at the FCGR2/3 locus in prediction models for IVIg resistance or CAA risk in KD patients of European descent is unlikely.
The lack of a direct link between polymorphisms in either the activating or inhibitory FcγRs indicates that other factors likely play a role. The mechanism of action of IVIg could be explained by additional interactions with multiple other components of the immune system (e.g., dendritic cells, natural killer cells, T cells, and neutralization of autoantibodies) (49). For instance, it has been suggested that IVIg contains antibodies capable of neutralizing cytokines, further strengthened by the beneficial effects of anti-inflammatory treatment with TNF antagonists (50) and IL-1 antagonists (51, 52). Furthermore, it is known that the glycosylation of antibodies significantly impacts their effector functions and immunogenicity (53). Specifically, the proportion of anti-inflammatory sialylated IgG antibodies (up to 15% of healthy serum IgG) (54)) is reduced in inflammatory diseases (55), including KD (56). It has been suggested that α-2,6-sialylated IgG may be the biologically active component for the immunomodulatory effects of IVIg (57, 58), possibly via the induction of IL-33 and the increase of the inhibitory FcγRIIb surface expression on macrophages, dendritic cells, and B cells following IVIg infusion (59, 60) although such mechanisms do not occur in all circumstances (61, 62). In the setting of KD, treatment response has previously been shown to only be associated with lower sialylation levels of endogenous IgG, but not therapeutic IVIg (56). In addition, previous studies have indicated distinctive metabolic profiles in IVIg-resistant KD (63, 64) and KD patients with CAAs (65–67), indicating that the mechanisms behind IVIg resistance and CAA formation are multifaceted and require further elucidation.
Although we captured all SNPs and CNVs that are known to date in our current study, future whole-genome sequencing techniques may reveal new functionally relevant variations at the FCGR2/3 locus, which could not be captured in the current study.
One of the limitations of our study was that our cohorts mainly included European patients. The underrepresentation of Asian patients, in particular, increases the risk of overlooking ethnicity-specific associations of genetic variations at the FCGR2/3 locus with clinical outcomes in KD. A second limitation was the low number of studies that could be included in our meta-analysis, partially explained by the rarity of Kawasaki disease, making it challenging to obtain cohorts powered for investigating IVIg resistance and CAA risk. Consequently, we suspect a certain degree of publication bias when no significant effects are identified in (underpowered) patient populations. However, a major strength of our study was our extensive and homogeneous study population, which included only complete KD cases, ensuring sufficient power to investigate the associations with IVIg resistance and CAA risk (see Materials and methods). Secondly, we investigated ethnic differences by primarily separating the ethnic groups in the case–control analysis and then performing a meta-analysis including the complete dataset, which indicated minimal heterogeneity (except for the FCGR3B-NA2 haplotype, I2 = 73%).
In conclusion, our current study does not support the hypothesis that variations within the FCGR2/3 locus predict resistance to IVIg and risk of CAA (17). Although it is impossible to fully exclude an association of genetic variations at the FCGR2/3 locus with clinical outcomes in KD, we determined that our patient population offers sufficient statistical power, and our combined meta-analysis indicates that such a correlation is unlikely and that genotyping FCGR2/3 variants in patients with acute KD to guide initial treatment is not useful.
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/SNP/snp_viewBatch.cgi?sbid=1063642.
The studies involving humans were approved by the Medical Ethical Board of the AMC with the reference number 2012_155 (no. NL41023.018.12) Human Research Protection Program of the University of California San Diego (IRB 170790) Bioethics Committee at the Children’s Memorial Health Institute Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
PU: Data curation, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. SN: Data curation, Formal analysis, Investigation, Project administration, Writing – original draft, Writing – review & editing. MT: Formal analysis, Methodology, Supervision, Validation, Writing – review & editing. JG: Data curation, Writing – review & editing. PB: Data curation, Project administration, Writing – review & editing. MK-D: Data curation, Project administration, Writing – review & editing. MO-N: Data curation, Project administration, Writing – review & editing. DvS: Data curation, Project administration, Writing – review & editing. CT: Data curation, Project administration, Writing – review & editing. DB: Data curation, Project administration, Writing – review & editing. CS: Data curation, Project administration, Writing – review & editing. JB: Data curation, Writing – review & editing. IK: Data curation, Project administration, Writing – review & editing. TK: Conceptualization, Data curation, Supervision, Writing – review & editing. SN: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a grant from the Landsteiner Foundation for Blood Transfusion Research (LSBR 2022) awarded to SQN. In addition, the Kawasaki study is funded by the Dutch Foundation Kind and Handicap and an anonymous donor. The collection of DNA samples from the US KD Genetics Consortium was funded in part by a grant to JB from the National Institutes of Health, National Heart Blood Lung Institute R01HL140898. DB is supported by a National Health and Medical Research Council (Australia) Investigator Grant APP1175744. Research at Murdoch Children’s Research Institute is supported by the Victorian Government’s Operational Infrastructure Support Program. The sponsors had no role in the study design, analysis, or decision for publication.
We thank all children and parents who participated in this study and gratefully acknowledge the Kawasaki Study Group of collaborating institutes for their dedication to this project. We thank the members of the US Kawasaki Disease Consortium: Jane W. Newburger, Anne H. Rowley, Marian E. Melish, and Adriana H. Tremoulet.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1323171/full#supplementary-material
KD, Kawasaki disease; CAA, coronary artery aneurysm; IVIg, intravenous immunoglobulin; FcγRs, Fc-gamma receptors; AHA, American Heart Association; SHARE, Single Hub and Access point for pediatric Rheumatology in Europe; SNPs, single-nucleotide polymorphisms; CNVs, copy number variations; MAFs, minor allele frequencies; LD, linkage disequilibrium; MLPA, multiplex ligation-dependent probe amplification; TDT, transmission disequilibrium test; FBAT, family-based association test.
1. Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Arerugi. (1967) 16:178–222.
2. Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. (2004) 114:1708–33. doi: 10.1542/peds.2004-2182
3. Newburger JW, Takahashi M, Burns JC, Beiser AS, Chung KJ, Duffy CE, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. (1986) 315:341–7. doi: 10.1056/NEJM198608073150601
4. Nadig PL, Joshi V, Pilania RK, Kumrah R, Kabeerdoss J, Sharma S, et al. Intravenous immunoglobulin in Kawasaki disease-evolution and pathogenic mechanisms. Diagnost (Basel). (2023) 13(14):2338. doi: 10.3390/diagnostics13142338
5. Lo MS, Newburger JW. Role of intravenous immunoglobulin in the treatment of Kawasaki disease. Int J Rheum Dis. (2018) 21:64–9. doi: 10.1111/1756-185X.13220
6. Bayry J, Ahmed EA, Toscano-Rivero D, Vonniessen N, Genest G, Cohen CG, et al. Intravenous immunoglobulin: mechanism of action in autoimmune and inflammatory conditions. J Allergy Clin Immunol Pract. (2023) 11:1688–97. doi: 10.1016/j.jaip.2023.04.002
7. Shock A, Humphreys D, Nimmerjahn F. Dissecting the mechanism of action of intravenous immunoglobulin in human autoimmune disease: Lessons from therapeutic modalities targeting Fcγ receptors. J Allergy Clin Immunol. (2020) 146:492–500. doi: 10.1016/j.jaci.2020.06.036
8. Tremoulet AH, Best BM, Song S, Wang S, Corinaldesi E, Eichenfield JR, et al. Resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatr. (2008) 153:117–21. doi: 10.1016/j.jpeds.2007.12.021
9. Burns JC, Capparelli EV, Brown JA, Newburger JW, Glode MP. Intravenous gamma-globulin treatment and retreatment in Kawasaki disease. US/Canadian Kawasaki Syndrome Study Group. Pediatr Infect Dis J. (1998) 17:1144–8. doi: 10.1097/00006454-199812000-00009
10. Newburger JW, Takahashi M, Beiser AS, Burns JC, Bastian J, Chung KJ. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N Engl J Med. (1991) 324:1633–9. doi: 10.1056/NEJM199106063242305
11. Kobayashi T, Inoue Y, Takeuchi K, Okada Y, Tamura K, Tomomasa T. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation. (2006) 113:2606–12. doi: 10.1161/CIRCULATIONAHA.105.592865
12. Kobayashi T, Saji T, Otani T, Takeuchi K, Nakamura T, Arakawa H. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. Lancet. (2012) 379:1613–20. doi: 10.1016/S0140-6736(11)61930-2
13. Lin M-T, Chang C-H, Sun L-C, Liu H-M, Chang H-W, Chen C-A. Risk factors and derived formosa score for intravenous immunoglobulin unresponsiveness in Taiwanese children with Kawasaki disease. J Formosan Med Assoc. (2016) 115:350–5. doi: 10.1016/j.jfma.2015.03.012
14. Egami K, Muta H, Ishii M, Suda K, Sugahara Y, Iemura M, et al. Prediction of resistance to intravenous immunoglobulin treatment in patients with Kawasaki disease. J Pediatr. (2006) 149:237–40. doi: 10.1016/j.jpeds.2006.03.050
15. Sleeper LA, Minich LL, McCrindle BM, Li JS, Mason W, Colan SD, et al. Evaluation of Kawasaki disease risk-scoring systems for intravenous immunoglobulin resistance. J Pediatr. (2011) 158:831–835.e3. doi: 10.1016/j.jpeds.2010.10.031
16. Fu P, Du Z, Pan Y. Novel predictors of intravenous immunoglobulin resistance in chinese children with Kawasaki disease. Pediatr Infect Dis J. (2013) 32:e319–23. doi: 10.1097/INF.0b013e31828e887f
17. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: A scientific statement for health professionals from the American heart association. Circulation. (2017) 135(17):e927–e999. doi: 10.1161/CIR.0000000000000484
18. de Graeff N, Groot N, Ozen S, Eleftheriou D, Avcin T, Bader-Meunier B, et al. European consensus-based recommendations for the diagnosis and treatment of Kawasaki disease - the SHARE initiative. Rheumatol (Oxford). (2019) 58:672–82. doi: 10.1093/rheumatology/key344
19. Feldstein LR, Rose EB, Horwitz SM, Collins JP, Newhams MM, Son MBF, et al. Multisystem inflammatory syndrome in U.S. children and adolescents. New Engl J Med. (2020) 383:334–46. doi: 10.1056/NEJMoa2021680
20. Whittaker E, Bamford A, Kenny J, Kaforou M, Jones CE, Shah P, et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA. (2020) 324:259. doi: 10.1001/jama.2020.10369
21. Piram M. Epidemiology of Kawasaki disease in Europe. Front Pediatr. (2021) 9:673554. doi: 10.3389/fped.2021.673554
22. Harnden A, Mayon-White R, Perera R, Yeates D, Goldacre M, Burgner D. Kawasaki disease in England: ethnicity, deprivation, and respiratory pathogens. Pediatr Infect Dis J. (2009) 28:21–4. doi: 10.1097/INF.0b013e3181812ca4
23. Gardner-Medwin JMM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. (2002) 360:1197–202. doi: 10.1016/S0140-6736(02)11279-7
24. Fujita Y, Nakamura Y, Sakata K, Hara N, Kobayashi M, Nagai M, et al. Kawasaki disease in families. Pediatrics. (1989) 84:666–9. doi: 10.1542/peds.84.4.666.
25. Uehara R, Yashiro M, Nakamura Y, Yanagawa H. Kawasaki disease in parents and children. Acta Paediatr. (2003) 92:694–7. doi: 10.1080/08035320310002768
26. Kumrah R, Vignesh P, Rawat A, Singh S. Immunogenetics of Kawasaki disease. Clin Rev Allergy Immunol. (2020) 59:122–39. doi: 10.1007/s12016-020-08783-9
27. Nagelkerke SQ, Schmidt DE, de Haas M, Kuijpers TW. Genetic variation in low-to-medium-affinity Fcγ Receptors: functional consequences, disease associations, and opportunities for personalized medicine. Front Immunol. (2019) 10:2237. doi: 10.3389/fimmu.2019.02237
28. Biezeveld M, Geissler J, Merkus M, Kuipers IM, Ottenkamp J, Kuijpers T. The involvement of Fc gamma receptor gene polymorphisms in Kawasaki disease. Clin Exp Immunol. (2007) 147:106–11. doi: 10.1111/j.1365-2249.2006.03266.x
29. Tsang-A-Sjoe MWP, Nagelkerke SQ, Bultink IEM, Geissler J, Tanck MWT, Tacke CE, et al. Fc-gamma receptor polymorphisms differentially influence susceptibility to systemic lupus erythematosus and lupus nephritis. Rheumatol (United Kingdom). (2016) 55:939–48. doi: 10.1093/rheumatology/kev433
30. Nagelkerke SQ, Tacke CE, Breunis WB, Tanck MWT, Geissler J, Png E, et al. Extensive ethnic variation and linkage disequilibrium at the FCGR2/3 locus: Different genetic associations revealed in Kawasaki disease. Front Immunol. (2019) 10:185. doi: 10.3389/fimmu.2019.00185
31. Shrestha S, Wiener H, Shendre A, Kaslow RA, Wu J, Olson A, et al. Role of activating FcR gene polymorphisms in Kawasaki disease susceptibility and intravenous immunoglobulin response. Circ Cardiovasc Genet. (2012) 5(3):309–16. doi: 10.1161/CIRCGENETICS.111.962464/-/DC1
32. Taniuchi S, Masuda M, Teraguchi M, Ikemoto Y, Komiyama Y, Takahashi H, et al. Polymorphism of Fcγ RIIa may affect the efficacy of γ-globulin therapy in Kawasaki disease. J Clin Immunol. (2005) 25:309–13. doi: 10.1007/s10875-005-4697-7
33. Khor CC, Davila S, Breunis WB, Lee Y-C, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. (2011) 43:1241–6. doi: 10.1038/ng.981
34. Shrestha S, Wiener HW, Olson AK, Edberg JC, Bowles NE, Patel H, et al. Functional FCGR2B gene variants influence intravenous immunoglobulin response in patients with Kawasaki disease. J Allergy Clin Immunol. (2011) 128:677–680.e1. doi: 10.1016/j.jaci.2011.04.027
35. Makowsky R, Wiener HW, Ptacek TS, Silva M, Shendre A, Edberg JC, et al. FcγR gene copy number in Kawasaki disease and intravenous immunoglobulin treatment response. Pharmacogenet Genomics. (2013) 23:455–62. doi: 10.1097/FPC.0b013e328363686e
36. Duan J, Lou J, Zhang Q, Ke J, Qi Y, Shen N, et al. A genetic variant rs1801274 in FCGR2A as a potential risk marker for Kawasaki disease: A case-control study and meta-analysis. PloS One. (2014) 9:e103329. doi: 10.1371/journal.pone.0103329
37. Gauderman WJ. Sample size requirements for association studies of gene-gene interaction. Am J Epidemiol. (2002) 155:478–84. doi: 10.1093/aje/155.5.478
38. Breunis WB, van Mirre E, Bruin M, Geissler J, de Boer M, Peters M, et al. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood. (2008) 111:1029–38. doi: 10.1182/blood-2007-03-079913
39. Flesch BK, Reil A. Molecular genetics of the human neutrophil antigens. Transfus Med Hemother. (2018) 45:300–9. doi: 10.1159/000491031
40. Yan Y, Ma Y, Liu Y, Hu H, Ying Y, Zhang S, et al. Combined analysis of genome-wide-linked susceptibility loci to Kawasaki disease in Han Chinese. Hum Genet. (2013) 132:669–80. doi: 10.1007/s00439-013-1279-2
41. Chatzikyriakidou A, Aidinidou L, Giannopoulos A, Papadopoulou-Legbelou K, Kalinderi K, Fidani L. Absence of association of FCGR2A gene polymorphism rs1801274 with Kawasaki disease in Greek patients. Cardiol Young. (2015) 25:681–3. doi: 10.1017/S1047951114000626
42. Wang Z, Geng P-L. CD32a polymorphism rs1801274 affects the risk of Kawasaki disease. Artif Cells Nanomed Biotechnol. (2020) 48:620–6. doi: 10.1080/21691401.2019.1645156
43. Lin L, Wang SY, Yang SB, Xiao FC. Association between the FCGR2A gene H131R polymorphism and risk of Kawasaki disease: a meta-analysis. Genet Mol Res. (2015) 14:6256–64. doi: 10.4238/2015.June.9.12
44. Ferdosian F, Dastgheib SA, Hosseini-Jangjou SH, Nafei Z, Lookzadeh MH, Noorishadkam M, et al. Association of TNF- α rs1800629, CASP3 rs72689236 and FCGR2A rs1801274 Polymorphisms with Susceptibility to Kawasaki Disease: A Comprehensive Meta-Analysis. Fetal Pediatr Pathol. (2021) 40:320–36. doi: 10.1080/15513815.2019.1707917
45. Warmerdam PA, van de Winkel JG, Vlug A, Westerdaal NA, Capel PJ. A single amino acid in the second Ig-like domain of the human Fc gamma receptor II is critical for human IgG2 binding. J Immunol. (1991) 147:1338–43. doi: 10.4049/jimmunol.147.4.1338.
46. Maxwell KF, Powell MS, Hulett MD, Barton PA, McKenzie IF, Garrett TP, et al. Crystal structure of the human leukocyte Fc receptor, Fc gammaRIIa. Nat Struct Biol. (1999) 6:437–42. doi: 10.1038/8241
47. Burckhardt JJ, Gardi A, Oxelius VA, Preud'homme JL, Scherz R, Skvaril F, et al. Immunoglobulin G subclass distribution in three human intravenous immunoglobulin preparations. Vox Sang. (1989) 57:10–4. doi: 10.1111/j.1423-0410.1989.tb04976.x
48. Nagelkerke SQ. A field guide to human Fc-gamma receptors: Genetics, cellular expression and interaction with immunoglobulins (2017). Amsterdam: AMC-UvA. Available online at: https://hdl.handle.net/11245.1/d6aaf779-527d-4352-8378-96e4683ba49c (Accessed Jul. 27, 2023).
49. Burns JC, Franco A. The immunomodulatory effects of intravenous immunoglobulin therapy in Kawasaki disease. Expert Rev Clin Immunol. (2015) 11:819–25. doi: 10.1586/1744666X.2015.1044980
50. Tremoulet AH, Jain S, Jaggi P, Jimenez-Fernandez S, Pancheri JM, Sun X, et al. Infliximab for intensification of primary therapy for Kawasaki disease: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet. (2014) 383:1731–8. doi: 10.1016/S0140-6736(13)62298-9
51. Yang J, Jain S, Capparelli EV, Best BM, Son MB, Baker A, et al. Anakinra treatment in patients with acute Kawasaki disease with coronary artery aneurysms: A phase I/IIa trial. J Pediatr. (2022) 243:173–180.e8. doi: 10.1016/j.jpeds.2021.12.035
52. Cohen S, Tacke CE, Straver B, Meijer N, Kuipers IM, Kuijpers TW. A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygenation. Ann Rheum Dis. (2012) 71:2059–61. doi: 10.1136/annrheumdis-2012-201658
53. Vattepu R, Sneed SL, Anthony RM. Sialylation as an important regulator of antibody function. Front Immunol. (2022) 13:818736. doi: 10.3389/fimmu.2022.818736
54. Huffman JE, Pučić-Baković M, Klarić L, Hennig R, Selman MHJ, Vučković F, et al. Comparative performance of four methods for high-throughput glycosylation analysis of immunoglobulin G in genetic and epidemiological research. Mol Cell Proteomics. (2014) 13:1598–610. doi: 10.1074/mcp.M113.037465
55. Gudelj I, Lauc G, Pezer M. Immunoglobulin G glycosylation in aging and diseases. Cell Immunol. (2018) 333:65–79. doi: 10.1016/j.cellimm.2018.07.009
56. Ogata S, Shimizu C, Franco A, Touma R, Kanegaye JT, Choudhury BP, et al. Treatment response in Kawasaki disease is associated with sialylation levels of endogenous but not therapeutic intravenous immunoglobulin G. PloS One. (2013) 8:e81448. doi: 10.1371/journal.pone.0081448
57. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. (2006) 313:670–3. doi: 10.1126/science.1129594
58. Washburn N, Schwab I, Ortiz D, Bhatnagar N, Lansing JC, Medeiros A, et al. Controlled tetra-Fc sialylation of IVIg results in a drug candidate with consistent enhanced anti-inflammatory activity. Proc Natl Acad Sci U.S.A. (2015) 112:E1297–306. doi: 10.1073/pnas.1422481112
59. Fiebiger BM, Maamary J, Pincetic A, Ravetch JV. Protection in antibody- and T cell-mediated autoimmune diseases by antiinflammatory IgG Fcs requires type II FcRs. Proc Natl Acad Sci U.S.A. (2015) 112:E2385–94. doi: 10.1073/pnas.1505292112
60. Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. (2001) 291:484–6. doi: 10.1126/science.291.5503.484
61. Campbell IK, Miescher S, Branch DR, Mott PJ, Lazarus AH, Han D, et al. Therapeutic effect of IVIG on inflammatory arthritis in mice is dependent on the Fc portion and independent of sialylation or basophils. J Immunol. (2014) 192:5031–8. doi: 10.4049/jimmunol.1301611
62. Nagelkerke SQ, Dekkers G, Kustiawan I, van de Bovenkamp FS, Geissler J, Plomp R, et al. Inhibition of FcγR-mediated phagocytosis by IVIg is independent of IgG-Fc sialylation and FcγRIIb in human macrophages. Blood. (2014) 124:3709–18. doi: 10.1182/blood-2014-05-576835
63. Chen Z, Sai S, Nagumo K, Wu Y, Chiba H, Hui S-P. Distinctive serum lipidomic profile of IVIG-resistant Kawasaki disease children before and after treatment. PloS One. (2023) 18:e0283710. doi: 10.1371/journal.pone.0283710
64. Rambabu N, Mathew MJ, Kaveri SV, Bayry J. Boolean analysis of the transcriptomic data to identify novel biomarkers of IVIG response. Autoimmun Rev. (2021) 20:102850. doi: 10.1016/j.autrev.2021.102850
65. Nakashima Y, Sakai Y, Mizuno Y, Furuno K, Hirono K, Takatsuki S, et al. Lipidomics links oxidized phosphatidylcholines and coronary arteritis in Kawasaki disease. Cardiovasc Res. (2021) 117:96–108. doi: 10.1093/cvr/cvz305
66. Qian G, Xu L, Qin J, Huang H, Zhu L, Tang Y, et al. Leukocyte proteomics coupled with serum metabolomics identifies novel biomarkers and abnormal amino acid metabolism in Kawasaki disease. J Proteomics. (2021) 239:104183. doi: 10.1016/j.jprot.2021.104183
Keywords: Kawasaki disease, IVIg, CAA, FCGR2, FCGR3, genetics, FCGR2Ap.His166Arg
Citation: Uittenbogaard P, Netea SA, Tanck MWT, Geissler J, Buda P, Kowalczyk-Domagała M, Okarska-Napierała M, van Stijn D, Tacke CE, US Kawasaki Disease Genetics Consortium, Burgner DP, Shimizu C, Burns JC, Kuipers IM, Kuijpers TW and Nagelkerke SQ (2024) FCGR2/3 polymorphisms are associated with susceptibility to Kawasaki disease but do not predict intravenous immunoglobulin resistance and coronary artery aneurysms. Front. Immunol. 15:1323171. doi: 10.3389/fimmu.2024.1323171
Received: 17 October 2023; Accepted: 08 February 2024;
Published: 18 September 2024.
Edited by:
Durga Prasanna Misra, Sanjay Gandhi Post Graduate Institute of Medical Sciences (SGPGI), IndiaReviewed by:
Jagadeesh Bayry, Indian Institute of Technology Palakkad, IndiaCopyright © 2024 Uittenbogaard, Netea, Tanck, Geissler, Buda, Kowalczyk-Domagała, Okarska-Napierała, van Stijn, Tacke and US Kawasaki Disease Genetics Consortium, Burgner, Shimizu, Burns, Kuipers, Kuijpers and Nagelkerke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sietse Q. Nagelkerke, cy5xLm5hZ2Vsa2Vya2VAYW1zdGVyZGFtdW1jLm5s
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.