 Rulan Wang
Rulan Wang Zhenkun Liu1,2†
Zhenkun Liu1,2† Jiewei Liu
Jiewei Liu Qinghua Zhou
Qinghua Zhou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 January 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1320244
This article is part of the Research Topic Treatment Strategies for Lung Cancer: A Progress Update and Future Perspectives View all 6 articles
Lung cancer poses a global threat to human health, while common cancer treatments (chemotherapy and targeted therapies) have limited efficacy. Immunotherapy offers hope of sustained remission for many patients with lung cancer, but a significant proportion of patients fail to respond to treatment owing to immune resistance. There is extensive evidence to suggest the immunosuppressive microenvironment as the cause of this treatment failure. Numerous studies have suggested that the adenosine (ADO) pathway plays an important role in the formation of an immunosuppressive microenvironment and may be a key factor in the development of immune resistance in EGFR-mutant cell lung cancer. Inhibition of this pathway may therefore be a potential target to achieve effective reversal of ADO pathway-mediated immune resistance. Recently, an increasing number of clinical trials have begun to address the broad prospects of using the ADO pathway as an immunotherapeutic strategy. However, few researchers have summarized the theoretical basis and clinical rationale of the ADO pathway and immune checkpoint dual blockade in a systematic and detailed manner, particularly in lung cancer. As such, a timely review of the potential value of the ADO pathway in combination with immunotherapy strategies for lung cancer is warranted. This comprehensive review first describes the role of ADO in the formation of a lung tumor-induced immunosuppressive microenvironment, discusses the key mechanisms of ADO inhibitors in reversing lung immunosuppression, and highlights recent evidence from preclinical and clinical studies of ADO inhibitors combined with immune checkpoint blockers to improve the lung cancer immunosuppressive microenvironment.
Lung cancer is a malignant tumor that seriously threatens human life and health worldwide, with a high incidence and mortality rate, making it one of the most common malignant tumors in recent years (1). Chemotherapy and targeted therapies have limited efficacy in lung cancer; even after effective chemotherapy, the 5-year survival rate of patients with advanced stages is only approximately 10% (2, 3). Moreover, the emergence of resistance to targeted therapy inevitably occurs within a short period (4). In recent years, immunotherapy has led to breakthroughs in the field of lung cancer (5). The combination of platinum-based chemotherapy and immunotherapy has resulted in an improved 5-year overall survival rate in patients with advanced non-small cell lung cancer (NSCLC), reaching 19.3% for non-squamous NSCLC (2) and 18.4% for squamous NSCLC (3). However, bottlenecks have inevitably been encountered. Due to the development of immune resistance, a significant proportion of patients show almost no benefit from immunotherapy (6). Further, there is some data to suggest that the objective efficacy rate for patients treated with PD-1/PD-L1 inhibitors as a monotherapy is only approximately 12.5%, with poor efficacy in the remaining 87.5% of the population (7). As such, expanding the beneficiary population of immunotherapy and enhancing its therapeutic effect have become important topics of research in the field of immunotherapy.
Studies have shown that the presence of a tumor-induced immunosuppressive microenvironment is a critical bottleneck limiting the development of immunotherapy (8, 9); this environment is related to the number and status of tumor-infiltrating lymphocytes (TILs) in the tumor microenvironment (TME) (10). As such, the effective reversal of the immunosuppressive microenvironment and enhanced efficacy of immunotherapy remain pressing issues (11). The adenosine (ADO) pathway is known to be critical for the formation of an immunosuppressive microenvironment. As such, inhibiting the activity of this pathway may be a potential mechanism to effectively reverse ADO pathway-mediated immune resistance (12). In addition, ADO inhibitors in combination with immune checkpoint blockers may be effective as a potential new oncological treatment option to expand the beneficial population for immunotherapy. Several recent studies have addressed the broad prospects of the ADO pathway as an immunotherapeutic strategy (13). However, few researchers have systematically summarized the theoretical basis and clinical rationale for ADO pathway and immune checkpoint dual blockade in lung cancer.
Tumor cells, as well as other cells in the TME, actively secrete ATP in response to cell death, hypoxia, nutrient depletion, and chronic inflammation. Hypoxia and transforming growth factor-β promote solid tumors and immunosuppressive cells in the TME to express high levels of exonucleosidases (14). ATP is involved in numerous metabolic processes through various intracellular and extracellular pathways, ultimately leading to its conversion into ADO (Figure 1). In brief, the exonucleosidase CD39 hydrolyzes ATP to generate ADP and AMP, which are further converted to ADO by the exonucleosidase CD73 (13). The adenosine diphosphate ribose/cyclic adenosine diphosphate ribose generated by nicotinamide-adenine dinucleotide (catalyzed by exonuclease CD38) undergoes further metabolism by extracellular nucleotide pyrophosphatase/phosphodiesterase-1 to AMP, which serves as a substrate for ADO generated by exonuclease CD73. Consequently, CD38 plays a pivotal role in establishing an immunosuppressive tumor microenvironment in solid tumors. Notably, enzymes such as adenosine deaminase and adenosine kinase regulate the final metabolic conversion of ADO and the activation level of ADO receptors; however, their role in tumor development is unclear, making this a therapeutic target worth exploring in the ADO pathway (13).
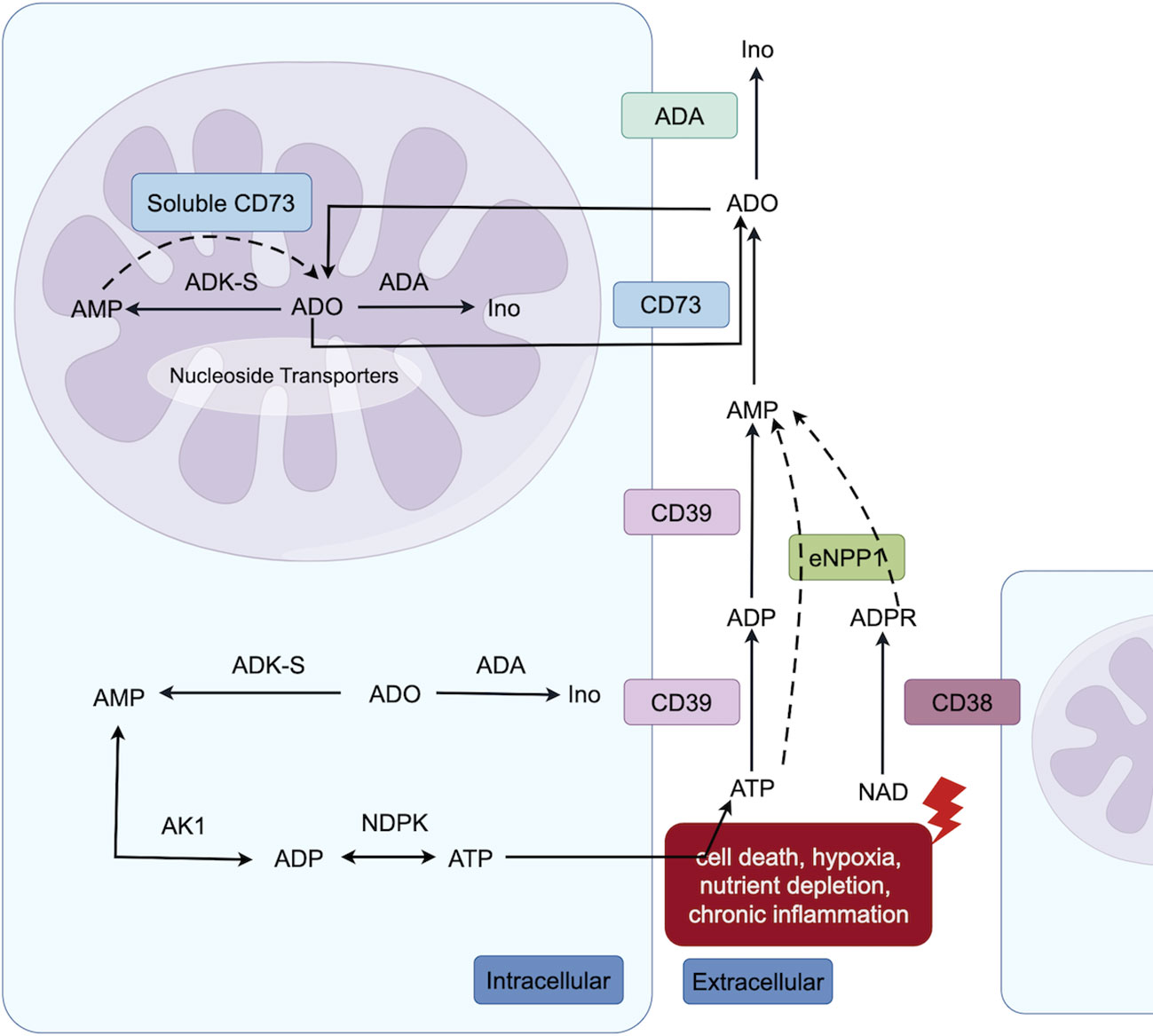
Figure 1 Intracellular (both cytoplasmic and nuclear) and extracellular adenosine metabolic pathways. Extracellular ATP undergoes conversion to ADO via the classical catabolic pathway, catalyzed by CD39 and CD73 enzymes. Additionally, nicotinamide-adenine dinucleotide is degraded by CD38 to generate adenosine diphosphate ribose, which can be further metabolized into AMP through the action of Extracellular nucleotide pyrophosphatase/phosphodiesterase-1, ultimately leading to ADO formation. Within the nucleus, AMP can also be converted into ADO by soluble CD73. Furthermore, both in nuclear and cytoplasmic, adenosine kinase can facilitate the synthesis of AMP from ADO, thereby participating in ATP metabolism (The figure is made by Figdraw).
Currently, the mechanisms of immunotherapy resistance include a lack of neoantigens or abnormal antigen presentation, low tumor load, low PD-L1 expression, T-cell infiltration disorder or T-cell exhaustion, presence of immunosuppressive cells or factors, and abnormal signaling pathways (15). This study therefore focused on elucidating the underlying mechanisms of resistance to immune checkpoint inhibitors associated with the ADO signaling pathway (Figure 2) as well as exploring the potential of ADO pathway inhibitors in overcoming immune resistance in lung cancer.
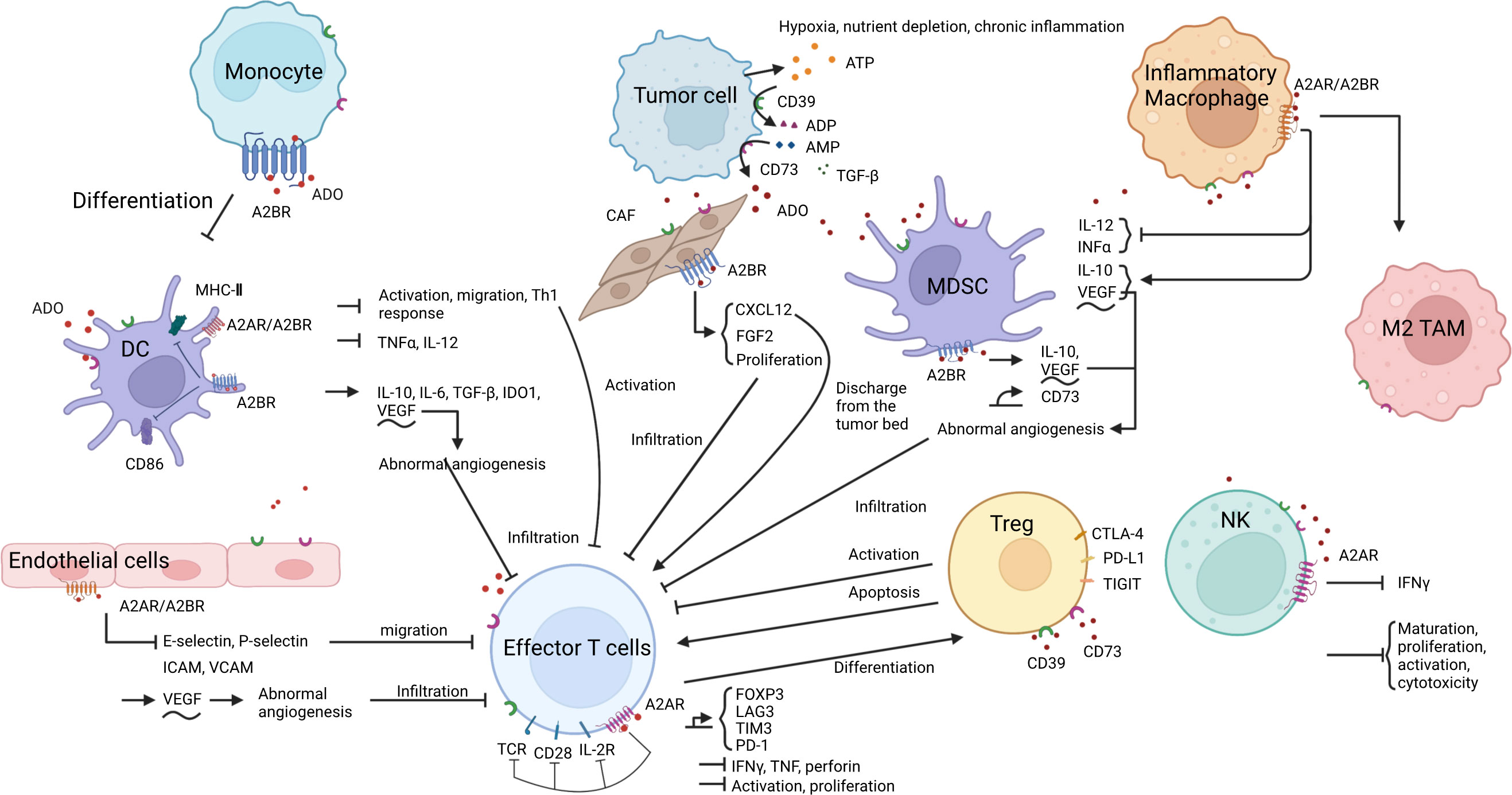
Figure 2 ADO and the formation of an immunosuppressive microenvironment. 1. ADO metabolism: Under specific conditions, ATP in the TME is converted into ADO by CD39 and CD73. 2. ADO-induced immunosuppression: ADO binds to A2AR or A2BR on the surface of various immune cells, endothelial cells, and fibroblasts, thereby eliciting diverse biological effects. These effects encompass inhibition of antigen presentation, impediment of immune effector cell activation, infiltration and function as well as NK cell activity, promotion of immunosuppressive cell proliferation and functional expression, induction of aberrant angiogenesis, ultimately culminating in the establishment of an inhibitory immune microenvironment.
The ADO produced in the TME binds to A2 receptors (A2R) on monocytes (16), dendritic cells (DCs) (17), myeloid-derived suppressor cells (MDSCs) (18), regulatory T cells (Tregs) (19, 20), and macrophages (21, 22), thereby modulating their differentiation and function (16, 18–20, 23–26), ultimately leading to the induction of an immunosuppressive microenvironment conducive to lung cancer development (27, 28). Various inhibitors targeting the ADO pathway can improve the immunosuppressive microenvironment by inhibiting the process by which ADO acts, or by directly inhibiting ADO production (14). In the RAS mutant NSCLC mouse model, the novel CD73 antibody Ab001/Ab002 and the humanized antibody Hu001/Hu002 were found to effectively regulate the TME, reduce the infiltration level of M2 tumor-associated macrophages and MDSCs, induce the accumulation of mature DCs, promote effector T cells (Teff) proliferation and interferon γ (IFN-γ) secretion, enhance T-cell-mediated cytotoxicity, and ultimately inhibit tumor growth in mice (29). The proportion of Tregs in a co-culture system of lung adenocarcinoma cells and peripheral blood mononuclear cells was reduced following CD73 knockdown (30). The addition of AMP to the T cell in vitro culture system inhibited T cell proliferation and division, and this inhibition was alleviated by the addition of the anti-CD73 antibody oleclumab (30).
The ADO receptor signaling pathway inhibits the activation of antigen-presenting cells (APCs) to hinder antigen presentation, thereby limiting the opportunity for T cell activation by antigens (16, 25, 31–33). ADO further impedes the migration of DCs (16), thereby preventing the encounter between antigen-carrying DCs and naïve T cells, and consequently regulating the immune response (34). Previous studies have demonstrated that myeloid ADO receptor A2 (A2AR) or ADO receptor B2 (A2BR) deficient mice exhibit elevated expression levels of costimulatory molecules CD86 and major histocompatibility complex II (MHC II, markers of the activation and maturation of antigen-presenting cells) on APCs, as well as increased CD8+ T cell activation and proliferation, higher levels of IFN-γ secretion on APCs, and slower tumor growth (25, 35). Additionally, A2R antagonists can reverse the impaired CD86 and MHC II expression in APCs (25, 32).
ADO induces T cell infiltration disorder mainly by impairing the antigen presentation process (as described in the previous subsection) (10, 36) and inhibiting the secretion of various adhesion molecules such as e-selectin, p-selectin, and intercellular adhesion molecules (ICAMs) of endothelial cells to block T-cell migration (37) (38). Additionally, ADO can also promote abnormal angiogenesis by inducing vascular endothelial growth factor secretion, resulting in abnormal tumor vascular structure and function that hinder immune cell infiltration (14) (39). Furthermore, ADO in the TME binds to TILs (40, 41) and APCs (35), blocking effector T-cell activation, proliferation, and secretion of various cytokines such as IFNγ, tumor necrosis factor (TNF), and perforin (13, 38). ADO inhibitors direct normal T cell activation by improving antigen presentation and promoting the formation of a normal circulatory system, allowing activated T cells to enter the tumor bed with the assistance of the normal circulatory system (41). There is also evidence to indicate that Tregs in the TME inhibit the transendothelial migratory capacity of Teff by inducing high expression of CD39, promoting ADO production, and reducing monocyte-induced expression of the adhesion molecule ICAM-1 on endothelial cells (42). CD39 or ADO inhibitors effectively restore the migratory capacity of Teffs (42). ADO has also been shown to inhibit the chemotactic properties of CD3+ and CD8+ T cells by decreasing KCa3.1 channels. A2AR blockers or KCa3.1 channel activators can block this phenomenon and promote the migration and infiltration of T cells (43).
ADO has further been shown to block IFN-γ-induced STAT1 phosphorylation, inhibit the inflammatory response induced by macrophage activation (44), and eliminate the increased production of IL-12, IFN-γ, and TNF-α mediated by IL-18 (45). Treatment of activated CD4+ T cells with ADO resulted in a significant decrease in A2AR-mediated IFN-γ release (46). In contrast, the production of IFN-γ, TNF-α, and granzyme B was found to be increased in CD73-deficient cells, indicating an augmented cytotoxic potential (47). CD4+ T lymphocytes co-incubated with CD73 monoclonal antibody have increased IFN-γ production (48). Mutations in key genes in the IFN-γ signaling pathway result in loss of PD-L1-responsive expression, making such patients less likely to respond to PD-1 blockade therapy (49). It has been shown that anti-CD73 monoclonal antibodies can enhance the antitumor effects of PD-1 antibodies by promoting CD8+ T-cell infiltration and IFN-γ secretion (50). Caffeine is an A2AR inhibitor (50), and prior research has shown that the combination of caffeine and anti-PD-1 monoclonal antibodies can significantly increase the levels of TNF-α and IFN-γ in tumors, thus exerting stronger antitumor activity (51).
Cases of NSCLC with epidermal growth factor receptor (EGFR) mutations are known to respond poorly to immune checkpoint inhibitor therapy (52). Although it has been shown that PD-L1, tumor mutation burden (TMB), and CD8+ TILs are all significantly higher in patients with resistance to EGFR-tyrosine kinase inhibitor (TKI) therapy (53, 54), this elevation does not seem to translate into a benefit in immune checkpoint blockade (ICB) treatment (55–58). The poor outcomes of these patients appear to be linked to Treg cell-mediated immunosuppression (59, 60). Le et al. (12) previously analyzed NSCLC samples from multiple databases at multiple levels, including immune-related resistance patterns and genomic and gene mapping, to explore the mechanisms underlying immune resistance in EGFR-mutated (mEGFR) NSCLC. These results suggest that the immunologically inert phenotype (low PD-L1 expression, low TMB, and low CD8+ T cells) of mEGFR NSCLC may be attributed to the upregulation of the NT5E (encoding the exonucleosidase CD73) and ADO A1 receptor genes in the ADO pathway. More notably, NT5E was shown to be highly expressed in tumor samples relative to normal lung epithelial cells (59), as well as in TKI-resistant tumor samples compared to untreated tumor samples (12). Griesing et al. (61) and Han et al. (62) previously confirmed the higher expression of CD73 in mEGFR NSCLC using a similar approach. Further, Jin et al. found that CD73 is commonly upregulated in NSCLC. Interestingly, several studies have found that CD73 expression in NSCLC positively correlates with PD-L1 expression (63). In addition, CD73 expression was found to be reduced in EGFR TKI-sensitive cell lines after EGFR TKI treatment (61, 62, 64). However, in EGFR TKI-resistant cell lines, CD73 expression increased and was no longer affected by EGFR TKI treatment (61). This phenomenon has also been observed in clinical specimens (53). In addition, in vitro experiments by Le et al. revealed that CD73 is highly expressed on the surface of lung adenocarcinoma cell lines carrying EGFR mutations (12). In a co-culture system of lung adenocarcinoma cells and peripheral blood mononuclear cells, the combination of anti-CD73 and anti-PD-1 antibodies was found to enhance the IFN-γ-mediated antitumor effects of T cells (30). In addition, the antitumor activity of an anti-CD73 antibody combined with an anti-PD-1 antibody has been validated in transgenic mice carrying mEGFR (12). Specifically, another study showed that the combination of the anti-CD73 antibody olecumab and the anti-PD-L1 antibody durvalumab significantly reduced tumor volume in an NSG mouse model carrying mEGFR NSCLC cells and showed that neither olecumab nor durvalumab alone significantly induced antitumor effects. In addition, the combination of oleclumab and durvalumab was found to significantly increase the proportion and number of infiltrating CD8+ T cells in tumors, while also increasing the levels of IFN-γ and TNF-α (64). A2R has also been proposed as a target of interest. Using a homozygous lung cancer mouse model, Chen et al. demonstrated that A2BR-deficient mice exhibited slower tumor growth and a higher frequency of total tumor-infiltrating CD8+ T cells and tumor antigen-specific CD8+ T cells than wild-type mice. In addition, a higher proportion of IFN-γ-secreting CD8+ T cells was identified in the tumors of A2BR-deficient mice (35).
There are currently two main classes of ADO-related drugs specifically developed for the treatment of tumors (65) (Table 1). The first attenuates the effect of ADO on the immune microenvironment by decreasing its concentration in the TME. The mechanisms of action include the inhibition of ADO synthesis and promotion of ADO metabolism. These are predominantly enzyme inhibitors of the ADO production pathway, including CD39, CD73, and CD38. The second category ameliorates immunosuppression by inhibiting ADO function in the TME. These primarily include ADO receptor inhibitors, such as somatic A2AR and A2BR inhibitors. The ADO-related drugs that have entered the clinical study phase are shown in Table 1, in which * indicates drugs that have been studied for lung cancer. As shown in Table 1, CD39 inhibitors have not been studied independent of lung cancer. In particular, oleculumabs have recently made breakthroughs in the field of lung cancer.
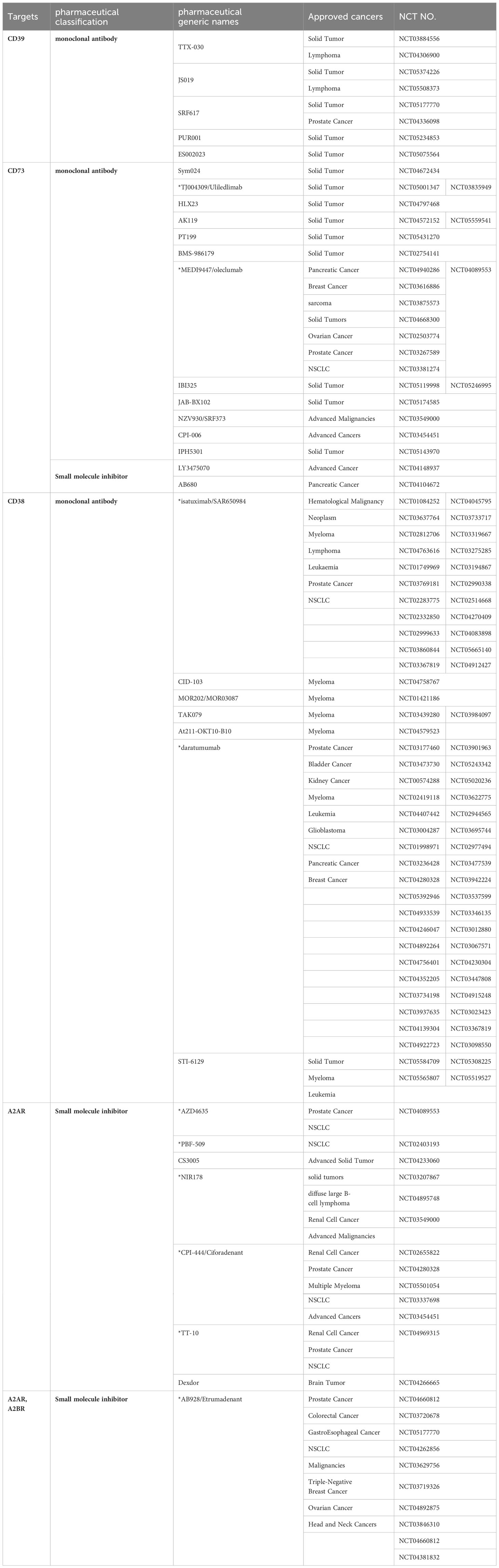
Table 1 ADO-related drugs approved for oncology-related clinical trials.
The Hudson study (NCT03334617) was the first to explore the efficacy of the CD73 monoclonal antibody oleclumab in combination with durvalumab in patients with metastatic NSCLC following failure of PD-1/PD-L1 maintenance therapy (66). The results of this study showed a median progression-free survival (mPFS) of 2.63 months and an overall survival (OS) of 12.08 months in patients with acquired resistance to immunotherapy. Although this data are relatively less favorable compared to those of albumin paclitaxel and docetaxel second-line chemotherapy (mPFS: 4.2 and 3.4 months and mOS: 16.2 and 13.6 months, respectively) (67), this study nevertheless indicates a new direction for patients who are intolerant to second-line chemotherapy. Interim data from the COAST study (68), published in April 2022 are also encouraging. For the first time, this study showed that consolidation therapy with a PD-L1 inhibitor (durvalumab) in combination with a CD73 inhibitor (oleclumab) further improved the clinical outcomes in patients with unresectable stage III NSCLC after radiotherapy. Compared to patients maintained on durvalumab alone, the durvalumab combined with oleclumab group showed a significantly increased objective remission rate (ORR) (17.9% vs. 30.0%), disease control rate (DCR) (16-week DCR rate of 58.2% vs. 81.7%, respectively), and prolonged mPFS (6.3 months vs. not achieved). However, OS data have not yet been published. The incidence of emergency adverse events (EAEs) was similar between the combination and single-agent arms. In the combination group, grade 3 EAEs included coughing (1.7%) and dyspnea (1.7%) (68). The COAST study demonstrated, for the first time, that the clinical efficacy of immunotherapy could be improved by immunomodulation. The increased ORR and prolonged mPFS in the combination arm further provide data to support the further development of the world’s first CD73-related phase III clinical study, PACIFIC-9 (NCT05221840). The phase 2 NeoCOAST study further explored the efficacy and safety of durvalumab in combination with olecumab in the neoadjuvant treatment of NSCLC (69). These findings are highly promising (70, 71). The combination therapy group had significantly higher major pathological remission rates (MPR) (11% vs. 19%) and pathological complete remission rates (PCR) (3.7% vs. 9.5%) than the durvalumab monotherapy group. Regarding safety, the incidence of grade 3 or higher treatment-related adverse events (TRAEs) was 0% and 4.8%, respectively. No AE-related deaths occurred in any of the patients. Based on the results of this study, NeoCOAST-2 (NCT05061550) was applied to assess the safety and efficacy of neoadjuvant durvalumab treatment in combination with chemotherapy, olecumab, and adjuvant therapy in patients with resectable early stage NSCLC. Recruitment is currently underway in Japan. Uliledumab is another CD73 inhibitor with efficacy in NSCLC. A previous trial (NCT04322006) was conducted to evaluate the safety, tolerability, and efficacy of uliledlimumab alone or in combination with the PD-1 inhibitor toripalimab in advanced solid tumors. Of the 19 patients with advanced NSCLC who were not candidates for standard therapy, five achieved partial remission and nine had stable disease, with an ORR of 26% and a DCR of 73.7% (72, 73).
CD38 inhibitors are primarily used for the treatment of myeloma and have shown promising results. CD38 inhibitors do not appear to have a definitive efficacy in lung cancer. Two prior trials (NCT03023423 and NCT03367819) have thus far evaluated the efficacy of the CD38 inhibitors isatuximab and daratumumab, respectively, in combination with PD-1/PD-L1 inhibitors in NSCLC but did not achieve satisfactory results. The NCT03023423 trial investigated the safety and efficacy of atezolizumab alone or in combination with daratumumab in patients with advanced or metastatic NSCLC who did not received immunotherapy (74). These studies found no significant improvement in ORR, clinical benefit rate (CBR), mPFS, or mOS in the combination group compared with the single-agent group (ORR 13% vs 4.3%, respectively; CBR 43.5% vs 52.2%, respectively; mPFS 1.5 months vs 1.7 months, respectively; and mOS not achieved vs 7.1 months, respectively) (74). In terms of safety, 38.6% and 56.8% of patients in both groups experienced grade 3 or higher adverse events. In terms of biomarkers, CD38 expression was generally low in both groups, with mean h-scores of only 26.1 and 28.3, respectively (74). The high level of CD38 expression by immune cells in patients with myeloma may be a possible reason for the difference in the efficacy of CD38 inhibitors in NSCLC. NCT03367819 was discontinued after the interim analysis because of a limited treatment response. The results showed that, in terms of efficacy, no patients with NSCLC treated with a combination of isatuximab and cemiplegia achieved complete remission (CR) or partial remission (PR), 65% maintained stable disease (SD), and the mPFS was 4.01 months (75). Interestingly, combination therapy resulted in a decrease in CD38+ immune cells in the TME and an increase in peripherally activated and cytolytic T cells; however, no significant antitumor activity was observed (75). Overall, 70% of the patients developed TRAEs, 20% of which were grade 3 or higher, indicating that the safety of combination therapy is manageable (75).
The Morpheus study compared objective remission rates and safety of second-line therapy with atezolizumab plus CPI-444 and docetaxel in NSCLC patients who showed disease progression during or after treatment with platinum-based regimens and PD-L1/PD-1 checkpoint inhibitors (76). The ORR was 6.7% and 21.4%, respectively, and the mPFS was 2.3 months and 3.2 months, respectively. mOS has not been reported previously (76). In terms of safety, patients receiving atezolizumab and CPI-444 did not experience grade 5 adverse events or adverse events leading to drug (76). This study demonstrated that atezolizumab plus CPI-444 has a controlled safety profile and preliminary antitumor activity.
PBF-509 is another A2AR inhibitor assessed in clinical studies of NSCLC. One phase I/II study investigated the safety, tolerability, and feasibility of the oral immunosuppressant PBF-509 alone or in combination with the PD-1 inhibitor PDR001 for the treatment of NSCLC (77, 78). The DCRs were 42.9% and 66.7% in the single-agent and combination groups, respectively, with ORRs of 9.5% and 8.3%, mOS of 9.7 and 5.4 months, and mPFS of 3.9 and 2.8 months, respectively (78). The incidences of grade 3 or higher TRAEs in the single-agent and combination groups were 16% and 36%, respectively. The most common TRAE in both groups was nausea (44.0% vs. 28%) (78). This study suggests that PBF-509 has preliminary antitumor activity in NSCLC; however, the efficacy of PBF-509 in combination with PD-1 inhibitors needs to be further confirmed.
From the data published in the above studies (as shown in Table 2), it is clear that the combination of ADO inhibitors and PD-1/PD-L1 inhibitors has significant potential to improve the prognosis of NSCLC, with obleclumab, a CD73 monoclonal antibody, being the most promising. These studies provided strong evidence that ADO inhibitors can improve or reverse PD-L1 resistance in these patients.
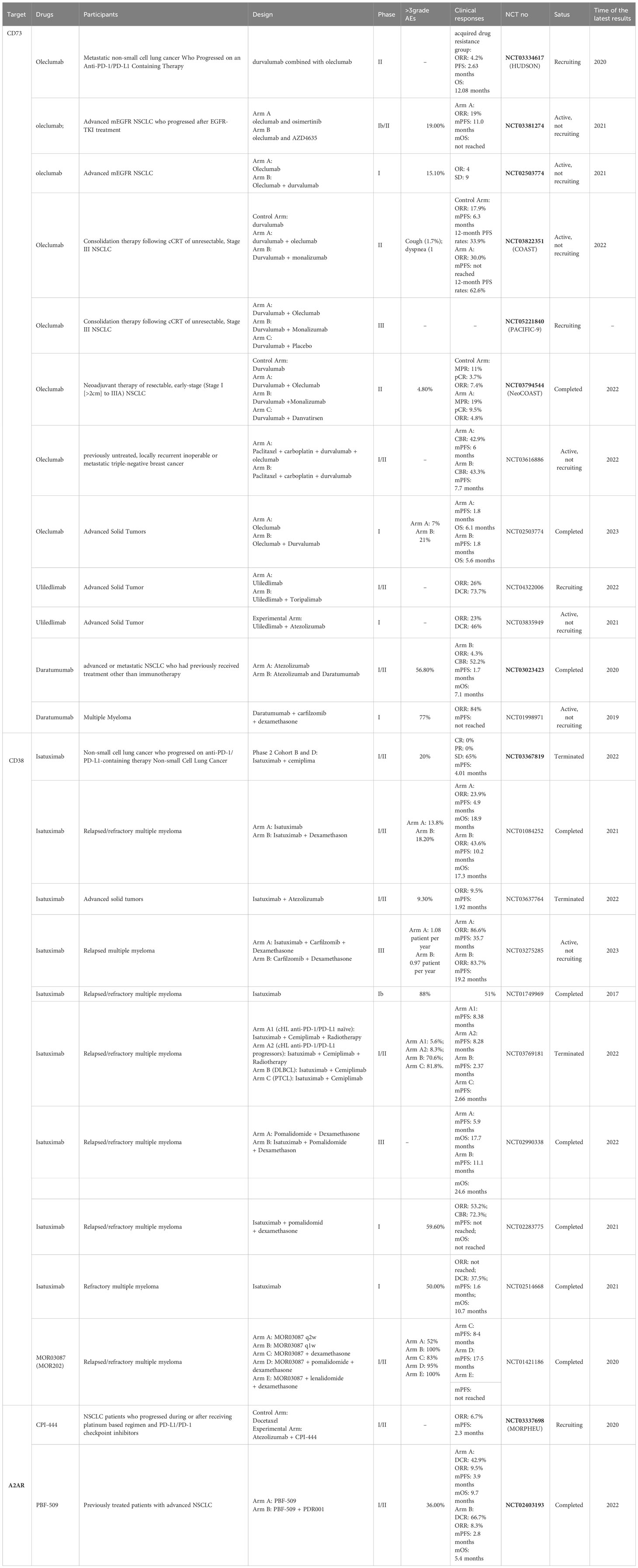
Table 2 Clinical study of ADO-related drugs for lung cancer.
Biomarkers that can predict the clinical efficacy of ICBs in combination with ADO inhibitors are still being explored. Many studies have shown that high CD73 expression in tumor tissues is an indicator of poor prognosis in NSCLC (79–84). The expression levels of CD39 and CD38 were also similar significance (85–88). CD73 expression has been found to be positively correlated with histopathological grade, tumor invasion, and lymph node metastasis (63, 89). Of particular interest is the correlation between CD73 and PD-1/PD-L1 expression (63, 90, 91). The expression of both PD-L1 and CD73 is elevated in drug-resistant NSCLC following treatment with EGFR-TKIs (53). Previous studies have shown that CD73 expression can predict the efficacy of immune checkpoint inhibitors (92). High CD73 expression in NSCLC cells appears to be associated with a better response to ICB treatment (84, 93). In addition, it has also been shown that the ratio of MDSCs to CD39+CD8+ T cells could serve as a potential biomarker to predict the blocking effect of immune checkpoint inhibitors in patients with NSCLC (67, 85). The results of one prior trial (NCT04322006) indicated that co-expression of CD73 and PD-L1 may be a potential biomarker for predicting the efficacy of CD73 inhibitors in combination with ICBs (72). In a study that included 19 patients with advanced NSCLC, the clinical response to uliledolimumab and toripalimab treatment was significantly correlated with CD73 expression in the tumor. Four of 5 PR patients had high CD73 expression (tumor cell or immune cell expression level ≥35%), and 4 of 9 SD patients had high CD73 expression (72). Similar findings have been reported in Neocoast study. In the durvalumab and olecularab-combination group, higher baseline CD73 expression was associated with fewer tumor cells surviving surgery and pathological remission. Upregulation of genes related to B cell activation and antigen presentation was also detected in the combination group of patients with MPR. Combination treatment with durvalumab and oleculum also increased the density of immune effector NKG2A+ cells in the tumor immune microenvironment (70). A phase I study, NCT02503774, also found that the frequencies of CD8+ T cells, PD-L1, and granzyme B were upregulated in five of the six patients treated with the combination of durvalumab and olecumab, in whom biomarkers were detected by biopsy (94). However, the evidence provided by these studies was limited. In the future, randomized controlled studies with larger sample sizes should be performed to explore the benefits of combination therapy. Accurate detection of TME components (e.g., with immune cell fraction assay, immunoreactive molecule assay, tumor cell and immune cell surface molecule expression assay, and TMB), complemented by combination proteomics, genomics, single-cell sequencing, next-generation sequencing, and other technologies, will help to further clarify their potential biomarkers and thus guide the application of combination strategies to more appropriate populations and achieve optimal clinical benefits.
Recently, the specific mechanisms of ADO in the formation of an immunosuppressive microenvironment in lung cancer have been revealed. Current evidence suggests that ADO can cause immune resistance in lung cancer by inducing the formation of an immunosuppressive microenvironment, thereby affecting the antigen presentation process, promoting T-cell rejection and T-cell failure, and interfering with IFN-γ signaling pathway. In contrast, ADO inhibitors play critical roles in the above mentioned segments reversing immune resistance. In the field of lung cancer, ADO inhibitors in combination with ICBs have achieved staged progress, and several phase III clinical trials are currently underway. The importance of ADO inhibitors in inhibiting tumor progression and improving the immunosuppressive microenvironment is becoming clear, especially for the CD73 monoclonal antibody olecumab. However, related research is still nascent and there are still many controversies regarding ADO inhibitors. First, the mechanisms underlying the ADO pathway in lung tumorigenesis and development have not yet been fully elucidated. Secondly, the exact efficacy and adverse effects of ADO inhibitors in NSCLC have not yet been demonstrated in phase III clinical studies. Third, the available clinical data were not sufficient to accurately identify the beneficiary population and the scope of use of ADO inhibitors. Finally, more multicenter, randomized, controlled studies are needed to explore the usage strategies of ADO inhibitors, such as which ADO inhibitors are most effective and how they are combined with ICBs. Future studies should address these questions and explore a broader and brighter future for the development and use of ADO inhibitors to provide valuable opportunities for systemic treatment of NSCLC.
RW: Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. ZL: Data curation, Writing – original draft, Writing – review & editing. TW: Formal analysis, Writing – original draft, Writing – review & editing. JZ: Formal analysis, Visualization, Writing – review & editing. JL: Conceptualization, Formal analysis, Funding acquisition, Writing – review & editing. QZ: Conceptualization, Formal analysis, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Wu Jieping Medical Foundation, China (312150082) and the Regional Innovation Cooperation Project of Sichuan Science and Technology Program (2021YFQ0029).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
NSCLC, non-small cell lung cancer; TILs, tumor-infiltrating lymphocytes; TME, tumor microenvironment; ADO, adenosine; DCs, dendritic cells; A2R, ADO receptor 2; MDSCs, myeloid-derived suppressor cells; Tregs, regulatory T cells; Teff, effector T cells; IFN γ, interferon γ; APCs, antigen-presenting cells; A2AR, ADO receptor A2; A2BR, ADO receptor B2; MHC II, major histocompatibility complex II; ICAMs, intercellular adhesion molecules; TNF, tumor necrosis factor; EGFR, epidermal growth factor receptor; TMB, tumor mutation burden; TKI, tyrosine kinase inhibitors; ICB, immune checkpoint blockade; mEGFR, EGFR mutation; mPFS, median progression-free survival; OS, overall survival; ORR, objective remission rate; DCR, disease control rate; EAEs, emergency adverse events; MPR, major pathologic remission rates; PCR, pathologic complete remission rates; TRAEs, treatment-related adverse events; CBR, clinical benefit rate; CR, Complete Remission; PR, Partial Remission; SD, stable disease.
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
2. Garassino MC, Gadgeel S, Speranza G, Felip E, Esteban E, Domine M, et al. Pembrolizumab plus pemetrexed and platinum in nonsquamous non-small-cell lung cancer: 5-year outcomes from the phase 3 KEYNOTE-189 study. J Clin Oncol (2023) 41(11):1992–8. doi: 10.1200/JCO.22.01989
3. Novello S, Kowalski DM, Luft A, Gumus M, Vicente D, Mazieres J, et al. Pembrolizumab plus chemotherapy in squamous non-small-cell lung cancer: 5-year update of the phase III KEYNOTE-407 study. J Clin Oncol (2023) 41(11):1999–2006. doi: 10.1200/JCO.22.01990
4. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. (2017) 17(11):637–58. doi: 10.1038/nrc.2017.84
5. Reck M, Remon J, Hellmann MD. First-line immunotherapy for non-small-cell lung cancer. J Clin Oncol (2022) 40(6):586–97. doi: 10.1200/JCO.21.01497
6. Frisone D, Friedlaender A, Addeo A, Tsantoulis P. The landscape of immunotherapy resistance in NSCLC. Front Oncol (2022) 12:817548. doi: 10.3389/fonc.2022.817548
7. Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open (2019) 2(5):e192535. doi: 10.1001/jamanetworkopen.2019.2535
8. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
9. Yang L, He YT, Dong S, Wei XW, Chen ZH, Zhang B, et al. Single-cell transcriptome analysis revealed a suppressive tumor immune microenvironment in EGFR mutant lung adenocarcinoma. J Immunother Cancer (2022) 10(2):e003534. doi: 10.1136/jitc-2021-003534
10. Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell (2016) 29(3):285–96. doi: 10.1016/j.ccell.2016.02.004
11. Blomberg OS, Spagnuolo L, de Visser KE. Immune regulation of metastasis: mechanistic insights and therapeutic opportunities. Dis Model Mech (2018) 11(10):dmm036236. doi: 10.1242/dmm.036236
12. Le X, Negrao MV, Reuben A, Federico L, Diao L, McGrail D, et al. Characterization of the immune landscape of EGFR-mutant NSCLC identifies CD73/adenosine pathway as a potential therapeutic target. J Thorac Oncol (2021) 16(4):583–600. doi: 10.1016/j.jtho.2020.12.010
13. Boison D, Yegutkin GG. Adenosine metabolism: emerging concepts for cancer therapy. Cancer Cell (2019) 36(6):582–96. doi: 10.1016/j.ccell.2019.10.007
14. Augustin RC, Leone RD, Naing A, Fong L, Bao R, Luke JJ. Next steps for clinical translation of adenosine pathway inhibition in cancer immunotherapy. J Immunother Cancer. (2022) 10(2):e004089. doi: 10.1136/jitc-2021-004089
15. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell (2020) 37(4):443–55. doi: 10.1016/j.ccell.2020.03.017
16. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood (2008) 112(5):1822–31. doi: 10.1182/blood-2008-02-136325
17. Panther E, Corinti S, Idzko M, Herouy Y, Napp M, la Sala A, et al. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood (2003) 101(10):3985–90. doi: 10.1182/blood-2002-07-2113
18. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol (2011) 187(11):6120–9. doi: 10.4049/jimmunol.1101225
19. Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol (2014) 5:304. doi: 10.3389/fimmu.2014.00304
20. Zhang T, Fu W, Liu D, He Y, Wang J, Ma T. Adenosine influences Foxp3 expression of Tregsvia the A2aR/CREB pathway in a mouse model of sepsis. Shock (2023). doi: 10.1097/SHK.0000000000002281
21. Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J (2012) 26(1):376–86. doi: 10.1096/fj.11-190934
22. Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation (2013) 36(4):921–31. doi: 10.1007/s10753-013-9621-3
23. Zhang H, Han K, Li H, Zhang J, Zhao Y, Wu Y, et al. hPMSCs regulate the level of TNF-alpha and IL-10 in th1 cells and improve hepatic injury in a GVHD mouse model via CD73/ADO/fyn/nrf2 axis. Inflammation (2024) 47(1):244–63. doi: 10.1007/s10753-023-01907-1
24. Garcia-Rocha R, Monroy-Garcia A, Hernandez-Montes J, Weiss-Steider B, Gutierrez-Serrano V, Del Carmen Fuentes-Castaneda M, et al. Cervical cancer cells produce TGF-beta1 through the CD73-adenosine pathway and maintain CD73 expression through the autocrine activity of TGF-beta1. Cytokine (2019) 118:71–9. doi: 10.1016/j.cyto.2018.09.018
25. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res (2014) 74(24):7250–9. doi: 10.1158/0008-5472.CAN-13-3583
26. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget (2015) 6(29):27478–89. doi: 10.18632/oncotarget.4393
27. Pico de Coana Y, Poschke I, Gentilcore G, Mao Y, Nystrom M, Hansson J, et al. Ipilimumab treatment results in an early decrease in the frequency of circulating granulocytic myeloid-derived suppressor cells as well as their Arginase1 production. Cancer Immunol Res (2013) 1(3):158–62. doi: 10.1158/2326-6066.CIR-13-0016
28. Santegoets SJ, Stam AG, Lougheed SM, Gall H, Jooss K, Sacks N, et al. Myeloid derived suppressor and dendritic cell subsets are related to clinical outcome in prostate cancer patients treated with prostate GVAX and ipilimumab. J Immunother Cancer. (2014) 2:31. doi: 10.1186/s40425-014-0031-3
29. Jin R, Liu L, Xing Y, Meng T, Ma L, Pei J, et al. Dual mechanisms of novel CD73-targeted antibody and antibody-drug conjugate in inhibiting lung tumor growth and promoting antitumor immune-effector function. Mol Cancer Ther (2020) 19(11):2340–52. doi: 10.1158/1535-7163.MCT-20-0076
30. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology (2016) 5(8):e1208875. doi: 10.1080/2162402X.2016.1208875
31. Xaus J, Mirabet M, Lloberas J, Soler C, Lluis C, Franco R, et al. IFN-gamma up-regulates the A2B adenosine receptor expression in macrophages: a mechanism of macrophage deactivation. J Immunol (1999) 162(6):3607–14. doi: 10.4049/jimmunol.162.6.3607
32. Wilson JM, Ross WG, Agbai ON, Frazier R, Figler RA, Rieger J, et al. The A2B adenosine receptor impairs the maturation and immunogenicity of dendritic cells. J Immunol (2009) 182(8):4616–23. doi: 10.4049/jimmunol.0801279
33. Cekic C, Linden J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res (2014) 74(24):7239–49. doi: 10.1158/0008-5472.CAN-13-3581
34. Hofer S, Ivarsson L, Stoitzner P, Auffinger M, Rainer C, Romani N, et al. Adenosine slows migration of dendritic cells but does not affect other aspects of dendritic cell maturation. J Invest Dermatol (2003) 121(2):300–7. doi: 10.1046/j.1523-1747.2003.12369.x
35. Chen S, Akdemir I, Fan J, Linden J, Zhang B, Cekic C. The expression of adenosine A2B receptor on antigen-presenting cells suppresses CD8(+) T-cell responses and promotes tumor growth. Cancer Immunol Res (2020) 8(8):1064–74. doi: 10.1158/2326-6066.CIR-19-0833
36. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
37. Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics (2021) 11(11):5365–86. doi: 10.7150/thno.58390
38. Vigano S, Alatzoglou D, Irving M, Menetrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol (2019) 10:925. doi: 10.3389/fimmu.2019.00925
39. Mediavilla-Varela M, Luddy K, Noyes D, Khalil FK, Neuger AM, Soliman H, et al. Antagonism of adenosine A2A receptor expressed by lung adenocarcinoma tumor cells and cancer associated fibroblasts inhibits their growth. Cancer Biol Ther (2013) 14(9):860–8. doi: 10.4161/cbt.25643
40. Mastelic-Gavillet B, Navarro Rodrigo B, Decombaz L, Wang H, Ercolano G, Ahmed R, et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8(+) T cells. J Immunother Cancer. (2019) 7(1):257. doi: 10.1186/s40425-019-0719-5
41. Mediavilla-Varela M, Castro J, Chiappori A, Noyes D, Hernandez DC, Allard B, et al. A novel antagonist of the immune checkpoint protein adenosine A2a receptor restores tumor-infiltrating lymphocyte activity in the context of the tumor microenvironment. Neoplasia (2017) 19(7):530–6. doi: 10.1016/j.neo.2017.02.004
42. Sundstrom P, Stenstad H, Langenes V, Ahlmanner F, Theander L, Ndah TG, et al. Regulatory T cells from colon cancer patients inhibit effector T-cell migration through an adenosine-dependent mechanism. Cancer Immunol Res (2016) 4(3):183–93. doi: 10.1158/2326-6066.CIR-15-0050
43. Chimote AA, Balajthy A, Arnold MJ, Newton HS, Hajdu P, Qualtieri J, et al. A defect in KCa3.1 channel activity limits the ability of CD8(+) T cells from cancer patients to infiltrate an adenosine-rich microenvironment. Sci Signal (2018) 11(527):eaaq1616. doi: 10.1126/scisignal.aaq1616
44. Barnholt KE, Kota RS, Aung HH, Rutledge JC. Adenosine blocks IFN-gamma-induced phosphorylation of STAT1 on serine 727 to reduce macrophage activation. J Immunol (2009) 183(10):6767–77. doi: 10.4049/jimmunol.0900331
45. Takahashi HK, Iwagaki H, Hamano R, Wake H, Kanke T, Liu K, et al. Effects of adenosine on adhesion molecule expression and cytokine production in human PBMC depend on the receptor subtype activated. Br J Pharmacol (2007) 150(6):816–22. doi: 10.1038/sj.bjp.0707126
46. Romio M, Reinbeck B, Bongardt S, Huls S, Burghoff S, Schrader J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. Am J Physiol Cell Physiol (2011) 301(2):C530–9. doi: 10.1152/ajpcell.00385.2010
47. Briceno P, Rivas-Yanez E, Rosemblatt MV, Parra-Tello B, Farias P, Vargas L, et al. CD73 ectonucleotidase restrains CD8+ T cell metabolic fitness and anti-tumoral activity. Front Cell Dev Biol (2021) 9:638037. doi: 10.3389/fcell.2021.638037
48. Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol (2005) 174(2):1073–80. doi: 10.4049/jimmunol.174.2.1073
49. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discovery (2017) 7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223
50. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res (2013) 19(20):5626–35. doi: 10.1158/1078-0432.CCR-13-0545
51. Tej G, Neogi K, Nayak PK. Caffeine-enhanced anti-tumor activity of anti-PD1 monoclonal antibody. Int Immunopharmacol. (2019) 77:106002. doi: 10.1016/j.intimp.2019.106002
52. Qiao M, Jiang T, Liu X, Mao S, Zhou F, Li X, et al. Immune checkpoint inhibitors in EGFR-mutated NSCLC: dusk or dawn? J Thorac Oncol (2021) 16(8):1267–88. doi: 10.1016/j.jtho.2021.04.003
53. Isomoto K, Haratani K, Hayashi H, Shimizu S, Tomida S, Niwa T, et al. Impact of EGFR-TKI treatment on the tumor immune microenvironment in EGFR mutation-positive non-small cell lung cancer. Clin Cancer Res (2020) 26(8):2037–46. doi: 10.1158/1078-0432.CCR-19-2027
54. Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol (2015) 10(6):910–23. doi: 10.1097/JTO.0000000000000500
55. Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (2016) 387(10027):1540–50. doi: 10.1016/S0140-6736(15)01281-7
56. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med (2015) 373(17):1627–39. doi: 10.1056/NEJMoa1507643
57. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet (2017) 389(10066):255–65. doi: 10.1016/S0140-6736(16)32517-X
58. Lee CK, Man J, Lord S, Links M, Gebski V, Mok T, et al. Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer-A meta-analysis. J Thorac Oncol (2017) 12(2):403–7. doi: 10.1016/j.jtho.2016.10.007
59. Giatromanolaki A, Kouroupi M, Pouliliou S, Mitrakas A, Hasan F, Pappa A, et al. Ectonucleotidase CD73 and CD39 expression in non-small cell lung cancer relates to hypoxia and immunosuppressive pathways. Life Sci (2020) 259:118389. doi: 10.1016/j.lfs.2020.118389
60. Madeddu C, Donisi C, Liscia N, Lai E, Scartozzi M, Maccio A. EGFR-mutated non-small cell lung cancer and resistance to immunotherapy: role of the tumor microenvironment. Int J Mol Sci (2022) 23(12):6489. doi: 10.3390/ijms23126489
61. Griesing S, Liao BC, Yang JC. CD73 is regulated by the EGFR-ERK signaling pathway in non-small cell lung cancer. Anticancer Res (2021) 41(3):1231–42. doi: 10.21873/anticanres.14880
62. Han Y, Lee T, He Y, Raman R, Irizarry A, Martin ML, et al. The regulation of CD73 in non-small cell lung cancer. Eur J Cancer. (2022) 170:91–102. doi: 10.1016/j.ejca.2022.04.025
63. Rocha P, Salazar R, Zhang J, Ledesma D, Solorzano JL, Mino B, et al. CD73 expression defines immune, molecular, and clinicopathological subgroups of lung adenocarcinoma. Cancer Immunol Immunother. (2021) 70(7):1965–76. doi: 10.1007/s00262-020-02820-4
64. Tu E, McGlinchey K, Wang J, Martin P, Ching SL, Floc’h N, et al. Anti-PD-L1 and anti-CD73 combination therapy promotes T cell response to EGFR-mutated NSCLC. JCI Insight (2022) 7(3):e142843. doi: 10.1172/jci.insight.142843
65. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. (2018) 6(1):57. doi: 10.1186/s40425-018-0360-8
66. Besse B, Awad M, Forde P, Thomas M, Park K, Goss G, et al. Hudson: an open-label, multi-drug, biomarker-directed, phase II platform study in patients with NSCLC, who progressed on anti-PD(L)1 therapy. J Of Thorac Oncol (2021) 16(3):S118–S9. doi: 10.1016/j.jtho.2021.01.299
67. Yoneshima Y, Morita S, Ando M, Nakamura A, Iwasawa S, Yoshioka H, et al. Phase 3 trial comparing nanoparticle albumin-bound paclitaxel with docetaxel for previously treated advanced NSCLC. J Thorac Oncol (2021) 16(9):1523–32. doi: 10.1016/j.jtho.2021.03.027
68. Herbst RS, Majem M, Barlesi F, Carcereny E, Chu Q, Monnet I, et al. COAST: an open-label, phase II, multidrug platform study of durvalumab alone or in combination with oleclumab or monalizumab in patients with unresectable, stage III non-small-cell lung cancer. J Clin Oncol (2022) 40(29):3383–93. doi: 10.1200/JCO.22.00227
69. Campelo RG, Forde P, Weder W, Spicer J, He P, Hamid O, et al. NeoCOAST: neoadjuvant durvalumab alone or with novel agents for resectable, early-stage (I-IIIA) non-small cell lung cancer. J OF Thorac Oncol (2019) 14(10):S719–S. doi: 10.1016/j.jtho.2019.08.1533
70. Cascone T G-CR, Spicer J. NeoCOAST: open-label, randomized, phase 2, multidrug platform study of neoadjuvant durvalumab alone or combined with novel agents in patients (pts) with resectable, early-stage non-small-cell lung cancer (NSCLC). AACR Annu Meeting (2022). doi: 10.1158/1538-7445.AM2022-CT011
71. Spicer JC, Kar G, Zheng Y, Blando J, Tan TH, et al. Platform study of neoadjuvant durvalumab (D) alone or combined with novel agents in patients (pts) with resectable, early-stage non-small cell lung cancer (NSCLC): Pharmacodynamic correlates and circulating tumor DNA (ctDNA) dynamics in the NeoCOAST study. Ann Of Oncol (2022) 33(7):S971–S. doi: 10.1016/j.annonc.2022.07.1055
72. Zhou Q, Wu L, Cui J, Jiang B, Yao Y, Zhang J, et al. Safety, efficacy, pharmacokinetics of uliledlimab alone or combined with toripalimab in advanced solid tumor: Initial results of a phase I/II study. J Clin Oncol (2022) 40(16_suppl):e21123–e. doi: 10.1200/JCO.2022.40.16_suppl.e21123
73. Robert F, Dumbrava EE, Xing Y, Mills E, Freddo JL, Theuer CP, et al. Preliminary safety, pharmacokinetics (PK), pharmacodynamics (PD) and clinical efficacy of uliledlimab (TJ004309), a differentiated CD73 antibody, in combination with atezolizumab in patients with advanced cancer. J Clin Oncol (2021) 39(15_suppl):2511. doi: 10.1200/JCO.2021.39.15_suppl.2511
74. Pillai RN, Ramalingam SS, Thayu M, Lorenzini P, Alvarez Arias DA, Moy C, et al. Daratumumab plus atezolizumab in previously treated advanced or metastatic NSCLC: brief report on a randomized, open-label, phase 1b/2 study (LUC2001 JNJ-54767414). JTO Clin Res Rep (2021) 2(2):100104. doi: 10.1016/j.jtocrr.2020.100104
75. Zucali PA, Lin CC, Carthon BC, Bauer TM, Tucci M, Italiano A, et al. Targeting CD38 and PD-1 with isatuximab plus cemiplimab in patients with advanced solid Malignancies: results from a phase I/II open-label, multicenter study. J Immunother Cancer (2022) 10(1):e003697. doi: 10.1136/jitc-2021-003697
76. Felip E, Lim FL, Johnson M, O’Brien M, Barlesi F, Mazieres J, et al. Phase Ib/II open-label, randomised evaluation of atezolizumab (atezo) + CPI-444 vs docetaxel as second/third-line therapy in MORPHEUS-NSCLC (non-small cell lung cancer). Ann OF Oncol (2020) 31:S850–S. doi: 10.1016/j.annonc.2020.08.1629
77. Chiappori A, Creelan B, Tanvetyanon T, Gray JE, Haura EB, Thapa R, et al. Phase I/II study of the A2AR antagonist NIR178 (PBF-509), an oral immunotherapy, in patients (pts) with advanced NSCLC. J OF Clin Oncol (2018) 36(15):S538–S538. doi: 10.1200/JCO.2018.36.15_suppl.9089
78. Chiappori AA, Creelan B, Tanvetyanon T, Gray JE, Haura EB, Thapa R, et al. Phase I study of taminadenant (PBF509/NIR178), an adenosine 2A receptor antagonist, with or without spartalizumab (PDR001), in patients with advanced non-small cell lung cancer. Clin Cancer Res (2022) 28(11):2313–20. doi: 10.1158/1078-0432.CCR-21-2742
79. Inoue Y, Yoshimura K, Kurabe N, Kahyo T, Kawase A, Tanahashi M, et al. Opposing prognostic roles of CD73 and A2A adenosine receptor in non-small-cell lung cancer. J Of Thorac Oncol (2017) 12(1):S625–S6. doi: 10.1016/j.jtho.2016.11.804
80. Koh J, Lee KY, Kim B, Kim MS, Cho HJ, Sun JM, et al. CD39 increase on cytotoxic T-cell induced by myeloid-derived suppressor cell correlated with poor prognosis in patients with non-small cell lung cancer. Cancer Immunol Res (2019) 7(2):Supplement S. doi: 10.1158/2326-6074.CRICIMTEATIAACR18-A138
81. Gao ZW, Liu C, Yang L, Chen HC, Yang LF, Zhang HZ, et al. CD73 severed as a potential prognostic marker and promote lung cancer cells migration via enhancing EMT progression. Front Genet (2021) 12:728200. doi: 10.3389/fgene.2021.728200
82. Zhang H, Cao Y, Tang J, Wang R. CD73 (NT5E) promotes the proliferation and metastasis of lung adenocarcinoma through the EGFR/AKT/mTOR pathway. BioMed Res Int (2022) 2022:9944847. doi: 10.1155/2022/9944847
83. Inoue Y, Yoshimura K, Kurabe N, Kahyo T, Kawase A, Tanahashi M, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget (2017) 8(5):8738–51. doi: 10.18632/oncotarget.14434
84. Ishii H, Azuma K, Kinoshita T, Matsuo N, Naito Y, Tokito T, et al. Predictive value of CD73 expression in EGFR-mutation positive non-small-cell lung cancer patients received immune checkpoint inhibitors. J Of Clin Oncol (2018) 36(15):Supplement S. doi: 10.1200/JCO.2018.36.15_suppl.9065
85. Koh J, Kim Y, Lee KY, Hur JY, Kim MS, Kim B, et al. MDSC subtypes and CD39 expression on CD8(+) T cells predict the efficacy of anti-PD-1 immunotherapy in patients with advanced NSCLC. Eur J Immunol (2020) 50(11):1810–9. doi: 10.1002/eji.202048534
86. Su D, Wu G, Xiong R, Sun X, Xu M, Mei Y, et al. Tumor immune microenvironment characteristics and their prognostic value in non-small-cell lung cancer. Front Oncol (2021) 11:634059. doi: 10.3389/fonc.2021.634059
87. Qi Y, Xia Y, Lin Z, Qu Y, Qi Y, Chen Y, et al. Tumor-infiltrating CD39(+)CD8(+) T cells determine poor prognosis and immune evasion in clear cell renal cell carcinoma patients. Cancer Immunol Immunother. (2020) 69(8):1565–76. doi: 10.1007/s00262-020-02563-2
88. Ni X, Wan W, Ma J, Liu X, Zheng B, He Z, et al. A novel prognostic biomarker of luminal breast cancer: high CD39 expression is related to poor survival. Front Genet (2021) 12:682503. doi: 10.3389/fgene.2021.682503
89. Tahkola K, Ahtiainen M, Kellokumpu I, Mecklin JP, Laukkarinen J, Laakkonen J, et al. Prognostic impact of CD73 expression and its relationship to PD-L1 in patients with radically treated pancreatic cancer. Virchows Arch (2021) 478(2):209–17. doi: 10.1007/s00428-020-02888-4
90. Martin P, Spitzmueller A, Wu S, Widmaier M, Korn R, Altharnmer S, et al. Mutually exclusive expression of CD73 and PDL1 in tumors from patients (pt) with NSCLC, gastroesophageal (GE) and urothelial bladder carcinoma (UBC). . J Of Clin Oncol (2017) 35:Supplement 15. doi: 10.1200/JCO.2017.35.15_suppl.3079
91. Ono K, Shiozawa E, Ohike N, Fujii T, Shibata H, Kitajima T, et al. Immunohistochemical CD73 expression status in gastrointestinal neuroendocrine neoplasms: A retrospective study of 136 patients. Oncol Lett (2018) 15(2):2123–30. doi: 10.3892/ol.2017.7569
92. Streicher K, Brandon W, Wu S, Coffman K, Damera G, Durham N, et al. Increased CD73 and reduced IFNG signature expression in relation to response rates to anti-PD-1(L1) therapies in EGFR-mutant NSCLC. . J Of Clin Oncol (2017) 35:Supplement 15. doi: 10.1200/JCO.2017.35.15_suppl.11505
93. Ishii H, Azuma K, Kawahara A, Kinoshita T, Matsuo N, Naito Y, et al. Predictive value of CD73 expression for the efficacy of immune checkpoint inhibitors in NSCLC. Thorac Cancer. (2020) 11(4):950–5. doi: 10.1111/1759-7714.13346
94. Bendell J, LoRusso P, Overman MJ, Noonan AM, Kim DW, Strickler J, et al. Safety and efficacy of the anti-CD73 monoclonal antibody (mAb) oleclumab +/- durvalumab in patients (pts) with advanced colorectal cancer (CRC), pancreatic ductal adenocarcinoma (PDAC), or EGFR-mutant non-small cell lung cancer (EGFRm NSCLC). J Of Clin Oncol (2021) 39(15):Supplement S. doi: 10.1200/JCO.2021.39.15_suppl.9047
Keywords: adenosine, CD73, non-small cell lung cancer, immunosuppressive microenvironment, immunotherapy, drug resistance
Citation: Wang R, Liu Z, Wang T, Zhang J, Liu J and Zhou Q (2024) Landscape of adenosine pathway and immune checkpoint dual blockade in NSCLC: progress in basic research and clinical application. Front. Immunol. 15:1320244. doi: 10.3389/fimmu.2024.1320244
Received: 12 October 2023; Accepted: 10 January 2024;
Published: 29 January 2024.
Edited by:
Yulong Sun, University of New South Wales, AustraliaReviewed by:
Howard Donninger, University of Louisville, United StatesCopyright © 2024 Wang, Liu, Wang, Zhang, Liu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiewei Liu, amVycnlfbGp3QDE2My5jb20=; Qinghua Zhou, cHJvZl9xaF96aG91QDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.