 Yanxin Xu1†
Yanxin Xu1† Bo Shao
Bo Shao Yafeng Zhang
Yafeng Zhang- 1Department of Colorectal Surgery, The First Affiliated Hospital of Zhengzhou University, Henan, Zhengzhou, China
- 2Institute for Hospital Management of Henan Province, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Lysosomes are intracellular digestive organelles that participate in various physiological and pathological processes, including the regulation of immune checkpoint molecules, immune cell function in the tumor microenvironment, antigen presentation, metabolism, and autophagy. Abnormalities or dysfunction of lysosomes are associated with the occurrence, development, and drug resistance of tumors. Lysosomes play a crucial role and have potential applications in tumor immunotherapy. Targeting lysosomes or harnessing their properties is an effective strategy for tumor immunotherapy. However, the mechanisms and approaches related to lysosomes in tumor immunotherapy are not fully understood at present, and further basic and clinical research is needed to provide better treatment options for cancer patients. This review focuses on the research progress related to lysosomes and tumor immunotherapy in these
1 Introduction
For an extended period, lysosomes have primarily been considered integral components of cellular degradation and recycling due to their inherent acidity and rich complement of hydrolytic enzymes, facilitating the breakdown of cellular constituents and extracellular molecules into their constituent components (1). However, with the deepening of research, it has become evident that lysosomes play a broader role in various cellular processes. This is mediated through multiple mechanisms, including the mammalian target of rapamycin complex 1 (mTOR1) (2, 3), Ca2+ ion efflux, transcription factor EB (TFEB) nuclear translocation (4), as well as the differential localization of lysosomal membrane proteins and associated proteins (5–7). Lysosomes are now recognized as crucial mediators of cellular material transport, encompassing processes such as autophagy, endocytosis, and phagocytosis, while also significantly contributing to proliferation, differentiation, metabolic regulation, secretion activities, and the quality control of protein aggregates and damaged organelles (1, 8, 9). They have emerged as multifaceted hubs for immune regulation, nutrient sensing, and information trafficking. Any alterations or functional impairments in lysosomes have the potential to disrupt the intrinsic equilibrium of cells and tissues, leading to or exacerbating human diseases, including cancer (10, 11).
The process of tumor initiation and progression is closely intertwined with the contribution of the tumor microenvironment (TME) (7, 12). The TME constitutes a complex biological system surrounding tumor cells, including non-tumor cells such as immune cells, fibroblasts, endothelial cells, extracellular matrix (ECM), blood vessels, lymphatic vessels, signaling molecules, and metabolites, among others (7, 12). Among these components, lysosomes play an indispensable role in regulating immune functions, antigen presentation, and immune evasion (13). Antigen-presenting cells (APCs) primarily rely on lysosomes to engulf exogenous antigens, process them into peptides, and transport them to the major histocompatibility complex class II (MHC-II) loading compartment (14). Alternatively, they can also utilize autophagosomes to target cytoplasmic proteins for lysosomal degradation, thereby activating CD4+ helper T cell responses and coordinating specific immune reactions (15). However, the major histocompatibility complex class I (MHC-I), when bound to the autophagy cargo receptor NBR1, undergoes autophagic/lysosomal co-localization degradation, impairing the immune surveillance function of CD8+ T cells (16). Additionally, lysosomes are involved in the intracellular trafficking, presentation, and degradation of co-stimulatory molecules and immune checkpoint proteins (17). For instance, the co-localization of CKLF-like marvel transmembrane domain-containing protein 6 (CMTM6) with programmed cell death ligand 1 (PD-L1) on the plasma membrane and recycling endosomes of cancer cells prevents PD-L1 lysosomal degradation, thereby inhibiting the activity of tumor-specific T cells (18). CD47, widely expressed on the surface of various cancer cells, interacts with the macrophage-expressed SIRPα, generating a “don’t eat me” signal (19). Lysosomal protein RAGA drives the endocytosis of CD47 and relocalizes it to lysosomes for degradation, thus restoring macrophage phagocytic signaling (20). Furthermore, lysosomes influence immune responses by modulating the lineage differentiation pathways of immune cells. After antigen stimulation, the mTOR signaling pathway becomes activated, guiding immune cells towards various functional lineage differentiations. Lack of mTOR in precursor cells, even when activated and maintaining IL-2 levels, fails to differentiate into Th1, Th2, or Th17 effector cells, instead favoring the differentiation into regulatory T cells (Tregs) (21). However, sustained activation of the mTOR signal may lead to T cell functional exhaustion, resulting in terminal differentiation and reduced proliferative potential (22), potentially weakening the anti-tumor response. Furthermore, evolutionarily conserved autophagy-related genes such as ATG5 are involved in the renewal, differentiation, and homeostasis maintenance of most immune cells (23). For instance, macrophages differentiated from peripheral blood monocytes, stimulated by granulocyte-macrophage colony-stimulating factor (GM-CSF), release BECL1 and undergo ATG5 cleavage, triggering autophagic signaling to promote the survival of differentiated macrophages (24). However, some studies have found that inhibiting autophagy can promote macrophage differentiation from myeloid progenitor cells (25). Additionally, efficient B cell development in the bone marrow and the maintenance of the number of peripheral B-1a B cells, independent of antigen influence, rely on the autophagy gene ATG5 (25). Activated B cells entering the germinal center require a transition from classical autophagy to non-canonical LC3-associated autophagy to differentiate into long-lived plasma cells and memory B cells (26). In summary, these findings suggest that lysosomes play a multifaceted role in shaping the immune response and its intensity regulation, contributing significantly to the modulation of the tumor immune microenvironment.
Tumor immunotherapy is a treatment approach that harnesses the body’s own immune system to recognize and eliminate cancer cells. It offers advantages such as high specificity, long-lasting effects, and minimal toxicity. It represents a new generation of cancer treatment methods that have rapidly developed following traditional treatments like surgery, radiation therapy, and chemotherapy (27, 28). Currently, immunotherapy methods applied in clinical practice primarily include immune checkpoint inhibitors (ICIs), adoptive cell therapy (ACT), and cancer vaccines, among others (29–31). In recent years, the pivotal role of lysosomes in supporting cancer cell survival and regulating the TME has garnered increasing attention. This has led to lysosomes being considered a promising therapeutic intervention target (32–34). Combining emerging technologies, various targeted lysosome-based combination immunotherapies have been developed. For instance, Tang et al. reported a pH-gated nano-adjuvant (PGN) that selectively targets lysosomes in tumor-associated macrophages (TAMs), mitigating lysosomal acidity and protease activity to polarize them towards an M1 phenotype, thereby enhancing antigen cross-presentation (35). Targeting lysosome degradation functions, on one hand, synergizes with ICIs. Lysosome-targeting chimeric molecules (LYTACs) have been designed to degrade cell membrane proteins (34), with one end binding to the target protein through antibodies or small molecules, and the other end being an oligosaccharide peptide, effectively targeting CI-M6PR through the endocytic/lysosomal pathway to successfully degrade epidermal growth factor receptor (EGFR) and PD-L1 (36). On the other hand, it can also synergize with tumor vaccines to improve vaccine specificity and enrichment. For instance, Deng et al. designed mRNA vaccines based on sialic acid-cholesterol derivatives, ensuring both dendritic cells (DCs) targeting and endosomal escape (37).
Targeting lysosome trafficking processes inhibits the endosomal sorting complex required for transport (ESCRT)-mediated repair of small membrane wounds in cancer cells, promoting the release of granular enzymes from tumor-infiltrating T cells (TILs) into the cytoplasmic solute (33). Targeting lysosome toll-like receptors (TLRs) activates innate immunity and modulates the responsiveness of TME to immunotherapy. For example, when the cancer vaccine NY-ESO-1 is combined with the TLR3 agonist poly-ICLC, the immune response against the cancer/testis antigen NY-ESO-1 is enhanced (38). Additionally, chloroquine, as the only autophagy inhibitor currently used in clinical cancer therapy, has been shown to significantly improve the therapeutic efficacy of combined ICIs in various cancers (16, 39, 40).
In summary, lysosome targeting, a novel cancer immunotherapy strategy, disrupts tumor-immune interactions in the TME through multiple mechanisms, thereby enhancing or synergizing with immunotherapy outcomes. Although the aforementioned is appealing and full of potential, it currently faces several challenges such as the need to overcome issues with selective drug delivery, safety, and resistance, as well as to explore further the precise underlying mechanisms.
2 Basic characteristics of lysosomes
Lysosomes, as the principal degradative component of eukaryotic cells, are responsible for the primary and ultimate degradation as well as recycling processes within the cell (1). In addition to this, lysosomes, in coordination with other organelles, also participate in various physiological processes such as cholesterol homeostasis, membrane repair, bone and tissue remodeling, pathogen defense, cell death, and cell signaling (4). These multifaceted functions render lysosomes a central and dynamic organelle, not merely confined to the terminal point of endocytic pathways (41). Lysosomes are encapsulated by a double-layered membrane, forming vesicles rich in specific soluble hydrolytic enzymes, integral membrane proteins, and lysosome-associated proteins. The lumen of lysosomes contains over 60 types of acidic hydrolytic enzymes, along with enzyme activators. They are responsible not only for intracellular degradation and proprotein processing but also play roles in antigen processing, degradation of the extracellular matrix, and initiation of apoptosis (5). The function of these hydrolytic enzymes is contingent upon the acidic pH (4.0-5.0) within the lumen, maintained by the vacuolar ATPase embedded in the limiting membrane (42). Furthermore, more than 30 known integral membrane proteins are highly glycosylated and serve functions such as protecting the lysosomal membrane from degradation, importing proteins from the cytoplasm, membrane fusion, and transporting degradation products to the cytoplasm, ion exchange, etc. Examples include lysosome-associated membrane proteins 1 and 2 (LAMP1/2), lysosome integral membrane protein 2 (LIMP2), and SNAREs (43, 44). Specific lysosome-related molecules are detailed in Table 1. Most importantly, the recruitment of the mechanistic target of mTOR1 to lysosomes is facilitated by RAG-GTPases, coupling amino acid supply with cell growth and autophagic signals (3). Triggered in a manner dependent or independent of mTOR1, TFEB undergoes nucleocytoplasmic shuttling, regulated by phosphorylation. This promotes the formation of autophagosomes, autolysosomes, and lysosome-dependent cell death, among other processes (45). Rab GTPases recruit and activate tethering factors for contact with other organelles, coupling lysosomes with motor complexes, such as dynein-dynactin and kinesin motors, which drive vesicle movement along microtubules (46).
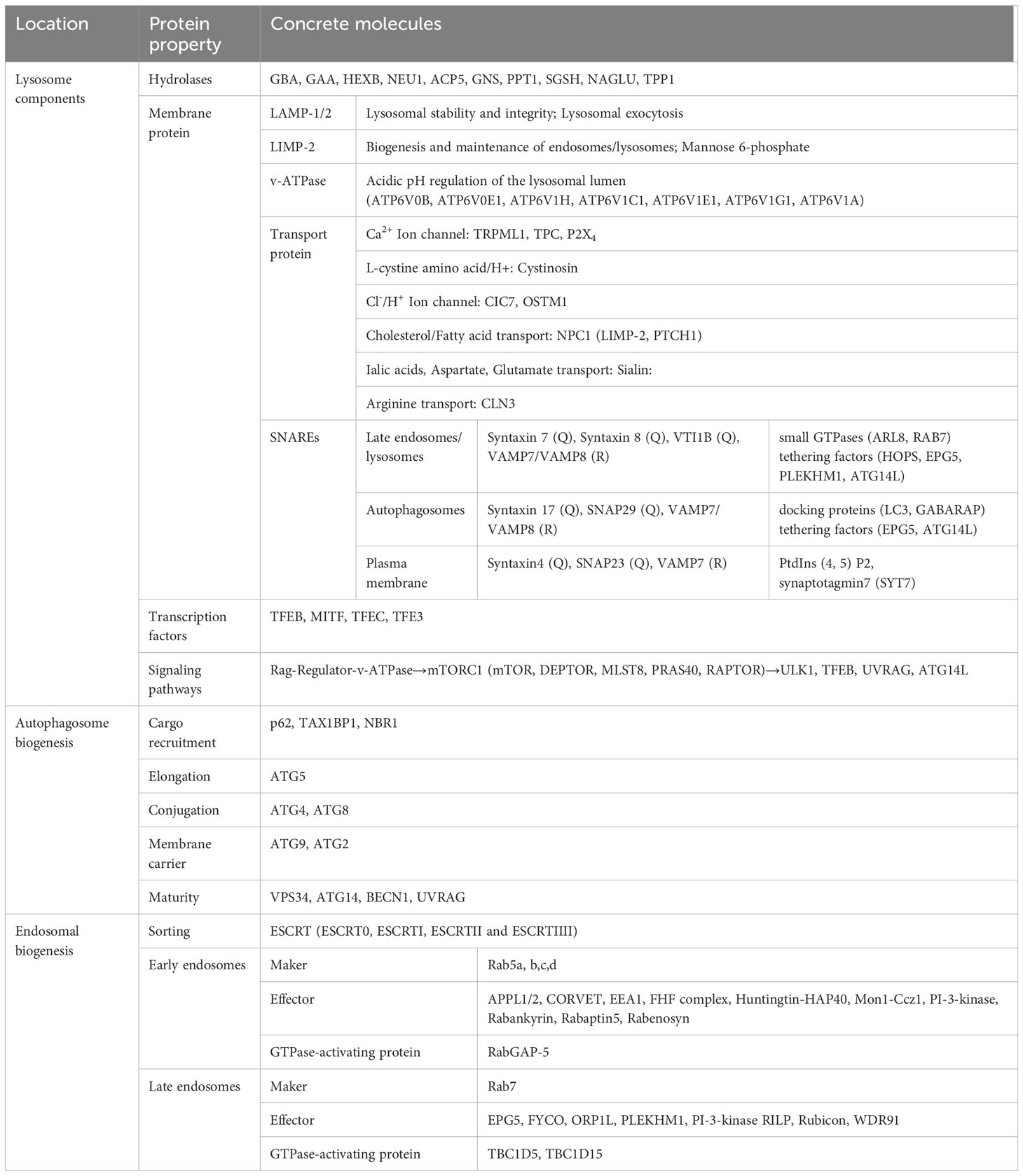
Table 1 Lysosomal component functions.
Currently, the majorly reported lysosomal transport pathways encompass autophagy, endocytosis, and phagocytosis. Lysosomes play a crucial role in autophagy. Autophagic transport pathways are cellular mechanisms that rely on lysosomal degradation to metabolize, clear, and recycle damaged proteins and organelles, thereby preserving cellular and metabolic homeostasis. These pathways encompass micro-autophagy, macro-autophagy, and chaperone-mediated autophagy.
Micro-autophagy: In micro-autophagy, cargo is directly sequestered onto lysosomes with the assistance of HSC70, ESCRT, and SNARE assembly. Following internalization via lysosomal membrane protrusions, invagination, or endosome invagination, the cargo undergoes degradation facilitated by lysosomal hydrolases (7).
Macro-autophagy: Macro-autophagy is responsible for degrading various subcellular materials and ubiquitin-labeled targets. During cellular stress, mTORC1 is inhibited, promoting the phosphorylation of the ULK1-FIP200-ATG13 initiation complex. This subsequently activates the Beclin-1-PI3KC3-VPS15/p150 complex, leading to phagophore elongation, which subsequently closes to form mature double-membrane-bound autophagosomes (12, 47–49). Autophagosomes are transported closer to lysosomes through the STX17-SNAP29-VAMP8 complex and recruit HOPS for membrane fusion with lysosomes, forming autolysosomes (50, 51). Within the autophagic flux process, cargo undergoes degradation and recycling.
Chaperone-mediated autophagy (CMA): Certain selected proteins containing KFERQ-like amino acid sequences or modified structures can also be targeted for degradation via CMA (52). KFERQ-like sequences facilitate protein binding to Hsc70, forming a complex with other co-chaperones such as HSP40, HIP, HOP, and HSP90. This complex then binds to lysosome-associated membrane protein 2A (LAMP2A) on lysosomal membranes (53). When the GXXG motif in the structural membrane remains intact, LAMP2A forms multimers, resulting in the formation of a translocation site that provides a transmembrane channel for the attached protein (52, 54).
The internalization of extracellular fluids or particles into cells via the formation of small or large vesicles derived from the plasma membrane is known as endocytosis and phagocytosis, respectively. These processes represent unique ways in which lysosomes contribute to lysosomal biogenesis (8, 55). Proteins originating from the plasma membrane or the trans-golgi network (TGN), such as M6PRs carrying lysosomal hydrolases, form early endosomes through the constitutive secretory pathway and are marked by Rab5 and EEA1 (56). During this process, lysosome-associated proteins from the endoplasmic reticulum (ER) can directly or indirectly join in (57). Early endosomes contain structures known as tubular sorting endosomes (TSE) that transport recycling proteins back to the plasma membrane or TGN (58). Meanwhile, ESCRT sorting mechanisms primarily guide the degradation of ubiquitin-tagged proteins in endosomes and regulate the formation of early endosomes, as exemplified by the EGFR (6, 59). Additionally, there exist ESCRT-independent sorting mechanisms that enrich proteins like CD63 and MHC-II in exosomes secreted into the extracellular space (60, 61). Mon1-Ccz1 and Rab11 mediate fusion and fission events, giving rise to late endosomes. Upon activation of the Rab7 GTPase enzyme, late endosome positioning is regulated through microtubule-dependent motor proteins RILP and FYCO1, and membrane fusion occurs through SNARE complexes, leading to heterotypic or homotypic fusion and the formation of lysosome-related organelles (57). Ultimately, this process results in the degradation of cargo within lysosomes. The relevant summary is shown in Table 1.
3 Lysosome-associated autophagy and tumor immunity
Autophagy is a process in which cellular contents are encapsulated in double-membrane vesicles and fused with lysosomes for degradation, which removes damaged or redundant organelles, proteins, and other materials from cells (62). The process of autophagy includes the formation of autophagosomes, the fusion of autophagosomes with lysosomes, and the degradation of autophagic substances in lysosomes. Lysosomes are the end point of autophagy and where autophagic substances are recycled or excreted from the cell (63). Autophagy and lysosomes have important effects on cellular metabolism, signal transduction, immune response, and death. The balance between autophagy and lysosomes is important for maintaining cellular homeostasis and coping with various stresses. Imbalance between autophagy and lysosomes may lead to various diseases such as neurodegenerative diseases, infectious diseases and tumors (64). Autophagy has a double-edged role in tumorigenesis and progression, inhibiting tumor formation as well as promoting tumor survival and drug resistance. Autophagy also affects tumor immunity in a variety of ways, including regulating tumor cell immunogenicity, antigen presentation, cytokine secretion, and immune checkpoint protein expression may be positive or negative, depending on the different cell types, tumor types, and treatment conditions (65).
Among the negative effects of autophagy on tumor immunity, on the one hand, autophagy reduces the expression of MHC-I molecules on the surface of tumor cells by targeting them to the lysosome for degradation or by blocking their assembly in the endoplasmic reticulum, thus reducing the visibility and sensitivity of tumor cells to CD8+ T cells (66). Alec C. Kimmelman et al. found that in pancreatic ductal adenocarcinoma (PDAC) cells, MHC-I molecules are selectively targeted to lysosomes for degradation rather than presenting antigen at the cell surface. This process is dependent on autophagy, specifically the autophagy cargo receptor NBR1, which binds to MHC-I and delivers it to autophagosomes and lysosomes. Knockdown or pharmacological inhibition of autophagy-related genes or proteins reduces the degradation of MHC-I in lysosomes and restores the level of MHC-I on the surface of PDAC cells. In an mouse model in vivo, autophagy inhibition reduces PDAC cell degradation in lysosomes and enhances PDAC cell killing by CD8+ T cells (16). On the other hand, autophagy can regulate the function of immune cells by affecting their differentiation, polarization, activation, migration, and other processes, thereby attenuating their killing effect on tumor cells or enhancing their promoting effect on tumor cells. Panayotis Verginis et al. found that lysosomes function in myeloid-derived suppressor cells (MDSCs) not only in degradation and recycling, but also in the regulation of signal transduction and gene expression (67). The mTORC1 is an important regulator of cell growth and metabolism. When the amino acid level in lysosomes is high, the receptor on the surface of lysosomes activates mTORC1 and promotes the proliferation and maturation of MDSCs. When autophagy is enhanced, the amino acid level in lysosomes decreases, leading to inhibition of mTORC1, which affects the function of MDSCs (67). Xu et al. found that in renal cell carcinoma (RCC) patients, high expression of LAPTM4B was associated with a significant increase in M2-type macrophages in the TME, which is thought to be associated with tumor metastasis. LAPTM4B is a lysosome-associated gene that regulates the number and function of lysosomes and affects the level and efficiency of autophagy, and it was found that LAPTM4B was expressed at different levels in different types of renal cell carcinoma cells and was associated with lysosomal and autophagic pathway activity. The positive effect of autophagy on tumor immunity can also be attributed to the modulation of immune cell function, which can weaken the killing effect of immune cells on tumor cells or enhance the promotion effect of immune cells on tumor cells (68). The role of autophagy in nuclear factor-kappa B (NF-κB) regulation was explored in a study by Lei et al. by using a macrophage differentiation model. The authors found that lysosome-dependent pathways lead to the translocation of NF-κB p65 from the nucleus to aggregates (ALS) and degradation through selective autophagy. The lysosomal inhibitor bafilomycin A1 prevented the degradation of NF-κB p65, allowing M2 macrophages to produce high levels of proinflammatory factors. In addition lysosomal dysfunction leads to accumulation of NF-κB p65 in ALS, increasing immunosuppression in M2 macrophages (69).
4 Lysosome-related metabolism and tumor immunity
4.1 Lysosomal lipid peroxidation
Lipid peroxidation is a biochemical reaction that refers to the formation of peroxides when unsaturated fatty acids or their derivatives lose hydrogen atoms in response to oxidative stress. Lipid peroxides are highly reactive and toxic and can damage cell membranes, proteins, DNA and other biomolecules (70). Lysosomes are intracellular digestive organs that degrade various biomolecules, including lipids. lysosomal lipid peroxidation (LLP) and cell death induced by the lysosomal inhibitor DC661 was found to be independent of known programmed cell death pathways (autophagy, apoptosis, necroptosis, iron-death, and inflammatory death) as well as iron, calcium, or proteolytic enzymes in lysosomes in a study by Ravi K. Amaravadi et al. LLP is a mechanism that drives lysosomal cell death and a unique form of immunogenic cell death (ICD). ICD refers to a mode of cell death that activates an anti-tumor immune response and is characterized by the surface or release of a number of immune-stimulating molecules, such as calreticulin (CRT), HMGB1, and ATP, from tumor cells. LLP-induced ICD enhances the immunogenicity of tumor cells, allowing them to be recognized and cleared by the immune system. LLP also activates terminally differentiated T cells and enhances T cell-mediated killing. Tumor cells treated with the LLP inducer DC661 can serve as an effective tumor vaccine, inducing an adaptive immune response and achieving tumor rejection in “immunologically hot “ tumors (71). Hence lysosomal inhibitors can be used as a novel ICD inducer that works synergistically with other immunotherapeutic tools, such as ICIs and CAR-T cells, to improve the efficacy and breadth of tumor immunotherapy.
4.2 Hypoxia and lysosomes
Hypoxia or inadequate tissue oxygenation is a critical factor in the difficulty of eradicating most cancers (72). In hypoxic conditions, tumor cells activate the hypoxia-inducible factor (HIF)-1α, which induces many downstream target genes, including vascular endothelial growth factor (VEGF), and promotes tumor angiogenesis, gene expression, exosome secretion, etc., resulting in stronger proliferation, invasion, and metastasis of tumor cells (73). The hypoxic microenvironment also affects the level of lysosomal function and thus tumor immunity. The research by Manran Liu et al. suggests that under hypoxic conditions in breast cancer’s Cancer-Associated Fibroblasts (CAFs), lysosomal dysfunction occurs where autophagosomes fail to fuse with lysosomes but instead merge with multivesicular bodies (MVBs), promoting exosome release and subsequently enhancing cancer cell invasion and metastasis (74). Specifically, the dysregulated ATM is oxidatively activated in CAFs and phosphorylates a key lysosomal proton pump, ATP6V1G1, leading to impaired lysosomal acidification. Concurrently, the oxidized ATM phosphorylates BNIP3, inducing autophagosome accumulation and exosome release. Additionally, Zhang et al. found that hypoxia significantly increases the number of extracellular vesicles (EVs) secreted by HNSCC cells (75). In a hypoxic environment, HIF1a negatively regulates the expression of a V-ATPase member, ATP6V1A, disrupting the acidic environment within lysosomal lumen and lysosomal integrity, thus reducing MVB and lysosome fusion and promoting the release of intraluminal vesicles as EVs (76). Tumor EVs can interact with immune cells, affecting immune responses. Tumor EVs surface PD-L1 can bind to CD8+ T cell surface programmed cell death protein 1 (PD-1), inhibiting CD8+ T cell activation and proliferation; surface ICAM-1 can interact with T cell surface LFA-1, promoting EVs PD-L1 mediated immune suppression; tumor EVs can also induce immune cell apoptosis or differentiation into immunosuppressive cells (77, 78). These studies indicate that hypoxia in tumor cells can affect lysosomal function and regulate EVs secretion, making lysosome-EVs a novel target for tumor immune therapy.
Additionally, HIF1a is a critical transcriptional regulator that enables metabolic adaptation in cells under low oxygen levels (79). The stability and activity levels of HIF1a are controlled by various mechanisms. Previous studies have discovered that HIF1a can bind to HSC70 and LAMP2A for transport and become destabilized in lysosomes via the CMA pathway (2). Using lysosomal inhibitors can rescue lysosomal expression of HIF1a in epithelial tumor cells. However, recent research indicates that in melanocytes, the degradation of HIF1a protein is not sensitive to chloroquine but instead is associated with the Golgi apparatus. Authors using Brefeldin A to disrupt endoplasmic reticulum-Golgi transport found that HIF1a is localized to the nucleus and its transcriptional activity is decreased. These results suggest that in cells undergoing different degradation pathways, there may exist several distinct pools of HIF1a (80). In tumor cells, HIF1a might act not only as a molecular sensor for oxygen levels but also to monitor protein-folding capacities, ensuring cancer cells adapt to hypoxic conditions and avoid the detrimental effects of low oxygen. Therefore, the relationship between HIF1a and various organelles remains complex and necessitates further, more definitive research.
4.3 Glucose metabolism and lysosomes in tumor immune cells
The relationship between lysosomes and glucose metabolism is a newly discovered area of research with important physiological and pathological implications. Lysosomes are not only able to utilize glucose as an energy source, but also sense glucose availability and regulate cell metabolism, growth and survival through signaling pathways (81). Cao et al.’s study found that M2-type TAMs had the highest individual uptake of intra-tumor glucose, and its increased glucose uptake promoted hexosamine biosynthesis pathway (HBP)-dependent O-GlcNAcylation modification, which enhanced the pro-cancer function of TAMs. Glucose metabolism mediates O-GlcNAcylation modification of Cathepsin B at the Ser210 site via lysosomal OGT and increases the level of mature Cathepsin B in macrophages and its secretion in the TME (82). Cathepsin B is a lysosomal protease that degrades the extracellular matrix and promotes tumor cell invasion and migration (83). This suggests that lysosomal OGT, a key molecule of glucose metabolism in regulating the pro-cancer function of TAM, affects lysosomal function and tumor biological behavior by modifying Cathepsin B via O-GlcNAcylation. This finding provides new insights and ideas for tumor immunotherapy.
5 Important influence of lysosomes on antigen presentation process
Antigen presentation refers to a process in the immune system that involves a number of APCs capturing, processing, and presenting antigenic fragments for recognition by T cells (84). Antigen presentation is categorized into two types, which are MHC-I and MHC-II (85). MHC is a polymorphic protein that binds antigenic fragments and transports them to the cell surface (86). MHC-I antigen presentation refers to the presentation of endogenous antigens, i.e., antigens originating from within the cell, such as viral or tumor proteins, by all nucleated cells (as well as platelets) via MHC-I molecules. These antigens are degraded by the proteasome into small peptide segments, which are then transported through the transporter protein associated with antigen presentation (TAP) to the ER, where they bind to MHC-I molecules and are transported to the cell surface. This approach allows CD8+ T cells to recognize and kill infected or mutated cells (85, 86). MHC-II antigen presentation refers to the presentation of exogenous antigens, i.e., antigens originating from outside the cell, such as bacteria or parasites, by some specialized APCs via MHC-II (87). These antigens, after being phagocytosed or endocytosed by APCs, are hydrolyzed into small peptide fragments in endosomes or lysosomes, which then bind to MHC-II molecules and are transported to the cell surface. This approach allows CD4+ T cells to recognize and provide assistance signals to other immune cells (88, 89).
Lysosomes provide raw material for antigen presentation by breaking down phagocytosed or endocytosed antigens into small peptide fragments via hydrolytic enzymes. Lysosomes play an important role in tumor immunity, on the one hand, they can participate in the MHC-I inhibitory axis of tumor cells, thus reducing the expression of MHC-I on the cell surface and allowing tumor cells to evade recognition and clearance by T cells. Jun Wang et al. identified a pan-tumor cell surface MHC-I inhibitory axis (STW axis) dominated by the membrane proteins SUSD6 and TMEM127. The two membrane proteins, SUSD6 and TMEM127, can bind to MHC-I and recruit the E3 ubiquitin ligase WWP2 to ubiquitinate MHC-I, which in turn promotes the endocytosis of MHC-I from the cell surface. After endocytosis, MHC-I is transported to early endosomes and then degraded in lysosomes by fusion with lysosomes. And the lysosomal inhibitor chloroquine inhibits SUSD6 and TMEM127-mediated MHC-I degradation, thereby increasing MHC-I expression on the cell surface (87). Li et al. also found that PCSK9 physically binds to MHC I and promotes its degradation in lysosomes, thereby reducing its recycling and reutilization on the cell surface, and confirmed that the degradation of MHC-I by PCSK9 occurs via the lysosomal pathway using drug intervention experiments (90). Keisuke Yamamoto et al. found that MHC-I in PDAC cells is mainly located in autophagosomes and lysosomes rather than on the cell surface (16). MHC-I is recognized by the autophagy receptor NBR1 and selectively transported to lysosomes for degradation via the autophagy pathway. Inhibition of autophagy or lysosomes restores MHC-I expression on the surface of PDAC cells, which enhances antigen presentation, activates CD8+ T cells and DCs, and inhibits tumor growth (16).Zou et al. found that optineurin deficiency impaired the integrity of the IFN-γ and MHC-I signaling pathways through palmitoylation-dependent IFNGR1 lysosomal transport and degradation, which drove immune escape and endogenous immunotherapy resistance in rectal cancer (91). On the other hand, lysosomes can promote tumor cell MHC-II antigen presentation, thereby enhancing the effects of immune surveillance and immune checkpoint blockade therapy. Merkel cell carcinoma (MCC) is a rare and highly aggressive neuroendocrine skin cancer, with most cases associated with Merkel cell polyomavirus (MCPyV) infection (92). Teri Heiland et al. showed that a cancer vaccine was designed to promote a potent, antigen-specific CD4+ T-cell response to MCPyV-LT. The authors utilized their nucleic acid platform UNITE™ (UNiversal Intracellular Targeted Expression), which fuses tumor-associated antigens to LAMP1 (93). LAMP1 is a glycoprotein localized on lysosomal and endosomal membranes that transports antigen from the cytoplasm to the lysosome, thereby enhancing antigen presentation on MHC-II (94). This lysosomal targeting technology enhances antigen presentation and balanced T-cell responses. LAMP1-MCPyV-LT induced stronger CD4+ T cell responses and higher levels of anti-MCPyV-LT antibodies in mice compared to MCPyV-LT without LAMP1 fusion (93). Yifan Ma et al. designed a self-adjuvanted nanovaccine (SANV) in a study that constructed an individualized tumor immunotherapy strategy using a pH-sensitive galactosylated dextran-retinoid (GDR) nanogel as a carrier and the patient’s own tumor cell lysates as antigens. The nanogel is pH-sensitive and capable of releasing antigens and retinoids in acidic lysosomes, the latter of which activates the retinoic acid receptor (RAR) signaling pathway and promotes DCs maturation. In addition, it induces lysosomal cleavage, leading to intracellular reactive oxygen species (ROS) production, which enhances proteasome activity and MHC-I molecule-mediated antigen cross-presentation, activates the CTL response, and inhibits the differentiation and function of Tregs and TAMs (95).
Furthermore, lysosomes regulate the efficiency and quality of antigen presentation, by controlling the extent and timing of antigen degradation, as well as influencing the stability and expression levels of MHC molecules. A study conducted by Sanjay Garg et al. designed a novel microencapsulated cancer vaccine (egg white-loaded pH/redox dual-sensitive microencapsulated vaccine, OLM-D) that combines an antigen with a homemade amphiphilic poly(l-histidine)-poly(ethylene glycol) (PLH-PEG) via cleavable covalent binding. Lysosomal escape is the process by which endocytosed material is released from the lysosome into the cytoplasm, which enhances antigen processing and presentation within the cell. OLM-D rapidly releases egg white under acidic or reducing conditions, leading to lysosomal rupture and ROS production, which triggers lysosomal escape. OLM-D enhances antigen processing and presentation by DCs through lysosomal escape, thereby activating the immune response of T cells (96).
6 The crucial impact of lysosomes on immune checkpoint molecules
Immune checkpoints are certain molecules that can regulate the activation and function of immune cells, maintaining immune homeostasis and self-tolerance (97). Several immune checkpoint molecules, such as PD-L1, can be present on the cell surface or intracellularly and inhibit T cell responses by binding to receptors. There are interactions and regulation between immune checkpoint molecules and lysosomes (98). On one hand, lysosomes can regulate their expression levels and functions by degrading immune checkpoint molecules. On the other hand, immune checkpoint molecules can also influence the formation and function of lysosomes. There exists a dynamic balance and mutual regulation between immune checkpoint molecules and lysosomes, which is significant for the proper functioning of the immune system and cancer immunotherapy (99, 100).
6.1 PD-1 and PD-L1
PD-L1 is an immune checkpoint molecule that can bind to the PD-1 receptor, inhibiting the activation and proliferation of T cells, thereby allowing tumor cells to evade immune surveillance (101). Antibodies targeting PD-L1 can disrupt the interaction between PD-L1 and PD-1, activating T cells to exert cytotoxic effects on tumors. This is an effective cancer immunotherapy approach. However, PD-L1 is not only present on the cell surface but also within cells, with its expression levels dynamically regulated through dynamic transport and degradation mechanisms (102). Therefore, merely blocking the surface molecules of PD-L1 with antibodies may not be sufficient to fully inhibit PD-L1’s function. PD-L1 is expressed on the cell membrane but can also be internalized and transported to lysosomes for degradation. The degradation process involves various molecular modifications, including glycosylation, phosphorylation, and palmitoylation (99, 102).
A study by Xu et al. discovered a novel mechanism regulating PD-L1 expression and function, namely, palmitoylation modification. Palmitoylation is a lipid modification, which refers to the covalent attachment of palmitic acid to cysteine residues on proteins via a thioester bond. This modification alters the protein’s hydrophobicity, stability, subcellular localization, and function, allowing the protein to anchor to the cell membrane (103). The authors demonstrated the presence of a palmitoylation site in the cytoplasmic domain of PD-L1, which can undergo palmitoylation modification catalyzed by the enzyme DHHC3. This modification inhibits the ubiquitination of PD-L1, thereby preventing its degradation in the lysosome, enhancing the stability and function of PD-L1. Inhibiting the palmitoylation of DHHC3 or PD-L1 through pharmacological or genetic methods can reduce the expression of PD-L1 in tumor cells, enhancing the cytotoxic effect of T cells against the tumor. The authors also designed a competitive inhibitor that can selectively target the palmitoylation site of PD-L1, thereby inhibiting the expression and function of PD-L1 and enhancing the immune response of T cells against tumors. Hence, lysosomes play a crucial role in regulating the expression and function of PD-L1 (102).
The glycosylation of PD-L1 on the cell membrane is a crucial regulatory factor for its stability and function, affecting the affinity and signal transduction between PD-L1 and PD-1 (104, 105). Zhou et al.’ research also investigated how glycosylation modifications of programmed cell death ligand 2 (PD-L2) promote immune evasion and resistance to anti-EGFR therapy in tumor cells (106). PD-L2 is an immune checkpoint molecule capable of binding to PD-1, and its expression on the surface of tumor cells can inhibit the activity and function of T cells (107, 108). The authors have discovered a novel enzyme, FUT8, which can perform N-glycosylation modifications on PD-L2 and enhance its binding to EGFR. Glycosylation-modified PD-L2 activates the EGFR/STAT3 signaling pathway while reducing the affinity of cetuximab for EGFR. Glycosylation modifications also stabilize the protein levels of PD-L2 by blocking the ubiquitin-dependent lysosomal degradation pathway, thereby enhancing the binding of PD-L2 to PD-1 and the immune evasion effect (106).
The phosphorylation of PD-L1 is a crucial step in its intracellular trafficking and degradation, involving various kinases and phosphatases (109). The known phosphorylation sites of PD-L1 currently include S195, S197, and Y248. The phosphorylation site primarily involved in the lysosomal degradation pathway is S197 (S195 phosphorylation site is associated with PD-L1 degradation pathway dependent on the endoplasmic reticulum, while Y248 is located in the extracellular region of PD-L1, primarily enhancing the affinity and signal transduction between PD-L1 and PD-1) (109). GSK3β is a multifunctional kinase that can participate in various signaling pathways and biological processes (110). The phosphorylation of PD-L1 by GSK3β or AMPK promotes its translocation from the cell membrane to lysosomes. Therefore, GSK3β or AMPK can reduce the stability and function of PD-L1 on the cell membrane in this manner, enhancing T cell-mediated cytotoxicity against tumor cells (111).
Additionally, a study by Mark A. Dawson et al. also found the significant role of lysosomes in the degradation of PD-L1. They utilized CRISPR-Cas9 technology to perform a whole-genome knockout screen in the pancreatic cancer cell line BxPC-3 and discovered an unknown protein, CMTM6. CMTM6 is a recently discovered regulator of PD-L1 expression. It does not affect the maturation of PD-L1 but co-localizes with PD-L1 on the cell membrane and recycling endosomes, preventing PD-L1 from being transported to lysosomes for degradation, thus maintaining its stability on the cell surface (18, 112). Additionally, a study by Zhang et al. investigated how targeting the free fatty acid receptor 4 (GPR84) in MDSCs can overcome resistance to PD-1 immune therapy in esophageal cancer (113). These studies have all revealed the crucial role of the lysosomal degradation pathway of PD-L1 in regulating immune evasion by tumor cells, providing new targets and mechanisms for the development of novel immunotherapy strategies. On the other hand, immune checkpoint molecules can also influence the formation and function. A study by Deng et al. found that tubeimoside-1 (TBM-1) can significantly reduce the expression of PD-L1 on the surface of various cancer cells (113). TBM-1 can activate TFEB, a key regulator of lysosome biogenesis, by inhibiting the activation of mTORC1. TFEB can transcriptionally activate various lysosome-related genes, including LAMP1, LAMP2, CTSD, etc., thereby increasing the quantity and functionality of lysosomes (114). TFEB can also promote the lysosomal degradation of PD-L1 by binding to the promoter region of its encoding gene CD274, inhibiting its transcription. In this way, TFEB can reduce the surface levels of PD-L1 on cancer cells, enhancing the recognition and killing of cancer cells by immune cells. In addition, TBM-1 can also enhance the TME through TFEB, increasing the quantity and activity of TILs while reducing the number and functionality of immunosuppressive cells (MDSCs and Tregs). These effects are associated with the degradation of PD-L1 by TBM-1, as the impact of TBM-1 on the TME disappears in PD-L1 knockout cancer cells (113) (Figure 1).
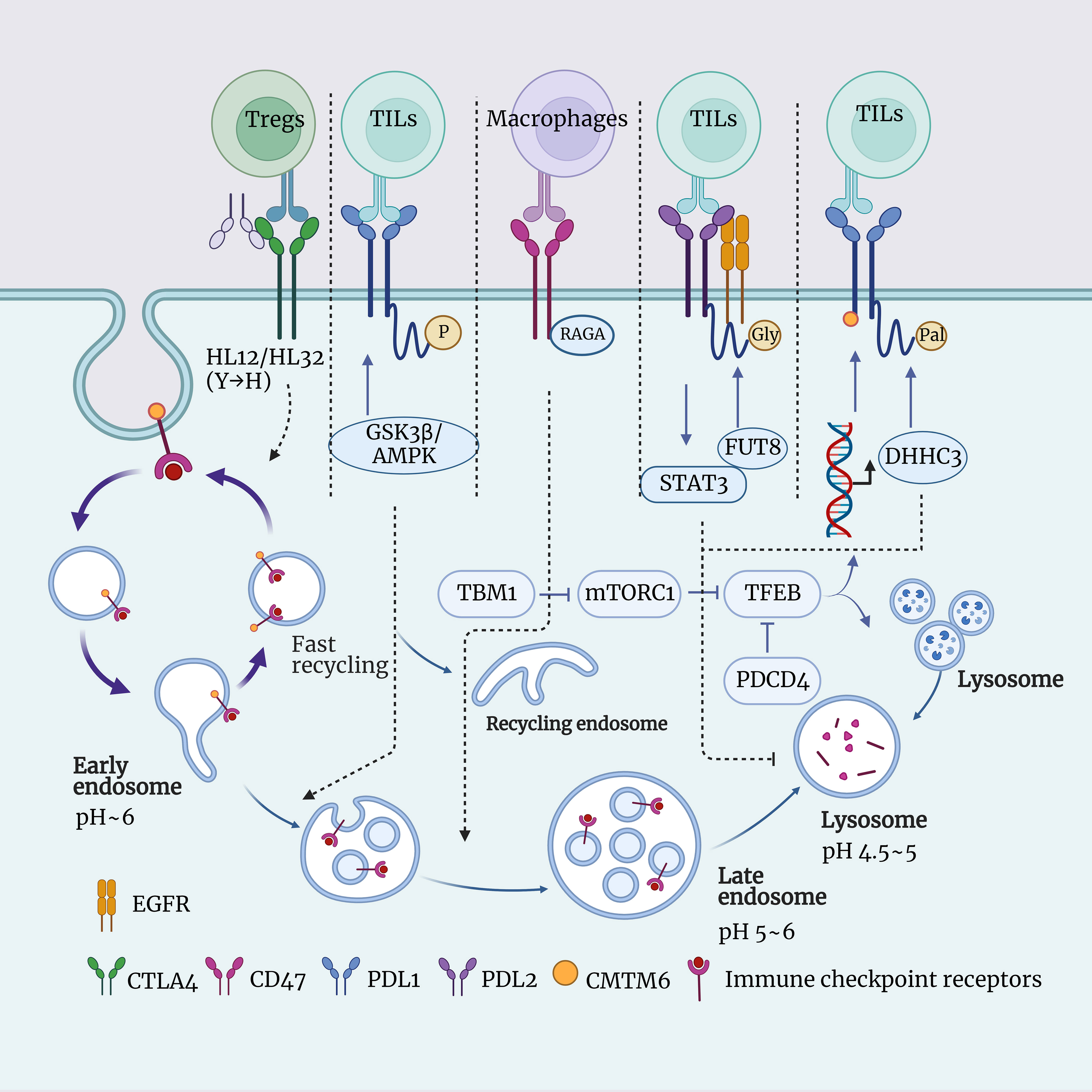
Figure 1 Schematic representation of the interaction between lysosomes and immune checkpoints. This figure illustrates the role of lysosomes in immune checkpoint signaling, including CTLA4, PDL1, PDL2, CD47, and more. Lysosomes are intracellular digestive organelles capable of degrading materials from phagocytosis or autophagy. Lysosomes also have the ability to modulate the expression and function of immune checkpoints, thus influencing the balance of immune responses. The aim is to highlight the significant role of lysosomes in immune checkpoint signal transduction and their potential as therapeutic targets.
6.2 Cytotoxic t lymphocyte-associated protein-4
CTLA-4 is another common immune checkpoint molecule that regulates the activity of T cells by binding to CD80/CD86, preventing the occurrence of autoimmune diseases (115). The expression of CTLA-4 on the surface of T cells is dynamically regulated. It can be internalized into intracellular vesicles through endocytosis, and then it can either return to the cell surface through recycling or be sent to lysosomes for degradation (116, 117). When CTLA-4 is transported to the lysosome, it is hydrolyzed by enzymes into smaller fragments, thereby reducing the quantity of CTLA-4 on the surface of T cells, which impacts the function of T cell (118). The anti-CTLA-4 monoclonal antibody (mAb) is a means of cancer immunotherapy that can enhance the cytotoxicity of T cells against tumor cells by blocking the interaction between CTLA-4 and CD80/CD86 (119). The anti-CTLA-4 mAb currently used in clinical practice, such as Ipilimumab and Tremelimumab, all lead to the degradation of CTLA-4 in lysosomes, thereby reducing the function of Tregs and increasing the risk of immune-related adverse events (irAEs). However, this also results in an improvement in cancer immunotherapy effectiveness (119, 120). Yang Liu et al.’ research has identified a novel anti-CTLA-4 mAb, HL12 or HL32. These antibodies can dissociate from CTLA-4 after endocytosis and, through an LRBA-dependent mechanism, facilitate the recycling of CTLA-4 to the cell surface. This, in turn, preserves the functionality of Treg cells, reducing the occurrence of irAEs, but simultaneously limiting the improvement in cancer immunotherapy effectiveness. Furthermore, to enhance the bioavailability of antibodies, the authors introduced a mutation from histidine (H) to tyrosine (Y). This mutation increased the pH sensitivity of the anti-CTLA-4 mAbs, allowing it to dissociate from CTLA-4 in acidic environments, thus avoiding lysosomal degradation. These pH-sensitive anti-CTLA-4 mAbs have been shown to more effectively deplete Tregs within tumors and eradicate established tumors (120). This is a new paradigm in cancer research, wherein altering the pH sensitivity and recyclability of anti-CTLA-4 mAbs through lysosomal characteristics can simultaneously enhance its safety and efficacy (Figure 1).
6.3 Others
Programmed cell death factor 4 (PDCD4) is a tumor suppressor associated with cell cycle and apoptosis, capable of modulating various cellular processes such as autophagy, inflammation, transformation, and invasion by inhibiting the function of eukaryotic initiation factor 4A (eIF4A), thereby influencing protein translation (121). PDCD4 is downregulated or lost in various tumors and is associated with the occurrence, progression, and prognosis of cancer (122). The research by Zhang et al. found that PDCD4 can reduce the overall levels of TFEB, thereby inhibiting its accumulation and transcriptional activity within the cell nucleus, ultimately leading to a decrease in the number and function of lysosomes (123). TFEB is a transcription factor that regulates various cellular processes, including lysosome biogenesis, autophagy, lysosomal exocytosis, lipid metabolism, and energy metabolism. Its activity is tightly controlled by multiple post-translational modifications, protein interactions, and spatial distribution (124). And it has been demonstrated that the inhibition of lysosome function by PDCD4 depends on TFEB, and in the TME, PDCD4 deficiency can promote the anti-tumor effect of macrophages by enhancing TFEB expression (123) (Figure 1).
CD47 is an immune checkpoint, which is a molecule capable of regulating immune responses (125). CD47 primarily inhibits the phagocytosis of tumor cells by macrophages by binding to the surface receptor SIRPα and sending a “don’t eat me” signal (125). In this way, tumor cells can evade the clearance of the immune system, leading to immune escape. Therefore, drugs targeting CD47 or SIRPα can block this signal, restoring the phagocytic function of macrophages, thereby achieving anti-tumor effects (19). Currently, there are various forms of drugs under development, including CD47 antibodies, SIRPα fusion proteins, SIRPα antibodies, and CD47 bispecific antibodies, among others. These drugs can not only enhance the phagocytosis of tumor cells by macrophages but also activate other immune cells such as T cells and NK cells, inducing apoptosis in tumor cells, and more (19, 126). The research by Jin et al. found that RAGA can bind to CD47 and promote the transport of CD47 from late endosomes to lysosomes for degradation (20). The downregulation of RAGA leads to the accumulation of CD47 on the surface of LUAD cells, enhancing the binding of CD47 to SIRPα and inhibiting macrophage phagocytosis of LUAD cells (20). The current relationship between CD47 and lysosomes has not been fully elucidated. However, in the future, by exploring the impact of CD47 degradation in lysosomes on other signaling pathways and biological functions, the development of interventions targeting the CD47 lysosomal degradation process holds significant promise for enhancing the efficacy of cancer immunotherapy (Figure 1).
7 Lysosomal effects on different immune cells in the TME
7.1 TAMs
TAMs are the most abundant immune cells in the TME and can be classified into M1 and M2 types based on their function and phenotype (127). M1 macrophages exhibit anti-tumor activity, secreting pro-inflammatory cytokines and cytotoxic molecules, activating T cells and natural killer cells. M2 macrophages, on the other hand, promote tumor growth, secretion of anti-inflammatory cytokines and growth factors, facilitating tumor growth, metastasis, angiogenesis, and immune evasion (128).
Lysosomes play a crucial role in regulating the polarization and function of TAMs. On one hand, lysosomes can influence the signaling pathways of TAMs, thereby affecting their functional outputs. Wei et al.’ research revealed that LAMP2a is upregulated in TAMs and plays a significant role in tumor progression (129). Inducing LAMP2a inactivation through shRNA or CRISPR/Cas9 can impede TAMs activation and tumor growth. LAMP2a degradation leads to the promotion of tumor-activating macrophages by degrading peroxiredoxin 1 (PRDX1) and CREB-regulated transcription coactivator 1 (CRTC1). In addition, extracellular vesicles released by tumor cells (T-MPs) can also influence macrophage polarization by affecting the characteristics of lysosomes (129). Lysosomes are vesicles containing acidic hydrolytic enzymes that can degrade engulfed substances. When T-MPs enter lysosomes, they disrupt lysosomal function, leading to an increase in lysosomal pH and the release of calcium. The molecular events enable T-MPs to induce the transformation of M2 macrophages into M1 type through the lysosome-dependent pathway. The DNA signals carried by T-MPs can activate the cGAS-STING-TBK1-STAT6 signaling pathway within macrophages, thereby inducing macrophages to express anti-inflammatory factors such as IL-10 and TGF-β, forming M2-type macrophages (129). However, when T-MPs are delivered into lysosomes, they release DNA fragments and bind with cGAS, thereby activating the STING-TBK1-IRF3 signaling pathway, inducing macrophages to express pro-inflammatory cytokines such as IFN-β and TNF-α, leading to the formation of M1-type macrophages. Furthermore, the long-chain non-coding RNA signals carried by T-MPs can also induce macrophages to release pro-inflammatory cytokines, such as IL-1β, leading to the formation of M1-type macrophages. This effect is mediated by the long-chain non-coding RNA within T-MPs binding to TLR7 or TLR8 and being recognized and activated within lysosomes (129).
On the other hand, lysosomes can influence the metabolic state of TAMs, thereby affecting their phenotypic transition. Fan et al.’ research found that TFEB upregulates the expression of cytokine signaling suppressor protein 3 (SOCS3) and peroxisome proliferator-activated receptor gamma (PPARγ), as well as autophagic/lysosomal activity. This inhibition suppresses the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome and HIF-1α-mediated hypoxic responses, thereby inhibiting a range of effector molecules in TAMs, including arginase-1, interleukin IL-10, IL-1β, IL-6, and prostaglandin E2 (130). The research has identified that TFEB is the primary regulatory factor of TAMs in breast cancer. TFEB controls TAMs’ gene expression and functions through various autophagy/lysosome-dependent and non-dependent pathways (130).
Furthermore, TAM can activate CD8+ T cells by cross-presenting antigens, thereby effectively eliminating tumors (131, 132). However, the cross-presentation ability of TAM is inhibited by the overactive cysteine proteases within its lysosomes, resulting in the inability to activate CD8+ T cells (133, 134). A study by Lev Becker et al. developed a DNA nanodevice (E64-DNA) that can target the lysosomes of mouse TAMs, inhibiting cysteine proteases within them, thereby enhancing TAMs cross-presentation capabilities, boosting CD8+ T cell responses, and cytotoxicity. E64-DNA can also alter the phenotype and function of TAMs, shifting them from the tumor-promoting M2 type to the tumor-inhibiting M1 type (133).
Multiple Myeloma (MM) is a malignant hematological disease caused by the clonal proliferation of plasma cells. The occurrence and development of MM are closely associated with immune cells in the TME, among which TAMs are one of the main cell types (135). The NLRP3 inflammasome is a multiprotein complex composed of NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), and caspase-1. It can recognize various endogenous and exogenous danger signals, activate caspase-1, and promote the maturation and secretion of IL-1β and IL-18 (136, 137). Beta-2 microglobulin (β2m) is a small molecular protein that primarily exists as the light chain of MHC-I molecules on the surface of all nucleated cells (136). In MM, elevated β2m levels are associated with tumor burden, renal impairment, and poor prognosis (138, 139). In a study conducted by Heiko Bruns et al., it was found that β2m can be engulfed by TAMs, leading to the formation of β-amyloid-like fibers within lysosomes. These β-amyloid-like fibers can trigger lysosomal rupture, releasing NLRP3 and ASC from within the lysosome. After lysosome rupture, NLRP3 and ASC can bind to caspase-1 in the cytoplasm, forming the NLRP3 inflammasome. The NLRP3 inflammasome can activate caspase-1, promoting the maturation and secretion of IL-1β and IL-18 (136).
The relationship between lysosomes and TAMs is complex and bidirectional, with different lysosomal functions potentially having different effects on TAMs. Modulating lysosomal function may be an effective strategy for regulating TAMs’ phenotype and function, thereby achieving tumor immunotherapy (Figure 2).
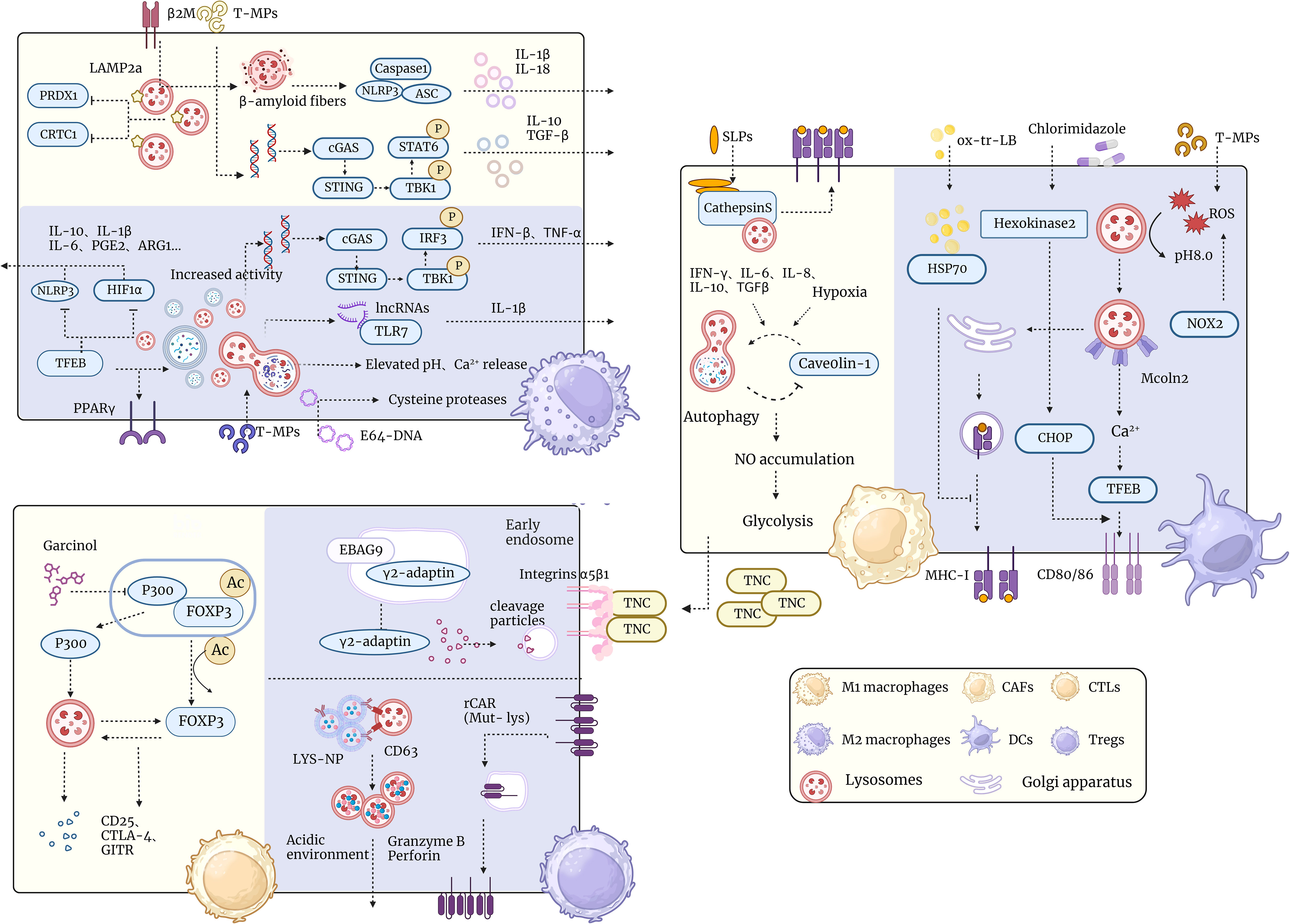
Figure 2 Schematic representation of lysosomal interactions with tumor microenvironmental immune cells. The diagram demonstrates the relationship of lysosomes in the tumor microenvironment with different types of immune cells such as TAM, T cells, CAF, DC cells, etc. Lysosomes have multiple functions in the tumor microenvironment, such as regulating immune cell polarization and activation, influencing tumor cell metabolism and proliferation, and participating in tumor-associated inflammation and immune escape. The diagram shows a number of lysosome-associated factors and pathways that can affect the functions and interactions of immune cells in the tumor microenvironment through different mechanisms, such as promoting or inhibiting immune responses and altering the phenotype and plasticity of tumor cells. This diagram aims to illustrate the important role of lysosomes in the tumor microenvironment and potential therapeutic targets.
7.2 Relationship between lysosomes and T cell activation in the TME
T cells are crucial immune cells capable of recognizing and eliminating infected or mutated cells, including cancer cells (140). However, cancer cells can evade or inhibit T cell attack through a variety of mechanisms, resulting in a microenvironment conducive to tumor growth and metastasis (141). In the TME, T cells can be classified into different subpopulations based on their function, phenotype, and differentiation status, each having distinct roles and fates (142). In general, T cells in the TME can be divided into two major categories: tumor-specific T cells and non-tumor-specific T cells. Tumor-specific T cells refer to T cells capable of recognizing tumor antigens and initiating an immune response against them, including CD8+ cytotoxic T cells and CD4+ helper T cells (143). Non-tumor-specific T cells refer to T cells that cannot recognize tumor antigens or are unresponsive to them, including Tregs, double-negative (DN) T cells, gamma-delta (γδ) T cells (141, 144, 145). The non-tumor-specific T cells typically possess immunosuppressive or regulatory functions, which can inhibit the activity or proliferation of tumor-specific T cells.
The functionality of TILs may be influenced by their impact on the endolysosomal pathway’s biosynthesis and membrane fusion processes. The formation and function of lysosomes are closely related to endosomes. Endosomes are vesicular structures involved in the process of endocytosis. They can envelop extracellular substances or liquids into cells. Subsequently, they fuse with lysosomes to form endolysosomes, where hydrolytic degradation takes place (146). The research by Armin Rehm et al. discovered that Estrogen receptor-binding fragment-associated gene 9 (EBAG9) is an intracellular membrane protein that can bind with γ2-adaptin to form an intracellular membrane complex (147). The binding of EBAG9 to γ2-adaptin inhibits the recognition and binding of γ2-adaptin to the progranulin precursor protein, thereby reducing the transport efficiency of the progranulin precursor protein. In the absence of EBAG9 in Cytotoxic T Lymphocytes (CTLs), the lytic granules become smaller, possibly due to the interaction between EBAG9 and γ2-adaptin, which also affects the membrane fusion process of lysosomes, thereby regulating the adaptive immune response function mediated by CTLs (147).
Tregs are a type of lymphocyte that can suppress immune responses and play a crucial role in maintaining immune tolerance and preventing autoimmune diseases (148, 149). However, Treg cells can also suppress anti-tumor immunity, thereby promoting tumor growth and metastasis (150). Hence, it is an important scientific question how to effectively regulate the activity of Treg, both to treat autoimmune diseases and to enhance anti-tumor immunity. Forkhead box p3 (FOXP3) is a transcription factor that regulates Treg development and function (151). Zhang et al. found that the natural p300 inhibitor Garcinol dissociates p300 from the FOXP3 complex and undergoes lysosome-dependent degradation (152). Interaction between p300 and FOXP3 promotes their lysosomal transport and degradation. When p300 is inhibited by Garcinol, it dissociates from the FOXP3 complex and is delivered to the lysosome for hydrolysis. This leads to a decrease in the level of FOXP3 acetylation, which allows FOXP3 to be degraded by the lysosome as well. This process could not be rescued by the proteasome inhibitor MG132, suggesting that the lysosome is the major pathway for p300 and FOXP3 degradation. Garcinol is able to affect the function of Treg cells by inhibiting p300 and decreasing the level of FOXP3 acetylation. Garcinol is able to reduce the inhibitory effect of Tregs on effector T cells (Teff) by decreasing the expression of some key molecules by Treg cells, such as CD25, CTLA-4, and glucocorticoid-induced TNF receptor (GITR). In addition, Garcinol was able to increase the expression of some pro-inflammatory factors such as IL-17 and IFN-γ by Tregs, thus enhancing the killing effect of Tregs on tumor cells (152). Accordingly, it is possible that targeting lysosomes could limit Treg function and enhance the efficacy of tumor-targeted therapies.
7.3 DCs
DCs are specialized antigen-presenting cells capable of presenting foreign or self-antigens to T cells to initiate and regulate the immune response (153). There is a close relationship between DC cells and lysosomes. above all, lysosomes are important sites for antigen processing and presentation by DC cells. DC cells transport antigen to the lysosome by phagocytosis, autophagy, or cross-presentation, where hydrolysis, modification, and loading of the antigen onto MHC molecules occurs to form MHC-antigen peptide complexes, which are then transported to the cell surface via vesicles and presented to T cells (154). Huang et al. found that T-MP contain tumor antigenic profiles and innate signals, and that after endocytosis of T-MP by DC, T-MP is transported to the lysosome, where RO production catalyzed by NADPH oxidase 2 (NOX2) peaks the lysosomal pH from 5.0 to 8.5. This increase in pH, coupled with T-MP-driven lysosomal migration to the center, promotes the formation of MHC I-tumor antigen peptide complexes (155). Peter Cresswell et al. found that TNF-α, CpG, and LPS-induced cross-presentation of mature DCs was significantly reduced, whereas CD40L-induced cross-presentation of mature DCs was maintained relatively (156). This difference could not be explained by a decrease in antigen uptake or translocation to the cytoplasm, but was associated with an increase in endosomal/lysosomal acidification. In addition, the authors found that inhibition of endosomal/lysosomal acidification restored or enhanced cross-presentation of mature DCs (157). Thereby confirming that endosomal/lysosomal acidification is an important factor in regulating the cross-presentation capacity of mature DCs.
Additionally, lysosomes are critical regulators of DCs maturation and activation. DC cells have efficient antigen uptake but inefficient antigen presentation in the immature state DC cells have efficient antigen uptake but inefficient antigen presentation in the immature state. When DC cells encounter hazardous signals, maturation and activation occur, expressing high levels of MHC molecules and co-stimulatory molecules that enhance antigen presentation. Lysosomes play an important role in this process, and Bo Huang et al. found that T-MP activates Mcoln2 channels in lysosomes, releasing calcium ions, which in turn activates TFEB transcription factors, promoting the expression of CD80 and CD86, and enhancing DCs maturation and activation (158). Xia et al. found that clomidazole, an antifungal drug, promotes DC-mediated antigen presentation and enhances T-cell responses. Chlormidazole acts on hexokinase 2, regulates lactate metabolites, and enhances lysosomal pathway and Chop expression in DCs, thereby inducing DCs maturation and T cell activation (159).
Lysosomes are potential targets for immune tolerance and immunoresistance in DC cells. DC cells are immune-tolerant or immune-resistant in certain situations, such as TMEs, autoimmune diseases, etc. losing the ability to effectively activate T cells (160). DCs associated with tumors have been reported to be defective in ability to cross-present antigens (161). This is associated with abnormalities in lysosomal function. Dmitry I. Gabrilovich et al. found a high accumulation of liposomes (LBs) containing oxidatively truncated (ox-tr) lipids in tumor-associated DCs, but not in normal DCs. These ox-tr-LB are mainly derived from exosomes released by tumor cells after apoptosis. The ox-tr lipid in ox-tr-LB was able to bind to HSP70 via covalent bonding, thus blocking the interaction of HSP70 with the peptide-MHC I complex (pMHC). This leads to accumulation of pMHC in late endosomes/lysosomes rather than translocation to the cell surface. Consequently, tumor-associated DCs do not efficiently present antigen to CD8+ T cells, which reduces the intensity and quality of T cell responses (161). Maria G. Masucci et al. used a candidate specific antibody (idiotype) vaccine, IGKV3-20, as a model in their study, fusing it to the glycine-alanine repeat sequence (GAr) of the Epstein-Barr virus nuclear antigen (EBNA)-1 to inhibit its degradation in the proteasome and target it to lysosomal for processing. The fusion-type IGKV3-20 was more stable in mDCs and localized more to the lysosome than wild-type IGKV3-20. Transduction of fusion-type IGKV3-20 by mDCs efficiently induces CD4+ and CD8+ CTL responses, and these CTLs are able to kill autologous mother cells expressing IGKV3-20 or pulsed IGKV3-20-synthesized peptides or HLA-matched IGKV3-20-positive tumor cell lines (162). Hence our reasoning that fusion of a candidate specific antibody vaccine with GAr could broaden the immune response by facilitating the presentation of antigenic epitopes that require lysosome-dependent processing steps (Figure 2).
7.4 CAFs
CAFs are a distinct type of stromal cell found within the TME, acting as a supportive scaffold for the extracellular matrix of TME cells (163). In addition, CAFs secrete cytokines (such as IL-6, IL-8, IL-10, and IFN-γ), growth factors (like basic fibroblast growth factor (FGFβ) and TGFβ2), chemokines, and extracellular matrix proteins [such as tenascin-c (TNC)], which regulate angiogenesis, lymph-angiogenesis, and immune responses (163). Cytokines including IFN-γ, IL-6, IL-8, and IL-10, along with TGF-β, can induce autophagy in CAFs (164) Under chronic hypoxia, CAFs utilize autophagy to degrade caveolin-1. The reduced expression of caveolin-1 positively feeds back to drive autophagy upregulation, leading to NO accumulation, causing CAFs to adopt a glycolytic phenotype, producing a large amount of lactate, dynamically co-evolving with cancer cells, and becoming metabolically coupled (2, 165). ECM proteins from CAFs, notably TNC, interact with α5β1 integrins on the surface of T cells, inhibiting the reorganization of the actin cytoskeleton necessary for T cell activation, thereby preventing T cell proliferation and activation, and ultimately overcoming immune surveillance (166). TNC is degraded via the Skp2-p62 dependent autophagy-lysosomal system. In TNBC cells with impaired autophagy, accumulated TNC can lead to resistance against T cell-mediated immune attacks. Utilizing anti-TNC antibodies makes cells more sensitive to T cell-mediated tumor killing and enhances the anti-tumor efficacy of PD-1 blockade in autophagy-deficient TNBC tumors (32). Additionally, CAFs can suppress tumor-specific T cell functionality by expressing various immune-suppressive molecules (such as PD-L1, IDO, and TGF-β) or through the cross-presentation of exogenous antigens (163, 167). In research by Els ME Verdegaal and others, human colorectal cancer (CRC)-derived CAFs were used to investigate their capacity for cross-presenting novel antigen-derived synthetic long peptides (SLPs), i.e., tumor-derived mutated peptides, and the impact of this on tumor-specific T cell functionality (168). Human CRC-derived CAFs possess a greater capability to cross-present novel antigen-derived SLPs compared to normal colonic fibroblasts. The presentation of antigens by fibroblasts involves the lysosomal protease Cathepsin S. Lysosomes contain various hydrolases, among which Cathepsin S is a cysteine protease capable of cleaving proteins to generate peptides suitable for binding to HLA-I molecules. Cathepsin S is highly expressed in CAFs within human CRC tissues, enabling them to effectively cross-present novel antigen-derived SLPs, thereby inducing the activation of tumor-specific T cells (169).
8 The utilization of the acidic properties of lysosomes for the action of artificially synthesized drugs
We know that the regulation of TME is considered a method to enhance the effectiveness of cancer treatment. During the process of tumor drug therapy, the main obstacles leading to low drug delivery efficiency are the premature leakage of drugs and low cellular uptake efficiency. Therefore, based on the low pH characteristics of lysosomes, some novel nano/micromaterials have been designed to target lysosomes to enhance immunotherapy.
Wang et al. used lipid-coated calcium phosphate (LCP) nanoparticles (NPs) to co-deliver TRP2 mRNA encoding cancer antigen and PD-L1 siRNA to dendritic cells in lymph nodes. After internalization by dendritic cells, the dissolution of the NP core in the acidic compartment of lysosomes results in high osmotic pressure, thereby disrupting endosomal membranes to achieve effective endosomal escape and nucleic acid release. This promotes antigen presentation by dendritic cells and induces robust antigen-specific activation and proliferation of CD4+ and CD8+ T cells (13). VEGF and placental growth factor (PLGF) are two important pro-angiogenic factors that are overexpressed in breast cancer cells and M2-type TAMs. They promote tumor growth, metastasis, and immune suppression either synergistically or independently (170–172). Yin et al. developed a dual pH-sensitive multifunctional NPs for simultaneous delivery of VEGF siRNA and PLGF siRNA to M2-TAMs and breast cancer cells, achieving gene silencing of VEGF and PLGF. These NPs are composed of polyethylene glycol (PEG) and mannose dual-modified trimethyl chitosan (PEG-MT) and citraconic anhydride-grafted poly (PC). They possess characteristics such as prolonged circulation time in the bloodstream, enhanced accumulation in tumor tissues, active and passive targeting of M2-TAMs and breast cancer cells, endosomal/lysosomal escape, and intracellular siRNA release. These NPs exhibit a significant charge conversion under pH 5.0 and 6.5 conditions, promoting endocytosis and endosomal/lysosomal escape. This process is achieved through the dual pH-sensitivity of PC, where it undergoes hydrolysis at pH 5.0, resulting in a change in surface charge from negative to positive, increasing interaction with negatively charged endosomal membranes. At pH 6.5, PC undergoes protonation, causing a change in surface charge from positive to negative, reducing interaction with negatively charged lysosomal membranes (173). Xia et al. developed a cancer vaccine based on porous silicon microparticles (PSM) loaded with growth factor receptor 2 (HER2) antigen. After being engulfed by dendritic cells, these particles exhibited prolonged early endosomal localization and enhanced cross-presentation. They induced type I interferon responses through TRIF and MAVS-dependent pathways, resulting in potent CD8+ T cell-dependent anti-tumor immunity in HER2-positive breast tumor-bearing mice (174).
Polymer/metal organic frameworks (MOFs) are a class of crystalline materials characterized by the coordination of metal ions with multidentate organic ligands. They possess high porosity and flexible functionality, making them ideal candidates for biomedical applications such as drug delivery and magnetic resonance imaging (175). Duan et al. engineered MOFs to load tumor-associated antigens. Due to the relatively unstable metal-ligand bonds, they can degrade in the acidic environment of endosomes/lysosomes, thereby releasing enhanced antigen cross-presentation. Additionally, MOFs introduce immunostimulatory CpG through Watson-Crick base pairing, further enhancing cytotoxic T lymphocyte responses in B16-OVA melanoma (176). In a study by Ma et al., a self-adjuvanted nano-vaccine was designed. This vaccine utilized a pH-sensitive galactosylated dextran-retinaldehyde (GDR) nanogel as a carrier and the patient’s own tumor cell lysate as an antigen, creating a personalized tumor immunotherapy strategy. The nanogel exhibits pH sensitivity, allowing it to release antigens and retinaldehyde within acidic lysosomes. The retinaldehyde activates the retinoic acid receptor (RAR) signaling pathway, promoting DCs maturation. Additionally, it induces lysosome rupture, leading to intracellular ROS generation, thereby enhancing proteasomal activity and MHC-I-mediated antigen cross-presentation, activating CTL responses, and suppressing the differentiation and function of Tregs and TAMs (95).
9 Clinical strategies for lysosome-based cancer therapy derived from fundamental research
Cancer vaccines are an immunotherapeutic approach that harnesses the immune system to effectively eliminate malignant cells. However, the current efficacy of cancer vaccines is suboptimal, primarily due to imprecise and uncontrollable antigen and adjuvant delivery and release (177).
The use of nucleic acids for cancer therapy. However, primary immune cells present inherent challenges in terms of efficiency and low activity compared to tumor cells. For targeting TAMs, recombinant bacterial ghosts have been developed to carry plasmid DNA and linear double-stranded DNA, localized within the lumen of the bacterial envelope rather than on the outer surface (178, 179). Loading ghosts with shLAMP2a allows for the stable modulation of peroxiredoxin 1 (PRDX1) and CREB-regulated transcription coactivator 1 (CRTC1) within TAMs, both of which serve as responsive factors activating the ROS signaling pathway, thereby inducing macrophage inflammation and controlling tumor growth (180). Unfortunately, strategies based on nucleic acid transfection have not yet been applied in clinical experiments and require extensive preliminary exploration. A summary of these preclinical experiments is provided in Table 2.
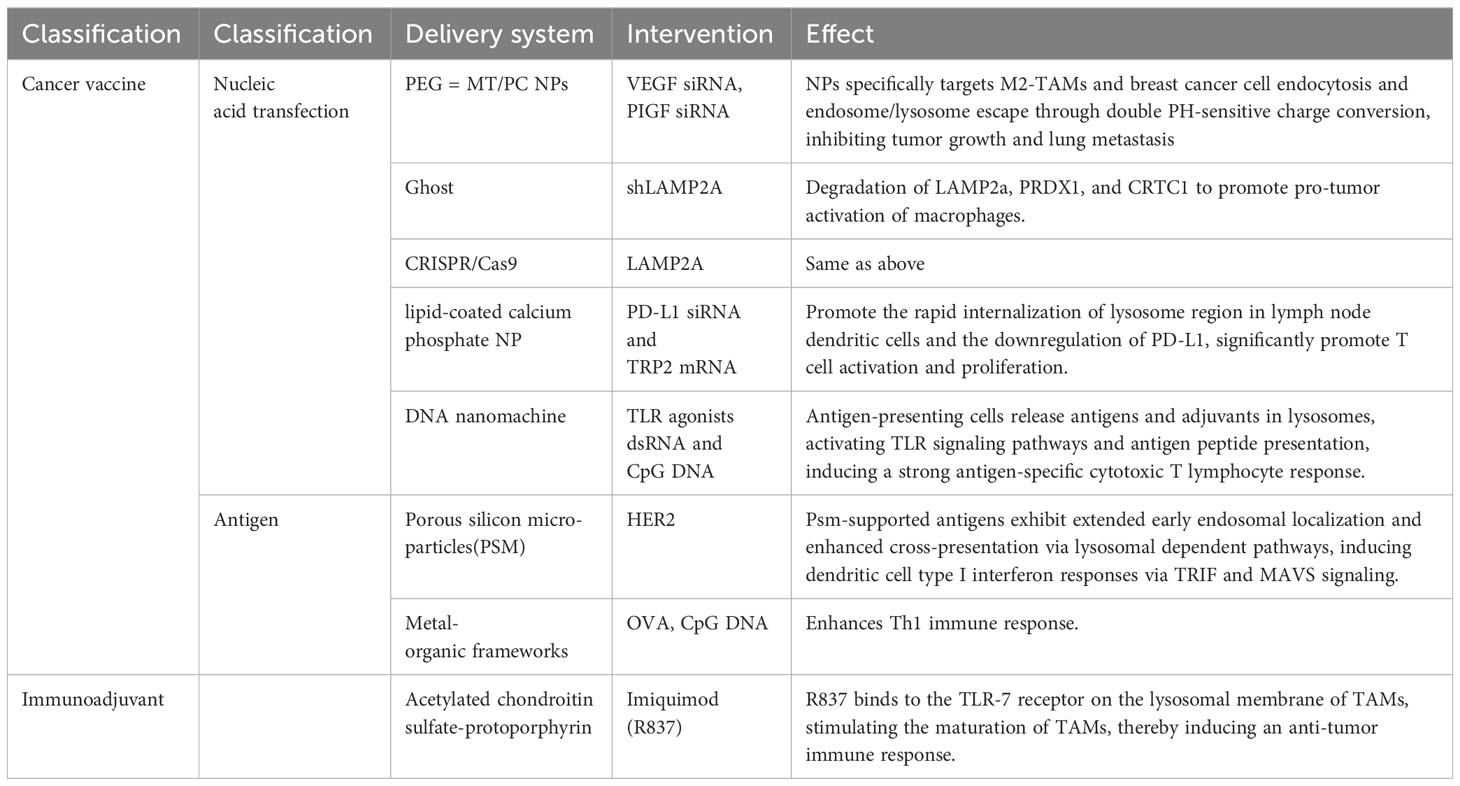
Table 2 Preclinical experiments targeting lysosomes.
TLRs primarily serve as sentinels for detecting and identifying various distinct molecular patterns associated with diseases, known as pathogen-associated molecular patterns (PAMPs). Upon TLR activation, they can induce signaling pathways dependent on either myeloid differentiation primary response 88 (MyD88) or TIR domain-containing adapter-inducing interferon-β (TRIF), ultimately leading to the activation of NF-κB, the secretion of cytokines and chemokines, and the initiation of both innate and adaptive immune responses (181). In the context of cancer, the presence of damage-associated molecular patterns (DAMPs) within tumor cells stimulates the activation of TLRs on immune cells within the TME, resulting in chronic inflammation. These alterations are interconnected with changes in the progression of tumors, inhibition of apoptosis, and the resistance of tumors to immune responses (182).
As a result, applying TLR agonists in cancer immunotherapy holds promise for transforming “cold” tumors into “hot” ones. This approach addresses the challenge of low response rates to single ICI and enhances the overall effectiveness of immunotherapy (177). Members of the TLR family, including TLR3, TLR7, TLR9, and TLR10, are situated within the endosomal/lysosomal compartment, and their ligands encompass double-stranded RNA (dsRNA), single-stranded RNA (ssRNA), CpG DNA, and others. Ligands for TLR agonists employ the innate immune pathways to modulate the type of immune response generated by vaccines (183).
Currently, several clinical trials are either underway or have been completed involving TLR agonists, mTOR inhibitors, and Hydroxychloroquine (lysosome inhibitors) in combination with cancer immunotherapy, as outlined in Table 3. For instance, Xiao et al. have developed polymer micelles designed to target TLR7. These micelles enable the specific delivery of R837 to TAMs, prompting their maturation. This, in turn, triggers anti-tumor immune responses and mitigates immune suppression within the TME (184).
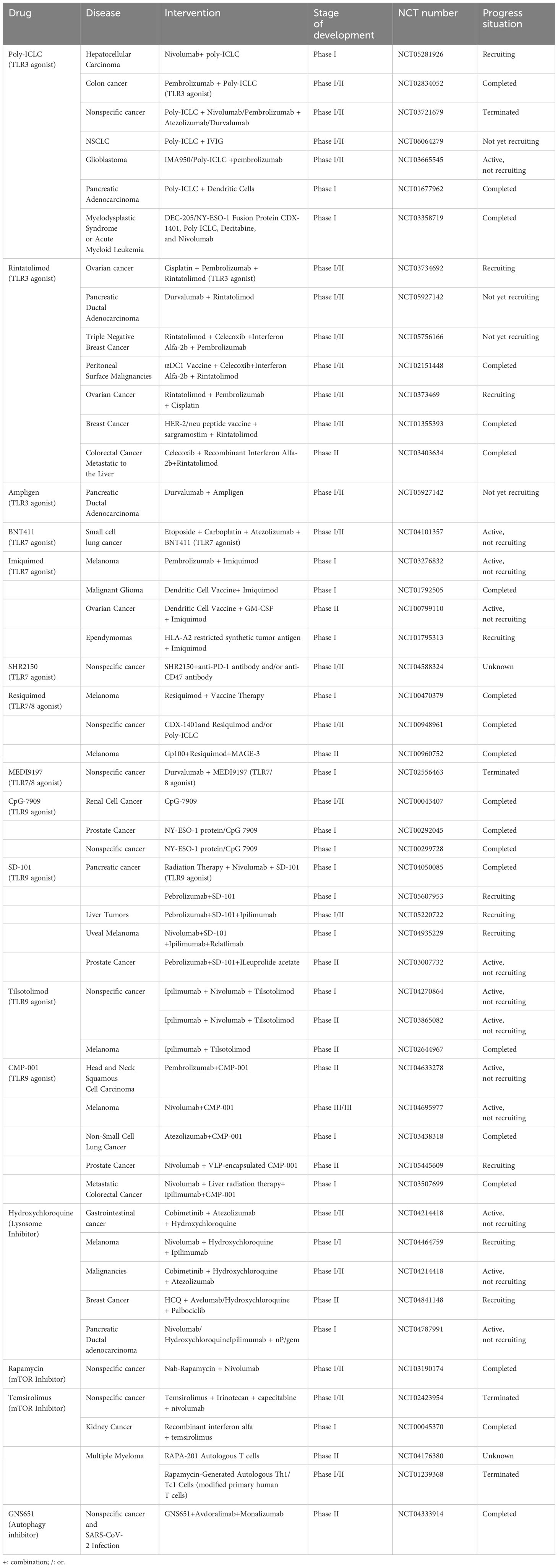
Table 3 Interventions targeting lysosomes.
Furthermore, Ding et al. have designed and produced a cancer vaccine based on DNA nanomachines. This vaccine is created through the precise assembly of two types of molecular adjuvants (TLR agonists dsRNA and CpG DNA) and antigen peptides within the interior of tubular DNA nanostructures. The DNA nanomachine can be activated by the low pH within lysosomes of APCs, leading to the unfolding of its structure and the release of antigens and adjuvants. This activation subsequently triggers the TLR signaling pathways and the presentation of antigen peptides, resulting in a potent antigen-specific CTL response. The vaccine induces noticeable maturation of APCs, antigen-specific CTL responses, and subsequently, tumor regression. Additionally, the vaccine establishes long-lasting T-cell memory responses that effectively safeguard mice against tumor rechallenge (185).
Adoptive T-cell therapy, including chimeric antigen receptor T-cell therapy, TILs therapy, and endogenous T-cell therapy, involves the isolation of T-cells from a patient’s peripheral blood.
These T-cells are then activated and enhanced for tumor specificity through biological or genetic engineering techniques before being expanded and reintroduced into the peripheral blood. While adoptive T-cell immunotherapy has been approved for the treatment of B-cell lymphomas, its success rates in solid tumors are comparatively lower.
CAR is an artificially designed molecule that enables T-cells to recognize and eliminate tumor cells expressing specific antigens.
CAR-T cell therapy is an immunotherapeutic approach that utilizes genetic engineering to introduce CARs into a patient’s own T-cells. It has demonstrated significant efficacy in certain types of cancer (186). However, CAR-T cell therapy still faces limitations regarding its persistence and functionality within the body, resulting in tumor relapse or resistance in some patients (187).
Lysosomes play a negative regulatory role in CAR-T cells by degrading CAR, thereby reducing CAR-T cell activity and persistence. Research by Wang et al. revealed that after binding with tumor antigens, CAR undergoes ubiquitination and subsequent internalization, leading to degradation in lysosomes, which diminishes CAR-T cell activity and persistence. To counteract this process, they designed a modified CAR (rCAR) by mutating all lysine residues in the cytoplasmic region of CAR, making it less susceptible to ubiquitination and downregulation. Compared to traditional CARs, rCAR can reappear on the cell surface after internalization and continue to function within intracellular compartments. In mouse tumor models, rCAR-T cells exhibited higher surface CAR levels, enhanced cytotoxicity, prolonged survival, and improved anti-tumor effects (188). This innovative rCAR design holds promise for enhancing the safety and effectiveness of clinical CAR-T cell therapy.
Perforin and granzymes are two essential weaponry components required for T-cell-mediated killing of tumor cells or infected cells (189, 190).
Lysosomes are responsible for storing and transporting perforin and granzymes. When T cells form an immune synapse with target cells, lysosomes release perforin and granzymes at the synapse. Perforin can form holes in the cell membrane of the target cells, and granzyme can enter the cytoplasm of the target cells through these holes and trigger apoptosis of the target cells (189, 191). Adoptive T-cell therapy faces a challenge in solid tumors due to their low responsiveness, possibly due to suppressed T-cells displaying weak tumor targeting and reduced activation in the immunosuppressive TME, leading to a sharp reduction in the delivery of potent toxins to the tumor region. Therefore, harnessing the synthetic and release capabilities of lysosomes is crucial to enhance the effectiveness of T-cell immunotherapy. Zhang et al. have developed an adoptive T-cell vehicle (ATV) loaded with lysosome-reactive nanoparticles (LYS-NP). LYS-NP consists of a MOF as a substrate structure and a biotin-modified lysosome-targeting aptamer (CD63-aptamer). Perforin and granzyme B can be loaded onto the MOF, which is then engulfed by T-cells and localized within lysosomes. In an acidic environment, the MOF degrades, releasing perforin, granzyme B, and Ca2+, leading to their substantial accumulation within lysosomes. When the T-cell receptor (TCR) binds with MHC to form an immune synapse, lysosomes autonomously release their contents, thereby enhancing the killing capability of T cells (192). This novel strategy of reprogramming T-cell lysosomes to augment their anti-tumor effect provides a new, efficient, safe, controllable, and adjustable approach for solid tumor immunotherapy, offering significant clinical application potential.
10 Conclusion
This article provides an overview of the physiological functions of lysosomes, their role in the TME, and potential tumor immunotherapy approaches targeting lysosomes. Lysosomes serve as not only the cellular degradation centers but also play crucial roles in immune regulation, nutrient sensing, and cellular communication processes. Recent research has shown that lysosomal homeostasis has a dual role in tumor initiation and progression. It can activate immune surveillance by processing and presenting tumor antigens and regulating inflammation, while also supporting cancer cell metabolism and resistance to stress, facilitating tumor progression.
Current evidence highlights the critical importance of lysosomes in disease. A deeper understanding of lysosome-related mechanisms will enhance our ability to leverage their biological properties, thereby increasing the efficacy of tumor immunotherapy across various stages of immune response. During the antigen presentation stage, lysosomes are responsible for antigen processing within APCs, binding to MHC-II, and presenting them on the cell membrane. In tumor cells, the use of lysosomes or autophagy inhibitors can restore the membrane expression of MHC-I. In the T-cell activation process, lysosomes can enhance the degradation or transport of immune checkpoint molecules like PD-1, PD-L1, and CTLA4, reducing immune evasion by tumor cells. In the recruitment and infiltration of immune cells, lysosomes participate in the release of various adhesion factors, chemokines, and the regulation of selectins. Additionally, lysosomes can promote tumor nutrient adaptation and immune suppression by modulating cellular autophagy and metabolism.
In tumor therapy, various approaches targeting lysosomes have shown promising results. When combined with immunotherapy, these methods may further improve treatment outcomes. Examples include using lysosomal acidic environments for pH-sensitive nanoparticle drug delivery and release, avoiding lysosomal degradation of CAR in ACT therapy, and using lysosome or autophagy inhibitors, TLR agonists in combination with DCs vaccines or ICIs, among others. It’s important to note that the role of autophagy in tumor progression varies at different stages and in different cell types. Therefore, interventions to inhibit or stimulate autophagy should be carefully chosen and implemented.
In conclusion, while targeting lysosomes or harnessing their properties represents a potential effective strategy for tumor immunotherapy, the mechanisms and methods related to lysosomes in tumor immunotherapy are not fully understood at present. Further basic and clinical research is needed to address issues related to lysosomal drug delivery, specificity, resistance, and personalized treatment plans, with the aim of providing better treatment options for cancer patients.
Author contributions
YX: Writing – original draft. BS: Writing – original draft. YZ: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol (2020) 21:101–18. doi: 10.1038/s41580-019-0185-4
2. Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival. Cell Cycle (2010) 9:3515–33. doi: 10.4161/cc.9.17.12928
3. Jewell JL, Russell RC, Guan K-L. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol (2013) 14:133–9. doi: 10.1038/nrm3522
4. Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol (2019) 21:133–42. doi: 10.1038/s41556-018-0244-7
5. Conus S, Simon H-U. Cathepsins: Key modulators of cell death and inflammatory responses. Biochem Pharmacol (2008) 76:1374–82. doi: 10.1016/j.bcp.2008.07.041
6. Saksena S, Sun J, Chu T, Emr S. ESCRTing proteins in the endocytic pathway. Trends Biochem Sci (2007) 32:561–73. doi: 10.1016/j.tibs.2007.09.010
7. Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy (2021) 17:2680–8. doi: 10.1080/15548627.2020.1823124
8. Meng Y, Heybrock S, Neculai D, Saftig P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol (2020) 30:452–66. doi: 10.1016/j.tcb.2020.02.007
9. Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol (2005) 6:439–48. doi: 10.1038/nrm1660
10. Cao M, Luo X, Wu K, He X. Targeting lysosomes in human disease: from basic research to clinical applications. Signal Transduction Targeted Ther (2021) 6:379. doi: 10.1038/s41392-021-00778-y
11. Zhang Z, Yue P, Lu T, Wang Y, Wei Y, Wei X. Role of lysosomes in physiological activities, diseases, and therapy. J Hematol Oncol (2021) 14:79. doi: 10.1186/s13045-021-01087-1
12. Solsona R, Sanchez AMJ. Ribosome biogenesis and resistance training volume in human skeletal muscle. J Physiol (2020) 598:1121–2. doi: 10.1113/JP279490
13. Wang Y, Zhang L, Xu Z, Miao L, Huang L. mRNA vaccine with antigen-specific checkpoint blockade induces an enhanced immune response against established melanoma. Mol Ther (2018) 26:420–34. doi: 10.1016/j.ymthe.2017.11.009
14. Ammanathan V, Vats S, Abraham IM, Manjithaya R. Xenophagy in cancer. Semin Cancer Biol (2020) 66:163–70. doi: 10.1016/j.semcancer.2020.02.015
15. Germic N, Frangez Z, Yousefi S, Simon H-U. Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ (2019) 26:715–27. doi: 10.1038/s41418-019-0297-6
16. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature (2020) 581:100–5. doi: 10.1038/s41586-020-2229-5
17. Feng M, Jiang W, Kim BY, Zhang C, Fu Y-X, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer (2019) 19:568–86. doi: 10.1038/s41568-019-0183-z
18. Burr ML, Sparbier CE, Chan Y-C, Williamson JC, Woods K, Beavis PA, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature (2017) 549:101–5. doi: 10.1038/nature23643
19. Veillette A, Chen J. SIRPα-CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol (2018) 39:173–84. doi: 10.1016/j.it.2017.12.005
20. Zhang L, Yu J, Zheng M, Zhen H, Xie Q, Zhang C, et al. RAGA prevents tumor immune evasion of LUAD by promoting CD47 lysosome degradation. Commun Biol (2023) 6:211. doi: 10.1038/s42003-023-04581-z
21. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30:832–44. doi: 10.1016/j.immuni.2009.04.014
22. Völkl S, Rensing-Ehl A, Allgäuer A, Schreiner E, Lorenz MR, Rohr J, et al. Hyperactive mTOR pathway promotes lymphoproliferation and abnormal differentiation in autoimmune lymphoproliferative syndrome. Blood (2016) 128:227–38. doi: 10.1182/blood-2015-11-685024
23. Clarke AJ, Simon AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol (2019) 19:170–83. doi: 10.1038/s41577-018-0095-2
24. Jacquel A, Obba S, Boyer L, Dufies M, Robert G, Gounon P, et al. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood (2012) 119:4527–31. doi: 10.1182/blood-2011-11-392167
25. Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy (2008) 4:309–14. doi: 10.4161/auto.5474
26. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol (2013) 14:298–305. doi: 10.1038/ni.2524
27. Kirchhammer N, Trefny MP, Auf der Maur P, Läubli H, Zippelius A. Combination cancer immunotherapies: Emerging treatment strategies adapted to the tumor microenvironment. Sci Transl Med (2022) 14:eabo3605. doi: 10.1126/scitranslmed.abo3605
28. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
29. He M, Yang T, Wang Y, Wang M, Chen X, Ding D, et al. Immune checkpoint inhibitor-based strategies for synergistic cancer therapy. Adv Healthc Mater (2021) 10:e2002104. doi: 10.1002/adhm.202002104
30. Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science (2018) 359:1355–60. doi: 10.1126/science.aar7112
31. Wang Z, Wu Z, Liu Y, Han W. New development in CAR-T cell therapy. J Hematol Oncol (2017) 10:53. doi: 10.1186/s13045-017-0423-1
32. Li Z-L, Zhang H-L, Huang Y, Huang J-H, Sun P, Zhou N-N, et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat Commun (2020) 11:3806. doi: 10.1038/s41467-020-17395-y
33. Bonam SR, Wang F, Muller S. Lysosomes as a therapeutic target. Nat Rev Drug Discovery (2019) 18:923–48. doi: 10.1038/s41573-019-0036-1
34. Ahn G, Riley NM, Kamber RA, Wisnovsky S, Moncayo von Hase S, Bassik MC, et al. Elucidating the cellular determinants of targeted membrane protein degradation by lysosome-targeting chimeras. Science (2023) 382:eadf6249. doi: 10.1126/science.adf6249
35. Tang M, Chen B, Xia H, Pan M, Zhao R, Zhou J, et al. pH-gated nanoparticles selectively regulate lysosomal function of tumour-associated macrophages for cancer immunotherapy. Nat Commun (2023) 14:5888. doi: 10.1038/s41467-023-41592-0
36. Pei J, Wang G, Feng L, Zhang J, Jiang T, Sun Q, et al. Targeting lysosomal degradation pathways: new strategies and techniques for drug discovery. J Med Chem (2021) 64:3493–507. doi: 10.1021/acs.jmedchem.0c01689
37. Tang X, Zhang J, Sui D, Yang Q, Wang T, Xu Z, et al. Simultaneous dendritic cells targeting and effective endosomal escape enhance sialic acid-modified mRNA vaccine efficacy and reduce side effects. J Controlled Release (2023) 364:529–45. doi: 10.1016/j.jconrel.2023.11.008
38. Zhu S, Zhang T, Zheng L, Liu H, Song W, Liu D, et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol Oncol (2021) 14:156. doi: 10.1186/s13045-021-01164-5
39. Ouyang Z, Gao Y, Shen S, Jia B, Yu H, Wang H, et al. A minimalist dendrimer nanodrug for autophagy inhibition-amplified tumor photothermo-immunotherapy. Nano Today (2023) 51:101936. doi: 10.1016/j.nantod.2023.101936
40. Zhao X, Hu S, Zeng L, Liu X, Song Y, Zhang Y, et al. Irradiation combined with PD-L1-/- and autophagy inhibition enhances the antitumor effect of lung cancer via cGAS-STING-mediated T cell activation. iScience (2022) 25:104690. doi: 10.1016/j.isci.2022.104690
41. Perera RM, Zoncu R. The lysosome as a regulatory hub. Annu Rev Cell Dev Bi (2016) 32:223–53. doi: 10.1146/annurev-cellbio-111315-125125
42. Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev (2016) 32:75–88. doi: 10.1016/j.arr.2016.05.004
43. Hesketh GG, Wartosch L, Davis LJ, Bright NA, Luzio JP. The Lysosome and Intracellular Signalling. In: Lamaze C, Prior I, editors. Endocytosis and Signaling, vol. 57 . Cham: Springer International Publishing (2018). p. 151–80. doi: 10.1007/978-3-319-96704-2_6
44. Lübke T, Lobel P, Sleat DE. Proteomics of the lysosome. Biochim Biophys Acta (BBA) - Mol Cell Res (2009) 1793:625–35. doi: 10.1016/j.bbamcr.2008.09.018
45. Tan A, Prasad R, Lee C, Jho E-H. Past, present, and future perspectives of transcription factor EB (TFEB): mechanisms of regulation and association with disease. Cell Death Differ (2022) 29:1433–49. doi: 10.1038/s41418-022-01028-6
46. Langemeyer L, Fröhlich F, Ungermann C. Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol (2018) 28:957–70. doi: 10.1016/j.tcb.2018.06.007
47. Yamamoto H, Zhang S, Mizushima N. Autophagy genes in biology and disease. Nat Rev Genet (2023) 24:382–400. doi: 10.1038/s41576-022-00562-w
48. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer (2020) 19:12. doi: 10.1186/s12943-020-1138-4
49. Zhen Y, Spangenberg H, Munson MJ, Brech A, Schink KO, Tan K-W, et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy (2020) 16:826–41. doi: 10.1080/15548627.2019.1639301
50. Yu S, Melia TJ. The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr Opin Cell Biol (2017) 47:92–8. doi: 10.1016/j.ceb.2017.03.010
51. Liu R, Zhi X, Zhong Q. ATG14 controls SNARE-mediated autophagosome fusion with a lysosome. Autophagy (2015) 11:847–9. doi: 10.1080/15548627.2015.1037549
52. Kaushik S, Kiffin R, Cuervo AM. Chaperone-mediated autophagy at a glance. J Cell Sci (2011) 124:495–9. doi: 10.1242/jcs.073874
53. Kaushik S, Kiffin R, Cuervo AM. Chaperone-mediated autophagy and aging: A novel regulatory role of lipids revealed. Autophagy (2007) 3:387–9. doi: 10.4161/auto.4246
54. Kaushik S, Cuervo AM. The coming of age of chaperonemediated autophagy. Nat Rev Mol Cell Biol (2018) 19:365–81. doi: 10.1038/s41580-018-0001-6
55. Nguyen JA, Yates RM. Better together: current insights into phagosome-lysosome fusion. Front Immunol (2021) 12:636078. doi: 10.3389/fimmu.2021.636078
56. Waguri S, Dewitte F, Le Borgne R, Rouillé Y, Uchiyama Y, Dubremetz J-F, et al. Visualization of TGN to endosome trafficking through fluorescently labeled MPR and AP-1 in living cells. Mol Biol Cell (2003) 14:142–55. doi: 10.1091/mbc.e02-06-0338
57. Pankiv S, Alemu EA, Brech A, Bruun J-A, Lamark T, Overvatn A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol (2010) 188:253–69. doi: 10.1083/jcb.200907015
58. Meel E, Klumperman J. Imaging and imagination: understanding the endo-lysosomal system. Histochem Cell Biol (2008) 129:253–66. doi: 10.1007/s00418-008-0384-0
59. Janvier K, Bonifacino JS. Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol Biol Cell (2005) 16:4231–42. doi: 10.1091/mbc.e05-03-0213
60. Théry C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, et al. Molecular characterization of dendritic cell-derived exosomes. J Cell Biol (1999) 147:599–610. doi: 10.1083/jcb.147.3.599
61. Escola J-M, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem (1998) 273:20121–7. doi: 10.1074/jbc.273.32.20121
62. Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell (2019) 177:1682–99. doi: 10.1016/j.cell.2019.05.026
63. Mahapatra KK, Mishra SR, Behera BP, Patil S, Gewirtz DA, Bhutia SK. The lysosome as an imperative regulator of autophagy and cell death. Cell Mol Life Sci (2021) 78:7435–49. doi: 10.1007/s00018-021-03988-3
64. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell (2008) 132:27–42. doi: 10.1016/j.cell.2007.12.018
65. Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther (2022) 7:196. doi: 10.1038/s41392-022-01046-3
66. Yamamoto K, Venida A, Perera RM, Kimmelman AC. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy (2020) 16:1524–5. doi: 10.1080/15548627.2020.1769973
67. Alissafi T, Hatzioannou A, Mintzas K, Barouni RM, Banos A, Sormendi S, et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J Clin Invest (2018) 128:3840–52. doi: 10.1172/JCI120888
68. Wang H, Wang Q, Wu Y, Lou J, Zhu S, Xu Y. Autophagy-related gene LAPTM4B promotes the progression of renal clear cell carcinoma and is associated with immunity. Front Pharmacol (2023) 14:1118217. doi: 10.3389/fphar.2023.1118217
69. Chang C-P, Su Y-C, Hu C-W, Lei H-Y. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ (2013) 20:515–23. doi: 10.1038/cdd.2012.146
70. Barrera G, Pizzimenti S, Dianzani MU. Lipid peroxidation: control of cell proliferation, cell differentiation and cell death. Mol Aspects Med (2008) 29:1–8. doi: 10.1016/j.mam.2007.09.012
71. Bhardwaj M, Lee JJ, Versace AM, Harper SL, Goldman AR, Crissey MAS, et al. Lysosomal lipid peroxidation regulates tumor immunity. J Clin Invest (2023) 133:e164596. doi: 10.1172/JCI164596
72. Mennerich D, Kubaichuk K, Kietzmann T. DUBs, hypoxia, and cancer. Trends Cancer (2019) 5:632–53. doi: 10.1016/j.trecan.2019.08.005
73. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer (2019) 18:157. doi: 10.1186/s12943-019-1089-9
74. Xi L, Peng M, Liu S, Liu Y, Wan X, Hou Y, et al. Hypoxia-stimulated ATM activation regulates autophagy-associated exosome release from cancer-associated fibroblasts to promote cancer cell invasion. J Extracell Vesicles (2021) 10:e12146. doi: 10.1002/jev2.12146
75. Wang X, Wu R, Zhai P, Liu Z, Xia R, Zhang Z, et al. Hypoxia promotes EV secretion by impairing lysosomal homeostasis in HNSCC through negative regulation of ATP6V1A by HIF-1α. J Extracell Vesicles (2023) 12:e12310. doi: 10.1002/jev2.12310
76. van Niel G, Carter DRF, Clayton A, Lambert DW, Raposo G, Vader P. Challenges and directions in studying cell-cell communication by extracellular vesicles. Nat Rev Mol Cell Biol (2022) 23:369–82. doi: 10.1038/s41580-022-00460-3
77. Meng W, Hao Y, He C, Li L, Zhu G. Exosome-orchestrated hypoxic tumor microenvironment. Mol Cancer (2019) 18:57. doi: 10.1186/s12943-019-0982-6
78. Morrissey SM, Yan J. Exosomal PD-L1: roles in tumor progression and immunotherapy. Trends Cancer (2020) 6:550–8. doi: 10.1016/j.trecan.2020.03.002
79. Wu Q, You L, Nepovimova E, Heger Z, Wu W, Kuca K, et al. Hypoxia-inducible factors: master regulators of hypoxic tumor immune escape. J Hematol Oncol (2022) 15:77. doi: 10.1186/s13045-022-01292-6
80. Lakhter AJ, Lahm T, Broxmeyer HE, Naidu SR. Golgi associated HIF1a serves as a reserve in melanoma cells. J Cell Biochem (2016) 117:853–9. doi: 10.1002/jcb.25381
81. Gu J, Geng M, Qi M, Wang L, Zhang Y, Gao J. The role of lysosomal membrane proteins in glucose and lipid metabolism. FASEB J (2021) 35:e21848. doi: 10.1096/fj.202002602R
82. Shi Q, Shen Q, Liu Y, Shi Y, Huang W, Wang X, et al. Increased glucose metabolism in TAMs fuels O-GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer Cell (2022) 40:1207–1222.e10. doi: 10.1016/j.ccell.2022.08.012
83. Man SM, Kanneganti T-D. Regulation of lysosomal dynamics and autophagy by CTSB/cathepsin B. Autophagy (2016) 12:2504–5. doi: 10.1080/15548627.2016.1239679
84. Van Kaer L, Parekh VV, Postoak JL, Wu L. Role of autophagy in MHC class I-restricted antigen presentation. Mol Immunol (2019) 113:2–5. doi: 10.1016/j.molimm.2017.10.021
85. Sari G, Rock KL. Tumor immune evasion through loss of MHC class-I antigen presentation. Curr Opin Immunol (2023) 83:102329. doi: 10.1016/j.coi.2023.102329
86. Jongsma MLM, Neefjes J, Spaapen RM. Playing hide and seek: Tumor cells in control of MHC class I antigen presentation. Mol Immunol (2021) 136:36–44. doi: 10.1016/j.molimm.2021.05.009
87. Chen X, Lu Q, Zhou H, Liu J, Nadorp B, Lasry A, et al. A membrane-associated MHC-I inhibitory axis for cancer immune evasion. Cell (2023) 186:3903–3920.e21. doi: 10.1016/j.cell.2023.07.016
88. Axelrod ML, Cook RS, Johnson DB, Balko JM. Biological consequences of MHC-II expression by tumor cells in cancer. Clin Cancer Res (2019) 25:2392–402. doi: 10.1158/1078-0432.CCR-18-3200
89. Heuberger CE, Janney A, Ilott N, Bertocchi A, Pott S, Gu Y, et al. MHC class II antigen presentation by intestinal epithelial cells fine-tunes bacteria-reactive CD4 T cell responses. Mucosal Immunol (2023), S1933-0219(23)00032–6. doi: 10.1016/j.mucimm.2023.05.001
90. Liu X, Bao X, Hu M, Chang H, Jiao M, Cheng J, et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature (2020) 588:693–8. doi: 10.1038/s41586-020-2911-7
91. Du W, Hua F, Li X, Zhang J, Li S, Wang W, et al. Loss of optineurin drives cancer immune evasion via palmitoylation-dependent IFNGR1 lysosomal sorting and degradation. Cancer Discovery (2021) 11:1826–43. doi: 10.1158/2159-8290.CD-20-1571
92. Becker JC, Stang A, DeCaprio JA, Cerroni L, Lebbé C, Veness M, et al. Merkel cell carcinoma. Nat Rev Dis Primers (2017) 3:17077. doi: 10.1038/nrdp.2017.77
93. Buchta Rosean C, Leyder EC, Hamilton J, Carter JJ, Galloway DA, Koelle DM, et al. LAMP1 targeting of the large T antigen of Merkel cell polyomavirus results in potent CD4 T cell responses and tumor inhibition. Front Immunol (2023) 14:1253568. doi: 10.3389/fimmu.2023.1253568
94. Eskelinen E-L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med (2006) 27:495–502. doi: 10.1016/j.mam.2006.08.005
95. Wang C, Li P, Liu L, Pan H, Li H, Cai L, et al. Self-adjuvanted nanovaccine for cancer immunotherapy: Role of lysosomal rupture-induced ROS in MHC class I antigen presentation. Biomaterials (2016) 79:88–100. doi: 10.1016/j.biomaterials.2015.11.040
96. Jiang D, Mu W, Pang X, Liu Y, Zhang N, Song Y, et al. Cascade cytosol delivery of dual-sensitive micelle-tailored vaccine for enhancing cancer immunotherapy. ACS Appl Mater Interfaces (2018) 10:37797–811. doi: 10.1021/acsami.8b09946
97. Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol (2020) 17:725–41. doi: 10.1038/s41571-020-0413-z
98. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity (2018) 48:434–52. doi: 10.1016/j.immuni.2018.03.014
99. Gou Q, Dong C, Xu H, Khan B, Jin J, Liu Q, et al. PD-L1 degradation pathway and immunotherapy for cancer. Cell Death Dis (2020) 11:955. doi: 10.1038/s41419-020-03140-2
100. Zhu Q, Wang H, Chai S, Xu L, Lin B, Yi W, et al. O-GlcNAcylation promotes tumor immune evasion by inhibiting PD-L1 lysosomal degradation. Proc Natl Acad Sci U.S.A. (2023) 120:e2216796120. doi: 10.1073/pnas.2216796120
101. Chen S, Crabill GA, Pritchard TS, McMiller TL, Wei P, Pardoll DM, et al. Mechanisms regulating PD-L1 expression on tumor and immune cells. J Immunother Cancer (2019) 7:305. doi: 10.1186/s40425-019-0770-2
102. Yao H, Lan J, Li C, Shi H, Brosseau J-P, Wang H, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat BioMed Eng (2019) 3:306–17. doi: 10.1038/s41551-019-0375-6
103. Ko P-J, Dixon SJ. Protein palmitoylation and cancer. EMBO Rep (2018) 19:e46666. doi: 10.15252/embr.201846666
104. Wang Y-N, Lee H-H, Hsu JL, Yu D, Hung M-C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J BioMed Sci (2020) 27:77. doi: 10.1186/s12929-020-00670-x
105. Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol (2015) 10:473–510. doi: 10.1146/annurev-pathol-012414-040438
106. Xu Y, Gao Z, Hu R, Wang Y, Wang Y, Su Z, et al. PD-L2 glycosylation promotes immune evasion and predicts anti-EGFR efficacy. J Immunother Cancer (2021) 9:e002699. doi: 10.1136/jitc-2021-002699
107. Fan Z, Wu C, Chen M, Jiang Y, Wu Y, Mao R, et al. The generation of PD-L1 and PD-L2 in cancer cells: From nuclear chromatin reorganization to extracellular presentation. Acta Pharm Sin B (2022) 12:1041–53. doi: 10.1016/j.apsb.2021.09.010
108. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2:261–8. doi: 10.1038/85330
109. Zhang R, Yang Y, Dong W, Lin M, He J, Zhang X, et al. D-mannose facilitates immunotherapy and radiotherapy of triple-negative breast cancer via degradation of PD-L1. Proc Natl Acad Sci U.S.A. (2022) 119:e2114851119. doi: 10.1073/pnas.2114851119
110. Lin J, Song T, Li C, Mao W. GSK-3β in DNA repair, apoptosis, and resistance of chemotherapy, radiotherapy of cancer. Biochim Biophys Acta Mol Cell Res (2020) 1867:118659. doi: 10.1016/j.bbamcr.2020.118659
111. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen M-K, Hsu J-M, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res (2017) 23:3711–20. doi: 10.1158/1078-0432.CCR-16-3215
112. Liang J, Li S, Li W, Rao W, Xu S, Meng H, et al. CMTM6, a potential immunotherapy target. J Cancer Res Clin (2022) 148:47–56. doi: 10.1007/s00432-021-03835-9
113. Liu X, Yin M, Dong J, Mao G, Min W, Kuang Z, et al. Tubeimoside-1 induces TFEB-dependent lysosomal degradation of PD-L1 and promotes antitumor immunity by targeting mTOR. Acta Pharm Sin B (2021) 11:3134–49. doi: 10.1016/j.apsb.2021.03.039
114. Sha Y, Rao L, Settembre C, Ballabio A, Eissa NT. STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J (2017) 36:2544–52. doi: 10.15252/embj.201796699
115. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood (2018) 131:58–67. doi: 10.1182/blood-2017-06-741033
116. Cui J, Yu J, Xu H, Zou Y, Zhang H, Chen S, et al. Autophagy-lysosome inhibitor chloroquine prevents CTLA-4 degradation of T cells and attenuates acute rejection in murine skin and heart transplantation. Theranostics (2020) 10:8051–60. doi: 10.7150/thno.43507
117. Iida T, Ohno H, Nakaseko C, Sakuma M, Takeda-Ezaki M, Arase H, et al. Regulation of cell surface expression of CTLA-4 by secretion of CTLA-4-containing lysosomes upon activation of CD4+ T cells. J Immunol (2000) 165:5062–8. doi: 10.4049/jimmunol.165.9.5062
118. Hou TZ, Verma N, Wanders J, Kennedy A, Soskic B, Janman D, et al. Identifying functional defects in patients with immune dysregulation due to LRBA and CTLA-4 mutations. Blood (2017) 129:1458–68. doi: 10.1182/blood-2016-10-745174
119. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res (2013) 19:5626–35. doi: 10.1158/1078-0432.CCR-13-0545
120. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell (2018) 33:649–663.e4. doi: 10.1016/j.ccell.2018.02.010
121. Matsuhashi S, Manirujjaman M, Hamajima H, Ozaki I. Control mechanisms of the tumor suppressor PDCD4: expression and functions. Int J Mol Sci (2019) 20:2304. doi: 10.3390/ijms20092304
122. Lu K, Chen Q, Li M, He L, Riaz F, Zhang T, et al. Programmed cell death factor 4 (PDCD4), a novel therapy target for metabolic diseases besides cancer. Free Radical Bio Med (2020) 159:150–63. doi: 10.1016/j.freeradbiomed.2020.06.016
123. Chen X, Guan Y, Zhang Y, Jia Y, Li W, Guo C, et al. Programmed cell death 4 modulates lysosomal function by inhibiting TFEB translation. Cell Death Differ (2021) 28:1237–50. doi: 10.1038/s41418-020-00646-2
124. Jiang T-Y, Shi Y-Y, Cui X-W, Pan Y-F, Lin Y-K, Feng X-F, et al. PTEN deficiency facilitates exosome secretion and metastasis in cholangiocarcinoma by impairing TFEB-mediated lysosome biogenesis. Gastroenterology (2023) 164:424–38. doi: 10.1053/j.gastro.2022.11.025
125. Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPα Immune checkpoint. Immunity (2020) 52:742–52. doi: 10.1016/j.immuni.2020.04.011
126. Jia X, Yan B, Tian X, Liu Q, Jin J, Shi J, et al. CD47/SIRPα pathway mediates cancer immune escape and immunotherapy. Int J Biol Sci (2021) 17:3281–7. doi: 10.7150/ijbs.60782
127. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
128. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
129. Tang K, Ma J, Huang B. Macrophages’ M1 or M2 by tumor microparticles: lysosome makes decision. Cell Mol Immunol (2022) 19:1196–7. doi: 10.1038/s41423-022-00892-z
130. Li Y, Hodge J, Liu Q, Wang J, Wang Y, Evans TD, et al. TFEB is a master regulator of tumor-associated macrophages in breast cancer. J Immunother Cancer (2020) 8:e000543. doi: 10.1136/jitc-2020-000543
131. Ma S, Sun B, Duan S, Han J, Barr T, Zhang J, et al. YTHDF2 orchestrates tumor-associated macrophage reprogramming and controls antitumor immunity through CD8+ T cells. Nat Immunol (2023) 24:255–66. doi: 10.1038/s41590-022-01398-6
132. Fang W, Zhou T, Shi H, Yao M, Zhang D, Qian H, et al. Progranulin induces immune escape in breast cancer via up-regulating PD-L1 expression on tumor-associated macrophages (TAMs) and promoting CD8+ T cell exclusion. J Exp Clin Cancer Res (2021) 40:4. doi: 10.1186/s13046-020-01786-6
133. Cui C, Chakraborty K, Tang XA, Schoenfelt KQ, Hoffman A, Blank A, et al. A lysosome-targeted DNA nanodevice selectively targets macrophages to attenuate tumours. Nat Nanotechnol (2021) 16:1394–402. doi: 10.1038/s41565-021-00988-z
134. Mohamed HT, El-Ghonaimy EA, El-Shinawi M, Hosney M, Götte M, Woodward WA, et al. IL-8 and MCP-1/CCL2 regulate proteolytic activity in triple negative inflammatory breast cancer a mechanism that might be modulated by Src and Erk1/2. Toxicol Appl Pharmacol (2020) 401:115092. doi: 10.1016/j.taap.2020.115092
135. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20:3328. doi: 10.3390/ijms20133328
136. Hofbauer D, Mougiakakos D, Broggini L, Zaiss M, Büttner-Herold M, Bach C, et al. β2-microglobulin triggers NLRP3 inflammasome activation in tumor-associated macrophages to promote multiple myeloma progression. Immunity (2021) 54:1772–1787.e9. doi: 10.1016/j.immuni.2021.07.002
137. Shao B-Z, Xu Z-Q, Han B-Z, Su D-F, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol (2015) 6:262. doi: 10.3389/fphar.2015.00262
138. Han JH, Jeong S-H, Kim SH, Yuk HD, Jeong CW, Kwak C, et al. Increased urinary B2-microglobulin is associated with poor prognosis of upper tract urothelial carcinoma. Front Oncol (2022) 12:1008763. doi: 10.3389/fonc.2022.1008763
139. Shang Y, Fu X, Chang Y, Li Y, Zhang M. B2 microglobulin is a novel prognostic marker of Angioimmunoblastic T-cell lymphoma. Sci Rep-uk (2018) 8:12907. doi: 10.1038/s41598-018-31212-z
140. Basu A, Ramamoorthi G, Albert G, Gallen C, Beyer A, Snyder C, et al. Differentiation and regulation of TH cells: A balancing act for cancer immunotherapy. Front Immunol (2021) 12:669474. doi: 10.3389/fimmu.2021.669474
141. Shan F, Somasundaram A, Bruno TC, Workman CJ, Vignali DAA. Therapeutic targeting of regulatory T cells in cancer. Trends Cancer (2022) 8:944–61. doi: 10.1016/j.trecan.2022.06.008
142. Bawden E, Gebhardt T. The multifaceted roles of CD4+ T cells and MHC class II in cancer surveillance. Curr Opin Immunol (2023) 83:102345. doi: 10.1016/j.coi.2023.102345
143. St Paul M, Ohashi PS. The roles of CD8+ T cell subsets in antitumor immunity. Trends Cell Biol (2020) 30:695–704. doi: 10.1016/j.tcb.2020.06.003
144. Lo Presti E, Pizzolato G, Corsale AM, Caccamo N, Sireci G, Dieli F, et al. γδ T cells and tumor microenvironment: from immunosurveillance to tumor evasion. Front Immunol (2018) 9:1395. doi: 10.3389/fimmu.2018.01395
145. Wu Z, Zheng Y, Sheng J, Han Y, Yang Y, Pan H, et al. CD3+CD4-CD8- (Double-negative) T cells in inflammation, immune disorders and cancer. Front Immunol (2022) 13:816005. doi: 10.3389/fimmu.2022.816005
146. Halcrow PW, Lynch ML, Geiger JD, Ohm JE. Role of endolysosome function in iron metabolism and brain carcinogenesis. Semin Cancer Biol (2021) 76:74–85. doi: 10.1016/j.semcancer.2021.06.013
147. Rüder C, Höpken UE, Wolf J, Mittrücker H-W, Engels B, Erdmann B, et al. The tumor-associated antigen EBAG9 negatively regulates the cytolytic capacity of mouse CD8+ T cells. J Clin Invest (2009) 119:2184–203. doi: 10.1172/JCI37760
148. Göschl L, Scheinecker C, Bonelli M. Treg cells in autoimmunity: from identification to Treg-based therapies. Semin Immunopathol (2019) 41:301–14. doi: 10.1007/s00281-019-00741-8
149. Ohkura N, Sakaguchi S. Transcriptional and epigenetic basis of Treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res (2020) 30:465–74. doi: 10.1038/s41422-020-0324-7
150. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27:109–18. doi: 10.1038/cr.2016.151
151. Dong Y, Yang C, Pan F. Post-translational regulations of foxp3 in treg cells and their therapeutic applications. Front Immunol (2021) 12:626172. doi: 10.3389/fimmu.2021.626172
152. Du T, Nagai Y, Xiao Y, Greene MI, Zhang H. Lysosome-dependent p300/FOXP3 degradation and limits Treg cell functions and enhances targeted therapy against cancers. Exp Mol Pathol (2013) 95:38–45. doi: 10.1016/j.yexmp.2013.04.003
153. Marciscano AE, Anandasabapathy N. The role of dendritic cells in cancer and anti-tumor immunity. Semin Immunol (2021) 52:101481. doi: 10.1016/j.smim.2021.101481
154. Bretou M, Sáez PJ, Sanséau D, Maurin M, Lankar D, Chabaud M, et al. Lysosome signaling controls the migration of dendritic cells. Sci Immunol (2017) 2:eaak9573. doi: 10.1126/sciimmunol.aak9573
155. Parenky AC, Akalkotkar A, Mulla NS, D’Souza MJ. Harnessing T-cell activity against prostate cancer: A therapeutic microparticulate oral cancer vaccine. Vaccine (2019) 37:6085–92. doi: 10.1016/j.vaccine.2019.08.033
156. Wagner CS, Grotzke J, Cresswell P. Intracellular regulation of cross-presentation during dendritic cell maturation. PloS One (2013) 8:e76801. doi: 10.1371/journal.pone.0076801
157. Ma J, Wei K, Zhang H, Tang K, Li F, Zhang T, et al. Mechanisms by which dendritic cells present tumor microparticle antigens to CD8+ T cells. Cancer Immunol Res (2018) 6:1057–68. doi: 10.1158/2326-6066.CIR-17-0716
158. Wang Z, Xu F, Hu J, Zhang H, Cui L, Lu W, et al. Modulation of lactate-lysosome axis in dendritic cells by clotrimazole potentiates antitumor immunity. J Immunother Cancer (2021) 9:e002155. doi: 10.1136/jitc-2020-002155
159. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol (2016) 37:855–65. doi: 10.1016/j.it.2016.09.006
160. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
161. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun (2017) 8:2122. doi: 10.1038/s41467-017-02186-9
162. Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev (2021) 101:147–76. doi: 10.1152/physrev.00048.2019
163. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer (2021) 20:131. doi: 10.1186/s12943-021-01428-1
164. Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle (2011) 10:1784–93. doi: 10.4161/cc.10.11.15674
165. Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution. Cell Cycle (2010) 9:3256–76. doi: 10.4161/cc.9.16.12553
166. Jachetti E, Caputo S, Mazzoleni S, Brambillasca CS, Parigi SM, Grioni M, et al. Tenascin-C protects cancer stem-like cells from immune surveillance by arresting T-cell activation. Cancer Res (2015) 75:2095–108. doi: 10.1158/0008-5472.CAN-14-2346
167. Pei L, Liu Y, Liu L, Gao S, Gao X, Feng Y, et al. Roles of cancer-associated fibroblasts (CAFs) in anti- PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer (2023) 22:29. doi: 10.1186/s12943-023-01731-z
168. Lee MY, Jeon JW, Sievers C, Allen CT. Antigen processing and presentation in cancer immunotherapy. J Immunother Cancer (2020) 8:e001111. doi: 10.1136/jitc-2020-001111
169. Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol (2011) 11:823–36. doi: 10.1038/nri3084
170. Kong X, Bu J, Chen J, Ni B, Fu B, Zhou F, et al. PIGF and Flt-1 on the surface of macrophages induces the production of TGF-β1 by polarized tumor-associated macrophages to promote lung cancer angiogenesis. Eur J Pharmacol (2021) 912:174550. doi: 10.1016/j.ejphar.2021.174550
171. Siveen KS, Prabhu K, Krishnankutty R, Kuttikrishnan S, Tsakou M, Alali FQ, et al. Vascular endothelial growth factor (VEGF) signaling in tumour vascularization: potential and challenges. Curr Vasc Pharmacol (2017) 15:339–51. doi: 10.2174/1570161115666170105124038
172. Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to anti-angiogenic therapy in cancer-alterations to anti-VEGF pathway. Int J Mol Sci (2018) 19:1232. doi: 10.3390/ijms19041232
173. Song Y, Tang C, Yin C. Combination antitumor immunotherapy with VEGF and PIGF siRNA via systemic delivery of multi-functionalized nanoparticles to tumor-associated macrophages and breast cancer cells. Biomaterials (2018) 185:117–32. doi: 10.1016/j.biomaterials.2018.09.017
174. Xia X, Mai J, Xu R, Perez JET, Guevara ML, Shen Q, et al. Porous silicon microparticle potentiates anti-tumor immunity by enhancing cross-presentation and inducing type I interferon response. Cell Rep (2015) 11:957–66. doi: 10.1016/j.celrep.2015.04.009
175. Giliopoulos D, Zamboulis A, Giannakoudakis D, Bikiaris D, Triantafyllidis K. Polymer/metal organic framework (MOF) nanocomposites for biomedical applications. Molecules (2020) 25:185. doi: 10.3390/molecules25010185
176. Duan F, Feng X, Yang X, Sun W, Jin Y, Liu H, et al. A simple and powerful co-delivery system based on pH-responsive metal-organic frameworks for enhanced cancer immunotherapy. Biomaterials (2017) 122:23–33. doi: 10.1016/j.biomaterials.2017.01.017
177. Morse MA, Gwin WR, Mitchell DA. Vaccine therapies for cancer: then and now. Target Oncol (2021) 16:121–52. doi: 10.1007/s11523-020-00788-w
178. Paukner S, Kudela P, Kohl G, Schlapp T, Friedrichs S, Lubitz W. DNA-loaded bacterial ghosts efficiently mediate reporter gene transfer and expression in macrophages. Mol Ther (2005) 11:215–23. doi: 10.1016/j.ymthe.2004.09.024
179. Szostak M, Wanner G, Lubitz W. Recombinant bacterial ghosts as vaccines. Res Microbiol (1990) 141:1005–7. doi: 10.1016/0923-2508(90)90141-C
180. Wang R, Liu Y, Liu L, Chen M, Wang X, Yang J, et al. Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. Ebiomedicine (2019) 40:118–34. doi: 10.1016/j.ebiom.2019.01.045
181. Xun Y, Yang H, Kaminska B, You H. Toll-like receptors and toll-like receptor-targeted immunotherapy against glioma. J Hematol Oncol (2021) 14:176. doi: 10.1186/s13045-021-01191-2
182. Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer (2009) 9:57–63. doi: 10.1038/nrc2541
183. Kaur A, Baldwin J, Brar D, Salunke DB, Petrovsky N. Toll-like receptor (TLR) agonists as a driving force behind next-generation vaccine adjuvants and cancer therapeutics. Curr Opin Chem Biol (2022) 70:102172. doi: 10.1016/j.cbpa.2022.102172
184. Wei X, Liu L, Li X, Wang Y, Guo X, Zhao J, et al. Selectively targeting tumor-associated macrophages and tumor cells with polymeric micelles for enhanced cancer chemo-immunotherapy. J Control Release (2019) 313:42–53. doi: 10.1016/j.jconrel.2019.09.021
185. Liu S, Jiang Q, Zhao X, Zhao R, Wang Y, Wang Y, et al. A DNA nanodevice-based vaccine for cancer immunotherapy. Nat Mater (2021) 20:421–30. doi: 10.1038/s41563-020-0793-6
186. Haslauer T, Greil R, Zaborsky N, Geisberger R. CAR T-cell therapy in hematological Malignancies. Int J Mol Sci (2021) 22:8996. doi: 10.3390/ijms22168996
187. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11:69. doi: 10.1038/s41408-021-00459-7
188. Li W, Qiu S, Chen J, Jiang S, Chen W, Jiang J, et al. Chimeric antigen receptor designed to prevent ubiquitination and downregulation showed durable antitumor efficacy. Immunity (2020) 53:456–470.e6. doi: 10.1016/j.immuni.2020.07.011
189. Lettau M, Kabelitz D, Janssen O. Lysosome-related effector vesicles in T lymphocytes and NK cells. Scand J Immunol (2015) 82:235–43. doi: 10.1111/sji.12337
190. Dowling SD, Macian F. Autophagy and T cell metabolism. Cancer Lett (2018) 419:20–6. doi: 10.1016/j.canlet.2018.01.033
191. Fischer A, Latour S, de Saint Basile G. Genetic defects affecting lymphocyte cytotoxicity. Curr Opin Immunol (2007) 19:348–53. doi: 10.1016/j.coi.2007.04.006
Keywords: lysosomes, tumor immunity, immunotherapy, tumor microenvironment, lysosomal autophagy
Citation: Xu Y, Shao B and Zhang Y (2024) The significance of targeting lysosomes in cancer immunotherapy. Front. Immunol. 15:1308070. doi: 10.3389/fimmu.2024.1308070
Received: 05 October 2023; Accepted: 22 January 2024;
Published: 02 February 2024.
Edited by:
Tao Sun, Nankai University, ChinaReviewed by:
Qiong Gao, University of Michigan, United StatesFilomena De Nigris, University of Campania Luigi Vanvitelli, Italy
Copyright © 2024 Xu, Shao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yafeng Zhang, aHlkemhhbmd5YWZlbmdAMTYzLmNvbQ==; Bo Shao, enplZHVzaGFvQGZveG1haWwuY29t
†These authors have contributed equally to this work